 Zhicheng Jin
Zhicheng Jin Xuefeng Sun
Xuefeng Sun Yaoyao Wang
Yaoyao Wang Chao Zhou
Chao Zhou Haihua Yang
Haihua Yang Suna Zhou
Suna Zhou
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 10 October 2022
Sec. Immunological Tolerance and Regulation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1018903
This article is part of the Research Topic Autophagy and Immunoregulation View all 5 articles
Immunotherapies, such as immune checkpoint inhibitors (ICIs) and chimeric antigen receptor (CAR) T cells, have revolutionized the treatment of patients with advanced and metastatic tumors resistant to traditional therapies. However, the immunosuppressed tumor microenvironment (TME) results in a weak response to immunotherapy. Therefore, to realize the full potential of immunotherapy and obstacle barriers, it is essential to explore how to convert cold TME to hot TME. Autophagy is a crucial cellular process that preserves cellular stability in the cellular components of the TME, contributing to the characterization of the immunosuppressive TME. Targeted autophagy ignites immunosuppressive TME by influencing antigen release, antigen presentation, antigen recognition, and immune cell trafficking, thereby enhancing the effectiveness of cancer immunotherapy and overcoming resistance to immunotherapy. In this review, we summarize the characteristics and components of TME, explore the mechanisms and functions of autophagy in the characterization and regulation of TME, and discuss autophagy-based therapies as adjuvant enhancers of immunotherapy to improve the effectiveness of immunotherapy.
Current prognoses for individuals with advanced cancer are generally poor, making cancer the second most common cause of global deaths (1). Immunotherapy has recently improved survival advantages over traditional treatments for various tumor types, especially advanced non-small cell lung cancer (NSCLC) and melanoma. However, how to raise response rates is still an urgent issue (2). Although immunotherapy combined with chemotherapy, targeted medication, or radiotherapy has been confirmed to finitely improve the anti-tumor effect, it is more necessary to explore other safe and efficient immunosensitizers or combination regimens (3, 4).
Tumor immunogenicity deficiency and immunosuppressive tumor microenvironment (TME) formation are the major causes of immunotherapy ineffectiveness or resistance (5). TME is a complex ecosystem made up of extracellular soluble compounds, stromal cells, immune cells, aberrant vascular networks, tumor cells, and dynamic oxygen content (6). Died tumor cells release antigens, triggering traditional antigen-presentation dendritic cells (DC) to catch and present major histocompatibility complex (MHC) class I-antigens to T cells, followed by immune activation mediated by the activated CD8+T cells (7). Effector CD8+ T cells, also named cytotoxic T lymphocytes (CTL), play the most significant adaptive tumor-killing effects in TME by releasing cytotoxic perforins, granzymes, and cytokines such as interferon (IFN)-γ and tumor necrosis factor-α (8). Natural killer (NK) cells act as major MHC I-independent tumor-killing immune cells and are the important complement of T cell-mediated antitumor immunity (9). However, the anti-tumor immunity will be weakened by immunosuppressive TME formed by recruiting immunosuppressive cells, such as Myeloid-derived suppressor cells (MDSC) and regulatory T cells (Treg) (10, 11). Tumor-associated macrophages (TAMs), accounting for 50% of infiltrating tumor stromal cells, are major immune cells in TME with phenotypically heterogeneous and functionally diverse. M1 macrophages can secrete proinflammatory cytokines, increase tumor antigen presentation as well as directly kill tumor cells through phagocytosis, resulting in immune activation (12). Moreover, M2 macrophages exert pro-tumor function by producing cytokines, such as interleukin (IL)-10 and transforming growth factor (TGF)-β, and play an immunosuppressive effect by the expression of programmed cell death ligand (PD-L1) and PD-L2 directly inhibiting cytotoxic T cell functions (12). As shown in Figure 1, TME could be shifted between “Hot” and “Cold” TME by recruiting immunosuppressive or immunostimulatory cells, which affect the response of tumors to immunotherapy (13). “Hot” TME is characterized by a prominent infiltration of CTLs, high expression of PD-L1 on tumor cells, upregulation of antigen-presenting cells (APC) markers, and activation of type 1IFN responses that all help to enhance the response to immunotherapy (13, 14). “Cold” TME is also known as an “immune desert” or “immune rejection” TME, and is characterized by rare CTLs infiltration, extensive fibrosis, abnormal vessel structure, and redundant immunosuppressive cells infiltrated, as well as low MHC I expression (14). The main mechanisms of TME regulation include antigen release, presentation, recognition of antigens, and recruitment and outflow of immune cells. Reprogramming the immunosuppressive TME into the immunostimulatory phenotype may enhance the response sensitivity of tumors to immunotherapy (6, 14, 15).
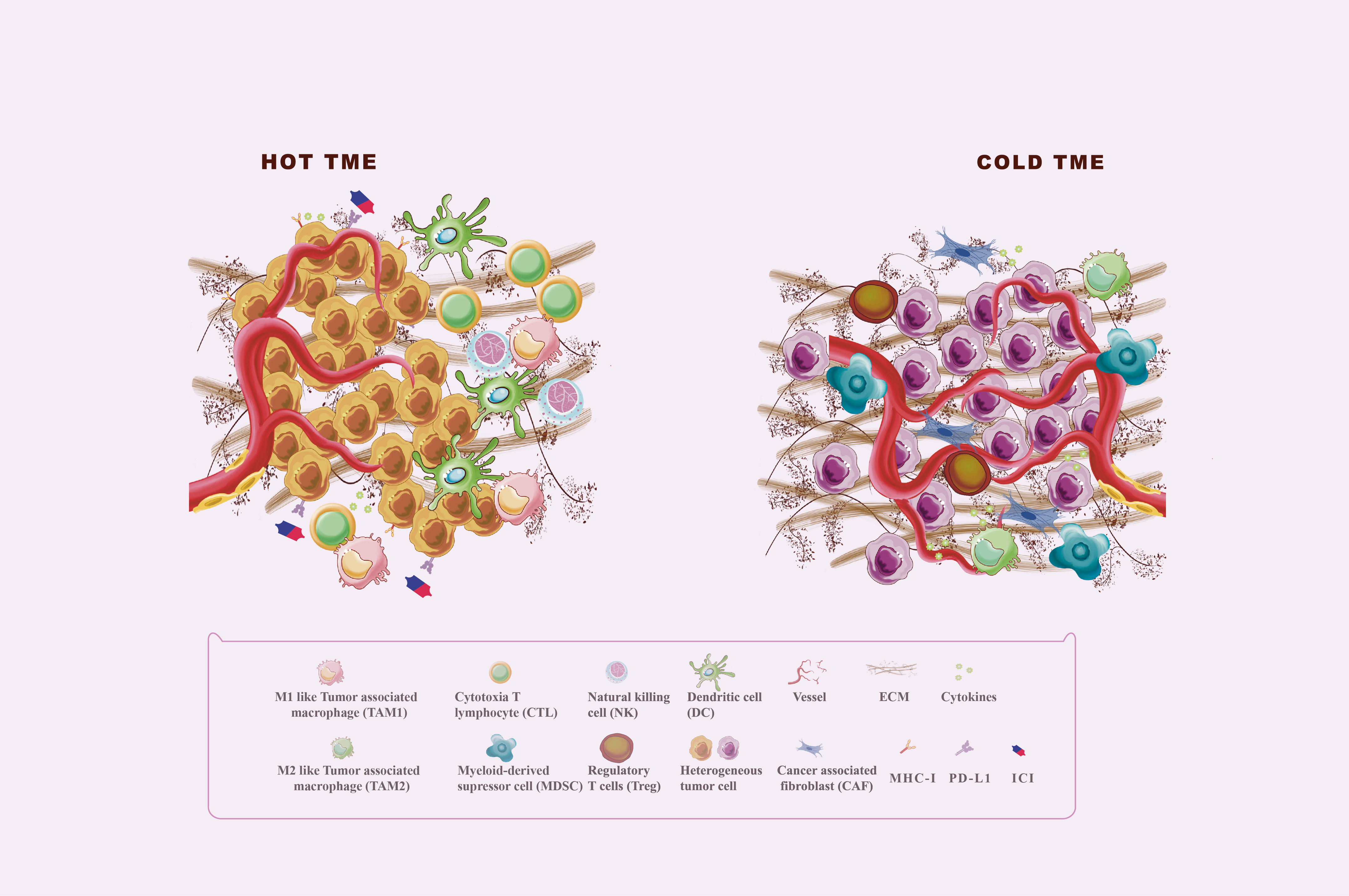
Figure 1 Hot and cold TME. The tumor microenvironment (TME) is divided into the immune desert (cold) and immunoinflammatory (hot) phenotypes. In the immune desert phenotype, the absence of T cells in the tumor may be due to the lack of tumor antigens and antigen-presenting cells (APC), the secretion of immunosuppressive molecules such as TGF-β and IL-10, and the infiltration of immunosuppressive cells including MDSC, M2 macrophages, and Treg. In addition, abnormal angiogenesis and excessive ECM also play an important role in the formation of cold TME. The immunoinflammatory phenotype is thought to be a prominent infiltration of cytotoxic T lymphocytes (CTLs) in the core of the TME, with activated antigen-presenting cell (APC) markers and type 1 interferon (IFN) responses.
Autophagy is a process by which either unfolded/misfolded/damaged protein aggregates or organelles are eradicated for the maintenance of cellular homeostasis (16). US Food and Drug Administration (FDA)-approved autophagy inhibitors hydroxychloroquine (HCQ) and chloroquine (CQ) have recently shown clinical benefits in the treatment of cancer patients when combined with chemotherapy, radiotherapy, or monotherapy (17). Altered autophagy in cancer, immune or stromal cells can regulate tumor-immune interactions to remodel the TME (18). Some studies have manifested that combination treatments of autophagy-targeted medicines and immunotherapy boost anti-tumor immune efficacy and enhance clinical benefits (18). Although autophagy-targeted medicines combined with immunotherapy are potential treatments, the precise mechanisms of which are still in the exploratory phase.
This study provides a comprehensive and in-depth review of the role of autophagy in TME and tumor immunotherapy. It gives insights into the role of autophagy in TME regulation, suggesting that immunotherapy will be improved through the exploration of either autophagy-based immunosensitizers or combination therapies.
Immunotherapies exhibit CTLs-dependent tumor-killing effects and are popularly applied in clinical treatment. TME without infiltration of T cells or with T cells exhaustion is the biggest obstacle to response to immunotherapy for cancer patients. Hypoxia, abnormal vasculature, and alteration in the three-dimensional stromal environment are the three most important characteristics of TME. These characteristics mainly impair the priming and infiltration of T cells, resulting in the exhaustion of T cells forming an immunosuppressive TME.
Currently, cancer immunotherapies fully utilize the immune system to eradicate tumor cells. Two notable and successful immunotherapies are immune checkpoint inhibitors (ICIs) and chimeric antigen receptor (CAR) T cells. Due to factors such as the extracellular matrix (ECM), current CAR-T cells are only effective against hematological tumors, toxic at high doses, and cannot simultaneously target multiple antigens (19). The failure of T cells to penetrate the TME and their concomitant exhaustion significantly impact ICI therapies (20). Either the innate or acquired immunosuppressive TME seriously impairs the efficacies of immunotherapies (21).
The TME comprises tumor cells, immune cells, stromal cells, vascular endothelial cells (ECs), and their non-cellular components such as the extracellular soluble molecules and ECM, along with vascular networks (6). These complex networks of cells and non-cellular components regulate the functions of immune cells within the tumor, consequently impacting the efficacy of immunotherapies (6). Furthermore, the TME is a dynamic network structure that changes with either cancer progression or the administration of various treatments (6). Hypoxia-caused three-dimensional stromal environment alteration and aberrant vascular construction affect the communication among tumor, immune and stromal cells, dynamically altering the TME into a cold state.
As the tumor grows, the pre-existing vasculature cannot satisfy its perfusion, leading to declining oxygen levels and the formation of a hypoxic environment (22). Hypoxia-inducible factor-1 (HIF-1), produced by either tumor or TME-associated cells, is a significant regulator of hypoxia. It then stimulates the expression of numerous genes involved in establishing immunosuppressive TME (22). Activated HIF-1 signaling attenuates MHC-I antigen presentation and hampers the infiltration and cytotoxic functions of the effector T cells and NK cells (22, 23). And, enhanced HIF-1 promotes the development and recruitment of immunosuppressive cells including M2-type macrophages, MDSC, and Treg (22, 23). Furthermore, HIF-1-mediated elevated expression of PD-L1 and CD47 contributes to the exhaustion of CTL and inactivation of phagocytosis (22, 23). Negative regulatory genes of the anti-tumor immune were found to be largely elevated in hypoxia TME, associated with the malignant phenotype of the tumors and poor patient prognosis (24). Interestingly, HIF-1 signaling stimulates angiogenesis by boosting pro-angiogenic factors, influencing another significant characteristic of TME (25).
The harsh TME often results in disrupted blood flow and oxygen-/nutrient-poor blood perfusion. Hypoxia demarcates the start of angiogenesis. HIF-1 is an upstream regulatory molecule of vascular endothelial growth factor (VEGF), by which and its receptor on Ecs, VEGFR2, regulates angiogenesis (25). Therefore, cancer cells initiate VEGF-mediated angiogenesis to satisfy their increased demand for oxygen and nutrients, resulting in the proliferation of tumor Ecs (26). However, the nascent blood vessels are often structurally and morphologically abnormal and thus fail to adequately supply oxygen and nutrients. This abnormal vasculature also curtails T cell infiltration, whereas the VEGF-mediated pathways prevent the maturation of DC and enhance the recruitment of immunosuppressive cell populations —Treg, M2-TAMs, and MDSC— into the tumor site, resulting in an immunosuppressive TME (26, 27). And, blocking the VEGF-VEGFR axis may promote the accumulation of effector T cells within TME to fire up anti-tumor immunity (28).
Rapidly growing tumors destabilize the structures and functions of surrounding tissues, contributing to structural alterations of the ECM (29). Stromal cells are connective tissue cells of organs and cancer-associated fibroblasts (CAF) are the most common stromal cells in the TME (30, 31). Cancer cells release TGF-β that activates CAF, which then secretes various immunomodulatory chemicals, including IL-11, IL-6, and TGF-β, to not only inhibit anti-tumor immunity but also deposit increasing amounts of ECM (30, 31). Thus, the rigid and dense ECM of the tumor stroma, activated CAF, and pro-fibrotic soluble substances are physical and functional barriers to the infiltration of immune effector cells, ultimately impeding immunotherapy (32).
TME is a dynamic and changing process during cancer development and anti-cancer treatment. Autophagy plays important role in TME remodeling. Hypoxia-induced autophagy in tumor cells or immune cells results in various outcomes, contributing to the formation of the immunosuppressive TME that fuels tumor growth. Autophagy modulation in ECs and stromal cells can affect tumor vascular and three-dimensional stromal environments to regulate immune cell recruitment and function through complicated mechanisms.
