 D. Alejandro Canaria1
D. Alejandro Canaria1 Maia G. Clare1
Maia G. Clare1 Bingyu Yan2
Bingyu Yan2 Charlotte B. Campbell1
Charlotte B. Campbell1 Zachariah A. Ismaio1Nicole L. Anderson1
Zachariah A. Ismaio1Nicole L. Anderson1 Sungtae Park1
Sungtae Park1 Alexander L. Dent3
Alexander L. Dent3 Majid Kazemian2,4
Majid Kazemian2,4 Matthew R. Olson1*
Matthew R. Olson1*- 1Department of Biological Sciences, Purdue University, West Lafayette, IN, United States
- 2Department of Biochemistry, Purdue University, West Lafayette, IN, United States
- 3Department of Microbiology and Immunology, Indiana University School of Medicine, Indianapolis, IN, United States
- 4Department of Computer Science, Purdue University, West Lafayette, IN, United States
IL-9-producing CD4+ T helper cells, termed Th9 cells, differentiate from naïve precursor cells in response to a combination of cytokine and cell surface receptor signals that are elevated in inflamed tissues. After differentiation, Th9 cells accumulate in these tissues where they exacerbate allergic and intestinal disease or enhance anti-parasite and anti-tumor immunity. Previous work indicates that the differentiation of Th9 cells requires the inflammatory cytokines IL-4 and TGF-β and is also dependent of the T cell growth factor IL-2. While the roles of IL-4 and TGF-β-mediated signaling are relatively well understood, how IL-2 signaling contributes to Th9 cell differentiation outside of directly inducing the Il9 locus remains less clear. We show here that murine Th9 cells that differentiate in IL-2-limiting conditions exhibit reduced IL-9 production, diminished NF-kB activation and a reduced NF-kB-associated transcriptional signature, suggesting that IL-2 signaling is required for optimal NF-kB activation in Th9 cells. Interestingly, both IL-9 production and the NF-kB transcriptional signature could be rescued by addition of the NF-kB-activating cytokine IL-1β to IL-2-limiting cultures. IL-1β was unique among NF-kB-activating factors in its ability to rescue Th9 differentiation as IL-2 deprived Th9 cells selectively induced IL-1R expression and IL-1β/IL-1R1 signaling enhanced the sensitivity of Th9 cells to limiting amounts of IL-2 by suppressing expression of the Th9 inhibitory factor BCL6. These data shed new light on the intertwined nature of IL-2 and NF-kB signaling pathways in differentiating Th cells and elucidate the potential mechanisms that promote Th9 inflammatory function in IL-2-limiting conditions.
Introduction
CD4+ T helper (Th) cells differentiate from a pool of naïve quiescent precursors into cytokine-producing effector cells in response to a complex set of cell surface receptor and cytokine cues. For IL-9-secreting Th cells, termed Th9 cells, this involves induction of TNF receptor family member co-stimulatory signals (i.e. CD28, OX40, GITR) along with specific cytokines including IL-4, TGF-β (1–4). The combination of these signals induces a downstream transcription factor network centered around STAT6, IRF4, BATF, and PU.1 that endows these cells with a unique proliferative capacity and the production of high levels of IL-9 (2). Th9 cells provide optimal control of parasite infection and potent anti-cancer immunity after adoptive cell therapy (5–8). However, when dysregulated, these cells can also contribute to autoimmunity in the form of allergic airway disease and ulcerative colitis (9, 10). Therefore, understanding the factors and cellular pathways that drive Th9 cell differentiation is critical to develop novel therapies that enhance anti-tumor immunity or thwart autoimmunity.
IL-2 is rapidly produced by T cells after T cell receptor activation/CD28 co-stimulation and is a potent activator of STAT5 and T cell proliferation (11, 12). Beyond its role in proliferation, STAT5 promotes the differentiation of Th9 cells by directly binding and transactivating the Il9 locus and allowing for epigenetic remodeling of upstream regulatory regions (13). IL-2/STAT5 signaling also contributes to Th9 cell differentiation through transcriptional repression where STAT5 represses expression of BCL6 that competes for STAT5 binding at the Il9 locus and limits Th9 differentiation (14, 15). Further, we demonstrated that STAT5 suppresses a Th17-like phenotype in differentiating Th9 cells (16). Despite the importance of IL-2 in the differentiation process, we and others have noted that IL-9-producing Th cells often develop and act to induce protective or auto-immunity in IL-2-limiting environments. For example, Th9 cells maintain potent inflammatory activity despite IL-2 sequestration by tumor- or tissue-resident T regulatory cells (17–19). Similarly, Th9 cells co-develop with IL-2/STAT5-inhibited Th17 cells in settings of chronic fungal- or house dust mite-driven allergic disease and maintain a strong Th9-associated gene signature in HDM allergic patients (20–22). Together, these data suggest that additional signals may be involved in promoting Th9 cell differentiation when IL-2 is limiting.
Heightened NF-kB activity is another hallmark of activated T cells and is thought to precede IL-2 production upon initial T cell priming. During the process of T cell activation, both canonical (p50/c-rel/p65) and non-canonical (p52/Relb) NF-kB-activating pathways are induced after ligation of co-stimulatory receptors (i.e. CD28) (23–27). Co-receptor ligation leads to the nuclear localization of NF-kB subunits where they pair with NFAT to drive the production of IL-2 and several other early activation-associated genes (27). During T helper cell differentiation, NF-kB generally promotes the differentiation of all Th cell lineages by cooperating with lineage-associated STAT factors and “master” TFs to activate gene transcription (27). In Th1, Th2 and Th17 differentiation programs, the canonical NF-kB pathway plays a predominant role (28–30) whereas Th9 cells exhibit activation of both canonical and non-canonical pathways (3). NF-kB activation is prolonged in Th9 cells as compared to other Th lineages and suggests a unique role for this pathway in Th9 differentiation (31). Indeed, enhancing the activation of NF-kB via cytokines (TNF, TL1A) or TNFR family members (OX40, GITR, DR3) augments Th9 cell differentiation and enhances Th9-mediated inflammatory potential (3, 32–34). Of note, many NF-kB-activating factors are often elevated in tumors and inflamed tissues where IL-2 availability is thought to be limiting (3, 35–38) and suggests that engagement of the NF-kB pathway may act to induce or maintain the Th9 phenotype in these IL-2-limited environments.
While both IL-2 signaling and NF-kB are induced by co-stimulatory signals and required for optimal Th9 cell differentiation, the interplay between IL-2 and NF-kB signaling in this process is not well understood. In this report, we established a critical link between IL-2/STAT5 signaling and the induction of the NF-kB transcriptional signature in differentiating Th9 cells. When Th9 cells were cultured in the absence of IL-2 or under IL-2-limiting conditions, we observed a coordinate loss of both IL-9 production and an NF-kB-targeted transcriptome, both of which could be rescued by addition of the exogenous NF-kB-activating cytokine IL-1β. Interestingly, IL-1β was unique among the NF-kB-activating factors we tested for its ability to rescue IL-9 production due to the specific upregulation of the IL-1 receptor (IL-1R1) in the absence of IL-2. IL-1β additionally repressed the expression of the Th9 inhibitory transcription factor BCL6 and ectopic BCL6 expression greatly diminished the IL-1β-mediated rescue of IL-9. These data implicate a feedback loop where IL-2-deprived Th9 cells upregulate IL-1R1 and enhance their capacity to respond to IL-1β. In turn, IL-1β-mediated activation of NF-kB limits the expression of the anti-Th9 factor BCL6 which results in enhanced IL-2 responsiveness and IL-9 production. This model addresses the seeming paradox in which IL-2 is required for in vitro Th9 differentiation, but Th9 cells are often found in inflamed tissues in vivo where IL-2 is limited.
Materials and methods
Mice
C57BL/6 and OT-II transgenic mice were originally acquired from Jackson laboratories and bred and housed in our AAALAC-accredited Purdue University animal housing facilities under a 12 hour on, 12 hour off light cycle. All mice received autoclaved T.2018SC.15 18% protein chow (Envigo) and autoclaved drinking water ad libitum. The animal protocol was established according to Purdue University Institutional Animal Care and Use Committee (IACUC) guidelines. The experiments done here were done in compliance with a Purdue Animal Care and Use Committee-approved protocol.
Naïve CD4 T cell isolation and cell culture
Spleens and lymph nodes from C57BL/6 and OT-II mice were mechanically dissociated with frosted slides to generate a single cell suspension for CD44-negative naïve CD4 T cell isolation, using the Naïve CD4+ T cell Isolation Kit (Miltenyi Biotec, Auburn, CA) according to manufacturer’s protocol. Naïve CD4 T cell (CD4+, CD44low, CD62Lhi) purity was >90% as measured by an Attune NxT Flow cytometer (Thermo Fisher Scientific). After isolation, a total of 1x106 cells/mL were cultured in a 1:1 combination of RPMI and DMEM supplemented with 1% penicillin/streptomycin (Life Technologies), 10% FBS, and 50 mM 2-ME (Life Technologies). The cells were cultured at 37°C for 4 days in plates coated with anti-CD3-(2μg/mL, 2C11, Bio X cell), with soluble aCD28 (4μg/mL, 37.51, Bio X cell), and the Th9-polarizing conditions: anti-IFNγ(10μg/mL), IL-4(20ng/mL), TGFβ(4ng/mL), anti-IL-10R(10μg/mL). IL-2 was neutralized using anti-IL-2 and anti-CD25 antibodies as previously published (16). In some conditions, we supplemented the cultures with NF-kB activators IL-1β (20ng/mL), IL-33 (20ng/mL), PAM3CSK4 (1μg/mL), DTA1 (5μg/mL) and OX40L (5μg/mL), and we used the NF-kB inhibitors QNZ (2nM, Cayman Chemical) and BAY 11-7082 (0.5μM, Cayman Chemical) with the respective dilution of DMSO as vehicle control. After 4 days in culture, we assessed cell number and cell viability in each well using trypan blue cell counting on a hemocytometer and the cytokine and TF expression through flow cytometry as previously described (16).
Intracellular cytokine and transcription factor staining
After culture, 1X106 cells/mL in each condition were used for intracellular cytokine staining (ICS) and/or transcription factor (TF) staining. For ICS, harvested differentiated Th9 cells were stimulated with PMA (0.5μg/mL) and ionomycin (0.5ug/ml, Sigma-Aldrich) for 2.5h at 37°C using U-bottom 96-well plates. After 2.5 hours, monensin (2μM final concentration, BioLegend) was added to each well for an additional 2.5-hour incubation. After that, the cells were collected and stained with fixable viability dye (eFluor780, Thermo Fisher Scientific) and mouse anti-CD4 antibody (RM4-5, 1μg/mL, Thermo Fisher Scientific), for retroviral transductions experiments the cells were also stained with anti-H2-Kk antibody (36-7-5, 5μg/mL, BioLegend). The cells were subsequently fixed with 3% formaldehyde for 15 min. After fixation, the cells were fixed again with True-Nuclear Fixation Buffer (BioLegend) for 45 min at room temperature and subsequently permeabilized with True-Nuclear Permeabilization Buffer (BioLegend) as per manufacturer’s instructions. The cells were then stained with antibodies targeting the cytokines: IL-9 (RM9A4, 1μg/mL, BioLegend) IL-17A (TC11-18H10.1, 1μg/mL, BioLegend) IL-2 (JES6-5II4, 1μg/mL BioLegend) and transcription factors BCL6 (K112-91, 1μL/test, BD Pharmingen), and IRF4 (IRF4.3E4, 0.5μg/ml, BioLegend) for 30 min, washed, and resuspended in FACS buffer for performing flow cytometry analyses.
