 Jérémy Alloo
Jérémy Alloo Ines Leleu
Ines Leleu Corinne Grangette
Corinne Grangette Sylviane Pied
Sylviane Pied- Center for Infection and Immunity of Lille-CIIL, Centre National de la Recherche Scientifique-CNRS UMR 9017-Institut National de la Recherche Scientifique et Médicale-Inserm U1019, Institut Pasteur de Lille, Univ. Lille, Lille, France
Many parasitic diseases (including cerebral malaria, human African trypanosomiasis, cerebral toxoplasmosis, neurocysticercosis and neuroschistosomiasis) feature acute or chronic brain inflammation processes, which are often associated with deregulation of glial cell activity and disruption of the brain blood barrier’s intactness. The inflammatory responses of astrocytes and microglia during parasite infection are strongly influenced by a variety of environmental factors. Although it has recently been shown that the gut microbiota influences the physiology and immunomodulation of the central nervous system in neurodegenerative diseases like Alzheimer’s disease and Parkinson’s, the putative link in parasite-induced neuroinflammatory diseases has not been well characterized. Likewise, the central nervous system can influence the gut microbiota. In parasite infections, the gut microbiota is strongly perturbed and might influence the severity of the central nervous system inflammation response through changes in the production of bacterial metabolites. Here, we review the roles of astrocytes and microglial cells in the neuropathophysiological processes induced by parasite infections and their possible regulation by the gut microbiota.
Introduction
Many of the protozoans and metazoans associated with high-mortality, high-morbidity diseases (such as Plasmodium (P.) falciparum, Toxoplasma (T.) gondii and Trypanosoma (T.) brucei, Taenia (T.) solium, and Schistosoma (S.) mansoni) can invade the brain and induce neuropathological disorders (1, 2). The latter are often associated with systemic or local, acute or chronic neuroinflammatory processes with a variety of clinical outcomes (Supplementary Tables 1, 2). Parasite-induced brain inflammatory disorders mostly damage the central nervous system (CNS), with life-threatening consequences (3–5). In most cases, the causative neuroinflammatory mechanisms have yet to be determined. However, the pro-inflammatory cytokines and chemokines released by activated states of astrocytes and microglial cells have been identified as key factors in these neuropathophysiological processes.
Astrocytes and microglial cells are major components of the CNS, where they help to regulate homeostasis and maintain the intactness of the blood-brain barrier (BBB) (6–9). They serve as the CNS’s resident immune cells and so have a major role in the local innate immune response and inflammatory processes – particularly during pathogen invasion or tissue damage (7, 10–12). Astrocytes and microglia also function as antigen-presenting cells (13).
Deregulation of glial cell activity is commonly observed in CNS inflammatory parasitic diseases. This deregulation is often associated with the cytotoxic effects of nitric oxide, reactive oxygen species, and pro-inflammatory mediators. Nevertheless, deregulation can also be associated with a neuroprotective response (7). In fact, astrocytes secrete neuroactive molecules (including nerve growth factor, glioma-derived growth factor, ciliary neurotrophic factor, and neurotrophic factors [like leukemia inhibitory factor]) that have an important role not only in neuroregeneration but also in the attenuation of neurotoxic phenomena (13, 14). Furthermore, microglial cells can adopt an alternative anti-inflammatory phenotype characterized by the production of cytokines/chemokines that protect neurons (10) and also regulate the level of fatty acids and neurotrophic or angiogenic factors (15). Accordingly, glial cells are key regulators of pro/anti-inflammatory responses induced in the brain during parasitic diseases. These inflammatory responses are dependent on numerous parameters associated to the parasite specie, their life cycle and biology, the targeted cells, induced-immune responses, antigenic variation and immune escape strategy elicited by the parasite (16). Here, we review the various response patterns associated with acute or chronic parasite-induced brain inflammation.
During a host-parasite interaction, the inflammatory response of astrocytes and glial cells is strongly influenced by various environmental factors, including host genetic factors, immune experience, and the intestinal microbiota (11, 17). The gut microbiota (GM) is known to have a major role in the immunomodulation of the CNS in general and during neuroinflammatory and neurodegenerative diseases (such as Parkinson’s disease, Alzheimer’s disease, depression, and multiple sclerosis) in particular (17–21). We shall also review (i) the roles of astrocyte and microglial cells in the neuropathophysiological processes induced by parasite infections and (ii) how these roles are possibly influenced by the GM (12).
Parasites that trigger acute neuroinflammation
Cerebral malaria
Cerebral malaria (CM) is the deadliest complication of Plasmodium falciparum infection and causes approximately 500,000 deaths per year worldwide (2). Patients with CM generally develop acute neurologic manifestations: a combination of diffuse encephalopathy and decreased consciousness progresses to deep coma, seizures and, in some cases, death (22). Children who survive CM show transient or permanent neurologic sequelae and cognitive impairments (23). Along with parasite sequestration within the brain microvessels, CM is associated with an impaired immune response and excessive, uncontrolled neuroinflammation resulting from the production of pro-inflammatory cytokines and chemokines by activated astrocytes and microglial cells (3, 24, 25). We recently reported that this pro-inflammatory response is promoted by (i) the phagocytosis of infected red blood cells by microglial cells and (ii) the transfer of parasite microvesicles to astrocytes via microtubule-associated protein 1 light chain 3 (LC3)-dependent autophagy. The LC3-associated phagocytosis results in the production of high levels of chemokine (C-X-C motif) ligand 10 (CXCL10), chemokine (C-C motif) ligand 2 (CCL2), tumor necrosis factor alpha (TNF-α), and interferon gamma (IFN-γ)) known to be involved in the pathogenesis of CM (26, 27). As shown in Figures 1, 2, these cytokines and chemokines promote disruption of the BBB and the brain infiltration of pathological αβ CD8+ T lymphocytes expressing chemokine (C-X-C motif) receptor 3 (CXCR3), leading to neuronal damage (25, 28–32).
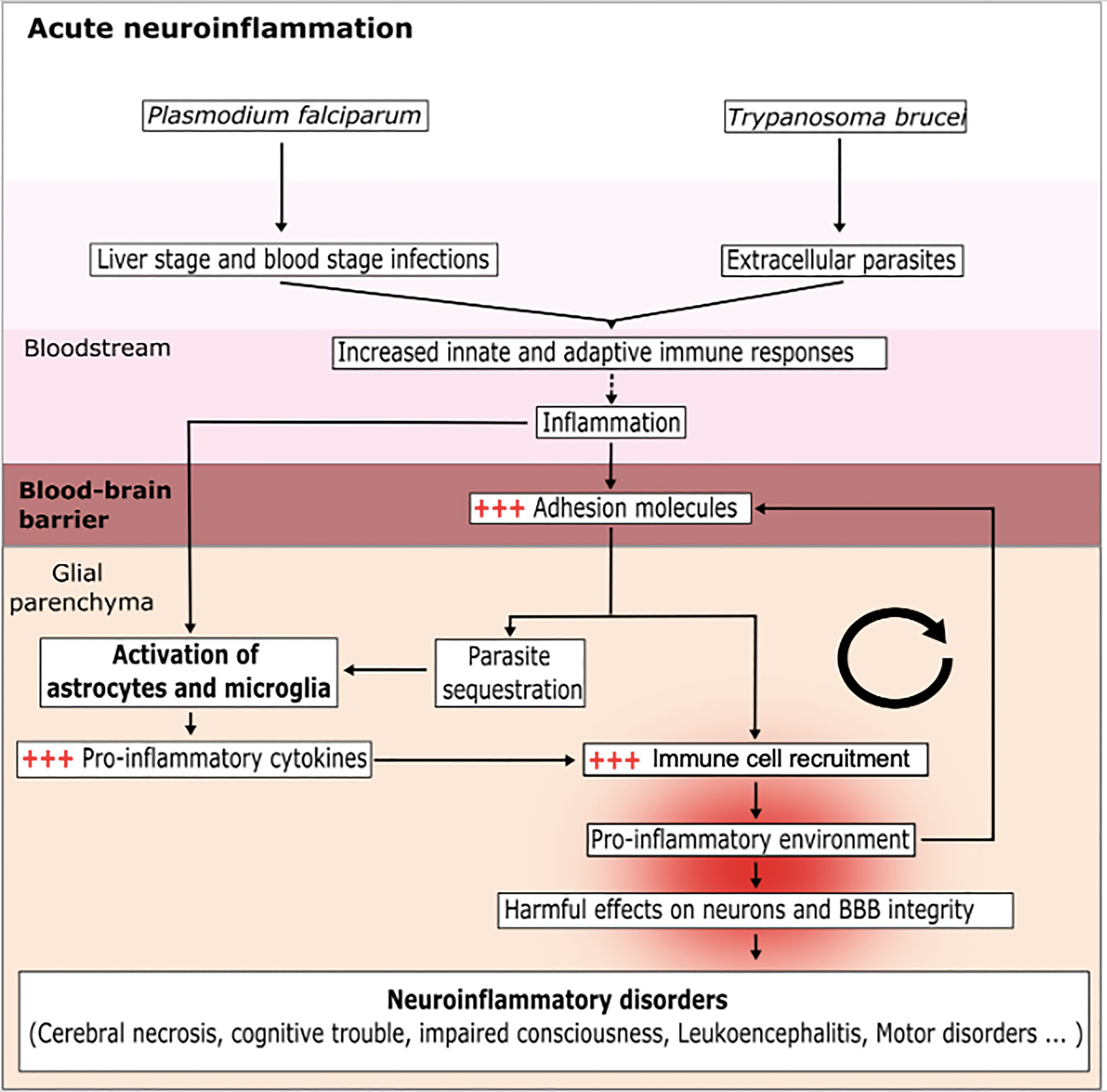
Figure 1 Mechanisms of the acute CNS inflammation induced by a protozoan parasite infection. The parasites P. falciparum and T. brucei enter the blood and then infect various cell types, in order to escape the immune system and splenic clearance. During this phase, the parasites activate circulating immune cells; in turn, this induces inflammation and favors the expression of adhesion molecules (ICAM-1 and VCAM-1) by endothelial cells and the activation of glial cells (astrocytes and microglia). This results in a vicious circle because the inflammation makes it easier for parasites to enter and accumulate in the glial parenchyma. The sensing of parasites by the glial cells induces the production of pro-inflammatory cytokines, allows the recruitment of immune cells and creates a pro-inflammatory environment. The brain inflammation disturbs the BBB and helps the parasite to invade the glial parenchyma. The accumulation of parasites exacerbates the activation of astrocytes and microglia cells and leads to a harmful, pro-inflammatory environment.
