
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 23 September 2022
Sec. Alloimmunity and Transplantation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1020889
This article is part of the Research TopicEffects of cell interactions at the endothelium in leukocyte differentiation and polarization during inflammationView all 13 articles
 Guiyu Song1,2†Shaoxun Wang1,3†
Guiyu Song1,2†Shaoxun Wang1,3† Mahsa Nouri Barkestani1†
Mahsa Nouri Barkestani1† Clancy Mullan3,4Matthew Fan1
Clancy Mullan3,4Matthew Fan1 Bo Jiang3,5Quan Jiang1Xue Li1,6
Bo Jiang3,5Quan Jiang1Xue Li1,6 Dan Jane-wit1,4,7*
Dan Jane-wit1,4,7*Endothelial cells (ECs) form a critical immune interface regulating both the activation and trafficking of alloreactive T cells. In the setting of solid organ transplantation, donor-derived ECs represent sites where alloreactive T cells encounter major and minor tissue-derived alloantigens. During this initial encounter, ECs may formatively modulate effector responses of these T cells through expression of inflammatory mediators. Direct allorecognition is a process whereby recipient T cells recognize alloantigen in the context of donor EC-derived HLA molecules. Direct alloresponses are strongly modulated by human ECs and are galvanized by EC-derived inflammatory mediators.
Complement are immune proteins that mark damaged or foreign surfaces for immune cell activation. Following labeling by natural IgM during ischemia reperfusion injury (IRI) or IgG during antibody-mediated rejection (ABMR), the complement cascade is terminally activated in the vicinity of donor-derived ECs to locally generate the solid-phase inflammatory mediator, the membrane attack complex (MAC). Via upregulation of leukocyte adhesion molecules, costimulatory molecules, and cytokine trans-presentation, MAC strengthen EC:T cell direct alloresponses and qualitatively shape the alloimmune T cell response. These processes together promote T cell-mediated inflammation during solid organ transplant rejection.
In this review we describe molecular pathways downstream of IgM- and IgG-mediated MAC assembly on ECs in the setting of IRI and ABMR of tissue allografts, respectively. We describe work demonstrating that MAC deposition on ECs generates ‘signaling endosomes’ that sequester and post-translationally enhance the stability of inflammatory signaling molecules to promote EC activation, a process potentiating EC-mediated direct allorecognition. Additionally, with consideration to first-in-human xenotransplantation procedures, we describe clinical therapeutics based on inhibition of the complement pathway. The complement cascade critically mediates EC activation and improved understanding of relevant effector pathways will uncover druggable targets to obviate dysregulated alloimmune T cell infiltration into tissue allografts.
Human ECs comprise a barrier interface regulating activation and recruitment of lymphocytes. In the setting of solid organ transplantation, donor ECs represent sites for initial alloantigen encounter by recipient alloreactive T cells. Donor ECs in humans express both MHC class I and II molecules as well as sufficient costimulatory molecules to enable direct allorecognition. In direct allorecognition, alloimmune T cells recognize antigen in the context of EC-derived HLA molecules. Major alloantigens including class I and II HLA molecules mediate strong type 1 responses, and the strength and quality of these responses are modulated by inflammatory mediators expressed by cognate ECs.
Complement-derived mediators are well known to modulate the direct alloresponse. Complement are evolutionarily ancient immune proteins that allow host recognition of foreign surfaces like donor ECs. In solid organ transplantation, complement proteins become targeted for activation on donor ECs during perioperative ischemia reperfusion injury (IRI) and antibody-mediated rejection (ABMR). When activated on ECs, complement proteins undergo a series of proteolytic cleavages, resulting in the formation of heterodimeric protein complexes with novel enzymatic activities enabling formation of inflammatory mediators. Anaphylatoxins, C3a and C5a, as well as membrane attack complexes (MAC) are inflammatory mediators generated following complement activation. Complement-derived inflammatory mediators promote EC activation and enhance EC-mediated TCR : MHC interactions, costimulatory processes, and T cell recruitment. Improved understanding of the immune mechanisms surrounding complement-mediated EC activation in the setting of direct allorecognition will inform therapies to block EC:lymphocyte interactions and will improve outcomes for transplant recipients.
This review will principally focus on MAC and its attendant molecular mechanisms inducing EC activation to modulate direct allorecognition. We first review complement activation pathways forming MAC, modes of allorecognition, and mechanisms for resisting MAC-induced cell death. We then describe experimental methodologies for studying the biological effects of MAC and MAC-induced signaling pathways. We focus on recent work elucidating MAC-induced ‘signaling endosomes’ that sequester and enhance the stability of pro-inflammatory mediators to promote EC activation. In light of recent first-in-human xenotransplant procedures, we close with a description of therapies focused on complement inhibition and their clinical applications in solid organ transplantation.
The complement system is an evolutionarily conserved system of effectors, split products, and regulatory proteins enabling host recognition of endogenously altered, damaged, or foreign surfaces (1). Following activation by donor ECs, the complement system generates inflammatory mediators, anaphylatoxins, and membrane attack complexes (MAC) that modulate direct allorecognition. We review the 3 established pathways for complement activation below (Figure 1).
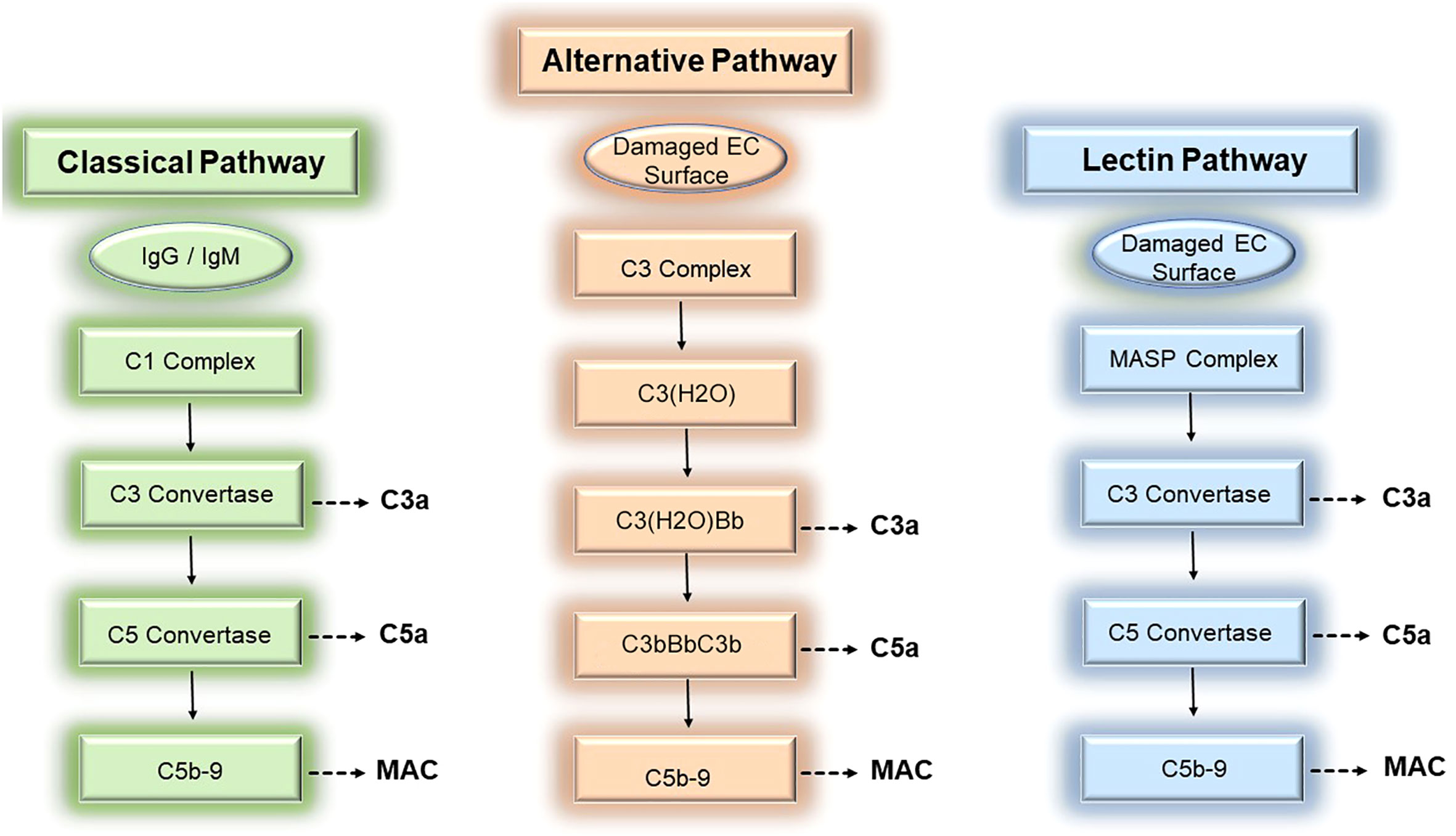
Figure 1 Three pathways of complement activation. The complement cascade may be activated by donor endothelial cells via 3 separable pathways, the classical (left), alternative (middle), and lectin (right) pathways, to mediate rejection of tissue allografts.
The complement system is comprised of ~30 soluble- and surface-bound proteins, 9 of which are complement effector proteins. Complement effector proteins, termed C1-C9, are principally produced by the liver and circulate as inactive zymogens. Initially identified as a heat-labile component of normal plasma, complement effectors were renamed C1 to C9 by the World Health Organization in 1968 to reflect the order by which these proteins become activated rather than the order whereby these proteins were discovered (2). Complement effectors, C1-C9, circulate as inactive zymogens and become activated on donor ECs during ischemia reperfusion injury (IRI) and antibody-mediated rejection (ABMR). During these conditions, surface-bound Abs including IgM or IgG bind to post-ischemic neo-antigens (3–5), MHC molecules (6, 7), or non-HLA alloantigens (8, 9) expressed on donor ECs. EC-bound Abs subsequently trigger activation of circulating complement effectors via the classical complement pathway.
