 Jing Jin
Jing Jin Jian Duan
Jian Duan Leiya Du4
Leiya Du4 Qijie Zhao
Qijie Zhao
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 23 November 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1027756
Intracranial aneurysm subarachnoid hemorrhage (SAH) is a cerebrovascular disorder associated with high overall mortality. Currently, the underlying mechanisms of pathological reaction after aneurysm rupture are still unclear, especially in the immune microenvironment, inflammation, and relevant signaling pathways. SAH-induced immune cell population alteration, immune inflammatory signaling pathway activation, and active substance generation are associated with pro-inflammatory cytokines, immunosuppression, and brain injury. Crosstalk between immune disorders and hyperactivation of inflammatory signals aggravated the devastating consequences of brain injury and cerebral vasospasm and increased the risk of infection. In this review, we discussed the role of inflammation and immune cell responses in the occurrence and development of aneurysm SAH, as well as the most relevant immune inflammatory signaling pathways [PI3K/Akt, extracellular signal-regulated kinase (ERK), hypoxia-inducible factor-1α (HIF-1α), STAT, SIRT, mammalian target of rapamycin (mTOR), NLRP3, TLR4/nuclear factor-κB (NF-κB), and Keap1/nuclear factor (erythroid-derived 2)-like 2 (Nrf2)/ARE cascades] and biomarkers in aneurysm SAH. In addition, we also summarized potential therapeutic drugs targeting the aneurysm SAH immune inflammatory responses, such as nimodipine, dexmedetomidine (DEX), fingolimod, and genomic variation-related aneurysm prophylactic agent sunitinib. The intervention of immune inflammatory responses and immune microenvironment significantly reduces the secondary brain injury, thereby improving the prognosis of patients admitted to SAH. Future studies should focus on exploring potential immune inflammatory mechanisms and developing additional therapeutic strategies for precise aneurysm SAH immune inflammatory regulation and genomic variants associated with aneurysm formation.
Intracranial aneurysms are common and have a high incidence of occurring in 1% to 2% of the population, wherein the incidence of rupture is nearly 16.4 in 100,000 persons per year (1, 2). Subarachnoid hemorrhage (SAH) is a serious clinical condition that is usually caused by a ruptured intracranial aneurysm, which caused nearly 85% of SAH (3, 4). SAH was reported to occur at a fairly young age, and the mortality rate of aneurysmal hemorrhage is nearly 50% (5). Numerous clinical trials have been conducted to improve outcomes for patients with SAH, whereas there are still challenges in the aneurysm SAH prevention and lower-risk treatment development (4).
SAH can be separated into traumatic and spontaneous, where the spontaneous SAH is known to have the highest incidence and is most often attributed to a ruptured intracranial aneurysm (6). Cerebral aneurysm-acquired lesions that develop at the major arterial branch point of the Willis circle result in hemodynamic stress-induced retrogradation of the internal elastic lamina with loss of the tunica media (4). Intracranial aneurysm SAH is a critical cerebrovascular accident with high mortality and high disability among survivors (7). Some pathophysiological factors are independent of angiographic vasospasm and are related to poor clinical prognosis, such as blood–brain barrier (BBB) disruption, inflammation, immune cell activation, and oxidative cascades, ultimately contributing to cell death (8, 9). Among which milieu, microglial-induced immune responses like macrophage were positively associated with neuroinflammation development and neuronal necrosis after SAH (10, 11). Injured neurons and dying cells will release inflammatory molecules to the extracellular milieu, which was associated with poor clinical outcomes in patients with aneurysm SAH (12, 13). These dangerous molecules may further drive the neuroinflammation and brain injury after SAH (14). Brain injury following intracranial aneurysm SAH is multimodal and serious, as early brain injury (EBI), but is also secondary to the development of immune-inflammation events (9, 10, 15, 16). Crosstalk between immune cell populations, active substances, and inflammation responses may aggravate the symptoms of SAH and contribute to poor prognosis (9, 17, 18). Treatment of immune-inflammation disorders has great potential to attenuate EBI and devastating secondary damage and ameliorate outcomes in patients with SAH. Therefore, the identification of immune-inflammation mechanisms of intracranial aneurysm SAH and its associated sequelae could be beneficial for these patients (19).
In the past decade, several treatable risk factors (cigarettes, alcohol, hypertension) and untreatable risk factors (age, sex, genetics) have been reported to increase the incidence of aneurysms (2, 20). The mechanisms of intracranial aneurysm occurrence and rupture are complex, especially immune microenvironmental and genetic factors (21). Abundant evidence supported that the etiology of intracranial aneurysms is related to genetic factors (22, 23). Genetic syndromes associated with intracranial aneurysms have been identified as an increased risk compared with the general population (2, 21). Meanwhile, individual genetic variations proposed higher aneurysm SAH and worse neuronal injury, such as THSD1 and EDN1 gene variants which were highly enriched in aneurysm SAH patients (24, 25). Specific biomarkers for intracranial aneurysm provide a potential therapeutic avenue for intracranial aneurysms, such as platelet-derived growth factor receptor β gene (PDGFRB) (26). Despite the knowledge of genetic and inflammatory mechanisms of brain injury caused by intracranial aneurysm SAH which is currently understood, the complexity of the immune cell responses and the crosstalk of the above factors in this process have not been described in detail.
In this review, we summarized the immune cells and inflammation-related mechanisms during the occurrence and development of aneurysm SAH, as well as several pivotal signaling pathways related to immune inflammatory, vasospasm, EBI, and therapeutic potential after aneurysm SAH. In addition, we discussed potential therapeutic drugs targeting the immune and inflammatory response, as well as prophylactic agents for aneurysm SAH. Understanding the specific pathological mechanisms of aneurysm SAH is important for developing strategies to prevent disease development and brain injury.
Intracranial hemorrhage (ICH) refers to any bleeding within the intracranial vault, such as brain parenchyma and meningeal spaces (27). SAH is regarded as bleeding into the space between the pia and the arachnoid membranes (28). Non-traumatic causes of hemorrhages include ruptured aneurysms, arteriovenous malformations, tumors, and vasculopathies (29–31). Previously, brain tumors like glioma/glioblastoma have a direct compressive or invasive function on the cerebral vessel, and it is observed to have a high incidence of ICH and fatal outcomes (32, 33). In both primary or metastatic brain tumors with ICH, angiogenesis mediators vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs) were associated with vascular rupture hemorrhage (31). More recently, in acute leukemia-related ICH, early ICH is characterized by leukostasis associated with abnormal hemostasis, whereas late ICH has systemic inflammation (34). On the other hand, aneurysmal SAH is due to rupture of an aneurysm in the subarachnoid space, which is commonly seen at the bifurcation of the basal cerebral artery, especially near the circle of Willis (35, 36). To date, the risk factors of aneurysmal growth and rupture remain complex; for example, size (>7 mm), inflammation, genetic syndromes, and hypertension can contribute to the aneurysmal rupture and SAH (37, 38).
For ICH, secondary brain injury following ICH is closely associated with hematoma toxicity, oxidative stress, and inflammation, among which hematoma toxicity and oxidative stress are mediators of cell death (39–41). Aronowski et al. indicated that hematoma will contribute to direct mechanical injury to the brain parenchyma, as well as perihematomal edema (42). The porphyrin derivatives were observed to inhibit heme oxygenase 1 (HO-1) and reduce the ICH damage (43). HO-1, an enzyme involved in biliverdin, carbon monoxide, and iron conversion (44), was observed with an increase in endothelial cells and microglial/macrophages after ICH (45). Of note, HO-1 deficiency mice showed ameliorated ICH-mediated brain damage, which was different from the ability to aggravate injury in many other brain injury models (44). Recently, in this aspect, low HO-1 expression in early SAH patients has been associated with vasospasm, whereas delayed cerebral ischemia (DCI) showed higher HO-1 levels (46, 47). In addition, under ICH pathological status, an overproduction of reactive oxygen species (ROS) was observed, where bivalent iron (Fe2+) promotes hydrogen peroxide (H2O2) disintegration (39, 48) and oxidase enzyme participates in the ROS biological generation process (41, 49). Meanwhile, mice with a generically deleted NADPH enzyme showed reduced damage after ICH (41). Recently, ROS accumulation after SAH has been considered to be a by-product of oxidative phosphorylation in the mitochondria, which is a major target of ROS-induced damage in SAH patients (50). Similarly, both early stages of SAH and ICH were accompanied by ROS generation, which impaired antioxidant defense systems and signal cascade responses (49, 51, 52).
Hematoma formation after ICH usually stimulates inflammatory reaction through microglial/macrophages and/or inflammatory signaling pathways, thereby contributing to immune cascade activation and pro-inflammatory cytokine secretion (53–55). Activated microglia were previously reported to recruit hematogenous inflammatory cells to the ICH injury areas by cytokines and chemotactic factors (56). Meanwhile, microglial/macrophage-mediated phagocytosis facilitates brain cleanup after the early inflammatory responses after ICH, where multicellular surface receptors (CD36, CD91, and SLC) assist in reducing cellular debris following ICH (57, 58). With inflammatory signaling coordination, oxidative stress can enhance the inflammation response after ICH, such as nuclear factor-κB (NF-κB), TNFα, and matrix metalloproteinase-9 (MMP-9) (42). In the chronic phase of ICH, inflammatory stress will impair the white matter tracts and contribute to severe neurological dysfunctions, especially motor and memory functions (59). Evidence indicated that NF-κB is activated in ICH-related brain injury as early as 15 min after the hemorrhage, which will induce nitric oxide synthase (iNOS), TNFα, interleukin, and cyclooxygenase-2 inflammatory cytokines (60, 61). Expression of these genes will lead to neuroinflammation and BBB hyperpermeability (62).
Inflammation is correlated with various neurodegenerative diseases, including SAH, Alzheimer’s disease, and Parkinson’s disease (63). Inflammation is an important mechanism that has been implicated in the pathogenesis of SAH, where cellular inflammation- and molecular inflammation-elicited neuronal injuries have been detected in the subarachnoid space (15). Innate cell immunity obviously generates inflammation responses in the subarachnoid space in an inside-out form. Molecular agents of inflammation were proposed to be increased within posthemorrhagic aneurysms (Figure 1), where factors such as IL-6 and TNF-α are correlated with poor clinical prognosis (15, 19). In addition, within brain injury and the related inflammatory responses, lysis of erythrocytes after SAH showed a positive correlation with increased levels of IL-6 and TNF-α in the brain cortex (64). Following aneurysmal SAH, the increase in pro-inflammatory cytokine IL-6 has been recently presented with neutrophil accumulation in the brain and local and peripheral inflammation responses (65, 66). Immune cell infiltration showed a target therapeutic potential in patients with aneurysmal SAH (65). Moreover, IL-6 has been further defined as a contributing factor to brain injury and is related to poor clinical prognosis (67), wherein IL-6 involved in neuroinflammation response is closely associated with EBI after aneurysmal SAH (68–70). Of note, soluble gp130 (sgp130) and IL-6 receptor (IL-6R) represented the IL-6-transducing antagonist and agonist receptors, respectively (71). With the development of SAH, the level of the IL-6 antagonist gp130 is increased to antagonize the elevated levels of IL-6, which decreases within a few days, presumably resulting in cerebral vasospasm and neuroinflammatory injury (71). Recent studies showed that full-length gp130 is the most potent inhibitor of IL-6 trans-signaling (72). Most recently, a study has shown that through IL-6 signaling, tissue-specific sgp130 can trigger the upregulation of innate immune system components (73), where sgp130-related immune cell and chemokine recruitment might protect against neuroinflammation (74, 75). The molecular weight of the main sgp130 isoforms ranges from 50 to 110 kDa, which has a high affinity (1 mM) for the IL-6/IL-6R complex to neutralize its pro-inflammatory functions (76).
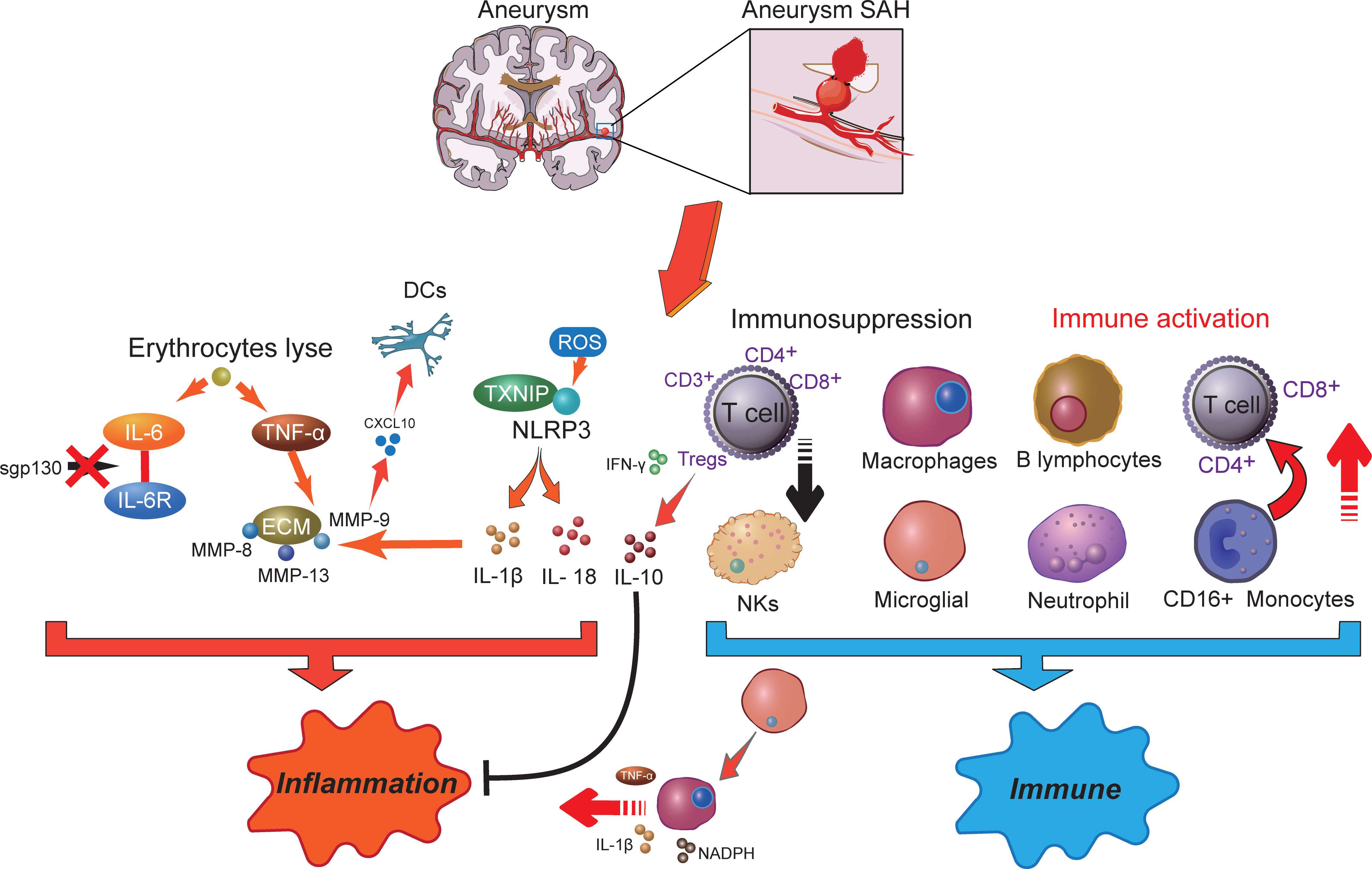
Figure 1 The potential molecular mechanisms of inflammatory effects (right) and immune responses (left) in aneurysm-related subarachnoid hemorrhage. SAH, subarachnoid hemorrhage; DCs, dendritic cells; IL-6, interleukin 6; ROS, reactive oxygen species; MMP, matrix metalloproteinases; NLRP3, NOD-like receptor family pyrin domain-containing 3; TXNIP, thioredoxin-interacting protein; TNF-α, tumor necrosis factor-α; INF-γ, interferon-γ; NKs, natural killer cells; NADPH, nicotinamide adenine dinucleotide phosphate oxidase; Tregs, regulatory T cells.
Although numerous histochemical alterations occur during the development of an aneurysm up to the point of rupture, the release of blood into the subarachnoid space afterward contributes to more serious histological and inflammatory changes (77). Another important inflammation activation-related element, thioredoxin-interacting protein (TXNIP) that interacts with the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome to induce interleukin IL-1β secretion, was previously demonstrated to connect with tumorigenesis and insulin resistance (78). Meanwhile, inflammasomes are part of the innate immune system. The NLRP3 inflammasome is a multiprotein complex that orchestrates innate immune responses, whereas unregulated NLRP3 inflammasome activation in pathology responses can lead to unintended immune and inflammatory pathological conditions, such as mitochondrial metabolism and ROS accumulation (79). Recently, the NLRP3 inflammasome has been extensively studied and observed to be associated with the release of IL-1β and IL-18, which exacerbated the inflammation response after SAH and promoted the occurrence of EBI (80, 81). The intervention of the NLRP3 signaling cascade can alleviate neuroinflammatory responses and restore neurobehavioral function (82). NLRP3 activators can produce ROS, which subsequently activate the inflammasome (83). A strong antioxidant melatonin has been shown to protect against EBI and inflammatory response after SAH (84) and to improve aneurysm SAH clinical outcomes (85). Among which, melatonin suppressed pro-inflammatory cytokine levels in the cortical levels, such as IL-1β, IL-6, and TNF-α (84). Upregulation of these cytokines has been demonstrated to exacerbate brain disorders after SAH (86).
In addition, during aneurysm rupture and consequent SAH, extracellular matrix (ECM) remodeling plays an important role in inflammation. TNF-α has been demonstrated as an upstream regulator for MMP-9 (87). MMP gene expression is upregulated after SAH, where MMP-8, MMP-9, and MMP-13 were observed to accumulate in the vascular wall via the p38 kinase signaling pathways (88). Among them, MMP-9 has been recently reported to have a consistently higher level in aneurysm SAH patients, which may cause cerebral vasospasm, DCI, and neuronal death by promoting neuroinflammation (89). MMPs also participate in the inflammation regulation of pro-inflammatory cytokines and chemokines, especially the function of MMP-9 on CXCL10 and CCL2 (90, 91). Of note, CXCL10 is associated with intrathecal immune activation and dendritic cell (DC) accumulation after aneurysm SAH (92). On the other hand, MMPs are a family of zinc endopeptidases that can open the BBB by degrading tight-junction proteins (93). Melatonin treatment reduces VEGF expression to prevent BBB disruption following SAH (84). Furthermore, VEGF is modulated by several extracellular stimuli, including pro-inflammatory cytokines like IL-6 and TNF-α (94). IL-1β is also an important regulator of MMP-9 and causes BBB disruption after SAH (95). Thus, a variety of immune inflammation processes occur in different compartments following aneurysm SAH and are possibly associated with inflammatory cytokines and immunomodulatory molecule generation.
Although accumulating evidence supports the function of inflammation in aneurysmal SAH, the exact immune mechanisms remain to be elucidated. It has been postulated that SAH following aneurysm rupture induces immune responses including secretion of active substances with vasoactive and pro-inflammatory functions, ultimately contributing to EBI (18, 96–98). Immunosuppression following nervous system injury is a critical issue clinically (Figure 1), because more than 50% of brain-injury patients develop infection (99). In symptomatic aneurysmal SAH patients, poor outcome is associated with symptoms of impaired local immune competence (100). Substantial evidence suggested that temporary impairment of the immune system is an important risk factor in the emergence of infection after aneurysmal SAH (9). Furthermore, pronounced SAH-induced immunosuppression is detected in the early stages of injury after aneurysmal SAH, where a reversed correlation between IL-6 level and CD3+ T cells was observed (101). Among which, the high incidence of bacterial pneumonia in symptomatic aneurysmal SAH patients may be attributed to impaired immune responses and reduced T-cell count. In a previous study, clinical investigations suggested that the risk of subsequent SAH was associated with immune-mediated diseases, such as autoimmune hemolytic anemia, Crohn’s disease, and hyperthyroid conditions (102). In addition, patients with aneurysmal SAH undergoing surgical treatment experienced a transient deterioration in immune functions, especially immunosuppression (9). Decreased immune cell subgroups were significantly associated with aneurysmal SAH, such as the downregulation of CD3+, CD4+, CD8+ T cells, natural killer cells (NKs), and regulatory T cells (Tregs), leading to an unfavorable postoperative prognosis. Nevertheless, following stroke, activated T cells infiltrated the brain, consequently releasing cytokines and ROS, which may result in brain injury, where ROS likely contributed to neuronal inflammation, neuronal cell death, and poor outcomes (103). After that, increased neuroantigens could further induce adaptive immune response and cause additional T-cell activation and brain injury. However, in the middle/late stages in DCI patients, aneurysm SAH-induced immunosuppression was observed to decrease the T-cell population, resulting in an increased risk of infectious complications (103, 104). In a more recent study, following aneurysm SAH, immunosuppressive Tregs were significantly increased and presented a different activation status in the EBI and DCI phases (105). In patients with DCI, CD3+ Tregs showed a higher population compared with EBI and were closely associated with infections. Meanwhile, CD3- Tregs were significantly reduced in patients with EBI. In the EBI phase, low-dose IL-2 treatment significantly prevented the Treg population and suppressed neuroinflammation following SAH, wherein the decreased proinflammatory factors and peripheral neutrophils improved neuronal injury and neurological functions (106). Plausibly, activated Tregs have the effective ability to inhibit the conventional T-cell proliferation and readily produce cytokines (107). Herein, under these circumstances, immunosuppressive Tregs act as modulators of the immune system, resulting in suppression of inflammation by affecting the pro-inflammatory (TNF-α and IFN-γ) and anti-inflammatory (IL-10) factor generation (108–110). Moreover, Tregs also suppress the peripheral MMP-9 production, thereby preventing BBB damage and neuroinflammation (111), which showed the neuroprotective effect and therapeutic potential for aneurysm SAH.
On the other hand, immune activation after aneurysmal SAH has been shown to play a pivotal role in host defense against infection (9). Shortly after the aneurysmal rupture, damage to the brain tissue and blood components led to the exposure of antigens that stimulated innate immune function, which might contribute to its activation and induction of acute immune-inflammatory responses (112). Subsequently, innate immune responses generate molecules that deliver signals, resulting in activation of T cells, effector cells, and B lymphocytes to attach in proinflammatory vessels with release of various adhesion factors and cytokines (112–114). The release of pro-inflammatory cytokines directly eliminates damaged cells, induces and regulates inflammation, and destroys microbes (104). Moreover, the increase in the M1/M2 macrophage ratio plays an important role in both intracranial aneurysm and SAH (115, 116). CXCL1 antibody intervention may give potential to increase the macrophage proportion and anti-inflammatory function. Macrophages can eliminate dead cells and debris and provide defense against infection (117, 118), which will decrease the EBI and complications in aneurysm SAH (119). However, M1 polarization of macrophages is a proinflammatory phenotype associated with reduced debris removal capability and enhanced production of proinflammatory cytokines like TNFα, IL-1β, and NADPH, ultimately contributing to nervous system inflammation (120). It is becoming increasingly clear that the dual role of immune cells needs to be further explored.
