 Sebastian Wurster
Sebastian Wurster Stephanie S. Watowich
Stephanie S. Watowich Dimitrios P. Kontoyiannis
Dimitrios P. Kontoyiannis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 25 October 2022
Sec. Microbial Immunology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1018202
This article is part of the Research TopicImmune intervention avenues to control fungal infections in immunocompromised individualsView all 5 articles
Opportunistic fungal infections have high mortality in patients with severe immune dysfunction. Growing evidence suggests that the immune environment of invasive fungal infections and cancers share common features of immune cell exhaustion through activation of immune checkpoint pathways. This observation gave rise to several preclinical studies and clinical case reports describing blockade of the Programmed Cell Death Protein 1 and Cytotoxic T-Lymphocyte Antigen 4 immune checkpoint pathways as an adjunct immune enhancement strategy to treat opportunistic fungal infections. The first part of this review summarizes the emerging evidence for contributions of checkpoint pathways to the immunopathology of fungal sepsis, opportunistic mold infections, and dimorphic fungal infections. We then review the potential merits of immune checkpoint inhibitors (ICIs) as an antifungal immunotherapy, including the incomplete knowledge of the mechanisms involved in both immuno-protective effects and toxicities. In the second part of this review, we discuss the limitations of the current evidence and the many unknowns about ICIs as an antifungal immune enhancement strategy. Based on these gaps of knowledge and lessons learned from cancer immunology studies, we outline a research agenda to determine a “sweet spot” for ICIs in medical mycology. We specifically discuss the importance of more nuanced animal models, the need to study ICI-based combination therapy, potential ICI resistance, the role of the immune microenvironment, and the impact of ICIs given as part of oncological therapies on the natural immunity to various pathogenic fungi.
Conventional antifungal therapy for the treatment of invasive fungal infections (IFIs) remains challenging due to a limited number of available drugs, considerable toxicities, drug-drug interactions, and the increasing global spread of resistant pathogens such as azole-resistant Aspergillus fumigatus and echinocandin-resistant Candida species (1, 2). Investigational antifungal agents currently studied in phase 2 or 3 trials continue to have major gaps in their therapeutic spectrum, especially limited activity against Mucorales and Fusarium species (3). Furthermore, despite improved clinical management and the introduction of new antifungal agents, outcomes of IFIs remain dismal in patients with profound immune dysfunction, e.g., patients with persistent neutropenia or refractory leukemia (4, 5). Therefore, the development of facile immune enhancement strategies to potentiate the efficacy of conventional antifungals remains an important pursuit (6).
Over the past 20 years, many groups designed cellular immune therapeutics to restore and augment antifungal immunity, including adoptive T cell transfer, chimeric antigen receptor T cells, ex-vivo-pulsed dendritic cells (DCs), and azole-loaded neutrophils (7–12). However, despite a multitude of promising preclinical data, only few of these approaches eventually entered small-scale clinical studies, which is not surprising, as cellular immune therapeutics are costly, difficult to scale, time- and labor-intensive, logistically challenging, and subject to considerable regulatory hurdles (11, 12). These obstacles for clinical translation of cellular immune therapeutics have reinvigorated the interest in non-cellular immune enhancement strategies to bolster host defense against opportunistic pathogens (12). For instance, recombinant interferon-gamma (IFN-γ), a cytokine that is considered a pivotal driver of protective antifungal immunity, has been studied as an adjunct immune-stimulatory therapy in patients with IFIs (13). Furthermore, recombinant hematopoietic growth factors (GM-CSF and G-CSF) were shown to improve fungicidal immunity in mouse models (14, 15), are commonly used in the management of congenital and acquired neutropenia (16), and have been studied as an adjunct immunotherapeutic approach in patients with IFIs (17, 18).
Increasing evidence pointing to similarities in the immune pathogenesis of cancers and infections has led to the exploration of existing oncological immune therapeutics, such as immune checkpoint inhibitors (ICIs), as adjunct treatments in infectious diseases (19, 20). As extensively reviewed elsewhere, ICIs were studied in case series or early-stage clinical trials in patients with HIV infection, hepatitis C, progressive multifocal leukoencephalopathy, and bacterial sepsis (19). Furthermore, dozens of preclinical studies have suggested that ICIs could become a facile and effective adjunct immunotherapeutic approach in a broad spectrum of infectious diseases including IFIs (19).
The first part of this review (section 2) examines the evidence from in-vitro studies, animal research, and clinical case reports supporting a role of checkpoint pathways as potential therapeutic targets in fungal sepsis, opportunistic mold infections, and dimorphic fungal infections. In the second part (section 3), we discuss the many unknowns about antifungal ICI therapy and outline a research agenda to better understand the “sweet spot” for ICIs in medical mycology based on lessons learned from immuno-oncology studies.
Activation of T cells by antigen-presenting cells (APCs) and non-lymphoid cells (i.e., infected tissues and tumor cells) is tightly controlled by a balance of co-stimulatory and co-inhibitory signals (Figure 1A). Immune checkpoint pathways, that is, co-inhibitory pathways expressed as feedback loops after immune activation or in response to chronic antigen exposure and inflammation, are crucial to induce tolerance to autoantigens and thereby limit autoimmunity (21). However, checkpoint pathways also promote immune exhaustion in scenarios of excessive or chronic antigen exposure due to inflammation, infection, or cancer (21). Although the exact definition of immune exhaustion is debated, the term commonly refers to a hypofunctional state of immune cells, especially T cells, characterized by reduced cytokine production, limited proliferation, epigenetic and metabolic changes, and upregulation of inhibitory receptors (22).
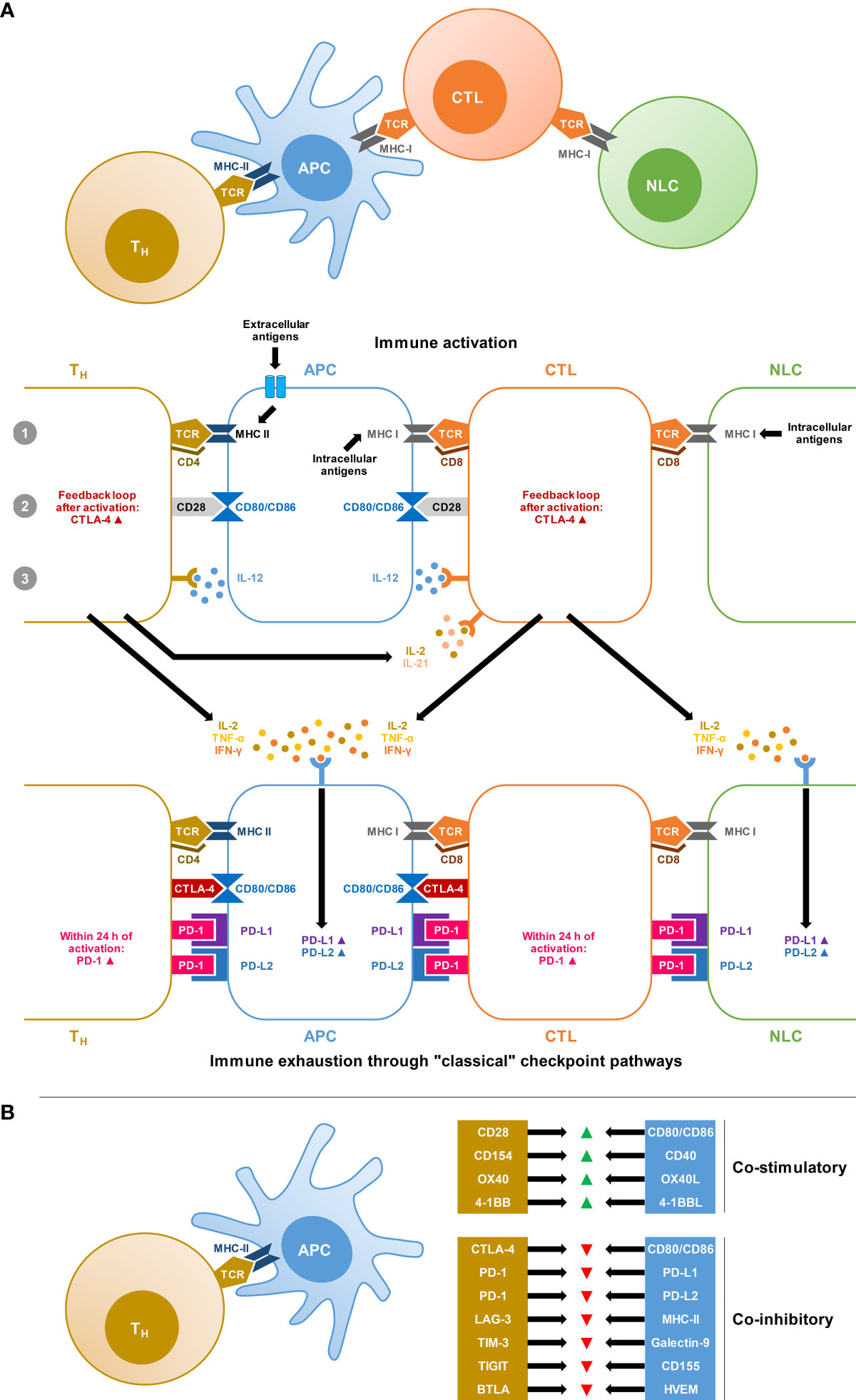
Figure 1 Co-stimulatory and co-inhibitory signals involved in T-cell activation and immune exhaustion. (A) Simplified schematic of the “3-signal concept” of T-cell activation by professional antigen-presenting cells (APCs) and non-lymphatic cells (NLCs), i.e., infected tissue or tumor cells. Signal 1: Antigen-loaded Major Histocompatibility Complex (MHC) is recognized by T cells with a corresponding T-cell receptor (TCR) and the interaction is stabilized by CD4 expressed on T-helper (Th) cells or CD8 expressed on cytotoxic T lymphocytes (CTLs). Signal 2: T-cell-expressed CD28 interacts with the B7 complex (CD80 and CD86) on APCs. Signal 3: Additional cytokine signals, especially interleukin-12 (IL-12) produced by APCs are required for full stimulation. This system is equipped with negative feedback loops, resulting in the upregulation of Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) on T cells, which outcompetes the interaction of CD28 with the B7 complex. Furthermore, chronic antigen exposure and cytokine signals, especially interferon gamma (IFN-γ) stimulate the expression of the Programmed Death Ligands PD-L1 and PD-L2 on APCs and NLCs. These ligands interact with Programmed Cell Death Protein 1 (PD-1), which is upregulated within 24 hours of T-cell activation and initiates inhibitory signaling cascades after ligand binding. In reality, this system is highly complex and further tine-tuned by additional co-stimulatory and co-inhibitory signals and the immune environment. (B) Selection of co-stimulatory and co-inhibitory signals in the T-cell/APC interplay undergoing preclinical and/or clinical evaluation as potential targets for oncological immunotherapies. Abbreviations: BTLA, B and T lymphocyte Attenuator; CD, cluster of differentiation; HVEM, Herpes Virus Entry Mediator; LAG-3, Lymphocyte-Activation Gene 3; TIGIT, T Cell Immunoreceptor With Immunoglobulin and ITIM Domain; TIM-3, T cell Immunoglobulin and Mucin Domain-Containing Protein 3.
The two major classical checkpoint pathways associated with immune exhaustion are the Cytotoxic T-Lymphocyte Antigen 4 (CTLA-4) pathway and the Programmed Cell Death Protein 1 (PD-1) pathway (21). The CTLA-4 pathway is predominantly involved in the activation of naïve T-cells at the priming stage (21, 23). The most potent co-stimulatory signal for naïve T-cell activation is the interaction of the T-cellular CD28 surface protein with CD80 and CD86 on APCs. To prevent overzealous activation and autoreactivity, the CD28 pathway is equipped with a negative feedback loop through the expression and exocytosis of CTLA-4, which then outcompetes the interaction of CD28 with CD80/CD86 due to its greater affinity, resulting in inhibitory signals (21, 23) (Figure 1A).