Phospho-p65 staining
Naïve CD4 T cells were cultured as above using Th9 polarizing conditions with or without anti-IL-2/CD25. After culture, cells were collected and stained with anti-CD4 antibody (RM4-5, 1μg/mL, Thermo Fisher Scientific) and fixable viability dye (eFluor780, Thermo Fisher Scientific) for 15 minutes in 1xPBS at 4°C. Cells were washed with 200μL of PBS and fixed with 1.5% formaldehyde at room temperature for 10 minutes. Cells were washed with 1xPBS and treated with 100% ice-cold methanol for 1 hour at 4°C. Subsequently, the cells were pelleted at 1200xg for 10 min at 4°C, washed with 1xPBS and stained in FACS buffer for 1h at 4°C with Phospho-NF-kB p65(ser529) antibody (NfkBp65S529-H3, Invitrogen ref MA5-37414) as per manufacturer’s recommended amount per test (5μl/1X106 cells). Cells where then washed, resuspended in FACS buffer for flow cytometric analyses.
RNA isolation and qRT-PCR
After in vitro polarization in the conditions listed above, we isolated RNA using TRIzol™ Reagent (Thermo Fisher) as per manufacturer’s instructions. After quantifying RNA concentration using a NanoDrop™ ONE-W (Thermo Fisher Scientific) spectrophotometer, 1000ng of RNA were reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) and used for quantitative PCR analysis using theTaqMAN™ Fast Advanced Master Mix (Thermo Fisher Scientific) and the probes: Il9(Mm00433405_m1), Batf3(mM01318274_m1), Sgk1(Mm00441380_m1), Atf3(Mm00476033_m1), Csf1(Mm00432686_m1), Bcl6(Mm00477633_m1), Il1r1(Mm00434237_m1), Il1rap(Mm00492638_m1), Il1rl1(Mm00516117_m1), Tlr2(Mm01213946_g1), Tnfrsf4,(Mm00442039_m1), Tnfrsf18(Mm00437136_m1), (all from Thermo Fisher Scientific), and a Bio-rad CFX96 system as per manufacturer’s instructions. Relative expression analysis was performed using the 2-ΔΔct method and data were quantified and plotted using GraphPad Prism V9.4.0 software.
Ovalbumin-induced allergy in vivo model
Naïve CD4 T cells where isolated from OT-II TCR-transgenic mice as per above, and the resulting cells were cultured under Th9 conditions, with or without the addition of IL-1β and with or without IL-2 neutralization as mentioned above. At 4 days after culture, the cells were rested in media for 24 hours, followed by subsequent washing (2 washes with 10 mL sterile PBS), counted via hemocytometer and 0.5 X106 cells were retro-orbitally injected into lightly isoflurane-anesthetized C57BL/6 mice. At 24 hours post-adoptive transfer, the mice were treated intranasally with recombinant chicken ovalbumin protein (100 μg in 25μl of 1xPBS, Sigma Aldrich) and TSLP (200 ng in 25 μl of 1xPBS, Sino Biological) daily for 5 consecutive days. Mice treated with IL-9 neutralizing antibody (9C1, BioXcell) were treated daily with 100μg in 50μL of PBS before the OVA/TSLP challenge. Twenty-four hours after the last intranasal treatment, the mice were euthanized using cervical dislocation and dissected for lung tissue collection.
Lung digestion, cell isolation and stimulation
Lungs from OVA-induced allergy mice were perfused with 1X PBS and dissected. The lungs were placed in FACS tubes with digestion buffer (1.5mg/ml type III collagenase (Worthington), 100ng/mL DNase I, RPMI media and 10% Newborn Calf serum) and sliced in 2mm2 tissue pieces using dissecting scissors. Minced lung tissue was then digested for 50 minutes in a rotor incubator at 37°C. After digestion, the digested tissue was transferred to a 70μm nylon cell strainer (USA Scientific) to remove debris. The cell suspension was then treated with 155mM Ammonium Chloride, 12mN Sodium Bicarbonate, 0.1mM EDTA buffer to lyse red blood cells and mononuclear cells were enriched from this suspension via centrifugation (500xg for 10 min) through 40% Percoll (GE Healthcare). Pelleted cells were resuspended in complete RPMI media and counted before staining for the inflammatory cell markers: CD45(30-F11, 0.2μg/mL, BioLegend), CD3(17A2, 1μg/mL, BioLegend), CD19 (6D5, 1μg/mL, BioLegend), NK1.1(PK136 1μg/mL, BioLegend), Siglec-F (1RNM44N, 1 μg/mL, Invitrogen), Ly6G(RB6-8C5, 1μg/mL, BioLegend), CD11c(N418, 1 μg/mL, BioLegend), CD11b (M1/70, 1 μg/mL, BioLegend) for assessing immune cells infiltrating the lungs using flow cytometry. For stimulated panel: 1X106 cells were stimulated using PMA (0.5ng/mL) and ionomycin (0.5ug/ml, Sigma-Aldrich) for 4h at 37°C using U-bottom 96-well plates. After the 4h, monensin (2μM final concentration, BioLegend) was added to each well for an additional 2-hour incubation. After that, the cells were stained as per above using the surface markers: anti-CD90.2(53-2.1, 1.5μg/mL, BioLegend), anti-CD4(RM4-5, 1μg/mL, Thermo Fisher Scientific), anti-CD45(30-F11, 0.2μg/mL, BioLegend), anti-mouse-TCR Vα2 (B20.1, 1μg/mL, BioLegend), anti-mouse-TCR Vβ5 (MR9-4, 2μg/mL, BioLegend), fixable viability dye (eFluor780, Thermo Fisher Scientific) and intracellular anti-IL-9 (RM9A4, 1μg/mL, BioLegend). Stained cells were analyzed using an Attune NxT Flow cytometer (Thermo Fisher Scientific).
Histology
Lung tissue was collected on day 6 after adoptive cell transfer and OVA treatment and stored in buffered formalin (Formal-Fixx, Thermo Fisher Scientific) as per manufacturer’s instructions. Then the samples were transferred to 70% ethanol, and submitted to HistoWiz (https://home.histowiz.com) for tissue sectioning, Hematoxylin and Eosin (H&E) staining and imaging.
Retroviral transduction
The vector MSCV-H2-Kk (gifted by Mark Kaplan, Indiana School of Medicine) was utilized for generating viral particles for ectopic expression BCL6 as we have previously described (16). Briefly, Plat-E cells were transfected with 10μg of plasmid and their empty vector controls, and with 5μg of the packaging plasmid pCL-Eco (gifted by Dr. Mark Kaplan, Indiana School of Medicine) using Lipofectamine 2000 (Thermo Fisher Scientific) per the supplier’s instruction. At 48 hours post-transfection, the supernatant containing viral particles was harvested and cleared of cellular debris by centrifugation at 300xg for 10 minutes. Viral particle-containing supernatants were used for spin infection of 24 hour-activated Th9 cells in the presence of 8μg/ml of polybrene at 500xg for 1.5h at 37°C. Infected cells were harvested at 72 hours post-viral transduction for intracellular staining and flow cytometry as above.
RNA-seq analysis
The raw files of sequencing read are downloaded from GSE41317. Gene expression levels in each library were quantified by “rsem-calculate-expression” by RSEM v1.3.0 (39) with default parameters except “—bowtie-n 1 –bowtie-m 100 –seed-length 28”. The bowtie index (GRCm38) required by RSEM software was prepared by “rsem-prepare-reference” on all RefSeq genes, downloaded from UCSC table browser in April 2017. EdgeR v3.24.3 (40) package was utilized to identify differentially expressed genes (DEGs) between IL-2 knock-out and wildtype samples. Gene set enrichment analysis (GSEA) was performed using GSEA “investigate Gene Sets” (41) with top 500 differentially expressed genes (DEGs) in all hallmark gene sets.
Results
IL-2/STAT5 signaling is required for optimal NF-kB signaling in Th9 cells
We, and others, have previously demonstrated that IL-2 signaling and downstream STAT5 activation was required for the differentiation of Th9 cells and IL-9 production (15, 16, 42, 43). While STAT5 directly binds a number of conserved regions upstream of the Il9 locus, STAT5 also indirectly promotes the Th9 phenotype by suppressing a Th17-like phenotype and limiting expression of the repressive factor BCL6 (14, 15). However, these indirect mechanisms are incompletely understood. To address this, we initially assessed cellular pathways that were altered in WT and IL-2-deficient Th9 cells (15). Surprisingly, the top pathway that was altered in IL-2-deficient Th9 cells was related to NF-kB signaling, followed by IL-2/STAT5 signaling (Figure 1A). These data are meaningful as recent studies identified an important role for NF-kB in promoting IL-9 expression in various conditions and enhancing their inflammatory activity in vivo (3, 32–34). Consistent with this role in promoting the Th9 phenotype, the majority (64%) of genes within the 50 identified NF-kB signature exhibited reduced expression as compared to WT cells (i.e. Sgk1, Atf3, Batf3) whereas relatively fewer NF-kB-related genes were enhanced (i.e. Csf1) in the absence of IL-2 (Figures 1B, C). We validated these data via qPCR in our laboratory with Th9 cells that were cultured normally or under IL-2-limiting conditions (i.e. IL-2 and CD25 blockade) and observed significantly decreased expression of Il9, Sgk1, Atf3, and Batf3 when IL-2 was limiting (Figure 1D). In agreement with our RNA-seq analysis, we also observed significantly increased expression of Csf1 and Bcl6 after IL-2 blockade (Figures 1C, D). Canonical NF-kB transcriptional regulation is mediated by the p50/p65 complex (RelA) which, under inactive conditions, is sequestered in the cytoplasm by IkBa. Activation of NF-kB requires degradation of IkBα and translocation of p65/p50 to the nucleus, both processes require phosphorylation (44, 45). To test whether the activity of NF-kB was altered in IL-2-sufficient or IL-2-depleted Th9 cells, we detected phosphorylated p65 in the residue Serine 529 using flow cytometry, which has been associated with increased transcriptional activity of NF-kB (46–48).We observed that phospho-S529-p65 was significantly reduced in IL-2-deprived Th9 cells after 4 days of culture (Figure 1E) indicating that IL-2 signaling is required for the induction or maintenance of active NF-kB in differentiating Th9 cells. Together, these data indicate that IL-2 signaling is critical for the activity of NF-kB and the induction of its target genes in Th9 cells.
