
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 11 November 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1035532
 Aidan T. Hanrath1,2†
Aidan T. Hanrath1,2† Catherine F. Hatton1,2†Florian Gothe1Cathy Browne3Jane Vowles3
Catherine F. Hatton1,2†Florian Gothe1Cathy Browne3Jane Vowles3 Peter Leary4Simon J. Cockell4,5
Peter Leary4Simon J. Cockell4,5 Sally A. Cowley3
Sally A. Cowley3 William S. James3
William S. James3 Sophie Hambleton1,6
Sophie Hambleton1,6 Christopher J. A. Duncan1,2,4*
Christopher J. A. Duncan1,2,4*Macrophages are key target cells of Zika virus (ZIKV) infection, implicated as a viral reservoir seeding sanctuary sites such as the central nervous system and testes. This rests on the apparent ability of macrophages to sustain ZIKV replication without experiencing cytopathic effects. ZIKV infection of macrophages triggers an innate immune response involving type I interferons (IFN-I), key antiviral cytokines that play a complex role in ZIKV pathogenesis in animal models. To investigate the functional role of the IFN-I response we generated human induced pluripotent stem cell (iPSC)-derived macrophages from a patient with complete deficiency of IFNAR2, the high affinity IFN-I receptor subunit. Accompanying the profound defect of IFN-I signalling in IFNAR2 deficient iPS-macrophages we observed significantly enhanced ZIKV replication and cell death, revealing the inherent cytopathicity of ZIKV towards macrophages. These observations were recapitulated by genetic and pharmacological ablation of IFN-I signalling in control iPS-macrophages and extended to a model of iPS-microglia. Thus, the capacity of macrophages to support noncytolytic ZIKV replication depends on an equilibrium set by IFN-I, suggesting that innate antiviral responses might counterintuitively promote ZIKV persistence via the maintenance of tissue viral reservoirs relevant to pathogenesis.
Zika virus (ZIKV) is a neurotropic flavivirus of global public health importance owing to its pandemic potential (1). Effective vaccines and therapeutics for ZIKV remain elusive, although intensive efforts to develop them are ongoing. ZIKV infection is associated with pregnancy loss and neurological syndromes including microcephaly following intrauterine infection, as well as meningoencephalitis and Guillain-Barré syndrome in adults (2). Although predominantly mosquito borne, ZIKV is also capable of spreading via sexual transmission in semen (3). Important uncertainties concerning ZIKV pathogenesis remain, including how it accesses the central nervous system (CNS) of the developing foetus to cause microcephaly and what defines protective versus pathogenic immune responses. Addressing these questions should identify therapeutic targets and inform protective strategies to limit transmission (4).
Macrophages, immune cells that play an essential role in vertebrate host defence against microbes and in tissue repair and homeostasis (5), have been implicated in several aspects of ZIKV pathogenesis. Macrophages are key target cells of ZIKV and have been implicated both as a cellular reservoir (6), and as a vector in transmitting ZIKV to sanctuary sites such as the CNS, testes and placenta (7–10). This mechanism of viral subversion appears to rely on the ability of macrophages to support ZIKV replication without being susceptible to cytopathic effects (7–9), contrasting with other cell types such as neural progenitor cells (11–13) and epithelial cells (14). Other important viral pathogens, such as HIV, exploit a similar strategy of noncytolytic macrophage replication to form long-lived reservoirs that aid transmission to other permissive cell types (15, 16) and facilitate pathogenesis (17). ZIKV infection of human monocyte-derived macrophages is associated with induction of a robust innate immune response, dominated by type I interferons (IFN-I) (18) and proinflammatory cytokines (8, 19, 20). This appears to contrast with human monocytes (21, 22) and other myeloid cells such as dendritic cells (23, 24), which reportedly fail to induce IFNs upon ZIKV infection. Given that macrophages are permissive and ZIKV deploys multiple strategies to evade IFN-I responses (4, 25), there remains uncertainty about the functional role played by IFN-I.
Type I interferons (IFN-I) are essential to human antiviral immunity (26, 27) and patients deficient in the IFN-I receptor components IFNAR1 or IFNAR2 are susceptible to severe viral disease following exposure to both live-attenuated viral vaccines and wild-type viruses (26–33). Signalling through IFNAR activates a JAK-STAT signalling pathway culminating in the expression of hundreds of interferon-stimulated genes (ISGs) that govern its antiviral properties (27). Yet IFN-I plays a poorly defined and apparently complex role in ZIKV pathogenesis (4). Like other human viral pathogens, ZIKV has evolved multiple strategies to subvert IFN-I restriction in human cells (4), most notably through degradation of STAT2 by the ZIKV NS5 protein (4, 25, 34). Failure to bind murine STAT2 limits ZIKV host range (35) and prior to the development of transgenic mice encoding human STAT2 (35), most in vivo studies were undertaken in Ifnar1 deficient mice, preventing formal assessment of the confrontation between ZIKV and the IFN-I system. Whilst several ISGs have been identified that are capable of restricting ZIKV in an exogenous expression context (36–38), and pre-treatment with recombinant IFN-I provokes an antiviral state capable of restricting ZIKV infection (39), studies in human cells have yielded inconsistent findings concerning whether ZIKV induces an IFN-I response (4, 7–9, 14, 22–25, 40, 41). Adding complexity to this issue, IFN-I signalling contributed to pregnancy loss in an immunocompetent mouse model (42), suggesting that IFN-I may also be pathogenic in certain situations. To our knowledge, no study has hitherto assessed the functional impact of endogenous IFN-I responses in pathogenically relevant human cells. Progress has been in part limited by the resistance of primary human target cells such as macrophages to genetic manipulation.
Here we assess the role of IFN-I in macrophages derived from human induced pluripotent stem cells (iPSC), a valid model of yolk-sac derived, MYB-independent tissue macrophages (43–45). Generating iPSC from a patient deficient in the high-affinity type I IFN receptor subunit IFNAR2 (26), we identify a critical role for autocrine and paracrine IFN-I signalling in controlling ZIKV replication and protecting macrophages from ZIKV-mediated cell death. These findings were recapitulated by genetic and pharmacological ablation of IFN-I signalling in control iPS-macrophages and were also observed in a model of iPS-derived microglia (46), indicating that robust IFN-I immunity underlies the apparent resistance of tissue macrophages to ZIKV-mediated cytotoxicity and may sustain persistent infection with implications for intra-host spread and seeding of viral reservoirs.
To address the impact of IFN-I signalling on macrophage responses to ZIKV infection, we derived induced pluripotent stem cells (iPSC) from dermal fibroblasts from a patient bearing a homozygous nonsense mutation of IFNAR2 (26), the high-affinity subunit of the IFN-I receptor (herein termed IFNAR2PT). Following quality control assays, consisting of assessment of pluripotency marker expression by flow cytometry, elimination of Sendai vector by RT-PCR and SNP array confirmation of genome integrity (Figures 1A–C), we selected two clones (6 and 11) alongside two previously described healthy control lines (IFNAR2WT1-2) (47, 48). We differentiated iPSCs to human macrophages (iPS-Mϕ) from embryoid bodies using a well-characterised, validated model (45). We quantified the expression of well-defined cell surface markers associated with macrophage differentiation including human leukocyte antigen CD45, lipopolysaccharide receptor CD14, and the major histocompatibility complex II cell surface receptor (HLA-DR) by flow cytometry (Figure 1D). Phagocytic function was confirmed by measuring uptake of zymosan yeast particles (pHrodo), which fluoresce in a pH-sensitive manner to demonstrate acidification in the phagolysosome (Figure 1E). By immunoblotting of whole cell lysates, we verified that IFNAR2 protein was absent in IFNAR2PT iPS-Mϕ (Figure 1F). Accordingly, these cells exhibited a profound defect of IFN-I signalling both at rest (as reflected in the reduction in ISG15 and STAT1 in unstimulated IFNAR2PT iPS-Mϕ) and upon treatment with recombinant IFNα in comparison to IFNAR2WT iPS-Mϕ (Figure 1E). Responses to IFNγ were preserved (Figure S1), consistent with previous findings in other cell types (26, 29, 30).
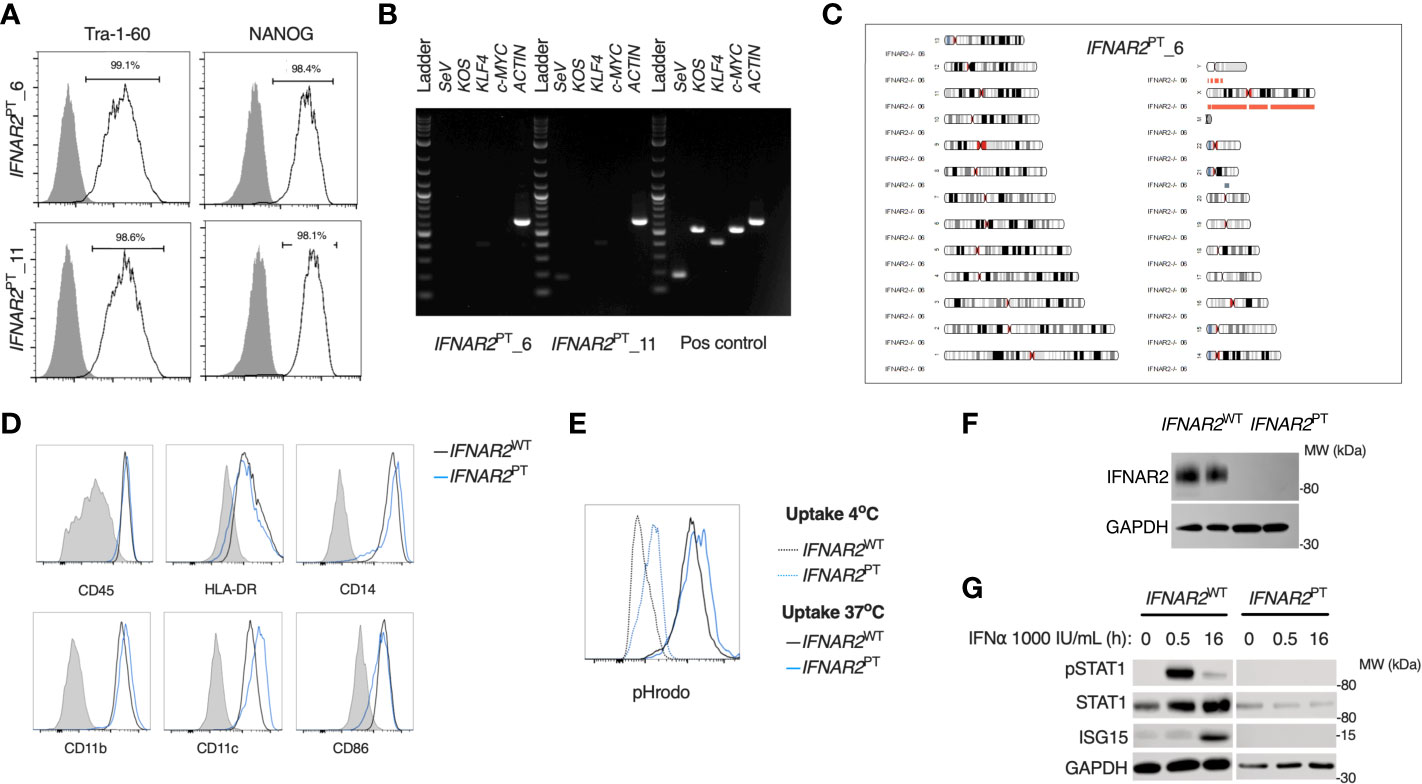
Figure 1 A model of IFNAR2 deficient human iPS-macrophages (IFNAR2PT iPS-Mϕ). (A) Expression of pluripotency markers by IFNAR2PT iPSC clones 6 and 11 by flow cytometry.(B) PCR showing clearance of Sendai virus vector from IFNAR2PT iPSC clones 6 and 11. (C) Karyogram produced from SNP array showing no gross abnormalities in the previously unpublished IFNAR2PT iPSC clone 6. Red bars indicate loss or single copy, grey indicates loss of heterozygosity on chromosome 21 in the region of IFNAR2 (representative of data in IFNAR2PT clone 11). (D) Expression of macrophage surface markers in IFNAR2PT (clone 6) and IFNAR2WT (WT1) iPS-Mϕ by flow cytometry, representative of repeat experiments in IFNAR2PT clone 11 and WT2. (E) Phagocytic uptake of Zymosan pHrodo particles in IFNAR2PT (clone 6) and IFNAR2WT (WT1) iPS-Mϕ, representative of repeat experiments in clone 11 and WT2. (F) Immunoblot of IFNAR2 and GAPDH in IFNAR2WT (WT2) and IFNAR2PT (clone 11) iPS-Mϕ, representative of repeat experiments in WT1 and clone 6. (G) Immunoblot of IFN-I signalling in IFNα2b (1000 IU/mL) treated IFNAR2PT (clone 11) and IFNAR2WT (WT1) iPS-Mϕ, representative of n = 3 independent experiments.
