 Linfeng Zhu
Linfeng Zhu Guoqin Tong
Guoqin Tong Fan Yang
Fan Yang Yijun Zhao
Yijun Zhao Guangjie Chen
Guangjie Chen- 1Department of Urology, The Children’s Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, Hangzhou, China
- 2Department of Neurology, The First People’s Hospital of XiaoShan District, Hangzhou, China
Uremic neuropathy in children encompasses a wide range of central nervous system (CNS), peripheral nervous system (PNS), autonomic nervous system (ANS), and psychological abnormalities, which is associated with progressive renal dysfunction. Clinically, the diagnosis of uremic neuropathy in children is often made retrospectively when symptoms improve after dialysis or transplantation, due to there is no defining signs or laboratory and imaging findings. These neurological disorders consequently result in increased morbidity and mortality among children population, making uremia an urgent public health problem worldwide. In this review, we discuss the epidemiology, potential mechanisms, possible treatments, and the shortcomings of current research of uremic neuropathy in children. Mechanistically, the uremic neuropathy may be caused by retention of uremic solutes, increased oxidative stress, neurotransmitter imbalance, and disturbance of the blood-brain barrier (BBB). Neuroimmune, including the change of inflammatory factors and immune cells, may also play a crucial role in the progression of uremic neuropathy. Different from the invasive treatment of dialysis and kidney transplantation, intervention in neuroimmune and targeted anti-inflammatory therapy may provide a new insight for the treatment of uremia.
1 Introduction
Uremia, a term used to describe the presenting syndrome patients experience in end-stage renal disease (ESRD), is characteristic for a variety of clinical and metabolic disorders with progressive kidney failure, which usually caused by chronic kidney disease (CKD) or acute kidney injury (AKI) (1). It can cause the accumulation of organic waste products in plasma, referred to as uremic solutes or uremic toxins, which are not yet fully identified (2). Besides uremia toxins, oxidative stress and inflammation also persist in uremia. These risk factors may involve in generalized organ dysfunction and consequently result in a significant decrease in quality of life and increased mortality, which has made uremia rapidly become an urgent public health problem worldwide. Due to the disturbance of the internal environment, uremia can cause a series of complications. Generally, nervous system dysfunction is quite common in patients with uremia, knowns as uremic neuropathy. Patients with uremic neuropathy may experience insomnia, agitation, paranoia, cognitive impairment, and coma for weeks or months or even years, followed by exacerbation and ultimately a poor prognosis (3). It has been also reported that sleep disorders and restless legs syndrome are common in dialysis patients with neurological complications. The pathogenesis of uremic neuropathy remains unclear.
Children as a special population are gradually arousing concern among patients with uremia. Pediatric uremia differs from adult uremia in that the largest diagnosed group in children are associated with congenital abnormalities and inherited disorders, whereas glomerular and renal diseases in the context of diabetes are relatively rare. At present, the treatment of children with uremia is still mainly dialysis and kidney transplantation (4, 5). Additionally, since children are in a critical period of growth and development, the therapy of recombinant human growth hormone is required for the complications of growth retardation caused by uremia (6, 7). Further, uremia in children can also cause neurological complications that not only lead to substantial disability and mortality but also incite medical issues beyond childhood. It is generally known that the first year after birth is a critical period for brain development, with the proliferation of glial cells and the rapid formation of myelin sheaths (8). Therefore, any factor that interferes with these two processes could have a major impact on brain development. Multiple neurological and developmental abnormalities have been identified in children who develop chronic renal failure during the first year of life. These abnormalities include microcephaly, retardation, cerebral edema, recurrent seizures and ataxia (9–11). Moreover, children between 3 and 16 years old with chronic renal impairment are more likely to have hearing impairment, decreased hearing sensitivity (12). Children born by mothers with chronic kidney disease are more likely to develop to cerebral palsy and intellectual disability (13). This may suggest that the CNS of children is more susceptible to uremia. However, studies on the epidemiology and corresponding mechanisms of uremia-induced neuropathic disorders in children are scarce.
The aim of this review was to investigate the epidemiology, clinical features, underlying mechanism and possible treatments of the neuropathic disorders in pediatric uremia, in order to provide new insights for pediatricians.
2 Epidemiology of pediatric uremia
Epidemiological researches of uremia on growing children are scarce. Nonetheless, given that uremia is usually caused by CDK and AKI, the epidemiology of CDK and AKI indirectly reflects the prevalence of uremia. According to a report from the United States, the incidence of CDK in adults is approximately 11%, which is 50 times greater than that of ESRD (14). Besides, a review of 15-year admissions in Nigeria estimated the prevalence of severe pediatric CKD to be 15 cases per million (15). The average incidence of CDK in Kuwaiti children was 38.2 cases per million per year (16). The Swedish national survey reported a prevalence of advanced CKD was 21 cases per million per year (17). A population-based data from Italy reported a point prevalence of CKD of 74.7 cases per million per year (18). Taken together, compared with adults, the prevalence of CKD in children has been reported at 15-74.7 per million, while the incidence of AKI ranged from 37% to 51% (19). Such variation in data is likely to be influenced by geography, culture and the medical resources. Moreover, the prevalence of ESRD in children is less than 2% of the total ESRD population (20). Nonetheless, pediatric CKD is a progressive disease, and the mortality rate among children with ESRD who receive dialysis is 30 to 150 times higher than the general pediatric population (21). During 2012, the prevalence of ESRD in children treated with dialysis in a Brazilian cohort was 20 cases per million (22), while a nation-wide survey from China between the 2007 to 2012 showed that 45.9% children with ESRD were on dialysis (23). The 1-, 2-, and 5- year overall survival rates of children on dialysis were 96.9%, 94.5%, 89.5% respectively (24). Furthermore, uremic neuropathy is quite prevalent in children with CKD on continuous hemodialysis. The prevalence of uremic neuropathy in children is not available, however, a small sample research reported that it was approximately 22% (25). Thus, the diagnosis and treatment of pediatric CKD and AKI must emphasize early detection, primary prevention, and active management to avoid further progression to ESRD and uremia. A population-based epidemiology of uremia in children is urgently needed.
3 Uremic neuropathy
3.1 Background
Uremic neuropathy encompasses a wide range of neurological abnormalities associated with poor renal function. It can involve the central nervous system (CNS), peripheral nervous system (PNS), autonomic nervous system (ANS) as well as psychological abnormalities (Table 1). CNS complications include cognitive impairment (26), seizures (27, 28), cranial neuropathy (29, 30), extrapyramidal disorders such as Parkinson’s disease (31) and chorea (32). Pediatric patients are more prone to the CNS damage caused by uremia, including microcephaly, retardation, cerebral edema, recurrent seizures and ataxia (9–11). Moreover, Hurkx et al. found delayed peak of brainstem auditory evoked potentials and unusual sensory evoked potentials in younger children, suggesting unusual myelination and synapse formation in children with chronic renal failure (CRF) (39).

Table 1 The symptoms of uremic neuropathy.
The symptoms of peripheral neuropathy include loss of tendon reflexes and muscle atrophy (33). The ANS complications is manifested by defective function of sweat glands (34), abnormal response to the valsalva maneuver (35), decreased baroreflex sensitivity (36), and hypotension (37). Psychological abnormalities include depression emotional changes, disturbing and depression (28, 38). Thus, the symptoms of uremic neuropathy are very complex and lack clear clinical, laboratory, or radiographic findings. The diagnosis of uremic neuropathy is frequent retrospective, only when symptoms improve after dialysis or kidney transplantation as well as other causes of neuropathy have been excluded (40). Although the prevalence of uremic neuropathy in children is not available, it has been reported it is quite common in children with CKD on maintenance hemodialysis (25). Neurologic complications contribute largely to the disability and mortality in patients with kidney failure. The age of onset and duration of CDK may influence neurocognitive and functional outcomes (41). Early life is an important period for the rapid proliferation and myelination of glial cells, and any significant insult can have enduring effects on neural structure and function (42, 43). Further knowledge of the potential link between uremia and neurological symptoms is needed, especially in children, to better understand this complication and to exert early intervention.
3.2 The general underlying mechanisms of uremic neuropathy
3.2.1 Uremic toxin accumulation
The development of uremic syndrome is usually caused by the retention of retained solutes that are filtered by healthy kidneys and have deleterious effects on biological function, known as uremic toxins (44). The European uremic toxin working group proposed a classification of 90 uremic solutes in 2003 (2). By reviewing the literature, Flore et al. identified 88 uremic toxins (45). According to their physicochemical properties, uremic toxins can be classified into small water-soluble compound, middle molecular compounds and protein-bound compounds (Table 2). Due to the difference in hydrophobicity, small molecular weight uremic toxins can exist as a free water-soluble form or reversibly bind to serum proteins, thereby altering protein function (75). Several studies suggest that these toxins may originate from diet or microbial metabolism as they can also be produced in the gut (76, 77). Recently, mitochondria have been implicated as contributors to uremic toxin production. Mitochondria may directly impact the synthesis of uremic toxins, especially the oxidation products or peroxidation products of cellular components, and uremic toxins in turn can cause damage to mitochondria, thereby producing more uremic toxins, forming a positive feedback loop that leads to increased production of uremic toxins (78). Studies also have reported that uremic toxins contribute to uremic neuropathy (79). Understanding the relationship between uremic toxins and neurological disorders contribute to better comprehend the pathogenesis of uremic neuropathy.

Table 2 The uremic toxins affecting neurogoical disorder.
3.2.1.1 Small water-soluble compounds
3.2.1.1.1 Guanidine compounds
Guanidine compounds (GCs), such as guanidinosuccinic acid (GSA), guanidine and methylguanidine, are considered to be the major cause of cognitive dysfunction in uremic syndrome of CKD (46–48). The accumulation of guanidine compounds can trigger oxidative stress (49) and lead to neuronal damage (50). Study in uremic animal models reported that GSA impaired synaptic function in CA1 region and affected memory process by blocking gamma aminobutyric acid (GABA) and glycine receptor-associated ion channels (51). It was also found an elevation of GSA and methylguanidine in the epileptic brain in animal models (52). Moreover, methylguanidine may promote uremia-related neurodegeneration by enhancing the pro-apoptotic effects of H2O2 and altering mitochondrial calcium homeostasis in glial cells (53).