Autophagy is a core intracellular degradation system that transports cytoplasmic components to lysosomes for degradation and renewal —this is a double-edged sword for tumor progression (16). Autophagy suppresses carcinogenesis in the early stages of tumors by removing damaged organelles and DNA. Interestingly, Autophagy is a cytoprotective mechanism for tumors at advanced stages, which increases cancer cell survival and resistance to stresses. This then sustains tumor metabolism, growth, and survival to mediate tumor promotion and development, ultimately promoting tumorigenesis (16). Autophagy is induced by cellular or environmental stress and participates in several intricate biological processes. Initiation, nucleation, elongation, fusion with the lysosome, and destruction are necessary steps of autophagy involving more than thirteen autophagy-related genes (ATG) and proteins (33). Briefly, inhibition of the mammalian target of rapamycin (mTOR) and activation of AMP-activated protein kinase (AMPK) can stimulate the unc-51-like autophagy-activated kinase 1(ULK1) complex, and the class III phosphatidylinositol 3-kinase (PtdIns3K) complex in order, followed by the formation of the phagophore (33). ULK 1 complex includes ULK1, ATG13, FIP200, and ATG101. And PtdIns3K complex contains Beclin1, VPS 34/PIK3C3, VPS 15, ATG 14, and AMBRA-1. In addition, two ubiquitin-like coupling cascades, including autophagy-related 5 (ATG5)-ATG12 and microtubule-associated light chain 3 (MAP-LC3/ATG8/LC3) coupling systems, are required for phagophore elongation. Then, the phagophore grows and fuses on its own to form a double-membrane autophagosome with the LC3-II. And portions of the cytoplasm are gradually engulfed in the autophagosome. Finally, the unions of autophagosomes with lysosomes degrade the cargo and release decomposition products into the cytoplasm for reuse. In addition, substrates can be selectively degraded by various selective autophagy receptors, such as p62/SQSTM1(sequestosome-1) and NBR1. LC3-II acts as a docking site for cargo adaptors that enable cargo loading into autophagosomes (16, 33).
Moreover, cancer cells can communicate with neighboring cells via secretory autophagy. This autophagy-dependent secretion system affects immune cell function and accelerates tumor growth (34, 35). Although secretory and degradative autophagy both utilize various chemicals and activities (e.g., autophagosome formation, ubiquitin), secretory autophagy does not degrade its cargo via lysosomes. Multivesicular bodies and autophagosomes combine to form amphiboles, which then fuse either with secretory lysosomes or directly with the plasma membrane to secrete proteins (34, 36). Autophagosome trafficking depends on outer membrane proteins. For example, LC3-II identifies the secretory and degradative routes, STX17 fuses degradative autophagosomes to lysosomes, and TRIM16 and SEC22B control autophagosome secretion (36). Despite secretory autophagy mediating the secretion of IL-1, IL-8, CXCL6, CXCL8, TGF-β, high-mobility group box 1 (HMGB1), and autophagic vacuoles, its regulation remains unclear (36, 37). These suggest that selectively targeting specific stage of autophagy profoundly affects its accompanied secretory pathways. Furthermore, late and early autophagy inhibitors have contrasting effects on secretion —for example, Spautin-1 and CQ are both autophagy inhibitors but have opposing secretory effects (37). The stage appliance of autophagy inhibitors during the autophagy process was elicited in Figure 2.
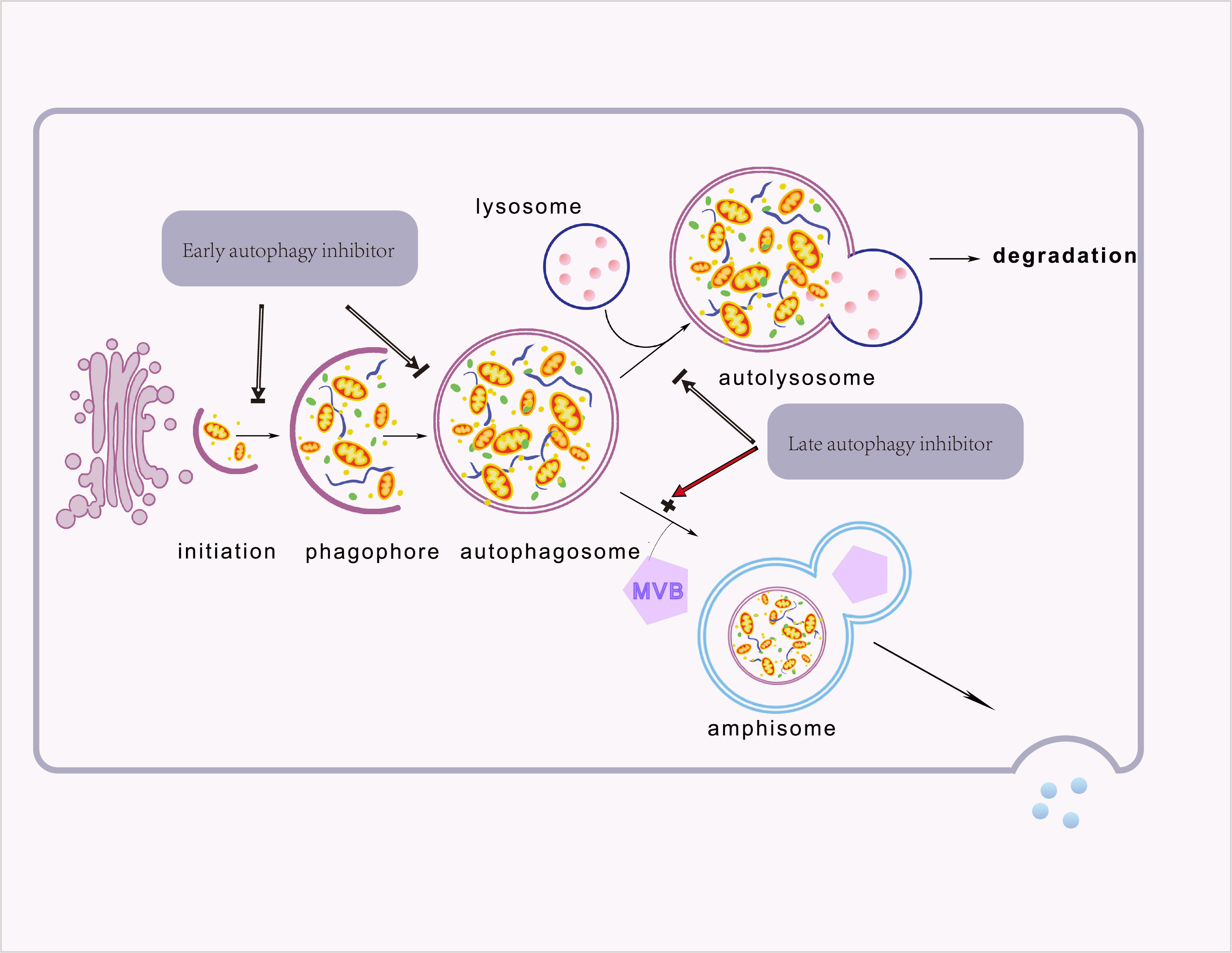
Figure 2 The different types of autophagy. Proteins, organelles, and other cellular components are sequestered in a newly formed isolation membrane. This isolation membrane then swells and seals to form a double membrane-bound vesicle, the autophagosome. Degradation of the autophagosome occurs when the autophagosome fuses with the lysosome. In secretory autophagy, Autophagosomes fuse with multivesicular bodies to produce double-membrane bodies that can fuse with the plasma membrane and secrete cargo into the extracellular space. Furthermore, late and early autophagy inhibitors have different effects on secretion.
Hypoxic, alteration in the three-dimensional stromal environment, and abnormal angiogenesis are characteristics of TME, which can induce autophagy in various constituent cells. Autophagy then promotes reshaping of the ECM, remodeling of the cellular composition, and reprogramming of interactions between tumor and stromal cells —this ultimately redefines the TME and thus alters the efficacious of immunotherapies (18, 38). We now review the relationships between some characteristics of TME and autophagy (Figure 3).
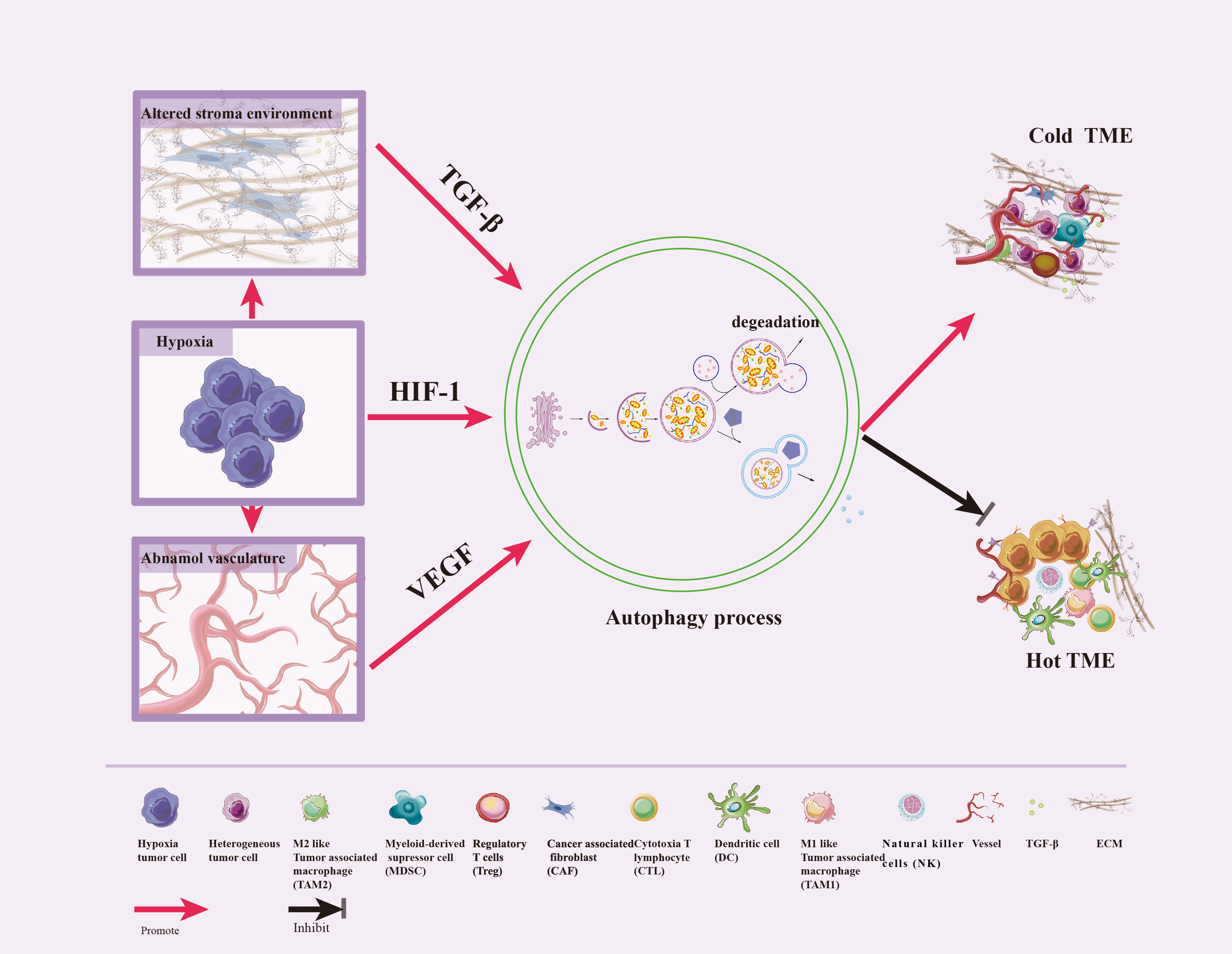
Figure 3 Crosstalk between autophagy and TME features. Hypoxic stress is a typical feature of TME, which triggers the abnormal vasculature and the alteration of the three-dimensional stromal environment. In response to these stresses, autophagy can be induced by HIF-1, VEGF, and TGF-β to promote tumor cell survival, enhance differentiation of normal fibroblasts to CAF, promote dense ECM formation, inhibit immune cells infiltration and vascular normalization, impair CTL and NK cell-mediated anti-tumor immune responses, and convert hot TME to cold.
Hypoxia is the widely accepted stimulator of autophagy induction via HIF-1-mediated expression regulation of key genes associated with autophagy, including adenovirus E1B 19 kDa-interacting protein 3 (BNIP3), BNIP3-like (BNIP3L), ATG9A, PIK3C3, Beclin 1, ATG5 and ATG7 (39–43). Increased BNIP3 and BNIP3L expression induced by HIF-1, can disrupt the Bcl-2-Beclin1 complex to initiate autophagy (41). In addition, hypoxia activates the AMP/AMPK/mTOR pathway and inhibits the PI3K/AKT/mTOR pathway to initiate autophagy (44, 45). Through intrinsic cytoprotective pathways, hypoxia-induced autophagy enhances tumor cell survival and contributes to the formation of the immunosuppressive TME that fuels tumor growth (39, 40, 46, 47). For example, HIF-1-dependent autophagy was crucial in inhibiting CTL and NK cell-mediated anti-tumor immune responses (47, 48). In head and neck squamous cell carcinomas, defective autophagy led to increased tumor sensitivity to treatments and lower tolerance to hypoxia (49). In addition, it is noteworthy that autophagy is found to induce HIF-1degradation (50). Further study will be needed to confirm whether autophagy-mediated HIF-1 degradation can consequently affect TME and immune cells. Hypoxia-induced autophagy plays a key role in tumor progression and immunotherapy resistance. How to reduce hypoxia-induced autophagy may be the future research point.
