 Zefan Liu
Zefan Liu Yucheng Xiang1
Yucheng Xiang1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 20 October 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1027124
This article is part of the Research Topic Community Series in Immunotherapy with Checkpoint Inhibitors for Non-small Cell Lung Cancer, Colon Cancer and Esophageal Cancer, volume II View all 19 articles
Immune checkpoint blockade (ICB) has gained unparalleled success in the treatment of colorectal cancer (CRC). However, undesired side effects, unsatisfactory response rates, tumor metastasis, and drug resistance still hinder the further application of ICB therapy against CRC. Advancing ICB with nanotechnology can be game-changing. With the development of immuno-oncology and nanomaterials, various nanoplatforms have been fabricated to enhance the efficacy of ICB in CRC treatment. Herein, this review systematically summarizes these recent nano-strategies according to their mechanisms. Despite their diverse and complex designs, these nanoplatforms have four main mechanisms in enhancing ICB: 1) targeting immune checkpoint inhibitors (ICIs) to tumor foci, 2) increasing tumor immunogenicity, 3) remodeling tumor microenvironment, and 4) pre-sensitizing immune systems. Importantly, advantages of nanotechnology in CRC, such as innovating the mode-of-actions of ICB, modulating intestinal microbiome, and integrating the whole process of antigen presentation, are highlighted in this review. In general, this review describes the latest applications of nanotechnology for CRC immunotherapy, and may shed light on the future design of ICB platforms.
Colorectal cancer (CRC) is one of the leading causes of cancer-related deaths worldwide (1, 2). CRC induced 0.94 million deaths all over the world in 2020 (3). Moreover, more than 3.0 million new CRC cases are predicted in 2040 (2–4). To date, the standard treatment for CRC patients still remains surgical resection, but one-third of them are suffering from post-operative diseases. Challenges in the treatment of CRC are the formation of distant metastasis and the development of drug resistance (4). CRC gradually shows no response to traditional chemotherapeutics, thus novel therapies are urgently needed. Recently, therapy strategies that harness the host immune system against CRC seem to be beneficial for patients, especially those with high mutations (5, 6).
Tumor cells utilize immune checkpoint pathways to dampen T cell activation and evade attack by tumor-specific T cells (7). Immune checkpoint inhibitors (ICIs) competitively bind to checkpoint molecules and block the checkpoint-mediated suppression of the immune system (8). Monoclonal antibodies against checkpoint molecules such as programmed death 1 (PD-1)/programmed death ligand 1 (PD-L1) and cytotoxic T lymphocyte antigen 4 (CTLA-4) have yielded unprecedent success in CRC patients (9, 10). Some small-molecule compounds that directly inhibit PD-1/PD-L1 interaction (11, 12) and its regulatory proteins, such as bromodomain and extra-terminal domain (BET) (13, 14) and Src homology 2 domain containing protein tyrosine phosphatase (SHP2) (15, 16), as well as inhibitors of other immune checkpoints (CD47, CTLA-4, V-domain Ig suppressor of T-cell activation (VISTA)) (17, 18), are also under pre-clinical investigations. However, only highly mutated CRC patients (about 15% of total cases) that are mismatch repair deficient (dMMR) or exhibit high levels of microsatellite instability (MSI-H) can benefit from ICIs. In contrast, majority of CRC patients which are mismatch-repair-proficient (pMMR) or microsatellite instability-low (MSI-L) show negligible response to ICIs. Low tumor mutation and lack of immune cell infiltration are hypothesized as underlying mechanisms in these tumors (19–21). To date, nanotechnology provides powerful devices to detect, diagnose, and treat cancer (22, 23), and is considered as a potential strategy to reverse the immune resistance of CRC (24).
Compared with conventional chemotherapeutics, nanomedicines not only exhibit superior tumoricidal ability and less side effects, but also present the potential to enhance immune checkpoint blockage (ICB) therapies (25) (1): By advancing the delivery of ICIs to CRC tumor sites, nanotechnology can directly enhance checkpoint blockade. During blood circulation, ICIs, especially small molecules, are difficult to accumulate in tumor beds. Macrophage-mediated phagocytosis system attenuates the delivery efficiency of ICIs (13). Even after entering CRC tissues, the amphiphilic cell membranes, lysosome degradation, and subcellular barriers (such as nuclear membranes, mitochondrial membranes, and endoplasmic reticulum membranes) also hinder the efficacy of ICIs (26). On the contrary, micro and nanosized particles can target to tumor tissue passively via the leaky tumor vasculature or actively via binding to receptors on tumor cell surface (27). Some systems can even deliver ICIs to certain subcellular compartments (28) (2). By regulating the cell death pathways, nanotechnology can transform immunologically tolerant cell corpse into immunogenic tumor vaccines, therefore amplifying ICIs efficacy (29, 30). Different from apoptosis, some engineered nano-systems can induce immunogenic cell death (ICD). In contrast to immune escape, tumor cells undergoing ICD will recruit antigen presenting cells (APCs), accelerate immune cells maturation, and initiate tumor antigen specific immune response via releasing various cytokines (31). Some other types of cell death such as ferroptosis, pyroptosis, and necroptosis might also be beneficial for increasing tumor immunogenicity (32–34) (3). By reprogramming immune-suppressive tumor microenvironment (TME), nanotechnology can revive the functions of ICIs (35–37). In order to respond to ICB, sufficient tumoricidal immune cell infiltration is necessary (38). In the progress of tumorigenesis, CRC tissues constantly release chemokines, cytokines, and exosomes as systemic factors to remold extracellular matrix (ECM) and recruit immunosuppressive cells, creating an immune desert milieu, which also terms as “cold” tumors (39, 40). Nanoparticles (NPs) can delivery agents that cut off the immune suppressive pathways, reversing malignant hallmarks in TME and reducing tumor-reside immunosuppressive cells (35, 41) (4). By facilitating the immune response against tumor-exclusive antigen pulses, NPs can potently provoke antigen specific immunity against CRC. As prophylactic or therapeutic interventions, NPs co-deliver antigens and immune-boosting adjuvants to host, pre-sensitizing immune system and systematically generating cytotoxic CD8+ T lymphocytes (CTLs) (42, 43). These four mechanisms are schematically illustrated in Figure 1.
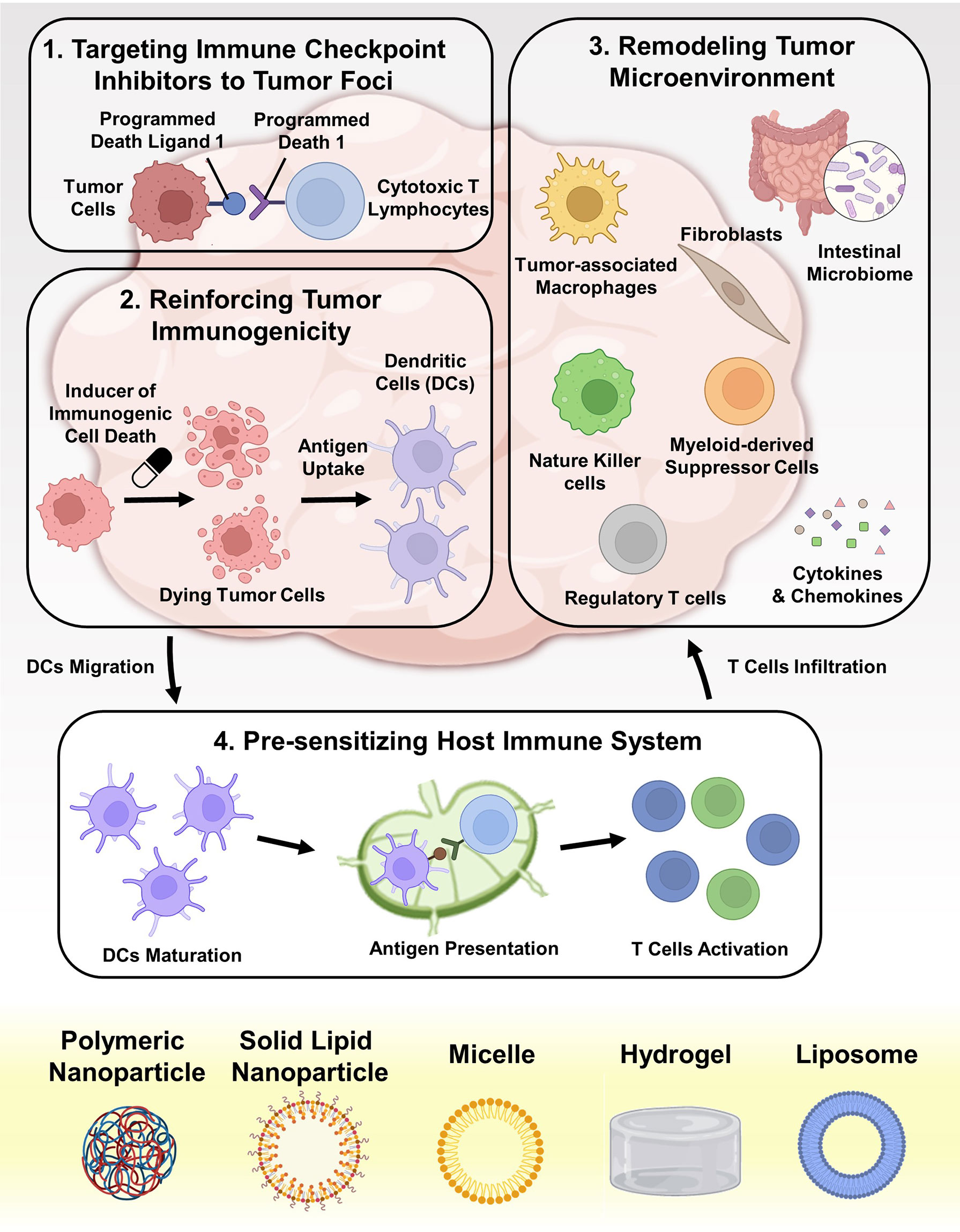
Figure 1 Schematic depiction of advancing ICB in CRC therapy with nanotechnology. The underlying mechanisms can be divided into four aspects: (i) targeted delivering various ICIs (such as antibodies, small molecules, peptides, and SiRNAs) into tumor foci; (ii) reinforcing the immunogenicity of dying tumor cells by cytotoxic agents and other drugs; (iii) remolding the immunosuppressive TME, including eliminating immunosuppressive factors and depleting immunosuppressive cells; (iv) pre-sensitizing the host immune system by delivering tumor vaccines and adjuvants to APCs.
In this review, we summarize the progress of nanotechnology applied to ICB-based CRC treatment in recent three years according to their underlying mechanisms. Particular advantages of nanotechnology in CRC immunotherapy, such as innovating the mode-of-actions of ICB, modulating intestinal microbiome, and integrating the whole process of antigen presentation, are highlighted. This review is expected to clarify the cross-interactions among drugs, materials, and organisms in CRC immunotherapy, and further improve the future design of ICB nanoplatforms.
Although ICIs have shown considerable clinical potency in prolonging survival of patients (44), growing evidences indicate that systemic administration of checkpoint blockade antibodies such as anti-PD-1 antibodies (αPD-1), anti-PD-L1 antibodies (αPD-L1), and anti-CTLA-4 antibodies (αCTLA-4) may cause undesirable autoimmune and inflammatory responses, such as colitis, dermatitis, and hypophysitis (45–47). Once happened, these unbearable adverse effects would seriously weaken therapeutic outcomes, or even fail the whole treatment (48, 49). Nanotechnology offers an attractive approach to bypass these side effects. Antibodies can be conjugated on or encapsulated in natural/artificial drug carriers, therefore avoiding antibody exposure in blood circulation (50, 51). In addition, nanosized drug delivery systems (DDSs) can passively/actively accumulate in solid tumors post systemic administration. Some DDSs can even be locally applied within tumor tissues (52, 53). Collectively, these formulations would remarkably elevate the selectivity of ICIs to tumors.
Checkpoint antibody-loaded NPs have been extensively studied. Early in 2010, Hellstrom et al. leverage functionalized mesoporous silica (FMS) to entrap αCTLA-4 (54–56). High drug loading and sustained drug release were achieved via adjusting the pore size of FMS, which minimize the risk of autoimmunologic toxicity (56). In addition to serving as drug reservoirs, antibody-conjugated NPs play significant roles in tumor theranostics. In colon tumor-bearing mice models, Popovtzer et al. demonstrated that the accumulation level of αPD-L1 conjugated gold NPs in tumors is an important parameter to predict the response of ICB therapy (57). Kang et al. attached methoxy poly (ethylene glycol) (MePEG) and chlorin e6 (Ce6) to Atezolizumab, a PD-L1 antibody, with a cathepsin B responsive linker (58). This immune checkpoint inhibitor nanocomposites (ICI NCs) avoided the ICI exposure in normal tissues, and exhibited tumor-activated fluorescence imaging (FI) and photodynamic therapy (PDT) on murine colon tumor #26 (CT26) tumor xenografts. Schneck et al. developed immuno-switch NPs that modified with αPD-L1 and anti 4-1BB antibodies on their surface (59). These dual-targeting NPs exhibited prolonged tumor retention than soluble free antibodies. After administration, immuno-switch NPs inhibited PD-L1 signal in tumor cells, and concurrently activated 4-1BB signal in CD8+ T cells, activating immune response against murine colon carcinoma 38 (MC38) in a two-pronged pathway.
Solid tumors exhibited higher vascular density than normal tissues, and the wall of blood capillaries are highly leaky. Therefore, blood-circulating macromolecules (above 40 kDa) and NPs tend to extravasate and retain in tumor tissues (60). Moreover, the lymphatic drainage system is dysfunctional in tumors, which prevents the clearance of intra-tumoral NPs (61). This phenomenon is termed as the enhanced permeability and retention (EPR) effect. In general, the tumor accumulation efficiency depends on the blood circulation time of NPs and the tumor volume (62). NPs with prolonged blood circulation as well as decreased clearance by liver and kidney have more opportunity to be transported into tumor capillaries. And larger tumors have more disorganized vasculatures for NPs accumulation. Although this effect has been widely-acknowledged in mice models, its contribution to drug delivery in human is still controversial (62). The carrier with active tumor-homing capability is a better choice for ICI delivery. Wang et al. developed platelets as the carrier for αPD-L1 delivery (63, 64). Platelets have inflammation-targeting ability, and can secret various chemokines to boost T cell immunity, which is very favorable for delivering αPD-L1 into residual microtumors. Platelets binding with αPD-L1 (P-αPD-L1) aggregated in tumor tissue, turning into platelet-derived microparticles (PMPs) for tumor-specific antibody release (64). Treatment of P-αPD-L1 effectively prevented tumor metastasis and recurrence in incomplete tumor resection and thermal ablation (TA) models (Figure 2A).
