
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 20 October 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1024755
This article is part of the Research Topic Targeted Therapies for Aggressive Cancers View all 5 articles
 Yu-Xiang Ma1†
Yu-Xiang Ma1† Fu-Rong Liu1†
Fu-Rong Liu1† Yang Zhang1†Qun Chen1Zhi-Qiang Chen1Qian-Wen Liu1Yan Huang2
Yang Zhang1†Qun Chen1Zhi-Qiang Chen1Qian-Wen Liu1Yan Huang2 Yun-Peng Yang2
Yun-Peng Yang2 Wen-Feng Fang2Ning Xi3Ning Kang3Yu-Lei Zhuang3Qi Zhang3Ying-Zhi Jiang3Li Zhang2*
Wen-Feng Fang2Ning Xi3Ning Kang3Yu-Lei Zhuang3Qi Zhang3Ying-Zhi Jiang3Li Zhang2* Hong-Yun Zhao1*
Hong-Yun Zhao1*Background: CT053PTSA is a novel tyrosine kinase inhibitor that targets MET, AXL, VEGFR2, FLT3 and MERTK. Here, we present preclinical data about CT053PTSA, and we conducted the first-in-human (FIH) study to evaluate the use of CT053PTSA in adult patients with pretreated advanced solid tumors.
Methods: The selectivity and antitumor activity of CT053PTSA were assessed in cell lines in vitro through kinase and cellular screening panels and in cell line-derived tumor xenograft (CDX) and patient-derived xenograft (PDX) models in vivo. The FIH, phase I, single-center, single-arm, dose escalation (3 + 3 design) study was conducted, patients received at least one dose of CT053PTSA (15 mg QD, 30 mg QD, 60 mg QD, 100 mg QD, and 150 mg QD). The primary objectives were to assess safety and tolerability, to determine the maximum tolerated dose (MTD), dose-limiting toxicity (DLT), and the recommended dose of CT053PTSA for further study. Secondary objectives included pharmacokinetics, antitumor activity.
Results: CT053 (free-base form of CT053PTSA) inhibited MET, AXL, VEGFR2, FLT3 and MERTK phosphorylation and suppressed tumor cell angiogenesis by blocking VEGF and HGF, respectively, in vitro. Moreover, cell lines with high MET expression exhibited strong sensitivity to CT053, and CT053 blocked the MET and AXL signaling pathways. In an in vivo study, CT053 significantly inhibited tumor growth in CDX and PDX models. Twenty eligible patients were enrolled in the FIH phase I trial. The most common treatment-related adverse events were transaminase elevation (65%), leukopenia (45%) and neutropenia (35%). DLTs occurred in 3 patients, 1/6 in the 100 mg group and 2/4 in the 150 mg group, so the MTD was set to 100 mg. CT053PTSA was rapidly absorbed after the oral administration of a single dose, and the Cmax and AUC increased proportionally as the dose increased. A total of 17 patients in this trial underwent tumor imaging evaluation, and 29.4% had stable disease.
Conclusions: CT053PTSA has potent antitumor and antiangiogenic activity in preclinical models. In this FIH phase I trial, CT053PTSA was well tolerated and had a satisfactory safety profile. Further trials evaluating the clinical activity of CT053PTSA are ongoing.
Cellular signaling networks and interactions regulate the biological capabilities of cancer cells (1). One of the most important signaling pathways in the cell signal transduction network is the tyrosine kinase receptor pathway. Receptor tyrosine kinases (RTKs) are a subclass of tyrosine kinases that are involved in mediating cell-to-cell communication and controlling a wide range of complex biological functions, including cell growth, motility, differentiation, metabolism and immune responses (2, 3).Dysregulation of RTK signaling leads to many human diseases, especially cancer, making these proteins promising drug targets for cancer therapy (4). Cellular-mesenchymal to epithelial transition factor (c-MET, MET) is an RTK with one known ligand, hepatocyte growth factor (HGF), and it is expressed by endothelial cells, epithelial cells, hepatocytes, neurons, and hematopoietic cells (5). Aberrant activation of the MET/HGF pathway is involved in the development and metastatic progression of various tumors, and this pathway constitutes the mechanism by which cancer patients acquire resistance to targeted treatment and attenuates tumor response to immunotherapy (6–11). In addition, overexpression of MET is observed in many cancers and is a poor prognostic factor for survival (12–16). AXL is an RTK that belongs to the TAM (TYRO3, AXL, and MERTK) family, and activation of AXL has been implicated in promoting cell proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT); mediating drug resistance; affecting the immune response; and indicating poor prognosis in several cancers (17–21). Aberrant expression of MERTK, another TAM RTK, also contributes to chemotherapy resistance, colony-forming potential, migration, EMT induction and involves in main process of anti-tumoral immunity in tumor cells (3, 22–25). Vascular endothelial growth factors (VEGFs) regulate vascular development, angiogenesis and lymphangiogenesis by binding to a number of receptors during the process of cancer angiogenesis. Recently, VEGFs are found immunosuppressive by inhibiting functional T cells, increasing the recruitment of suppressive immune cells, and hindering dendritic cells differentiation and activation (26). VEGFR-2 is the major RTK that regulates VEGF-induced vascular endothelial function (27). Moreover, MET, VEGFR-2 and AXL transactivation and synergistic interactions promote the activation of downstream signaling (28–30). RTKs that target MET, VEGFR-2 and AXL significantly increase the tumor suppressive effect and reduce the chance of tumor resistance to treatments.
CT053PTSA is a potent inhibitor of MET, AXL, VEGFR-2, FMS-like tyrosine kinase 3 (FLT3) and MERTK, all of which have been implicated in tumor pathogenesis. CT053PTSA (CT053 in a free-base form) was synthesized by HEC R&D Center, Sunshine Lake Pharma Co., Ltd. The nuclear structure and target of CT053PTSA are similar to those of cabozantinib, an antitumor drug marketed by Exelixis in the United States. The molecular formula of CT053PTSA is C31H29FN4O5 • C7H8O3S, and the molar mass is 728.29. The objectives of this study were to assess the antitumor activity of CT053PTSA in vivo and in MET-dependent tumor xenografts. Moreover, a FIH, phase I, single-center, single-arm, dose escalation (3 + 3 design) study was conducted in patients with advanced solid tumors who had previously received treatment. The primary objectives were to assess safety and tolerability as well as to determine the maximum tolerated dose (MTD), dose-limiting toxicity (DLT), and the dose of CT053PTSA recommended for further study. The secondary objectives included elucidating the pharmacokinetics and antitumor activity of CT053PTSA. The study is registered with ClinicalTrials.gov as NCT04577703.
CT053PTSA (Figure 1A) was synthesized by HEC R&D Center, Sunshine Lake Pharma Co., Ltd. The results of biochemical kinase binding assays and kinase inhibition assays are shown in the Supplemental Materials.