PD-1 is more broadly expressed on T cells, B cells, natural killer (NK) cells, and mononuclear phagocytes, and is rapidly activated following T-cell receptor (TCR) stimulation (21, 24). PD-1 signaling inhibits T-cell survival and proliferation, suppresses the release of T-cell effector cytokines, and interferes with TCR signaling (21). PD-1 interacts with two main ligands, PD-L1 and PD-L2, that are widely expressed on professional APCs as well as nonlymphoid cells and tissues (Figure 1A). IFN-γ, secreted by activated T cells, is considered the most potent stimulus of PD-L1 upregulation (21, 24).
Besides these classical immune checkpoints, additional molecules with co-inhibitory functions in the interplay between T cells and innate immune cells or non-lymphoid tissues are increasingly studied (25) (Figure 1B). The best-characterized emerging checkpoint molecules are Lymphocyte-Activation Gene 3 (LAG-3), T cell Immunoglobulin and Mucin Domain-Containing Protein 3 (TIM-3), and T Cell Immunoreceptor With Immunoglobulin and ITIM Domain (TIGIT) (25).
Several studies documented the activation of these immune checkpoint pathways during invasive yeast infections (19). After initial hyperinflammation, sepsis patients often experience a profound anti-inflammatory response with functional impairment of lymphocytes and phagocytes (26–30). For instance, circulating T cells and NK cells from patients and mice with Candida sepsis displayed increased expression of immunosuppressive signals, especially PD-1 and PD-L1, and concomitant downregulation of co-stimulatory molecules (31, 32). This sustained immunoinhibitory response contributes to poor outcomes despite appropriate antifungal therapy, including increased risk of secondary infections, and long-lasting immune impairment in survivors (28–30). A recent study expanded these findings by showing strong co-induction of PD-1, LAG-3, and TIM-3 on peripheral blood mononuclear cells (PBMCs) from patients with invasive C. albicans infection (29). Notably, the immune environment in patients with invasive candidiasis (IC) partially resembled patterns of immune dysfunction and exhaustion seen in tumor-infiltrating lymphocytes (29). Among the upregulated checkpoint markers, PD-1 showed the strongest association with poor mortality outcomes in IC patients (29). In-vitro blockade of PD-1 on patient cells enhanced C. albicans-induced Th-cell effector responses, especially the production of the type 1 T-helper cell (Th1) signature cytokine IFN-γ (29). Single-cell RNA sequencing after stimulation of PBMCs from healthy donors with C. albicans antigens further corroborated the co-induction of the PD-1 axis, CTLA-4, LAG-3, and other co-inhibitory molecules on Th cells, cytotoxic T lymphocytes (CTLs), and mononuclear phagocytes (33).
Checkpoint activation in non-albicans Candida sepsis was demonstrated by Spec and colleagues who assayed co-stimulatory and inhibitory signals on circulating T cells from patients with bloodstream infection (BSI) due to various Candida species (28). Compared with non-septic critically ill patients, Th cells and CTLs from patients with Candida BSIs showed significantly upregulated PD-1 expression, whereas CD28 was suppressed (28). Furthermore, our recent pilot study of checkpoint induction in an immunocompetent mouse model of C. auris sepsis revealed elevated expression of PD-1 on T cells and PD-L1 on macrophages from infected mice (34). PD-L1 expression on macrophages strongly correlated with C. auris yeast cell burden in kidney tissue, suggesting that PD-1/PD-L1 checkpoint induction might hamper fungal clearance (34). Similarly, persistent cryptococcal lung infection induced robust and sustained upregulation of PD-1 on Th cells and its ligands PD-L1 and PD-L2 on DCs and alveolar macrophages in a murine infection model (35).
Further, two studies demonstrated profound and sustained immune exhaustion after Pneumocystis jirovecii pneumonia (PJP). T and B cells from non-HIV patients with PJP displayed strongly elevated transcription and surface expression of PD-1 and PD-L1 on T and B cells (36). The authors confirmed these findings in a murine PJP model and showed that upregulation of PD-1 on Th cells and CTLs persisted for up to 6 weeks after Pneumocystis infection (36). Others found increased PD-1 expression on alveolar macrophages from mice with PJP and pulmonary accumulation of Myeloid-Derived Suppressor Cells (MDSCs) that strongly expressed PD-L1 (37). Furthermore, alveolar macrophages from uninfected mice showed increased PD-1 expression and weaker phagocytic capacity after co-culture with MDSCs from mice with PJP (37). This effect was partially reversible by pretreatment of ex-vivo MDSCs with anti-PD-L1 (37).
In vitro data and animal studies also support a role of checkpoint upregulation after mold infection or sensitization to mold antigens. Balb/c mice sensitized via intraperitoneal and intranasal administration of Aspergillus fumigatus allergens showed elevated expression of CTLA-4 with concomitant downregulation of CD28 on pulmonary Th cells (38). This mechanism is thought to protect the host against tissue damage during persistent anti-Aspergillus responses, e.g., through regulation of the balance between regulatory T cells (Treg) and proinflammatory type 17 T-helper (Th17) cells or reversal of the proinflammatory phenotype of Treg cells (39, 40). Likewise, others found that A. fumigatus cell wall α-(1,3)-glucan stimulated the expression of PD-L1 on human DCs, which in turn promoted polarization of naïve T cells toward Treg cells while suppressing Th1 responses (41). Antibody-mediated blockade of PD-L1 in vitro led to significant inhibition of α- (1, 3)-glucan-induced Treg cells while enhancing IFN-γ production by Th1 cells. These findings suggested that PD-L1 on DCs orchestrates the balance between Treg and Th1 responses to Aspergillus antigens (41).
Two studies provided insights into the activation of checkpoint pathways during dimorphic fungal infections (42, 43). T-cells from patients with active paracoccidioidomycosis showed increased expression of CTLA-4 and decreased proliferative capacity after stimulation with the strong T-cell activator phytohemagglutinin. Antibody-mediated blockade of CTLA-4 enhanced IFN-γ responses of ex-vivo PBMCs from patients with paracoccidioidomycosis (42). Mice infected with Histoplasma capsulatum showed broadly upregulated PD-L1 expression on multiple splenic immune cell subsets (43). The suppressive phenotype of macrophages after infection with H. capsulatum strongly inhibited proliferation and cytokine secretion by activated T-cells during in-vitro co-culture (43).
Collectively, these studies provide solid evidence that co-inhibitory checkpoint pathways are induced by a broad spectrum of pathogenic fungi, such as yeasts, molds, and dimorphic fungi. Moreover, several of these studies revealed a link between upregulated checkpoint pathway expression and signs of impaired antifungal immunity, pointing to a role of checkpoint pathways as therapeutic targets in medical mycology.
In 2000, McGaha & Murphy published the first comprehensive in-vivo study demonstrating a therapeutic potential of ICIs in experimental mycology. The authors showed that antibody-mediated blockade of CTLA-4 prolonged survival of mice intravenously infected with the highly virulent C. neoformans strain NU-2 (44). Additionally, CTLA-4 blockade augmented cell-mediated responses to immunization with the cryptococcal antigen CneF and enhanced immunization-induced protection from subsequent C. neoformans infection (44). In another study, treatment with a PD-1 antibody (anti-PD-1) promoted fungal clearance from lungs of mice with persistent C. neoformans infection and reduced fungal dissemination to brain and spleen tissue (35). However, in both cryptococcal infection models, attainment of favorable outcomes required high-dose ICI treatment given twice weekly over a course of several weeks (35, 44).
In a sub-lethal PJP mouse model, anti-PD-1 treatment every 3 days for 3 weeks resulted in faster weight gain of both immunocompetent and corticosteroid-immunosuppressed Pneumocystis-infected mice (36). However, improved fungal clearance after anti-PD-1 treatment was only seen in immunocompetent mice (36).
Several studies suggested a therapeutic benefit of ICIs alone or in combination with antifungals in mice with primary or secondary Candida sepsis. Spec and colleagues compared blockade of PD-1, PD-L1, and CTLA-4 inhibition in both single-hit C. albicans sepsis and secondary candidiasis after peritonitis induced by cecal ligation and puncture (CLP) (32). Although the PD-1 pathway inhibitors tended to provide a stronger early survival benefit than anti-CTLA-4 in mice with single-hit candidiasis, improvement in 12-day mortality was comparable between the ICI treatments (32). Similarly, all three inhibitors yielded comparable survival benefits in mice with secondary C. albicans sepsis compared with the corresponding isotype controls (32). Notably, all experiments in this study were conducted with concomitant fluconazole therapy in order to document the efficacy of immunotherapy when added to routine antifungal therapy. Others additionally confirmed a mono-therapeutic benefit of CTLA-4 blockade in mice with single-hit C. albicans sepsis or secondary candidiasis after CLP (45). Interestingly, an inverse relationship between the anti-CTLA-4 dose and survival was noted. High doses of anti-CTLA-4, as used in preclinical cancer immunotherapy studies (“oncological dosing”), worsened survival, whereas a 4- to 6-fold lower dose of anti-CTLA-4 improved survival outcomes (45). As a complementary approach to classical antibody-mediated checkpoint blockade, administration of a short-acting peptide (Compound 8) that inhibits PD-1/PD-L1 signaling also provided a significant survival benefit in mice with secondary C. albicans sepsis after CLP (31).
The CLP mouse model was further utilized to study the role of PD-1 inhibitors in post-sepsis aspergillosis. While antifungal treatment with amphotericin B alone was not effective to mitigate secondary aspergillosis after CLP, adjunct anti-PD-1 treatment significantly improved 20-day survival and strongly reduced the fungal burden in brain, lung, and kidney tissues (46). However, a limitation of this study was the rather artificial intravenous route of infection with a high inoculum of A. fumigatus. Therefore, our group evaluated ICI therapy in neutropenic mice with invasive pulmonary aspergillosis (IPA), the predominant clinical manifestation of invasive aspergillosis in immunocompromised patients (47). Oncological dosing of anti-CTLA-4 or dual checkpoint blockade yielded no significant therapeutic benefit, likely due to exuberant toxicity in a background of acute infection (47). Despite signs of hyperinflammatory toxicity, oncological dosing of anti-PD-1 significantly improved 8-day survival outcomes of mice with IPA. Furthermore, an 8-fold lower dose of anti-PD-1 had considerably attenuated toxicity, provided a significant survival advantage, and improved fungal clearance in mice with IPA compared with isotype- and mock-treated controls (47). Moreover, combination therapy with anti-PD-1 and caspofungin (CAS), a drug that has modest activity against A. fumigatus but has been associated with protective immunomodulatory activity (48–50), resulted in a significant additive survival benefit in A. funigatus-infected mice compared with either CAS or anti-PD-1 treatment alone (47). Using the same low-dose regimen as in the IPA study, monotherapy with PD-1 pathway inhibitors also improved morbidity and mortality and enhanced fungal clearance in neutropenic mice with invasive pulmonary mucormycosis (IPM) (51). While early protection was comparable between anti-PD-1 and anti-PD-L1, the latter provided more consistent and sustained improvement of morbidity and mortality in mice with IPM (51). Additionally, both genetic PD-1 deficiency and antibody-mediated PD-1 blockade strongly improved survival of mice infected with an otherwise lethal Histoplasms capsulatum strain (43).
Altogether, these preclinical studies make a compelling case that ICIs can improve the outcomes of various IFIs in diverse backgrounds of underlying immune dysfunction (i.e., post-sepsis immune exhaustion or pharmacologically induced neutropenia). However, several of these studies also revealed signs of ICI-related immunotoxicities in a setting of acute infection, hyperinflammation, and high antigenic load, especially with high-dose ICIs or dual checkpoint blockade.
There are very limited clinical data on antifungal ICI therapy, consisting of five case reports:
The first case of adjunct ICI treatment as salvage therapy for an invasive fungal infection was reported in 2017 in a patient with antifungal therapy-refractory mucormycosis (52). A previously healthy 30-year old woman developed gastrosplenic mucormycosis after suffering a polytrauma and second-degree burns, complicated by sepsis and deep bacterial wound infections. Despite initiation of liposomal amphotericin B and posaconazole and extensive surgical revision with gastrectomy and splenectomy, the patient developed surgically intractable Mucoralean lesions of peritoneal and vascular structures. Because of the poor prognosis and the patient’s state of severe immune paralysis with lymphocytopenia, high expression of PD-1 on Th cells, and signs of poor monocytic maturation, combination immunotherapy consisting of five doses of IFN-γ and a single dose of the PD-1 inhibitor nivolumab was initiated. PD-1 expression reverted to the levels of healthy controls within four days of nivolumab administration. In addition, monocyte maturation and lymphocytopenia improved over a course of two weeks. The patient eventually recovered after several weeks (52).