The classical pathway involves successive formation of 4 heterodimeric complexes to generate complement-derived inflammatory mediators. The first complex, the C1 complex, contains the pattern recognition receptor, C1q, whose globular head binds to the Fc region of pentameric IgM and monomeric IgG (10, 11) during IRI and ABMR. In addition to Abs, C1q also binds a plethora of EC-derived molecules including various damage-associated molecular patterns (DAMPs, 12, 13), apoptotic cells (14, 15), and altered self-proteins (16–20). Upon ligand binding, C1q becomes complexed with the C1r and C1s proteases to form a C1q-binding tetramer, C1r2C1s2 (21–23). The activated C1 recognition complex subsequently promotes proteolytic cleavage of C4 and C2 whose split products form a second protein complex comprised of C4bC2a heterodimers. C4bC2a heterodimers are a C3 convertase that acquires the ability to proteolytically cleave C3. Cleavage of C3 by the C3 convertase generates the soluble inflammatory mediator, C3a, and a split product, C3b. C3b binds to the C3 convertase to form a third protein complex, C4bC2aC3b, which becomes a C5 convertase with the ability to cleave C5. The C5 convertase mediates cleavage of C5 to generate the potent anaphylatoxin, C5a, as well as the split product, C5b. C5b heterodimerizes with terminal complement proteins C6, C7, to form a ring-like substrate that interacts with the surface lipid bilayer of target cells like donor ECs (24). C5b-7 is a MAC initiator complex that subsequently recruits C8. C8 in turn binds to and accelerates the polymerization of C9 monomers (24). Following binding to C8, C9 monomers recruited from soluble pools in sera undergo unidirectional, clockwise polymerization to form a pore-like structure of 90-110Å in diameter and permeable to molecules of 10-17 kD (25–28). Successful insertion of the C5b-9 complex into donor EC membranes results in formation of MAC, the fourth complement protein complex. MAC are composed of 13-18 C9 molecules (27); trimeric C8 composed of α, β, and γ subunits (29); and the initiator C5b-7 ring which becomes a stoichiometrically minor component of the completed transmembrane structure (27, 28). Following MAC insertion into EC surfaces, MAC cause non-cytolytic EC activation as described in the subsequent section.
Complement activation may be triggered by 2 additional pathways, the lectin pathway and the alternative pathway, both of which have been implicated in vascular rejection of tissue allografts and will be described briefly here. In the lectin pathway, a recognition complex containing mannose-binding lectins (MBLs) or ficolins act as pattern recognition receptors canonically sensing carbohydrate and molecules containing an acetyl determinant like N-acetylglucosamine (GlcNAc) and N-acetylgalactosamine (GalNAc, 30–32). MBLs/ficolins become complexed with mannose-associated serine proteases (MASP1/2/3, 33, 34) that, analogous to C1r/s, sequentially cleave C4 and C2 to generate the second protein complex, C4b2a, that functions as a C3 convertase. Following activation by the MASP recognition complex, the lectin pathway proceeds to generate the same downstream complexes as the classical pathway above including the C5 convertase (C4bC2aC3b) and MAC (C5b-9). Activation of the lectin pathway has been linked to IRI (35, 36), and polymorphisms in the MBL2 pattern recognition receptor, is significantly associated with development of ABMR (37).
The third complement activation pathway, the alternative pathway, has been widely implicated in IRI (38–41). C3 is the most abundant complement protein in plasma, circulating at a concentration of ~1.2mg/mL, and various C3 cleavage products centrally mediate inflammation. C3 molecules undergo tick-over, or constitutive spontaneous hydrolysis, to form biologically active C3(H2O) molecules at a basally low rate of ~1-2 molecules per hour (42). Foreign surfaces like donor ECs dramatically accelerate tick-over, allowing formation of threshold quantities of C3(H2O) that bind to and cleave factor B, forming a C3(H2O)Bb complex that functions as a C3 convertase (43–46). The C3 convertase activity of C3(H2O)Bb cleaves C3 to generate C3a and the split product, C3b. C3b subsequently binds to Bb to form new a protein complex, C3bBbC3b, containing 2 molecules of C3b complexed with Bb. The stability of these complexes is enhanced by properdin/factor D. The C3bBbC3b complex acquires C5 convertase activity and cleaves C5 to form C5a and C5b, the latter of which becomes part of the MAC initiator complex, C5b-7. Similar to the classical or lectin pathways above, C3a, C5a, and MAC are generated as inflammatory mediators through successive proteolytic amplification. Similar to the classical and lectin pathways where gene polymorphisms have been linked to patient outcomes polymorphic variation in C3 has shown associations with ABMR in numerous studies (47–50).
Complement effectors are highly conserved among metazoans of both invertebrate and vertebrate species. Among the most ancient complement proteins are the early complement effectors, C1-C5, whose evolutionary ages range between 500-1000 million years and whose split products show biological activity. Split products derived from early complement effectors include soluble-phase anaphylatoxins, C3a and C5a, and solid-phase opsonins, iC3b, C3b, and C3d, which covalently bind to donor ECs during IRI and ABMR. A large body of evidence ascribes an inflammatory role for soluble complement mediators in immune cell activation which includes EC:T cell interactions. In addition to opsonizing foreign material, C3a and C5a bind to cognate GPCRs, C3aR and C5aR, respectively, to transmit inflammatory signals to target cells like donor ECs. C3a binds to C3aR, and C5a binds to 2 C5a receptor isoforms, C5aR1 and C5aR2, showing distinct cellular localization and having opposing signaling effects. Knockout mouse models involving C3 and C5 have demonstrated a strong role for anaphylatoxins in mediating myeloid cell recruitment to transplanted tissues as well as roles in recruitment of CD4+ T cells. It is worth noting that there is substantial evidence that C3 and C5, may be also produced by parenchymal and immune cells and that these proteins may become cleaved intracellularly to mediate immune signaling. The roles of intracellularly-derived C3a and C5a and their roles in T cell activation have been comprehensively reviewed elsewhere (51).
Humans express 4 complement receptors, termed CR1-CR4 which variably bind to C3 cleavage products including C3b, iC3b, C3d, C3dg. In particular, iC3b/C3b/C3d form covalent attachments to host proteins, damaged erythrocytes, and Fc receptors of Abs and importantly show binding to CR2. CR2 is heavily expressed on follicular dendritic cells and B cells and mediates pathological B cell activation in various disease settings including viral infection and ABMR (52).
As opposed to early complement components, the terminal complement effectors forming MAC, C6-C9, emerged later in evolution (1). Genetic deficiencies of terminal complement components are widely recognized as conferring increased risk for infection by encapsulated bacterial, in particular Neisseriae, and has strongly influenced clinical guidelines regarding vaccination strategies involving multiple pathogens (53). Genetic loss of C9 confers a 700-fold increased risk for Neisserial meningitis (54), suggesting evolutionary development of MAC as a defense mechanism against infection by such pathogens.
While seminally studied for their roles in mediating cytolysis (2), MAC formed from autologous complement proteins from the same species elicit EC activation without substantial EC death in vivo (reviewed in 55, 56). In contrast to in vitro models showing that MAC may induce cell death of up to ≥50% of treated cells, complement activation in patients, even during severe or terminal instances of ABMR, show disproportionately low frequencies of donor EC cell death in relation to the degree of complement activation. This process also holds true for SARS-CoV-2 infection where ECs show non-cytopathic infection (57) despite strong MAC deposition on target tissues (58, 59); and systemic lupus erythematosus (SLE) where complement-mediated endothelitis occurs without widespread vessel rarefaction (60).
Seminal studies characterizing the physiologic function of MAC employed anucleated red blood cells or xenogeneic substrates like bacteria, as target cells (2). In these studies, the pore-forming capability of MAC which since have been well characterized ultrastructurally and on an atomic scale (24–29) were definitively demonstrated, an effect causing target cell lysis. However, resistance to MAC-induced cell death in nucleated, autologous cells was initially recognized in the 1980s (61–64), and since this time, at least 4 molecular mechanisms enabling target cell survival have been described.
In contrast to anucleated cells and foreign pathogens, autologous cells may constrain MAC-induced cytolysis in a species-specific manner. This phenomenon, known as homologous restriction, is mediated by complement regulatory proteins (65). Many proteins within the complement system contain complement control protein (CCP) domains consisting of conserved repeats that direct interactions among proteins in the complement system. The GPI-anchored proteins, CD55 and CD59, blocking generation of C5b and polymerization of C9, respectively, contain recursive CCP domains that limit assembly and facilitate removal of MAC from the cell surface (66). CCP domains, also known as “Sushi” domains were recently exploited in a bioinformatics approach to identify a new complement regulator, CSMD1, which was found to block MAC assembly at the level of C7 (67) and was implicated in complement activity and infertility in vivo and in a patient cohort carrying a non-synonymous CSDM1 mutation (68). MAC regulatory proteins expressed in autologous, nucleated cells thus act as scavengers for limiting the assembly and surface removal of MAC, enabling autonomous cells to resist osmotic lysis. As described below, transgenic knockins of CD46 and CD55, CCP-containing proteins, were used in porcine renal and cardiac xenografts implanted in humans (69, 70).
As a second MAC regulatory mechanism, C5b-9 insertion into target EC membranes is blocked via binding to blood-based chaperones, clusterin (71–73, aka, apolipoprotein J) and vitronectin (74, aka S protein). The C5b-7 ring interacting with target surfaces like donor ECs (24) promotes oligomerization of at least three C9 molecules (75) to form the terminal complement complex, C5b-9. Excess C5b-9 molecules bind to C9-binding chaperones and become trapped in a transition state that is non-permissive for their insertion into target membranes, thereby resulting solubilization (55, 76). Soluble terminal complement complexes, called, Sc5b-9, spatially constrains the radius of MAC assembly to the vicinity of Ab-bound donor ECs. Levels of Sc5b-9 are dramatically elevated during immune responses, and Sc5b-9 has been widely investigated as a biomarker reflecting tissue-bound levels of MAC (77). The biophysical underpinnings for the cell non-autonomous constraint of MAC activity by C9-binding chaperones has been widely investigated, in part due to the notion that the endogenous MAC inactivators, clusterin or vitronectin, could be therapeutically exploited to subvert excess complement activity (reviewed in 78–80).
Exovesiculation or shedding of surface-bound MAC represents a third mechanism by which autologous, nucleated cells survive MAC-induced lysis (81–83). Extracellular vesicles (EVs) are a heterogeneous group of cell-derived vesicles including exosomes and microvesicles. Exosomes and microvesicles are vesicular structures released extracellularly from living cells following MAC deposition on EC surfaces. Exosomes are spherical vesicles of 30-120nm in size that are produced via inward invagination of late endosomal membranes to form multivesicular bodies (83). Multivesicular bodies subsequently fuse with the plasma membrane to release their intraluminal contents including exosomes extracellularly. In contrast, microvesicles are ~100-1000nm in diameter, are derived from cell membranes, and contain high levels of surface-derived EC molecules including integrins, CD40L, and complement regulatory proteins CD55 and CD59 (83, 84). EVs including exosomes and microvesicles can be released by ECs (85, 86) and PMNs (87) and efficiently activate complement in vitro (85, 87) and thus it is postulated that EV membranes may serve as decoy substrates to limit MAC deposition on host tissues.
Finally, elimination of MAC from donor EC surfaces may reduce MAC to sub-lytic levels, thereby enabling intracellular coping pathways to restore homeostasis. As reviewed elsewhere (88), various intracellular pathways promote resistance to MAC-induced cytolysis, a process that has been explored in the setting of tumor cell evasion from complement-dependent cytotoxicity. Intracellular pathways conferring resistance to MAC-induced cytolysis include pathways related to intracellular trafficking of MAC (81, 82), calcium handling (89), MAPKs (82, 90), heat shock proteins (91), and NF-κB (92, 93). Together, the 4 regulatory mechanisms above contribute to survival of autologous, nucleated cells from MAC-induced lysis.