Previously, Balboa et al. reported that high levels of CD16+ monocytes stimulated T-cell proliferation and predicted higher antigen-presenting cell (APC) activity in peripheral blood (PB) (121). Indeed, a threefold increase in the potency of APCs was observed in CD16+ monocytes compared with CD16− monocytes (122). Recently, PB analysis in aneurysm SAH patients observed the activation of some immune cell subpopulations such as CD4+/CD8+ T cells, CD16+ monocytes, and neutrophils (123). These results highlight the participation of innate immunity in aneurysmal SAH. The increased proportion of CD16+ monocytes potentially indicated that stimulation of the innate immune system was resided in aneurysmal SAH patients. In addition, the expression of cell-based CD28 in the adaptive immune system induced greater activation of aneurysmal SAH CD4+ and CD8+ T cells in PB than in the cerebrospinal fluid (CSF) (123). CD28 is also the B7 receptor expressed on naïve T cells and provides costimulatory signals that are required for T-cell activation (124). Moreover, CD28 stimulation induces T-cell activation of potential co-stimulatory signals, consequently leading to the generation of various interleukins (125). In terms of this, increased IL-2 receptor and CD8 levels in SAH patients have shown the vital function of immune response in SAH pathogenesis (126). Thus, these findings indicated the participation of innate and adaptive immune responses in the immunopathogenesis of aneurysmal SAH.
The results of pilot studies may be various, but distinguishing the mechanisms of immune suppression and hyperactivation will facilitate the provision of personalized patient treatment to regulate the immune function and protect against aneurysmal SAH. Dysregulation of the immune cell subgroup is closely associated with the clinical prognosis of aneurysmal SAH patients (101, 123), which might be a candidate biomarker to predict patient diagnosis as well as the development of effective therapeutic strategies to eliminate the complications in aneurysmal SAH. Further research on the immunosuppression induced by aneurysm SAH (especially Tregs) and its relationship with inflammatory factors can provide new ideas for the treatment of aneurysm SAH.
Signaling pathway dysfunction can lead to poor outcomes after aneurysmal SAH, which is closely related to primary and secondary injuries in disease development. A precise signaling pathway regulation that triggers both immune modulation and inflammatory responses hold great promise in elucidating pathological mechanisms after SAH. A more comprehensive understanding of SAH-related immune inflammation underlying mechanisms will boost our ability to develop novel therapeutic options. Herein, based on current knowledge, we discussed immune cell and inflammatory function relevant signaling pathway modulation in the context of aneurysmal SAH (Table 1).
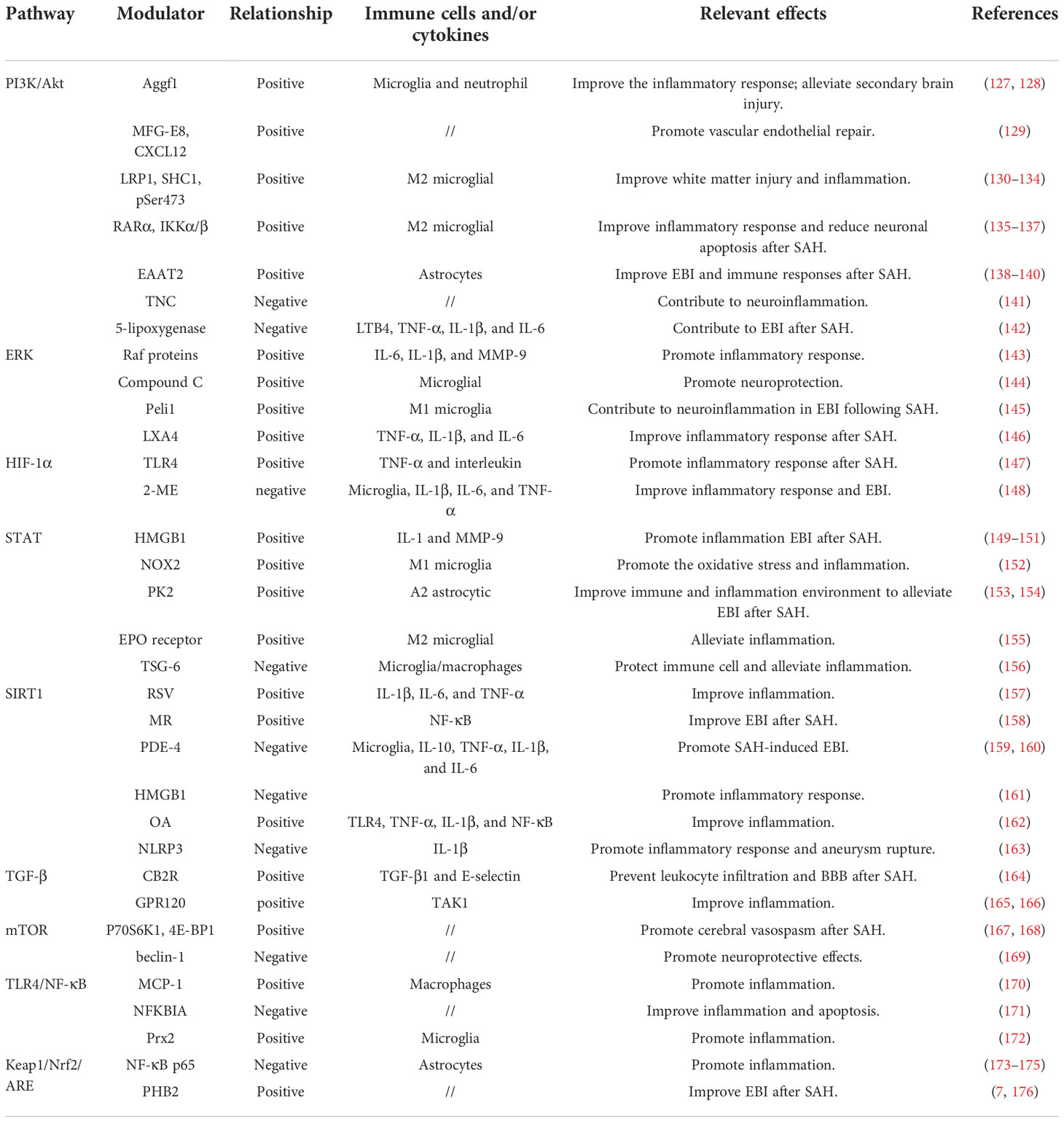
Table 1 Different pathways relevant modulators and effects in aneurysm SAH.
PI3K/Akt signaling pathway dysregulation was previously demonstrated to be associated with various SAH sequela, such as EBI, vasospasm, and neurological injury. The onset of the Akt cascade is activated by tyrosine kinases, immune cell receptors, cytokine receptors, G-protein-coupled receptors, and stimulation of PIP3 generation by PI3K that potentially further influences the immune inflammatory response (177, 178). In SAH, the upregulated Aggf1 expression will provoke the PI3K/Akt signaling and decrease upstream NF-κB activation to improve the inflammation response (127) (Figure 2A). Of note, the above interactions were presented with decreased neutrophil infiltration and microglial activation. The suppression of neutrophils showed potential to improve the immunosuppression response by decreased immune cell monocyte recruitment, thus alleviating secondary brain injury (128). On the other hand, proper immune boosting may also be beneficial in protecting nerves in the brain. The CXCL12 chemotaxis for T cells, lymphocytes, and macrophages had previously been considered to maintain the immune environment in injured blood vessels (179) and played a pivotal role in neuroprotection and against neuroinflammation in recent studies (180, 181). Moreover, Wang et al. indicated that milk fat globule–epidermal growth factor 8 (MFG-E8) exhibited vascular endothelium protection effects through promoting the PI3K/Akt/CXCL12 cascade (129). In the brain of SAH, MFG-E8 directly enhanced PI3K expression and CXCL12 to promote vascular endothelial repair, wherein PI3K activation is causative for increased CXCL12 expression (129). However, the underlying mechanisms of immune cell regulation remain obscure. Furthermore, low-density lipoprotein receptor-related protein-1 (LRP1) activation was reported to attenuate white matter injury (WMI) in SAH patients via the PI3K/Akt pathway, wherein the intracellular adaptor protein SHC1 was required for LRP1 transduction (130). M2 microglial polarization was found to be associated with inflammation-induced functions, and the LPR1 ligand mediated anti-inflammatory M2 microglial phenotypes after SAH (130, 131). The morphological changes of microglia, as the immune cells of the brain, are closely related to their functions (132). Importantly, Akt was appeared to play a crucial role in M1 to M2 polarization via regulation of Ser473 phosphorylation after WMI (133). The activated microglia do not merely modulate the endogenous immune response of brain injury but also alleviate inflammation (134). Similarly, retinoic acid receptor α (RARα) was demonstrated to promote M1 to M2 microglial phenotypic polarization and has anti-inflammatory effects after SAH, relying mainly on regulating the PI3K/Akt pathway (135). The activation of Akt was involved in the phosphorylation of the inflammation-related proteins IKKα/β, through activating the ubiquitin/protease system to promote IKK degradation (136). As a result, this cascade is shown to reduce neuronal apoptosis after SAH (136). On the other hand, the inhibition of PI3K was accompanied by an elevated bim protein level, which is important for cell apoptosis (137). In the mouse model, the bim gene was found to be regulated by IKK and was positively associated with SAH-induced EBI (182, 183).
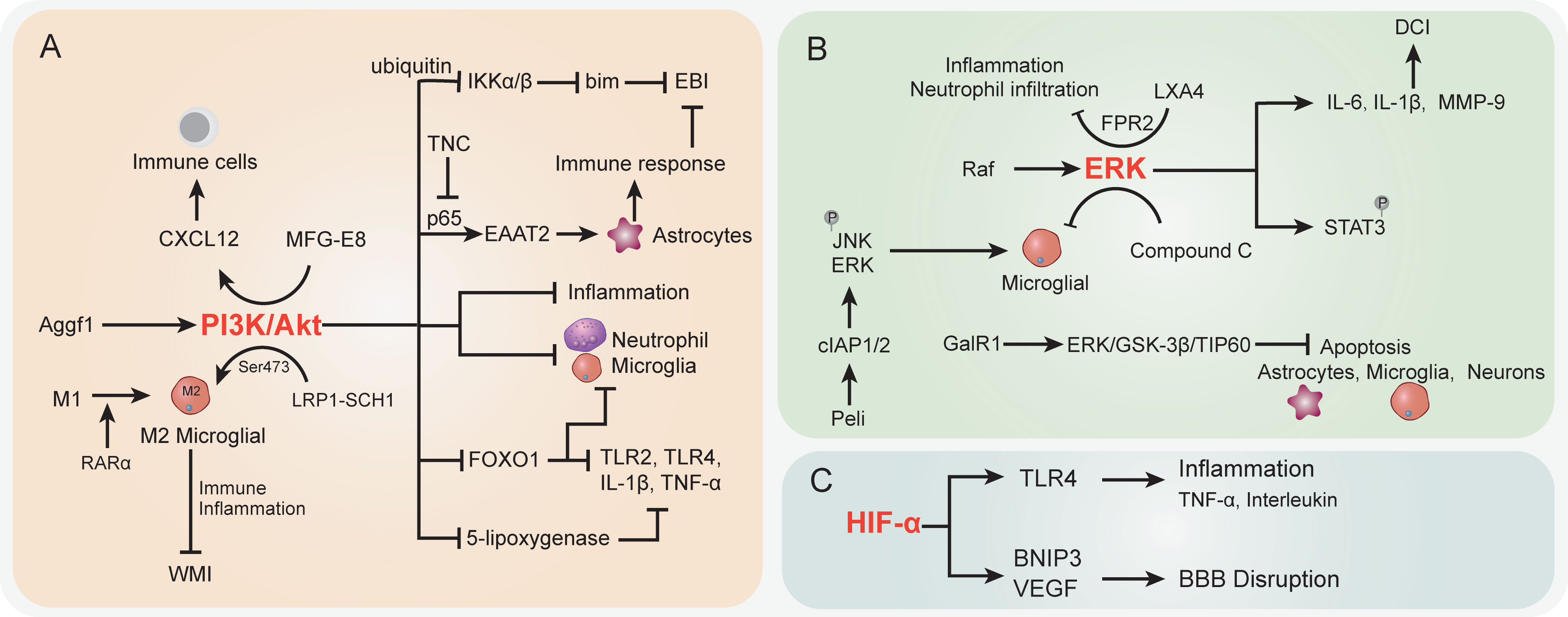
Figure 2 The specific role played by PI3K/Akt (A), ERK (B), and HIF-α (C) pathways in disease development following SAH. CI, delayed cerebral ischemia; EBI, early brain injury; BBB, blood–brain barrier; WMI, white matter injury.
In more downstream candidates of the PI3K/Akt cascade, the transcription factor forkhead box protein o1 (FOXO1) was negatively associated with PI3K/Akt signaling activation; it showed the ability to regulate downstream pro-inflammatory molecules (TLR2, TLR4, IL-1β, and TNF-α) and different types of immune cells (neutrophils, macrophages, DCs, and Tregs) (184). Whether the decrease in FOXO1 protein here affects immune response in SAH needs further analysis. Intriguingly, the EBI after SAH was characterized by the reduction of EAAT2 in astrocytes, which was directly regulated by Akt signaling (138). Data indicated that excitatory amino acid transporter 2 (EAAT2) deficiency in astrocytes was closely associated with innate and adaptive immune pathway disorder. In a mouse model of SAH, Akt activity was observed to be decreased in the brain, thereby leading to a lower EAAT2 level (138), whereas the reactivation of Akt signaling will promote the p65 phosphorylation and significantly improve EAAT2 expression in astrocytes (139, 140), ultimately ameliorating EBI after SAH. The activation of the PI3K/Akt signaling pathway represents a promising positive effect on EBI after SAH (185), as well as a neuroprotective effect (186, 187). In an oxygen hemoglobin-induced SAH mouse model, the upregulated tenascin-C (TNC) after SAH impaired the PI3K/Akt/p65 cascade, thereby leading to neuroinflammation (141). Moreover, the PI3K/Akt cascade was reported to participate in the alleviation of inflammation by inhibiting inflammatory mediators in stroke and promoting tight-junction proteins to protect BBB integrity (188, 189). In terms of neuroinflammation and BBB disruption caused by SAH, PI3K/Akt cascade activation attenuated the above symptoms (127). Recently, Liu et al. reported that increased 5-lipoxygenase in cytoplasm of cortical neurons along with expression of upregulated inflammatory factors LTB4, TNF-α, IL-1β, and IL-6 contributed to EBI after SAH (142). In this process, activation of PI3K/Akt signaling significantly suppressed the 5-lipoxygenase-induced SAH pathologic manifestation (142). Taken together, the activation of the PI3K/Akt signaling pathway potentially improves the immune inflammatory response, eventually resulting in protection against the damage following SAH.
The Raf-mitogen-activated protein kinase kinase (MEK)1/2-extracellular signal-regulated kinase (ERK) 1/2 pathway is one of the components of six MAPK signal transduction pathways that are widely involved in cell regulation (190) (Figure 2B). SAH increased P38 MAPK phosphorylation and attenuated the phosphorylation of ERK (191). Phosphorylation proteomic analysis suggested that the STAT3 pathway was activated upon SAH induction, most likely downstream of ERK1/2, because STAT3 phosphorylation was suppressed by MEK1/2 inhibition (192). Transcriptional overexpression of inflammatory molecules (cytokines and metalloproteinases) in cerebral arteries is caused by SAH-induced activation of the MEK/ERK pathway (143, 193). According to a previous study, cytokine (IL-6 and IL-1β) and MMP-9 upregulation can be prevented by specific blockade of the MEK/ERK pathway via inhibiting upstream Raf proteins after SAH (143), indicating that the MEK/ERK pathway plays a crucial role in DCI following SAH and the cerebrovascular inflammatory response. Synchronously, another research by Maddahi et al. indicated that inhibition of the MEK1/2 pathway only within the time window of 6–24 h after SAH can change cerebrovascular inflammatory response and neurological prognosis later following SAH (194). The underlying mechanism is that IL-1β, IL-6, MMP-9, and pERK1/2 protein expression levels in cerebral artery walls increased with time and increased at the early stage of 6 h after SAH and reached the peak at the late stage of 48–72 h. At the early time points (1 to 24 h) post-SAH, TNFα immunoreactivity in the brain tissue was remarkably enhanced, which is colocalized with glial fibrillary acidic protein (GFAP), a marker of astrocytes and glial cells in perivascular and brain tissues (194).
In addition, the effect of compound C (a classical inhibitor of MAPK) on microglial shape change was mediated by activated ERK1/2, PI3K/Akt signaling, or small Rho GTPase, which provided evidence for the neuroprotective role of compound C in SAH (144). As a clue, Peli is an adaptor protein that interacts with Pelle, which is a Drosophila homologue of the mammalian interleukin-1 receptor-associated kinase (195), whereas, as an E3 ubiquitin ligase, Peli was also upregulated in TLR4-dependent microglial activation post-SAH in a time-dependent manner and induced proinflammatory cytokine IL-6 in microglia (145, 196). Peli1 induced microglia-mediated neuroinflammation in EBI following SAH by enhancing the phosphorylation levels of ERK and JNK via cIAP1/2 activation. Meanwhile, Peli1 also encouraged M1 microglia to exhibit the polarization markers CD16/32 and iNOS after SAH, indicating that the inhibition of Peli1 might generate neuroprotective effects during EBI after SAH (145). Following SAH, the expression of lipoxin A4 (LXA4), an important endogenous lipid, is suppressed, whereas pro-inflammatory cytokine (TNF-α, IL-1β, IL-6) and factor (NF-κB, MMP9, ICAM-1, MPO) expressions were upregulated. Application of LXA4 in mice after SAH attenuates the above inflammatory response and neutrophil infiltration through the LXA4/FPR2/ERK1/2 signaling pathway (146). Moreover, EBI after SAH has been proved to be significantly pathologically influenced by neuronal apoptosis in pathological aspects (197). Activation of galanin receptor 1 (GalR1) has an anti-apoptotic effect in ischemic stroke. More recently, Shi et al. indicated that GalR1 is expressed in some astrocytes and microglia, but mainly in neurons, and activation of GalR1 is reported to recede neuronal apoptosis via the ERK/GSK-3β/TIP60 pathway after SAH (198). In summary, the ERK pathway plays an important role in inflammatory response after SAH, and early inhibition of ERK signaling after SAH may be effective in neuroprotection.
In the context of ischemic stroke and cerebral hemorrhage, hypoxia-inducible factor-1 (HIF-1) is reported to have a dual function by stimulating both pro-survival and pro-death pathways in the central nervous system (CNS) (199, 200) (Figure 2C). HIF-1 protein expression was upregulated at 12 h and reached the peak at 24 h after SAH (201), and HIF-1 stimulation may be detrimental at an early stage after SAH, whereas activation of HIF-1 could be neuroprotective at a later stage post-SAH, suggesting that HIF-1 also performs pro-survival and pro-death roles following SAH (202). In a rat model of SAH utilizing endovascular perforation, HIF-1α can cause cell apoptosis, BBB disruption, and brain edema in EBI after SAH by upregulating the activation of BNIP3 and VEGF expression (203, 204). Simultaneously, as a target of miR-675, HIF-1α improved TLR4 expression via increasing the TLR4 promoter’s transcriptional activity, whereas TLR4 is essential for pro-inflammatory cytokine (TNF-α and interleukin) release following SAH. Nevertheless, these pro-inflammatory cytokines participate in cell apoptosis which play a crucial manifestation of post-SAH EBI (205). In addition, 2-methoxyestradiol (2-ME), a natural endogenous metabolite of 17-β estradiol, has antitumor, anti-angiogenic, and anti-inflammatory abilities (147, 206). Research data showed that 2-ME can reduce inflammatory factor (IL-1β, IL-6, and TNF-α) expression levels; downregulate brain water content, microglial activation, BBB permeability, and cell apoptosis; and enhance neurological dysfunction in rats. However, the mechanism of this protective effect is that 2-ME inhibits the expressions of HIF-1α, MMP-9, and VEGF, which is related to BBB disruption after SAH and inflammatory response to EBI (148). The HIF-1α signaling pathway as a regulatory target of inflammatory response after SAH needs to be further investigated.
In the SAH case, the activation of STAT-related signaling potentially contributed to morphological changes in cerebral arteries (207). The STAT pathway has been largely studied in vascular diseases (208). Recent studies demonstrated that STAT signaling was also involved in inflammation and immune cell balance during SAH (Figure 3A). JAK2/STAT3 signaling was regarded as an important inflammatory signaling pathway in mediating immune responses, which has a critical role in keeping the balance between pro-inflammation and anti-inflammation (209). For STAT3, a pivotal part of the STAT signaling cascade is known to regulate gene expression. The phosphorylation of STAT3 activated pro-inflammatory gene expression and influenced the pathologic progression of SAH (210). Among which, JAK2 is the essential component of STAT3 activation. Recently, An et al. reported that the activated JAK2/STAT3 cascade after mouse SAH was positively associated with pro-inflammatory molecular HMGB1 expression in both nucleus and cytoplasm (149), which subsequently promoted the pro-inflammatory cytokines like IL-1 and MMP-9 and contributed to EBI after SAH (150, 151). Simultaneously, many studies highlighted the HMGB1 function in brain injury and vasospasm, and inhibition of acetylation and release of HMGB1 paved a way to decrease inflammation after SAH (211, 212). It should be noted that the JAK2/STAT3 cascade was involved in immune cell microglial regulation after SAH. Pang et al. indicated that the JAK2/STAT3 cascade acted as the upstream of NADPH oxidase 2 (NOX2) expression in M1 microglia, which is the basis for oxidative stress and inflammatory cytokines in a SAH mouse model (152). Inhibition of the JAK2/STAT3/NOX2 cascade significantly suppressed the M1 microglial activation, subsequently improving the oxidative stress and inflammation. Moreover, targeting the STAT3 pathway showed potential to prevent BBB disruption following SAH (213) and exerted neuroprotective effects (214, 215). On the other hand, prokineticin 2 (PK2) was demonstrated to promote an anti-inflammatory A2 astrocytic phenotype and prevent neuronal injury (153). Ma et al. indicated that the effect of PK2 on the formation of A2 astrocytes of SAH was linked to STAT3 phosphorylation (154). Accumulation of A2 astrocytes potentially improved the immune cell, BBB, and neuron damage after SAH (216). Thus, the activated PK2/STAT3 cascade might promote the A2 astrocytes and improve the immune and inflammation environment to alleviate EBI after SAH (154). On the contrary, in the A1 pro-inflammatory astrocytic phenotype, activated STAT3 after SAH was deemed to be responsible for A1 activity, whereas the inhibition of STAT3 significantly abolished the astrocytic A1 polarization (217). The overactivation of STAT3 in A1 astrocytes is detrimental during SAH (217).