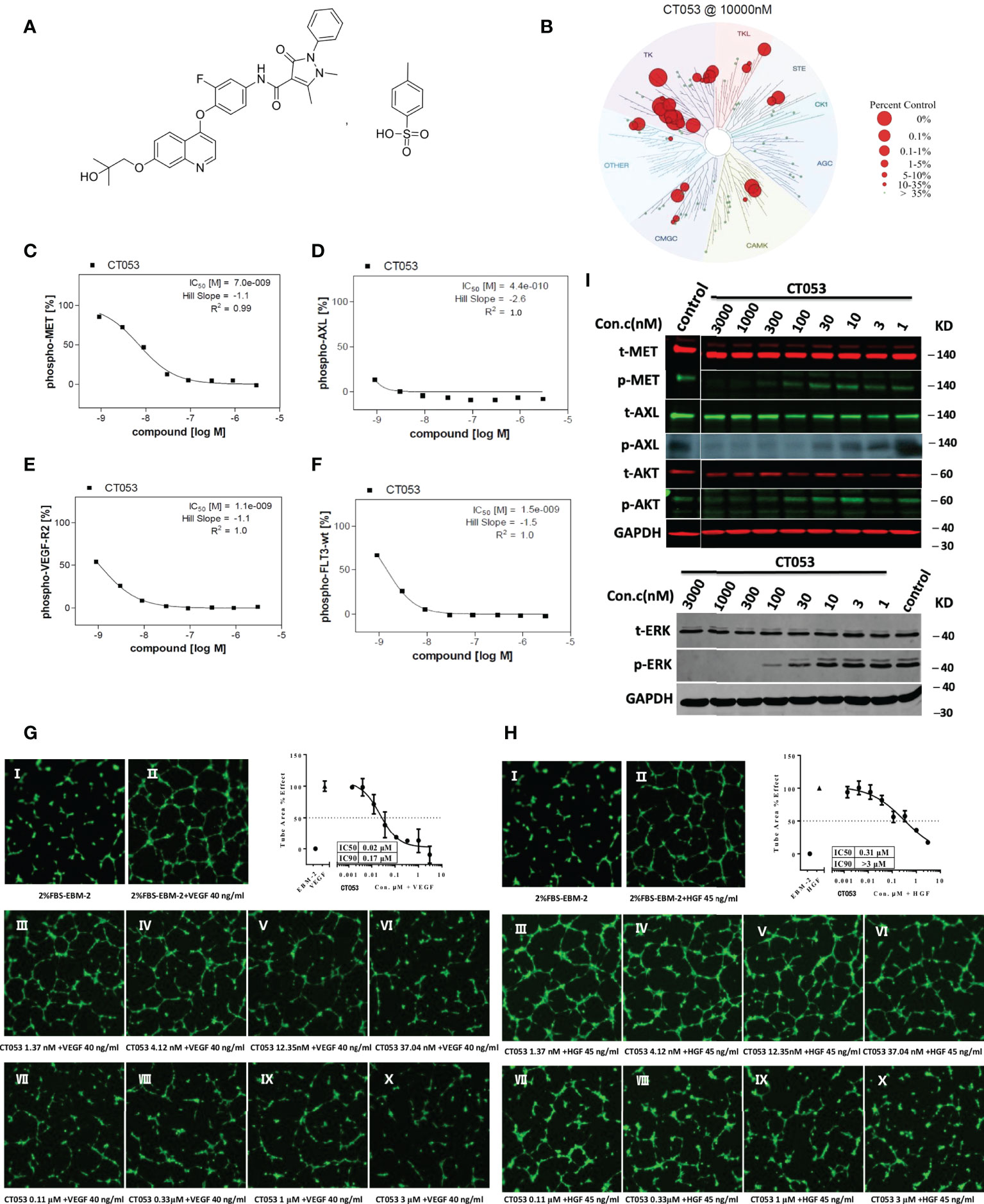
Figure 1 Chemical structure, cellular inhibitory effect and tubule formation effect of CT053PTSA in vivo. (A) Chemical structure of CT053PTSA. (B) Small molecule kinase interaction maps for CT053. CT053 was screened against a KinomeScan (http://www.kinomescan.com) panel of 95 kinase assays. Red circles indicate kinases bound, and circle size indicates binding affinity. Cellular inhibitory effect of CT053 on kinase phosphorylation: CT053 inhibited the phosphorylation of MET in MKN45 cells, and the IC50 value was 7.0 nM (C). AXL was examined in AXL-MEFs cells, and the IC50 value was 0.44 nM (D). VEGFR-2 and FLT3 were examined in HUVECs and FLT-wt-MEFs, and the IC50 values were 1.1 nM (E) and 1.5 nM (F). CT053 inhibits VEGF-induced (G) and HGF-induced (H) tubule formation. Tubule formation was visualized in HUVECs incubated with 2% FBS-EBM-2 as a control and with VEGF at a concentration of 40 ng/ml and HGF at a concentration of 45 ng/ml. Elevated concentrations of CT053 (1.37 nm (III), 4.12 nm (IV), 12.35 nm (V), 37.04 nm (VI), 0.11 μM (VII), 0.33 μM (VIII), 1 μM (IX), and 3 μM (X)) were coincubated with VEGF/HGF, the percentage area of tube formation stimulated by VEGF/HGF decreased, and the IC50 values were 0.02 μM and 0.31 μM. (I) Inhibition of the phosphorylation of MET, AXL and signaling molecules (AKT and ERK) in SNU-5 cells. SNU-5 cells were treated with different dilutions of CT053 (3000 nm, 1000 nm, 300 nm, 100 nm, 30 nm, 10 nm, 3 nm, and 1 nm), and MET autophosphorylation, AXL phosphorylation, AKT phosphorylation, and ERK phosphorylation were all inhibited by CT053. AXL phosphorylation was completely suppressed by 100 nM CT053, and the phosphorylation of MET and its downstream protein ERK was inhibited by 1000 nM CT053. A higher concentration of CT053 was required to suppress the induced activity of AKT.
Phosphorylation of the receptors MET, AXL, VEGFR2, and FLT3 was assessed in the transgenic human MKN45 gastric tumor cell line overexpressing MET, mouse embryonic fibroblasts (MEFs) expressing full-length human AXL (AXL-MEFs), human umbilical vein endothelial cells (HUVECs) overexpressing VEGFR-2, and MEFs expressing full-length human FLT3 (FLT3-wt-MEFs), respectively. The cells were serum starved overnight and then incubated with the compound CT053 diluted in gradient concentration in serum-free medium for 90 min at 37C. Then, HUVECs and FLT3-wt-MEFs were stimulated with the ligands VEGF-A (100 ng/ml, for 3 min) and FLT3-ligand (50 ng/ml, for 5 min). MKN45 (overexpressing MET) and AXL-MEFs cells had no stimulation. Cells treated with staurosporine (AXL, FLT3-wt, c-Kit, FLT3, TIE2, VEGF-R2; [1,0E-05 M]) or BMS777607 (MET; [1,0E-06 M]) or sunitinib (VEGF-R3; [1,0E-06 M]) or crizotinib (RON; [1,0E-05 M] were defined as low control (n = 8). The median value of those wells represented the background and was set to 0%. Cells treated with solvent alone were defined as high control (n = 8). The median value of those wells was set to 100%. Test sample-treated cells as well as high and low control were stimulated in identical manner. Quantification of receptor tyrosine kinase autophosphorylation was assessed in 96-well plates via sandwich ELISA using a receptor specific capture antibody and an anti-phosphotyrosine detection antibody. Raw data were converted into percent substrate phosphorylation relative to high controls, which were set to 100%. IC50 values were determined using GraphPad Prism 5 software with constrain of bottom to 0 and top to 100 using a nonlinear regression curve fit with variable hill slope.
HUVECs were incubated with serial dilutions of the compound CT053 (1.37 nM, 4.12 nM, 12.35 nM, 37.04 nM, 0.11 μM, 0.33 μM, 1 μM, or 3 μM) for 8 hours in the presence or absence of VEGF (40 ng/ml, R&D, MN, USA) or HGF (45 ng/ml, R&D, MN, USA). Then, after staining with Calcein AM (ThermoFisher, CA, USA), images of tubule formation were captured, and the total tube area was quantified with ImageXpress HCS (Molecular Devices, CA, USA).
Human gastric tumor cell lines with high MET expression levels, namely, the GTL-16, MKN45, SNU638, Hs746T, SNU-620, and SNU-5 cell lines, were used in the assay. These cell lines were obtained from American Type Culture Collection (ATCC), Japanese Collection of Research Bioresources (JCRB), Shanghai Institutes of Biological Sciences, CAS (SIBS), Korean Cell Line Bank (KCLB) or National Institutes of Health (NIH). The cell lines were cultured overnight in 96-well plates and then treated with serial dilutions of the compound CT053. Cell viability was measured after 72 hours using a CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega, WI, USA) according to the manufacturer’s instructions.
The MET mRNA expression level in each human gastric cell lines was evaluated with real-time PCR. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was considered as the internal control. The amplification was performed in a 20 μl mixture containing 10 μl of TaqMan Universal PCR Master Mix (Invitrogen, CA, USA) with UNG, 1 μl of the MET (Hs01565584_m1) or GAPDH (Hs02786624_g1) TaqMan Gene Expression Assay (Thermo, CA, USA), and 5 μl of cDNA solution. Each sample was analyzed in triplicate on an Agilent Technologies Stratagenes Mx3005P (CA, USA). Relative MET mRNA expression is presented as the fold-change, which was calculated using the 2^ (-ΔΔCt) method.
The SNU-5 cell line was selected for western blotting analysis of signaling molecules. After CT053 treatment, proteins were extracted by protein lysis, and the process of western blotting analysis included adding the samples to SDS-PAGE gels, transferring the proteins to polyvinylidene fluoride membranes (Millipore, MA, USA), blocking the membranes with 5% nonfat milk in TBST, incubating the membranes with the primary antibody, probing the membranes with a peroxidase-linked secondary antibody, and visualizing the protein bands. Immunoreactive proteins (p-AXL, t-ERK and p-ERK) were visualized using the ECL system (Pierce Chemical, TX, USA). Other proteins were detected by Odyssey (LI-COR, NE, USA) at a wavelength of 700 nm or 800 nm. In addition, we also examined the effects of CT053 on the phosphorylation of MET in the GA3121 PDX model by western blotting. Tumors were harvested after 21 days of treatment with vehicle or CT053. Tumor lysates were subjected to western blotting. The following antibodies were purchased from Cell Signaling Technology (Cell Signaling Technology Cat#3148, RRID : AB_1031042): anti-MET (Cell Signaling Technology Cat# 3148, RRID : AB_1031042), anti-p-MET Y1234/1235 (Antibodies-Online Cat# ABIN461461, RRID : AB_10786128), anti-AXL (Cell Signaling Technology Cat# 4566, RRID : AB_2062563), anti-p-AXL Y702 (5724S), anti-ERK (9107S), anti-p-ERK Thr202/Tyr204 (4370S), anti-p-AKT Ser473 (4060S), and anti-AKT (Cell Signaling Technology Cat# 2920, RRID : AB_1147620).
Human tumor xenografts were established in female BALB/cA nude mice (5-6 weeks old) by administering the SNU-5 and MKN-45 cells, which have high MET expression levels. For efficacy studies in patient-derived tumor xenograft (PDX) mouse models, MET-overexpressing GA0046 and GA3121 tumors (obtained from Crownbio, CA, USA) were established by directly implanting surgical tissues into immunodeficient mice. Tumor-bearing mice were randomized into groups (8 animals per group) according to their tumor sizes, and when the average tumor volume reached approximately 150 mm3, the mice were treated with vehicle or CT053 at doses of 1, 3, 10 or 20 mg/kg once per day by oral gavage for 16 or 21 days. The subcutaneous tumor size and body weight were measured twice weekly. The tumor volumes were determined by measuring two perpendicular diameters with calipers and calculated as (length × width2)/2. Tumor growth inhibition (TGI: TGI% = (1 - tumor volume (treatment)/tumor volume (control) ×100) was calculated to evaluate the effect of the treatment on tumor growth inhibition. Tumors were harvested from the GA3121 PDX model mice after 21 days of treatment with vehicle or CT053 at a dose of 20 mg/kg to perform further experiment.