The second case of antifungal ICI therapy (53) was a 51-year-old woman with relapsed acute myeloid leukemia (AML) after allogeneic hematopoietic stem cell transplantation, who developed fever and sinusitis during chemotherapy-induced pancytopenia. The patient was diagnosed with pan-sinusitis caused by Lichtheimia ramosa and A. fumigatus and developed progressive mucormycosis with orbital invasion despite initiation of antifungal therapy with liposomal amphotericin B and isavuconazole, several surgical interventions, and daily filgrastim (G-CSF). After initiation of nivolumab (four doses in total, every other week) and IFN-γ (ten doses in total, thrice weekly), mucormycosis partially regressed and the patient was discharged. Under nivolumab treatment, markers of lymphocyte activation improved while the expression of PD-1 and CTLA-4 decreased. Although the fungal infection was stable for another month with continued antifungal therapy, the patient developed AML progression and eventually died from septic shock with disseminated intravascular coagulation (53).
In another recent case report (54), combined immunotherapy with nivolumab and IFN-γ was used to treat an extensive polymicrobial soft tissue infection secondary to a minor thoracic trauma in a previously healthy 38-year-old female patient. Pathogens identified by culture and molecular techniques included Enterococci, multiple gram-negative bacteria, Aspergillus spp., Mucor spp., L. ramosa, and Rhizopus arrhizus. The patient was managed with multiple antibiotics, high-dose liposomal amphotericin B (10 mg/kg/day), triazoles, aggressive surgical debridement, hyperbaric oxygen therapy, and daily IFN-γ injections. Due to organ failures, coagulopathy, persistent detection of L. ramosa and R. arrhizus in blood and tissue cultures, severe monocyte deactivation, and high T-cellular PD-1 expression, a single dose of nivolumab was given on day 27 after admission. PD-1 expression became negative on the following day, fungal cultures and PCR became negative over the course of one week, and the patient’s clinical status improved rapidly. The patient was discharged after 5.5 months following extensive reconstructive surgery (54).
Similarly, combinatorial immunotherapy with nivolumab and IFN-γ was reported in a 56-year-old diabetic patient with COVID-19 who had received dexamethasone and tocilizumab (anti-IL-6) and developed invasive pulmonary aspergillosis one week after intensive-care unit (ICU) admission, later complicated by two Mucoralean brain abscesses and ethmoidal sinusitis (55). The brain lesions were insufficiently responsive to combined antifungal therapy with liposomal amphotericin B and triazoles, and only partially accessible for surgical debridement. Neutrophil and lymphocyte counts were normal to high, but both Th cells and CTLs displayed strong PD-1 expression. The patient received two doses of nivolumab 4 weeks apart and IFN-γ thrice weekly for four weeks. Subsequently, PD-1 expression decreased, CTL proliferation modestly increased, and cerebral abscesses decreased in size. However, the patient developed ventilation-associated pneumonia with septic shock, oliguric renal failure, and hepatic cytolysis, necessitating discontinuation of liposomal amphotericin B and immunotherapy. Under isavuconazole only, cerebral mucormycosis progressed and the patient deteriorated. The authors discussed that earlier salvage immunotherapy might have had a stronger immunological and clinical impact and underscored the need to study optimal timing of antifungal ICI therapy in the setting of underlying antifungal therapy (55).
Additionally, the first case of nivolumab treatment in a patient with invasive fusariosis was reported recently (56). A 55-year old male AML patient undergoing high-dose chemotherapy developed neutropenic fever and pneumonia and was diagnosed with disseminated fusariosis caused by Fusarium solani. The patient received aggressive antifungal therapy with liposomal amphotericin B and voriconazole and was treated on the ICU for several weeks. Eleven weeks later, the patient developed hepatosplenic fungal lesions. As renal toxicities limited amphotericin B dosing, voriconazole remained sub-therapeutic, and the hepatosplenic lesions showed high expression of PD-L1, nivolumab was initiated as salvage treatment (four doses in total, every other week). PD-1 expression on T cells strongly declined after initiation of nivolumab, whereas T-helper-cell concentrations (especially Th1 cells) in peripheral blood increased. Furthermore, signals of enhanced CTL and NK-cell activation were seen. Radiological, histopathological, and microbiological examination confirmed resolution of the fungal lesions and showed no evidence of ongoing fusariosis. Liposomal amphotericin was continued on an outpatient basis for twelve months and no recurrence of fungal infection was seen. However, the patient developed adrenal insufficiency, likely as a side effect of the ICI treatment (56).
While these case reports suggest a role of ICIs as a “last resort” immunotherapeutic intervention, therapeutic success relied on a combination with extensive surgery and/or aggressive conventional antifungal treatment. Additionally, combination immunotherapy with other immunomodulators was used in four out of five case reports. Furthermore, there is a possibility of “publication bias”. Larger case series or interventional trials with more thorough immune phenotyping would be needed to gauge the clinical benefit of ICIs in medical mycology. Notably, the few published cases suggested a trend of more favorable outcomes in a priori immunocompetent patients, i.e., those with trauma-related infections. This early observation highlights the importance to study the impact of underlying malignant diseases and other comorbidities on infection-induced immune exhaustion, responses to antifungal ICI therapy, and ICI toxicities (Table 1).

Table 1 Future research objectives to address the many unanswered questions about antifungal ICI therapy.
Mechanistic studies of ICI-driven immune enhancement in fungal infection models mostly focused on exhaustion markers and cytokine responses. Although CTLA-4 blockade inhibited T-cell exhaustion and apoptosis in a murine model of Candida sepsis after CLP, no significant impact of ICI therapy on the cytokine environment was found (45). In contrast, others showed increased IFN-γ production by stimulated splenocytes from ICI-treated mice with primary or secondary Candida albicans sepsis (32). Interestingly, the authors found concomitant induction of the immunosuppressive Treg cytokine IL-10, especially in anti-PD-1-treated animals, along with strong elevations of the proinflammatory cytokine IL-6 (32), which has been linked to both protective anti-yeast responses (57) and immunotherapy-related immunotoxicities (58).
The dualism of presumably protective and adverse cytokine responses after ICI therapy was confirmed in mice with IPM, where blockade of the PD-1/PD-L1 pathway led to modest elevations of proinflammatory cytokines and chemokines associated with fungal clearance (e.g., GM-CSF, TNF-α), paralleled by signals of enhanced Th2 (IL-5, IL-13) and Treg (IL-10) cytokine responses after ICI treatment (51). Additionally, anti-PD-1-treated mice with IPM had massively elevated serum levels of IL-6 and other proinflammatory cytokines that could contribute to both protective immune cell recruitment and immunotoxicities (51).
Multiplex cytokine profiling in lung homogenates from anti-PD-1-treated mice with IPA revealed significant induction of key pro-inflammatory cytokines (e.g., TNF-α, IL-1β) and neutrophil-attracting chemokines (e.g., CXCL2) compared with isotype-treated controls (47). Consistent with these findings, hyphal invasion foci in anti-PD-1-treated mice with IPA were surrounded by more dense leukocyte infiltrates, which displayed an increased neutrophil-to-lymphoid cell ratio compared with isotype-treated infected mice (47).
Immune phenotyping in anti-PD-1-treated mice with PJP revealed heterogenous results. While global immune cell infiltration into the lung tissue increased after anti-PD-1 treatment, the percentage of neutrophils decreased in both the lung and bloodstream (36). However, these measurements were performed 3 weeks post-infection and might not have captured the full picture of immune cell kinetics, especially the early trends (59).
Few studies provided additional non-cytokine surrogates of APC activation. Both anti-PD-1 and anti-PD-L1 enhanced the fungal killing potential of ex-vivo splenocytes from mice with IPM compared with isotype treatments (51). APCs from anti-PD-1-treated mice with post-sepsis aspergillosis had significantly restored expression of the maturation marker CD83 compared with mock-treated infected controls, resembling expression levels in uninfected animals (46). Similarly, DCs and macrophages of anti-PD-L1-treated mice with secondary C. albicans sepsis showed increased expression of major histocompatibility (MHC) II molecules, which are pivotal for antigen presentation to Th cells (32). In both studies, ICI-mediated enhancement of APC activation and maturation was paralleled by reinvigoration of IFN-γ release from re-stimulated ex-vivo T cells, further supporting that ICIs can restore exhausted APC/T-cell feedback loops during fungal infections (32, 46). In contrast, anti-PD-1 did not alter the pulmonary accumulation or activation profile of myeloid cells in mice with persistent cryptococcal lung infection, but enhanced activation of Th1 and Th17 cells while dampening the production of Th2 and Treg cytokines (35). Similarly, genetically PD-1-deficient mice had significantly enhanced Th1 and Th17 responses during Pneumocystis infection, paralleled by signals of macrophage polarization toward a protective M1 phenotype and strong upregulation of both pro- and anti-inflammatory cytokines (36). However, these readouts were not studied after pharmacological PD-1 blockade (36).
Although some surrogates of adverse immune alterations (e.g., high serum levels of IL-6) have been described, the determinants of immunotoxicities after antifungal ICI therapy are largely unknown. The immunopathogenesis of IFIs often comprises a complex combination of immunosuppression and exhaustion with topical and systemic inflammation. Patients whose pathogenesis is predominantly driven by hyperinflammation rather than immune exhaustion might not benefit from ICIs but potentially experience worsening of immunopathology and progressive infection. For instance, a case of acute exacerbation and progression of chronic pulmonary aspergillosis has been described in a patient receiving nivolumab to treat lung cancer (60).
As a severe form, hyperinflammatory toxicities can manifest as an immune reconstitution inflammatory syndrome (IRIS), a clinical entity observed in immunocompromised patients who experience sudden restoration of immune activity, e.g., due to abrupt tapering of corticosteroids or initiation of highly active antiretroviral therapy in patients with HIV infection (61). IRIS has been described after ICI use in patients with underlying opportunistic infections (e.g., tuberculosis) and can result in severe systemic inflammation, overshooting immune responses to the underlying infection, and tissue damage (19, 62).
Additionally, cancer immunology studies have described a multitude of autoimmune toxicities of ICIs that can affect essentially any organ, as extensively reviewed elsewhere (63). While colitis, diarrhea, dermatological events, and endocrine toxicities are the commonest irAEs in patients receiving ICIs as cancer immunotherapy (63), manifestations of autoimmune toxicities after anti-infectious ICI therapy remain to be characterized. Notably, management of autoimmune toxicities after ICI therapy often requires corticosteroids and/or other immune-attenuating agents (e.g., TNF-α inhibitors) that may in turn aggravate an underlying IFI (19).
In summary, while toxicities are poorly understood, limited data from preclinical models and patients with IFIs suggest that the immune environment created by ICI treatment could promote clearance of opportunistic fungi through a combination of increased innate immune cell recruitment, restored APC/T-cell interactions, and enhanced maturation and fungicidal activity of APCs (Figure 2). However, the detailed underpinnings of ICI-mediated changes to the immune environment at the site of infection are still poorly understood, as are the long-term effects of antifungal ICI therapy and the determinants of immunotoxicities. These gaps of knowledge hamper the identification of much-needed (immune) biomarkers to identify patient cohorts who would benefit most from antifungal ICI therapy.
