 Rui Mao
Rui Mao Wanqing Kong2
Wanqing Kong2 Yukai He
Yukai He- 1Georgia Cancer Center, Medical College of Georgia, Augusta University, Augusta, GA, United States
- 2South Carolina Governors School for Science and Math, Hartsville, SC, United States
- 3Department of Medicine, Medical College of Georgia, Augusta University, Augusta, GA, United States
The overall efficacy of chimeric antigen receptor modified T cells (CARTs) remain limited in solid tumors despite intensive studies that aim at targeting multiple antigens, enhancing migration, reducing tonic signaling, and improving tumor microenvironment. On the other hand, how the affinity and engaging kinetics of antigen-binding domain (ABD) affects the CART’s efficacy has not been carefully investigated. In this article, we first analyzed 38 published solid tumor CART trials and correlated the response rate to their ABD affinity. Not surprisingly, majority (25 trials) of the CARTs utilized high-affinity ABDs, but generated merely 5.7% response rate. In contrast, 35% of the patients treated with the CARTs built from moderate-affinity ABDs had clinical responses. Thus, CARTs with moderate-affinity ABDs not only have less off-target toxicity, but also are more effective. We then reviewed the effects of ABD affinity on the biology and function of CARTs, providing further evidence that moderate-affinity ABDs may be better in CART development. In the end, we propose that a fast-on/fast-off (high Kon and Koff) kinetics of CART-target engagement in solid tumor allow CARTs to generate sufficient signaling to kill tumor cells without being driven to exhaustion. We believe that studying the ABD affinity and the kinetics of CART-tumor interaction may hold a key to designing effective CARTs for solid tumors.
Introduction
Immunotherapy is now the 4th pillar of cancer treatment (1, 2), and its efficacy relies on the tumor-infiltrating T cells (3), which, unfortunately, many solid tumors do not have (4). Engineering patients T cells with a T cell receptor (TCR) (5–8) or chimeric antigen receptor (CAR) (9) provides the much-needed tumor-specific T cells. CAR combines the antibody specificity and TCR signaling apparatus, which can activate T cells upon engaging with tumor surface antigen (9). The CAR-modified T cells (CARTs) thus recognize and kill tumor cells independent of MHC that is frequently downregulated, a common cause of tumor escape. CARTs have generated remarkable antitumor responses in treating hematological cancers (9–12), which results in 7 FDA-approved CARTs (13), but also ignites tremendous effort to develop solid tumor CARTs (14, 15). However, despite intensive studies, by far, the clinical efficacy of solid tumor CARTs remains limited (13, 16–18). A meta-analysis of 22 solid tumor CART trials (268 patients) reveals merely ~9% response rate (19). Evidently, the current CARTs do not work well for solid tumors. However, since 90% of cancers are solid tumors (20), investigators have been diligently working on and looking forward to a breakthrough in designing effective solid tumor CARTs.
To generate antitumor effects, CARTs need to migrate into a solid tumor mass, undergo antigen-driven activation and expansion, exert their effector function on target cells, persist sufficiently long enough to eradicate the entire tumor mass, and then form immune memory to monitor and prevent tumor relapse. Several excellent reviews (18, 21–23) have discussed the multiple strategies to improve each of these steps in the hopes of enhancing the efficacy of solid tumor CARTs. These approaches include 1) targeting multiple antigens to prevent tumor escape and to avoid off-tumor toxicity, 2) enhancing CART trafficking and infiltration into solid tumors (24–28), 3) improving the tumor microenvironment (TME) (29, 30). In addition, scientists have been studying strategies to improve CART fitness and persistence by selecting proper T cell subset (31), by reducing tonic signaling (such as utilizing 4-1BB (32) or single ITAM CD28 (33) as co-stimulatory domain), and by co-expressing C-Jun (34) or constitutive STAT5 (35). Furthermore, CAR expression under an inducible promoter (36, 37) or with a SynNotch switch (38) could diminish tonic signaling and exhaustion. These efforts have resulted in some incremental improvement. However, the overall efficacy of solid tumor CARTs still remains low. Thus, it is imperative to explore and study other components in the CAR in order to improve the efficacy of solid tumor CARTs.
A typical CAR is composed of the antigen-binding domain (ABD), hinge and transmembrane domain (TM), and intracellular signaling domain that normally consists of 1-2 co-stimulatory domains (CD) and the ζ chain (13). Each component contributes to the CART’s function and antitumor efficacy (39, 40). For example, the CD plays an important role in CART activation and persistence, and the consensus view is that CD28 generates stronger CART activation, but the 4-1BB CD renders CARTs longer persistence (32, 41). On the other hand, although the ABD is critical by rendering CAR specificity, how ABD affinity affects the activation and expansion, survival, and persistence of CARTs remained largely unknown until recently. In addition, the effect of ABD affinity on clinical efficacy of CARTs has not been studied. In this article, we will first review the response rate of solid tumor CART trials and correlate their efficacy to ABD affinity. Then, we analyze the effect of ABD affinity on CART biology and function, including activation, expansion, function, and exhaustion. In the end, we propose that CARTs with moderate-affinity ABD and fast-on/fast-off “fly-kiss” engaging kinetics will likely generate better effects in treating solid tumors.
Correlation of ABD affinity and clinical efficacy of CARTs: Moderate is better
The ABD, most of which are the single chain variable fragment (scFv) of monoclonal antibodies (mAbs), allows CARTs to specifically bind and kill tumor cells. However, thus far, the ABD affinity was not rationally considered in most CAR designs. This is reflected by the incomplete data of ABDs, the lack of Kon and KOff, or inconsistent use of KD (dissociation constant) and EC50 (half-maximal effective concentration), which will be discussed later in detail. For clarity, we refer to mAb binding strength as “affinity” (single pair of molecules) and the CAR or CART binding strength as “avidity” (multiple pairs of molecules). Over last few decades, affinity enhancement has been the main goal in antibody drug development. Approximately 100 therapeutic mAbs have been approved by the FDA (42), and many are high-affinity. Naturally, these clinically safe high-affinity mAbs, such as Cetuximab (KD=1.8nM) (43) and Herceptin (KD=5nM) (44), were used to create CARTs. High-affinity mAbs are preferred for CAR construction also because they may induce strong T cell activation and detect low levels of antigen (45–47). However, it is unclear how the ABD affinity affects the antitumor efficacy of solid tumor CARTs. Recently, we analyzed 14 solid tumor CART trials and found a trend that moderate affinity ABDs correlate to better efficacy (13). Thus, our first goal in this article is to verify the correlation of ABD affinity and clinical efficacy by expanding analysis to more solid tumor CART trials.
Based on the latest counting, there are 292 solid tumor CART trials in the world (48, 49), most of them are Phase I studies and have not completed. We were able to find 38 published solid tumor CART trials (total 453 patients). We analyzed and summarized the clinical response (partial and complete response, PR and CR) of each trial in Table 1 and Supplemental Table 1. We found that, among the 453 patients in the 38 trials, 57 (12.58%) patients had PR or CR. This is seemingly higher than the response rate of ~9% reported in another meta-analysis (19), which is likely due to the latest addition of Claudin 18.2 CART trials that demonstrated 44.64% in 56 gastrointestinal (GI) cancer patients (143–145). Importantly, from the 38 CART trials, we traced back to the original CART development and found the ABD affinity (KD) (Table 1 and Supplemental Table 1). The correlation of ABD affinity and response rate was also presented in Figure 1. We arbitrarily divided the ABDs as high- (KD<20nM), moderate- (KD=20-100nM), and low- (KD>100nM) affinity. Not surprisingly, 2/3 of the trials (25/38, 65.79%) utilized high-affinity ABDs in their CARTs. The response rate in the high-affinity group is merely 5.70% (17 out of 298 patients) (Figure 1 and Supplemental Table 1). Only 9 of the 25 trials generated low response. The other 16 CARTs built from high-affinity ABDs showed no responses (the best result is stable diseases). In contrast, 8 out of 10 trials of CARTs with moderate-affinity ABDs showed an impressive response rate (18.18%-75%). The overall response rate of moderate-affinity ABD CARTs reaches 34.78% (40 out of 115 patients). Thirdly, when the ABD affinity is too low (KD>100nM), the CARTs demonstrated no clinical responses (Figure 1, Table 1, and Supplemental Table 1), suggesting that when the ABD affinity is below a certain threshold, the CARTs will not have adequate avidity to engage and kill tumor cells. These 38 trial data demonstrated that the affinity of ABDs is critical in determining the efficacy of solid tumor CARTs. ABDs with proper moderate-affinity may have the optimal engagement for CARTs to kill tumor cells inside tumor mass. Currently, there is no available data on the optimal ABD affinities in different CARs. However, it is likely that the optimal affinity of ABDs may vary among different CARTs and may depend on the engagement modes of CART-tumor cells in hematological cancers vs. solid tumors (see sections below).
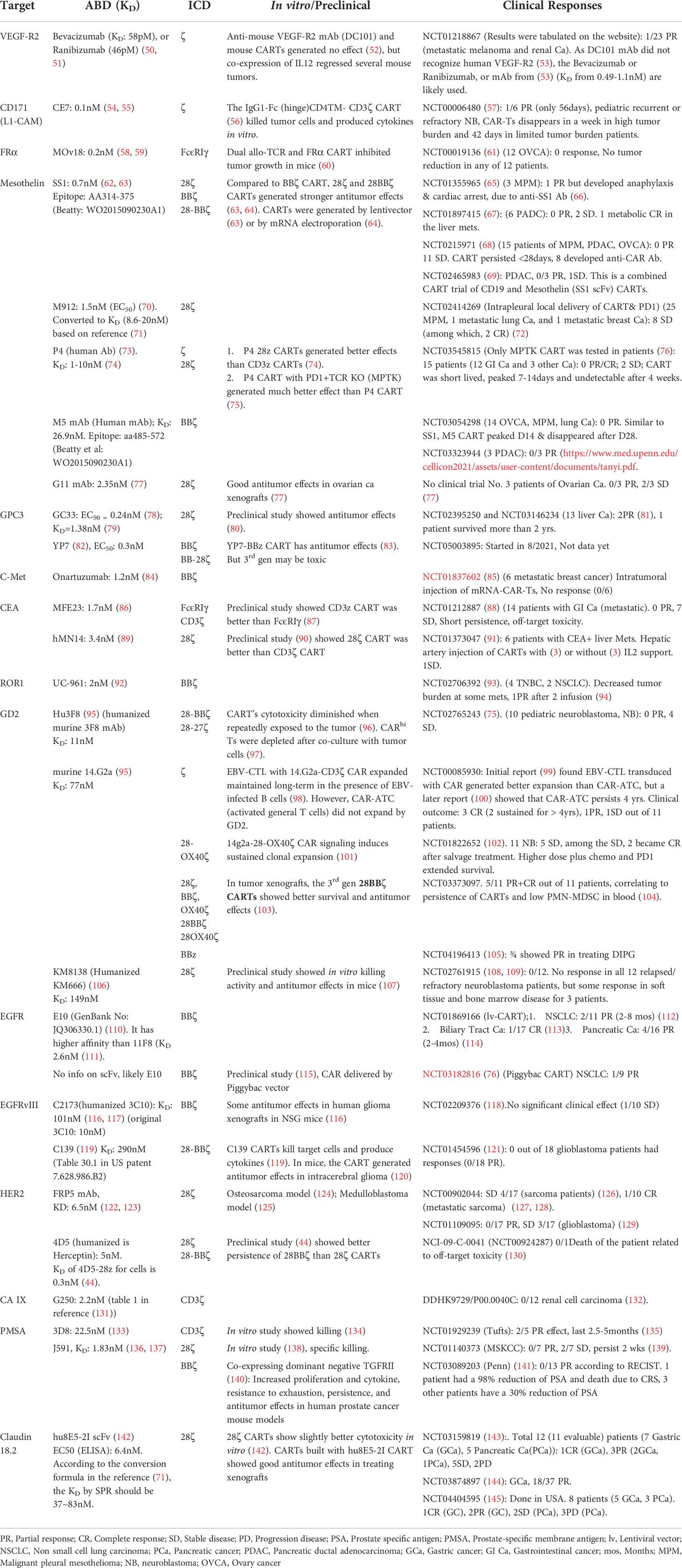
Table 1 Summary of solid tumor CAR-T clinical trials: Affinity of antigen binding domains vs. clinical efficacy.
