 Jiqi Shan1†
Jiqi Shan1† Yi Zhang
Yi Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 September 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1016646
This article is part of the Research Topic Gastrointestinal Cancer Immunotherapy: from Drug Resistance Mechanisms to Overcoming Strategies View all 16 articles
Colorectal cancer (CRC) is the third most common cancer in the world. Although there are standard treatment options for CRC, most patients respond poorly to these treatments. Immunotherapies have gradually emerged due to the increasing awareness and understanding of tumor immunity, exhibiting good therapeutic efficacy in various cancers. Immunotherapies include cytokines, immune checkpoint inhibitors (ICIs), and adoptive cell therapies. In particular, ICIs, which are antibodies against cytotoxic T lymphocyte-associated protein 4 (CTLA-4), programmed cell death 1 (PD-1), or its ligand PD-L1, have been successfully applied clinically for solid tumors, relieving the inhibitory effect of the tumor microenvironment on T cells. However, only a minority of patients with cancer achieve a durable clinical response during immunotherapy. Several factors restrict the efficacy of immunotherapy, leading to the development of drug resistance. In this review, we aimed to discuss the current status of immunotherapy for CRC and elaborate on the mechanisms that mediate resistance to immunotherapy and other potential therapeutic strategies.
Colorectal cancer (CRC) has a high morbidity rate and poor prognosis. The five-year survival rate for patients with advanced CRC is around 14%, and metastasis occurs in more than 50% of patients with CRC (1, 2). Immunotherapies comprise a novel and effective therapeutic strategy for patients with various cancers. With recent developments in cancer immunotherapy, both hematological and solid tumors respond to this treatment. In the last decade, immunotherapy has become popular as an alternative to surgery, chemotherapy, and radiotherapy for treating various tumors (3, 4).
Immune checkpoints are a class of molecules expressed on the surface of immune cells that regulate the level of immune activation. They prevent autoimmune abnormalities and launch immune attacks on normal cells. However, in tumors, immune checkpoints, such as programmed cell death 1 (PD-1) and cytotoxic T lymphocyte antigen 4 (CTLA-4), are abnormally activated, resulting in a weakened tumor immune response (5–7). As a result of its ability to interfere with the interaction between immune checkpoints and their receptors, immune checkpoint blockade (ICB) therapy has shown impressive therapeutic effects on a wide variety of tumor types. For CRC, two PD-1-blocking antibodies, pembrolizumab and nivolumab, which are already approved by the Food and Drug Administration (FDA), have shown efficacy in patients with mCRC(metastatic colorectal cancer) that are mismatch-repair-deficient and have high microsatellite instability (dMMR–MSI-H). The ICB drug ipilimumab, a fully humanized monoclonal antibody, blocks the CTLA-4 receptor and has also been approved by the FDA in combination with nivolumab to treat dMMR-MSI-H CRC (8–10). In a study of patients with locally advanced mismatch repair-deficient (dMMR) CRC, 14 patients achieved complete clinical remission after six months of treatment with the anti-PD-1 drug dostarlimab-gxly alone. The exciting result is that dostarlimab has saved all patients in that study from chemotherapy, radiation, or surgery (11). However,immunotherapy is effective in some cases of dMMR-MSI-H CRC but is minimally effective in pMMR-MSI-L CRC. Many colon cancer patients show resistance to immunotherapy. Hence, improving the efficacy of immunotherapy for dMMR-MSI-H and exploring new mechanisms for the treatment of pMMR-MSI-L CRC is key to improving the prognosis of patients with tumors.
Colorectal cancer can be classified through a mismatch repair/microsatellite instability system. Microsatellites represent a kind of tandem repeats including 1–6 nucleotides that frequently occur in nuclear genomes (12). Microsatellite instability (MSI) refers to the insertion/deletion of repeated DNA nucleotide units in microsatellites. During DNA replication, the mismatch repair (MMR) system corrects insertions, deletions, or mismatched bases and identifies and repairs DNA damage (13). MMR deficiency results in the failure to detect and correct microsatellite replication errors, resulting in diffuse MSI. High microsatellite instability usually gives rise to the accumulation of somatic mutations. Of these, frameshift mutations are highly positively correlated with the frequency of neoantigens, MSI, and MMR deficiency. They are usually detected in CRC via immunohistochemistry as the loss of MMR proteins (MSH2, MSH6, MLH1 and PMS2) or by testing MSI via PCR (14, 15). Based on the mutations of MMR proteins and MSI, CRC can be classified into three main types: dMMR-MSI-H tumors with a higher overall mutation burden (>12 mutations per 106 DNA bases), pMMR–MSI-L tumors with a much lower mutation burden (<8.24 mutations per 106 DNA bases), and pMMR-MSS tumors lacking MSI features (13, 16). The MSS/MSI-L subtypes occupy a large proportion (85%) of CRC cases, whereas the dMMR/MSI-H patients accounts for only approximately 15% of all CRC cases and 5% of the mCRCs (17).
Immune cell function and classification are key factors in immunotherapy, in which tumor-infiltrating lymphocytes (TILs), as the main force of adaptive immune response, play a pivotal role in defense against tumors (18, 19). And powerful immune cytolytic activity (CYT) badgers deeply with many factors including tumor mutation burden and deregulated immune checkpoint (20, 21). Compared to those harboring pMMR-MSI-L/MSS, dMMR-MSI-H patients usually showed satisfactory prognosis and responded better to ICIs, since they owned a high mutational burden whose accumulation might produce more neoantigens for immune recognition. Because of the higher neoantigen load, dMMR–MSI-H tumors are usually heavily infiltrated by functional TILs, which quickly start it activation program and release a large number of cytokines when receiving stimulus from antigen-presenting cells (4, 22). Furthermore, it proves that MSI-CRC has higher expression levels of immune checkpoints, such as PD-L1, CTLA-4 as well as LAG-3, compared with MSS-CRC, which may explain the positive response of dMMR–MSI-H subtype to immunotherapies (18). A phase 2 clinical study (NCT03206073) confirmed that patients with MSI-CRC show much better progression-free survival (PFS) and objective response rate (ORR) than MSS ones when treated with pembrolizumab. Nevertheless, there are few functional TILs and many immunosuppressive cells, consisting of Tregs, MDSCs, and TAMs, infiltrated in the pMMR-MSI-L tumor microenvironment (Figure 1).
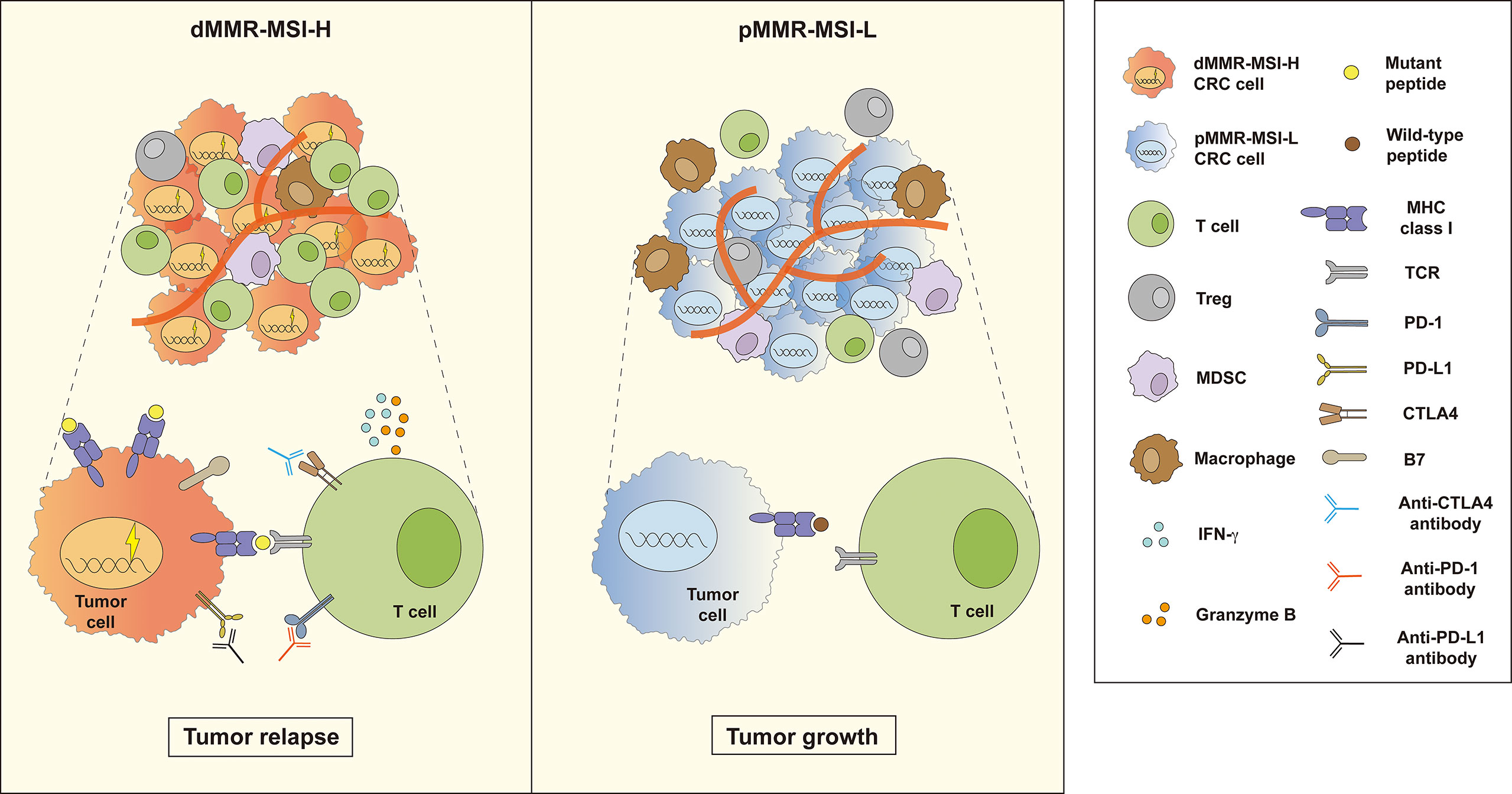
Figure 1 Two outcomes of immunotherapy in dMMR–MSI-H and pMMR–MSI-L CRC. Compared with pMMR–MSI-L, patients with dMMR–MSI-H experience better tumor reduction after treatment with immune checkpoint inhibitors (ICIs). Many functional tumor infiltrating lymphocytes (TILs) release a large number of cytokines such as IFN-γ and granzyme B in the dMMR–MSI-H tumor microenvironment (TME). However, the TME of pMMR–MSI-L CRC contains fewer functional TILs and more immunosuppressive cells, such as Tregs, MDSCs, and TAMs, which inhibit TIL function.