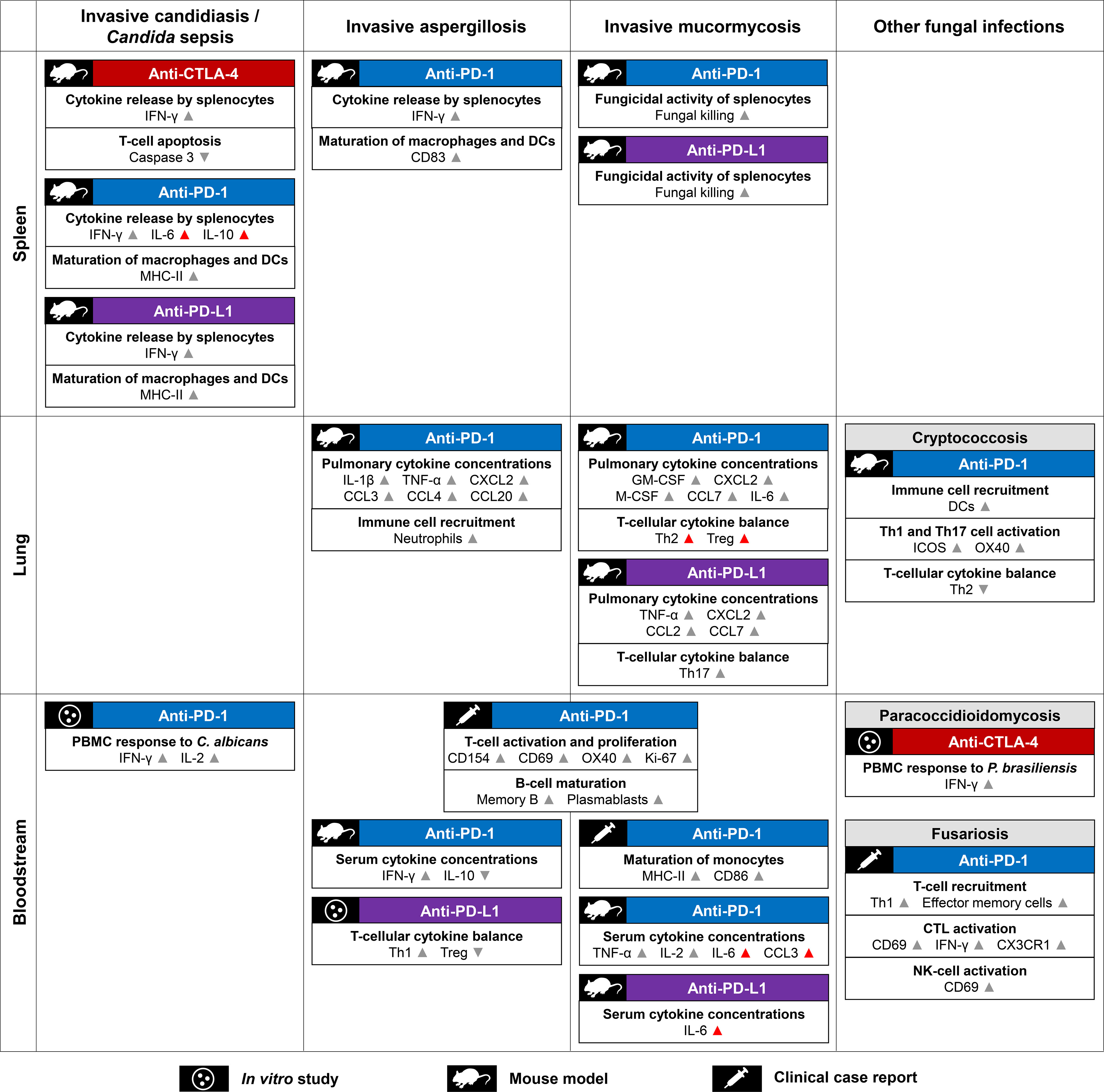
Figure 2 Summary of proposed mechanisms contributing to checkpoint inhibitor-mediated immune enhancement against opportunistic fungal pathogens. Mechanistic readouts are grouped by type of infection and the body site of specimen collection. Grey arrowheads indicate changes to key readouts likely associated with antifungal immune enhancement. Unfavorable responses (e.g., Th2 and Treg activation) and immunological changes potentially linked to toxicities (e.g., high concentrations of serum IL-6) are indicated by red arrowheads. Abbreviations: CD, cluster of differentiation; CTL, cytotoxic T lymphocyte; CTLA-4, Cytotoxic T-Lymphocyte Antigen 4; C(X)CL, C-(X-)C-motif chemokine receptor ligand; CX3CR1, C-X3-C-motif Chemokine Receptor 1; DC, dendritic cell; (G)M-CSF, (Granulocyte) Macrophage Colony Stimulating Factor; IFN, interferon; IL, interleukin; MHC, Major Histocompatibility Complex; NK cell, natural killer cell; PBMCs, peripheral blood mononuclear cells; PD-1, Programmed Cell Death Protein 1; PD-L1, Programmed Death Ligand 1; Th(1/2/17), (type 1/2/17) T-helper cells; Treg, regulatory T cells.
Pharmacologically immunosuppressed but otherwise healthy rodent models are widely used for preclinical fungal immunology studies (64, 65), including several studies on ICI therapy discussed above. While being relatively simple, cost-efficient, and generally well reproducible, these models often fail to adequately reflect the underlying immune dysfunction of high-risk patients, e.g., those with acute leukemia. Consequently, results cannot be extrapolated to other host environments without additional experimental validation (64, 65), creating a major unmet need to study the impact of ICIs on the outcomes and immunopathology of opportunistic mycoses in pathophysiologically relevant preclinical models, e.g., leukemic mice (66).
Although most models of Candida sepsis do not rely on pharmacological immunosuppression, there are three common limitations. On the one hand, preclinical sepsis models often utilize intravenous inocula of millions or billions of yeast cells per kg body weight, especially when assessing pathogens with relatively low virulence such as Candida auris (34, 67). Such unnaturally high inocula can result in overwhelming infection (68) and are a considerable confounder of immunological responses and exhaustion phenotypes compared to real-life clinical scenarios. Additionally, the published studies on ICI therapy in single-hit or CLP Candida sepsis models utilized a priori healthy mice. As particularly high mortality of Candida sepsis has been reported in immunocompromised patients and in patients with prior antibiotic treatment, co-infections, obesity, preexisting cardiovascular diseases, diabetes mellitus, or major surgery (69, 70), recapitulation of these conditions in more nuanced mammalian models would be needed. Lastly, fungal sepsis is frequently associated with indwelling catheters, resulting in immune-resistant yeast biofilms (71). As such biofilms stimulate inflammatory host responses while immunological clearance is inefficient, catheter-associated infections could be particularly prone to immune exhaustion (71). However, studies supporting that ICIs can promote a meaningful immune response against yeasts engulfed in catheter-associated biofilms are lacking.
Fungal infection models are often designed to produce an acutely lethal infection (68), which might underestimate the role of protracted immune exhaustion while exaggerating hyperinflammatory toxicities during the initial inflammatory phase of infection. Thus, it would be desirable to validate the efficacy of ICIs in preclinical models across a broader range of inocula and by using different fungal isolates with varying virulence. Another major limitation of all published animal studies is the initiation of antifungal ICI therapy within few hours to days after fungal infection, aiming to intercept immune exhaustion rather than attempting to revert an exhausted immune landscape. Although yielding promising results, this approach is rather artificial in view of the often difficult and delayed diagnosis of IFIs. Additionally, early ICI therapy in a background of high fungal burden and inflammation likely contributed to the reports of considerable toxicities, especially at full oncological dosing of ICIs (45, 47). In contrast, all clinical case reports described late-stage application of ICIs in patients with IMIs, a strategy that might have suboptimal efficacy in a setting of severely exhausted and terminally differentiated effector cells (55). Therefore, the optimal timing of ICI therapy needs to be thoroughly studied in preclinical models. Other common limitations of most published preclinical studies on antifungal ICI therapy included the use of a single dosing regimen (19), the omission of first-line antifungal therapy as the “standard of care”, the utilization of inbred mice, and performance of studies in semi-sterile facilities without natural mold exposure (72, 73). The latter two limitations bear additional significance in view of the increasingly recognized modulation of treatment responses to oncological ICI therapy by variations in the host microbiome (74, 75).
Despite these limitations, there has been considerable agreement among the published studies in transgenic (e.g., PD-1-deficient) mice, immunotherapeutic studies in murine infection models, assessment of ex-vivo human and murine immune cells, and clinical case reports regarding the effects of checkpoint induction and inhibition in a broad spectrum of IFIs. Nonetheless, more refined models with increasing incorporation of underlying conditions such as leukemia (66) or co-infections (76) would not only provide a more representative host environment but could also play a major role in defining specific host populations that might particularly benefit from antifungal ICI therapy.
Beyond the PD-1/PD-L1 and CTLA-4 pathways, an increasing number of preclinical and clinical immunooncology studies explore the merits of additional checkpoint targets to mount broad anti-tumor immunity and mitigate treatment resistance. Co-inhibitory pathways can be subdivided depending on their involvement in i) interactions of APCs and Th cells, ii) interactions of APCs and CTLs, and iii) interactions of CTLs and somatic, non-lymphoid cells, such as tumor cells or infected tissue (77, 78). We would hypothesize that molecules involved in the Th/APC interplay (Figure 1B) are the most promising targets to treat IFIs, since the role of CTLs in natural antifungal immunity is less clearly defined than the multifaceted Th responses (39, 79). Notably, checkpoint pathways with a known role in the APC/Th interplay, such as TIM-3, LAG-3, and TIGIT, are among the most promising checkpoint targets in the cancer immunotherapy pipeline (80). Furthermore, there is some evidence for a role of these targets in the host response to (viral) infections, including potential synergies with conventional checkpoint targets such as PD-1 (81, 82). Therefore, detailed exploration of these targets in preclinical models of fungal infections and in ex-vivo samples from infected patients would be warranted.
Beyond their role as independent therapeutic targets, other checkpoint pathways were shown to drive resistance to PD-1- or CTLA-4-targeted oncological ICI therapy (83). Data about resistance formation to antifungal ICI therapy are scarce, as most of the published preclinical studies used relatively short follow-up periods and detailed information on checkpoint marker expression beyond PD-1/PD-L1 was provided only in a single clinical case report (53). Nonetheless, there are signals suggesting potential paths to ICI resistance in fungal infections. For instance, immune phenotyping of ex-vivo T cells from patients with IC suggested co-induction of multiple checkpoint pathways, posing potential limitations to single-agent ICI therapy (29). PD-1/PD-L1 pathway blockade in a murine mucormycosis model successfully suppressed the targeted molecules and co-suppressed CTLA-4 but led to rapid upregulation of TIM-3 within 4 days of ICI initiation (51). Likewise, nivolumab therapy in a patient with aspergillosis and mucormycosis caused an initial upregulation of other checkpoint pathways, such as TIM-3 and LAG-3 (53). Both pathways are well-known drivers of resistance to oncological ICI therapy (84). These findings deserve further study in preclinical infection models in order to better understand the kinetics and implications of compensatory checkpoint induction after antifungal ICI therapy.
As an alternative strategy to releasing the “immunological brake” through ICIs, agonists of co-stimulatory molecules such as CD28, CD134/OX40, CD137, or CD154/CD40L have been investigated in preclinical oncology studies and are currently undergoing early-phase clinical evaluation (85, 86). These molecules have pleiotropic effects on early immune activation and can potentially synergize with ICIs (85, 86). Of note, most of these targets have well-defined functions in the immune response to pathogenic fungi. For instance, CD154 is crucial for the initiation and enhancement of antifungal host defense and has been studied as a target for investigational immune diagnostics in medical mycology (87, 88). Additionally, both CD137 and CD154 have been previously proposed as reliable selection markers for the enrichment of mold-reactive T-cells for adoptive transfer (8, 89). It is therefore conceivable that co-stimulation-enhancing agents could confer increased protection against fungal pathogens. However, these agents are associated with significant toxicities and are prone to causing cytokine release syndrome (85, 86). Although new fusion proteins or topical application could reduce these side effects (85), the narrow therapeutic index of co-stimulation-enhancing agents is concerning for their use in an acute infection setting. Nonetheless, as these agents are increasingly introduced into the oncological treatment landscape, it will be important to investigate their effects on host defense against opportunistic pathogens and their potential for immunotherapeutic applications in medical mycology.