Recent studies demonstrate that autophagy is essential for endothelial differentiation and survival of ECs. Abnormal angiogenesis causes hypoxia, which promotes autophagy in tumor-associated ECs and possibly mediates resistance to hypoxia-induced cell death (51). In addition, autophagy was required for VEGF-mediated endothelial differentiation in breast cancer stem-like cells (52). VEGF treatment activated AMPK-ULK1 axis in breast cancer stem-like cells initiating autophagy (52). In addition, autophagy was a key process for immune sensing and vascular normalization of ECs resulting in governing immune cell recruitment in tumors (53, 54). Autophagy activation in tumor cells may adversely affect numerous angiogenic proteins (such as VEGFR2 and HIF-1) and impede the tumor angiogenic vascular system (55). Soluble decorin, as a partial agonist of VEGFR2, induced autophagic degradation of intracellular VEGFA in ECs to suppress angiogenesis via VEGFR2/AMPK/PEG3 axis (56). Administration of autophagic inhibitors such as chloroquine or bafilomycin A1, or depletion of ATG5, results in the accumulation of intracellular VEGFA (56). Furthermore, CQ, an autophagy inhibitor, induces tumor vascular normalization by inhibiting VEGF-A mediated phosphorylation of VEGFR 2 (57). In addition, TME angiogenesis modifies pre-existing vascular characteristics and restricts endogenous T cell migration, impacting both the efficacy and utility of CAR T cell therapy for solid tumors (58). Some studies support that modulation of autophagy in tumor ECs can sensitize immunotherapy. For instance, Endostar prevents angiogenesis by blocking the VEGF-related signaling pathway. In a murine model, anti-PD-1, in combination with Endostar, dramatically enhance PI3K/AKT/mTOR-mediated autophagy, leading to the reversing of immunosuppressive TME (59). And, autophagy in tumor cells impairs T cell survival and function to maintain an immunosuppressive TME. Thus, targeting autophagy may reverse the abnormal vasculature and fire up the immunosuppressive TME, which may enhance the immunotherapy response.
Altering the autophagic activity of stromal cells —mainly fibroblasts—, can recreate the three-dimensional stromal environment and induce reprogramming in the TME. Previous reports suggest that autophagy in fibroblasts is upregulated in the TME and plays a role in promoting tumor progression. Firstly, the hypoxic TME induces differentiation of normal fibroblasts into CAF via the p62/SQSTM1-autophagy-Nrf2-ATF6 axis (60). Hypoxia-induced TGF-β regulates the activation and function of CAF. And, CAF secretes TGF-β and promotes dense ECM formation, boosting T-cell exclusion via the chemical and physical barriers (31). Secondly, autophagy in CAF support pro-fibroproliferative responses, including type 1 collagen deposition and tissue stiffness, resulting in a rigid desmoplastic stroma that impedes CTL infiltration and activation (61). Furthermore, autophagy in CAF regulates immune cell recruitment through the secretion of inflammatory factors such as IL-6 (61). In addition, autophagy activated by TGF-β1 was necessary for the development of myofibroblast and CAF phenotypes, which was associated with enhanced migration and invasion of oral squamous cell carcinomas (62).
Altered autophagy can recreate the immunostimulating TME to promote anti-tumor immune by enhanced immunogenic cell death (ICD), tumor antigen releasement, antigen presentation, promoted antigen recognization, effector immune cells infiltration, and immunosuppressive cell outflow, which may be applied to improve the efficacy of immunotherapy for tumors. With few exceptions, autophagy inhibition is favor influence for burning up “cold” TME. To gain an efficient prospective benefit, we should focus on the targeted cells, targeting autophagy type, effective stages in the autophagy process, and the immune-signal regulatory point of the autophagy modulators. Figure 4 showed the main process and mechanisms involved in autophagy-mediated TME remodeling. These mechanisms are explicated in Table 1.
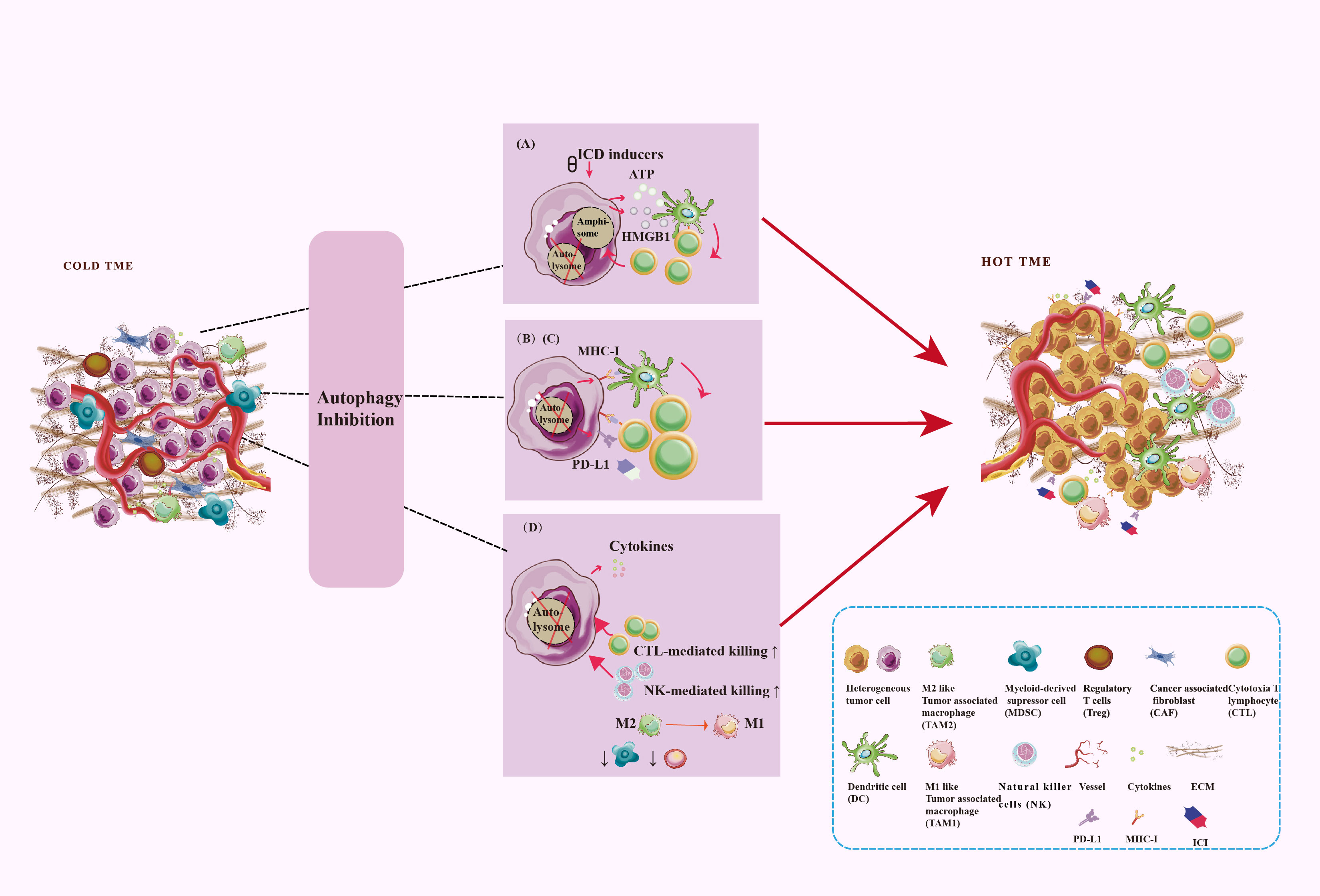
Figure 4 Targeting autophagy burns the cold TME. TME regulation mechanism includes antigen signal release, antigen presentation, antigen recognization, as well as immune cell infiltration. Autophagy plays a key role in these processes. (A) Secretory autophagy promotes ICD to burn the cold TME. (B) Autophagy inhibits the MHC-I expression on the surface of tumor cells and DC to inhibit antigen presentation. (C) Autophagy inhibits PD-L1 expression on the surfaces of tumor cells. (D) Autophagy inhibits CD8+ T cells and NK cells infiltration and tumor-killing effect and promotes the survival and development of Treg and MDSC. Autophagy promotes TAM polarize to M2 and enhances the immunosuppressive function of macrophages.
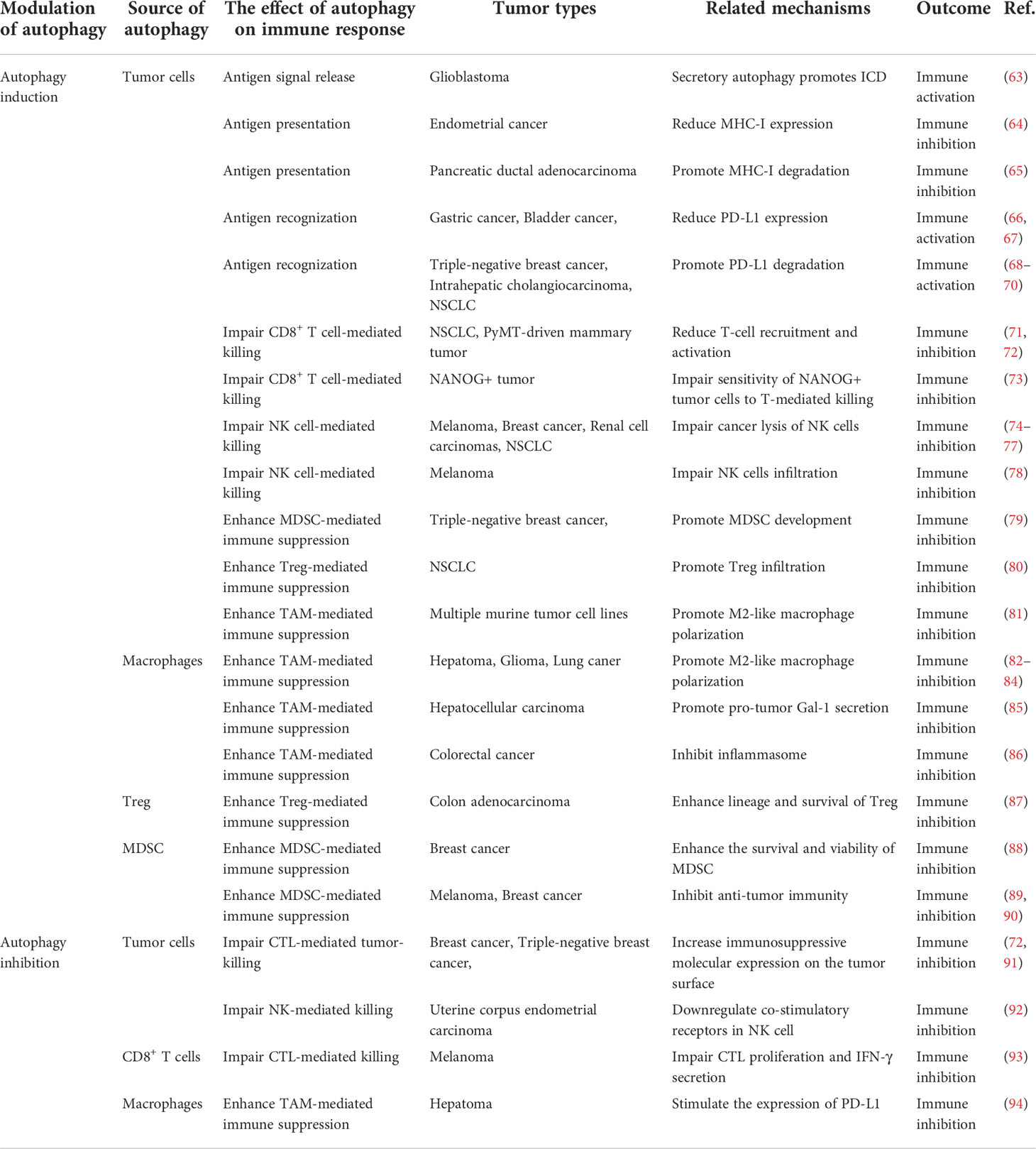
Table 1 The role of autophagy in the transformation of hot TME to cold TME.
By releasing damage-associated molecular patterns (DAMPs) such as HMGB1, calreticulin, and adenosine triphosphate (ATP) into the TME, systemic chemotherapy, localized radiotherapy, and oncolytic viruses enhance the immunogenicity of tumor cells. These DAMPs aid development of DC, activate cytotoxic T cells, produce inflammatory chemokines and cytokines, and polarize TAMs into M1 states, leading to tumor cell death (95). This regulated cell death type is called ICD and contributes to reprogramming immunostimulatory TME (95). Unconventional autophagy−based secretions such as secretory autophagy participated in the release of ICD-related DAMPs, and this could be impaired by ATG5 knockdown (63, 96). Autophagy enables ICD-associated ATP secretion by the preservation of lysosomal ATP pools and secretory autophagy pathway-mediated releases (97). Upon autophagic activation, an ATP-loaded amphisome fused with the plasma membrane via the VAMP7-SNARE complex, releasing ATP into the extracellular medium (98). The release of HMGB1 is similar (63). Upon entry into the outer cell membrane, ATP and HMGB1 bind to P2RX7 and TLR4 receptors on the DC (96). However, autophagy also results in antigen damage, which impairs the immune response. DAMPs and pathogen-associated molecular patterns can initiate autophagy, then autophagosomes will eradicate these molecules to maintain immunological homeostasis (99). Based on its role in the phagocytosis of antigens, autophagy is a potential protective mechanism for tumor cells against ICD-induced immune responses (99). HMGB1 secretion was inhibited by early autophagy inhibitors. Conversely, the late autophagy inhibitors CQ and Bafilomycin A1 increased HMGB1 secretion (37). Therefore, a current research focus is on how to regulate this pathway to attenuate degradative autophagy and enhance secretory autophagy, thereby promoting ICD and igniting cold TMEs. Combining late autophagy inhibitors with ICD inducers to potentiate cancer immunotherapy is a current innovative strategy.
Autophagy degrades proteins of tumor cells such as tumor-specific and tumor-associated antigens. When proteasomes and lysosomes are both inhibited, SLiPs, DRiPs, and misfolded proteins form protein aggregates (ALIS/aggresomes), activating autophagy via interactions between p62/SQSTM1 and ATG8/LC3. Peptide intermediates linked to HSP90 are in autophagosomes and through secretory autophagy, they are secreted from tumor cells as cross-presenting immunogenic substrates. These tumor cell-derived autophagosomes are called Defective Ribosomal Products-containing Blebs (DRibbles). As demonstrated in clinical and experimental models, DRibbles are efficient carriers of tumor antigens that induced robust cross-presentation by APCs (100, 101). DRibbles-pulsed-bone marrow cells or DC were peri-tumorally titrated with GITR agonists and PD-1 blocking antibodies, increasing the cytotoxicity activity of CD8+ T cells via an antigen-presenting independent mechanism (102). In addition, tumor cell-released autophagosomes (TRAPs) converted macrophages to an immunosuppressive M2-like phenotype that is defined by PD-L1 and IL-10 expression, resulting in T cell inactivation and cold TME formation (81). Thus, secretory autophagy plays a key role in tumor antigen releasement, evoking the CD8+ T cells priming.
