
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 06 October 2022
Sec. Viral Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1031608
This article is part of the Research Topic Immunotherapeutics Development against Hantaviruses View all 6 articles
 Fatima Noor1
Fatima Noor1 Usman Ali Ashfaq1*
Usman Ali Ashfaq1* Muhammad Asif1
Muhammad Asif1 Muhammad Muzammal Adeel2
Muhammad Muzammal Adeel2 Abdulrahman Alshammari3
Abdulrahman Alshammari3 Metab Alharbi3
Metab Alharbi3Hemorrhagic fever with renal syndrome (HFRS) is an acute zoonotic disease transmitted through aerosolized excrement of rodents. The etiology of HFRS is complex due to the involvement of viral factors and host immune and genetic factors. The viral species that dominantly cause HFRS are Puumala virus (PUUV), Seoul virus (SEOV), Dobrava-Belgrade virus (DOBV), and Hantaan virus (HTNV). Despite continuous prevention and control measures, HFRS remains a significant public health problem worldwide. The nucleocapsid protein of PUUV, SEOV, DOBV, and HTNV is a multifunctional viral protein involved in various stages of the viral replication cycle. However, the exact role of nucleoproteins in viral pathogenesis is yet to be discovered. Targeting a universal host protein exploited by most viruses would be a game-changing strategy that offers broad-spectrum solutions and rapid epidemic control. The objective of this study is to understand the replication and pathogenesis of PUUV, SEOV, DOBV, and HTNV by targeting tyrosine-based motif (YXXΦ[I/L/M/F/V]) and YXXΦ-like tetrapeptides. In the light of the current study, in silico analysis uncovered many different YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides within nucleoproteins of PUUV, SEOV, DOBV, and HTNV. Following that, the 3D structures of nucleoproteins were predicted using AlphaFold2 to map the location of YXXΦ[I/L/M/F/V] motif and YXXΦ-like tetrapeptides in a 3D environment. Further, in silico analysis and characterization of Post Translational Modifications (PTMs) revealed multiple PTMs sites within YXXΦ[I/L/M/F/V] motif and YXXΦ-like tetrapeptides, which contribute to virulence and host immune regulation. Our study proposed that the predicted YXXΦ[I/L/M/F/V] motif and YXXΦ-like tetrapeptides may confer specific functions such as virulence, host immune regulation, and pathogenesis to nucleoproteins of PUUV, SEOV, DOBV, and HTNV. However, in vivo and in vitro studies on YXXΦ[I/L/M/F/V] motif and YXXΦ-like tetrapeptides will assign new biological roles to these antiviral targets.
Hemorrhagic fever with renal syndrome (HFRS) is a major rodent-borne zoonosis (1, 2). About 60,000–100,000 cases of human HFRS were reported annually in more than 70 countries around the world (3). HFRS is characterized by hypotension, fever, kidney damage, and bleeding. HFRS is an acute zoonotic viral disease and a distinct clinical syndrome endemic in Europe and Asia (4–6). The spatial heterogeneity of HFRS may be influenced by geographical differences and socioeconomic status (7, 8). Moreover, the outbreak of HFRS can occur in a short time and infect a large community of people (9, 10). Until now, effective clinical treatment is lacking for HFRS and consists of symptomatic and supportive care, mainly focusing on maintaining blood pressure and balancing fluid and electrolyte composition (11). Therefore, there is an urgent need to find an effective treatment for HFRS.
HFRS is caused by different species of hantaviruses including Puumala virus (PUUV) (12), Seoul virus (SEOV) (13), Dobrava-Belgrade virus (DOBV) (14), and Hantaan virus (HTNV) (15). Hantaviruses are membrane-enveloped viruses. Hantavirus is a genus of negative-sense, single-stranded, enveloped RNA viruses in the family Hantaviridae within the order Bunyavirales (16–18). The hantavirus genome is composed of a three segments coding for G1 and G2 glycoproteins, nucleocapsid protein, and viral polymerase. Comparatively to non-pathogenic strains, pathogenic hantaviruses significantly alter the transcriptional activity of many cellular genes (19, 20). Recent studies provide solid evidence that hantaviruses’ nucleocapsid proteins have a key role in virus transcription, replication, and assembly (21, 22). The nucleoprotein, encoded by the S segment, of hantaviruses consists of 429 to 433 amino acids (23). This nucleoprotein interacts with the host proteins and limits the activation of the major antiviral signaling pathways in affected cells. Furthermore, hantaviruses do not appear to have any pathogenic effects on their natural hosts. However, transmission to humans can result in deadly illnesses, particularly in South America, where the lethality rate of the hantavirus exceeds 40% (24, 25).
YXXΦ[I/L/M/F/V] is a tyrosine-based sorting motif in which Φ is an amino acid with bulky hydrophobic side chain, and X can be any residues (26). YXXΦ[I/L/M/F/V] motif was crucial for endocytosis from the cell surface and intracellular protein transport (27). YXXΦ[I/L/M/F/V] motif has a significant role in the internalization process and is typically found in the cytoplasmic domains of various transmembrane receptors, including the transferrin and asialoglycoprotein receptors (28). In the secretory and endosomal pathways, YXXΦ[I/L/M/F/V] signal motifs have dual specificity in the form of an endocytotic functional motif and a trafficking signal (29). Some viruses have incorporated these signaling motifs into their proteins and then utilize it to regulate the immune system (30–32). Karamichali et al. (33) reported that YXXΦ motif plays a pivotal role in the events following Hepatitis C virus (HCV) non-enveloped capsid-like particles clathrin-mediated endocytosis, which affects the HCV life cycle by enhancing viral replication.
The C terminus of Gn-cytoplasmic tail contains a highly conserved YxxL motif, and Hantavirus Cardiopulmonary Syndrome-causing hantaviruses are claimed to have two such motifs that form an immunoreceptor tyrosine-based activation motif (ITAM) (34). However, no study todate provide evidences about the existence of YXXΦ[I/L/M/F/V] motifs in HFRS-causing hantavirus. In the current work, YXXΦ[I/L/M/F/V] motif and YXXΦ-like tetrapeptides were mapped within nucleoprotein sequences of PUUV, SEOV, DOBV, and HTNV. To our knowledge, this is the first study that uncovered the putative YXXΦ[I/L/M/F/V] motif and YXXΦ-like tetrapeptides as well as their potential role in the host immune regulation, pathogenesis, and virulence. Moreover, these findings have sparked a new interest in the search treatment options for HFRS by targeting YXXΦ[I/L/M/F/V] motif and YXXΦ-like tetrapeptides. Additionally, we believe that targeting YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides with pharmaceutical drugs that will result in PTMs with appropriate detrimental effects on immunological qualities and structural integrity of nucleoproteins, may contribute to design of effective anti-HFRS treatments.
Retrieval of protein sequences is a preliminary step for identifying the tyrosine-based motifs to predict the host-viral interaction. The nucleoprotein sequences of Puumala virus (PUUV), Seoul virus (SEOV) Dobrava-Belgrade virus (DOBV), and Hantaan virus (HTNV) were retrieved from UniProt (35) with the following UniProt identifiers: P27313 (PUUV), W0LUE3 (SEOV), Q805Q9 (DOBV), and P05133 (HTNV) respectively. These sequences were then analyzed for putative YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides.
Aligning multiple sequences assess highly conserved regions as compared to other regions. Thus, similar regions may help classify sequences or inform experiment design. Regarding this, the obtained nucleoprotein sequences of PUUV, SEOV, DOBV, and HTNV were then subjected to Clustal Omega (36) for Multiple Sequence Alignment (MSA) with default parameters (includes single distance matrix calculation along with clustering and one set of N‐1 pairwise alignments). Clustal Omega is MSA software that can accurately align thousands of sequences in a short time.
The nucleoprotein sequences of PUUV, SEOV, DOBV, and HTNV were scanned with Eukaryotic Linear Motif (ELM) (37) web server for predicting the tyrosine-based motif (YXXΦ[I/L/M/F/V]) based on contextual information, including taxonomy, evolutionary conservation, cellular compartment, and structural features. ELM is a freely available repository of short linear motifs that have been experimentally verified and manually curated. The motif probability cutoff was set to 100 and other parameters were set to the default values. Extensive literature search was also made to map the YXXΦ[I/L/M/F/V] motif and YXXΦ-like tetrapeptides within nucleoprotein sequences.
The topology of a protein structure provides a brief description of its fold, including only the sequence of secondary structure elements along with their approximate orientations and relative spatial coordinates. This information can be embodied in a two-dimensional (2D) diagram of protein topology. Protter (38) web server was used to generate a pictorial representation of the 2D topology of YXXΦ[I/L/M/F/V] motifs along with YXXΦ-like tetrapeptides in nucleoprotein sequences. Protter predicts transmembrane sites via experimental proteomic data as well as annotated sequence information. 2D structure prediction showed the N and C terminal topology of nucleoprotein sequences.
Predicting the secondary structure of the proteins provide a meaningful insight into the functions of proteins which serve as initial steps toward the tertiary structure prediction. In the current study, DeepGSH (39) web server was used for predicting secondary structural elements including α-helices, coils, and, β-sheets of nucleoprotein sequences within PUUV, SEOV, DOBV, and HTNV. This in turn provides a snapshot of the location of YXXΦ[I/L/M/F/V] motifs, including YXXΦ-like tetrapeptides within viral sequences. DeepGSH showed excellent robustness and better performance than existing tools, thus, DeepGSH was suitable for predicting secondary structural features.
Internal disordered regions exhibit different flavors or types of disorder. PONDR (Predictor of Natural Disordered Regions) (40) was used to calculate the per-residue disorder distribution in nucleoprotein sequences of PUUV, SEOV, DOBV, and HTNV. In PONDR webserver, XL1_XT predictor was selected for predicting disordered regions while other parameters were kept by default. The PONDR web server uncovers long and short disordered regions that might differ in their amino acid characteristics.
Post-translational modifications (PTMs) can happen at any step of the protein lifespan and have a key role in many different biological processes by influencing dynamics and structure of proteins. By modifying inter- and intramolecular interactions, PTMs not only add new functionalities but also dynamically regulate the activity of proteins. Current study used various tools to predict PTM sites in nucleoprotein sequences of PUUV, SEOV, DOBV, and HTNV. NetPhos-3.1 server (41) was used to prefigure Ser, Tyr, and Thr phosphorylations with default settings. NetPhos-3.1 server used ensembles of neural networks to predict phosphorylation sites in nucleoprotein sequences. Similarly, iNitro-Tyr (42) web server was used for Tyr nitration. Generating a complete and accurate dataset is required to evaluate the Tyr sulfation and its interaction with other Tyr variations. Unfortunately, the experimental identification of sulfation sites is a time- and labor-consuming. As an alternate, computational prediction of sulfation sites using protein primary sequences might provide a helping hand in the identification of putative sulfated residues. Regarding this, GPS-TSP (43) web server was employed to predict the probability of Tyr sulfation within YXXΦ[I/L/M/F/V] motifs.