3.2.1.1.2 Hypoxanthine
Hypoxanthine, a metabolite of purines, has been shown to have effects on the CNS (54). Hypoxanthine damages rat hippocampus and striatum. These effects are mediated by elevated levels of free radicals and uric acid, which lead to changes in acetylcholinesterase and butyrylcholinesterase activity (55). Hypoxanthine can be converted to xanthine by xanthine oxidase (XO) and subsequently to uric acid and have been shown to be positively associated with lipid peroxidation (56). Consistent with this result, XO inhibition ameliorates brain injury and renal dysfunction (57, 58).
3.2.1.2 Middle molecular compounds
3.2.1.2.1 Parathyroid hormone
Excessive parathyroid hormone (PTH) is mainly caused by hyperparathyroidism due to hyperphosphatemia that occurs in CDK (59, 60). Elevated parathyroid hormone reduces the conduction velocity of motor nerve in uremic patients (79). Hyperparathyroidism increases calcium entry into the brain and interferes with nerve transmission, thereby inducing neurotoxicity (61), while removal of parathyroid glands can prevent calcium excess in uremic brain (62). Furthermore, parathyroidectomy can prevent electroencephalogram (EEG) abnormalities in uremic animals, whereas administration of parathyroid extract to normal animals can induces similar EEG changes to uremic animals (63). It has also been suggested a harmful effect of PTH on cognitive function in CKD patients (64). However, there is still a lack of research supporting this view, which needs further investigation.
3.2.1.2.2 β2-microglobulin
Limited information is available on the relationship between β2-microglobulin and neuropathy. It has been reported that β2-microglobulin is cytotoxic for the SH-SY5Y neuroblastoma cells at concentrations readily attainable in the plasma of hemodialysis patients (65). Despite the potential neurotoxicity of β2-microglobulin, the concentration of β2-microglobulin in CSF is maintained at a low level due to the protective effect of the blood-brain barrier (BBB). However, BBB may be damaged in uremic patients. Furthermore, β2-microglobulin has been also identified as a pro-aging factor that impairs hippocampal cognition and neurogenesis (66).
3.2.1.3 Protein-bound compounds
3.2.1.3.1 Indoxyl sulfate
The serum indoxyl sulfate concentration in uremic patients was markedly increased (80). The average serum level of total indoxyl sulfate in patients with uremia was approximately 43 times higher than that in normal subjects (45). The accumulation of indoxyl sulfate within brain structures may be related to the expression of organic anion transporter 3 (OAT3) efflux transporter (67). Indoxyl sulfate involve in human astrocyte apoptosis through oxidative stress and protein kinase inhibition (68). Bartłomiej et al. also reported that the level of indoxyl sulfate in the cerebrospinal fluid of patients with Parkinson’s disease was higher than expected, which may affect the progression of Parkinson’s disease (69). Furthermore, intraperitoneal injection of indoxyl sulfate leads to emotional disturbance in mice unilateral nephrectomy, which is associated with altered neuronal cell function and neuroinflammation (70).
3.2.1.3.2 Homocysteine
The serum level of total homocysteine in uremic patients was significantly higher than that in the healthy people (81). Homocysteine has been demonstrated to display direct neurotoxic effects (71). Indeed, homocysteine causes direct neurotoxicity in cortical neurons by activating the glutamate N-methyl-d-aspartate (NMDA) receptors (72). Overactivation of these receptors may mediate ischemic damage in the brain (73). Thus, homocysteine may be involved not only in cerebrovascular injury, but also in neurotoxicity. A prospective cohort study reported plasma homocysteine as an independent risk factor that may play a persistent role in dementia (74). Further studies are needed to assess the impact of hyperhomocysteinemia in uremia-related neurological damage.
3.2.2 Oxidative stress
Oxidative stress can be regarded as an imbalance between reactive oxygen species (ROS) production and antioxidant defense. This disturbance can impact cellular function and damage nucleic acids, proteins and lipids (82). Numerous studies have demonstrated increased oxidative stress in uremic patients. Low-density lipoproteins (LDL) from uremic patients are more prone to copper-induced lipid peroxidation than plasma LDL from healthy subjects in vitro (83). In addition to lipid peroxidation, uremic oxidative stress is also biochemically characterized by a state of accumulation of reactive aldehydes and oxidative thiols and depletion of reductive thiols (84). Reactive aldehydes can be regarded as end products of various oxidation reactions, such as the oxidation of alcohol and amino (85). Several reactive aldehyde compounds, including acrolein, malondialdehyde, methylglyoxal, glyoxal, and hydroxynonenal have been demonstrated to be 10-fold higher concentrations in uremic plasma than that in healthy individuals (86, 87). In particular, α, β-unsaturated aldehydes are important oxidation products with capable of forming advanced glycation end products (AGEs) (88). In uremia, the accumulation of reactive aldehydes is primarily due to reduced renal catabolism and increased production by myeloperoxidase-catalyzed activation of phagocytes (84). Thiols, important antioxidant component, are found to be depleted in hemodialysis patients and thus is unavailable to participate in antioxidant defense (87). Furthermore, plasma glutathione levels decreased in hemodialysis patients and accompanied by a significant decrease in glutathione peroxidase function (89). Mitochondria are also involved in the production of ROS. Impaired mitochondrial respiratory system in CKD patients is deemed as a consequence of enhanced oxidative stress, which may explain the abnormal energy metabolism in this population. Many uremic toxins such as indoxyl sulfate, p-cresyl sulfate, kynurenic acid and hippurate can inhibit the mitochondrial respiratory chain and lead to the overproduction of ROS (78).
3.2.2.1 The role of oxidative stress in uremic neuropathy
Neuronal tissue is quite vulnerable to oxidative stress (90). Oxidative stress not only causes direct neurotoxicity through phospholipid membrane peroxidation (91), but also incites excitotoxicity by promoting glutamate release (92, 93). This leads to the activation of NMDA and non-NMDA receptors, resulting in severely elevated intracellular Ca2+ levels, neuronal nitric oxide synthase (NOS) activation, peroxynitrite formation, protein nitration, mitochondrial damage, and ultimately causing neuronal damage or death (84). As mentioned above, lipid peroxidation, increased AGEs, and mitochondrial dysfunction are present in uremic patients. Lipid peroxidation can cause several deleterious effects during the development of neurodegenerative diseases (94). Since lipid peroxidation can increase the production of N-carboxymethyl lysine, which is a major form of AGEs (95). It has reported that the expression of AGEs receptors increased in human peripheral neuropathy (96). AGEs induce hypertrophy of the endoneurial microvascular basement membrane by stimulating pericytes to release TGF-β and vascular endothelial growth factor (VEGF), thereby destroying the BBB (97). In addition, AGEs exert deleterious effects on cells by upregulating pro-inflammatory cytokines and triggering inflammation (98). Similarly, reduced energy due to mitochondrial dysfunction can lead to neuronal damage, death and neuropathy (99). Additionally, oxidative stress is associated with an increased inflammatory response. Indeed, oxidative stress and inflammation can form a feed-forward cycle, leading to neurodegeneration (100). Oxidative stress can directly or indirectly induce the production of pro-inflammatory cytokines by activating tumor necrosis factor-α (TNF-α) and nuclear factor-kappaB (NF-κB). These factors can trigger the inflammatory mechanisms in neurodegeneration (100, 101). As a stimulator, oxidative stress can also cause the dysfunction of proteasome, thereby enhancing free radical generation and protein oxidation, as well as inhibiting ubiquitination. Consequently, unwanted proteins will accumulate in the cytoplasm and induce the formation of senile plaques (102), which is closely related to cognitive impairment and Alzheimer’s disease (103).
3.2.3 Neurotransmitter imbalance
Neurotransmitters are defined as substances that carry messages between neurons. When neurotransmitters bind to neurotransmitter receptors, it can transmit either excitatory or inhibitory signals, thereby triggering a series of responses. However, when neurotransmitters become dysregulated, it can result in neurological disorders (104). Franz et al. found that both the extracellular concentrations of neuroexcitatory and neuroinhibitory amino acids increased in medial preoptic area (MPOA) of chronic renal failure rats (105). Moreover, Schaefer et al. found that basal outflow of synthetic g-aminobutyric acid (GABA), glutamate and aspartate was increased in the uremic rat brain (105). Consistent with this result, Biasoli et al. demonstrated altered transport of amino acids across the BBB and resulted in a high phenylalanine-tyrosine ratio and low glutamine level in the brain (106). Similarly, some other researchers found the phenomenon of plasma/cerebrospinal fluid amino acid imbalance in infants with CRF. The accumulation of abnormal amino acids in CSF was detrimental to the function of Na-K pump in synaptosomes, and might affect the development of neurons and astroglia, which may partially explain the potential mechanisms of impairments caused by CRF in pediatric patients (107).
Glutamine is the main source of synthetic GABA, and low levels of glutamine in the brain suggest the deficiency of GABA (108). Perry et al. dissected 10 patients with ESRD and dialysis encephalopathy and found decreased GABA levels in many brain regions, with reductions of more than 40% in the cortex and thalamus. Clinical consequences of GABA deficiency may be associate with cognitive impairment (109). This may suggest that cognitive impairment in uremia may be related with reduced GABA. As a well-known neurotransmitter, acetylcholine plays a multifaceted role (110). However, renal failure results in a disorder of acetylcholine metabolism, which leads to its accumulation and release in brain synaptosomes. This is mainly due to decreased activity of choline kinase, to some extent, mediated by the secondary hyperparathyroidism of renal failure (111). Thus, behavioral and motor changes in uremic patients may be due in part to disturbances in acetylcholine metabolism.
3.2.4 Blood brain barrier disruption
BBB is a highly complex and dynamic structure involving endothelial cells, basement membranes, astrocyte foot processes, and pericytes that separates the CNS from the peripheral blood circulation (112). The BBB can tightly regulate the exchange of substances such as molecules and ions between the blood and the CNS. This is critical for strictly controlling the CNS homeostasis, as well as protecting the CNS from pathogens, inflammation, toxins, injury and disease (113). Recent studies have shown that BBB disruption play an essential role in neurodegenerative processes (114, 115). Meanwhile, some animal models of acute and chronic renal failure show BBB disruption in the context of uremia, but the mechanism remains unclear (116, 117). It has been reported that uremia increases the permeability of BBB as assessed by radioisotopes (118). Some uremic toxins, such as indoxyl sulfate, has been reported to mediate the activation of the transcription factor aryl hydrocarbon receptor (AhR) to induce BBB disruption and uremia-related cognitive impairment. Uremia activates inflammatory and oxidative pathways and inhibits antioxidant and cytoprotective systems, which erode the brain’s capillary-junction complexes (119). This suggests a possible disruption of the integrity of the blood-brain barrier in uremic patients.