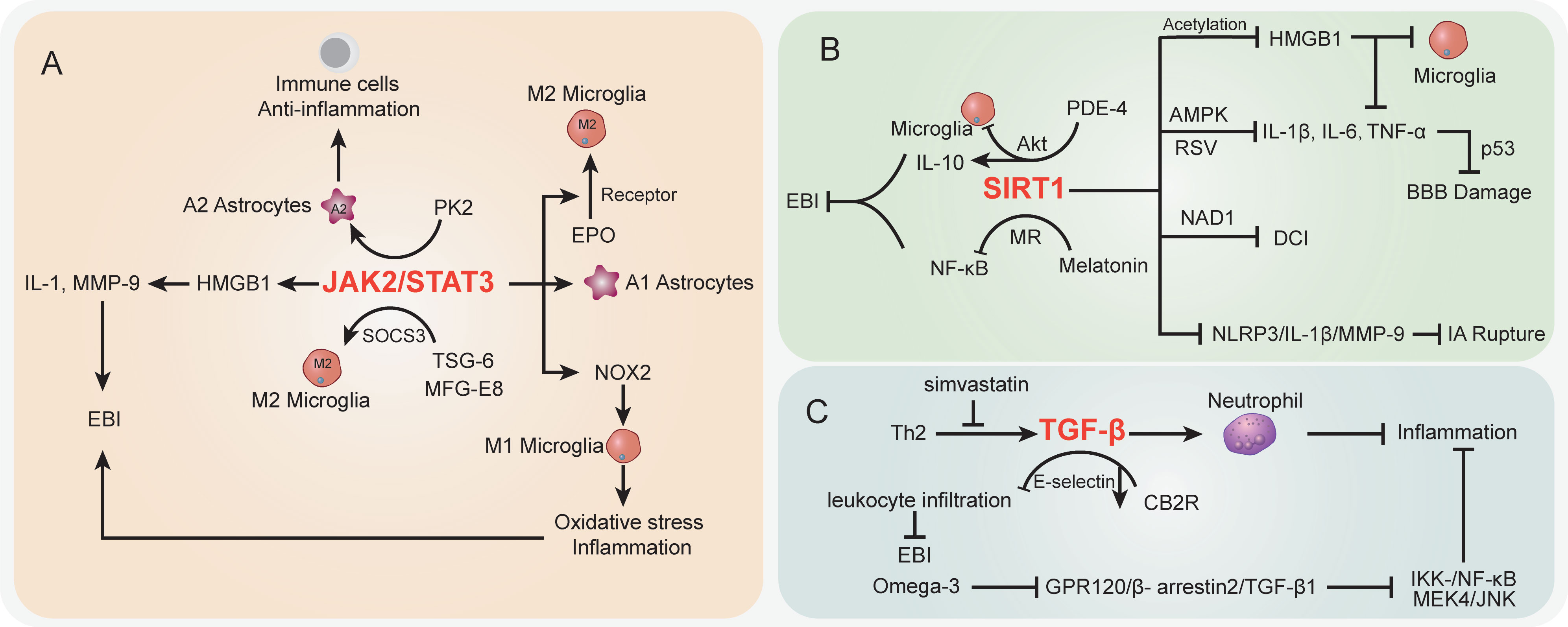
Figure 3 The specific role played by STAT (A), SIRT1 (B), and TGF-β (C) pathways in disease development following SAH.
Interestingly, the activated JAK2/STAT3 cascade represented a severe condition after SAH, whereas its phosphorylation was observed to promote microglial M2 polarization and alleviate inflammation (155). Among which, the erythropoietin (EPO) treatment SAH model has upregulated EPO receptor (EPOR) expression along with the JAK2/STAT3 cascade to enhance the M2 polarization, whereas interfering with any of the above node will abolish the polarization process (155). Thus, the EPOR/JAK2/STAT3 cascade plays an important role in microglial functions and EBI after SAH. Furthermore, in microglial polarization, the SAH-protective molecule TNF-stimulated gene-6 (TSG-6) was deemed to play an important role in anti-inflammatory M2 phenotype transformation via the SOCS3/STAT3 cascade, wherein TSG-6 could decrease the STAT3 expression and increase SOCS3 expression (156). The TSG-6 protective effects in immune cell infiltration and inflammation have been wildly studied in the brain, especially the function in inhibiting the activation of microglia/macrophages (218). Recently, Gao et al. indicated that the interaction between milk fat globule-epidermal growth factor-8 (MFG-E8) and integrin β3 receptor could stimulate the SOCS3/STAT3 cascade then participate in the microglial M2 polarization and relieve the neuroinflammation after SAH (219). Of note, the STAT3 absence is the core to trigger the microglial morphological polarization process after SAH (220). The low abundance of TSG-6 will lead to the attenuated innate immunity response and elevate M1 microglia after SAH concomitant with inflammation and poor outcomes (156). Herein, the different cascades might contribute to distinct results after SAH and the therapeutic strategies targeting the STAT pathway should be carefully considered.
SIRT1 is a class III histone deacetylase that controls a number of physiological processes, such as DNA damage repair, oxidative stress, inflammation, energy consumption, and cell death (221, 222). SIRT1 activity is dependent on and adjusted by nicotinamide adenine dinucleotide (NAD1) (223) (Figure 3B). NF-κB, p53, nuclear factor (erythroid-derived 2)-like 2 (Nrf2), forkhead box o (FOXO), hypoxia-inducible factors (HIFs), and liver X receptor (LXR) are histone and non-histone substrates that SIRT1 deacetylates (224, 225). The increased expression of SIRT1 has been reported to have a neuroprotective effect on brain edema and endogenous protection against DCI after SAH, as well as inducing the attenuation of neurovascular dysfunction following SAH (226–228), and the p53 pathway regulated by endogenous SIRT1 can crucially affect BBB permeability and brain edema after SAH (226). Concomitant with FOXO1, NF-кB, and p53 decreased acetylation, activation of SIRT1 pathways after SAH markedly reduced the levels of IL-1β, IL-6, and TNF-α; decreased Bax and cleaved caspase-3 levels and microglial activation; and increased Bcl-2 expression (229, 230). In EBI after SAH, pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) and neural apoptosis were also suppressed by resveratrol (RSV) via the AMPK/SIRT1 cascade (157). Meanwhile, melatonin has the ability to downregulate Ac-NF-κB and Bax expression and upregulate SIRT1 expression, suggesting that melatonin improved EBI following SAH through the melatonin receptor (MR)/SIRT1/NF-κB signaling pathway (158). Phosphodiesterase-4 (PDE-4) is crucial in a variety of injuries to the CNS, and PDE4 inhibition can inhibit neuronal apoptosis through the SIRT1/Akt pathway and ultimately protect rats from SAH-induced EBI (159). As a PDE4 inhibitor, rolipram significantly enhanced SIRT1 expression, whereas NF-κB activation is repressed in EBI after SAH. Mechanically, rolipram can upregulate protective cytokine IL-10 expression and inhibit pro-inflammatory cytokine (TNF-α, IL-1ß, and IL-6) expression as well as downregulate microglial activation (160). Moreover, the robust cerebral inflammation following SAH was linked to a considerable activation of the HMGB1/NF-kB pathway (14, 161). Accumulating evidence has shown that SIRT1 regulates HMGB1 hyperacetylation and suppresses HMGB1 translocation release (161). Zhang et al. indicated that enhanced SIRT1 expression can inhibit the inflammatory response mediated by HMGB1/NF-KB activation after SAH. As a selective SIRT1 inhibitor, ex527 reversed berberine-induced SIRT1 activation and attenuated berberine anti-inflammatory and neuroprotective effects on SAH, as illustrated by upregulated TNF-a, IL-1β, IL-6, and ICAM-1 release and microglial activation (161). Han and colleagues reported that oleanolic acid (OA) enhanced the expression of SIRT1 rather than suppressed the JAK/STAT3 pathway to lower the acetylation level of HMGB1. OA displays an anti-inflammatory effect by regulating TLR4, TNF-α, IL-1β, and NF-κB expression via SIRT1 signaling. HMGB1 is mostly expressed in neurons in EBI after SAH, which is associated with apoptosis, whereas HMGB1 is primarily expressed in microglia in DCI following SAH, which is associated with immunological activation (162). Moreover, as a multiprotein oligomer, the nucleotide-binding oligomerization domain–like receptor family pyrin domain–containing 3 (NLRP3) inflammasome is responsible for inflammatory response activation, which can promote IL-1β maturation and induce IL-1β release, ultimately leading to inflammation and tissue damage (231). More recently, in an aneurysm model under estrogen-deficient conditions, ERα and SIRT1 depletion may promote the activation of the NLRP3/IL-1β/MMP-9 pathway and enhance intracranial aneurysm rupture leading to SAH (163). Overall, targeting the SIRT1 pathway is a promising method to attenuate EBI and DCI after SAH via regulating inflammatory response.
Transforming growth factor (TGF)-β1 signaling plays an important regulatory role in endothelial cell differentiation, maintaining vascular wall integrity and the vascular network (232). The cortical and brainstem levels of TGF-β1 after SAH were remarkably enhanced in rats with the high-dose simvastatin group, which also repressed immunosuppressive cytokine TGF-β1 expression by lymphocytes and IL-1β expression post-SAH (233) (Figure 3C). Simvastatin triggers a Th2 immunological transition in these animals, and infiltrating Th2 cells are to blame for the observed rise in TGF-β1 production in the brain after therapy, ultimately providing neuroprotection against the neurological impairment following SAH (233). In addition, TGF-β1 can also inhibit neutrophil recruitment by reducing endothelial E-selectin expression (234). Cannabinoid-type 2 receptor (CB2R) agonism is reported to downregulate neuroinflammation (164). Fujii and colleagues suggested that CB2R stimulation prevents leukocyte infiltration into the brain by upregulating TGF-β1 and downregulating E-selectin, which protects the BBB after SAH and reduces neurological outcomes and brain edema (164). Meanwhile, omega-3 fatty acids are also able to exert effective anti-inflammatory effects through the G protein-coupled receptor 120 (GPR120) signaling pathway (165). Omega-3 fatty acids inhibited SAH-mediated inflammatory responses and apoptosis by the GPR120/β-arrestin2/TGF-β1 binding protein-1 (TAK1) anti-inflammatory pathway, eventually suppressing IKK-/NF-κB and MEK4/JNK downstream pathways (166), whereas fingolimod (FTY720), an immunomodulatory agent, enhanced Tregs and attenuated NKs in SAH mice treated with fingolimod after 3 days. Inflammatory cytokine IL-6 and TNF-α expressions were also decreased, whereas IL-10 and TGF-β1 were upregulated in serum with fingolimod post-SAH (235, 236). In summary, further research into drugs capable of modulating the TGF-1β pathway may provide new ideas for improving the post-SAH inflammatory response.
The mechanisms underlying poor prognosis following SAH are complex and multifactorial. The mammalian target of rapamycin (mTOR) is an atypical serine/threonine kinase involved in regulating major cellular functions, including growth, proliferation, survival, and protein synthesis (237). Through reducing excessive mitochondrial fission, mTOR inhibition protects against neuronal damage in EBI following SAH, indicating that mTOR activation additionally aggravated the neuronal and mitochondrial injury (238, 239) (Figure 4). As the core of the pathway–pathway interaction network, mTOR signaling is also associated with genes related to intracranial aneurysms (240). Circular RNAs (circRNAs) are closely related to many vascular diseases (241). Of note, by regulating the mTOR signaling pathway, circRNAs have been implicated in the formation of intracranial aneurysms (240). Moreover, mTOR has the ability to shape the immune system like immune cell migration, cytokine generation, antigen presentation, and macrophage polarization, further influencing the immune and inflammatory responses (242). On the other hand, previous studies have demonstrated that the mTOR signaling pathway plays a vital role in cerebral vasospasm following SAH (243). The increased levels of mTOR, P70S6K1, and 4E-BP1 (167) in basilar arteries were significantly associated with SAH and potentially mediated the activation of cerebral vasospasm. As a member of the PI3K family, mTOR orchestrates the phosphorylation of key downstream proteins P70S6K1 and 4E-BP1, both of which promote the proliferation of key vasculature wall cells (168). The mTOR/P70S6K1/4E-BP1 signaling pathway is significantly activated following SAH injury, and inhibition of mTOR is implicated as an attractive potential therapeutic strategy for vasospasm following SAH (243). Intriguingly, in a mouse brain ischemia model, inhibition of mTOR upstream suppressor PTEN observed that mTOR activation was directly involved in cortical neuron proliferation and enhanced neuronal axon densities (244). The mTOR activation improved long-term functional recovery after stroke rather than the acute phase, which may be beneficial for improving DCI after SAH (244). Moreover, the delayed cerebral vasospasm caused by bilirubin oxidation end products (BOXes) may have contributed to neurological impairment (167, 245). The end products of heme metabolism Z-BOX B significantly upregulated the phosphorylation of Akt, mTOR, and p70S6K, whereas rapamycin was able to counteract Z-BOX B’s effects. Recently, in a CoCl2-induced oxidative neuronal injury model, Z-BOX B dramatically reversed the hypoxia-induced neuronal injuries and stopped the apoptosis of primary cortical neurons through the Akt/mTOR/p70S6K signaling pathway (246). Meanwhile, the rapamycin specificity inhibited the expression of mTOR and upregulated the beclin-1 level to improve neuroprotective effects in ischemia reperfusion injury (169), where beclin-1 will stimulate the macrophage autophagy in the brain (247, 248). These data underscore the idea that targeting the mTOR signaling pathway can efficiently prevent macrophage function and suppress neuroinflammation in SAH patients (249). Hence, we need more studies to further confirm that targeting the mTOR signaling pathway modulates neuroinflammation after SAH.
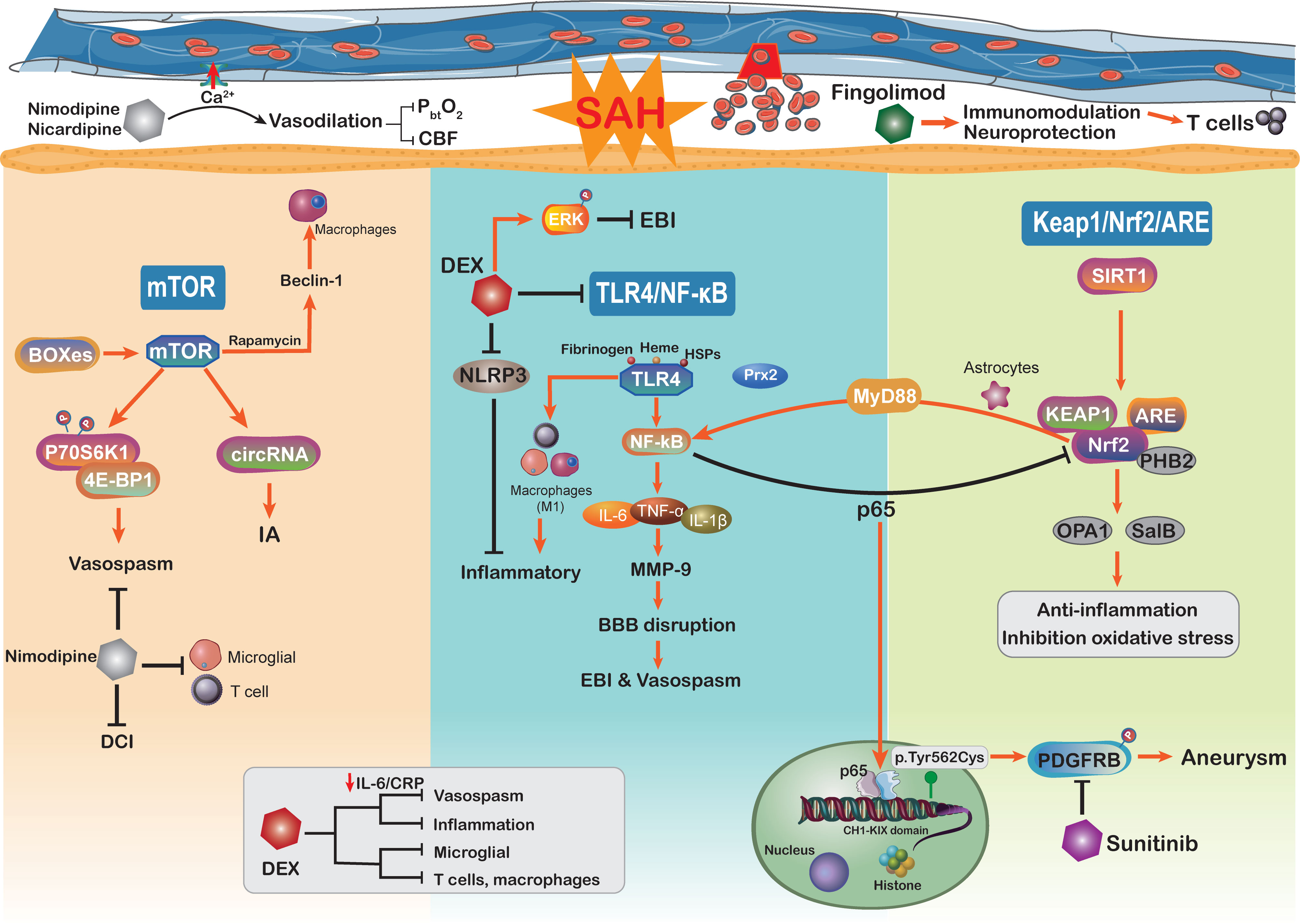
Figure 4 Schematic diagram of the mTOR, TLR4/NF-kB, Keap1/Nrf2/ARE signaling pathways and related therapy strategies in the pathophysiology of subarachnoid hemorrhage. CBF, cerebral blood flow; DEX, dexmedetomidine; CRP, C-reactive protein.
The TLR4/NF-κB signaling pathway plays an important role in the secretion of inflammatory factors such as IL-1β, TNF-α, and IL-6, which has been proved to be involved in the EBI following SAH (86, 250). TLRs are a family of receptors that play an essential role in brain innate immunity and inflammatory responses (251, 252). TLR-4 activation potentially acts as a costimulatory molecule for T-cell activation, where the cytokines IFN-γ and IL-1 participate in this process within the brain immune microenvironment and constitute the neuroinflammation (253–255). NF-κB is a putative inflammation regulator in multiple pro-inflammatory functions (256), which influences the DNA transcription and immune response in aneurysm SAH (171). Pro-inflammatory factors TNF-α, IL-1β, COX, and MMP-9 gene expressions were reported to be regulated by NF-κB (119, 257). The inhibition of NF-κB may alleviate MCP-1-induced macrophages infiltrating inflammation and reduce aneurysm formation and rupture (170). The aneurysm wall is characterized by brain immune cell population alteration, such as NKs, T cells, mast cells, and macrophages (258). On the other hand, the expression of NF-κB inhibitor gene NFKBIA in SAH patients has an anti-inflammatory effect and is associated with apoptosis and neurotrophin signaling (171). Activated microglia were attributed to roles as antigen-presenting cells and respond to TLR-4, thus shaping the adaptive immune response in the neuroinflammation (259). The activation of the NF-κB cascade is associated with increased microglial and macrophage populations in aneurysm (260). Moreover, the activation of TLR-4/NF-κB signaling can transfer macrophage into the M1 phenotype (261, 262). In aneurysm, macrophages do not merely influence the post-SAH inflammatory responses but also are related to intracranial aneurysm formation and rupture (257). The upregulation of M1 macrophage population showed the ability to mediate inflammation and promote the risk of rupture (257). Immune cell dysregulation and inflammation represent the cornerstones of aneurysm SAH occurrence; it seems to be the promising therapeutic target to the aneurysm SAH.
Some physiological derangements such as raised intracranial pressure and global cerebral ischemia after SAH have been shown to be mediated by inflammation and oxidative stress (8, 263). Accumulating evidence indicated that inflammatory cascades are involved in EBI after aneurysm SAH, especially the vasospasm (263). TLR4 activation is modulated by a variety of endogenous ligands including ROS, fibrinogen, heme, and heat shock proteins, all of which will be released following SAH (264). Moreover, patients with SAH are reported to express higher levels of TLR4 on PB cells, which is related to worse functional recovery and more serious SAH (265). The TLR4/NF-κB signaling pathway partly participated in cerebral vasospasm COX-1 upregulation (264). Moreover, activation of TLR4 on mononuclear cells is closely associated with cerebral vasospasm and DCI after aneurysm SAH, resulting in worse neurological function recovery (265). The inhibition of TLR4/NF-κB signaling decreased the EBI and cerebral vasospasm via improving the MyD88- (early phase) and TRIF- (late phase) dependent inflammatory response, which also protected against DCI and prevented poor outcomes (266). The efficient mechanism of TLR4 expression in regulating NF-κB activation has become a focus of research. In a recent study, damage-associated molecule peroxiredoxin 2 (Prx2) can regulate TLR4 function on microglia and subsequently stimulate the TLR4/NF-κB pathway following aneurysm SAH (172). Furthermore, TLR4/NF-κB signaling pathway activation may be involved in the mechanism by which neuroinflammation is exacerbated by releasing a multitude of inflammatory factors, such as IL-6, IL-β, TNF-α, and CD86 (267). Targeting the TLR4/NF-κB cascade not only downregulates pro-inflammatory cytokines levels but also alleviates the number of macrophages, neutrophil infiltration, and cell death. There is a great deal of evidence showing that the development of SAH is correlated with TLR4/NF-κB pathway activation, and this pathway may be a potential therapeutic target (86). In recent years, inhibition of the NF-κB cascade can efficiently alleviate SAH-related EBI, where the novel drug Netrin-1 shows the ability to improve the neurological deficits and brain injury via the regulation of the NF-κB signaling pathway (268).
Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) is an essential transcription factor that regulates the antioxidative system, which reduces the progression of various oxidative stress-related disorders (269, 270). It binds to a specific DNA site, antioxidant response element (ARE), to regulate the transcription of detoxifying or antioxidant enzymes (271). Nrf2 and ARE are key modulators in reducing inflammatory damage and oxidative stress, both of which are involved in SAH (272). Nrf2 also downregulates haptoglobin (Hp), hemopexin, red blood cells, and hemoglobin (Hb) after SAH (273). The haptoglobin (Hp; α and β peptide chains) phenotype determines outcomes in SAH and binds to hemoglobin (Hb) via a strong extracellular interaction (274). Previously, within the p62 catalyzing after SAH, the oxidized intracellular redox sensor Keap1 has been proved to directly accelerate Nrf2 release and activation (275, 276). Thus, activation of the Keap1/Nrf2/ARE pathway by its inducer may reduce the inflammatory response and ameliorate EBI after SAH. A recent study has shown that deletion of Nrf2 was associated with an increased inflammatory response and cell death (238). Continuous activation of the NF-κB pathway induces pro-inflammatory cytokine production and exacerbates inflammation (238, 273). Moreover, some studies have indicated that there is crosstalk between Nrf2 and the NF-κB signaling pathway in inflammation and injury (277, 278), which may influence the innate immune cell function. The NF-κB p65 subunit suppresses the Keap1/Nrf2/ARE pathway at the transcriptional level through competitively acting on the local histone hypoacetylation CH1-KIX domain (173). Furthermore, stimulation of the Keap1/Nrf2/ARE pathway after SAH can significantly downregulate the inflammatory response and oxidative stress (279). In addition to NF-κB upregulation, downstream inflammatory cytokines such as TNF-α, IL-1β, IL-6, and MMP9 are upregulated in astrocytes after SAH (174). Activated astrocytes play an important role in the neuro-immune axis and have the ability to modulate intracranial innate and adaptive immune response, like T cells, Tregs, and macrophages (175).