The three-dimensional (3D) structures of nucleoproteins were predicted using AlphaFold2 (44) to map the positions of both YXXΦ[I/L/M/F/V] motifs as well as YXXΦ-like tetrapeptides of PUUV, SEOV, DOBV, and HTNV in a 3D environment. AlphaFold2 is a neural network-based protein structure prediction program created by Google DeepMind. The AlphaFold2-based prediction was run with the “single sequence” mode. This algorithm first searches for homologous sequences with existing structures to use as a scaffold for placing the new sequence. Additionally, we also specified that the algorithm should run an Amber relaxation procedure to repair any structural violations in the predicted model. AlphaFold2 generated a list of models, but the top-ranked model was selected on the basis of the highest overall pLDDT scores. AlphaFold produces a per-residue estimate of its confidence on a scale from 0 - 100. This confidence measure is called pLDDT and corresponds to the model’s predicted score. Regions with pLDDT > 90 are expected to be modelled to high accuracy. Regions with pLDDT between 70 and 90 are expected to be modelled well (a generally good backbone prediction). While, regions with pLDDT between 50 and 70 are low confidence and should be treated with caution. Lastly, the final predicted structures of PUUV, SEOV, DOBV, and HTNV were superimposed using Chimera X (45) to measure the structural similarity of proteins.
Mapping immune epitopes can provide a helping hand in the identification of antigenic regions within motifs of pathogenic proteins (46, 47). Following that, Immune Epitope Database (IEDB) resource was used to predict immune epitopes within YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides as they might activate B cells by functioning as linear antigenic epitopes (48). In IEDB web server, Chou & Fasman β-turn predictor was used for antibody epitope prediction. Lastly, the Immunoreceptor Tyrosine-based Inhibition Motifs (ITIMs) were predicted using the ELM webserver. ITIM-containing molecules regulate a wide range of biological processes, mainly but not exclusively associated with immunity. The workflow of the present study is displayed in Figure 1.

Figure 1 Graphical representation of methodology adopted to uncover YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides within nucleoprotein sequences of PUUV, SEOV, DOBV, and HTNV.
MSA highlighted a high homology between the nucleoprotein sequence of PUUV, SEOV DOBV, and HTNV (Figure 2). HTNV has 82.98% similarity with DOBV, 79.95% similarity with SEOV, and 61.07% similarity with PUUV. These four nucleoprotein sequences were then mapped for potential YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides using ELM web server. It is noteworthy that PUUV, DOBV, and HTNV shared identical YXXΦ[I/L/M/F/V] motif present at position (98–101) and were later named as “Conserved” (YGNV) motif (Table 1). Similarly, PUUV has one unique YXXΦ[I/L/M/F/V] motif named as “PUUV-upstream 1”(YVSM) that is located in (178–181) amino acids. SEOV has two unique YXXΦ[I/L/M/F/V] motifs named as “SEOV-upstream 1”(YEDF) and ‘SEOV-upstream 2”(YVPM) motifs which is present at (165–168) and (178–181) amino acids respectively. DOBV has only “Conserved” (YGNV) motif. On the other hand, HTNV has one unique motif named as “HTNV-upstream” which is present at (178–181) amino acids.

Figure 2 MSA of PUUV, SEOV, DOBV, and HTNV nucleoprotein sequences. Stars: amino acid sequence similarity; colons: amino acid sequence difference.
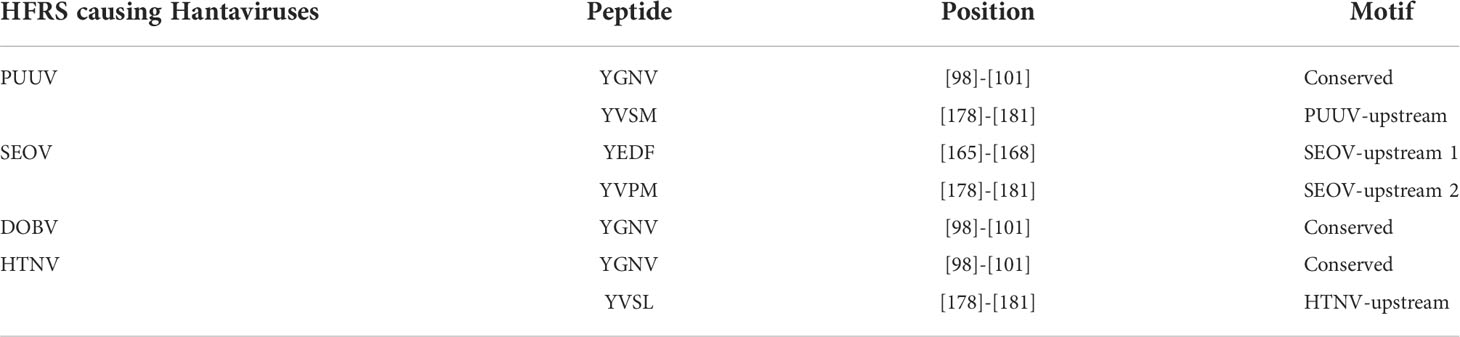
Table 1 Canonical YXXΦ[I/L/M/F/V] motifs within nucleoprotein sequences of HFRS causing Hantaviruses.
Following that, some YXXΦ-like tetrapeptides were also predicted in nucleoprotein sequences (Figure 3) in which YXXΦ[I/L/M/F/V] motif is “distorted” owing to residual substitutions. In PUUV, DOBV, and HTNV, the “SEOV-upstream 1” has residual substitutions. Therefore, the “SEOV-upstream 1” converted into FEDI (165-168 amino acids) YXXΦ-like tetrapeptides in PUUV, FEDV (165-168 amino acids) YXXΦ-like tetrapeptides in DOBV and HTNV.

Figure 3 Illustrative representation of YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides within viral sequences. YXXΦ[I/L/M/F/V] motifs are highlighted with yellow color and the YXXΦ-like tetrapeptides are highlighted with green color. Stars, amino acid sequence similarity; colons, amino acid sequence difference.
The two-dimensional (2D) topology of motifs and intracellular and extracellular regions were visualized with the Protter web server (Figure 4). The 2D topology prediction using Protter demonstrated that nucleoprotein sequences have their C-termini located intracellularly while N termini is extracellularly, confirming Nin-Cout topology. Moreover, the Protter web server predicts that “Conserved” (YGNV) motif is present in extracellular regions while other “PUUV-upstream”, “SEOV-upstream 1”, “SEOV-upstream 2”, and “HTNV-upstream” motifs and YXXΦ-like tetrapeptides present in the intracellular region.
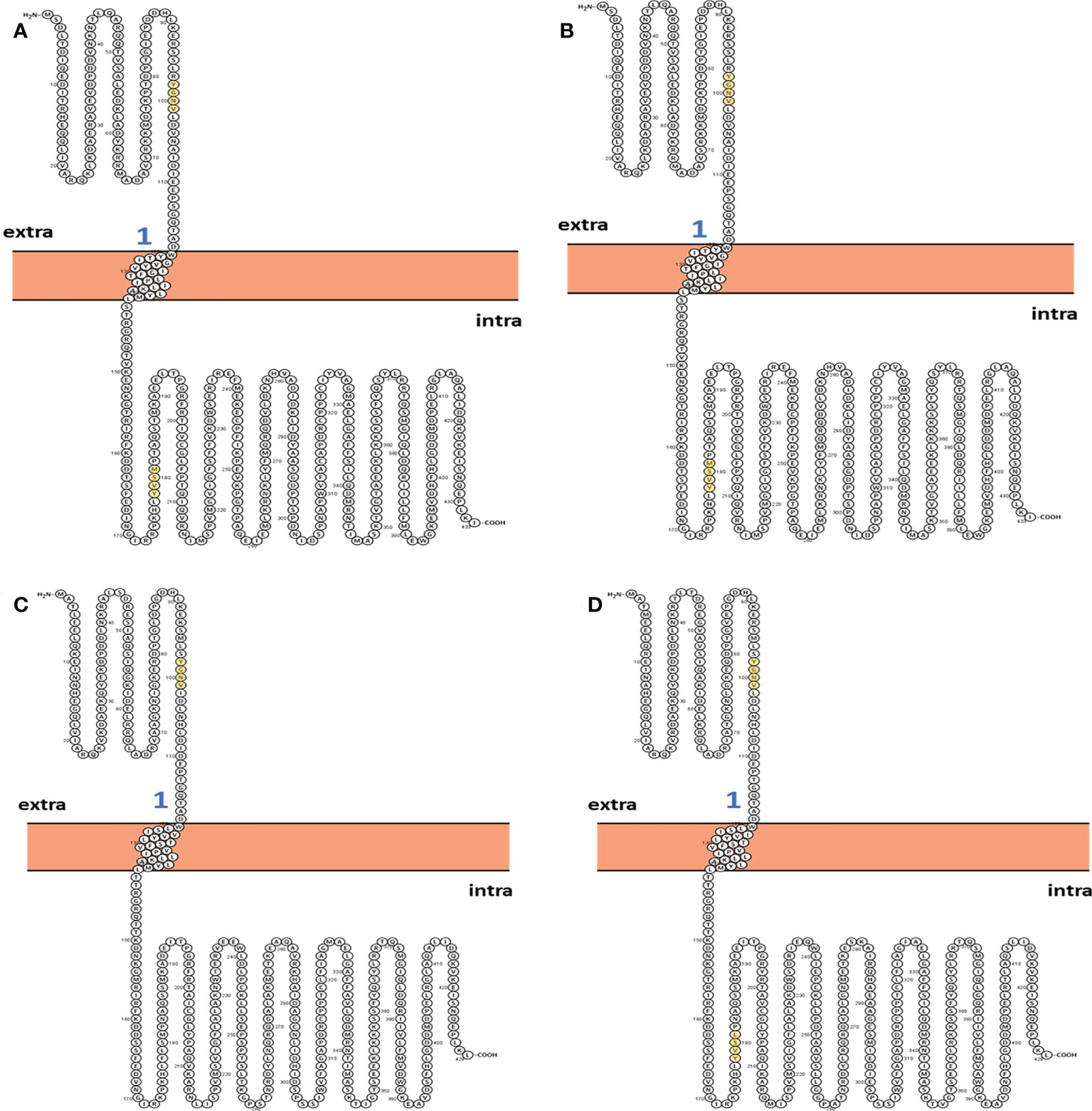
Figure 4 2D Topology of (A) PUUV, (B) SEOV, (C) DOBV, and (D) HTNV motifs.