Evolutionary development of mechanisms for resisting MAC-induced cytolysis implies separable, non-cytolytic effects of MAC on target cells and provides a basis for studying inflammatory signaling in MAC-respondent cells like donor ECs. To study effects of non-cytolytic MAC, at least 5 experimental models for inducing MAC assembly have been developed. A widely used system for assembling MAC on nucleated cells involves the use of xenogeneic antisera, e.g., rabbit sera overlay on mouse target cells. This widely-used protocol has allowed discovery of various MAC-induced inflammatory pathways operative in immune cells including neutrophils, dendritic cells, and ECs (88) including NLRP3 inflammasome activation (94, 95). A second model for eliciting MAC assembly on nucleated cells incorporates stepwise addition of terminal complement proteins at defined stoichiometric ratios to allow formation of MAC in cell culture (96). This protocol, theoretically recapitulating homologous restriction, has been adapted for use in vivo (97). A third protocol for experimental MAC assembly involves the use of zymosan-activated serum. Zymosan is primarily composed of the highly cross-linked polysaccharides alpha-D-mannan, beta-D-glucan, and other minor polysaccharide polymers derived from yeast (S. cerevisiae). Yeast-derived zymosan potently activates all 3 complement pathways and has been widely used to study downstream effects of anaphylatoxins on immune cells such as neutrophils and macrophages (28).
Recently, human sera have been exploited as a source for complement proteins to enable formation of autologous MAC on human ECs. In a model of ABMR, human ‘high’ panel reactive antibody (PRA) sera taken from allo-sensitized transplant candidates were used to elicit autologous MAC deposition on human ECs (98). PRA sera induced non-cytolytic MAC on human ECs in an IgG-dependent manner in vitro and in vivo, facilitating identification of MAC signaling pathways including non-canonical NF-κB (98–100), NLRP3 inflammasome (101, 102), and canonical NF-κB (101). These pathways collectively contributed to EC activation and enhanced EC-mediated direct allorecognition. In a model of IRI, human sera used as a source for complement proteins deposited non-cytolytic MAC on human ECs in an IgM-dependent manner following in vitro anoxia of human ECs or coronary artery segments (102). Using this model, EC-mediated direct alloresponses promoted selective expansion of T peripheral helper (TPH) cells but not T follicular helper (TFH) cells in an IL-18-dependent manner to promote B cell activation and de novo DSA within donor tissues. Various model systems have been developed to assemble non-cytolytic MAC in vitro and in vivo and have shown utility in defining MAC signaling effects across numerous clinical settings.
Alloantigen encounter may occur via 3 pathways including direct allorecognition, indirect allorecognition, and semi-direct allorecognition (Figure 2; 103). In direct allorecognition, the antigen presenting cells (APCs) are the donor-derived cells that present donor MHC: peptide complexes to the host T cells. In indirect allorecognition, the donor-derived antigens are acquired and processed by the recipient APCs. The recipient APCs then present the donor antigens to host T cells. Semidirect allorecognition occurs when MHC: peptide complexes shed from donor-derived cells become captured by recipient APCs (2). The recipient APCs then present MHC: peptide complexes to host T cells. T cell activation is restricted by the donor MHC molecules in direct allorecognition, whereas the T cell activation is restricted by recipient MHC molecules in indirect allorecognition. Because the intact donor-derived MHC molecules are presented by the recipient APCs in semidirect allorecognition, T cell activation is restricted by donor MHC molecules in this context (104).
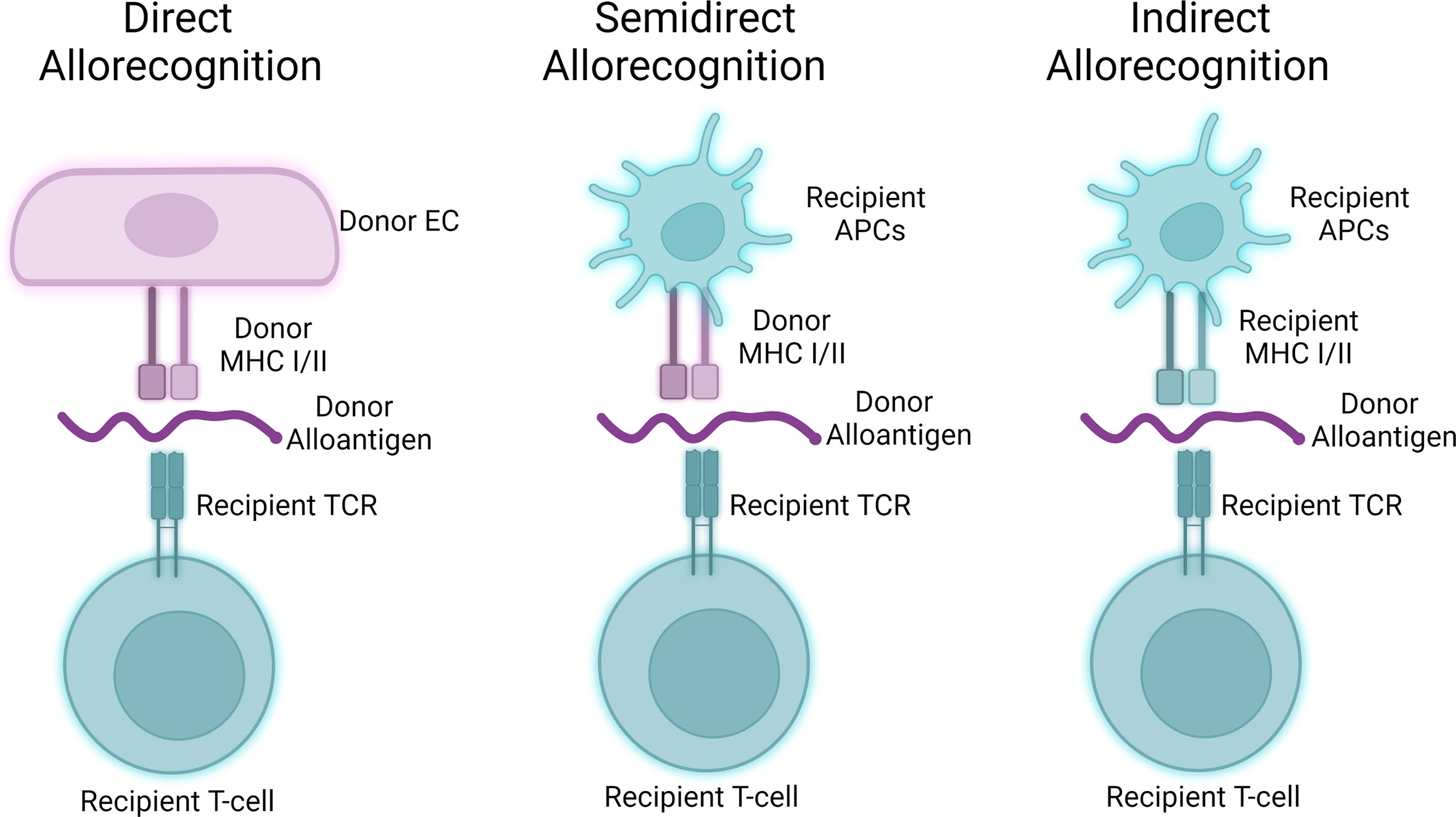
Figure 2 Three pathways of allorecognition. Donor-derived alloantigens may activate allogeneic T cells via three separable pathways. Human endothelial cells may act as antigen presenting cells to participate in direct allorecognition responses (left), while murine hosts primarily participate in semidirect and indirect allorecognition (middle, right) through interactions involving professional antigen presenting cells.
Human ECs are functionally different from mouse ECs in allograft rejection as they are capable to provide costimulatory signaling to activate T cells via direct allorecognition. This is because of the unique expression pattern in antigen-presenting molecules and co-stimulators in human endothelial cells. Human ECs not only express both MHC I and MHC II but also constitutively express co-stimulators inducing LFA-3, ICOS ligand, programmed cell death protein Ligand-1 (PDL1), 4-1BB ligand, and OX40-ligand. Murine ECs unlike human ECs lack the ability to strongly elicit direct alloresponses due to the lack of expression of key costimulatory molecules, most saliently CD58/LFA3 (105). These co-stimulators in human ECs allow direct recognition and activation of central and effector memory T cells (106–108). The primary vascular cell type responsible for direct allorecognition in graft arteries is ECs as vascular smooth muscle cell expresses rather a low level of HLA molecules (105). Following direct allorecognition, humoral- and surface-derived signals cause EC-mediated allostimulation. While MHC class I/II strongly induce type 1, i.e., IFN-γ, responses from T cells, EC:T cell crosstalk results in EC elaboration of various cytokine factors including TGF-β and IL-6 that may further influence differentiation of T cells activated through direct allorecognition. Allostimulation following direct allorecognition is further enhanced by processes affecting ECs including local complement activity as described in the following section. In ABMR, the major cell infiltrate in graft artery intima are T cells, most of which demonstrate a type 1 effector profile characterized by interferon-gamma (IFN-γ) production. There is a significant amount of IFN-γ production in the recovered T cells isolated from lesions in allograft vasculopathy, and IFN-γ is necessary and sufficient to drive cardiac allograft vasculopathy, a condition characterized by pathologic vascular changes in transplant patients with chronic ABMR (109).
Major or minor alloantigen mismatch in a heterotopic heart transplantation model in mice elicits T cell-mediated rejection principally via indirect allorecognition. To test EC-mediated direct allorecognition, a response operative in human tissues, our group has developed humanized mouse models. In a human artery xenograft model, human coronary artery segments are subjected to various treatments simulating IRI (99, 110, 111) or ABMR (98–102, 112) prior to surgical implantation into descending aortae of immunodeficient SCID/beige mice engrafted with human T and B cells. Human artery xenografts subjected to IRI potentiate the T cell-meditated allograft injury in the human artery xenograft model. Because the vascular smooth muscle cannot activate allogeneic T cells and the absence of leukocytes in the donor grafts activated human ECs appears to be the dominant APCs in this system (99, 110, 111). Indeed, human artery xenografts subjected to IRI or ABMR treatments show non-cytolytic MAC assembly on intimal ECs, a process causing EC activation and resulting in potentiated alloimmune T cell activation. As myeloid cells including antigen presenting cells like dendritic cells do not engraft in SCID/beige mice, the potentiated T cell activation in this model is primarily driven by direct allorecognition and formation of de novo donor specific antibody in this model occurs within allograft tissues and not the spleen (110). The grafted artery shows intimal expansion characterized by an infiltration of CD45RO+ T cells, the formation of a human EC-lined microvessel, and the presence of VMSCs. These observations highly mimic the clinical features of graft arteriosclerosis in allograft vasculopathy (109). To further uncover the underlying mechanisms, we found that formation of MAC (99) and MAC-induced inflammatory mediators in human ECs including ZFYVE21 (100), NLRP3 (101, 102), IL-15 (112), and IL-18 (110) potentiates frequencies of CD4+ T cells producing IFN-γ.