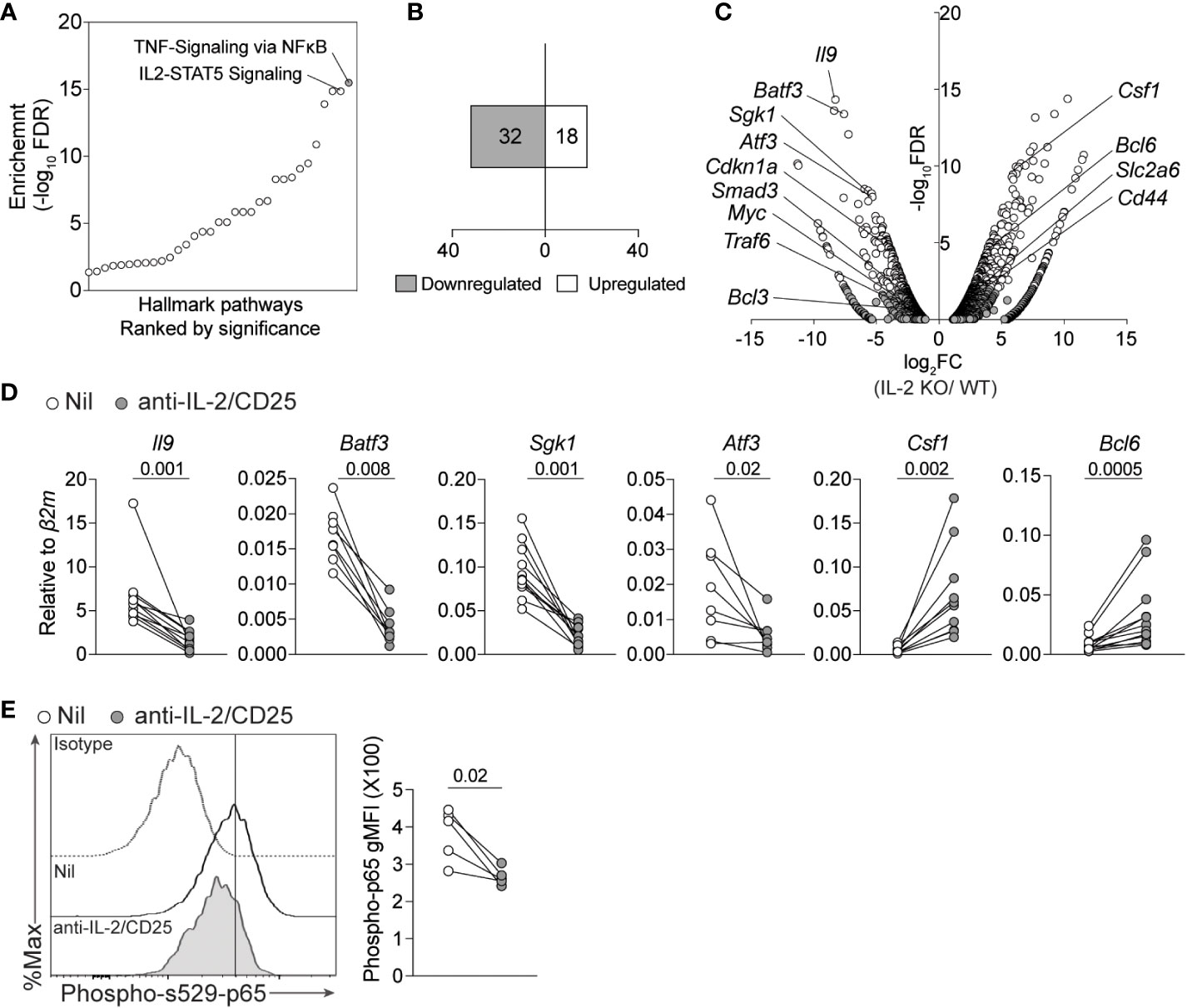
Figure 1 IL-2 deficiency results in a reduced NF-kB-associated gene signature in Th9 cells. (A) Pathway analysis from bulk-RNA-seq analysis of Th9 cells polarized from WT or IL-2-KO mice previously published (GSE41317). (B) Number of up- and down- regulated NF-kB signature genes (Fold change >2, p-value<0.05). (C) Volcano plot depicting selected genes regulated by NF-kB that are differentially expressed in IL-2 KO vs WT Th9 cells. (D) Th9 cells were cultured in standard or IL-2-limiting conditions, and 4 days after initial activation total RNA was isolated for qPCR validation of selected NF-kB-associated genes obtained from panel (A) Data in panel (D) are representative of naive T cell cultures from 8-11 individual mice pooled from 3 individual experiments. (E). Representative histograms and quantified geometric Mean Fluorescence Intensity (gMFI) of phospho- s529-p65 staining in cells cultured as in (D) Each data point represents values obtained from T cells isolated from one mouse, for a total of n=5 mice. Data were considered significantly different when paired t-test p value was ≤0.05.
IL-1β signaling rescues IL-9 production in IL-2 limiting conditions via activation of NF-kB
As IL-2 signaling was required for optimal induction of IL-9 and the NF-kB transcriptional signature, we hypothesized that restoration of NF-kB activity would rescue IL-9 production by these cells under IL-2 limiting conditions. To test this, we cultured naïve CD4 T cells under standard Th9 conditions with and without IL-2 blockade and further added NF-kB-activating cytokines (IL-1β, IL-33) or triggered the signaling of NF-kB-inducing receptors (TLR2 via PAM3CSK4, OX40 or GITR via crosslinking antibodies) that have been previously shown to enhance Th9 cell differentiation (3, 34, 49). Consistent with these previous reports, addition of IL-1β, IL-33 and crosslinking antibodies enhanced Th9 cell differentiation under IL-2 replete conditions. However, treatment with OX40 crosslinking antibodies only modestly enhanced IL-9 production and was not statistically significant (Figure 2A). Under IL-2-limiting conditions, only the addition of 10ng/ml of IL-1β significantly and consistently enhanced IL-9 production as compared to the untreated (e.g. Nil) control while all others NF-kB activators exhibited a limited capacity to rescue IL-9 production in these initial screening experiments (Figure 2A). For this reason, we have focused on IL-1β signaling for the remainder of our studies.
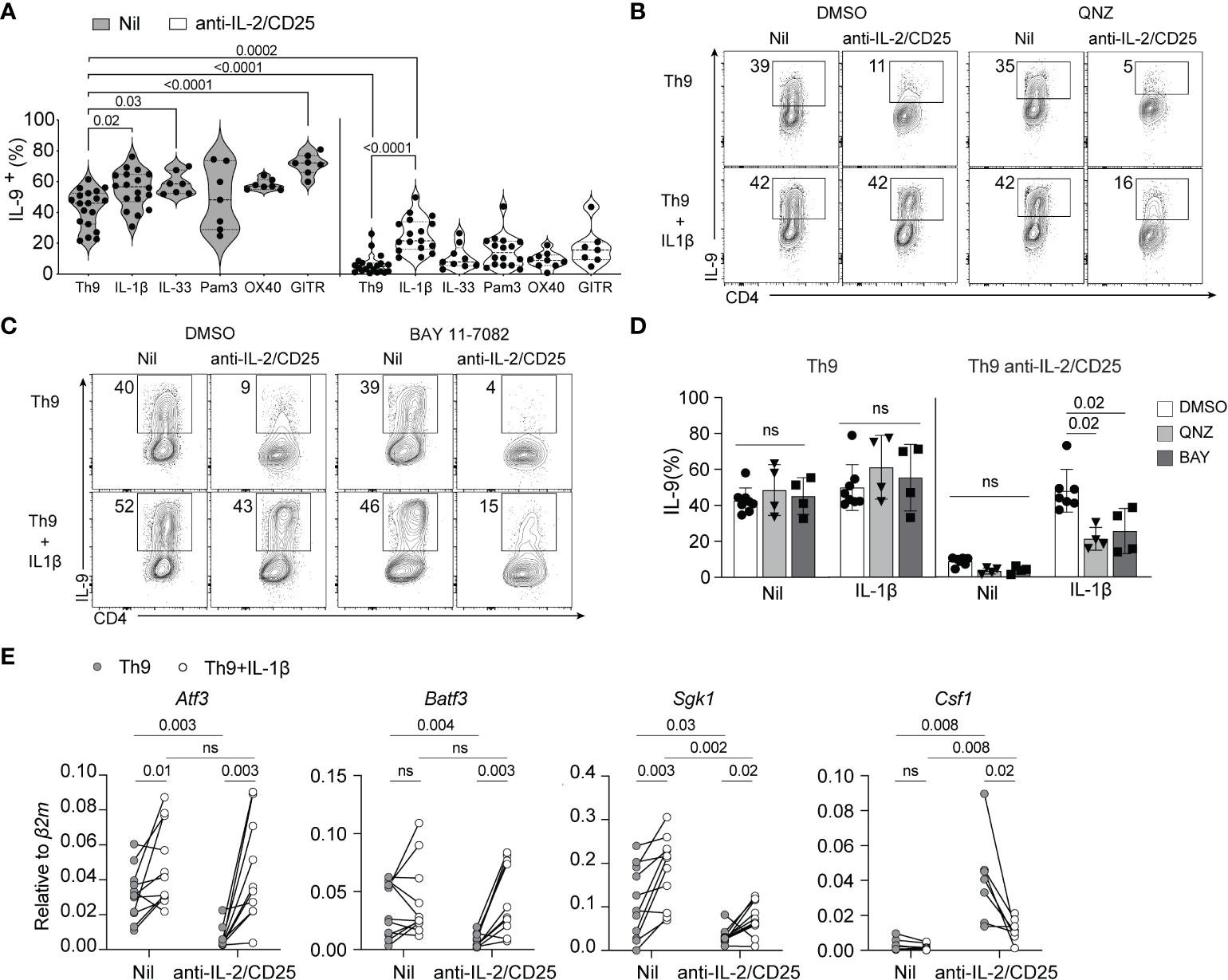
Figure 2 IL-1β-induced NF-kB rescues Th9 differentiation in IL-2-limiting conditions. (A) Th9 cells were cultured in standard or IL-2-limiting conditions in the presence of the indicated NF-kB-activating cell surface receptors and cytokines, and intracellular IL-9 was measured at day 5 of culture after restimulation of cells with PMA and ionomycin. Each data point represents cells from at least n=7 mice or more per group. (B, C) Representative contour plots of IL-9 production of Th9 and IL-2-deprived Th9 cells in the presence or absence of IL-1β and the NF-kB inhibitors QNZ (1nm) or BAY 11-7082 (2.5μM), depicting the NF-kB-dependency of IL-1β-mediated rescue of Th9 cell differentiation. (D) Quantification of IL-9+ Th cells in culture with NF-kB inhibitors, each data point represents naïve T cell cultures from individual mice n= 4 mice from drug-treated cells and n=7 for DMSO controls. (E) NF-kB-associated gene expression at day 5 of culture from non-restimulated Th9 and IL-2-deprived Th9 cells cultured in the presence or absence of IL-1β. Each point represents data from T cells isolated from at least 8 or more mice per group. Data are considered significant if p value ≤0.05 in a one-way ANOVA analysis with a Tukey post-test. Error bars represent standard deviation. ns, non-significant.
IL-1β binds its receptor, IL-1R1, and signals through MyD88 and TRAF6 which leads to the downstream activation of NF-kB and the MAPK pathway (50–55). As our RNA-seq analysis indicated that the NF-kB-associated transcriptional signature is affected by limited IL-2, we asked if NF-kB signaling was required for IL-1β-mediated rescue of IL-9 production. To this end, we cultured cells under standard or IL-2-limiting conditions as above in the presence or absence of an increased dose of IL-1β (20ng/ml, to enhance IL-9 rescue potential) and the NF-kB inhibitors QNZ or BAY110782 and assessed IL-9 production at the end of culture. Th9 cells cultured under IL-2 limiting conditions exhibited a similar reduction of IL-9 production and this was significantly rescued with addition of IL-1β in our vehicle control (DMSO)-treated cells now to standard Th9 levels, likely due to the increased amount of IL-1β used. Cells under IL-2 limiting conditions treated with IL-1β and QNZ or BAY117082 exhibited a 50% loss of IL-9 production, indicating that NF-kB activity was at least partially required for the rescuing effect of IL-1β (Figures 2B-D). Of note, the concentrations of drugs used in these studies was below that which has been published in other studies as we found higher inhibitor concentrations caused enhanced cell death under IL-2-limiting conditions. Therefore, the degree of NF-kB dependency we have measured here is likely underestimated.