Multiple clinical trials have been conducted to estimate the effects of inhibiting PD-1, PD-L1, or CTLA-4 in dMMR–MSI-H and pMMR–MSI-L CRC. NCT02460198 is a phase II study conducted on 124 previously treated patients with locally advanced unresectable or metastatic dMMR/MSI-H CRC (cohort A: standard therapies including fluoropyrimidine, oxaliplatin, and irinotecan; cohort B: fluoropyrimidine + oxaliplatin or fluoropyrimidine + irinotecan, with or without anti-VEGF/EGFR monoclonal antibody) (23). Pembrolizumab (200 mg) was intravenously administrated to these patients every three weeks until approximately 52 cycles. ORR was the primary endpoint, and disease control rate, PFS, overall survival (OS), tolerability, safety and response duration were the secondary endpoints. The results showed that the ORR was 32.8% (95% CI, 21.3% to 46%) and 34.9% (95% CI, 23.3% to 48.0%) in cohorts A and B, respectively. The median PFS was 2.3 months (95% CI, 2.1 to 8.1 months) and the median OS was 31.4 months (95% CI, 21.4 to 58.0 months) for cohort A. In contrast, the median PFS was 4.1 months (95% CI, 2.1 to 18.9 months) and the median OS was 47.0 months (95% CI, 19.2 months to not reached) for cohort B. Although most of the patients suffered different adverse events during the trial, only five patients withdrew from either cohort because of adverse events. Finally, pembrolizumab proved effective and safe in dMMR/MSI-H CRCs. Compared with blocking PD-1, the effectiveness of strategies to block PDL1 or CTLA-4 is subtle and seems to be weaker in clinical trials. Another phase II trial (NCT02870920) utilizes durvalumab combined with tremelimumab for treating patients with advanced CRC receiving the best supportive care, the treatment group showed a longer OS (median OS: 6.6 months vs. 4.1 months; P-value = 0.07) than the control group (24). However, superior PFS in treatment group was not observed, with a 95% CI ranging from 1.8 to 1.9 months. In addition, the ORR was only 0.8% (one patient) in the anti-PD-L1/CTLA-4 therapy group. ICIs have a significant impact on tumor treatment, nevertheless, only partial patients benefit from this regimen. This motivated us to deepen understanding of tumor immunity and explore new solutions for this disease. To date, the FDA approved three immunotherapeutic drugs between 2017 and 2018 (pembrolizumab, nivolumab, and ipilimumab) to treat MSI-H/dMMR CRC (25). Pembrolizumab has become the first-line regimen for treating CRC cases owning an MSI-H/dMMR or unresectable phenotype. Nevertheless, due to the immunosuppressive TME and the significantly low ratio of the MSI-H/dMMR subset in all patients with CRC, patients show resistance to immunotherapy and obtain limited improvement from these immunotherapies. Thus, developing new immunotherapeutic targets and combined therapeutic strategies has become a hot topic (Table 1 and Table 2). And numerous trials have focused on these issues through different mechanisms, such as remolding the antibody structure (Tislelizumab, KN035), combining with other targets (anti-VEGF) or plus with chemoradiotherapy, etc. For example, KN035 is a novel anti-PD-L1 antibody with remolded structure empowering it with superior solubility and stability, which makes KN035 the first checkpoint inhibitor administered subcutaneously. A single-arm, phase II study NCT03667170 found that KN035 therapy showed a pretty good ORR of 43.1% (95% CI, 30.8% to 56.0%) in a cohort of 65 advanced CRC patients during a 28-day treatment cycle (26). Since most of the newly admitted clinical trials are still in recruiting status, the results of these strategies are in urgent to see.
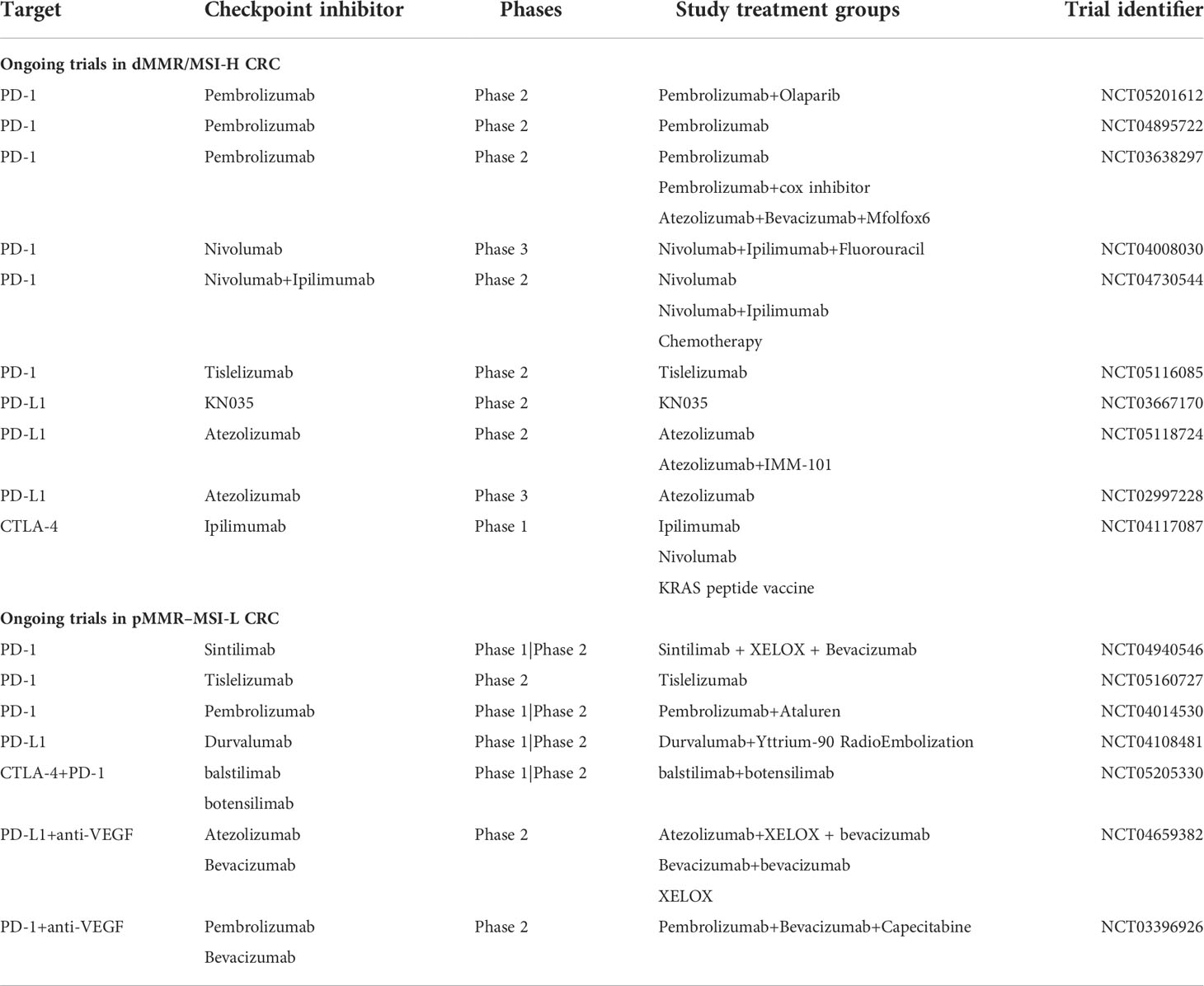
Table 1 Table.
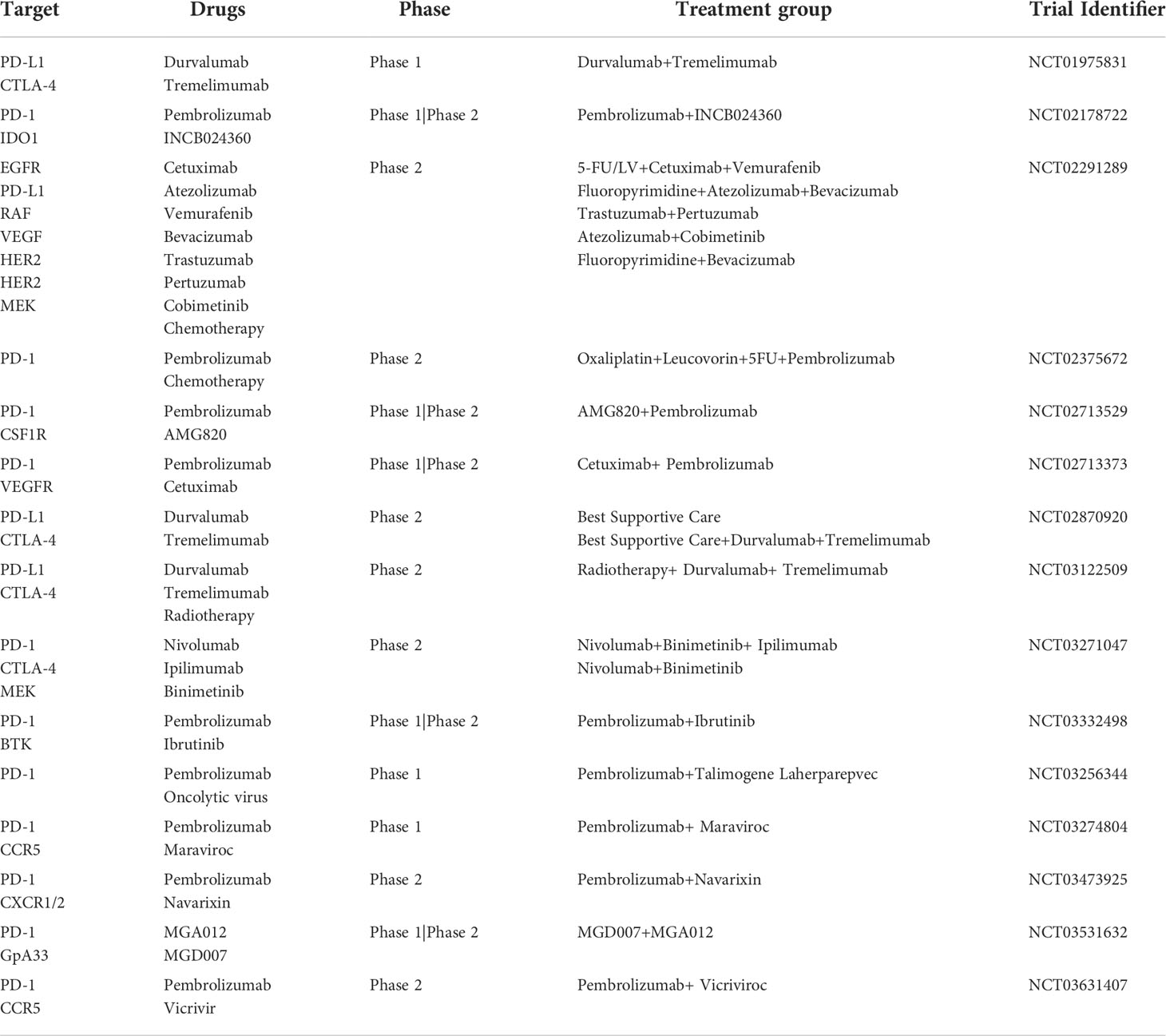
Table 2 Trials using combination immunotherapies for CRC.
Multiple mechanisms are involved in the process of immunotherapy resistance. Immunotherapy resistance can divided into Tumor-intrinsic and -extrinsic (Figure 2).