Human African trypanosomiasis
Human African trypanosomiasis (HAT) results from infection by either T. brucei (T.b), T. gambiense, or T. brucei rhodesiense. Infection by T. brucei starts with a hematolymphatic phase and ends with a meningoencephalitic stage. The neuropathology affected approximately 3000 people in Africa in 2015 (5) and is characterized by headaches, psychological changes, sleep disturbances, sensorimotor problems, psychiatric disturbances, and (in some cases) death (33). The parasite colonizes the brain parenchyma in several steps (34). HAT is an immune process that results from the excessive activation of perivascular macrophages, astrocytes, and microglia cells (35, 36). For example, the formation of microglial nodules and astrocytic hypertrophy has been observed in T.b-infected brains (10, 11). Some parasites are phagocytosed by innate immune system cells (such as perivascular macrophages) but most escape immune clearance by expressing a new variant surface glycoprotein during each humoral immune system attack (37). Activation of glial cells in the brain promotes the production of pro-inflammatory factors (TNF-α, IFN-γ, CXCL10, and CXCL9), which is associated with local functional disturbance. In Trypanosoma-infected brains, macrophage activation via the toll-like receptor 9 (TLR9) pathway and the MyD88 innate immune signal transduction adaptor (Myd88) leads to the release of TNF-α, IFN-α/ß/γ and interleukin (IL)-1ß (38). These inflammatory mediators promote the endothelial expression of intercellular cell adhesion molecule 1 (ICAM1) and vascular cell adhesion molecule 1 (VCAM1). Limited production of CXCL10 by astrocytes has also been observed but is enough to trigger the recruitment of T lymphocytes and then their infiltration into the CNS parenchyma (11). The recruited T lymphocytes are sensitized by Trypanosoma antigen presented by macrophages. Further massive T lymphocyte recruitment is triggered by the CXCL10 produced by activated astrocytes. In turn, this promotes the release of matrix metalloproteinase-2 and -9 (MMP2/9), which are involved in loss of the BBB’s integrity and thus create a gateway for parasite dissemination (11) (Figures 1, 2).
In both CM and trypanosomiasis, the elevated production of pro-inflammatory cytokines and chemokines (by astrocytes) and the subsequent T lymphocyte recruitment are harmful. It is noteworthy that CXCL10 (produced mainly by astrocytes) might have a major neuropathogenic role; for example, CXCL10-/- and CXCR3-/- mice challenged with P.berghei ANKA or T.brucei control the infection more readily and are resistant to the neurologic disorder (11).
Parasites that drive chronic neuroinflammatory processes
Neurotoxoplasmosis
Neurotoxoplasmosis is caused by the protozoan parasite T. gondii. The disease is contracted by ingestion of either excreted oocysts or cysts located in the muscle and nervous tissues of infected mammals (39). Toxoplasmosis is one of the world’s major food-borne diseases and is widespread in a third of the world's population (12). Primary infection with T. gondii is asymptomatic in most hosts but tends to be symptomatic in immunocompromised individuals and pregnant women. T. gondii can replicate within a wide variety of cell types, including brain cells (40). T. gondii can persist throughout the host’s lifetime in the brain as cysts, which are located principally in neurons and glial cells (39). The parasite’s initial contact with the host cells engages cell surface molecules, such as surface antigen-1, laminin, ICAM1, VCAM1, and activated leukocyte cell adhesion molecule (41, 42). The effectiveness of the human immune response to T. gondii is evidenced by low incidence of symptomatic disease, despite an overall seroprevalence of about 30%. It is clear that T cell trafficking and migration into the infected brain are critical factors in damage to the CNS. The crosstalk between T. gondii and the host involves a wide range of proteins and signaling networks. If an individual becomes immunocompromised, T. gondii cysts in the brain can reactivate and produce potentially lethal neurotoxoplasmosis. Activation of glial cells by interaction with T. gondii favors the production of pro-inflammatory cytokines and chemokines such as IL-1α, IL-6, granulocyte-macrophage colony-stimulating factor, CXCL10, CCL2, CCL3, CCL4, and IFN-γ (39, 43). Astrocytes that lack the IL-6 receptor or glial fibrillary acidic protein lose their ability to control the parasite. As a result, the inflammation spreads throughout the brain and can be fatal (44) (Figures 2, 3). Excessive brain inflammation and neuronal damage are prevented by the autoregulation of astrocytes and neurons via the secretion of anti-inflammatory cytokines, such as IL-27 and transforming growth factor beta (TGF-β); these cytokines have immunosuppressive activity and influence T cell functions (11, 39, 43, 45).
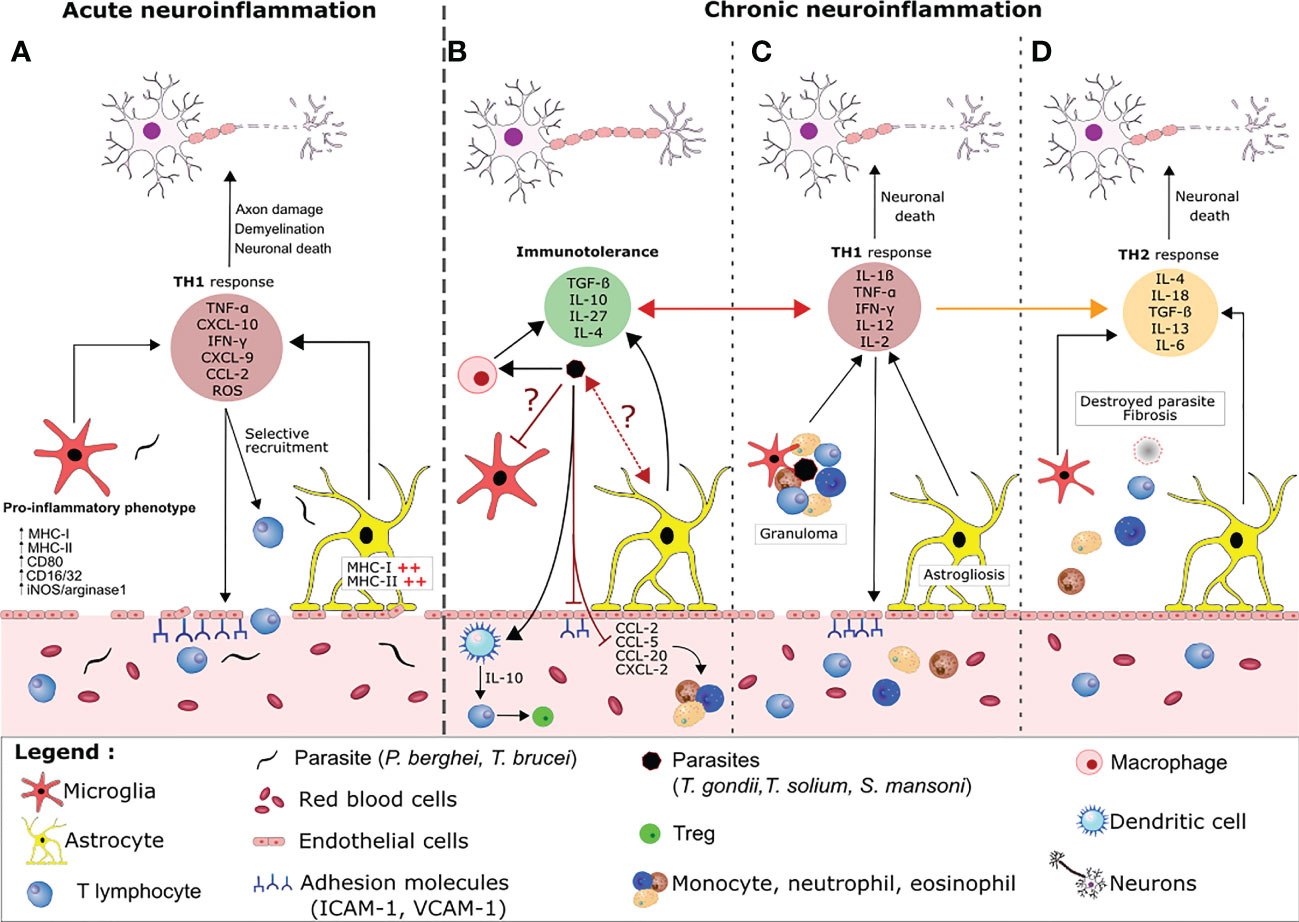
Figure 2 Host regulation of neuroimmune processes during acute versus chronic parasite infection. (A) During an acute parasite infection (e.g by P. falciparum or T.brucei), parasites cross the BBB and activate astrocytes and microglia, which produce large amounts of pro-inflammatory cytokine/chemokines (e.g CXCL-10) that recruit T lymphocytes. Together, CD8+ and CD4+ T lymphocytes favor a pro-inflammatory environment by releasing molecules like perforin, granzyme, reactive oxygen species and IFN-γ. This release leads to disruption of the BBB, which favors parasite entry, aggravates the brain inflammation, and causes collateral damage to neurons. (B) In contrast, immune tolerance and pro-inflammatory responses are balanced during a chronic parasite infection. The cysts release compounds that inhibit granuloma formation and the activation of resident glial cells. For example, the parasites polarize pro-inflammatory macrophages into anti-inflammatory macrophages, which suppress the production of adhesion molecules and the local TH1 response via TGF-β and IL-10 production. Other cyst-derived compounds polarize CD4+ cells into regulatory T cells (Tregs) by modulating the maturation of dendritic cells and preventing the infiltration and migration of neutrophils, eosinophils and monocytes from the peripheral system into the brain via the production of immunomodulatory cytokines and the blockade of chemokines and adhesion molecules. Through an as-yet unknown mechanism, the parasite also inhibits the activation of microglia and astrocytes. Nevertheless, some of the material released by the cysts elicits an inflammatory reaction (mainly characterized by the secretion of IFN-γ and TNF-α by the activated glial cells and leukocytes). The chemoattractants lead to the recruitment of neutrophils, eosinophils and monocytes, which form a granuloma. Formation of a granuloma limits the collateral damage caused by the TH1 response and enables the parasite to be contained and destroyed. The astrocytes and microglia become activated and the BBB is damaged (C). Progressively, the TH1 response is replaced by a TH2 response and fibrosis occurs where the cyst was located. This fibrosis is associated with neuronal damage (D).