IL-1β normalizes the expression of selected NF-kB genes in IL-2 limiting conditions
We showed above that IL-1β rescues IL-9 production in IL-2-limiting conditions, but how this impacts other NF-kB-regulated genes in the signature we identified remains unclear. To address this, we performed qPCR of select NF-kB-associated genes (Batf3, Atf3, Sgk1, and Csf1) that were differentially regulated in IL-2-deficient Th9 cells as depicted above (Figures 1C, D). IL-1β was able to normalize the expression of genes such as, Batf3, Atf3 and Csf1 (Figure 2D). However, the expression of Sgk1, was only marginally increased as compared to controls (Figure 2D). These data indicate that IL-1β, at least in part, normalizes the IL-2-dependent NF-kB gene signature in Th9 cells.
IL-1β rescues the in vivo inflammatory activity of Th9 cells
Given the role of Th9 cells during allergic inflammation (2), we asked whether IL-1β could rescue the inflammatory potential of Th9 cells differentiated under IL-2-limiting conditions. To this end, we employed an ovalbumin (OVA)-induced lung allergy model in which the adoptive transfer of ovalbumin-specific CD4 T cells (OT-II cells) and intranasal treatment with ovalbumin and TSLP results in the induction of lung allergy in a Th9-dependent manner as previously shown (13). In our experiments, OT-II cells were cultured under various Th9-polarizing conditions (Th9, Th9+IL-1β, Th9 anti-IL-2/CD25, Th9 anti-IL-2/CD25+IL-1β) and transferred intravenously (i.v.) prior to intranasal OVA exposure (Figure 3A). An additional group of mice received anti-IL-9 to determine the requirement of IL-9 for pulmonary inflammation. After OVA treatment, all OT-II Th9 recipient mice had similar numbers of Vα2/Vβ5.1+ OT-II cells (p>0.05 for all groups), indicating that the in vitro polarizing conditions had minimal impact on Th cell proliferation in vivo after OVA exposure (Figure 3B). Similar to our in vitro data, IL-2-deprived Th9 cells exhibited reduced capacity to produce IL-9 as compared to standard Th9 cells after in vivo intranasal OVA challenge. Again consistent with our previous findings, the frequency and total numbers of pulmonary IL-9-producing OT-II cells were increased in mice that received IL-1β-treated IL-2-deprived Th9 cells as compared to mice that received untreated IL-2-deprived cells (Figures 3C, D), indicating that IL-1β rescues the in vivo capacity of Th9 cells to maintain IL-9 production when primed in IL-2-limiting conditions. In terms of Th9-induced eosinophilic inflammation, mice receiving Th9 cells cultured under IL-2 limiting conditions had reduced pulmonary eosinophil numbers as compared to IL-2 replete Th9 and Th9+IL-1β controls (Figure 3E). In contrast, mice receiving Th9 anti-IL-2/CD25+IL-1β cells exhibited a fully rescued eosinophil response, and this effect was abrogated by in vivo neutralization of IL-9 (Figure 3E). This is consistent with the lung histology data obtained after hematoxylin and eosin (H&E) staining, which show that Th9 cells induce pulmonary inflammation as compared to PBS controls and this is similar in mice that received Th9 cells cultured with IL-1β (Figure 3F). These data likely suggest that our standard Th9 conditions already exhibit maximal NF-kB activity and that their in vivo inflammatory capacity is not enhanced by addition of exogenous IL-1β. In contrast, mice that received IL-2-deprived Th9 cells exhibited dramatically reduced airway pathology that was similar to PBS controls. Transfer of IL-2 deprived Th9 cells treated with IL-1β, however, had similar levels of pathology as compared to standard Th9 recipient mice. Importantly, the group of mice that received IL-1β-rescued Th9 cells with in vivo anti-IL-9 blockade exhibited reduced levels of inflammation (Figure 3F) indicating that both lung eosinophilia and airway inflammation are IL-9-dependent. Together, our data suggest that IL-1β restores the in vivo IL-9-dependent inflammatory capacity of IL-2-deprived Th9 cells.
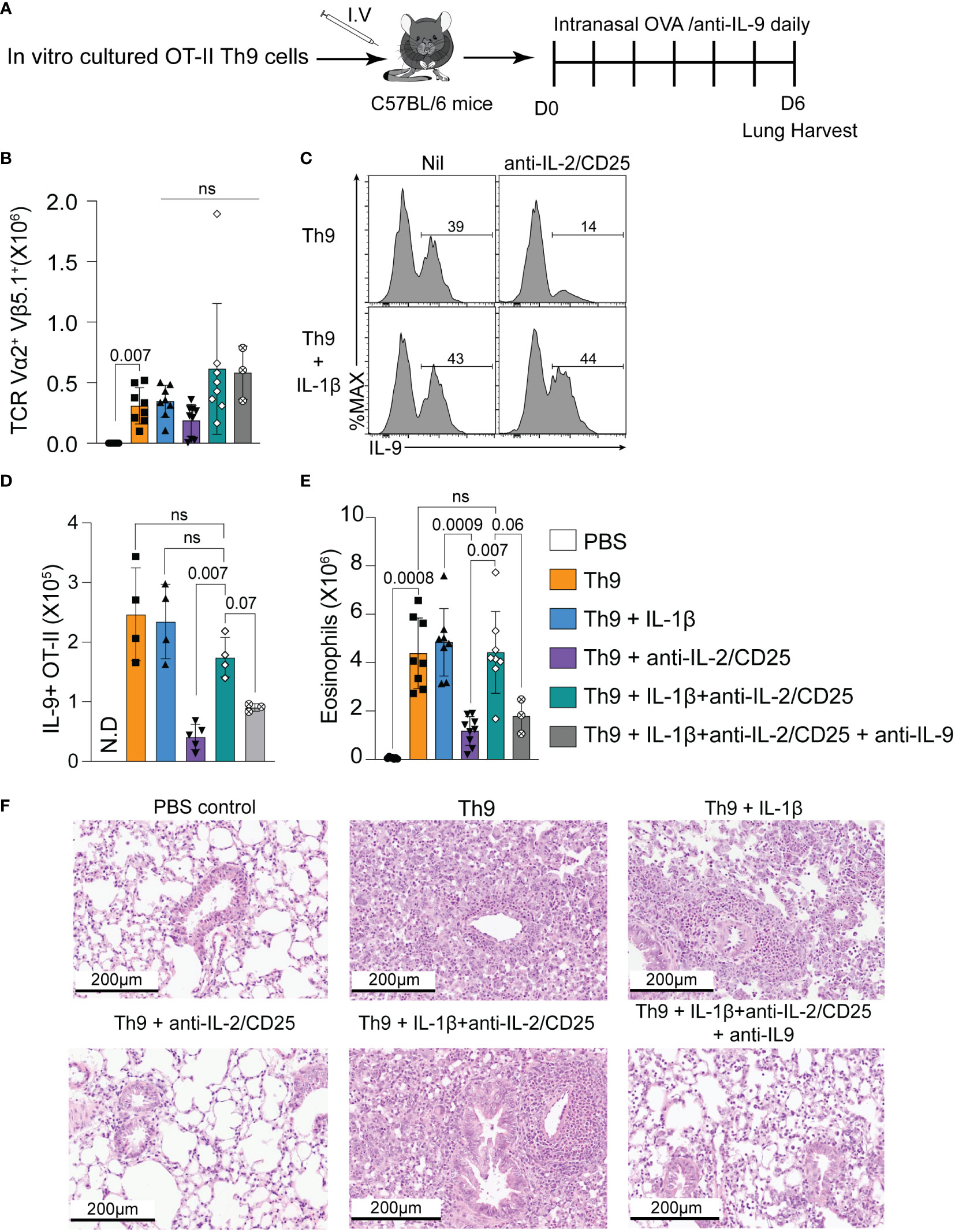
Figure 3 IL-1β rescues the in vivo IL-9-dependent inflammatory capacity of IL-2-deprived Th9 cells. (A) Schematic of the experimental design of the Ovalbumin (OVA)-induced allergy model after adoptive transfer of indicated Th9 cells. (B) Total number of Vα2/Vβ5+ OT-II CD4 T cells in the lung 24 hours post-final intranasal OVA exposure. (C) Representative histograms of IL-9 staining from Vα2/Vβ5+ OT-II CD4 T cells isolated from lungs, gating indicates percentage of OT-II cells expressing IL-9. (D) Total cell number of IL-9- producing Vα2/Vβ5+ OT-II CD4 T cells in each group. Each data point represents one mouse. n= 4 for Th9, Th9+ Il-1β, and Th9+ IL-1β anti-IL-2/CD25. n=5 for Th9+ anti-IL-2/CD25, and n=3 for Th9+ IL-1β anti-IL-2/CD25 + anti-IL-9. N.D: non determined (E) Total pulmonary eosinophil numbers in indicated recipient mice. Data are representative of 3-7 mice per group from 2-3 pooled experiments for every group except the anti-IL-9 treated mice. This group was only included in one experiment where there were three mice. (F) Representative H&E images of recipient mice lung tissue harvested on day 6 after OVA treatment and adoptive OT-II cell transfer. Data are considered significant if p value ≤0.05 in a one-way ANOVA analysis with a Tukey post-test. Error Bars indicate standard deviation. ns, non-significant.
IL-2/STAT5 represses IL-1R1 expression
IL-1β was uniquely able to rescue Th9 differentiation when IL-2 was limited as compared to other NF-kB-activating cytokines or receptors. As many of these receptors share common downstream signaling pathways (i.e., MyD88, TRAF6 and NF-kB), we reasoned that this may be due to differences in cytokine or TNF-family receptor expression under IL-2 limiting conditions. Indeed, we observed that mRNA expression of components of the IL-1β receptor (Il1r1 and Il1rap) was upregulated when IL-2 was limiting whereas the IL-33 receptor (Il1rl1), Tlr2, and GITR (Tnfrsf18) were unchanged in these conditions (Figure 4A). Interestingly, OX40 (Tnfrsf4) was significantly reduced, which potentially explains why it was not consistently able to rescue Th9 differentiation under these conditions (Figure 2). Importantly, we also observed enhanced cell surface protein expression of IL-1R1 when IL-2 was limiting (Figures 4B, C). These data together support a model where cells that differentiate in IL-2-limiting conditions upregulate IL-1R1 that, in turn, promotes Th9 differentiation via NF-kB.