Secondary structure predictions are increasingly becoming the working horse for predicting the function of motifs. Furthermore, the secondary structural elements particularly α-helices, coils, and, β-sheets of nucleoprotein sequences were uncovered with the DeepGSH server (Figure 5). DeepGSH server revealed that “Conserved” (YGNV) motif, “PUUV-upstream” (YVSM), “SEOV-upstream 1”(YEDF), and “SEOV-upstream 2”(YVPM), “HTNV-upstream” (YVSL) are situated in coil segments. While YXXΦ-like tetrapeptides were also found to be located on short coil segments. No other differences were observed.
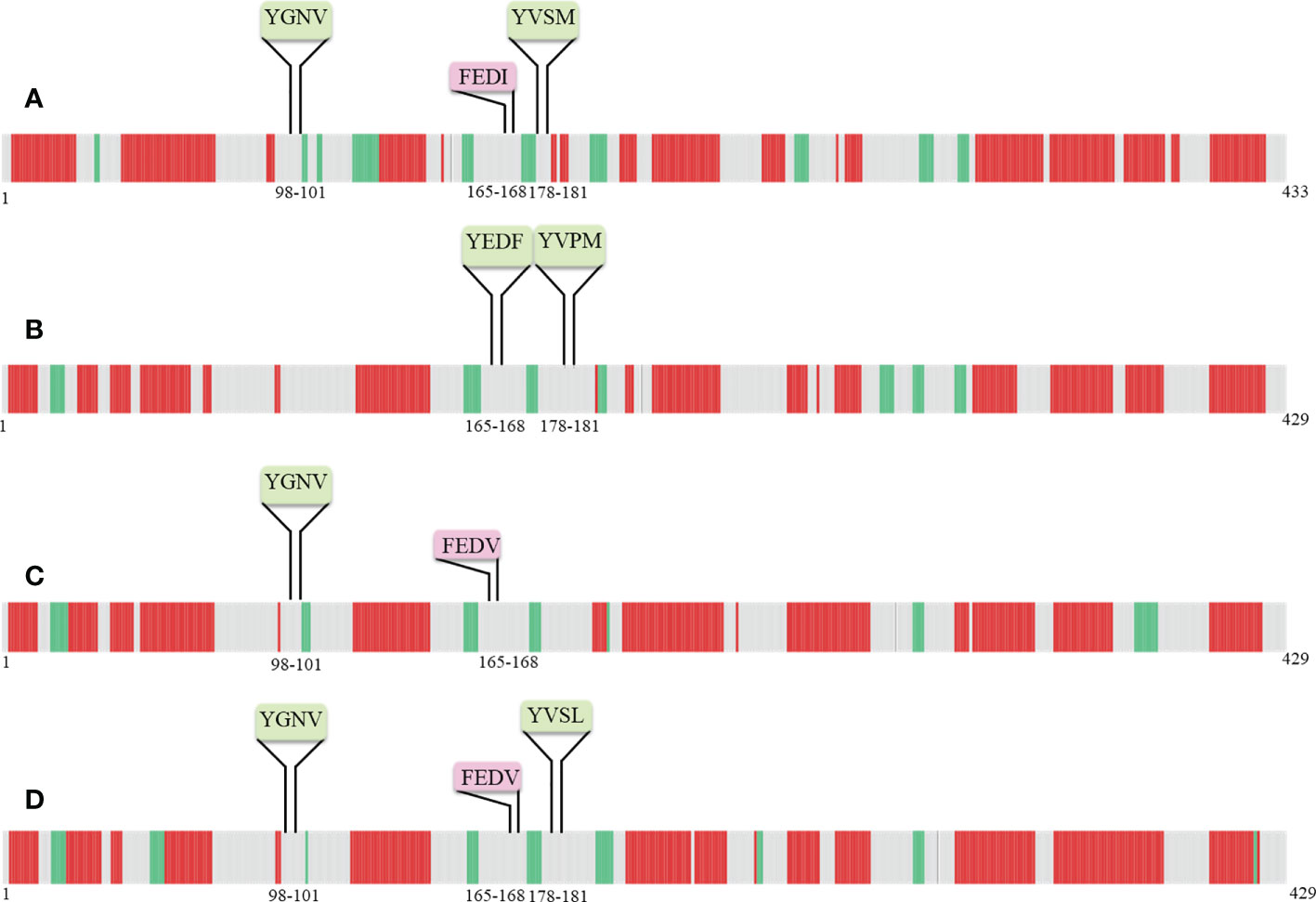
Figure 5 Pictorial representation of secondary structure of (A) PUUV, (B) SEOV, (C) DOBV, and (D) HTNV nucleoprotein sequences. α-helices represented with red color, β-strand represented with green color, while grey color indicated coils.
Internal disordered regions in proteins are a class of proteins that do not adopt stable secondary or tertiary structure but instead exist in a heterogeneous ensemble of conformations. Such regions give viral proteins a great flexibility and enable fast adaptability to the host environment, evasion of host defense mechanisms, and survival. Regarding this, PONDR-XL1_XT predictor was employed, revealing the disordered and ordered regions of nucleoprotein sequences. It is worth noting that all YXXΦ-like tetrapeptides and YXXΦ[I/L/M/F/V] motifs are present in the ordered region of viral proteins (Table 2).
Table 2 Enlists internal disordered regions of PUUV, SEOV, DOBV, and HTNV.
Post-Translational Modifications (PTMs), are among the most crucial mechanisms for enhancing, modifying, or suppressing the functions of proteins. Although analyzing these changes poses difficult problems, its identification yields valuable insights about their active role in biological processes. As PTMs has a key role to play in the regulation of biological processes, therefore these modifications may be conserved across orthologous proteins. At the same time, the divergence of PTMs might contribute to phenotypic diversity. In the framework of the present study, different tools and databases were used to identify PTM sites within both YXXΦ-like tetrapeptides and YXXΦ[I/L/M/F/V] motifs.
Phosphorylation cascades involve a series of mechanisms for controlling the cell-division cycle, two-component systems, and signal transductions. Prediction of phosphorylated residues within the YXXΦ[I/L/M/F/V] motifs of nucleoproteins using NetPhos3.1 web server uncovered that Tyr98 of YGNV (conserved YXXΦ motif) can be phosphorylated in PUUV, DOB, and HTNV (Figure 6). Similarly, Tyr178 and Ser 180 of the “PUUV-upstream motif” (YVSM) can be phosphorylated. In case of SEOV, the Tyr165 and Tyr 178 of the ‘SEOV-upstream 1” motif (YEDF)and SEOV-upstream 2 motif (YVPM) can be phosphorylated. In the same vein, Tyr178 and Ser 180 of “HTNV-upstream” (YVSL) was found to be phosphorylated in the HTNV.

Figure 6 Representation of phosphorylated amino acids within the YXXΦ[I/L/M/F/V] motifs of viral sequences. Stars, amino acid sequence similarity; colons, amino acid sequence difference.
Protein Tyr nitration is a PTM mediated by nitric oxide-derived molecules. Tyr nitration is a free radical [•NO2 (or •NO or ONOO+)] process involving the intermediacy of tyrosyl radicals; in spite of being a nonenzymatic process, nitration is selectively directed toward a limited subset of Tyr residues. Prediction with iNitro-Tyr server revealed that Tyr98 of “Conserved” (YGNV) motif in HTNV is nitrated (Figure 7). It is noteworthy that, Leu (at -2 position) increases the probability of Tyr nitration of “Conserved” motif. Similarly, Tyr165 of “SEOV-upstream 1” (YEDF) motif in SEOV is nitrated. No other Tyr was found to be nitrated in PUU and DOBV.
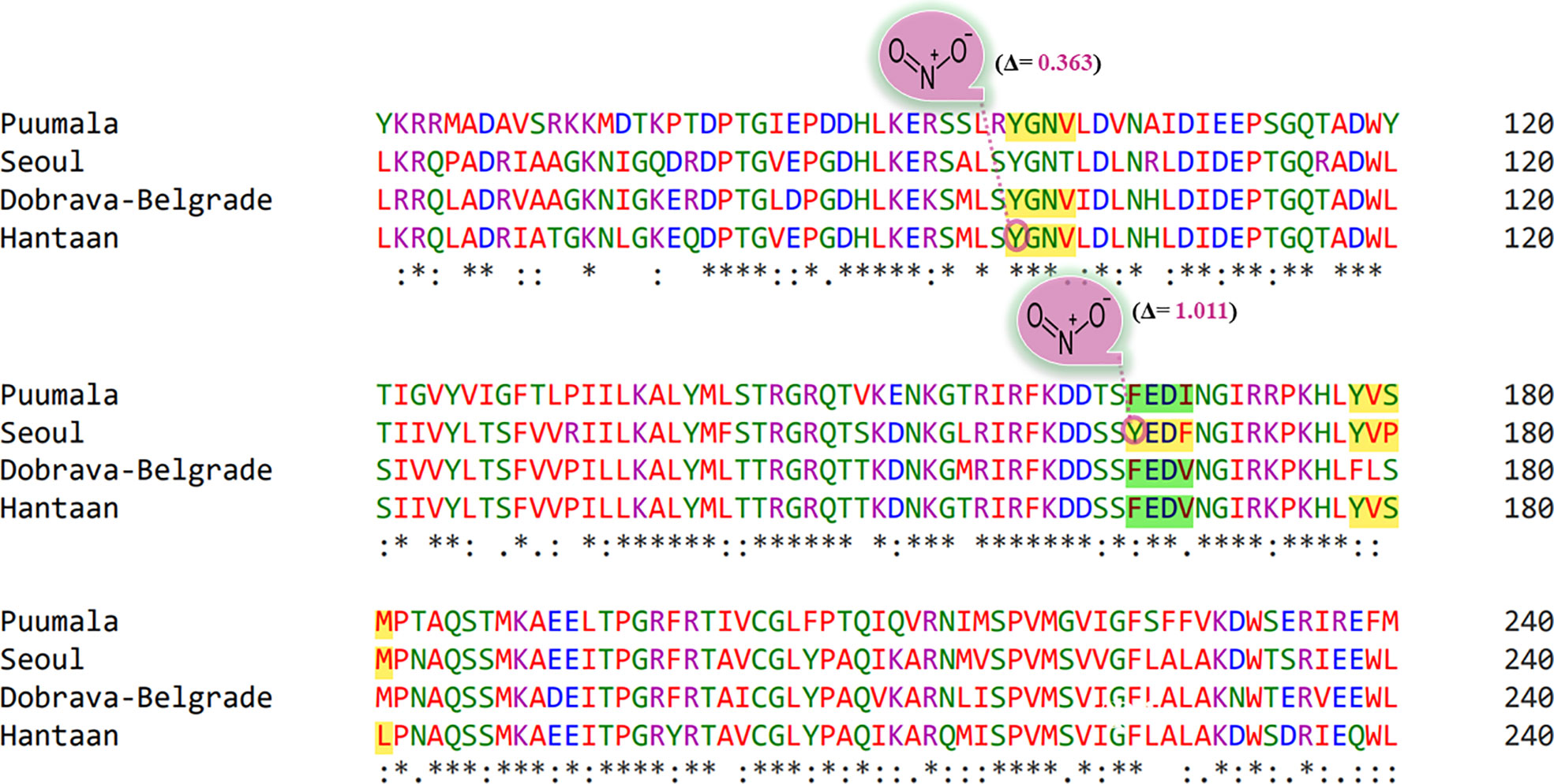
Figure 7 Graphical representation of putative nitrated residues within viral sequences. Stars, amino acid sequence similarity; colons, amino acid sequence difference.