Initially we sought to characterise the innate immune response to infection with a clinical isolate of epidemic Asian lineage ZIKV H/FP/2013 (ZIKVFP) in IFNAR2PT and IFNAR2WT iPS-Mϕ. By RT-PCR analysis of cell lysates at 24 hours post-infection (h.p.i.) we observed the robust induction of IFNA1, IFNB and IFNL1 (Figure 2A). Alongside the induction of IFN genes we observed robust expression of proinflammatory cytokines, including IL6, TNF and IL1B (Figure 2B). Only IL6 was significantly reduced in IFNAR2PT iPS-Mϕ, in keeping with recent data indicating that IL6 is induced by STAT2 and IRF9 in cooperation with NF-κB (49). Consistent with the robust induction of IFN genes, immunoblot analysis of lysates prepared at 48h.p.i. demonstrated tyrosine phosphorylation of STAT1, accompanied by expression of the ISG proteins RSAD2 and ISG15 in IFNAR2WT iPS-Mϕ, indicative of IFN signalling (Figure 2C). This was absent in IFNAR2PT iPS-Mϕ, reflecting the defect of IFN-I signalling in the latter and demonstrating that their induction by ZIKV infection was IFNAR dependent.
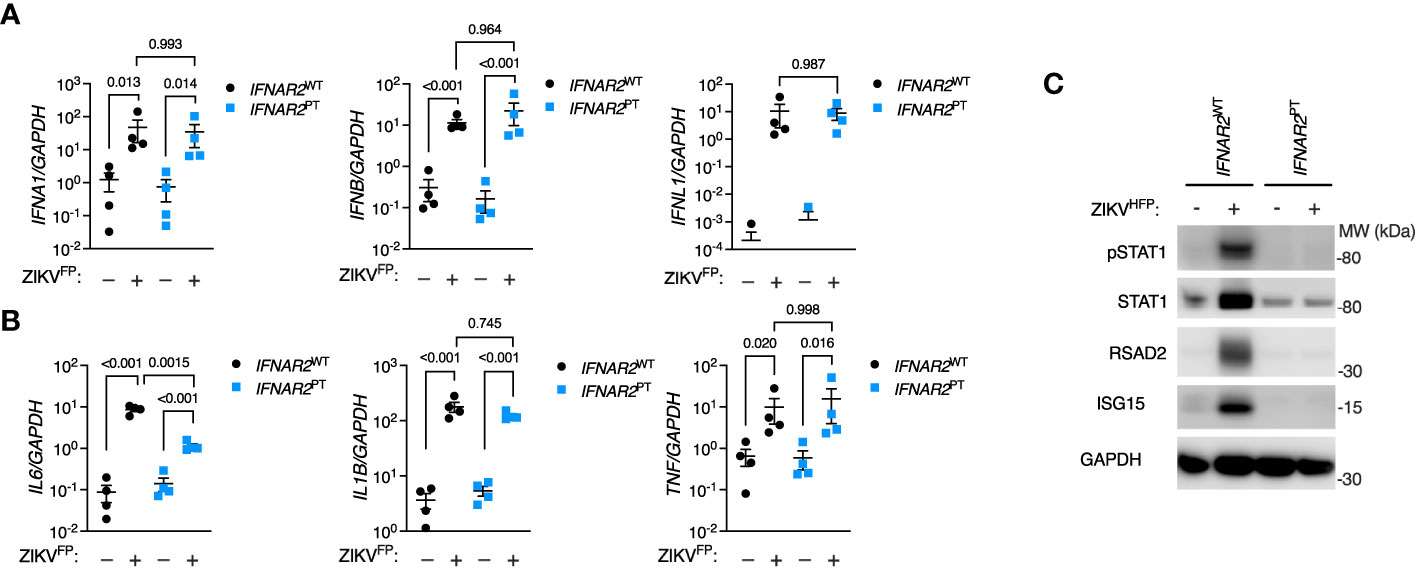
Figure 2 IPS-macrophages mount a robust IFN-I response to ZIKV infection. (A) RT-PCR quantification of IFNA1, IFNB and IFNL1 relative to GAPDH (24 h.p.i. ZIKVFP MOI = 10.0, n = 4 independent experiments in IFNAR2WT [WT2] and IFNAR2PT [clone 6]). Mean ± SD, ANOVA with Sidak’s test for multiple comparisons. (B) RT-PCR quantification of IL6, TNF and IL1B relative to GAPDH (24 h.p.i. ZIKVFP MOI=10.0, n = 4 independent experiments in IFNAR2WT [WT2] and IFNAR2PT [clone 6]). Mean ± SD, ANOVA with Sidak’s test for multiple comparisons. (C) Immunoblot of pSTAT1, STAT1, RSAD2, GAPDH and ISG15 in IFNAR2WT (WT2) and IFNAR2PT (clone 11) iPS-Mϕ (48 h.p.i. ZIKVFP MOI=1.0), representative of n = 3 independent experiments including IFNAR2PT (clone 6) and IFNAR2WT (WT1) iPS-Mϕ.
We next sought to assess the impact of the loss of IFN-I signalling on replication of ZIKVFP by analysis of viral Envelope (ENV) protein expression by immunoblot of whole cell lysates at 72 h.p.i., or by immunofluorescence staining of fixed and permeabilised cells at 24 h.p.i., alongside plaque assay on permissive Vero cells of infectious particle release in supernatants (Figures 3A–C). These analyses demonstrated productive ZIKV infection that was significantly enhanced in IFNAR2PT iPS-Mϕ across a range of multiplicities of infection (MOI). This phenotype was independent of viral origin, as it was also seen with African lineage MP1751 infection (ZIKVMP Figure 3A). These data were consistent with a study employing JAK inhibitors in primary placental macrophages (50), demonstrating that the robust IFN-I response of macrophages is functional and acts to limit productive ZIKV replication.
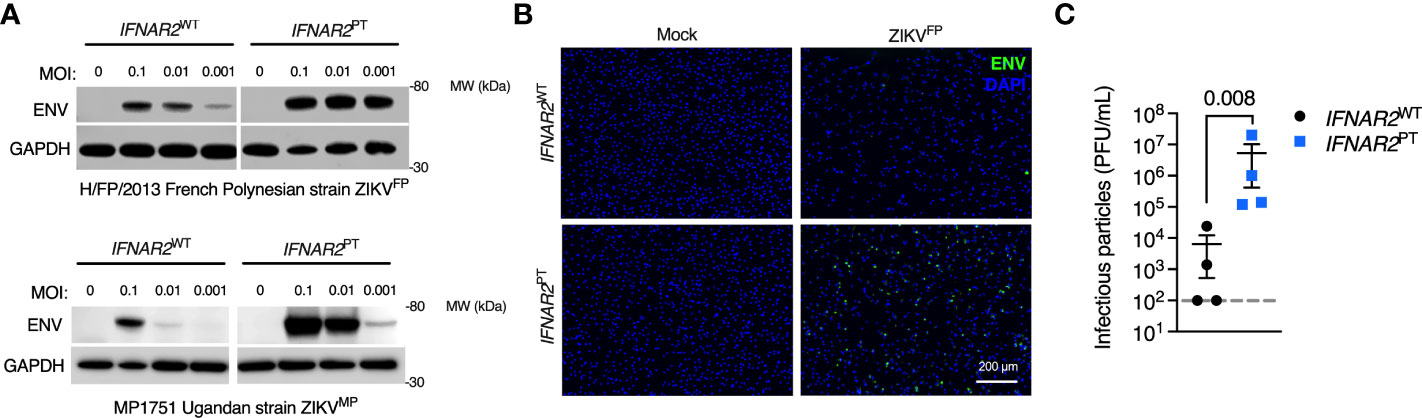
Figure 3 Enhanced ZIKV replication in IFNAR2 deficient iPS-macrophages. (A) Immunoblot of ENV and GAPDH expression in IFNAR2WT (WT2) and IFNAR2PT (clone 11) iPS-Mϕ, 72 h.p.i. with Asian lineage (ZIKVFP) and African lineage (ZIKVMP) at the MOI demonstrated, representative of n = 3 independent experiments. (B) Immunofluorescence analysis of ENV expression in IFNAR2WT (WT2) and IFNAR2PT (clone 11) iPS-Mϕ (24 h.p.i. with ZIKVFP MOI=1.0), representative of n = 3 independent experiments. (C) Plaque assay on Vero cells of ZIKV infectious particles in supernatants (48 h.p.i. ZIKVFP MOI = 0.001, n = 4 independent experiments in IFNAR2WT [clones WT1 & WT2] and IFNAR2PT [clones 6 & 11]). Mean ± SD, t test.
To explore this interaction at the single cell level, we undertook immunofluorescence analysis of ZIKV ENV and ISG expression via immunostaining for IFITM3 in cells at 24 h.p.i. with ZIKVFP, infecting with MOI 10 in order to achieve detectable ENV expression in WT iPS-Mϕ (Figure 4A). This analysis revealed a significant increase of ENV expression in IFNAR2PT iPS-Mϕ (approximately 60%, compared to an average of 10-15% in IFNAR2WT iPS-Mϕ), accompanied by a failure of both basal and inducible IFITM3 expression in IFNAR2PT iPS-Mϕ (Figure 4B), consistent with our earlier findings. Upon ZIKV exposure of IFNAR2WT iPS-Mϕ, IFITM3 expressing cells were frequently seen surrounding infected cells, the latter defined by ZIKV ENV expression (see inset box, Figure 4A), suggesting paracrine signalling. IFITM3 positive bystander cells were not seen in IFNAR2PT iPS-Mϕ cultures. Using the image analysis tool CellProfiler, we compared expression of these markers to define the relationship between infection and ISG expression by genotype at the single cell level. Analysis of >2000 cells per genotype revealed that upon ZIKV infection in IFNAR2WT iPS-Mϕ, IFITM3 was predominantly expressed in uninfected bystander (ENV-) cells, consistent with paracrine regulation of IFITM3 expression. By contrast, IFNAR2PT iPS-Mϕ lacked IFITM3 expression in bystander cells, reflecting the profound loss of paracrine IFN-I response. Interestingly, in infected (ENV+) IFNAR2WT iPS-Mϕ, IFITM3 was largely undetectable (Figure 4C), suggesting that ZIKV infection prevented IFN-I signalling in infected cells, consistent with prior studies (25, 34).