As a result of ROS generation, mitochondrial dysfunction is also involved in the pathological mechanism of EBI following SAH (280). The dynamic processes of mitochondrial function are closely associated with the Keap1/Nrf2/ARE signaling pathway (7). A previous study of hepatocellular carcinoma suggested that the binding of Nrf2 and prohibitin 2 (PHB2) is required for efficient expression (281). PHB2, an inner mitochondrial membrane protein, is a crucial receptor related to mitochondrial function (176). The synergistic expression of the downstream protein optic atrophy 1 (OPA1) not merely protects nerves after SAH but also is involved in the Nrf2-mediated signaling pathway (282). Treatments with increased levels of Nrf2, PHB2, and OPA1 have shown the ability to attenuate EBI (7). Therefore, attributing to its interaction with PHB2 in mitochondrial dysfunction, the Keap1/Nrf2/ARE pathway may play an important role in pathological mechanism underlying SAH.
In terms of oxidative stress, oxidative damage amelioration is associated with suppression of ROS generation and superoxide dismutase activity (270). The Keap1/Nrf2/ARE pathway has been demonstrated to be an antioxidant target in the SAH model of oxidative stress responses (279). Numerous recent studies have shown that SIRT1 exerts potent antioxidant effects by enhancing the expression and activity of the Keap1/Nrf2/ARE pathway, which can improve SAH-induced oxidative damage (229, 270, 283, 284). In addition, the Nrf2-ARE cascade is activated in the brain after SAH to prevent the brain from EBI, which probably inhibits cerebral oxidative stress by inducing antioxidant and detoxifying enzymes (285). A study in a mouse model of SAH showed that Nrf2 expression is upregulated in the arteries as a compensatory mechanism (286). Similarly, in another early SAH mouse model, Nrf2 expression was increased in the cortex in a time-dependent (12, 24, and 48 h) manner compared with the expression observed in the control group (285). Nrf2 knockout significantly reversed the antioxidant effects of salvianolic acid B (SalB) in SAH-induced oxidative damage (270). To date, substantial evidence demonstrated that the Keap1/Nrf2/ARE pathway is activated in SAH. Taken together, these results suggest that the Keap1/Nrf2/ARE pathway can be a target for immune modulation and anti-inflammatory and antioxidative therapy after SAH.
The management of patients with aneurysmal SAH remains a highly demanding challenge in critical care medicine. According to the above immune cells and inflammatory regulation, several therapeutic and preventive drugs from current preclinical and animal experiments were discussed in our review (Table 2). We summarized drug types with great potential to influence the immune inflammatory regulation after aneurysmal SAH like dexmedetomidine, nicardipine, nimodipine, and fingolimod, as well as prophylactic drugs for development of aneurysms like sunitinib (Figure 4).
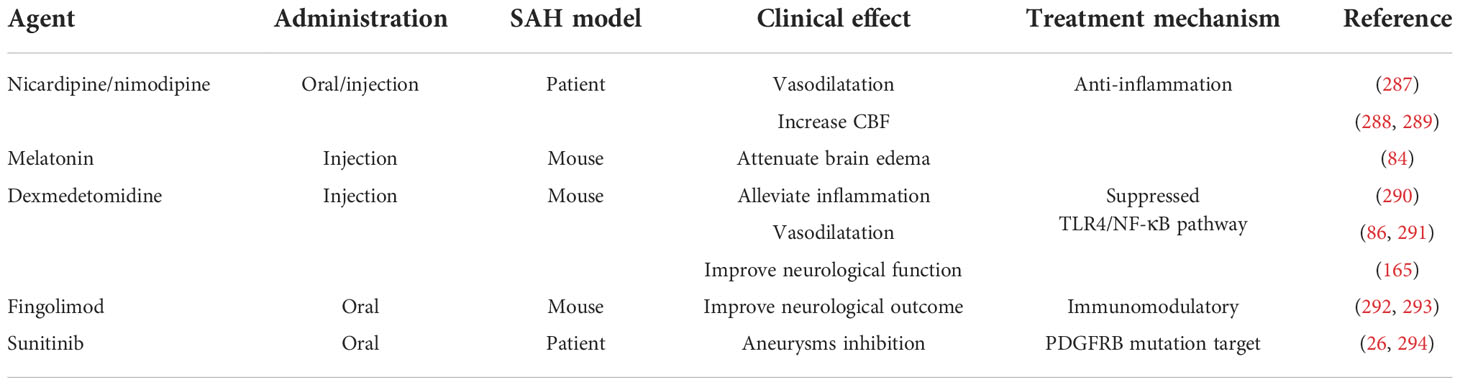
Table 2 Therapy strategies in aneurysm SAH.
Retrospective and prospective studies have shown that intrathecal nicardipine can improve outcome, decrease angiographic vasospasm, and downregulate mean blood flow velocity in SAH. The analogue of calcium channel antagonist nicardipine, oral nimodipine, remains the only FDA-approved medication to improve aneurysmal SAH (295). Nimodipine is lipophilic and intersects with intact BBB to achieve bioavailability (296). The average bioavailability of oral nimodipine (45 mg/4 h) was only approximately 16% of the maximal plasma concentration 1 h after ingestion in SAH patients (287). Hepatic metabolism by cytochrome P450 may contribute to the low plasma concentration. For the T cells, nimodipine can inhibit the Ca2+ influx to suppress T-cell function and related inflammation (297). In addition to modulating the immune inflammatory response, nimodipine also suppressed the T-cell energy metabolism, proliferation, and Th1 differentiation (297), which played a potential role in shaping the immune responses after aneurysm SAH. The previous in vitro studies demonstrated that L-type calcium channel blocking has a positive role in microglial activation suppression and further contributes to neuroprotection and pro-inflammatory factor inhibition (298). However, the dearth of dependable evidence for nimodipine effect in the clinical SAH immune microenvironment still limited the horizon of its application. Generally, appropriate microglial activation in the early stage is essential for harmful substance clearance after SAH, whereas the hyperactivated microglial will aggravates brain damage and promotes the release of pro-inflammatory cytokines, chemokines, and cytotoxic substances (299). Targeting the T-cell and microglial activation might be a novel therapeutic alternative for nimodipine in aneurysm SAH.
Moreover, pleiotropic mechanisms of nimodipine activation are associated with L-type calcium channel-mediated vascular smooth muscle cell vasodilation (300). Nimodipine has been shown to be an efficient vasodilator in vitro and in vivo, resulting in improvement in the outcomes after aneurysmal SAH (288). The inhibition of calcium influx into the cellular compartment may downregulate smooth muscle contractility, thereby causing vasodilatation (301). As a result, oral nimodipine administration decreases average arterial blood pressure and cerebral perfusion pressure. Similarly, monitoring of physiologic parameters after nimodipine administration revealed decreased brain tissue oxygenation (PbtO2) and poor cerebral blood flow (CBF) in aneurysmal SAH patients. Different physiologic changes in the brain oxygenation and pressure indexes were improved by nimodipine administration (301, 302). Of note, intra-arterial and intravenous nimodipine therapy after aneurysmal SAH was reported to attenuate cerebral artery constriction and increase CBF (289, 303). Previous angiographic studies showed significant and immediate clinical improvement (reaching 75%) in macro-vasoconstriction in 50%–65% of SAH patients following intra-arterial administration of nimodipine (304). In addition, through regulating the pressure reactivity index (PRx), the risks of rebound ischemia in patients at risk of recurrent vasospasm can be minimized by infusion with intra-arterial nimodipine (301, 305, 306). Nimodipine can also enhance lymphatic system function and mitigate neurological defects and cerebral edema in SAH mice by activating the cAMP/PKA pathway (307). Thus, nimodipine is not merely involved in immune cell regulation but also has shown a great potential in improving the SAH relevant vascular lesion.
However, there are studies on the efficacy of intravenous or oral administration of nimodipine and its use as an adjunct treatment option in aneurysmal SAH patients. In current applications, nimodipine provides less than optimal efficacy and causes dose limitation in a number of SAH patients (287, 300). Among the different routes of delivery, lipid-based drug delivery systems have attracted increasing attention due to their solubility, bioavailability, and stability (308). Recently, local administration of vasoactive drugs (nimodipine) and prolonged-release pellets has been shown to reduce the incidence of cerebral vasospasm and delayed ischemic deficits after severe aneurysmal SAH (309, 310). Of note, nicardipine prolonged-release implants (NPRIs) formulated in copoly(lactic/glycolic acid) (PLGA) have been developed as an approach to local delivery (310, 311). NPRIs have a good safety and tolerability profile with no complications and no signs of neuronal toxicity in aneurysm-clipped patients (310). Moreover, nanotechnology development has revealed that different structural modifications presented with great potential to improve the drugs’ therapeutic effect. Among these, nanostructured lipid carrier lactoferrin-modified PEGylated NLC (Lf-NLC) exhibits a high loading content and uniform particle size biodistribution, which was designed and constructed for the efficient delivery of nimodipine in treating strokes in the brain (296). It was previously observed that epithelial cells overexpressing low-density lipoprotein (LDL) provide a unique opportunity for therapeutic agent delivery by Lf (312). By crossing the BBB, Lf-NLC can be internalized into cytoplasm via the Lf-receptor-related endocytosis pathway to deliver nimodipine to brain tissues (296). Moreover, nimodipine dose reduction or discontinuation influenced by arterial blood pressure is a frequent occurrence, which is also related to poor clinical outcome (295). In contrast, the multivariate analysis showed that full dosage of nimodipine decreased the risk of unfavorable clinical outcome (OR 0.895, P = 0.029). The chemical modification of nimodipine significantly increased the local concentration and decreased the adverse reaction. In these studies, a more favorable clinical outcome, decreased mortality, improved cerebral vasospasm, and lower delayed ischemic lesion incidences have been reported.
NicaPlant (BIT Pharma), a novel sustained nicardipine release system composed of a mixture of two completely degradable polymers, has been developed to provide pharmaceutical equivalence and improve ease of manufacturing compared with NPRIs. In a chronic cranial window model, the application of NicaPlant for more than 3 weeks, with a higher arterial vessel diameter due to vessel dilatation (21.6 ± 2.6 µm vs. 17.8 ± 1.5 µm in controls, P < 0.01 vs. the control group), was observed by using in vivo epifluorescence video microscopy (313). The active ingredients in NicaPlant do not stimulate local tissue reaction, vessel leakage, or the leukocyte–endothelial cell interaction, which improve the safety of this delivery system. Maintaining a stable blood drug concentration is important for SAH patients’ treatment. Therefore, NicaPlant showed a good safety and efficacy profile in aneurysmal SAH patients compared with NPRI and improved patient outcomes while avoiding systemic side effects (313)
More recently, the results of the randomized, open-label, phase I/IIa dose-escalation trial of NEWTON (Nimodipine micro particles to enhance recovery while reducing toxicity after SAH) showed good safety, tolerability, pharmacokinetics, and clinical effects in aneurysmal SAH (314). NEWTON has been shown to be a safe and well-tolerated nimodipine microparticular formulation with a significant reduction in systemic side effects when compared with oral nimodipine (315). Furthermore, patients treated with NEWTON showed obvious reductions in DCI (31% NEWTON vs. 61% enteral nimodipine) and the need for rescue therapy (24% vs. 56%) (314). A new phase 3 double-blind, double-dummy, randomized NEWTON trial in aneurysmal SAH patients is currently underway in 374 participants (NTC02790632). However, no public interim trial data are available in clinicaltrails.gov. This study design might be helpful for the construction of new drug carriers and reduced vasospasm after aneurysmal SAH.
Dexmedetomidine (DEX), a highly selective α2 receptor agonist, showed protective effects in many neurological diseases, including inflammation inhibition and lower sympathetic activity (316). As a potent antioxidant and anti-inflammatory drug, recent studies have shown that DEX exerts a neuroprotective effect in traumatic brain injury (317, 318). One study demonstrated that post-SAH treatment with DEX attenuated disease-related damage through activation of the extracellular signal-regulated kinase (phospho-ERK) (319). DEX (25 µg/kg) administered for SAH obviously decreased neutrophil infiltration, microglial activation, and pro-inflammatory factor release and improved the neurological scores and tight-junction proteins (290). In the first 24 h after SAH, hyperactivated microglial and several brain blood immune cells (T cells, macrophages, and neutrophils) were significantly reduced by DEX, potentially providing neuroprotection for brain injury (290, 320). DEX relieves microglial pyroction in post-SAH EBI by activating the PI3K/Akt/GSK3β pathway and inhibits SAH-induced release of pro-inflammatory cytokines (321). In terms of neuroprotection, this study also showed that DEX alleviated SAH-induced neuroinflammation in the context of the NLRP3 inflammasome and inhibited the TLR4/NF-κB pathway. The NLRP3 inflammasome is the most common inflammasome and is related to IL-1β and IL-18 secretion, which exacerbates the inflammatory response, apoptosis, and BBB disruption (80). Previous studies also demonstrated that inhibiting the NLRP3 inflammasome activation provides effective neuroprotection against EBI after SAH, suggesting that the NLRP3 inflammasome is a therapeutic target for SAH (322). In addition, there is compelling evidence indicating that TLR4/NF-κB pathway suppression may be a potential target for SAH therapy (86). A recent study showed that activation of the NLRP3 inflammasome occurs in two major steps. The first step involves microbial infection-related pathogen-associated molecular pattern (PAMP) signaling, and the second step involves inflammasome oligomerization and recruitment of apoptosis-associated speck-like protein (ASC) (290, 323). These processes convert pro-IL-1β and pro-IL-18 to mature IL-1β and IL-18. Meanwhile, DEX treatment significantly suppresses the expression of inflammatory factors IL-1β, TNF-α, and IL-6 in SAH (290). Thus, the anti-inflammatory effects of DEX in SAH may be mediated through suppression of the TLR4/NF-κB pathway and NLRP3 inflammasome activation.
The inflammatory response stimulated by SAH is involved in the process of vasospasm (19). The inflammatory response indicator, C-reactive protein (CRP), is produced by hepatocytes and is related to increased IL-6, both of which are closely linked to vasospasm after SAH (291, 324). Increased IL-6 and CRP levels after SAH may be a consequence of vasospasm. Conversely, DEX administration decreased the serum IL-6 and CRP levels in the SAH, eventually attenuating cerebral vasospasm and improving neurological deficit outcomes (324). DEX administration could attenuate SAH-induced vasospasm and improve the SAH rat activity score (325). Recently, DEX was also used as an adjunct therapy for brain injury and is related to sympathetic nervous system activity in the acute phase (326). Intriguingly, low-dose DEX contributed to a significant reduction in serum lactate levels 24 h after administration, which is associated with favorable clinical outcomes during the early phase in SAH patients (326, 327). However, the standard dosage response of DEX is associated with adverse events. Several studies demonstrated that serum lactate levels are regulated by multiple factors, and the sympathetic activity-related catecholamine release has been identified as a main factor in the acute phase of SAH (328, 329). Therefore, the DEX auxiliary role in SAH microenvironment cytokines and etiological factor regulation may ameliorate early-phase SAH symptoms.
Some pharmacological treatments for aneurysmal SAH are limited by the occurrence of hypotension (324). Consequently, novel and effective approaches for the treatment of aneurysmal SAH patients are urgently needed. As an oral immunomodulatory agent applied for the treatment of multiple sclerosis and common nervous inflammatory disorders, fingolimod (FTY720) was approved by the United States FDA in 2010 as a first-line drug for multiple sclerosis (236). FTY720 is a sphingosine-1-phosphate (S1P) analog, the therapeutic activity of which could be due to regulation of movement across the BBB, critical cellular processes, and lymphocyte subset migration (236, 330). Several studies have shown that FTY720 treatment of cerebral ischemia and hemorrhage improves brain edema, infarct size, stroke-related neuroinflammation, neuronal death, and clinical outcome (331, 332). Recently, an investigation of FTY720 for the aneurysmal SAH treatment in a rat model (292) showed that this intervention restricted intravascular leukocyte adhesion to pial venules and improved neurological outcomes. Simultaneously, by activating the PI3K/Akt/eNOS pathway, FTY720 was able to promote nitric oxide (NO) production, and the anti-apoptotic and anti-inflammatory effects of FTY720 can relieve cerebral vasospasm (333). In addition, FTY720 exhibited widespread distribution and long-term behavioral changes with a half-life of approximately 10 days (236, 331). FTY720 has also been shown to improve both innate and adaptive immunity in animal models. However, the immune-related molecular effects are species-specific (334). The different FTY720 regulatory mechanisms between mouse and human immune systems should be taken with more consideration. Current data indicate that various effects of FTY720 can influence critical elements of aneurysmal SAH, such as BBB permeability, neuroinflammation, and microvascular dysregulation (331, 335). Furthermore, FTY720 is known to retain CD4+/CD8+ T cells and central memory T cells in lymph nodes, which also has a partial effect on peripheral effector memory T cells and protection against infections (236). Previous studies have demonstrated that FTY720 can restrict circulating leukocytes and immune depression and improve outcome without increasing the risk of lung bacterial infections in a mouse model of cerebral ischemic stroke (293). There are, however, some similarities between aneurysmal SAH and transient cerebral ischemia (292). Accordingly, immunomodulation has emerged as a potential therapeutic strategy to alleviate brain injury and improve clinical outcome after aneurysmal SAH. Despite this, results obtained from experimental models require further investigation to confirm the long-term effects in humans.
Recent research has demonstrated that p.Tyr562Cys somatic genomic mutation (g.149505130T>C [GRCh37/hg19]; c.1685A>G) in the platelet-derived growth factor receptor β gene (PDGFRB) coding region might be a novel mechanism in the pathophysiology of intracranial aneurysms and suggest a potentially effective role of sunitinib in targeted therapy (26). Sunitinib, a targeted receptor agent used for tyrosine kinase inhibitors (TKIs) with anti-angiogenic and antitumor activity, is approved by the FDA for gastrointestinal stromal tumor (GIST) therapy (294). Overexpression or mutation studies have shown that the PDGF and PDGFR families play an important role in tumor cell growth and survival regulation. Moreover, PDGF functions mainly through two different receptor tyrosine kinases (336), PDGFR-α and PDGFR-β, and activates major signal transduction cascades such as the PI3K/Akt and phospholipase C-gamma pathways (337, 338). Sunitinib is a known inhibitor of PDGFR-β kinases (339). A recent study has shown that the p.R561C mutation in PDGFRB is associated with infantile myofibromatosis (340). In this study, sunitinib treatment significantly reduced PDGFRB phosphorylation and tumor cell proliferation without changing the phosphorylation of MEK1/2, ERK1/2, and several other protein kinases. Western blot analysis has shown that expression of the Tyr562Cys variant is consistent with higher basal levels of pPDGFRB, pSRC, pAKT, and pERK1/2, and PDGFRB phosphorylation can activate downstream signaling (26). PDGFRB phosphorylation contributed to cerebrovascular dilation and lesions, which is observed to be expressed in vascular smooth muscle cells (341). More recently, the individual with intracranial aneurysm was observed to have PDGFRB alteration, where the vascular complications like SAH may be associated with PDGFRB hyperactivation (342, 343). Interestingly, sunitinib-mediated inhibition of PDGFRB phosphorylation in intracranial aneurysms patients with the (p.Tyr562Cys) variation is more significant compared those with wild-type PDGFRB, which provides the potential to prevent intracranial aneurysm formation and rupture (26). However, the p.Asp850Tyr variation exhibited marked resistance to sunitinib under the same conditions. Therefore, appropriate sunitinib intervention in the early diagnosis of candidate variation and/or intracranial aneurysms should be taken into consideration. A similar study in abdominal aortic aneurysm demonstrated that PDGFRB imatinib inhibition obviously alleviated the deteriorated aneurysm (344). The identification sunitinib to PDGFRB phosphorylation inhibition provides a novel avenue for target therapeutic strategies in intracranial aneurysm and potentially prevents aneurysm-related malignant complications like SAH.
Intracranial aneurysm SAH is a devastating disease with a high fate ratio and limited prevention and treatment approaches. Accumulating evidence suggests that immune inflammatory responses, like different immune cells and inflammatory factors, potentially contribute to aneurysm SAH pathological events, such as immunosuppression, infection, cerebral vasospasm, EBI, and DCI. Moreover, the crosstalk between immune cells and inflammation regulation mechanism in the occurrence and development of aneurysm SAH cannot be ignored, especially the microenvironment changes and brain injury. Interconnected inflammation and immune cells, placed in the vicinity of the SAH region, elicited the pathogenic generation of molecules (IL-1, IL-6, TNF-α, MMPs, NLRP3) and have a critical role in local immune function deterioration. Importantly, the immunopathogenesis of SAH is also characterized by different signaling pathways, wherein compelling evidence revealed their functions in inflammation and immune cell regulation. The PI3K/Akt, ERK, STAT, and SIRT1 cascades seem to have an essential role in microglial and astrocyte-related immune regulation and inflammation response in SAH. Meanwhile, the HIF-α, mTOR, and TLR4/NF-κB cascades have been linked to macrophage immune regulation and brain injury following SAH and are possible to be the promising therapeutic targets. Distinguishing the mechanisms of immune suppression and hyperactivation will facilitate our understanding of personalized aneurysmal SAH treatment. However, due to a controversial signaling interaction between inflammation and immune cells and complex SAH pathological conditions, the underlying regulation mechanisms are still largely unknown. The pros and cons function of immune inflammatory modulation signaling activation in aneurysm SAH and neuroprotection should be further elucidated.
Therapeutic strategies targeting immune inflammation regulation showed a promising future in some preclinical studies. Aneurysmal SAH carries a high mortality and requires emergency treatment. However, there is still no robust evidence that anti-immune/inflammatory treatment can be apply to aneurysm SAH patients. Nimodipine targeting the calcium channel and/or cAMP/PKA pathway and its optimized chemical modifications improved aneurysm SAH therapy effects and decreased the adverse reaction and exhibited the ability for immune cell regulation. Through intervention of pathways like PI3K/Akt pathway, DEX-modulated ERK and TLR4/NF-κB cascade, and FTY720-related immune inflammation regulation, aneurysm SAH was likely to be prevented from more severe development. These available treatment options broaden the current horizons for aneurysm SAH therapy. However, heterogeneity of the aneurysm SAH immune inflammation and controversial drug-regulating mechanisms limited the clinical effects. The efficacy and safety of these drug and derivatives require further exploration. On the other hand, the drugs that prevent patients from aneurysm rupture and SAH should be another favorable direction. Understanding the basis of aneurysm development and rupture is important for early diagnosis and intervention. Although the aneurysm development and rupture development are still poorly understood, the immune inflammatory and genetic factors have non-negligible roles for SAH. Of note, genomic variations associated with aneurysm formation and rupture provide potential target treatment strategies for aneurysm SAH patients, such as sunitinib targeting the PDGFRB variant. The early aneurysm diagnosis and management showed great potential to reduce the harmful events. Taken together, further study is necessary to clarifying the immune and inflammatory regulation mechanisms, thus developing innovative drugs and target/systematic therapy strategies to improve clinical outcomes.
JJ and JD conceived and drafted the manuscript. LD performed all literature searches. WX prepared and adjusted the figures. QZ and XP gave the concepts of the manuscript. QZ and XP approved the version to be submitted. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Brown RD Jr., Broderick JP. Unruptured intracranial aneurysms: epidemiology, natural history, management options, and familial screening. Lancet Neurol (2014) 13:393–404. doi: 10.1016/S1474-4422(14)70015-8
2. Müller TB, Sandvei MS, Kvistad KA, Rydland J, Håberg A, Vik A, et al. Unruptured intracranial aneurysms in the Norwegian nord-trøndelag health study (HUNT): risk of rupture calculated from data in a population-based cohort study. Neurosurgery (2013) 73:256–261. doi: 10.1227/01.neu.0000430295.23799.16.