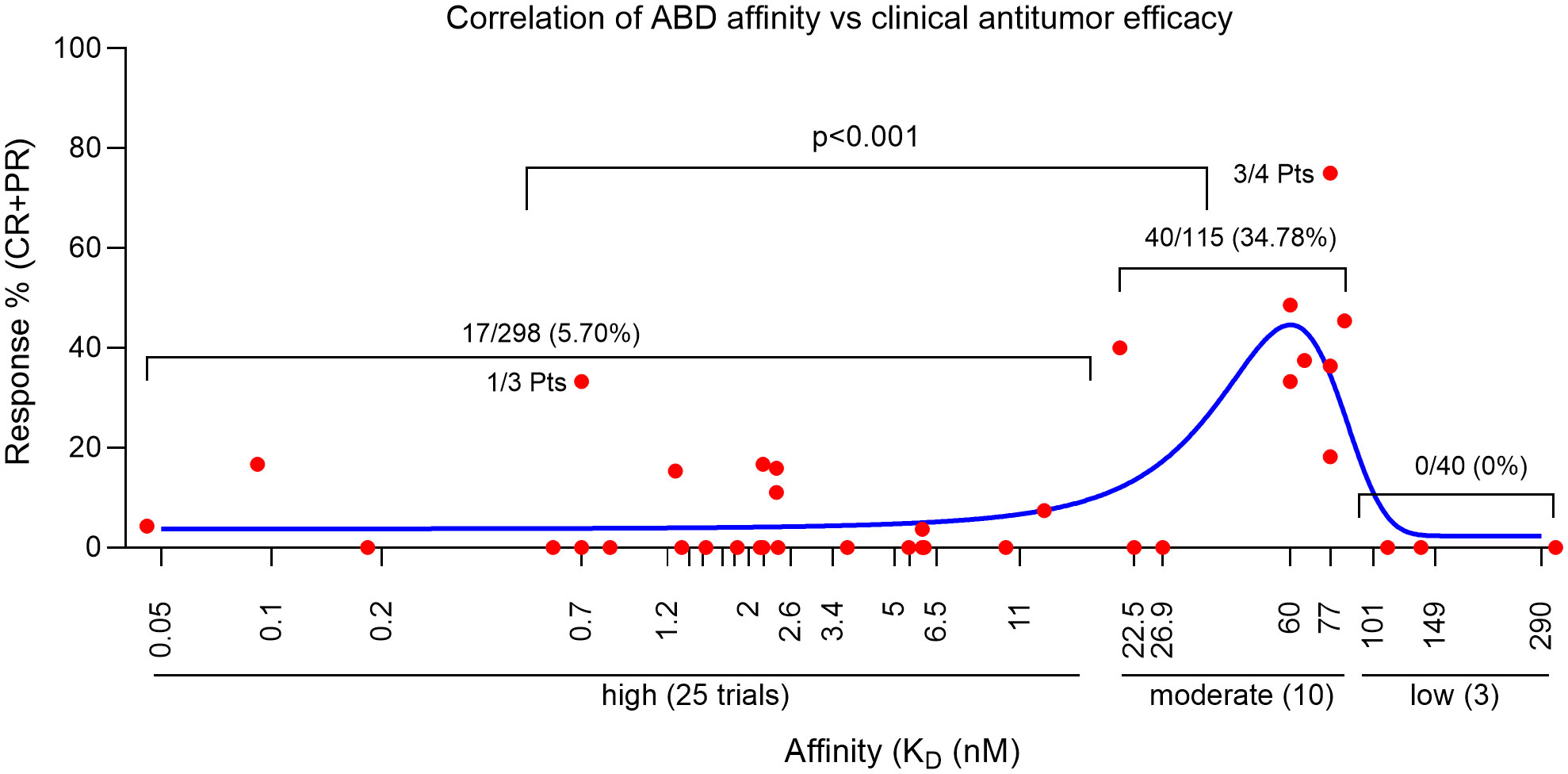
Figure 1 Correlation of CART clinical efficacy and their ABD affinity. Each red dot represents the % of responses of one clinical trial. The blue line is the Gaussian regression. Statistics was done with NCSS II software (Kaysville, Utah).
The 38 solid tumor CART trials in Table 1 target different antigens and epitopes. It is known that the epitope location (relative to the cell membrane) plays an important role in deciding CART’s functions and antitumor efficacy in preclinical tumor models (27). To minimize the effects of epitope location and to analyze the correlation of ABD affinity more precisely to the antitumor efficacy of CARTs, we compared the clinical response of three GD2 CARTs. Disialoganglioside GD2, a major ganglioside, is a carbohydrate antigen expressed on the tumors of neuroectodermal origin, including melanoma, neuroblastoma, sarcoma, and small cell lung cancer (146). GD2 has a hydrophobic ceramide tail inserted into the cell membrane and a pentasaccharide moiety head on the outside of membrane (Figure 2A) (147). Multiple anti-GD2 mAbs are developed for cancer therapies (148), and some are approved by FDA (149). The three anti-GD2 mAbs used to develop CARTs have different affinities (Table 1) but target the same membrane-proximal sugar moiety (147, 150). Thus, the effect of epitope location can be neglected when the CART’s efficacy is compared. In four clinical trials using the GD2 CARTs made with moderate-affinity mAb 14.G2a (KD=77nM), 13 out of 37 patients had PR or CR (35.16% response rate) (Table 1, Supplemental Table 1, and Figure 2B). Although the 14.G2a-based GD2 CARTs in different trials utilized different CDs, they all generated good clinical responses, further suggesting that ABDs may play a deciding role in the antitumor outcome of CARTs. In contrast, the GD2 CARTs built with high-affinity Hu3F8 mAb (KD=11nM) or with low-affinity KM8138 (humanized KM666) mAb (KD=149nM) did not generate clinical response (Figure 2B, Table 1, and Supplemental Table 1).
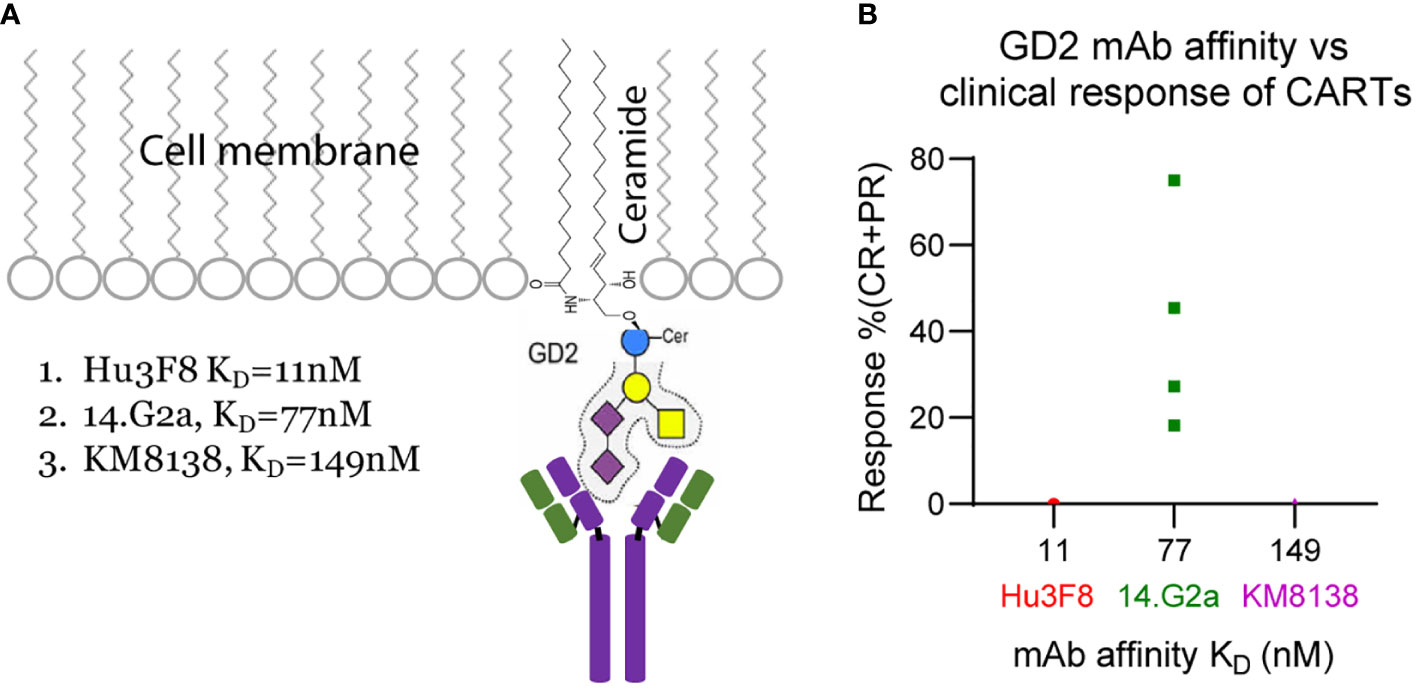
Figure 2 GD2 sugar moiety (A) and correlation of anti-GD2 mAb affinity with clinical efficacy (B) of the GD2 CARTs.