In a second humanized model, human umbilical vein endothelial cells (HUVECs) are suspended on collagen-fibronectin gel matrices and implanted subcutaneously in SCID/beige mice. Three to four weeks post-implantation, HUVECs self-organize into perfused microvascular networks anastomosing with murine vasculature (113). By transducing HUVECs with various overexpression, dominant negative constructs (101), or CRISPR/Cas9 gene edits (114) prior to implantation, this model allows interrogation into how immune molecules including MAC-induced signaling proteins in ECs affect direct allorecognition. Murine and humanized models enable the study of indirect and direct allorecognition responses by ECs, respectively.
Donor ECs are a principal cell type affected by MAC in ABMR and IRI. via activation on donor ECs, MAC centrally governs EC:T cell interactions, and MAC formation on ECs is prognostic for, diagnostic for, and a therapeutic target for lymphocyte-mediated injury of tissue allografts in these settings. The use of antisera or human sera to assemble MAC on nucleated cells as described above has enabled elucidation of MAC signaling pathways and their effects on direct allorecognition.
MHC-derived alloantigens strongly mediate type 1 responses, i.e., IFN-γ production, and the strength and quality of this response was found to be modulated by MAC. Non-cytolytic MAC deposition on human ECs upregulated adhesion molecules including VCAM-1 and E-selectin, and inflammatory gene transcripts encoding chemokines like CCL5 and CCL20 and cytokines like IL-6 (98). In EC:T cell cocultures, MAC-treated ECs increased frequencies of adherent CD4+CD45RO+ memory T cells (Tmem) under conditions of postcapillary venular shear stress and non-specifically enhanced bulk cytokine effector responses involving IFN-γ, IL-4, and IL-17 in EC:T cell cocultures. In vivo, human coronary artery segments exposed to PRA and implanted in immunodeficient mice engrafted with human T and B cells showed increased infiltration of intimal IFN-γ+ T cells causing neointimal expansion, i.e., cardiac allograft vasculopathy (98, 99). Thus, MAC broadly enhances the recruitment and strength of effector activity of Tmem following EC-mediated direct allorecognition.
In two separable processes, MAC may qualitatively shape allostimulation, the immune response occurring following direct allorecognition. First, MAC may induce differential expansion of T cell subsets following direct allorecognition. A survey of costimulatory molecules in MAC-treated ECs showed that MAC upregulated PD-L2 and ICOS-L, cognate receptors for PD-1 and ICOS, respectively (110). PD-1 and ICOS are heavily expressed on 2 recently described subsets, T peripheral helper (TPH) and T follicular helper (TFH) cells. These subsets express PD-1 and ICOS and elaborate IL-21 to promote B cell maturation and Ab responses. MAC-treated ECs were found to selectively enhance expansion of TPH cells in an IL-18-dependent manner, a process eliciting production of de novo DSA within inflamed peripheral vessels. Mechanistically, MAC induced NLRP3 inflammasome activity in ECs, resulting in IL-18 release. IL-18 binds to its cognate receptor, IL-18R, which was selectively expressed on TPH cells co-expressing CCR2 but not TFH cells co-expressing CXCR5. This differential expression of IL-18R caused selective expansion of TPH but not TFH cells. CCR2+ TPH cells infiltrated into IRI tissues to mediate B cell maturation and formation of de novo DSA in human artery xenografts in vivo, and mass cytometry (CyTOF) analysis of Tmem in renal transplant recipients with IRI showed selective expansion of TPH but not TFH cells in peripheral circulation (110). This study demonstrated that MAC could qualitatively shape the T cell response by promoting differential expansion of a defined T cell subset following direct allorecognition.
Secondly, MAC-induced IL-1β resulting from inflammasome activity enhances allostimulation by expanding cytotoxic T lymphocytes. In an autocrine fashion, MAC-induced IL-1β was found to increase production of IL-15 which became associated with the IL-15 receptor α chain (IL-15Rα) at the EC surface (112). This complex trans-presented IL-15 to effector memory CD8+ T cells to induce their proliferation and to allow these cells to become cytotoxic T lymphocytes (CTLs), characterized by production of granzyme B and perforin. Trans-presentation of IL-15 by ECs to T cells additionally increased trans-endothelial migration by activating CD11a/CD18 and motility. While there is a propensity for activation and recruitment of CD8+ T cells relative to CD4+ T cells by EC trans-presentation of IL-15, both populations respond to some degree to surface IL-15/IL-15Rα complexes (112). With IL-15 blockade by monoclonal antibody, co-cultured CD4+ and CD8+ T cells lose polyfunctionality, an effect which is only partially reproduced by IL-1 receptor antagonism. A likely explanation for this is the evolution of other cytokines by the co-cultured T cells themselves that signal through canonical NF-κB, such as tissue necrosis factor α, and are sufficient to produce surface IL-15/IL-15Rα (115). This study shows that MAC assembly on ECs could induce sequential production of IL-1β and IL-15 to qualitatively modulate the direct alloresponse. Accumulated data show that MAC may modulate both the strength and quality of direct alloresponses of alloimmune T cells.
Companion studies have elucidated inflammatory signaling pathways induced by MAC that modulated the direct alloresponses above. Using human PRA sera to assemble autologous MAC on human ECs (Figure 3), it was found that MAC and not IgG or anaphylatoxins, activated non-canonical NF-κB, a pathway marked by NF-κB-inducing kinase (NIK). Activation of non-canonical NF-κB by MAC occurred within 15-30 min (98, 100) in a TRAF3-independent manner (99), features starkly contrasting with described pathways of non-canonical NF-κB involving ligand:receptor interactions occurring in 18-24 hr and requiring TRAF3 degradation. The unusual pattern of non-canonical NF-κB activity induced by MAC, coupled with the observation that MAC became internalized to form a MAC+Rab5+ intracellular compartment (100), led to the hypothesis of the ‘signaling endosome’ enabling MAC-induced inflammatory signaling.
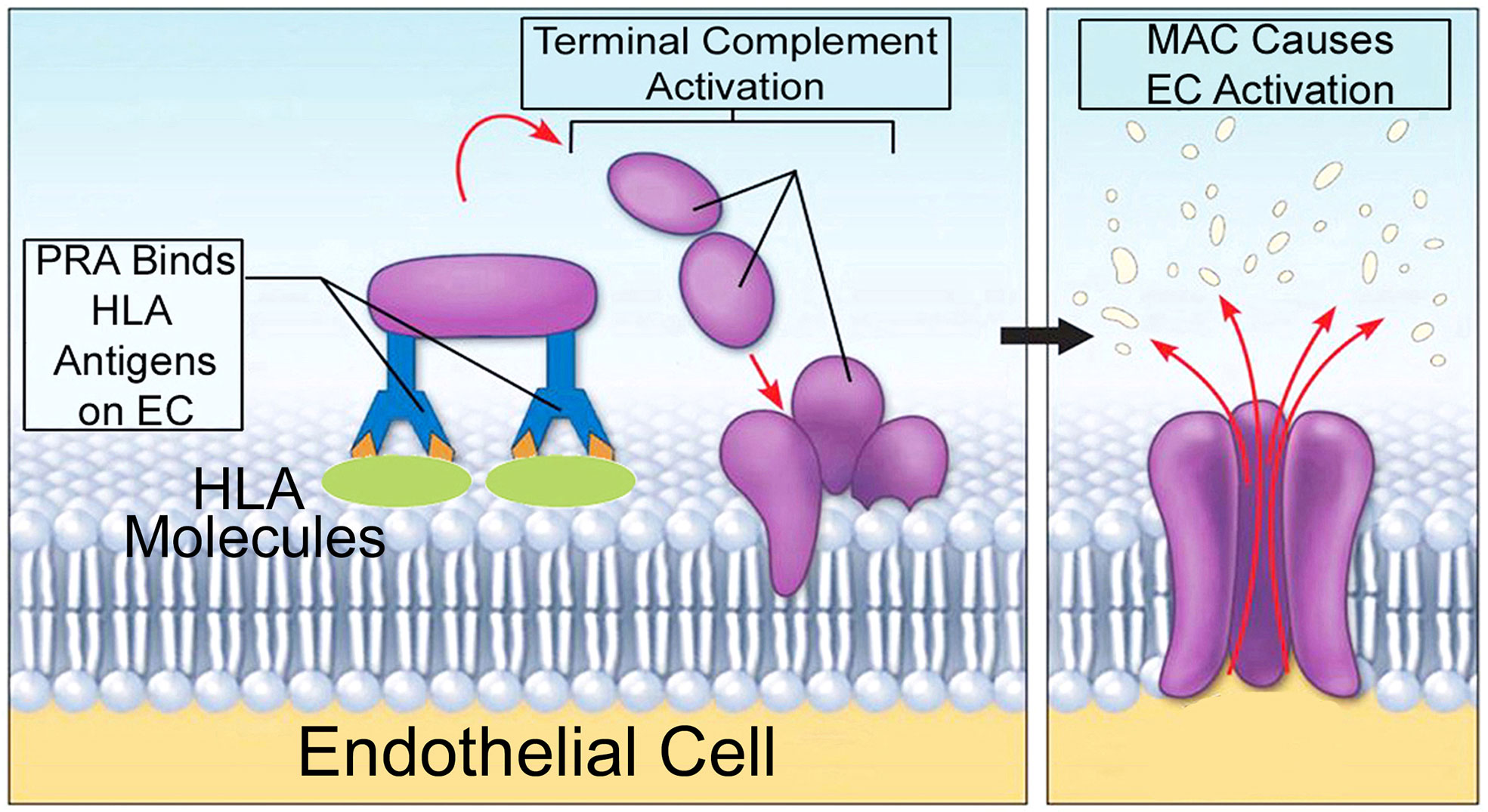
Figure 3 A model for autologous alloantibody-induced MAC assembly. 'High' panel reactive antibody (PRA) sera is obtained from transplant candidates and contain high titers of alloantibodies binding to many HLA specificities. PRA mediates binding of donor specific alloantibody to surface MHC I/II molecules on human ECs. This activates complement components within PRA sera, causing terminal activation of the classical complement pathway and formation of non-cytolytic MAC on target ECs. Human-derived MAC on human ECs causes EC activation without inducing cell lysis.
Drug, siRNA, and dominant negative treatments blocking MAC+Rab5+ endosome formation reduced inflammatory gene transcripts and attenuated EC-mediated generation of IFN-γ+ Tmem in EC:T cell cocultures (100). This remarkable finding indicated that MAC did not elicit NF-κB activity or EC activation from the cell surface. Rather, MAC rapidly underwent clathrin-mediated endocytosis and transfer to Rab5+ vesicles, forming MAC+Rab5+ endosomes. This compartment contained ‘signaling endosomes’ which were found to sequester and post-translationally enhance the stability of signal-activating molecules by protecting these same molecules from proteasome degradation (100).