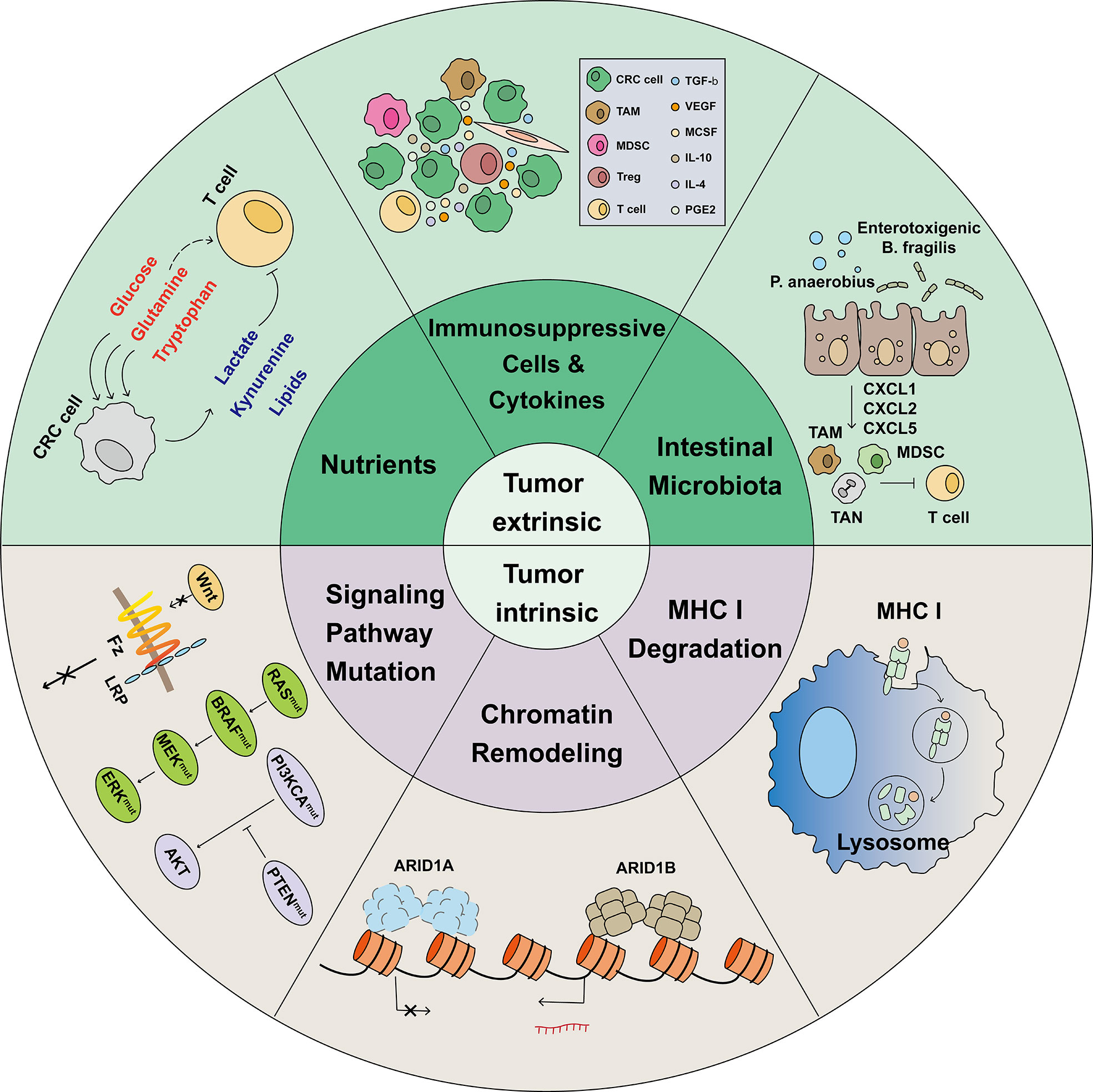
Figure 2 Mechanisms associated with immunotherapy resistance. The limited effectiveness of immunotherapy is primarily due to various mechanisms of immunotherapy resistance. In the heterogeneous tumor microenvironment, cells such as Tregs, MDSCs, and TAMs, combined with tumor-released immunosuppressive cytokines, induce tumor infiltrating lymphocytes (TILs) exhaustion. In the TME, tumor cells have a greater ability to compete for nutrients, such as glucose, glutamine, and tryptophan, which are necessary for proper cellular function. Meanwhile, tumor cells release lactate, kynurenine, and Oxidized low density lipoprotein (oxLDLs), which are harmful to TILs. In CRC, intestinal microbiota such as P. anaerobius and enterotoxigenic B ragilis induce tumor cells to release CXCL1, CXCL2, and CXCL5 and recruit immunosuppressive cells. Before identifying and killing TILs, tumor cells activate the lysosomal degradation pathway of MHC class 1 to escape T cell killing. The switching/sucrose non-fermentable (SWI/SNF) complex has been identified as a tumor suppressor gene in CRC; AT-Rich Interactive Domain-containing protein 1A (ARID1A) is the most frequent target of SWI/SNF mutations. However, ARID1A mutations was found correlated with markedly higher level of immune infiltrates in colon cancer. Gene and signal pathway mutations, such as those in WNT, RAS, BRAF, MEK, ERK, PI3KCA, and PTEN, were reported to be associated with immunotherapy resistance.
Intercellular interaction is a well-known immunosuppressive mechanism in the TME, with the classic PD-L1/PD-1 signaling between tumor cells and CD8+ T cells being proven in different cancers. Nevertheless, many suppressive cells other than tumor cells also profoundly inhibit T-cell cytotoxicity, including Tregs, MDSCs, and TAMs. These cell subsets differentiate improperly and upregulate immunosuppressant-associated proteins, which usually form an anti-inflammatory environment and result in CD8+ T cell dysfunction and immune resistance.
Regulatory T cells (Tregs) are CD25+ FOXP3+ CD4 T cells originating from the thymus or peripheral blood (27). Tregs function as a tumor-promoting component in the TME because they inhibit the immune response against CRC through cell-to-cell contact or the secretion of cytokines and metabolites. Studies report that Tregs regulate the immune response mainly through several mechanisms (27–29). First, they can secrete cytokines such as IL-10, TGF-β, and IL-35, which facilitate Treg proliferation and suppress effector T cell activity (29–31). In addition, the surface marker CD25, which is the α-chain of the IL-2 receptor, can deprive IL-2 because of its high affinity (32, 33). Second, Tregs can exert anti-inflammatory effects by catalyzing the conversion of pro-inflammatory adenosine triphosphate to anti-inflammatory adenosine through the CD39-CD73 axis (34). Third, Tregs constitutively express immune checkpoints, such as CTLA-4, which impair APC maturation and cause T cell exhaustion (35). Finally, it has been reported that Tregs utilize granzyme and perforin to lyse DCs or T cells (36, 37). Tregs also inhibit T cells from exerting anti-tumor functions in various ways. Hence, thinking about how to reduce Treg populations or inhibit their function is one of the key methods for improving the effect of immunotherapy.
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells that originate from the bone marrow. The immunosuppressive effects of MDSCs mainly depend on two enzymes: inducible Nitric Oxide Synthase (iNOS) and Arginase 1 (ARG1). High ARG1 catalyzes L-arginine to urea and ornithine,causes a depletion of L-arginine in the TME. iNOS uses L-arginine to produce NO. The depletion of L-arginine impairs T cell proliferation and activity by decreasing CD3ζ expression (38). In addition, NO, in cooperation with O2-, can result in T cell apoptosis through the tyrosine phosphorylation of the TCR/CD3 complex (39, 40).
Tumor-associated macrophages (TAMs) are key immune components of the TME. Based on their polarization fate, they can be divided into the M1 (pro-inflammatory) and M2 (anti-inflammatory) phenotypes. Interestingly, M1 and M2 cells can repolarize to each other depending on the interventions administered (for example, by targeting mitochondrial metabolic pathways) (41–43). Numerous studies have reported the supportive effects of TAMs on malignant cells in the CRC TME. TAMs can induce tumor angiogenesis by secreting VEGF and MCP-1, MIP-1α, and MIP-2α to promote tumor growth and invasion (44, 45). TAMs impair T cell function in multiple ways, including restricting infiltration, blocking proliferation and activation, and suppressing cytotoxicity. For instance, IL-10 produced by TAMs can decrease CD8 protein levels and impair TCR signaling (46). Other factors such as iNOS, ARG1, and PD-L1 expression also cause T cell dysfunction. Therefore, transforming TAMs from the M2 phenotype to the M1 phenotype is key to restoring T cell function.
Cytokines enriched in the TME are another important pathway that interferes with the efficiency of immunotherapy against tumors. Various studies have concluded the vital role of immunosuppressive cytokines (such as TGF-β, VEGF, IL-4, and IL-10) in the disruption of CD8+ T cell function. In brief, immunosuppressive cytokines are well defined to recruit, polarize, or activate immunosuppressive cells and indirectly interfere with T cell function, although some directly inhibit T cell cytotoxicity. These effects restrict the efficiency of immunotherapy.
TGF-β is a multipotent cytokine that inhibits tumor formation at early stages but promotes tumor progression at advanced stages. Most cells in the TME can produce TGF-β, including cancer cells, fibroblasts, TAMs, Tregs, and even platelets (47, 48). Bardeesy et al. reported that TGF-β/SMAD4 signaling results in premalignant pancreatic cell apoptosis harboring KRAS mutations (49). In addition, primary mesenchymal stromal cells in the TME can induce CXCL5 overexpression in CRC, promoting tumor metastasis and angiogenesis via the CCL7/CCR1/KLF5 pathway; TGF-β/SMAD4 signaling can reverse this effect (50). However, the pro-tumor and immunosuppressive effects of TGF-β are more important in tumor development and are thought to be potential therapeutic targets. For example, TGF-β enhances EMT in CRC through the USF2/S100A8 axis (51). The TGF-β/SMAD pathway decreases CTL cytotoxicity by decreasing perforin, granzyme A, granzyme B and IFN-γ expression (52). TGF-β is also vital for recruiting suppressive immune cells such as Tregs and TAMs.
Vascular endothelial growth factor (VEGF) is a family of cytokines consisting of five members: VEGF-A, VEGF-B, VEGF-C, VEGF-D, and PlGF. This family of proteins is mainly expressed in CRC tissues and the hypoxic TME and is highly involved in tumor progression and metastasis. The classical function of VEGF is to promote tumor angiogenesis via commonly known receptors (VEGFR-1, VEGFR-2, and VEGFR-3) expressed on adjacent vascular endothelia (53, 54). Despite their angiogenic effect, they also strongly influence inflammatory cell infiltration and function. A recent study showed that VEGF-A directly influences T cell exhaustion by inducing the expression of the transcription factor TOX in T cells in MSS CRC (55). Anti-VEGF therapy can also indirectly augment the ability of CD8+ T cells to produce IFN-γ and TNF-α by modulating the hypoxic TME (56).
IL-4 and IL-10 are cytokines classically expressed by Th2 T lymphocytes and are involved in infection and autoimmune diseases. However, they are also highly enriched in multiple cell types in the CRC TME (42, 57–60). Both are important pro-tumor and immune regulatory factors that target IL4R and IL10R receptors, respectively. Koller, F. L. et al. and Liu, H. et al. reported that IL4 could promote HT-29 cell proliferation (61, 62), while Mantilla-Rojas, C. et al. reported that IL-10/SRC contributes to CRC progression (63). IL-4 and IL-10 can suppress CD8+ tumor-infiltrating T cell function by recruiting immunosuppressive components, such as Tregs, M2 TAMs, and MDSCs. However, IL-10 has recently been reported to enhance the anti-tumor effects of CD8+ T cells directly and indirectly (64, 65).