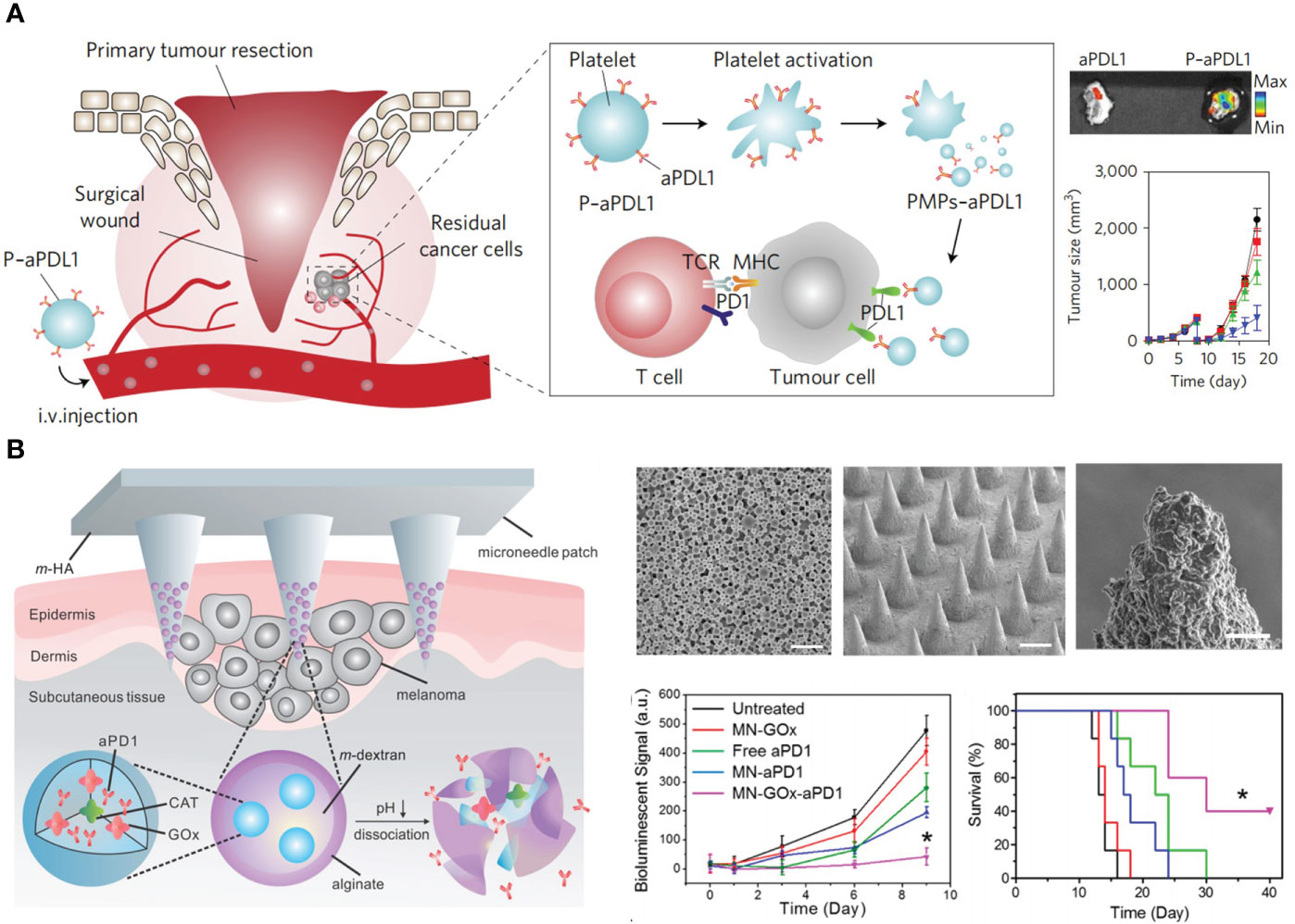
Figure 2 Nanotechnology targets ICIs to tumor foci to advance ICB in CRC therapy. (A) Left: schematic depiction of delivering αPD-L1 (aPDL1) to postsurgical tumor bed by platelets (P-aPDL1). Right top: P-aPDL1 sustainedly accumulated in tumor tissues. Right bottom: P-aPDL1 (blue curve) effectively inhibited tumor growth as compared with PBS-treated group (black curve). Reprinted with permission from reference (64). (B). Left: schematic depiction of the composition of αPD-1 loaded microneedle (MN-GOx-aPD1). Right top: scanning electron microscope (SEM) images of αPD-1 loaded nanoparticles, microneedle patch, and magnified microneedle apex (from left to right). Right bottom: MN-GOx-aPD1 significantly inhibited tumor growth and prolonged mice survival. Reprinted with permission from reference (65). *p<0.05 versus untreated.
Rather than systemic delivery, local application of antibodies within tumoral and peritumoral regions is an excellent approach to obviate the overactivation of the immune system (66). Melief et al. formulated αCTLA-4 into a water in oil emulsion composed of Montanide ISA-51 for subcutaneous (s.c.) injection in the tumor area (67, 68). This sustained-release platform had similar therapeutic consequences with systemic administration, but the dosage was only one-eighth of intravenous injection (i.v.), which contrastingly decreased antibody titers in serum and improved therapeutic safety. Similarly, Hubbell et al. prepared peptide-functionalized ICB antibodies for peritumoral injection (69, 70). A peptide derived from placenta growth factor-2 (PlGF-2123–144) showed super affinity with ECM. They showed the conjugation of PlGF-2123–144 elevated the tissue retention and decreased the plasma concentration of therapeutic antibodies (αPD-L1 and αCTLA-4), reducing the risk of systemic adverse effects, such as autoimmune diabetes. PlGF-2123–144 functionalized antibodies facilitated the infiltration of CD8+ and CD4+ T cells into tumors, resulting in delayed growth of primary and distance tumors. Gu et al. reported a microneedle (MN) patch for the transdermal delivery of αPD-1. Glucose oxidase (GOx) and αPD-1 were co-encapsulated into pH-sensitive dextran NPs, and these NPs were further loaded into hyaluronic acid MN arrays (65). GOx catalyzed the conversion of blood glucose to gluconic acid, forming a local acidic milieu for the self-disintegration of dextran NPs, leading to a sustained release of αPD-1 for three days. This simple and biocompatible platform could also be applied to co-deliver αPD-1 and αCTLA-4, achieving synergic antitumor effects (Figure 2B).
As the biological drugs, administration of antibodies may lead to infusion reactions and anti-drug antibodies (71, 72). Peptide-based ICIs, especially antagonists against PD-1 and PD-L1, are more preferable options. In comparison with antibodies, peptide drugs have deeper tumor penetration due to their low molecule weight (Mw). Given the lacking of Fc fragments, peptides exhibited lower immunogenicity and better safety. Moreover, peptide ICIs are more stable in structure, and cost less in manufacture, storage, and drug administration (73, 74). Even though, peptide ICIs have weaknesses including insufficient affinity, short body circulation, and a lack of tumor selectivity, which requires to be solved by suitable delivery systems. Kim et al. designed ferritin nanocages (PpNF) that displayed PD-L1- binding peptide (CLQKTPKQC) with multivalency on their surface (75). PpNF specifically accumulated in tumor tissues, and restored the antitumor activities of T cells. Notably, PpNF loaded with doxorubicin (DOX) had better tumor inhibition effect than αPD-L1 in CT26 tumor models (75). Huang et al. synthesized a liner polymer-drug conjugate of PD-L1 antagonistic peptide MSP (CPLGVRGSGQYASYHCWCWRDPGRSGGSK) (76). This polymer (P-MSP-DMA) intertwined with a mitochondria-targeted polymer-drug conjugate (P-D-R8MTS) via electrostatic interaction to form a nanocomplex (SNV). SNV specifically dissociated in tumors in response to the charge reversal of dimethylmaleic anhydride (DMA) group triggered by acidic TME. This nanoplatform integrated PD-L1 blockade with mitochondria-targeted induction of ICD, resulting in considerable inhibition of tumor growth and metastasis (76). NPs incorporated with PD-L1-binding peptides can also be combined with photothermal therapy (PTT), which was exemplified by Zhang et al. and You et al. (50, 52). Zhang et al. conjugated a PD-L1 antagonistic peptide (NYSKPTDRQYHF) on the surface of IR780-loaded NPs (aNP@IR780) (50). And You et al. co-encapsulated aN anti-PD-1 peptide ((SNTSESF)2 KFRVTQLAPKQIKE-NH2) and the hollow gold nanoshell (HAuNS) into NPs (AA@PN) (52). Both strategies simultaneously triggered tumor ablation and blocked PD-1/PD-L1 interaction between tumor cells and T cells, exhibiting an abscopal effect to suppress distant tumor growth in a bilateral CT26 tumor model.
Besides peptides, nuclei acid-based therapeutics against checkpoint molecules is another therapeutic alternative for ICB therapy. Wang et al. used poly (ethylene glycol)-block-poly (d,l-lactide) (PEG-PLA) and N-bis(2-hydroxyethly)-N-methyl-N-(2-cholesteryloxycarbonyl aminoethyl) ammonium bromide (BHEM-Chol) to encapsulate CTLA-4 siRNA (77). The prepared NPs (NPsiCTLA-4) were capable to deliver siRNA cargos to both CD8+ and CD4+ T cells in vivo, facilitating their activation and proliferation. Ahn et al. prepared poly (lactic-co-glycolic acid) (PLGA) NPs to co-loading PD-1 siRNA and PD-L1 siRNA (siRNA@PLGA) (78). In the MC38 tumor model, they found the concurrent silencing of PD-1 and PD-L1 by siRNA@PLGA had better antitumor effect than single silencing of each one. Han et al. reported a nanoplatform with a novel PD-L1 binding aptamer, PL1 (51). In their design, PL1 single-stranded oligonucleotides were hybridized with folic acid (FA) and siRNA against proprotein convertase subtilisin-kexin type 9 (PCSK9) to obtain DNA tetrahedral nanoparticles (TDN-FA/PL1/Pcsk9-siRNA). TDN-FA/PL1/Pcsk9-siRNA were guided to CT26 CRC cells by FA, ensuring the synergy between PD-L1 blockade and Pcsk9 downregulation.
More than merely serving as a carrier for ICIs delivery and a platform for combinatory tumor immunotherapy, nanotechnology provides an opportunity to innovate the mode-of-action of ICB therapeutics. For example, Yang et al. leveraged lysosome-mediated receptor degradation to realize a durable PD-L1 downregulation. PD-L1 peptide antagonists (PPA, NYSKPTDRQYHF) were conjugated to the linear polymer composed of N-2 hydroxypropyl methacrylamide (HPMA) (79). By this way, PPA were transformed into a multivalent polymer-peptide antagonist against (MPPA). MPPA could gather and crosslink PD-L1 on tumor cell surface, biasing their trafficking to lysosome degradation and preventing their recycling to cell surface. This polymer-assisted receptor crosslinking strategy produced a long-lasting elimination of PD-L1 checkpoint, and strongly facilitated polymer-epirubicin (EPI) conjugate (KT-1) mediated chemo-immunotherapy. Nanotechnology may foster the druggability of ICI-like agents (79). Huang et al. designed a engineered PD-L1 trap as a novel ICB protein (53). In order to address the side toxicities of systemic PD-L1 blockade, the coding plasmid of PD-L1 trap was encapsulated in lipid-protamine-DNA nanoparticles (LPD). This system specifically distributed in tumor tissues, enabling the local production of the PD-L1 trap. In a CT26 murine colon tumor model, this strategy not only improved the tolerance of ICB therapy without inducing Th17 cells accumulation in spleen, but also achieved potent PD-L1 inhibition to potentiate oxaliplatin (OXA)-mediated chemotherapy (53).
In CRC, highly immunogenic tumors showed relatively good response to ICB-based immunotherapy (1, 80, 81). In contrast, the effect of ICIs in tumors with low immunogenicity requires further improvement (82). Recently, ICD has been reported to transform originally immunotolerant cell debris into immunogenic vaccines (83). ICD induced by anthracyclines was first reported by Guido Kroemer et al. in 2007 (84). Different from immune tolerant apoptosis, ICD can provoke the immune system to generate response against antigens from dead tumor cells, which is also known as “bystander effect” (85). Briefly, tumor cells undergoing ICD expose calreticulin (CRT) on the outer leaflet of the cell membrane, secreting adenosine triphosphate (ATP) and releasing high mobility group box 1 (HMGB1) into extracellular microenvironment (29, 30, 86). These markers facilitate APCs recruitment, antigen engulfment and presentation during immune initiation (87).
DOX is often administrated along with other chemotherapeutics to elevate efficacy. Although DOX is found to facilitate effector T cells infiltration and synergize with ICIs via ICD induction, applications of free DOX are still hindered by cardiac toxicity and unsatisfactory tumor accumulation (29). Jeffrey A. Hubbell et al. reported a collagen-binding serum albumin platform for advanced colon carcinoma therapy (88). Serum albumin (SA) based carrier can passively deliver drug to tumor sites via the extravasation through pathological vasculature. To further endow SA active targeting capacity, a collagen-binding domain (CBD) was fused recombinantly to give CBD-SA. Lastly, DOX was loaded to CBD-SA via a pH-sensitive linker (DOX-CBD-SA). Surprisingly, when combing with αPD-1, a complete eradication of MC38 colon carcinoma was observed. To understand the underlying mechanism, T cells and natural killer (NK) cells in treated tumors were extracted. The numbers of CD8+ T cells, CD4+ T cells and NK cells per unit tumor mass increased after DOX-CBD-SA treatment. The increased tumor-infiltrating lymphocytes subsequently potentiated therapeutic efficiency of immune-checkpoint blockade. Generally, DOX-CBD-SA can potently kill tumor cells and simultaneously stimulate host antitumor immunity, decreasing adverse events. To further improve the immunogenicity, subcellular level targeting strategy was considered. Mitochondria is one of the most important organelles and serves as the source of damage associated molecular patterns (DMAPs) such as ATP, heat shock protein 70 (HSP70), and HSP90. The released DMAPs facilitate the presentation of tumor-associated antigens. Zhan et al. engineered a mitochondria-targeted polymeric nanoparticle (R848@cRGD-PDCS) (89). Under near-infrared irradiation exposure, mitochondria were destroyed by photothermal-mediated hyperthermia, causing the release of tumor-associated antigens and DMAPs. αPD-L1 therapy showed limited inhibitory effects in tumor growth, but the combination with R848@cRGD-PDCS (under irradiation) exhibited favorable ability to eradicate primary tumors and prevent metastasis (Figure 3A).