Tyr sulfation is a PTM in which a sulfonate group from the donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS) is transferred to the hydroxyl group of a peptidyltyrosine residue. In this regard, GPS-TSP prediction server uncovered that Tyr98 of “Conserved” (YGNV) motif is sulfonated in PUUV, DOBV, and HTNV (Figure 8). In case of SEOV, Tyr165 of “SEOV-upstream 1” (YEDF) motif is also sulfonated. However, no other YXXΦ motifs were found to be sulfonated. In short, PTMs facilitate virus proliferation by accelerating viral assembly and replication during infection. Thus, PTM prediction is a beneficial method to understand viral pathogenesis and replication better.
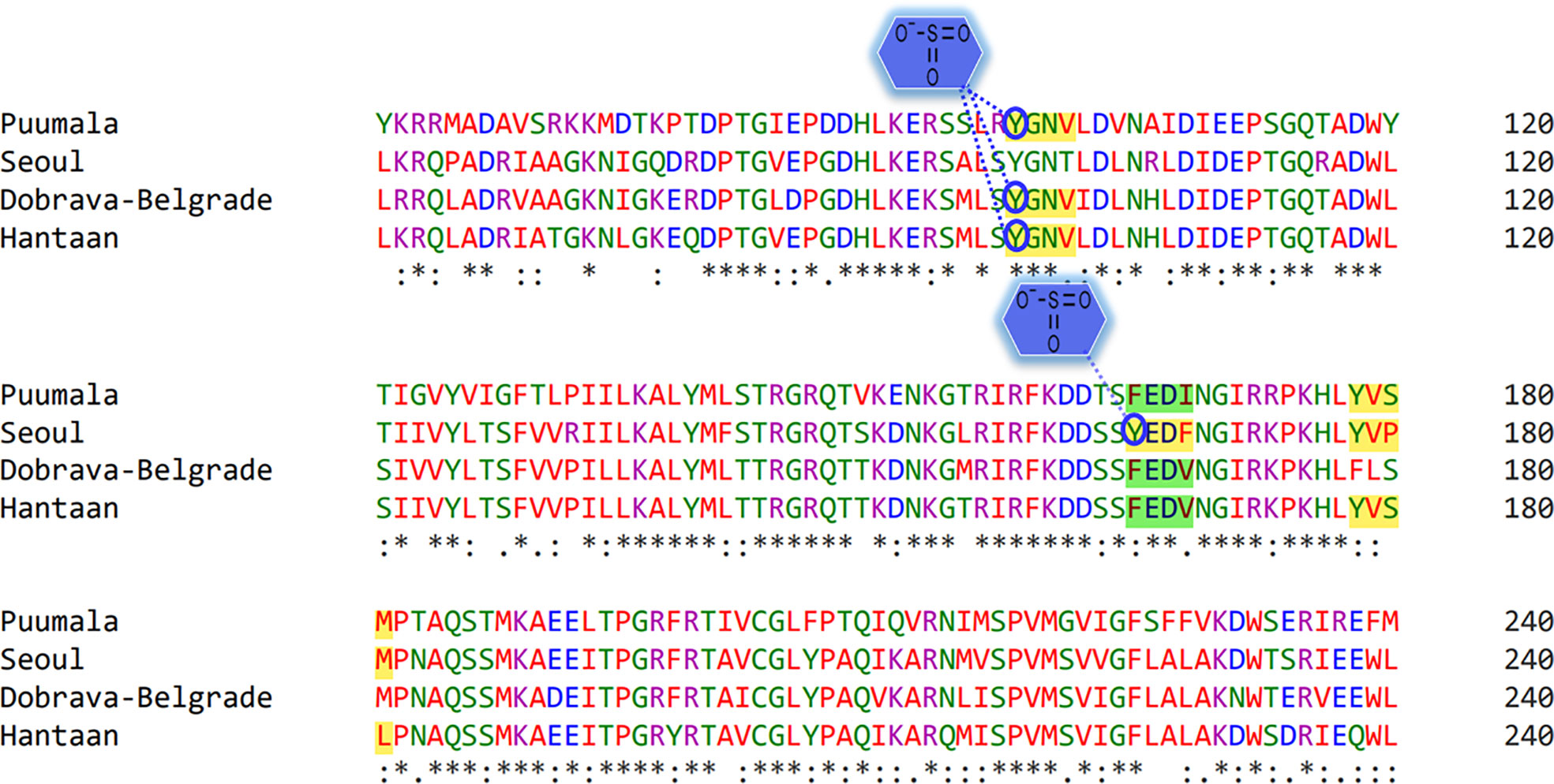
Figure 8 Representation of sulfonated sites within the YXXΦ[I/L/M/F/V] motifs of viral sequences. Stars, amino acid sequence similarity; colons, amino acid sequence difference.
3D structural models of the nucleoproteins were generated using Alphafold2. The AlphaFold2-based prediction was run with the “mmseqs2” mode by ColabFold. Furthermore, to further improve protein structure quality, we used amber force fields to fix structural violations in the 3D structure of nucleoproteins. Later, AlphaFold2 generated five models for each input sequence. In all five models predicted by AlphaFold2, the top-ranked models were selected based on the pLDDT values. The 3D structures of nucleoproteins provide a comprehensive snapshot of YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides in a 3D environment. Figure 9 shows the 3D structure of nucleoproteins and the position of motifs and motif-like sequences which is indicated with black arrows. Furthermore, the structure of PUUV, SEOV, DOBV, and HTNV were then superimposed using ChimeraX to unravel the structural homology between motifs of viral sequences (Figure 10).
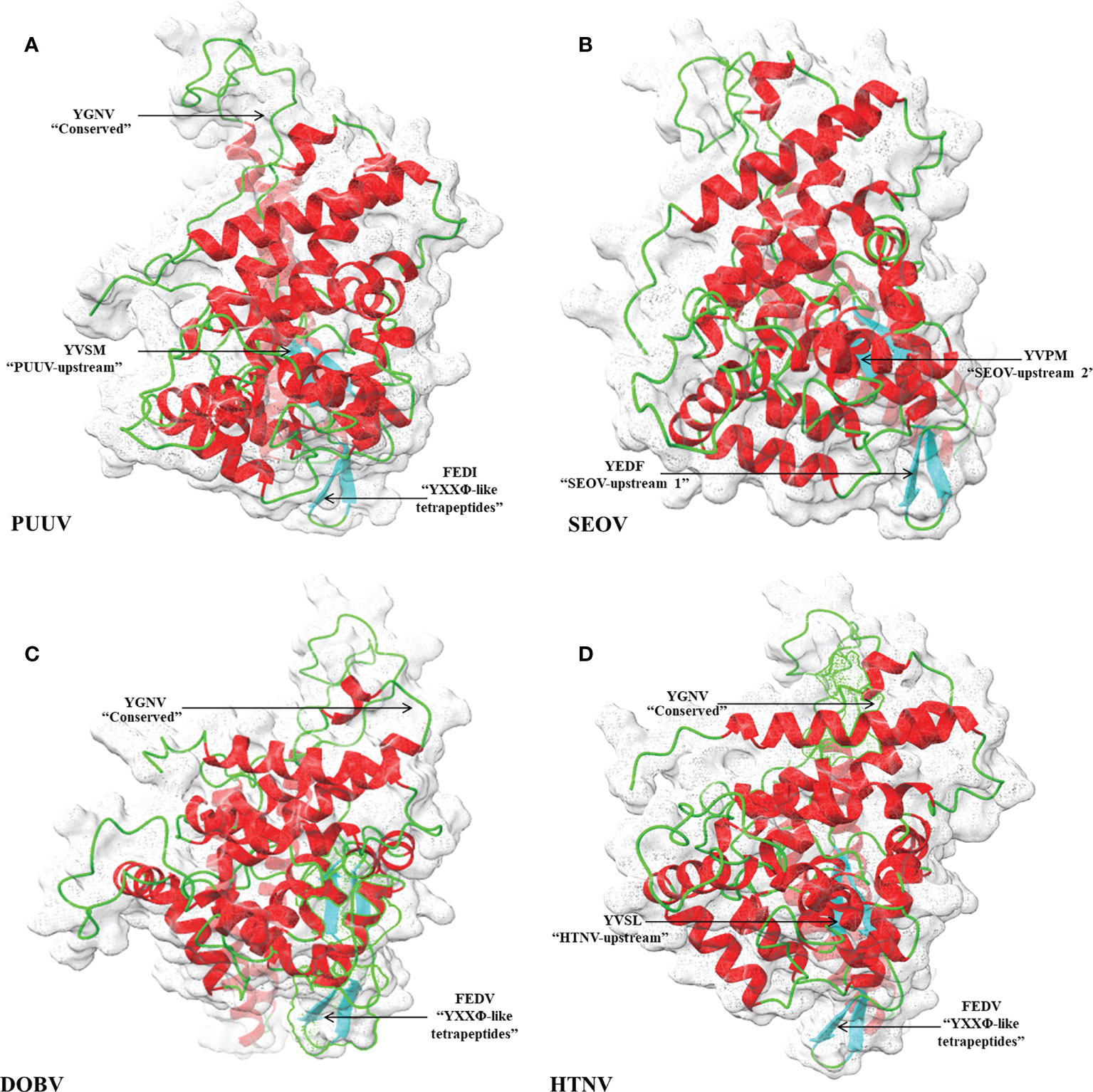
Figure 9 Structural representation of 3D structure (A) PUUV (B) SEOV (C) DOBV (D) HTNV. In structure, green represented coils, red represented α-helices and cyanrepresented β-strand. The position of YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides is represented with arrows.