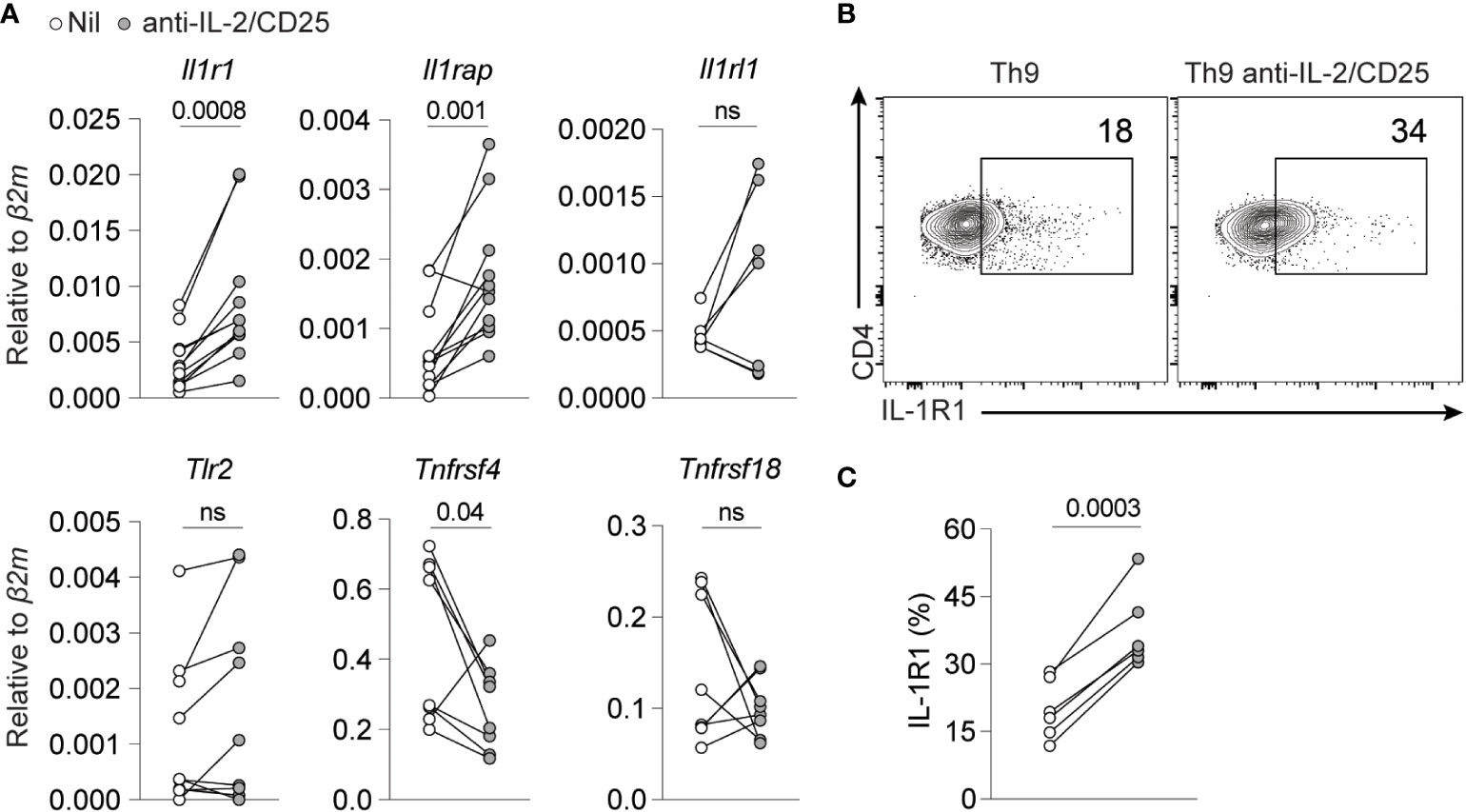
Figure 4 IL-2 limiting conditions enhance IL-1R1 expression on Th9 cells. (A) NF-KB-inducing cell surface and cytokine receptor gene expression in standard Th9 or IL-2-deprived Th9 cells. Each point represents data from one mouse. The combined data represent at least a n=7 mice. (B) Representative contour plots of cell surface IL-1R1 expression on Th9 and IL-2-deprived Th9 cells on day 5 of culture. (C) Quantification of IL-1R expression from panel (B) Data are representative of Th9 cultures from 6-7 mice pooled from 3 individual experiments. Data were considered significantly different from paired t-test at p value ≤0.05. ns, non-significant.
IL-1β enhances Th9 IL-2 responsiveness
IL-1 signaling has been previously shown to enhance IL-6/STAT3 signaling in Th17 cells by repressing suppressor of cytokine signaling (SOCS3) (56). SOCS3 has also been recently implicated in repressing IL-2 or IL-7-driven STAT5 signaling in Tregs (57). Based on these previous studies, we hypothesized that IL-1β may enhance Th9 IL-2 responsiveness via a similar mechanism. To test this, we initially titrated anti-IL-2 and anti-CD25 antibodies to generate culture conditions with decreasing degrees of IL-2 availability and subsequently measured IL-9 production from these cells. With standard Th9 conditions we observed an expected dose-dependent decrease in IL-9 (Figures 5A, B) and a dose-dependent increase in IL-17A with limiting IL-2 availability that was consistent with what we have previously shown (16) (Figure 5A). In Th9 cells cultured with IL-1β, we still observed a dose-dependent decrease in IL-9 production, however, these cells were more resistant to limited IL-2 availability (e.g. it required ~2-fold more IL-2/CD25 blocking antibodies to mitigate IL-9 production) than cells cultured without IL-1β (Figures 5A, B). We then asked if this enhanced resistance to limited IL-2 availability coincided with reduced SOCS3 expression and/or elevated phospho-STAT5 (pSTAT5). However, we did not observe changes in either Socs3 mRNA or pSTAT5 in the presence or absence of IL-1β in standard or IL-2-deprived Th9 conditions (Figures 5C, D). These data suggest that while IL-1β enhances the sensitivity of Th9 cells to limiting amounts of IL-2, this likely occurs through SOCS3-/pSTAT5-independent mechanisms.
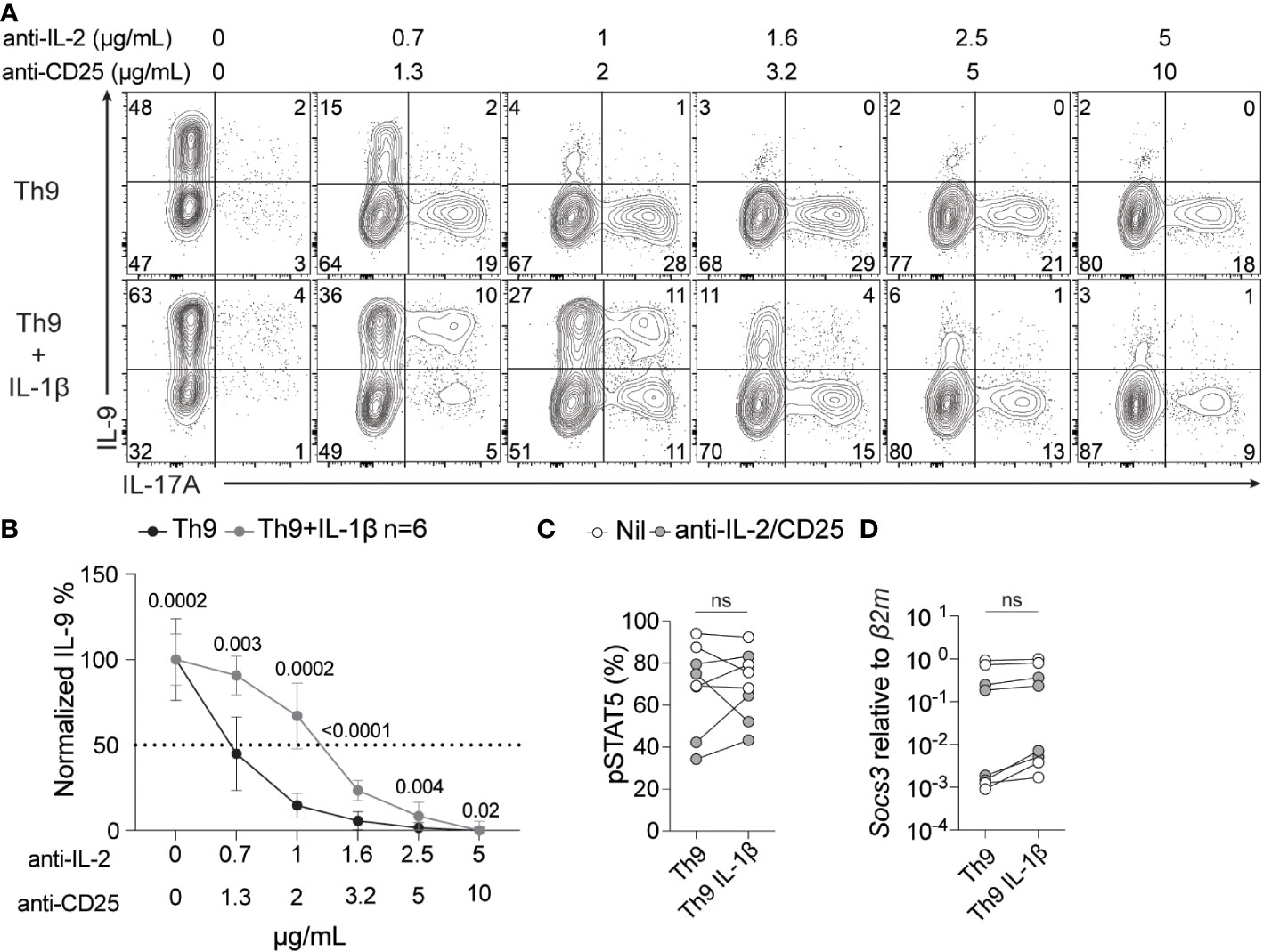
Figure 5 IL-1β signaling enhances IL-2 sensitivity during Th9 differentiation. Th9 cells were cultured in the presence or absence of IL-1β and increasing amounts of anti-IL-2/CD25 and IL-9 and IL-17A were measured by ICS at day 5 of culture. (A) Representative contour plots of intracellular IL-9 and IL-17A after restimulation with PMA and ionomycin. (B) Quantification of %IL-9+ Th cells at day 5 of culture from panel A, p<0.05 by paired Student’s t-test was considered significant, error bars represent SD. pSTAT5 protein (C) and Socs3 mRNA (D) levels of Th9 cells with or without IL-1β at no IL-2/CD25 blockade (Nil) or at 1μg/ml anti-IL-2/2μg/ml anti-CD25 (anti-IL-2/CD25). Data in panel A and B are representative of naïve T cell cultures from 6 mice and pSTAT5 and Socs3 data are representative of cultures from 4-6 mice from 2 pooled experiments. ns, non-significant.
IL-1β promotes Th9 differentiation in IL-2-limiting conditions by suppressing the expression of BCL6
Previous work has shown that BCL6 expression is repressed by IL-2/STAT5 signaling and BCL6 binding to the Il9 locus results in reduced Il9 expression and Th9 cell differentiation (14, 15). In line with these findings, we showed that Bcl6 mRNA was also elevated in Th9 cells cultured under IL-2-limiting conditions (Figure 1D). Here, we questioned whether IL-1β-mediated rescue of IL-9 production correlated with changes in BCL6 expression. As expected, BCL6 protein levels increased in Th9 cells cultured under IL-2 deprivation, validating our mRNA analyses (Figures 6A-C). Further, addition of IL-1β to IL-2-deprived Th9 cells resulted in a significant reduction of BCL6 mRNA and protein as compared to IL-2-deprived Th9 cells (Figures 6A-C). These data suggest that IL-1β suppresses BCL6 that would normally inhibit IL-9 production when IL-2 is limiting. To test this possibility, we asked if maintaining high levels of BCL6 expression in IL-1β-treated cells would prevent the rescue of Th9 differentiation. To this end, we transduced CD4 T cells with an empty vector retrovirus (EV RV) or BCL6-expressing retrovirus (Bcl6 RV) at 24 hours post-initiating Th9 anti-IL-2/CD25 cultures in the presence or absence of IL-1β. As expected, Th9 cells transduced with BCL6 RV had heightened BCL6 expression as compared to EV controls (Figure 6D) and reduced production of IL-9 in IL-2-limiting conditions (Figures 6E, F), validating the suppressive role of BCL6 in Th9 cell differentiation (14, 15). Interestingly, while IL-1β enhanced IL-9 production in EV RV-transduced cells, this was dramatically reduced in cells transduced with BCL6 RV (Figures 6E, F). Together, these data strongly suggest that IL-1β promotes IL-9 production through its repressive effects on BCL6.