Endogenous antigens of tumor cells are presented to the anti-tumor immune response in two distinct ways. First, the antigens are directly presented by MHC-I of the tumor cells for recognition and elimination by CD8+ T cells. Second, DC recognize tumor antigens via pattern recognition receptors, process them, and cross-present antigens to T cells, resulting in the activation of anti-tumor CD8+ T-cell immunity (7). Some researches demonstrate the role of autophagy in antigen presentation. Autophagosome membranes are possibly produced by the endoplasmic reticulum, and thus peptide-sensitive MHC class I molecules may be present therein (7). Furthermore, autophagy inhibition possibly enhances MHC-I expression on the surface of DC and tumor cells for reducing endocytosis and degradative function, thereby promoting activation and migration of DC, and attraction of CD8+ T cells. Ultimately, these ignite the immunosuppressive TME and overcome resistance to the immune checkpoint blockade (ICB) (99, 103). Inhibiting the selective autophagy of MHC-I —this was mediated by the autophagic cargo receptor NBR1— increased MHC-I expression in tumor cells (65). In endometrial cancer, overexpression of LC3 constrained the MHC-I transactivator nucleotide-binding oligomerization domain-like receptor family caspase-containing recruitment domain 5 (NLRC5), a transcriptional regulator of the MHC-I gene, which then decreased the expression of MHC-I (99). Blocking tumor-derived progranulin destroys autophagosomes, restoring MHC-I expression. Then, progranulin antibody therapy increases levels of granzyme B, TNF, IFN-γ, and CD8+ T cells, reviving CD8+ T cell anti-tumor cytotoxicity (64). Adaptor-associated kinase 1 (AAK1) mediated endocytosis and autophagic degradation of MHC-1 in DC, leading to inhibition of antigen presentation and CD8+ T cell initiation. However, DC with ATG5/ATG7 deficiency showed increased MHC-I expression (103). DC are the most efficient specialized APCs. DC may store antigen in endolysosomal compartments for extended periods, and this preserves MHC-I antigen cross-presentation to CD8+ T cells. However, autophagic processes compromise DC antigen storage and presentation. For example, DC from ATG5-deficient mice stored antigen in storage compartments for prolonged periods, consequently curtailing late MHC-I cross-presentation and boosting antigen cross-presentation into CD8+ T cells (104). DC from VPS34-deficient animals had homeostatic maintenance along with a partially activated phenotype, spontaneously generated cytokines, and displayed the increased activity of conventional MHC class I and class II antigen-presentation pathways (105). Autophagy inhibition combined with anti-PD-1 therapy increase tumor neoantigen presentation in LKB1-inactivated high-TMB tumor and achieve a greater anti-tumor effect (106). Thus, autophagy inhibition may enhance antigen presentation through elevated MHC-1 expression and activated DC phenotypes, leading to the priming of T cells and enhancement of the immunotherapy effect.
Immune checkpoints are pathways with inhibitory or stimulatory properties, modifying immune cell activity. The most well-known inhibitory checkpoints are PD-1 and PD-L1, which inhibit T cell activation, proliferation, and function (107). ICIs are a breakthrough in treating tumors and applied to block the binding between PD-L1 and PD-1, which can re-activate CTLs and NK cells (107). Links between autophagy and immune checkpoints have recently been established. For example, ROS generated by andrographolide inhibits the JAK2/STAT3 pathway in NSCLC, leading to the activation of p62-dependent selective autophagy and promoting the degradation of PD-L1 (68). Sigma1 is a ligand-operated integral membrane chaperone —scaffolding protein— that is abundant in endoplasmic reticulums of various cancer cell lines and generally binds to glycosylated PD-L1 to preserve protein stability (69). Pharmacological suppression or deletion of the Sigma1 decreases PD-L1 expression on the tumor surface via selective autophagy (69). KRAS-mutated intrahepatic cholangiocarcinoma cells had activated ERK signaling. Blocking of ERK signaling induced autophagy to degrade PD-L1, while genetically silencing the ATG7 expression partially reversed degradation (70). Furthermore, ATG7 overexpression enhanced the steadiness and expression of PD-L1 mRNA by autophagy-induced FOXO3a/miR-145 degradation in bladder cancer models (66). In gastric cancer cells, inhibiting autophagy causes the buildup of p62/SQSTM1 and the activation of nuclear factor (NF)- κB, resulting in the induction of PD-L1 (67). Controversially, another study showed that 5-HT activated p-STAT3/autophagy axis via 5-HT1aR, leading to upregulated PD-L1 expression and an immunosuppressive environment, which remains to be confirmed in the further steps (108). Taken together, autophagy modulators may be a new immunosensitizer, for it directly or indirectly downregulates PD-L1 expression.
Another newly discovered immunological checkpoint CD155 expressing on tumor cells functions as a ligand for the costimulatory receptor CD226 and the co-inhibitory receptor TIGIT of natural killer cells and T cells. ATG5-dependent autophagy induced by artesunate could enhance CD155 overexpression on uterine somatic endometrial carcinoma (UCEC) cells. CD155 overexpression upregulated CD226 and downregulated TIGIT, hence enhancing the cytotoxicity of NK cells (109). However, the precise mechanism is unknown. Other immune checkpoints SIRPα and CD47 release a “don’t eat me” signal to prevent the recognization and phagocytosis of cancer cells by immune cells. The interaction of CD47 on cancer cells with SIRPα on macrophages could suppress the phagocytosis of macrophages (110). By blocking the CD47/SIRPα axis, SIRPαD1-Fc selectively targets NSCLC cells and activated macrophages to recognize and phagocytose tumor cells (110). But it also causes protective autophagy in cancer cells, which may be a reaction to cellular stress. Concurrently targeting CD47 and autophagy improves macrophage-mediated phagocytosis and cytotoxicity against NSCLC cells (110). Zhang et al. discovered that autophagy in glioblastoma cells could impair the immunotherapeutic benefits of anti-CD47-SIRPα therapy through decreased phagocytosis of macrophages and decreased cytotoxicity of CD8+ T cells (111).
Thus, autophagy regulates antigen recognization mainly by the degradation of immune checkpoint proteins and the regulation of intracellular signaling pathways thereby influencing antitumor immune responses and immunotherapy efficacy. Combining autophagy modulators with ICIs may be a promising anti-tumor therapy to improve the effectiveness of ICIs and need to be further explored in clinical application.
Tumor cell eradication depends on the attraction and subsequent infiltration of CTLs and NK cells into the TME, the absence of which contributes to immunotherapy resistance and tumor progression (38). Relatedly, autophagy affects CTL activity and infiltration (112). For example, FIP200, an essential autophagy gene, is responsible for restricting T cell recruitment and activation in the TBK1/IRF/IFN signaling axis. In immunocompetent breast cancer models, impairment of this noncanonical autophagic function of FIP200, in combination with immune checkpoint blockade therapy, yielded good responses (71). SKIL promoted the growth of tumors and prevented the entry of CD8+ T cells into NSCLC cells by upregulating the TAZ/autophagy axis and downregulating the STING pathway (72). In addition, alteration of autophagy can increase tumor cell sensitivity to T cell-mediated tumor killing. For instance, NANOG was a major transcription factor that enhances secretory autophagy in tumor cells via promoting LC3B expression, leading to EGF autocrine (73). EGF subsequently upregulated the EGFR-AKT signaling pathway and then led to tumor cell resistance to CTL killing (73). Early autophagy inhibitors combined with ICI can reverse tumor refractory phenotype (73). However, ATG7/ATG5 deficient triple-negative breast cancer cells were less vulnerable to T cell-mediated death due to the p62-mediated selective autophagy of Tenascin-C, a candidate immunosuppressor (113). Similarily, B7H3 expressed on tumor cell surface inhibited not only activation and proliferation of T cells but also the production of immunostimulation cytokine (114). And, autophagy has been affirmed to participate in the degradation of B3H7 (91). In breast cancer models, V9302 decreased B7H3 expression and increases CD8+ T cell activation by the autophagy-lysosome pathway (91). In addition, anti-PD-1/PD-L1 mAb combined with B7H3 blockers (anti-B7H3 mAb or V9302) could transform “immune desert” tumors into “hot”, improve the curative benefits in metastatic or advanced breast cancer (91). Furthermore, autophagy is also involved in regulating T-cell immune activity. ATG3-, ATG7-, or ATG5- T cells cannot proliferate efficiently (115). DeVorkin et al. showed that loss of autophagy triggered T cells into a glycolytic phenotype along with reduced S-adenosylmethionine levels. Thus, ATG5-/- CD8+T cells gained an effector memory state that promoted CD8+ T cell-mediated tumor rejection and INF-γ release (116). In brief, autophagy plays a “double-edged” regulation in CTL cell-dependent immunotherapy. It is required to consider that autophagy modulators affect which target cells in the combination of immunotherapy.
NK cells express a range of stimulatory and inhibitory receptors that determine whether to kill tumor cells by binding to specific ligands on tumor cells (9). Autophagy involves the cytolytic activities, and memory responses of NK cells and essentially participates in the downregulation and activation of effector molecules and receptors respectively (117). Activated NK cells form synapses with the tumor cells and release two cytotoxic effectors (perforin and granzyme) to mediate the death of tumor cells. The gap-junctional connexin 43 is required for the synapse (74). Hypoxia-induced connexin 43 overexpression could selectively induce autophagy to hinder its localization on the immunological synapse to promote tumor cells’ evasion of NK cell-mediated death. However, ATG5 siRNA-mediated autophagy inhibition can reverse these processes (74). Moreover, connexin 43 channel is also an essential component of CTL cytotoxic immunosynaptic-mediated tumor cell death (118). In addition, autophagy selectively destroys the NK-derived granzyme B in cancer cells, reducing tumor cell sensitivity to natural killer-mediated lysis. Targeting BECN1/Beclin1 and ULK1 can revive the cytotoxicity of granzyme B (75–77). Likewise, the dysfunctional autophagy of cancer cells enhances the recruitment of NK cells to the tumor periphery. Becn1/Beclin1-deficient melanoma cells in the TME expressed high levels of CCL5 via the activation of the MAPK8/JNK-JUN/c-Jun signaling pathway, which boosted the infiltration of functional NK cells into the TME and curtailed tumor progression (119). Furthermore, when autophagy was inhibited in melanoma cells by either ATG5 or p62/SQSTM1 deficiency or CQ treatment, melanoma cells could recruit NK cells into the tumor site by CCL5 releasing (119). Blocking autophagy of tumor cells promote NK cell-mediated aggregation and killing, converting TMEs from cold to hot. Autophagy inhibitors possibly potentiate CTL-based or NK cell-based immunotherapies.
Malignant tumors recruit immunosuppressive cells —such as TAM with an anti-inflammatory M2 phenotype, MDSCs, and Tregs— to suppress T cell functions and form the immunosuppressive TME. Conversely, effective T and NK cells are rare in “cold” TME. Autophagy essentially contributes to the activation and infiltration of these immunosuppressive cells, which may impair T-mediated tumor killing, reshaping the TME into a cold state and ultimately causing resistance to immunotherapy (92).
Tumor cells can actively attract circulating monocytes to the tumor site by secreting CCL2, and then the recruited monocytes developed into TAMs through granulocyte-macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (MCSF) (120, 121). These chemokines and cytokines induced autophagy of monocytes to keep them alive and allow them to differentiate into TAM (120, 121). In response to stimuli from TMEs, various TAMs are polarized to M2-like phenotypes and this results in immunosuppression and tumor progression. Macrophages took up Beclin1-dependent tumor cell-released autophagosomes and activated MyD88-p38-STAT3 axis via TLR4. This resulted in overexpression of PD-L1 and IL-10, which limited CD8+ T cell recruitment and promoted tumor formation (81). In addition, hepatocellular carcinoma-derived HMGB1 triggered M2 macrophage polarization via a TLR2/NOX2/autophagy axis (82, 83). Furthermore, Inhibition of autophagy in macrophages reprogrammed pro-tumor M2-like TAMs to a tumor-suppressing M1 phenotype that exerted anti-tumor effects by regulating not only NF-κB p65 protein homeostasis but also an IL-6-pSTAT3-miR-155-3p-autophagy-pSTAT3 positive feedback loop (82, 83). In addition, Galectin-1 was a soluble tumor-promoting factor secreted by TAMs through TLR2-dependent secretory autophagy, —it was associated with poor outcomes (85). However, a recent study suggested that autophagy invalidation in macrophages induces an immunosuppressive phenotype to alter the antitumoral immune response, leading to hepatocarcinogenesis (94). The phagocytosis of TAMs is efficient in the early stages of tumor-specific antigen processing and innate tumor killing. Autophagy is a requirement for TAMs phagocytosis (122). Through IDO1 expression and kynurenine metabolism, IFN-γ promoted autophagy and macrophage phagocytosis in cervical cancer cells (122). In summary, autophagy functions as a regulatory process in macrophages, maintaining cellular homeostasis and regulating specific immune functions such as recruitment, differentiation, polarization, phagocytosis, and pro-tumor factors production. Targeting autophagy in macrophages may thus be a novel and practical anti-cancer approach.
Tumor-infiltrating Tregs suppress anti-tumor immune responses and promote tumor immune escape (11). Autophagy in tumor cells linked to Treg infiltration and immunosuppressive activities of TME. Results from the NSCLC xenograft model showed that tissue-specific knockdown of ATG5 attracted Treg migration to the TME (80). Except for this, the autophagy pathway also participated in Treg cell lineage differentiation and function. As the transcription factor FOXP3 is required for the differentiation and immunosuppressive activity of Treg cells, Treg cells with the autophagy-related genes AMBRA1 and ATG7 deletion fail to express FOXP3, leading to the malfunction of Treg cells (123). In addition, ATG5-/ATG7- Treg cells displayed functional deficiency via increasing mTORC1 expression, c-Myc expression, and glycolytic activity (87).
MDSCs are immunosuppressive cells derived from bone marrow progenitor cells and immature bone marrow cells (12). To meet their bioenergetics and biosynthesis requirements, tumor cells reroute metabolic pathways one of which, is glycolysis, a hallmark of cancer (79). Glycolysis enhanced granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) expression via the AMPK-ULK1 and autophagic pathways, which then promote MDSC development (79). MDSCs activate autophagy to survive in the harsh TME induced by cellular stresses such as nutrition and hypoxia. Under these stressful conditions, MDSCs released HMGB1 to maintain their viability via initiating autophagy (88). Autophagy-deficient MDSCs displayed reduced lysosomal degradation, which promoted the surface expression of MHC class II molecules and resulted in the effective activation of tumor-specific CD4+ T cells (89). In addition, tumor growth increased β2-AR expression in MDSC leading to enhanced autophagy and activation of arachidonic acid via pressure-activated signals. These increased the release of PGE2, an immunosuppressive mediator (90). Taken together, autophagy promotes the immunosuppressive function of MDSC, thereby facilitating the formation of an immunosuppressive TME.
The effectiveness of immunotherapy is significantly influenced by the cold TME, which is also regulated by autophagy. Therefore, strategies targeting autophagy should be exploited to develop efficient immunotherapy sensitizers. However, the physiological processes involved in autophagy are complex and there is no consensus on whether autophagy should be activated or suppressed. Almost all stages of autophagy, including vesicle nucleation, maturation, fusion, and lysosomal destruction, have been identified as potential therapeutic targets. Understanding these regulatory pathways may lead to the development of new cancer treatment options.