3.3 The alteration of neuroimmune
Recent studies have uncovered the interaction between the nervous and immune systems, which can modulate immune function and inflammation, known as neuroimmune (120). One of the well- explored neuroimmune pathways is the cholinergic anti-inflammatory pathway (CAP). Chemicals released in the inflammatory bind to receptors on sensory nerve terminals and transmit neuronal signals to the brain via the afferents of vagus (121). This signal activates the vagal efferent nerve through the nucleus of the solitary tract and the dorsal vagus motor nucleus. The activated efferent vagal nerve stimulates the splenic nerve, although whether there is a direct connection between the efferent nerve of vagus and the splenic nerve remains controversial (122). Subsequently, norepinephrine is released from splenic nerve terminals and bind to β2-adrenergic receptors expressed by CD4+T cells in the spleen to mediate acetylcholine release from these cells (123). Acetylcholine interacts with the α7 nicotinic acetylcholine receptors (α7nAChRs) expressed on macrophages, which are key mediators to the regulation of neuroimmune, thus leading to inhibition of NF-κB and activation of Janus kinase 2 signal transducer and activator of transcription 3 (JAK2-STAT3) pathway, and ultimately inhibiting the production of pro-inflammatory cytokines, such as TNFα, and suppressing inflammation (124–126). Notably, the neuroimmune cross-talk between the nervous system and the kidney to maintain a normal physiological state. However, pathological state, such as uremia, can disrupt this interaction, further leading to disturbances in homeostasis that may ultimately result in neuropathy (127).
3.3.1 Inflammatory cytokines and cells
Multiple factors can contribute to immune dysregulation and inflammatory activation in uremic patients. Uremia exhibits a persistent low-grade inflammatory environment, involving impaired inflammatory cell function and dysregulated cytokine networks. Some of these factors may be related to the primary disease, and some may be due to the highly proinflammatory oxidative stress generated by the uremic environment (128). Indeed, a feed-forward loop is formed between oxidative stress and inflammation, which lead to neurodegeneration (100). Oxidative stress can induce production of pro-inflammatory cytokines directly or indirectly via activating TNF-α and NF-κB (129–131). Available data suggest that the anti-inflammatory cytokine interleukin-10 (IL-10) and the major pro-inflammatory cytokines IL-6 (IL-6) and TNF-α may play critical roles in uremia (132). The cerebrorenal interactions suggested that IL-6 and TNF-α are likely to have an effect on the CNS, although the mechanisms are yet to be identified (133). NALP3, the best characterized inflammasome, act as a connection between inflammation and immunity. The NALP3 is activated in immune peripheral cells isolated from uremic patients. These cells show higher levels of mRNA of NALP3, IL-1β and IL-18 compared to healthy subjects (134). Interestingly, metabolic acidosis is another cause of inflammation among chronic hemodialysis patients, which develop with a decrease in glomerular filtration rate (GFR) (135, 136). Uremic toxins may contribute to gut dysbiosis in CKD and facilitate the translocation of gut bacteria, which in turn activates systemic inflammatory responses (137). Furthermore, protein-bound uremic toxins, such as indoxyl sulfate exert a proinflammatory effect by stimulating crosstalk between leukocytes and blood vessels, resulting in vascular injury (138). In addition, leukocyte oxidative burst, that is, the production of large amounts of ROS via the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex (139), may further aggravate the inflammatory response (140, 141).
3.3.2 Immune cells
3.3.2.1 Innate immune system
3.3.2.1.1 Polymorphonuclear neutrophils
Polymorphonuclear neutrophils (PMNLs), named after their lobulated nuclei, contain multiple granules in their cytoplasm and are particularly important in nonspecific cellular immunity (142). In patients with ESRD, the expression of toll-like receptor 2 (TLR2), TLR4 and integrins on PMNLs is increased (143). This increased expression is a key mediator of oxidative stress and inflammation associated with renal failure, which contribute to tissue damage in this population (144). The level of ROS production, degranulation, and extracellular trap formation in PMNLs from dialysis patients are significantly elevated, suggesting spontaneous activation (145). Synergistic elimination of activated PMNLs is critical for resolution of inflammation (146).
3.3.2.1.2 Monocytes and macrophages
Monocytes are produced in the bone marrow and distributed in all body tissues as macrophages (147). Circulating monocytes were widely expanded in ESRD patients, especially CD14+CD16+ subsets (143). This may be due to the increased expression of integrins, TLRs and proinflammatory cytokines in uremic condition. The possibility therefore might lead to the release of CD14+CD16+ monocytes from the bone marrow (148). These abnormalities suggest spontaneous activation of monocytes and contribute to the prevailing oxidative stress and inflammation in ESRD. Thus, the number of circulating monocytes in ESRD patients, particularly the CD14+CD16+ subsets might be related to the degree of systemic inflammation and/or uremia.
3.3.2.1.3 Dendritic cells
Dendritic cells are considered to be the major professional antigen presenting cell in the immune system (149). Uremia has a negative effect on dendritic cells. Study have shown that monocyte-derived dendritic cells from CKD patients are less able to stimulate T cells than healthy controls (150). This may be related to the decreased expression of the key costimulatory molecule CD86 in the uremic situation (151). Dendritic cells are also present in the renal and involve in the progression of kidney failure by binding to glomerular antigens (152).
3.3.2.2 Adaptive immune cells
3.3.2.2.1 T cells
Exposure of naive T cells to antigen leads to clonal expansion and differentiation of memory T cells and effector T cells, which play a central role in cell-mediated immune responses (152). Patients with ESRD showed significantly reduced naïve and CD4+ and CD8+ T cells (153). Furthermore, the proliferation of T cell in ESRD patients have been reported to be impaired when detect in uremic serum (154). This is associated with increased sensitivity to apoptosis in both naive and memory T cells from uremic patients (155).
3.3.2.2.2 B cells
B lymphocytes are mainly produced by hematopoietic stem cells in the bone marrow and mediate humoral immunity by producing antigen-specific antibodies (156). Similar to T cells, B cell lymphopenia in ESRD patients is associated with progressive loss of renal function (157). Additionally, CD5+ innate B cells and CD27+ memory B cells are reduced in childhood chronic renal failure (158). Fernández et al. reported that uremic environment may sensitize B cells to apoptosis in ESRD patients (159). Madeleine et al. reported that the uremic environment may interfere with the differentiation and survival of B cells by down-regulating the BAFF receptor in transitional B cells in ESRD patients (157). Thus, the deficiency and dysfunction of B cells in advanced CKD may be mediated simultaneously by increased B cell apoptosis and impaired differentiation and maturation of transitional B cells.
4 Potential treatment for uremia
Hemodialysis and renal transplant are two common treatments for uremia. Efficient solute removal usually improves CNS symptoms over multiple dialysis sessions. However, dialysis is relatively effective only for some small water-soluble solutes, and some protein-bound solutes cannot be effectively removed. Kidney transplants are also effective at removing uremic toxins. Patients with kidney transplant are given immunosuppressants and prophylactic antibiotics, which cause changes in the gut microbiota (160).
4.1 Anti-inflammatory therapy
As mentioned above, pro-inflammatory factors play an important role in uremia, therefore, targeted anti-inflammatory therapy is recommended for these patients. Statins, a typical lipid-lowering drug, has been demonstrated to have an anti-inflammatory effect in hemodialysis patients (161). Angiotensin-converting enzyme (ACE) inhibitors can suppress TNF-α in advanced chronic renal (162). In patients with chronically uremia, pentoxifylline was confirmed to reduce the whole-body proteolysis, a characteristic of systemic inflammation activation (163).
4.2 Neuroimmunomodulatory therapy
Besides, neuroimmunomodulation of organ function has attracted extensive attention as a new treatment for diseases. Considering the anti-inflammatory effect of CAP pathway, stimulation of vagal nerve, has been shown to protect the kidney from AKI (164). However, electrical stimulation of the vagus nerve requires invasive surgery. In clinical application, ultrasound, a non-invasive imaging technique, can be used as a candidate for the activation of CAGigliotti et al. reported that ultrasound can suppresses inflammation in renal by stimulating the CAP pathway (165). This result might lead to a powerful nonpharmacological approach to block the inflammation of kidney and preserve function. Further studies are needed to explore the underlying mechanisms of neuroimmunomodulatory in inflammation and organ damage.
5 Limitations of current researches and perspectives
The paucity of data on pediatric uremia is a thorny problem. Due to the differences in region, culture and detection methods, the heterogeneity of the existing data is relatively high. For this reason, we discuss a general underlying mechanism of uremia. Therefore, a unified standard is urgently needed to register and follow up these patients. Besides, the association between uremic toxins and uremic neuropathy has not been fully elucidated, especially in children, which may require large prospective studies. Finding biostatistical associations between specific clinical signs and solute concentrations by combining high-throughput analytical methods with clinical databases represents an intriguing but underexplored track to unravel the pathophysiology of uremic neuropathy.
Moreover, the roles of immune cells and inflammatory factors in the uremic immune microenvironment and the inflammatory microenvironment remain to be explored, and the temporal and spatial distribution of immune cells and inflammatory factors needs to be further investigated. New methods such as single-cell sequencing and temporal-spatial omics need to be used to further explore the underlying mechanisms. In addition, non-invasive treatments, neuroimmune interventions, and targeted anti-inflammatory treatments in uremia, such as dose gradients or time gradients, need to be further explored.
Going forward, technological innovations and the combined efforts of the global scientific community may help elucidate some fundamental questions about the causes and treatment of uremic neuropathy in children, and provide new insights for this disease that can help pediatricians prescribe tailored treatment for children.
6 Conclusion
Pediatric uremic neuropathy is an extremely complex complication that leads to morbidity and mortality in children. In this review, we discuss the epidemiology of childhood uremic neuropathy, the underlying mechanisms, possible treatments, and current research gaps (Figure 1). Uremic neuropathy is associated with accumulation of toxins, oxidative stress, neurotransmitter imbalance, and disturbance of the blood-brain barrier. Among them, neuroimmune, including the change of inflammatory factors and immune cells, play a crucial role in the progression of uremic neuropathy. Different from traditional dialysis and kidney transplantation, treatments that focus on anti-inflammation and neuroimmune intervention show promising prospects.