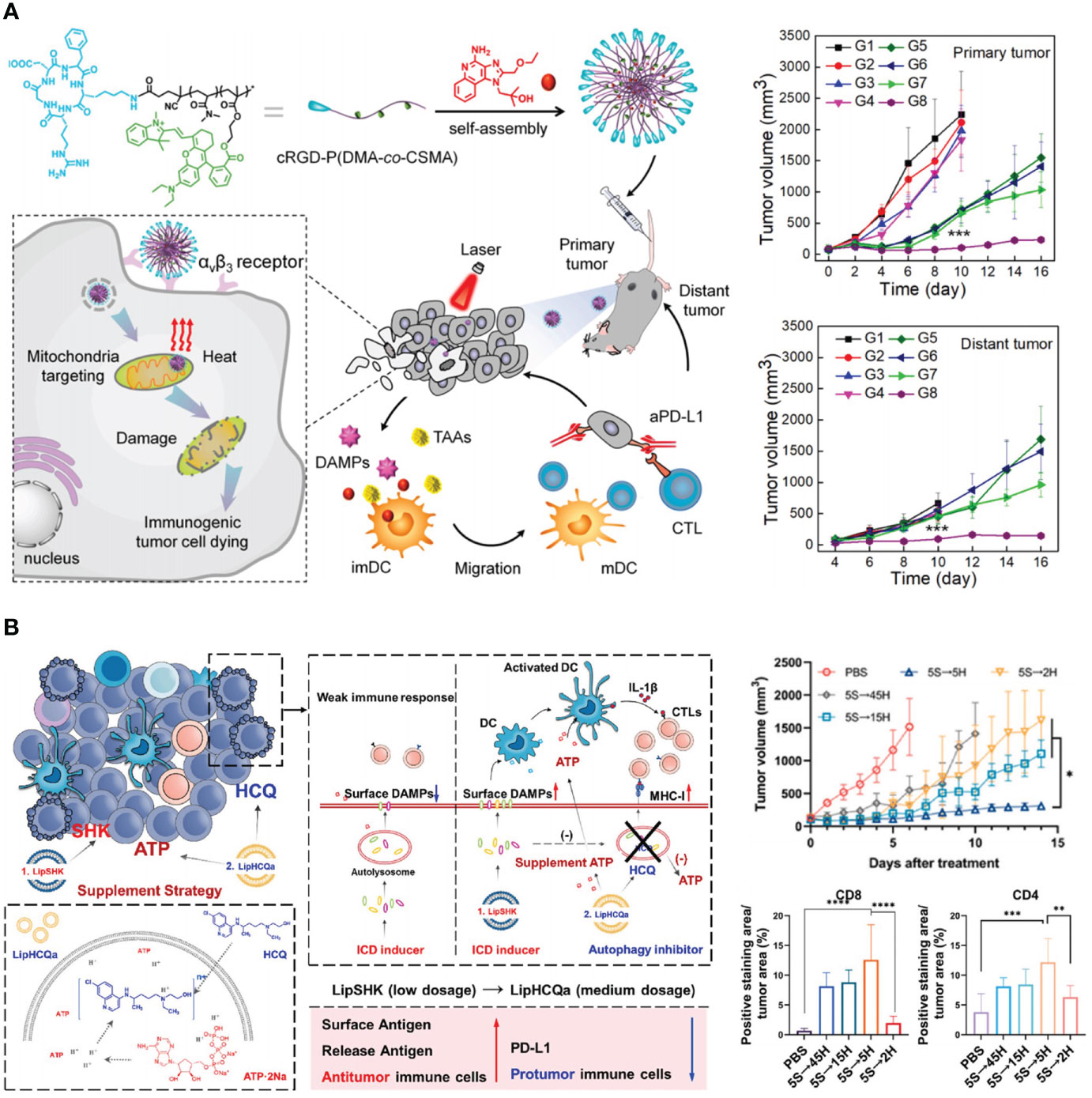
Figure 3 Nanotechnology reinforces tumor immunogenicity to advance ICB in CRC therapy. (A) Left: schematic depiction of mitochondria-targeted and photo-activated nanoparticles (R848@cRGD-PDCS) that triggered ICD to potentiate ICB therapy. Right: R848@cRGD-PDCS (G8) inhibited the growth of both primary tumor and distant tumor in combination with αPD-L1. Reprinted with permission from reference (89). ***p<0.001 versus G1. (B) Left: schematic depiction of the mechanism of recovering tumor immunogenicity by autophagy inhibition. Right: impacts of different autophagy inhibition therapies on tumor growth and the percentage of CD8+ and CD4+ T cells. Reprinted with permission from reference (90). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Chemotherapeutics (such as OXA and DOX) and PDT were reported to induce ICD synergistically. Lin et al. proposed a core-shell nanoscale coordination polymer (NCP@pyrolipid) which not only directly eliminated tumor cells but also promoted the checkpoint blockade immunotherapy (91). In the study, OXA in the core and the photosensitizer pyropheophorbide-lipid conjugate (pyrolipid) in the shell synergistically eradicate cancer cells, resulting in robust ICD and subsequent abscopal effects. Moreover, after integrating PD-L1 blockade with NCP@pyrolipid, tumor regression was observed in both light-irradiated primary tumors and distant tumors without light-irradiation, indicating that a potent tumor-specific immunity was evoked. The authors observed increased portions of antigen-specific CTLs in the CRC-bearing mice injected with NCP@pyrolipid (with irradiation) plus αPD-L1. The immunogenic environment induced by both OXA and PDT remarkably enhanced PD-L1 therapy via spurring systemic antitumor immune response. Yu et al. reported an prodrug-based polymeric nanoparticle to realize optimal administration of OXA combining with PTT (92). Besides, fluorescence -guided PTT can further enhance tumor immunogenicity and release drug in a spatiotemporally controllable way. A donor-spacer-acceptor-space-donor (D-S-A-S-D) type fluorophore was farther inserted to improve immunogenicity and amplify the efficacy of αPD-L1 therapy. This combinatory chemo/photothermal therapy with PD-L1 blockade (PBOXA@TQTCD+L-αPDL1) was tested in vivo. Results revealed that the combinatory therapy not only inhibit tumor growth much more potently than αPD-L1 therapy alone, but also improve the survival rate of mice with tumor. When it comes to the tumor recurrence inhibition, the central memory CD8+ T cells (TCM) in spleen representing long-term immune memory was analyzed. TCM ratio in the combinatory therapy was at least two times higher than that of αPD-L1 alone, indicating the activation of a long-term immune surveillance against tumor recurrence. This combinatorial therapy might enlighten clinical CRC management.
However, the PDT or PTT is a localized therapy and restricted by light penetration. Alternatively, Lin et al. reported another tactic that using reactive oxygen species (ROS) based chemotherapeutic to induce potent ICD and synergize with OXA. The author engineered self-assembled coordination polymer nanoparticles (OxPt/DHA) loading OXA in the core and ROS-generating dihydroartemisinin (DHA) in the shell for CRC treatment (93). In a tumor rechallenge experiment, mice vaccinated with OxPt/DHA-treated cells showed a potent immune resistance against live MC38 cells and no tumor formation was observed. The efficacy of OxPt/DHA combining with α-PD-L1 blockade therapy was tested in tumor models of CT26 and MC38 on immunocompetent BALB/c and C57BL/6 mice, separately. In both CT26 and MC38 models, the α-PD-L1 therapy alone failed to control tumor growth. In the contrary, all of the tumors treated with OxPt/DHA plus α-PD-L1 regressed and ultimately disappeared on days 40~50. Until 120 days, no recurrence was found. Results revealed that OxPt/DHA is a potential clinical candidate to synergize with ICIs.
Besides inducing ICD in situ in tumor sites to enhance immunogenicity and amplify immune checkpoint blockade (ICB) therapy, immunogenically dying tumors cells themselves can also be transformed into a powerful platform for cancer vaccination. Moon et al. have manufactured dying tumor cells surface-modified with adjuvant-contained NPs (94). Results revealed that dying tumor cells undergoing ICD could be further filled with adjuvant nano-depots to successfully initiate antigen cross-presentation by dendritic cells and activate potent antigen-specific CD8α+ T cells in mice model bearing CRC. Additionally, the combinatory regimen using this whole tumor-cell vaccination and immune checkpoint inhibition resulted in a complete tumor eradication in about 78% of mice inoculated with CT26. A long-term immunity was also observed, indicating the potential to prevent tumor recurrence. This strategy might shed light on “personalized” therapy which is tailored according host’s own tumor cells. The inflammatory microenvironment after surgery and residual tumor “seeds” were responsible for post-operative metastasis. To solve this dilemma, Li et al. embedded autologous cancer cells succumbing to ICD and anti-inflammatory drug dexamethasone in hydrogel, the hydrogel could be injected into a resection site, in which it was rapidly solidified and gradually degraded (95). The dying cells provided a whole array of tumor-associated antigens and became highly immunogenic vaccines which enabled antigen specific immunization. After combining with αPD-L1 therapy, a complete tumor regression was observed, which might be attributed to their complementary functions to evocate and unleash tumoricidal T cells. This strategy provides a novel option for inhibiting metastasis after surgery.
Autophagy refers to the process by which cells degrade their constituents by autophagosomes. Autophagy is necessary in sustaining and modulating cell homeostasis. In addition, autophagy facilitates the release of ATP from lysosome in ICD inducing, promoting antitumor immune response. However, autophagy can destroy tumor-associated antigens, therefore attenuating antitumor immunity. To overcome this difficulty, Wang et al. designed a liposome named as LipHCQa which encapsulated shikonin (ICD inducer), hydroxychloroquine (autophagy inhibitor), and ATP for the treatment of colon cancer (90). This compensatory liposome showed enhanced immune infiltration when compared with shikonin loaded liposome alone, indicating the importance of blocking autophagy on ICB amplification (Figure 3B).
Specific series of intracellular suicide process was named as programmed cell death (96, 97). In the past few decades, apoptosis had been assumed as the sole modality of programmed cell death (84, 98). Recently, several other pathways of programmed cell death were identified, such as ferroptosis and pyroptosis (99, 100). These particular cell death pathways might be used to enhance the immunogenicity of tumor cells and synergize with ICB (101). Since firstly proposed in 2012 by Stockwell and co-workers, ferroptosis has attracted numerous attentions in the field of oncology and biochemistry (102, 103). Ferroptosis is an iron and ROS dependent dell death. Cells undergoing ferroptosis showed increased lipid peroxidation products and ROS that is derived from iron metabolism. Han et al. designed core-shell nanoparticles (ZnP@DHA/Pyro-Fe) loaded with a cholesterol derivative of dihydroartemisinin and pyropheophorbide-iron (Pyro-Fe) to potentiate CRC immunotherapy via inducing ferroptosis. ZnP@DHA/Pyro-Fe treated cancer cells showed increased DAMPs release and result into intra-tumoral immune cell infiltration (104). Further combination with αPD-L1 checkpoint blockade led to better therapeutic effect. Different from caspase-dependent apoptosis, necroptosis is featured by expanded cell volume, organelle swelling, cell membrane fracture, and leaking of intracellular components. Nowadays, mixed-lineage kinase domain like protein (MLKL), receptor interacting protein-1 (RIPK1), and RIPK3 pathways were thought to be essential in tumor necrosis factor-α (TNF-α) mediated necroptosis. Compared with poorly immunogenic apoptosis failing to activating antitumor immunity, necrosis showed great potential in priming immune response due to the increase immunogenicity (105). Sun et al. prepared dimethyl fumarate loaded star-PCL-azo-mPEG (sPCEG-azo) polymeric micelles (106). The micelles are colon-targeted and induce necroptosis in colon cancer cells via a mechanism characterized with increased ROS. The elevated ROS generation result in immunogenicity and contribute to antitumor immunity, which may further augment ICB. Another necroptosis-inducible nanoparticle was reported by Park et al. The nanobubbles (NBs) contains Ce6 as the sonosensitizer and perfluoropentane as the gas precursor (107). After ultrasound exposure, NBs could disintegrate plasma membrane and lead to damage-associated molecular patterns release, inducing acoustic cavitation mediated necroptosis and ROS-mediated tumor regression. The NBs promoted antitumor immunity via accelerating dendritic cells maturation and CD8+ T cells activation. Further combinatory regimen including PD-L1 blockade plus NBs even led to complete eradication of primary CT26 tumor and metastasis.
TME is an intricate milieu including tumor and immune cells, bacteria, as well as multiple soluble signal mediators (108, 109). All of them contribute to the distinct physiological characteristics (hypoxia, acidity, inflammation, and immune escape) of TME. Mounting evidence reveal that the heterogeneity of TME is an important factor for the low responsiveness of ICB therapies, and reversing immunosuppressive TME is very promising to overcome ICI resistance (110). Considering the close interaction between components in TME, nanoplatforms that counteract these suppressors in multi-pronged ways are very promising (36, 111, 112). Nanoplatforms that potentiated ICB against CRC by modulating TME are summarized in Table 1.