As luminal tract organs communicate with the outside environment, the colon and rectum are exposed to more than 100 trillion microorganisms, including bacteria, fungi, protozoa, and viruses, from their proximal to distal ends. Physically, the gut microbiota is vital for host homeostasis by providing important nutrients such as vitamins and essential amino acids. However, dysbiosis facilitates many pathological processes such as inflammation, dysplasia, and cancer (66–68). Patients with CRC showed significantly different abundances and reduced diversity of the gut microbiota. For example, Fusobacterium nucleatum and Peptostreptococcus anaerobius are highly enriched in CRC tissues; however, Clostridium butyricum and Bifidobacterium animalis are depleted (66, 69). The gut microbiota and its metabolites are widely known to participate in tumor growth and immune responses. F. nucleatum promotes HCT116 and LoVo cell proliferation by activating TLR4 signaling (70). It polarizes TAMs to the M2 phenotype through diverse pathways contributing to CRC progression and metastasis (71, 72). The Fap2 protein produced by F. nucleatum hinders T cell activation by interacting with TIGIT (73). On the contrary, probiotics, a huge commensal bacterial family in the intestine, are of great importance for tumor regression and improving the effects of immunotherapies. Clostridium butyricum releases butyrate into the TME and hinders CRC proliferation by inhibiting HDAC activity (74, 75). Recent studies have shown that the administration of Lactobacillus rhamnosus GG and Lactobacillus casei can facilitate CD8+ TIL infiltration and cytotoxic cytokine secretion, improving the anti-tumor effect of anti-PD-1 agents (76, 77). As a newly identified TME factor in CRC, the influence of the microbiota on tumor cells and CD8+ T cells is complex and requires further research. Patients with CRC generally show an unbalanced bias towards pro-tumor microbiota enrichment, which indicates that recovering intestinal microbiota homeostasis or the exogenous administration of anti-tumor microbiota and its metabolites is closely related to the prognosis associated with CRC immunotherapy.
Altered nutrient metabolism is a hallmark of many tumors. As members of the hypoxic and resource-limited TME, tumor cells and CD8+ T cells urgently require large amounts of nutrients to maintain their survival and biological activities. Tumor cells usually exhibit higher glucose consumption and stronger competitive uptake of essential amino acids, exacerbating the shortage of important nutrients and hindering T cell survival and cytotoxicity. In addition, increased lipid production and the generation of transformation products from glucose and some amino acids are also critical immunosuppressive factors that impair T cell immune responses.
Distinct from the normal colorectal epithelium, CRC cells consume large amounts of glucose to maintain rapid expansion. However, they are prone to glycolysis rather than OXPHOS even in ample oxygen conditions, classically called the Warburg effect. In this process, glucose is converted into pyruvate and ATP together with an important pro-tumor byproduct, lactate, which further contributes to acidosis in the CRC TME (78, 79). Different mechanisms have been reported to regulate glucose uptake by CRC cells. For example, Tang et al. and Wang et al. concluded that the lncRNAs GLCC1 and LINRIS promote tumor glycolysis and facilitate CRC progression by stabilizing c-MYC transcription in mouse or PDX models, correlating with poor prognosis in patients with CRC (80, 81). Moreover, enhanced glycolysis also induced the resistance of CRC cells to chemotherapies such as 5-FU (82). In addition to competitive uptake, malignant cells can directly impair glucose metabolism in T cells by expressing CD155 and combining with TIGIT on the T cell surface, resulting in dysfunction in T cell energy utilization (83–85). The byproduct lactate is also a versatile factor that promotes CRC progression and metastasis through various mechanisms. Deng et al. recently reported that lactate stimulates the tube formation of endothelial cells and eventually angiogenesis in the CRC TME (86). Moreover, several articles have reported the effect of lactate on M2 phenotype polarization in TAMs (87–89). Therefore, eliminating lactate and improving the acidic TME restore CD8+ T cell anti-tumor immunity in many cancers (90–92).
Tryptophan is an essential amino acid that participates extensively in tumor immunity in different cancer types. Three enzymes, IDO1, IDO2, and TDO2, are responsible for tryptophan catabolism, transforming more than 95% of tryptophan to kynurenine, leading to the deficiency of tryptophan in the TME (93, 94). The transformed product, kynurenine, is a profoundly immunosuppressive metabolite. Tumor-derived kynurenine induces PD-1 expression in CD8+ T cells by activating the transcription factor AHR both in a mouse model and in patient samples (95). In addition, kynurenine is negatively correlated with CD8+ T cell infiltration in the CRC TME. The induction of FOXP3 and Treg polarization by kynurenine may contribute to this process (96, 97).
Unlike tryptophan, glutamine is a nonessential amino acid that is highly consumed in many malignancies. It renders many vital biological processes, including nutrient exchange and the biosynthesis of other amino acids, lipids, and nucleotides, by providing nitrogen and carbon (98, 99). As the second most abundant nutrient after glucose in the TME, glutamine catabolism is an indispensable method of energy generation via the TCA cycle (99, 100). Because of its role as a substrate for many bioactive elements and ATP production, glutamine is required in malignant cells and other immune components. Turowski et al. first confirmed the ability of glutamine to stimulate the proliferation of human colon cancer cell lines Caco-2 and SW620 (101). Further studies have shown that KRAS mutations heighten glutamine uptake in CRC cells by upregulating SLC1A5 expression. Moreover, it was found that glutaminolysis-derived succinate promotes CRC cell proliferation and stemness by upregulating LRG5 expression and enhancing Wnt/β-catenin signaling (102, 103). Lack of glutamine is seriously affect T cell proliferation after activation (104).
Lipids are a family of hydrophobic or amphipathic molecules that consist of fatty acids, glycerophospholipids, sphingolipids, and sterols. Although high lipid accumulation in the CRC TME has been recognized, their profound impact on tumor-infiltrated CD8+ T cells has only been noticed in recent years. CRC cells can produce various lipids, such as fatty acids, obliging T cells to uptake these lipids, resulting in T cell dysfunction (105–107). Mechanistically, CD8+ T cells in the TME showed the increased expression of CD36, a scavenger receptor for fatty acid uptake. The abnormal accumulation of fatty acids inside T cells further initiates lipid peroxidation, induces T cell ferroptosis, and reduces the production of cytokines, such as IFN-γ and TNF-α (107).
The T cell antigen receptor (TCR) is a multimolecular structure expressed on the T cell membrane that recognizes peptide/MHC complexes on the tumor cell surface (108). The expression of MHC class I molecules is crucial for an effective adaptive immune response. A low expression or deletion of MHC class I molecules frequently occurs in colon tumors resistant to immunotherapy (109–111). The oncoprotein SND1 promotes the ER-associated degradation of MHC class I, resulting in disordered CD8+ T cell function and decreased anti-tumor ability (112). A rational combination of systemic chemotherapy and DC i.t. injection has been shown to induce a complete anti-tumor response in MC38 murine adenocarcinoma cells (113). Moreover, increased mitophagy in intestinal epithelial cells can trigger lysosomal membrane permeability, and the subsequent release of proteases into the cytoplasm increases MHC class I presentation and activation of CD8+ T cells via cross-modification of dendritic cells (114).
The occurrence and development of CRC result from multiple gene interactions and the involvement of various signaling pathways. However, gene mutations and defects in signaling pathways also affect the immunotherapy sensitivity and prognosis of patients with colon cancer (115). As a key cascade that regulates cell development and stemness, Wnt/β-catenin pathway plays a prominent role in the occurrence of colon cancer (116). The aberrant activation of the Wnt/β-catenin pathway is a key driver in the maintenance and proliferation of gastrointestinal stem cells (117). The Wnt/β-catenin signaling pathway is also considered an important carcinogenic signaling pathway related to immune evasion. Activation of Wnt/β-catenin pathway in tumor leads to the decreasing production of CCL4 released by CD103+ dendritic cells. This inhibit CD8+ T cell activation and infiltration (118, 119). The Cancer Genome Atlas Network found that 55% of non-hypermutated tumors had alterations in KRAS, NRAS, or BRAF, which have significant mutually exclusive mutation patterns (12). PIK3CA mutations are some of the most common genetic alterations in solid tumors, inducing defects in the phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) pathway, which is frequently dysregulated. PIK3CA mutations lead to the attenuation of tumor apoptosis and improvement of tumor invasion. Treatment with the PI3K inhibitor LY294002 has been shown to downregulate PIK3CA signaling and inhibit the progression of PIK3CA-mutant colon cancer (120). Meanwhile, mutations of PIK3CA could increase total mutation burden (TMB) which may improve immunotherapy sensitivity (121).
The switching/sucrose non-fermentable (SWI/SNF) complex regulates transcription via nucleosome topology modulation and has been identified as a cancer suppressor gene in human malignancies (122, 123). SWI/SNF exhibits a broad mutation pattern associated with the progression and invasion of tumor cells. AT-Rich Interactive Domain-containing protein 1A (ARID1A), as one of the components making up the largest SWI/SNF subunit together with AT-Rich Interactive Domain-containing protein 1B (ARID1B), is the most frequent target of SWI/SNF mutations. mutations in ARID1A impair enhancer-mediated gene regulation and prognosis in patients with colon cancer (124, 125). Interestingly, SWI/SNF complex stability is also vital for tumor cell viability, which might be impaired by ARID1A mutation. In that situation, ARID1B is usually functionally normal and enable to partially compensate for ARID1A function as its homolog. But simultaneous mutation of ARID1A and ARID1B is synthetically lethal for colon cancer. ARID1A mutations was found correlated with markedly higher level of immune infiltrates in colon cancer. MSH2, a Mismatch repair protein, was found to interact with ARID1A. Low expression of ARID1A impair DNA mismatch repair, resulting in increases in TMB and cytotoxic T cell infiltration (126).
Immunotherapy resistance of decrease immunotherapy sensitivity in CRC. Targeting these tumor intrinsic and extrinsic resistance mechanisms is the key to improving the effectiveness of immunotherapy (Figure 3).