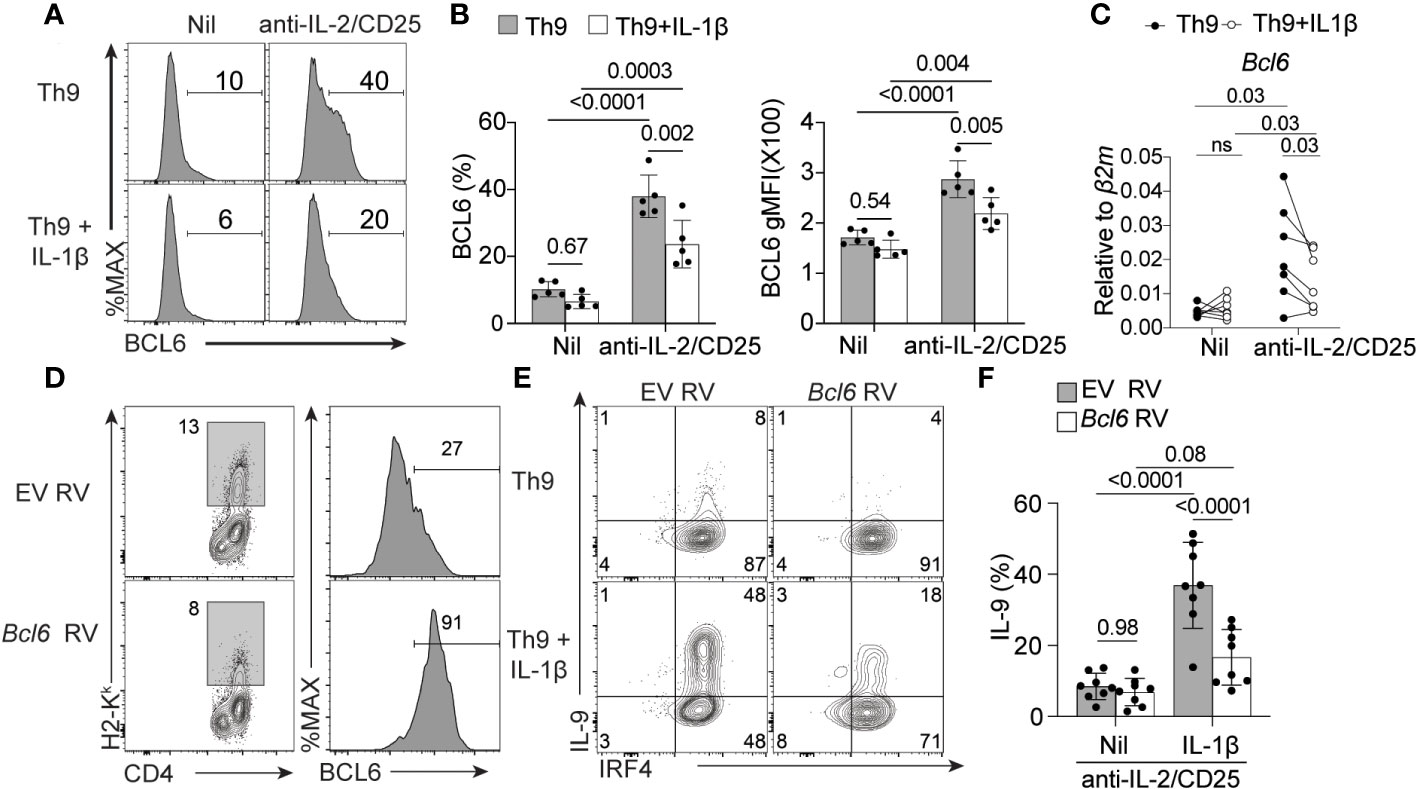
Figure 6 IL-1β reduces the expression of BCL6 to rescue IL-9 production in IL-2-limiitng conditions. (A) Representative histograms of BCL6 expression in Th9 cells with and without IL-2 deprivation and with and without IL-1β as determined by flow cytometry. (B) Quantification of %BCL6+ Th cells (left panel) and relative BCL6 protein expression (geometric mean fluorescence intensity (MFI), right panel).(C)Bcl6 mRNA relative expression in Th9 cells cultured as in panel (A). (D) Representative histograms depicting empty vector retrovirus (EV RV) or Bcl6-expressing retrovirus (Bcl6 RV) transduction efficiency of Th9 cells cultured under IL-2 limiting conditions and associated BCL6 protein expression from transduced H2-Kk+ cells). (E) Representative contour plots of intracellular IRF4 and IL-9 staining of IL-2-deprived Th9 cells in the presence or absence of IL-1β that have been transduced with empty vector or Bcl6-expressing retrovirus. (F) Quantification of data in panel (E) Data represent naïve T cell cultures from 5-8 mice per group from 2-3 individual experiments. Statistical significance (p<0.05) was determined by one-way ANOVA. Error bars represent SD. ns, non-significant.
Discussion
The in vitro differentiation of IL-9-producing Th9 cells requires IL-2/STAT5 signaling as IL-2 deficiency or IL-2 blockade results in a complete loss of IL-9 producing cells (15, 16, 43), and the induction of a population of IL-17-producing Th17-like cells (16). Paradoxically, previous work indicates that Th9 cells are often prevalent, or exert their functions, in inflammatory environments with poor IL-2 bioavailability (i.e. tumors, chronically inflamed tissues) and co-develop with IL-2-sequestering Tregs and IL-2-suppressed Th17 cells (17–22). These data indicate that other signals may promote the Th9 phenotype and function in IL-2-limiting environments in vivo.
In this work, we identify a novel circuit that acts to enhance or maintain Th9 cell function within IL-2-limited environments driven by the inflammatory cytokine IL-1β. In this circuit, we show that IL-2 deprivation results in a dramatic reduction of the Th9 NF-kB transcriptional signature and a reciprocal increase in the expression of the IL-1 receptor (IL-1R1) that enhances Th9 cell responsiveness to exogenous IL-1β. Reinvigoration of the NF-kB pathway in IL-2-deprived Th9 cells with IL-1β restores their capacity to produce IL-9 by repressing the expression of the Th9 inhibitory transcription factor BCL6. These data suggest that Th9 cells may compensate for limited IL-2/STAT5 signaling in inflamed tissues by enhancing their responsiveness to IL-1β.
Our data indicate that IL-2/STAT5 signaling is critical for inducing the NF-kB pathway during Th9 cell differentiation. However, how IL-2/STAT5 signaling interfaces with NF-kB activation is not well understood. Our current understanding of these pathways in T cells largely comes from studies interrogating NF-kB/IL-2 synergy in T cell activation after T cell receptor (TCR) engagement. Within this setting, TCR/CD28 engagement results in the downstream activation of NF-kB which then directly transactivates the IL-2 gene and induced T cell proliferation and differentiation (27). Examples of how IL-2 may impact NF-kB signaling, however, are more limited. Stimulation of human monocytes with IL-2 resulted in enhanced NF-kB DNA binding capacity (58) and IL-2-induced NK cell cytolytic activity was largely abrogated after treatment of NF-kB inhibitors (59, 60). These studies suggest that IL-2-mediated activation of NF-kB may be common between lymphocyte populations and conserved between mouse and human immune cells. Defining the mechanisms of this interaction will provide further insight into how IL-2 promotes cell proliferation and function, and our work suggests that NF-kB activity could potentially be compensated for by NF-kB-activating cytokines when IL-2 is limiting.
NF-kB-activating cellular receptors and cytokines are often elevated in inflamed tissues and have been previously shown to enhance Th9 cell differentiation and function. For example, the NF-kB-activating receptors OX40 and GITR drive the conversion of Tregs to the Th9 phenotype which is consistent with the role of these receptors in driving allergic disease and anti-tumor immunity (3, 34). Additionally, the NF-kB-inducing cytokines TNF, TL1A and IL-1β all promote Th9 cell differentiation and specifically enhance the anti-tumor efficacy of Th9 adoptive cellular therapy (ACT) (31–33, 61). Despite the fact that NF-kB-activating receptors and cytokines almost universally promote the Th9 phenotype, we found that only IL-1β was able to consistently rescue Th9 cell differentiation under IL-2 limiting conditions in vitro and IL-9-mediated allergic inflammation in vivo. Mechanistically, we showed that IL-2 limiting conditions enhanced expression of IL-1R1 and the co-receptor IL-1RAP at both the mRNA levels and IL-1R1 at the cell surface protein level, implicating that IL-2 deprivation uniquely enhances the sensitivity of Th9 cells to IL-1β. The differentiation of both T follicular helper cells (TFH) and Th17 cells is suppressed by IL-2/STAT5 signaling and correlates with high levels of IL-1R1 expression (56, 62). IL-1β/IL-1R1-signaling also enhances the capacity of these Th subsets to produce their signature functions or cytokines. For example, T follicular helper cells express minimal IL-2Rα (CD25) and high levels of IL-1R1 expression and treatment with exogenous IL-1β after protein vaccination enhances TFH function in mice (62). For Th17 cells, IL-1R1 is induced through IL-6 signaling (63), and we and others showed that IL-6/STAT3-signaling in Th cells results in diminished IL-2/STAT5 responsiveness (42, 64). Addition of exogenous IL-1β to Th cell cultures enhanced in vitro Th17 cell differentiation and IL-1R1-deficient T cells failed to induce Th17-driven disease in murine experimental autoimmune encephalomyelitis (EAE) (63). These findings suggest that IL-2-limiting conditions may universally promote IL-1R1 expression and enhance Th cell functionality to enhance protective immunity or drive disease.
Similar to IL-1β, IL-1α also signals through IL-1R1, induces NF-kB activation via MyD88 (65, 66) and enhances IL-9 production in standard Th9 cells (67). These findings suggest that IL-1α may also be able to rescue IL-9 production under IL-2 limiting conditions. However, in a pilot experiment, we found that IL-1α did not consistently rescue IL-9 production in IL-2-deprived Th9 cells (data not shown). These data suggest that IL-1β and IL-1α may have differing capacities to rescue IL-9 based on differences in their intracellular signaling. Previous work has shown that IL-1β and IL-1α interact differently with the IL-1R due to their differential lectin capacity, which may result in differences in cell signaling despite using the same receptor (68). Further, IL-1α and IL-1β exhibit divergent roles in several in vivo models of inflammation (66, 69, 70). Thus, it is possible that these related cytokines may also differentially regulate IL-9 production in IL-2 limiting conditions. However, the mechanisms that may regulate these differences are not directly forthcoming. Additional work is required to determine how these different IL-1 family members elicit distinct responses in this setting.