This was a single-center, single-arm, dose-escalation phase I study in patients with advanced solid tumors who failed to achieve an effect with or who had not received standard systemic anticancer treatment. Eligible patients had at least one measurable lesion per RECIST v 1.1, Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, and adequate hematological, renal, cardiac and hepatic functions. The primary objectives were to assess safety and tolerability as well as to determine the MTD, DLT, and dose recommended for further study. The secondary objectives were to assess the PK and preliminary efficacy of CT053PTSA.
CT053PTSA was administered orally, and this dose-escalation trial included 5 dose levels: 15 mg QD, 30 mg QD, 60 mg QD, 100 mg QD, and 150 mg QD. An initial accelerated titration design was used in the 15 and 30 mg QD cohorts, and the “3+3” schema was used starting in the 60 mg QD cohort. DLT was defined as any of the following events occurring during cycle 0 (7-day wash-out period after a single dose) and cycle 1 (multiple doses, including a 7-day observation period after 28 days of administration) per Common Terminology Criteria for Adverse Events (CTCAE) version 4.0: G4 neutropenia lasting ≥ 7 days; febrile neutropenia with fever ≥ 38.5°C; G3 thrombocytopenia associated with bleeding or G4 thrombocytopenia lasting ≥ 4 days; grade ≥ 3 nausea/vomiting/diarrhea/hypertension not alleviated after 4 days of supportive care; any other grade ≥ 3 nonhematological toxicities (excluding alopecia, hyperuricemia without gout). The study protocol was approved by an institutional review board at Sun Yat-sen University Cancer Center. Written informed consent was obtained from all the patients before enrollment.
A clinical pharmacokinetic study of CT053PTSA was performed for both single doses and multiple doses. Blood samples were collected from each subject at prespecified times before and after single-dose administration (systemic blood samples: before treatment and 0.5, 1, 2, 3, 4, 5, 8, 12, 24, 36, 48, 72, 120 and 168 hours after treatment) and during multiple dosing (cycle 1 before treatment on Days 8, 15 and 22, as well as systemic blood samples on Day 28).
Descriptive statistics of safety are presented per CTCAE v4.0. Subjects who received at least one dose of CT053PTSA were considered eligible to be evaluated and included in safety analyses. Single-dose and multiple-dose PK parameters included peak concentration (Cmax); time to reach Cmax (Tmax); the area under the plasma concentration time curve from 0 to 168 hours, at 24 hours, and infinity (AUC0-168, AUC0-24, AUC0-inf); and oral clearance (CL/F) and apparent volume of distribution (Vz/F) for both CT053PTSA and its metabolites: CT053-M1. According to the RECIST 1.1 criteria, imaging examinations were performed at each specified time point in the protocol. The antitumor activity was measured by RECIST1.1.
Statistical analysis of CT053-treated tumors versus vehicle-treated tumors or versus nontreated tumors was performed by repeated-measures ANOVA. The data from the tumor xenograft models were expressed as the means ± standard error of the mean (SEM) and were plotted as a function of time. Noncompartmental pharmacokinetic analysis was performed using Phoenix WinNonlin Version 6.2.1 (Certara Inc., NJ, USA). Demographics and laboratory results of all the subjects were summarized using descriptive statistics. The pharmacokinetic parameters Tmax and T1/2 were shown as the median (min, max) and mean ± SD, respectively. A two-sided P < 0.05 was considered to indicate statistical significance. Statistical analysis was performed by using GraphPad Prism 5.0 (GraphPad Prism, RRID : SCR_002798) and SPSS software version 19.0 (SPSS, RRID : SCR_002865).
To assess the potential off-target inhibitory activity of CT053 on other kinases, we screened the compounds against a KinomeScan panel of 95 kinase binding assays. The highest affinity target identified for CT053 was the tyrosine kinase family (Figure 1B), which indicated that the kinase interaction pattern for CT053 was highly specific.
A follow-up IC50 calculation was performed with 8 concentrations of CT053 against 27 kinases selected from the first screening step. CT053 is a potent inhibitor of AXL, with an IC50 value of 3.4 nM. CT053 strongly inhibited several kinases, including MET, VEGFR2, MERTK and FLT3 (IC50 = 68, 46, 14, 8.6 nM, respectively) (Supplementary Table 1). In cellular assays, CT053 inhibited the phosphorylation of MET, AXL, VEGFR2, and FLT3 with IC50 values of 7.0, 0.44, 1.1, and 1.5 nM, respectively (Figures 1C-F).
We further investigated the effect of CT053 on angiogenesis in vitro. The results showed that as the concentration of CT053 increased (1.37 nM, 4.12 nM, 12.35 nM, 37.04 nM, 0.11 μM, 0.33 μM, 1 μM, or 3 μM), the percentage area of tube formation stimulated by VEGF and HGF decreased, and the IC50 values were 0.02 and 0.31 μM, respectively (Figures 1G, H). These data suggest that CT053 inhibits tumor cell angiogenesis in vitro.
CT053 potently inhibits the tyrosine kinase activity of recombinant MET and the phosphorylation of MET in in vitro cell-based assays, with IC50 values of 68 nM and 7.0 nM, respectively (Supplementary Table 1 and Figure 1C). To investigate biomarkers that could help identify tumors that are sensitive to MET kinase inhibition, an in vitro antiproliferative screen was performed using a gastric tumor cell line panel. Six cell lines (SNU-5, SNU638, SNU-620, GTL-16, MKN45, and Hs746T cells) showed high MET mRNA levels with a median ratio value of 18.75 (range: 5.64-28.80), whereas the other eleven cell lines showed significantly lower MET mRNA levels with a median ratio value of 1.01 (range: 0.01-3.27) (P < 0.001) (Supplementary Table 2). Cell lines with high MET mRNA levels exhibited strong sensitivity to CT053, with an average IC50 value of 0.095 μM. However, CT053 had a very weak inhibitory effect on cell lines with low MET mRNA levels, with an average IC50 value of more than 10 μM (P < 0.001) (Supplementary Table 2). Previous studies reported that MET gene expression has been linked to the sensitivity of cell lines to MET inhibitors (31, 32). Our results demonstrate that sensitivity to CT053PTSA is associated with MET transcription and mRNA expression in vitro.
To show the effect of CT053 on the MET and AXL signaling pathways, SNU-5 cells were treated with serial dilutions of CT053. The results showed that CT053 inhibited MET autophosphorylation. Moreover, at 100 nM, CT053 completely suppressed AXL phosphorylation, and at 1000 nM, CT053 completely suppressed the phosphorylation of MET and its downstream protein ERK. However, a much higher concentration of CT053 was required to suppress the induced AKT activity (Figure 1I).
We performed two in vivo efficacy studies in cell line-derived tumor xenograft (CDX) models (derived from the SNU-5 and MKN45 cell lines) and two in vivo antitumor efficacy studies in PDX models (GA0046 and GA3121). CT053 significantly inhibited tumor growth compared with vehicle, exhibiting TGI values of 32% (P = 0.88), 87% (P = 0.018) and 187% (P = 0.017) at doses of 1, 3, and 10 mg/kg in the SNU-5 CDX model (Figure 2A) and TGI values of 31% (P = 0.014), 85% (P < 0.001), 97% (P < 0.001) at doses of 3, 10 and 20 mg/kg in the MKN-45 CDX model, respectively (Figure 2B). Furthermore, CT053 also had a potent inhibitory effect on tumor growth, with TGI values of 18% (P = 0.88), 57% (P = 0.01), and 75% (P = 0.002) at doses of 3, 10, and 20 mg/kg in the GA0046 PDX model, respectively (Figure 2C), and a TGI value of 172% (P = 0.001) at 20 mg/kg CT053 in the GA3121 model (Figure 2D). In addition, we found that the phosphorylation of MET was significantly inhibited in the CT053-treated group compared with the vehicle group in the GA3121 PDX model (Figure 2E). These results indicated that CT053 suppressed tumor growth in the CDX and PDX models with high MET expression in a dose-dependent manner, and the attenuation of tumor growth by CT053 most likely occurs due to its inhibition of MET signaling activity. Body weight was measured to monitor the toxicity and there was no significant change in body weight for all groups.