As an extension of the signaling endosome hypothesis, we reasoned that these same structures were enriched in signal-activating proteins. We devised a protocol for proteomic analysis of FACS-sorted MAC+Rab5+ endosomes, and this protocol facilitated unbiased identification of ZFYVE21 which we found was a novel Rab5 effector (100). ZFYVE21 is a conserved molecule localized to early endosomes initially implicated in tumor cell migration (116) but whose functions remain poorly understood. Our group found that ZFYVE21 regulated lipid and protein remodeling of MAC+Rab5+ endosomes to cause sequential pAkt recruitment and enhanced NIK stability. Post-translationally stabilized levels of NIK promoted recruitment of NLRP3 from the endoplasmic reticulum and caspase-1 from the cytosol to appose these molecules on signaling endosomes. As a result, inflammasome activity characterized by cleaved caspase-1, occurred, resulting IL-1β-dependent activation of canonical NF-κB. In sum, MAC sequentially activated 3 inflammatory pathways in an inter-dependent fashion. MAC caused early-phase activation of non-canonical NF-κB (Figure 4). Non-canonical NF-κB caused assembly of NLRP3 inflammasomes on signaling endosomes within 30 min (101, 102). Endosome-associated caspase-1 activity generated IL-1β, causing late-phase activation of canonical NF-κB at the ≥4 hr timepoint (101). Inhibition of MAC via a monoclonal anti-C5 Ab (110), pharmacologic blockade of ZFYVE21 induction (100), or drug-induced attenuation of NLRP3 oligomerization (112) reduced EC-mediated direct allorecognition and EC injury in a humanized mouse model. These studies collectively advanced the hypothesis that non-cytolytic MAC forms signaling endosomes that sequester and enhance the stability of inflammatory signaling proteins.
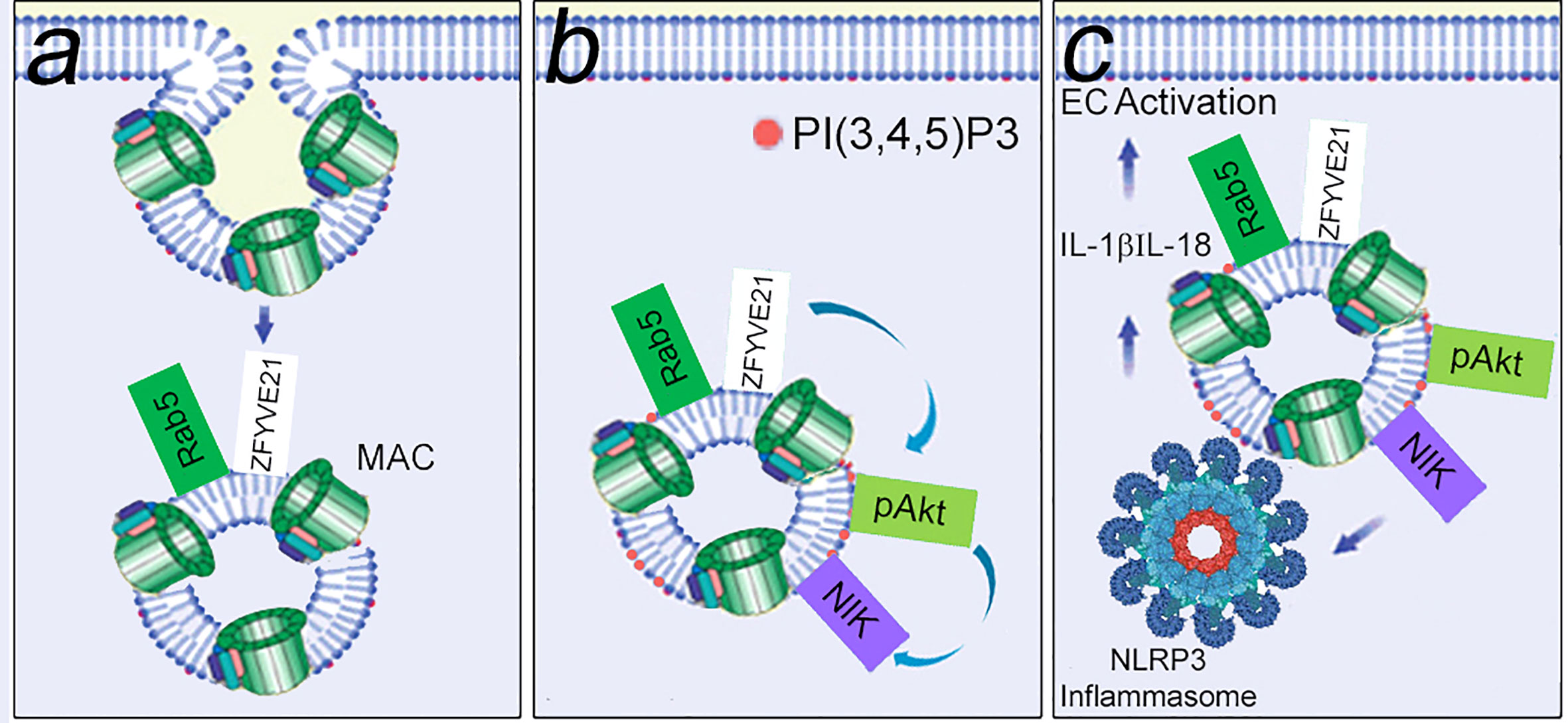
Figure 4 MAC-induced signaling endosomes cause EC activation. Non-cytolytic MAC assembled on EC surfaces rapidly undergoes clathrin-mediated endocytosis to form MAC+Rab5+ signaling endosomes that sequester signal-activating elements. In a Rab5-dependent manner, signaling endosomes post-translationally stabilize ZFYVE21, a Rab5 effector. (A) ZFYVE21 recruits SMURF2, an E3 ubiquitin ligase, to mediate degradative removal of PTEN from signaling endosomes, causing enrichment for PI(3,4,5)P3 and recruitment of phosphorylated Akt (pAkt, B). This process sequentially activates non-canonical NF-κB [marked by NIK (NF-kB Inducing Kinase)], NLRP3 inflammasomes, and IL-1β- mediated canonical NF-kB (C).
Recent studies have shown remarkable clinical utility of complement-based therapeutics. Drug development efforts have focused on attenuating salient steps in the complement pathway including protease activation of the C1 recognition complex, early pathway amplification at the level of C3, and terminal pathway activation at the level of C5. We highlight FDA-approved drug inhibitors acting at each of the steps in the complement pathway above. Sutimlimab is a humanized monoclonal antibody targeting C1s, the protease component of the classical complement pathway mediating cleavage of C2 and C4. Sutimlimab recently received FDA approval in Feb 2022 for the indication of cold agglutinin disease, a form of autoimmunehemolytic anemia involving MAC-induced hemolysis (117). Compstatins are cyclic peptides originally derived from a peptide isolated via phage display library (118) and iteratively modified to selectively bind C3 and to inhibit its cleavage by C3 convertases (119). A derivative of this original peptide, pegcetacoplan, received fast-track and orphan drug approval in July 2022 for the indication of paroxysmal nocturnal hemoglobinuria (PNH), a rare genetic disorder caused by aberrant glycosylphosphatidylinositol (GPI) conformation causing deficient expression of CD55 and CD59 and unconstrained MAC-induced hemolysis. Eculizumab is a humanized murine monoclonal antibody against C5, which prevents C5 cleavage and the formation of MAC by any of the three complement pathways. Like pegcetacoplan, eculizumab was granted orphan drug approval for PNH along with atypical hemolytic uremic syndrome, a condition also characterized by MAC-induced hemolytic anemia. Since its approval, eculizumab has gained an additional indication for blocking B cell activation in myasthenia gravis and off-label usage for blocking direct alloresponses in ABMR (120). Recently, a second and presumably more cost-effective long-acting monoclonal C5 Ab, ravulizumab, has also received FDA approval for the indications above. The risk:benefit profiles for the drugs above appear reasonable, and the complement pathway has been shown to be a viable drug target for multiple clinical conditions.
Highlighting its fundamental importance in solid organ transplantation, the complement pathway was identified as a key genetic target in first-in-human porcine xenotransplants. We briefly describe this exciting and rapidly evolving clinical application. Galactose-α-1,3-galactose (α-gal) is a terminal carbohydrate modification present in porcine but not human tissues, and human subjects frequently show high titers of circulating xeno-Abs against this epitope. Xeno-Abs against α-gal cause vascular rejection via hyperacute ABMR, a process mediated by MAC, and this catastrophic condition has limited the utility of xenografts in patients. Formation of α-gal is mediated by the porcine enzyme, α-1,3-galactosyltransferase. In December 2020, the FDA granted a first-of-its-kind approval for intentional genomic alterations (IGA) targeting porcine 1,3-galactosyltransferase. In porcine xenografts, CRISPR-based methods knocking out porcine 1,3-galactosyltransferase was combined with subcapsular autologous thymic tissue implants in a proof-of-principle study to prevent hyperacute ABMR. Two porcine-to-human xenotransplant procedures were performed in two brain-dead patients with end-stage renal failure, and following surgical implantation, over a period of ~2 days the implanted xenografts carrying the α-gal IGA demonstrated improvements in renal function including decreasing creatinine, increasing estimated glomerular filtration rate, and increasing urine production. These improved parameters occurred in the absence of hyperacute ABMR (69). This initial study demonstrated proper functioning of porcine tissue within a human environment in the absence of complement-mediated ABMR.
In a follow-up study involving an orthotopic heart xenograft (70), further IGAs were made to the porcine xenograft to promote xenograft acceptance. CRISPR-mediated gene edits were made to knock out 3 highly immunogenic xenoantigens (galactose-a-1,3-galactose, Sda blood group antigen, and N-glycolylneuraminic acid) and growth hormone receptor to prevent intrinsic xenograft growth. To block complement-mediated injury, knockins were performed for the complement regulatory proteins, CD46 and CD55, as well as EC-derived proteins facilitating direct allorecognition, CD47 and heme oxygenase-1. Complement proteases intersect and show activity against proteins related to coagulation (121), and coagulation factors including thrombomodulin and protein C were knocked in to prevent hypercoagulability linked to inefficient thromboregulation via porcine proteins. Excitingly, the porcine xenograft containing the gene edits above survived 60 days following organ implantation and showed various signs of proper functioning including normal sinus rhythm, cardiac output, longitudinal strain, and left ventricular ejection fraction. The patient’s post-operative course was complicated by oligoanuric renal failure requiring renal replacement therapy, bacterial and fungal peritonitis, and increased titers of the porcine CMV/suid herpesvirus 2, indicating a possible zoonotic infection. Endomyocardial biopsies, though showing signs of injury following the patient’s demise at day post-op day 60, did not show evidence of acute ABMR. These preliminary findings appeared to validate the efficacy of the immunomodulatory IGAs made to the porcine xenograft which included complement regulatory proteins. Genetic supplementation for endogenous complement inhibition through IGA is a newly emerging therapeutic, and the clinical application for this technology in patients appears very promising at the moment.
In solid organ transplantation, autologous MAC are non-cytolytic and modulate adaptive immunity by enhancing the strength and quality of direct allorecognition by ECs. Highlighting its clinical relevance, MAC formation on ECs is prognostic for, diagnostic for, and a therapeutic target for vascular rejection. Blockade of MAC via pharmaceutical and genetic approaches are emerging as new treatment modalities. Future work elucidating the immune roles of MAC may uncover gene targets enabling blockade of its formation on donor tissues and/or attenuation of its downstream inflammatory pathways.