The benefit of moderate-affinity ABDs in solid tumor CARTs was further demonstrated in 3 latest trials (two were in China and one was in USA) of the same Claudin 18.2 CARTs (143–145). Claudin 18.2, a member of the tight junction protein family, is considered a gastric-specific isoform with higher expression on cancers than normal tissue. Claudin 18.2 specific mAbs and CARTs are being developed to treat GI cancers. In the latest trial of Claudin 18.2 CARTs, 18 out of 37 GI cancer patients demonstrated an overall response rate of 48.64% (144). A similar response rate was also reported in other two recent trials (143, 145). The overall response rate of this Claudin 18.2 CARTs reached an impressive 44.64% in 3 trials (25 out of 56 patients). Unfortunately, the KD of Claudin 18.2 mAb, 8E5, was not reported. However, investigators did measure the EC50 of 8E5 mAb binding to Claudin 18.2 + 293 cells, which is 49.19nM (142). After humanization and optimization, the final mAb Hu8E5-2I used in the Claudin 18.2 CARTs has EC50 6.4nM for binding Claudin 18.2 + 293 cells (142), which is 20x lower than GC33 (EC50 = 0.24nM) and YP7 (EC50 = 0.3nM). According to a comparative study (71), the value of EC50 determined by ELISA is 5.76-13 folds lower than the KD value measured by surface plasmon resonance (SPR). Based on this factor, we calculated the KD value of Hu8E5-2I is likely between 36 to 83nM, which falls in the moderate-affinity range. We thus used the average 60nM to do the plot in Figure 1.
Moderate-affinity ABD is also good for blood cancer CARTs. While the original CD19 CARTs built with FMC63 mAb (KD=0.328nM) generated remarkable antitumor efficacy and have been approved by FDA, recent studies showed that CD19 CARTs made with a new mAb CAT with lower affinity (KD=14nM) generated enhanced expansion and prolonged persistence in treating refractory AML compared to the FMC63-based CD19 CART (151).
In summary, although multiple factors may contribute to CART’s function, the data analysis of 38 solid tumor CART clinical trials demonstrate that the ABD affinity is possibly the most important one in deciding the CART’s antitumor efficacy. Moderate affinity ABD not only allows CARTs to distinguish the antigenhigh tumor cells from antigenlow normal cells (see section “the ABD affinity and CART’s on-target/off-tumor toxicity”), but also enable them to generate stronger antitumor efficacy. Thus, different from antibody drugs, in the CART development, moderate ABD affinity may be better.
One exception to the “moderate-affinity” role is the mesothelin targeting M5 CART. The M5 mAb is moderate affinity (KD: 26.9nM), but the M5 CARTs had no antitumor effects in treating multiple solid tumors (https://www.med.upenn.edu/cellicon2021/assets/user-content/documents/tanyi.pdf). Further analysis showed that the Koff of M5-mesothelin is low, thus the dwell time (T1/2) of M5-mesothelin is much longer than that of the moderate affinity mAbs of 3D8 and 14.G2a (Table 2). The T1/2 of M5-meosthelin is 613 seconds, while the T1/2 of 3D8-PMSA and 14.G2a-GD2 complex is 5 and 62 seconds, respectively. Thus, even though they have similar moderate affinity, their binding kinetics of targets are different (see the section of “The KD, Kon, Koff, and T1/2 of ABDs and their effects on CARTs”). Due to the limited examples, it remains to be verified whether the higher Koff (thus shorter dwell time) of ABD-antigen complex is indeed important in deciding the efficacy of solid tumor CARTs.
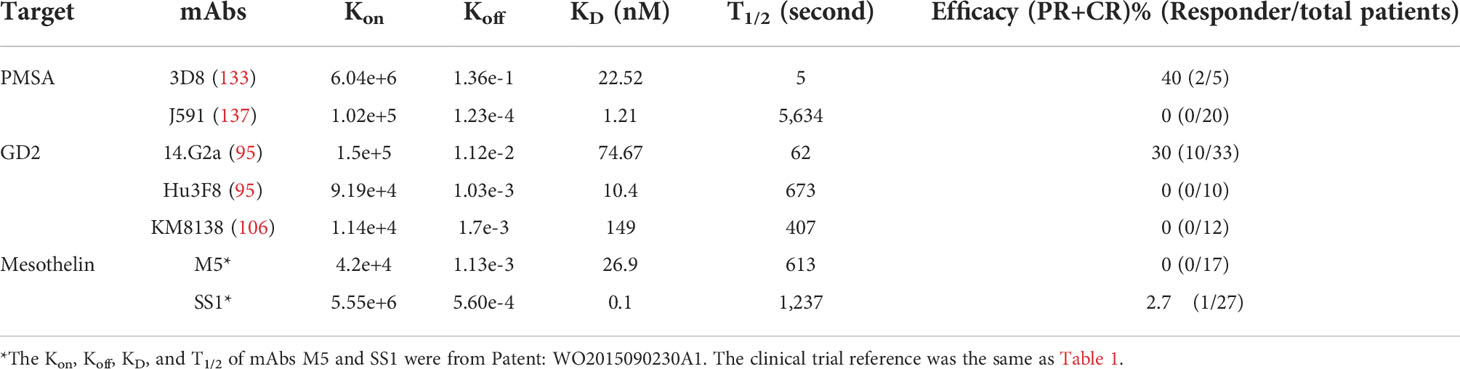
Table 2 Relationship of Koff and T1/2 of ABDs and clinical efficacy of CARTs.
The effects of ABD affinity on the biology and function of CARTs
Different from conventional small molecule and antibody medicines, CARTs are living drugs, i.e., they multiply and expand, and must be alive and activated to be functional. In general, soluble antigen does not induce CART activation and expansion (152), suggesting that oligomerization of CARs on cell surface is important in CART activation although the immunological synapse of CAR is nonclassical and not well defined (153, 154). The engaging avidity between CART and target cell is determined by the ABD affinity, CAR level, and antigen level (155). In this article, we focus on the effect of ABD affinity on CART’s biology and function, such as activation, function, persistence, and antitumor effects, especially in solid tumors, where the engagement between CART and tumor is multi-dimensional, persistent, and intense.
The ABD affinity needs to reach a threshold for CARTs to have a productive engagement with tumor cells, which generates sufficient signaling to activate and expand CARTs and to kill tumor cells. An increase of ABD affinity within a range may enhance CART activation and function (156). However, ABD affinity beyond a certain level will not further enhance CART function (157), but may be harmful. The ABD affinity can affect CART biology and function in the following ways. 1) When the CART-tumor cell engagement is too strong, the CARTs are difficult to dissociate from the killed or dying tumor cells. The occupied CARTs will be unable to re-engage with different target cells and induce serial killing of tumor cells. 2) A strong CART-tumor cell engagement may allow CAR to nibble a piece of the target cell membrane and the associated antigen (158). This process of trogocytosis will tag the CARTs to become the target and victim of other CARTs (fratricide). Trogocytosis also cause tumor escape due to antigen loss on target cells. For example, the CD19 CARTs based on high-affinity FMC63 mAb had higher trogocytosis and fratricide than the CD19 CARTs from a lower-affinity CAT mAb (158, 159). 3) The strong and persistent engagement of high avidity CARTs with tumor cells may drive CARTs to exhaustion and activation-induced cell death (AICD). We recently found that, compared to the CARTs with from high-affinity GC33 mAb (KD=1.38nM), our GPC3-specific CARTs derived from a novel moderate-affinity 8F8 mAb (KD=23nM) are less exhausted and less apoptotic inside tumor lesions (79). 4) The ABD affinity affects the polyfunctionality of CARTs. Using CyTOF technology, Michelozzi et al. compared the FMC63 (high-affinity) and CAT (moderate-affinity) CD19 CARTs and found that, after engaging with CD19+ leukemia cells, the CAT CD19 CARTs contained significantly more polyfunctional T cells than the FMC63-derived CARTs (160). This suggests proper moderate-affinity ABD may allow CARTs to preserve their polyfunctionality, which is important for antitumor effect (161). Similarly, we also observed that the low-affinity 8F8 CARTs maintain better cytokines of IL2 and IFNγ production inside solid tumor lesions (79). 5) The ABD affinity affects the formation of memory T cells. Previous studies showed that reduction of TCR functional avidity via lowering Lck expression (162) or by TCR downregulation (163) increased memory T cells. Similarly, our recent study (79) showed that, compared to GC33 CARTs, our 8F8 CARTs contained more memory T cells and persisted longer in the solid tumors. Importantly, the moderate-affinity 8F8 derived CARTs also maintained better function in the tumors, resulting in durable antitumor effects in treating human tumor xenografts. 6) The ABD with reduced affinity allow CARTs to differentiate tumor cells from normal cells based on quantitative antigen difference, which will broaden the targetable tumor-associated surface antigens that can benefit tumor selectivity (164) (also seem below).
The ABD affinity on CART’s antigen sensitivity and on-target/off-tumor toxicity
Moderate-affinity ABD may be good for CARTs to maintain function. However, lowering ABD affinity may reduce CART’s sensitivity of detecting the antigenlow tumor cells. For example, compared to the EGFR CARTs derived from the high-affinity Cetuximab (KD=1.8nM), the CARTs derived from the low-affinity Nimotuzumab (KD=21nM) could distinguish antigenhigh vs. antigenlow target cells, but showed less control of antigenlow human tumor xenografts in mouse (43). Fortunately, affinity is not the only factor that affect antigen sensitivity. The affinity of TCR is much lower than CARs, but is able to detect single molecule of pMHC complex (165), while CARTs need 200 molecules of antigen for activation (166). Even with the same affinity, the sensitivity of TCR 10-100 times higher than CAR (167), suggesting that the signaling apparatus of TCR complex also play an important role in deciding the antigen sensitivity. Along this line, it was reported that manipulation of CD domain and ITAM enhanced the antigen sensitivity of CARTs (168). Thus, it is possible to lower the ABD affinity while maintaining the antigen sensitivity.
A positive side effect of losing antigen sensitivity is the reduction of on-target/off-tumor toxicity because most tumor antigens are not unique to tumor cells, but rather are the shared self-antigens that are also present in normal cells albeit at lower levels. In fact, the initial studies of utilizing low-affinity ABDs in CART development were intended to distinguish the antigenhigh tumors from antigenlow normal cells to avoid off-tumor toxicity (43, 47, 169–173). Some recent preclinical in vivo studies further illustrated that the CARTs derived from low-affinity ABDs were indeed less toxic. Using the transgenic mice that express different levels of HER2 antigen, Castellarin et al. showed that CARTs built with low-affinity HER2 mAbs had less in vivo toxicity, but also generated better antitumor effects compared to high-avidity CARTs because they are less likely be trapped in the antigenlow normal tissues (173). In another latest report, Giardino et al. developed a pair of new GPC3-specific mAbs, GPC3-1 (KD=73nM) and GPC3-2 (KD=11nM), which could bind both human GPC3 and mouse GPC3. They demonstrated that GPC3-1 and GPC3-2 CARTs generated similar antitumor effects in mouse models. However, the low-avidity GPC3-1 CARTs demonstrated much lower toxicity in mice than the GPC3-2 CARTs (174). Thus, it is important to find an optimal moderate-affinity ABD to construct CARTs that maximize its effects on target tumor cells, while minimizing off-tumor toxicity. Different targets may need different optimal affinities. For example, in our meta-analysis of clinical trial data, we found that the CARTs built with the KD of ABDs between 20-100nM generated effective CARTs (Figure 1). However, in the ICAM-1 targeted CARTs, the KD of LFA binding ICAM-1 is at micromolar (KD=20µM) to generate the most effective antitumor effects with reduced toxicity in preclinical tumor model (171).