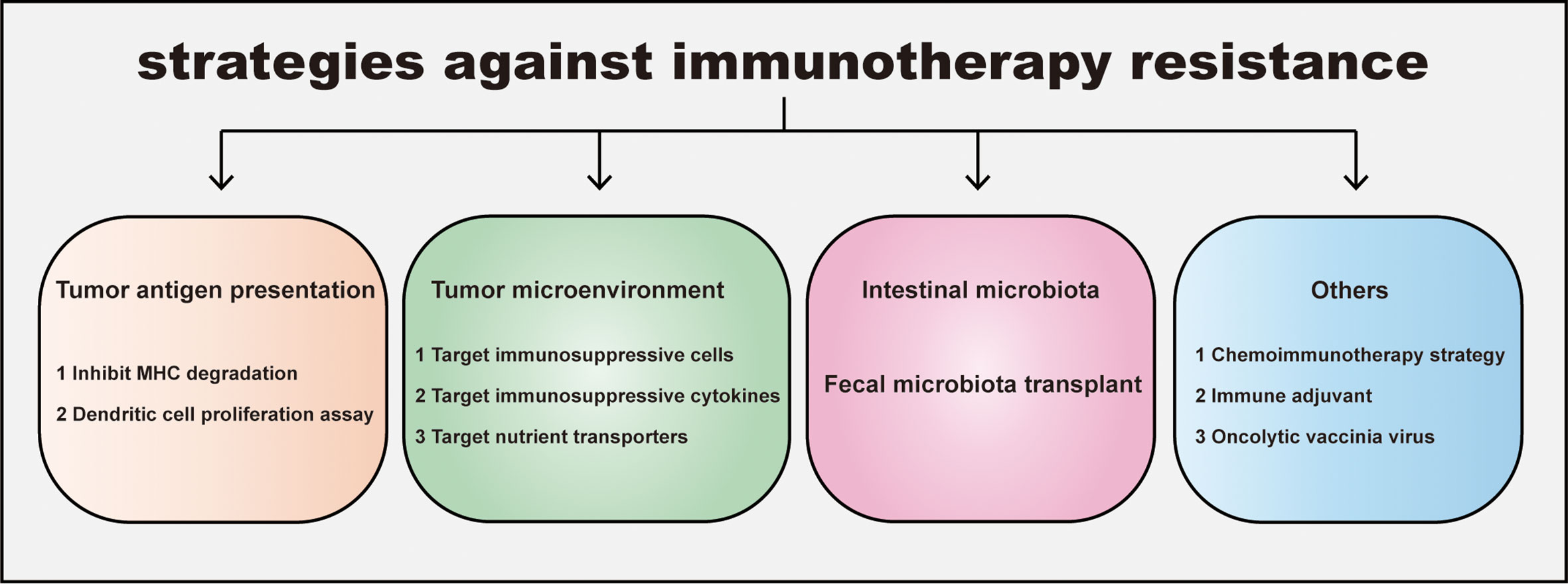
Figure 3 Strategies for overcoming immunotherapy resistance in CRC. The resistance leads to an ineffective immunotherapy and tumor progression. Four distinct strategies against immunotherapy resistance are listed: promoting tumor antigen presentation, regulating tumor immunosupprssive microenvironment, manipulating intestinal microbiota and others including combination therapy etc. Inhibiting MHC degradation and boosting dendritic cell proliferation could elevate tumor antigen presentation; Targeting immunosuppressive cells, cytokines in Tumor microenvironment (TME) and nutrient transporters help CD8+T escape the inhibition of TME. In addition, gut microbiota and its metabolite are widely proven to participate in tumor growth and immune response. Consistantly, fecal microbiota transplantation makes patients sensitive to immunotherapy. Other strategies for overcoming immunotherapy resistance, such as the administration of chemoimmunotherapy, immune adjuvant, oncolytic vaccinia virus can also improve the sensitivity of immunotherapy in CRC.
Chemotherapy was previously thought to be immunosuppressive due to its concomitant severe myelosuppression and leukopenia (127). However, numerous studies have reported its synergistic effects to immunotherapy, which makes the chemoimmunotherapy come back to clinical practice and become standard care for selected patients (128–130). Indeed, chemotherapy is found to induce tumor immunogenic cell death (ICD), which elicits extensive innate and adaptive immune response. For example, the classical chemotherapy regimen FOLFOX or FOLFIRI is reported to induce DAMPs (damage‐associated molecular patterns) in both mouse and human tumor cell lines. And DAMPs is a strong signaling to promote DC maturation and tumor antigen presentation, thus leading to tumor ICD and subsequent enhanced T cell anti-tumor response (131). According to the current conclusions, seeking the dose balance of chemotherapy and immunotherapy may be an attractive strategy to avoid the side effects and boost the therapeutic effects. To illustrate the combined efficacy of immunotherapy and chemotherapy in colorectal cancer, NCT02375672 administrated Pembrolizumab (MK-3475) together with standard-dose mFOLFOX6 regimen to treat 30 advanced CRC patients. And the median PFS is 8.8 months (95% CI, 7.7 to 11.3 months) with ORR being 56.7%, which showed exciting potential of combined medical strategies (132).
Before enhancing T cell function with various exogenous elements, the first step for T cells in killing tumor cells is to recognize tumor antigen epitopes, in which endogenous DCs play a key role. However, due to insufficient DC infiltration and abnormal maturation affected by the TME, tumor antigens are usually not well-presented. Thus T cells cannot differentiate and be activated normally. Thus, promoting DC expansion and maturation is a promising method for boosting T cell-based therapy. Fms-like tyrosine kinase 3 ligand (Flt3L) is a growth factor that expands dendritic cells, increasing their number (133). A recent study developed a strategy called an in situ vaccine, which combined Flt3L, radiotherapy, and a TLR3 agonist (Flt3L for DC expansion, radiotherapy for loading DCs with the antigen, and TLR3 agonist for DC activation) to test its efficacy in a murine lymphoma model (134). Mechanistically, this strategy expanded and activated intra-tumoral DCs, which further promoted TAA presentation and anti-tumor T cell activation. With an exciting long-term tumor regression of at least 3 months, researchers conducted a clinical trial on patients with low-grade B-cell lymphoma using rhuFlt3L and poly-ICLC combined with low-dose radiotherapy. Preliminary results showed good tolerance and an ORR of 72.7%. This has aroused our interest in the relevance of dendritic cells to ICB therapy and revealed the possibility of developing novel therapies that effectively control drug-resistant CRC, such as the evaluation of combinations of Flt3L and ICIs in clinical trials.
Targeting the TME is another potential way to boost the effect of immunotherapy. For example, a clinical trial (NCT04126733) that began in 2019 focused on the efficacy of regorafenib, a VEGFR inhibitor targeting TME angiogenesis, in combination with nivolumab in patients with pMMR-MSS CRC. The primary endpoint ORR was only 7.1% (95% CI, 2.4% to 15.9%), and the median PFS was 8.00 weeks (95%CI, 7.86 to 10.57 weeks). In another trial (NCT02713529) that used pembrolizumab combination with AMG820, a CSF1R inhibitor repolarizing TMAs from M2 to M1 type (135), the ORR for patients with pMMR CRC was quite low (4.9%; 95% CI, 0.60% to 16.53%).
Dysbiosis in CRC has multiple influences on the TME, which establishes an environment that favors tumor immune escape. Since patients with tumors always harbor altered microbiota characteristics, rebuilding intestinal microbiota homeostasis may be an attractive way to overcome treatment resistance in these patients. One plausible method is to transplant the microbiota of immunotherapy responders to non-responders. In the clinical trial NCT03341143 that tested this hypothesis, researchers carefully screened two patients with metastatic melanoma whose tumors had completely disappeared after prior PD-1 therapy (136). Then transplant the microbiota of screened two patients to non-responders.Miraculously, the recipients showed tumor reduction or stable disease for more than a year, with an ORR of 20% and a median PFS of 3 months (median follow-up of 7 months). Microbiota transplantation reversed the response to PD-1 drugs in patients with anti-PD-1-refractory melanoma (137, 138). This reveals the potential value and significance of gut microbiota in immunotherapy. In addition, through the adjusted daily diet and the intake of prebiotics, the gut microbiota can influence the pre-existing commensal microbes in the gut. In the future, improving the gut microbiota via gut microbiota transplantation or other methods is likely to improve the effectiveness of immunotherapy in patients with CRC.
Due to the low immunogenicity of various tumor cells and the immunosuppressive TME, natural T cells usually have a limited anti-tumor response even in the presence of ICIs. Engineered chimeric antigen receptor T (CAR-T) cells armored with various molecules have recently been developed to resolve this issue (134). Nevertheless, the medical efficacy of CAR-T therapy is still limited because of the low recruitment ratio and the presence of only a few desirable epitopes (139, 140). The use of oncolytic viruses (OVs) and engineered bacteria are promising methods to overcome the deficiencies of CAR-T cells because not only can they get into tumor tissues unimpeded and directly kill tumor cells, but they can also release or amplify tumor immunogenicity to stimulate immune cell responses, enhance immune cell infiltration, and result in effective anti-tumor immunity (141). A recent study reported that an oncolytic virus called CD19t could induce tumor cells to express CD19 on their surface before killing them, subsequently redirecting CD19-CAR-T cytotoxicity and enhancing the anti-tumor response in mouse models (132). In another research of murine CRC model, a kind of engineered Brucella melitensis (BmΔvjbR) in short of virulence was developed to assist CAR-T efficacy. The team found that intravenous administration of BmΔvjbR could significantly promote intratumoral M1-macrophage polarization and expansion, which further enhanced CAR-T infiltration and activity (142). However, no studies have focused on combining OVs/engineered bacteria and CAR-T cells in clinical patients, which is an urgent need.
Immunoadjuvants are non-specific non-immunogenic immunostimulatory molecules that can elicit rapid and strong immune responses when administered before or simultaneously with pathogen antigens. However, currently proven clinical immunoadjuvants are usually highly involved in activating humoral immunity instead of cellular immunity, limiting their application mostly in bacterial and some viral infections, like in various vaccines (140). This provoked our interest in seeking adjuvants that prefer the activation of cellular immunity to treat tumors since T cells play a key role in the anti-tumor response. Fortunately, recent studies have shown that manganese salt is an excellent adjuvant in eliciting both NK and T cell anti-tumor responses that mainly promoted DC maturation and antigen cross-presentation in preclinical models, a major breakthrough in the field. In particular, combination therapies comprising anti-PD-1 agents and manganese chloride have shown great preliminary clinical efficacy, with a median ORR of 45.5% in a phase I clinical study, NCT03991559 (143). Thus, manganese salt administration is anticipated for the future development of immunotherapies and tumor vaccines.
The number and function of TILs determine tumor prognosis in tumor immunotherapy. Hence, improving the number and function of TILs is the key to addressing immunotherapy resistance. By further understanding the heterogeneity of the tumor microenvironment and the defense and escape mechanisms of the gut microbiota and tumor cells against the attack of the immune system, we can target these drug resistance mechanisms to reduce immunotherapy resistance and improve the prognosis of patients with cancer. It is believed that with ICIs as the primary therapeutic backbone, targeting the tumor microenvironment and gut microbiota using combination treatments comprising ICIs, radiotherapy, chemotherapy, and various new therapeutic modalities will be continuously carried out, resulting in a higher percentage of patients that would benefit from immunotherapy.