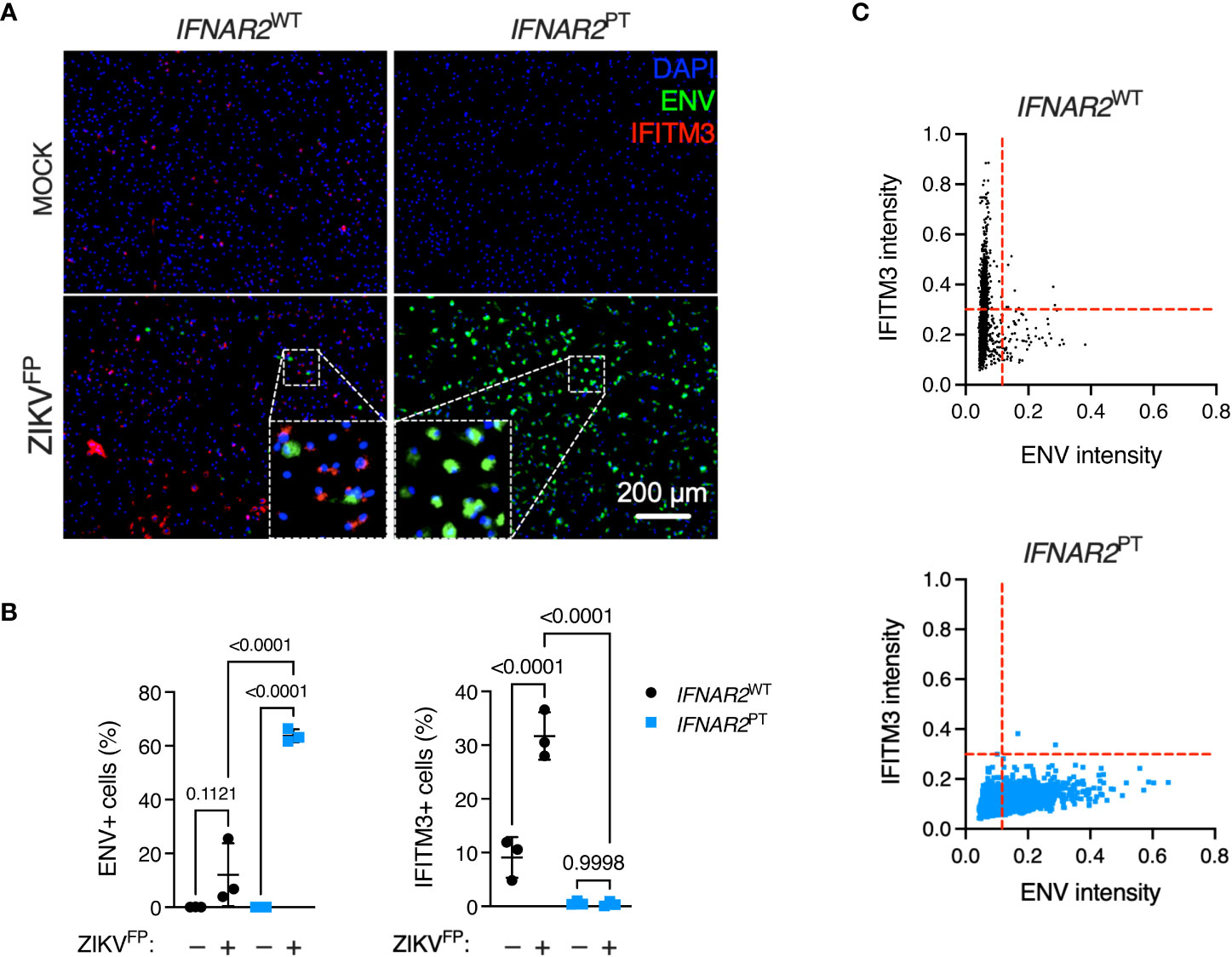
Figure 4 IFN-I mediates paracrine protection of iPS-macrophages. (A) Immunofluorescence analysis of ZIKV ENV and IFITM3 expression in IFNAR2PT (clone 11) and IFNAR2WT (WT2) iPS-Mϕ (24 h.p.i. ZIKVFP MOI = 10.0). Representative images from one of three independent experiments are shown. Scale bar = 200 μm. (B) CellProfiler quantification of images in (A) showing proportion of cells expressing ZIKV ENV (left panel) or the ISG IFITM3 (right panel). Mean ± SD of n = 3 independent experiments, ANOVA with Sidak’s test for multiple comparisons. (C) CellProfiler analysis of single cell expression of ENV and IFITM3 in IFNAR2WT and IFNAR2PT iPS-Mϕ from images in (A), n = 2,434 (WT) cells and n = 2,202 (PT) cells respectively. Red dotted lines represent gating. Representative data from one of three independent experiments.
Accompanying heightened ZIKV replication in IFN-I incompetent cells, we observed morphological abnormalities suggestive of cell death, with progressive membrane blebbing and cell shrinkage from 48 h.p.i. (Figure 5A) accompanied by immunodetection of cleaved caspase 3 (Figure 5B), indicative of apoptosis and in keeping with previous reports of the mechanism of ZIKV-induced cell death in other cell types (11, 14). These data indicated that failure of IFN-I mediated control rendered IFNAR2PT iPS-Mϕ vulnerable to cytopathic effects (CPE), contrasting with the resistance of WT macrophages to ZIKV CPE observed by ourselves and previously reported (7–9). We confirmed this using an imaging based live-cell viability assay which showed cell death in approximately 60% of IFNAR2PT iPS-Mϕ at 72 h.p.i. (Figure 5C). Quantification of these data using CellProfiler demonstrated that ZIKV CPE was significantly enhanced in IFNAR2PT but not IFNAR2WT iPS-Mϕ, a phenotype that was independent of viral lineage, as it was observed equally following ZIKVFP and ZIKVMP infections (Figure 5D). Prior treatment with IFNα2b (1000 IU/mL) was unable to protect IFNAR2PT iPS-Mϕ from cytopathic effects, consistent with their defect of endogenous IFN-I signalling. By contrast, treatment of IFNAR2PT iPS-Mϕ with IFNγ partially rescued ZIKV-induced cell death (Figure 5D), in keeping with their intact response to IFNγ (Figure S1), indicating that IFNAR2PT iPS-Mϕ were not globally immunodeficient.
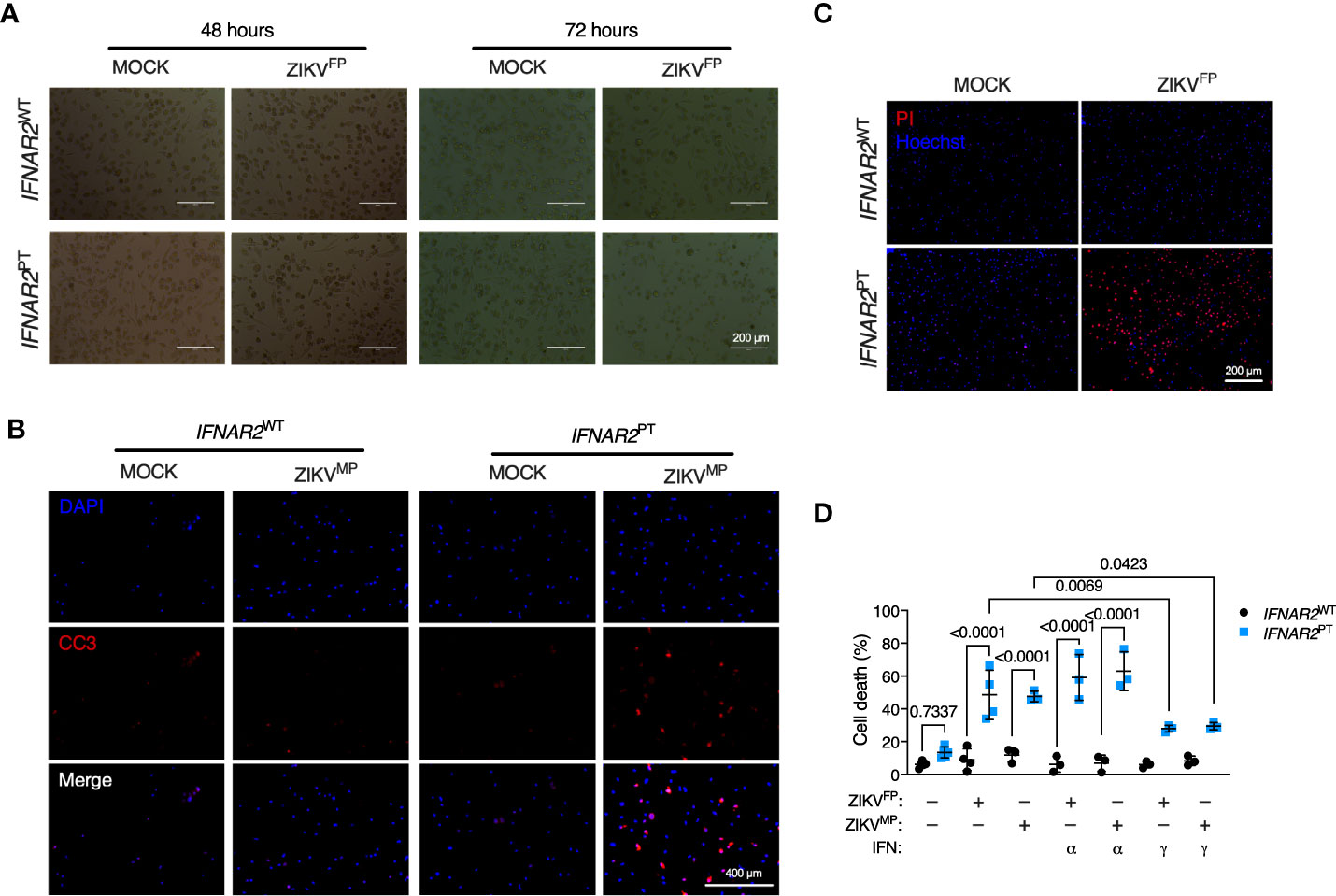
Figure 5 IFNAR2-deficient iPS-macrophages are vulnerable to ZIKV cytopathic effects. (A) Progressive cytopathicity in IFNAR2PT (clone 6) but not IFNAR2WT (WT2) iPS-Mϕ following infection with ZIKVFP MOI = 1.0, showing morphological features of cell shrinkage, membrane blebbing and cell fragmentation. Scale bar, 200 μm. (B) Immunofluorescence analysis of cleaved caspase 3 (CC3) at 48 h.p.i. ZIKVMP MOI = 1.0. Representative images of two independent experiments. Scale bar, 400 μm. (C) Immunofluorescence analysis of cell viability showing representative images of cell death at 72 h.p.i. ZIKVFP MOI = 1.0 in IFNAR2PT (clone 11) but not IFNAR2WT (WT2) iPS-Mϕ, representative of n = 4 independent experiments. Scale bar, 200 μm. (D) CellProfiler quantification of cell viability assay in IFNAR2PT (clone 11) and IFNAR2WT (WT1 and WT2) iPS-Mϕ with or without recombinant IFNα2b or IFNγ (1000 IU/mL) pretreatment. 72 h.p.i. ZIKVFP MOI = 1.0 (n = 4 independent experiments) or ZIKVMP MOI = 1.0 (n = 3 independent experiments). Mean ± SD, ANOVA with Sidak’s test for multiple comparisons.
These observations implied that defective IFN-I signalling accounted for heightened ZIKV replication and vulnerability to cell death in IFNAR2PT iPS-Mϕ. However we could not rule out the possibility that these findings related to another aspect of the genetic background of this IFNAR2 deficient patient. To validate our findings, we adopted two complementary approaches of IFNAR blockade to determine whether this phenotype could be recapitulated in control iPS-Mϕ. First, we used the JAK inhibitor ruxolitinib (RUX) at a dose (10 μM) previously shown to ablate IFN-I signalling (51). Treatment of IFNAR2WT iPS-Mϕ with RUX for 16 h prior to infection led to a significant enhancement of ZIKV-induced cell death compared to vehicle (DMSO) treatment (Figure 6), reproducing our previous observations in IFNAR2PT iPS-Mϕ and suggesting that loss of the inducible signalling response to ZIKV infection is the major mechanism by which IFNAR2 deficiency contributes to macrophage susceptibility. By contrast, treatment of IFNAR2WT iPS-Mϕ with RUX prior to infection did not further enhance cell death, indicating that other IFN-I independent, JAK dependent signalling pathways (including IFNλs) made little contribution to protection against cytopathic effects.
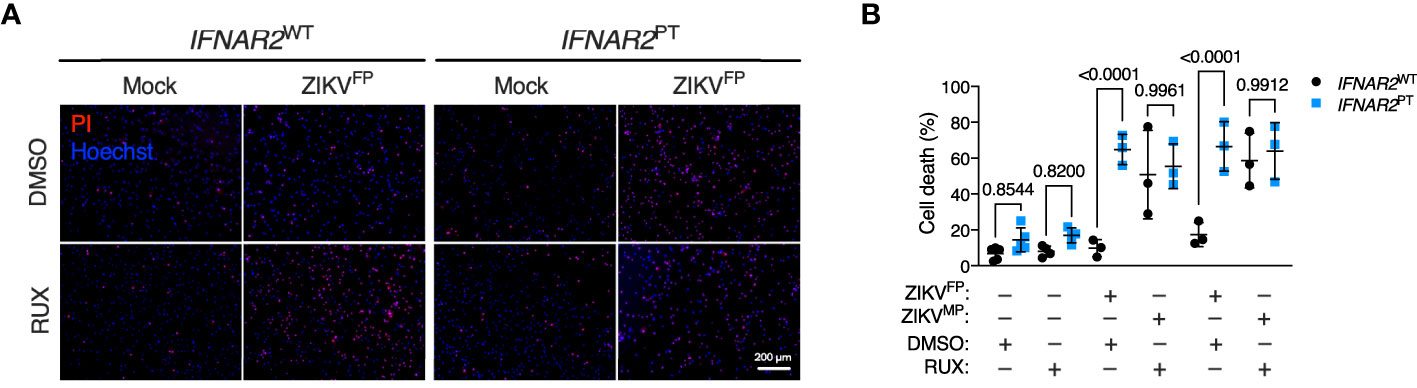
Figure 6 JAK inhibition recapitulates ZIKV cytopathicity in wild-type iPS-macrophages. (A) Immunofluorescence analysis of cell viability showing representative images of cell death at 72 h.p.i. in IFNAR2WT (WT1) and IFNAR2PT (clone 11) iPS-Mϕ treated with RUX (10 uM) or DMSO control (ZIKVFP MOI = 1.0, representative images of n = 4 independent experiments). Scale bar = 200 μm. PI = propidium iodide. (B) CellProfiler quantification of cell viability assay in IFNAR2PT (clone 11) and IFNAR2WT (WT1 and WT2) iPS-Mϕ treated with RUX (10 uM) or DMSO control (72 h.p.i. ZIKVFP MOI = 1.0, n = 4 independent experiments and ZIKVMP MOI = 1.0, n = 3 independent experiments). Mean ± SD, ANOVA with Sidak’s test for multiple comparisons. .