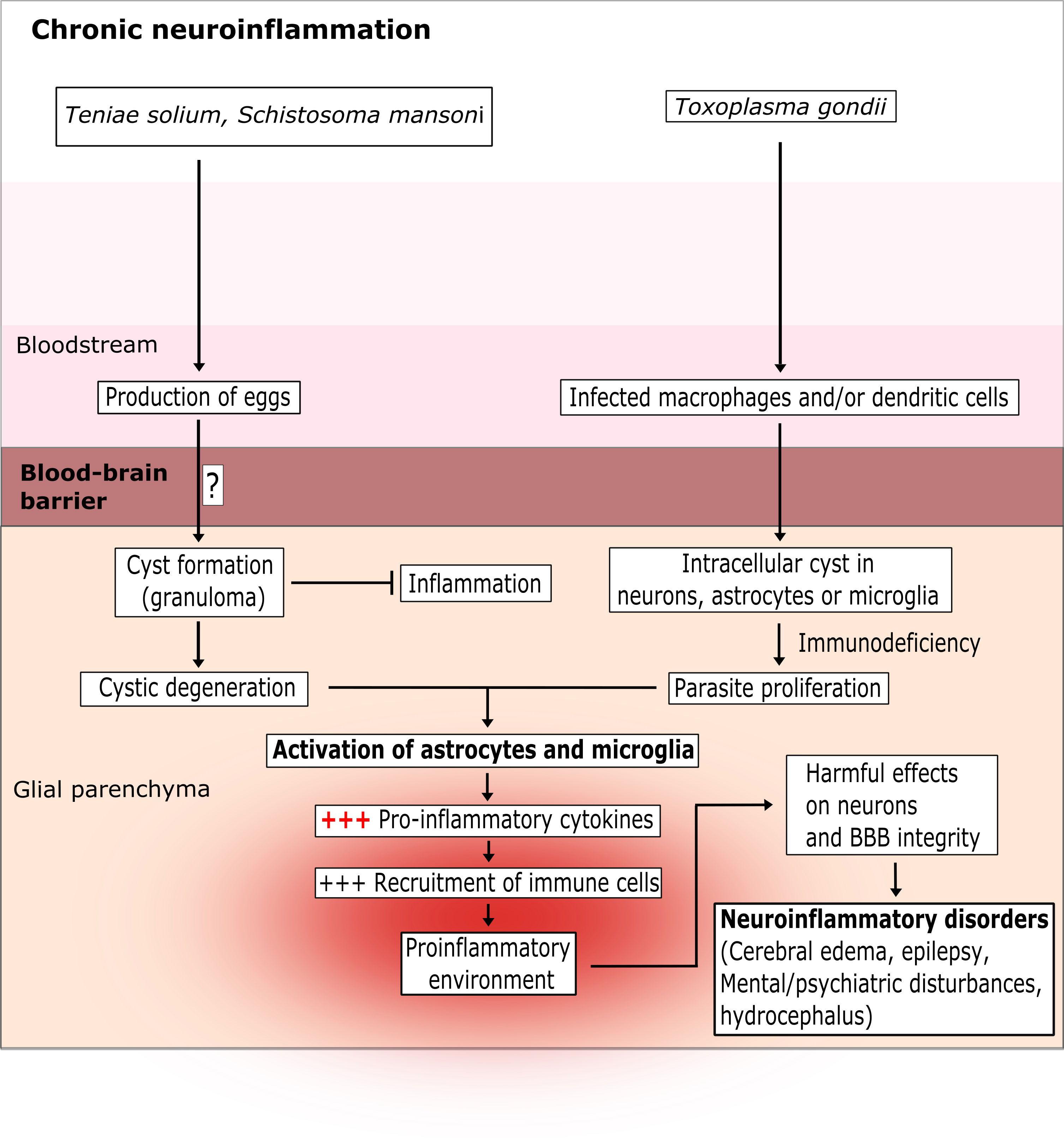
Figure 3 The chronic latent brain inflammation induced by parasite infection. T. solium and S. mansoni are intestinal parasites. Their eggs pass into the blood and then cross the BBB via as-yet unknown mechanisms. T. gondii infects leukocytes and crosses the BBB via a “trojan horse” mechanism or via a paracellular or transcellular route. Once inside the glial parenchyma, the parasites form extracellular cysts (for T. solium and S. mansoni) or intracellular cysts (for T. gondii) in neurons and microglia cells. The cysts can survive for several years and induce a low-level pro-inflammatory response. However, after few years or in an immunocompromised state, the cysts degenerate (for T. solium and S. mansoni) or proliferate (for T. gondii) and strongly activate astrocytes and microglia cells. In turn, this excessive activation creates a pro-inflammatory environment that damages neurons and the BBB.
Neurocysticercosis
Neurocysticercosis (NCC) is a CNS disease caused by T. solium. It was responsible for around 28,000 deaths in 2010. NCC is a neglected tropical disease and is mainly found in countries with ineffective healthcare systems (46). The clinical manifestations associated with NCC include seizures, epilepsy, focal neurologic impairments, elevated intracranial pressure, and cognitive decline. The disease manifestations depend on the number and size of the cysticercus or the parasitic stage development (47). NCC is thought to result from the neuroimmune processes induced by the parasite’s eggs, which are produced in the intestine. Once the eggs have entered the bloodstream, they disseminate to various tissues - including the brain. After crossing the BBB, T. solium forms a cyst within the brain and can persist for several month or years - probably through active evasion and immunosuppression (47–49). After 3 to 10 years, the larvae degenerate (via as-yet unknown mechanisms) and lose their ability to control the CNS inflammation (47, 50). A TH1 pro-inflammatory response is then engaged; the TNF-α, IFN-γ, IL-1ß, IL-18, and IL12 released by microglia and macrophages trigger the expression of the adhesion molecules ICAM1 and VCAM1 and the chemoattractants and chemokines CXCL2, CXCL8, CCL5, CCL2, and CCL20 by astrocytes and endothelial cells. This damages the BBB and leads to cerebrospinal fluid leakage, greater leukocyte migration, and granuloma formation in the CNS (47, 50). The granulomas primarily comprise multinucleated giant cells, macrophages, T and B lymphocytes, plasma cells, neutrophils, eosinophils, and microglia around a degenerated cyst. The TH1 response is counterbalanced by a TH2 response characterized by IFN-γ, IL-18, IL-4, IL-10, IL-13 and TGF-β production and that leads to progressive fibrosis (47, 49, 51). Neuroinflammation and granuloma in NCC constitute a “double-edged sword”: the granuloma protects the adjacent CNS tissue from the parasite but leads to damaging fibrosis and seizures (52) (Figures 2, 3).
Neuroschistomiasis
Neuroschistomiasis (NS) is a poorly characterized neuroinflammatory disease remaining caused by S. mansoni, S. japonicum, and S. haematobium. The prevalence of NS has not been reliably determined, although at least 600 million people in at least 79 countries are thought to be at risk of infection (53). About 2 to 4% of individuals infected by S.japonicum develop neurological manifestations (54). Patients suffering from NS generally have headaches, visual disturbances, delirium, seizures, motor impairments, ataxia, and encephalopathy (55). NS results from an immune process associated with the ectopic deposition of eggs in the leptomeninges and the cerebral cortex, via as-yet unknown mechanisms (53). By analogy with NCC, it has been suggested that eggs in the brain escape the immune response and survive there for weeks. However, larval degeneration triggers the release of antigens and a TH1 pro-inflammatory response favoring (i) the recruitment of CD4+ T cells, eosinophils, macrophages and monocytes, and (ii) granuloma formation, leading to brain tissue necrosis (53). In fact, the TH1 response (characterized by IFN-γ, IL-2, IL-4, IL-5, IL-9 and IL-13 production) is gradually replaced by a TH2 anti-inflammatory response with overproduction of IL-10 (limiting the degree of neurologic damage) (54) (Figures 1, 2). However, many questions remain with regard to the pathophysiological mechanisms associated with NS, including how the parasite interacts with glial cells, how it crosses the BBB, and how it survives in the brain.
Indeed, the current lack of effective treatments and the emergence of resistant parasites highlight the urgent need for novel biomarkers and potentially curative therapeutics for these neuroinflammatory diseases. Targeting the GM (in order to reduce the glial cells’ pro-inflammatory activity and thus counteract the brain inflammation) is an interesting strategy, as already demonstrated for Alzheimer’s disease, Parkinson's disease or multiple sclerosis (17, 19, 20). However, researchers are only now starting to investigate the GM’s effect on parasite-induced neuroinflammatory diseases and to develop prebiotics, probiotics or fecal transplantation techniques for preventing and dampening the disease process (56–58).
Involvement of the GM in parasite-driven CNS inflammation
The host’s microbiota is a consortium of bacteria, viruses, fungi, archaea, and protozoa. These microorganisms co-evolved with the host and colonize the skin and the respiratory, urogenital and gastrointestinal tracts (59). Commensal communities are essential for the body’s development and functioning. The GM harbors trillions of microbes (mostly bacteria); an adult human harbors around 200 to 300 different bacterial species, most of which are located in the ileum and the colon (60) and interact with each other (61). Whereas the human genome contains about 23,000 genes, the GM provides up to 10 million genes in total (62) and about 600,000 in a given individual (63). Even though the GM is highly resilient, its composition changes and can be modulated by factors like diet, age, sex, geographic location, ethnicity, exercise, and drug intake (63). The GM’s crucial role in human health comes through a variety of physiological mechanisms: modulation of the gut barrier and the host’s metabolism (64, 65), local and systemic immune regulation (66, 67), neural development, and emotional development (65, 68–70). Abnormal changes in the GM’s composition or activity – a so-called state of dysbiosis – are thought to be involved in the development of many diseases. It has recently been shown that dysbiosis contributes to extra-intestinal diseases and notably those affecting the CNS (71). Dysbiosis is involved in the development of the brain (68) but also in mental illnesses, such as eating disorders (72), autism (73), schizophrenia, anxiety disorders, mood disorders (74), and neurodegenerative diseases associated with neuroinflammation (such as Alzheimer’s and Parkinson’s diseases) (21, 75–79). In patients suffering from neurodegenerative diseases, a growing body of evidence indicates an association between GM dysbiosis and leakage of the intestinal barrier. This leakage favors inflammatory responses and the release of compounds into the systemic circulation, which worsen the brain inflammatory process (21, 80, 81).