3. Zeyu Z, Yuanjian F, Cameron L, Sheng C. The role of immune inflammation in aneurysmal subarachnoid hemorrhage. Exp Neurol (2021) 336:113535. doi: 10.1016/j.expneurol.2020.113535
4. Macdonald RL, Schweizer TA. Spontaneous subarachnoid haemorrhage. Lancet (London England) (2017) 389:655–66. doi: 10.1016/S0140-6736(16)30668-7
5. van Gijn J, Kerr RS, Rinkel GJ. Subarachnoid haemorrhage. Lancet (London England) (2007) 369:306–18. doi: 10.1016/S0140-6736(07)60153-6
6. Mashaly HA, Provencio JJ. Inflammation as a link between brain injury and heart damage: the model of subarachnoid hemorrhage. Cleveland Clinic J Med (2008) 75 Suppl 2:S26–30. doi: 10.3949/ccjm.75.Suppl_2.S26
7. Zhang T, Xu S, Wu P, Zhou K, Wu L, Xie Z, et al. Mitoquinone attenuates blood-brain barrier disruption through Nrf2/PHB2/OPA1 pathway after subarachnoid hemorrhage in rats. Exp Neurol (2019) 317:1–9. doi: 10.1016/j.expneurol.2019.02.009
8. Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Trans Stroke Res (2013) 4:432–46. doi: 10.1007/s12975-013-0257-2
9. Zhou Y, Jiang Y, Peng Y, Zhang M. The quantitative and functional changes of postoperative peripheral blood immune cell subsets relate to prognosis of patients with subarachnoid hemorrhage: A preliminary study. World Neurosurg (2017) 108:206–15. doi: 10.1016/j.wneu.2017.08.091
10. Xie Y, Guo H, Wang L, Xu L, Zhang X, Yu L, et al. Human albumin attenuates excessive innate immunity via inhibition of microglial Mincle/Syk signaling in subarachnoid hemorrhage. Brain Behav Immunity (2017) 60:346–60. doi: 10.1016/j.bbi.2016.11.004
11. Lucke-Wold BP, Logsdon AF, Manoranjan B, Turner RC, McConnell E, Vates GE, et al. Aneurysmal subarachnoid hemorrhage and neuroinflammation: A comprehensive review. Int J Mol Sci (2016) 17:497. doi: 10.3390/ijms17040497
12. Sokół B, Woźniak A, Jankowski R, Jurga S, Wąsik N, Shahid H, et al. HMGB1 level in cerebrospinal fluid as a marker of treatment outcome in patients with acute hydrocephalus following aneurysmal subarachnoid hemorrhage. J Stroke Cerebrovascular Dis Off J Natl Stroke Assoc (2015) 24:1897–904. doi: 10.1016/j.jstrokecerebrovasdis.2015.05.002
13. Coulibaly AP, Provencio JJ. Aneurysmal subarachnoid hemorrhage: an overview of inflammation-induced cellular changes. Neurother J Am Soc Exp NeuroTherapeutics (2020) 17:436–45. doi: 10.1007/s13311-019-00829-x
14. Sun Q, Wu W, Hu YC, Li H, Zhang D, Li S, et al. Early release of high-mobility group box 1 (HMGB1) from neurons in experimental subarachnoid hemorrhage in vivo and in vitro. J Neuroinflamm (2014) 11:106. doi: 10.1186/1742-2094-11-106
15. Schneider UC, Xu R, Vajkoczy P. Inflammatory events following subarachnoid hemorrhage (SAH). Curr Neuropharmacol (2018) 16:1385–95. doi: 10.2174/1570159X16666180412110919
16. Geraghty JR, Lung TJ, Hirsch Y, Katz EA, Cheng T, Saini NS, et al. Systemic immune-inflammation index predicts delayed cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery (2021) 89:1071–9. doi: 10.1093/neuros/nyab354
17. Kurz K, Teerlink T, Sarcletti M, Weiss G, Zangerle R, Fuchs D. Plasma concentrations of the cardiovascular risk factor asymmetric dimethylarginine (ADMA) are increased in patients with HIV-1 infection and correlate with immune activation markers. Pharmacol Res (2009) 60:508–14. doi: 10.1016/j.phrs.2009.07.009
18. Lindgren C, Hultin M, Koskinen LO, Lindvall P, Borota L, Naredi S. ADMA levels and arginine/ADMA ratios reflect severity of disease and extent of inflammation after subarachnoid hemorrhage. Neurocritical Care (2014) 21:91–101. doi: 10.1007/s12028-013-9945-8
19. Penn DL, Witte SR, Komotar RJ, Sander Connolly E Jr. Pathological mechanisms underlying aneurysmal subarachnoid haemorrhage and vasospasm. J Clin Neurosci Off J Neurosurg Soc Australasia (2015) 22:1–5. doi: 10/j.2014.05.025.
20. Jabbarli R, Dinger TF, Darkwah Oppong M, Pierscianek D, Dammann P, Wrede KH, et al. Risk factors for and clinical consequences of multiple intracranial aneurysms: A systematic review and meta-analysis. Stroke (2018) 49:848–55. doi: 10.1161/STROKEAHA.117.020342
21. Rinkel GJ, Djibuti M, Algra A, van Gijn J. Prevalence and risk of rupture of intracranial aneurysms: a systematic review. Stroke (1998) 29:251–6. doi: 10.1161/01.STR.29.1.251
22. Zhou S, Dion PA, Rouleau GA. Genetics of intracranial aneurysms. Stroke (2018) 49:780–7. doi: 10.1161/STROKEAHA.117.018152
23. Yasuno K, Bilguvar K, Bijlenga P, Low SK, Krischek B, Auburger G, et al. Genome-wide association study of intracranial aneurysm identifies three new risk loci. Nat Genet (2010) 42:420–5. doi: 10.1038/ng.563
24. Santiago-Sim T, Fang X, Hennessy ML, Nalbach SV, DePalma SR, Lee MS, et al. THSD1 (Thrombospondin type 1 domain containing protein 1) mutation in the pathogenesis of intracranial aneurysm and subarachnoid hemorrhage. Stroke (2016) 47:3005–13. doi: 10.1161/STROKEAHA.116.014161
25. Foreman PM, Starke RM, Hendrix P, Harrigan MR, Fisher WSR, Vyas NA, et al. Endothelin polymorphisms as a risk factor for cerebral aneurysm rebleeding following aneurysmal subarachnoid hemorrhage. Clin Neurol Neurosurg (2017) 157:65–9. doi: 10.1016/j.clineuro.2017.04.007
26. Karasozen Y, Osbun JW, Parada CA, Busald T, Tatman P, Gonzalez-Cuyar LF, et al. Somatic PDGFRB activating variants in fusiform cerebral aneurysms. Am J Hum Genet (2019) 104:968–76. doi: 10.1016/j.ajhg.2019.03.014
27. Naidech AM. Intracranial hemorrhage. Am J Respir Crit Care Med (2011) 184:998–1006. doi: 10.1164/rccm.201103-0475CI
28. Roethlisberger M, Achermann R, Bawarjan S, Stienen MN, Fung C, D'Alonzo D, et al. Predictors of occurrence and anatomic distribution of multiple aneurysms in patients with aneurysmal subarachnoid hemorrhage. World Neurosurg (2018) 111:e199–205. doi: 10.1016/j.wneu.2017.12.046
29. Corovic A, Kelly S, Markus HS. Cerebral amyloid angiopathy associated with inflammation: A systematic review of clinical and imaging features and outcome. Int J Stroke Off J Int Stroke Society (2018) 13:257–67. doi: 10.1177/1747493017741569
30. Ziai WC, Thompson CB, Mayo S, McBee N, Freeman WD, Dlugash R, et al. Intracranial hypertension and cerebral perfusion pressure insults in adult hypertensive intraventricular hemorrhage: Occurrence and associations with outcome. Crit Care Med (2019) 47:1125–34. doi: 10.1097/CCM.0000000000003848
31. Weinstock MJ, Uhlmann EJ, Zwicker JI. Intracranial hemorrhage in cancer patients treated with anticoagulation. Thromb Res (2016) 140 Suppl 1:S60–5. doi: 10.1016/S0049-3848(16)30100-1
32. Kreisl TN, Toothaker T, Karimi S, DeAngelis LM. Ischemic stroke in patients with primary brain tumors. Neurology (2008) 70:2314–20. doi: 10.1212/01.wnl.0000314648.82924.6f
33. van Baarsen K, Roth J, Serova N, Packer RJ, Shofty B, Thomale UW, et al. Optic pathway-hypothalamic glioma hemorrhage: a series of 9 patients and review of the literature. J Neurosurg (2018) 129:1407–15. doi: 10.3171/2017.8.JNS163085
34. Intusoma U, Nakorn CN, Chotsampancharoen T. Intracranial hemorrhage in childhood acute leukemia: Incidence, characteristics, and contributing factors. Pediatr Neurol (2019) 99:23–30. doi: 10.1016/j.pediatrneurol.2019.06.005
35. Lazzaro MA, Ouyang B, Chen M. The role of circle of Willis anomalies in cerebral aneurysm rupture. J neurointerv Surg (2012) 4:22–6. doi: 10.1136/jnis.2010.004358
36. Lindner SH, Bor AS, Rinkel GJ. Differences in risk factors according to the site of intracranial aneurysms. J Neurol Neurosurg Psychiatry (2010) 81:116–8. doi: 10.1136/jnnp.2008.163063
37. Tawk RG, Hasan TF, D'Souza CE, Peel JB, Freeman WD. Diagnosis and treatment of unruptured intracranial aneurysms and aneurysmal subarachnoid hemorrhage. Mayo Clinic Proc (2021) 96:1970–2000. doi: 10.1016/j.mayocp.2021.01.005
38. Korja M, Kaprio J. Controversies in epidemiology of intracranial aneurysms and SAH. Nat Rev Neurol (2016) 12:50–5. doi: 10.1038/nrneurol.2015.228
39. Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2003) 23:629–52. doi: 10.1097/01.WCB.0000073905.87928.6D
40. Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol (2006) 5:53–63. doi: 10.1016/S1474-4422(05)70283-0
41. Tang J, Liu J, Zhou C, Ostanin D, Grisham MB, Neil Granger D, et al. Role of NADPH oxidase in the brain injury of intracerebral hemorrhage. J Neurochem (2005) 94:1342–50. doi: 10.1111/j.1471-4159.2005.03292.x
42. Aronowski J, Zhao X. Molecular pathophysiology of cerebral hemorrhage: secondary brain injury. Stroke (2011) 42:1781–6. doi: 10.1161/STROKEAHA.110.596718
43. Wagner KR, Dwyer BE. Hematoma removal, heme, and heme oxygenase following hemorrhagic stroke. Ann New York Acad Sci (2004) 1012:237–51. doi: 10.1196/annals.1306.020
44. Wang J, Doré S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain J Neurol (2007) 130:1643–52. doi: 10.1093/brain/awm095
45. Wang J, Doré S. Heme oxygenase 2 deficiency increases brain swelling and inflammation after intracerebral hemorrhage. Neuroscience (2008) 155:1133–41. doi: 10.1016/j.neuroscience.2008.07.004
46. Frase S, Steimer M, Selzner L, Kaiser S, Foit NA, Niesen WD, et al. Temporal expression pattern of hemoxygenase-1 expression and its association with vasospasm and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neurocritical Care (2022) 36:279–91. doi: 10.1007/s12028-021-01299-w
47. Suzuki H, Muramatsu M, Kojima T, Taki W. Intracranial heme metabolism and cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Stroke (2003) 34:2796–800. doi: 10.1161/01.STR.0000103743.62248.12
48. Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke (2003) 34:2964–9. doi: 10.1161/01.STR.0000103140.52838.45
49. Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signaling (2008) 10:179–206. doi: 10.1089/ars.2007.1672
50. Yang S, Chen X, Li S, Sun B, Hang C. Melatonin treatment regulates SIRT3 expression in early brain injury (EBI) due to reactive oxygen species (ROS) in a mouse model of subarachnoid hemorrhage (SAH). Med Sci Monit Int Med J Exp Clin Res (2018) 24:3804–14. doi: 10.12659/MSM.907734
51. Li J, Chen J, Mo H, Chen J, Qian C, Yan F, et al. Minocycline protects against NLRP3 inflammasome-induced inflammation and P53-associated apoptosis in early brain injury after subarachnoid hemorrhage. Mol Neurobiol (2016) 53:2668–78. doi: 10.1007/s12035-015-9318-8
52. Khey KMW, Huard A, Mahmoud SH. Inflammatory pathways following subarachnoid hemorrhage. Cell Mol Neurobiol (2020) 40:675–93. doi: 10.1007/s10571-019-00767-4
53. Imai T, Matsubara H, Hara H. Potential therapeutic effects of Nrf2 activators on intracranial hemorrhage. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2021) 41:1483–500. doi: 10.1177/0271678X20984565
54. Shao Z, Tu S, Shao A. Pathophysiological mechanisms and potential therapeutic targets in intracerebral hemorrhage. Front Pharmacol (2019) 10:1079. doi: 10.3389/fphar.2019.01079
55. Ziai WC. Hematology and inflammatory signaling of intracerebral hemorrhage. Stroke (2013) 44:S74–8. doi: 10.1161/STROKEAHA.111.000662
56. Wang J, Doré S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2007) 27:894–908. doi: 10.1038/sj.jcbfm.9600403
57. Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol (2007) 61:352–62. doi: 10.1002/ana.21097
58. Kourtzelis I, Hajishengallis G, Chavakis T. Phagocytosis of apoptotic cells in resolution of inflammation. Front Immunol (2020) 11:553. doi: 10.3389/fimmu.2020.00553
59. Tao C, Hu X, Li H, You C. White matter injury after intracerebral hemorrhage: Pathophysiology and therapeutic strategies. Front Hum Neurosci (2017) 11:422. doi: 10.3389/fnhum.2017.00422
60. Zhao X, Zhang Y, Strong R, Zhang J, Grotta JC, Aronowski J. Distinct patterns of intracerebral hemorrhage-induced alterations in NF-kappaB subunit, iNOS, and COX-2 expression. J Neurochem (2007) 101:652–63. doi: 10.1111/j.1471-4159.2006.04414.x
61. Wen J, Yang CY, Lu J, Wang XY. Ptprj-as1 mediates inflammatory injury after intracerebral hemorrhage by activating NF-κB pathway. Eur Rev Med Pharmacol Sci (2018) 22:2817–23. doi: 10.26355/eurrev_201805_14981
62. Tschoe C, Bushnell CD, Duncan PW, Alexander-Miller MA, Wolfe SQ. Neuroinflammation after intracerebral hemorrhage and potential therapeutic targets. J Stroke (2020) 22:29–46. doi: 10.5853/jos.2019.02236
63. Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell (2010) 140:918–34. doi: 10.1016/j.cell.2010.02.016
64. Zhang ZH, Han YL, Wang CX, Zhou CH, Wu LY, Zhang HS, et al. The effect of subarachnoid erythrocyte lysate on brain injury: a preliminary study. Biosci Rep (2016) 36(4):e00359. doi: 10.1042/BSR20160100
65. Gris T, Laplante P, Thebault P, Cayrol R, Najjar A, Joannette-Pilon B, et al. Innate immunity activation in the early brain injury period following subarachnoid hemorrhage. J Neuroinflamm (2019) 16:253. doi: 10.1186/s12974-019-1629-7
66. McMahon CJ, Hopkins S, Vail A, King AT, Smith D, Illingworth KJ, et al. Inflammation as a predictor for delayed cerebral ischemia after aneurysmal subarachnoid haemorrhage. J neurointerv Surg (2013) 5:512–7. doi: 10.1136/neurintsurg-2012-010386
67. Zhang J, Xu X, Zhou D, Li H, You W, Wang Z, et al. Possible role of raf-1 kinase in the development of cerebral vasospasm and early brain injury after experimental subarachnoid hemorrhage in rats. Mol Neurobiol (2015) 52:1527–39. doi: 10.1007/s12035-014-8939-7
68. Xu H, Li J, Wang Z, Feng M, Shen Y, Cao S, et al. Methylene blue attenuates neuroinflammation after subarachnoid hemorrhage in rats through the Akt/GSK-3β/MEF2D signaling pathway. Brain Behav Immunity (2017) 65:125–39. doi: 10.1016/j.bbi.2017.04.020
69. Luo Y, Fang Y, Kang R, Lenahan C, Gamdzyk M, Zhang Z, et al. Inhibition of EZH2 (Enhancer of zeste homolog 2) attenuates neuroinflammation via H3k27me3/SOCS3/TRAF6/NF-κB (Trimethylation of histone 3 lysine 27/Suppressor of cytokine signaling 3/Tumor necrosis factor receptor family 6/Nuclear factor-κB) in a rat model of subarachnoid hemorrhage. Stroke (2020) 51:3320–31. doi: 10.1161/STROKEAHA.120.029951
70. Chaudhry SR, Stoffel-Wagner B, Kinfe TM, Güresir E, Vatter H, Dietrich D, et al. Elevated systemic IL-6 levels in patients with aneurysmal subarachnoid hemorrhage is an unspecific marker for post-SAH complications. Int J Mol Sci (2017) 18(12):2580. doi: 10.3390/ijms18122580
71. Nakura T, Osuka K, Inukai T, Takagi T, Takayasu M. Soluble gp130 regulatess interleukin-6 in cerebrospinal fluid after subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry (2011) 82:952–4. doi: 10.1136/jnnp.2009.197244
72. Wolf J, Waetzig GH, Chalaris A, Reinheimer TM, Wege H, Rose-John S, et al. Different soluble forms of the interleukin-6 family signal transducer gp130 fine-tune the blockade of interleukin-6 trans-signaling. J Biol Chem (2016) 291:16186–96. doi: 10.1074/jbc.M116.718551
73. Schumacher N, Yan K, Gandraß M, Müller M, Krisp C, Häsler R, et al. Cell-autonomous hepatocyte-specific GP130 signaling is sufficient to trigger a robust innate immune response in mice. J Hepatol (2021) 74:407–18. doi: 10.1016/j.jhep.2020.09.021
74. Burton MD, Johnson RW. Interleukin-6 trans-signaling in the senescent mouse brain is involved in infection-related deficits in contextual fear conditioning. Brain Behav Immunity (2012) 26:732–8. doi: 10.1016/j.bbi.2011.10.008
75. Fielding CA, McLoughlin RM, McLeod L, Colmont CS, Najdovska M, Grail D, et al. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J Immunol (Baltimore Md 1950) (2008) 181:2189–95. doi: 10.4049/jimmunol.181.3.2189
76. Bonomi A, Veglia F, Baldassarre D, Strawbridge RJ, Golabkesh Z, Sennblad B, et al. Analysis of the genetic variants associated with circulating levels of sgp130. Results IMPROVE Study Genes Immunity (2020) 21:100–8. doi: 10.1038/s41435-019-0090-z
77. Penn DL, Witte SR, Komotar RJ, Sander Connolly E Jr. The role of vascular remodeling and inflammation in the pathogenesis of intracranial aneurysms. J Clin Neurosci Off J Neurosurg Soc Australasia (2014) 21:28–32. doi: 10.1016/j.jocn.2013.07.004
78. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol (2010) 11:136–40. doi: 10.1038/ni.1831
79. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev (2015) 265:35–52. doi: 10.1111/imr.12286
80. Gaidt MM, Hornung V. The NLRP3 inflammasome renders cell death pro-inflammatory. J Mol Biol (2018) 430:133–41. doi: 10.1016/j.jmb.2017.11.013
81. Li JR, Xu HZ, Nie S, Peng YC, Fan LF, Wang ZJ, et al. Fluoxetine-enhanced autophagy ameliorates early brain injury via inhibition of NLRP3 inflammasome activation following subrachnoid hemorrhage in rats. J Neuroinflamm (2017) 14:186. doi: 10.1186/s12974-017-0959-6
82. Xu P, Tao C, Zhu Y, Wang G, Kong L, Li W, et al. TAK1 mediates neuronal pyroptosis in early brain injury after subarachnoid hemorrhage. J Neuroinflamm (2021) 18:188. doi: 10.1186/s12974-021-02226-8
83. Xu X, Zhang L, Ye X, Hao Q, Zhang T, Cui G, et al. Nrf2/ARE pathway inhibits ROS-induced NLRP3 inflammasome activation in BV2 cells after cerebral ischemia reperfusion. Inflammation Res Off J Eur Histamine Res Soc [et al] (2018) 67:57–65. doi: 10.1007/s00011-017-1095-6
84. Chen J, Chen G, Li J, Qian C, Mo H, Gu C, et al. Melatonin attenuates inflammatory response-induced brain edema in early brain injury following a subarachnoid hemorrhage: a possible role for the regulation of pro-inflammatory cytokines. J Pineal Res (2014) 57:340–7. doi: 10.1111/jpi.12173
85. Zhan CP, Zhuge CJ, Yan XJ, Dai WM, Yu GF. Measuring serum melatonin concentrations to predict clinical outcome after aneurysmal subarachnoid hemorrhage. Clinica Chimica Acta; Int J Clin Chem (2021) 513:1–5. doi: 10.1016/j.cca.2020.12.006
86. Liu H, Yang M, Pan L, Liu P, Ma L. Hyperbaric oxygen intervention modulates early brain injury after experimental subarachnoid hemorrhage in rats: Possible involvement of TLR4/NF-x03BA; b-mediated signaling pathway. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol (2016) 38:2323–36. doi: 10.1159/000445586
87. Xue H, Sun K, Xie W, Hu G, Kong H, Wang Q, et al. Etanercept attenuates short-term cigarette-smoke-exposure-induced pulmonary arterial remodelling in rats by suppressing the activation of TNF-a/NF-kB signal and the activities of MMP-2 and MMP-9. Pulmon Pharmacol Ther (2012) 25:208–15. doi: 10.1016/j.pupt.2012.02.006
88. Vikman P, Ansar S, Edvinsson L. Transcriptional regulation of inflammatory and extracellular matrix-regulating genes in cerebral arteries following experimental subarachnoid hemorrhage in rats. Lab Invest J Neurosurg (2007) 107:1015–22. doi: 10.3171/JNS-07/11/1015
89. Roa JA, Sarkar D, Zanaty M, Ishii D, Lu Y, Karandikar NJ, et al. Preliminary results in the analysis of the immune response after aneurysmal subarachnoid hemorrhage. Sci Rep (2020) 10:11809. doi: 10.1038/s41598-020-68861-y
90. Vafadari B, Salamian A, Kaczmarek L. MMP-9 in translation: from molecule to brain physiology, pathology, and therapy. J Neurochem (2016) 139 Suppl 2:91–114. doi: 10.1111/jnc.13415
91. Nissinen L, Kähäri VM. Matrix metalloproteinases in inflammation. Biochim Biophys Acta (2014) 1840:2571–80. doi: 10.1016/j.bbagen.2014.03.007
92. Mohme M, Sauvigny T, Mader MM, Schweingruber N, Maire CL, Rünger A, et al. Immune characterization in aneurysmal subarachnoid hemorrhage reveals distinct monocytic activation and chemokine patterns. Trans Stroke Res (2020) 11:1348–61. doi: 10.1007/s12975-019-00764-1
93. Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2007) 27:697–709. doi: 10.1038/sj.jcbfm.9600375
94. Tzeng HE, Tsai CH, Chang ZL, Su CM, Wang SW, Hwang WL, et al. Interleukin-6 induces vascular endothelial growth factor expression and promotes angiogenesis through apoptosis signal-regulating kinase 1 in human osteosarcoma. Biochem Pharmacol (2013) 85:531–40. doi: 10.1016/j.bcp.2012.11.021
95. Sozen T, Tsuchiyama R, Hasegawa Y, Suzuki H, Jadhav V, Nishizawa S, et al. Role of interleukin-1beta in early brain injury after subarachnoid hemorrhage in mice. Stroke (2009) 40:2519–25. doi: 10.1161/STROKEAHA.109.549592
96. Wan H, AlHarbi BM, Macdonald RL. Mechanisms, treatment and prevention of cellular injury and death from delayed events after aneurysmal subarachnoid hemorrhage. Expert Opin Pharmacother (2014) 15:231–43. doi: 10.1517/14656566.2014
98. Provencio JJ. Inflammation in subarachnoid hemorrhage and delayed deterioration associated with vasospasm: a review. Acta Neurochirurgica Supplement (2013) 115:233–8. doi: 10.1007/978-3-7091-1192-5_42.