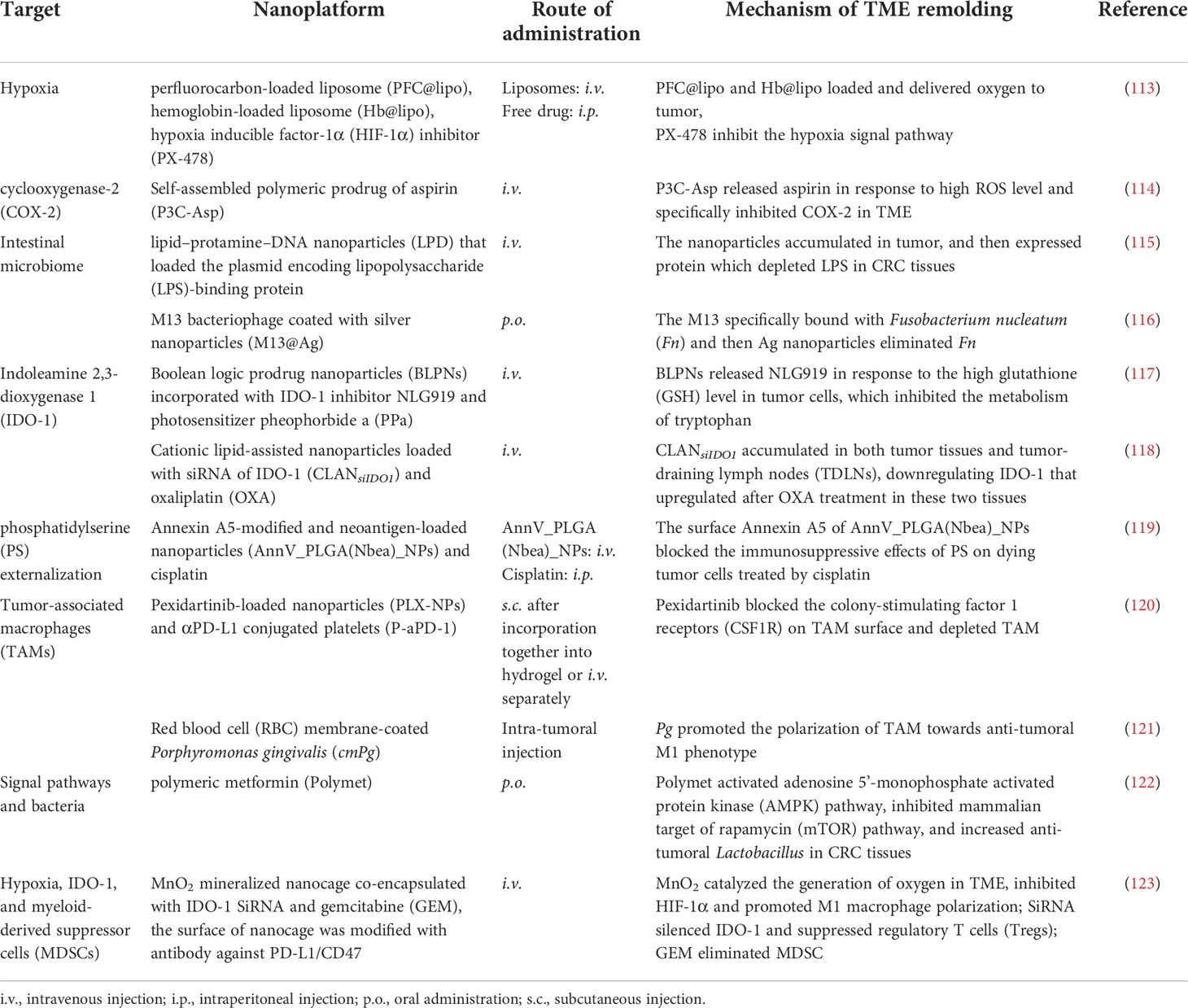
Table 1 Summary of nanoplatforms that remolds TME to advance ICB in CRC therapy.
Hypoxia reduced the therapeutic efficiency of ICIs in many aspects. It increased the expression of PD-L1 and CTLA-4 via hypoxia inducible factor-1α (HIF-1α) pathway, and weakened antigen presentation of APCs (124, 125). You et al. adopted three strategies to alleviate hypoxia (113): (i) directly deliver oxygen into tumors by a perfluorocarbon-loaded liposome (PFC@lipo); (ii) directly deliver oxygen into tumors by a hemoglobin-loaded liposome (Hb@lipo); and (iii) indirectly inhibit HIF-1α in tumors by a small molecular inhibitor PX-478. They found systematic administration of Hb@lipo was the most ideal strategy to combine with αPD-1 for the treatment of murine triple-negative breast cancer (4T1) and CT26 tumors. Due to the hypoxia milieu, tumor cells are glycolytic and produce plenty of lactate as the metabolite, resulting in anergy of tumor-infiltrated immune cells. Dhar et al. developed a mitochondria-targeted NPs (T-Mito-DCA-NPs) for the delivery of dichloroacetate (DCA), an inhibitor of pyruvate dehydrogenase kinase 1 (PDK1) (126). This formulation selectively inhibited tumoral glycolysis without affecting immune cells. As a result, T-Mito-DCA-NPs significantly elevated the intra-tumoral infiltration of CD8+ and CD4+ T cells and downregulated the expression of checkpoint molecules including PD-1, CTLA-4, LAG3, and Tim3 on their surface. Combination of mitochondria-targeted DCA and αPD-1 effectively improved the infiltration of CD8+ and CD4+ T cells in CT26 tumors.
Inflammation is an important hallmark in TME that promote tumorigenesis, expansion, metastasis, and immune escape (127). Tumor cells highly expressed cyclooxygenase-2 (COX-2) and secreted a large amount of prostaglandin E2 (PGE2) to recruit myeloid-derived suppressor cells (MDSCs) (128, 129). MDSCs generate several immunosuppressive cytokines, such as ROS, transforming growth factor-β (TGF-β), and interleukin 10 (IL-10), leading to failure of ICB therapy (130–132). Chen et al. synthesized a self-assembled polymeric prodrug (P3C-Asp) of aspirin, a classical non-steroidal anti-inflammatory drug (NSAID) (114). P3C-Asp released aspirin in response to high ROS level in tumor tissues, decreasing PGE2 secretion and reversing tumor immunosuppression. Combination therapy of P3C-Asp+αPD-L1 eradicated CT26 tumors in 100% of mice.
Different from other cancers, the progress of CRC is closely associated with intestinal microbiome. The species and abundance of intestinal microbiome highly affected the balance in GI track, contributing to several gut diseases, such as colitis, fibrosis and CRC (133). Fusobacterium nucleatum, Bacteroides fragilis, and Escherichia coli are the main pathogenic bacteria in gut that promote tumor progression and hinder the responsiveness of CRC to αPD-L1 therapy (134–136). Bacteria colonized in cancerous GI tracts produced massive endotoxin, also known as lipopolysaccharide (LPS). LPS accelerated the growth and liver metastasis of colorectal tumors via Toll-like receptor 4 (TLR4) and nuclear factor kappa-B (NF-κB) pathway (137–139). Moreover, chemotherapeutic agents are able to disrupt the mucus barrier in gastrointestinal (GI) track, facilitating the colonization and invasion of gut bacteria (140). Huang et al. designed an LPS-binding fusion protein as the trap to deplete LPS in orthotopic CRC tissues (115). For tumor selectivity, they encapsulated the coding sequence of LPS trap protein into lipid–protamine–DNA nanoparticles (LPD). This system passively accumulated in CT26 tumors, enabling LPS trap protein expressed within malignant tissues. They found LPS trap treatment elevated the infiltration of T cells, which favored ICB therapy. Combinatory treatment of the LPS trap and αPD-L1 not only retarded the growth of orthotopic CT26-FL3 tumors but also inhibited their spontaneous liver metastasis. Zhang et al. identified Fusobacterium nucleatum (Fn) as pro-tumoral gut bacteria that restricted T cell infiltration and enriched MDSCs in CRC tissues (116). They screened a Fn-binding M13 bacteriophage by phage display technology and modified antibacterial silver nanoparticles (AgNP) on its surface. The obtained M13@Ag could specifically eliminate Fn in GI track to reduce MDSCs in TME, and facilitate antigen presentation due to its intrinsic immunogenicity. In orthotopic CRC models, combination therapy of M13@Ag and αPD-1 significantly improved the overall survival of mice (Figure 4A).
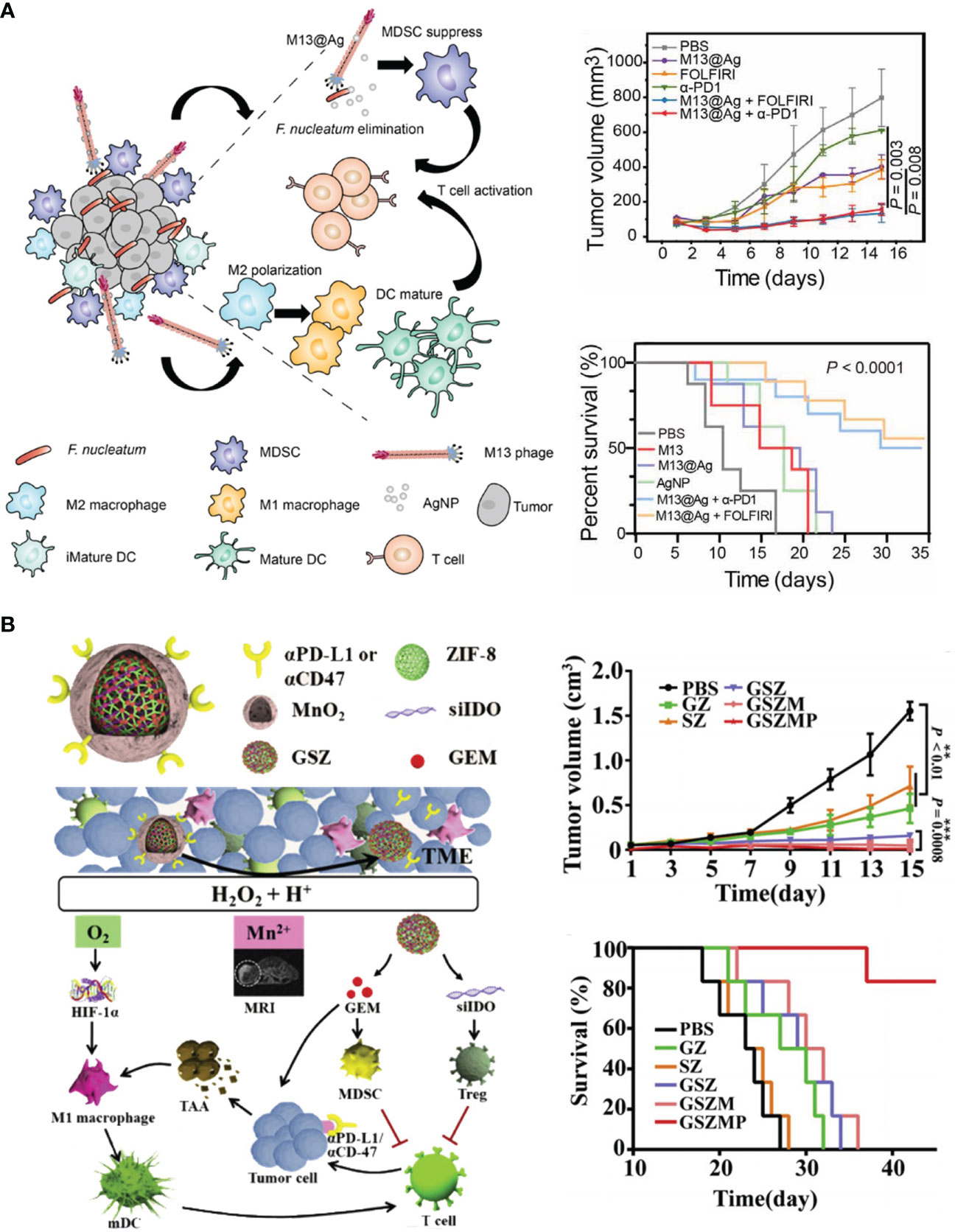
Figure 4 Nanotechnology remolds TME to advance ICB in CRC therapy. (A) Left: schematic depiction of engineered bacteriophage (M13@Ag) that regulated intestinal microbiome to modulate TME against CRC. Right: M13@Ag significantly inhibited tumor growth and prolonged mice survival in combination with αPD-1. Reprinted with permission from reference (116). (B) Left: schematic depiction of the composition of versatile nano-modulator (GSZMP) and its mechanism in potentiating ICB therapy. Right: GSZMP potently inhibited tumor growth and prolonged mice survival. Reprinted with permission from reference (123).
Indoleamine 2,3-dioxygenase 1 (IDO-1) is highly overexpressed on tumor cells (141, 142). It is a rate-limiting enzyme in the kynurenine pathway that convert tryptophan to kynurenine (143). The accumulation of kynurenine in TME contributed to dendritic cells (DCs) deactivation, CTL apoptosis, and regulatory T cells (Tregs) increment (144, 145). Due to its key role in tumor immunosuppression, IDO-1 is termed as the “metabolic immune checkpoint” (118). Inhibiting IDO-1 have been demonstrated to potentiate ICD-based chemo/photo-immunotherapy and PD-1/PD-L1 blockade therapy, but the therapeutic consequence is strongly relied on well-designed delivery strategies. Yu et al. developed boolean logic prodrug nanoparticles (BLPNs) that logically gated by matrix metalloproteinase (MMP), acid, and glutathione (GSH) to release photosensitizer pheophorbide a (PPa) and IDO-1 inhibitor NLG919 to treat CT26 tumors (117). PPa induced ICD to trigger T cell response, which was further amplified by NLG919. Wang et al. accomplished the concurrent inhibition of IDO-1 in both tumors and tumor-draining lymph nodes (TDLNs) by siRNA-loaded cationic lipid-assisted nanoparticles (CLANsiIDO1) (118). They found OXA treatment aggravated IDO-1 overexpression in tumors and TDLNs, which was in accordance with other immune checkpoints like PD-L1 and CTLA-4. CLANsiIDO1 accumulated in TDLNs and tumors, downregulated IDO-1 in both tissues, and improved tumor inhibition by OXA in the CT26 colon cancer model.
Chemotherapy triggers phosphatidylserine (PS) externalization on the surface of tumor cells undergoing apoptosis (146, 147). In line with canonical immune checkpoints, the exposed PS consolidated the immunosuppressive TME, restricted the phagocytosis of APCs and upregulated the expression of PD-L1 (148, 149). Recently, Park et al. developed annexin A5-labeled NPs (AnnV_PLGA_NPs) as the inhibitor to this innate immune checkpoint. Mutant neoantigen peptides (Nbea, PAPRAVLTGHDHEIVCVSVCAELGLVI) were loaded in NPs (AnnV_PLGA(Nbea)_NPs) to elicit antigen-specific antitumor immunity (119). In company with cisplatin (Cis)-mediated chemotherapy, AnnV_PLGA(Nbea)_NPs spurred the infiltration of immune-activate cells and the secretion of pro-inflammatory cytokines, while depleting immune-suppressive MDSCs and Tregs and decreased the production of anti-inflammatory cytokines at the same time. The immunostimulatory effect of AnnV_PLGA(Nbea)_NPs can be amplified by αPD-L1, and the triple-therapy of Cis + AnnV_PLGA(Nbea)_NPs + αPD-L1 resulted in noticeable rejection of CT26 tumor growth.