As discussed above, dosing of ICIs during acute infection and inflammation is limited by toxicity concerns and immunomodulators are unlikely to be applied without concomitant conventional antifungal therapy. All three major classes of modern antifungals – lipid amphotericin B, azoles, and echinocandins – have known immunopharmacological effects that could be implicated in potential synergies with ICIs. These immunomodulatory effects are best-characterized for echinocandins (48, 49). Echinocandins disturb the equilibrium of the fungal cell wall and expose beta-glucans that are considered the most potent fungal stimulus of innate immune cell activation (48, 49, 90). Echinocandins can thereby enhance fungicidal activity of mononuclear phagocytes and neutrophils and induce proinflammatory cytokine secretion (48–50, 91). We have previously described a significant additive survival advantage of PD-1 blockade and CAS in our murine IPA model despite the poor monotherapeutic activity of CAS against A. fumigatus (47). Several promising investigational antifungal drugs, especially rezafungin and ibrexafungerp, have known cell well-altering properties (3) and are therefore likely to exert similar immunomodulatory effects that might provide synergies with ICIs. Triazoles can induce upregulation of the pattern recognition receptors TLR2 and TLR4 (48) that are pivotal for activation of neutrophils and mononuclear phagocytes by fungal antigens (79). Although mechanistic synergies with ICIs remain to be defined, anti-PD-1 provided an added benefit when combined with fluconazole in a C. albicans sepsis model (32). Immunomodulatory properties of lipid amphotericin B formulations are predominantly associated with the liposomal packaging (92, 93). Liposomes can stimulate APCs, modulate TLR expression, and promote nonoxidative fungal killing by neutrophils (92, 93). Consequently, pretreatment with empty liposomes improved survival and attenuated immune injury in a murine IPA model (92). Given the presumed effects of ICIs on neutrophil recruitment and enhancement of phagocytic activity of APCs, immunological synergism with liposomal amphotericin B is conceivable and deserves further study.
Four clinical reports of antifungal ICI therapy further suggested that combinatorial immunomodulation strategies, especially the combination of ICIs and the Th1-polarizing cytokine IFN-γ, could provide additional benefits (52–55). IFN-γ has been previously used as salvage therapy in patients with fungal infections in a background of dysfunctional immunity (13, 94). For instance, IFN-γ was well-tolerated in patients with C. albicans sepsis and partially restored MHC II expression and secretion of TNF-α and IL-1β (13). Oncological studies are exploring synergies between ICIs and IFN-γ in preclinical models and in patients with advanced solid tumors (95). Notably, ICIs alone did not enhance Th1 cytokine release in our IPA and IPM models (47, 51), supporting a potential independent benefit of IFN-γ in ICI-based combination immunotherapy. Additionally, hematopoietic growth factors such as G-CSF and GM-CSF (6, 11, 14–18), whose endogenous levels in mice were only modestly induced by antifungal ICI therapy alone (47, 51), are potentially interesting targets for antifungal combination immunotherapy. However, thorough preclinical studies are needed to determine the risk of additive immunotoxicities when combining ICIs with additional immunomodulators.
Additionally, combinations of ICIs with adoptive T-cell transfer and chimeric antigen receptor T cells have been studied as investigational oncological salvage treatments (96). Ex-vivo expansion of cellular products, as proposed for immunotherapeutic applications in medical mycology (9), often requires prolonged antigen challenge and/or the use of stimulatory cytokine cocktails. These manufacturing-related factors and subsequent encounters with a suppressive in-vivo environment can promote exhaustion of the infused cells (97, 98). Although PD-1 pathway blockade was shown to augment the efficacy of CAR T-cell therapy against various malignancies (99, 100), CAR T-cell exhaustion might eventually be overcome by engineering of exhaustion-resistant cells (98). In contrast, concomitant checkpoint blockade could have merits for adoptive transfer of unmodified, enriched, allogenic antigen-reactive T cells (8, 89), a methodology developed to clinical scale and tested in small, early-phase studies for mycological applications (101, 102).
The strategies discussed above predominantly focus on strengthening ICI-mediated immune augmentation through synergistic immune interventions. Additionally, oncological trials are increasingly exploring combinatorial treatment strategies that aim to decouple protective anti-tumor responses from detrimental off-target toxicities. IL-6 emerged as an interesting target in this context (103). Given that studies of ICI therapy in preclinical models of fungal infections showed strong induction of IL-6 (32, 51), such approaches warrant further study in medical mycology. On the other hand, IL-6 inhibitors are associated with an increased fungal infection risk, especially in COVID-19 patients (55, 104). Therefore, dosing and timing relative to the ICI treatment will likely determine the impact of IL-6 inhibitors on the delicate balance between fungal clearance, suppression of antifungal immunity, and the level of immunotoxicities. A similar dualism could be seen with inhibitors of CXCR1 activation by CCL3. This chemokine showed strongly elevated serum levels in mice with IPM receiving anti-PD-1 and could be implicated in immunotoxicities (51). Prior research by our group showed improved survival and attenuation of hyperinflammatory tissue injury in mice with disseminated candidiasis receiving a CXCR1/CCL3 inhibitor during the initial inflammatory stage (105); however, combined application with ICIs during fungal infections remains to be studied.
Both the general rationale for a merit of ICIs in medical mycology and potential future strategies for combination immunotherapy adapted from oncological research partially rely on commonalities between the immune environment of infected tissue and cancers (106). As extensively reviewed elsewhere, these commonalities include similar triggers (e.g., nucleic acids and other damage-induced molecular patterns), receptor signaling pathways (e.g., a major role of TLR signaling), and features of immune exhaustion (19, 106).
Some features of the tumor microenvironment that are hallmarks of protective responses to immunotherapy (77, 107) are also known to be crucial for host defense against fungal pathogens. For example, DC and macrophage polarization can shape the ICI-induced antitumor response (107). Cancer immunology studies suggested that M1 macrophages favor antitumor immunity through NK-cell and CTL activation and are associated with therapeutic response to ICIs, whereas most subsets of M2 macrophages elicit pro-angiogenetic and immunosuppressive effects (107–109). Similarly, M1-polarization is considered beneficial in antifungal immunity and has been linked to increased NK-cell activation, phagocytic capacity, and secretion of IFN-γ in the response to A. fumigatus (110) and C. albicans (111).
On the other hand, we need to be aware of major differences between the tumor and infection environment. Clearance of tumor cells strongly relies on CTL effector responses and the density of functional CTLs is a major predictor of ICI efficacy against various tumors (112, 113). In contrast, little is known about the contributions of CTLs during antifungal ICI therapy. Additionally, there are major differences between the myeloid cell repertoire associated with fungal elimination (e.g., polymorphonuclear neutrophils or alveolar macrophages) and the mostly immunosuppressive myeloid-derived cell populations dominating the tumor environment (106).
Another distinct difference between host immunity to many fungal pathogens and cancers is the constant exposure to fungal commensals and aero-antigens. For instance, chronic occupational mold exposure has been shown to modulate the T-cell repertoire and cytokine responses to Aspergillus antigens (114, 115). Similarly, fungal commensals, especially in the intestinal microbiome, are powerful modulators of the antifungal T-cell repertoire (116, 117). It remains unclear and difficult to study in the currently used animal models how prior encounters with fungal antigens and the mycobiome of fungal commensals impact responses to antifungal ICI therapy.
Studies describing potential mechanisms of ICI-mediated antifungal immune enhancement (section 2.4) were restricted to analyses of host biomarkers and did not assess changes in fungal responses to the treatment. However, immune-oncologists described the ICI-enhanced immune environment as a Darwinian scenario for tumor cells, which are often not entirely eradicated and either become more aggressive or deploy immunoinhibitory mechanisms to undermine host defense (106). Tumor cells possess a panoply of strategies to suppress inflammatory responses and evade ICI-augmented host immunity, e.g., through suppression of antigen expression or modulation of the cytokine environment (106). Although similar scenarios would be conceivable during fungal infection in view of the many known immune-evasive mechanisms of opportunistic fungi (118, 119), specific changes in fungal metabolism during ICI-induced immune stress remain to be identified. Such effects might also be triggered by biochemical features of the surrounding tissue. For instance, hypoxia is a known contributor to oncological ICI resistance (120) and has pleiotropic effects on host-pathogen interplay during IFIs (121), including potential attenuation of antifungal DC responses (122), fungal cell wall alterations associated with immune evasion (123), and homeostatic changes in angiogenesis (124). As these processes are highly dynamic, serial application of increasingly powerful “omic” technologies including dual host-pathogen transcriptomics and spatial proteomics should be employed to shed light into both the versatile host immune environment and fungal reprogramming during ICI therapy.
This review focused on therapeutic-intent checkpoint blockade after a fungal infection has been contracted or even as a last resort after unsuccessful conventional antifungal therapy. In contrast, the impact of oncological ICI therapy on the epidemiology and immunopathology of IFIs is largely unexplored. After their tremendous success in the treatment of various solid tumors, ICIs are now increasingly investigated as an immunotherapeutic option in patients with hematological malignancies who are at the highest risk for fungal infections, especially patients with AML (125–127). Although the mono-therapeutic activity of ICIs against AML is relatively poor (126), ICIs might have a promising role as part of combination therapy with hypomethylating agents, especially in elderly patients or those with co-morbidities who cannot tolerate high-intensity cytotoxic chemotherapy (125–127). For instance, a single-arm phase 2 trial of 5-azacytidine (5-AC) plus nivolumab showed encouraging response rates and survival outcomes in patients with relapsed/refractory AML and randomized phase 3 trials are in progress (126, 128).
We have previously proposed the “double-hit hypothesis” that ICIs given as part of AML remission-induction therapy might provide an additional “off-target” benefit by improving the outcomes of opportunistic fungal infections in leukemic patients (125). Specifically, anti-leukemia ICI therapy could reduce the risk and severity of opportunistic infections through facilitation of less-cytotoxic conventional chemotherapy (125), improved remission rates (126, 127), and direct enhancement of antifungal immune responses (see section 2.4). However, this hypothesis has not been studied in clinical trials or animal models. Therefore, there is a need to employ preclinical leukemia models (see section 3.1) to study the effects of different chemotherapy regimens with and without ICIs on the outcomes of opportunistic mold infections during remission-induction chemotherapy (RIC). Furthermore, recently developed, holistic (whole blood-based) ex-vivo assays of antigen-specific immunity (129, 130) should be employed to compare innate and adaptive immunity to pathogenic fungi in preclinical models and leukemia patients receiving RIC with and without concomitant ICIs. Given the rapid emergence of new targeted anti-leukemia therapies, such studies should be further expanded to CD47 macrophage checkpoint inhibitors (131) and the growing number of anti-leukemic small molecules with potential implications for antifungal immunity (132).
A growing body of studies has documented a role of immune exhaustion and the induction of immune checkpoint pathways during opportunistic infections due to various classes of pathogenic fungi. In addition, several preclinical studies suggested protective therapeutic activity of classical immune checkpoint inhibitors (i.e., PD-1, PD-L1, and/or CTLA-4 inhibitors) in murine models of fungal sepsis, invasive mold infections, or dimorphic fungal infections. Positive effects of antifungal ICI therapy on innate immune cell recruitment, activation and maturation of innate immune cells, T-cell activation and exhaustion, and cytokine production have been described. Although some commonalities between the tumor and infection environment have been established, many unknowns remain regarding global, pathogen-specific, and organ-specific changes to the infection microenvironment during antifungal ICI therapy as well as the fungal response to ICI-induced immune enhancement (Table 1). Furthermore, the narrow therapeutic index and risk of detrimental immunotoxicities remain major challenges for broader clinical translation of antifungal ICI therapy. Systematic preclinical studies of timing, dosing, synergies with first-line antifungals and other immunomodulators, and evaluation of emerging ICIs in more nuanced animal models are needed to expand our data framework to inform future clinical applications of antifungal ICI therapy (Table 1). Furthermore, the development of host biomarkers will be pivotal to guide patient stratification and predict successful augmentation of antifungal immunity and potential immunotoxicities. Beyond further studies of the immunotherapeutic merits and implementation of ICIs in opportunistic fungal infections, we need to expand our understanding of the effect of ICIs given as part of oncological (e.g., anti-leukemia) therapies on antifungal immunity and the course of opportunistic IFIs in high-risk patients.