Given that JAK inhibition can impact a broad range of IFN and cytokine signalling pathways we sought to provide conclusive genetic evidence of the relevance of IFN-I to this phenotype, using CRISPR/Cas9 to delete IFNAR2 in WT1 and WT2 control iPSC lines (Figure 7A), selecting IFNAR2-/- and isogenic IFNAR2+/+ single cell iPSC clones for differentiation into iPS-Mϕ. IFNAR2 knockout was verified by PCR (Figure 7B) and confirmed by immunoblotting of IFNAR2-/- iPS-Mϕ lysates (Figure 7C). Macrophage phenotyping was undertaken as previously described for IFNAR2PT iPS-Mϕ and, as before, was unaffected by IFNAR2 expression status (Figures 7D, E). In comparison to IFNAR2+/+ iPS-Mϕ, IFNAR2-/- iPS-Mϕ exhibited a profound defect of IFN-I signalling both at rest - reflected in the reduction in basal STAT1 and STAT2 in unstimulated lysates - and upon treatment with recombinant IFNα, where phosphorylation of the signalling intermediates STAT1 and STAT2 and induction of RSAD2 protein was absent (Figure 7F). This mirrored our earlier observations in IFNAR2PT iPS-Mϕ. Consequently, infection of IFNAR2-/- iPS-Mϕ with ZIKVFP recapitulated the enhanced permissiveness to infection and CPE previously observed in IFNAR2PT iPS-Mϕ (Figures 7G, H), confirming the association of the observed phenotypes with defective IFNAR signalling.
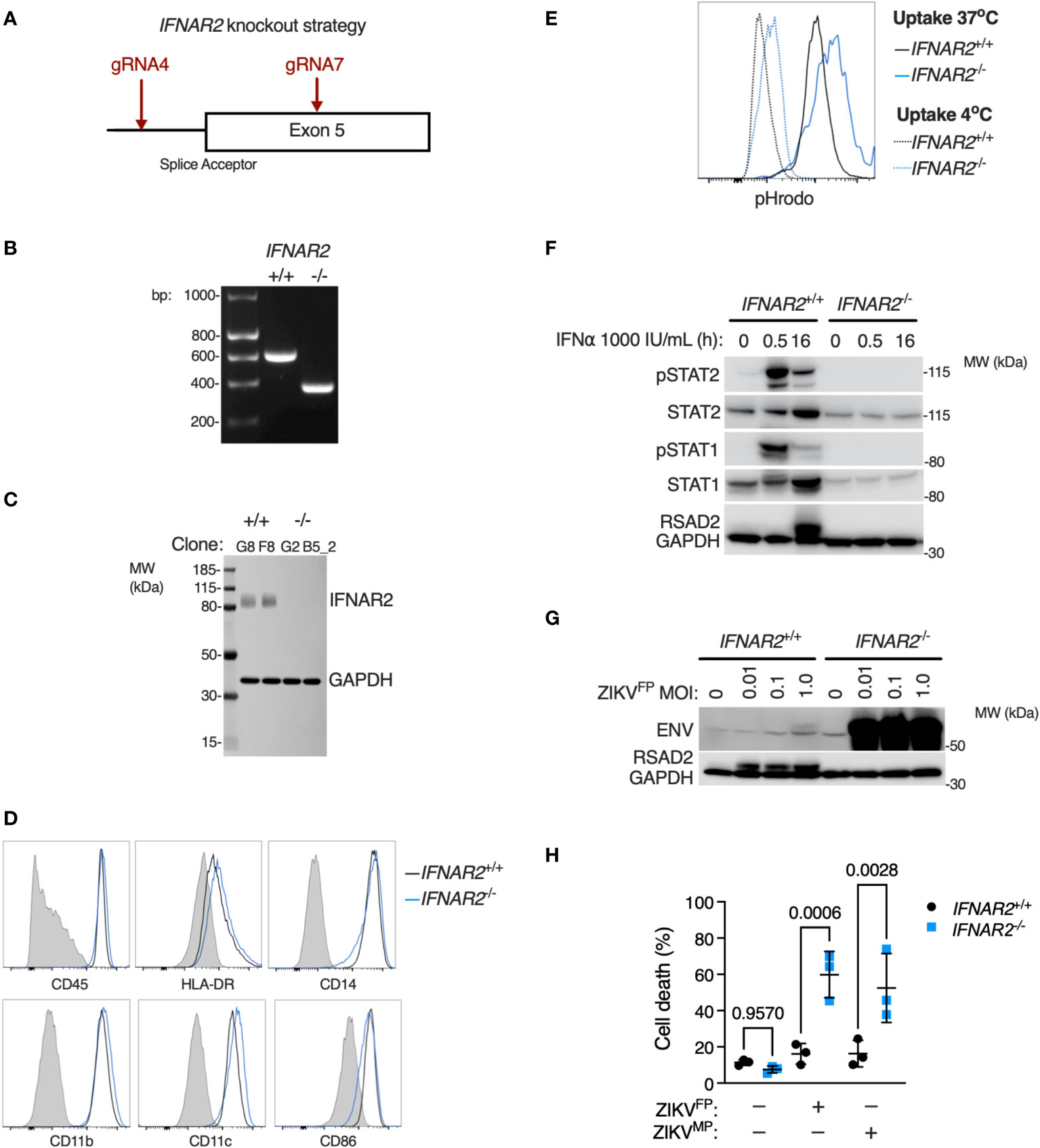
Figure 7 IFNAR2 knockout in wild-type iPS-macrophages recapitulates heightened ZIKV replication and cytopathicity. (A) CRISPR/Cas9 guide design. (B) PCR of IFNAR2 amplicon demonstrating exon 5 splice acceptor site excision in iPSC clones (G8 and G2). Representative of experiments in clones F8 and B5_2. (C) Immunoblot of IFNAR2 and GAPDH expression, demonstrating IFNAR2 ablation in iPS-Mϕ clones (G8, F8, G2, B5_2). Representative of n=2 independent experiments. (D) Expression of macrophage surface markers in IFNAR2-/- (B5_2) and IFNAR2+/+ (F8) iPS-Mϕ by flow cytometry, representative of repeat experiments in F8/B5_2. (E) Phagocytic uptake of Zymosan pHrodo particles in IFNAR2-/- (B5_2) and IFNAR2+/+ (F8) iPS-Mϕ by flow cytometry, representative of repeat experiments in F8/B5_2. (F) Immunoblot of IFN-I signalling in IFNα2b (1000 IU/mL) treated IFNAR2-/- (B5_2) and IFNAR2+/+ (F8) iPS-Mϕ, representative of n = 3 independent experiments. (G) Immunoblot of ENV, RSAD2 and GAPDH expression in IFNAR2-/- (B5_2) and IFNAR2+/+ (F8) iPS-Mϕ, 72 h.p.i. post infection, representative of n=3 independent experiments. (H) CellProfiler quantification of cell viability assay in IFNAR2-/- (G2, B5_2) and isogenic IFNAR2+/+ (G8, F8) iPS-Mϕ (72 h.p.i. ZIKVFP or ZIKVMP MOI = 1.0, n = 3 independent experiments). Mean ± SD, ANOVA with Sidak’s test for multiple comparisons.
An advantage of the CRISPR/Cas9 approach is that it provides IFNAR2-/- and IFNAR2+/+ iPS-Mϕ on an isogenic background, overcoming donor-donor variation in gene expression that can potentially confound differential expression (DE) analysis (52). To explore the global transcriptional response to ZIKV in an unbiased manner, we undertook RNA-sequencing of libraries prepared from ZIKVFP and mock-infected IFNAR2-/- and isogenic IFNAR2+/+ iPS-Mϕ at 24 h.p.i (clones B5_ 2 and F8 respectively). Principal component and pathway analyses revealed the dominant contribution of IFNAR signalling to the ZIKV response, consistent with our prior findings, and also revealed substantial differences in the transcriptome of uninfected IFNAR2-/- and isogenic IFNAR2+/+ iPS-Mϕ, consistent with the loss of ‘tonic’ IFN-I signalling in IFNAR2-/- iPS-Mϕ (Figures 8A, B). Of 492 genes significantly DE in IFNAR2+/+ compared to IFNAR2-/- iPS-Mϕ upon ZIKV infection, 467 (95%) were ISGs based on interrogation of the gene lists in the Interferome database (53) (http://interferome.its.monash.edu.au/interferome). Consistent with our prior findings, a broad range of IFN, chemokine and cytokine genes were induced upon ZIKV infection in both IFNAR2-/- and isogenic IFNAR2+/+ iPS-Mϕ, with IFNs predominating (Figure 8C). ZIKV reads were also significantly enriched in IFNAR2-/- iPS-Mϕ (Figure 8D) at this relatively early timepoint, consistent with the findings of our earlier analyses. To complement this analysis, we visualised the expression of a subset of context-specific ISGs induced in human macrophages by IFN-I treatment (54), annotating significantly DE genes between IFNAR2-/- and isogenic IFNAR2+/+ iPS-Mϕ upon ZIKV exposure (Figure 8E). This emphasised the profound alteration to the transcriptome of IFNAR2-/- iPS-Mϕ at rest and upon ZIKV infection. It also revealed the more modest induction of a subset of ISGs in IFNAR2-/- iPS-Mϕ, presumably secondary to IFNAR-independent induction of ISGs by IRF3 (55). Regardless of the mechanism, IFNAR-independent ISG induction was insufficient to have a functional impact on antiviral protection under the experimental conditions studied.