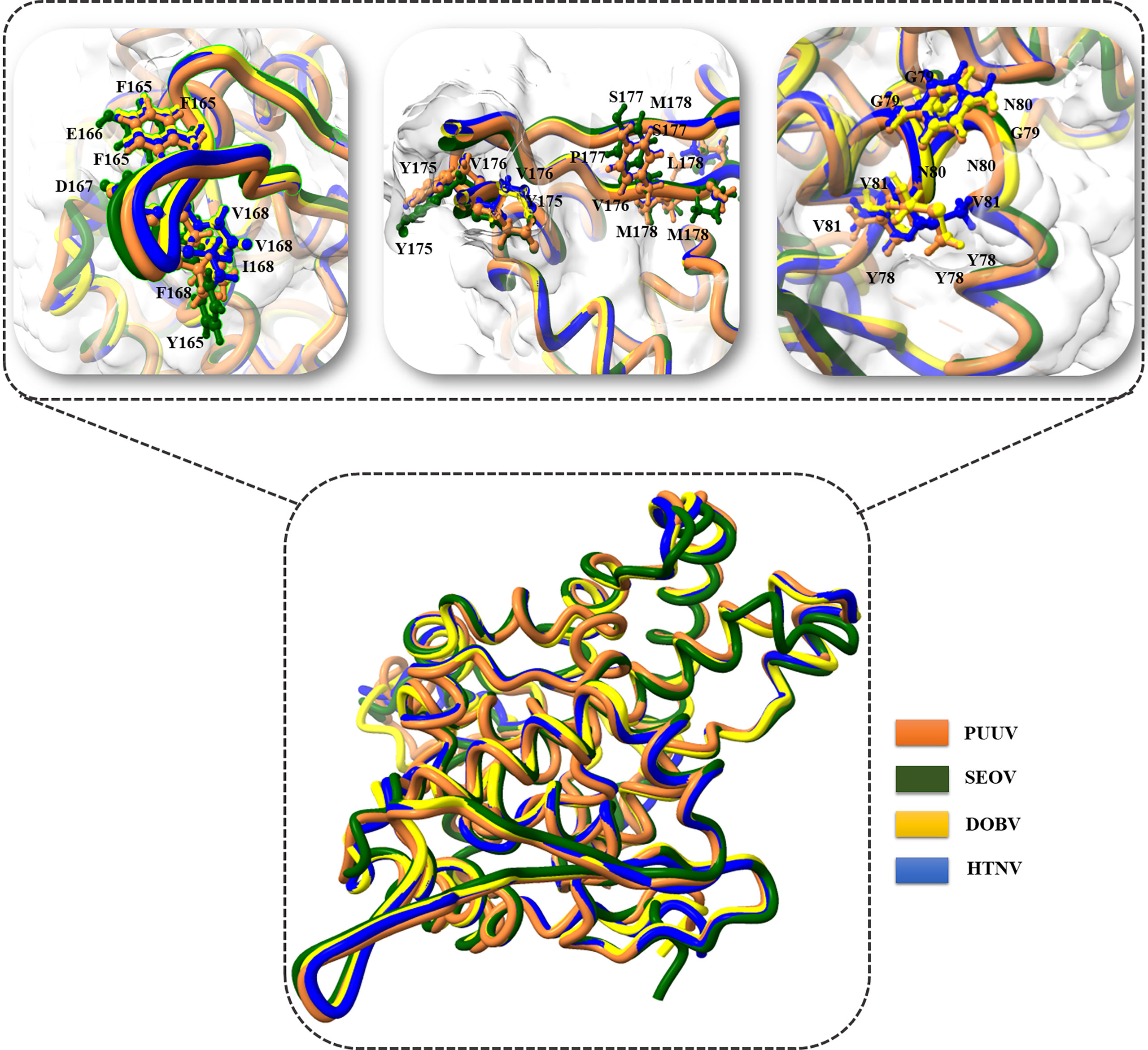
Figure 10 Superimposition of PUUV, SEOV, DOBV, and HTNV.
The nucleoproteins of hantaviruses have immunodominant antigen and these nucleoproteins has conserved antigenicity in contrast to glycoproteins of hantaviruses. Nucleoprotein has thus been employed for serological diagnosis and seroepidemiological research. Consequently, a comprehensive understanding of nucleoproteins is mandatory to stop the process of replication in hantavirus (49). Regarding this, the immune epitopes of YXXΦ-like tetrapeptides and YXXΦ[I/L/M/F/V] motifs were predicted using Chou & Fasman β-turn predictor available in IEDB server. The Chou & Fasman β-turn predictor evaluates the antigenic properties on the basis of β-turn structure scale. It implies that the epitope has always been turned in a β shape (50). The analysis with Chou & Fasman β-turn predictor revealed that, predicted motifs and motif-like tetrapeptides belong to antigenic regions of nucleoproteins (Figure 11; Supplementary Table S1). Those who met the precise criteria of average score >0.95 were considered epitopes (Table 3).
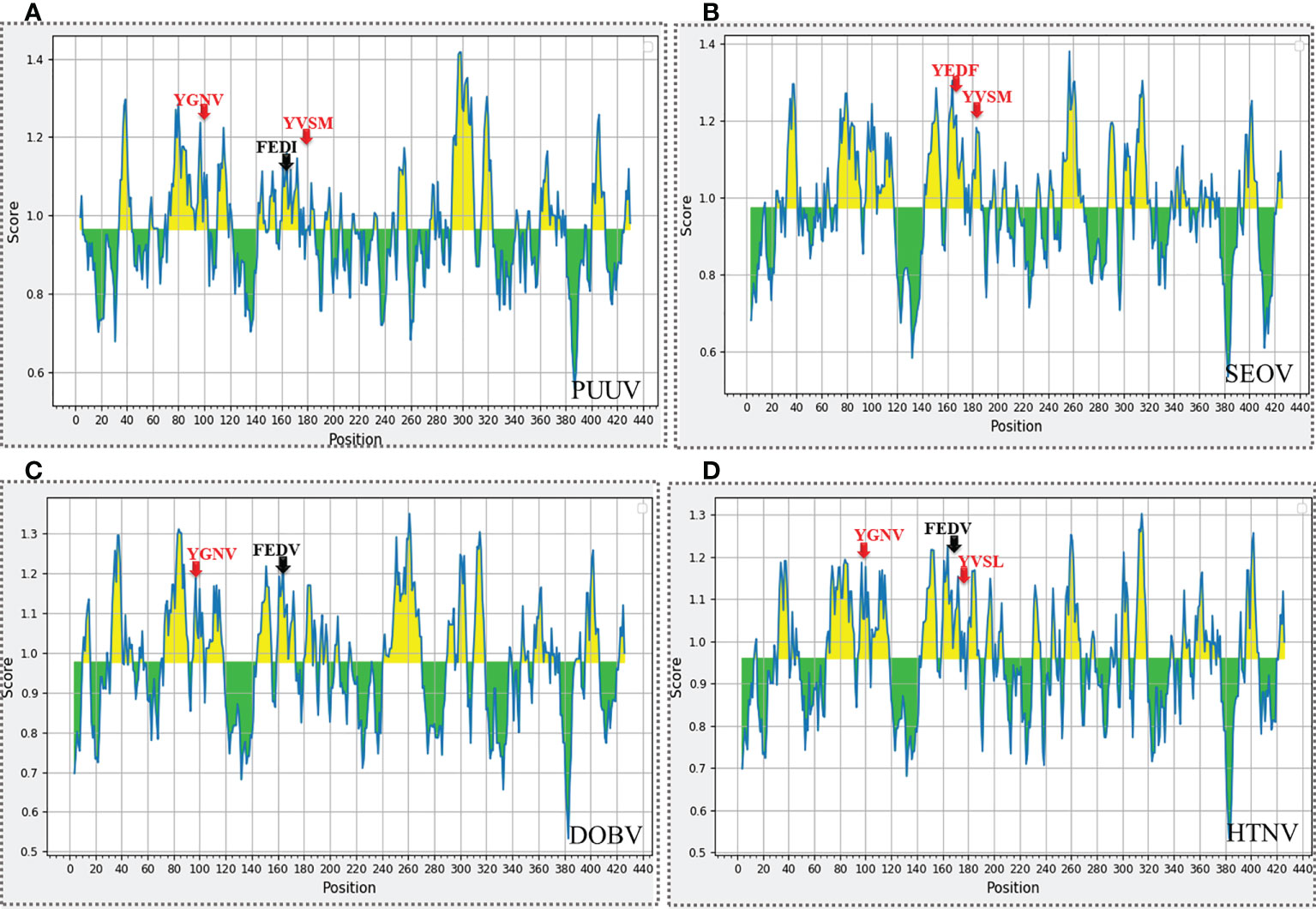
Figure 11 Immune epitope prediction of YXXΦ[I/L/M/F/V] motifs (in red color) and YXXΦ-like tetrapeptides (in black color) via Chou & Fasman method of β-turn prediction. (A) PUUV, (B) SEOV, (C) DOBV, and (D) HTNV. Those amino acids having scores >0.95 are highlighted with yellow while other amino acids were represented with green.