MB, SW, CM, MF, BJ, QJ, GS, XL, and DJ drafted the text of this review. MB and DJ produced figures for the manuscript. All authors contributed to the article and approved the submitted version.
DJ was supported by grants from the NIH (R01HL141137-01, R01HL141137-04S2), the Veterans Hospital Administration (I01 BX005117), and the American Lung Association (ETRA 736563). JP was supported by grants from the NIH (R01HL-51014).
Work supporting development of humanized mouse models was supported in part through a research grant from Alexion Pharmaceuticals (JP). Work supporting identification of ZFYVE21 was supported in part through a research grant from Abbvie Pharmaceutical Research and Development (JP, DJ).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Elvington M, Liszewski MK, Atkinson JP. Evolution of the complement system: from defense of the single cell to guardian of the intravascular space. Immunol Rev (2016) 274(1):9–15. doi: 10.1111/imr.12474
2. Nesargikar PN, Spiller B, Chavez R. The complement system: history, pathways, cascade and inhibitors. Eur J Microbiol Immunol (2012) . 2(2):103–11. doi: 10.1556/EuJMI.2.2012.2.2
3. Zhang M, Austen WG, Chiu I, Alicot EM, Humg R, Ma M, et al. Identification of a specific self-reactive IgM antibody that initiates intestinal ischemia/reperfusion injury. Proc Natl Acad Sci USA (2004) 101(11):3886–91. doi: 10.1073/pnas.0400347101
4. Gadjeva MG, Rouseva MM, Zlatarova AS, Reid KB, Kishore U, Kojouharova MS. Interaction of human C1q with IgG and IgM: revisited. Biochemistry (2008) 47(49):13093–102. doi: 10.1080/08916930701262986
5. Kulik L, Fleming SD, Moratz C, Reuter JW, Novikov A, Chen K, et al. Pathogenic natural antibodies recognizing annexin IV are required to develop intestinal ischemia-reperfusion injury. J Immunol (2009) 182(9):5363–73. doi: 10.4049/jimmunol.0803980
6. Valenzuela NM, Reed EF. Antibody-mediated rejection across solid organ transplants: manifestations, mechanisms, and therapies. J Clin Invest (2017) 127(7):2492–504. doi: 10.1172/JCI90597
7. Clerkin KJ, Farr MA, Restaino SW, Zorn E, Latif F, Vasilescu ER, et al. Donor-specific anti-HLA antibodies with antibody-mediated rejection and long-term outcomes following heart transplantation. J Heart Lung Transplant (2017) 36(5):540–5. doi: 10.1016/j.healun.2016.10.016
8. Zou Y, Stastny P, Süsal C, Döhler B, Opelz G. Antibodies against MICA antigens and kidney-transplant rejection. N Engl J Med (2007) 357(13):1293–300. doi: 10.1056/NEJMoa067160
9. Lammerts RGM, Altulea D, Hepkema BG, Sanders JS, van den Born J, Berger SP. Antigen and cell-based assays for the detection of non-HLA antibodies. Front Immunol (2022) 3:864671. doi: 10.3389/fimmu.2022.864671
10. Zlatarova AS, Rouseva M, Roumenina LT, Gadjeva M, Kolev M, Dobrev I, et al. Existence of different but overlapping IgG- and IgM-binding sites on the globular domain of human C1q. Biochemistry (2006) 45:9979–88. doi: 10.1021/bi060539v
11. Marqués G, Antón LC, Barrio E, Sánchez A, Ruiz S, Gavilanes F, et al. Arginine residues of the globular regions of human C1q involved in the interaction with immunoglobulin G. J Biol Chem (1993) 268(14):10393–402. doi: 10.1016/S0021-9258(18)82214-1
12. Tissot B, Daniel R, Place C. Interaction of the C1 complex of complement with sulfated polysaccharide and DNA probed by single molecule fluorescence microscopy. Eur J Biochem (2003) 270:4714–20. doi: 10.1046/j.1432-1033.2003.03870
13. Terrasse R, Tacnet-Delorme P, Moriscot C, Pérard J, Schoehn G, Vernet T, et al. Human and pneumococcal cell surface glyceraldehyde-3-phosphate dehydrogenase (GAPDH) proteins are both ligands of human C1q protein. J Biol Chem (2012) 287:42620–33. doi: 10.1074/jbc.M112.423731
14. Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, et al. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med (2000) 192(3):359–66. doi: 10.1084/jem.192.3.359
15. Navratil JS, Watkins SC, Wisnieski JJ, Ahearn JM. The globular heads of C1q specifically recognize surface blebs of apoptotic vascular endothelial cells. J Immunol (2001) 166(5):3231–9. doi: 10.4049/jimmunol.166.5.3231
16. Erlich P, DUmestre-Perard C, Ling WL, Lemaire-Vieille C, Schoehn G, Arlaud GJ, et al. Complement protein C1q forms a complex with cytotoxic prion protein oligomers. J Biol Chem (2010) 285(25):19267–76. doi: 10.1074/jbc.M109.071860
17. Klein MA, Kaeser PS, Schwarz P, Weyd H, Xenarios I, Zinkernagel RM, et al. Complement facilitates early prion pathogenesis. Nat Med (2001) 7:488–92. doi: 10.1038/86567
18. Tacnet-Delorme P, Chevallier S, Arlaud GJ. Beta-amyloid fibrils activate the C1 complex of complement under physiological conditions: evidence for a binding site for a beta on the C1q globular regions. J Immunol (2001) 167(11):6374–81. doi: 10.4049/jimmunol.167.11.6374
19. Gregersen E, Betzer C, Kim WS, Kovacs G, Reimer L, Halliday GM, et al. Alpha-synuclein activates the classical complement pathway and mediates complement-dependent cell toxicity. J Neuroinflamm (2021) 18(1):177. doi: 10.1186/s12974-021-02225-9
20. Biró A, Thielens NM, Cervenák L, Prohászka Z, Füst G, Arlaud GJ. Modified low density lipoproteins differentially bind and activate the C1 complex of complement. Mol Immunol (2007) 44(6):1169–77. doi: 10.1016/j.molimm.2006.06.013
21. Arlaud GJ, Gaboriaud C, Thielens NM, Budayova-Spano M, Rossi V, Fontecilla-Camps JC. Structural biology of the C1 complex of complement unveils the mechanisms of its activation and proteolytic activity. Mol Immunol (2002) 39:383–94. doi: 10.1016/S0161-5890(02)00143-8
22. Budayova-Spano M, Grabarse W, Thielens NM, Hillen H, Lacroix M, Schmidt M, et al. Monomeric structures of the zymogen and active catalytic domain of complement protease c1r: Further insights into the c1 activation mechanism. Structure (2002) 1993(10):1509–19. doi: 10.1093/emboj/21.3.231
23. Gaboriaud C, Frachet P, Thielens NM, Arlaud GJ. The human c1q globular domain: structure and recognition of non-immune self ligands. Front Immunol (2011) 2:92. doi: 10.3389/fimmu.2011.00092
24. Parsons ES, Stanley GJ, Pyne ALB, Hodel AW, Nievergelt AP, Menny A, et al. Single-molecule kinetics of pore assembly by the membrane attack complex. Nat Commun (2019) 10(1):2066. doi: 10.1038/s41467-019-10058-7
25. Serna M, Giles JL, Morgan BP, Bubeck D. Structural basis of complement membrane attack complex formation. Nat Commun (2016) 7:10587. doi: 10.1038/ncomms10587
26. Dudkina NV, Spicer BA, Reboul CF, Conroy PJ, Lukoyanova N, Elmlund H, et al. Structure of the poly-C9 component of the complement membrane attack complex. Nat Commun (2016) 7:10588. doi: 10.1038/ncomms10588
27. Sharp TH, Koster AJ, Gros P. Heterogeneous MAC initiator and pore structures in a lipid bilayer by phase-plate cryo-electron tomography. Cell Rep (2016) 15(1):1–8. doi: 10.1016/j.celrep.2016.03.002
28. Hadders MA, Bubeck D, Roversi P, Hakobyan S, Forneris F, Morgan BP, et al. Assembly and regulation of the membrane attack complex based on structures of C5b6 and sC5b9. Cell Rep (2012) 1(3):200–7. doi: 10.1016/j.celrep.2012.02.003
29. Lovelace LL, Cooper CL, Sodetz JM, Lebioda L. Structure of human C8 protein provides mechanistic insight into membrane pore formation by complement. J Biol Chem (2011) 286(20):17585–92. doi: 10.1074/jbc.M111.219766
30. Ma YG, Cho MY, Zhao M, Park JW, Matsushita M, Fujita T, et al. Human mannose-binding lectin and l-ficolin function as specific pattern recognition proteins in the lectin activation pathway of complement. J Biol Chem (2004) 279(24):25307–12. doi: 10.1074/jbc.M400701200
31. Matsushita M, Fujita T. Activation of the classical complement pathway by mannose-binding protein in association with a novel C1s-like serine protease. J Exp Med (1992) 176(6):1497–502. doi: 10.1084/jem.176.6.1497
32. Teillet F, Dublet B, Andrieu J-P, Gaboriaud C, Arlaud GJ, Thielens NM. The two major oligomeric forms of human mannan-binding lectin: chemical characterization, carbohydrate-binding properties, and interaction with MBL-associated serine proteases. J Immunol (2005) 1950(174):2870–7. doi: 10.4049/jimmunol.174.5.2870
33. Thiel S, Vorup-Jensen T, Stover CM, Schwaeble W, Laursen SB, Poulsen K, et al. A second serine protease associated with mannan-binding lectin that activates complement. Nature (1997) 386:506–10. doi: 10.1038/386506a0
34. Matsushita M, Endo Y, Fujita T. Cutting edge: complement-activating complex of ficolin and mannose-binding lectin-associated serine protease. J Immunol (2000) 164(5):2281–4. doi: 10.4049/jimmunol.164.5.2281
35. de Vries B, Walter SJ, Peutz-Kootstra CJ, Wolfs TG, van Heurn LW, Buurman WA. The mannose-binding lectin-pathway is involved in complement activation in the course of renal ischemia-reperfusion injury. Am J Pathol (2004) 165(5):1677–88. doi: 10.1016/S0002-9440(10)63424-4