A link between the GM and the brain has been evidenced by experiments in germ-free mice, in which damage to the BBB can be partially restored by fecal transplants (82, 83). Microglia from germ-free mice or mice treated with an antibiotic presented an immature profile and an impaired immune response. This defect of the GM is associated with changes in microglial mRNA profiles in the germ-free mice: genes involved in cell activation, pathogen recognition, and host defense were downregulated in the animals’ microglia, whereas genes encoding survival factors (which are usually suppressed in conventional animals) were upregulated (84, 85). Furthermore, other experiments in animal models showed that antibiotic-associated dysbiosis reduced neurogenesis in the hippocampus and thereby induced memory loss (83, 86). On the same lines, many research studies have highlighted the GM’s impact on the BBB’s integrity and the activity of CNS cells – either by modulating metabolites produced by bacterial species (82, 87–91), modulating the immune response (86, 92–96) or even by influencing the activity of the vagal nerve (97).
Even if Toxoplasma, Plasmodium, Trypanosoma and Schistosoma are all known to disturb the GM, a link with brain inflammation resulting from infection is not clearly established (56, 98–101). However, modification of the GM’s composition has been described during toxoplasmosis since gram-negative bacteria like the enterobacteria aggravated the ileitis induced by T. gondii infection by perturbing tryptophan metabolism, dopamine level and decreasing Treg counts (66, 102, 103). Ileitis in T. gondii-infected mice was associated with elevated intestinal permeability, greater bacterial translocation, mild-to-moderate meningitis, behavior and cognitive disorder in wild-type mice (relative to germ-free mice) (102, 104–107).
GM dysbiosis has also been observed in P. yoelii- and P.berghei ANKA-infected C57BL/6 mice. Dysbiosis was generally associated with a low Firmicutes count and elevated Proteobacteria and Verrucomicrobiae counts (98, 108). These preclinical results were confirmed in a study of P. falciparum-infected children living in a rural village in Mali: the GM was enriched in Bacteroidetes and depleted in Firmicutes, relative to the GM in European children. We are investigating the possible association between the development of cerebral malaria and the composition of the GM. Our preliminary results suggest a role for GM dysbiosis in the brain inflammatory process leading to cerebral malaria in C57BL/6 susceptible mice infected with P. berghei ANKA by modifying the pro-inflammatory response of glial cells. Moreover, dysbiosis induced by antibiotics or probiotics protect against cerebral malaria These data corroborate previous published work showing that the P. berghei ANKA infection induced changes in the GM which in turn impact malaria severity (56, 98). GM dysbiosis has also been observed during cerebral trypanosomiasis gut microbiota in mice and human (101). GM dysbiosis was associated with alterations in fatty acids and bile acids metabolism in infected mice (109). In addition, it has been recently described that the increase of BBB permeability correlates with increased levels of IL-17, IL-22 and brain infiltration by bacterial metabolites, notably butyrate (110). This results in a rise in glutamate, excitotoxicity and cell death in the brain parenchyma (111). In neuroschistosomiasis, a recent study has shown that CNS macrophages including microglia play a role in forming granulomas around the parasite eggs (55). Indeed, in mice infected with S. japonicum it has been recently shown alterations in the intestinal barrier and GM composition upon infection with a possible link to microglial cells phenotype (112, 113).
The consequences of GM modification by a parasite infection
Parasite infections can induce dysbiosis and thus impact the host’s inflammatory response and susceptibility to infection (114). Indeed, it has been demonstrated that the GM influences the pathogenicity of certain parasites or helminths directly (through bacteria-parasite interactions) or indirectly (by providing essential metabolites and therefore a more conducive environment for parasite proliferation and growth) (88). Of note, in vivo imaging of P. berghei-expressing luciferase has shown high amount of parasites in the mesentery and intestinal lumen which perturbated GM by favouring interactions and nutritional competition with gut bacteria (98, 115–119). Parasites can also modulate the GM composition indirectly through immunoregulatory factors. It is the case for T. gondii, IFN-γ and NO released during infection can cause the loss of Paneth cell and inhibit the production of antimicrobial peptides (116–119).
In some cases, the GM is enriched in certain bacterial phyla that reduce the severity of the disease associated with protozoan, fungal, helminth or bacterial infections (103). This is the case for P. berghei ANKA infections: the GM of "resistant" mice had a higher proportion of Firmicutes (56). It has also been shown that mice experimentally colonized with the α-galactosyl-expressing intestinal pathobiont E. coli O86: B7 produced anti-α-galactosyl antibodies, which prevent invasion of the liver by sporozoites (120).
Could modulation of the GM help to prevent the neuroinflammation induced by parasite infections?
It has been suggested that a eubiotic GM helps to maintain and/or restore homeostasis of the BBB and to prevent inflammatory processes in the brain. Various signaling molecules produced by the GM enable microbes to communicate with the host’s neuro-immune system, and cytokines are produced by the host in response to microbes (121). Most of these molecules act on glial cells (Figures 4, 5) and might modulate inflammation. Indeed, short-chain fatty acids (SCFAs) can influence the gut–brain axis via various mechanisms, including epigenetic modifications. SCFAs can inhibit the TLR4/TBK1/NF-κB/TNF-α pathway in astrocytes and microglial cells by inhibiting histone deacetylase, which results in downregulation of pro-inflammatory cytokine/chemokines production (20, 115, 124–127). Acetate can cross the BBB, and butyrate can restore the barrier’s intactness. SCFAs can also reverse dysfunctional microglial phenotypes in GF mice (82). Interestingly, aryl hydrocarbon receptor agonists inhibit VEGF-ß production and stimulate TGF-α production by the microglia and thereby limit the astrocyte’s inflammatory response, disturbance of the BBB, and the infiltration of leukocytes into the CNS (116, 117) (Figure 5).
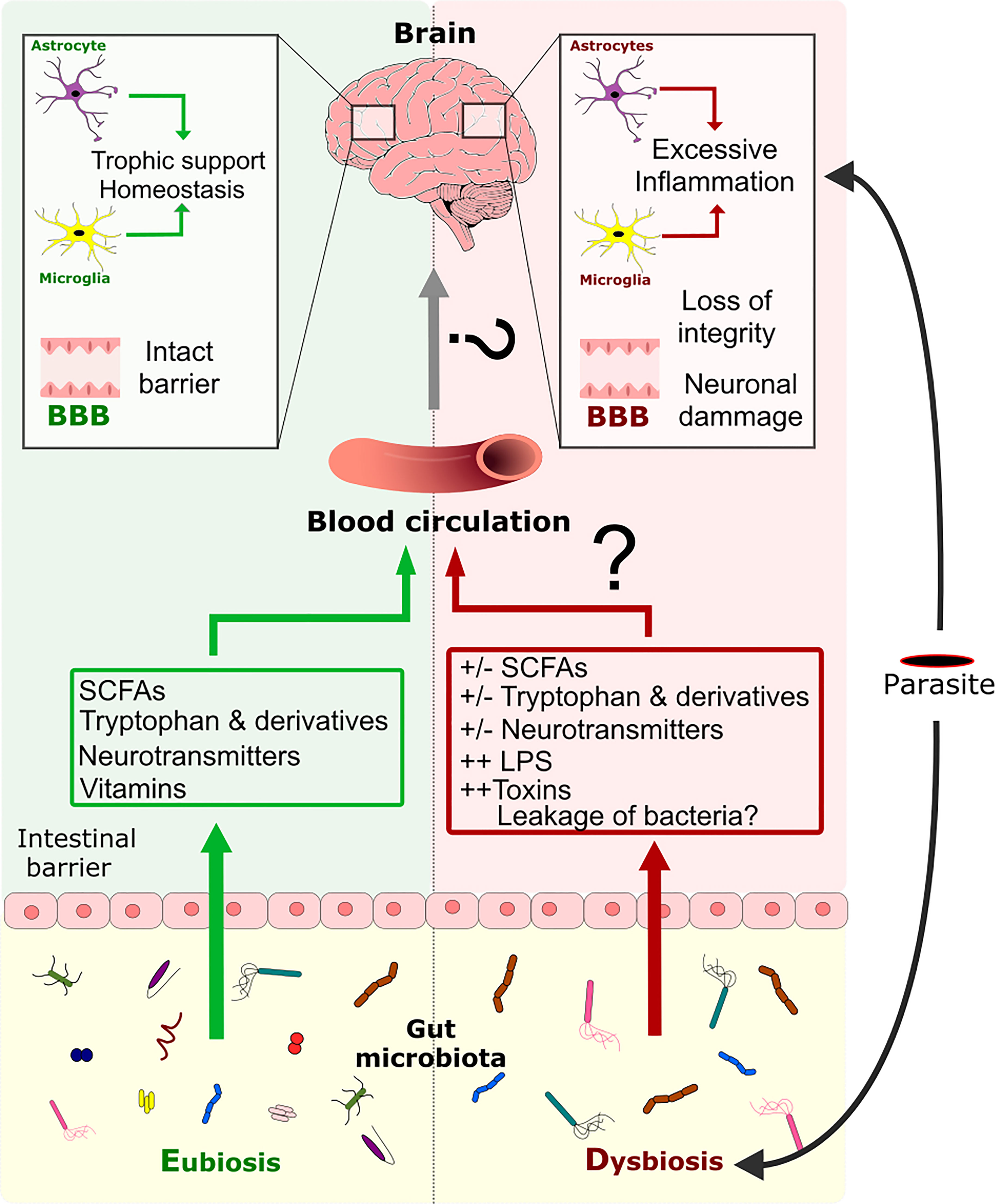
Figure 4 Modulation of the CNS inflammation response by the GM during a parasite infection. Eubiosis of GM favors the maintenance of the gut barrier’s intactness (122, 123). Moreover, the GM can produce metabolites like SCFAs, tryptophan, tryptophan derivatives, neurotransmitters, and vitamins, which are disseminated through the host’s circulation. These metabolites are known to have an impact on the BBB’s intactness and on CNS cells like astrocytes and microglia. Infection by a parasite induces dysbiosis of the GM directly or indirectly, which perturbs metabolite production, impairs gut barrier intactness and allows the possible translocation of bacteria throughout the organism. Dysbiosis is associated with an impairment of glial cell activity and loss of the BBB’s intactness. Dysbiosis might also favor the excessive pro-inflammatory response of glial cells induced by the parasite and that leads to neuronal damage.