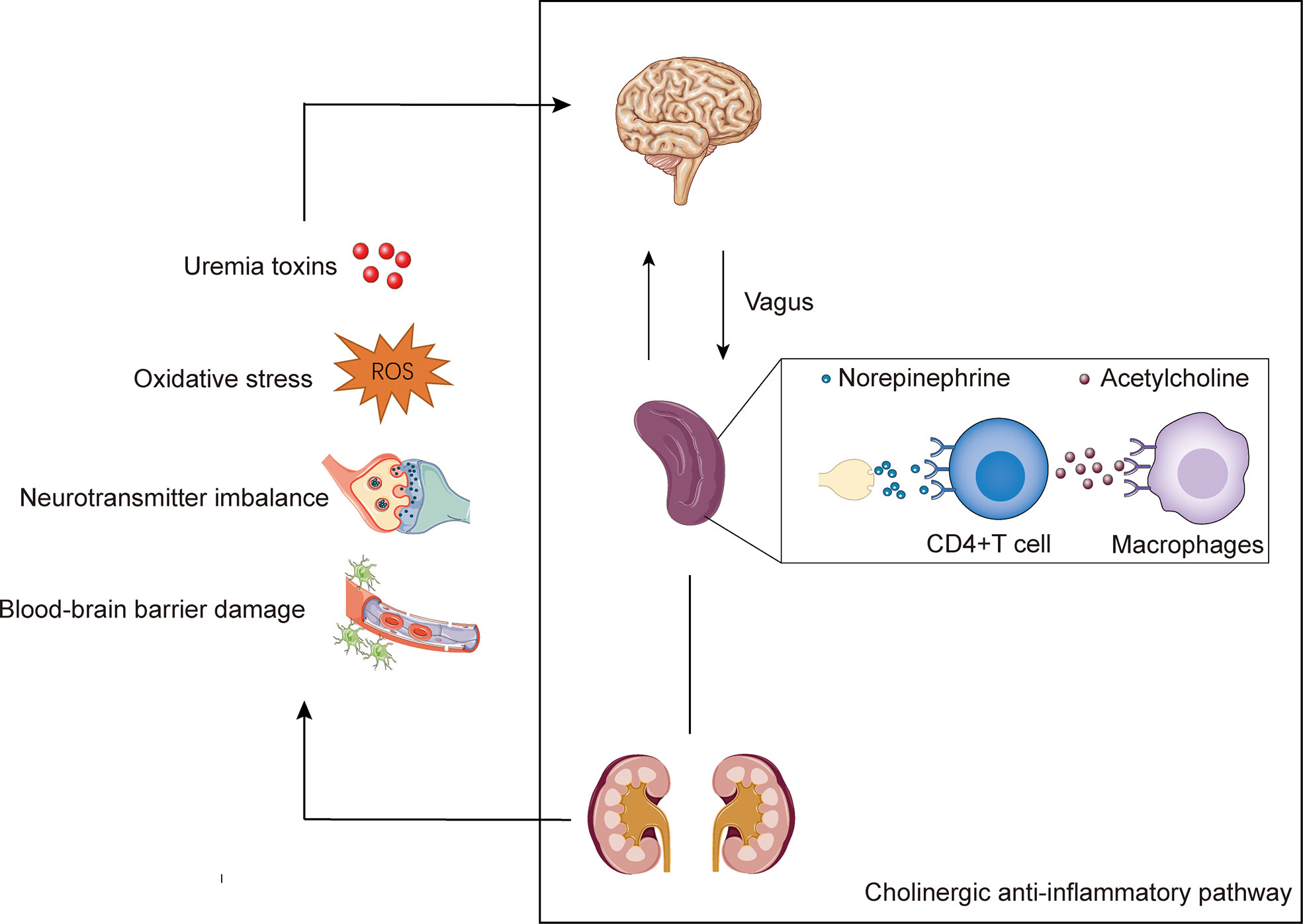
Figure 1 The potential mechanisms of uremic neuropathy and the cholinergic anti-inflammatory pathway. Uremic neuropathy is associated with accumulation of toxins, oxidative stress, neurotransmitter imbalance, and disturbance of the blood-brain barrier. These factors may lead to the release of inflammatory factors and stimulate the sensory afferent vagus nerve endings. The cholinergic anti-inflammatory pathway is starting with vagal activation. Signals transmits to the brain via the afferents of vagus. The activated efferent vagal nerve stimulates the splenic nerve and release norepinephrine. The norepinephrine binds to β2-adrenergic receptors expressed by CD4+T cells in the spleen and mediate the of release acetylcholine. Acetylcholine interacts with the α7 nicotinic acetylcholine receptors (α7nAChRs) expressed on macrophages, and ultimately inhibiting the production of pro-inflammatory.
Author contributions
LZ and GT wrote the main manuscript text. GC revised the manuscript. FY and YZ prepared figures and table. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Basic Public Welfare Research Project of Zhejiang Province (LGF22H050004) and Medical and Health Technology Program of Zhejiang Province (2022481900).
Acknowledgments
We thank all the authors of the original work and reviewers for their time and kindness in reviewing this paper. The authors would like to thank servier medical art for providing open access to the picture materials.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ESRD, end-stage renal disease; CDK, chronic kidney disease; AKI, acute kidney injury; CNS, central nervous system; PNS, peripheral nervous system; ANS, autonomic nervous system; GCs, guanidine compounds; GSA, guanidinosuccinic acid; XO, xanthine oxidase; PTH, parathyroid hormone; EEG, electroencephalogram; BBB, blood-brain barrier; OTA3, organic anion transporter 3; NMDA, N-methyl-d-aspartat; ROS, reactive oxygen species; LDL, low-density lipoproteins; AGES, advanced glycation end products; NOS, nitric oxide synthase; MPOA, medial preoptic area; GABA, gaminobutyric acid; AhR, aryl hydrocarbon receptor; CAP, cholinergic anti-inflammatory pathway; α7nAChRs, α7 nicotinic acetylcholine receptors; JAK2-STAT3, Janus kinase 2 signal transducer and activator of transcription 3; NF-κB, nuclear factor-kappaB; TNF-α, tumor necrosis factor-α; GFR, glomerular filtration rate; PMNLs, polymorphonuclear leukocytes; TLR2, toll-like receptor; ACE, angiotensin-converting enzyme.
References
2. Vanholder R, De Smet R, Glorieux G, Argilés A, Baurmeister U, Brunet P, et al. Review on Uremic Toxins: Classification, Concentration, and Interindividual Variability. Kidney Int (2003) 63(5):1934–43. doi: 10.1046/j.1523-1755.2003.00924.x
3. Hamed SA. Neurologic Conditions and Disorders of Uremic Syndrome of Chronic Kidney Disease: Presentations, Causes, and Treatment Strategies. Expert Rev Clin Pharmacol (2019) 12(1):61–90. doi: 10.1080/17512433.2019.1555468
4. Potter D, Larsen D, Leumann E, Perin D, Simmons J, Piel CF, et al. Treatment of Chronic Uremia in Childhood. Ii. Hemodialysis. Pediatr (1970) 46(5):678–89.
5. Potter D, Belzer FO, Rames L, Holliday MA, Kountz SL, Najarian JS. The Treatment of Chronic Uremia in Childhood. I. Transplantation. Pediatr (1970) 45(3):432–43.