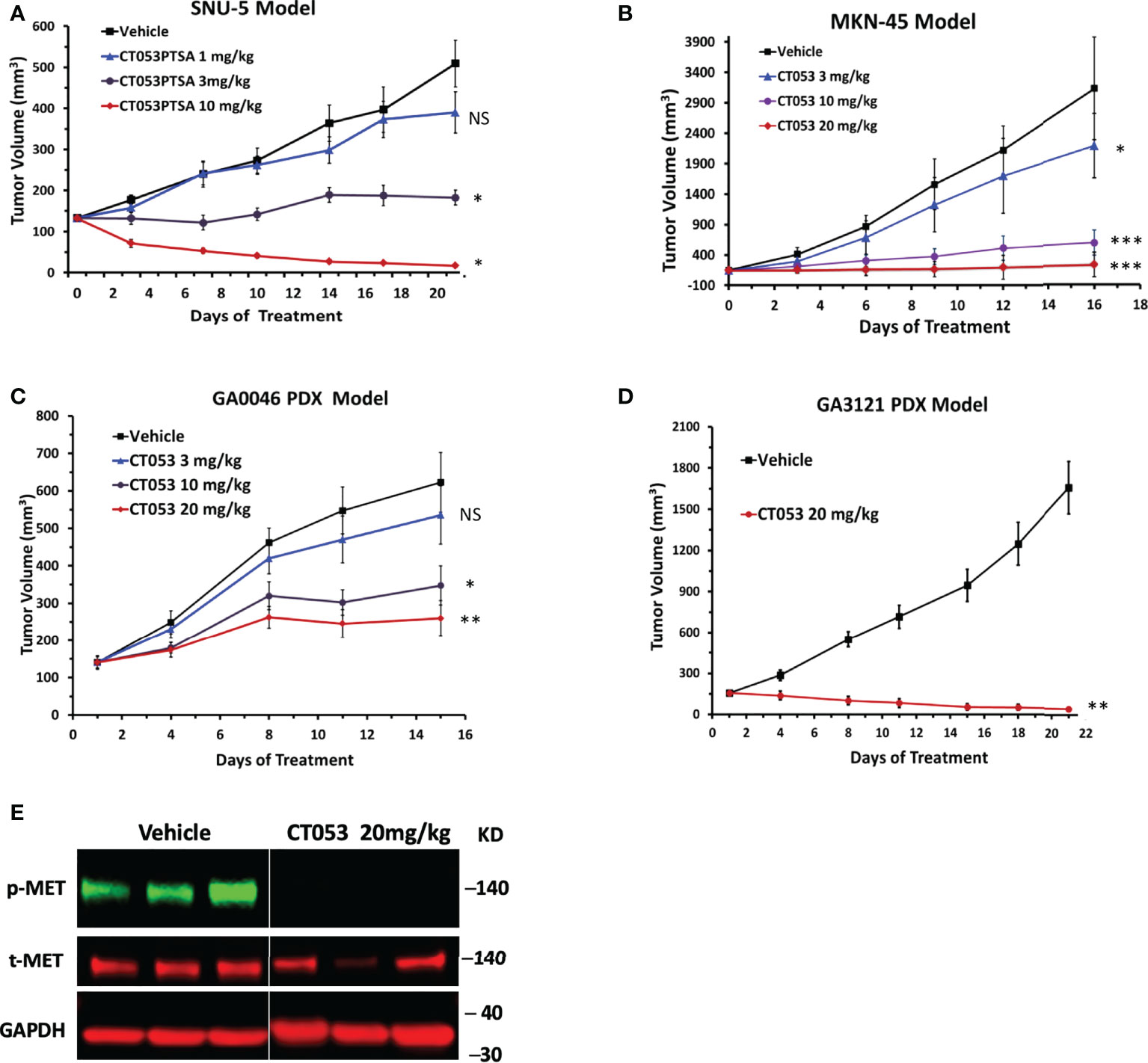
Figure 2 CT053 inhibits tumor growth in a MET-dependent manner in tumor xenograft models. Mice bearing SNU-5 CDX xenografts (A), MKN45 CDX xenografts (B), GA0046 PDX xenografts (C) or GA3121 PDX xenografts (D) were treated orally once daily with vehicle or CT053 at doses of 1, 3, 10 or 20 mg/kg. The tumor volume was measured twice per week, and the data represent (n = 8) the mean ± standard error of the mean (SEM) for each group. An adjusted value of P < 0.05 was considered statistically significant. Each treatment group was compared to the control group (*, P < 0.05; **, P < 0.01; ***, P < 0.001; NS = not significant). Statistical significance was determined by repeated-measures ANOVA. (E) Effects of CT053 treatment on the phosphorylation of MET in the PDX model. Samples were harvested from the GA3121 PDX model mice after 21 days of treatment with vehicle or CT053 at a dose of 20 mg/kg.
A total of 20 patients were enrolled in the study (Figure 3). The median age was 47.3 years (range: 28-66 years), and all of the patients received at least one dose of CT053PTSA and were included in the safety analysis set from February 2014 to December 2015. The baseline characteristics are summarized in Table 1.
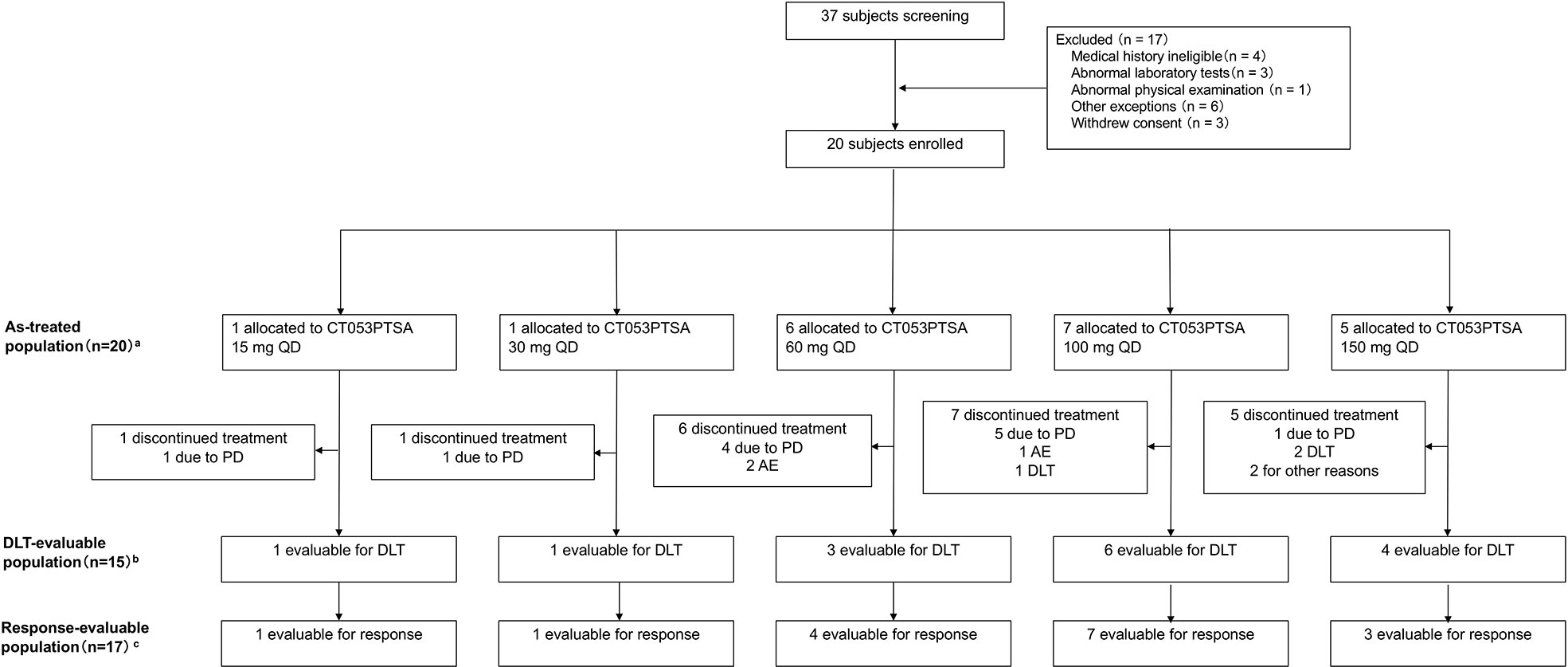
Figure 3 Flow chart of patients enrolled in the study. A total of 20 patients received at least one dose of CT053PTSA. aThe as-treated population included patients who received any dose of CT053PTSA, grouped according to actual dose received; bThe population that was eligible to for DLT assessment included all the patients enrolled in the dose-escalation phase who received one full dose of CT053PTSA and completed the safety follow-up through the DLT evaluation period, defined as the time during cycle 0 (7 days after a single dose) and cycle 1 (multiple doses, including a 7-day observation period after 28 days of administration), or who experienced any DLT during this period; cThe population that was eligible for response assessment included all patients in the as-treated population who had ≥ 1 postbaseline tumor assessment or alternatively who died from any cause or who discontinued treatment due to clinical PD prior to any postbaseline tumor assessment. AE, adverse event; PD, progressive disease; DLT, dose-limiting toxicity.

Table 1 Baseline patient demographics and clinical characteristics.
Nineteen (95%) patients experienced at least one adverse event (AE), and of these patients, treatment-related adverse events (TRAEs) were reported in 16 (80%). The most frequently reported (≥ 20%) TRAEs of any grade included transaminase elevation (65%), leukopenia (45%), neutropenia (35%), decreased platelet count (30%), decreased appetite (30%), and hypertension (20%) (Table 2). Most AEs were grade 1-2 and easily managed. Only 3 patients (15%, 100 mg QD: n = 1; 150 mg QD: n = 2) experienced grade 3 TRAEs, including decreased platelet count (10%), elevated transaminase (5%), leukopenia (5%), decreased blood sodium (5%), decreased appetite (5%), hypertension (5%) and stomatitis (5%), during treatment. One serious adverse event (SAE) was reported at 150 mg, which was grade 3 stomatitis (also a DLT) leading to hospitalization. Regarding DLTs, one out of six patients who were eligible for DLT assessment experienced a DLT (grade 3 transaminase elevation) at 100 mg QD. Two of four patients who were eligible for DLT assessment at 150 mg QD experienced DLTs, namely, grade 3 blood sodium decreased, grade 3 decreased appetite, grade 3 hypertension and grade 3 stomatitis. Thus, the MTD was 100 mg, and the recommended dose for further study of CT053PTSA was determined to be 60~100 mg QD.