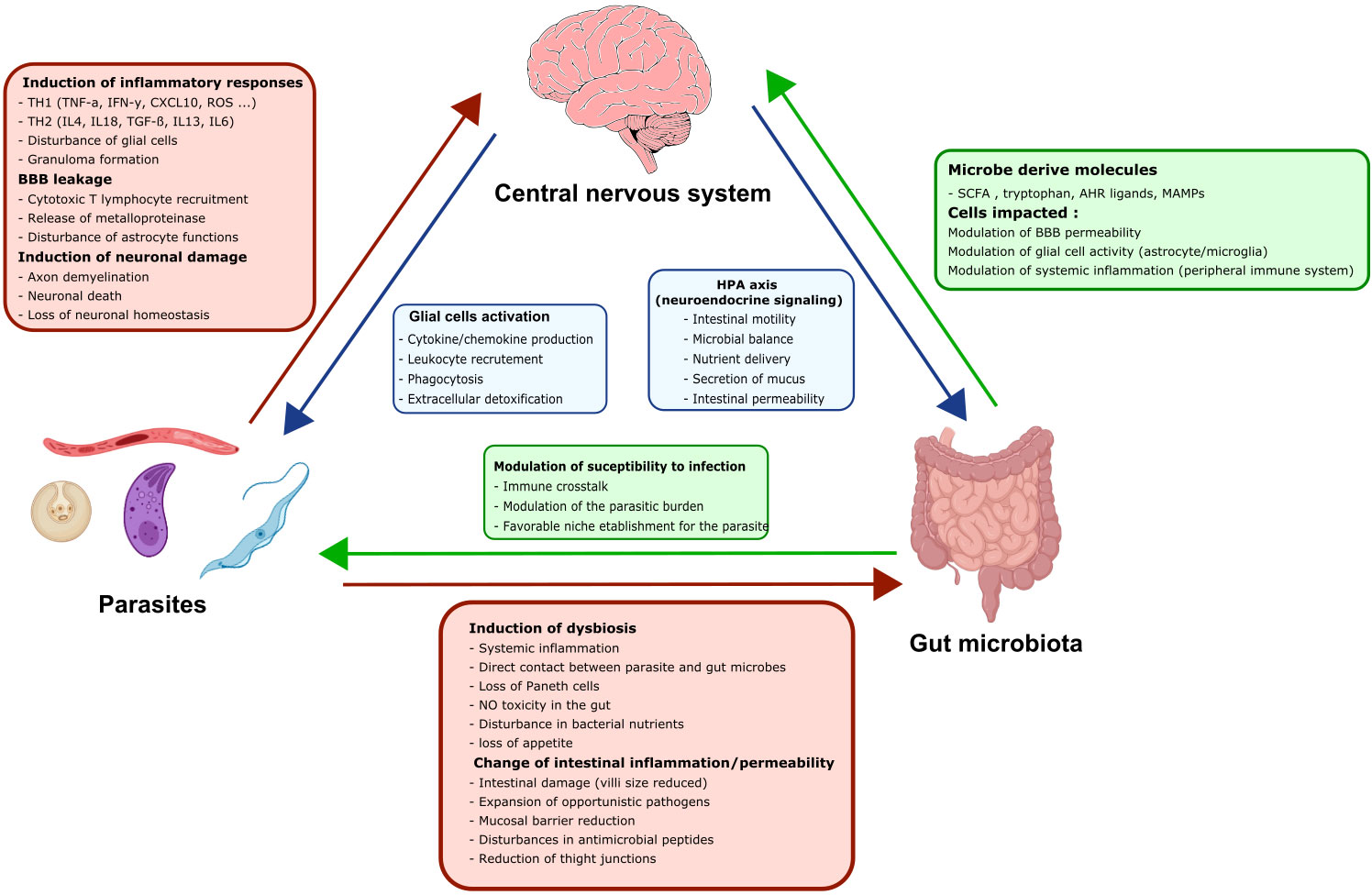
Figure 5 The three-way dialogue between parasites, the gut microbiota, and glial cells. A complex bidirectional dialogue exists between the gut microbiota, the CNS and the parasite. Green boxes and arrows summarize gut microbiota interactions with the CNS and the parasites. Red boxes and arrows represent parasites interactions with the CNS and the gut microbiota. Blue boxes and arrows indicate CNS interactions with the gut microbiota and the parasites.
Furthermore, many researchers have reported on the GM’s influence through the synthesis of various neurotransmitters and neuromodulators with crucial roles in gut-brain communication (Figure 5). Indeed, some bacteria are able to produce amino acids like gamma-aminobutyric acid (the main inhibitory neurotransmitter in the CNS). The GM can also regulate the level of serotonin, which has central roles in anxiety and depression and mediates changes in hippocampal levels of 5-hydroxyindoleacetic acid and brain-derived neurotrophic factor and plasma levels of tryptophan (118). The GM can notably control neurotransmitter production through the regulation of glutamate, which can be neurotoxic at high levels (119, 128, 129). Recently, bacterial-surface-derived compounds like peptidoglycans have emerged as potential key regulators of GM–brain interactions (130).
Several strategies can be used to restore the GM, such as (i) the administration of specific nutrients that promote the growth of certain bacterial species (i.e. prebiotics), (ii) the introduction or expansion of "beneficial" bacteria species (i.e. probiotics), and (iii) the wholesale or selective transplantation of a donor GM (i.e. fecal transplantation) (57). Many probiotics reportedly exhibit anti-inflammatory properties, notably in the context of inflammatory bowel diseases (122, 131). Several probiotics have been shown to communicate with the brain and influence behavior through vagal nerve signaling. Promising results have also been reported in the context of autoimmune neurodegenerative diseases like Parkinson's disease and Alzheimer's disease. Lactobacillus reuteri is able to metabolize tryptophan into indoles, which bind to the aryl hydrocarbon receptor expressed by astrocytes and microglia; this modulates the production of pro-inflammatory chemokine/cytokines and the cells’ ability to respond to lipopolysaccharide by limiting NFκB translocation (90, 117, 132–134) (Figure 4). Probiotics might also interact with enteroendocrine cells, i.e. sensory cells that form synapses with vagal afferents (neuropodia) (135). Lastly, we have shown that some probiotics exhibit anti-inflammatory properties via the interaction between peptidoglycan and NOD2 (136). We speculate that probiotic-derived peptidoglycans might constitute a novel means of treating neuro-inflammation.
Along with antiparasitic drugs, the value of fecal transplants and probiotics was recently evaluated in the context of infections by protozoans (such as Giardia duodenalis, Cryptosporidium parvum, Eimeria tenella) and nematodes (such as Toxocara canis and Strongyloides venezuelensis) (58). Fecal transplantation appears to be the most effective way of restoring the GM and combating neuroinflammatory diseases but is used on critical cases only. A better understanding of the molecular crosstalk between the GM and the brain should lead to the development of novel therapies for CNS inflammation caused by parasite infections.
Conclusion
The brain inflammation processes induced during parasite infections are not well understood and cannot easily be treated. Therefore, any treatment that can reduce this neuroinflammation will have a considerable public health impact. In this context, strategies that target the GM (notably by the development of food supplements able to regulate acute, harmful inflammation in particular and/or neuroinflammatory diseases in general) should be taken into consideration, along with antiparasite drugs.
Author contributions
SP designed, revised and supervised the work. JA reviewed the literature, created the figures, and wrote the manuscript. IL and CG participated to the analysis, drafting and revising the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by REGION HAUTS-DE-FRANCE, Innovation program START AIRR “PROMAL” and LabEx PARAFRAP ANR-11-LABX-0024i.
Acknowledgments
We wish to thank all the authors who have participated in this Research Topic and the reviewers for their insightful comments and suggestions. AJ was funded by the College Doctoral Nord Lille de France. University of Lille. We also thank Mr David FRASER at Biotech Communication SARL for language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1024998/full#supplementary-material
References
1. Casadevall A, Pirofski L. The damage-response framework of microbial pathogenesis. Nat Rev Microbiol (2003) 1:17–24. doi: 10.1038/nrmicro732
2. Organisation mondiale de la santé. World malaria report 2018. (Geneva: World Health Organization) (2018).
3. Mung’Ala-Odera V, Snow RW, Newton CRJC. The burden of the neurocognitive impairment associated with plasmodium falciparum malaria in sub-saharan Africa. Am J Trop Med Hyg (2004) 71:64–70. doi: 10.4269/ajtmh.2004.71.64
4. Lundkvist GB, Kristensson K, Bentivoglio M. Why trypanosomes cause sleeping sickness. Physiology (2004) 19:198–206. doi: 10.1152/physiol.00006.2004
5. Büscher P, Cecchi G, Jamonneau V, Priotto G. Human African trypanosomiasis. Lancet (2017) 390:2397–409. doi: 10.1016/S0140-6736(17)31510-6
6. Subhramanyam CS, Wang C, Hu Q, Dheen ST. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol (2019) 94:112–20. doi: 10.1016/j.semcdb.2019.05.004
7. Jha MK, Jo M, Kim J-H, Suk K. Microglia-astrocyte crosstalk: An intimate molecular conversation. Neurosci (2019) 25:227–40. doi: 10.1177/1073858418783959
8. Yang Q, Zhou J. Neuroinflammation in the central nervous system: Symphony of glial cells. Glia (2019) 67:1017–35. doi: 10.1002/glia.23571
9. Linnerbauer M, Wheeler MA, Quintana FJ. Astrocyte crosstalk in CNS inflammation. Neuron (2020) 108:608–22. doi: 10.1016/j.neuron.2020.08.012
10. Bentivoglio M, Mariotti R, Bertini G. Neuroinflammation and brain infections: Historical context and current perspectives. Brain Res Rev (2011) 66:152–73. doi: 10.1016/j.brainresrev.2010.09.008
11. Masocha W, Kristensson K. Passage of parasites across the blood-brain barrier. Virulence (2012) 3:202–12. doi: 10.4161/viru.19178
12. Mallewa M, Wilmshurst JM. Overview of the effect and epidemiology of parasitic central nervous system infections in African children. Semin Pediatr Neurol (2014) 21:19–25. doi: 10.1016/j.spen.2014.02.003
13. Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C, et al. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci (2002) 202:13–23. doi: 10.1016/S0022-510X(02)00207-1
14. Escartin C, Pierre K, Colin A, Brouillet E, Delzescaux T, Guillermier M, et al. Activation of astrocytes by CNTF induces metabolic plasticity and increases resistance to metabolic insults. J Neurosci (2007) 27:7094–104. doi: 10.1523/JNEUROSCI.0174-07.2007
15. Singhal G, Baune BT. Microglia: An interface between the loss of neuroplasticity and depression. Front Cell Neurosci (2017) 11. doi: 10.3389/fncel.2017.00270
16. Luis Muñoz-Carrillo J, Contreras-Cordero JF, Gutiérrez-Coronado O, Villalobos-Gutiérrez Trinidad P, Ramos-Gracia LG, Hernández-Reyes VE. Cytokine profiling plays a crucial role in activating immune system to clear infectious pathogens. In: Tyagi RK, Bisen PS, editors. Immune response activation and immunomodulation. (London: IntechOpen) (2019).
17. Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci (2017) 20:145–55. doi: 10.1038/nn.4476
18. Cekanaviciute E, Yoo BB, Runia TF, Debelius JW, Singh S, Nelson CA, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci (2017) 114:10713–8. doi: 10.1073/pnas.1711235114
19. Westfall S, Lomis N, Kahouli I, Dia SY, Singh SP, Prakash S, et al. Microbiome, probiotics and neurodegenerative diseases: deciphering the gut brain axis. Cell Mol Life Sci (2017) 74:3769–87. doi: 10.1007/s00018-017-2550-9
20. Sun M-F, Zhu YL, Zhou ZL, Jia XB, Xu YD, Yang Q, et al. Neuroprotective effects of fecal microbiota transplantation on MPTP-induced parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-α signaling pathway. Brain Behav Immun (2018) 70:48–60. doi: 10.1016/j.bbi.2018.02.005
21. Kowalski K, Mulak A. Brain-Gut-Microbiota axis in alzheimer’s disease. J Neurogastroenterol Motil (2019) 25:48–60. doi: 10.5056/jnm18087
22. Ghazanfari N, Mueller SN, Heath WR. Cerebral malaria in mouse and man. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.02016
23. Schiess N, Villabona-Rueda A, Cottier KE, Huether K, Chipeta J, Stins MF, et al. Pathophysiology and neurologic sequelae of cerebral malaria. Malar J (2020) 19:266. doi: 10.1186/s12936-020-03336-z
24. Hunt NH, Golenser J, Chan-Ling T, Parekh S, Rae C, Potter S. Immunopathogenesis of cerebral malaria. Int J Parasitol (2006) 36:569–82. doi: 10.1016/j.ijpara.2006.02.016
25. Baptista FG, Pamplona A, Pena AC, Mota MM, Pied S, Vigario AM, et al. Accumulation of Plasmodium berghei -infected red blood cells in the brain is crucial for the development of cerebral malaria in mice. Infect Immun (2010) 78:4033–9. doi: 10.1128/IAI.00079-10
26. Shrivastava SK, Dalko E, Delcroix-Genete D, Herbert F, Cazenave P-A, Pied S, et al. Uptake of parasite-derived vesicles by astrocytes and microglial phagocytosis of infected erythrocytes may drive neuroinflammation in cerebral malaria. Plasmodium Interact Glial Cells Glia (2017) 65:75–92. doi: 10.1002/glia.23075
27. Leleu I, Genete D, Desnoulez SS, Saidi N, Brodin P, Lafont F, et al. A noncanonical autophagy is involved in the transfer of Plasmodium -microvesicles to astrocytes. Autophagy (2021) 18(7):1583–98. doi: 10.1080/15548627.2021.1993704
28. Mazier D, Nitcheu J, Idrissa-Boubou M. Cerebral malaria and immunogenetics. Parasit Immunol (2000) 22:613–23. doi: 10.1046/j.1365-3024.2000.00342.x
29. Belnoue E, Kayibanda M, Vigario AM, Deschemin J-C, Rooijen van N M, et al. On the pathogenic role of brain-sequestered αβ CD8 + T cells in experimental cerebral malaria. J Immunol (2002) 169:6369–75. doi: 10.4049/jimmunol.169.11.6369
30. Bagot S, Idrissa Boubou M, Campino S, Behrschmidt C, Gorgette O, Guénet JL, et al. Susceptibility to experimental cerebral malaria induced by Plasmodium berghei ANKA in inbred mouse strains recently derived from wild stock. Infect Immun (2002) 70:2049–56. doi: 10.1128/IAI.70.4.2049-2056.2002
31. Bagot S, Nogueira F, Collette A, do Rosario V, Lemonier F, Cazenave PA, et al. Comparative study of brain CD8 + T cells induced by sporozoites and those induced by blood-stage Plasmodium berghei ANKA involved in the development of cerebral malaria. Infect Immun (2004) 72:2817–26. doi: 10.1128/IAI.72.5.2817-2826.2004
32. Campanella GSV, Tager AM, El Khoury JK, Thomas SY, Abrazinski TA, Manice LA, et al. Chemokine receptor CXCR3 and its ligands CXCL9 and CXCL10 are required for the development of murine cerebral malaria. Proc Natl Acad Sci (2008) 105:4814–9. doi: 10.1073/pnas.0801544105
33. World Health Organization, WHO Expert Committee on the Control and Surveillance of Human African Trypanosomiasis Control and surveillance of human African trypanosomiasis: report of a WHO expert committee. (Geneva, Switzerland: World Health Organization) (2013). Available at: https://apps.who.int/iris/handle/10665/95732
34. Kristensson K, Nygård M, Bertini G, Bentivoglio M. African Trypanosome infections of the nervous system: Parasite entry and effects on sleep and synaptic functions. Prog Neurobiol (2010) 91:152–71. doi: 10.1016/j.pneurobio.2009.12.001
35. Sharafeldin A, Eltayeb R, Pashenkov M, Bakhiet M. Chemokines are produced in the brain early during the course of experimental African trypanosomiasis. J Neuroimmunol (2000) 103:165–70. doi: 10.1016/S0165-5728(99)00238-6
36. Kennedy PGE. Human African trypanosomiasis of the CNS: current issues and challenges. J Clin Invest (2004) 113:496–504. doi: 10.1172/JCI200421052
37. Moreno C, Temporão A, Torres T, Sousa Silva M. Trypanosoma brucei interaction with host: Mechanism of VSG release as target for drug discovery for African trypanosomiasis. Int J Mol Sci (2019) 20:1484. doi: 10.3390/ijms20061484
38. Rodgers J, Steiner I, Kennedy PGE. Generation of neuroinflammation in human African trypanosomiasis. Neurol - Neuroimmunol Neuroinflamm (2019) 6:e610. doi: 10.1212/NXI.0000000000000610
39. Schlüter D, Barragan A. Advances and challenges in understanding cerebral toxoplasmosis. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00242
40. Persson CM, Lambert H, Vutova PP, Dellacasa-Lindberg I, Nederby J, Yagita H, et al. Transmission of Toxoplasma gondii from infected dendritic cells to natural killer cells. Infect Immun (2009) 77:970–6. doi: 10.1128/IAI.00833-08
41. Mineo JR, Kasper LH. Attachment of toxoplasma gondii to host cells involves major surface protein, SAG-1 (P-30). Exp Parasitol (1994) 79:11–20. doi: 10.1006/expr.1994.1054
42. Silva NM, Manzan RM, Carneiro WP, Milanezi CM, Silva JS, Ferro Vieira EA, et al. Toxoplasma gondii: The severity of toxoplasmic encephalitis in C57BL/6 mice is associated with increased ALCAM and VCAM-1 expression in the central nervous system and higher blood–brain barrier permeability. Exp Parasitol (2010) 126:167–77. doi: 10.1016/j.exppara.2010.04.019
43. Elsheikha HM, Marra CM, Zhu X-Q. Epidemiology, pathophysiology, diagnosis, and management of cerebral toxoplasmosis. Clin Microbiol Rev (2020) 34:e00115-19. doi: 10.1128/CMR.00115-19
44. Drögemüller K, Helmuth U, Brunn A, Sakowicz-Burkiewicz M, Gutmann DH, Mueller W, et al. Astrocyte gp130 expression is critical for the control of Toxoplasma encephalitis. J Immunol (2008) 181:2683–93. doi: 10.4049/jimmunol.181.4.2683
45. Cekanaviciute E, Dietrich HK, Axtell RC, Williams AM, Egusquiza R, Wai KM, et al. Astrocytic TGF-β signaling limits inflammation and reduces neuronal damage during central nervous system. Toxoplasma Infect J Immunol (2014) 193:139–49. doi: 10.4049/jimmunol.1303284
46. Mewara A, Goyal K, Sehgal R. Neurocysticercosis: A disease of neglect. Trop Parasitol (2013) 3:106–13. doi: 10.4103/2229-5070.122111
47. Fleury A, Cardenas G, Adalid-Peralta L, Fragoso G, Sciutto E. Immunopathology in. Taenia solium neurocysticercosis Parasit Immunol (2016) 38:147–57. doi: 10.1111/pim.12299
48. Adalid-Peralta L, Arce-Sillas A, Fragoso G, Cárdenas G, Rosetti M, Casanova-Hernández D, et al. Cysticerci drive dendritic cells to promote In vitro and In vivo tregs differentiation. Clin Dev Immunol (2013) 2013:1–9. doi: 10.1155/2013/981468
49. Garcia HH, O’Neal SE, Noh J, Handali S, The Cysticercosis Working Group in Peru. Laboratory diagnosis of neurocysticercosis (Taenia solium). J Clin Microbiol (2018) 56::e00424-18. doi: 10.1128/JCM.00424-18
50. Prodjinotho UF, Lema J, Lacorcia M, Schmidt V, Vejzagic N, Sikasunge C, et al. Host immune responses during taenia solium neurocysticercosis infection and treatment. PloS Negl Trop Dis (2020) 14:e0008005. doi: 10.1371/journal.pntd.0008005
51. Restrepo BI, Alvarez JI, Castaño JA, Arias LF, Restrepo M, Trujillo J, et al. Brain granulomas in neurocysticercosis patients are associated with a Th1 and Th2 profile. Infect Immun (2001) 69:4554–60. doi: 10.1128/IAI.69.7.4554-4560.2001
52. Giorgio S, Gallo-Francisco PH, Roque GAS, Flóro e Silva M. Granulomas in parasitic diseases: the good and the bad. Parasitol Res (2020) 119:3165–80. doi: 10.1007/s00436-020-06841-x
53. Carod-Artal FJ. Neurological complications of schistosoma infection. Trans R Soc Trop Med Hyg (2008) 102:107–16. doi: 10.1016/j.trstmh.2007.08.004
54. Ferrari TCA, Gazzinelli G, Corrêa-Oliveira R. Immune response and pathogenesis of neuroschistosomiasis mansoni. Acta Trop (2008) 108:83–8. doi: 10.1016/j.actatropica.2008.02.010
55. Tan Z, Lei Z, Zhang Z, Zhang H, Shu K, Hu F, et al. Identification and characterization of microglia/macrophages in the granuloma microenvironment of encephalic schistosomiasis japonicum. BMC Infect Dis (2019) 19:1088. doi: 10.1186/s12879-019-4725-5
56. Villarino NF, LeCleir GR, Denny JE, Dearth SP, Harding CL, Sloan SS, et al. Composition of the gut microbiota modulates the severity of malaria. Proc Natl Acad Sci (2016) 113:2235–40. doi: 10.1073/pnas.1504887113
57. Hyland N, Stanton C. The gut-brain axis: dietary, probiotic, and prebiotic interventions on the microbiota. (Amsterdam: Elsevier/Academic Press) (2016).