6. Tönshoff B, Mehis O, Heinrich U, Blum WF, Ranke MB, Schauer A. Growth-Stimulating Effects of Recombinant Human Growth Hormone in Children with End-Stage Renal Disease. J Pediatr (1990) 116(4):561–6. doi: 10.1016/s0022-3476(05)81603-8
7. Fine RN. Growth Hormone and the Kidney: The Use of Recombinant Human Growth Hormone (Rhgh) in Growth-Retarded Children with Chronic Renal Insufficiency. J Am Soc Nephrol (1991) 1(10):1136–45. doi: 10.1681/asn.V1101136
8. Terashima M, Ishikawa A, Männer J, Yamada S, Takakuwa T. Early Development of the Cortical Layers in the Human Brain. J Anat (2021) 239(5):1039–49. doi: 10.1111/joa.13488
9. Geary DF, Fennell RS, Andriola M, Gudat J, Rodgers BM, Richard GA. Encephalopathy in Children with Chronic Renal Failure. J Pediatr (1980) 97(1):41–4. doi: 10.1016/s0022-3476(80)80127-2
10. Rotundo A, Nevins TE, Lipton M, Lockman LA, Mauer SM, Michael AF. Progressive Encephalopathy in Children with Chronic Renal Insufficiency in Infancy. Kidney Int (1982) 21(3):486–91. doi: 10.1038/ki.1982.50
11. McGraw ME, Haka-Ikse K. Neurologic-Developmental Sequelae of Chronic Renal Failure in Infancy. J Pediatr (1985) 106(4):579–83. doi: 10.1016/s0022-3476(85)80075-5
12. Fadel FI, Yamamah GAN, Hasanin RM, Mostafa EA, Abdalgeleel SA, Salah MM, et al. Hearing Assessment in Egyptian Children with Chronic Renal Failure on Regular Hemodialysis and Renal Transplantation Children. Ther Apher Dial (2021). doi: 10.1111/1744-9987.13783
13. Tsuchiyama F, Makino Y, Hirasawa K, Nagata S, Matsui H. Cerebral Palsy and Intellectual Disability in the Children Of women with Chronic Kidney Disease. Pediatr Neurol (2017) 73:71–7. doi: 10.1016/j.pediatrneurol.2016.03.007
14. Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS. Prevalence of Chronic Kidney Disease and Decreased Kidney Function in the Adult Us Population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis (2003) 41(1):1–12. doi: 10.1053/ajkd.2003.50007
15. Anochie I, Eke F. Chronic Renal Failure in Children: A Report from Port Harcourt, Nigeria (1985-2000). Pediatr Nephrol (2003) 18(7):692–5. doi: 10.1007/s00467-003-1150-0
16. Al-Eisa A, Naseef M, Al-Hamad N, Pinto R, Al-Shimeri N, Tahmaz M. Chronic Renal Failure in Kuwaiti Children: An Eight-Year Experience. Pediatr Nephrol (2005) 20(12):1781–5. doi: 10.1007/s00467-005-2000-z
17. Esbjörner E, Berg U, Hansson S. Epidemiology of Chronic Renal Failure in Children: A Report from Sweden 1986-1994 Swedish Pediatric Nephrology Association. Pediatr Nephrol (1997) 11(4):438–42. doi: 10.1007/s004670050312
18. Ardissino G, Daccò V, Testa S, Bonaudo R, Claris-Appiani A, Taioli E, et al. Epidemiology of Chronic Renal Failure in Children: Data from the Italkid Project. Pediatrics (2003) 111(4 Pt 1):e382–7. doi: 10.1542/peds.111.4.e382
19. Sutherland SM, Kwiatkowski DM. Acute Kidney Injury in Children. Adv Chronic Kidney Dis (2017) 24(6):380–7. doi: 10.1053/j.ackd.2017.09.007
20. Warady BA, Chadha V. Chronic Kidney Disease in Children: The Global Perspective. Pediatr Nephrol (2007) 22(12):1999–2009. doi: 10.1007/s00467-006-0410-1
21. Groothoff JW. Long-Term Outcomes of Children with End-Stage Renal Disease. Pediatr Nephrol (2005) 20(7):849–53. doi: 10.1007/s00467-005-1878-9
22. Konstantyner T, Sesso R, de Camargo MF, de Santis Feltran L, Koch-Nogueira PC. Pediatric Chronic Dialysis in Brazil: Epidemiology and Regional Inequalities. PloS One (2015) 10(8):e0135649. doi: 10.1371/journal.pone.0135649
23. Jiang Y, Shen Y, Lau KK. Survey of Chronic Haemodialysis in Children between 2007 and 2012 in China. Nephrol (Carlton) (2014) 19(7):375–8. doi: 10.1111/nep.12229
24. Chesnaye NC, Schaefer F, Groothoff JW, Bonthuis M, Reusz G, Heaf JG, et al. Mortality Risk in European Children with End-Stage Renal Disease on Dialysis. Kidney Int (2016) 89(6):1355–62. doi: 10.1016/j.kint.2016.02.016
25. Abd El Naby SA, Bahbah WA, Kasemy ZA, Mahmoud AA. Neurophysiological and Neuroradiological Changes in Children with Chronic Kidney Disease. Front Pediatr (2020) 8:570708. doi: 10.3389/fped.2020.570708
26. Murray AM, Bell EJ, Tupper DE, Davey CS, Pederson SL, Amiot EM, et al. The Brain in Kidney Disease (Brink) Cohort Study: Design and Baseline Cognitive Function. Am J Kidney Dis (2016) 67(4):593–600. doi: 10.1053/j.ajkd.2015.11.008
27. Lacerda G, Krummel T, Hirsch E. Neurologic Presentations of Renal Diseases. Neurol Clin (2010) 28(1):45–59. doi: 10.1016/j.ncl.2009.09.003
28. Seifter JL, Samuels MA. Uremic Encephalopathy and Other Brain Disorders Associated with Renal Failure. Semin Neurol (2011) 31(2):139–43. doi: 10.1055/s-0031-1277984
29. Jegatheswaran J, Torres C, Clark E. Uremic Cranial Neuropathy. Kidney Int (2018) 93(4):1021. doi: 10.1016/j.kint.2017.10.011
30. Korzets A, Marashek I, Schwartz A, Rosenblatt I, Herman M, Ori Y. Ischemic Optic Neuropathy in Dialyzed Patients: A Previously Unrecognized Manifestation of Calcific Uremic Arteriolopathy. Am J Kidney Dis (2004) 44(6):e93–7. doi: 10.1053/j.ajkd.2004.08.035
31. Cupidi C, Piccoli F, La Bella V. Acute Reversible Parkinsonism in a Diabetic-Uremic Patient. Clin Neurol Neurosurg (2006) 108(6):601–3. doi: 10.1016/j.clineuro.2005.04.002
32. Kiryluk K, Khan F, Valeri A. Acute Chorea and Bilateral Basal Ganglia Lesions in a Hemodialysis Patient. Kidney Int (2008) 73(9):1087–91. doi: 10.1038/sj.ki.5002768
33. Hurkx W, Hulstijn-Dirkmaat I, Pasman J, Rotteveel J, Visco Y, Schröder C. Evoked Potentials in Children with Chronic Renal Failure, Treated Conservatively or by Continuous Ambulatory Peritoneal Dialysis. Pediatr Nephrol (1995) 9(3):325–8. doi: 10.1007/bf02254201
34. Yu XZ, Lu S, Gou W, Wang W, Zou SH, Han YX, et al. Assessment of the Characteristics and Quality of Life of Patients with Uremic Peripheral Neuropathy. Clin Nephrol (2017) 87(2017):134–9. doi: 10.5414/cn108913
35. Ewing DJ, Winney R. Autonomic Function in Patients with Chronic Renal Failure on Intermittent Haemodialysis. Nephron (1975) 15(6):424–9. doi: 10.1159/000180525
36. Soriano G, Eisinger RP. Abnormal Response to the Valsalva Maneuver in Patients on Chronic Hemodialysis. Nephron (1972) 9(4):251–6. doi: 10.1159/000180157
37. Lazarus JM, Hampers CL, Lowrie EG, Merrill JP. Baroreceptor Activity in Normotensive and Hypertensive Uremic Patients. Circulation (1973) 47(5):1015–21. doi: 10.1161/01.cir.47.5.1015
38. Kersh ES, Kronfield SJ, Unger A, Popper RW, Cantor S, Cohn K. Autonomic Insufficiency in Uremia as a Cause of Hemodialysis-Induced Hypotension. New Engl J Med (1974) 290(12):650–3. doi: 10.1056/nejm197403212901203
39. Kogon AJ, Matheson MB, Flynn JT, Gerson AC, Warady BA, Furth SL, et al. Depressive Symptoms in Children with Chronic Kidney Disease. J Pediatr (2016) 168:164–70.e1. doi: 10.1016/j.jpeds.2015.09.040
40. Fraser CL, Arieff AI. Nervous System Complications in Uremia. Ann Intern Med (1988) 109(2):143–53. doi: 10.7326/0003-4819-109-2-143
41. Popel J, Joffe R, Acton BV, Bond GY, Joffe AR, Midgley J, et al. Neurocognitive and Functional Outcomes at 5 years of Age after Renal Transplant in Early Childhood. Pediatr Nephrol (2019) 34(5):889–95. doi: 10.1007/s00467-018-4158-1
42. Lawry KW, Brouhard BH, Cunningham RJ. Cognitive Functioning and School Performance in Children with Renal Failure. Pediatr Nephrol (1994) 8(3):326–9. doi: 10.1007/bf00866349
43. Dubois J, Dehaene-Lambertz G, Kulikova S, Poupon C, Hüppi PS, Hertz-Pannier L. The Early Development of Brain White Matter: A Review of Imaging Studies in Fetuses, Newborns and Infants. Neuroscience (2014) 276:48–71. doi: 10.1016/j.neuroscience.2013.12.044
44. Vanholder R, Argilés A, Baurmeister U, Brunet P, Clark W, Cohen G, et al. Uremic Toxicity: Present State of the Art. Int J Artif Organs (2001) 24(10):695–725.
45. Duranton F, Cohen G, De Smet R, Rodriguez M, Jankowski J, Vanholder R, et al. Normal and Pathologic Concentrations of Uremic Toxins. J Am Soc Nephrol (2012) 23(7):1258–70. doi: 10.1681/asn.2011121175
46. Sindhu KK. Uremic Toxins: Some Thoughts on Acrolein and Spermine. Ren Fail (2016) 38(10):1755–8. doi: 10.1080/0886022x.2016.1229990
47. Schepers E, Glorieux G, Vanholder R. The Gut: The Forgotten Organ in Uremia? Blood Purif (2010) 29(2):130–6. doi: 10.1159/000245639
48. Meijers B, Glorieux G, Poesen R, Bakker SJ. Nonextracorporeal Methods for Decreasing Uremic Solute Concentration: A Future Way to Go? Semin Nephrol (2014) 34(2):228–43. doi: 10.1016/j.semnephrol.2014.02.012
49. Popkov VA, Silachev DN, Zalevsky AO, Zorov DB, Plotnikov EY. Mitochondria as a Source and a Target for Uremic Toxins. Int J Mol Sci (2019). doi: 10.3390/ijms20123094
50. Avram MM, Feinfeld DA, Huatuco AH. Search for the Uremic Toxin Decreased Motor-Nerve Conduction Velocity and Elevated Parathyroid Hormone in Uremia. New Engl J Med (1978) 298(18):1000–3. doi: 10.1056/nejm197805042981805
51. De Deyn PP, Vanholder R, Eloot S, Glorieux G. Guanidino Compounds as Uremic (Neuro)Toxins. Semin Dial (2009) 22(4):340–5. doi: 10.1111/j.1525-139X.2009.00577.x
52. Torremans A, Marescau B, Van Dam D, Van Ginneken C, Van Meir F, Van Bogaert PP, et al. Gsa: Behavioral, Histological, Electrophysiological and Neurochemical Effects. Physiol Behav (2005) 84(2):251–64. doi: 10.1016/j.physbeh.2004.12.001
53. D'Hooge R, Pei YQ, Marescau B, De Deyn PP. Convulsive Action and Toxicity of Uremic Guanidino Compounds: Behavioral Assessment and Relation to Brain Concentration in Adult Mice. J Neurol Sci (1992) 112(1-2):96–105. doi: 10.1016/0022-510x(92)90138-b
54. de Jonge WJ, Marescau B, D'Hooge R, De Deyn PP, Hallemeesch MM, Deutz NE, et al. Overexpression of Arginase Alters Circulating and Tissue Amino Acids and Guanidino Compounds and Affects Neuromotor Behavior in Mice. J Nutr (2001) 131(10):2732–40. doi: 10.1093/jn/131.10.2732
55. De Deyn PP, Macdonald RL. Guanidino Compounds That Are Increased in Cerebrospinal Fluid and Brain of Uremic Patients Inhibit Gaba and Glycine Responses on Mouse Neurons in Cell Culture. Ann Neurol (1990) 28(5):627–33. doi: 10.1002/ana.410280505
56. D'Hooge R, Manil J, Colin F, De Deyn PP. Guanidinosuccinic Acid Inhibits Excitatory Synaptic Transmission in Ca1 Region of Rat Hippocampal Slices. Ann Neurol (1991) 30(4):622–3. doi: 10.1002/ana.410300419