Tumor-associated macrophages (TAMs) represented a large population in intra-tumoral immunosuppressive cells (150). Generally, TAM can be divided into anti-tumoral M1 phenotype and pro-tumoral M2 phenotype via their different markers. M1 macrophages not only killed tumors in an innate manner, but also presented tumor antigen to T cells and activated adaptive tumor immunity (151). M2 macrophages are the major TAMs in immunosuppressive tumors, such as triple-negative breast cancer (TNBC) and CRC (152). They affected the function of tumor and immune cells in TME by secreting vascular endothelial growth factor (VEGF), IL-10, TGF-β, and arginase-1 (153). There are three paradigms for targeting TAMs: (i) inhibiting TAMs recruitment, (ii) depleting pre-existing TAMs, and (iii) re-educating TAMs from pro-tumoral M2 macrophages to anti-tumoral M1 phenotype (151, 152). Results of Hu et al. revealed that locally depleting TAMs in postsurgical tumor beds by Pexidartinib-loaded nanoparticles (PLX-NPs) created an appreciable condition for the local and systemic PD-1 blockade therapy (120). Pexidartinib eliminated TAMs by blocking colony-stimulating factor 1 receptors (CSF1R) on TAM surface. Alginate hydrogel incorporated with PLX-NP (PLX-NP@Gel) decreased F4/80+ macrophages and increased IFN+CD8+ CTLS in tumor beds. αPD-1 conjugated platelets (P-aPD-1) were co-encapsulated into hydrogel for local implantation (PLX-NP-P-aPD-1@Gel) or systematically injected (PLX-NP- @Gel+P-aPD-1). Both regimes considerably inhibited post-surgery tumor recurrence in murine melanoma (B16F10), CT26, and 4T1 tumor models. Conventional TAM-modulating strategies employed small molecules, peptide, antibodies, and nuclei acids, while Zhou et al. reported a bacteria-based approach to repolarize TAMs (121). They prepared a red blood cell (RBC) membrane-coated formulation of Porphyromonas gingivalis (cmPg). Porphyromonas gingivalis (Pg) not only promoted the conversion of TAMs to M1 phenotype, but also secreted melanin for tumor PTT. With the help of PD-L1, cmPg retarded the growth of primary and secondary CT26 colon tumors.
Because of the intricate crosstalk between various immune cells, it is plausible to manipulate versatile targets by one nanoplatform. Huang et al. developed an orally delivered polymeric metformin (Polymet) that notably reinforced αPD-L1 therapy (122). The underlying mechanism of Polymet involved reprograming the immunosuppressive TME via adenosine 5′-monophosphate activated protein kinase (AMPK) pathway and mammalian target of rapamycin (mTOR) pathway, as well as lifting the abundance of anti-tumoral Lactobacillus in CRC tissues. In comparison with this single-mode therapy, it is more preferable to design multi-modular nanodrugs that counteracted several immune suppressors in TME. To this end, Jiang et al. developed a versatile nano-modulator, GSZMP. SiRNA targeting IDO-1 (siIDO) and gemcitabine (GEM) were co-encapsulated in a nanocage composed of Zinc 2-methylimidazole (ZIF-8) metal organic frameworks (MOFs) (123). The surface of drug-loaded nanocage was further tattooed with MnO2 mineralization and electrostatically modified with αPD-L1 or anti-CD47 antibody (αCD47) for the treatment of TNBC and colon adenocarcinoma (COAD), respectively. MnO2 catalytically generated O2 to alleviate hypoxia in TME, which promoted the repolarization of TAMs into M1 phenotype. GEM selectively depleted MDSCs, and siIDO inhibited the activity of Tregs. Overall, GSZMP reversed the “cold” TME in a multi-pronged pathway, which effectively potentiate ICB therapy (Figure 4B).
Tumor vaccines exhibit unique prophylactic effect against tumorigenesis and have showed combinatory therapeutic effect with immune checkpoint therapies (154–156). The aim of them is to pre-sensitize the immune system before tumor expansion and generate sufficient antigen-specific T cells, which creates a condition for subsequent invigoration of these T cells (157). Advantages of tumor vaccines can be summarized as follows: (i) tumor vaccines greatly decrease the risk of tumorigenesis; (ii) tumor vaccines induce systemic antitumor immunity that is able to attack undetectable tumor foci and metastasis (158); (iii) tumor vaccines elicit durable immune surveillance that against tumor recurrence for a long time (159); (iv) tumor vaccines can be personalized by using autologous components, which is more favorable to address the mutation of tumor antigens (158, 160). Nowadays, several tumor-exclusive neoantigens have been identified for vaccine design, but their immunostimulatory efficiency are still limited in in vivo studies (161). Codelivery with adjuvants will potently enhance T-cell-spurring by tumor vaccines, wherein the contrasting different drug properties between antigen and adjuvant should be concerned (159, 162). Most tumor-specific antigens are water-soluble macromolecules, such as peptides, proteins, and long chain ribonucleic acids. TLR agonists are a class of well-studied adjuvants with multiple drug forms: polyinosinic-polycytidylic acid (poly I:C, double-stranded RNA analogue, TLR3 agonist), monophosphoryl lipid A (MPLA, lipid, TLR4 agonist), Resiquimod (R848, hydrophobic small molecule, TLR7/8 agonist), and cytosine phosphate guanidine (CpG, oligodeoxynucleotides, TLR9 agonist) (163, 164). Therefore, it is highly challenging to synchronize the pharmacokinetics of antigens and adjuvants in their codelivery.
Chen et al. synthesized a bi-adjuvant neoantigen nano-vaccine (banNV) that co-loaded peptide neoantigen Adpgk and two adjuvants R848 and CpG in one nanoplatform (165). Because of the activation of two TLR pathways, immunization with bi-adjuvant banNV elicited stronger T cell response than single adjuvant vaccines. As a result of immune activation, PD-1 was profoundly upregulated on Adpgk-specific CD8+ T cells, which led to the incomplete MC38 tumor regression after banNV treatments. Coordination withαPD-1 significantly improved the therapeutic outcomes of this bi-adjuvant vaccine therapy (Figure 5A). Lee et al. demonstrated the synergism of PD-L1 blockade with DC vaccine (168). They developed an immunoadjuvant nanocomplex (PSPEI-PIC) consisted of polysorbitol-co-PEI (PSPEI) polymer and poly(I:C). PSPEI-PIC assisted DC vaccines to activate tumor-specific T cells, but undesirably increased PD-L1 expression in tumor beds. Accordingly, combination of PDPEI-PIC, DC vaccine, and αPD-L1 achieved considerable therapeutic efficacy on MC38 tumor model.
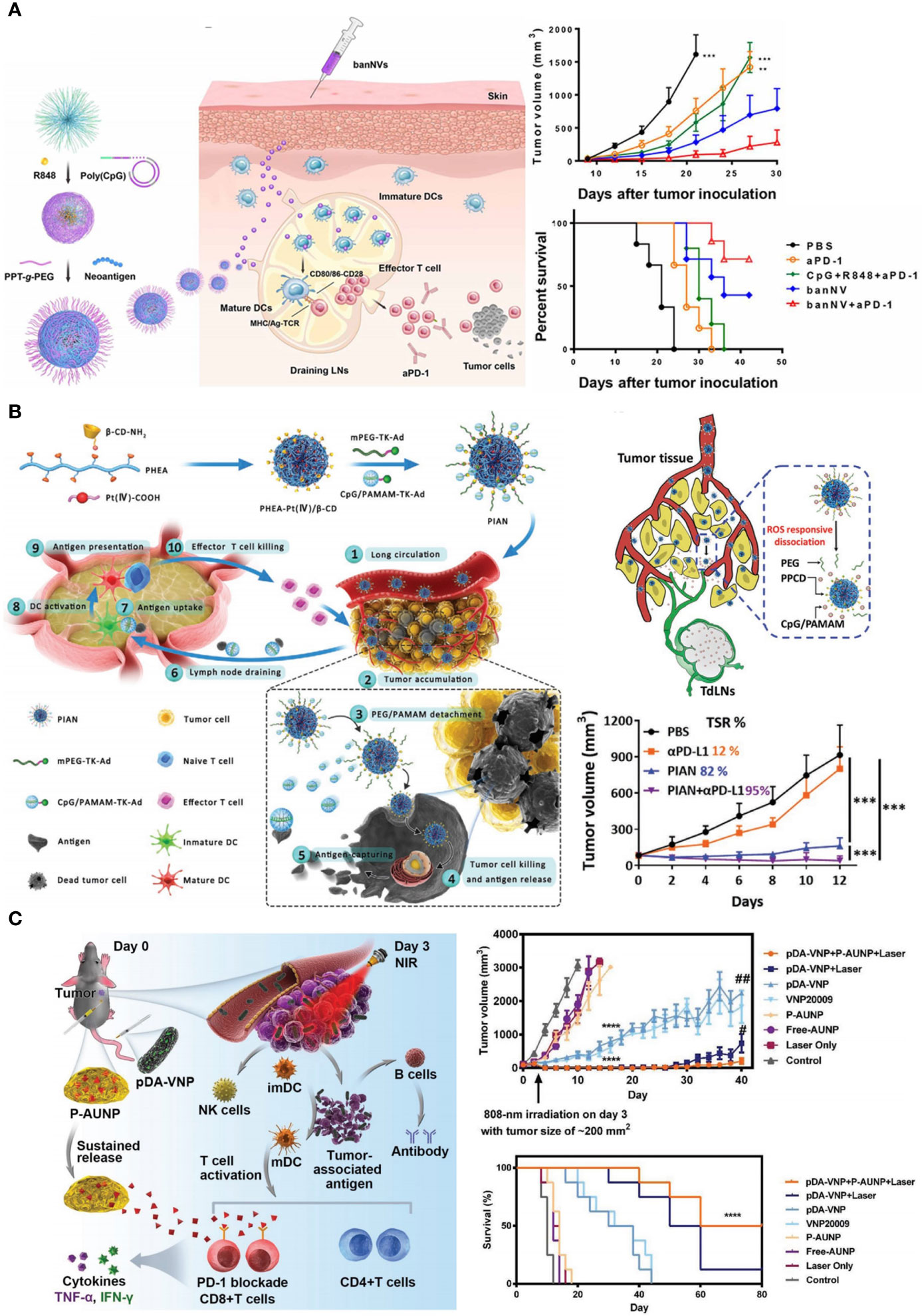
Figure 5 Nanotechnology pre-sensitizes host immune system to advance ICB in CRC therapy. (A) Left: schematic depiction of bi-adjuvant nano-vaccine (banNV) that sensitized antitumor T cells. Right: banNV remarkably facilitated αPD-1 in tumor growth inhibition and survival improvement. Reprinted with permission from reference (165). **p<0.01, ***p<0.001 versus banNV+αPD-1. (B) Left: schematic depiction of the programmable immune activation nanomedicine (PIAN) that generated tumor antigens in situ and transported these antigens to TDLNs. Right bottom: the combination of PIAN and αPD-L1 achieved considerable tumor inhibition with a tumor suppression rate (TSR) of 95%. Reprinted with permission from reference (166). ***p<0.001. (C) Left: schematic depiction of the combination therapy of anti-PD-1 peptide depot and bacteria-based PTT. Right: the combination therapy notably inhibited tumor growth and prolonged mice survival. Reprinted with permission from reference (167). ****p<0.0001 versus Control. #p<0.05, ##p<0.01 versus pDA-VNP+P-AUNP+Laser.
Co-delivery of cytotoxic agents and immune adjuvants provides another template for tumor immunization. Chemical drugs and photosensitizers that can trigger ICD are usually used as cytotoxic agents in these nanomedicines, such as DOX, OXA, and Ce6 (169, 170). After administration, tumor cells succumbed to ICD inducers and released plenty of whole-cell antigens. These in-situ generated tumor antigens were immediately captured and presented by APCs with the help of adjuvant, such as imiquimod (R837, TLR7 agonist). It should be noted that cytotoxic agents worked within tumor tissues but immune adjuvants stimulated APCs in TDLNs, raising a paradox in drug delivery (41, 171). To simultaneously fulfill the site-of-actions of cytotoxic agents and adjuvants, Chen et al. developed a programmable immune activation nanomedicine (PIAN) (166). PIAN initially accumulated in tumor tissues, releasing Pt (IV) compounds (PPCD) in response to ROS for tumor killing and antigen release. Concurrently, CpG-loaded dendrimers (CpG/PAMAM) also released, capturing antigen and then transferred into TDLNs to facilitate antigen presentation. PIAN resulted in strong antitumor immune response, and completely cured 40% of colorectal tumor bearing mice in combination with PD-L1 blockade (Figure 5B).
Except for co-delivery with tumor antigens or antigen inducers, a sole-delivery of TLR agonists also functioned in tumor beds and exhibited synergistic effect with checkpoint inhibitors. Researches from Liu et al. revealed that intra-tumoral injection of NPs loaded with R837 or MPLA (PLGA-R837 or PLGA-MPLA) promoted DC maturation after surgery or TA of tumors, while consolidated the immunosuppressive TME (42). The anti-CTLA4 antibody (αCTLA4) were employed to inhibit Tregs. Triple therapy of TA, PLGA-R837 and αCTLA4 exerted abscopal effect to eradicate the secondary CT26 tumors and saved 100% mice from death. After primary tumor ablation, immune memory against CT26 tumors was able to lasted for 80 days. Zhang et al. prepared platelet membrane-coated nanoparticles (PNP-R848) to locally deliver R848 into tumors (172). They found the coating of platelet membrane prolonged tumor retention, improving the binding and uptake of NPs by tumor-resided immune cells. Treatment with PNP-R848 thoroughly eliminated MC38 murine colorectal adenocarcinoma and triggered a long-term immune memory that allowed mice to reject tumor rechallenge for twice.
In addition to TLR agonists, some materials derived from bacteria and virus intrinsically have adjuvant-like effects (164). Moreover, their particulate properties enable drug encapsulation. For example, Steinmetz et al. combined cowpea mosaic virus (CPMV) with αPD-1 or agonistic OX40-specific antibodies (αOX40) to combat several immunocompetent tumor models (173, 174). CPMV upregulated PD-1 on CD4+ and CD8+ effector T cells and OX40 on Tregs, which sensitized tumors to OX40 agonists and PD-1 inhibitors. In the CT26 colon cancer model, combination of αOX40 and CPMV realized better therapeutic outcomes than αPD-1+CPMV. Similar results were obtained in ovarian tumor and B16F10 melanoma models. Sun et al. are devoted to exploit Salmonella Typhimurium as the drug carrier for tumor immunotherapy (167, 175, 176). Salmonella has intrinsic tumor-homing capability, and it can release several pathogen-associated molecular patterns (PAMPs) such as flagellin and LPS to stimulate immune cells via TLR pathways (175). Many strategies have been developed to engineer Salmonella as a nanocarrier, including genetic modulation, surface modification with therapeutic agents or NPs (Figure 5C) (167), as well as extracting their bacterial outer membrane vesicles (OMVs) as the adjuvant to coat nano-vaccines (177, 178). These pathogen-mimicking strategies offer a simple method to achieve colocalization of antigen and adjuvant in drug delivery, and strongly correlate with the peptide-based local PD-1 blockade therapy.