SW and DK reviewed the fungal immunology literature, wrote and edited the paper. SSW provided additional input regarding immuno-oncological studies and their potential implications for antifungal ICI therapy. All authors contributed to the article and approved the submitted version.
This manuscript was supported by the MD Anderson Cancer Center, Division of Internal Medicine Research and Quality Improvement Award (to SW), an MD Anderson Cancer Center Institutional Research Grant (award number 2022-00060729-Y1 to SW), NIH funding (R01 AI133822 and R56 AI109294 to SSW; R03 AI166285 to DK), and the Robert C. Hickey Chair for Clinical Care endowment (to DK).
We thank Jordan Pietz (MD Anderson Cancer Center, Creative Communications) for input regarding data visualization.
SSW reports consultant fees from Asylia Therapeutics Inc and Cellino Biotech Inc. DK reports honoraria and research support from Gilead Sciences and Astellas Pharma. He received consultant fees from Astellas Pharma, Merck, and Gilead Sciences, and is a member of the Data Review Committee of Cidara Therapeutics, AbbVie, Scynexis, and the Mycoses Study Group.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Roemer T, Krysan DJ. Antifungal drug development: challenges, unmet clinical needs, and new approaches. Cold Spring Harb Perspect Med (2014) 4(5):a019703. doi: 10.1101/cshperspect.a019703
2. Novak AR, Bradley ME, Kiser TH, Mueller SW. Azole-resistant aspergillus and echinocandin-resistant candida - what are the treatment options? Curr Fungal Infect Rep (2020) 14(2):141–52. doi: 10.1007/s12281-020-00379-2
3. Lamoth F, Lewis RE, Kontoyiannis DP. Investigational antifungal agents for invasive mycoses: a clinical perspective. Clin Infect Dis (2022) 75(3):534–44. doi: 10.1093/cid/ciab1070
4. Axell-House DB, Wurster S, Jiang Y, Kyvernitakis A, Lewis RE, Tarrand JJ, et al. Breakthrough mucormycosis developing on mucorales-active antifungals portrays a poor prognosis in patients with hematologic cancer. J Fungi (2021) 7(3):217. doi: 10.3390/jof7030217
5. van de Peppel RJ, Dekkers OM, von dem Borne PA, de Boer MG. Relapsed and secondary disease drive the risk profile for invasive aspergillosis prior to stem cell transplantation in patients with acute myeloid leukemia or myelodysplastic syndrome. Med Mycol (2014) 52(7):699–705. doi: 10.1093/mmy/myu036
6. Sam QH, Yew WS, Seneviratne CJ, Chang MW, Chai LYA. Immunomodulation as therapy for fungal infection: Are we closer? Front Microbiol (2018) 9:1612. doi: 10.3389/fmicb.2018.01612
7. Deo SS, Virassamy B, Halliday C, Clancy L, Chen S, Meyer W, et al. Stimulation with lysates of aspergillus terreus, candida krusei and rhizopus oryzae maximizes cross-reactivity of anti-fungal T cells. Cytotherapy; (2016) 18(1):65–79. doi: 10.1016/j.jcyt.2015.09.013
8. Bacher P, Jochheim-Richter A, Mockel-Tenbrink N, Kniemeyer O, Wingenfeld E, Alex R, et al. Clinical-scale isolation of the total aspergillus fumigatus-reactive T-helper cell repertoire for adoptive transfer. Cytotherapy; (2015) 17(10):1396–405. doi: 10.1016/j.jcyt.2015.05.011
9. Kumaresan PR, Manuri PR, Albert ND, Maiti S, Singh H, Mi T, et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci USA (2014) 111(29):10660–5. doi: 10.1073/pnas.1312789111
10. Baistrocchi SR, Lee MJ, Lehoux M, Ralph B, Snarr BD, Robitaille R, et al. Posaconazole-loaded leukocytes as a novel treatment strategy targeting invasive pulmonary aspergillosis. J Infect Dis (2017) 215(11):1734–41. doi: 10.1093/infdis/jiw513
11. Lauruschkat CD, Einsele H, Loeffler J. Immunomodulation as a therapy for aspergillus infection: Current status and future perspectives. J Fungi (Basel) (2018) 4(4):137. doi: 10.3390/jof4040137
12. Karavalakis G, Yannaki E, Papadopoulou A. Reinforcing the immunocompromised host defense against fungi: Progress beyond the current state of the art. J Fungi (Basel) (2021) 7(6):451. doi: 10.3390/jof7060451
13. Delsing CE, Gresnigt MS, Leentjens J, Preijers F, Frager FA, Kox M, et al. Interferon-gamma as adjunctive immunotherapy for invasive fungal infections: a case series. BMC Infect Dis (2014) 14:166. doi: 10.1186/1471-2334-14-166
14. Roilides E, Blake C, Holmes A, Pizzo PA, Walsh TJ. Granulocyte-macrophage colony-stimulating factor and inter-feron-gamma prevent dexamethasone-induced immunosuppression of antifungal monocyte activity against aspergillus fumigatus hyphae. J Med Vet Mycol (1996) 34(1):63–9. doi: 10.1080/02681219680000101
15. Sionov E, Mendlovic S, Segal E. Experimental systemic murine aspergillosis: treatment with polyene and caspofungin combination and G-CSF. J Antimicrob Chemother (2005) 56(3):594–7. doi: 10.1093/jac/dki252
16. Mehta HM, Malandra M, Corey SJ. G-CSF and GM-CSF in neutropenia. J Immunol (1995) 195(4):1341–9. doi: 10.4049/jimmunol.1500861
17. Wan L, Zhang Y, Lai Y, Jiang M, Song Y, Zhou J, et al. Effect of granulocyte-macrophage colony-stimulating fac-tor on prevention and treatment of invasive fungal disease in recipients of allogeneic stem-cell transplantation: a prospective multicenter randomized phase IV trial. J Clin Oncol (2015) 33(34):3999–4006. doi: 10.1200/JCO.2014.60.5121
18. Safdar A, Rodriguez G, Zuniga J, Al Akhrass F, Georgescu G, Pande A. Granulocyte macrophage colony-stimulating factor in 66 patients with myeloid or lymphoid neoplasms and recipients of hematopoietic stem cell transplantation with invasive fungal disease. Acta Haematol (2013) 129(1):26–34. doi: 10.1159/000342121
19. Abers MS, Lionakis MS, Kontoyiannis DP. Checkpoint inhibition and infectious diseases: A good thing? Trends Mol Med (2019) 25(12):1080–93. doi: 10.1016/j.molmed.2019.08.004
20. Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nat Rev Immunol (2018) 18(2):91–104. doi: 10.1038/nri.2017.112
21. Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am J Clin Oncol (2016) 39(1):98–106. doi: 10.1097/COC.0000000000000239
22. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining 'T cell exhaustion'. Nat Rev Immunol (2019) 19(11):665–74. doi: 10.1038/s41577-019-0221-9
23. Gardner D, Jeffery LE, Sansom DM. Understanding the CD28/CTLA-4 (CD152) pathway and its implications for costimulatory blockade. Am J Transplant (2014) 14(9):1985–91. doi: 10.1111/ajt.12834
24. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
25. Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer (2019) 18(1):155. doi: 10.1186/s12943-019-1091-2
26. van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol (2017) 17(7):407–20. doi: 10.1038/nri.2017.36
27. Patricio P, Paiva JA, Borrego LM. Immune response in bacterial and candida sepsis. Eur J Microbiol Immunol (Bp) (2019) 9(4):105–13. doi: 10.1556/1886.2019.00011
28. Spec A, Shindo Y, Burnham CA, Wilson S, Ablordeppey EA, Beiter ER, et al. T Cells from patients with candida sepsis display a suppressive immunophenotype. Crit Care (2016) 20:15. doi: 10.1186/s13054-016-1182-z
29. Mellinghoff SC, Thelen M, Bruns C, Garcia-Marquez M, Hartmann P, Lammertz T, et al. T-Cells of invasive candidiasis patients show patterns of T-cell-exhaustion suggesting checkpoint blockade as treatment option. J Infect (2022) 84(2):237–47. doi: 10.1016/j.jinf.2021.12.009
30. Winters BD, Eberlein M, Leung J, Needham DM, Pronovost PJ, Sevransky JE. Long-term mortality and quality of life in sepsis: a systematic review. Crit Care Med (2010) 38(5):1276–83. doi: 10.1097/CCM.0b013e3181d8cc1d
31. Shindo Y, McDonough JS, Chang KC, Ramachandra M, Sasikumar PG, Hotchkiss RS. Anti-PD-L1 peptide improves survival in sepsis. J Surg Res (2017) 208:33–9. doi: 10.1016/j.jss.2016.08.099
32. Chang KC, Burnham CA, Compton SM, Rasche DP, Mazuski RJ, McDonough JS, et al. Blockade of the negative co-stimulatory molecules PD-1 and CTLA-4 improves survival in primary and secondary fungal sepsis. Crit Care (2013) 17(3):R85. doi: 10.1186/cc12711
33. Deng W, Su Z, Liang P, Ma Y, Liu Y, Zhang K, et al. Single-cell immune checkpoint landscape of PBMCs stimulated with candida albicans. Emerg Microbes Infect (2021) 10(1):1272–83. doi: 10.1080/22221751.2021.1942228
34. Wurster S, Albert ND, Kontoyiannis DP. Candida auris bloodstream infection induces upregulation of the PD-1/PD-L1 immune checkpoint pathway in an immunocompetent mouse model. mSphere; (2022) 7(2):e0081721. doi: 10.1128/msphere.00817-21
35. Roussey JA, Viglianti SP, Teitz-Tennenbaum S, et al. Anti-PD-1 antibody treatment promotes clearance of persistent cryptococcal lung infection in mice. J Immunol (2017) 199(10):3535–46. doi: 10.4049/jimmunol.1700840
36. Zhang C, Rong HM, Li T, Zhai K, Tong ZH. PD-1 deficiency promotes macrophage activation and T-helper cell type 1/T-helper cell type 17 response in pneumocystis pneumonia. Am J Respir Cell Mol Biol (2020) 62(6):767–82. doi: 10.1165/rcmb.2019-0234OC
37. Lei GS, Zhang C, Lee CH. Myeloid-derived suppressor cells impair alveolar macrophages through PD-1 receptor ligation during pneumocystis pneumonia. Infect Immun (2015) 83(2):572–82. doi: 10.1128/IAI.02686-14
38. Barrios CS, Johnson BD, Henderson JD, Fink JN, Kelly KJ, Kurup VP. Enhanced expression of CTLA-4 and concurrent downregulation of CD28 on lung cells of mice exposed to aspergillus antigen. J Allergy Clin Immunol (2005) 15(2):S258. doi: 10.1016/j.jaci.2004.12.1042
39. Dewi IMW, van de Veerdonk FL, Gresnigt MS. The multifaceted role of T-helper responses in host defense against aspergillus fumigatus. J Fungi (Basel) (2017) 3(4):55. doi: 10.3390/jof3040055
40. Raijmakers RPH, Sprenkeler EGG, Aleva FE, Jacobs CWM, Kanneganti TD, Joosten LAB, et al. Toll-like receptor 2 induced cytotoxic T-lymphocyte-associated protein 4 regulates aspergillus-induced regulatory T-cells with pro-inflammatory characteristics. Sci Rep (2017) 7(1):11500. doi: 10.1038/s41598-017-11738-4
41. Stephen-Victor E, Karnam A, Fontaine T, Beauvais A, Das M, Hegde P, et al. Aspergillus fumigatus cell wall α-(1,3)-Glucan stimulates regulatory T-cell polarization by inducing PD-L1 expression on human dendritic cells. J Infect Dis (2017) 216(10):1281–94. doi: 10.1093/infdis/jix469