The KD, Kon, Koff, and T1/2 of ABDs and their effects on CARTs
The affinity can be measured by SPR and ELISA. ELISA measures the EC50 (the concentration required to obtain a 50% maximum protein-ligand binding), whereas SPR measures the association (Kon) and dissociation rate (Koff) for the calculation of equilibrium dissociation constant KD(equal Koff/Kon), a more widely used parameter for binding affinity. Individual Kon (Number/M*S) and Koff (Number/S) value can represent ligand binding kinetics much better in a time-dependent manner (175): A higher number of Kon means faster ligand binding whereas a higher Koff indicates that the complex dissociates faster. As both Kon and Koff determined the ligand binding affinity (KD), 2 ligands with same or similar affinity (KD) might have different Kon and Koff value changing in the same direction (either increase or decrease), and thus show completely different binding kinetics. In this case, the ABD binding kinetics (Kon and Koff) may be even more important than KD or EC50 in determining the CART’s efficacy. Another important parameter in comparing the ligand binding is the half-life of the complex, T1/2, which relates to Koff by the formula T1/2=Ln2 (0.693)/Koff (175). Thus, T1/2 indicates the stability or dwell time of the complex.
The effect of Kon and Koff (0r related dwell time (T1/2) of TCR-pMHC complex on T cell activation has been well-studied (165, 176, 177). If the TCR and pMHC have a fast on-rate (higher Kon), the TCR-pMHC complex with a higher Koff (a short dwell time) can be highly stimulatory (165) because the pMHCs can bind and rebind the same TCR (178) or multiple TCRs (175) several times, creating an effective longer dwell time than a single TCR-pMHC encounter (165). This may contribute to the high sensitivity of TCR that can detect one pMHC complex on target cells (178). On the other hand, if the Kon is low, the dissociated ligand will not easily rebind a TCR. Under such circumstances, the outcome of the TCR-pMHC engagement will likely depend on the dwell time of the TCR-pMHC complex. In other words, if the Kon is low, the complex needs to be stable (lower Koff) to generate sufficient signaling for activation.
A similar principle may apply to the CART-tumor engagement. The affinity (KD) of ABD-antigen engagement is in the range of pM-nM (179), which is ~3 logs lower than that of TCR-pMHC (176). Thus, the dwell time of antibody-antigen complex is in the range of hours or even day (180), much longer than that of TCR-pMHC (normally in seconds) (176). Such long stable engagement may result in persistent activation of CARTs that can drive them into exhaustion and AICD. A long engagement may not be necessary, but rather be harmful in solid tumors. Such argument is in agreement with the fact that moderate-affinity 14.G2a-based GD2 CARTs generated much better clinical efficacy than the high-affinity 3F8-based CARTs (Supplemental Table 1 and Figure 2B). The anti-GD2 mAb 14.G2a has similar Kon as 3F8, but has 10x higher Koff (95). Thus, the engagement dwell time of 14.G2a CARTs is 10x shorter than the 3F8 CARTs. This fast-on/fast-off “fly-kiss” mode of engagement by 14.G2a CARTs allows CARTs to have a shorter intermittent disengagement in the solid tumors. Such transient break during “off” time may rejuvenate and preserve CART function (37). Similarly, the moderate-affinity CAT mAb has similar Kon as high-affinity FMC63, but has much higher Koff (151), which may contribute to the formation of memory T cells and polyfunctionality of CAT CARTs (160) and durable antitumor effects (151). Thus, the dwell time and kinetics of ABD-target engagement may be more important than affinity (KD) in deciding the outcome of CARTs. For example, the M5 mAb has a T1/2 of 613 seconds (Table 2), which may be the reason why M5 CARTs did not generate therapeutic effects in clinical trials. In contrast, the 3D8 and 14.G2a CARTs that generated impressive clinical responses have the ABDs with higher Koff. Their T1/2 is 5 and 62 seconds, respectively (Table 2). A high Kon allow ABD bind target quickly, a higher Koff may benefit for CART survival because of faster dissociation. This bind/off/rebind “fly-kiss” style of engagement may be optimal for CART to exert their function while avoiding being-driven into exhaustion, especially in solid tumor mass. Along this line of analysis, measuring the Kon and Koff of the Claudin 18.2 mAb Hu8E5-2I should help verify if the fast-on/fast-off “fly-kiss” intermittent engagement mode indeed enhances CART’s antitumor efficacy. Similarly, it will be very interesting to know whether the recently developed low-affinity GPC3 specific mAb GPC3-1 (174) that has a high Koff will generate clinical efficacy in future trials.
A fast-on/fast-off “fly-kiss” mode of engagement may be required for effective solid tumor CARTs
A fundamental anatomical difference between blood cancers and solid tumors is the tumor mass, in which CART-tumor cell engagement is intense and persistent. In hematological cancers, tumor cells are in the blood and do not aggregate together to form tumor mass, and thus CARTs have immediate access to target tumor cells after infusion. Importantly, the engagement of CART-tumor cells in the blood is individualized in 1-on-1 mode and intermittent (Figure 3). CART can “enjoy a temporary break” after each killing before finding the next target. On the other hand, in solid tumors, CARTs first need to migrate into a tumor mass. Once CARTs infiltrate a tumor lesion, they are surrounded by tumor cells from every possible direction. Thus, the engagement of CART-tumor cells in a solid tumor is multi-dimensional 1-on-N or N-on-N mode and persistent (Figure 3). There is no intermittent break for the CARTs unless they can spontaneously be disengaged due to higher Koff or until the tumor mass is eliminated. Furthermore, a solid tumor has a complex extracellular matrix stroma that further restrains CART movement and aggravates the antigen assault on them. Such constant and intense engagement with antigens will drive CARTs exhaustion or AICD. Thus, due to different mode and intensity of CART-tumor engagement, the ABD affinity requirement for solid tumor CARTs is likely different from the CD19 and other blood cancer CARTs. CARTs with high-affinity ABDs will be more prone to exhaustion and AICD in solid tumors than in blood cancers.
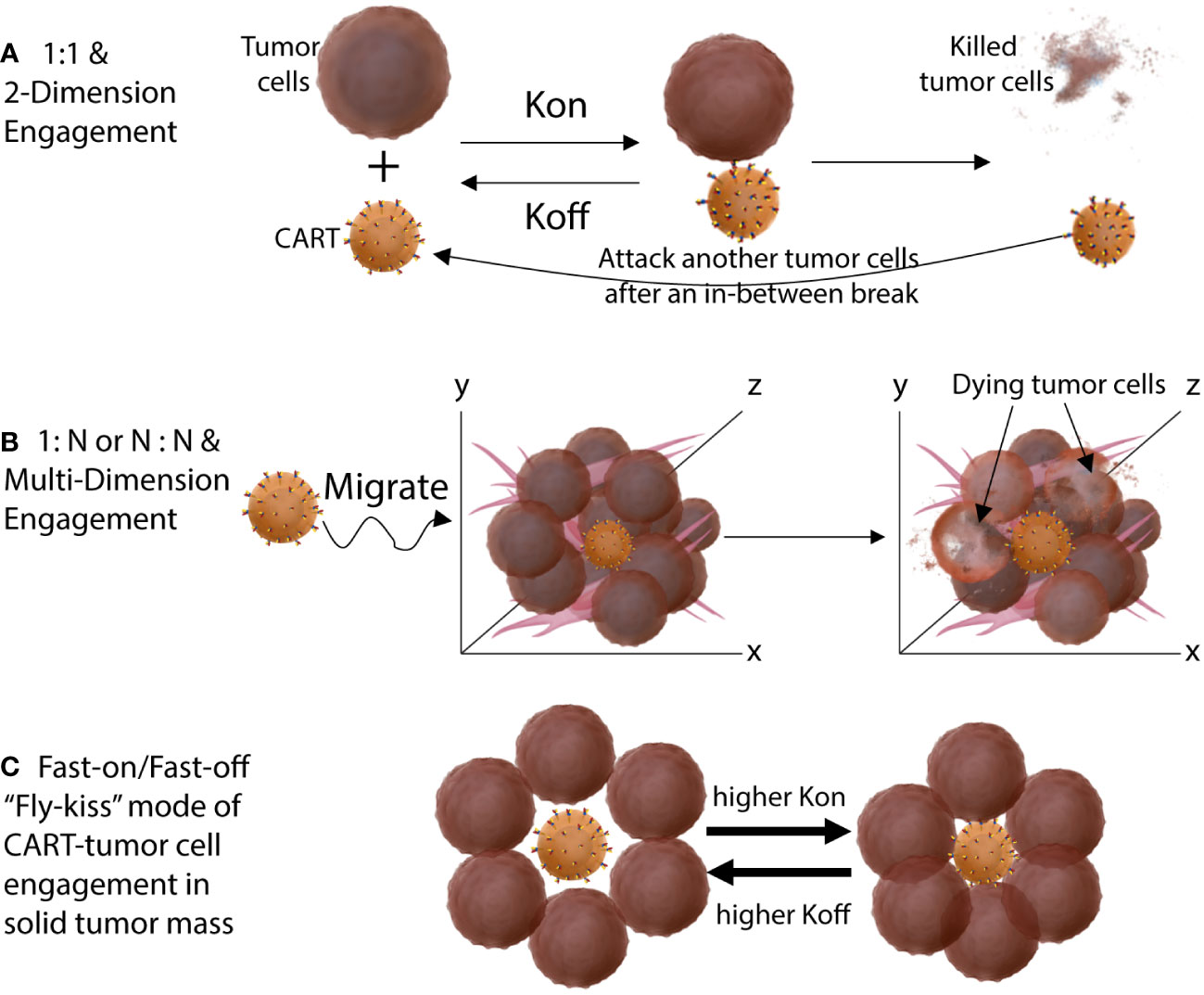
Figure 3 Different engagement modes of CART-tumor cells in hematological cancers (A) and in solid tumors (B). The CARTs engage tumor cells in a solid tumor mass in 1-on-N or N-on-N multi-dimensional mode. (C) The fast-on/fast-off (bind-off-rebind) “fly-kiss” style of CART-tumor engagement when both the Kon and Koff is high. High Koff allows CARTs to disengage from tumor cells even they are surrounded in solid tumors. High Kon make sure that CART will re-engage tumor cells even when tumor antigen level is level.