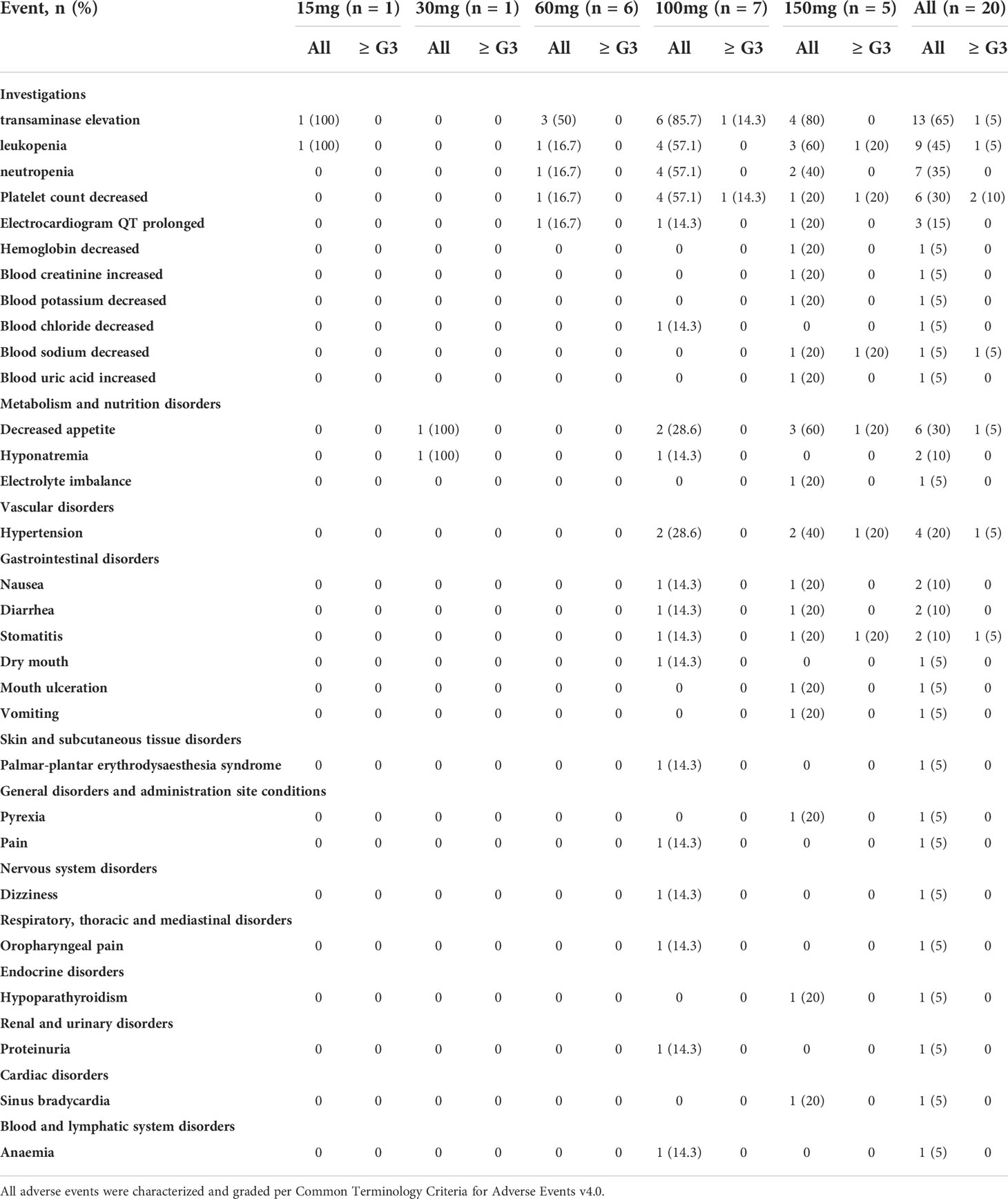
Table 2 Treatment-related adverse events reported in all patients.
The PK parameters of CT053PTSA and its active metabolite CT053-M1 were evaluated after single- and multiple-dose administration. Prior to initiating a continuous daily treatment cycle, patients participated in a single-dose PK treatment period with a 7-day washout. Concentration-time plots of the plasma CT053PTSA and CT053-M1 levels in each cohort after a single dose (Day 1) or after multiple oral doses (Day 28) are shown in Figure 4, and the pharmacokinetic parameter summary is provided in Supplementary Table 3. CT053PTSA was rapidly absorbed after oral administration with a median Tmax of 2~3 h for a single dose and 2 h for multiple doses. The observed mean half-life ranged from 24.7 to 33.9 hours for a single oral dose and from 13.8 to 38.4 hours for multiple oral doses. A steady state was reached by Day 8 following daily dosing. One or multiple peaks were observed after the Cmax peak in the plasma concentration-time profiles within 48 h in most subjects. After a single oral dose, CT053 followed a general linear PK profile for doses from 15 to 150 mg, and a linear PK profile was also observed for doses from 60-100 mg after multiple doses. After oral administration, CT053-M1 was the most abundant metabolite in the plasma.
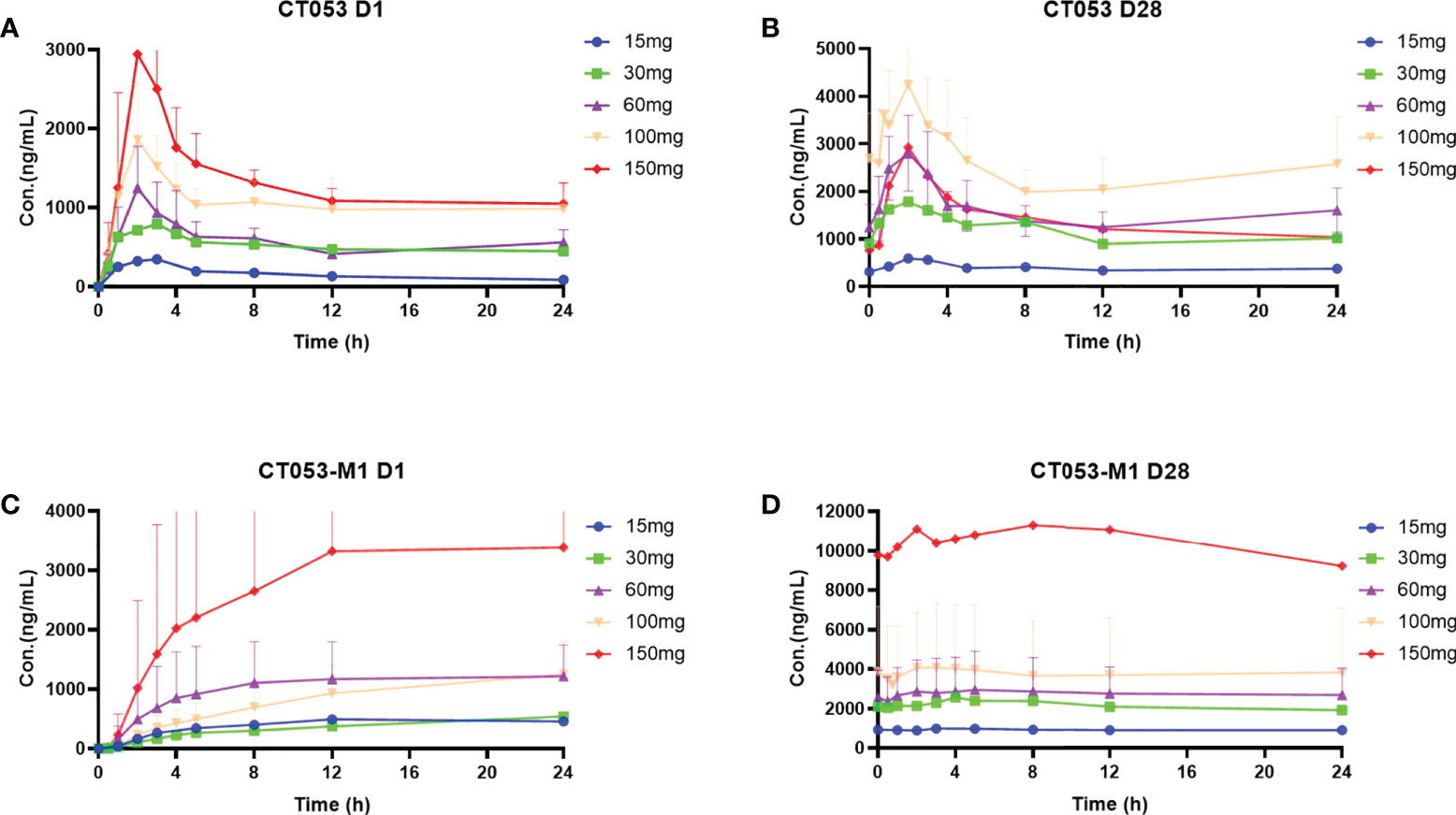
Figure 4 Concentration-time profiles (mean ± SD) of plasma CT053PTSA and CT053-M1 levels by cohort after a single dose (Day 1) after multiple oral doses (Day 28). (A) CT053PTSA levels after a single dose. (B) CT053PTSA levels after multiple oral doses. (C) CT053-M1 levels after a single dose. (D) CT053-M1 levels after multiple oral doses.
We also evaluated the preliminary efficacy of CT053PTSA. A total of 17 patients underwent tumor imaging evaluation after taking CT053PTSA, and none of them achieved CR or PR. Five patients (29.4%) achieved SD. The DCR was 29.4%. Among these patients, two who received 60 mg QD CT053PTSA achieved SD for 16 weeks and 24 weeks. One patient in the 100 mg dose group achieved SD for 8 weeks.
Moreover, we analyzed the patient who achieved SD for 24 weeks. The patient was a 47-year-old female with EGFR exon 19-21 wild type, ALK negative non-small cell lung cancer (NSCLC). She had failed 1 line of platinum-based chemotherapy with a PFS of 27 months. She underwent a biopsy on neck lymph node before CT053PTSA treatment (Figure 5A), results showed that tumor tissue was MET overexpressed by immunohistochemistry (IHC) (Figure 5B). Baseline CT imaging showed a left supraclavicular lymph node (target lesion, Figure 5C) and multiple lung metastases (all < 1 cm, nontarget lesions, Figure 5D). The plasma of the patient was prospectively collection on the first day of cycle 1, 3, 6, whole exon sequencing of 416 genes was performed to assess circulating tumor DNA (ctDNA) with Illumina platform. As showed in Supplementary Table 4, targeted genes of CT053PTSA including AXL A79T mutation, FLT3 P893L mutation were eliminated compared baseline after CT053PTSA treatment. Other truncating mutations of tumor related genes (APC, BRCA1, EGFR, FLT4, etc) were also decreased rapidly during treatment. The patient received 6-time tumor assessments, the response was classified as SD (Figure 5E), and the nontarget lesions remained unchanged (Figure 5F). However, the patient experienced tumor progression after 6 cycles of CT053PTSA administration, and which point, the target lesion enlarged.