42. Campanelli AP, Martins GA, Souto JT, Pereira MS, Livonesi MC, Martinez R, et al. Fas-fas ligand (CD95-CD95L) and cytotoxic T lymphocyte antigen-4 engagement mediate T cell unresponsiveness in patients with paracoccidioidomycosis. J Infect Dis (2003) 187(9):1496–505. doi: 10.1086/374646
43. Lázár-Molnár E, Gácser A, Freeman GJ, Almo SC, Nathenson SG, Nosanchuk JD, et al. The PD-1/PD-L costimulatory pathway critically affects host resistance to the pathogenic fungus histoplasma capsulatum. Proc Natl Acad Sci USA (2008) 105(7):2658–63. doi: 10.1073/pnas.0711918105
44. McGaha T, Murphy JW. CTLA-4 down-regulates the protective anticryptococcal cell-mediated immune response. Infect Immun (2000) 68(8):4624–30. doi: 10.1128/IAI.68.8.4624-4630.2000
45. Inoue S, Bo L, Bian J, Unsinger J, Chang K, Hotchkiss RS. Dose-dependent effect of anti-CTLA-4 on survival in sepsis. Shock; (2011) 36(1):38–44. doi: 10.1097/SHK.0b013e3182168cce
46. Vu CTB, Thammahong A, Yagita H, Azuma M, Hirankarn N, Ritprajak P, et al. Blockade of PD-1 attenuated postsepsis aspergillosis Via the activation of IFN-γ and the dampening of IL-10. Shock; (2020) 53(4):514–24. doi: 10.1097/SHK.0000000000001392
47. Wurster S, Robinson P, Albert ND, Tarrand JJ, Goff M, Swamydas M, et al. Protective activity of programmed cell death protein 1 blockade and synergy with caspofungin in a murine invasive pulmonary aspergillosis model. J Infect Dis (2020) 222(6):989–94. doi: 10.1093/infdis/jiaa264
48. Simitsopoulou M, Roilides E, Walsh TJ. Immunomodulatory properties of antifungal agents on phagocytic cells. Immunol Invest (2011) 40(7-8):809–24. doi: 10.3109/08820139.2011.615877
49. Ben-Ami R, Lewis RE, Kontoyiannis DP. Immunocompromised hosts: immunopharmacology of modern antifungals. Clin Infect Dis (2008) 47(2):226–35. doi: 10.1086/589290
50. Lamaris GA, Lewis RE, Chamilos G, May GS, Safdar A, Walsh TJ, et al. Caspofungin-mediated beta-glucan unmasking and enhancement of human polymorphonuclear neutrophil activity against aspergillus and non-aspergillus hyphae. J Infect Dis (2008) 198(2):186–92. doi: 10.1086/589305
51. Wurster S, Albert ND, Bharadwaj U, Kasembeli MM, Tarrand JJ, Daver N, et al. Blockade of the PD-1/PD-L1 immune checkpoint pathway improves infection outcomes and enhances fungicidal host defense in a murine model of invasive pulmonary mucormycosis. Front Immunol (2022) 13:838344. doi: 10.3389/fimmu.2022.838344
52. Grimaldi D, Pradier O, Hotchkiss RS, Vincent JL. Nivolumab plus interferon-γ in the treatment of intractable mucormycosis. Lancet Infect Dis (2017) 17(1):18. doi: 10.1016/S1473-3099(16)30541-2
53. Banck JC, Mueller N, Mellinghoff SC, Thelen M, Fraccaroli A, Blumenberg V, et al. Immune checkpoint blockade for aspergillosis and mucormycosis coinfection. Hemasphere; (2021) 5(3):e530. doi: 10.1097/HS9.0000000000000530
54. Lukaszewicz AC, Venet F, Boibieux A, Lherm M, Devigne B, Monneret G. Nivolumab and interferon-γ rescue therapy to control mixed mould and bacterial superinfection after necrotizing fasciitis and septic shock. Med Mycol Case Rep (2022) 37:19–22. doi: 10.1016/j.mmcr.2022.06.003
55. Serris A, Ouedrani A, Uhel F, Gazzano M, Bedarida V, Rouzaud C, et al. Case report: Immune checkpoint blockade plus interferon-Γ add-on antifungal therapy in the treatment of refractory covid-associated pulmonary aspergillosis and cerebral mucormycosis. Front Immunol (2022) 13:900522. doi: 10.3389/fimmu.2022.900522
56. Khatamzas E, Mellinghoff SC, Thelen M, Schlößer HA, Kunz WG, Buerkle C, et al. Nivolumab induces long-term remission in a patient with fusariosis. Eur J Cancer (2022) 173:91–4. doi: 10.1016/j.ejca.2022.06.035
57. van Enckevort FH, Netea MG, Hermus AR, Sweep CG, Meis JF, Van der Meer JW, et al. Increased susceptibility to systemic candidiasis in interleukin-6 deficient mice. Med Mycol (1999) 37(6):419–26. doi: 10.1046/j.1365-280X.1999.00247.x
58. Hommes JW, Verheijden RJ, Suijkerbuijk KPM, Hamann D. Biomarkers of checkpoint inhibitor induced immune-related adverse events-a comprehensive review. Front Oncol. (2021) 10:585311. doi: 10.3389/fonc.2020.585311
59. Better J, Matt U. Pneumocystis pneumonia: Checkpoint inhibition to the rescue? Am J Respir Cell Mol Biol (2020) 62(6):674–5. doi: 10.1165/rcmb.2020-0051ED
60. Uchida N, Fujita K, Nakatani K, Mio T. Acute progression of aspergillosis in a patient with lung cancer receiving nivolumab. Respirol Case Rep (2017) 6(2):e00289. doi: 10.1002/rcr2.289
61. Barber DL, Andrade BB, Sereti I, Sher A. Immune reconstitution inflammatory syndrome: the trouble with immunity when you had none. Nat Rev Microbiol (2012) 10(2):150–6. doi: 10.1038/nrmicro2712
62. Lasagna A, Cassaniti I, Sacchi P, Baldanti F, Bruno R, Pedrazzoli P. Infectious complications and immunotherapy: old pitfalls and new horizons. Future Oncol (2022) 18(22):2377–81. doi: 10.2217/fon-2022-0277
63. Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol (2019) 16(9):563–80. doi: 10.1038/s41571-019-0218-0
64. Hohl TM. Overview of vertebrate animal models of fungal infection. J Immunol Methods (2014) 410:100–12. doi: 10.1016/j.jim.2014.03.022
65. Desoubeaux G, Cray C. Rodent models of invasive aspergillosis due to aspergillus fumigatus: Still a long path toward standardization. Front Microbiol (2017) 8:841. doi: 10.3389/fmicb.2017.00841
66. Leiva-Juárez MM, Ware HH, Kulkarni VV, Zweidler-McKay PA, Tuvim MJ, Evans SE. Inducible epithelial resistance protects mice against leukemia-associated pneumonia. Blood; (2016) 128(7):982–92. doi: 10.1182/blood-2016-03-708511
67. Xin H, Mohiuddin F, Tran J, Adams A, Eberle K, et al. Experimental mouse models of disseminated candida auris infection. mSphere; (2019) 4(5):e00339–19. doi: 10.1128/mSphere.00339-19
68. Capilla J, Clemons KV, Stevens DA, Adams A, Eberle K. Animal models: an important tool in mycology. Med Mycol (2007) 45(8):657–84. doi: 10.1080/13693780701644140
69. Cortegiani A, Misseri G, Fasciana T, Giammanco A, Giarratano A, Chowdhary A. Epidemiology, clinical characteristics, resistance, and treatment of infections by candida auris. J Intensive Care (2018) 6:69. doi: 10.1186/s40560-018-0342-4
70. Thomas-Rüddel DO, Schlattmann P, Pletz M, Kurzai O, Bloos F. Risk factors for invasive candida infection in critically ill patients: A systematic review and meta-analysis. Chest; (2022) 161(2):345–55. doi: 10.1016/j.chest.2021.08.081
71. Garcia-Perez JE, Mathé L, Humblet-Baron S, Braem A, Lagrou K, Van Dijck P, et al. A framework for understanding the evasion of host immunity by candida biofilms. Front Immunol (2018) 9:538. doi: 10.3389/fimmu.2018.00538
72. Huggins MA, Sjaastad FV, Pierson M, Kucaba TA, Swanson W, Staley C, et al. Microbial exposure enhances immunity to pathogens recognized by TLR2 but increases susceptibility to cytokine storm through TLR4 sensitization. Cell Rep (2019) 28(7):1729–1743.e5. doi: 10.1016/j.celrep.2019.07.028
73. Zhang C, Franklin CL, Ericsson AC. Consideration of gut microbiome in murine models of diseases. Microorganisms; (2021) 9(5):1062. doi: 10.3390/microorganisms9051062
74. Lee KA, Thomas AM, Bolte LA, Björk JR, de Ruijter LK, Armanini F, et al. Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Nat Med (2022) 28(3):535–44. doi: 10.1038/s41591-022-01695-5
75. Wu J, Wang S, Zheng B, Qiu X, Wang H, Chen L. Modulation of gut microbiota to enhance effect of checkpoint inhibitor immunotherapy. Front Immunol (2021) 12:669150. doi: 10.3389/fimmu.2021.669150
76. Tobin JM, Nickolich KL, Ramanan K, Pilewski MJ, Lamens KD, Alcorn JF, et al. Influenza suppresses neutrophil recruitment to the lung and exacerbates secondary invasive pulmonary aspergillosis. J Immunol (2020) 205(2):480–8. doi: 10.4049/jimmunol.2000067
77. Cogdill AP, Andrews MC, Wargo JA. Hallmarks of response to immune checkpoint blockade. Br J Cancer (2017) 117(1):1–7. doi: 10.1038/bjc.2017.136
78. Jiang X, Liu G, Li Y, Pan Y. Immune checkpoint: The novel target for antitumor therapy. Genes Dis (2019) 8(1):25–37. doi: : 10.1016/j.gendis.2019.12.004
79. Romani L. Immunity to fungal infections. Nat Rev Immunol (2011) 11(4):275–88. doi: 10.1038/nri2939
80. Pant A, Medikonda R, Lim M. Alternative checkpoints as targets for immunotherapy. Curr Oncol Rep (2020) 22(12):126. doi: 10.1007/s11912-020-00983-y
81. Holder KA, Grant MD. TIGIT blockade: A multipronged approach to target the HIV reservoir. Front Cell Infect Microbiol (2020) 10:175. doi: 10.3389/fcimb.2020.00175
82. Jin HT, Anderson AC, Tan WG, West EE, Ha SJ, Araki K, et al. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc Natl Acad Sci USA (2010) 107(33):14733–8. doi: 10.1073/pnas.1009731107
83. Thommen DS, Schreiner J, Müller P, Herzig P, Roller A, Belousov A, et al. Progression of lung cancer is associated with increased dysfunction of T cells defined by coexpression of multiple inhibitory receptors. Cancer Immunol Res (2015) 3:1344–55. doi: 10.1158/2326-6066.CIR-15-0097
84. Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR. Resistance to checkpoint inhibition in cancer immunotherapy. Transl Oncol (2020) 13(3):100738. doi: 10.1016/j.tranon.2019.12.010
85. Richards DM, Sefrin JP, Gieffers C, Hill O, Merz C. Concepts for agonistic targeting of CD40 in immuno-oncology. Hum Vaccin Immunother (2020) 16(2):377–87. doi: 10.1080/21645515.2019.1653744
86. Linch SN, McNamara MJ, Redmond WL, Hill O, Merz C. OX40 agonists and combination immunotherapy: Putting the pedal to the metal. Front Oncol (2015) 5:34. doi: 10.3389/fonc.2015.00034
87. Koehler FC, Cornely OA, Wisplinghoff H, Schauss AC, Salmanton-Garcia J, Ostermann H, et al. Candida-reactive T cells for the diagnosis of invasive candida infection-a prospective pilot study. Front Microbiol (2018) 9:1381. doi: 10.3389/fmicb.2018.01381
88. Steinbach A, Cornely OA, Wisplinghoff H, Schauss AC, Vehreschild JJ, Rybniker J, et al. Mould-reactive T cells for the diagnosis of invasive mould infection-a prospective study. Mycoses; (2019) 62(7):562–9. doi: 10.1111/myc.12919
89. Stuehler C, Nowakowska J, Bernardini C, Topp MS, Battegay M, Passweg J, et al. Multispecific aspergillus T cells selected by CD137 or CD154 induce protective immune responses against the most relevant mold infections. J Infect Dis (2015) 211(8):1251–61. doi: 10.1093/infdis/jiu607