Autophagy acts as a guardian to promote tumor cell survival in response to stress in the TME. Nowadays, various autophagy inhibitors are accessible drugs being applied to target autophagy, being cataloged as early-stage inhibitors (SAR405, 3MA, and SBI-0206965) and late-stage inhibitors (CQ, HCQ) (124). Early-stage inhibitors regulate the nucleation of the autophagy process by targeting ULK1/ULK2 or VPS34, while late-stage inhibitors target lysosome disorder the degradation stage of the autophagy process (124). So far, secretory autophagy-based clinical studies are in the preliminary stage, and late and early-stage inhibitors of autophagy have diverse effects on secretory autophagy (37). In addition, mTOR inhibitors also play an important role in antitumor therapy as enhancers of autophagy initiation (125). Autophagy targeting compounds used for cancer treatment are listed in Table 2.
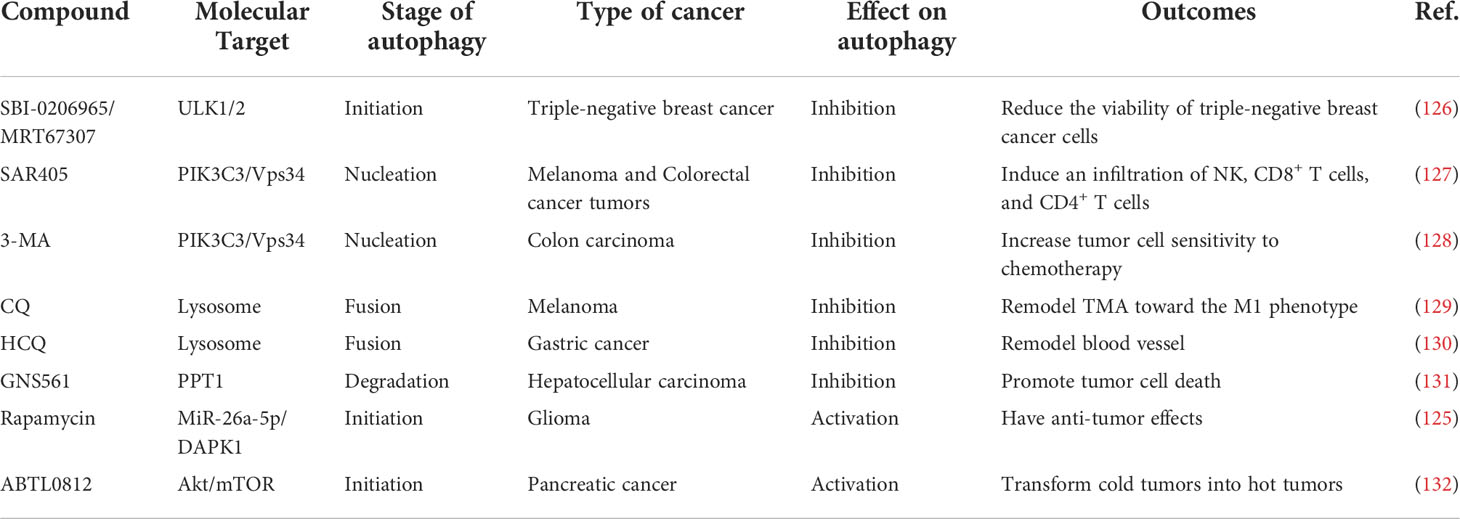
Table 2 The applied autophagy targeting agents in treatments in vivo and vitro.
Evidence from preclinical studies suggests that targeting autophagy can improve the efficacy of numerous cancer treatments. Several US Food and Drug Administration (FDA)-approved medications, including the inhibitor CQ and its derivative HCQ, as well as inducers like rapamycin, have been identified as effective autophagy modulators (125, 130). A previous meta-analysis reported that autophagy inhibitors, such as CQ and HCQ alone or combined with other anti-cancer drugs were well tolerant and significantly improved cancer patients’ overall response (133). Rapamycin exerts anti-tumor effects by promoting autophagy in glioma cells, which was dependent on the miR-26a-5p/DAPK1 pathway activation (125). Although many small molecule therapeutics have been identified as effective anti-cancer drugs, further clinical investigations are being carried out. The compound GNS561 inhibits the enzyme palmitoyl protein thioesterase 1 (PPT1), causing Zn2+ accumulation in the lysosomes and reducing autophagic flux. Therefore, it is regarded as a promising anti-cancer therapy (131). By activating cytotoxic autophagy in tumor cells, the autophagy inducer ABTL0812 increases the death of cancerous cells. The first human Phase I/Ib dose-escalation clinical trial demonstrated that ABTL0812 was safe, tolerable, and has powerful anti-cancer properties (134).
Immunotherapy is widely used to treat advanced tumor patients. However, the efficacy of immunotherapy is impeded by cold TME. Autophagy regulates intracellular homeostasis, cell survival, cell activation, cell proliferation, and differentiation. Therefore, modulating autophagy can remodel the immunosuppressive TME into immunostimulatory TME. The accumulated evidence of clinical trials demonstrated that an autophagy-targeting strategy can improve the efficacy of immunotherapy. Table 3 describes the ongoing trials about the combination of autophagy inhibitors and immunotherapy for cancer treatment registered on ClinicalTrials.gov (https://clinicaltrials.gov/). Due to their positive effects on tumor cells and animal models, PD-L1 inhibitors and autophagy inhibitors CQ are the focus of most ongoing clinical trials. Other combination therapies including TME targeting are also being investigated in preclinical studies, and the results are encouraging.
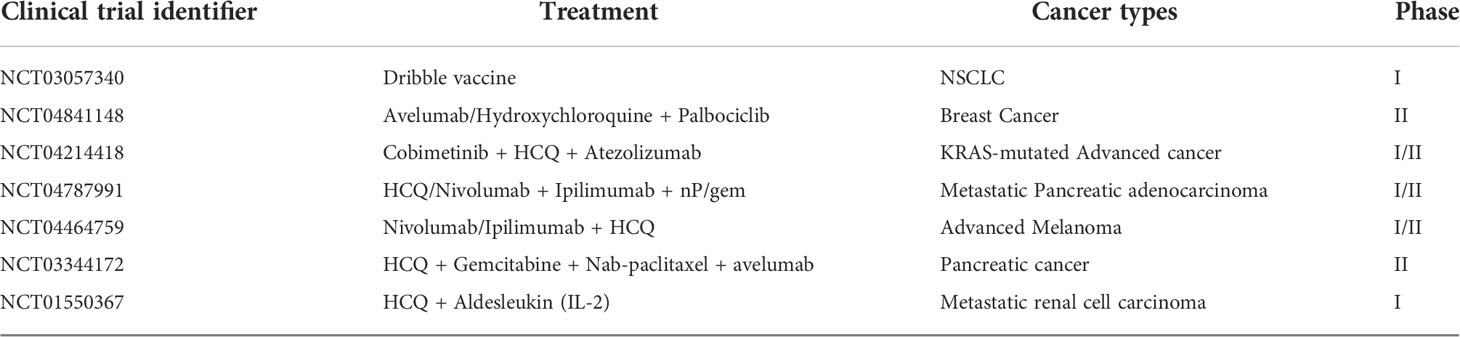
Table 3 The ongoing clinical trials about the combinations of immunotherapy with autophagy targeted drugs.
In recent research, researchers have focused on developing strategies to increase autophagy to improve ICD effects. Local administration of low-dose chemotherapeutic medicines plus the autophagy enhancer rapamycin (CAER) led to systemic anti-tumor T-cell immunity in vivo. In addition to enhancing the mortality of B16F10 and 4T1 tumor cells and increasing the levels of autophagy in vitro, the low-dose CAER treatment promoted neoantigen-specific T-cell responses. It also modified the TME by lowering systemic toxicity (135). In MDA-MB-231 and CT26 cancer cells, brucine enhanced the effects of ICD, such as CRT exposure and HMGB1 release, and inhibited autophagy by hindering the destruction of autolysosomes. However, ATG5 knockdown significantly decreased the release of HMGB1 and CRT exposure caused by brucine (136). And, early autophagy inhibition can reduce ICD but late autophagy inhibition can increase it, which is dependent on the secretory autophagy response generated by the autophagy inhibitor (137). In contrast, an optimal dose of combined LipSHK (ICD inducer) and LipHCQa (autophagy inhibitor) could maximize ICD-based antitumor immunity in colon cancer (138). Thus, combining ICD inducers with autophagy targeting drugs may be an innovative strategy to improve cancer immunotherapy. Moreover, the secretion of HMGB1 and ATP may be effective markers for treatment response prediction.
The absence of tumor-infiltrating T cells and recruitment of diverse immunosuppressive cells are the significant features of “cold” TME, causing resistance to ICls. And, targeting autophagy has been found to regulate anti-tumor immune response by remodeling the immune TME. Therefore, combination treatment approaches may enhance the response to ICIs in cancer patients. For example, the autophagic cargo receptor NBR1 plays a key role in the targeting of MHC-I molecules for lysosomal degradation. In pancreatic ductal adenocarcinoma cells, MHC-I is less visible on the cell surface but is more pronounced in autophagosomes and lysosomes. CQ and dual ICB treatment (anti-PD1 and anti-CTLA4 antibodies) synergize to enhance the immune system’s ability to fight tumors (139). Researchers demonstrated that combined SIRPα-Fc and CQ treatment interrupted the CD47/SIRPα axis and disrupted the protective autophagy in tumor cells, enhanced phagocytosis of macrophages, and then activated CD8+ T cell-mediated anti-tumor immune (111). In addition, HCQ and rapamycin treatment reduced autophagic flux and expression levels of CD47 and SIRPα, thereby enhancing the phagocytosis of TAM, which has strong phagocytic activity (140). HCQ and rapamycin treatment could also improve anti-PD-1 therapy by reprogramming M2-like TAM to M1-like phenotype and enhancing T cell-mediated cytotoxicity (141). Combined with the anti-PD-1 therapy, CQ/HCQ targeting palmitoyl protein thioesterase 1 (PPT1), a novel regulator of cancer cell autophagy, enhances the anti-tumor immune response by switching the macrophage M2 to M1 phenotype, lowering MDSCs, and increasing T cell-mediated cytotoxicity (142). In addition, silencing the autophagy-associated protein BECLIN1 or VPS34 promotes the release of pro-inflammatory CCL5 and CXCL10 in the TME of melanoma and CRC tumor cells via activation of STAT1/IRF7 axis, resulting in increased infiltration of central immune effector cells (NK, CD8+ and CD4+ T cells, DC, and M1 macrophages). And, SB02024 or SAR405 (VPS34 inhibitors) converts cold immune deserts to hot immune TMEs and reverses anti-PD-1/PD-L1 treatment resistance in melanoma and CRC tumor models (78, 143). ESK981, a novel autophagy inhibitor, was identified to increase the susceptibility of cold tumors to ICIs by producing CXC10 that draws T cells by targeting the autophagy-associated protein Pikfyve in prostate cancer cells (144, 145). Although Pemetrexed and cisplatin (PEM/CDDP) chemotherapy combined with ICIs did not have the synergic effect in patients with metastatic NSCLC, the combination regimen with MEK inhibitors (MEKi) blocking autophagy could trigger the CXCL10 secretion and CD8+ T cell recruitment to enhance the tumor-killing effect (146). Furthermore, in LKB1 mutant tumor models, ULK1 suppression and PD-1 antibody inhibition act as co-promoters of the effector T cell growth and tumor regression. Mechanistically, LKB1 deficiency inhibits the production of immune peptides by reducing the expression of the immunoproteasome component. This state can be changed by inhibiting the autophagy regulator ULK1, which increases the level of immunoproteasome expression and then increases lung tumor infiltration in CD4+ and CD8+ T cells (106). By modulation of immune recognition, immune effector cell chemotaxis, immune suppressor cell reduction, and antigen presentation in the TME, autophagy inhibitors can increase the infiltration of CTL cells in the TME, contributing to enhancing ICIs treatment.
Currently, the most advanced forms of cancer immunotherapy are CAR-T cell therapy. Immunotherapy known as CAR T cells uses T cells taken from patients, and genetically altered to express receptors that identify cancer-specific antigens, and then transfused (147). Due to the hostile solid TME, which acts as a barrier to CAR T cell infiltration and activity, CAR T cell treatment has been clinically successful in treating hematologic tumors but unsuccessful in treating solid malignancy (147). Because autophagy can effectively control the immunosuppressive TME, it may be beneficial in patients with solid tumors receiving CAR T cells. In mice with gliomas, autophagy significantly alerts the persistence of CAR-T and acts as an antagonist to CAR-induced trogocytosis and immune checkpoint activation (148). This suggests that autophagy stimulation may promote CAR T cell tumor fitness and survival in TME (148).
Because of the elevated levels of the tumor-secreting chemokines CCL5 and CXCL10, autophagy inhibition increases the formation of NK cells in glioblastoma in vivo. Furthermore, suppression of autophagy alters NK cell phenotypes to improve NK cell function and promotes NK cell-mediated cytotoxicity against glioblastoma cells. In one study, the combination of CQ and multifunctional genetically engineered NK cells outperformed multifunctional genetically engineered NK cells alone, inhibiting the growth of GBM tumors (149).
Some cancer vaccines have been suggested to be effective immunotherapy for several malignancies, including glioma, breast cancer, and liver cancer. They deliver high-quality antigens to active APCs and induce strong CD4+ T helper cell and cytotoxic T lymphocyte responses (150). Inducing autophagy in DC promotes peptide presentation to CD4 T cells, which is a novel strategy for increasing vaccine efficacy (151). Professional APCs transmit tumor proteins to T lymphocytes for activation via the MHC-I after consuming and degrading them with proteases during cross-presentation. Defective ribosome products (DRiPs) and short-lived proteins (SLiPs), two putative tumor-associated proteins, are produced in large amounts by tumor cells but are intrinsically uns` and only momentarily expressed under physiological conditions before being polyubiquitinated and broken down by tumor cell proteasomes. Inhibiting proteasomal degradation and altering the cellular autophagic pathway results in the production of the DRibbles vaccine product, which stabilizes the DRiPs/SLiPs proteins and induces the formation of autophagosomes that contain the proteins mentioned above as well as other protein products that are known to facilitate cross-presentation (152). A previous study use proteasome and lysosome inhibitors to prepare CMV-autophagosomes (DRibbles) (150). They show that IFN-DC loaded with DRibbles activated CMV-specific T cells (150).
It has been acknowledged that by boosting the anti-tumor immune response, immunotherapy improves the prognosis of patients with advanced cancer. However, only a tiny percentage of individuals with advanced disease have a satisfactory and sustained response to immunotherapy. Immunotherapy fosters further development of the TME. All TME components either negatively or positively influence the response to immunotherapy. By regulating antigen release, antigen presentation, antigen recognition, and immune cell trafficking, targeted autophagy therapy can create hot TME to improve the efficacy of cancer immunotherapy. In addition, immunological characteristics of TME are important factors to be considered in the strategies for immunotherapies improvement.