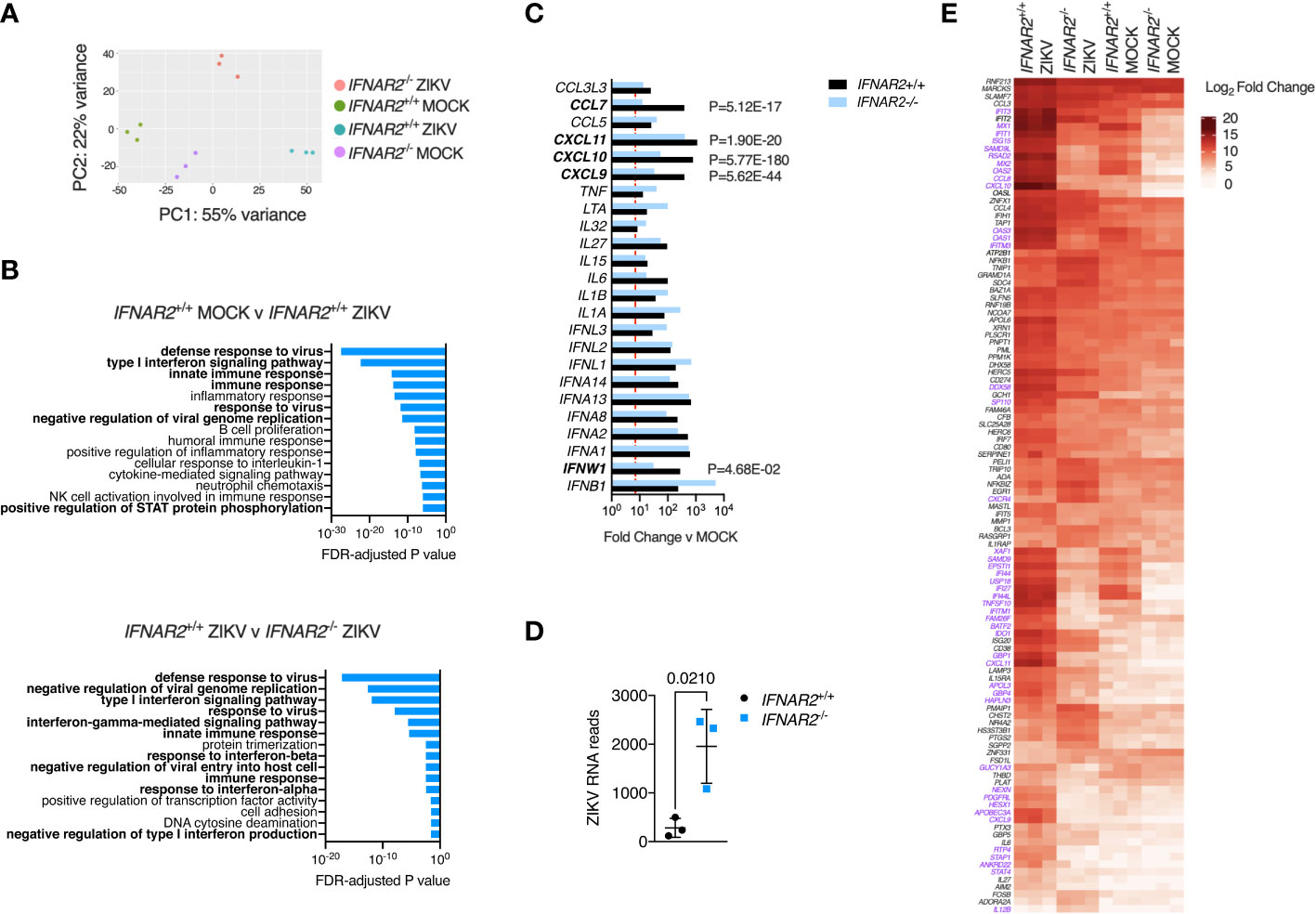
Figure 8 IFN-I signalling dominates the transcriptional response to ZIKV infection. (A) Principal component analysis of RNA-seq data (24 h.p.i. ZIKVFP MOI = 1.0, n=3 biological replicates in isogenic IFNAR2-/- [B5_2] and IFNAR2+/+ [F8] iPS-Mϕ). (B) Gene ontology analysis (FDR<5%) of RNA-seq data, comparing mock v infected IFNAR2+/+ iPS-Mϕ (top) and IFNAR2-/- v IFNAR2+/+ iPS-Mϕ (bottom). Selected pathways highlighted in bold. (C) Significantly differentially expressed DE IFN, chemokine and cytokine genes, comparing mock v ZIKV exposed conditions for IFNAR2+/+ (black bars) or IFNAR2-/- genotype (blue bars). Genes not reaching the DE threshold (Log2 FC ≥ 3, FDR < 5%) in both genotypes are not displayed. Red dotted line represents FC threshold (Log2 FC ≥ 3). FDR-adjusted P values are also included for significantly DE genes (in bold) comparing IFNAR2-/- and IFNAR2+/+ ZIKV exposed datasets. (D) Aligned ZIKV reads from data in (A), mean ± SD, t test. (E) Heatmap displaying expression of annotated macrophage-specific ISGs. Colour intensity reflects Log2 FC. Significantly DE genes (Log2 FC ≥ 3, FDR < 5%) are shown in purple text for the comparison of IFNAR2-/- and IFNAR2+/+ ZIKV exposed datasets.
According to our current understanding, human microglia originate from yolk-sac progenitors which enter the CNS early in development, prior to closure of the blood-brain barrier, and are maintained there by local proliferation (56). As the main brain-resident macrophage population, microglia have key roles in homeostasis and pathogen defence (56). However, these cells have also been implicated as a ‘Trojan horse’, potentially transmitting ZIKV to the developing brain during vertical transmission in early pregnancy and contributing to the development of microcephaly (9, 10, 57). To understand the relevance of our observations to microglia, we employed a validated method for differentiating microglia-like cells from iPSC (iPS-MGL) (46). We differentiated IFNAR2PT and IFNAR2WT iPSC, in addition to IFNAR2-/- and isogenic IFNAR2+/+ iPSC, to iPS-MGL, confirming by immunofluorescence analysis expression of the microglial marker TMEM119 (58, 59), alongside increased expression of IBA1 compared to iPS-Mϕ (Figure 9A). Upon infection with ZIKVFP and ZIKVMP we observed the same phenotypes in iPS-MGL that were previously seen in IFNAR2-deficient iPS-Mϕ, including a robust pro-inflammatory response, significantly heightened ZIKV replication, increased infectious particle release and vulnerability to CPE (Figures 9B–E), indicating that IFN-I mediated restraint of ZIKV replication and cytopathicity is also key to the response of CNS-resident human microglia to ZIKV infection.
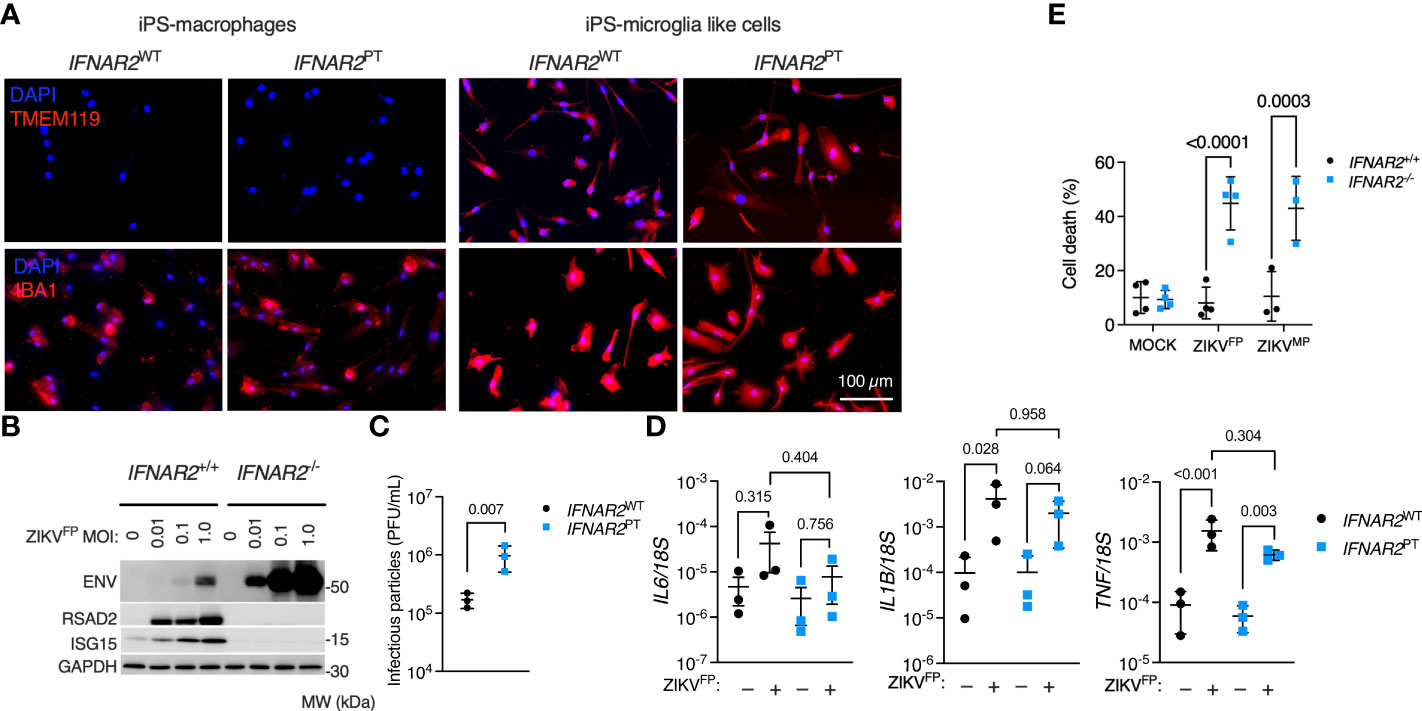
Figure 9 Enhanced ZIKV infection and CPE in IFNAR2-deficient iPS-microglia-like cells. (A) Immunofluorescence analysis of microglial markers IBA1 and TMEM119 in IFNAR2PT (PT clone 11) and IFNAR2WT (WT2) iPS-Mϕ and iPS-microglia-like cells (iPS-MGLs), representative of repeat experiments in G2, B5_2, G8 and F8 lines. Scale bar = 100 μm. (B) Immunoblot of ENV, RSAD2, ISG15, GAPDH in ZIKVFP-infected IFNAR2+/+ (F8) and IFNAR2-/- (B5_2) iPS-MGLs at 72 h.p.i., representative of n = 3 independent experiments in IFNAR2PT [clones 6 and 11] and IFNAR2WT [WT2]). (C) Plaque assay on Vero cells of ZIKV infectious particles in supernatants (48 h.p.i. ZIKVFP MOI = 1, n = 3 independent experiments in IFNAR2WT (WT2) and IFNAR2PT (clone 6) iPS-microglia-like cells. Mean ± SD, t test. (D) RT-PCR quantification of IL6, IL1B and TNF relative to 18S (24 h.p.i. ZIKVFP MOI = 1, n = 3 independent experiments in IFNAR2WT [WT2] and IFNAR2PT [clones 6 and 11]) microglia-like cells. Mean ± SD, ANOVA with Sidak’s test for multiple comparisons. (E) Cell viability assay (72 h.p.i. ZIKVFP MOI = 1.0 [n = 4 independent experiments] or ZIKVMP MOI = 1.0 [n = 3 independent experiments]) in IFNAR2-/- (B5_2), IFNAR2PT (clone 6), IFNAR2+/+ (F8) and IFNAR2PT (WT2) iPS-MGLs. Mean ± SD, ANOVA with Sidak’s test for multiple comparisons.
Many successful human pathogens have evolved sophisticated mechanisms to subvert tissue macrophages - exploiting this cellular niche for survival and/or transmission (14). Here we employed genetically tractable models of human macrophages and microglia derived from wild-type and IFNAR2 deficient iPSCs to interrogate interactions of ZIKV, an RNA viral pathogen of global public health importance, with the innate immune system of macrophages. We observe that the ability of wild-type iPS-macrophages to resist cytopathic effects of ZIKV depends on robust autocrine and paracrine IFN-I signalling that acts to contain viral replication and spread. ZIKV is highly human-adapted, relying on myriad methods to evade restriction by antiviral IFN signalling, especially via the targeting of human (but not mouse) STAT2 protein (4, 25, 34). Our data resolve an open question concerning the extent to which human endogenous IFN-I responses are capable of restricting ZIKV replication in the face of these viral countermeasures, providing a framework in which to explore host susceptibility to ZIKV disease and identify targets for host-directed antiviral therapy.
Although the question of the functional role of IFN-I in clinically relevant ZIKV target cells had not been addressed directly, there is evidence that IFN-I participates in ZIKV resistance of target cells. For instance, genetic ablation of AXL attenuated ZIKV replication in an astrocytic cell line, seemingly by enhancing endogenous IFN-I signalling (40). AXL is a recognised negative regulator of IFNAR, indicating that loss of AXL may have promoted IFN-I signalling. Similarly, deletion of the ISG ISG15 reduced ZIKV replication in iPS-derived neural progenitor cells (iPS-NPCs) (41). Whilst this result might at first glance appear paradoxical, ISG15 has divergent species-specific function, acting in mouse as an antiviral effector but in human as a negative regulator of IFNAR signalling (60). Consistent with these data, blockade of IFN-I signalling with JAK inhibitors or IFNAR antibodies was reported to enhance ZIKV replication in placental macrophages (50) and iPS-microglia (9). Importantly, the consequences of IFN-I inhibition for macrophage survival were not assessed.
Our data, relying on a robust genetic approach involving use of IFNAR2 deficient patient iPSCs and validated by precise CRISPR/Cas9 IFNAR2 knockout in control iPSCs, confirm and substantially extend this indirect evidence, showing that IFN-I is decisive in controlling ZIKV replication despite extensive viral countermeasures (4), in turn protecting iPS-macrophages and iPS-microglia from profound cytopathicity. Thus macrophage lineage cells are not as a population intrinsically resistant to cytopathic effects of ZIKV, as is widely reported (7–9) but instead mount a robust paracrine IFN-I response that restrains viral replication and maintains viability of the majority of bystander cells. This IFN-I restriction presumably explains why experimental manipulation, such as the use of flavivirus immune serum to enhance macrophage ZIKV uptake (61), is necessary to enhance infection efficiency of primary monocyte-derived macrophages (18).