36. Castellano G, Melchiorre R, Loverre A, Ditonno P, Montinaro V, Rossini M, et al. Therapeutic targeting of classical and lectin pathways of complement protects from ischemia-reperfusion-induced renal damage. Am J Pathol (2010) 176(4):1648–59. doi: 10.2353/ajpath.2010.090276
37. Marrón-Liñares GM, Núñez L, Crespo-Leiro MG, Barge-Caballero E, Pombo J, Paniagua-Martin MJ, et al. Polymorphisms in genes related to the complement system and antibody-mediated cardiac allograft rejection. J Heart Lung Transplant (2018) 37(4):477–85. doi: 10.1016/j.healun.2017.07.006
38. Huang Y, Qiao F, Atkinson C, Holers VM, Tomlinson S. A novel targeted inhibitor of the alternative pathway of complement and its therapeutic application in ischemia/reperfusion injury. J Immunol (2008) 181(11):8068–76. doi: 10.4049/jimmunol.181.11.8068
39. He S, Atkinson C, Qiao F, Cianflone K, Chen X, Tomlinson S. A complement-dependent balance between hepatic ischemia/reperfusion injury and liver regeneration in mice. J Clin Invest (2009) 119(8):2304–16. doi: 10.1172/JCI38289
40. Park P, Haas M, Cunningham PN, Bao L, Alexander JJ, Quigg RJ. Injury in renal ischemia-reperfusion is independent from immunoglobulins and T lymphocytes. Am J Physiol Renal Physiol (2002) 282(2):F352–7. doi: 10.1152/ajprenal.00160.2001
41. Lin T, Zhou W, Farrar CA, Hargreaves RE, Sheerin NS, Sacks SH. Deficiency of C4 from donor or recipient mouse fails to prevent renal allograft rejection. Am J Pathol (2006) 168(4):1241–8. doi: 10.2353/ajpath.2006.050360
42. Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med (1981) 154:856–67. doi: 10.1084/jem.154.3.856
43. Isenman DE, Kells DI, Cooper NR, Müller-Eberhard HJ, Pangburn MK. Nucleophilic modification of human complement protein C3: correlation of conformational changes with acquisition of C3b-like functional properties. Biochemistry (1981) 20:4458–67. doi: 10.1021/bi00518a034
44. Li K, Gor J, Perkins SJ. Self-association and domain rearrangements between complement C3 and C3u provide insight into the activation mechanism of C3. Biochem J (2010) 431:63–72. doi: 10.1042/BJ20100759
45. Nishida N, Walz T, Springer TA. Structural transitions of complement component C3 and its activation products. Proc Natl Acad Sci USA (2006) 103:19737–42. doi: 10.1073/pnas.0609791104
46. Rodriguez E, Nan R, Li K, Gor J, Perkins SJ. A revised mechanism for the activation of complement C3 to C3b: a molecular explanation of a disease-associated polymorphism. J Biol Chem (2014) 290(4):2334–50. doi: 10.1074/jbc.M114.605691
47. Damman J, Daha MR, Leuvenink HG, van Goor H, Hillebrands JL, Dijk MC, et al. Association of complement C3 gene variants with renal transplant outcome of deceased cardiac dead donor kidneys. Am J Transplant (2012) 12(3):660–8. doi: 10.1111/j.1600-6143.2011.03880.x
48. Varagunam M, Yaqoob MM, Döhler B, Opelz G. C3 polymorphisms and allograft outcome in renal transplantation. N Engl J Med (2009) 360(9):874–80. doi: 10.1056/NEJMoa0801861
49. Andrews PA, Finn JE, Mathieson PW, Sacks SH. Molecular analysis of C3 allotypes related to transplant outcome in human renal allografts. Transplantation (1995) 60(11):1342–6. doi: 10.1097/00007890-199512000-00025
50. Brown KM, Kondeatis E, Vaughan RW, Kon SP, Farmer CK, Taylor JD, et al. Influence of donor C3 allotype on late renal-transplantation outcome. N Engl J Med (2006) 354(19):2014–23. doi: 10.1056/NEJMoa052825
51. West EE, Kolev M, Kemper C. Complement and the regulation of T cell responses. Annu Rev Immunol (2018) 36:309–38. doi: 10.1146/annurev-immunol-042617-053245
52. Nauser CL, Farrar CA, Sacks SH. Complement recognition pathways in renal transplantation. J Am Soc Nephrol (2017) 28(9):2571–8. doi: 10.1681/ASN.2017010079
53. Sobh A, Bonilla FA. Vaccination in primary immunodeficiency disorders. J Allergy Clin Immunol Pract (2016) 4(6):1066–75. doi: 10.1016/j.jaip.2016.09.012
54. Nagata M, Hara T, Aoki T, Mizuno Y, Akeda H, Inaba S, et al. Inherited deficiency of ninth component of complement: an increased risk of meningococcal meningitis. J Pediatr (1989) 114(2):260–4. doi: 10.1016/s0022-3476(89)80793-0
55. Cross AR, Glotz D, Mooney N. The role of the endothelium during antibody-mediated rejection: From victim to accomplice. Front Immunol (2018) 9:106. doi: 10.3389/fimmu.2018.00106
56. Piotti G, Palmisano A, Maggiore U, Buzio C. Vascular endothelium as a target of immune response in renal transplant rejection. Front Immunol (2014) 5:505. doi: 10.3389/fimmu.2014.00505
57. Liu F, Han K, Blair R, Kenst K, Qin Z, Upcin B, et al. SARS-CoV-2 infects endothelial cells In vivo and In vitro. Front Cell Infect Microbiol (2021) 11:701278. doi: 10.3389/fcimb.2021.701278
58. Elsoukkary SS, Mostyka M, Dillard A, Berman DR, Ma LX, Chadburn A, et al. Autopsy findings in 32 patients with COVID-19: A single-institution experience. Pathobiology (2021) 88(1):56–68. doi: 10.1159/000511325
59. Borczuk AC, Salvatore SP, Seshan SV, Patel SS, Bussel JB, Mostyka M, et al. COVID-19 pulmonary pathology: a multi-institutional autopsy cohort from Italy and new York city. Mod Pathol (2020) 33(11):2156–68. doi: 10.1038/s41379-020-00661-1
60. Guillevin L, Dörner T. Vasculitis: mechanisms involved and clinical manifestations. Arthritis Res Ther (2007) 9(Suppl 2):S9. doi: 10.1186/ar2193
61. Carney DF, Hammer CH, Shin ML. Elimination of terminal complement complexes in the plasma membrane of nucleated cells: influence of extracellular Ca2+ and association with cellular Ca2+. J Immunol (1986) 137:263–70.
62. Morgan BP, Dankert JR, Esser AF. Recovery of human neutrophils from complement attack: removal of the membrane attack complex by endocytosis and exocytosis. J Immunol (1987) 138:246–53.
63. Kerjaschki D, Schulze M, Binder S, Kain R, Ojha PP, Susani M, et al. Transcellular transport and membrane insertion of the C5b-9 membrane attack complex of complement by glomerular epithelial cells in experimental membranous nephropathy. J Immunol (1989) 143:546–52. doi: 10.1111/j.1399-3089.2005.00237.x
64. Scolding NJ, Houston WA, Morgan BP, Campbell AK, Compston DA. Reversible injury of cultured rat oligodendrocytes by complement. Immunology (1989) 67:441–6. doi: 10.1038/s42003-019-0529-9
65. Morgan BP, Berg CW, Harris CL. ''Homologous restriction'' in complement lysis: roles of membrane complement regulators. Xenotransplantation (2005) 12(4):258–65. doi: 10.1111/j.1399-3089.2005.00237.x
66. Ojha H, Ghosh P, Singh Panwar H, Shende R, Gondane A, Mande SC, et al. Spatially conserved motifs in complement control protein domains determine functionality in regulators of complement activation-family proteins. Commun Biol (2019) 2:290. doi: 10.1038/s42003-019-0529-9
67. Escudero-Esparza A, Kalchishkova N, Kurbasic E, Jiang WG, Blom AM. The novel complement inhibitor human CUB and sushi multiple domains 1 (CSMD1) protein promotes factor I-mediated degradation of C4b and C3b and inhibits the membrane attack complex assembly. FASEB J (2013) 27:5083–93. doi: 10.1096/fj.13-230706
68. Lee AS, Rusch J, Lima AC, Usmani A, Huang N, Lepamets M, et al. Rare mutations in the complement regulatory gene CSMD1 are associated with male and female infertility. Nat Commun (2019) 10(1):4626. doi: 10.1038/s41467-019-12522-w
69. Montgomery RA, Stern JM, Lonze BE, Tatapudi VS, Mangiola M, Wu M, et al. Results of two cases of pig-to-Human kidney xenotransplantation. N Engl J Med (2022) 386(20):1889–98. doi: 10.1056/NEJMoa2120238
70. Griffith BP, Goerlich CE, Singh AK, Rothblatt M, Lau CL, Shah A, et al. Genetically modified porcine-to-Human cardiac xenotransplantation. N Engl J Med (2022) 387(1):35–44. doi: 10.1056/NEJMoa2201422
71. Choi NH, Mazda T, Tomita M. A serum protein SP40,40 modulates the formation of membrane attack complex of complement on erythrocytes. Mol Immunol (1989) 26(9):835–40. doi: 10.1016/0161-5890(89)90139-9
72. McDonald JF, Nelsestuen GL. Potent inhibition of terminal complement assembly by clusterin: characterization of its impact on C9 polymerization. Biochemistry (1997) 36:7464–73. doi: 10.1021/bi962895r
73. Wilson MR, Zoubeidi A. Clusterin as a therapeutic target. Expert Opin Ther Targets (2017) 21(2):201–13. doi: 10.1080/14728222.2017.1267142
74. Dahlbäck B, Podack ER. Characterization of human s protein, an inhibitor of the membrane attack complex of complement. Demonstration Free reactive thiol Group Biochem (1985) 24(9):2368–74. doi: 10.1021/bi00330a036
75. Tschopp J, Podack ER, Muller-Eberhard HJ. The membrane attack complex of complement: C5b-8 complex as accelerator of C9 polymerization. J Immunol (1985) 134:495–9.
76. Podack ER, Muller-Eberhard HJ. Binding of desoxycholate, phosphatidylcholine vesicles, lipoprotein and of the s-protein to complexes of terminal complement components. J Immunol (1978) 121:1025–30.