Although ICB-based immunotherapy has revolutionized CRC treatment, challenges such as side effects, tumor metastasis, low response rate, and therapy resistance remain in clinic. Application of nanotechnology might be ground-breaking. This review systematically discusses the current strategies that utilize nanotechnology to potentiate CRC therapy in combination with ICIs, wherein four main mechanisms are involved, including increasing delivery efficiency of ICIs, reinforcing tumor immunogenicity, reprogramming TME, and directly initiating immunity. In coming decades, we hope to witness the progress of more advanced CRC immunotherapies. Further researches are required to establish regimens that can benefit more CRC patients.
ZL, YX, and YZ searched literatures and drafted the manuscript. ZL and XK designed the topic and revised the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol (2019) 16(6):361–75. doi: 10.1038/s41575-019-0126-x
2. Boland PM, Ma WW. Immunotherapy for colorectal cancer. Cancers (2017) 9(5):50. doi: 10.3390/cancers9050050
3. Xi Y, Xu P. Global colorectal cancer burden in 2020 and projections to 2040. Trans Oncol (2021) 14(10):101174. doi: 10.1016/j.tranon.2021.101174
4. Kopetz S. New therapies and insights into the changing landscape of colorectal cancer. Nat Rev Gastroenterol Hepatol (2019) 16(2):79–80. doi: 10.1038/s41575-018-0100-z
5. Xiang B, Snook AE, Magee MS, Waldman SA. Colorectal cancer immunotherapy. Discovery Med (2013) 15(84):301. doi: 10.1590/S0100-879X2012007500152
6. Johdi NA, Sukor NF. Colorectal cancer immunotherapy: options and strategies. Front Immunol (2020) 11:1624. doi: 10.3389/fimmu.2020.01624
7. He M, Yang T, Wang Y, Wang M, Chen X, Ding D, et al. Immune checkpoint inhibitor-based strategies for synergistic cancer therapy. Adv Healthc Mater (2021) 10(9):2002104. doi: 10.1002/adhm.202002104
8. Sahin IH, Akce M, Alese O, Shaib W, Lesinski GB, El-Rayes B, et al. Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br J Cancer (2019) 121(10):809–18. doi: 10.1038/s41416-019-0599-y
9. Adachi K, Tamada K. Immune checkpoint blockade opens an avenue of cancer immunotherapy with a potent clinical efficacy. Cancer Sci (2015) 106(8):945–50. doi: 10.1111/cas.12695
10. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol (2018) 62:29–39. doi: 10.1016/j.intimp.2018.06.001
11. Yang X, Cheng B, Xiao Y, Xue M, Liu T, Cao H, et al. Discovery of novel CA-4 analogs as dual inhibitors of tubulin polymerization and PD-1/PD-L1 interaction for cancer treatment. Eur J Med Chem (2021) 213:113058. doi: 10.1016/j.ejmech.2020.113058
12. OuYang Y, Gao J, Zhao L, Lu J, Zhong H, Tang H, et al. Design, synthesis, and evaluation of o-(Biphenyl-3-ylmethoxy)nitrophenyl derivatives as PD-1/PD-L1 inhibitors with potent anticancer efficacy. In Vivo. J Med Chem (2021) 64(11):7646–66. doi: 10.1021/acs.jmedchem.1c00370
13. Chen L, Liu C, Xiang Y, Lyu J, Zhou Z, Gong T, et al. Exocytosis blockade of endoplasmic reticulum-targeted nanoparticle enhances immunotherapy. Nano Today (2022) 42:101356. doi: 10.1016/j.nantod.2021.101356
14. Andrieu GP, Shafran JS, Smith CL, Belkina AC, Casey AN, Jafari N, et al. BET protein targeting suppresses the PD-1/PD-L1 pathway in triple-negative breast cancer and elicits anti-tumor immune response. Cancer Lett (2019) 465:45–58. doi: 10.1016/j.canlet.2019.08.013
15. Liu C, Lu H, Wang H, Loo A, Zhang X, Yang G, et al. Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clin Cancer Res (2021) 27(1):342–54. doi: 10.1158/1078-0432.CCR-20-2718
16. Fan Z, Tian Y, Chen Z, Liu L, Zhou Q, He J, et al. Blocking interaction between SHP2 and PD-1 denotes a novel opportunity for developing PD-1 inhibitors. EMBO Mol Med (2020) 12(6):e11571. doi: 10.15252/emmm.201911571
17. Li K, Tian H. Development of small-molecule immune checkpoint inhibitors of PD-1/PD-L1 as a new therapeutic strategy for tumour immunotherapy. J Drug Target (2019) 27(3):244–56. doi: 10.1080/1061186X.2018.1440400
18. Sasikumar PG, Ramachandra M. Small-molecule immune checkpoint inhibitors targeting PD-1/PD-L1 and other emerging checkpoint pathways. BioDrugs (2018) 32(5):481–97. doi: 10.1007/s40259-018-0303-4
19. de Miguel M, Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell (2020) 38(3):326–33. doi: 10.1016/j.ccell.2020.07.004
20. Ciardiello D, Vitiello PP, Cardone C, Martini G, Troiani T, Martinelli E, et al. Immunotherapy of colorectal cancer: Challenges for therapeutic efficacy. Cancer Treat Rev (2019) 76:22–32. doi: 10.1016/j.ctrv.2019.04.003
21. Golshani G, Zhang Y. Advances in immunotherapy for colorectal cancer: a review. Ther Adv Gastroenterol (2020) 13:1756284820917527. doi: 10.1177/1756284820917527
22. Shim MK, Yang S, Sun I-C, Kim K. Tumor-activated carrier-free prodrug nanoparticles for targeted cancer immunotherapy: Preclinical evidence for safe and effective drug delivery. Adv Drug Deliv Rev (2022) 183:114177. doi: 10.1016/j.addr.2022.114177
23. Cremolini C, Vitale E, Rastaldo R, Giachino C. Advanced nanotechnology for enhancing immune checkpoint blockade therapy. Nanomaterials (2021) 11(3):661. doi: 10.3390/NANO11030661
24. Cabeza L, Perazzoli G, Mesas C, Jiménez-Luna C, Prados J, Rama AR, et al. Nanoparticles in colorectal cancer therapy: latest in vivo assays, clinical trials, and patents. AAPS PharmSciTech (2020) 21(5):1–15. doi: 10.1208/s12249-020-01731-y
25. Kim J, Hong J, Lee J, Lahiji SF, Kim Y-H. Recent advances in tumor microenvironment-targeted nanomedicine delivery approaches to overcome limitations of immune checkpoint blockade-based immunotherapy. J Controlled Release (2021) 332:109–26. doi: 10.1016/j.jconrel.2021.02.002
26. Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol (2015) 33(9):941–51. doi: 9780429027819
27. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat mater (2013) 12(11):991–1003. doi: 10.1038/nmat3776
28. Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS nano (2009) 3(1):16–20. doi: 10.1021/nn900002m
29. Xiang Y, Chen L, Liu C, Yi X, Li L, Huang Y. Redirecting chemotherapeutics to the endoplasmic reticulum increases tumor immunogenicity and potentiates anti-PD-L1 therapy. Small (2022) 18(6):2104591. doi: 10.1002/smll.202104591
30. Liu C, Li L, Lyu J, Xiang Y, Chen L, Zhou Z, et al. Split bullets loaded nanoparticles for amplified immunotherapy. J Controlled Release (2022) 347:199–210. doi: 10.1016/j.jconrel.2022.05.011
31. Sun Y, Feng X, Wan C, Lovell JF, Jin H, Ding J, et al. Role of nanoparticle-mediated immunogenic cell death in cancer immunotherapy. Asian J Pharmaceutical Sci (2021) 16(2):129–32. doi: 10.1016/j.ajps.2020.05.004
32. Li JY, Yao YM, Tian YP. Ferroptosis: A trigger of proinflammatory state progression to immunogenicity in necroinflammatory disease. Front Immunol (2021) 12:701163. doi: 10.3389/fimmu.2021.701163
33. Li J, Anraku Y, Kataoka K. Self-boosting catalytic nanoreactors integrated with triggerable crosslinking membrane networks for initiation of immunogenic cell death by pyroptosis. Angewandte Chemie (2020) 132(32):13628–32. doi: 10.1002/anie.202004180
34. Wang Y-Y, Liu X-L, Zhao R. Induction of pyroptosis and its implications in cancer management. Front Oncol (2019) 9:971. doi: 10.3389/fonc.2019.00971
35. Dewitte H, Verbeke R, Breckpot K, De Smedt SC, Lentacker I. Nanoparticle design to induce tumor immunity and challenge the suppressive tumor microenvironment. Nano Today (2014) 9(6):743–58. doi: 10.1016/j.nantod.2014.10.001
36. Gao S, Yang D, Fang Y, Lin X, Jin X, Wang Q, et al. Engineering nanoparticles for targeted remodeling of the tumor microenvironment to improve cancer immunotherapy. Theranostics (2019) 9(1):126. doi: 10.7150/thno.37568
37. Yu Q, Tang X, Zhao W, Qiu Y, He J, Wan D, et al. Mild hyperthermia promotes immune checkpoint blockade-based immunotherapy against metastatic pancreatic cancer using size-adjustable nanoparticles. Acta Biomater (2021) 133:244–56. doi: 10.1016/j.actbio.2021.05.002
38. Quatrini L, Mariotti FR, Munari E, Tumino N, Vacca P, Moretta L. The immune checkpoint PD-1 in natural killer cells: expression, function and targeting in tumour immunotherapy. Cancers (2020) 12(11):3285. doi: 10.3390/cancers12113285
39. Tran T-H, Mattheolabakis G, Aldawsari H, Amiji M. Exosomes as nanocarriers for immunotherapy of cancer and inflammatory diseases. Clin Immunol (2015) 160(1):46–58. doi: 10.1016/j.clim.2015.03.021
40. Nawaz M, Shah N, Zanetti BR, Maugeri M, Silvestre RN, Fatima F, et al. Extracellular vesicles and matrix remodeling enzymes: the emerging roles in extracellular matrix remodeling, progression of diseases and tissue repair. Cells (2018) 7(10):167. doi: 10.3390/cells7100167
41. Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials (2014) 35(2):814–24. doi: 10.1016/j.biomaterials.2013.10.003
42. Han X, Wang R, Xu J, Chen Q, Liang C, Chen J, et al. In situ thermal ablation of tumors in combination with nano-adjuvant and immune checkpoint blockade to inhibit cancer metastasis and recurrence. Biomaterials (2019) 224:119490. doi: 10.1016/j.biomaterials.2019.119490
43. Islam MA, Rice J, Reesor E, Zope H, Tao W, Lim M, et al. Adjuvant-pulsed mRNA vaccine nanoparticle for immunoprophylactic and therapeutic tumor suppression in mice. Biomaterials (2021) 266:120431. doi: 10.1016/j.biomaterials.2020.120431
44. Kubli SP, Berger T, Araujo DV, Siu LL, Mak TW. Beyond immune checkpoint blockade: emerging immunological strategies. Nat Rev Drug Discovery (2021) 20(12):899–919. doi: 10.1038/s41573-021-00155-y
45. Kourie HR, Klastersky J. Immune checkpoint inhibitors side effects and management. Immunotherapy (2016) 8(7):799–807. doi: 10.2217/imt-2016-0029
46. Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the immune-related adverse effects of immune checkpoint inhibitors: a review. JAMA Oncol (2016) 2(10):1346–53. doi: 10.1001/jamaoncol.2016.1051
47. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol (2019) 16(9):563–80. doi: 10.1038/s41571-019-0218-0
48. Spain L, Diem S, Larkin J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev (2016) 44:51–60. doi: 10.1016/j.ctrv.2016.02.001
49. Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. New Engl J Med (2018) 378(2):158–68. doi: 10.1056/NEJMra1703481
50. Yu Y, Li J, Song B, Ma Z, Zhang Y, Sun H, et al. Polymeric PD-L1 blockade nanoparticles for cancer photothermal-immunotherapy. Biomaterials (2022) 280:121312. doi: 10.1016/j.biomaterials.2021.121312
51. Guo W, Gao H, Li H, Ge S, Zhang F, Wang L, et al. Self-assembly of a multifunction DNA tetrahedron for effective delivery of aptamer PL1 and Pcsk9 siRNA potentiate immune checkpoint therapy for colorectal cancer. ACS Appl Mater Interf (2022) 14(28):31634–44. doi: 10.1021/acsami.2c06001
52. Luo L, Yang J, Zhu C, Jiang M, Guo X, Li W, et al. Sustained release of anti-PD-1 peptide for perdurable immunotherapy together with photothermal ablation against primary and distant tumors. J Controlled Release (2018) 278:87–99. doi: 10.1016/j.jconrel.2018.04.002
53. Song W, Shen L, Wang Y, Liu Q, Goodwin TJ, Li J, et al. Synergistic and low adverse effect cancer immunotherapy by immunogenic chemotherapy and locally expressed PD-L1 trap. Nat Commun (2018) 9(1):1–11. doi: 10.1038/s41467-018-04605-x
54. Yu M, Gu Z, Ottewell T, Yu C. Silica-based nanoparticles for therapeutic protein delivery. J Mater Chem B (2017) 5(18):3241–52. doi: 10.1039/C7TB00244K
55. Francis DM, Thomas SN. Progress and opportunities for enhancing the delivery and efficacy of checkpoint inhibitors for cancer immunotherapy. Adv Drug deliv Rev (2017) 114:33–42. doi: 10.1016/j.addr.2017.04.011
56. Lei C, Liu P, Chen B, Mao Y, Engelmann H, Shin Y, et al. Local release of highly loaded antibodies from functionalized nanoporous support for cancer immunotherapy. J Am Chem Soc (2010) 132:6906–7. doi: 10.1021/ja102414t
57. Meir R, Shamalov K, Sadan T, Motiei M, Yaari G, Cohen CJ, et al. Fast image-guided stratification using anti-programmed death ligand 1 gold nanoparticles for cancer immunotherapy. ACS Nano (2017) 11(11):11127–34. doi: 10.1021/acsnano.7b05299
58. Jeong Y-I, Yoo SY, Heo J, Kang DH. Chlorin e6-conjugated and PEGylated immune checkpoint inhibitor nanocomposites for pulmonary metastatic colorectal cancer. ACS omega (2019) 4(20):18593–9. doi: 10.1021/acsomega.9b02386
59. Kosmides AK, Sidhom JW, Fraser A, Bessell CA, Schneck JP. Dual targeting nanoparticle stimulates the immune system to inhibit tumor growth. ACS Nano (2017) 11(6):5417–29. doi: 10.1021/acsnano.6b08152