The single cell imaging analyses presented here indicate that IFN-I mediated protection of wild-type iPS-macrophages derives largely from paracrine IFN-I signalling, suggesting that IFN-I produced by macrophages might also confer protection on neighbouring stromal cell types present in tissues in natura. This idea that macrophages might act in a protective capacity as antiviral sentinels promoting IFN-mediated defence of neighbouring cells is supported by recent findings in animal models (62, 63). However, our data are also consistent with a model in which innate antiviral IFN responses counterintuitively promote ZIKV persistence by preventing macrophage depletion, facilitating the maintenance of macrophage reservoirs in sanctuary sites such as the brain or testes (7–10). Similar findings were reported for human fetal astrocytes, which supported long-term ZIKV replication in the face of a robust innate immune response (64). Thus our data imply a relatively complex role for IFN-I signalling in ZIKV pathogenesis, contributing on the one hand to protection of macrophages against unrestrained ZIKV replication and cytopathic effects, whilst on the other maintaining an infected reservoir in immune sanctuary sites. Examples from paramyxovirus models indicate that innate immune responses may contribute to RNA virus persistence (65, 66). The specific contribution of IFN-I responses to viral persistence in macrophages in other relevant families, such as the Retroviridae and Togaviridae, will be an important line of investigation (67).
Our focus on iPS-macrophages and iPS-microglia is the principal limitation of this study. It is likely that co-culture and/or organoid models will be needed in future studies to model the collective impact of microglial IFN-I signalling on viral behaviour in neighbouring CNS target cells such as astrocytes and neural progenitor cells. Nevertheless, the use of this system provided unambiguous genetic evidence by enabling the analysis of macrophages derived from patients with ultra-rare genetic disorders where cellular material is limited and simultaneously provided a means of overcoming the resistance of macrophages to genetic manipulation by CRISPR/Cas9. Our studies also employed the two major ZIKV lineages (African and Asian), lending generalisability to the findings.
In summary, the data presented here show that human macrophages and microglia-like cells respond to ZIKV infection with robust paracrine IFN-I signalling that restricts viral replication and promotes cell survival in the face of ongoing infection. These data establish the functional role of IFN-I in control of ZIKV replication in a disease-relevant cell type and shed light on the complex role played by macrophages in ZIKV pathogenesis. Given that microcephaly and encephalitis are relatively rare outcomes of ZIKV infection, an important question for future studies will be the extent to which defects in host IFN-I immunity [via genetic lesions (26–33) or neutralising IFN-I autoantibodies (68)] might contribute to the development of CNS complications of ZIKV disease. Another question for future studies is whether the mode of IFN-I dependent resistance to viral cytopathic effects we report applies more generally to RNA viruses that target and exploit macrophages (5).
Human induced pluripotent stem cells (iPSC) were reprogrammed from skin dermal fibroblasts from a patient carrying a homozygous nonsense variant in IFNAR2 (c.A311del, p.Gly104fs110X, termed IFNAR2PT). Written informed parental consent was provided (Newcastle and North Tyne Research Ethics Committee Ref: 16/NE/0002). Briefly, fibroblasts were reprogrammed at passage 3-5, using Cytotune 2 Sendai virus reprogramming kit (Invitrogen) according to the manufacturer’s instructions, with modifications as described (47). QC assays on bulk banked stocks (at or near to passage 10) included flow cytometry assessment of pluripotency marker expression, RT-PCR to check clearance of CytoTune vectors and Illumina SNP array to check genome integrity and identity/tracking back to the source fibroblasts (Figures S1A–C). Clones 6 and 11 were used for experiments.
The IFNAR2WT control iPSC lines SFC841-03-01 (48) (WT1, Ref: STBCi044-A) and SFC856-03-04 (47) (WT2, Human Pluripotent Stem Cell Registry Ref: STBCi063-A) were previously generated using the same methodology, at the James Martin Stem Cell Facility at the University of Oxford. These lines are available from the European Bank for Induced pluripotent Stem Cells (EBISC). iPSCs were cultured feeder-free on Geltrex (Gibco) coated plates in mTeSR1 (Stem Cell Technologies, Canada) media, exchanged daily. Cultures were passaged using 0.5 mM EDTA (Gibco). Cryopreserved master stocks of iPSC were used and passaged no more than three times prior to differentiation to macrophages or microglia.
The control iPSC lines SFC856-03-04 and SFC841-03-01 were used for gene editing. Two guide RNAs (gRNAs) were used to target the IFNAR2 exon 5 splice acceptor (reference transcript: NM_001289125.3). This was predicted to cause out of frame skipping of exon 5 from all processed IFNAR2 transcripts, leading to nonsense mediated RNA decay and absence of IFNAR2 protein expression. The guide RNA (gRNA) sequences were: CATTTTCAATAAGATGGTTG (gRNA4) and ACCGTCCTAGAAGGATTCAG (gRNA7). These gRNAs were purchased from Integrated DNA Technologies (IDT, USA) and were complexed to tracRNA, before mixing with 1.5 μg Alt-R HiFi Cas9 Nuclease (IDT, USA) to form a ribonucleoprotein (RNP) complex, according to manufacturer’s instructions. The gRNA : Cas9 RNP complex was transfected into 2 x 105 iPSCs in single-cell suspension by electroporation (Neon transfection system, ThermoFisher Scientific) in 10 μL using the ‘HiTrans’ settings: 1400 v, 20 ms width, 1 pulse. iPSC were treated with 10 μL RHO kinase inhibitor Y-27632 (AbCam Biochemicals) for 3 h prior to transfection. After 3 d iPSC were single-cell plated onto irradiated mouse embryonic fibroblast feeders. Single-cell colonies were subsequently picked, expanded under feeder-free conditions and screened by PCR gel electrophoresis (primers available on request). One IFNAR2-/- and isogenic IFNAR2+/+ pair was selected from each parental line for further validation by PCR (Figure 7B) and capillary sequencing (not shown). SFC856-03-04 clones were B5_2 (IFNAR2-/-) and F8 (IFNAR2+/+); SFC841-03-01 clones were G2 (IFNAR2-/-) and G8 (IFNAR2+/+). IFNAR2 expression was examined by immunoblot.
Vero cells were cultured in complete Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 10% foetal calf serum (FCS; Gibco), 1% penicillin/streptomycin (Gibco) and 1% L-glutamine (Gibco). All cells were incubated in a humidified atmosphere with 5% CO2 at 37°C. Cytokines/inhibitors were used at the following concentrations: human recombinant IFN-α2b (1000 IU/ml; Intron A, Schering-Plough, USA); IFN-γ (1000 IU/ml; Immunikin, Boehringer Ingelheim, Germany) and Ruxolitinib (10 μM; Calbiochem, USA).
iPSCs were differentiated to macrophage precursor cells via embryoid bodies (EBs) using AggreWells (Stem Cell Technologies) using a published methodology with no alterations (45). Media for embryoid body generation consisted of mTeSR with 50 mg/mL BMP4, 50 mg/mL VEGF and 20 mg/mL SCF. Approx. 300 EBs were split between two T175 flasks in 15 mL of medium per flask. ‘Factory’ medium comprised XVIVO15 (Lonza, Basel, Switzerland) with 100 ng/mL recombinant human M-CSF (Gibco), 25 ng/mL recombinant human IL-3 (Gibco), 2 nM GlutaMAX (Gibco), 50 μM 2-mercaptoethanol and 100 units/mL penicillin with 100 µg/mL streptomycin (Gibco). Precursor cells were harvested and plated into final experimental format for final differentiation to either iPS-Mϕ in macrophage differentiation medium (XVIVO15 with 100ng/mL recombinant human M-CSF (Gibco), 2 mM GlutaMAX (Gibco) and 100 units/mL penicillin with 100 µg/mL streptomycin) for 7 days prior to use, or iPS-microglia-like cells (iPS-MGLs) in microglia differentiation medium (Advanced DMEM/F12 + N2 Supplement (Gibco) with 100 ng/mL recombinant human IL-34 (PeproTech), 10 ng/mL recombinant human GM-CSF (Gibco), 2 mM Glutamax, 50 μM 2-mercaptoethanol and 100 units/mL penicillin with 100 µg/mL streptomycin) for 14 days, changing medium twice weekly, prior to use in experiments.
Two strains of ZIKV were used in this study, the epidemic Asian lineage strain H/FP/2013 (patient isolate, French Polynesia, 2013, herein ZIKVFP) was obtained from the European Virus Archive (Ref: 001v-EVA1545) and the African lineage strain MP1751 (mosquito isolate, Uganda, 1962, herein ZIKVMP) was obtained from the UK National Collection of Pathogenic Viruses (Ref: 1308258v). Viral stocks were propagated at low MOI in Vero cells (obtained from Professor R Randall, St Andrew’s University), and viral titre determined by serial dilution and standard plaque assay. Stocks were aliquoted, clarified by centrifugation and clarified supernatants were aliquoted and frozen at -80°C, and thawed for single use. The same stocks of each virus were used for all experiments.
For experimental infection, MOI was approximated as the ratio of PFU to cells. Cells were exposed to a known titre of virus in a standard volume (50 μL for 96 well plates, 250 μL for 24 well plates, 1000 μL for 6 well plates) in macrophage or microglia medium. In parallel, cells were mock infected with medium in the same volume but without virus. At 2 hours post-infection, the inoculum was removed and replaced with fresh medium (macrophage or microglia), with or without treatments (IFNs, RUX etc) as indicated, until the time required for the experiment.
Supernatant from infected cell cultures were harvested and stored at the indicated time points. Aliquots of supernatant were thawed and serially diluted in DMEM with 1% FCS (Gibco), and 250 μL added to 24-well plates of confluent Vero cells. After 2 hours of incubation, 1.5% methylcellulose in DMEM with 1% FCS (Gibco) was gently added. At 72 hours post-infection, media was aspirated and cells fixed with 4% formaldehyde, before being stained with 0.25% crystal violet stain, washed and plaques counted on a lightbox to determine plaque-forming units per mL (PFU/mL).
iPS-Mϕ were lysed in BL buffer at 24h.p.i. ZIKVFP MOI=10, iPS-MGL were lysed in BL buffer at 24h.p.i. ZIKVFP MOI=1. T otal RNA was extracted using the ReliaPrepTM RNA Cell Miniprep System (Promega, USA) and treated with DNase I according to the manufacturer’s instructions. Quantity of purified RNA was measured spectrophotometrically (A260/A280) using a NanoDrop OneC Microvolume UV-Vis Spectrophotometer (Thermo Fisher, MA, USA). RT-PCR was performed using the SensiFAST™ Probe No-ROX One-Step Kit (Bioline, USA) with AriaMx Real-time PCR System (Agilent Technologies, CA, USA) according to manufacturer’s instructions. The following TaqMan gene expression assay (Thermo Fisher) was used: IFNA1 (Hs03044218_g1). The primers were designed using the Roche Universal ProbeLibrary Assay Design tool (Roche, Basel, Switzerland) with the indicated UPL probes. Further details, including additional primer/probe information, are summarised in Table S1. Target gene expression was normalised to the housekeeper GAPDH or 18S as indicated. Each sample was run in technical duplicate. Cycling conditions were as follows: reverse transcription at 50°C for 15 min, followed by initial polymerase activation at 95°C for 10 min, and then 40 cycles of denaturation at 95°C for 15 sec and annealing/extension at 60°C for 1 min.
RNA was extracted from iPS-Mϕ at 24h.p.i. ZIKVFP MOI=10 using BL buffer and ReliaPrepTM RNA Cell Miniprep System (Promega) according to the manufacturer’s instructions. Sequencing was performed over four lanes of a single flow cell on an Illumina NextSeq (Illumina, USA) over 75 cycles. Sequencing was single ended. Raw FASTQ files were first inspected for quality using FastQC (69) and MultiQC (70). All FASTQ files were of a very high quality, so no filtering or trimming was performed. Pseudo-alignment and transcript counting was performed and generated for the four FASTQ files for each sample using Salmon (71). These counts are then imported into R for subsequent analysis. Transcripts were agglomerated into gene counts. Individual sample counts were merged into a single count table of samples as columns and genes as rows, with cells representing individual raw counts. Library normalisation, dispersion estimation, and log transformation was performed on the count table using the DESeq2 pipeline (72), set as default. RNA-seq data were uploaded to the Gene Expression Online (GEO) repository (accession no. GSE198542).