58. Ribeiro CdM, Zorgi NE, Meireles LR, Garcia JL, de Andrade Junior HF. CD19 LYMPHOCYTE PROLIFERATION INDUCED BY bifidobacterium animalis subsp. lactis IN C57BL/6 MICE EXPERIMENTALLY INFECTED WITH toxoplasma gondii. Rev Inst Med Trop São Paulo (2016) 58:26. doi: 10.1590/S1678-9946201658026
59. Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? revisiting the ratio of bacterial to host cells in humans. Cell (2016) 164:337–40. doi: 10.1016/j.cell.2016.01.013
60. MetaHIT Consortium, Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature (2010) 464:59–65. doi: 10.1038/nature08821
61. Gerhardt S, Mohajeri M. Changes of colonic bacterial composition in parkinson’s disease and other neurodegenerative diseases. Nutrients (2018) 10:708. doi: 10.3390/nu10060708
62. MetaHIT Consortium, Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol (2014) 32:834–41. doi: 10.1038/nbt.2942
63. Dogra SK, Doré J, Damak S. Gut microbiota resilience: Definition, link to health and strategies for intervention. Front Microbiol (2020) 11:572921. doi: 10.3389/fmicb.2020.572921
64. LeBlanc JG, Milani C, Giori GS, Sesma F, Sinderen van D, Ventura M, et al. Bacteria as vitamin suppliers to their host: a gut microbiota perspective. Curr Opin Biotechnol (2013) 24:160–8. doi: 10.1016/j.copbio.2012.08.005
65. Flint HJ, Scott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol (2012) 9:577–89. doi: 10.1038/nrgastro.2012.156
66. Dodd D, Spitzer MH, Treuren Van W, Merrill BD, Hryckowian AJ, Higginbottom SK, Le A, et al, et al. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature (2017) 551:648–52. doi: 10.1038/nature24661
67. Geuking MB, Cahenzli J, Lawson MAE, Ng DCK, Slack E, Hapfelmeier S, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity (2011) 34:794–806. doi: 10.1016/j.immuni.2011.03.021
68. Cox LM, Weiner HL. Microbiota signaling pathways that influence neurologic disease. Neurotherapeutics (2018) 15:135–45. doi: 10.1007/s13311-017-0598-8
69. Denny JE, Powers JB, Castro HF, Zhang J, Joshi-Barve S, Campagna SR, et al. Differential sensitivity to plasmodium yoelii infection in C57BL/6 mice impacts gut-liver axis homeostasis. Sci Rep (2019) 9:3472. doi: 10.1038/s41598-019-40266-6
70. Mayer EA, Tillisch K, Gupta A. Gut/brain axis and the microbiota. J Clin Invest (2015) 125:926–38. doi: 10.1172/JCI76304
71. Rutsch A, Kantsjö JB, Ronchi F. The gut-brain axis: How microbiota and host inflammasome influence brain physiology and pathology. Front Immunol (2020) 11:604179. doi: 10.3389/fimmu.2020.604179
72. Kleiman SC, Watson HJ, Bulik-Sullivan EC, Huh EY, Tarantino LM, Bulik CM, et al. The intestinal microbiota in acute anorexia nervosa and during renourishment: Relationship to depression, anxiety, and eating disorder psychopathology. Psychosom Med (2015) 77:969–81. doi: 10.1097/PSY.0000000000000247
73. Kang D-W, Park JG, Ilhan ZE, Wallstrom G, LaBaer J, Adams JB, et al. Reduced incidence of prevotella and other fermenters in intestinal microflora of autistic children. PloS One (2013) 8:e68322. doi: 10.1371/journal.pone.0068322
74. Jiang H, Ling Z, Zhang Y, Mao H, Ma Z, Yin Y, et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun (2015) 48:186–94. doi: 10.1016/j.bbi.2015.03.016
75. Roy Sarkar S, Banerjee S. Gut microbiota in neurodegenerative disorders. J Neuroimmunol (2019) 328:98–104. doi: 10.1016/j.jneuroim.2019.01.004
76. Braak H, de Vos RAI, Bohl J, Del Tredici K. Gastric α-synuclein immunoreactive inclusions in meissner’s and auerbach’s plexuses in cases staged for parkinson’s disease-related brain pathology. Neurosci Lett (2006) 396:67–72. doi: 10.1016/j.neulet.2005.11.012
77. Kieburtz K, Wunderle KB. Parkinson’s disease: Evidence for environmental risk factors: Evidence for environmental risk factors. Mov Disord (2013) 28:8–13. doi: 10.1002/mds.25150
78. Savica R, Carlin JM, Grossardt BR, Bower JH, Ahlskog JE, Maraganore DM, et al. Medical records documentation of constipation preceding Parkinson disease: A case-control study. Neurology (2009) 73:1752–8. doi: 10.1212/WNL.0b013e3181c34af5
79. Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH. Is alpha-synuclein in the colon a biomarker for premotor parkinson’s disease? evidence from 3 cases: Is alpha-synuclein a biomarker for premotor PD? Mov Disord (2012) 27:716–9. doi: 10.1002/mds.25020
80. Keshavarzian A, Green SJ, Engen PA, Voigt RM, Naqib A, Forsyth CB, et al. Colonic bacterial composition in parkinson’s disease: COLONIC MICROBIOTA IN PARKINSON’S DISEASE. Mov Disord (2015) 30:1351–60. doi: 10.1002/mds.26307
81. Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, et al. Gut microbiome alterations in alzheimer’s disease. Sci Rep (2017) 7:13537. doi: 10.1038/s41598-017-13601-y
82. Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med (2014) 6:263ra158. doi: 10.1126/scitranslmed.3009759
83. Luczynski P, Neufeld KAM, Oriach CS, Clarke G, Dinan TG, Cryan JF, et al. Growing up in a bubble: Using germ-free animals to assess the influence of the gut microbiota on brain and behavior. Int J Neuropsychopharmacol (2016) 19:pyw020. doi: 10.1093/ijnp/pyw020
84. Khosravi A, Yáñez A, Price JG, Chow A, Merad M, Goodridge HS, et al. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe (2014) 15:374–81. doi: 10.1016/j.chom.2014.02.006
85. Megur A, Baltriukienė D, Bukelskienė V, Burokas A. The microbiota–Gut–Brain axis and alzheimer’s disease: Neuroinflammation is to blame? Nutrients (2020) 13:37. doi: 10.3390/nu13010037
86. Parker A, Fonseca S, Carding SR. Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes (2020) 11:135–57. doi: 10.1080/19490976.2019.1638722
87. Dinan TG, Stanton C, Cryan JF. Psychobiotics: A novel class of psychotropic. Biol Psychiatry (2013) 74:720–6. doi: 10.1016/j.biopsych.2013.05.001
88. Giau V, Wu SY, Jamerlan A, An SSA, Kim SY, Hulme J, et al. Gut microbiota and their neuroinflammatory implications in alzheimer’s disease. Nutrients (2018) 10:1765. doi: 10.3390/nu10111765
89. Matt SM, Allen JM, Lawson MA, Mailing LJ, Woods JA, Johnson RW, et al. Butyrate and dietary soluble fiber improve neuroinflammation associated with aging in mice. Front Immunol (2018) 9. doi: 10.3389/fimmu.2018.01832
90. Rothhammer V, Mascanfroni ID, Bunse L, Takenaka MC, Kenison JE, Mayo L, et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat Med (2016) 22:586–97. doi: 10.1038/nm.4106
91. Stone TW, Darlington LG. The kynurenine pathway as a therapeutic target in cognitive and neurodegenerative disorders: Kynurenines and CNS disorders. Br J Pharmacol (2013) 169:1211–27. doi: 10.1111/bph.12230
92. Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Human gut microbes associated with obesity. Nature (2006) 444:1022–3. doi: 10.1038/4441022a
93. Lin P, Ding B, Feng C, Yin S, Zhang T, Qi X, et al. Prevotella and klebsiella proportions in fecal microbial communities are potential characteristic parameters for patients with major depressive disorder. J Affect Disord (2017) 207:300–4. doi: 10.1016/j.jad.2016.09.051
94. O’Brien SM, Scott LV, Dinan TG. Cytokines: abnormalities in major depression and implications for pharmacological treatment. Hum Psychopharmacol Clin Exp (2004) 19:397–403. doi: 10.1002/hup.609
95. Di Meo F, Donato S, Pardo Di A, Maglione V, Filosa S, Crispi S, et al. New therapeutic drugs from bioactive natural molecules: The role of gut microbiota metabolism in neurodegenerative diseases. Curr Drug Metab (2018) 19:478–89. doi: 10.2174/1389200219666180404094147
96. Mayer EA, Knight R, Mazmanian SK, Cryan JF, Tillisch K. Gut microbes and the brain: Paradigm shift in neuroscience. J Neurosci (2014) 34:15490–6. doi: 10.1523/JNEUROSCI.3299-14.2014
97. Forsythe P, Bienenstock J, Kunze WA. Vagal pathways for microbiome-Brain-Gut axis communication. In: Lyte M, Cryan JF, editors. Microbial endocrinology: The microbiota-Gut-Brain axis in health and disease. (New York, NY: Springer) (2014) 817:115–33.