In the anti-tumor setting, culture of Th9 cells with IL-1β or cells with heightened NF-kB activity exhibited enhanced expression of the CD4 cytotoxic lymphocyte transcription factor Eomes and enhanced granzyme/perforin-associated killing after adoptive transfer (31, 61). In addition, IL-1β-treated Th9 cells also induced the expression of the transcription factor IRF1 which enhanced their capacity to produce both IL-9 and IL-21. Interestingly, anti-tumor activity of Th9 cells in these studies was dependent on IL-21, but not IL-9. In this tumor model, the enhanced anti-tumor effects of IL-1β-treated Th9 cells were indirect and required IL-21 priming of CD8 T cells and natural killer (NK) cells (51). In contrast, we found that IL-1β treatment rescued the in vivo inflammatory potential of Th9 cells in an allergic airway model of disease in an IL-9-dependent manner. These differences in IL-9 vs. IL-21-dependency likely stem from the model system used (i.e. allergic airway disease in our study vs. cancer), especially as CD8 T cells and NK cells are minimally involved in the Th9-mediated allergic response. Indeed, Th cell-derived IL-9 is critical for the induction of the allergic response through the recruitment of mast cells and eosinophils to the lung via an airway macrophage intermediate (71). These data suggest that IL-1β may differentially modulate Th9 activity during allergy (via maintaining IL-9 production) and cancer (by induction of IL-21).
Mechanistically, we show that IL-1β rescues the production of IL-9 by IL-2-deprived Th9 cells by repressing the expression of the Th9 inhibitory transcription factor BCL6. As BCL6 competes for STAT5 binding at the Il9 locus, our data suggest that IL-1β-mediated repression of BCL6 enhances the capacity of STAT5 to bind the locus when IL-2/STAT5 signaling is limited. However, how IL-1β mediates repression of BCL6 in these settings remains unclear. In macrophages, BCL6 is a suppressor of inflammatory activity where BCL6-deficient macrophages exhibit heightened and dysregulated cytokine production after stimulation via NF-kB activating receptors (i.e. LPS/TLR4) (72). Macrophage NF-kB and BCL6-induced regulomes intersected and exhibited potential antagonism where NF-kB activation limited the BCL6 regulome. Intriguingly, this may also be through direct suppression of BCL6 as NF-kB p65 directly bound several conserved non-coding regions surrounding the Bcl6 gene (72). While IL-1β-induced NF-kB suppresses BCL6 expression, BCL6 also appears to reciprocally suppress NF-kB activation. Macrophage BCL6 and its co-repressor binding is enriched surrounding NF-kB-regulated genes and disruption of BCL6/co-repressor interactions results in elevated inflammatory response gene expression (72, 73). Based on these data, it is intriguing to speculate that the reduced NF-kB signature that we observed in IL-2-deprived Th9 cells may be regulated by BCL6. These data suggest that BCL6 inhibitors may be beneficial in enhancing NF-kB signaling and Th9 function, especially in response to chronic antigens and in tumor draining lymph nodes tumors where Th cell BCL6 expression is elevated (74, 75).
Together, our data define an IL-1β-NF-kB-BCL6 axis that governs Th9 differentiation in IL-2-limiting conditions. As Th9 cells play important roles in auto- and anti-tumor immunity, our studies suggest that therapeutic targeting IL-1β/IL-1R1 signaling may be beneficial in dampening Th9-mediated inflammation or may be bolstered to enhance tumor clearance.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE41317.
Ethics statement
The animal study was reviewed and approved by Purdue Animal Care and Use Committee.
Author contributions
DC and MC performed the in vitro experiments. DC performed in vivo experiments. BY completed the bioinformatic analyses. DC, CC and ZI performed q-RT-PCR. NA and SP assisted with tissue preparation. DC analyzed the results. AD provided reagents and insight in BCL6 studies. MK and MO contributed to the writing of the paper and conceived the study. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by startup funds (Purdue University) and a Showalter Trust Award (41000747) to MO, and an NIGMS R35GM138283 to MK. DC was supported by a Purdue University Ross-Lynn Graduate Student Fellowship and a Purdue College of Science Bilsland Fellowship. BY was supported by the SIRG Graduate Research Assistantships Award from the Purdue University Center for Cancer Research, P30CA023168 and a Purdue College of Agriculture Bilsland Fellowship.
Acknowledgments
The authors would like to acknowledge Dr. Mark H. Kaplan for critical review of the manuscript and Ms. Tripti Bera for her technical assistance in these studies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Li J, Chen S, Xiao X, Zhao Y, Ding W, Li XC. IL-9 and Th9 cells in health and diseases–from tolerance to immunopathology. Cytokine Growth Factor Rev (2017) 37:47–55. doi: 10.1016/j.cytogfr.2017.07.004
2. Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol (2015) 15:295–307. doi: 10.1038/nri3824
3. Xiao X, Balasubramanian S, Liu W, Chu X, Wang H, Taparowsky EJ, et al. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nat Immunol (2012) 13:981–90. doi: 10.1038/ni.2390
4. Smith SE, Hoelzinger DB, Dominguez AL, Van Snick J, Lustgarten J. Signals through 4-1BB inhibit T regulatory cells by blocking IL-9 production enhancing antitumor responses. Cancer Immunol Immunother CII (2011) 60:1775–87. doi: 10.1007/s00262-011-1075-6
5. Licona-Limón P, Henao-Mejia J, Temann AU, Gagliani N, Licona-Limón I, Ishigame H, et al. Th9 cells drive host immunity against gastrointestinal worm infection. Immunity (2013) 39:744–57. doi: 10.1016/j.immuni.2013.07.020
6. Lu Y, Hong S, Li H, Park J, Hong B, Wang L, et al. Th9 cells promote antitumor immune responses. Vivo J Clin Invest (2012) 122:4160–71. doi: 10.1172/JCI65459
7. Xue G, Zheng N, Fang J, Jin G, Li X, Dotti G, et al. Adoptive cell therapy with tumor-specific Th9 cells induces viral mimicry to eliminate antigen-loss-variant tumor cells. Cancer Cell (2021) 39:1610–1622.e9. doi: 10.1016/j.ccell.2021.09.011
8. Zhao Y, Chu X, Chen J, Wang Y, Gao S, Jiang Y, et al. Dectin-1-activated dendritic cells trigger potent antitumour immunity through the induction of Th9 cells. Nat Commun (2016) 7:12368. doi: 10.1038/ncomms12368
9. Gerlach K, Hwang Y, Nikolaev A, Atreya R, Dornhoff H, Steiner S, et al. TH9 cells that express the transcription factor PU.1 drive T cell-mediated colitis via IL-9 receptor signaling in intestinal epithelial cells. Nat Immunol (2014) 15:676–86. doi: 10.1038/ni.2920
10. Sehra S, Yao W, Nguyen ET, Glosson-Byers NL, Akhtar N, Zhou B, et al. TH9 cells are required for tissue mast cell accumulation during allergic inflammation. J Allergy Clin Immunol (2015) 136:433–440.e1. doi: 10.1016/j.jaci.2015.01.021
11. Jones DM, Read KA, Oestreich KJ. Dynamic roles for IL-2-STAT5 signaling in effector and regulatory CD4+ T cell populations. J Immunol Baltim Md (2020) 205:1721–30. doi: 10.4049/jimmunol.2000612
12. Li P, Mitra S, Spolski R, Oh J, Liao W, Tang Z, et al. STAT5-mediated chromatin interactions in superenhancers activate IL-2 highly inducible genes: Functional dissection of the Il2ra gene locus. Proc Natl Acad Sci U.S.A. (2017) 114:12111–9. doi: 10.1073/pnas.1714019114
13. Fu Y, Wang J, Panangipalli G, Ulrich BJ, Koh B, Xu C, et al. STAT5 promotes accessibility and is required for BATF-mediated plasticity at the Il9 locus. Nat Commun (2020) 11:4882. doi: 10.1038/s41467-020-18648-6
14. Bassil R, Orent W, Olah M, Kurdi AT, Frangieh M, Buttrick T, et al. BCL6 controls Th9 cell development by repressing Il9 transcription. J Immunol Baltim Md (2014) 193:198–207. doi: 10.4049/jimmunol.1303184
15. Liao W, Spolski R, Li P, Du N, West EE, Ren M, et al. Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression. Proc Natl Acad Sci U.S.A. (2014) 111:3508–13. doi: 10.1073/pnas.1301138111
16. Canaria DA, Yan B, Clare MG, Zhang Z, Taylor GA, Boone DL, et al. STAT5 represses a STAT3-independent Th17-like program during Th9 cell differentiation. J Immunol (2021) 207:1265–74. doi: 10.4049/jimmunol.2100165
17. Busse D, de la Rosa M, Hobiger K, Thurley K, Flossdorf M, Scheffold A, et al. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc Natl Acad Sci U.S.A. (2010) 107:3058–63. doi: 10.1073/pnas.0812851107
18. Carmenate T, Ortíz Y, Enamorado M, García-Martínez K, Avellanet J, Moreno E, et al. Blocking IL-2 signal In vivo with an IL-2 antagonist reduces tumor growth through the control of regulatory T cells. J Immunol Baltim Md (2018) 200:3475–84. doi: 10.4049/jimmunol.1700433
19. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8:1353–62. doi: 10.1038/ni1536
20. Kerzerho J, Maazi H, Speak AO, Szely N, Lombardi V, Khoo B, et al. Programmed cell death ligand 2 regulates TH9 differentiation and induction of chronic airway hyperreactivity. J Allergy Clin Immunol (2013) 131:1048–57. doi: 10.1016/j.jaci.2012.09.027
21. Olson MR, Ulrich BJ, Hummel SA, Khan I, Meuris B, Cherukuri Y, et al. Paracrine IL-2 is required for optimal type 2 effector cytokine production. J Immunol Baltim Md 1950 (2017) 198:4352–9. doi: 10.4049/jimmunol.1601792
22. Seumois G, Ramírez-Suástegui C, Schmiedel BJ, Liang S, Peters B, Sette A, et al. Single-cell transcriptomic analysis of allergen-specific T cells in allergy and asthma. Sci Immunol (2020) 5:eaba6087. doi: 10.1126/sciimmunol.aba6087
23. Rao S, Gerondakis S, Woltring D, Shannon MF. C-rel is required for chromatin remodeling across the IL-2 gene promoter. J Immunol Baltim Md 1950 (2003) 170:3724–31. doi: 10.4049/jimmunol.170.7.3724
24. Chen X, Wang J, Woltring D, Gerondakis S, Shannon MF. Histone dynamics on the interleukin-2 gene in response to T-cell activation. Mol Cell Biol (2005) 25:3209–19. doi: 10.1128/MCB.25.8.3209-3219.2005
25. Gerondakis S, Siebenlist U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb Perspect Biol 2:1–29. doi: 10.1101/cshperspect.a000182
27. Oh H, Ghosh S. NF-κB: roles and regulation in different CD4(+) T-cell subsets. Immunol Rev (2013) 252:41–51. doi: 10.1111/imr.12033
28. Aronica MA, Mora AL, Mitchell DB, Finn PW, Johnson JE, Sheller JR, et al. Preferential role for NF-kappa B/Rel signaling in the type 1 but not type 2 T cell-dependent immune response in vivo. J Immunol Baltim Md (1999) 163:5116–24.