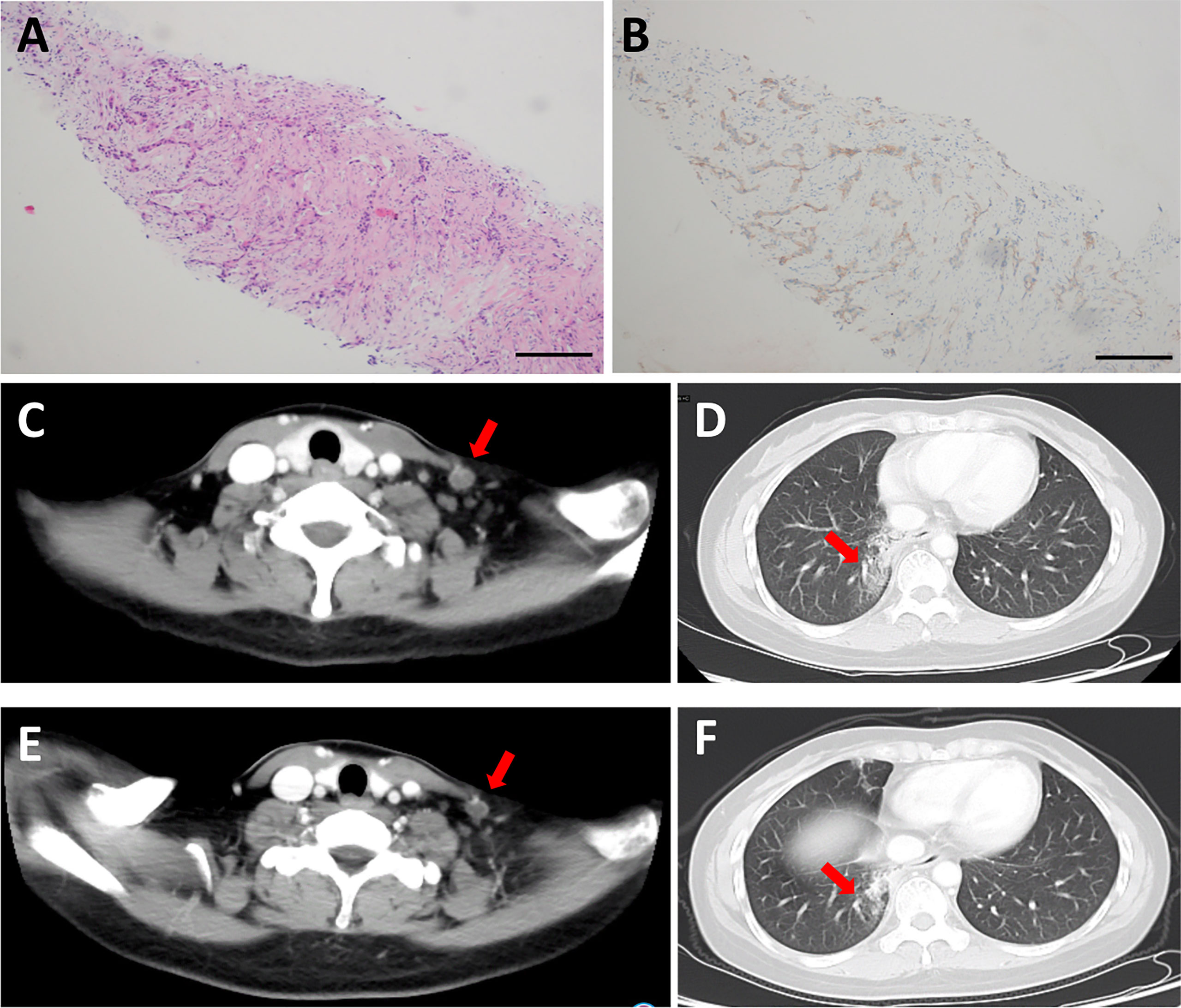
Figure 5 Representative H&E- and IHC-stained biopsy sections and CT images from a patient in the 60 mg CT053PTSA treatment group with advanced non-small cell lung cancer (EGFR, ALK negative) who achieved a durable stable response. (A) Representative H&E image of a stained biopsy from the left supraclavicular lymph node at baseline. (B) Representative IHC image of MET overexpression in a stained biopsy from the left supraclavicular lymph node at baseline. Baseline CT images (2014-11-24): (C) target lesion: left supraclavicular lymph node (red arrow) on enhanced CT. and (D), nontarget lesion: lung metastases on enhanced CT. (E, F), Cycle 2 Day 28 CT image (2015-02-04): target lesion and nontarget lesion at stable response.
CT053PTSA is a novel multikinase inhibitor with activity against MET, AXL, VEGFR2, FLT3 and MERTK, among others. In our study, CT053PTSA demonstrated potent inhibition of the phosphorylation of these kinases and tubule formation of tumor cells. Moreover, CT053PTSA showed significant antitumor efficacy in all models in the short term in vivo. Then, we conducted a FIH, phase I, single-center, single-arm, dose escalation clinical study, and the results indicated that CT053PTSA was well tolerated and safe from 15 mg QD to 100 mg QD. In addition, 100 mg QD was determined to be the MTD, and the recommended dose for further study of CT053PTSA was determined to be 60~100 mg QD.
Small molecule tyrosine kinase inhibitors (TKIs) that target tyrosine kinase receptor pathways have shown remarkable results in terms of specific therapeutic effects on various cancers. Inhibitors that target MET, AXL, VEGFR-2, FLT3 and MERTK are becoming research and development hotspots owing to their effect on promoting tumor development and progression. Cabozantinib is a multitarget RTK inhibitor that inhibits several RTKs, including MET, RET, VEGFR-1-3, KIT, FLT-3, TIE-2 (TEK tyrosine kinase, endothelial), tropomyosin-related kinase B (TRKB) and AXL, and it has been clinically approved for the treatment of medullary thyroid cancer (MTC) (33), renal cell cancer (RCC) (34), and hepatocellular carcinoma (HCC) (35).
Although CT053PTSA has a higher IC50 for MET and VEGFR-2 than cabozantinib, in cellular assays, CT053 inhibited the phosphorylation of MET, AXL, VEGFR2, and FLT3 with IC50 values of 7.0, 0.44, 1.1, and 1.5 nM, respectively, which were much lower than those of cabozantinib, with IC50 values of 7.8, 42, 1.9, and 7.5 mmol/L, respectively (36). Moreover, CT053PTSA inhibits the proliferation of HUVECs stimulated by HGF and VEGF to an extent that is comparable to cabozantinib. Crizotinib is a small molecule TKI that targets anaplastic lymphoma kinase (ALK), ROS1 and MET (37, 38). In cellular assays, crizotinib inhibited the phosphorylation of MET in NCI-H69 and HOP92 cells with IC50 values of 13 nM and 16 nM, respectively, which are higher than the values of CT053PTSA. Furthermore, the inhibition of anti-proliferation sensitivity and endothelial cell tubule formation by CT053PTSA in vitro was consistent with crizotinib (37). The results of in vivo and in vitro studies of CT053PTSA are comparable to those of cabozantinib and crizotinib, indicating that CT053PTSA could be a potent promising agent for treating tumors.
In the current FIH phase I study, the most common TRAEs were also commonly reported in studies of other MET inhibitors (39, 40). A phase I study of cabozantinib in Japanese patients with NSCLC demonstrated that 70% (16/23) of patients experienced ≥ grade 3 AEs, and the most common AEs included palmar-plantar erythrodysesthesia (100%), increased alanine aminotransferase (95%), increased aspartate aminotransferase (95%), hypertension (87%), and diarrhea (78%) (41). Merestinib is a multikinase inhibitor that targets MET, AXL and FLT3. A phase I study of merestinib showed that 40% (20/50) of patients experienced ≥ grade 3 TRAEs (42). Even though these were also observed in patients treated with CT053PTSA, they were much less frequently reported in our study, in which 15% of patients experienced ≥ grade 3 AEs. Twenty percent of patients experience hypertension, which is considered to be associated with the inhibition of VEGF-mediated angiogenesis (43). Hematologic toxicity included leukopenia (45%), neutropenia (35%), and decreased platelet count (30%), which are believed to be related to the inhibition of FLT3 (44). The TRAEs reported in this study were predominantly grade 1 and 2, and they were managed with supportive care. Three patients experienced DLTs, 2 patients treated with 150 mg QD, and one patient treated with 100 mg QD. CT053PTSA had a better safety profile, with no unexpected adverse reactions reported in this study. Therefore, according to the results of the study, the MTD was 150 mg, which was confirmed as the recommended dose for further study. Pharmacokinetic analysis indicated that CT053PTSA was rapidly absorbed after oral administration to patients with advanced solid tumors, with a median Tmax of 2~3 h; this value was 4 h for cabozantinib (45). Exposure (Cmax and AUC0-24 h) to CT053PTSA increased with dose. The PK data supported the QD regimen, and the half-life was reported to be 24.7-33.9 h, while it was 108-126 h for cabozantinib (45). Therefore, the QD regimen for CT053PTSA was determined for further development. We further investigated the preliminary efficacy of CT053PTSA. No CR or PR was observed, and 5 out of 17 patients achieved SD. The efficacy was acceptable because the patients who were enrolled all had heavily treated advanced solid tumors, accounted for a small number of patients, and were not selected based on MET, AXL, VEGFR-2, and FLT3 expression. We retrospectively found that the patient with MET overexpression had durable stable disease for approximately six months. In this case, the overexpression of MET was confirmed by IHC, and in our preclinical study, in vivo and in vitro studies were also performed on cells and models that overexpressed MET. The results indicated the consistency of the antitumor efficacy of CT053PTSA preclinical and clinical settings. Currently, ctDNA has been used for detecting early cancer occurrence (46) and predicting clinical response after treatment (47, 48). In the case report, after CT053PTSA administration, the amount of ctDNA gradually decreased and was undetectable at cycle 6. Furthermore, the abundance of truncation mutations in tumor suppressor genes was decreased after cycle 0 administration of CT053PTSA. Therefore, it is expected that CT053PTSA will have exhibit performance in cancer patients with MET, AXL, VEGFR-2 and FLT3 overexpression.
Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) provide a favorable treatment outcome in EGFR mutation-positive NSCLC patients. However, their prognosis remains unfavorable because of the occurrence of either intrinsic or acquired resistance (49). Activation of alternate or bypass pathways occurs in 5-22% of patients with high MET expression (50, 51) and in 20% of patients with upregulated AXL expression (52), and these events account for acquired resistance to EGFR TKIs via the activation of downstream effectors, including NF-kappaB and AKT, or the promotion of EMT (53, 54). Our results demonstrated that CT053PTSA inhibited the phosphorylation of MET and AXL and blocked the MET, AXL and downstream signaling pathways. CT053PTSA combined with an EGFR-TKI will be a better treatment strategy for NSCLC. Based on the preclinical data and the results of this phase I study, subsequent clinical trials are ongoing and registered with ClinicalTrials.gov (NCT03758287).
Zhang Y et al. (11) found that lung cancer patients with MET amplification were resistant to immune checkpoint inhibitor and had a poor survival by down-regulating STING levels and antitumor T-cell infiltration. In addition, the study conducted by Li Q et al. (55) found that low-dose VEGFR2 blockade led to more infiltration and activation of immune cell, promoted generation of TGF-β, that in turn upregulated PD-1 expression on immune cells. Moreover, TAM (TYRO3, AXL, and MERTK) family also had negative influence on cancer immunotherapy by mediating efferocytosis, negative regulating of dendritic cell activity, and dysregulating production of chemokines (3). The studies indicated that targeting MET, AXL, and VEGFR2 may enhance the efficacy of immune checkpoint inhibitor for cancer patients, which can be explored in further study.
Our study also has several limitations. First, in the preclinical study, the tumor cell lines we used had high MET mRNA and/or protein overexpression levels, and we did not investigate the effect of CT053PTSA on models with MET mutations. Second, the phase I trial was performed in a single center, and the participants were all of Chinese descent, which may reduce the generalizability of the findings. Third, we did not evaluate the effect of food intake on the pharmacokinetics of CT053PTSA.
In conclusion, CT053PTSA has potent antitumor and antiangiogenic activity in preclinical models. In this phase I, FIH clinical trial, CT053PTSA was generally well tolerated and safe for patients treated with the recommended dose. The results support the ongoing evaluation of the clinical activity of CT053PTSA in the treatment of cancer patients.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Sun Yat-sen University Cancer Center Ethics Committee. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Institutional Animal Care and Use Committee (IACUC) of HEC Pharma. Co. Ltd. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
H-YZ and LZ conceived and designed this study. Y-XM, F-RL and YZ collected and analysed the data, writing-original draft. NX and NK carried out the preclinical experiments. QC, Z-QC, Q-WL collected the clinical data. Y-XM, YH, Y-PY, W-FF enrolled patients. Y-LZ, QZ and Y-ZJ assisted the implementation of the clinical trial. All authors reviewed or revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Nature Science Foundation of China (82073396 and 81872201 for H-YZ, 82002409 for Y-XM, 81872449 for LZ), Guangdong Basic and Applied Basic Research Foundation (2018A0303130243 for H-YZ, 2020A1515010020 for Y-XM), and HEC R&D Center, Sunshine Lake Pharma Co., Ltd.
We thank all patients and their family for participating in the trial.
Authors NX, NK, YLZ, QZ and YZJ are employed by Sunshine Lake Pharma Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1024755/full#supplementary-material
1. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. Peng S, Wang R, Zhang X, Ma Y, Zhong L, Li K, et al. Egfr-tki resistance promotes immune escape in lung cancer Via increased pd-L1 expression. Mol Cancer (2019) 18(1):165. doi: 10.1186/s12943-019-1073-4
3. Akalu YT, Rothlin CV, Ghosh S. Tam Receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol Rev (2017) 276(1):165–77. doi: 10.1111/imr.12522
4. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer (2018) 17(1):58. doi: 10.1186/s12943-018-0782-4
5. Fasolo A, Sessa C, Gianni L, Broggini M. Seminars in clinical pharmacology: An introduction to met inhibitors for the medical oncologist. Ann Oncol (2013) 24(1):14–20. doi: 10.1093/annonc/mds520
6. Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan C, et al. Function of the c-met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer (2018) 17(1):45. doi: 10.1186/s12943-018-0796-y
7. Ko B, He T, Gadgeel S, Halmos B. Met/Hgf pathway activation as a paradigm of resistance to targeted therapies. Ann Transl Med (2017) 5(1):4. doi: 10.21037/atm.2016.12.09
8. Lai GGY, Lim TH, Lim J, Liew PJR, Kwang XL, Nahar R, et al. Clonal met amplification as a determinant of tyrosine kinase inhibitor resistance in epidermal growth factor receptor-mutant non-Small-Cell lung cancer. J Clin Oncol (2019) 37(11):876–84. doi: 10.1200/JCO.18.00177
9. Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, et al. Amplification of the met receptor drives resistance to anti-egfr therapies in colorectal cancer. Cancer Discovery (2013) 3(6):658–73. doi: 10.1158/2159-8290.CD-12-0558
10. Novoplansky O, Fury M, Prasad M, Yegodayev K, Zorea J, Cohen L, et al. Met activation confers resistance to cetuximab, and prevents Her2 and Her3 upregulation in head and neck cancer. Int J Cancer (2019) 145(3):748–62. doi: 10.1002/ijc.32170
11. Zhang Y, Yang Q, Zeng X, Wang M, Dong S, Yang B, et al. Met amplification attenuates lung tumor response to immunotherapy by inhibiting sting. Cancer Discovery (2021) 11(11):2726–37. doi: 10.1158/2159-8290.CD-20-1500
12. Lee SJ, Lee J, Park SH, Park JO, Lim HY, Kang WK, et al. C-met overexpression in colorectal cancer: A poor prognostic factor for survival. Clin Colorectal Cancer (2018) 17(3):165–9. doi: 10.1016/j.clcc.2018.02.013
13. Cappuzzo F, Marchetti A, Skokan M, Rossi E, Gajapathy S, Felicioni L, et al. Increased met gene copy number negatively affects survival of surgically resected non-Small-Cell lung cancer patients. J Clin Oncol (2009) 27(10):1667–74. doi: 10.1200/JCO.2008.19.1635
14. Mao ZY, Zhu GQ, Ren L, Guo XC, Su D, Bai L. Prognostic value of c-met expression in cholangiocarcinoma. Technol Cancer Res Treat (2016) 15(2):227–33. doi: 10.1177/1533034615578959
15. Chen S, Zhu Y, Cui J, Wang Y, Xia Y, Song J, et al. The role of c-met in prognosis and clinicopathology of renal cell carcinoma: Results from a single-centre study and systematic review. Urol Oncol (2017) 35(8):532 e15– e23. doi: 10.1016/j.urolonc.2017.03.027
16. Li L, Sun Z, Huang X, Li X, Sun L, Zhang L, et al. Role of c-met expression on prognosis of head and neck cancer: A literature review and meta-analysis. Head Neck (2019) 41(6):1999–2006. doi: 10.1002/hed.25655
17. Gay CM, Balaji K, Byers LA. Giving axl the axe: Targeting axl in human malignancy. Br J Cancer (2017) 116(4):415–23. doi: 10.1038/bjc.2016.428
18. Rankin EB, Fuh KC, Taylor TE, Krieg AJ, Musser M, Yuan J, et al. Axl is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res (2010) 70(19):7570–9. doi: 10.1158/0008-5472.CAN-10-1267
19. Goyette MA, Duhamel S, Aubert L, Pelletier A, Savage P, Thibault MP, et al. The receptor tyrosine kinase axl is required at multiple steps of the metastatic cascade during Her2-positive breast cancer progression. Cell Rep (2018) 23(5):1476–90. doi: 10.1016/j.celrep.2018.04.019
20. Zhu C, Wei Y, Wei X. Axl receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol Cancer (2019) 18(1):153. doi: 10.1186/s12943-019-1090-3
21. Cardone C, Blauensteiner B, Moreno-Viedma V, Martini G, Simeon V, Vitiello PP, et al. Axl is a predictor of poor survival and of resistance to anti-egfr therapy in ras wild-type metastatic colorectal cancer. Eur J Cancer (2020) 138:1–10. doi: 10.1016/j.ejca.2020.07.010
22. Graham DK, DeRyckere D, Davies KD, Earp HS. The Tam family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer (2014) 14(12):769–85. doi: 10.1038/nrc3847