60. Wu J. The enhanced permeability and retention (EPR) effect: The significance of the concept and methods to enhance its application. J Personal Med (2021) 11(8):771. doi: 10.3390/jpm11080771
61. Nel A, Ruoslahti E, Meng H. New insights into “Permeability” as in the enhanced permeability and retention effect of cancer nanotherapeutics. ACS Nano (2017) 11(10):9567–9. doi: 10.1021/acsnano.7b07214
62. Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics (2014) 4(1):81–9. doi: 10.7150/thno.7193
63. Han X, Chen J, Chu J, Liang C, Ma Q, Fan Q, et al. Platelets as platforms for inhibition of tumor recurrence post-physical therapy by delivery of anti-PD-L1 checkpoint antibody. J Controlled Release (2019) 304:233–41. doi: 10.1016/j.jconrel.2019.05.008
64. Wang C, Sun W, Ye Y, Hu Q, Bomba HN, Gu Z. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nat Biomed Eng (2017) 1(2):1–10. doi: 10.1038/s41551-016-0011
65. Wang C, Ye Y, Hochu GM, Sadeghifar H, Gu Z. Enhanced cancer immunotherapy by microneedle patch-assisted delivery of anti-PD1 antibody. Nano Lett (2016) 16(4):2334–40. doi: 10.1021/acs.nanolett.5b05030
66. Zhou M, Zuo Q, Huang Y, Li L. Immunogenic hydrogel toolkit disturbing residual tumor "seeds" and pre-metastatic "soil" for inhibition of postoperative tumor recurrence and metastasis. Acta Pharm Sin B (2022) 12(8):3383–97. doi: 10.1016/j.apsb.2022.02.017
67. Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-Cell–dependent tumor eradication and decreases risk of toxic side EffectsLow-dose CTLA-4 blocking antibody treatment. Clin Cancer Res (2013) 19(19):5381–9. doi: 10.1158/1078-0432.CCR-12-0781
68. Fransen M, Ossendorp F, Arens R, Melief CJ. Local immunomodulation for cancer therapy: providing treatment where needed. Oncoimmunology (2013) 2(11):e26493. doi: 10.4161/onci.26493
69. Ishihara J, Fukunaga K, Ishihara A, Larsson HM, Potin L, Hosseinchi P, et al. Matrix-binding checkpoint immunotherapies enhance antitumor efficacy and reduce adverse events. Sci Trans Med (2017) 9(415):eaan0401. doi: 10.1126/scitranslmed.aan0401
70. Ishihara J, Ishihara A, Potin L, Hosseinchi P, Fukunaga K, Damo M, et al. Improving efficacy and safety of agonistic anti-CD40 antibody through extracellular matrix AffinityECM-binding character improves anti-CD40 antibody. Mol Cancer Ther (2018) 17(11):2399–411. doi: 10.1158/1535-7163.MCT-18-0091
71. Warnke C, Hermanrud C, Lundkvist M, Fogdell-Hahn A. Anti-drug antibodies. Drugs Ther Stud (2012) 2(1):e11–1. doi: 10.4081/dts.2012.e11
72. Hsu L, Armstrong AW. Anti-drug antibodies in psoriasis: a critical evaluation of clinical significance and impact on treatment response. Expert Rev Clin Immunol (2013) 9(10):949–58. doi: 10.1586/1744666X.2013.836060
73. Zyla E, Musielak B, Holak TA, Dubin G. Structural characterization of a macrocyclic peptide modulator of the PD-1/PD-L1 immune checkpoint axis. Molecules (2021) 26(16):4848. doi: 10.3390/MOLECULES26164848
74. Sasikumar PG, Ramachandra M. Peptide and peptide-inspired checkpoint inhibitors: protein fragments to cancer immunotherapy. Med Drug Discov (2020) 8:100073. doi: 10.1016/j.medidd.2020.100073
75. Jeon IS, Yoo JD, Gurung S, Kim M, Lee C, Park EJ, et al. Anticancer nanocage platforms for combined immunotherapy designed to harness immune checkpoints and deliver anticancer drugs. Biomaterials (2021) 270:120685. doi: 10.1016/j.biomaterials.2021.120685
76. Li Q, Chen C, Kong J, Li L, Li J, Huang Y. Stimuli-responsive nano vehicle enhances cancer immunotherapy by coordinating mitochondria-targeted immunogenic cell death and PD-L1 blockade. Acta Pharm Sin B (2022) 12(5):2533–49. doi: 10.1016/j.apsb.2021.11.005
77. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer (2016) 16(5):275–87. doi: 10.1038/nrc.2016.36
78. Kwak SY, Lee S, Han HD, Chang S, Kim KP, Ahn HJ. PLGA nanoparticles codelivering siRNAs against programmed cell death protein-1 and its ligand gene for suppression of colon tumor growth. Mol Pharm (2019) 16(12):4940–53. doi: 10.1021/acs.molpharmaceut.9b00826
79. Li L, Li Y, Yang CH, Radford DC, Wang J, Janat-Amsbury M, et al. Inhibition of immunosuppressive tumors by polymer-assisted inductions of immunogenic cell death and multivalent PD-L1 crosslinking. Adv Funct Mater (2020) 30(12):1908961. doi: 10.1002/adfm.201908961
80. Kamatham S, Shahjehan F, Kasi PM. Immune checkpoint inhibitors in metastatic colorectal cancer: current status, recent advances, and future directions. Curr Colorect Cancer Rep (2019) 15(4):112–21. doi: 10.1007/s11888-019-00437-z
81. Oliveira AF, Bretes L, Furtado I. Review of PD-1/PD-L1 inhibitors in metastatic dMMR/MSI-h colorectal cancer. Front Oncol (2019) 9:396. doi: 10.3389/fonc.2019.00396
82. Lynch D, Murphy A. The emerging role of immunotherapy in colorectal cancer. Ann Trans Med (2016) 4(16):305. doi: 10.21037/atm.2016.08.29
83. Bezu L, Gomes-da-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, et al. Combinatorial strategies for the induction of immunogenic cell death. Front Immunol (2015) 6:275. doi: 10.3389/fimmu.2015.00275
84. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med (2007) 13(1):54–61. doi: 10.1038/nm1523
85. Xiang Y, Chen L, Li L, Huang Y. Restoration and enhancement of immunogenic cell death of cisplatin by coadministration with digoxin and conjugation to HPMA copolymer. ACS Appl Mater Interf (2020) 12(1):1606–16. doi: 10.1021/acsami.9b19323
86. Li Y, Yang L, Xu X, Li M, Zhang Y, Lin Q, et al. Multifunctional size-expandable nanomedicines enhance tumor accumulation and penetration for synergistic chemo-photothermal therapy. ACS Appl Mater Interf (2021) 13(39):46361–74. doi: 10.1021/acsami.1c14170
87. Alzeibak R, Mishchenko TA, Shilyagina NY, Balalaeva IV, Vedunova MV, Krysko DV. Targeting immunogenic cancer cell death by photodynamic therapy: past, present and future. J immunother Cancer (2021) 9(1):e001926. doi: 10.1136/jitc-2020-001926
88. Sasaki K, Ishihara J, Ishihara A, Miura R, Mansurov A, Fukunaga K, et al. Engineered collagen-binding serum albumin as a drug conjugate carrier for cancer therapy. Sci Adv (2019) 5(8):eaaw6081. doi: 10.1126/sciadv.aaw6081
89. Zhang Y, He X, Zhang Y, Zhao Y, Lu S, Peng Y, et al. Native mitochondria-targeting polymeric nanoparticles for mild photothermal therapy rationally potentiated with immune checkpoints blockade to inhibit tumor recurrence and metastasis. Chem Eng J (2021) 424:130171. doi: 10.1016/j.cej.2021.130171
90. Li J, Cai W, Yu J, Zhou S, Li X, He Z, et al. Autophagy inhibition recovers deficient ICD-based cancer immunotherapy. Biomaterials (2022) 287:121651. doi: 10.1016/j.biomaterials.2022.121651
91. He C, Duan X, Guo N, Chan C, Poon C, Weichselbaum RR, et al. Core-shell nanoscale coordination polymers combine chemotherapy and photodynamic therapy to potentiate checkpoint blockade cancer immunotherapy. Nat Commun (2016) 7(1):1–12. doi: 10.1038/ncomms12499
92. Zhu Q, Sun F, Li T, Zhou M, Ye J, Ji A, et al. Engineering oxaliplatin prodrug nanoparticles for second near-infrared fluorescence imaging-guided immunotherapy of colorectal cancer. Small (2021) 17(13):2007882. doi: 10.1002/smll.202007882
93. Duan X, Chan C, Han W, Guo N, Weichselbaum RR, Lin W. Immunostimulatory nanomedicines synergize with checkpoint blockade immunotherapy to eradicate colorectal tumors. Nat Commun (2019) 10(1):1–15. doi: 10.1038/s41467-019-09221-x
94. Fan Y, Kuai R, Xu Y, Ochyl LJ, Irvine DJ, Moon JJ. Immunogenic cell death amplified by co-localized adjuvant delivery for cancer immunotherapy. Nano Lett (2017) 17(12):7387–93. doi: 10.1021/acs.nanolett.7b03218
95. Li J, Zhang P, Zhou M, Liu C, Huang Y, Li L. Trauma-responsive scaffold synchronizing oncolysis immunization and inflammation alleviation for post-operative suppression of cancer metastasis. ACS Nano (2022) 6(4):6064–79. doi: 10.1021/acsnano.1c11562
96. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol (2007) 35(4):495–516. doi: 10.1080/01926230701320337
97. Cohen JJ, Duke RC, Fadok VA, Sellins KS. Apoptosis and programmed cell death in immunity. Annu Rev Immunol (1992) 10(1):267–93. doi: 10.1146/annurev.iy.10.040192.001411
98. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol (2001) 9(3):113–4. doi: 10.1016/S0966-842X(00)01936-3
99. Dong T, Liao D, Liu X, Lei X. Using small molecules to dissect non-apoptotic programmed cell death: necroptosis, ferroptosis, and pyroptosis. Chembiochem (2015) 16(18):2557–61. doi: 10.1002/cbic.201500422
100. Ye J, Zhang R, Wu F, Zhai L, Wang K, Xiao M, et al. Non-apoptotic cell death in malignant tumor cells and natural compounds. Cancer Lett (2018) 420:210–27. doi: 10.1016/j.canlet.2018.01.061
101. Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol (2020) 13(1):1–18. doi: 10.1186/s13045-020-00946-7
102. Dixon Scott J, Lemberg Kathryn M, Lamprecht Michael R, Skouta R, Zaitsev Eleina M, Gleason Caroline E, et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
103. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
104. Han W, Duan X, Ni K, Li Y, Chan C, Lin W. Co-Delivery of dihydroartemisinin and pyropheophorbide-iron elicits ferroptosis to potentiate cancer immunotherapy. Biomaterials (2022) 280:121315. doi: 10.1016/j.biomaterials.2021.121315
105. Zang X, Song J, Li Y, Han Y. Targeting necroptosis as an alternative strategy in tumor treatment: From drugs to nanoparticles. J Controlled Release (2022) 349:213–26. doi: 10.1016/j.jconrel.2022.06.060
106. Ma Z-G, Ma R, Xiao X-L, Zhang Y-H, Zhang X-Z, Hu N, et al. Azo polymeric micelles designed for colon-targeted dimethyl fumarate delivery for colon cancer therapy. Acta Biomater (2016) 44:323–31. doi: 10.1016/j.actbio.2016.08.021
107. Um W, Ko H, You DG, Lim S, Kwak G, Shim MK, et al. Necroptosis-inducible polymeric nanobubbles for enhanced cancer sonoimmunotherapy. Adv Mater (2020) 32(16):e1907953. doi: 10.1002/adma.201907953
108. Mbeunkui F, Johann DJ. Cancer and the tumor microenvironment: A review of an essential relationship. Cancer chemother Pharmacol (2009) 63(4):571–82. doi: 10.1007/s00280-008-0881-9
109. Ma S, Qin L, Wang X, Wang W, Li J, Wang H, et al. The expression of VISTA on CD4+ T cells associate with poor prognosis and immune status in non-small cell lung cancer patients. Bosn J Basic Med Sci (2022) 22(5):707–15. doi: 10.17305/bjbms.2021.6531
110. Vesely MD, Zhang T, Chen L. Resistance mechanisms to anti-PD cancer immunotherapy. Annu Rev Immunol (2022) 40:45–74. doi: 10.1146/annurev-immunol-070621-030155
111. Musetti S, Huang L. Nanoparticle-mediated remodeling of the tumor microenvironment to enhance immunotherapy. ACS nano (2018) 12(12):11740–55. doi: 10.1021/acsnano.8b05893
112. Son S, Shin JM, Shin S, Kim CH, Lee JA, Ko H, et al. Repurposing macitentan with nanoparticle modulates tumor microenvironment to potentiate immune checkpoint blockade. Biomaterials (2021) 276:121058. doi: 10.1016/j.biomaterials.2021.121058
113. Jiang M, Qin B, Luo L, Li X, Shi Y, Zhang J, et al. A clinically acceptable strategy for sensitizing anti-PD-1 treatment by hypoxia relief. J Controlled Release (2021) 335:408–19. doi: 10.1016/j.jconrel.2021.06.001
114. Ma S, Song W, Xu Y, Si X, Zhang Y, Tang Z, et al. A ROS-responsive aspirin polymeric prodrug for modulation of tumor microenvironment and cancer immunotherapy. CCS Chem (2020) 2(6):390–400. doi: 10.31635/ccschem.020.202000140
115. Song W, Tiruthani K, Wang Y, Shen L, Hu M, Dorosheva O, et al. Trapping of lipopolysaccharide to promote immunotherapy against colorectal cancer and attenuate liver metastasis. Adv Mater (2018) 30(52):1805007. doi: 10.1002/adma.201805007
116. Dong X, Pan P, Zheng D-W, Bao P, Zeng X, Zhang X-Z. Bioinorganic hybrid bacteriophage for modulation of intestinal microbiota to remodel tumor-immune microenvironment against colorectal cancer. Sci Adv (2020) 6(20):eaba1590. doi: 10.1126/sciadv.aba1590
117. Hou B, Zhou L, Wang H, Saeed M, Wang D, Xu Z, et al. Engineering stimuli-activatable boolean logic prodrug nanoparticles for combination cancer immunotherapy. Adv Mater (2020) 32(12):1907210. doi: 10.1002/adma.201907210
118. Huang H, Jiang CT, Shen S, Liu A, Gan YJ, Tong QS, et al. Nanoenabled reversal of IDO1-mediated immunosuppression synergizes with immunogenic chemotherapy for improved cancer therapy. Nano Lett (2019) 19(8):5356–65. doi: 10.1021/acs.nanolett.9b01807