Even though several methods have been developed to influence the immune system and improve clinical outcomes, further investigation is needed to determine the mechanisms involved. Although few clinical studies have been conducted, several cellular and animal experiments combining autophagy targeting with immunotherapy have been conducted. Even though the majority of cellular and animal trial outcomes demonstrate that combination therapies are superior to immunotherapy alone, it is not clear whether the efficacy is similar across species. Notably, only CQ or HCQ are used as autophagy inhibitors in cancer therapy. However, their therapeutic outcomes are limited by their high toxicity and poor selectivity. Consequently, further research should be carried out to explore the target genes associated with autophagy. From a clinician’s standpoint, autophagy-targeted medicines should be customized to treat specific stages and grades of cancer.
Currently, there is no consensus on the best time to administer autophagy-targeted medications—before, during, or after immunotherapy. Autophagy-targeting drugs can alter the tumor’s immune milieu, hence influencing immunotherapy. To promote the effectiveness of immune-oncology medications, effective dosing schedules of autophagy modulators alone or in combination should be explored to modify the immunologic activity of TME in time and make a balance between side effects and tumor response benefits. Moreover, research is needed to determine multiple modality-sensitive indicators for predicting the patients’ future responses to anti-tumor alone or combination therapies. For improving clinical appliance, we can also focus on the affecting process, stage, and sequence of autophagy in TME remodeling, and how to conduct precise modulation of autophagy in the appropriate target cells of TME. Although suppressing tumors by targeting autophagy is currently thought to be a good therapeutic strategy, further research is required to determine how the host will be affected in the long run. Future studies are also needed to explain the “occasionality” interaction between autophagy modulators and immunological response. Additionally, the distinct cell populations that control autophagy and hence remodel the immune milieu should be explored, as well as whether the association is causative or accidental.
Given that both autophagy activation and inhibition have been found to increase the effectiveness of anti-cancer medications, the role of autophagy in cancer therapy requires further clarification. Therefore, the key question is whether we should strive to increase or decrease autophagy while treating cancer. Additionally, exploring the regulatory mechanisms of autophagy needs to consider that degradative autophagy blocking is followed by induction and regulation of secretory autophagy. These investigations may help to coordinate the effect of autophagy modulators better. To maximize patient benefit and enhance cancer treatment, precision-targeted medications should be further studied. Of note, the currently utilized autophagy inhibitors are not especially effective at enhancing anti-tumor immunity. Thus, research is needed to clarify the appropriate dose, target, time, marker, and tumor type to maximize their anti-tumor activity and sensitize immunotherapy.
ZJ, XS contributed to the data collection and writing the manuscript. YW, CZ made the figures and tables. SZ, and HY designed and guided this study and edited the manuscript. All authors listed made a substantial, direct, and intellectual contribution to the work and approved it for publication. All authors contributed to the article and approved the submitted version.
This study was supported by National Natural Science Foundation of China (NSFC 81872458), and Natural Science Foundation of Zhejiang Province (LY19H160017).
We appreciate the researchers and study participants for their contributions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
2. Wang F, Wang S, Zhou Q. The resistance mechanisms of lung cancer Immunotherapy. Front Oncol (2020) 10:568059. doi: 10.3389/fonc.2020.568059
3. Li T, Pan S, Gao S, Xiang W, Sun C, Cao W, et al. Diselenide-pemetrexed assemblies for combined cancer immuno-, radio-, and chemotherapies. Angewandte Chemie (International ed English) (2020) 59(7):2700–4. doi: 10.1002/anie.201914453
4. Herrera FG, Ronet C, Ochoa de Olza M, Barras D, Crespo I, Andreatta M, et al. Low-dose radiotherapy reverses tumor immune desertification and resistance to Immunotherapy. Cancer Discov (2022) 12(1):108–33. doi: 10.1158/2159-8290.Cd-21-0003
5. Qiu Q, Lin Y, Ma Y, Li X, Liang J, Chen Z, et al. Exploring the emerging role of the gut microbiota and tumor microenvironment in cancer Immunotherapy. Front Immunol (2020) 11:612202. doi: 10.3389/fimmu.2020.612202
6. Duan Q, Zhang H, Zheng J, Zhang L. Turning cold into hot: Firing up the tumor microenvironment. Trends Cancer (2020) 6(7):605–18. doi: 10.1016/j.trecan.2020.02.022
7. Wang Y, Xiang Y, Xin VW, Wang XW, Peng XC, Liu XQ, et al. Dendritic cell biology and its role in tumor Immunotherapy. J Hematol Oncol (2020) 13(1):107. doi: 10.1186/s13045-020-00939-6
8. Robins E, Zheng M, Ni Q, Liu S, Liang C, Zhang B, et al. Conversion of effector CD4+ T cells to a CD8+ MHC II-recognizing lineage. Cell Mol Immunol (2021) 18(1):150–61. doi: 10.1038/s41423-019-0347-5
9. Huntington ND, Cursons J, Rautela J. The cancer-natural killer cell immunity cycle. Nat Rev Cancer (2020) 20(8):437–54. doi: 10.1038/s41568-020-0272-z
10. Ostrand-Rosenberg S, Beury DW, Parker KH, Horn LA. Survival of the fittest: how myeloid-derived suppressor cells survive in the inhospitable tumor microenvironment. Cancer Immunol Immunother (2020) 69(2):215–21. doi: 10.1007/s00262-019-02388-8
11. Dees S, Ganesan R, Singh S, Grewal IS. Regulatory T cell targeting in cancer: Emerging strategies in Immunotherapy. Eur J Immunol (2021) 51(2):280–91. doi: 10.1002/eji.202048992
12. Lopez-Yrigoyen M, Cassetta L, Pollard JW. Macrophage targeting in cancer. Ann New York Acad Sci (2021) 1499(1):18–41. doi: 10.1111/nyas.14377
13. Sadeghi Rad H, Monkman J, Warkiani ME, Ladwa R, O’Byrne K, Rezaei N, et al. Understanding the tumor microenvironment for effective Immunotherapy. Med Res Rev (2021) 41(3):1474–98. doi: 10.1002/med.21765
14. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov (2019) 18(3):197–218. doi: 10.1038/s41573-018-0007-y
15. Ollauri-Ibáñez C, Ayuso-Íñigo B, Pericacho M. Hot and cold tumors: Is endoglin (CD105) a potential target for vessel normalization? Cancers (2021) 13(7):1552. doi: 10.3390/cancers13071552
16. Zada S, Hwang JS, Ahmed M, Lai TH, Pham TM, Elashkar O, et al. Cross talk between autophagy and oncogenic signaling pathways and implications for cancer therapy. Biochim Biophys Acta Rev Cancer (2021) 1876(1):188565. doi: 10.1016/j.bbcan.2021.188565
17. Mohsen S, Sobash PT, Algwaiz GF, Nasef N, Al-Zeidaneen SA, Karim NA. Autophagy agents in clinical trials for cancer therapy: A brief review. Curr Oncol (2022) 29(3):1695–708. doi: 10.3390/curroncol29030141
18. Luo X, Qiu Y, Dinesh P, Gong W, Jiang L, Feng X, et al. The functions of autophagy at the tumour-immune interface. J Cell Mol Med (2021) 25(5):2333–41. doi: 10.1111/jcmm.16331
19. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov (2020) 19(3):185–99. doi: 10.1038/s41573-019-0051-2
20. Kubli SP, Berger T, Araujo DV, Siu LL, Mak TW. Beyond immune checkpoint blockade: emerging immunological strategies. Nat Rev Drug Discov (2021) 20(12):899–919. doi: 10.1038/s41573-021-00155-y
21. Donlon NE, Power R, Hayes C, Reynolds JV, Lysaght J. Radiotherapy, Immunotherapy, and the tumour microenvironment: Turning an immunosuppressive milieu into a therapeutic opportunity. Cancer Lett (2021) 502:84–96. doi: 10.1016/j.canlet.2020.12.045
22. Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer (2019) 18(1):1–15. doi: 10.1186/s12943-019-1089-9
23. Zahedi-Amiri A, Malone K, Beug ST, Alain T, Yeganeh B. Autophagy in tumor immunity and viral-based immunotherapeutic approaches in cancer. Cells (2021) 10(10):2672. doi: 10.3390/cells10102672
24. Lin W, Wu S, Chen X, Ye Y, Weng Y, Pan Y, et al. Characterization of hypoxia signature to evaluate the tumor immune microenvironment and predict prognosis in glioma groups. Front Oncol (2020) 10:796. doi: 10.3389/fonc.2020.00796
25. Morfoisse F, Kuchnio A, Frainay C, Gomez-Brouchet A, Delisle M-B, Marzi S, et al. Hypoxia induces VEGF-c expression in metastatic tumor cells via a HIF-1α-independent translation-mediated mechanism. Cell Rep (2014) 6(1):155–67. doi: 10.1016/j.celrep.2013.12.011
26. Verhoeven J, Baelen J, Agrawal M, Agostinis P. Endothelial cell autophagy in homeostasis and cancer. FEBS Lett (2021) 595(11):1497–511. doi: 10.1002/1873-3468.14087
27. Townsend KN, Spowart JE, Huwait H, Eshragh S, West NR, Elrick MA, et al. Markers of T cell infiltration and function associate with favorable outcome in vascularized high-grade serous ovarian carcinoma. PloS One (2013) 8(12):e82406. doi: 10.1371/journal.pone.0082406
28. Tada Y, Togashi Y, Kotani D, Kuwata T, Sato E, Kawazoe A, et al. Targeting VEGFR2 with ramucirumab strongly impacts effector/activated regulatory T cells and CD8(+) T cells in the tumor microenvironment. J Immunotherapy Cancer (2018) 6(1):106. doi: 10.1186/s40425-018-0403-1
29. Guo H, Zhang T, Yu Y, Xu F. Cancer physical hallmarks as new targets for improved Immunotherapy. Trends Cell Biol (2021) 31(7):520–4. doi: 10.1016/j.tcb.2021.03.011
30. Chandra Jena B, Sarkar S, Rout L, Mandal M. The transformation of cancer-associated fibroblasts: Current perspectives on the role of TGF-β in CAF mediated tumor progression and therapeutic resistance. Cancer Lett (2021) 520:222–32. doi: 10.1016/j.canlet.2021.08.002
31. Desbois M, Wang Y. Cancer-associated fibroblasts: Key players in shaping the tumor immune microenvironment. Immunol Rev (2021) 302(1):241–58. doi: 10.1111/imr.12982
32. Ying L, Yazdani M, Koya R, Zhao R. Engineering tumor stromal mechanics for improved T cell therapy. Biochim Biophys Acta Gen Subj (2022) 1866(4):130095. doi: 10.1016/j.bbagen.2022.130095
33. Ma R, Yu D, Peng Y, Yi H, Wang Y, Cheng T, et al. Resveratrol induces AMPK and mTOR signaling inhibition-mediated autophagy and apoptosis in multiple myeloma cells. Acta Biochim Biophys Sin (2021) 53(6):775–83. doi: 10.1093/abbs/gmab042
34. Bustos SO, Leal Santos N, Chammas R, Andrade L. Secretory autophagy forges a therapy resistant microenvironment in melanoma. Cancers (2022) 14(1):234. doi: 10.3390/cancers14010234
35. Tzeng HT, Yang JL, Tseng YJ, Lee CH, Chen WJ, Chyuan IT. Plasminogen activator inhibitor-1 secretion by autophagy contributes to melanoma resistance to chemotherapy through tumor microenvironment modulation. Cancers (2021) 13(6):1253. doi: 10.3390/cancers13061253
36. New J, Thomas SM. Autophagy-dependent secretion: mechanism, factors secreted, and disease implications. Autophagy (2019) 15(10):1682–93. doi: 10.1080/15548627.2019.1596479
37. Kim YH, Kwak MS, Lee B, Shin JM, Aum S, Park IH, et al. Secretory autophagy machinery and vesicular trafficking are involved in HMGB1 secretion. Autophagy (2021) 17(9):2345–62. doi: 10.1080/15548627.2020.1826690
38. Jiang T, Chen X, Ren X, Yang JM, Cheng Y. Emerging role of autophagy in anti-tumor immunity: Implications for the modulation of Immunotherapy resistance. Drug Resist Update (2021) 56:100752. doi: 10.1016/j.drup.2021.100752
39. Qureshi-Baig K, Kuhn D, Viry E, Pozdeev VI, Schmitz M, Rodriguez F, et al. Hypoxia-induced autophagy drives colorectal cancer initiation and progression by activating the PRKC/PKC-EZR (ezrin) pathway. Autophagy (2020) 16(8):1436–52. doi: 10.1080/15548627.2019.1687213
40. He Z, Cai K, Zeng Z, Lei S, Cao W, Li X. Autophagy-associated circRNA circATG7 facilitates autophagy and promotes pancreatic cancer progression. Cell Death Dis (2022) 13(3):233. doi: 10.1038/s41419-022-04677-0
41. Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol (2009) 29(10):2570–81. doi: 10.1128/mcb.00166-09
42. Dowdell AS, Cartwright IM, Goldberg MS, Kostelecky R, Ross T, Welch N, et al. The HIF target ATG9A is essential for epithelial barrier function and tight junction biogenesis. Mol Biol Cell (2020) 31(20):2249–58. doi: 10.1091/mbc.E20-05-0291
43. Wu J, Lei Z, Yu J. Hypoxia induces autophagy in human vascular endothelial cells in a hypoxia-inducible factor 1−dependent manner. Mol Med Rep (2015) 11(4):2677–82. doi: 10.3892/mmr.2014.3093