Thus, we propose that moderate-affinity ABD and fast-on/fast-off engaging kinetics are especially necessary for solid tumor CARTs to be effective. A higher Kon of the ABD will make sure that CARTs will bind to target cell fast even when the antigen level is low; a higher Koff will allow CARTs to disengage even if they are surrounded by tumor cells in a solid tumor mass. Such a fast-on/fast-off “fly-kiss” style of engagement allows CARTs to kill tumor cells without being-driven into exhaustion and AICD. In addition, currently, we have little knowledge on how the different mode and kinetics of CART-tumor engagement may affect the epigenetics, gene expression, metabolism, and thus the fitness of CARTs. Further investigation into these mechanisms will likely help design more effective CARTs for solid tumors. We think that solid tumor CART development should focus more on the ABD affinity and the engaging kinetics of CART and tumor cells. Recently, strategies have been discussed to tune the ABD affinity for better and effective CART development (181) even though there is no obvious approach to select ABDs with particular Kon and Koff yet. This intentional and rational design of CARs with tuning ABD affinity and binding dynamics in mind will likely generate more effective solid tumor CARTs that can potentially match the remarkable success observed in hematological cancers.
Author contributions
RM and YH analyzed the clinical responses of 38 clinical trials and searched the ABD affinity in the CARTs. WK drew the CART-tumor engagement mode presented in Figure 3. All three authors wrote the manuscript together and approved the submitted version.
Acknowledgments
The CART research work in YH’s laboratory is partially funded by Paceline Cancer Research Award grant from Georgia Cancer Center, Augusta University. We thank Dr. Ramses Sadek for performing the statistical analysis of the clinical trial data. We also thank current and former members for their contributions in improving the antitumor efficacy of engineered T cells.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1032403/full#supplementary-material
References
1. Greten TF, Lai CW, Li G, Staveley-O'Carroll KF. Targeted and immune-based therapies for hepatocellular carcinoma. Gastroenterology. (2019) 156(2):510–24. doi: 10.1053/j.gastro.2018.09.051
2. Hato T, Goyal L, Greten TF, Duda DG, Zhu AX. Immune checkpoint blockade in hepatocellular carcinoma: current progress and future directions. Hepatology. (2014) 60(5):1776–82. doi: 10.1002/hep.27246
3. Li F, Li C, Cai X, Xie Z, Zhou L, Cheng B, et al. The association between CD8+ tumor-infiltrating lymphocytes and the clinical outcome of cancer immunotherapy: A systematic review and meta-analysis. EClinicalMedicine. (2021) 41:101134. doi: 10.1016/j.eclinm.2021.101134
4. Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, et al. Cold tumors: A therapeutic challenge for immunotherapy. Front Immunol (2019) 10:168. doi: 10.3389/fimmu.2019.00168
5. Docta RY, Ferronha T, Sanderson JP, Weissensteiner T, Pope GR, Bennett AD, et al. Tuning T-cell receptor affinity to optimize clinical risk-benefit when targeting alpha-Fetoprotein-Positive liver cancer. Hepatology. (2019) 69(5):2061–75. doi: 10.1002/hep.30477
6. Zhu W, Peng Y, Wang L, Hong Y, Jiang X, Li Q, et al. Identification of alpha-fetoprotein-specific T-cell receptors for hepatocellular carcinoma immunotherapy. Hepatology. (2018) 68(2):574–89. doi: 10.1002/hep.29844
7. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. (2006) 314(5796):126–9. doi: 10.1126/science.1129003
8. Shafer P, Kelly LM, Hoyos V. Cancer therapy with TCR-engineered T cells: Current strategies, challenges, and prospects. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.835762
9. June CH, Sadelain M. Chimeric antigen receptor therapy. New Engl J Med (2018) 379(1):64–73. doi: 10.1056/NEJMra1706169
10. Kershaw MH, Westwood JA, Darcy PK. Gene-engineered T cells for cancer therapy. Nat Rev Cancer. (2013) 13(8):525–41. doi: 10.1038/nrc3565
11. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. New Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
12. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. New Engl J Med (2011) 365(8):725–33. doi: 10.1056/NEJMoa1103849
13. Mao R, Hussein MS, He Y. Chimeric antigen receptor engineered T cells and their application in the immunotherapy of solid tumours. Expert Rev Mol Med (2022) 24:e7. doi: 10.1017/erm.2021.32
14. Bagley SJ, O'Rourke DM. Clinical investigation of CAR T cells for solid tumors: Lessons learned and future directions. Pharmacol Ther (2020) 205:107419. doi: 10.1016/j.pharmthera.2019.107419
15. Castellarin M, Watanabe K, June CH, Kloss CC, Posey AD Jr. Driving cars to the clinic for solid tumors. Gene Ther (2018) 25(3):165–75. doi: 10.1038/s41434-018-0007-x
16. D’Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T cells: the long and winding road to solid tumors. Cell Death Disease. (2018) 9(3):282. doi: 10.1038/s41419-018-0278-6
17. Schaft N. The landscape of CAR-T cell clinical trials against solid tumors–a comprehensive overview. Cancers (Basel). (2020) 12(9):2567. doi: 10.3390/cancers12092567
18. Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T cell therapy for solid tumors: Bright future or dark reality? Mol Ther (2020) 28(11):2320–39. doi: 10.1016/j.ymthe.2020.09.015
19. Hou B, Tang Y, Li W, Zeng Q, Chang D. Efficiency of CAR-T therapy for treatment of solid tumor in clinical trials: A meta-analysis. Dis Markers. (2019) 2019:3425291. doi: 10.1155/2019/3425291
20. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
21. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. (2021) 21(3):145–61. doi: 10.1038/s41568-020-00323-z
22. Majzner RG, Mackall CL. Clinical lessons learned from the first leg of the CAR T cell journey. Nat Med (2019) 25(9):1341–55. doi: 10.1038/s41591-019-0564-6
23. Newick K, O'Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med (2017) 68(1):139–52. doi: 10.1146/annurev-med-062315-120245
24. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol (2018) 36(4):346–51. doi: 10.1038/nbt.4086
25. Foeng J, Comerford I, McColl SR. Harnessing the chemokine system to home CAR-T cells into solid tumors. Cell Rep Med (2022) 3(3):100543. doi: 10.1016/j.xcrm.2022.100543
26. Tian Y, Wen C, Zhang Z, Liu Y, Li F, Zhao Q, et al. CXCL9-modified CAR T cells improve immune cell infiltration and antitumor efficacy. Cancer Immunol Immunother (2022) 71(11):2663–2675. doi: 10.1007/s00262-022-03193-6
27. Zhang Z, Jiang D, Yang H, He Z, Liu X, Qin W, et al. Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death Disease. (2019) 10(7):476. doi: 10.1038/s41419-019-1711-1
28. Guerra E, Di Pietro R, Basile M, Trerotola M, Alberti S. Cancer-homing CAR-T cells and endogenous immune population dynamics. Int J Mol Sci (2021) 23(1):405. doi: 10.3390/ijms23010405
29. Rodriguez-Garcia A, Palazon A, Noguera-Ortega E, Powell DJ Jr., Guedan S. CAR-T cells hit the tumor microenvironment: Strategies to overcome tumor escape. Front Immunol (2020) 11:1109. doi: 10.3389/fimmu.2020.01109
30. van den Ende T, van den Boorn HG, Hoonhout NM, van Etten-Jamaludin FS, Meijer SL, Derks S, et al. Priming the tumor immune microenvironment with chemo(radio)therapy: A systematic review across tumor types. Biochim Biophys Acta (BBA) - Rev Cancer. (2020) 1874(1):188386. doi: 10.1016/j.bbcan.2020.188386
31. Bailey SR, Nelson MH, Majchrzak K, Bowers JS, Wyatt MM, Smith AS, et al. Human CD26(high) T cells elicit tumor immunity against multiple malignancies via enhanced migration and persistence. Nat Commun (2017) 8(1):1961. doi: 10.1038/s41467-017-01867-9
32. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21(6):581–90. doi: 10.1038/nm.3838
33. Feucht J, Sun J, Eyquem J, Ho Y-J, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med (2019) 25(1):82–8. doi: 10.1038/s41591-018-0290-5
34. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. C-jun overexpression in CAR T cells induces exhaustion resistance. Nature. (2019) 576(7786):293–300. doi: 10.1038/s41586-019-1805-z
35. Ding Z-C, Shi H, Aboelella NS, Fesenkova K, Park E-J, Liu Z, et al. Persistent STAT5 activation reprograms the epigenetic landscape in CD4+ T cells to drive polyfunctionality and antitumor immunity. Sci Immunol (2020) 5(52):eaba5962. doi: 10.1126/sciimmunol.aba5962
36. Labanieh L, Majzner RG, Klysz D, Sotillo E, Fisher CJ, Vilches-Moure JG, et al. Enhanced safety and efficacy of protease-regulated CAR-T cell receptors. Cell. (2022) 185(10):1745–63.e22. doi: 10.1016/j.cell.2022.03.041
37. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science (2021) 372(6537):eaba1786. doi: 10.1126/science.aba1786
38. Hyrenius-Wittsten A, Su Y, Park M, Garcia JM, Alavi J, Perry N, et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Trans Med (2021) 13(591):eabd8836. doi: 10.1126/scitranslmed.abd8836
39. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res (2015) 3(2):125–35. doi: 10.1158/2326-6066.CIR-14-0127
40. Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T design: Elements and their synergistic function. EBioMedicine. (2020) 58:102931. doi: 10.1016/j.ebiom.2020.102931
41. Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Trans Immunol (2019) 8(5):e1049. doi: 10.1002/cti2.1049
42. Mullard A. FDA Approves 100th monoclonal antibody product. Nat Rev Drug Discovery (2021) 20(7):491–5. doi: 10.1038/d41573-021-00079-7
43. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res (2015) 75(17):3505–18. doi: 10.1158/0008-5472.CAN-15-0139
44. Zhao Y, Wang QJ, Yang S, Kochenderfer JN, Zheng Z, Zhong X, et al. A herceptin-based chimeric antigen receptor with modified signaling domains leads to enhanced survival of transduced T lymphocytes and antitumor activity. J Immunol (2009) 183(9):5563–74. doi: 10.4049/jimmunol.0900447
45. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res (2013) 19(12):3153–64. doi: 10.1158/1078-0432.CCR-13-0330
46. Lynn RC, Feng Y, Schutsky K, Poussin M, Kalota A, Dimitrov DS, et al. High-affinity FRβ-specific CAR T cells eradicate AML and normal myeloid lineage without HSC toxicity. Leukemia. (2016) 30(6):1355–64. doi: 10.1038/leu.2016.35
47. Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res (2018) 6(1):36–46. doi: 10.1158/2326-6066.CIR-17-0211
48. Barros LRC, Couto SCF, da Silva Santurio D, Paixão EA, Cardoso F, da Silva VJ, et al. Systematic review of available CAR-T cell trials around the world. Cancers (Basel). (2022) 14(11):2667. doi: 10.3390/cancers14112667
49. Patel U, Abernathy J, Savani BN, Oluwole O, Sengsayadeth S, Dholaria B. CAR T cell therapy in solid tumors: A review of current clinical trials. eJHaem. (2022) 3(S1):24–31. doi: 10.1002/jha2.356
50. de Moraes Neto JE, Pereira F, Neves RL, de Barros NMT, Gil CD, Fernandes AG, et al. Preclinical assessment of intravitreal ramucirumab: in vitro and in vivo safety profile. Int J Retina Vitreous. (2020) 6(1):40. doi: 10.1186/s40942-020-00243-y
51. Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF trap, ranibizumab and bevacizumab. Angiogenesis. (2012) 15(2):171–85. doi: 10.1007/s10456-011-9249-6
52. Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res (2012) 18(6):1672–83. doi: 10.1158/1078-0432.CCR-11-3050
53. Witte L, Hicklin DJ, Zhu Z, Pytowski B, Kotanides H, Rockwell P, et al. Monoclonal antibodies targeting the VEGF receptor-2 (Flk1/KDR) as an anti-angiogenic therapeutic strategy. Cancer metastasis Rev (1998) 17(2):155–61. doi: 10.1023/A:1006094117427
54. Amstutz H, Rytz C, Novak-Hofer I, Spycher M, Schubiger PA, Blaser K, et al. Production and characterization of a mouse/human chimeric antibody directed against human neuroblastoma. Int J Cancer. (1993) 53(1):147–52. doi: 10.1002/ijc.2910530127
55. Meli ML, Carrel F, Waibel R, Amstutz H, Crompton N, Jaussi R, et al. Anti-neuroblastoma antibody chCE7 binds to an isoform of L1-CAM present in renal carcinoma cells. Int J Cancer. (1999) 83(3):401–8. doi: 10.1002/(SICI)1097-0215(19991029)83:3<401::AID-IJC17>3.0.CO;2-A
56. Gonzalez S, Naranjo A, Serrano LM, Chang W-C, Wright CL, Jensen MC. Genetic engineering of cytolytic T lymphocytes for adoptive T-cell therapy of neuroblastoma. J Gene Med (2004) 6(6):704–11. doi: 10.1002/jgm.489
57. Park JR, Digiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther J Am Soc Gene Ther (2007) 15(4):825–33. doi: 10.1038/sj.mt.6300104
58. Molthoff CF, Buist MR, Kenemans P, Pinedo HM, Boven E. Experimental and clinical analysis of the characteristics of a chimeric monoclonal antibody, MOv18, reactive with an ovarian cancer-associated antigen. J Nucl Med (1992) 33(11):2000–5.