99. Kox M, Pompe J, Hoedemaekers A, Pickkers P. Does subarachnoid haemorrhage affect the innate immune response? Intensive Care Med (2007) 33:1303. doi: 10.1007/s00134-007-0664-5.
100. Kim JY, Kawabori M, Yenari MA. Innate inflammatory responses in stroke: mechanisms and potential therapeutic targets. Curr Medicinal Chem (2014) 21:2076–97. doi: 10.1172/JCI135530
101. Zhong W, Zhang Z, Zhao P, Shen J, Li X, Wang D, et al. The impact of initial systemic inflammatory response after aneurysmal subarachnoid hemorrhage. Turkish Neurosurg (2017) 27:346–52. doi: 10.5137/1019-5149.
102. Ramagopalan SV, Pakpoor J, Seminog O, Goldacre R, Graham L, Goldacre MJ. Risk of subarachnoid haemorrhage in people admitted to hospital with selected immune-mediated diseases: record-linkage studies. BMC Neurol (2013) 13:176. doi: 10.1186/1471-2377-13-176
103. Brait VH, Arumugam TV, Drummond GR, Sobey CG. Importance of T lymphocytes in brain injury, immunodeficiency, and recovery after cerebral ischemia. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2012) 32:598–611. doi: 10.1038/jcbfm.2012.6
104. Sarrafzadeh A, Schlenk F, Meisel A, Dreier J, Vajkoczy P, Meisel C. Immunodepression after aneurysmal subarachnoid hemorrhage. Stroke (2011) 42:53–8. doi: 10.1161/STROKEAHA.110.594705
105. Chaudhry SR, Kahlert UD, Kinfe TM, Endl E, Dolf A, Niemelä M, et al. Differential polarization and activation dynamics of systemic T helper cell subsets after aneurysmal subarachnoid hemorrhage (SAH) and during post-SAH complications. Sci Rep (2021) 11:14226. doi: 10.1038/s41598-021-92873-x
106. Dong G, Li C, Hu Q, Wang Y, Sun J, Gao F, et al. Low-dose IL-2 treatment affords protection against subarachnoid hemorrhage injury by expanding peripheral regulatory T cells. ACS Chem Neurosci (2021) 12:430–40. doi: 10.1021/acschemneuro.0c00611
107. Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol (2010) 10:490–500. doi: 10.1038/nri2785
108. Mirlekar B, Patil S, Bopanna R, Chattopadhyay S. MAR binding protein SMAR1 favors IL-10 mediated regulatory T cell function in acute colitis. Biochem Biophys Res Commun (2015) 464:647–53. doi: 10.1016/j.bbrc.2015.07.028
109. Stern MJ, Gorman PA, Kaslow L. The group counseling v exercise therapy study. a controlled intervention with subjects following myocardial infarction. Arch Internal Med (1983) 143:1719–25. doi: 10.1001/archinte.1983.00350090097016
110. Saand AR, Yu F, Chen J, Chou SH. Systemic inflammation in hemorrhagic strokes - a novel neurological sign and therapeutic target? J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2019) 39:959–88. doi: 10.1177/0271678X19841443
111. Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y, et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Ann Neurol (2013) 74:458–71. doi: 10.1002/ana.23815
112. Chu HX, Kim HA, Lee S, Moore JP, Chan CT, Vinh A, et al. Immune cell infiltration in malignant middle cerebral artery infarction: comparison with transient cerebral ischemia. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2014) 34:450–9. doi: 10.1038/jcbfm.2013.217
113. Liesz A, Kleinschnitz C. Regulatory T cells in post-stroke immune homeostasis. Trans Stroke Res (2016) 7:313–21. doi: 10.1007/s12975-016-0465-7
114. de Vries HE, Blom-Roosemalen MC, van Oosten M, de Boer AG, van Berkel TJ, Breimer DD, et al. The influence of cytokines on the integrity of the blood-brain barrier in vitro. J Neuroimmunol (1996) 64:37–43. doi: 10.1016/0165-5728(95)00148-4
115. Nowicki KW, Hosaka K, Walch FJ, Scott EW, Hoh BL. M1 macrophages are required for murine cerebral aneurysm formation. J neurointerv Surg (2018) 10:93–7. doi: 10.1136/neurintsurg-2016-012911
116. Solár P, Klusáková I, Jančálek R, Dubový P, Joukal M. Subarachnoid hemorrhage induces dynamic immune cell reactions in the choroid plexus. Front Cell Neurosci (2020) 14:18. doi: 10.3389/fncel.2020.00018
117. Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol (2015) 33:643–75. doi: 10.1146/annurev-immunol-032414-112220
118. Gauthier T, Chen W. Modulation of macrophage immunometabolism: A new approach to fight infections. Front Immunol (2022) 13:780839. doi: 10.3389/fimmu.2022.780839
119. Kwan K, Arapi O, Wagner KE, Schneider J, Sy HL, Ward MF, et al. Cerebrospinal fluid macrophage migration inhibitory factor: a potential predictor of cerebral vasospasm and clinical outcome after aneurysmal subarachnoid hemorrhage. J Neurosurg (2019) 4:1–6. doi: 10.3171/2019.6.JNS19613
120. Minogue AM. Role of infiltrating monocytes/macrophages in acute and chronic neuroinflammation: Effects on cognition, learning and affective behaviour. Prog Neuropsychopharmacol Biol Psychiatry (2017) 79:15–8. doi: 10.1016/j.pnpbp.2017.02.008
121. Balboa L, Romero MM, Basile JI, Sabio y García CA, Schierloh P, Yokobori N, et al. Paradoxical role of CD16+CCR2+CCR5+ monocytes in tuberculosis: efficient APC in pleural effusion but also mark disease severity in blood. J Leukocyte Biol (2011) 90:69–75. doi: 10.1189/jlb.1010577
122. Grage-Griebenow E, Zawatzky R, Kahlert H, Brade L, Flad H, Ernst M. Identification of a novel dendritic cell-like subset of CD64(+) / CD16(+) blood monocytes. Eur J Immunol (2001) 31:48–56. doi: 10.1002/1521-4141(200101)31:1<48::AID-IMMU48>3.0.CO;2-5
123. Moraes L, Grille S, Morelli P, Mila R, Trias N, Brugnini A, et al. Immune cells subpopulations in cerebrospinal fluid and peripheral blood of patients with aneurysmal subarachnoid hemorrhage. SpringerPlus (2015) 4:195. doi: 10.1186/s40064-015-0970-2
124. Rohr J, Guo S, Huo J, Bouska A, Lachel C, Li Y, et al. Recurrent activating mutations of CD28 in peripheral T-cell lymphomas. Leukemia (2016) 30:1062–70. doi: 10.1038/leu.2015.357
125. Lim HS, Cordoba SP, Dushek O, Goyette J, Taylor A, Rudd CE, et al. Costimulation of IL-2 production through CD28 is dependent on the size of its ligand. J Immunol (Baltimore Md 1950) (2015) 195:5432–9. doi: 10.4049/jimmunol.1500707
126. Mathiesen T, Andersson B, Loftenius A, von Holst H. Increased interleukin-6 levels in cerebrospinal fluid following subarachnoid hemorrhage. J Neurosurg (1993) 78:562–7. doi: 10.3171/jns.1993.78.4.0562
127. Zhu Q, Enkhjargal B, Huang L, Zhang T, Sun C, Xie Z, et al. Aggf1 attenuates neuroinflammation and BBB disruption via PI3K/Akt/NF-κB pathway after subarachnoid hemorrhage in rats. J Neuroinflamm (2018) 15:178. doi: 10.1186/s12974-018-1211-8
128. Sansing LH, Harris TH, Kasner SE, Hunter CA, Kariko K. Neutrophil depletion diminishes monocyte infiltration and improves functional outcome after experimental intracerebral hemorrhage. Acta Neurochirurgica Supplement (2011) 111:173–8. doi: 10.1007/978-3-7091-0693-8_29
129. Wang J, Wang Y, Zuo Y, Duan J, Pan A, Li JM, et al. MFGE8 mitigates brain injury in a rat model of SAH by maintaining vascular endothelial integrity via TIGβ5/PI3K/CXCL12 signaling. Exp Brain Res (2021) 239:2193–205. doi: 10.1007/s00221-021-06111-x
130. Peng J, Pang J, Huang L, Enkhjargal B, Zhang T, Mo J, et al. LRP1 activation attenuates white matter injury by modulating microglial polarization through Shc1/PI3K/Akt pathway after subarachnoid hemorrhage in rats. Redox Biol (2019) 21:101121. doi: 10.1016/j.redox.2019.101121
131. Yu Z, Sun D, Feng J, Tan W, Fang X, Zhao M, et al. MSX3 switches microglia polarization and protects from inflammation-induced demyelination. J Neurosci Off J Soc Neurosci (2015) 35:6350–65. doi: 10.1523/JNEUROSCI.2468-14.2015
132. Eyo UB, Wu LJ. Microglia: Lifelong patrolling immune cells of the brain. Prog Neurobiol (2019) 179:101614. doi: 10.1016/j.pneurobio.2019.04.003
133. Wang G, Shi Y, Jiang X, Leak RK, Hu X, Wu Y, et al. HDAC inhibition prevents white matter injury by modulating microglia/macrophage polarization through the GSK3β/PTEN/Akt axis. Proc Natl Acad Sci U States A (2015) 112:2853–8. doi: 10.1073/pnas.1501441112
134. Xiong XY, Liu L, Yang QW. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog Neurobiol (2016) 142:23–44. doi: 10.1016/j.pneurobio.2016.05.001
135. Tian Y, Liu B, Li Y, Zhang Y, Shao J, Wu P, et al. Activation of RARα receptor attenuates neuroinflammation after SAH via promoting M1-to-M2 phenotypic polarization of microglia and regulating Mafb/Msr1/PI3K-Akt/NF-κB pathway. Front Immunol (2022) 13:839796. doi: 10.3389/fimmu.2022.839796
136. Sun J, Yang X, Zhang Y, Zhang W, Lu J, Hu Q, et al. Salvinorin a attenuates early brain injury through PI3K/Akt pathway after subarachnoid hemorrhage in rat. Brain Res (2019) 1719:64–70. doi: 10.1016/j.brainres.2019.05.026
137. Burmi RS, Maginn EN, Gabra H, Stronach EA, Wasan HS. Combined inhibition of the PI3K/mTOR/MEK pathway induces Bim/Mcl-1-regulated apoptosis in pancreatic cancer cells. Cancer Biol Ther (2019) 20:21–30. doi: 10.1080/15384047.2018.1504718
138. Feng D, Wang W, Dong Y, Wu L, Huang J, Ma Y, et al. Ceftriaxone alleviates early brain injury after subarachnoid hemorrhage by increasing excitatory amino acid transporter 2 expression via the PI3K/Akt/NF-κB signaling pathway. Neuroscience (2014) 268:21–32. doi: 10.1016/j.neuroscience.2014.02.053
139. Chen X, Nie X, Mao J, Zhang Y, Yin K, Sun P, et al. Perfluorooctane sulfonate mediates secretion of IL-1β through PI3K/AKT NF-кB pathway in astrocytes. Neurotoxicol Teratology (2018) 67:65–75. doi: 10.1016/j.ntt.2018.03.004
140. Wu X, Kihara T, Akaike A, Niidome T, Sugimoto H. PI3K/Akt/mTOR signaling regulates glutamate transporter 1 in astrocytes. Biochem Biophys Res Commun (2010) 393:514–8. doi: 10.1016/j.bbrc.2010.02.038
141. Tong X, Zhang J, Shen M, Zhang J. Silencing of tenascin-c inhibited inflammation and apoptosis Via PI3K/Akt/NF-κB signaling pathway in subarachnoid hemorrhage cell model. J Stroke Cerebrovascular Dis Off J Natl Stroke Assoc (2020) 29:104485. doi: 10.1016/j.jstrokecerebrovasdis.2019.104485
142. Liu L, Zhang P, Zhang Z, Liang Y, Chen H, He Z, et al. 5-lipoxygenase inhibition reduces inflammation and neuronal apoptosis via AKT signaling after subarachnoid hemorrhage in rats. Aging (2021) 13:11752–61. doi: 10.18632/aging.202869
143. Maddahi A, Ansar S, Chen Q, Edvinsson L. Blockade of the MEK/ERK pathway with a raf inhibitor prevents activation of pro-inflammatory mediators in cerebral arteries and reduction in cerebral blood flow after subarachnoid hemorrhage in a rat model. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2011) 31:144–54. doi: 10.1038/jcbfm.2010.62
144. Huang C, Lu X, Wang JL, Tong LJ, Ling Y, Jiang B, et al. Compound c induces the ramification of murine microglia in an AMPK-independent and small rhogtpase-dependent manner. Neuroscience (2016) 331:24–39. doi: 10.1016/j.neuroscience.2016.06.018
145. Huang XP, Peng JH, Pang JW, Tian XC, Li XS, Wu Y, et al. Peli1 contributions in microglial activation, neuroinflammatory responses and neurological deficits following experimental subarachnoid hemorrhage. Front Mol Neurosci (2017) 10:398. doi: 10.3389/fnmol.2017.00398
146. Liu L, Zhang P, Zhang Z, Hu Q, He J, Liu H, et al. LXA4 ameliorates cerebrovascular endothelial dysfunction by reducing acute inflammation after subarachnoid hemorrhage in rats. Neuroscience (2019) 408:105–14. doi: 10.1016/j.neuroscience.2019.03.038
147. Stubelius A, Andréasson E, Karlsson A, Ohlsson C, Tivesten A, Islander U, et al. Role of 2-methoxyestradiol as inhibitor of arthritis and osteoporosis in a model of postmenopausal rheumatoid arthritis. Clin Immunol (Orlando Fla) (2011) 140:37–46. doi: 10.1016/j.clim.2011.03.006
148. Hu Q, Du Q, Yu W, Dong X. 2-methoxyestradiol alleviates neuroinflammation and brain edema in early brain injury after subarachnoid hemorrhage in rats. Front Cell Neurosci (2022) 16:869546. doi: 10.3389/fncel.2022.869546
149. An JY, Pang HG, Huang TQ, Song JN, Li DD, Zhao YL, et al. AG490 ameliorates early brain injury via inhibition of JAK2/STAT3-mediated regulation of HMGB1 in subarachnoid hemorrhage. Exp Ther Med (2018) 15:1330–8. doi: 10.3892/etm.2017.5539
150. Qiu J, Xu J, Zheng Y, Wei Y, Zhu X, Lo EH, et al. High-mobility group box 1 promotes metalloproteinase-9 upregulation through toll-like receptor 4 after cerebral ischemia. Stroke (2010) 41:2077–82. doi: 10.1161/STROKEAHA.110.590463
151. Agnello D, Wang H, Yang H, Tracey KJ, Ghezzi P. HMGB-1, a DNA-binding protein with cytokine activity, induces brain TNF and IL-6 production, and mediates anorexia and taste aversion. Cytokine (2002) 18:231–6. doi: 10.1006/cyto.2002.0890
152. Pang J, Peng J, Matei N, Yang P, Kuai L, Wu Y, et al. Apolipoprotein e exerts a whole-brain protective property by promoting M1? microglia quiescence after experimental subarachnoid hemorrhage in mice. Trans Stroke Res (2018) 9:654–68. doi: 10.1007/s12975-018-0665-4
153. Neal M, Luo J, Harischandra DS, Gordon R, Sarkar S, Jin H, et al. Prokineticin-2 promotes chemotaxis and alternative A2 reactivity of astrocytes. Glia (2018) 66:2137–57. doi: 10.1002/glia.23467
154. Ma M, Li H, Wu J, Zhang Y, Shen H, Li X, et al. Roles of prokineticin 2 in subarachnoid hemorrhage-induced early brain injury via regulation of phenotype polarization in astrocytes. Mol Neurobiol (2020) 57:3744–58. doi: 10.1007/s12035-020-01990-7
155. Wei S, Luo C, Yu S, Gao J, Liu C, Wei Z, et al. Erythropoietin ameliorates early brain injury after subarachnoid haemorrhage by modulating microglia polarization via the EPOR/JAK2-STAT3 pathway. Exp Cell Res (2017) 361:342–52. doi: 10.1016/j.yexcr.2017.11.002
156. Li R, Liu W, Yin J, Chen Y, Guo S, Fan H, et al. TSG-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway. J Neuroinflamm (2018) 15:231. doi: 10.1186/s12974-018-1279-1
157. Li Z, Han X. Resveratrol alleviates early brain injury following subarachnoid hemorrhage: possible involvement of the AMPK/SIRT1/autophagy signaling pathway. Biol Chem (2018) 399:1339–50. doi: 10.1515/hsz-2018-0269
158. Zhao L, Liu H, Yue L, Zhang J, Li X, Wang B, et al. Melatonin attenuates early brain injury via the melatonin Receptor/Sirt1/NF-κB signaling pathway following subarachnoid hemorrhage in mice. Mol Neurobiol (2017) 54:1612–21. doi: 10.1007/s12035-016-9776-7
159. Li Q, Peng Y, Fan L, Xu H, He P, Cao S, et al. Phosphodiesterase-4 inhibition confers a neuroprotective efficacy against early brain injury following experimental subarachnoid hemorrhage in rats by attenuating neuronal apoptosis through the SIRT1/Akt pathway. Biomed Pharmacother = Biomed Pharmacother (2018) 99:947–55. doi: 10.1016/j.biopha.2018.01.093
160. Peng Y, Jin J, Fan L, Xu H, He P, Li J, et al. Rolipram attenuates early brain injury following experimental subarachnoid hemorrhage in rats: Possibly via regulating the SIRT1/NF-κB pathway. Neurochem Res (2018) 43:785–95. doi: 10.1007/s11064-018-2480-4
161. Zhang XH, Peng L, Zhang J, Dong YP, Wang CJ, Liu C, et al. Berberine ameliorates subarachnoid hemorrhage injury via induction of sirtuin 1 and inhibiting HMGB1/Nf-κB pathway. Front Pharmacol (2020) 11:1073. doi: 10.3389/fphar.2020.01073
162. Han Y, Tong Z, Wang C, Li X, Liang G. Oleanolic acid exerts neuroprotective effects in subarachnoid hemorrhage rats through SIRT1-mediated HMGB1 deacetylation. Eur J Pharmacol (2021) 893:173811. doi: 10.1016/j.ejphar.2020.173811
163. Yamaguchi T, Miyamoto T, Shikata E, Yamaguchi I, Shimada K, Yagi K, et al. Activation of the NLRP3/IL-1β/MMP-9 pathway and intracranial aneurysm rupture associated with the depletion of ERα and Sirt1 in oophorectomized rats. J Neurosurg (2022) 20:1–8. doi: 10.3171/2022.4.JNS212945
164. Fujii M, Sherchan P, Krafft PR, Rolland WB, Soejima Y, Zhang JH. Cannabinoid type 2 receptor stimulation attenuates brain edema by reducing cerebral leukocyte infiltration following subarachnoid hemorrhage in rats. J Neurological Sci (2014) 342:101–6. doi: 10.1016/j.jns.2014.04.034
165. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell (2010) 142:687–98. doi: 10.1016/j.cell.2010.07.041
166. Yin J, Li H, Meng C, Chen D, Chen Z, Wang Y, et al. Inhibitory effects of omega-3 fatty acids on early brain injury after subarachnoid hemorrhage in rats: Possible involvement of G protein-coupled receptor 120/β-arrestin2/TGF-β activated kinase-1 binding protein-1 signaling pathway. Int J Biochem Cell Biol (2016) 75:11–22. doi: 10.1016/j.biocel.2016.03.008
167. Rapoport RM. Bilirubin oxidation products and cerebral vasoconstriction. Front Pharmacol (2018) 9:303. doi: 10.3389/fphar.2018.00303
168. Liao Q, Shi DH, Zheng W, Xu XJ, Yu YH. Antiproliferation of cardamonin is involved in mTOR on aortic smooth muscle cells in high fructose-induced insulin resistance rats. Eur J Pharmacol (2010) 641:179–86. doi: 10.1016/j.ejphar.2010.05.024
169. Li XG, Du JH, Lu Y, Lin XJ. Neuroprotective effects of rapamycin on spinal cord injury in rats by increasing autophagy and akt signaling. Neural Regeneration Res (2019) 14:721–7. doi: 10.4103/1673-5374.247476
170. Aoki T, Frösen J, Fukuda M, Bando K, Shioi G, Tsuji K, et al. Prostaglandin E2-EP2-NF-κB signaling in macrophages as a potential therapeutic target for intracranial aneurysms. Sci Signaling (2017) 10(465):6037. doi: 10.1126/aah.6037
171. Leng W, Fan D, Ren Z, Li Q. Identification of upregulated NF-κB inhibitor alpha and IRAK3 targeting lncRNA following intracranial aneurysm rupture-induced subarachnoid hemorrhage. BMC Neurol (2021) 21:197. doi: 10.1186/s12883-021-02156-1
172. Lu Y, Zhang XS, Zhang ZH, Zhou XM, Gao YY, Liu GJ, et al. Peroxiredoxin 2 activates microglia by interacting with toll-like receptor 4 after subarachnoid hemorrhage. J Neuroinflamm (2018) 15:87. doi: 10.1186/s12974-018-1118-4
173. Liu GH, Qu J, Shen X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim Biophys Acta (2008) 1783:713–27. doi: 10.1016/j.bbamcr.2008.01.002
174. Pan H, Wang H, Zhu L, Mao L, Qiao L, Su X. Depletion of Nrf2 enhances inflammation induced by oxyhemoglobin in cultured mice astrocytes. Neurochem Res (2011) 36:2434–41. doi: 10.1007/s11064-011-0571-6
175. Priego N, Valiente M. The potential of astrocytes as immune modulators in brain tumors. Front Immunol (2019) 10:1314. doi: 10.3389/fimmu.2019.01314
176. Wei Y, Chiang WC, Sumpter R Jr., Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell (2017) 168:224–38.e10. doi: 10.1016/j.cell.2016.11.042
177. Kashiwada M, Lu P, Rothman PB. PIP3 pathway in regulatory T cells and autoimmunity. Immunol Res (2007) 39:194–224. doi: 10.1007/s12026-007-0075-2
178. Hawkins PT, Stephens LR. PI3K signalling in inflammation. Biochim Biophys Acta (2015) 1851:882–97. doi: 10.1016/j.bbalip.2014.12.006
179. Harno KS. Telemedicine in managing demand for secondary-care services. J Telemedicine Telecare (1999) 5:189–92. doi: 10.1258/1357633991933611
180. Roosen K, Scheld M, Mandzhalova M, Clarner T, Beyer C, Zendedel A. CXCL12 inhibits inflammasome activation in LPS-stimulated BV2 cells. Brain Res (2021) 1763:147446. doi: 10.1016/j.brainres.2021.147446
181. Guyon A. CXCL12 chemokine and its receptors as major players in the interactions between immune and nervous systems. Front Cell Neurosci (2014) 8:65. doi: 10.3389/fncel.2014.00065
182. Yang L, Dong X, Zhang W. Astragaloside IV alleviates the brain damage induced by subarachnoid hemorrhage via PI3K/Akt signaling pathway. Neurosci Lett (2020) 735:135227. doi: 10.1016/j.neulet.2020.135227
183. Inta I, Paxian S, Maegele I, Zhang W, Pizzi M, Spano P, et al. Bim and noxa are candidates to mediate the deleterious effect of the NF-kappa b subunit RelA in cerebral ischemia. J Neurosci Off J Soc Neurosci (2006) 26:12896–903. doi: 10.1523/JNEUROSCI.3670-06.2006
184. Graves DT, Milovanova TN. Mucosal immunity and the FOXO1 transcription factors. Front Immunol (2019) 10:2530. doi: 10.3389/fimmu.2019.02530
185. Hao XK, Wu W, Wang CX, Xie GB, Li T, Wu HM, et al. Ghrelin alleviates early brain injury after subarachnoid hemorrhage via the PI3K/Akt signaling pathway. Brain Res (2014) 1587:15–22. doi: 10.1016/j.brainres.2014.08.069