Macrophages were lifted using EDTA and washed with FACS buffer (PBS plus 2% FCS and 0.05% sodium azide). Cells were stained at room temperature for one hour with the following antibodies: CD45 (clone 2D1, APC-H7), CD14 (clone M5E2, BUV737), CD11c (clone B-ly6, BV421), CD86 (clone 2331, FITC, all from BD), HLA-DR (clone L243, BV650), CD11b (clone ICRF4, BV785, all from Biolegend) or the respective isotype controls. Cell viability was assessed using 7-AAD (Biolegend). After the final washing step samples were acquired on a Symphony A5 flow cytometer (BD) and results were analysed using FlowJo (Ashland, OR, USA). See Figure S2 for the gating strategy.
IPS-macrophages were lifted using EDTA and resuspended in fresh macrophage medium (XVIVO15 containing 1% Penicillin/Streptomycin, 1% GlutaMAX, 100 ng/mL MCSF). pHrodo red Zymosan A bioparticles (Invitrogen) were diluted at a concentration of 100,000 beads per ul and mixed with the iPS-macrophages at a ratio of 10:1 beads to cells. Cells were either placed on ice immediately (negative control) or incubated for 2 hours at 37°C in a rocking incubator. Afterwards cells were immediately placed on ice to stop phagocytosis and 4′,6-diamidin-2-phenylindol (DAPI; ThermoFisher) was added to assess viability. Samples were acquired without further washing steps on a Symphony A5 flow cytometer (BD Biosciences) and results were analyzed using FlowJo (Ashland, OR, USA).
Proteins from cell lysates were separated by 10% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) gel electrophoresis using MOPS running buffer (Thermo Fisher), and transferred to a nitrocellulose membrane (Millipore, USA) using NuPage Tris-Glycine Transfer Buffer (Thermo Fisher) for immunoblotting (antibodies see Table S2). Blots were developed with Pierce ECL Western blotting substrate (Thermo Fisher) and imaged on a LI-COR Odyssey Fc (LI-COR, NE, USA). Densitometry analysis was undertaken using ImageStudio software (version 5.2.5, Li-COR).
Cells were grown on eight-well chamber slides (Millipore). Following treatment and/or infection, cells were fixed with 4% paraformaldehyde in PBS for 20 minutes at room temperature before blocking/permeabilisation with 0.5% Triton X-100 (Sigma-Aldrich, USA)/10% goat serum (Abcam, UK) in PBS for 1 hour at room temperature. Cells were incubated with mouse anti-ZIKA ENV and rabbit anti-IFITM3 specific primary antibodies (see Table S2) overnight at 4°C, then washed three times in PBS and incubated with goat anti-mouse IgG Alexa fluor 488 or goat anti-rabbit IgG Alexa fluor 555 secondary antibody (both 1 μg/ml; Thermo Fisher) for 1 hour at RT. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; 0.2 μg/ml; Sigma-Aldrich). All fluorescent images were taken using an EVOS FL fluorescence microscope (Thermo Fisher) at 10X magnification using DAPI, GFP, RFP and bright-field filters. Image analysis was performed using CellProfiler software version 3.1.8 (Broad institute, USA) (73).
Images were converted to greyscale, and within DAPI images each object (nucleus) was identified using the IdentifyPrimaryObject module. In order to quantify proteins within the whole cell including the cytoplasm, cells were identified within bright-field images by propagating out from each nucleus using the IdentifySecondaryObjects module, to create a mask for each cell. The intensity of the defined cells was measured and then predefined thresholds for GFP (Env protein) and RFP (IFITM3) were used to classify Zika positive and/or ISG productive cells, and the average value of at least n=4 images per well was used for analysis. A total of 58 sets of images (since all biological replicates were made up of at least n=3 technical replicate wells) with a median (IQR) of 255.5 (162 – 513.25) cells per image processed.
Cells in 96 well plates were infected at the indicated MOI for 72 hours, with or without pre-treatment with the agents indicated for 16 hours, and imaged. Live cell imaging solution containing 3 drops/mL of ReadyProbes Cell Viability Blue/Red Imaging Kit (Invitrogen) was added 30 minutes before imaging to assess cell viability. Images were obtained using an EVOS FL fluorescence microscope (Thermo Fisher) using DAPI and RFP filters at 10x magnification. Image analysis was performed using CellProfiler version 3.1.8 (Broad institute, USA). DAPI and RFP images were converted to greyscale and objects identified using IdentifyPrimaryObject modules. DAPI objects (corresponding to Hoechst nuclear staining) overlapping with RFP objects (corresponding to propidium iodide staining) were classified as dead cells, while non-overlapping DAPI objects were counted as live cells. All conditions were performed in technical duplicate and the average value of n=6 images per well was used for analysis. All experiments were performed at least three times. The cell profiler pipelines used are available at https://github.com/aidanhanrath/Cellprofiler.
Statistical analysis was performed using GraphPad Prism 9 software (GraphPad Software, CA, USA). Values are presented as mean ± SD of at least three independent experimental replicates. Continuous data were normalised or log-transformed prior to analysis using parametric significance tests. Statistical significances between two groups of data were determined using unpaired Student’s t-test and between three or more groups were determined using one-way analysis of variance (ANOVA), with Sidak’s post-hoc test to account for multiple comparisons. All tests were two-sided and an alpha of < 0.05 was the threshold for statistical significance.
Source data are provided with this paper as a data supplement. The source data file includes uncropped blots, all raw quantitative data and PFU counts. The results of differential expression analysis of RNA-seq data are also included as a supplementary dataset. Raw RNA seq data are available on GEO (Accession: GSE198542).
AH, CH, FG, CB, JV, PL, SJC and CD did experiments and analysed data. SAC, WJ, SH and CD obtained funding and provided supervision. AH and CD conceived the study and with CH drafted the manuscript. All authors contributed to the article and approved the submitted version.
AH was funded by an NIHR Academic Clinical Fellowship (ACF-2018-01-004) and the British Medical Association Foundation. FG is supported by the Munich Clinician Scientist Program at LMU (FoeFoLeplus) and received fellowships from the Bubble Foundation as well as the Care-for-Rare Foundation. CH and CD are funded by the UK Medical Research Council [MR/N013840/1 and MR/X001598/1 respectively]. SH and CD are funded by the Wellcome Trust [207556/Z/17/Z and 211153/Z/18/Z respectively]. iPS cell derivation and gene editing was carried out at the James Martin Stem Cell Facility, which receives financial support from the Oxford Martin School (LC0910-004).
We are grateful to Jon Coxhead and Rafiq Ahmed for assistance with Illumina RNA-sequencing, and to Javier Gilbert Jaramillo for discussions.
SH declares honoraria from CSL Behring and Takeda for teaching and consultancy.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher..
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1035532/full#supplementary-material
1. Pannu J, Barry M. Global health security as it pertains to zika, Ebola, and COVID-19. Curr Opin Infect Dis (2021) 34:401–8. doi: 10.1097/QCO.0000000000000775
2. Pierson TC, Diamond MS. The emergence of zika virus and its new clinical syndromes. Nature (2018) 560:573–81. doi: 10.1038/s41586-018-0446-y
3. Russell K, Hills SL, Oster AM, Porse CC, Danyluk G, Cone M, et al. Male-to-Female sexual transmission of zika virus-united states, January-April 2016. Clin Infect Dis (2017) 64:211–3. doi: 10.1093/cid/ciw692
4. Pardy RD, Valbon SF, Richer MJ. Running interference: Interplay between zika virus and the host interferon response. Cytokine (2019) 119:7–15. doi: 10.1016/j.cyto.2019.02.009
5. Nikitina E, Larionova I, Choinzonov E, Kzhyshkowska J. Monocytes and macrophages as viral targets and reservoirs. Int J Mol Sci (2018) 19:2821. doi: 10.3390/ijms19092821
6. Hirsch AJ, Smith JL, Haese NN, Broeckel RM, Parkins CJ, Kreklywich C, et al. Zika virus infection of rhesus macaques leads to viral persistence in multiple tissues. PloS Pathog (2017) 13:e1006219. doi: 10.1371/journal.ppat.1006219
7. Jurado KA, Simoni MK, Tang Z, Uraki R, Hwang J, Householder S, et al. Zika virus productively infects primary human placenta-specific macrophages. JCI Insight (2016) 1:e88461. doi: 10.1172/jci.insight.88461
8. Lang J, Cheng Y, Rolfe A, Hammack C, Vera D, Kyle K, et al. An hPSC-derived tissue-resident macrophage model reveals differential responses of macrophages to ZIKV and DENV infection. Stem Cell Rep (2018) 11:348–62. doi: 10.1016/j.stemcr.2018.06.006
9. Muffat J, Li Y, Omer A, Durbin A, Bosch I, Bakiasi G, et al. Human induced pluripotent stem cell-derived glial cells and neural progenitors display divergent responses to zika and dengue infections. Proc Natl Acad Sci USA (2018) 115:7117–22. doi: 10.1073/pnas.1719266115
10. Figueiredo CP, Barros-Aragao FGQ, Neris RLS, Frost PS, Soares C, Souza INO, et al. Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat Commun (2019) 10:3890. doi: 10.1038/s41467-019-11866-7
11. Souza BS, Sampaio GL, Pereira CS, Campos GS, Sardi SI, Freitas LA, et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci Rep (2016) 6:39775. doi: 10.1038/srep39775
12. Ghezzi S, Cooper L, Rubio A, Pagani I, Capobianchi MR, Ippolito G, et al. Heparin prevents zika virus induced-cytopathic effects in human neural progenitor cells. Antiviral Res (2017) 140:13–7. doi: 10.1016/j.antiviral.2016.12.023
13. Cavalcante BRR, Aragao-Franca LS, Sampaio GLA, Nonaka CKV, Oliveira MS, Campos GS, et al. Betulinic acid exerts cytoprotective activity on zika virus-infected neural progenitor cells. Front Cell Infect Microbiol (2020) 10:558324. doi: 10.3389/fcimb.2020.558324
14. Schilling M, Bridgeman A, Gray N, Hertzog J, Hublitz P, Kohl A, et al. RIG-I plays a dominant role in the induction of transcriptional changes in zika virus-infected cells, which protect from virus-induced cell death. Cells (2020) 9:e1476. doi: 10.3390/cells9061476
15. Duncan CJ, Russell RA, Sattentau QJ. High multiplicity HIV-1 cell-to-cell transmission from macrophages to CD4+ T cells limits antiretroviral efficacy. AIDS (2013) 27:2201–6. doi: 10.1097/QAD.0b013e3283632ec4
16. Duncan CJ, Williams JP, Schiffner T, Gartner K, Ochsenbauer C, Kappes J, et al. High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. J Virol (2014) 88:2025–34. doi: 10.1128/JVI.03245-13
17. Sattentau QJ, Stevenson M. Macrophages and HIV-1: An unhealthy constellation. Cell Host Microbe (2016) 19:304–10. doi: 10.1016/j.chom.2016.02.013
18. Carlin AF, Vizcarra EA, Branche E, Viramontes KM, Suarez-Amaran L, Ley K, et al. Deconvolution of pro- and antiviral genomic responses in zika virus-infected and bystander macrophages. Proc Natl Acad Sci USA (2018) 115:E9172–81. doi: 10.1073/pnas.1807690115
19. Lum FM, Low DK, Fan Y, Tan JJ, Lee B, Chan JK, et al. Zika virus infects human fetal brain microglia and induces inflammation. Clin Infect Dis (2017) 64:914–20. doi: 10.1093/cid/ciw878
20. Wang W, Li G, De W, Luo Z, Pan P, Tian M, et al. Zika virus infection induces host inflammatory responses by facilitating NLRP3 inflammasome assembly and interleukin-1beta secretion. Nat Commun (2018) 9:106. doi: 10.1038/s41467-017-02645-3