98. Taniguchi T, Miyauchi E, Nakamura S, Hirai M, Suzue K, Imai T, et al. Plasmodium berghei ANKA causes intestinal malaria associated with dysbiosis. Sci Rep (2015) 5:15699. doi: 10.1038/srep15699
99. Jenkins TP, Peachey LE, Ajami NJ, MacDonald AS, Hsieh MH, Brindley PJ, et al. Schistosoma mansoni infection is associated with quantitative and qualitative modifications of the mammalian intestinal microbiota. Sci Rep (2018) 8:12072. doi: 10.1038/s41598-018-30412-x
100. Floudas A, Aviello G, Schwartz C, Jeffery IB PW, Fallon PG. Schistosoma mansoni worm infection regulates the intestinal microbiota and susceptibility to colitis. Infect Immun (2019) 87:e00275-19. doi: 10.1128/IAI.00275-19
101. Duarte-Silva E, Morais LH, Clarke G, Savino W, Peixoto C. Targeting the gut microbiota in chagas disease: What do we know so far? Front Microbiol (2020) 11. doi: 10.3389/fmicb.2020.585857
102. Nascimento BB, Cartelle CT, Noviello MdL , Pinheiro BV, Vitor Almeida RW, Souza DdG , et al. Influence of indigenous microbiota on experimental toxoplasmosis in conventional and germ-free mice. Int J Exp Pathol (2017) 98:191–202. doi: 10.1111/iep.12236
103. Santos LM, Commodaro AG, Vasquez ARR, Kohlhoff M, Guerra Paula DA, Coimbra RS, et al. Intestinal microbiota regulates tryptophan metabolism following oral infection with. Toxoplasma gondii Parasit Immunol (2020) 42:e12720. doi: 10.1111/pim.12720
104. Bereswill S, Kühl AA, Alutis M, Fischer A, Möhle L, Struck D, et al. The impact of toll-like-receptor-9 on intestinal microbiota composition and extra-intestinal sequelae in experimental toxoplasma gondii induced ileitis. Gut Pathog (2014) 6:19. doi: 10.1186/1757-4749-6-19
105. Craven M, Egan CE, Dowd SE, McDonough SP, Dogan B, Denkers EY, et al. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal crohn’s disease. PloS One (2012) 7:e41594.
106. Haag L-M, Fischer A, Otto B, Plickert R, Kühl AA, Göbel UB, et al. Intestinal microbiota shifts towards elevated commensal escherichia coli loads abrogate colonization resistance against campylobacter jejuni in mice. PloS One (2012) 7:e35988. doi: 10.1371/journal.pone.0035988
107. Egan CE, Cohen SB, Denkers EY. Insights into inflammatory bowel disease using Toxoplasma gondii as an infectious trigger. Immunol Cell Biol (2012) 90:668–75. doi: 10.1038/icb.2011.93
108. Mooney JP, Lokken KL, Byndloss MX, George MD, Velazquez EM, Faber F, et al. Inflammation-associated alterations to the intestinal microbiota reduce colonization resistance against non-typhoidal salmonella during concurrent malaria parasite infection. Sci Rep (2015) 5:14603. doi: 10.1038/srep14603
109. McCall L-I, Tripathi A, Vargas F, Knight R, Dorrestein PC, Siqueira-Neto JL, et al. Experimental chagas disease-induced perturbations of the fecal microbiome and metabolome. PloS Negl Trop Dis (2018) 12:e0006344. doi: 10.1371/journal.pntd.0006344
110. Luu M, Weigand K, Wedi F, Breidenbend C, Leister H, Pautz S, et al. Regulation of the effector function of CD8+ T cells by gut microbiota-derived metabolite butyrate. Sci Rep (2018) 8:14430. doi: 10.1038/s41598-018-32860-x
111. Duarte-Silva E, Maes M, Macedo D, Savino W, Peixoto CA. Shared neuroimmune and oxidative pathways underpinning chagas disease and major depressive disorder. Transl Psychiatry (2020) 10:419. doi: 10.1038/s41398-020-01105-9
112. Zhao Y, Yang S, Li B, Li W, Wang J, Chen Z, et al. Alterations of the mice gut microbiome via schistosoma japonicum ova-induced granuloma. Front Microbiol (2019) 10. doi: 10.3389/fmicb.2019.00352
113. Mossad O, Erny D. The microbiota–microglia axis in central nervous system disorders. Brain Pathol (2020) 30:1159–77. doi: 10.1111/bpa.12908
114. Sekirov I, Finlay BB. The role of the intestinal microbiota in enteric infection: Intestinal microbiota and enteric infections. J Physiol (2009) 587:4159–67. doi: 10.1113/jphysiol.2009.172742
115. Yamawaki Y, Yoshioka N, Nozaki K, Ito H, Oda K, Harada K, et al. Sodium butyrate abolishes lipopolysaccharide-induced depression-like behaviors and hippocampal microglial activation in mice. Brain Res (2018) 1680:13–38. doi: 10.1016/j.brainres.2017.12.004
116. Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-a drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest (2012) 122:2454–68. doi: 10.1172/JCI60842
117. Wang Y, Wang Z, Wang Y, Li F, Jia J, Song X, et al. The gut-microglia connection: Implications for central nervous system diseases. Front Immunol (2018) 9:2325. doi: 10.3389/fimmu.2018.02325
118. Clarke G, Grenham S, Scully P, Fitzgerald P, Moloney RD, Shanahan F, et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry (2013) 18:666–73. doi: 10.1038/mp.2012.77
119. Haroon E, Miller AH, Sanacora G. Inflammation, glutamate, and glia: A trio of trouble in mood disorders. Neuropsychopharmacology (2017) 42:193–215. doi: 10.1038/npp.2016.199
120. Yilmaz B, Portugal S, Tran TM, Gozzelino R, Ramos S, Gomes J, et al. Gut microbiota elicits a protective immune response against malaria transmission. Cell (2014) 159:1277–89. doi: 10.1016/j.cell.2014.10.053
121. Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M, et al. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav Immun (2011) 25:397–407. doi: 10.1016/j.bbi.2010.10.023
122. Zaylaa M, Kassaa Al I, Alard J, Peucelle V, Boutillier D, Desramaut J, et al. Probiotics in IBD: Combining in vitro and in vivo models for selecting strains with both anti-inflammatory potential as well as a capacity to restore the gut epithelial barrier. J Funct Foods (2018) 47:304–15. doi: 10.1016/j.jff.2018.05.029
123. Al-Rashidi HE. Gut microbiota and immunity relevance in eubiosis and dysbiosis. Saudi J Biol Sci (2022) 29:1628–43. doi: 10.1016/j.sjbs.2021.10.068
124. Erny D, Angelis ALH, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
125. Reddy DS, Wu X, Golub VM, Dashwood WM, Dashwood RH. Measuring histone deacetylase inhibition in the brain. Curr Protoc Pharmacol (2018) 81:e41. doi: 10.1002/cpph.41
126. Silva YP, Bernardi A, Frozza RL. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front Endocrinol (2020) 11:25. doi: 10.3389/fendo.2020.00025
127. Soliman ML, Puig KL, Combs CK, Rosenberger TA. Acetate reduces microglia inflammatory signaling. vitro J Neurochem (2012) 123:555–67. doi: 10.1111/j.1471-4159.2012.07955.x
128. Taylor DL. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor -induced neurotoxicity in concert with microglial-derived fas ligand. J Neurosci (2005) 25:2952–64. doi: 10.1523/JNEUROSCI.4456-04.2005
129. Matute C, Domercq M, Sánchez-Gómez M-V. Glutamate-mediated glial injury: Mechanisms and clinical importance. Glia (2006) 53:212–24. doi: 10.1002/glia.20275
130. Gonzalez-Santana A, Diaz Heijtz R. Bacterial peptidoglycans from microbiota in neurodevelopment and behavior. Trends Mol Med (2020) 26:729–43. doi: 10.1016/j.molmed.2020.05.003
131. Alard J, Peucelle V, Boutillier D, Breton J, Kuylle S, Pot B, et al. New probiotic strains for inflammatory bowel disease management identified by combining in vitro and in vivo approaches. Benef Microbes (2018) 9:317–31. doi: 10.3920/BM2017.0097
132. Zelante T, Iannitti RG, Cunha C, Luca De A, Giovannini G, Pieraccini G, et al. Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity (2013) 39:372–85. doi: 10.1016/j.immuni.2013.08.003
133. Liu T, Li J, Liu Y, Xiao N, Suo H, Xie K, et al. Short-chain fatty acids suppress lipopolysaccharide-induced production of nitric oxide and proinflammatory cytokines through inhibition of NF-κB pathway in RAW264.7 cells. Inflammation (2012) 35:1676–84. doi: 10.1007/s10753-012-9484-z
134. Lee Y-J, Park CE, Kim JH, Sohn HJ, Lee J, Jung SY, et al. Naegleria fowleri lysate induces strong cytopathic effects and pro-inflammatory cytokine release in rat microglial cells. Korean J Parasitol (2011) 49:285. doi: 10.3347/kjp.2011.49.3.285
135. Kaelberer MM, Rupprecht LE, Liu WW, Weng P, Bohórquez DV. Neuropod cells: The emerging biology of gut-brain sensory transduction. Annu Rev Neurosci (2020) 43:337–53. doi: 10.1146/annurev-neuro-091619-022657
Keywords: parasitic disease, brain-inflammation, immunopathophysiology, astrocytes, microglia, gut microbiota, immunoregulation
Citation: Alloo J, Leleu I, Grangette C and Pied S (2022) Parasite infections, neuroinflammation, and potential contributions of gut microbiota. Front. Immunol. 13:1024998. doi: 10.3389/fimmu.2022.1024998
Received: 22 August 2022; Accepted: 21 November 2022;
Published: 08 December 2022.
Edited by:
Joseli Lannes-Vieira, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Tatiana Maron-Gutierrez, Oswaldo Cruz Foundation (Fiocruz), BrazilErkin Seker, University of California, Davis, United States
Copyright © 2022 Alloo, Leleu, Grangette and Pied. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylviane Pied, c3lsdmlhbmUucGllZEBwYXN0ZXVyLWxpbGxlLmZy