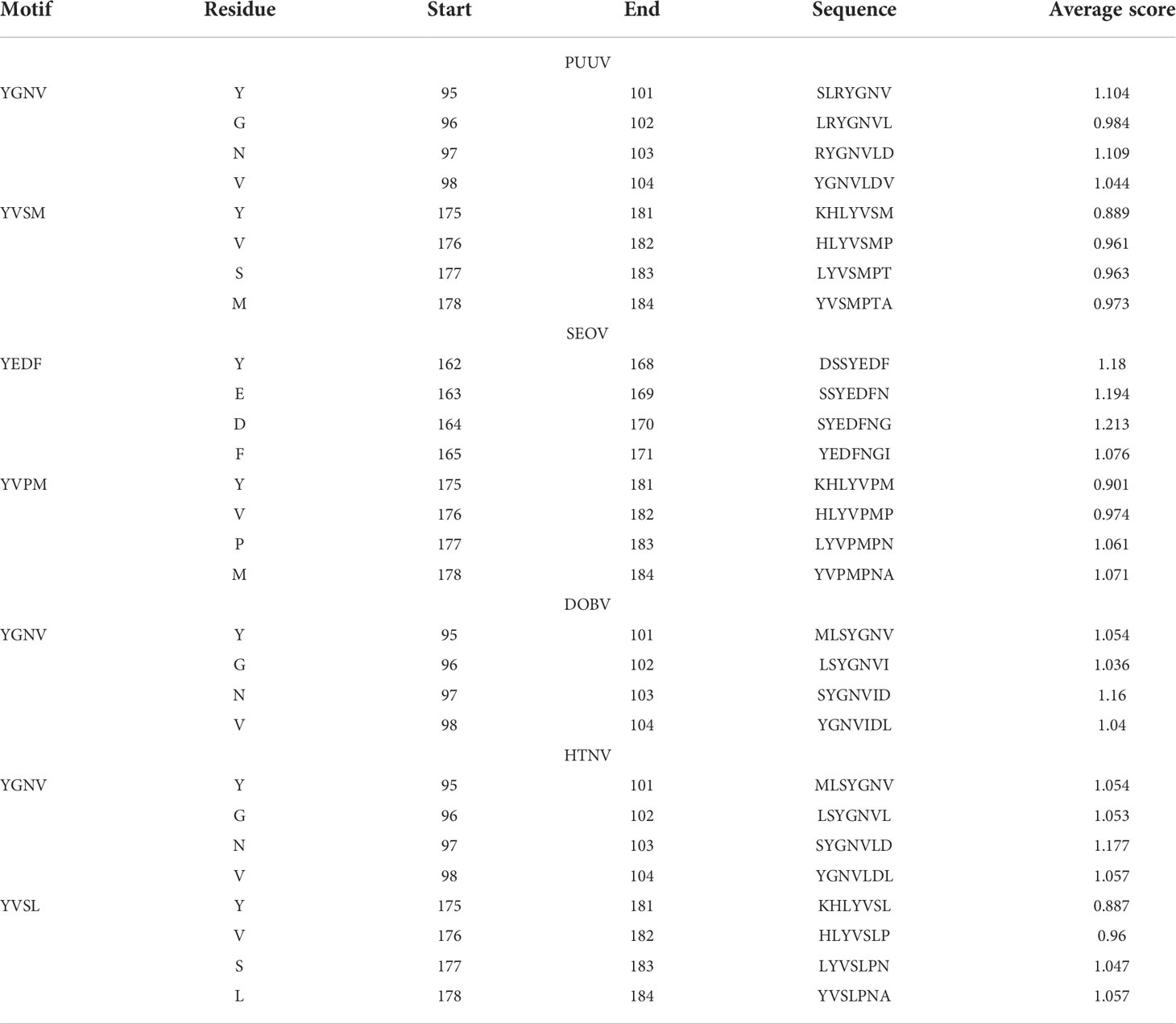
Table 3 Enlist immune epitopes within YXXΦ[I/L/M/F/V] motifs.
The nucleoprotein is predicted to contain multiple signaling motifs predicted by the ELM web server, including immunoreceptor tyrosine-based inhibitory motifs (ITIM). Finally, we searched whether YXXΦ[I/L/M/F/V] motifs or YXXΦ-like tetrapeptides of nucleoproteins could be an element of an ITIM motif, as recent studies provide strong shreds of evidences that ITIM motifs are responsible for the regulation and evasion of host immune responses in viruses (51). Intriguingly, nucleoprotein analysis using ELM server revealed that the “Conserved” (YGNV) motif belongs to ITIM (LRYGNV) motif located at position (96–101) amino acids (Figure 12).

Figure 12 Representation of ITIM motif within YXXΦ[I/L/M/F/V] motifs.
HFRS is a potentially fatal infectious disease with worldwide distribution. PUUV, SEOV, DOBV, and HTNV are the primary causative agents of HFRS (52). PUUV, SEOV, DOBV, and HTNV are a member of rodent-borne viruses called hantaviruses that cause life-threatening human diseases in Europe and Asia. The enveloped and single-stranded RNA genome of hantaviruses is tri-segmented into small, medium, and large (S, M and L) segments. The hantavirus nucleocapsid protein comprises 429 to 433 amino acids (about 50 kDa in size) (21, 53). It has been shown that the early immune response in people with hantavirus infections are mostly targeted toward the nucleoproteins. The nucleoproteins are highly conserved in the hantavirus genus and are one of the most abundant structural proteins in virus-infected cells (54, 55). Evidence suggests that substantial amounts of nucleoproteins are expressed early after infection. Nucleoproteins express in the cytoplasm of infected cell (56). Hantavirus nucleoprotein controls viral replication and assembly, encapsidates viral RNA, and plays a crucial part in the virus life cycle (57). Consequently, various diagnostics methods have now been developed based on recognizing the hantaviruses nucleoprotein or an anti-nucleoprotein antibody to halt the pathogenesis of viral infection at early stages (58, 59).
Fever and flu-like symptoms are the hallmarks of HFRS, which evolve into anemia, acute kidney damage with severe thrombocytopenia and coagulation disorders (60). Supportive therapy is the mainstay of care for patients with hantavirus infections. Care includes careful management of the patient’s fluid (hydration) and electrolyte (e.g., sodium, potassium, chloride) levels, maintenance of correct oxygen and blood pressure levels, and appropriate treatment of any secondary infections. There is currently no effective treatment available for either HFRS. Recently, Neveu et al. (61) identify YXXΦ motif within core proteins of hepatitis C virus and demonstrated that YXXΦ motif plays significant role in viral assembly. Further, Kakkanas et al. (29) used the same methodology and identified YXXΦ Motifs of the SARS Coronaviruses 1 and 2 ORF3a Peptides and proposed that targeting these YXXΦ Motifs might help in the prediction of by novel Host-virus interactions. This study revolves around the identification of tyrosine-based motif (YXXΦ[I/L/M/F/V]) and YXXΦ-like tetrapeptides within nucleoprotein sequences of PUUV, SEOV, DOBV, and HTNV to identify novel anti-viral targets. YXXΦ[I/L/M/F/V] motifs have key role to play in regulating immune responses. At first, homology between nucleoproteins of PUUV, SEOV, DOBV, and HTNV were determined which revealed high homology among these hantaviruses. Despite that, the protein sequences of PUUV, SEOV, DOBV, and HTNV shows several differences which highlighted that residual substitution generated YXXΦ-like tetrapeptides and these YXXΦ-like tetrapeptides are later prone to certain PTMs which ultimately affect the proper functioning of nucleoproteins. The other factor like disordered regions near YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides also affect the functionalities of these motifs.
Different PTMs sites were then observed within YXXΦ[I/L/M/F/V] motifs as well as YXXΦ-like tetrapeptides. Various biological and biomedical processes are governed mainly by protein phosphorylation. Esterification of residue by the addition of phosphate causes conformational changes in the protein, ultimately leading to protein activity or stability variations. Proteins can be phosphorylated on Ser, Thr, or Tyr residues. This PTM has the potential to alter the enzymatic activity, subcellular location, and stability of proteins with diverse roles in cells. Our study proposed phosphorylation of some Tyr and Ser residues within YXXΦ[I/L/M/F/V] motifs. In the light of recent literature, phosphorylation of Tyr within tyrosine-based motifs induce hinders the endocytosis of protein containing that particular motif (62, 63). While the phosphorylation of Ser influences the sorting of protein (64). If we compare our findings with published literature then it is worth noting that phosphorylation of Tyr and Ser residues within YXXΦ[I/L/M/F/V] motifs of PUUV, SEOV, DOBV, and HTNV hinders the endocytosis and affect the sorting process of nucleoproteins.
Tyr sulfation is expected to happen almost exclusively on secreted proteins and transmembrane-spanning proteins. According to available data, an organism’s total protein content can contain up to 1% sulfated Tyr residues. Therefore, the most frequent PTM on Tyr residues is Tyr sulfation (65, 66). Sulfated Tyr residues are currently engaged in several biological processes, mainly related to ligand binding and host–viral interactions (67). Our study predicts that sulfonated Tyr residues in YXXΦ[I/L/M/F/V] motifs. Considering that sulfation increases a protein’s molecular weight like phosphorylation, thus making the altered residues visible on the cell surface (68), it makes sense to assume that the nucleoprotein may engage in previously unrecognized interactions with extracellular host proteins upon sulfation.
Protein Tyr nitration is a covalent and specific modification of Tyr residues resulting in alteration of structure and function of the protein. It is a free radical process involving the reaction of a tyrosyl radical with •NO to form 3-NO-Tyr followed by sequential 2-electron oxidation to 3-NO-Tyr through Tyr iminoxyl radical. Importantly, not all Tyr residues within a protein sequence are amenable to nitration, therefore, Tyr nitration is not a random process, in fact, it is considered very specific. Recent studies reported that nitration of Tyr completely alters the function of protein which ultimately lead to viral pathogenesis (69). Thus, the nitration of Tyr residues in nucleoproteins of PUUV, SEOV, DOBV, and HTNV could significantly affect viral pathogenesis and survival.
Later, 3D structures of PUUV, SEOV, DOBV, and HTNV nucleoproteins were predicted using AlphaFold2 to map the location of YXXΦ [I/L/M/F/V] motifs and YXXΦ-like tetrapeptides in 3D environment. Finally, the Chou & Fasman β-turn predictor revealed that proposed motifs have good antigenic properties. Lastly, “Conserved” (YGNV) motif at position (96–101) amino acids found to be part of ITIM Tyr-based Immunoreceptor motif (LRYGNV) motif. This study also has some limitations as without any in vitro and in vivo validation, the proposed YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides can be questioned very easily because the predictive power of algorithms used in tools is relatively low. Fascinatingly, the ones that exist might instruct the tools to produce more accurate data. However, conformational research on the effect of PTMs on the functionality of proposed motifs is urgently needed to better understand their role in viral pathogenesis, virulence, and replication. This ultimately opens up new and exciting avenues for the treatment of HFRS.