29. Das J, Chen CH, Yang L, Cohn L, Ray P, Ray A. A critical role for NF-kappa b in GATA3 expression and TH2 differentiation in allergic airway inflammation. Nat Immunol (2001) 2:45–50. doi: 10.1038/83158
30. Ruan Q, Kameswaran V, Zhang Y, Zheng S, Sun J, Wang J, et al. The Th17 immune response is controlled by the rel-RORγ-RORγ T transcriptional axis. J Exp Med (2011) 208:2321–33. doi: 10.1084/jem.20110462
31. Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, et al. Th9 cells represent a unique subset of CD4+ T cells endowed with the ability to eradicate advanced tumors. Cancer Cell (2018) 33:1048–1060.e7. doi: 10.1016/j.ccell.2018.05.004
32. Jiang Y, Chen J, Bi E, Zhao Y, Qin T, Wang Y, et al. TNF-α enhances Th9 cell differentiation and antitumor immunity via TNFR2-dependent pathways. J Immunother Cancer (2019) 7:28. doi: 10.1186/s40425-018-0494-8
33. Richard AC, Tan C, Hawley ET, Gomez-Rodriguez J, Goswami R, Yang X-P, et al. The TNF-family ligand TL1A and its receptor DR3 promote T cell-mediated allergic immunopathology by enhancing differentiation and pathogenicity of IL-9-producing T cells. J Immunol Baltim Md (2015) 194:3567–82. doi: 10.4049/jimmunol.1401220
34. Xiao X, Shi X, Fan Y, Zhang X, Wu M, Lan P, et al. GITR subverts Foxp3(+) tregs to boost Th9 immunity through regulation of histone acetylation. Nat Commun (2015) 6:8266. doi: 10.1038/ncomms9266
35. Besnard A-G, Togbe D, Couillin I, Tan Z, Zheng SG, Erard F, et al. Inflammasome-IL-1-Th17 response in allergic lung inflammation. J Mol Cell Biol (2012) 4:3–10. doi: 10.1093/jmcb/mjr042
36. Gracias DT, Sethi GS, Mehta AK, Miki H, Gupta RK, Yagita H, et al. Combination blockade of OX40L and CD30L inhibits allergen-driven memory TH2 cell reactivity and lung inflammation. J Allergy Clin Immunol (2021) 147:2316–29. doi: 10.1016/j.jaci.2020.10.037
37. Mantovani A, Barajon I, Garlanda C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol Rev (2018) 281:57–61. doi: 10.1111/imr.12614
38. Sousa AR, Lane SJ, Nakhosteen JA, Lee TH, Poston RN. Expression of interleukin-1 beta (IL-1beta) and interleukin-1 receptor antagonist (IL-1ra) on asthmatic bronchial epithelium. Am J Respir Crit Care Med (1996) 154:1061–6. doi: 10.1164/ajrccm.154.4.8887608
39. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinf (2011) 12:323. doi: 10.1186/1471-2105-12-323
40. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinform Oxf Engl (2010) 26:139–40. doi: 10.1093/bioinformatics/btp616
41. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U.S.A. (2005) 102:15545–50. doi: 10.1073/pnas.0506580102
42. Olson MR, Verdan FF, Hufford MM, Dent AL, Kaplan MH. STAT3 impairs STAT5 activation in the development of IL-9-Secreting T cells. J Immunol Baltim Md (2016) 196:3297–304. doi: 10.4049/jimmunol.1501801
43. Schmitt E, Germann T, Goedert S, Hoehn P, Huels C, Koelsch S, et al. IL-9 production of naive CD4+ T cells depends on IL-2, is synergistically enhanced by a combination of TGF-beta and IL-4, and is inhibited by IFN-gamma. J Immunol Baltim Md (1994) 153:3989–96.
44. Wang D, Westerheide SD, Hanson JL, Baldwin AS. Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem (2000) 275:32592–7. doi: 10.1074/jbc.M001358200
45. Jefferies C, Bowie A, Brady G, Cooke EL, Li X, O’Neill LA. Transactivation by the p65 subunit of NF-kappaB in response to interleukin-1 (IL-1) involves MyD88, IL-1 receptor-associated kinase 1, TRAF-6, and Rac1. Mol Cell Biol (2001) 21:4544–52. doi: 10.1128/MCB.21.14.4544-4552.2001
46. Wang D, Baldwin AS. Activation of nuclear factor-kappaB-dependent transcription by tumor necrosis factor-alpha is mediated through phosphorylation of RelA/p65 on serine 529. J Biol Chem (1998) 273:29411–6. doi: 10.1074/jbc.273.45.29411
47. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol (2011) 12:695–708. doi: 10.1038/ni.2065
48. Maguire O, O’Loughlin K, Minderman H. Simultaneous assessment of NF-κB/p65 phosphorylation and nuclear localization using imaging flow cytometry. J Immunol Methods (2015) 423:3–11. doi: 10.1016/j.jim.2015.03.018
49. Karim AF, Reba SM, Li Q, Boom WH, Rojas RE. Toll like receptor 2 engagement on CD4+ T cells promotes TH9 differentiation and function. Eur J Immunol (2017) 47:1513–24. doi: 10.1002/eji.201646846
50. Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, et al. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity (1998) 9:143–50. doi: 10.1016/s1074-7613(00)80596-8
51. Végran F, Berger H, Boidot R, Mignot G, Bruchard M, Dosset M, et al. The transcription factor IRF1 dictates the IL-21-dependent anticancer functions of TH9 cells. Nat Immunol (2014) 15:758–66. doi: 10.1038/ni.2925
52. Kulawik A, Engesser R, Ehlting C, Raue A, Albrecht U, Hahn B, et al. IL-1β-induced and p38MAPK-dependent activation of the mitogen-activated protein kinase-activated protein kinase 2 (MK2) in hepatocytes: Signal transduction with robust and concentration-independent signal amplification. J Biol Chem (2017) 292:6291–302. doi: 10.1074/jbc.M117.775023
53. Kuno K, Matsushima K. The IL-1 receptor signaling pathway. J Leukoc Biol (1994) 56:542–7. doi: 10.1002/jlb.56.5.542
54. Jain A, Song R, Wakeland EK, Pasare C. T Cell-intrinsic IL-1R signaling licenses effector cytokine production by memory CD4 T cells. Nat Commun (2018) 9:3185. doi: 10.1038/s41467-018-05489-7
55. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) pathway. Sci Signal (2010) 3:cm1. doi: 10.1126/scisignal.3105cm1
56. Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, et al. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat Immunol (2015) 16:286–95. doi: 10.1038/ni.3099
57. Luckey MA, Kim T-H, Prakhar P, Keller HR, Crossman A, Choi S, et al. SOCS3 is a suppressor of γc cytokine signaling and constrains generation of murine Foxp3+ regulatory T cells. Eur J Immunol (2020) 50:986–99. doi: 10.1002/eji.201948307
58. Brach MA, Gruss HJ, Riedel D, Mertelsmann R, Herrmann F. Activation of NF-kappa b by interleukin 2 in human blood monocytes. Cell Growth Differ Mol Biol J Am Assoc Cancer Res (1992) 3:421–7.
59. Valle Blázquez M, Luque I, Collantes E, Aranda E, Solana R, Peña J, et al. Cellular redox status influences both cytotoxic and NF-kappa b activation in natural killer cells. Immunology (1997) 90:455–60. doi: 10.1111/j.1365-2567.1997.00455.x
60. Zhou J, Zhang J, Lichtenheld MG, Meadows GG. A role for NF-kappa b activation in perforin expression of NK cells upon IL-2 receptor signaling. J Immunol Baltim Md 1950 (2002) 169:1319–25. doi: 10.4049/jimmunol.169.3.1319
61. Xue G, Jin G, Fang J, Lu Y. IL-4 together with IL-1β induces antitumor Th9 cell differentiation in the absence of TGF-β signaling. Nat Commun (2019) 10:1376. doi: 10.1038/s41467-019-09401-9
62. Ritvo P-GG, Churlaud G, Quiniou V, Florez L, Brimaud F, Fourcade G, et al. Tfr cells lack IL-2Rα but express decoy IL-1R2 and IL-1Ra and suppress the IL-1-dependent activation of tfh cells. Sci Immunol (2017) 2:eaan0368. doi: 10.1126/sciimmunol.aan0368
63. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity (2009) 30:576–87. doi: 10.1016/j.immuni.2009.02.007
64. Oh H-M, Yu C-R, Golestaneh N, Amadi-Obi A, Lee YS, Eseonu A, et al. STAT3 protein promotes T-cell survival and inhibits interleukin-2 production through up-regulation of class O forkhead transcription factors. J Biol Chem (2011) 286:30888–97. doi: 10.1074/jbc.M111.253500
65. Voronov E, Dotan S, Krelin Y, Song X, Elkabets M, Carmi Y, et al. Unique versus redundant functions of IL-1α and IL-1β in the tumor microenvironment. Front Immunol (2013) 4:177. doi: 10.3389/fimmu.2013.00177
66. Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, et al. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol Baltim Md (2011) 187:4835–43. doi: 10.4049/jimmunol.1102048
67. Uyttenhove C, Brombacher F, Van Snick J. TGF-β interactions with IL-1 family members trigger IL-4-independent IL-9 production by mouse CD4(+) T cells. Eur J Immunol (2010) 40:2230–5. doi: 10.1002/eji.200940281
68. Vergoten G, Zanetta J-P. Structural differences between the putative carbohydrate-recognition domains of human IL-1 alpha, IL-1 beta and IL-1 receptor antagonist obtained by in silico modeling. Glycoconj J (2007) 24:183–93. doi: 10.1007/s10719-006-9021-0
69. Eislmayr K, Bestehorn A, Morelli L, Borroni M, Walle LV, Lamkanfi M, et al. Nonredundancy of IL-1α and IL-1β is defined by distinct regulation of tissues orchestrating resistance versus tolerance to infection. Sci Adv (2022) 8:eabj7293. doi: 10.1126/sciadv.abj7293
70. Benjamin JT, Moore DJ, Bennett C, van der Meer R, Royce A, Loveland R, et al. Cutting edge: IL-1α and not IL-1β drives IL-1R1-Dependent neonatal murine sepsis lethality. J Immunol Baltim Md (2018) 201:2873–8. doi: 10.4049/jimmunol.1801089
71. Fu Y, Wang J, Zhou B, Pajulas A, Gao H, Ramdas B, et al. An IL-9-pulmonary macrophage axis defines the allergic lung inflammatory environment. Sci Immunol (2022) 7:eabi9768. doi: 10.1126/sciimmunol.abi9768
72. Barish GD, Yu RT, Karunasiri M, Ocampo CB, Dixon J, Benner C, et al. Bcl-6 and NF-kappaB cistromes mediate opposing regulation of the innate immune response. Genes Dev (2010) 24:2760–5. doi: 10.1101/gad.1998010
73. Barish GD, Yu RT, Karunasiri MS, Becerra D, Kim J, Tseng TW, et al. The Bcl6-SMRT/NCoR cistrome represses inflammation to attenuate atherosclerosis. Cell Metab (2012) 15:554–62. doi: 10.1016/j.cmet.2012.02.012
74. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med (2011) 208:987–99. doi: 10.1084/jem.20101773
Keywords: Th cell differentiation, IL-9 cytokine, IL-2 signaling, nuclear factor-kB, Th9 cells
Citation: Canaria DA, Clare MG, Yan B, Campbell CB, Ismaio ZA, Anderson NL, Park S, Dent AL, Kazemian M and Olson MR (2022) IL-1β promotes IL-9-producing Th cell differentiation in IL-2-limiting conditions through the inhibition of BCL6. Front. Immunol. 13:1032618. doi: 10.3389/fimmu.2022.1032618
Received: 31 August 2022; Accepted: 19 October 2022;
Published: 01 November 2022.
Edited by:
Demin Wang, Versiti Blood Research Institute, United StatesReviewed by:
Haiyan Liu, National University of Singapore, SingaporeDebra K. Newman, Versiti Blood Research Institute, United States
Copyright © 2022 Canaria, Clare, Yan, Campbell, Ismaio, Anderson, Park, Dent, Kazemian and Olson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Matthew R. Olson, b2xzb24xMjZAcHVyZHVlLmVkdQ==