23. Linger RM, Cohen RA, Cummings CT, Sather S, Migdall-Wilson J, Middleton DH, et al. Mer or axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene (2013) 32(29):3420–31. doi: 10.1038/onc.2012.355
24. Lee-Sherick AB, Eisenman KM, Sather S, McGranahan A, Armistead PM, McGary CS, et al. Aberrant mer receptor tyrosine kinase expression contributes to leukemogenesis in acute myeloid leukemia. Oncogene (2016) 35(48):6270. doi: 10.1038/onc.2016.295
25. Brandao LN, Winges A, Christoph S, Sather S, Migdall-Wilson J, Schlegel J, et al. Inhibition of mertk increases chemosensitivity and decreases oncogenic potential in T-cell acute lymphoblastic leukemia. Blood Cancer J (2013) 3:e101. doi: 10.1038/bcj.2012.46
26. Yang J, Yan J, Liu B. Targeting Vegf/Vegfr to modulate antitumor immunity. Front Immunol (2018) 9:978. doi: 10.3389/fimmu.2018.00978
27. Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell Signal (2007) 19(10):2003–12. doi: 10.1016/j.cellsig.2007.05.013
28. Gujral TS, Karp RL, Finski A, Chan M, Schwartz PE, MacBeath G, et al. Profiling phospho-signaling networks in breast cancer using reverse-phase protein arrays. Oncogene (2013) 32(29):3470–6. doi: 10.1038/onc.2012.378
29. Salian-Mehta S, Xu M, Wierman ME. Axl and met crosstalk to promote gonadotropin releasing hormone (Gnrh) neuronal cell migration and survival. Mol Cell Endocrinol (2013) 374(1-2):92–100. doi: 10.1016/j.mce.2013.04.018
30. Ruan GX, Kazlauskas A. Axl is essential for vegf-a-Dependent activation of Pi3k/Akt. EMBO J (2012) 31(7):1692–703. doi: 10.1038/emboj.2012.21
31. Gavine PR, Ren Y, Han L, Lv J, Fan S, Zhang W, et al. Volitinib, a potent and highly selective c-met inhibitor, effectively blocks c-met signaling and growth in c-met amplified gastric cancer patient-derived tumor xenograft models. Mol Oncol (2015) 9(1):323–33. doi: 10.1016/j.molonc.2014.08.015
32. Du Z, Caenepeel S, Shen Y, Rex K, Zhang Y, He Y, et al. Preclinical evaluation of amg 337, a highly selective small molecule met inhibitor, in hepatocellular carcinoma. Mol Cancer Ther (2016) 15(6):1227–37. doi: 10.1158/1535-7163.MCT-15-0745
33. Elisei R, Schlumberger MJ, Muller SP, Schoffski P, Brose MS, Shah MH, et al. Cabozantinib in progressive medullary thyroid cancer. J Clin Oncol (2013) 31(29):3639–46. doi: 10.1200/JCO.2012.48.4659
34. Choueiri TK, Halabi S, Sanford BL, Hahn O, Michaelson MD, Walsh MK, et al. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: The alliance A031203 cabosun trial. J Clin Oncol (2017) 35(6):591–7. doi: 10.1200/JCO.2016.70.7398
35. Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med (2018) 379(1):54–63. doi: 10.1056/NEJMoa1717002
36. Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, et al. Cabozantinib (Xl184), a novel met and Vegfr2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther (2011) 10(12):2298–308. doi: 10.1158/1535-7163.MCT-11-0264
37. Christensen JG, Zou HY, Arango ME, Li Q, Lee JH, McDonnell SR, et al. Cytoreductive antitumor activity of pf-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-met, in experimental models of anaplastic Large-cell lymphoma. Mol Cancer Ther (2007) 6(12 Pt 1):3314–22. doi: 10.1158/1535-7163.MCT-07-0365
38. Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, et al. An orally available small-molecule inhibitor of c-met, pf-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res (2007) 67(9):4408–17. doi: 10.1158/0008-5472.CAN-06-4443
39. Puccini A, Marin-Ramos NI, Bergamo F, Schirripa M, Lonardi S, Lenz HJ, et al. Safety and tolerability of c-met inhibitors in cancer. Drug Saf (2019) 42(2):211–33. doi: 10.1007/s40264-018-0780-x
40. Shaw AT, Riely GJ, Bang YJ, Kim DW, Camidge DR, Solomon BJ, et al. Crizotinib in Ros1-rearranged advanced non-Small-Cell lung cancer (Nsclc): Updated results, including overall survival, from profile 1001. Ann Oncol (2019) 30(7):1121–6. doi: 10.1093/annonc/mdz131
41. Nokihara H, Nishio M, Yamamoto N, Fujiwara Y, Horinouchi H, Kanda S, et al. Phase 1 study of cabozantinib in Japanese patients with expansion cohorts in non-Small-Cell lung cancer. Clin Lung Cancer (2019) 20(3):e317–e28. doi: 10.1016/j.cllc.2018.12.018
42. He AR, Cohen RB, Denlinger CS, Sama A, Birnbaum A, Hwang J, et al. First-in-Human phase I study of merestinib, an oral multikinase inhibitor, in patients with advanced cancer. Oncologist (2019) 24(9):e930–e42. doi: 10.1634/theoncologist.2018-0411
43. Eskens FA, Verweij J. The clinical toxicity profile of vascular endothelial growth factor (Vegf) and vascular endothelial growth factor receptor (Vegfr) targeting angiogenesis inhibitors; a review. Eur J Cancer (2006) 42(18):3127–39. doi: 10.1016/j.ejca.2006.09.015
44. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting Flt3 mutations in aml: Review of current knowledge and evidence. Leukemia (2019) 33(2):299–312. doi: 10.1038/s41375-018-0357-9
45. Lacy SA, Miles DR, Nguyen LT. Clinical pharmacokinetics and pharmacodynamics of cabozantinib. Clin Pharmacokinet (2017) 56(5):477–91. doi: 10.1007/s40262-016-0461-9
46. Oliveira KCS, Ramos IB, Silva JMC, Barra WF, Riggins GJ, Palande V, et al. Current perspectives on circulating tumor DNA, precision medicine, and personalized clinical management of cancer. Mol Cancer Res (2020) 18(4):517–28. doi: 10.1158/1541-7786.MCR-19-0768
47. Yang Y, Huang J, Wang T, Zhou J, Zheng J, Feng J, et al. Decoding the evolutionary response to ensartinib in patients with alk-positive nsclc by dynamic circulating tumor DNA sequencing. J Thorac Oncol (2021) 16(5):827–39. doi: 10.1016/j.jtho.2021.01.1615
48. Song Y, Hu C, Xie Z, Wu L, Zhu Z, Rao C, et al. Circulating tumor DNA clearance predicts prognosis across treatment regimen in a Large real-world longitudinally monitored advanced non-small cell lung cancer cohort. Transl Lung Cancer Res (2020) 9(2):269–79. doi: 10.21037/tlcr.2020.03.17
49. Wu SG, Shih JY. Management of acquired resistance to egfr tki-targeted therapy in advanced non-small cell lung cancer. Mol Cancer (2018) 17(1):38. doi: 10.1186/s12943-018-0777-1
50. Guibert N, Hu Y, Feeney N, Kuang Y, Plagnol V, Jones G, et al. Amplicon-based next-generation sequencing of plasma cell-free DNA for detection of driver and resistance mutations in advanced non-small cell lung cancer. Ann Oncol (2018) 29(4):1049–55. doi: 10.1093/annonc/mdy005
51. Piotrowska Z, Isozaki H, Lennerz JK, Gainor JF, Lennes IT, Zhu VW, et al. Landscape of acquired resistance to osimertinib in egfr-mutant nsclc and clinical validation of combined egfr and ret inhibition with osimertinib and blu-667 for acquired ret fusion. Cancer Discovery (2018) 8(12):1529–39. doi: 10.1158/2159-8290.CD-18-1022
52. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the axl kinase causes resistance to egfr-targeted therapy in lung cancer. Nat Genet (2012) 44(8):852–60. doi: 10.1038/ng.2330
53. Wang Q, Yang S, Wang K, Sun SY. Met inhibitors for targeted therapy of egfr tki-resistant lung cancer. J Hematol Oncol (2019) 12(1):63. doi: 10.1186/s13045-019-0759-9
54. Blakely CM, Bivona TG. Resiliency of lung cancers to egfr inhibitor treatment unveiled, offering opportunities to divide and conquer egfr inhibitor resistance. Cancer Discovery (2012) 2(10):872–5. doi: 10.1158/2159-8290.CD-12-0387
Keywords: preclinical, phase I, tyrosine kinase inhibitor, MET, AXL, VEGFR2
Citation: Ma Y-X, Liu F-R, Zhang Y, Chen Q, Chen Z-Q, Liu Q-W, Huang Y, Yang Y-P, Fang W-F, Xi N, Kang N, Zhuang Y-L, Zhang Q, Jiang Y-Z, Zhang L and Zhao H-Y (2022) Preclinical characterization and phase I clinical trial of CT053PTSA targets MET, AXL, and VEGFR2 in patients with advanced solid tumors. Front. Immunol. 13:1024755. doi: 10.3389/fimmu.2022.1024755
Received: 22 August 2022; Accepted: 30 September 2022;
Published: 20 October 2022.
Edited by:
Geoffrey Brown, University of Birmingham, United KingdomReviewed by:
Sean Bong Lee, Tulane University, United StatesCopyright © 2022 Ma, Liu, Zhang, Chen, Chen, Liu, Huang, Yang, Fang, Xi, Kang, Zhuang, Zhang, Jiang, Zhang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong-Yun Zhao, emhhb2h5QHN5c3VjYy5vcmcuY24=; Li Zhang, emhhbmdsaUBzeXN1Y2Mub3JnLmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.