To sum up, our work explored the differences among nucleoprotein sequences of PUUV, SEOV, DOBV, and HTNV, especially within YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides. Overall, these studies highlight region of nucleoproteins that play critical roles in the biological function and where changes on a single residue have dramatic effects on virus replicative capacity. Furthermore, we envision that the targeting of the YXXΦ motifs with pharmacological reagents that will induce PTMs with suitable deleterious effects to the structural integrity and the immunological properties of nucleoproteins, could assist with the rational design of anti-HFRS treatments. We strongly believe that combining anti-HFRS drug with YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides may offer synergistic effects which ultimately produce fruitful outcome in controlling the viral infection worldwide.
HFRS is acute interstitial nephropathy characterized by high fever and varying degrees of renal insufficiency and hemorrhage. The nucleocapsid protein of hantaviruses represents an impressive example of a viral multifunctional protein. Despite that, nucleoproteins of PUUV, SEOV, DOBV, and HTNV are approximately similar in length; however, they exhibit considerable differences in their N-termini and residual substitutions throughout the length of nucleoprotein sequences, particularly within YXXΦ[I/L/M/F/V] motifs and YXXΦ-like tetrapeptides. The predicted 3D models and PTMs can attribute diverse functions to the nucleoproteins of PUUV, SEOV, DOBV, and HTNV. Thus, discovering such mechanisms can provide novel targets to develop potential antiviral drugs for the treatment of HFRS. Additional experimental research on this putative YXXΦ [I/L/M/F/V] motifs and YXXΦ-like tetrapeptides lead to an increase in our knowledge to fight against HFRS in the future by employing novel therapeutic approaches. Although it is difficult to draw conclusions about the biological functions of a protein from bioinformatically predicted structures, we hope that the results of our work will boost in vitro experimentation on this fascinating biomolecule, nucleoprotein that will ultimately assign new biological roles to it.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
FN drafted the manuscript, prepared illustrations, and discussed the content with the other authors. UA conceived the topic and revised the content of the manuscript. MAs and MM revised the manuscript. AA and MAl were involved in the final development of the project and funding acquisition. All authors contributed to the article and approved the submitted version.
Authors are thankful to the Researchers Supporting Project number (RSP2022R491), King Saud University, Riyadh, Saudi Arabia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1031608/full#supplementary-material
Supplementary Table 1 | Linear antigenic epitopes of nucleoproteins.
1. Hsieh C-J, Li C-W, Cheng C-A, Wu D-C, Wu W-C, Lin F-H, et al. Epidemiologic characteristics of domestic patients with hemorrhagic fever with renal syndrome in Taiwan: A 19-year retrospective study. Int J Environ Res (2020) 17(15):5291. doi: 10.3390/ijerph17155291
2. Xiang J, Hansen A, Liu Q, Tong MX, Liu X, Sun Y, et al. Impact of meteorological factors on hemorrhagic fever with renal syndrome in 19 cities in China, 2005–2014. Sci Total Environ (2018) 636:1249–56. doi: 10.1016/j.scitotenv.2018.04.407
3. Liang W, Gu X, Li X, Zhang K, Wu K, Pang M, et al. Mapping the epidemic changes and risks of hemorrhagic fever with renal syndrome in shaanxi province, China, 2005–2016. Sci Rep (2018) 8(1):1–10. doi: 10.1038/s41598-017-18819-4
4. Moolla N, Adler D, Blumberg L, Glass A, Grobbelaar A, le Roux C, et al. First report of an imported case of haemorrhagic fever with renal syndrome in south Africa. S Afr Med (2022) 112(8):29–31. doi: 10.7196/SAMJ.2022.v112i8.16540
5. Tariq M, Kim D-M. Chemotherapy. hemorrhagic fever with renal syndrome: Literature review, epidemiology, clinical picture and pathogenesis. JI (2022) 54(1):1. doi: 10.3947/ic.2021.0148
6. Oliveira S, Faccini-Martínez ÁA. Hantavirus infection and the renal syndrome Trop Nephrol (2020) 175–92. doi: 10.1007/978-3-030-44500-3_14
7. Bai X-H, Peng C, Jiang T, Hu Z-M, Huang D-S, Guan P. Distribution of geographical scale, data aggregation unit and period in the correlation analysis between temperature and incidence of hfrs in mainland China: A systematic review of 27 ecological studies. JPntd (2019) 13(8):e0007688. doi: 10.1371/journal.pntd.0007688
8. Guo B, Wang Y, Pei L, Yu Y, Liu F, Zhang D, et al. Determining the effects of socioeconomic and environmental determinants on chronic obstructive pulmonary disease (Copd) mortality using geographically and temporally weighted regression model across xi’an during 2014–2016. Sci Total Environ (2021) 756:143869. doi: 10.1016/j.scitotenv.2020.143869
9. Lovrić Z, Kolarić B, Ličina MK, Tomljenović M, Rode OĐ, Danis K, et al. An outbreak of haemorrhagic fever with renal syndrome linked with mountain recreational activities in Zagreb, Croatia, 2017. Epidemiol Infect (2018) 146(10):1236–9. doi: 10.1017/S0950268818001231
10. Ivanova A, Safronov V, Popov N, Kozhanova O, Matveeva N, Kresova U, et al. Epidemiological features of hfrs (Hemorrhagic fever with renal syndrome) outbreak in the saratov region in 2019. Problems of Particularly Dangerous Infections (2020) 2. doi: 10.21055/0370-1069-2020-2-78-85
11. Jiang H, Du H, Wang LM, Wang PZ, Bai XF. Hemorrhagic fever with renal syndrome: Pathogenesis and clinical picture. Front Cell infect Microbiol (2016) 6:1. doi: 10.3389/fcimb.2016.00001
12. Sironen T, Vaheri A, Plyusnin A. Molecular evolution of puumala hantavirus. JJov (2001) 75(23):11803–10. doi: 10.1128/JVI.75.23.11803-11810.2001
13. Lee HW, Baek LJ, Johnson KM. Isolation of hantaan virus, the etiologic agent of Korean hemorrhagic fever, from wild urban rats. JoID (1982) 146(5):638–44. doi: 10.1093/infdis/146.5.638
14. Papa A. Dobrava-Belgrade virus: Phylogeny, epidemiology, disease. JAR (2012) 95(2):104–17. doi: 10.1016/j.antiviral.2012.05.011
15. Lee HW, Lee PW, Johnson KM. Isolation of the etiologic agent of Korean hemorrhagic fever. Joid (1978) 137(3):298–308. doi: 10.1093/infdis/137.3.298
16. Liu R, Ma H, Shu J, Zhang Q, Han M, Liu Z, et al. Vaccines and therapeutics against hantaviruses. Front Microbiol (2020) 10:2989. doi: 10.3389/fmicb.2019.02989
17. Milholland MT, Castro-Arellano I, Suzán G, Garcia-Peña GE, Lee TE, Rohde RE, et al. Global diversity and distribution of hantaviruses and their hosts. Ecohealth (2018) 15(1):163–208. doi: 10.1007/s10393-017-1305-2
18. Klempa B. Reassortment events in the evolution of hantaviruses. JVg (2018) 54(5):638–46. doi: 10.1007/s11262-018-1590-z
19. Kaukinen P, Vaheri A, Plyusnin A. Hantavirus nucleocapsid protein: A multifunctional molecule with both housekeeping and ambassadorial duties. JAov (2005) 150(9):1693–713. doi: 10.1007/s00705-005-0555-4
20. Reuter M, Krüger DH. The nucleocapsid protein of hantaviruses: Much more than a genome-wrapping protein. JVG (2018) 54(1):5–16. doi: 10.1007/s11262-017-1522-3
21. Muyangwa M, Martynova EV, Khaiboullina SF, Morzunov SP, Rizvanov AA. Hantaviral proteins: Structure, functions, and role in hantavirus infection. JFim (2015) 6:1326. doi: 10.3389/fmicb.2015.01326
22. Hussein IT, Mir MA. How hantaviruses modulate cellular pathways for efficient replication? JFiB-E (2013) 5(1):154–66. doi: 10.2741/E604
23. Alminaite A, Halttunen V, Kumar V, Vaheri A, Holm L, Plyusnin A. Oligomerization of hantavirus nucleocapsid protein: Analysis of the n-terminal coiled-coil domain. Jov (2006) 80(18):9073–81. doi: 10.1128/JVI.00515-06
24. Jonsson CB, Figueiredo LTM, Vapalahti O. A global perspective on hantavirus ecology, epidemiology, and disease. JCmr (2010) 23(2):412–41. doi: 10.1128/CMR.00062-09
25. Sabino-Santos J, Maia FGM, Martins RB, Gagliardi TB, Souza W, Muylaert RL, et al. Natural infection of Neotropical bats with hantavirus in Brazil. Sci Rep (2018) 8(1):1–8. doi: 10.1038/s41598-018-27442-w
26. Aguilar RC, Boehm M, Gorshkova I, Crouch RJ, Tomita K, Saito T, et al. Signal-binding specificity of the M4 subunit of the adaptor protein complex ap-4. J Biol Chem (2001) 276(16):13145–52. doi: 10.1074/jbc.M010591200
27. Collawn JF, Stangel M, Kuhn LA, Esekogwu V, Jing S, Trowbridge IS, et al. Transferrin receptor internalization sequence yxrf implicates a tight turn as the structural recognition motif for endocytosis. Cell (1990) 63(5):1061–72. doi: 10.1016/0092-8674(90)90509-D
28. Berlioz-Torrent C, Shacklett BL, Erdtmann L, Delamarre L, Bouchaert I, Sonigo P, et al. Interactions of the cytoplasmic domains of human and simian retroviral transmembrane proteins with components of the clathrin adaptor complexes modulate intracellular and cell surface expression of envelope glycoproteins. J Virol (1999) 73(2):1350–61. doi: 10.1128/JVI.73.2.1350-1361.1999