186. Ma J, Wang Z, Liu C, Shen H, Chen Z, Yin J, et al. Pramipexole-induced hypothermia reduces early brain injury via PI3K/AKT/GSK3β pathway in subarachnoid hemorrhage rats. Sci Rep (2016) 6:23817. doi: 10.1038/srep23817
187. Xie Z, Enkhjargal B, Wu L, Zhou K, Sun C, Hu X, et al. Exendin-4 attenuates neuronal death via GLP-1R/PI3K/Akt pathway in early brain injury after subarachnoid hemorrhage in rats. Neuropharmacology (2018) 128:142–51. doi: 10.1016/j.neuropharm.2017.09.040
188. Dong L, Li YZ, An HT, Wang YL, Chen SH, Qian YJ, et al. The E3 ubiquitin ligase c-cbl inhibits microglia-mediated CNS inflammation by regulating PI3K/Akt/NF-κB pathway. CNS Neurosci Ther (2016) 22:661–9. doi: 10.1111/cns.12557
189. Li L, McBride DW, Doycheva D, Dixon BJ, Krafft PR, Zhang JH, et al. G-CSF attenuates neuroinflammation and stabilizes the blood-brain barrier via the PI3K/Akt/GSK-3β signaling pathway following neonatal hypoxia-ischemia in rats. Exp Neurol (2015) 272:135–44. doi: 10.1016/j.expneurol.2014.12.020
190. Krishna M, Narang H. The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci CMLS (2008) 65:3525–44. doi: 10.1007/s00018-008-8170-7
191. Gu X, Zheng C, Zheng Q, Chen S, Li W, Shang Z, et al. Salvianolic acid a attenuates early brain injury after subarachnoid hemorrhage in rats by regulating ERK/P38/Nrf2 signaling. Am J Trans Res (2017) 9:5643–52. doi: 10.1007/s00018-008-8170-7
192. Edvinsson L, Larsen SS, Maddahi A, Nielsen J. Plasticity of cerebrovascular smooth muscle cells after subarachnoid hemorrhage. Trans Stroke Res (2014) 5:365–76. doi: 10.1007/s12975-014-0331-4
193. Gatti S, Lonati C, Acerbi F, Sordi A, Leonardi P, Carlin A, et al. Protective action of NDP-MSH in experimental subarachnoid hemorrhage. Exp Neurol (2012) 234:230–8. doi: 10.1016/j.expneurol.2011.12.039
194. Maddahi A, Povlsen GK, Edvinsson L. Regulation of enhanced cerebrovascular expression of proinflammatory mediators in experimental subarachnoid hemorrhage via the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathway. J Neuroinflamm (2012) 9:274. doi: 10.1186/1742-2094-9-274
195. Grosshans J, Schnorrer F, Nüsslein-Volhard C. Oligomerisation of tube and pelle leads to nuclear localisation of dorsal. Mech Dev (1999) 81:127–38. doi: 10.1016/S0925-4773(98)00236-6
196. Medvedev AE, Murphy M, Zhou H, Li X. E3 ubiquitin ligases pellinos as regulators of pattern recognition receptor signaling and immune responses. Immunol Rev (2015) 266:109–22. doi: 10.1111/imr.12298
197. Cahill J, Calvert JW, Zhang JH. Mechanisms of early brain injury after subarachnoid hemorrhage. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2006) 26:1341–53. doi: 10.1038/sj.jcbfm.9600283
198. Shi H, Fang Y, Huang L, Gao L, Lenahan C, Okada T, et al. Activation of galanin receptor 1 with M617 attenuates neuronal apoptosis via ERK/GSK-3β/TIP60 pathway after subarachnoid hemorrhage in rats. Neurother J Am Soc Exp NeuroTherapeutics (2021) 18:1905–21. doi: 10.1007/s13311-021-01066-x
199. Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci Off J Soc Neurosci (2007) 27:6320–32. doi: 10.1523/JNEUROSCI.0449-07.2007
200. Jiang Y, Wu J, Keep RF, Hua Y, Hoff JT, Xi G. Hypoxia-inducible factor-1alpha accumulation in the brain after experimental intracerebral hemorrhage. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2002) 22:689–96. doi: 10.1097/00004647-200206000-00007
201. Hu Q, Wu C, Chen JY, Yan F, Li JR, Chen G. [The relationship between hypoxia-inducible factor-1α expression and apoptosis in early brain injury after subarachnoid hemorrhage]. Zhejiang da xue xue bao Yi xue ban = J Zhejiang Univ Med Sci (2014) 43:58–65. doi: 10.3785/j.issn.1008-9292.2014.01.009
202. Zhou Y, Martin RD, Zhang JH. Advances in experimental subarachnoid hemorrhage. Acta Neurochirurgica Supplement (2011) 110:15–21. doi: 10.1007/978-3-7091-0353
203. Wu C, Hu Q, Chen J, Yan F, Li J, Wang L, et al. Inhibiting HIF-1α by 2ME2 ameliorates early brain injury after experimental subarachnoid hemorrhage in rats. Biochem Biophys Res Commun (2013) 437:469–74. doi: 10.1016/j.bbrc.2013.06.107
204. Xu W, Xu R, Li X, Zhang H, Wang X, Zhu J. Downregulating hypoxia-inducible factor-1α expression with perfluorooctyl-bromide nanoparticles reduces early brain injury following experimental subarachnoid hemorrhage in rats. Am J Trans Res (2016) 8:2114–26.
205. Xu Z, Zhang F, Xu H, Yang F, Zhou G, Tong M, et al. Melatonin affects hypoxia-inducible factor 1α and ameliorates delayed brain injury following subarachnoid hemorrhage via H19/miR-675/HIF1A/TLR4. Bioengineered (2022) 13:4235–47. doi: 10.1080/21655979.2022.2027175
206. Solum EJ, Akselsen ØW, Vik A, Hansen TV. Synthesis and pharmacological effects of the anti-cancer agent 2-methoxyestradiol. Curr Pharm Design (2015) 21:5453–66. doi: 10.2174/1381612821666151002112511
207. Kusch A, Tkachuk S, Tkachuk N, Patecki M, Park JK, Dietz R, et al. The tight junction protein ZO-2 mediates proliferation of vascular smooth muscle cells via regulation of Stat1. Cardiovasc Res (2009) 83:115–22. doi: 10.1093/cvr/cvp117
208. Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG. Emerging translational approaches to target STAT3 signalling and its impact on vascular disease. Cardiovasc Res (2015) 106:365–74. doi: 10.1093/cvr/cvv103
209. Kim OS, Park EJ, Joe EH, Jou I. JAK-STAT signaling mediates gangliosides-induced inflammatory responses in brain microglial cells. J Biol Chem (2002) 277:40594–601. doi: 10.1074/jbc.M203885200
210. Samraj AK, Müller AH, Grell AS, Edvinsson L. Role of unphosphorylated transcription factor STAT3 in late cerebral ischemia after subarachnoid hemorrhage. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2014) 34:759–63. doi: 10.1038/jcbfm.2014.15
211. Muhammad S, Chaudhry SR, Kahlert UD, Lehecka M, Korja M, Niemelä M, et al. Targeting high mobility group box 1 in subarachnoid hemorrhage: A systematic review. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21082709
212. Paudel YN, Angelopoulou E, Piperi C, Othman I, Shaikh MF. HMGB1-mediated neuroinflammatory responses in brain injuries: Potential mechanisms and therapeutic opportunities. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21134609
213. Nishikawa H, Liu L, Nakano F, Kawakita F, Kanamaru H, Nakatsuka Y, et al. Modified citrus pectin prevents blood-brain barrier disruption in mouse subarachnoid hemorrhage by inhibiting galectin-3. Stroke (2018) 49:2743–51. doi: 10.1161/STROKEAHA.118.021757
214. Li S, Yang S, Sun B, Hang C. Melatonin attenuates early brain injury after subarachnoid hemorrhage by the JAK-STAT signaling pathway. Int J Clin Exp Pathol (2019) 12:909–15.
215. Yamamoto S, Mutoh T, Sasaki K, Mutoh T, Taki Y. Central action of rapamycin on early ischemic injury and related cardiac depression following experimental subarachnoid hemorrhage. Brain Res Bull (2019) 144:85–91. doi: 10.1016/j.brainresbull.2018.11.015
216. Liddelow SA, Barres BA. Reactive astrocytes: Production, function, and therapeutic potential. Immunity (2017) 46:957–67. doi: 10.1016/j.immuni.2017.06.006
217. Zhang L, Guo K, Zhou J, Zhang X, Yin S, Peng J, et al. Ponesimod protects against neuronal death by suppressing the activation of A1 astrocytes in early brain injury after experimental subarachnoid hemorrhage. J Neurochem (2021) 158:880–97. doi: 10.1111/jnc.15457
218. Zhang R, Liu Y, Yan K, Chen L, Chen XR, Li P, et al. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J Neuroinflamm (2013) 10:106. doi: 10.1186/1742-2094-10-106
219. Gao YY, Tao T, Wu D, Zhuang Z, Lu Y, Wu LY, et al. MFG-E8 attenuates inflammation in subarachnoid hemorrhage by driving microglial M2 polarization. Exp Neurol (2021) 336:113532. doi: 10.1016/j.expneurol.2020.113532
220. Zheng ZV, Chen J, Lyu H, Lam SYE, Lu G, Chan WY, et al. Novel role of STAT3 in microglia-dependent neuroinflammation after experimental subarachnoid haemorrhage. Stroke Vasc Neurol (2022) 7:62–70. doi: 10.1136/svn-2021-001028
221. Yuan B, Zhao XD, Shen JD, Chen SJ, Huang HY, Zhou XM, et al. Activation of SIRT1 alleviates ferroptosis in the early brain injury after subarachnoid hemorrhage. Oxid Med Cell Longev (2022) 2022:9069825. doi: 10.1155/2022/9069825
222. Hwang JW, Yao H, Caito S, Sundar IK, Rahman I. Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radical Biol Med (2013) 61:95–110. doi: 10.1016/j.freeradbiomed.2013.03.015
223. Hasegawa K, Yoshikawa K. Necdin regulates p53 acetylation via Sirtuin1 to modulate DNA damage response in cortical neurons. J Neurosci Off J Soc Neurosci (2008) 28:8772–84. doi: 10.1523/JNEUROSCI.3052-08.2008
224. Morris KC, Lin HW, Thompson JW, Perez-Pinzon MA. Pathways for ischemic cytoprotection: role of sirtuins in caloric restriction, resveratrol, and ischemic preconditioning. J Cereb Blood Flow Metab Off J Int Soc Cereb Blood Flow Metab (2011) 31:1003–19. doi: 10.1038/jcbfm.2010.229
225. Zhang F, Wang S, Gan L, Vosler PS, Gao Y, Zigmond MJ, et al. Protective effects and mechanisms of sirtuins in the nervous system. Prog Neurobiol (2011) 95:373–95. doi: 10.1016/j.pneurobio.2011.09.001
226. Zhou XM, Zhang X, Zhang XS, Zhuang Z, Li W, Sun Q, et al. SIRT1 inhibition by sirtinol aggravates brain edema after experimental subarachnoid hemorrhage. J Neurosci Res (2014) 92:714–22. doi: 10.1002/jnr.23359
227. Vellimana AK, Aum DJ, Diwan D, Clarke JV, Nelson JW, Lawrence M, et al. SIRT1 mediates hypoxic preconditioning induced attenuation of neurovascular dysfunction following subarachnoid hemorrhage. Exp Neurol (2020) 334:113484. doi: 10.1016/j.expneurol.2020.113484
228. Vellimana AK, Diwan D, Clarke J, Gidday JM, Zipfel GJ. SIRT1 activation: A potential strategy for harnessing endogenous protection against delayed cerebral ischemia after subarachnoid hemorrhage. Neurosurgery (2018) 65:1–5. doi: 10.1093/neuros/nyy201
229. Zhang XS, Wu Q, Wu LY, Ye ZN, Jiang TW, Li W, et al. Sirtuin 1 activation protects against early brain injury after experimental subarachnoid hemorrhage in rats. Cell Death Dis (2016) 7:e2416. doi: 10.1038/cddis.2016.292
230. Lin W, Yao H, Lai J, Zeng Y, Guo X, Lin S, et al. Cycloastragenol confers cerebral protection after subarachnoid hemorrhage by suppressing oxidative insults and neuroinflammation via the SIRT1 signaling pathway. Oxid Med Cell Longev (2022) 2022:3099409. doi: 10.1155/2022/3099409
231. Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, et al. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arteriosc Thromb Vasc Biol (2015) 35:127–36. doi: 10.1161/ATVBAHA.114.303763
232. Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev (1997) 8:21–43. doi: 10.1016/S1359-6101(96)00048-2
233. Ayer RE, Ostrowski RP, Sugawara T, Ma Q, Jafarian N, Tang J, et al. Statin-induced T-lymphocyte modulation and neuroprotection following experimental subarachnoid hemorrhage. Acta Neurochirurgica Supplement (2013) 115:259–66. doi: 10.1007/978-3-7091-1192-5_46
234. Gamble JR, Khew-Goodall Y, Vadas MA. Transforming growth factor-beta inhibits e-selectin expression on human endothelial cells. J Immunol (Baltimore Md 1950) (1993) 150:4494–503.
235. Wang Y, Zhou S, Han Z, Yin D, Luo Y, Tian Y, et al. Fingolimod administration improves neurological functions of mice with subarachnoid hemorrhage. Neurosci Lett (2020) 736:135250. doi: 10.1016/j.neulet.2020.135250
236. Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov (2010) 9:883–97. doi: 10.1038/nrd3248
237. Xu K, Liu P, Wei W. mTOR signaling in tumorigenesis. Biochim Biophys Acta (2014) 1846:638–54. doi: 10.1016/j.bbcan.2014.10.007
238. Sasaki K, Yamamoto S, Mutoh T, Tsuru Y, Taki Y, Kawashima R. Rapamycin protects against early brain injury independent of cerebral blood flow changes in a mouse model of subarachnoid haemorrhage. Clin Exp Pharmacol Physiol (2018) 45:859–62. doi: 10.1111/1440-1681.12950
239. Li Y, Wu P, Dai J, Zhang T, Bihl J, Wang C, et al. Inhibition of mTOR alleviates early brain injury after subarachnoid hemorrhage Via relieving excessive mitochondrial fission. Cell Mol Neurobiol (2020) 40:629–42. doi: 10.1007/s10571-019-00760-x
240. Ma Y, Zhang B, Zhang D, Wang S, Li M, Zhao J. Differentially expressed circular RNA profile in an intracranial aneurysm group compared with a healthy control group. Dis Markers (2021) 2021:8889569. doi: 10.1155/2021/8889569
241. Zhao M, Gao F, Zhang D, Wang S, Zhang Y, Wang R, et al. Altered expression of circular RNAs in moyamoya disease. J Neurological Sci (2017) 381:25–31. doi: 10.1016/j.jns.2017.08.011
242. Weichhart T, Hengstschläger M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol (2015) 15:599–614. doi: 10.1038/nri3901
243. Zhang W, Khatibi NH, Yamaguchi-Okada M, Yan J, Chen C, Hu Q, et al. Mammalian target of rapamycin (mTOR) inhibition reduces cerebral vasospasm following a subarachnoid hemorrhage injury in canines. Exp Neurol (2012) 233:799–806. doi: 10.1016/j.expneurol.2011.11.046
244. Mao L, Jia J, Zhou X, Xiao Y, Wang Y, Mao X, et al. Delayed administration of a PTEN inhibitor BPV improves functional recovery after experimental stroke. Neuroscience (2013) 231:272–81. doi: 10.1016/j.neuroscience.2012.11.050
245. Pyne-Geithman GJ, Nair SG, Stamper DN, Clark JF. Role of bilirubin oxidation products in the pathophysiology of DIND following SAH. Acta Neurochirurgica Supplement (2013) 115:267–73. doi: 10.1007/978-3-7091-1192-5_47
246. Liu J, Kong L, Chen D, Tang H, Lu Y, Yuan Y, et al. Bilirubin oxidation end product b prevents CoCl(2)-induced primary cortical neuron apoptosis by promoting cell survival Akt/mTOR/p70S6K signaling pathway. Biochem Biophys Res Commun (2022) 602:27–34. doi: 10.1016/j.bbrc.2022.02.063
247. Wang L, Xiong X, Zhang X, Ye Y, Jian Z, Gao W, et al. Sodium tanshinone IIA sulfonate protects against cerebral ischemia-reperfusion injury by inhibiting autophagy and inflammation. Neuroscience (2020) 441:46–57. doi: 10.1016/j.neuroscience.2020.05.054
248. Son Y, Cho YK, Saha A, Kwon HJ, Park JH, Kim M, et al. Adipocyte-specific Beclin1 deletion impairs lipolysis and mitochondrial integrity in adipose tissue. Mol Metab (2020) 39:101005. doi: 10.1016/j.molmet.2020.101005
249. Xian X, Ding Y, Dieckmann M, Zhou L, Plattner F, Liu M, et al. LRP1 integrates murine macrophage cholesterol homeostasis and inflammatory responses in atherosclerosis. eLife (2017) 6:e29292. doi: 10.7554/eLife.29292
250. Zhang XS, Li W, Wu Q, Wu LY, Ye ZN, Liu JP, et al. Resveratrol attenuates acute inflammatory injury in experimental subarachnoid hemorrhage in rats via inhibition of TLR4 pathway. Int J Mol Sci (2016) 17(8):1331. doi: 10.3390/ijms17081331
251. Keogh B, Parker AE. Toll-like receptors as targets for immune disorders. Trends Pharmacol Sci (2011) 32:435–42. doi: 10.1016/j.tips.2011.03.008
252. Guo M, Xu Y, Zhang CJ. TLR signaling in brain immunity. Handb Exp Pharmacol (2021) 276:213–37. doi: 10.1007/164_2021_542
253. Becher B, Spath S, Goverman J. Cytokine networks in neuroinflammation. Nat Rev Immunol (2017) 17:49–59. doi: 10.1038/nri.2016.123
254. Mellanby RJ, Cambrook H, Turner DG, O'Connor RA, Leech MD, Kurschus FC, et al. TLR-4 ligation of dendritic cells is sufficient to drive pathogenic T cell function in experimental autoimmune encephalomyelitis. J Neuroinflamm (2012) 9:248. doi: 10.1186/1742-2094-9-248
255. Béla SR, Dutra MS, Mui E, Montpetit A, Oliveira FS, Oliveira SC, et al. Impaired innate immunity in mice deficient in interleukin-1 receptor-associated kinase 4 leads to defective type 1 T cell responses, b cell expansion, and enhanced susceptibility to infection with toxoplasma gondii. Infect Immunity (2012) 80:4298–308. doi: 10.1128/IAI.00328-12
256. Harari OA, Liao JK. NF-κB and innate immunity in ischemic stroke. Ann New York Acad Sci (2010) 1207:32–40. doi: 10.1111/j.1749-6632.2010.05735.x
257. Muhammad S, Chaudhry SR, Dobreva G, Lawton MT, Niemelä M, Hänggi D. Vascular macrophages as therapeutic targets to treat intracranial aneurysms. Front Immunol (2021) 12:630381. doi: 10.3389/fimmu.2021.630381
258. Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, et al. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke (2004) 35:2287–93. doi: 10.1161/01.STR.0000140636.30204.da
259. Zuiderwijk-Sick EA, van der Putten C, Bsibsi M, Deuzing IP, de Boer W, Persoon-Deen C, et al. Differentiation of primary adult microglia alters their response to TLR8-mediated activation but not their capacity as APC. Glia (2007) 55:1589–600. doi: 10.1002/glia.20572
260. Theus MH, Brickler T, Meza AL, Coutermarsh-Ott S, Hazy A, Gris D, et al. Loss of NLRX1 exacerbates neural tissue damage and NF-κB signaling following brain injury. J Immunol (Baltimore Md 1950) (2017) 199:3547–58. doi: 10.4049/jimmunol.1700251
261. Liu CY, Guo SD, Yu JZ, Li YH, Zhang H, Feng L, et al. Fasudil mediates cell therapy of EAE by immunomodulating encephalomyelitic T cells and macrophages. Eur J Immunol (2015) 45:142–52. doi: 10.1002/eji.201344429
262. Liu L, Guo H, Song A, Huang J, Zhang Y, Jin S, et al. Progranulin inhibits LPS-induced macrophage M1 polarization via NF-кB and MAPK pathways. BMC Immunol (2020) 21:32. doi: 10.1186/s12865-020-00355-y
263. Sehba FA, Hou J, Pluta RM, Zhang JH. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol (2012) 97:14–37. doi: 10.1016/j.pneurobio.2012.02.003
264. Kawakita F, Fujimoto M, Liu L, Nakano F, Nakatsuka Y, Suzuki H. Effects of toll-like receptor 4 antagonists against cerebral vasospasm after experimental subarachnoid hemorrhage in mice. Mol Neurobiol (2017) 54:6624–33. doi: 10.1007/s12035-016-0178-7
265. Ma C, Zhou W, Yan Z, Qu M, Bu X. Toll-like receptor 4 (TLR4) is associated with cerebral vasospasm and delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage. Neurol Medico Chirurgica (2015) 55:878–84. doi: 10.2176/nmc.oa.2015-0077
266. Okada T, Suzuki H. Toll-like receptor 4 as a possible therapeutic target for delayed brain injuries after aneurysmal subarachnoid hemorrhage. Neural Regeneration Res (2017) 12:193–6. doi: 10.4103/1673-5374.200795
267. Liu FY, Cai J, Wang C, Ruan W, Guan GP, Pan HZ, et al. Fluoxetine attenuates neuroinflammation in early brain injury after subarachnoid hemorrhage: a possible role for the regulation of TLR4/MyD88/NF-κB signaling pathway. J Neuroinflamm (2018) 15:347. doi: 10.1186/s12974-018-1388-x
268. Chen J, Xuan Y, Chen Y, Wu T, Chen L, Guan H, et al. Netrin-1 alleviates subarachnoid haemorrhage-induced brain injury via the PPARγ/NF-KB signalling pathway. J Cell Mol Med (2019) 23:2256–62. doi: 10.1111/jcmm.14105
269. Lv H, Liu Q, Zhou J, Tan G, Deng X, Ci X. Daphnetin-mediated Nrf2 antioxidant signaling pathways ameliorate tert-butyl hydroperoxide (t-BHP)-induced mitochondrial dysfunction and cell death. Free Radical Biol Med (2017) 106:38–52. doi: 10.1016/j.freeradbiomed.2017.02.016
270. Zhang X, Wu Q, Lu Y, Wan J, Dai H, Zhou X, et al. Cerebroprotection by salvianolic acid b after experimental subarachnoid hemorrhage occurs via Nrf2- and SIRT1-dependent pathways. Free Radical Biol Med (2018) 124:504–16. doi: 10.1016/j.freeradbiomed.2018.06.035
271. Raghunath A, Sundarraj K, Nagarajan R, Arfuso F, Bian J, Kumar AP, et al. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol (2018) 17:297–314. doi: 10.1016/j.redox.2018.05.002
272. Zhang J, Zhu Y, Zhou D, Wang Z, Chen G. Recombinant human erythropoietin (rhEPO) alleviates early brain injury following subarachnoid hemorrhage in rats: possible involvement of Nrf2-ARE pathway. Cytokine (2010) 52:252–7. doi: 10.1016/j.cyto.2010.08.011
273. Zolnourian A, Galea I, Bulters D. Neuroprotective role of the Nrf2 pathway in subarachnoid haemorrhage and its therapeutic potential. Oxid Med Cell Longev (2019) 2019:6218239. doi: 10.1155/2019/6218239
274. Galea J, Cruickshank G, Teeling JL, Boche D, Garland P, Perry VH, et al. The intrathecal CD163-haptoglobin-hemoglobin scavenging system in subarachnoid hemorrhage. J Neurochem (2012) 121:785–92. doi: 10.1111/j.1471-4159.2012.07716.x
275. Smith EO, Byrd LD. Studying the behavioral effects of drugs in group-living nonhuman primates. Prog Clin Biol Res (1983) 131:1–31.
276. Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol (2010) 30:3275–85. doi: 10.1128/MCB.00248-10
277. Wahdan SA, Azab SS, Elsherbiny DA, El-Demerdash E. Piceatannol protects against cisplatin nephrotoxicity via activation of Nrf2/HO-1 pathway and hindering NF-κB inflammatory cascade. Naunyn-Schmiedeberg's Arch Pharmacol (2019) 392:1331–45. doi: 10.1007/s00210-019-01673-8
278. Lu Y, Yang YY, Zhou MW, Liu N, Xing HY, Liu XX, et al. Ketogenic diet attenuates oxidative stress and inflammation after spinal cord injury by activating Nrf2 and suppressing the NF-κB signaling pathways. Neurosci Lett (2018) 683:13–8. doi: 10.1016/j.neulet.2018.06.016
279. Liu Y, Qiu J, Wang Z, You W, Wu L, Ji C, et al. Dimethylfumarate alleviates early brain injury and secondary cognitive deficits after experimental subarachnoid hemorrhage via activation of Keap1-Nrf2-ARE system. J Neurosurg (2015) 123:915–23. doi: 10.3171/2014.11.JNS132348
280. Chen S, Feng H, Sherchan P, Klebe D, Zhao G, Sun X, et al. Controversies and evolving new mechanisms in subarachnoid hemorrhage. Prog Neurobiol (2014) 115:64–91. doi: 10.1016/j.pneurobio.2013.09.002
281. Kakehashi A, Ishii N, Shibata T, Wei M, Okazaki E, Tachibana T, et al. Mitochondrial prohibitins and septin 9 are implicated in the onset of rat hepatocarcinogenesis. Toxicol Sci an Off J Soc Toxicol (2011) 119:61–72. doi: 10.1093/toxsci/kfq307
282. Zhang T, Wu P, Zhang JH, Li Y, Xu S, Wang C, et al. Docosahexaenoic acid alleviates oxidative stress-based apoptosis Via improving mitochondrial dynamics in early brain injury after subarachnoid hemorrhage. Cell Mol Neurobiol (2018) 38:1413–23. doi: 10.1007/s10571-018-0608-3
283. Do MT, Kim HG, Choi JH, Jeong HG. Metformin induces microRNA-34a to downregulate the Sirt1/Pgc-1α/Nrf2 pathway, leading to increased susceptibility of wild-type p53 cancer cells to oxidative stress and therapeutic agents. Free Radical Biol Med (2014) 74:21–34. doi: 10.1016/j.freeradbiomed.2014.06.010
284. Zhang Y, Tao X, Yin L, Xu L, Xu Y, Qi Y, et al. Protective effects of dioscin against cisplatin-induced nephrotoxicity via the microRNA-34a/sirtuin 1 signalling pathway. Br J Pharmacol (2017) 174:2512–27. doi: 10.1111/bph.13862
285. Chen G, Fang Q, Zhang J, Zhou D, Wang Z. Role of the Nrf2-ARE pathway in early brain injury after experimental subarachnoid hemorrhage. J Neurosci Res (2011) 89:515–23. doi: 10.1002/jnr.22577
286. Wang Z, Chen G, Zhu WW, Zhou D. Activation of nuclear factor-erythroid 2-related factor 2 (Nrf2) in the basilar artery after subarachnoid hemorrhage in rats. Ann Clin Lab Sci (2010) 40:233–9.
287. Abboud T, Andresen H, Koeppen J, Czorlich P, Duehrsen L, Stenzig J, et al. Serum levels of nimodipine in enteral and parenteral administration in patients with aneurysmal subarachnoid hemorrhage. Acta Neurochirurgica (2015) 157:763–7. doi: 10.1007/s00701-015-2369-9
288. Choi HA, Ko SB, Chen H, Gilmore E, Carpenter AM, Lee D, et al. Acute effects of nimodipine on cerebral vasculature and brain metabolism in high grade subarachnoid hemorrhage patients. Neurocritical Care (2012) 16:363–7. doi: 10.1007/s12028-012-9670-8
289. Gaab MR, Haubitz I, Brawanski A, Korn A, Czech T. Acute effects of nimodipine on the cerebral blood flow and intracranial pressure. Neurochirurgia (1985) 28 Suppl 1:93–9. doi: 10.1055/s-2008-1054111
290. Yin D, Zhou S, Xu X, Gao W, Li F, Ma Y, et al. Dexmedetomidine attenuated early brain injury in rats with subarachnoid haemorrhage by suppressing the inflammatory response: The TLR4/NF-κB pathway and the NLRP3 inflammasome may be involved in the mechanism. Brain Res (2018) 1698:1–10. doi: 10.1016/j.brainres.2018.05.040
291. Schoch B, Regel JP, Wichert M, Gasser T, Volbracht L, Stolke D. Analysis of intrathecal interleukin-6 as a potential predictive factor for vasospasm in subarachnoid hemorrhage. Neurosurgery (2007) 60:828–36; discussion -36. doi: 10.1227/01.NEU.0000255440.21495.80
292. Xu HL, Pelligrino DA, Paisansathan C, Testai FD. Protective role of fingolimod (FTY720) in rats subjected to subarachnoid hemorrhage. J Neuroinflamm (2015) 12:16. doi: 10.1186/s12974-015-0234-7
293. Pfeilschifter W, Czech-Zechmeister B, Sujak M, Foerch C, Wichelhaus TA, Pfeilschifter J. Treatment with the immunomodulator FTY720 does not promote spontaneous bacterial infections after experimental stroke in mice. Exp Trans Stroke Med (2011) 3:2. doi: 10.1186/2040-7378-3-2
294. Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet (London England) (2006) 368:1329–38. doi: 10.1016/S0140-6736(06)69446-4
295. Sandow N, Diesing D, Sarrafzadeh A, Vajkoczy P, Wolf S. Nimodipine dose reductions in the treatment of patients with aneurysmal subarachnoid hemorrhage. Neurocritical Care (2016) 25:29–39. doi: 10.1007/s12028-015-0230-x
296. Zhao C, Zhang J, Hu H, Qiao M, Chen D, Zhao X, et al. Design of lactoferrin modified lipid nano-carriers for efficient brain-targeted delivery of nimodipine. Mater Sci Eng C Mater Biol Applications (2018) 92:1031–40. doi: 10.1016j.msec.2018.02.004
297. Hu Y, Chen G, Huang J, Li Z, Li Z, Xie Y, et al. The calcium channel inhibitor nimodipine shapes the uveitogenic T cells and protects mice from experimental autoimmune uveitis through the p38-MAPK signaling pathway. J Immunol (Baltimore Md 1950) (2021) 207(12):2933–43. doi: 10.4049/jimmunol.2100568.
298. Liu Y, Lo YC, Qian L, Crews FT, Wilson B, Chen HL, et al. Verapamil protects dopaminergic neuron damage through a novel anti-inflammatory mechanism by inhibition of microglial activation. Neuropharmacology (2011) 60:373–80. doi: 10.1016/j.neuropharm.2010.10.002
299. Zheng VZ, Wong GKC. Neuroinflammation responses after subarachnoid hemorrhage: A review. J Clin Neurosci Off J Neurosurg Soc Australasia (2017) 42:7–11. doi: 10.1016/j.jocn.2017.02.001
300. Towart R, Kazda S. The cellular mechanism of action of nimodipine (BAY e 9736), a new calcium antagonist [proceedings]. Br J Pharmacol (1979) 67:409p–10p. doi: 10.1111/j.1476-5381.1979.tb08695.x.
301. Hockel K, Diedler J, Steiner J, Birkenhauer U, Ernemann U, Schuhmann MU. Effect of intra-arterial and intravenous nimodipine therapy of cerebral vasospasm after subarachnoid hemorrhage on cerebrovascular reactivity and oxygenation. World Neurosurg (2017) 101:372–8. doi: 10.1016/j.wneu.2017.02.014
302. Albanna W, Weiss M, Conzen C, Clusmann H, Schneider T, Reinsch M, et al. Systemic and cerebral concentration of nimodipine during established and experimental vasospasm treatment. World Neurosurg (2017) 102:459–65. doi: 10.1016/j.wneu.2017.03.062
303. Scriabine A, van den Kerckhoff W. Pharmacology of nimodipine. A Rev Ann New York Acad Sci (1988) 522:698–706. doi: 10.1111/j.1749-6632.1988.tb33415.x
304. Biondi A, Ricciardi GK, Puybasset L, Abdennour L, Longo M, Chiras J, et al. Intra-arterial nimodipine for the treatment of symptomatic cerebral vasospasm after aneurysmal subarachnoid hemorrhage: preliminary results. AJNR Am J Neuroradiol (2004) 25:1067–76.
305. Ott S, Jedlicka S, Wolf S, Peter M, Pudenz C, Merker P, et al. Continuous selective intra-arterial application of nimodipine in refractory cerebral vasospasm due to aneurysmal subarachnoid hemorrhage. BioMed Res Int (2014) 2014:970741. doi: 10.1155/2014/970741
306. Hockel K, Diedler J, Steiner J, Birkenhauer U, Danz S, Ernemann U, et al. Long-term, continuous intra-arterial nimodipine treatment of severe vasospasm after aneurysmal subarachnoid hemorrhage. World Neurosurg (2016) 88:104–12. doi: 10.1016/j.wneu.2015.11.081
307. Hou C, Liu Q, Zhang H, Wang W, Wang B, Cui X, et al. Nimodipine attenuates early brain injury by protecting the glymphatic system after subarachnoid hemorrhage in mice. Neurochem Res (2022) 47:701–12. doi: 10.1007/s11064-021-03478-9
308. Nabi B, Rehman S, Baboota S, Ali J. Insights on oral drug delivery of lipid nanocarriers: a win-win solution for augmenting bioavailability of antiretroviral drugs. AAPS PharmSciTech (2019) 20:60. doi: 10.1208/s12249-018-1284-9
309. Schneider UC, Dreher S, Hoffmann KT, Schmiedek P, Kasuya H, Vajkoczy P. The use of nicardipine prolonged release implants (NPRI) in microsurgical clipping after aneurysmal subarachnoid haemorrhage: comparison with endovascular treatment. Acta Neurochirurgica (2011) 153:2119–25. doi: 10.1007/s00701-011-1129-8
310. Barth M, Capelle HH, Weidauer S, Weiss C, Münch E, Thomé C, et al. Effect of nicardipine prolonged-release implants on cerebral vasospasm and clinical outcome after severe aneurysmal subarachnoid hemorrhage: a prospective, randomized, double-blind phase IIa study. Stroke (2007) 38:330–6. doi: 10.1161/01.STR.0000254601.74596.0f
311. Kasuya H, Onda H, Takeshita M, Okada Y, Hori T. Efficacy and safety of nicardipine prolonged-release implants for preventing vasospasm in humans. Stroke (2002) 33:1011–5. doi: 10.1161/01.STR.0000014563.75483.22
312. Moreno-Navarrete JM, Serrano M, Sabater M, Ortega F, Serino M, Pueyo N, et al. Study of lactoferrin gene expression in human and mouse adipose tissue, human preadipocytes and mouse 3T3-L1 fibroblasts. association with adipogenic and inflammatory markers. J Nutr Biochem (2013) 24:1266–75. doi: 10.1016/j.jnutbio.2012.10.002
313. Bayerl SH, Ghori A, Nieminen-Kelhä M, Adage T, Breitenbach J, Vajkoczy P, et al. In vitro and in vivo testing of a novel local nicardipine delivery system to the brain: a preclinical study. J Neurosurg (2019) 132:465–72. doi: 10.3171/2018.9.JNS173085
314. Rustemi O. Letter by rustemi regarding article, "Randomized, open-label, phase 1/2a study to determine the maximum tolerated dose of intraventricular sustained release nimodipine for subarachnoid hemorrhage (NEWTON [Nimodipine microparticles to enhance recovery while reducing toxicity after subarachnoid hemorrhage])". Stroke (2017) 48:e113. doi: 10.1161/STROKEAHA.116.016512
315. Hänggi D, Etminan N, Macdonald RL, Steiger HJ, Mayer SA, Aldrich F, et al. NEWTON: Nimodipine microparticles to enhance recovery while reducing toxicity after subarachnoid hemorrhage. Neurocritical Care (2015) 23:274–84. doi: 10.1007/s12028-015-0112-2
316. Ren X, Ma H, Zuo Z. Dexmedetomidine postconditioning reduces brain injury after brain hypoxia-ischemia in neonatal rats. J Neuroimmune Pharmacol Off J Soc NeuroImmune Pharmacol (2016) 11:238–47. doi: 10.1007/s11481-016-9658-9
317. Akpınar O, Nazıroğlu M, Akpınar H. Different doses of dexmedetomidine reduce plasma cytokine production, brain oxidative injury, PARP and caspase expression levels but increase liver oxidative toxicity in cerebral ischemia-induced rats. Brain Res Bull (2017) 130:1–9. doi: 10.1016/j.brainresbull.2016.12.005
318. Shen M, Wang S, Wen X, Han XR, Wang YJ, Zhou XM, et al. Dexmedetomidine exerts neuroprotective effect via the activation of the PI3K/Akt/mTOR signaling pathway in rats with traumatic brain injury. Biomed Pharmacother = Biomed Pharmacother (2017) 95:885–93. doi: 10.1016/j.biopha.2017.08.125
319. Wang Y, Han R, Zuo Z. Dexmedetomidine post-treatment induces neuroprotection via activation of extracellular signal-regulated kinase in rats with subarachnoid haemorrhage. Br J Anaesthesia (2016) 116:384–92. doi: 10.1093/bja/aev549
320. Li Y, Yang H, Ni W, Gu Y. Effects of deferoxamine on blood-brain barrier disruption after subarachnoid hemorrhage. PloS One (2017) 12:e0172784. doi: 10.1371/journal.pone.0172784
321. Wei B, Liu W, Jin L, Guo S, Fan H, Jin F, et al. Dexmedetomidine inhibits gasdermin d-induced pyroptosis via the PI3K/AKT/GSK3β pathway to attenuate neuroinflammation in early brain injury after subarachnoid hemorrhage in rats. Front Cell Neurosci (2022) 16:899484. doi: 10.3389/fncel.2022.899484
322. Yang SJ, Shao GF, Chen JL, Gong J. The NLRP3 inflammasome: An important driver of neuroinflammation in hemorrhagic stroke. Cell Mol Neurobiol (2018) 38:595–603. doi: 10.1007/s10571-017-0526-9
323. Khan N, Kuo A, Brockman DA, Cooper MA, Smith MT. Pharmacological inhibition of the NLRP3 inflammasome as a potential target for multiple sclerosis induced central neuropathic pain. Inflammopharmacology (2018) 26:77–86. doi: 10.1007/s10787-017-0401-9
324. Rothoerl RD, Axmann C, Pina AL, Woertgen C, Brawanski A. Possible role of the c-reactive protein and white blood cell count in the pathogenesis of cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J Neurosurg Anesthesiol (2006) 18:68–72. doi: 10.1097/01.ana.0000181693.30750.af
325. Song Y, Lim BJ, Kim DH, Ju JW, Han DW. Effect of dexmedetomidine on cerebral vasospasm and associated biomarkers in a rat subarachnoid hemorrhage model. J Neurosurg Anesthesiol (2019) 31:342–9. doi: 10.1097/ANA.0000000000000504
326. Okazaki T, Hifumi T, Kawakita K, Shishido H, Ogawa D, Okauchi M, et al. Association between dexmedetomidine use and neurological outcomes in aneurysmal subarachnoid hemorrhage patients: A retrospective observational study. J Crit Care (2018) 44:111–6. doi: 10.1016/j.jcrc.2017.10.034
327. van Donkelaar CE, Dijkland SA, van den Bergh WM, Bakker J, Dippel DW, Nijsten MW, et al. Early circulating lactate and glucose levels after aneurysmal subarachnoid hemorrhage correlate with poor outcome and delayed cerebral ischemia: A two-center cohort study. Crit Care Med (2016) 44:966–72. doi: 10.1097/CCM.0000000000001569
328. Kraut JA, Madias NE. Lactic acidosis. New Engl J Med (2014) 371:2309–19. doi: 10.1056/NEJMra1309483
329. Benedict CR, Loach AB. Sympathetic nervous system activity in patients with subarachnoid hemorrhage. Stroke (1978) 9:237–44. doi: 10.1161/01.STR.9.3.237
330. Soliven B, Miron V, Chun J. The neurobiology of sphingosine 1-phosphate signaling and sphingosine 1-phosphate receptor modulators. Neurology (2011) 76:S9–14. doi: 10.1212/WNL.0b013e31820d9507
331. Wei Y, Yemisci M, Kim HH, Yung LM, Shin HK, Hwang SK, et al. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann Neurol (2011) 69:119–29. doi: 10.1002/ana.22186
332. Rolland WB 2nd, Manaenko A, Lekic T, Hasegawa Y, Ostrowski R, Tang J, et al. FTY720 is neuroprotective and improves functional outcomes after intracerebral hemorrhage in mice. Acta Neurochirurgica Supplement (2011) 111:213–7. doi: 10.1007/978-3-7091-0693-8_36
333. Kuai F, Zhou J, Qiu Y, Gao Y. FTY720 attenuates cerebral vasospasm after subarachnoid hemorrhage through the PI3K/AKT/eNOS and NF-κB pathways in rats. Neurol India (2022) 70:1517–24. doi: 10.4103/0028-3886.355128
335. Hasegawa Y, Suzuki H, Sozen T, Rolland W, Zhang JH. Activation of sphingosine 1-phosphate receptor-1 by FTY720 is neuroprotective after ischemic stroke in rats. Stroke (2010) 41:368–74. doi: 10.1161/STROKEAHA.109.568899
336. Heldin CH. Targeting the PDGF signaling pathway in tumor treatment. Cell Communication Signaling CCS (2013) 11:97. doi: 10.1186/1478-811X-11-97
337. Hoch RV, Soriano P. Roles of PDGF in animal development. Dev (Cambridge England) (2003) 130:4769–84. doi: 10.1242/dev.00721
338. Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev (2008) 22:1276–312. doi: 10.1101/gad.1653708
339. Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov (2007) 6:734–45. doi: 10.1038/nrd2380
340. Sramek M, Neradil J, Macigova P, Mudry P, Polaskova K, Slaby O, et al. Effects of sunitinib and other kinase inhibitors on cells harboring a PDGFRB mutation associated with infantile myofibromatosis. Int J Mol Sci (2018) 19(9):2599. doi: 10.3390/ijms19092599
341. Takenouchi T, Kodo K, Yamazaki F, Nakatomi H, Kosaki K. Progressive cerebral and coronary aneurysms in the original two patients with kosaki overgrowth syndrome. Am J Med Genet Part A. (2021) 185:999–1003. doi: 10.1002/ajmg.a.62027
342. Foster A, Chalot B, Antoniadi T, Schaefer E, Keelagher R, Ryan G, et al. Kosaki overgrowth syndrome: A novel pathogenic variant in PDGFRB and expansion of the phenotype including cerebrovascular complications. Clin Genet (2020) 98:19–31. doi: 10.1111/cge.13752
343. Chenbhanich J, Hu Y, Hetts S, Cooke D, Dowd C, Devine P, et al. Segmental overgrowth and aneurysms due to mosaic PDGFRB p.(Tyr562Cys). Am J Med Genet Part A (2021) 185:1430–6. doi: 10.1002/ajmg.a.62126
Keywords: subarachnoid hemorrhage (SAH), inflammation, immune cells, signaling pathways, therapeutic strategies
Citation: Jin J, Duan J, Du L, Xing W, Peng X and Zhao Q (2022) Inflammation and immune cell abnormalities in intracranial aneurysm subarachnoid hemorrhage (SAH): Relevant signaling pathways and therapeutic strategies. Front. Immunol. 13:1027756. doi: 10.3389/fimmu.2022.1027756
Received: 25 August 2022; Accepted: 31 October 2022;
Published: 23 November 2022.
Edited by:
Yongqing Li, School of Medicine, University of Michigan, United StatesReviewed by:
Morgan Salmon, Michigan Medicine, University of Michigan, United StatesCopyright © 2022 Jin, Duan, Du, Xing, Peng and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qijie Zhao, emhhb3FpamllNzdAMTYzLmNvbQ==; Xingchen Peng, cHh4MjAxNEAxNjMuY29t
†The authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.