57. Hiramatsu M. A Role for Guanidino Compounds in the Brain. Mol Cell Biochem (2003) 244(1-2):57–62.
58. Marzocco S, Popolo A, Bianco G, Pinto A, Autore G. Pro-Apoptotic Effect of Methylguanidine on Hydrogen Peroxide-Treated Rat Glioma Cell Line. Neurochem Int (2010) 57(5):518–24. doi: 10.1016/j.neuint.2010.06.016
59. Biasibetti-Brendler H, Schmitz F, Pierozan P, Zanotto BS, Prezzi CA, de Andrade RB, et al. Hypoxanthine Induces Neuroenergetic Impairment and Cell Death in Striatum of Young Adult Wistar Rats. Mol Neurobiol (2018) 55(5):4098–106. doi: 10.1007/s12035-017-0634-z
60. Wamser MN, Leite EF, Ferreira VV, Delwing-de Lima D, da Cruz JG, Wyse AT, et al. Effect of Hypoxanthine, Antioxidants and Allopurinol on Cholinesterase Activities in Rats. J Neural Transm (Vienna) (2013) 120(9):1359–67. doi: 10.1007/s00702-013-0989-x
61. Vida C, Corpas I, de la Fuente M, González EM. Age-Related Changes in Xanthine Oxidase Activity and Lipid Peroxidation, as Well as in the Correlation between Both Parameters, in Plasma and Several Organs from Female Mice. J Physiol Biochem (2011) 67(4):551–8. doi: 10.1007/s13105-011-0100-8
62. Hughes K, Flynn T, de Zoysa J, Dalbeth N, Merriman TR. Mendelian Randomization Analysis Associates Increased Serum Urate, Due to Genetic Variation in Uric Acid Transporters, with Improved Renal Function. Kidney Int (2014) 85(2):344–51. doi: 10.1038/ki.2013.353
63. Mink R, Johnston J. The Effect of Infusing Hypoxanthine or Xanthine on Hypoxic-Ischemic Brain Injury in Rabbits. Brain Res (2007) 1147:256–64. doi: 10.1016/j.brainres.2007.02.004
64. Duque EJ, Elias RM, Moysés RMA. Parathyroid Hormone: A Uremic Toxin. Toxins (Basel) (2020) 12(3. doi: 10.3390/toxins12030189
65. Craver L, Marco MP, Martínez I, Rue M, Borràs M, Martín ML, et al. Mineral Metabolism Parameters Throughout Chronic Kidney Disease Stages 1-5–Achievement of K/Doqi Target Ranges. Nephrol Dial Transplant (2007) 22(4):1171–6. doi: 10.1093/ndt/gfl718
66. Cogan MG, Covey CM, Arieff AI, Wisniewski A, Clark OH, Lazarowitz V, et al. Central Nervous System Manifestations of Hyperparathyroidism. Am J Med (1978) 65(6):963–70. doi: 10.1016/0002-9343(78)90748-9
67. Fraser CL, Sarnacki P, Arieff AI. Calcium Transport Abnormality in Uremic Rat Brain Synaptosomes. J Clin Invest (1985) 76(5):1789–95. doi: 10.1172/jci112170
68. Guisado R, Arieff AI, Massry SG, Lazarowitz V, Kerian A. Changes in the Electroencephalogram in Acute Uremia Effects of Parathyroid Hormone and Brain Electrolytes. J Clin Invest (1975) 55(4):738–45. doi: 10.1172/jci107984
69. Lourida I, Thompson-Coon J, Dickens CM, Soni M, Kuźma E, Kos K, et al. Parathyroid Hormone, Cognitive Function and Dementia: A Systematic Review. PloS One (2015) 10(5):e0127574. doi: 10.1371/journal.pone.0127574
70. Giorgetti S, Raimondi S, Cassinelli S, Bucciantini M, Stefani M, Gregorini G, et al. Beta2-Microglobulin Is Potentially Neurotoxic, but the Blood Brain Barrier Is Likely to Protect the Brain from Its Toxicity. Nephrol Dial Transplant (2009) 24(4):1176–81. doi: 10.1093/ndt/gfn623
71. Smith LK, He Y, Park JS, Bieri G, Snethlage CE, Lin K, et al. β 2-Microglobulin Is a Systemic Pro-Aging Factor That Impairs Cognitive Function and Neurogenesis. Nat Med (2015) 21(8):932–7. doi: 10.1038/nm.3898
72. Vanholder R, Schepers E, Pletinck A, Nagler EV, Glorieux G. The Uremic Toxicity of Indoxyl Sulfate and P-Cresyl Sulfate: A Systematic Review. J Am Soc Nephrol (2014) 25(9):1897–907. doi: 10.1681/asn.2013101062
73. Hosoya K, Tachikawa M. Roles of Organic Anion/Cation Transporters at the Blood-Brain and Blood-Cerebrospinal Fluid Barriers Involving Uremic Toxins. Clin Exp Nephrol (2011) 15(4):478–85. doi: 10.1007/s10157-011-0460-y
74. Lin YT, Wu PH, Tsai YC, Hsu YL, Wang HY, Kuo MC, et al. Indoxyl Sulfate Induces Apoptosis through Oxidative Stress and Mitogen-Activated Protein Kinase Signaling Pathway Inhibition in Human Astrocytes. J Clin Med (2019) 8(2). doi: 10.3390/jcm8020191
75. Sankowski B, Księżarczyk K, Raćkowska E, Szlufik S, Koziorowski D, Giebułtowicz J. Higher Cerebrospinal Fluid to Plasma Ratio of P-Cresol Sulfate and Indoxyl Sulfate in Patients with Parkinson's Disease. Clin Chim Acta (2020) 501:165–73. doi: 10.1016/j.cca.2019.10.038
76. Sun CY, Li JR, Wang YY, Lin SY, Ou YC, Lin CJ, et al. Indoxyl Sulfate Caused Behavioral Abnormality and Neurodegeneration in Mice with Unilateral Nephrectomy. Aging (Albany NY) (2021) 13(5):6681–701. doi: 10.18632/aging.202523
77. Perna AF, Satta E, Acanfora F, Lombardi C, Ingrosso D, De Santo NG. Increased Plasma Protein Homocysteinylation in Hemodialysis Patients. Kidney Int (2006) 69(5):869–76. doi: 10.1038/sj.ki.5000070
78. Ho PI, Ortiz D, Rogers E, Shea TB. Multiple Aspects of Homocysteine Neurotoxicity: Glutamate Excitotoxicity, Kinase Hyperactivation and DNA Damage. J Neurosci Res (2002) 70(5):694–702. doi: 10.1002/jnr.10416
79. Lipton SA, Kim WK, Choi YB, Kumar S, D'Emilia DM, Rayudu PV, et al. Neurotoxicity Associated with Dual Actions of Homocysteine at the N-Methyl-D-Aspartate Receptor. Proc Natl Acad Sci United States America (1997) 94(11):5923–8. doi: 10.1073/pnas.94.11.5923
80. Simon RP, Swan JH, Griffiths T, Meldrum BS. Blockade of N-Methyl-D-Aspartate Receptors May Protect against Ischemic Damage in the Brain. Sci (New York NY) (1984) 226(4676):850–2. doi: 10.1126/science.6093256
81. Seshadri S, Wolf PA, Beiser AS, Selhub J, Au R, Jacques PF, et al. Association of Plasma Total Homocysteine Levels with Subclinical Brain Injury: Cerebral Volumes, White Matter Hyperintensity, and Silent Brain Infarcts at Volumetric Magnetic Resonance Imaging in the Framingham Offspring Study. Arch Neurol (2008) 65(5):642–9. doi: 10.1001/archneur.65.5.642
82. Lushchak VI. Free Radicals, Reactive Oxygen Species, Oxidative Stress and Its Classification. Chem Biol Interact (2014) 224:164–75. doi: 10.1016/j.cbi.2014.10.016
83. Himmelfarb J. Oxidative Stress in Hemodialysis. Contrib Nephrol (2008) 161:132–7. doi: 10.1159/000130658
84. Himmelfarb J, Hakim RM. Oxidative Stress in Uremia. Curr Opin Nephrol Hypertens (2003) 12(6):593–8. doi: 10.1097/00041552-200311000-00004
85. Frijhoff J, Winyard PG, Zarkovic N, Davies SS, Stocker R, Cheng D, et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxidants Redox Signaling (2015) 23(14):1144–70. doi: 10.1089/ars.2015.6317
86. Miyata T, van Ypersele de Strihou C, Kurokawa K, Baynes JW. Alterations in Nonenzymatic Biochemistry in Uremia: Origin and Significance of "Carbonyl Stress" in Long-Term Uremic Complications. Kidney Int (1999) 55(2):389–99. doi: 10.1046/j.1523-1755.1999.00302.x
87. Himmelfarb J, McMonagle E, McMenamin E. Plasma Protein Thiol Oxidation and Carbonyl Formation in Chronic Renal Failure. Kidney Int (2000) 58(6):2571–8. doi: 10.1046/j.1523-1755.2000.00443.x