JS and DH contributed to conception and design of the study. JS organized the database. JS, DH and CS wrote the manuscript and created the figures and tables. JS, DH and QL wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by National Natural Science Foundation of China (grant nos. 81872333, U1804281, 91942314), Henan Provincial Medical Science and Technology Research Plan Provincial and Ministerial Joint Construction Project (grant no. SBGJ202101010).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Benson AB, Venook AP, Al-Hawary MM, Cederquist L, Chen YJ, Ciombor KK, et al. NCCN guidelines insights: Colon cancer, version 2.2018. J Natl Compr Canc Netw (2018) 16(4):359–69. doi: 10.6004/jnccn.2018.0021
2. Piawah S, Venook AP. Targeted therapy for colorectal cancer metastases: A review of current methods of molecularly targeted therapy and the use of tumor biomarkers in the treatment of metastatic colorectal cancer. Cancer (2019) 125(23):4139–47. doi: 10.1002/cncr.32163
3. Hu LF, Lan HR, Huang D, Li XM, Jin KT. Personalized immunotherapy in colorectal cancers: Where do we stand? Front Oncol (2021) 11:769305. doi: 10.3389/fonc.2021.769305
4. Leko V, Rosenberg SA. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell (2020) 38(4):454–72. doi: 10.1016/j.ccell.2020.07.013
5. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
6. Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell (1992) 71(7):1065–8. doi: 10.1016/s0092-8674(05)80055-8
7. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25(21):9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005
8. Lenz HJ, Van Cutsem E, Luisa Limon M, Wong KYM, Hendlisz A, Aglietta M, et al. First-line nivolumab plus low-dose ipilimumab for microsatellite instability-High/Mismatch repair-deficient metastatic colorectal cancer: The phase II CheckMate 142 study. J Clin Oncol (2022) 40(2):161–70. doi: 10.1200/JCO.21.01015
9. Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-Deficient/Microsatellite instability-high metastatic colorectal cancer. J Clin Oncol (2018) 36(8):773–9. doi: 10.1200/JCO.2017.76.9901
10. Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discovery (2022) 21(7):509–28. doi: 10.1038/s41573-021-00345-8
11. Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, et al. PD-1 blockade in mismatch repair-deficient, locally advanced rectal cancer. N Engl J Med (2022) 386(25):2363–76. doi: 10.1056/NEJMoa2201445
12. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487(7407):330–7. doi: 10.1038/nature11252
13. Ashktorab H, Smoot DT, Farzanmehr H, Fidelia-Lambert M, Momen B, Hylind L, et al. Clinicopathological features and microsatellite instability (MSI) in colorectal cancers from African americans. Int J Cancer (2005) 116(6):914–9. doi: 10.1002/ijc.21062
14. Gelsomino F, Barbolini M, Spallanzani A, Pugliese G, Cascinu S. The evolving role of microsatellite instability in colorectal cancer: A review. Cancer Treat Rev (2016) 51:19–26. doi: 10.1016/j.ctrv.2016.10.005
15. Hampel H, Frankel W, Panescu J, Lockman J, Sotamaa K, Fix D, et al. Screening for lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res (2006) 66(15):7810–7. doi: 10.1158/0008-5472.CAN-06-1114
16. Lubbe SJ, Webb EL, Chandler IP, Houlston RS. Implications of familial colorectal cancer risk profiles and microsatellite instability status. J Clin Oncol (2009) 27(13):2238–44. doi: 10.1200/JCO.2008.20.3364
17. Perucho M. Correspondence re: C.R. boland et al., a national cancer institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. cancer res., 58: 5248-5257, 1998. Cancer Res (1999) 59(1):249–56. doi: 10.1134/1.1601761
18. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov (2015) 5(1):43–51. doi: 10.1158/2159-8290.CD-14-0863
19. Mowat C, Mosley S, Namdar A, Schiller D, Baker K. Anti-tumor immunity in mismatch repair-deficient colorectal cancers requires type I IFN-driven CCL5 and CXCL10. Exp Med (2021) 218(9):e20210108. doi: 10.1084/jem.20210108
20. Zaravinos A, Roufas C, Nagara M, de Lucas Moreno B, Oblovatskaya M, Efstathiades C, et al. Cytolytic activity correlates with the mutational burden and deregulated expression of immune checkpoints in colorectal cancer. J Exp Clin Cancer Res (2019) 38(1):364. doi: 10.1186/s13046-019-1372-z
21. Roufas C, Georgakopoulos-Soares I, Zaravinos A. Molecular correlates of immune cytolytic subgroups in colorectal cancer by integrated genomics analysis. NAR Cancer (2021) 3(1):zcab005. doi: 10.1093/narcan/zcab005
22. Nagorsen D, Voigt S, Berg E, Stein H, Thiel E, Loddenkemper C. Tumor-infiltrating macrophages and dendritic cells in human colorectal cancer: relation to local regulatory T cells, systemic T-cell response against tumor-associated antigens and survival. J Transl Med (2007) 5:62. doi: 10.1186/1479-5876-5-62
23. Le DT, Kim TW, Van Cutsem E, Geva R, Jager D, Hara H, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-High/Mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol (2020) 38(1):11–9. doi: 10.1200/JCO.19.02107
24. Chen EX, Jonker DJ, Loree JM, Kennecke HF, Berry SR, Couture F, et al. Effect of combined immune checkpoint inhibition vs best supportive care alone in patients with advanced colorectal cancer: The Canadian cancer trials group CO.26 study. JAMA Oncol (2020) 6(6):831–8. doi: 10.1001/jamaoncol.2020.0910
25. Zhang X, Wu T, Cai X, Dong J, Xia C, Zhou Y, et al. Neoadjuvant immunotherapy for MSI-H/dMMR locally advanced colorectal cancer: New strategies and unveiled opportunities. Front Immunol (2022) 13:795972. doi: 10.3389/fimmu.2022.795972
26. Li J, Deng Y, Zhang W, Zhou AP, Guo W, Yang J, et al. Subcutaneous envafolimab monotherapy in patients with advanced defective mismatch repair/microsatellite instability high solid tumors. J Hematol Oncol (2021) 14(1):95. doi: 10.1186/s13045-021-01095-1
27. van Herk EH, Te Velde AA. Treg subsets in inflammatory bowel disease and colorectal carcinoma: Characteristics, role, and therapeutic targets. J Gastroenterol Hepatol (2016) 31(8):1393–404. doi: 10.1111/jgh.13342
28. Olguin JE, Medina-Andrade I, Rodriguez T, Rodriguez-Sosa M, Terrazas LI. Relevance of regulatory T cells during colorectal cancer development. Cancers (Basel) (2020) 12(7):1888. doi: 10.3390/cancers12071888
29. Collison LW, Pillai MR, Chaturvedi V, Vignali DA. Regulatory T cell suppression is potentiated by target T cells in a cell contact, IL-35- and IL-10-dependent manner. J Immunol (2009) 182(10):6121–8. doi: 10.4049/jimmunol.0803646
30. Damo M, Joshi NS. Treg cell IL-10 and IL-35 exhaust CD8(+) T cells in tumors. Nat Immunol (2019) 20(6):674–5. doi: 10.1038/s41590-019-0389-y
31. Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, et al. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J Immunol (2004) 172(2):834–42. doi: 10.4049/jimmunol.172.2.834
32. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8(12):1353–62. doi: 10.1038/ni1536
33. McNally A, Hill GR, Sparwasser T, Thomas R, Steptoe RJ. CD4+CD25+ regulatory T cells control CD8+ T-cell effector differentiation by modulating IL-2 homeostasis. Proc Natl Acad Sci USA (2011) 108(18):7529–34. doi: 10.1073/pnas.1103782108
34. Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol (2017) 18(12):1332–41. doi: 10.1038/ni.3868
35. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci U.S.A. (2021) 118(30). doi: 10.1073/pnas.2023739118
36. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity (2004) 21(4):589–601. doi: 10.1016/j.immuni.2004.09.002
37. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme b and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity (2007) 27(4):635–46. doi: 10.1016/j.immuni.2007.08.014
38. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res (2004) 64(16):5839–49. doi: 10.1158/0008-5472.CAN-04-0465
39. Brito C, Naviliat M, Tiscornia AC, Vuillier F, Gualco G, Dighiero G, et al. Peroxynitrite inhibits T lymphocyte activation and proliferation by promoting impairment of tyrosine phosphorylation and peroxynitrite-driven apoptotic death. J Immunol (1999) 162(6):3356–66.
40. Oberg HH, Lengl-Janssen B, Robertson MJ, Kabelitz D, Janssen O. Differential role of tyrosine phosphorylation in the induction of apoptosis in T cell clones via CD95 or the TCR/CD3-complex. Cell Death Differ (1997) 4(5):403–12. doi: 10.1038/sj.cdd.4400256
41. Yahaya MAF, Lila MAM, Ismail S, Zainol M, Afizan N. Tumour-associated macrophages (TAMs) in colon cancer and how to reeducate them. J Immunol Res (2019) 2019:2368249. doi: 10.1155/2019/2368249
42. Wang H, Tian T, Zhang J. Tumor-associated macrophages (TAMs) in colorectal cancer (CRC): From mechanism to therapy and prognosis. Int J Mol Sci (2021) 22(16):8470. doi: 10.3390/ijms22168470
43. Oyarce C, Vizcaino-Castro A, Chen S, Boerma A, Daemen T. Re-polarization of immunosuppressive macrophages to tumor-cytotoxic macrophages by repurposed metabolic drugs. Oncoimmunology (2021) 10(1):1898753. doi: 10.1080/2162402X.2021.1898753
44. Barbera-Guillem E, Nyhus JK, Wolford CC, Friece CR, Sampsel JW. Vascular endothelial growth factor secretion by tumor-infiltrating macrophages essentially supports tumor angiogenesis, and IgG immune complexes potentiate the process. Cancer Res (2002) 62(23):7042–9. doi: 10.1097/00002820-200212000-00014
45. Phinney BB, Ray AL, Peretti AS, Jerman SJ, Grim C, Pinchuk IV, et al. MK2 regulates macrophage chemokine activity and recruitment to promote colon tumor growth. Front Immunol (2018) 9:1857. doi: 10.3389/fimmu.2018.01857
46. Smith LK, Boukhaled GM, Condotta SA, Mazouz S, Guthmiller JJ, Vijay R, et al. Interleukin-10 directly inhibits CD8(+) T cell function by enhancing n-glycan branching to decrease antigen sensitivity. Immunity (2018) 48(2):299–312 e5. doi: 10.1016/j.immuni.2018.01.006
47. Itatani Y, Kawada K, Sakai Y. Transforming growth factor-beta signaling pathway in colorectal cancer and its tumor microenvironment. Int J Mol Sci (2019) 20(23):5822. doi: 10.3390/ijms20235822
48. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity (2019) 50(4):924–40. doi: 10.1016/j.immuni.2019.03.024
49. Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev (2006) 20(22):3130–46. doi: 10.1101/gad.1478706
50. Xu Z, Gao H, Zhang Y, Feng W, Miao Y, Xu Z, et al. CCL7 and TGF-beta secreted by MSCs play opposite roles in regulating CRC metastasis in a KLF5/CXCL5-dependent manner. Mol Ther (2022) 30(6):2327–41. doi: 10.1016/j.ymthe.2022.03.005
51. Li S, Zhang J, Qian S, Wu X, Sun L, Ling T, et al. S100A8 promotes epithelial-mesenchymal transition and metastasis under TGF-beta/USF2 axis in colorectal cancer. Cancer Commun (Lond) (2021) 41(2):154–70. doi: 10.1002/cac2.12130
52. Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell (2005) 8(5):369–80. doi: 10.1016/j.ccr.2005.10.012
53. Battaglin F, Puccini A, Intini R, Schirripa M, Ferro A, Bergamo F, et al. The role of tumor angiogenesis as a therapeutic target in colorectal cancer. Expert Rev Anticancer Ther (2018) 18(3):251–66. doi: 10.1080/14737140.2018.1428092
54. Dakowicz D, Zajkowska M, Mroczko B. Relationship between VEGF family members, their receptors and cell death in the neoplastic transformation of colorectal cancer. Int J Mol Sci (2022) 23(6):3375. doi: 10.3390/ijms23063375
55. Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H, et al. VEGF-a drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci Immunol (2019) 4(41):eaay0555. doi: 10.1126/sciimmunol.aay0555
56. de Almeida PE, Mak J, Hernandez G, Jesudason R, Herault A, Javinal V, et al. Anti-VEGF treatment enhances CD8(+) T-cell antitumor activity by amplifying hypoxia. Cancer Immunol Res (2020) 8(6):806–18. doi: 10.1158/2326-6066.CIR-19-0360
57. Shi J, Song X, Traub B, Luxenhofer M, Kornmann M. Involvement of IL-4, IL-13 and their receptors in pancreatic cancer. Int J Mol Sci (2021) 22(6):2998. doi: 10.3390/ijms22062998
58. Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med (2020) 217(1). doi: 10.1084/jem.20190418
59. Bankaitis KV, Fingleton B. Targeting IL4/IL4R for the treatment of epithelial cancer metastasis. Clin Exp Metastasis (2015) 32(8):847–56. doi: 10.1007/s10585-015-9747-9
60. Yin K, Xia X, Rui K, Wang T, Wang S. Myeloid-derived suppressor cells: A new and pivotal player in colorectal cancer progression. Front Oncol (2020) 10:610104. doi: 10.3389/fonc.2020.610104
61. Koller FL, Hwang DG, Dozier EA, Fingleton B. Epithelial interleukin-4 receptor expression promotes colon tumor growth. Carcinogenesis (2010) 31(6):1010–7. doi: 10.1093/carcin/bgq044
62. Liu H, Antony S, Roy K, Juhasz A, Wu Y, Lu J, et al. Interleukin-4 and interleukin-13 increase NADPH oxidase 1-related proliferation of human colon cancer cells. Oncotarget (2017) 8(24):38113–35. doi: 10.18632/oncotarget.17494
63. Mantilla-Rojas C, Yu M, Rinella ES, Lynch RM, Perry A, Jaimes-Alvarado J, et al. A molecular subtype of colorectal cancers initiates independently of epidermal growth factor receptor and has an accelerated growth rate mediated by IL10-dependent anergy. Oncogene (2021) 40(17):3047–59. doi: 10.1038/s41388-021-01752-2
64. Guo Y, Xie YQ, Gao M, Zhao Y, Franco F, Wenes M, et al. Metabolic reprogramming of terminally exhausted CD8(+) T cells by IL-10 enhances anti-tumor immunity. Nat Immunol (2021) 22(6):746–56. doi: 10.1038/s41590-021-00940-2
65. Qiao J, Liu Z, Dong C, Luan Y, Zhang A, Moore C, et al. Targeting tumors with IL-10 prevents dendritic cell-mediated CD8(+) T cell apoptosis. Cancer Cell (2019) 35(6):901–15 e4. doi: 10.1016/j.ccell.2019.05.005
66. Fong W, Li Q, Yu J. Gut microbiota modulation: a novel strategy for prevention and treatment of colorectal cancer. Oncogene (2020) 39(26):4925–43. doi: 10.1038/s41388-020-1341-1
67. Dalal N, Jalandra R, Bayal N, Yadav AK, Harshulika, Sharma M, et al. Gut microbiota-derived metabolites in CRC progression and causation. J Cancer Res Clin Oncol (2021) 147(11):3141–55. doi: 10.1007/s00432-021-03729-w
68. Hanus M, Parada-Venegas D, Landskron G, Wielandt AM, Hurtado C, Alvarez K, et al. Immune system, microbiota, and microbial metabolites: The unresolved triad in colorectal cancer microenvironment. Front Immunol (2021) 12:612826. doi: 10.3389/fimmu.2021.612826
69. Dai Z, Coker OO, Nakatsu G, Wu WKK, Zhao L, Chen Z, et al. Multi-cohort analysis of colorectal cancer metagenome identified altered bacteria across populations and universal bacterial markers. Microbiome (2018) 6(1):70. doi: 10.1186/s40168-018-0451-2
70. Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, et al. Fusobacterium nucleatum increases proliferation of colorectal cancer cells and tumor development in mice by activating toll-like receptor 4 signaling to nuclear factor-kappaB, and up-regulating expression of MicroRNA-21. Gastroenterology (2017) 152(4):851–66 e24. doi: 10.1053/j.gastro.2016.11.018
71. Hu L, Liu Y, Kong X, Wu R, Peng Q, Zhang Y, et al. Fusobacterium nucleatum facilitates M2 macrophage polarization and colorectal carcinoma progression by activating TLR4/NF-kappaB/S100A9 cascade. Front Immunol (2021) 12:658681. doi: 10.3389/fimmu.2021.658681
72. Xu C, Fan L, Lin Y, Shen W, Qi Y, Zhang Y, et al. Fusobacterium nucleatum promotes colorectal cancer metastasis through miR-1322/CCL20 axis and M2 polarization. Gut Microbes (2021) 13(1):1980347. doi: 10.1080/19490976.2021.1980347
73. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity (2015) 42(2):344–55. doi: 10.1016/j.immuni.2015.01.010
74. Stoeva MK, Garcia-So J, Justice N, Myers J, Tyagi S, Nemchek M, et al. Butyrate-producing human gut symbiont, clostridium butyricum, and its role in health and disease. Gut Microbes (2021) 13(1):1–28. doi: 10.1080/19490976.2021.1907272
75. Waby JS, Chirakkal H, Yu C, Griffiths GJ, Benson RS, Bingle CD, et al. Sp1 acetylation is associated with loss of DNA binding at promoters associated with cell cycle arrest and cell death in a colon cell line. Mol Cancer (2010) 9:275. doi: 10.1186/1476-4598-9-275
76. Owens JA, Saeedi BJ, Naudin CR, Hunter-Chang S, Barbian ME, Eboka RU, et al. Lactobacillus rhamnosus GG orchestrates an antitumor immune response. Cell Mol Gastroenterol Hepatol (2021) 12(4):1311–27. doi: 10.1016/j.jcmgh.2021.06.001
77. Aindelis G, Tiptiri-Kourpeti A, Lampri E, Spyridopoulou K, Lamprianidou E, Kotsianidis I, et al. Immune responses raised in an experimental colon carcinoma model following oral administration of lactobacillus casei. Cancers (Basel) (2020) 12(2):368. doi: 10.3390/cancers12020368
78. Wang G, Wang JJ, Yin PH, Xu K, Wang YZ, Shi F, et al. New strategies for targeting glucose metabolism-mediated acidosis for colorectal cancer therapy. J Cell Physiol (2018) 234(1):348–68. doi: 10.1002/jcp.26917
79. Zhou CB, Fang JY. The regulation of host cellular and gut microbial metabolism in the development and prevention of colorectal cancer. Crit Rev Microbiol (2018) 44(4):436–54. doi: 10.1080/1040841X.2018.1425671
80. Tang J, Yan T, Bao Y, Shen C, Yu C, Zhu X, et al. LncRNA GLCC1 promotes colorectal carcinogenesis and glucose metabolism by stabilizing c-myc. Nat Commun (2019) 10(1):3499. doi: 10.1038/s41467-019-11447-8
81. Wang Y, Lu JH, Wu QN, Jin Y, Wang DS, Chen YX, et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer (2019) 18(1):174. doi: 10.1186/s12943-019-1105-0
82. Dong S, Liang S, Cheng Z, Zhang X, Luo L, Li L, et al. ROS/PI3K/Akt and wnt/beta-catenin signalings activate HIF-1alpha-induced metabolic reprogramming to impart 5-fluorouracil resistance in colorectal cancer. J Exp Clin Cancer Res (2022) 41(1):15. doi: 10.1186/s13046-021-02229-6
83. Lin ZB, Long P, Zhao Z, Zhang YR, Chu XD, Zhao XX, et al. Long noncoding RNA KCNQ1OT1 is a prognostic biomarker and mediates CD8(+) T cell exhaustion by regulating CD155 expression in colorectal cancer. Int J Biol Sci (2021) 17(7):1757–68. doi: 10.7150/ijbs.59001
84. Shao Q, Wang L, Yuan M, Jin X, Chen Z, Wu C. TIGIT induces (CD3+) T cell dysfunction in colorectal cancer by inhibiting glucose metabolism. Front Immunol (2021) 12:688961. doi: 10.3389/fimmu.2021.688961
85. He W, Zhang H, Han F, Chen X, Lin R, Wang W, et al. CD155T/TIGIT signaling regulates CD8(+) T-cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res (2017) 77(22):6375–88. doi: 10.1158/0008-5472.CAN-17-0381
86. Deng F, Zhou R, Lin C, Yang S, Wang H, Li W, et al. Tumor-secreted dickkopf2 accelerates aerobic glycolysis and promotes angiogenesis in colorectal cancer. Theranostics (2019) 9(4):1001–14. doi: 10.7150/thno.30056
87. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature (2014) 513(7519):559–63. doi: 10.1038/nature13490
88. Wang X, Luo X, Chen C, Tang Y, Li L, Mo B, et al. The ap-2alpha/Elk-1 axis regulates sirpalpha-dependent tumor phagocytosis by tumor-associated macrophages in colorectal cancer. Signal Transduct Target Ther (2020) 5(1):35. doi: 10.1038/s41392-020-0124-z
89. Liu H, Liang Z, Zhou C, Zeng Z, Wang F, Hu T, et al. Mutant KRAS triggers functional reprogramming of tumor-associated macrophages in colorectal cancer. Signal Transduct Target Ther (2021) 6(1):144. doi: 10.1038/s41392-021-00534-2
90. Seth P, Csizmadia E, Hedblom A, Vuerich M, Xie H, Li M, et al. Deletion of lactate dehydrogenase-a in myeloid cells triggers antitumor immunity. Cancer Res (2017) 77(13):3632–43. doi: 10.1158/0008-5472.CAN-16-2938
91. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res (2012) 72(11):2746–56. doi: 10.1158/0008-5472.CAN-11-1272
92. Hermans D, Gautam S, Garcia-Canaveras JC, Gromer D, Mitra S, Spolski R, et al. Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8(+) T cell stemness and antitumor immunity. Proc Natl Acad Sci USA (2020) 117(11):6047–55. doi: 10.1073/pnas.1920413117
93. Santhanam S, Alvarado DM, Ciorba MA. Therapeutic targeting of inflammation and tryptophan metabolism in colon and gastrointestinal cancer. Transl Res (2016) 167(1):67–79. doi: 10.1016/j.trsl.2015.07.003
94. Peyraud F, Guegan JP, Bodet D, Cousin S, Bessede A, Italiano A. Targeting tryptophan catabolism in cancer immunotherapy era: Challenges and perspectives. Front Immunol (2022) 13:807271. doi: 10.3389/fimmu.2022.807271
95. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-repopulating cells induce PD-1 expression in CD8(+) T cells by transferring kynurenine and AhR activation. Cancer Cell (2018) 33(3):480–94 e7. doi: 10.1016/j.ccell.2018.02.005
96. Zhang X, Liu X, Zhou W, Du Q, Yang M, Ding Y, et al. Blockade of IDO-Kynurenine-AhR axis ameliorated colitis-associated colon cancer via inhibiting immune tolerance. Cell Mol Gastroenterol Hepatol (2021) 12(4):1179–99. doi: 10.1016/j.jcmgh.2021.05.018
97. Zhang R, Wang Y, Liu D, Luo Q, Du P, Zhang H, et al. Sodium tanshinone IIA sulfonate as a potent IDO1/TDO2 dual inhibitor enhances anti-PD1 therapy for colorectal cancer in mice. Front Pharmacol (2022) 13:870848. doi: 10.3389/fphar.2022.870848
98. Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci (2016) 73(2):377–92. doi: 10.1007/s00018-015-2070-4
99. Mates JM, Campos-Sandoval JA, Santos-Jimenez JL, Marquez J. Dysregulation of glutaminase and glutamine synthetase in cancer. Cancer Lett (2019) 467:29–39. doi: 10.1016/j.canlet.2019.09.011
100. Caniglia JL, Jalasutram A, Asuthkar S, Sahagun J, Park S, Ravindra A, et al. Beyond glucose: alternative sources of energy in glioblastoma. Theranostics (2021) 11(5):2048–57. doi: 10.7150/thno.53506
101. Turowski GA, Rashid Z, Hong F, Madri JA, Basson MD. Glutamine modulates phenotype and stimulates proliferation in human colon cancer cell lines. Cancer Res (1994) 54(22):5974–80. doi: 10.1002/1097-0142(19941115)74:10<2885::AID-CNCR2820741023>3.0.CO
102. Kandasamy P, Zlobec I, Nydegger DT, Pujol-Gimenez J, Bhardwaj R, Shirasawa S, et al. Oncogenic KRAS mutations enhance amino acid uptake by colorectal cancer cells via the hippo signaling effector YAP1. Mol Oncol (2021) 15(10):2782–800. doi: 10.1002/1878-0261.12999
103. Wong CC, Xu J, Bian X, Wu JL, Kang W, Qian Y, et al. In colorectal cancer cells with mutant KRAS, SLC25A22-mediated glutaminolysis reduces DNA demethylation to increase WNT signaling, stemness, and drug resistance. Gastroenterology (2020) 159(6):2163–80 e6. doi: 10.1053/j.gastro.2020.08.016
104. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity (2011) 35(6):871–82. doi: 10.1016/j.immuni.2011.09.021
105. Krauss D, Fari O, Sibilia M. Lipid metabolism interplay in CRC-an update. Metabolites (2022) 12(3):213. doi: 10.3390/metabo12030213
106. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell Metab (2021) 33(5):1001–12 e5. doi: 10.1016/j.cmet.2021.02.015
107. Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity (2021) 54(7):1561–77 e7. doi: 10.1016/j.immuni.2021.05.003
108. Trolle T, McMurtrey CP, Sidney J, Bardet W, Osborn SC, Kaever T, et al. The length distribution of class I-restricted T cell epitopes is determined by both peptide supply and MHC allele-specific binding preference. J Immunol (2016) 196(4):1480–7. doi: 10.4049/jimmunol.1501721
109. Gobin SJ, van Zutphen M, Westerheide SD, Boss JM, van den Elsen PJ. The MHC-specific enhanceosome and its role in MHC class I and beta(2)-microglobulin gene transactivation. J Immunol (2001) 167(9):5175–84. doi: 10.4049/jimmunol.167.9.5175
110. Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell (1993) 75(1):135–46. doi: 10.1016/S0092-8674(05)80090-X
111. Buus S, Sette A, Colon SM, Grey HM. Autologous peptides constitutively occupy the antigen binding site on ia. Science (1988) 242(4881):1045–7. doi: 10.1126/science.3194755
112. Wang Y, Wang X, Cui X, Zhuo Y, Li H, Ha C, et al. Oncoprotein SND1 hijacks nascent MHC-I heavy chain to ER-associated degradation, leading to impaired CD8(+) T cell response in tumor. Sci Adv (2020) 6(22):eaba5412. doi: 10.1126/sciadv.aba5412
113. Tanaka F, Yamaguchi H, Ohta M, Mashino K, Sonoda H, Sadanaga N, et al. Intratumoral injection of dendritic cells after treatment of anticancer drugs induces tumor-specific antitumor effect. Vivo Int J Cancer (2002) 101(3):265–9. doi: 10.1002/ijc.10597
114. Ziegler PK, Bollrath J, Pallangyo CK, Matsutani T, Canli O, De Oliveira T, et al. Mitophagy in intestinal epithelial cells triggers adaptive immunity during tumorigenesis. Cell (2018) 174(1):88–101 e16. doi: 10.1016/j.cell.2018.05.028
115. Ray K. Mapping the malignant transformation from polyps to CRC. Nat Rev Gastroenterol Hepatol (2022) 19(8):488. doi: 10.1038/s41575-022-00660-4
116. Zhang Y, Wang X. Targeting the wnt/beta-catenin signaling pathway in cancer. J Hematol Oncol (2020) 13(1):165. doi: 10.1186/s13045-020-00990-3
117. Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal (2014) 26(3):570–9. doi: 10.1016/j.cellsig.2013.11.032
118. Pai SG, Carneiro BA, Mota JM, Costa R, Leite CA, Barroso-Sousa R, et al. Wnt/beta-catenin pathway: modulating anticancer immune response. J Hematol Oncol (2017) 10(1):101. doi: 10.1186/s13045-017-0471-6
119. Spranger S, Gajewski TF. A new paradigm for tumor immune escape: beta-catenin-driven immune exclusion. J Immunother Cancer (2015) 3:43. doi: 10.1186/s40425-015-0089-6
120. Samuels Y, Diaz LA Jr., Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell (2005) 7(6):561–73. doi: 10.1016/j.ccr.2005.05.014
121. Voutsadakis IA. The landscape of PIK3CA mutations in colorectal cancer. Clin Colorectal Cancer (2021) 20(3):201–15. doi: 10.1016/j.clcc.2021.02.003
122. Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, Yaniv M. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep (2000) 1(6):500–6. doi: 10.1093/embo-reports/kvd129
123. Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet (2013) 45(6):592–601. doi: 10.1038/ng.2628
124. Peerapen P, Sueksakit K, Boonmark W, Yoodee S, Thongboonkerd V. ARID1A knockdown enhances carcinogenesis features and aggressiveness of caco-2 colon cancer cells: An in vitro cellular mechanism study. J Cancer (2022) 13(2):373–84. doi: 10.7150/jca.65511
125. Baldi S, Khamgan H, Qian Y, Wu H, Zhang Z, Zhang M, et al. Downregulated ARID1A by miR-185 is associated with poor prognosis and adverse outcomes in colon adenocarcinoma. Front Oncol (2021) 11:679334. doi: 10.3389/fonc.2021.679334
126. Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med (2018) 24(5):556–62. doi: 10.1038/s41591-018-0012-z
127. Lyman GH, Abella E, Pettengell R. Risk factors for febrile neutropenia among patients with cancer receiving chemotherapy: A systematic review. Crit Rev Oncol Hematol (2014) 90(3):190–9. doi: 10.1016/j.critrevonc.2013.12.006
128. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov (2019) 18(3):175–96. doi: 10.1038/s41573-018-0006-z
129. Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discovery (2019) 18(3):197–218. doi: 10.1038/s41573-018-0007-y
130. Heinhuis KM, Ros W, Kok M, Steeghs N, Beijnen JH, Schellens JHM. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann Oncol (2019) 30(2):219–35. doi: 10.1093/annonc/mdy551
131. Murao A, Aziz M, Wang H, Brenner M, Wang P. Release mechanisms of major DAMPs. Apoptosis (2021) 26(3-4):152–62. doi: 10.1007/s10495-021-01663-3
132. Park AK, Fong Y, Kim SI, Yang J, Murad JP, Lu J, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Transl Med (2020) 12(559):eaaz1863. doi: 10.1126/scitranslmed.aaz1863
133. Lin DS, Tian L, Tomei S, Amann-Zalcenstein D, Baldwin TM, Weber TS, et al. Single-cell analyses reveal the clonal and molecular aetiology of Flt3L-induced emergency dendritic cell development. Nat Cell Biol (2021) 23(3):219–31. doi: 10.1038/s41556-021-00636-7
134. Hammerich L, Marron TU, Upadhyay R, Svensson-Arvelund J, Dhainaut M, Hussein S, et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med (2019) 25(5):814–24. doi: 10.1038/s41591-019-0410-x
135. Kulkarni A, Chandrasekar V, Natarajan SK, Ramesh A, Pandey P, Nirgud J, et al. A designer self-assembled supramolecule amplifies macrophage immune responses against aggressive cancer. Nat BioMed Eng (2018) 2(8):589–99. doi: 10.1038/s41551-018-0254-6
136. Davar D, Dzutsev AK, McCulloch JA, Rodrigues RR, Chauvin JM, Morrison RM, et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science (2021) 371(6529):595–602. doi: 10.1126/science.abf3363
137. Baruch EN, Youngster I, Ben-Betzalel G, Ortenberg R, Lahat A, Katz L, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science (2021) 371(6529):602–9. doi: 10.1126/science.abb5920
138. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science (2018) 359(6371):97–103. doi: 10.1126/science.aan4236
139. Dagher O, King TR, Wellhausen N, Posey AD Jr. Combination therapy for solid tumors: Taking a classic CAR on new adventures. Cancer Cell (2020) 38(5):621–3. doi: 10.1016/j.ccell.2020.10.003
140. Wing A, Fajardo CA, Posey AD Jr., Shaw C, Da T, Young RM, et al. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res (2018) 6(5):605–16. doi: 10.1158/2326-6066.CIR-17-0314
141. Arenas-Gamboa AM, Rice-Ficht AC, Fan Y, Kahl-McDonagh MM, Ficht TA. Extended safety and efficacy studies of the attenuated brucella vaccine candidates 16 M(Delta)vjbR and S19(Delta)vjbR in the immunocompromised IRF-1-/- mouse model. Clin Vaccine Immunol (2012) 19(2):249–60. doi: 10.1128/CVI.05321-11
142. Guo F, Das JK, Kobayashi KS, Qin QM, AF T, Alaniz RC, et al. Live attenuated bacterium limits cancer resistance to CAR-T therapy by remodeling the tumor microenvironment. J Immunother Cancer (2022) 10(1):e003760. doi: 10.1136/jitc-2021-003760
Keywords: colorectal cancer, immunotherapy, immune checkpoint inhibitors, drug resistance, potential therapeutic strategies
Citation: Shan J, Han D, Shen C, Lei Q and Zhang Y (2022) Mechanism and strategies of immunotherapy resistance in colorectal cancer. Front. Immunol. 13:1016646. doi: 10.3389/fimmu.2022.1016646
Received: 11 August 2022; Accepted: 05 September 2022;
Published: 27 September 2022.
Edited by:
Jia Wei, Nanjing Drum Tower Hospital, ChinaReviewed by:
Apostolos Zaravinos, European University Cyprus, CyprusCopyright © 2022 Shan, Han, Shen, Lei and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Zhang, eWl6aGFuZ0B6enUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.