29. Kakkanas A, Karamichali E, Koufogeorgou EI, Kotsakis SD, Georgopoulou U, Foka P. Targeting the yxxφ motifs of the sars coronaviruses 1 and 2 Orf3a peptides by in silico analysis to predict novel virus–host interactions. JB (2022) 12(8):1052. doi: 10.3390/biom12081052
30. Humphrey MB, Lanier LL, Nakamura MC. Role of itam-containing adapter proteins and their receptors in the immune system and bone. JIr (2005) 208(1):50–65. doi: 10.1111/j.0105-2896.2005.00325.x
31. Heineman TC, Hall SL. Vzv Gb endocytosis and golgi localization are mediated by yxxφ motifs in its cytoplasmic domain. JV (2001) 285(1):42–9. doi: 10.1006/viro.2001.0930
32. Windheim M, Höning S, Leppard KN, Butler L, Seed C, Ponnambalam S, et al. Sorting motifs in the cytoplasmic tail of the immunomodulatory E3/49k protein of species d adenoviruses modulate cell surface expression and ectodomain shedding. J Biol Chem (2016) 291(13):6796–812. doi: 10.1074/jbc.M115.684787
33. Karamichali E, Serti E, Gianneli A, Papaefthymiou A, Kakkanas A, Foka P, et al. The unexpected function of a highly conserved yxxφ motif in hcv core protein. Infect Genet Evol (2017) 54:251–62. doi: 10.1016/j.meegid.2017.07.001
34. Hepojoki J, Strandin T, Lankinen H, Vaheri A. Hantavirus structure–molecular interactions behind the scene. J Gen Virol (2012) 93(Pt 8):1631–44. doi: 10.1099/vir.0.042218-0
35. Kalita P, Lyngdoh DL, Padhi AK, Shukla H, Tripathi T. Development of multi-epitope driven subunit vaccine against fasciola gigantica using immunoinformatics approach. Int J Biol macromol (2019) 138:224–33. doi: 10.1016/j.ijbiomac.2019.07.024
36. Sievers F, Higgins DG. The clustal omega multiple alignment package. Multiple Seq Alignment (2021) . p:3–16. doi: 10.1007/978-1-0716-1036-7_1
37. Kumar M, Gouw M, Michael S, Sámano-Sánchez H, Pancsa R, Glavina J, et al. Elm–the eukaryotic linear motif resource in 2020. Nucleic Acids Res (2020) 48(D1):D296–306. doi: 10.1093/nar/gkz1030
38. Omasits U, Ahrens CH, Müller S, Wollscheid B. Protter: Interactive protein feature visualization and integration with experimental proteomic data. JB (2014) 30(6):884–6. doi: 10.1093/bioinformatics/btt607
39. Li S, Yu K, Wang D, Zhang Q, Liu Z-X, Zhao L, et al. Deep learning based prediction of species-specific protein s-glutathionylation sites. Biochim Biophys Acta Proteins Proteom (2020) 1868(7):140422. doi: 10.1016/j.bbapap.2020.140422
40. Romero P, Obradovic Z, Dunker AK. Natively disordered proteins. JAb (2004) 3(2):105–13. doi: 10.2165/00822942-200403020-00005
41. Sevindik E. In silico sequence analysis of predicted beta-amylase 7-like protein in juglans regia l. EJoBR (2017) 7(2):148–53. doi: 10.5281/zenodo.583137
42. Xu Y, Wen X, Wen L-S, Wu L-Y, Deng N-Y, Chou K-C. Initro-tyr: Prediction of nitrotyrosine sites in proteins with general pseudo amino acid composition. JPo (2014) 9(8):e105018. doi: 10.1371/journal.pone.0105018
43. Wang B, Wang M, Li AJP. Prediction of post-translational modification sites using multiple kernel support vector machine. PeerJ (2017) 5:. doi: 10.7717/peerj.3261
44. Cramer PJNS, Biology M. Alphafold2 and the future of structural biology. Nat Struct Mol Biol (2021) 28(9):704–5. doi: 10.1038/s41594-021-00650-1
45. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, et al. Ucsf chimerax: Structure visualization for researchers, educators, and developers. Protein Sci (2021) 30(1):70–82. doi: 10.1002/pro.3943
46. Tahir Ul Qamar M, Ahmad S, Fatima I, Ahmad F, Shahid F, Naz A, et al. Designing multi-epitope vaccine against staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput Biol Med (2021) 132:104389. doi: 10.1016/j.compbiomed.2021.104389
47. Tahir Ul Qamar M, Rehman A, Tusleem K, Ashfaq UA, Qasim M, Zhu X, et al. Designing of a next generation multiepitope based vaccine (Mev) against sars-Cov-2: Immunoinformatics and in silico approaches. PloS One (2020) 15(12):e0244176. doi: 10.1371/journal.pone.0244176
48. Zhang Q, Wang P, Kim Y, Haste-Andersen P, Beaver J, Bourne PE, et al. Immune epitope database analysis resource (Iedb-ar). Nucleic Acids Res (2008) 36(suppl_2):W513–W8. doi: 10.1093/nar/gkn254
49. Yoshimatsu K, Arikawa J. Antigenic properties of n protein of hantavirus. JV (2014) 6(8):3097–109. doi: 10.3390/v6083097
50. Tahir ul Qamar M, Saleem S, Ashfaq UA, Bari A, Anwar F, Alqahtani SJ. Epitope-based peptide vaccine design and target site depiction against middle East respiratory syndrome coronavirus: An immune-informatics study. Jotm (2019) 17(1):1–14. doi: 10.1186/s12967-019-2116-8
51. Wang Z, Li R, Liu C, Qiao W, Tan J. Characterization of bovine foamy virus gag late assembly domain motifs and their role in recruiting escrt for budding. JV (2022) 14(3):522. doi: 10.3390/v14030522
52. Shid SJ, Sapate RB, Raut ID, Dhadde GS, Mali HS, Joshi S, et al. Hantavirus infection: An overview. Asian J Pharm res (2022) 12(2):150–4. doi: 10.52711/2231-5691.2022.00023
53. Hussein IT, Haseeb A, Haque A, Mir MA. Recent advances in hantavirus molecular biology and disease. Adv Appl Microbiol (2011) 74:35–75. doi: 10.1016/B978-0-12-387022-3.00006-9
54. Alminaite A. Structure and functions of hantavirus nucleocapsid protein. Doctoral Dissertation (2010). Helsinki University Press, University of Helsinki. Available at: http://ethesis.helsinki.fi.
55. Vapalahti O, Kallio-Kokko H, Närvänen A, Julkunen I, Lundkvist Å, Plyusnin A, et al. Human b-cell epitopes of puumala virus nucleocapsid protein, the major antigen in early serological response. J Med Virol (1995) 46(4):293–303. doi: 10.1002/jmv.1890460402
56. Van Regenmortel MH. Virus taxonomy. Classification and nomenclature of viruses. Seventh report of the International Committee on Taxonomy of Viruses. (2000), 395–480.
57. Meier K, Thorkelsson SR, Quemin ER, Rosenthal M. Hantavirus replication cycle–an updated structural virology perspective. JV (2021) 13(8):1561. doi: 10.3390/v13081561
58. Amada T, Yoshimatsu K, Yasuda SP, Shimizu K, Koma T, Hayashimoto N, et al. Rapid, whole blood diagnostic test for detecting anti-hantavirus antibody in rats. J Virol Methods (2013) 193(1):42–9. doi: 10.1016/j.jviromet.2013.04.021
59. Yoshimatsu K, Arikawa J. Serological diagnosis with recombinant n antigen for hantavirus infection. JVr (2014) 187:77–83. doi: 10.1016/j.virusres.2013.12.040
60. Avšič-Županc T, Saksida A, Korva M. Hantavirus infections. JCM Infect (2019) 21:e6–e16. doi: 10.1111/1469-0691.12291
61. Neveu G, Barouch-Bentov R, Ziv-Av A, Gerber D, Jacob Y, Einav S. Identification and targeting of an interaction between a tyrosine motif within hepatitis c virus core protein and Ap2m1 essential for viral assembly. PLoS Pathog (2012) 8(8):e1002845. doi: 10.1371/journal.ppat.1002845
62. Kittler JT, Chen G, Kukhtina V, Vahedi-Faridi A, Gu Z, Tretter V, et al. Regulation of synaptic inhibition by phospho-dependent binding of the Ap2 complex to a yecl motif in the gabaa receptor Γ2 subunit. Proc Natl Acad Sci U S A (2008) 105(9):3616–21. doi: 10.1073/pnas.0707920105
63. Lee H-J, Shieh C-K, Gorbalenya AE, Koonin EV, La Monica N, Tuler J, et al. The complete sequence (22 kilobases) of murine coronavirus gene 1 encoding the putative proteases and rna polymerase. Virology (1991) 180(2):567–82. doi: 10.1016/0042-6822(91)90071-I
64. Pallesen LT, Gustafsen C, Cramer JF, Petersen SV, Thirup SS, Madsen P, et al. Pak kinases target sortilin and modulate its sorting. Mol Cell Biol (2020) 40(3):e00411–19. doi: 10.1128/MCB.00411-19
65. Baeuerle PA, Huttner WJ. Tyrosine sulfation of yolk proteins 1, 2, and 3 in drosophila melanogaster J Biol Chem (1985) 260(10):6434–9. doi: 10.1016/S0021-9258(18)88991-8
66. Huttner WB. Sulphation of tyrosine residues–a widespread modification of proteins. JN (1982) 299(5880):273–6. doi: 10.1038/299273a0
67. Mehta AY, Heimburg-Molinaro J, Cummings RD, Goth CK. Emerging patterns of tyrosine sulfation and O-glycosylation cross-talk and Co-localization. JCoisb (2020) 62:102. doi: 10.1016/j.sbi.2019.12.002
68. Lind SB, Artemenko KA, Elfineh L, Zhao Y, Bergquist J, Pettersson U. Post translational modifications in adenovirus type 2. JV (2013) 447(1-2):104–11. doi: 10.1016/j.virol.2013.08.033
Keywords: hemorrhagic fever with renal syndrome, hantavirus, YXXΦ[I/L/M/F/V] motif, YXXΦ-like tetrapeptides, 3D structure, post-translational modifications
Citation: Noor F, Ashfaq UA, Asif M, Adeel MM, Alshammari A and Alharbi M (2022) Comprehensive computational analysis reveals YXXΦ[I/L/M/F/V] motif and YXXΦ-like tetrapeptides across HFRS causing Hantaviruses and their association with viral pathogenesis and host immune regulation. Front. Immunol. 13:1031608. doi: 10.3389/fimmu.2022.1031608
Received: 30 August 2022; Accepted: 20 September 2022;
Published: 06 October 2022.
Edited by:
Abbas Khan, Shanghai Jiao Tong University, ChinaReviewed by:
Muhammad Khurram, Abasyn University, PakistanCopyright © 2022 Noor, Ashfaq, Asif, Adeel, Alshammari and Alharbi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Usman Ali Ashfaq, YXNoZmFxdWFAZ2N1Zi5lZHUucGs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.