44. Lee YW, Cherng YG, Yang ST, Liu SH, Chen TL, Chen RM. Hypoxia induced by cobalt chloride triggers autophagic apoptosis of human and mouse drug-resistant glioblastoma cells through targeting the PI3K-AKT-mTOR signaling pathway. Oxid Med Cell Longevity (2021) 2021:5558618. doi: 10.1155/2021/5558618
45. Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ (2008) 15(10):1572–81. doi: 10.1038/cdd.2008.84
46. Gao L, Dou ZC, Ren WH, Li SM, Liang X, Zhi KQ. CircCDR1as upregulates autophagy under hypoxia to promote tumor cell survival via AKT/ERK(½)/mTOR signaling pathways in oral squamous cell carcinomas. Cell Death Dis (2019) 10(10):745. doi: 10.1038/s41419-019-1971-9
47. Messai Y, Noman MZ, Janji B, Hasmim M, Escudier B, Chouaib S. The autophagy sensor ITPR1 protects renal carcinoma cells from NK-mediated killing. Autophagy (2015). doi: 10.1080/15548627.2015.1017194
48. Noman MZ, Buart S, Van Pelt J, Richon C, Hasmim M, Leleu N, et al. The cooperative induction of hypoxia-inducible factor-1α and STAT3 during hypoxia induced an impairment of tumor susceptibility to CTL-mediated cell lysis. J Immunol (2009) 182(6):3510–21. doi: 10.4049/jimmunol.0800854
49. Keulers TG, Koch A, van Gisbergen MW, Barbeau LMO, Zonneveld MI, de Jong MC, et al. ATG12 deficiency results in intracellular glutamine depletion, abrogation of tumor hypoxia and a favorable prognosis in cancer. Autophagy (2022) 18(8):1898–914. doi: 10.1080/15548627.2021.2008690
50. DePavia A, Jonasch E, Liu XD. Autophagy degrades hypoxia inducible factors. Mol Cell Oncol (2016) 3(2):e1104428. doi: 10.1080/23723556.2015.1104428
51. Kang R, Zeh H, Lotze M, Tang D. The multifaceted effects of autophagy on the tumor microenvironment. Adv Exp Med Biol (2020) 1225:99–114. doi: 10.1007/978-3-030-35727-6_7
52. Yao Z, Yang Z, Chen F, Jiang Y, Fu C, Wang Y, et al. Autophagy is essential for the endothelial differentiation of breast cancer stem−like cells. Int J Mol Med (2020) 45(1):255–64. doi: 10.3892/ijmm.2019.4399
53. Chandra A, Rick J, Yagnik G, Aghi MK. Autophagy as a mechanism for anti-angiogenic therapy resistance. Semin Cancer Biol (2020) 66:75–88. doi: 10.1016/j.semcancer.2019.08.031
54. Dos Santos PK, Altei WF, Danilucci TM, Lino RLB, Pachane BC, Nunes ACC, et al. Alternagin-c (ALT-c), a disintegrin-like protein, attenuates alpha2beta1 integrin and VEGF receptor 2 signaling resulting in angiogenesis inhibition. Biochimie (2020) 174:144–58. doi: 10.1016/j.biochi.2020.04.023
55. Kumar S, Guru SK, Pathania AS, Kumar A, Bhushan S, Malik F. Autophagy triggered by magnolol derivative negatively regulates angiogenesis. Cell Death Dis (2013) 4(10):e889. doi: 10.1038/cddis.2013.399
56. Neill T, Chen CG, Buraschi S, Iozzo RV. Catabolic degradation of endothelial VEGFA via autophagy. J Biol Chem (2020) 295(18):6064–79. doi: 10.1074/jbc.RA120.012593
57. Schaaf MB, Houbaert D, Meçe O, To SK, Ganne M, Maes H, et al. Lysosomal pathways and autophagy distinctively control endothelial cell behavior to affect tumor vasculature. Front Oncol (2019) 9:171. doi: 10.3389/fonc.2019.00171
58. Andrea AE, Chiron A, Mallah S, Bessoles S, Sarrabayrouse G, Hacein-Bey-Abina S. Advances in CAR-T cell genetic engineering strategies to overcome hurdles in solid tumors treatment. Front Immunol (2022) 13:830292. doi: 10.3389/fimmu.2022.830292
59. Wu J, Zhao X, Sun Q, Jiang Y, Zhang W, Luo J, et al. Synergic effect of PD-1 blockade and endostar on the PI3K/AKT/mTOR-mediated autophagy and angiogenesis in Lewis lung carcinoma mouse model. BioMed Pharmacother (2020) 125:109746. doi: 10.1016/j.biopha.2019.109746
60. Kang JI, Kim DH, Sung KW, Shim SM, Cha-Molstad H, Soung NK, et al. p62-induced cancer-associated fibroblast activation via the Nrf2-ATF6 pathway promotes lung tumorigenesis. Cancers (2021) 13(4):864. doi: 10.3390/cancers13040864
61. Rudnick JA, Monkkonen T, Mar FA, Barnes JM, Starobinets H, Goldsmith J, et al. Autophagy in stromal fibroblasts promotes tumor desmoplasia and mammary tumorigenesis. Genes Dev (2021) 35(13-14):963–75. doi: 10.1101/gad.345629.120
62. Tan ML, Parkinson EK, Yap LF, Paterson IC. Autophagy is deregulated in cancer-associated fibroblasts from oral cancer and is stimulated during the induction of fibroblast senescence by TGF-β1. Sci Rep (2021) 11(1):584. doi: 10.1038/s41598-020-79789-8
63. Li Z, Fu W-J, Chen X-Q, Wang S, Deng R-S, Tang X-P, et al. Autophagy-based unconventional secretion of HMGB1 in glioblastoma promotes chemosensitivity to temozolomide through macrophage M1-like polarization. J Exp Clin Cancer Res (2022) 41(1):1–20. doi: 10.1186/s13046-022-02291-8
64. Cheung PF, Yang J, Fang R, Borgers A, Krengel K, Stoffel A, et al. Progranulin mediates immune evasion of pancreatic ductal adenocarcinoma through regulation of MHCI expression. Nat Commun (2022) 13(1):156. doi: 10.1038/s41467-021-27088-9
65. Yamamoto K, Venida A, Perera RM, Kimmelman AC. Selective autophagy of MHC-I promotes immune evasion of pancreatic cancer. Autophagy (2020) 16(8):1524–5. doi: 10.1080/15548627.2020.1769973
66. Zhu J, Li Y, Luo Y, Xu J, Liufu H, Tian Z, et al. A feedback loop formed by ATG7/Autophagy, FOXO3a/miR-145 and PD-L1 regulates stem-like properties and invasion in human bladder cancer. Cancers (2019) 11(3):349. doi: 10.3390/cancers11030349
67. Wang X, Wu WKK, Gao J, Li Z, Dong B, Lin X, et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J Exp Clin Cancer research: CR (2019) 38(1):140. doi: 10.1186/s13046-019-1148-5
68. Wang XR, Jiang ZB, Xu C, Meng WY, Liu P, Zhang YZ, et al. Andrographolide suppresses non-small-cell lung cancer progression through induction of autophagy and antitumor immune response. Pharmacol Res (2022) 179:106198. doi: 10.1016/j.phrs.2022.106198
69. Maher CM, Thomas JD, Haas DA, Longen CG, Oyer HM, Tong JY, et al. Small-molecule Sigma1 modulator induces autophagic degradation of PD-L1. Mol Cancer Res (2018) 16(2):243–55. doi: 10.1158/1541-7786.MCR-17-0166
70. Gao Z, Chen JF, Li XG, Shi YH, Tang Z, Liu WR, et al. KRAS acting through ERK signaling stabilizes PD-L1 via inhibiting autophagy pathway in intrahepatic cholangiocarcinoma. Cancer Cell Int (2022) 22(1):128. doi: 10.1186/s12935-022-02550-w
71. Okamoto T, Yeo SK, Hao M, Copley MR, Haas MA, Chen S, et al. FIP200 suppresses immune checkpoint therapy responses in breast cancers by limiting AZI2/TBK1/IRF signaling independent of its canonical autophagy function. Cancer Res (2020) 80(17):3580–92. doi: 10.1158/0008-5472.Can-20-0519
72. Ma F, Ding MG, Lei YY, Luo LH, Jiang S, Feng YH, et al. SKIL facilitates tumorigenesis and immune escape of NSCLC via upregulating TAZ/autophagy axis. Cell Death Dis (2020) 11(12):1028. doi: 10.1038/s41419-020-03200-7
73. Kim S, Cho H, Hong SO, Oh SJ, Lee HJ, Cho E, et al. LC3B upregulation by NANOG promotes immune resistance and stem-like property through hyperactivation of EGFR signaling in immune-refractory tumor cells. Autophagy (2021) 17(8):1978–97. doi: 10.1080/15548627.2020.1805214
74. Tittarelli A, Janji B, Van Moer K, Noman MZ, Chouaib S. The selective degradation of synaptic connexin 43 protein by hypoxia-induced autophagy impairs natural killer cell-mediated tumor cell killing. J Biol Chem (2015) 290(39):23670–9. doi: 10.1074/jbc.M115.651547
75. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme b degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci USA (2013) 110(43):17450–5. doi: 10.1073/pnas.1304790110
76. Messai Y, Noman MZ, Hasmim M, Janji B, Tittarelli A, Boutet M, et al. ITPR1 protects renal cancer cells against natural killer cells by inducing autophagy. Cancer Res (2014) 74(23):6820–32. doi: 10.1158/0008-5472.CAN-14-0303
77. Yao C, Ni Z, Gong C, Zhu X, Wang L, Xu Z, et al. Rocaglamide enhances NK cell-mediated killing of non-small cell lung cancer cells by inhibiting autophagy. Autophagy (2018) 14(10):1831–44. doi: 10.1080/15548627.2018.1489946
78. Noman MZ, Berchem G, Janji B. Targeting autophagy blocks melanoma growth by bringing natural killer cells to the tumor battlefield. Autophagy (2018) 14(4):730–2. doi: 10.1080/15548627.2018.1427398
79. Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab (2018) 28(1):87–103.e6. doi: 10.1016/j.cmet.2018.04.022
80. Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun (2014) 5:3056. doi: 10.1038/ncomms4056
81. Wen ZF, Liu H, Gao R, Zhou M, Ma J, Zhang Y, et al. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J Immunotherapy Cancer (2018) 6(1):151. doi: 10.1186/s40425-018-0452-5
82. Shiau D-J, Kuo W-T, Davuluri GVN, Shieh C-C, Tsai P-J, Chen C-C, et al. Hepatocellular carcinoma-derived high mobility group box 1 triggers M2 macrophage polarization via a TLR2/NOX2/autophagy axis. Sci Rep (2020) 10(1):1–14. doi: 10.1038/s41598-020-70137-4
83. Xu J, Zhang J, Zhang Z, Gao Z, Qi Y, Qiu W, et al. Hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction. Cell Death Dis (2021) 12(4):1–16. doi: 10.1038/s41419-021-03664-1
84. Jin Y, Pan Y, Zheng S, Liu Y, Xu J, Peng Y, et al. Inactivation of EGLN3 hydroxylase facilitates Erk3 degradation via autophagy and impedes lung cancer growth. Oncogene (2022) 41(12):1752–66. doi: 10.1038/s41388-022-02203-2
85. Davuluri GVN, Chen CC, Chiu YC, Tsai HW, Chiu HC, Chen YL, et al. Autophagy drives galectin-1 secretion from tumor-associated macrophages facilitating hepatocellular carcinoma progression. Front Cell Dev Biol (2021) 9:741820. doi: 10.3389/fcell.2021.741820
86. Tu CE, Hu Y, Zhou P, Guo X, Gu C, Zhang Y, et al. Lactate and TGF-β antagonistically regulate inflammasome activation in the tumor microenvironment. J Cell Physiol (2021) 236(6):4528–37. doi: 10.1002/jcp.30169
87. Wei J, Long L, Yang K, Guy C, Shrestha S, Chen Z, et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat Immunol (2016) 17(3):277–85. doi: 10.1038/ni.3365
88. Parker KH, Horn LA, Ostrand-Rosenberg S. High-mobility group box protein 1 promotes the survival of myeloid-derived suppressor cells by inducing autophagy. J leukocyte Biol (2016) 100(3):463–70. doi: 10.1189/jlb.3HI0715-305R
89. Alissafi T, Hatzioannou A, Mintzas K, Barouni RM, Banos A, Sormendi S, et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J Clin Invest (2018) 128(9):3840–52. doi: 10.1172/jci120888
90. Mohammadpour H, MacDonald CR, McCarthy PL, Abrams SI, Repasky EA. β2-adrenergic receptor signaling regulates metabolic pathways critical to myeloid-derived suppressor cell function within the TME. Cell Rep (2021) 37(4):109883. doi: 10.1016/j.celrep.2021.109883
91. Li Q, Zhong X, Yao W, Yu J, Wang C, Li Z, et al. Inhibitor of glutamine metabolism V9302 promotes ROS-induced autophagic degradation of B7H3 to enhance antitumor immunity. J Biol Chem (2022) 298(4):101753. doi: 10.1016/j.jbc.2022.101753
92. Wu Q, Tian AL, Li B, Leduc M, Forveille S, Hamley P, et al. IGF1 receptor inhibition amplifies the effects of cancer drugs by autophagy and immune-dependent mechanisms. J Immunotherapy Cancer (2021) 9(6):e002722. doi: 10.1136/jitc-2021-002722
93. Nishida M, Yamashita N, Ogawa T, Koseki K, Warabi E, Ohue T, et al. Mitochondrial reactive oxygen species trigger metformin-dependent antitumor immunity via activation of Nrf2/mTORC1/p62 axis in tumor-infiltrating CD8T lymphocytes. J Immunotherapy Cancer (2021) 9(9):e002954. doi: 10.1136/jitc-2021-002954
94. Deust A, Chobert M-N, Demontant V, Gricourt G, Denaës T, Thiolat A, et al. Macrophage autophagy protects against hepatocellular carcinogenesis in mice. Sci Rep (2021) 11(1):1–16. doi: 10.1038/s41598-021-98203-5
95. Hernández ÁP, Juanes-Velasco P, Landeira-Viñuela A, Bareke H, Montalvillo E, Góngora R, et al. Restoring the immunity in the tumor microenvironment: Insights into immunogenic cell death in onco-therapies. Cancers (2021) 13(11):2821. doi: 10.3390/cancers13112821
96. Gupta G, Borglum K, Chen H. Immunogenic cell death: A step ahead of autophagy in cancer therapy. J Cancer Immunol (2021) 3(1):47. doi: 10.33696/cancerimmunol.3.041
97. Wang Y, Martins I, Ma Y, Kepp O, Galluzzi L, Kroemer G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy (2013) 9(10):1624–5. doi: 10.4161/auto.25873
98. Fader CM, Aguilera MO, Colombo MI. ATP is released from autophagic vesicles to the extracellular space in a VAMP7-dependent manner. Autophagy (2012) 8(12):1741–56. doi: 10.4161/auto.21858
99. Zhan L, Zhang J, Zhang J, Liu X, Zhu S, Shi Y, et al. LC3 and NLRC5 interaction inhibits NLRC5-mediated MHC class I antigen presentation pathway in endometrial cancer. Cancer Lett (2022) 529:37–52. doi: 10.1016/j.canlet.2021.12.031