59. Miotti S, Canevari S, Ménard S, Mezzanzanica D, Porro G, Pupa SM, et al. Characterization of human ovarian carcinoma-associated antigens defined by novel monoclonal antibodies with tumor-restricted specificity. Int J Cancer. (1987) 39(3):297–303. doi: 10.1002/ijc.2910390306
60. Kershaw MH, Westwood JA, Hwu P. Dual-specific T cells combine proliferation and antitumor activity. Nat Biotechnol (2002) 20(12):1221–7. doi: 10.1038/nbt756
61. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12(20):6106–15. doi: 10.1158/1078-0432.CCR-06-1183
62. Li Q, Verschraegen CF, Mendoza J, Hassan R. Cytotoxic activity of the recombinant anti-mesothelin immunotoxin, SS1(dsFv)PE38, towards tumor cell lines established from ascites of patients with peritoneal mesotheliomas. Anticancer Res (2004) 24(3a):1327–35.
63. Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci United States America (2009) 106(9):3360–5. doi: 10.1073/pnas.0813101106
64. Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res (2010) 70(22):9053–61. doi: 10.1158/0008-5472.CAN-10-2880
65. Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce antitumor activity in solid malignancies. Cancer Immunol Res (2014) 2(2):112–20. doi: 10.1158/2326-6066.CIR-13-0170
66. Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T Cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res (2013) 1(1):26–31. doi: 10.1158/2326-6066.CIR-13-0006
67. Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. (2018) 155(1):29–32. doi: 10.1053/j.gastro.2018.03.029
68. Haas AR, Tanyi JL, O'Hara MH, Gladney WL, Lacey SF, Torigian DA, et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol Ther J Am Soc Gene Ther (2019) 27(11):1919–29. doi: 10.1016/j.ymthe.2019.07.015
69. Ko AH, Jordan AC, Tooker E, Lacey SF, Chang RB, Li Y, et al. Dual targeting of mesothelin and CD19 with chimeric antigen receptor-modified T cells in patients with metastatic pancreatic cancer. Mol Ther (2020) 28(11):2367–78. doi: 10.1016/j.ymthe.2020.07.017
70. Feng Y, Xiao X, Zhu Z, Streaker E, Ho M, Pastan I, et al. A novel human monoclonal antibody that binds with high affinity to mesothelin-expressing cells and kills them by antibody-dependent cell-mediated cytotoxicity. Mol Cancer Ther (2009) 8(5):1113–8. doi: 10.1158/1535-7163.MCT-08-0945
71. Heinrich L, Tissot N, Hartmann DJ, Cohen R. Comparison of the results obtained by ELISA and surface plasmon resonance for the determination of antibody affinity. J Immunol Methods (2010) 352(1):13–22. doi: 10.1016/j.jim.2009.10.002
72. Adusumilli PS, Zauderer MG, Rivière I, Solomon SB, Rusch VW, O'Cearbhaill RE, et al. A phase I trial of regional mesothelin-targeted CAR T-cell therapy in patients with malignant pleural disease, in combination with the anti–PD-1 agent pembrolizumab. Cancer Discovery (2021) 11(11):2748–63. doi: 10.1158/2159-8290.CD-21-0407
73. Bergan L, Gross JA, Nevin B, Urban N, Scholler N. Development and in vitro validation of anti-mesothelin biobodies that prevent CA125/Mesothelin-dependent cell attachment. Cancer letters (2007) 255(2):263–74. doi: 10.1016/j.canlet.2007.04.012
74. Lanitis E, Poussin M, Hagemann IS, Coukos G, Sandaltzopoulos R, Scholler N, et al. Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol Ther (2012) 20(3):633–43. doi: 10.1038/mt.2011.256
75. Yu L, Huang L, Lin D, Lai X, Wu L, Liao X, et al. GD2-specific chimeric antigen receptor-modified T cells for the treatment of refractory and/or recurrent neuroblastoma in pediatric patients. J Cancer Res Clin Oncol (2021) 148(10):2643–2652. doi: 10.21203/rs.3.rs-803629/v1
76. Wang Z, Li N, Feng K, Chen M, Zhang Y, Liu Y, et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell Mol Immunol (2021) 18(9):2188–98. doi: 10.1038/s41423-021-00749-x
77. Chen J, Hu J, Gu L, Ji F, Zhang F, Zhang M, et al. Anti-mesothelin CAR-T immunotherapy in patients with ovarian cancer. Cancer Immunology Immunother (2022). doi: 10.1007/s00262-022-03238-w
78. Nakano K, Orita T, Nezu J, Yoshino T, Ohizumi I, Sugimoto M, et al. Anti-glypican 3 antibodies cause ADCC against human hepatocellular carcinoma cells. Biochem Biophys Res Commun (2009) 378(2):279–84. doi: 10.1016/j.bbrc.2008.11.033
79. Caraballo Galva LD, Jiang X, Hussein MS, Zhang H, Mao R, Brody P, et al. Novel low-avidity glypican-3 specific CARTs resist exhaustion and mediate durable antitumor effects against HCC. Hepatology. (2022) 76(2):330–44. doi: 10.1002/hep.32279
80. Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res (2014) 20(24):6418–28. doi: 10.1158/1078-0432.CCR-14-1170
81. Shi D, Shi Y, Kaseb AO, Qi X, Zhang Y, Chi J, et al. Chimeric antigen receptor-Glypican-3 T-cell therapy for advanced hepatocellular carcinoma: Results of phase I trials. Clin Cancer Res an Off J Am Assoc Cancer Res (2020) 26(15):3979–89. doi: 10.1158/1078-0432.CCR-19-3259
82. Phung Y, Gao W, Man YG, Nagata S, Ho M. High-affinity monoclonal antibodies to cell surface tumor antigen glypican-3 generated through a combination of peptide immunization and flow cytometry screening. MAbs. (2012) 4(5):592–9. doi: 10.4161/mabs.20933
83. Li D, Li N, Zhang YF, Fu H, Feng M, Schneider D, et al. Persistent polyfunctional chimeric antigen receptor T cells that target glypican 3 eliminate orthotopic hepatocellular carcinomas in mice. Gastroenterology. (2020) 158(8):2250–65 e20. doi: 10.1053/j.gastro.2020.02.011
84. Merchant M, Ma X, Maun HR, Zheng Z, Peng J, Romero M, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci United States America (2013) 110(32):E2987–96. doi: 10.1073/pnas.1302725110
85. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res (2017) 5(12):1152–61. doi: 10.1158/2326-6066.CIR-17-0189
86. Rios X, Compte M, Gomez-Vallejo V, Cossio U, Baz Z, Morcillo MA, et al. Immuno-PET imaging and pharmacokinetics of an anti-CEA scFv-based trimerbody and its monomeric counterpart in human gastric carcinoma-bearing mice. Mol Pharm (2019) 16(3):1025–35. doi: 10.1021/acs.molpharmaceut.8b01006
87. Haynes NM, Snook MB, Trapani JA, Cerruti L, Jane SM, Smyth MJ, et al. Redirecting mouse CTL against colon carcinoma: Superior signaling efficacy of single-chain variable domain chimeras containing TCR-ζ vs FcϵRI-γ. J Immunol (2001) 166(1):182–7. doi: 10.4049/jimmunol.166.1.182
88. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunology Immunother (2017) 66(11):1425–36. doi: 10.1007/s00262-017-2034-7
89. Akamatsu Y, Murphy JC, Nolan KF, Thomas P, Kreitman RJ, Leung SO, et al. A single-chain immunotoxin against carcinoembryonic antigen that suppresses growth of colorectal carcinoma cells. Clin Cancer Res (1998) 4(11):2825–32.