77. Barnum SR, Bubeck D, Schein TN. Soluble membrane attack complex: Biochemistry and immunobiology. Front Immunol (2020) 11:585108. doi: 10.3389/fimmu.2020.585108
78. Rodríguez-Rivera C, Garcia MM, Molina-Álvarez M, González-Martín C, Goicoechea C. Clusterin: Always protecting. synthesis, function and potential issues. BioMed Pharmacother (2021) 134:111174. doi: 10.1016/j.biopha.2020.111174
79. Rea VE, Lavecchia A, Di Giovanni C, Rossi FW, Gorrasi A, Pesapane A, et al. Discovery of new small molecules targeting the vitronectin-binding site of the urokinase receptor that block cancer cell invasion. Mol Cancer Ther (2013) 12(8):1402–16. doi: 10.1158/1535-7163.MCT-12-1249
80. Rosenberg ME, Girton R, Finkel D, Chmielewski D, Barrie A 3rd, Witte DP, et al. Apolipoprotein j/clusterin prevents a progressive glomerulopathy of aging. Mol Cell Biol (2002) 22(6):1893–902. doi: 10.1128/MCB.22.6.1893-1902.2002
81. Pilzer D, Fishelson Z. Mortalin/GRP75 promotes release of membrane vesicles from immune attacked cells and protection from complement-mediated lysis. Int Immunol (2005) 17:1239–48. doi: 10.1074/jbc.M703742200
82. Moskovich O, Fishelson Z. Live cell imaging of outward and inward vesiculation induced by the complement c5b-9 complex. J Biol Chem (2007) 282:29977–86. doi: 10.3389/fimmu.2018.00721
83. Karasu E, Eisenhardt SU, Harant J, Huber-Lang M. Extracellular vesicles: Packages sent with complement. Front Immunol (2018) 9:721. doi: 10.3389/fimmu.2018.00721
84. Clayton A, Harris CL, Court J, Mason MD, Morgan BP. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur J Immunol (2003) 33:522–31. doi: 10.1016/S0021-9258(19)39666-8
85. Hamilton KK, Hattori R, Esmon CT, Sims PJ. Complement proteins C5b-9 induce vesiculation of the endothelial plasma membrane and expose catalytic surface for assembly of the prothrombinase enzyme complex. J Biol Chem (1990) 265(7):3809–14. doi: 10.1016/S0002-9440(10)64887-0
86. Taraboletti G, D’Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated components by endothelial cells. Am J Pathol (2002) 160:673–80. doi: 10.1016/S0002-9440(10)64887-0
87. Stein JM, Luzio JP. Ectocytosis caused by sublytic autologous complement attack on human neutrophils. the sorting of endogenous plasma-membrane proteins and lipids into shed vesicles. Biochem J (1991) 274(Pt 2):381–6. doi: 10.1042/bj2740381
88. Tegla CA, Cudrici C, Patel S, Trippe R 3rd, Rus V, Niculescu F, et al. Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol Res (2011) 51(1):45–60. doi: 10.1007/s12026-011-8239-5
89. Carney DF, Lang TJ, Shin ML. Multiple signal messengers generated by terminal complement complexes and their role in terminal complement complex elimination. J Immunol (1990) 145:623–9. doi: 10.1016/j.molimm.2009.09.016
90. Gancz D, Donin N, Fishelson Z. Involvement of the c-jun n-terminal kinases JNK1 and JNK2 in complement-mediated cell death. Mol Immunol (2009) 47:310–7. doi: 10.1038/s41419-017-0240-z
91. Rozenberg P, Ziporen L, Gancz D, Saar-Ray M, Fishelson Z. Cooperation between Hsp90 and mortalin/GRP75 in resistance to cell death induced by complement C5b-9. Cell Death Dis (2018) 9(2):150. doi: 10.1038/s41419-017-0240-z
92. Viedt C, Hansch GM, Brandes RP, Kubler W, Kreuzer J. The terminal complement complex C5b-9 stimulates interleukin-6 production in human smooth muscle cells through activation of transcription factors NF-kappa b and AP-1. FASEB J (2000) 14:2370–2. doi: 10.1096/fj.00-0468fje
93. Kilgore KS, Schmid E, Shanley TP, Flory CM, Maheswari V, Tramontini NL, et al. Sublytic concentrations of the membrane attack complex of complement induce endothelial interleukin-8 and monocyte chemoattractant protein-1 through nuclear factor-kappa b activation. Am J Pathol (1997) 150:2019–31. doi: 10.4049/jimmunol.1300489
94. Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, Kandasamy M, et al. Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β release. J Immunol (2013) 191(3):1006–10. doi: 10.4049/jimmunol.1300489
95. Triantafilou K, Hughes TR, Triantafilou M, Morgan BP. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J Cell Sci (2013) 126(Pt 13):2903–13. doi: 10.1242/jcs.124388
96. Tedesco F, Pausa M, Nardon E, Introna M, Mantovani A, Dobrina A. The cytolytically inactive terminal complement complex activates endothelial cells to express adhesion molecules and tissue factor procoagulant activity. J Exp Med (1997) 185(9):1619–27. doi: 10.1084/jem.185.9.1619
97. Dobrina A, Pausa M, Fischetti F, Bulla R, Vecile E, Ferrero E, et al. Cytolytically inactive terminal complement complex causes transendothelial migration of polymorphonuclear leukocytes. Vitro vivo Blood (2002) 99(1):185–92. doi: 10.1182/blood.v99.1.185
98. Jane-Wit D, Manes TD, Yi T, Qin L, Clark P, Kirkiles-Smith NC, et al. Alloantibody and complement promote T cell-mediated cardiac allograft vasculopathy through noncanonical nuclear factor-κB signaling in endothelial cells. Circulation (2013) 128(23):2504–16. doi: 10.1161/CIRCULATIONAHA.113.002972
99. Qin L, Li G, Kirkiles-Smith N, Clark P, Fang C, Wang Y, et al. Complement C5 inhibition reduces T cell-mediated allograft vasculopathy caused by both alloantibody and ischemia reperfusion injury in humanized mice. Am J Transplant (2016) 16(10):2865–76. doi: 10.1111/ajt.13834
100. Jane-wit D, Surovtseva YV, Qin L, Li G, Liu R, Clark P, et al. Complement membrane attack complexes activate noncanonical NF-κB by forming an akt+ NIK+ signalosome on Rab5+ endosomes. Proc Natl Acad Sci U.S.A. (2015) 112(31):9686–91. doi: 10.1073/pnas.1503535112
101. Fang C, Manes TD, Liu L, Liu K, Qin L, Li G, et al. ZFYVE21 is a complement-induced Rab5 effector that activates non-canonical NF-κB via phosphoinosotide remodeling of endosomes. Nat Commun (2019) 10(1):2247. doi: 10.1038/s41467-019-10041-2
102. Xie CB, Qin L, Li G, Fang C, Kirkiles-Smith NC, Tellides G, et al. Complement membrane attack complexes assemble NLRP3 inflammasomes triggering IL-1 activation of IFN-γ-Primed human endothelium. Circ Res (2019) 124(12):1747–59. doi: 10.1161/CIRCRESAHA.119.314845
103. Siu JHY, Surendrakumar V, Richards JA, Pettigrew GJ. T Cell allorecognition pathways in solid organ transplantation. Front Immunol (2018) 9:2548. doi: 10.3389/fimmu.2018.02548
104. Lin CM, Gill RG. Direct and indirect allograft recognition: pathways dictating graft rejection mechanisms. Curr Opin Organ Transplant (2016) 21(1):40–4. doi: 10.1097/MOT.0000000000000263
105. Pober JS, Jane-wit D, Qin L, Tellides G. Interacting mechanisms in the pathogenesis of cardiac allograft vasculopathy. Arterioscler Thromb Vasc Biol (2014) 8):1609–14. doi: 10.1161/ATVBAHA.114.302818
106. Hart DN, Fuggle SV, Williams KA, Fabre JW, Ting A, Morris PJ. Localization of HLA-ABC and DR antigens in human kidney. Transplantation (1981) 31:428–33. doi: 10.1097/00007890-198106000-00005
107. Hancock WW, Kraft N, Atkins RC. The immunohistochemical demonstration of major histocompatibility antigens in the human kidney using monoclonal antibodies. Pathology (1982) 14:409–14. doi: 10.3109/00313028209092120
108. Al-Lamki RS, Bradley JR, Pober JS. Endothelial cells in allograft rejection. Transplantation (2008) 86(10):1340–8. doi: 10.1097/TP.0b013e3181891d8b
109. Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, et al. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature (2000) 403(6766):207–11. doi: 10.1038/35003221
110. Liu L, Fang C, Fu W, Jiang B, Li G, Qin L, et al. Endothelial cell-derived interleukin-18 released during ischemia reperfusion injury selectively expands T peripheral helper cells to promote alloantibody production. Circulation (2020) 141(6):464–78. doi: 10.1161/CIRCULATIONAHA.119.042501
111. Yi T, Fogal B, Hao Z, Tobiasova Z, Wang C, Rao DA, et al. Reperfusion injury intensifies the adaptive human T cell alloresponse in a human-mouse chimeric artery model. Arterioscler Thromb Vasc Biol (2012) 32:353–60. doi: 10.1161/ATVBAHA.111.239285
112. Xie CB, Jiang B, Qin L, Tellides G, Kirkiles-Smith NC, Jane-Wit D, et al. Complement-activated interferon-γ-primed human endothelium transpresents interleukin-15 to CD8+ T cells. J Clin Invest (2020) 130(7):3437–52. doi: 10.1172/JCI135060
113. Schechner JS, Nath AK, Zheng L, Kluger MS, Hughes CC, Sierra-Honigmann MR, et al. In vivo formation of complex microvessels lined by human endothelial cells in an immunodeficient mouse. Proc Natl Acad Sci U.S.A. (2000) 97(16):9191–6. doi: 10.1073/pnas.150242297
114. Merola J, Reschke M, Pierce RW, Qin L, Spindler S, Baltazar T, et al. Progenitor-derived human endothelial cells evade alloimmunity by CRISPR/Cas9-mediated complete ablation of MHC expression. JCI Insight (2019) 4(20):e129739. doi: 10.1172/jci.insight.129739
115. Xie CB, Zhou J, Mackay S, Pober JS. Complement-activated human endothelial cells stimulate increased polyfunctionality in alloreactive T cells. Am J Transplant (2021) 21(5):1902–9. doi: 10.1111/ajt.16485
116. Nagano M, Hoshino D, Sakamoto T, Kawasaki N, Koshikawa N, Seiki M. ZF21 protein regulates cell adhesion and motility. J Biol Chem (2010) 285(27):21013–22. doi: 10.1074/jbc.M110.106443
117. Röth A, Barcellini W, D'Sa S, Miyakawa Y, Broome CM, Michel M, et al. Sutimlimab in cold agglutinin disease. N Engl J Med (2021) 384(14):1323–34. doi: 10.1056/NEJMoa2027760
118. Sahu A, Kay BK. J.D. lambris. inhibition of human complement by a C3-binding peptide isolated from a phage-displayed random peptide library. J Immunol (1996) 157:884–91. doi: 10.1016/j.clim.2021.108785
119. Mastellos DC, Ricklin D, Sfyroera G, Sahu A. From discovery to approval: A brief history of the compstatin family of complement C3 inhibitors. Clin Immunol (2022) 235:108785. doi: 10.1016/j.clim.2021.108785
120. Tan EK, Bentall A, Dean PG, Shaheen MF, Stegall MD, Schinstock CA. Use of eculizumab for active antibody-mediated rejection that occurs early post-kidney transplantation: A consecutive series of 15 cases. Transplantation (2019) 103(11):2397–404. doi: 10.1097/TP.0000000000002639
Keywords: complement, endothelial cell, allorecognition, antibody-mediated rejection, transplant
Citation: Song G, Wang S, Barkestani MN, Mullan C, Fan M, Jiang B, Jiang Q, Li X and Jane-wit D (2022) Membrane attack complexes, endothelial cell activation, and direct allorecognition. Front. Immunol. 13:1020889. doi: 10.3389/fimmu.2022.1020889
Received: 16 August 2022; Accepted: 07 September 2022;
Published: 23 September 2022.
Edited by:
Guido Moll, Charité Universitätsmedizin Berlin, GermanyReviewed by:
Jonathan Choy, Simon Fraser University, CanadaCopyright © 2022 Song, Wang, Barkestani, Mullan, Fan, Jiang, Jiang, Li and Jane-wit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Jane-wit, ZGFuLmphbmUtd2l0QHlhbGUuZWR1
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.