100. Li Y, Wang L-X, Pang P, Cui Z, Aung S, Haley D, et al. Tumor-derived autophagosome vaccine: mechanism of cross-presentation and therapeutic efficacy. Clin Cancer Res (2011) 17(22):7047–57. doi: 10.1158/1078-0432.CCR-11-0951
101. Xing Y, Cao R, Hu H. TLR and NLRP3 inflammasome-dependent innate immune responses to tumor-derived autophagosomes (DRibbles). Cell Death Dis (2016) 7(8):e2322–e. doi: 10.1038/cddis.2016.206
102. Patel JM, Cui Z, Wen ZF, Dinh CT, Hu HM. Peritumoral administration of DRibbles-pulsed antigen-presenting cells enhances the antitumor efficacy of anti-GITR and anti-PD-1 antibodies via an antigen presenting independent mechanism. J Immunotherapy Cancer (2019) 7(1):311. doi: 10.1186/s40425-019-0786-7
103. Loi M, Müller A, Steinbach K, Niven J, da Silva RB, Paul P, et al. Macroautophagy proteins control MHC class I levels on dendritic cells and shape anti-viral CD8+ T cell responses. Cell Rep (2016) 15(5):1076–87. doi: 10.1016/j.celrep.2016.04.002
104. Ho NI, Camps MGM, Verdoes M, Münz C, Ossendorp F. Autophagy regulates long-term cross-presentation by murine dendritic cells. Eur J Immunol (2021) 51(4):835–47. doi: 10.1002/eji.202048961
105. Parekh VV, Pabbisetty SK, Wu L, Sebzda E, Martinez J, Zhang J, et al. Autophagy-related protein Vps34 controls the homeostasis and function of antigen cross-presenting CD8α+ dendritic cells. Proc Natl Acad Sci (2017) 114(31):E6371–E80. doi: 10.1073/pnas.1706504114
106. Deng J, Thennavan A, Dolgalev I, Chen T, Li J, Marzio A, et al. ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1 mutant lung cancer. Nat Cancer (2021) 2(5):503–14. doi: 10.1038/s43018-021-00208-6
107. Labani-Motlagh A, Ashja-Mahdavi M, Loskog A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune responses. Front Immunol (2020) 11:940. doi: 10.3389/fimmu.2020.00940
108. Liu Y, Zhang H, Wang Z, Wu P, Gong W. 5-Hydroxytryptamine1a receptors on tumour cells induce immune evasion in lung adenocarcinoma patients with depression via autophagy/pSTAT3. Eur J Cancer (2019) 114:8–24. doi: 10.1016/j.ejca.2019.03.017
109. Zhang J, Zhou L, Xiang JD, Jin CS, Li MQ, He YY. Artesunate-induced ATG5-related autophagy enhances the cytotoxicity of NK92 cells on endometrial cancer cells via interactions between CD155 and CD226/TIGIT. Int Immunopharmacol (2021) 97:107705. doi: 10.1016/j.intimp.2021.107705
110. Zhang X, Fan J, Wang S, Li Y, Wang Y, Li S, et al. Targeting CD47 and autophagy elicited enhanced antitumor effects in non-small cell lung cancer. Cancer Immunol Res (2017) 5(5):363–75. doi: 10.1158/2326-6066.CIR-16-0398
111. Zhang X, Chen W, Fan J, Wang S, Xian Z, Luan J, et al. Disrupting CD47-SIRPalpha axis alone or combined with autophagy depletion for the therapy of glioblastoma. Carcinogenesis (2018) 39(5):689–99. doi: 10.1093/carcin/bgy041
112. Xu C, Zang Y, Zhao Y, Cui W, Zhang H, Zhu Y, et al. Comprehensive pan-cancer analysis confirmed that ATG5 promoted the maintenance of tumor metabolism and the occurrence of tumor immune escape. Front Oncol (2021) 11:652211. doi: 10.3389/fonc.2021.652211
113. Li ZL, Zhang HL, Huang Y, Huang JH, Sun P, Zhou NN, et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-c degradation. Nat Commun (2020) 11(1):3806. doi: 10.1038/s41467-020-17395-y
114. Lee YH, Martin-Orozco N, Zheng P, Li J, Zhang P, Tan H, et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res (2017) 27(8):1034–45. doi: 10.1038/cr.2017.90
115. Jia W, He MX, McLeod IX, Guo J, Ji D, He YW. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy (2015) 11(12):2335–45. doi: 10.1080/15548627.2015.1110666
116. DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, et al. Autophagy regulation of metabolism is required for CD8(+) T cell anti-tumor immunity. Cell Rep (2019) 27(2):502–13.e5. doi: 10.1016/j.celrep.2019.03.037
117. O’Sullivan TE, Johnson LR, Kang HH, Sun JC. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity (2015) 43(2):331–42. doi: 10.1016/j.immuni.2015.07.012
118. Hofmann F, Navarrete M, Alvarez J, Guerrero I, Gleisner MA, Tittarelli A, et al. Cx43-gap junctions accumulate at the cytotoxic immunological synapse enabling cytotoxic T lymphocyte melanoma cell killing. Int J Mol Sci (2019) 20(18):4509. doi: 10.3390/ijms20184509
119. Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, et al. Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci USA (2017) 114(44):E9271–e9. doi: 10.1073/pnas.1703921114
120. Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem (2009) 284(49):34342–54. doi: 10.1074/jbc.M109.042671
121. Jacquel A, Obba S, Boyer L, Dufies M, Robert G, Gounon P, et al. Autophagy is required for CSF-1-induced macrophagic differentiation and acquisition of phagocytic functions. Blood (2012) 119(19):4527–31. doi: 10.1182/blood-2011-11-392167
122. Yang SL, Tan HX, Niu TT, Liu YK, Gu CJ, Li DJ, et al. The IFN-γ-IDO1-kynureine pathway-induced autophagy in cervical cancer cell promotes phagocytosis of macrophage. Int J Biol Sci (2021) 17(1):339–52. doi: 10.7150/ijbs.51241
123. Becher J, Simula L, Volpe E, Procaccini C, La Rocca C, D’Acunzo P, et al. AMBRA1 controls regulatory T-cell differentiation and homeostasis upstream of the FOXO3-FOXP3 axis. Dev Cell (2018) 47(5):592–607.e6. doi: 10.1016/j.devcel.2018.11.010
124. Chaeichi-Tehrani N, Ferns GA, Hassanian SM, Khazaei M, Avan A. The therapeutic potential of targeting autophagy in the treatment of cancer. Curr Cancer Drug Targets (2021) 21(9):725–36. doi: 10.2174/1568009621666210601113144
125. Wang Z, Wang X, Cheng F, Wen X, Feng S, Yu F, et al. Rapamycin inhibits glioma cells growth and promotes autophagy by miR-26a-5p/DAPK1 axis. Cancer Manage Res (2021) 13:2691–700. doi: 10.2147/cmar.S298468
126. Ren H, Bakas NA, Vamos M, Chaikuad A, Limpert AS, Wimer CD, et al. Design, synthesis, and characterization of an orally active dual-specific ULK1/2 autophagy inhibitor that synergizes with the PARP inhibitor olaparib for the treatment of triple-negative breast cancer. J Medicinal Chem (2020) 63(23):14609–25. doi: 10.1021/acs.jmedchem.0c00873
127. Kocaturk NM, Akkoc Y, Kig C, Bayraktar O, Gozuacik D, Kutlu O. Autophagy as a molecular target for cancer treatment. Eur J Pharm Sci (2019) 134:116–37. doi: 10.1016/j.ejps.2019.04.011
128. Tan S, Peng X, Peng W, Zhao Y, Wei Y. Enhancement of oxaliplatin-induced cell apoptosis and tumor suppression by 3-methyladenine in colon cancer. Oncol Lett (2015) 9(5):2056–62. doi: 10.3892/ol.2015.2996
129. Chen M, Yang D, Sun Y, Liu T, Wang W, Fu J, et al. In situ self-assembly nanomicelle microneedles for enhanced photoImmunotherapy via autophagy regulation strategy. ACS Nano (2021) 15(2):3387–401. doi: 10.1021/acsnano.0c10396
130. Wang W, Liu L, Zhou Y, Ye Q, Yang X, Jiang J, et al. Hydroxychloroquine enhances the antitumor effects of BC001 in gastric cancer. Int J Oncol (2019) 55(2):405–14. doi: 10.3892/ijo.2019.4824
131. Brun S, Bestion E, Raymond E, Bassissi F, Jilkova ZM, Mezouar S, et al. GNS561, a clinical-stage PPT1 inhibitor, is efficient against hepatocellular carcinoma via modulation of lysosomal functions. Autophagy (2022) 18(3):678–94. doi: 10.1080/15548627.2021.1988357
132. Yoldi G, Munoz-Guardiola P, Megias E, Perez-Montoyo H, Yeste-Velasco M, Alfon J, et al. ABTL0812, a phase 2 clinical stage pro-autophagy anticancer drug exhibits immunomodulatory effects that modify tumor microenvironment in pancreatic cancer models. Cancer Res (2021) 81(13_Supplement):1805–.
133. Zhou W, Wang H, Yang Y, Chen Z-S, Zou C, Zhang J. Chloroquine against malaria, cancers and viral diseases. Drug Discovery Today (2020) 25(11):2012–22. doi: 10.1016/j.drudis.2020.09.010
134. Vidal L, Victoria I, Gaba L, Martín MG, Brunet M, Colom H, et al. A first-in-human phase I/Ib dose-escalation clinical trial of the autophagy inducer ABTL0812 in patients with advanced solid tumours. Eur J Cancer (Oxford Engl (2021) 1990):146. doi: 10.1016/j.ejca.2020.12.019
135. Yuan J, Yuan X, Wu K, Gao J. ‘A local and low-dose Chemotherapy/Autophagy-enhancing regimen treatment markedly inhibited the growth of established solid tumors through a systemic antitumor immune response. Front Oncol (2021) 11:658254. doi: 10.3389/fonc.2021.658254
136. Ishimwe N, Wei P, Wang M, Zhang H, Wang L, Jing M, et al. Autophagy impairment through lysosome dysfunction by brucine induces immunogenic cell death (ICD). Am J Chin Med (2020) 48(8):1915–40. doi: 10.1142/S0192415X20500962
137. Li DD, Xie B, Wu XJ, Li JJ, Ding Y, Wen XZ, et al. Late-stage inhibition of autophagy enhances calreticulin surface exposure. Oncotarget (2016) 7(49):80842–54. doi: 10.18632/oncotarget.13099
138. Li J, Cai W, Yu J, Zhou S, Li X, He Z, et al. Autophagy inhibition recovers deficient ICD-based cancer Immunotherapy. Biomaterials (2022) 287:121651. doi: 10.1016/j.biomaterials.2022.121651
139. Sharma G, Ojha R, Noguera-Ortega E, Rebecca VW, Attanasio J, Liu S, et al. PPT1 inhibition enhances the antitumor activity of anti-PD-1 antibody in melanoma. JCI Insight (2020) 5(17):e133225. doi: 10.1172/jci.insight.133225
140. Schulz D, Severin Y, Zanotelli VRT, Bodenmiller B. In-depth characterization of monocyte-derived macrophages using a mass cytometry-based phagocytosis assay. Sci Rep (2019) 9(1):1925. doi: 10.1038/s41598-018-38127-9
141. Hsu SPC, Chen YC, Chiang HC, Huang YC, Huang CC, Wang HE, et al. Rapamycin and hydroxychloroquine combination alters macrophage polarization and sensitizes glioblastoma to immune checkpoint inhibitors. J Neuro-Oncol (2020) 146(3):417–26. doi: 10.1007/s11060-019-03360-3
142. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature (2020) 581(7806):100–5. doi: 10.1038/s41586-020-2229-5
143. Janji B, Hasmim M, Parpal S, Berchem G, Noman MZ. Firing up the cold tumors by targeting Vps34. Oncoimmunology (2020) 9(1):1809936. doi: 10.1080/2162402x.2020.1809936
144. Qiao Y, Choi JE, Tien JC, Simko SA, Rajendiran T, Vo JN, et al. Autophagy inhibition by targeting PIKfyve potentiates response to immune checkpoint blockade in prostate cancer. Nat Cancer (2021) 2(9):978–93. doi: 10.1038/s43018-021-00237-1
145. Noman MZ, Parpal S, Van Moer K, Xiao M, Yu Y, Viklund J, et al. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 Immunotherapy. Sci Adv (2020) 6(18):eaax7881. doi: 10.1126/sciadv.aax7881
146. Limagne E, Nuttin L, Thibaudin M, Jacquin E, Aucagne R, Bon M, et al. MEK inhibition overcomes chemoImmunotherapy resistance by inducing CXCL10 in cancer cells. Cancer Cell (2022) 40(2):136–52 e12. doi: 10.1016/j.ccell.2021.12.009
147. Watanabe N, McKenna MK, Rosewell Shaw A, Suzuki M. Clinical CAR-T cell and oncolytic virotherapy for cancer treatment. Mol therapy: J Am Soc Gene Ther (2021) 29(2):505–20. doi: 10.1016/j.ymthe.2020.10.023
148. Liang J, Fang D, Heimberger AB. Chimeric antigen receptor (CAR) T cell donor dependent dysfunction modulation with activation of autophagy and inhibition of trogocytosis. Cancer Res (2021) 81(13_Supplement):1492.
149. Wang J, Toregrosa-Allen S, Elzey BD, Utturkar S, Lanman NA, Bernal-Crespo V, et al. Multispecific targeting of glioblastoma with tumor microenvironment-responsive multifunctional engineered NK cells. Proc Natl Acad Sci USA (2021) 118(45):e2107507118. doi: 10.1073/pnas.2107507118
150. Fan J, Wu Y, Jiang M, Wang L, Yin D, Zhang Y, et al. IFN-DC loaded with autophagosomes containing virus antigen is highly efficient in inducing virus-specific human T cells. Int J Med Sci (2019) 16(5):741–50. doi: 10.7150/ijms.31830
151. Jagannath C, Khan A, Wang J. Increased immunogenicity through autophagy. Immunology: Elsevier (2018) p:35–54.
152. Page DB, Hulett TW, Hilton TL, Hu HM, Urba WJ, Fox BA. Glimpse into the future: harnessing autophagy to promote anti-tumor immunity with the DRibbles vaccine. J Immunotherapy Cancer (2016) 4:25. doi: 10.1186/s40425-016-0130-4

Keywords: autophagy, tumor microenvironment, immune cells, immunogenic cell death, antigen presentation, immunotherapy
Citation: Jin Z, Sun X, Wang Y, Zhou C, Yang H and Zhou S (2022) Regulation of autophagy fires up the cold tumor microenvironment to improve cancer immunotherapy. Front. Immunol. 13:1018903. doi: 10.3389/fimmu.2022.1018903
Received: 14 August 2022; Accepted: 27 September 2022;
Published: 10 October 2022.
Edited by:
Jin Wang, Houston Methodist Research Institute, United StatesReviewed by:
Danfeng Guo, First Affiliated Hospital of Zhengzhou University, ChinaCopyright © 2022 Jin, Sun, Wang, Zhou, Yang and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suna Zhou, YW5ueXpob3UwOTEzQDE2My5jb20=; HaihuaYang, eWhoOTMxODFAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.