90. Emtage PC, Lo AS, Gomes EM, Liu DL, Gonzalo-Daganzo RM, Junghans RP. Second-generation anti-carcinoembryonic antigen designer T cells resist activation-induced cell death, proliferate on tumor contact, secrete cytokines, and exhibit superior antitumor activity in vivo: a preclinical evaluation. Clin Cancer Res (2008) 14(24):8112–22. doi: 10.1158/1078-0432.CCR-07-4910
91. Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor–modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res (2015) 21(14):3149–59. doi: 10.1158/1078-0432.CCR-14-1421
92. Choi MY, Widhopf GF, Wu CCN, Cui B, Lao F, Sadarangani A, et al. Pre-clinical specificity and safety of UC-961, a first-In-Class monoclonal antibody targeting ROR1. Clin Lymphoma Myeloma Leukemia (2015) 15:S167–S9. doi: 10.1016/j.clml.2015.02.010
93. Specht JM, Lee S, Turtle CJ, Berger C, Baladrishnan A, Srivastava S, et al. Abstract CT131: A phase I study of adoptive immunotherapy for advanced ROR1+ malignancies with defined subsets of autologous T cells expressing a ROR1-specific chimeric antigen receptor (ROR1-CAR). Cancer Res (2018) 78(13 Supplement):CT131–CT. doi: 10.1158/1538-7445.AM2018-CT131
94. Specht J, Lee S, Turtle C, Berger C, Balakrishnan A, Srivastava S, et al. Abstract P2-09-13: A phase I study of adoptive immunotherapy for ROR1+ advanced triple negative breast cancer (TNBC) with defined subsets of autologous T cells expressing a ROR1-specific chimeric antigen receptor (ROR1-CAR). Cancer Res (2019) 79(4_Supplement):P2–09-13-P2-09-13. doi: 10.1158/1538-7445.SABCS18-P2-09-13
95. Cheung NK, Guo H, Hu J, Tassev DV, Cheung IY. Humanizing murine IgG3 anti-GD2 antibody m3F8 substantially improves antibody-dependent cell-mediated cytotoxicity while retaining targeting in vivo. Oncoimmunology (2012) 1(4):477–86. doi: 10.4161/onci.19864
96. Sujjitjoon J, Sayour E, Tsao S-T, Uiprasertkul M, Sanpakit K, Buaboonnam J, et al. GD2-specific chimeric antigen receptor-modified T cells targeting retinoblastoma – assessing tumor and T cell interaction. Trans Oncol (2021) 14(2):100971. doi: 10.1016/j.tranon.2020.100971
97. Hoseini SS, Dobrenkov K, Pankov D, Xu XL, Cheung N-KV. Bispecific antibody does not induce T-cell death mediated by chimeric antigen receptor against disialoganglioside GD2. OncoImmunology. (2017) 6(6):e1320625. doi: 10.1080/2162402X.2017.1320625
98. Rossig C, Bollard CM, Nuchtern JG, Rooney CM, Brenner MK. Epstein-Barr Virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. (2002) 99(6):2009–16. doi: 10.1182/blood.V99.6.2009
99. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med (2008) 14(11):1264–70. doi: 10.1038/nm.1882
100. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. (2011) 118(23):6050–6. doi: 10.1182/blood-2011-05-354449
101. Pulè MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther (2005) 12(5):933–41. doi: 10.1016/j.ymthe.2005.04.016
102. Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther J Am Soc Gene Ther (2017) 25(9):2214–24. doi: 10.1016/j.ymthe.2017.05.012
103. Quintarelli C, Orlando D, Boffa I, Guercio M, Polito VA, Petretto A, et al. Choice of costimulatory domains and of cytokines determines CAR T-cell activity in neuroblastoma. Oncoimmunology. (2018) 7(6):e1433518. doi: 10.1080/2162402X.2018.1433518
104. Tumino N, Weber G, Besi F, Del Bufalo F, Bertaina V, Paci P, et al. Polymorphonuclear myeloid-derived suppressor cells impair the anti-tumor efficacy of GD2.CAR T-cells in patients with neuroblastoma. J Hematol Oncol (2021) 14(1):191. doi: 10.1186/s13045-021-01193-0
105. Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. (2022) 603(7903):934–41. doi: 10.1038/s41586-022-04489-4
106. Nakamura K, Tanaka Y, Shitara K, Hanai N. Construction of humanized anti-ganglioside monoclonal antibodies with potent immune effector functions. Cancer Immunol Immunother (2001) 50(5):275–84. doi: 10.1007/PL00006689
107. Thomas S, Straathof K, Himoudi N, Anderson J, Pule M. An optimized GD2-targeting retroviral cassette for more potent and safer cellular therapy of neuroblastoma and other cancers. PLos One (2016) 11(3):e0152196. doi: 10.1371/journal.pone.0152196
108. Straathof K, Flutter B, Wallace R, Jain N, Loka T, Depani S, et al. Antitumor activity without on-target off-tumor toxicity of GD2–chimeric antigen receptor T cells in patients with neuroblastoma. Sci Trans Med (2020) 12(571):eabd6169. doi: 10.1126/scitranslmed.abd6169
109. Straathof K, Flutter B, Wallace R, Thomas S, Cheung G, Collura A, et al. Abstract CT145: A cancer research UK phase I trial of anti-GD2 chimeric antigen receptor (CAR) transduced T-cells (1RG-CART) in patients with relapsed or refractory neuroblastoma. Cancer Res (2018) 78(13 Supplement):CT145–CT. doi: 10.1158/1538-7445.AM2018-CT145
110. Zhou Y, Zhang J, Jin H, Chen Z, Wu Q, Li W, et al. Prokaryotic expression and refolding of EGFR extracellular domain and generation of phage display human scFv against EGFR. Biomed Pharmacother (2013) 67(8):737–43. doi: 10.1016/j.biopha.2013.03.019
111. Lu D, Jimenez X, Witte L, Zhu Z. The effect of variable domain orientation and arrangement on the antigen-binding activity of a recombinant human bispecific diabody. Biochem Biophys Res Commun (2004) 318(2):507–13. doi: 10.1016/j.bbrc.2004.04.060
112. Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci (2016) 59(5):468–79. doi: 10.1007/s11427-016-5023-8
113. Guo Y, Feng K, Liu Y, Wu Z, Dai H, Yang Q, et al. Phase I study of chimeric antigen receptor-modified T cells in patients with EGFR-positive advanced biliary tract cancers. Clin Cancer Res (2018) 24(6):1277–86. doi: 10.1158/1078-0432.CCR-17-0432
114. Liu Y, Guo Y, Wu Z, Feng K, Tong C, Wang Y, et al. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy. (2020) 22(10):573–80. doi: 10.1016/j.jcyt.2020.04.088
115. Li H, Huang Y, Jiang D-Q, Cui L-Z, He Z, Wang C, et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis (2018) 9(2):177. doi: 10.1038/s41419-017-0238-6
116. Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Trans Med (2015) 7(275):275ra22. doi: 10.1126/scitranslmed.aaa4963
117. Nakayashiki N, Yoshikawa K, Nakamura K, Hanai N, Okamoto K, Okamoto S, et al. Production of a single-chain variable fragment antibody recognizing type III mutant epidermal growth factor receptor. Japanese J Cancer Res Gann. (2000) 91(10):1035–43. doi: 10.1111/j.1349-7006.2000.tb00882.x
118. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Trans Med (2017) 9(399):eaaa0984. doi: 10.1126/scitranslmed.aaa0984
119. Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther (2012) 23(10):1043–53. doi: 10.1089/hum.2012.041
120. Sampson JH, Choi BD, Sanchez-Perez L, Suryadevara CM, Snyder DJ, Flores CT, et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res (2014) 20(4):972–84. doi: 10.1158/1078-0432.CCR-13-0709
121. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J immunother (Hagerstown Md 1997). (2019) 42(4):126–35. doi: 10.1097/CJI.0000000000000260
122. Cao Y, Marks JW, Liu Z, Cheung LH, Hittelman WN, Rosenblum MG. Design optimization and characterization of Her2/neu-targeted immunotoxins: comparative in vitro and in vivo efficacy studies. Oncogene. (2014) 33(4):429–39. doi: 10.1038/onc.2012.612
123. Wels W, Harwerth IM, Mueller M, Groner B, Hynes NE. Selective inhibition of tumor cell growth by a recombinant single-chain antibody-toxin specific for the erbB-2 receptor. Cancer Res (1992) 52(22):6310–7.
124. Ahmed N, Salsman VS, Yvon E, Louis CU, Perlaky L, Wels WS, et al. Immunotherapy for osteosarcoma: genetic modification of T cells overcomes low levels of tumor antigen expression. Mol Ther J Am Soc Gene Ther (2009) 17(10):1779–87. doi: 10.1038/mt.2009.133
125. Ahmed N, Ratnayake M, Savoldo B, Perlaky L, Dotti G, Wels WS, et al. Regression of experimental medulloblastoma following transfer of HER2-specific T cells. Cancer Res (2007) 67(12):5957–64. doi: 10.1158/0008-5472.CAN-06-4309
126. Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol (2015) 33(15):1688–96. doi: 10.1200/JCO.2014.58.0225
127. Hegde M, Joseph SK, Pashankar F, DeRenzo C, Sanber K, Navai S, et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat Commun (2020) 11(1):3549. doi: 10.1038/s41467-020-17175-8
128. Navai SA, Derenzo C, Joseph S, Sanber K, Byrd T, Zhang H, et al. Abstract LB-147: Administration of HER2-CAR T cells after lymphodepletion safely improves T cell expansion and induces clinical responses in patients with advanced sarcomas. Cancer Res (2019) 79(13_Supplement):LB–147-LB. doi: 10.1158/1538-7445.AM2019-LB-147
129. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific chimeric antigen receptor–modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol (2017) 3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184
130. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther J Am Soc Gene Ther (2010) 18(4):843–51. doi: 10.1038/mt.2010.24
131. Lenferink AEG, McDonald PC, Cantin C, Grothe S, Gosselin M, Baardsnes J, et al. Isolation and characterization of monoclonal antibodies against human carbonic anhydrase-IX. MAbs. (2021) 13(1):1999194. doi: 10.1080/19420862.2021.1999194
132. Lamers CHJ, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: Clinical evaluation and management of on-target toxicity. Mol Ther (2013) 21(4):904–12. doi: 10.1038/mt.2013.17
133. Choi J, Kim M, Lee J, Seo Y, Ham Y, Lee J, et al. Antigen-binding affinity and thermostability of chimeric mouse-chicken IgY and mouse-human IgG antibodies with identical variable domains. Sci Rep (2019) 9(1):19242. doi: 10.1038/s41598-019-55805-4
134. Ma Q, Safar M, Holmes E, Wang Y, Boynton AL, Junghans RP. Anti-prostate specific membrane antigen designer T cells for prostate cancer therapy. Prostate (2004) 61(1):12–25. doi: 10.1002/pros.20073
135. Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo ASY, et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: Possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate (2016) 76(14):1257–70. doi: 10.1002/pros.23214
136. Smith-Jones PM, Vallabahajosula S, Goldsmith SJ, Navarro V, Hunter CJ, Bastidas D, et al. In vitro characterization of radiolabeled monoclonal antibodies specific for the extracellular domain of prostate-specific membrane antigen. Cancer Res (2000) 60(18):5237–43.
137. Nováková Z, Foss CA, Copeland BT, Morath V, Baranová P, Havlínová B, et al. Novel monoclonal antibodies recognizing human prostate-specific membrane antigen (PSMA) as research and theranostic tools. Prostate. (2017) 77(7):749–64. doi: 10.1002/pros.23311
138. Gong MC, Latouche JB, Krause A, Heston WD, Bander NH, Sadelain M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia (New York NY) (1999) 1(2):123–7. doi: 10.1038/sj.neo.7900018
139. Slovin SF, Wang X, Hullings M, Arauz G, Bartido S, Lewis JS, et al. Chimeric antigen receptor (CAR+) modified T cells targeting prostate-specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). J Clin Oncol (2013) 31(6_suppl):72.
140. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther J Am Soc Gene Ther (2018) 26(7):1855–66. doi: 10.1016/j.ymthe.2018.05.003
141. Narayan V, Barber-Rotenberg JS, Jung IY, Lacey SF, Rech AJ, Davis MM, et al. PSMA-targeting TGFβ-insensitive armored CAR t cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med (2022) 28(4):724–34. doi: 10.1038/s41591-022-01726-1
142. Jiang H, Shi Z, Wang P, Wang C, Yang L, Du G, et al. Claudin18.2-specific chimeric antigen receptor engineered T cells for the treatment of gastric cancer. JNCI: J Natl Cancer Institute (2019) 111(4):409–18. doi: 10.1093/jnci/djy134
143. Zhan X, Wang B, Li Z, Li J, Wang H, Chen L, et al. Phase I trial of claudin 18.2-specific chimeric antigen receptor T cells for advanced gastric and pancreatic adenocarcinoma. J Clin Oncol (2019) 37(15_suppl):2509.
144. Qi C, Gong J, Li J, Liu D, Qin Y, Ge S, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat Med (2022) 28(6):1189–98. doi: 10.1038/s41591-022-01800-8
145. Botta GP, Becerra CR, Jin Z, Kim DW, Zhao D, Lenz H-J, et al. Multicenter phase ib trial in the U.S. @ of salvage CT041 CLDN18.2-specific chimeric antigen receptor T-cell therapy for patients with advanced gastric and pancreatic adenocarcinoma. J Clin Oncol (2022) 40(16_suppl):2538.
146. Nazha B, Inal C, Owonikoko TK. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front Oncol (2020) 10:1000. doi: 10.3389/fonc.2020.01000
147. Horwacik I, Golik P, Grudnik P, Kolinski M, Zdzalik M, Rokita H, et al. Structural basis of GD2 ganglioside and mimetic peptide recognition by 14G2a antibody. Mol Cell Proteomics MCP. (2015) 14(10):2577–90. doi: 10.1074/mcp.M115.052720
148. Ahmed M, Cheung N-KV. Engineering anti-GD2 monoclonal antibodies for cancer immunotherapy. FEBS Letters (2014) 588(2):288–97. doi: 10.1016/j.febslet.2013.11.030
149. Perez Horta Z, Goldberg JL, Sondel PM. Anti-GD2 mAbs and next-generation mAb-based agents for cancer therapy. Immunotherapy. (2016) 8(9):1097–117. doi: 10.2217/imt-2016-0021
150. Ahmed M, Hu J, Cheung NK. Structure based refinement of a humanized monoclonal antibody that targets tumor antigen disialoganglioside GD2. Front Immunol (2014) 5:372. doi: 10.3389/fimmu.2014.00372
151. Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med (2019) 25(9):1408–14. doi: 10.1038/s41591-019-0549-5
152. Watanabe K, Kuramitsu S, Posey AD Jr., June CH. Expanding the therapeutic window for CAR T cell therapy in solid tumors: The knowns and unknowns of CAR T cell biology. Front Immunol (2018) 9:2486. doi: 10.3389/fimmu.2018.02486
153. Davenport AJ, Cross RS, Watson KA, Liao Y, Shi W, Prince HM, et al. Chimeric antigen receptor T cells form nonclassical and potent immune synapses driving rapid cytotoxicity. Proc Natl Acad Sci (2018) 115(9):E2068–E76. doi: 10.1073/pnas.1716266115
154. Li R, Ma C, Cai H, Chen W. The CAR T-cell mechanoimmunology at a glance. Advanced Science (2020) 7(24):2002628. doi: 10.1002/advs.202002628
155. Greenman R, Pizem Y, Haus-Cohen M, Goor A, Horev G, Denkberg G, et al. Shaping functional avidity of CAR T cells: Affinity, avidity, and antigen density that regulate response. Mol Cancer Ther (2021) 20(5):872–84. doi: 10.1158/1535-7163.MCT-19-1109
156. Abbott RC, Verdon DJ, Gracey FM, Hughes-Parry HE, Iliopoulos M, Watson KA, et al. Novel high-affinity EGFRvIII-specific chimeric antigen receptor T cells effectively eliminate human glioblastoma. Clin Trans Immunol (2021) 10(5):e1283. doi: 10.1002/cti2.1283
157. Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T Cell activation by antibody-like immunoreceptors: Increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. J Immunol (2004) 173(12):7647–53. doi: 10.4049/jimmunol.173.12.7647
158. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. (2019) 568(7750):112–6. doi: 10.1038/s41586-019-1054-1
159. Olson ML, Mause ERV, Radhakrishnan SV, Brody JD, Rapoport AP, Welm AL, et al. Low-affinity CAR T cells exhibit reduced trogocytosis, preventing rapid antigen loss, and increasing CAR T cell expansion. Leukemia. (2022) 36(7):1943–6. doi: 10.1038/s41375-022-01585-2
160. Michelozzi IM, Gomez-Castaneda E, Pohle RVC, Cardoso Rodriguez F, Sufi J, Puigdevall P, et al. The enhanced functionality of low-affinity CD19 CAR T cells is associated with activation priming and polyfunctional cytokine phenotype. Blood. (2020) 136:52–3. doi: 10.1182/blood-2020-141249
161. Rossi J, Paczkowski P, Shen YW, Morse K, Flynn B, Kaiser A, et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood. (2018) 132(8):804–14. doi: 10.1182/blood-2018-01-828343
162. Caserta S, Kleczkowska J, Mondino A, Zamoyska R. Reduced functional avidity promotes central and effector memory CD4 T cell responses to tumor-associated antigens. J Immunol (2010) 185(11):6545–54. doi: 10.4049/jimmunol.1001867
163. Wu S, Zhu W, Peng Y, Wang L, Hong Y, Huang L, et al. The antitumor effects of vaccine-activated CD8(+) T cells associate with weak TCR signaling and induction of stem-like memory T cells. Cancer Immunol Res (2017) 5(10):908–19. doi: 10.1158/2326-6066.CIR-17-0016
164. Sun S, Hao H, Yang G, Zhang Y, Fu Y. Immunotherapy with CAR-modified T cells: Toxicities and overcoming strategies. J Immunol Res (2018) 2018:2386187. doi: 10.1155/2018/2386187
165. Govern CC, Paczosa MK, Chakraborty AK, Huseby ES. Fast on-rates allow short dwell time ligands to activate T cells. Proc Natl Acad Sci (2010) 107(19):8724–9. doi: 10.1073/pnas.1000966107
166. Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, et al. Target antigen density governs the efficacy of anti–CD20-CD28-CD3 ζ chimeric antigen receptor–modified effector CD8+ T cells. J Immunol (2015) 194(3):911–20. doi: 10.4049/jimmunol.1402346
167. Harris DT, Hager MV, Smith SN, Cai Q, Stone JD, Kruger P, et al. Comparison of T cell activities mediated by human TCRs and CARs that use the same recognition domains. J Immunol (2018) 200(3):1088–100. doi: 10.4049/jimmunol.1700236
168. Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discovery (2020) 10(5):702–23. doi: 10.1158/2159-8290.CD-19-0945
169. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res (2015) 75(17):3596–607. doi: 10.1158/0008-5472.CAN-15-0159
170. Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, et al. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol Ther (2017) 25(8):1946–58. doi: 10.1016/j.ymthe.2017.04.024
171. Park S, Shevlin E, Vedvyas Y, Zaman M, Park S, Hsu YS, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep (2017) 7(1):14366. doi: 10.1038/s41598-017-14749-3
172. Salzer B, Schueller CM, Zajc CU, Peters T, Schoeber MA, Kovacic B, et al. Engineering AvidCARs for combinatorial antigen recognition and reversible control of CAR function. Nat Commun (2020) 11(1):4166. doi: 10.1038/s41467-020-17970-3
173. Castellarin M, Sands C, Da T, Scholler J, Graham K, Buza E, et al. A rational mouse model to detect on-target, off-tumor CAR T cell toxicity. JCI Insight (2020) 5(14):e136012. doi: 10.1172/jci.insight.136012
174. Giardino Torchia ML, Gilbreth R, Merlino A, Sult E, Monks N, Chesebrough J, et al. Rational design of chimeric antigen receptor T cells against glypican 3 decouples toxicity from therapeutic efficacy. Cytotherapy. (2022) 24(7):720–32. doi: 10.1016/j.jcyt.2022.03.008
175. Corzo J. Time, the forgotten dimension of ligand binding teaching. Biochem Mol Biol Educ (2006) 34(6):413–6. doi: 10.1002/bmb.2006.494034062678
176. Stone JD, Chervin AS, Kranz DM. T-Cell receptor binding affinities and kinetics: impact on T-cell activity and specificity. Immunology. (2009) 126(2):165–76. doi: 10.1111/j.1365-2567.2008.03015.x
177. Aleksic M, Dushek O, Zhang H, Shenderov E, Chen J-L, Cerundolo V, et al. Dependence of T cell antigen recognition on T cell receptor-peptide MHC confinement time. Immunity. (2010) 32(2):163–74. doi: 10.1016/j.immuni.2009.11.013
178. Siller-Farfán JA, Dushek O. Molecular mechanisms of T cell sensitivity to antigen. Immunol Rev (2018) 285:194–205. doi: 10.1111/imr.12690
179. Landry JP, Ke Y, Yu G-L, Zhu XD. Measuring affinity constants of 1450 monoclonal antibodies to peptide targets with a microarray-based label-free assay platform. J Immunol Methods (2015) 417:86–96. doi: 10.1016/j.jim.2014.12.011
180. Xu C, Rafique A, Potocky T, Paccaly A, Nolain P, Lu Q, et al. Differential binding of sarilumab and tocilizumab to IL-6Rα and effects of receptor occupancy on clinical parameters. J Clin Pharmacol (2021) 61(5):714–24. doi: 10.1002/jcph.1795
Keywords: Adoptive cell therapy, chimeric antigen receptors (CAR), CAR T cells, antigen-binding domain, T cell engineering, tumor immunotherapy, solid tumors
Citation: Mao R, Kong W and He Y (2022) The affinity of antigen-binding domain on the antitumor efficacy of CAR T cells: Moderate is better. Front. Immunol. 13:1032403. doi: 10.3389/fimmu.2022.1032403
Received: 30 August 2022; Accepted: 21 September 2022;
Published: 17 October 2022.
Edited by:
Zong Sheng Guo, University at Buffalo, United StatesReviewed by:
Bei Liu, The Ohio State University, United StatesYibo Yin, First Affiliated Hospital of Harbin Medical University, China
Bin Zhang, Northwestern University, United States
Copyright © 2022 Mao, Kong and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yukai He, eWhlQGF1Z3VzdGEuZWR1