88. Anderson MM, Hazen SL, Hsu FF, Heinecke JW. Human Neutrophils Employ the Myeloperoxidase-Hydrogen Peroxide-Chloride System to Convert Hydroxy-Amino Acids into Glycolaldehyde, 2-Hydroxypropanal, and Acrolein A Mechanism for the Generation of Highly Reactive Alpha-Hydroxy and Alpha,Beta-Unsaturated Aldehydes by Phagocytes at Sites of Inflammation. J Clin Invest (1997) 99(3):424–32. doi: 10.1172/jci119176
89. Ceballos-Picot I, Witko-Sarsat V, Merad-Boudia M, Nguyen AT, Thévenin M, Jaudon MC, et al. Glutathione Antioxidant System as a Marker of Oxidative Stress in Chronic Renal Failure. Free Radic Biol Med (1996) 21(6):845–53. doi: 10.1016/0891-5849(96)00233-x
90. Contestabile A. Oxidative Stress in Neurodegeneration: Mechanisms and Therapeutic Perspectives. Curr Top Med Chem (2001) 1(6):553–68. doi: 10.2174/1568026013394723
91. Farooqui AA, Ong WY, Horrocks LA. Biochemical Aspects of Neurodegeneration in Human Brain: Involvement of Neural Membrane Phospholipids and Phospholipases A2. Neurochem Res (2004) 29(11):1961–77. doi: 10.1007/s11064-004-6871-3
92. Savolainen KM, Loikkanen J, Naarala J. Amplification of Glutamate-Induced Oxidative Stress. Toxicol Lett (1995) 82-83:399–405. doi: 10.1016/0378-4274(95)03490-0
93. Beal MF. Aging, Energy, and Oxidative Stress in Neurodegenerative Diseases. Ann Neurol (1995) 38(3):357–66. doi: 10.1002/ana.410380304
94. Moreira PI, Smith MA, Zhu X, Nunomura A, Castellani RJ, Perry G. Oxidative Stress and Neurodegeneration. Ann N Y Acad Sci (2005) 1043:545–52. doi: 10.1196/annals.1333.062
95. Reddy S, Bichler J, Wells-Knecht KJ, Thorpe SR, Baynes JW. N Epsilon-(Carboxymethyl)Lysine Is a Dominant Advanced Glycation End Product (Age) Antigen in Tissue Proteins. Biochemistry (1995) 34(34):10872–8. doi: 10.1021/bi00034a021
96. Juranek JK, Kothary P, Mehra A, Hays A, Brannagan TH 3rd, Schmidt AM. Increased Expression of the Receptor for Advanced Glycation End-Products in Human Peripheral Neuropathies. Brain Behav (2013) 3(6):701–9. doi: 10.1002/brb3.176
97. Shimizu F, Sano Y, Haruki H, Kanda T. Advanced Glycation End-Products Induce Basement Membrane Hypertrophy in Endoneurial Microvessels and Disrupt the Blood-Nerve Barrier by Stimulating the Release of Tgf- β and Vascular Endothelial Growth Factor (Vegf) by Pericytes. Diabetologia (2011) 54(6):1517–26. doi: 10.1007/s00125-011-2107-7
98. Münch G, Schinzel R, Loske C, Wong A, Durany N, Li JJ, et al. Alzheimer's Disease—Synergistic Effects of Glucose Deficit, Oxidative Stress and Advanced Glycation Endproducts. J Neural Transm (1998) 105(4-5):439–61. doi: 10.1007/s007020050069
99. Yan MH, Wang X, Zhu X. Mitochondrial Defects and Oxidative Stress in Alzheimer Disease and Parkinson Disease. Free Radic Biol Med (2013) 62:90–101. doi: 10.1016/j.freeradbiomed.2012.11.014
100. Fischer R, Maier O. Interrelation of Oxidative Stress and Inflammation in Neurodegenerative Disease: Role of Tnf. Oxid Med Cell Longev (2015) 2015:610813. doi: 10.1155/2015/610813
101. Sivandzade F, Prasad S, Bhalerao A, Cucullo L. Nrf2 and Nf-κb Interplay in Cerebrovascular and Neurodegenerative Disorders: Molecular Mechanisms and Possible Therapeutic Approaches. Redox Biol (2019) 21:101059. doi: 10.1016/j.redox.2018.11.017
102. Andersen JK. Oxidative Stress in Neurodegeneration: Cause or Consequence? Nat Med (2004) 10 Suppl:S18–25. doi: 10.1038/nrn1434
103. Casanova MF, Carosella NW, Gold JM, Kleinman JE, Weinberger DR, Powers RE. A Topographical Study of Senile Plaques and Neurofibrillary Tangles in the Hippocampi of Patients with Alzheimer's Disease and Cognitively Impaired Patients with Schizophrenia. Psychiatry Res (1993) 49(1):41–62. doi: 10.1016/0165-1781(93)90029-g
105. Schaefer F, Vogel M, Kerkhoff G, Woitzik J, Daschner M, Mehls O. Experimental Uremia Affects Hypothalamic Amino Acid Neurotransmitter Milieu. J Am Soc Nephrol (2001) 12(6):1218–27. doi: 10.1681/asn.V1261218
106. Biasioli S, Chiaramonte S, Fabris A, Feriani M, Pisani E, Ronco C, et al. Neurotransmitter Imbalance in Plasma and Cerebrospinal Fluid During Dialytic Treatment. Trans Am Soc Artif Intern Organs (1983) 29:44–9.
107. Gerrits GP, Kamphuis S, Monnens LA, Trijbels JM, Schröder CH, Koster A, et al. Cerebrospinal Fluid Levels of Amino Acids in Infants and Young Children with Chronic Renal Failure. Neuropediatrics (1998) 29(1):35–9. doi: 10.1055/s-2007-973531
108. Holten AT, Gundersen V. Glutamine as a Precursor for Transmitter Glutamate, Aspartate and Gaba in the Cerebellum: A Role for Phosphate-Activated Glutaminase. J Neurochem (2008) 104(4):1032–42. doi: 10.1111/j.1471-4159.2007.05065.x
109. Tomoda T, Sumitomo A, Shukla R, Hirota-Tsuyada Y, Miyachi H, Oh H, et al. Bdnf Controls Gaba(a)R Trafficking and Related Cognitive Processes Via Autophagic Regulation of P62. Neuropsychopharmacology (2022) 47(2):553–63. doi: 10.1038/s41386-021-01116-0
111. Ni Z, Smogorzewski M, Massry SG. Derangements in Acetylcholine Metabolism in Brain Synaptosomes in Chronic Renal Failure. Kidney Int (1993) 44(3):630–7. doi: 10.1038/ki.1993.291
112. Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and Function of the Blood-Brain Barrier. Neurobiol Dis (2010) 37(1):13–25. doi: 10.1016/j.nbd.2009.07.030
113. Daneman R. The Blood-Brain Barrier in Health and Disease. Ann Neurol (2012) 72(5):648–72. doi: 10.1002/ana.23648
114. Noe CR, Noe-Letschnig M, Handschuh P, Noe CA, Lanzenberger R. Dysfunction of the Blood-Brain Barrier-a Key Step in Neurodegeneration and Dementia. Front Aging Neurosci (2020) 12:185. doi: 10.3389/fnagi.2020.00185
115. Xiang P, Chew WS, Seow WL, Lam BWS, Ong WY, Herr DR. The S1p(2) Receptor Regulates Blood-Brain Barrier Integrity and Leukocyte Extravasation with Implications for Neurodegenerative Disease. Neurochem Int (2021) 146:105018. doi: 10.1016/j.neuint.2021.105018
116. Jeppsson B, Freund HR, Gimmon Z, James JH, von Meyenfeldt MF, Fischer JE. Blood-Brain Barrier Derangement in Uremic Encephalopathy. Surgery (1982) 92(1):30–5.
117. Liu M, Liang Y, Chigurupati S, Lathia JD, Pletnikov M, Sun Z, et al. Acute Kidney Injury Leads to Inflammation and Functional Changes in the Brain. J Am Soc Nephrol (2008) 19(7):1360–70. doi: 10.1681/asn.2007080901
118. Bobot M, Thomas L, Moyon A, Fernandez S, McKay N, Balasse L, et al. Uremic Toxic Blood-Brain Barrier Disruption Mediated by Ahr Activation Leads to Cognitive Impairment During Experimental Renal Dysfunction. J Am Soc Nephrol (2020) 31(7):1509–21. doi: 10.1681/asn.2019070728
119. Jing W, Jabbari B, Vaziri ND. Uremia Induces Upregulation of Cerebral Tissue Oxidative/Inflammatory Cascade, Down-Regulation of Nrf2 Pathway and Disruption of Blood Brain Barrier. Am J Transl Res (2018) 10(7):2137–47.