21. Khaiboullina SF, Uppal T, Sarkar R, Gorzalski A, St Jeor S, Verma SC, et al. ZIKV infection regulates inflammasomes pathway for replication in monocytes. Sci Rep (2017) 7:16050. doi: 10.1038/s41598-017-16072-3
22. Colavita F, Quicke KM, Maddur MS, O'Neal JT, McDonald CE, Fedorova NB, et al. ZIKV infection induces an inflammatory response but fails to activate types I, II, and III IFN response in human PBMC. Mediators Inflammation (2018) 2018:2450540. doi: 10.1155/2018/2450540
23. Bowen JR, Quicke KM, Maddur MS, O'Neal JT, McDonald CE, Fedorova NB, et al. Zika virus antagonizes type I interferon responses during infection of human dendritic cells. PloS Pathog (2017) 13:e1006164. doi: 10.1371/journal.ppat.1006164
24. Vielle NJ, Zumkehr B, Garcia-Nicolas O, Blank F, Stojanov M, Musso D, et al. Silent infection of human dendritic cells by African and Asian strains of zika virus. Sci Rep (2018) 8:5440. doi: 10.1038/s41598-018-23734-3
25. Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, et al. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep (2016) 17:1766–75. doi: 10.15252/embr.201642627
26. Duncan CJ, Mohamad SM, Young DF, Skelton AJ, Leahy TR, Munday DC, et al. Human IFNAR2 deficiency: Lessons for antiviral immunity. Sci Transl Med (2015) 7:307ra154. doi: 10.1126/scitranslmed.aac4227
27. Duncan CJA, Randall RE, Hambleton S. Genetic lesions of type I interferon signalling in human antiviral immunity. Trends Genet (2021) 37:46–58. doi: 10.1016/j.tig.2020.08.017
28. Hernandez N, Bucciol G, Moens L, Le Pen J, Shahrooei M, Goudouris E, et al. Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reaction to measles and yellow fever live vaccines. J Exp Med (2019) 216:2057–70. doi: 10.1084/jem.20182295
29. Gothe F, Hatton CF, Truong L, Klimova Z, Kanderova V, Fejtkova M, et al. A novel case of homozygous IFNAR1 deficiency with haemophagocytic lymphohistiocytosis. Clin Infect Dis (2020) 74(1):136–9. doi: 10.1093/cid/ciaa1790
30. Duncan CJA, Skouboe MK, Howarth S, Hollensen AK, Chen R, Borresen ML, et al. Life-threatening viral disease in a novel form of autosomal recessive IFNAR2 deficiency in the Arctic. J Exp Med (2022) 219:e20212427. doi: 10.1084/jem.20212427
31. Bastard P, Manry J, Chen J, Rosain J, Seeleuthner Y, AbuZaitun O, et al. Herpes simplex encephalitis in a patient with a distinctive form of inherited IFNAR1 deficiency. J Clin Invest (2021) 131:e139980. doi: 10.1172/JCI139980
32. Bastard P, Hsiao KC, Zhang Q, Choin J, Best E, Chen J, et al. A loss-of-function IFNAR1 allele in Polynesia underlies severe viral diseases in homozygotes. J Exp Med (2022) 219:e20220028. doi: 10.1084/jem.20220028
33. Zhang Q, Matuozzo D, Le Pen J, Lee D, Moens L, Asano T, et al. Recessive inborn errors of type I IFN immunity in children with COVID-19 pneumonia. J Exp Med (2022) 219:e20220131. doi: 10.1084/jem.20220131
34. Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, et al. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe (2016) 19:882–90. doi: 10.1016/j.chom.2016.05.009
35. Gorman MJ, Caine EA, Zaitsev K, Begley MC, Weger-Lucarelli J, Uccellini MB, et al. An immunocompetent mouse model of zika virus infection. Cell Host Microbe (2018) 23:672–685 e676. doi: 10.1016/j.chom.2018.04.003
36. Van der Hoek KH, Eyre NS, Shue B, Khantisitthiporn O, Glab-Ampi K, Carr JM, et al. Viperin is an important host restriction factor in control of zika virus infection. Sci Rep (2017) 7:4475. doi: 10.1038/s41598-017-04138-1
37. Liao X, Xie H, Li S, Ye H, Li S, Ren K, et al. 2', 5'-oligoadenylate synthetase 2 (OAS2) inhibits zika virus replication through activation of type iota IFN signaling pathway. Viruses (2020) 12:418. doi: 10.3390/v12040418
38. Ren K, Sun H, Chen L, Chen N, Yu L. Myxovirus resistance protein a activates type I IFN signaling pathway to inhibit zika virus replication. Virus Res (2021) 306:198534. doi: 10.1016/j.virusres.2021.198534
39. Gobillot TA, Humes D, Sharma A, Kikawa C, Overbaugh J. The robust restriction of zika virus by type-I interferon in A549 cells varies by viral lineage and is not determined by IFITM3. Viruses (2020) 12:503. doi: 10.3390/v12050503
40. Chen J, Yang YF, Yang Y, Zou P, Chen J, He Y, et al. AXL promotes zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat Microbiol (2018) 3:302–9. doi: 10.1038/s41564-017-0092-4
41. Li Y, Omer Javed A, Keys HR, Lungjangwa T, Bosch I, et al. Genome-wide CRISPR screen for zika virus resistance in human neural cells. Proc Natl Acad Sci USA (2019) 116:9527–32. doi: 10.1073/pnas.1900867116
42. Yockey LJ, Jurado KA, Arora N, Millet A, Rakib T, Milano KM, et al. Type I interferons instigate fetal demise after zika virus infection. Sci Immunol (2018) 3:eaao1680. doi: 10.1126/sciimmunol.aao1680
43. Buchrieser J, James W, Moore MD. Human induced pluripotent stem cell-derived macrophages share ontogeny with MYB-independent tissue-resident macrophages. Stem Cell Rep (2017) 8:334–45. doi: 10.1016/j.stemcr.2016.12.020
44. Lee CZW, Kozaki T, Ginhoux F. Studying tissue macrophages in vitro: are iPSC-derived cells the answer? Nat Rev Immunol (2018) 18:716–25. doi: 10.1038/s41577-018-0054-y
45. van Wilgenburg B, Browne C, Vowles J, Cowley SA. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PloS One (2013) 8:e71098. doi: 10.1371/journal.pone.0071098
46. Haenseler W, Sansom SN, Buchrieser J, Newey SE, Moore CS, Nicholls FJ, et al. A highly efficient human pluripotent stem cell microglia model displays a neuronal-Co-culture-Specific expression profile and inflammatory response. Stem Cell Rep (2017) 8:1727–42. doi: 10.1016/j.stemcr.2017.05.017
47. Haenseler W, Zambon F, Lee H, Vowles J, Rinaldi F, Duggal G, et al. Excess alpha-synuclein compromises phagocytosis in iPSC-derived macrophages. Sci Rep (2017) 7:9003. doi: 10.1038/s41598-017-09362-3
48. Dafinca R, Scaber J, Ababneh N, Lalic T, Weir G, Christian H, et al. C9orf72 hexanucleotide expansions are associated with altered endoplasmic reticulum calcium homeostasis and stress granule formation in induced pluripotent stem cell-derived neurons from patients with amyotrophic lateral sclerosis and frontotemporal dementia. Stem Cells (2016) 34:2063–78. doi: 10.1002/stem.2388
49. Nan J, Wang Y, Yang J, Stark GR. IRF9 and unphosphorylated STAT2 cooperate with NF-kappaB to drive IL6 expression. Proc Natl Acad Sci USA (2018) 115:3906–11. doi: 10.1073/pnas.1714102115
50. Gavegnano C, Bassit LC, Cox BD, Hsiao HM, Johnson EL, Suthar M, et al. Jak inhibitors modulate production of replication-competent zika virus in human hofbauer, trophoblasts, and neuroblastoma cells. Pathog Immun (2017) 2:199–218. doi: 10.20411/pai.v2i2.190
51. Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF, et al. Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in STAT2. Sci Immunol (2019) 4:eaav7501. doi: 10.1126/sciimmunol.aav7501
52. Kim HS, Bernitz JM, Lee DF, Lemischka IR. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev (2014) 23:2673–86. doi: 10.1089/scd.2014.0167
53. Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res (2013) 41:D1040–6. doi: 10.1093/nar/gks1215
54. Greenwell-Wild T, Vazquez N, Jin W, Rangel Z, Munson PJ, Wahl SM. Interleukin-27 inhibition of HIV-1 involves an intermediate induction of type I interferon. Blood (2009) 114:1864–74. doi: 10.1182/blood-2009-03-211540
55. Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, et al. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol (2002) 76:5532–9. doi: 10.1128/jvi.76.11.5532-5539.2002
56. Prinz M, Jung S, Priller J. Microglia biology: One century of evolving concepts. Cell (2019) 179:292–311. doi: 10.1016/j.cell.2019.08.053
57. Xu P, Shan C, Dunn TJ, Xie X, Xia H, Gao J, et al. Role of microglia in the dissemination of zika virus from mother to fetal brain. PloS Negl Trop Dis (2020) 14:e0008413. doi: 10.1371/journal.pntd.0008413
58. Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci USA (2016) 113:E1738–46. doi: 10.1073/pnas.1525528113
59. Satoh J, Kino Y, Asahina N, Takitani M, Miyoshi J, Ishida T, et al. TMEM119 marks a subset of microglia in the human brain. Neuropathology (2016) 36:39–49. doi: 10.1111/neup.12235
60. Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun (2016) 7:11496. doi: 10.1038/ncomms11496
61. Bardina SV, Bunduc P, Tripathi S, Duehr J, Frere JJ, Brown JA, et al. Enhancement of zika virus pathogenesis by preexisting antiflavivirus immunity. Science (2017) 356:175–80. doi: 10.1126/science.aal4365
62. Winkler CW, Evans AB, Carmody AB, Peterson KE. Placental myeloid cells protect against zika virus vertical transmission in a Rag1-deficient mouse model. J Immunol (2020) 205:143–52. doi: 10.4049/jimmunol.1901289
63. Enlow W, Bordeleau M, Piret J, Ibanez FG, Uyar O, Venable MC, et al. Microglia are involved in phagocytosis and extracellular digestion during zika virus encephalitis in young adult immunodeficient mice. J Neuroinflamm (2021) 18:178. doi: 10.1186/s12974-021-02221-z
64. Limonta D, Jovel J, Kumar A, Airo AM, Hou S, Saito L, et al. Human fetal astrocytes infected with zika virus exhibit delayed apoptosis and resistance to interferon: Implications for persistence. Viruses (2018) 10:646. doi: 10.3390/v10110646
65. Xu J, Sun Y, Li Y, Ruthel G, Weiss SR, Raj A, et al. Replication defective viral genomes exploit a cellular pro-survival mechanism to establish paramyxovirus persistence. Nat Commun (2017) 8:799. doi: 10.1038/s41467-017-00909-6
66. Sun Y, Jain D, Koziol-White CJ, Genoyer E, Gilbert M, Tapia K, et al. Immunostimulatory defective viral genomes from respiratory syncytial virus promote a strong innate antiviral response during infection in mice and humans. PloS Pathog (2015) 11:e1005122. doi: 10.1371/journal.ppat.1005122
67. Griffin DE. Why does viral RNA sometimes persist after recovery from acute infections? PloS Biol (2022) 20:e3001687. doi: 10.1371/journal.pbio.3001687
68. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (2020) 370:eabd4585. doi: 10.1126/science.abd4585
69. de Sena Brandine G, Smith AD. Falco: high-speed FastQC emulation for quality control of sequencing data. F1000Res (2019) 8:1874. doi: 10.12688/f1000research.21142.2
70. Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics (2016) 32:3047–8. doi: 10.1093/bioinformatics/btw354
71. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods (2017) 14:417–9. doi: 10.1038/nmeth.4197
72. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol (2014) 15:550. doi: 10.1186/s13059-014-0550-8
Keywords: Zika virus, IFNAR2 deficiency, inborn errors of immunity, macrophages, type I interferons, cell death, antiviral state, interferon-stimulated genes
Citation: Hanrath AT, Hatton CF, Gothe F, Browne C, Vowles J, Leary P, Cockell SJ, Cowley SA, James WS, Hambleton S and Duncan CJA (2022) Type I interferon receptor (IFNAR2) deficiency reveals Zika virus cytopathicity in human macrophages and microglia. Front. Immunol. 13:1035532. doi: 10.3389/fimmu.2022.1035532
Received: 02 September 2022; Accepted: 21 October 2022;
Published: 11 November 2022.
Edited by:
Pei-Hui Wang, Shandong University, ChinaReviewed by:
Anthony Maxwell, Wayne State University, United StatesCopyright © 2022 Hanrath, Hatton, Gothe, Browne, Vowles, Leary, Cockell, Cowley, James, Hambleton and Duncan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher J. A. Duncan, Y2hyaXN0b3BoZXIuZHVuY2FuQG5jbC5hYy51aw==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.