119. Lee SE, Lee CM, Won JE, Jang G-Y, Lee JH, Park SH, et al. Enhancement of anticancer immunity by immunomodulation of apoptotic tumor cells using annexin A5 protein-labeled nanocarrier system. Biomaterials (2022) 288:121677. doi: 10.1016/j.biomaterials.2022.121677
120. Li Z, Ding Y, Liu J, Wang J, Mo F, Wang Y, et al. Depletion of tumor associated macrophages enhances local and systemic platelet-mediated anti-PD-1 delivery for post-surgery tumor recurrence treatment. Nat Commun (2022) 13(1):1–15. doi: 10.1038/s41467-022-29388-0
121. Chen Q, Liu C, Liu C, Zhong D, Hua S, He J, et al. Wrapping porphyromonas gingivalis for tumor microenvironment immunomodulation and photothermal immunotherapy. Nano Today (2021) 41:101311. doi: 10.1016/j.nantod.2021.101311
122. Xiong Y, Song W, Shen L, Wang Y, Zhang J, Hu M, et al. Oral metformin and polymetformin reprogram immunosuppressive microenvironment and boost immune checkpoint inhibitor therapy in colorectal cancer. Adv Ther (2020) 3(12):2000168. doi: 10.1002/adtp.202000168
123. Chen C, Li A, Sun P, Xu J, Du W, Zhang J, et al. Efficiently restoring the tumoricidal immunity against resistant malignancies via an immune nanomodulator. J Controlled Release (2020) 324:574–85. doi: 10.1016/j.jconrel.2020.05.039
124. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med (2014) 211(5):781–90. doi: 10.1084/jem.20131916
125. You L, Wu W, Wang X, Fang L, Adam V, Nepovimova E, et al. The role of hypoxia-inducible factor 1 in tumor immune evasion. Med Res Rev (2021) 41(3):1622–43. doi: 10.1002/med.21771
126. Kolb D, Kolishetti N, Surnar B, Sarkar S, Guin S, Shah AS, et al. Metabolic modulation of the tumor microenvironment leads to multiple checkpoint inhibition and immune cell infiltration. ACS Nano (2020) 14(9):11055–66. doi: 10.1021/acsnano.9b10037
127. Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metast Rev (2010) 29(2):285–93. doi: 10.1007/s10555-010-9224-5
128. Landskron G, de la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res (2014) 2014:149185. doi: 10.1155/2014/149185
129. Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, et al. The COX-2/PGE 2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis (2009) 30(3):377–86. doi: 10.1093/carcin/bgp014
130. Talmadge JE. Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin Cancer Res (2007) 13(18):5243–8. doi: 10.1158/1078-0432.CCR-07-0182
131. Umansky V, Sevko A. Melanoma-induced immunosuppression and its neutralization. Semin Cancer biol Elsevier (2012) 22(4):319–26. doi: 10.1016/j.semcancer.2012.02.003
132. Khaled YS, Ammori BJ, Elkord E. Myeloid-derived suppressor cells in cancer: recent progress and prospects. Immunol Cell Biol (2013) 91(8):493–502. doi: 10.1038/icb.2013.29
133. de Vos WM, de Vos EA. Role of the intestinal microbiome in health and disease: from correlation to causation. Nutr Rev (2012) 70 Suppl 1:S45–56. doi: 10.1111/j.1753-4887.2012.00505.x
134. Song W, Anselmo AC, Huang L. Nanotechnology intervention of the microbiome for cancer therapy. Nat Nanotechnol (2019) 14(12):1093–103. doi: 10.1038/s41565-019-0589-5
135. Vigneswaran J, Shogan BD. The role of the intestinal microbiome on colorectal cancer pathogenesis and its recurrence following surgery. J Gastrointest Surg (2020) 24(10):2349–56. doi: 10.1007/s11605-020-04694-4
136. Song M, Chan AT, Sun J. Influence of the gut microbiome, diet, and environment on risk of colorectal cancer. Gastroenterology (2020) 158(2):322–40. doi: 10.1053/j.gastro.2019.06.048
137. Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology (2007) 133(6):1869–81. doi: 10.1053/j.gastro.2007.09.008
138. Killeen SD, Wang JH, Andrews EJ, Redmond HP. Bacterial endotoxin enhances colorectal cancer cell adhesion and invasion through TLR-4 and NF-kappaB-dependent activation of the urokinase plasminogen activator system. Br J Cancer (2009) 100(10):1589–602. doi: 10.1038/sj.bjc.6604942
139. Hsu RY, Chan CH, Spicer JD, Rousseau MC, Giannias B, Rousseau S, et al. LPS-induced TLR4 signaling in human colorectal cancer cells increases beta1 integrin-mediated cell adhesion and liver metastasis. Cancer Res (2011) 71(5):1989–98. doi: 10.1158/0008-5472.CAN-10-2833
140. Salas-Benito D, Perez-Gracia JL, Ponz-Sarvise M, Rodriguez-Ruiz ME, Martinez-Forero I, Castanon E, et al. Paradigms on immunotherapy combinations with chemotherapy. Cancer Discovery (2021) 11(6):1353–67. doi: 10.1158/2159-8290.CD-20-1312
141. Munn DH, Mellor AL. IDO and tolerance to tumors. Trends Mol Med (2004) 10(1):15–8. doi: 10.1016/j.molmed.2003.11.003
142. Zhu MM, Dancsok AR, Nielsen TO. Indoleamine dioxygenase inhibitors: clinical rationale and current development. Curr Oncol Rep (2019) 21(1):1–13. doi: 10.1007/s11912-019-0750-1
143. Heng B, Lim CK, Lovejoy DB, Bessede A, Gluch L, Guillemin GJ. Understanding the role of the kynurenine pathway in human breast cancer immunobiology. Oncotarget (2016) 7(6):6506. doi: 10.18632/oncotarget.6467
144. Labadie BW, Bao R, Luke JJ. Reimagining IDO pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan–Kynurenine–Aryl hydrocarbon AxisTrp–Kyn–AhR immunotherapy. Clin Cancer Res (2019) 25(5):1462–71. doi: 10.1158/1078-0432.CCR-18-2882
145. Routy J-P, Routy B, Graziani GM, Mehraj V. The kynurenine pathway is a double-edged sword in immune-privileged sites and in cancer: implications for immunotherapy. Int J Tryptophan Res (2016) 9:IJTR. S38355. doi: 10.4137/IJTR.S38355
146. Segawa K, Nagata S. An apoptotic ‘eat me’signal: phosphatidylserine exposure. Trends Cell Biol (2015) 25(11):639–50. doi: 10.1016/j.tcb.2015.08.003
147. Mariño G, Kroemer G. Mechanisms of apoptotic phosphatidylserine exposure. Cell Res (2013) 23(11):1247–8. doi: 10.1038/CR.2013.115
148. Birge R, Boeltz S, Kumar S, Carlson J, Wanderley J, Calianese D, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ (2016) 23(6):962–78. doi: 10.1038/cdd.2016.11
149. Hutchins J. Antibody mediated blockade of phosphatidylserine improves immune checkpoint blockade by repolarizing immune suppressive mechanisms of the tumor microenvironment. Ann Oncol (2016) 27:vi368. doi: 10.1093/annonc/mdw378.28
150. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (2014) 6(3):1670–90. doi: 10.3390/cancers6031670
151. Anfray C, Ummarino A, Torres Andon F, Allavena P. Current strategies to target tumor-associated-macrophages to improve anti-tumor immune responses. Cells (2019) 9(1):46. doi: 10.3390/cells9010046
152. Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv Drug Deliv Rev (2017) 114:206–21. doi: 10.1016/j.addr.2017.04.010
153. Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metast Rev (2006) 25(3):315–22. doi: 10.1007/s10555-006-9001-7
154. Mougel A, Terme M, Tanchot C. Therapeutic cancer vaccine and combinations with antiangiogenic therapies and immune checkpoint blockade. Front Immunol (2019) 10:467. doi: 10.3389/fimmu.2019.00467
155. Zhao J, Chen Y, Ding Z-Y, Liu J-Y. Safety and efficacy of therapeutic cancer vaccines alone or in combination with immune checkpoint inhibitors in cancer treatment. Front Pharmacol (2019) 10:1184. doi: 10.3389/fphar.2019.01184
156. Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell (2015) 161(2):205–14. doi: 10.1016/j.cell.2015.03.030
157. Bender A, Karbach J, Neumann A, Jäger D, Al-Batran SE, Atmaca A, et al. LUD 00-009: phase 1 study of intensive course immunization with NY-ESO-1 peptides in HLA-A2 positive patients with NY-ESO-1-expressing cancer. Cancer Immun (2007) 7(1):16. doi: 10.1158/1424-9634.DCL-16.7.1
158. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol (2017) 18(3):168–82. doi: 10.1038/nri.2017.131
159. Tyagi P, Mirakhur B. MAGRIT: the largest-ever phase III lung cancer trial aims to establish a novel tumor-specific approach to therapy. Clin Lung Cancer (2009) 10(5):371–4. doi: 10.3816/CLC.2009.n.052
160. Rus Bakarurraini NAA, Ab Mutalib NS, Jamal R, Abu N. The landscape of tumor-specific antigens in colorectal cancer. Vaccines (2020) 8(3):371. doi: 10.3390/vaccines8030371
161. Schietinger A, Philip M, Schreiber H. Specificity in cancer immunotherapy. Semin immunol Elsevier (2008) 20(5):276–85. doi: 10.1016/j.smim.2008.07.001
162. Seremet T, Brasseur F, Coulie PG. Tumor-specific antigens and immunologic adjuvants in cancer immunotherapy. Cancer J (2011) 17(5):325–30. doi: 10.1097/PPO.0b013e3182326004
163. Luchner M, Reinke S, Milicic A. TLR agonists as vaccine adjuvants targeting cancer and infectious diseases. Pharmaceutics (2021) 13(2):142. doi: 10.3390/pharmaceutics13020142
164. Lahiri A, Das P, Chakravortty D. Engagement of TLR signaling as adjuvant: towards smarter vaccine and beyond. Vaccine (2008) 26(52):6777–83. doi: 10.1016/j.vaccine.2008.09.045
165. Ni Q, Zhang F, Liu Y, Wang Z, Yu G, Liang B, et al. A bi-adjuvant nanovaccine that potentiates immunogenicity of neoantigen for combination immunotherapy of colorectal cancer. Sci Adv (2020) 6(12):eaaw6071. doi: 10.1126/sciadv.aaw6071
166. Zhang Y, Ma S, Liu X, Xu Y, Zhao J, Si X, et al. Supramolecular assembled programmable nanomedicine as in situ cancer vaccine for cancer immunotherapy. Adv Mater (2021) 33(7):2007293. doi: 10.1002/adma.202007293
167. Chen W, Guo Z, Zhu Y, Qiao N, Zhang Z, Sun X. Combination of bacterial-photothermal therapy with an anti-PD-1 peptide depot for enhanced immunity against advanced cancer. Adv Funct Mater (2019) 30(1):1906623. doi: 10.1002/adfm.201906623
168. Vo M-C, Ahn S-Y, Chu T-H, Uthaman S, Pillarisetti S, Uong TNT, et al. A combination of immunoadjuvant nanocomplexes and dendritic cell vaccines in the presence of immune checkpoint blockade for effective cancer immunotherapy. Cell Mol Immunol (2021) 18(6):1599–601. doi: 10.1038/s41423-021-00666-z
169. Wang Q, Ma Y, Lu Z, Yu H, Li Z. Co-Delivery of chemotherapeutic drugs and immune adjuvants by nanoscale DNA tetrahedrons for synergistic cancer therapy. ACS Appl Nano Mater (2022) 5(1):101–6. doi: 10.1021/acsanm.1c03295
170. Da Silva CG, Camps MG, Li TM, Zerrillo L, Löwik CW, Ossendorp F, et al. Effective chemoimmunotherapy by co-delivery of doxorubicin and immune adjuvants in biodegradable nanoparticles. Theranostics (2019) 9(22):6485. doi: 10.7150/thno.34429
171. Yang X, Yu T, Zeng Y, Lian K, Zhou X, Li S, et al. Tumor-draining lymph node targeting chitosan micelles as antigen-capturing adjuvants for personalized immunotherapy. Carbohydr polymers (2020) 240:116270. doi: 10.1016/j.carbpol.2020.116270
172. Bahmani B, Gong H, Luk BT, Haushalter KJ, DeTeresa E, Previti M, et al. Intratumoral immunotherapy using platelet-cloaked nanoparticles enhances antitumor immunity in solid tumors. Nat Commun (2021) 12(1):1–12. doi: 10.1038/s41467-021-22311-z
173. Patel R, Czapar AE, Fiering S, Oleinick NL, Steinmetz NF. Radiation therapy combined with cowpea mosaic virus nanoparticle in situ vaccination initiates immune-mediated tumor regression. ACS Omega (2018) 3(4):3702–7. doi: 10.1021/acsomega.8b00227
174. Wang C. Steinmetz N.F. a combination of cowpea mosaic virus and immune checkpoint therapy synergistically improves therapeutic efficacy in three tumor models. Adv Funct Mater (2020) 30(27):2002299. doi: 10.1002/adfm.202002299
175. Chen W, Zhu Y, Zhang Z, Sun X. Advances in salmonella typhimurium-based drug delivery system for cancer therapy. Adv Drug Del Rev (2022) 185:114295. doi: 10.1016/j.addr.2022.114295
176. Wang Y, Zhou Z, Chen W, Qin M, Zhang Z, Gong T, et al. Potentiating bacterial cancer therapy using hydroxychloroquine liposomes. J Control Release (2018) 280:39–50. doi: 10.1016/j.jconrel.2018.04.046
177. Chen Q, Bai H, Wu W, Huang G, Li Y, Wu M, et al. Bioengineering bacterial vesicle-coated polymeric nanomedicine for enhanced cancer immunotherapy and metastasis prevention. Nano Lett (2020) 20(1):11–21. doi: 10.1021/acs.nanolett.9b02182
Keywords: colorectal cancer, immune checkpoint inhibitors, nanotechnology, drug delivery, tumor microenvironment
Citation: Liu Z, Xiang Y, Zheng Y and Kang X (2022) Advancing immune checkpoint blockade in colorectal cancer therapy with nanotechnology. Front. Immunol. 13:1027124. doi: 10.3389/fimmu.2022.1027124
Received: 24 August 2022; Accepted: 10 October 2022;
Published: 20 October 2022.
Edited by:
Udo S. Gaipl, University Hospital Erlangen, GermanyCopyright © 2022 Liu, Xiang, Zheng and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Kang, a2FuZ3hpbjIwMjIwNzI2QDE2My5jb20=; Zefan Liu, Nzc4Njg4MDhAcXEuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.