120. Huh JR, Veiga-Fernandes H. Neuroimmune Circuits in Inter-Organ Communication. Nat Rev Immunol (2020) 20(4):217–28. doi: 10.1038/s41577-019-0247-z
121. Hosoi T, Okuma Y, Matsuda T, Nomura Y. Novel Pathway for Lps-Induced Afferent Vagus Nerve Activation: Possible Role of Nodose Ganglion. Auton Neurosci (2005) 120(1-2):104–7. doi: 10.1016/j.autneu.2004.11.012
122. Bratton BO, Martelli D, McKinley MJ, Trevaks D, Anderson CR, McAllen RM. Neural Regulation of Inflammation: No Neural Connection from the Vagus to Splenic Sympathetic Neurons. Exp Physiol (2012) 97(11):1180–5. doi: 10.1113/expphysiol.2011.061531
123. Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-Synthesizing T Cells Relay Neural Signals in a Vagus Nerve Circuit. Sci (New York NY) (2011) 334(6052):98–101. doi: 10.1126/science.1209985
124. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic Acetylcholine Receptor Alpha7 Subunit Is an Essential Regulator of Inflammation. Nature (2003) 421(6921):384–8. doi: 10.1038/nature01339
125. Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, et al. Cholinergic Agonists Inhibit Hmgb1 Release and Improve Survival in Experimental Sepsis. Nat Med (2004) 10(11):1216–21. doi: 10.1038/nm1124
126. de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, et al. Stimulation of the Vagus Nerve Attenuates Macrophage Activation by Activating the Jak2-Stat3 Signaling Pathway. Nat Immunol (2005) 6(8):844–51. doi: 10.1038/ni1229
127. Tanaka S, Okusa MD. Crosstalk between the Nervous System and the Kidney. Kidney Int (2020) 97(3):466–76. doi: 10.1016/j.kint.2019.10.032
128. Kim HJ, Vaziri ND. Contribution of Impaired Nrf2-Keap1 Pathway to Oxidative Stress and Inflammation in Chronic Renal Failure. Am J Physiol Renal Physiol (2010). doi: 10.1152/ajprenal.00421.2009
129. Li J OW, Li W, Jiang ZG, Ghanbari HA. Oxidative Stress and Neurodegenerative Disorders. Int J Mol Sci (2013) 14(12):24438–75. doi: 10.3390/ijms141224438
130. Pentón-Rol G, Cervantes-Llanos M, Martínez-Sánchez G, Cabrera-Gómez JA, Valenzuela-Silva CM, Ramírez-Nuñez O, et al. Tnf-Alpha and Il-10 Downregulation and Marked Oxidative Stress in Neuromyelitis Optica. J Inflammation (Lond) (2009) 6:18. doi: 10.1186/1476-9255-6-18
131. Schweikl H, Birke M, Gallorini M, Petzel C, Bolay C, Waha C, et al. Hema-Induced Oxidative Stress Inhibits Nf- κ b Nuclear Translocation and Tnf Release from Lta- and Lps-Stimulated Immunocompetent Cells. Dent Mater (2021) 37(1):175–90. doi: 10.1016/j.dental.2020.10.029
132. Stenvinkel P, Ketteler M, Johnson RJ, Lindholm B, Pecoits-Filho R, Riella M, et al. Il-10, Il-6, and Tnf-Alpha: Central Factors in the Altered Cytokine Network of Uremia–the Good, the Bad, and the Ugly. Kidney Int (2005) 67(4):1216–33. doi: 10.1111/j.1523-1755.2005.00200.x
133. Watanabe K, Watanabe T, Nakayama M. Cerebro-Renal Interactions: Impact of Uremic Toxins on Cognitive Function. Neurotoxicology (2014) 44:184–93. doi: 10.1016/j.neuro.2014.06.014
134. Granata S, Masola V, Zoratti E, Scupoli MT, Baruzzi A, Messa M, et al. Nlrp3 Inflammasome Activation in Dialyzed Chronic Kidney Disease Patients. PloS One (2015) 10(3):e0122272. doi: 10.1371/journal.pone.0122272
135. Ori Y, Bergman M, Bessler H, Zingerman B, Levy-Drummer RS, Gafter U, et al. Cytokine Secretion and Markers of Inflammation in Relation to Acidosis among Chronic Hemodialysis Patients. Blood Purif (2013) 35(1-3):181–6. doi: 10.1159/000346689
136. Mihai S, Codrici E, Popescu ID, Enciu AM, Albulescu L, Necula LG, et al. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J Immunol Res (2018) 2018:2180373. doi: 10.1155/2018/2180373
137. Anders HJ, Andersen K, Stecher B. The Intestinal Microbiota, a Leaky Gut, and Abnormal Immunity in Kidney Disease. Kidney Int (2013) 83(6):1010–6. doi: 10.1038/ki.2012.440
138. Pletinck A, Glorieux G, Schepers E, Cohen G, Gondouin B, Van Landschoot M, et al. Protein-Bound Uremic Toxins Stimulate Crosstalk between Leukocytes and Vessel Wall. J Am Soc Nephrol (2013) 24(12):1981–94. doi: 10.1681/asn.2012030281
139. Harbort CJ, Soeiro-Pereira PV, von Bernuth H, Kaindl AM, Costa-Carvalho BT, Condino-Neto A, et al. Neutrophil Oxidative Burst Activates Atm to Regulate Cytokine Production and Apoptosis. Blood (2015) 126(26):2842–51. doi: 10.1182/blood-2015-05-645424
140. Ward RA, McLeish KR. Polymorphonuclear Leukocyte Oxidative Burst Is Enhanced in Patients with Chronic Renal Insufficiency. J Am Soc Nephrol (1995) 5(9):1697–702. doi: 10.1681/asn.V591697
141. Neirynck N, Glorieux G, Schepers E, Dhondt A, Verbeke F, Vanholder R. Pro-Inflammatory Cytokines and Leukocyte Oxidative Burst in Chronic Kidney Disease: Culprits or Innocent Bystanders? Nephrol Dial Transplant (2015) 30(6):943–51. doi: 10.1093/ndt/gfu409
142. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M, et al. Human Neutrophil Kinetics: Modeling of Stable Isotope Labeling Data Supports Short Blood Neutrophil Half-Lives. Blood (2016) 127(26):3431–8. doi: 10.1182/blood-2016-03-700336
143. Gollapudi P, Yoon JW, Gollapudi S, Pahl MV, Vaziri ND. Leukocyte Toll-Like Receptor Expression in End-Stage Kidney Disease. Am J Nephrol (2010) 31(3):247–54. doi: 10.1159/000276764
144. Sela S, Shurtz-Swirski R, Cohen-Mazor M, Mazor R, Chezar J, Shapiro G, et al. Primed Peripheral Polymorphonuclear Leukocyte: A Culprit Underlying Chronic Low-Grade Inflammation and Systemic Oxidative Stress in Chronic Kidney Disease. J Am Soc Nephrol (2005) 16(8):2431–8. doi: 10.1681/asn.2004110929
145. Kim JK, Hong CW, Park MJ, Song YR, Kim HJ, Kim SG. Increased Neutrophil Extracellular Trap Formation in Uremia Is Associated with Chronic Inflammation and Prevalent Coronary Artery Disease. J Immunol Res (2017) 2017:8415179. doi: 10.1155/2017/8415179
146. Bratton DL, Henson PM. Neutrophil Clearance: When the Party Is over, Clean-up Begins. Trends Immunol (2011) 32(8):350–7. doi: 10.1016/j.it.2011.04.009
147. Norden DM, Faw TD, McKim DB, Deibert RJ, Fisher LC, Sheridan JF, et al. Bone Marrow-Derived Monocytes Drive the Inflammatory Microenvironment in Local and Remote Regions after Thoracic Spinal Cord Injury. J Neurotrauma (2019) 36(6):937–49. doi: 10.1089/neu.2018.5806
148. Lim WH, Kireta S, Leedham E, Russ GR, Coates PT. Uremia Impairs Monocyte and Monocyte-Derived Dendritic Cell Function in Hemodialysis Patients. Kidney Int (2007) 72(9):1138–48. doi: 10.1038/sj.ki.5002425
149. Collin M, Bigley V. Human Dendritic Cell Subsets: An Update. Immunology (2018) 154(1):3–20. doi: 10.1111/imm.12888
150. Verkade MA, van Druningen CJ, Vaessen LM, Hesselink DA, Weimar W, Betjes MG. Functional Impairment of Monocyte-Derived Dendritic Cells in Patients with Severe Chronic Kidney Disease. Nephrol Dial Transplant (2007) 22(1):128–38. doi: 10.1093/ndt/gfl519
151. Verkade MA, van Druningen CJ, Op de Hoek CT, Weimar W, Betjes MG. Decreased Antigen-Specific T-Cell Proliferation by Modc among Hepatitis B Vaccine Non-Responders on Haemodialysis. Clin Exp Med (2007) 7(2):65–71. doi: 10.1007/s10238-007-0127-x
152. Kitching AR. Dendritic Cells in Progressive Renal Disease: Some Answers, Many Questions. Nephrol Dial Transplant (2014) 29(12):2185–93. doi: 10.1093/ndt/gfu076
153. Yoon JW, Gollapudi S, Pahl MV, Vaziri ND. Naïve and Central Memory T-Cell Lymphopenia in End-Stage Renal Disease. Kidney Int (2006) 70(2):371–6. doi: 10.1038/sj.ki.5001550
154. Meier P, Dayer E, Ronco P, Blanc E. Dysregulation of Il-2/Il-2r System Alters Proliferation of Early Activated Cd4+ T Cell Subset in Patients with End-Stage Renal Failure. Clin Nephrol (2005) 63(1):8–21. doi: 10.5414/cnp63008
155. Meier P, Dayer E, Blanc E, Wauters JP. Early T Cell Activation Correlates with Expression of Apoptosis Markers in Patients with End-Stage Renal Disease. J Am Soc Nephrol (2002) 13(1):204–12. doi: 10.1681/asn.V131204
156. Mauri C, Bosma A. Immune Regulatory Function of B Cells. Annu Rev Immunol (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
157. Pahl MV, Gollapudi S, Sepassi L, Gollapudi P, Elahimehr R, Vaziri ND. Effect of End-Stage Renal Disease on B-Lymphocyte Subpopulations, Il-7, Baff and Baff Receptor Expression. Nephrol Dial Transplant (2010) 25(1):205–12. doi: 10.1093/ndt/gfp397
158. Bouts AH, Davin JC, Krediet RT, Monnens LA, Nauta J, Schröder CH, et al. Children with Chronic Renal Failure Have Reduced Numbers of Memory B Cells. Clin Exp Immunol (2004) 137(3):589–94. doi: 10.1111/j.1365-2249.2004.02571.x
159. Fernández-Fresnedo G, Ramos MA, González-Pardo MC, de Francisco AL, López-Hoyos M, Arias M. B Lymphopenia in Uremia Is Related to an Accelerated in Vitro Apoptosis and Dysregulation of Bcl-2. Nephrol Dial Transplant (2000) 15(4):502–10. doi: 10.1093/ndt/15.4.502
160. Wang W, Xu S, Ren Z, Jiang J, Zheng S. Gut Microbiota and Allogeneic Transplantation. J Transl Med (2015) 13:275. doi: 10.1186/s12967-015-0640-8
161. Vernaglione L, Cristofano C, Muscogiuri P, Chimienti S. Does Atorvastatin Influence Serum C-Reactive Protein Levels in Patients on Long-Term Hemodialysis? Am J Kidney Dis (2004) 43(3):471–8. doi: 10.1053/j.ajkd.2003.11.008
162. Stenvinkel P, Andersson P, Wang T, Lindholm B, Bergström J, Palmblad J, et al. Do Ace-Inhibitors Suppress Tumour Necrosis Factor-Alpha Production in Advanced Chronic Renal Failure? J Intern Med (1999) 246(5):503–7. doi: 10.1046/j.1365-2796.1999.00560.x
163. Biolo G, Ciocchi B, Bosutti A, Situlin R, Toigo G, Guarnieri G. Pentoxifylline Acutely Reduces Protein Catabolism in Chronically Uremic Patients. Am J Kidney Dis (2002) 40(6):1162–72. doi: 10.1053/ajkd.2002.36864
164. Inoue T, Rosin DL, Okusa MD. Caping Inflammation and Acute Kidney Injury. Kidney Int (2016) 90(3):462–5. doi: 10.1016/j.kint.2016.07.009
Keywords: uremia, neuroimmune, inflammation, oxidative stress, pediatric
Citation: Zhu L, Tong G, Yang F, Zhao Y and Chen G (2022) The role of neuroimmune and inflammation in pediatric uremia-induced neuropathy. Front. Immunol. 13:1013562. doi: 10.3389/fimmu.2022.1013562
Received: 07 August 2022; Accepted: 01 September 2022;
Published: 15 September 2022.
Edited by:
Xiangsheng Zhang, Affiliated Beijing Friendship Hospital, Capital Medical University, ChinaReviewed by:
Zhen-Ni Guo, First Affiliated Hospital of Jilin University, ChinaLingyun Wu, Nanjing University, China
Copyright © 2022 Zhu, Tong, Yang, Zhao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangjie Chen, ZHIuY2hlbmd1YW5namllQHpqdS5lZHUuY24=
†These authors have contributed equally to this work