90. Wheeler RT, Kombe D, Agarwala SD, Fink GR. Dynamic, morphotype-specific candida albicans beta-glucan exposure during infection and drug treatment. PloS Pathog (2008) 4(12):e1000227. doi: : 10.1371/journal.ppat.1000227
91. Salvenmoser S, Seidler MJ, Dalpke A, Müller FM. Effects of caspofungin, candida albicans and aspergillus fumigatus on toll-like receptor 9 of GM-CSF-stimulated PMNs. FEMS Immunol Med Microbiol (2010) 60(1):74–7. doi: 10.1111/j.1574-695X.2010.00720.x
92. Lewis RE, Chamilos G, Prince RA, Kontoyiannis DP. Pretreatment with empty liposomes attenuates the immunopathology of invasive pulmonary aspergillosis in corticosteroid-immunosuppressed mice. Antimicrob Agents Chemother (2007) 51(3):1078–81. doi: 10.1128/AAC.01268-06
93. Bellocchio S, Gaziano R, Bozza S, Rossi G, Montagnoli C, Perruccio K, et al. Liposomal amphotericin b activates antifungal resistance with reduced toxicity by diverting toll-like receptor signalling from TLR-2 to TLR-4. J Antimicrob Chemother (2005) 55(2):214–22. doi: 10.1093/jac/dkh542
94. Abzug MJ, Walsh TJ. Interferon-gamma and colony-stimulating factors as adjuvant therapy for refractory fungal infections in children. Pediatr Infect Dis J (2004) 23(8):769–73. doi: 10.1097/01.inf.0000134314.65398.bf
95. Ding G, Shen T, Yan C, Zhang M, Wu Z, Cao L. IFN-γ down-regulates the PD-1 expression and assist nivolumab in PD-1-blockade effect on CD8+ T-lymphocytes in pancreatic cancer. BMC Cancer (2019) 19(1):1053. doi: 10.1186/s12885-019-6145-8
96. Kverneland AH, Pedersen M, Westergaard MCW, Nielsen M, Borch TH, Olsen LR, et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget; (2020) 11(22):2092–105. doi: 10.18632/oncotarget.27604
97. Poorebrahim M, Melief J, Pico de Coaña Y, Wickström S L, Cid-Arregui A, Kiessling R, et al. Counteracting CAR T cell dysfunction. Oncogene; (2021) 40(2):421–35. doi: 10.1038/s41388-020-01501-x
98. Gumber D, Wang LD. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine; (2022) 77:103941. doi: 10.1016/j.ebiom.2022.103941
99. Serganova I, Moroz E, Cohen I, Moroz M, Mane M, Zurita J, et al. Enhancement of PSMA-directed CAR adoptive immunotherapy by PD-1/PD-L1 blockade. Mol Ther Oncolytics (2016) 4:41–54. doi: 10.1016/j.omto.2016.11.005
100. John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res (2013) 19(20):5636–46. doi: 10.1158/1078-0432.CCR-13-0458
101. Papadopoulou A, Kaloyannidis P, Yannaki E, Cruz CR. Adoptive transfer of aspergillus-specific T cells as a novel anti-fungal therapy for hematopoietic stem cell transplant recipients: Progress and challenges. Crit Rev Oncol Hematol (2016) 98:62–72. doi: 10.1016/j.critrevonc.2015.10.005
102. Tramsen L, Schmidt S, Boenig H, Latgé JP, Lass-Flörl C, Roeger F, et al. Clinical-scale generation of multi-specific anti-fungal T cells targeting candida, aspergillus and mucormycetes. Cytotherapy; (2013) 15(3):344–51. doi: 10.1016/j.jcyt.2012.11.014
103. Dimitriou F, Hogan S, Menzies AM, Dummer R, Long GV. Interleukin-6 blockade for prophylaxis and management of immune-related adverse events in cancer immunotherapy. Eur J Cancer (2021) 157:214–24. doi: 10.1016/j.ejca.2021.08.031
104. Arastehfar A, Carvalho A, van de Veerdonk FL, Jenks JD, Koehler P, Krause R, et al. COVID-19 associated pulmonary aspergillosis (CAPA)-from immunology to treatment. J Fungi (Basel) (2020) 6(2):91. doi: 10.3390/jof6020091
105. Lionakis MS, Albert ND, Swamydas M, Lee CR, Loetscher P, Kontoyiannis DP. Pharmacological blockade of the chemokine receptor CCR1 protects mice from systemic candidiasis of hematogenous origin. Antimicrob Agents Chemother (2017) 61(3):e02365–16. doi: 10.1128/AAC.02365-16
106. Goldszmid RS, Dzutsev A, Trinchieri G. Host immune response to infection and cancer: unexpected commonalities. Cell Host Microbe (2014) 15(3):295–305. doi: 10.1016/j.chom.2014.02.003
107. Petitprez F, Meylan M, de Reyniès A, Sautès-Fridman C, Fridman WH. The tumor microenvironment in the response to immune checkpoint blockade therapies. Front Immunol (2020) 11:784. doi: 10.3389/fimmu.2020.00784
108. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA (2015) 112:6140–5. doi: 10.1073/pnas.1417320112
109. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol (2019) 106(2):345–58. doi: 10.1002/JLB.3RU1018-378RR
110. Zhang X, He D, Gao S, Wei Y, Wang L. Aspergillus fumigatus enhances human NK cell activity by regulating M1 macrophage polarization. Mol Med Rep (2019) 20(2):1241–9. doi: 10.3892/mmr.2019.10365
111. Zheng XL, Liang GZ, Shi DM, Yao HP, Zhang L, Liu WD, et al. Itraconazole promotes macrophage M1 polarization and phagocytic capacity of macrophage to candida albicans. Int J Dermatol Venereology (2019) 2(4):193–201. doi: 10.1097/JD9.0000000000000044
112. Chen X, Xu R, He D, Zhang Y, Chen H, Zhu Y, et al. CD8+ T effector and immune checkpoint signatures predict prognosis and responsiveness to immunotherapy in bladder cancer. Oncogene; (2021) 40(43):6223–34. doi: 10.1038/s41388-021-02019-6
113. Nojima Y, Shimizu K, Saisho S, Maeda AI, Kurosaki T, Kurose K, et al. Tumor PD-L1 and VEGF expression, and CD8 T cell infiltration predict clinical response to immune checkpoint inhibitors in non-small cell lung cancer. Anticancer Res (2021) 41(11):5469–75. doi: 10.21873/anticanres.15359
114. Lauruschkat CD, Etter S, Schnack E, Ebel F, Schäuble S, Page L, et al. Chronic occupational mold exposure drives expansion of aspergillus-reactive type 1 and type 2 T-helper cell responses. J Fungi (Basel) (2021) 7(9):698. doi: 10.3390/jof7090698
115. Page L, Weis P, Müller T, Dittrich M, Lazariotou M, Dragan M, et al. Evaluation of aspergillus and mucorales specific T-cells and peripheral blood mononuclear cell cytokine signatures as biomarkers of environmental mold exposure. Int J Med Microbiol (2018) 308(8):1018–26. doi: 10.1016/j.ijmm.2018.09.002
116. Scheffold A, Bacher P. Anti-fungal T cell responses in the lung and modulation by the gut-lung axis. Curr Opin Microbiol (2020) 56:67–73. doi: 10.1016/j.mib.2020.06.006
117. Bacher P, Hohnstein T, Beerbaum E, Röcker M, Blango MG, Kaufmann S, et al. Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against candida albicans. Cell; (2019) 176(6):1340–1355.e15. doi: 10.1016/j.cell.2019.01.041
118. Hernández-Chávez MJ, Pérez-García LA, Niño-Vega GA, Mora-Montes HM. Fungal strategies to evade the host immune recognition. J Fungi (Basel) (2017) 3(4):51. doi: 10.3390/jof3040051
119. Marcos CM, de Oliveira HC, de Melo WC, da Silva JF, Assato PA, Scorzoni L, et al. Anti-immune strategies of pathogenic fungi. Front Cell Infect Microbiol (2016) 6:142. doi: 10.3389/fcimb.2016.00142
120. Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J Clin Invest (2018) 128:5137–49. doi: 10.1172/JCI96268
121. Chung H, Lee YH. Hypoxia: A double-edged sword during fungal pathogenesis? Front Microbiol (2020) 11:1920. doi: 10.3389/fmicb.2020.01920
122. Fliesser M, Wallstein M, Kurzai O, Einsele H, Löffler J. Hypoxia attenuates anti-aspergillus fumigatus immune responses initiated by human dendritic cells. Mycoses; (2016) 59(8):503–8. doi: 10.1111/myc.12498
123. Pradhan A, Avelar GM, Bain JM, Childers DS, Larcombe DE, Netea MG, et al. Hypoxia promotes immune evasion by triggering β-glucan masking on the candida albicans cell surface via mitochondrial and cAMP-protein kinase a signaling. mBio; (2018) 9(6):e01318–18. doi: 10.1128/mBio.01318-18
124. Ben-Ami R, Lewis RE, Leventakos K, Kontoyiannis DP, et al. Aspergillus fumigatus inhibits angiogenesis through the production of gliotoxin and other secondary metabolites. Blood; (2009) 114(26):5393–9. doi: 10.1182/blood-2009-07-231209
125. Daver N, Kontoyiannis DP. Checkpoint inhibitors and aspergillosis in AML: the double hit hypothesis. Lancet Oncol (2017) 18(12):1571–3. doi: 10.1016/S1470-2045(17)30852-5
126. Jimbu L, Mesaros O, Popescu C, Neaga A, Berceanu I, Dima D, et al. Is there a place for PD-1-PD-L blockade in acute myeloid leukemia? Pharm (Basel) (2021) 14(4):288. doi: 10.3390/ph14040288
127. Daver N, Boddu P, Garcia-Manero G, Yadav SS, Sharma P, Allison J, et al. Hypomethylating agents in combination with immune checkpoint inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Leukemia; (2018) 32(5):1094–105. doi: 10.1038/s41375-018-0070-8
128. Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in Relapsed/Refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discov (2019) 9(3):370–83. doi: 10.1158/2159-8290.CD-18-0774
129. Lauruschkat CD, Page L, White PL, Etter S, Davies HE, Duckers J, et al. Development of a simple and robust whole blood assay with dual Co-stimulation to quantify the release of T-cellular signature cytokines in response to aspergillus fumigatus antigens. J Fungi (Basel) (2021) 7(6):462. doi: 10.3390/jof7060462
130. Hünniger K, Lehnert T, Bieber K, Martin R, Figge MT, Kurzai O. A virtual infection model quantifies innate effector mechanisms and candida albicans immune escape in human blood. PloS Comput Biology (2014) 10(2):e1003479. doi: 10.1371/journal.pcbi.1003479
131. Chao MP, Takimoto CH, Feng DD, McKenna K, Gip P, Liu J, et al. Therapeutic targeting of the macrophage immune checkpoint CD47 in myeloid malignancies. Front Oncol (2020) 9:1380. doi: 10.3389/fonc.2019.01380
Keywords: immunotherapy, fungal sepsis, candidiasis, aspergillosis, mucormycosis, checkpoint inhibitors, T cells, immune exhaustion
Citation: Wurster S, Watowich SS and Kontoyiannis DP (2022) Checkpoint inhibitors as immunotherapy for fungal infections: Promises, challenges, and unanswered questions. Front. Immunol. 13:1018202. doi: 10.3389/fimmu.2022.1018202
Received: 12 August 2022; Accepted: 11 October 2022;
Published: 25 October 2022.
Edited by:
Mario César Salinas-Carmona, Autonomous University of Nuevo León, MexicoReviewed by:
Mark S. Gresnigt, Hans Knöll Institute, GermanyCopyright © 2022 Wurster, Watowich and Kontoyiannis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sebastian Wurster, c3R3dXJzdGVyQG1kYW5kZXJzb24ub3Jn; Dimitrios P. Kontoyiannis, ZGtvbnRveWlAbWRhbmRlcnNvbi5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.