 Deyi Sun1,2
Deyi Sun1,2 Jiqian Xu
Jiqian Xu Yajun He
Yajun He You Shang
You Shang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 20 October 2022
Sec. Molecular Innate Immunity
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1012459
NLRC3 is a member of the pattern recognition receptors nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) family, and plays a pivotal regulatory role in modulating the activation of immune cells. In macrophages, NLRC3 inhibits the activation of the NF-κB signaling pathway, the STING/TBK1 signaling pathway, and the formation of the inflammasome. In the context of T cells immune response, NLRC3 prevents the activation of T cells by regulating the function of dendritic cells and directly influencing the function of T cells. Different from other pattern recognition receptors, NLRC3 is more closely associated with regulatory activity than pathogens recognition, it influences the fates of cells, for example, prevents proliferation, promotes apoptosis and inhibits pyroptosis. These cellular functions regulated by NLRC3 are involved in the development processes of a variety of diseases, such as infectious disease, sterile inflammatory diseases, and cancer. However, its characteristics, function and regulatory mechanism in immune response and immune-related diseases have not been addressed fully. In this review, we elaborate the potential roles of NLRC3 from several different levels, include molecular mechanism, cellular functions in the immune-related diseases.
NLRC3 is one of the members of NLR family. NLRs belong to the pattern recognition receptors (PRRs) family, which includes five sub-families, the Toll-like receptors (TLRs), C-type lectin receptors (CLRs), Rig-I-like helicase receptors (RLRs), nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), and absent-in-melanoma (AIM)-like receptors (ALRs) (1). TLR and CLR are membrane-bound receptors (2, 3), while NLR, RLR, and ALR localize to the cytoplasm (4–6). In general, once they recognize pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs), the PRRs trigger a set of inflammatory signal cascades. Interestingly, plenty of evidence has demonstrated that NLRs exerted many other functions in addition to their pattern recognition role during the immune response (7–9).
The NLR family comprises 22 kinds of protein in human and 34 in mice. NLRs are mainly composed of three domains, a series of leucine rich repeats (LRRs) form the C-terminal domain, which is responsible for the ligand recognizing. The intermediate domain nucleotide binding/oligomerization domain (NBD) is responsible for self-oligomerization and some regulatory activities. The N-terminal domains of different NLR subfamilies also differ, such as PYRIN domain (PYD), caspase activation and recruitment domain (CARD), and acidic transactivation or a baculoviral inhibitory repeats (BIR) domain. The N-terminal domain of NLRs serve as a bridge to link the NLRs with downstream effector molecules or adopter proteins (10).
NOD-like receptor (NLR) family CARD domain containing-3 (NLRC3) is one of the members of NLRs which serves as a negatively regulatory molecule in cytoplasm. NLRC3, also known as CLR16.2 or NOD3, was first detected by Conti et al. (11) in human T lymphocytes in 2005. NLRC3 mainly expressed in immune system, and the expression of NLRC3 is highest in CD4+ and CD8+ T cells, with moderate expression in the thymus, lymph node, and other tissues of the immune system (11). And NLRC3 also can be detected in other issues, in some immune-related diseases also observed the expression changes of NLRC3. Under both stimulated and unstimulated states, NLRC3 is only detected in the cytoplasm (11). In mammals, the N-terminal domain of NLRC3 is composed by 6-α-helical bundle, such constructure is similarly to CARD domain and PYD domain, but it doesn’t quite fit the definition of either domain. Subsequently, the N-terminal domain of NLRC3 was defined as the CARD domain (12) Figure 1A, different from other NLRs such as NLRX1, NLRC4, and NOD2, the LRR domain of NLRC3 contains a series of continuous positive charges, which are responsible for the affinity between NLRC3 and ligands (13). In fish, the structure of NLRC3 also differs in different fish. It can be divided into three structures. Type 1: Composed of CARD domain, NBD domain and LRR domain Figure 1B, the CARD domain of type I-NLRC3 is closer to the CARD domain of the human NLRC family. Type 2: Composed of FISNA domain, NBD domain, LRR domain and PRY/SPRI domain Figure 1C, the LRR domain of type 2-NLRC3 is shorter than that of type 1. Type 3: Consists of PYD domain, FISNA domain, and NBD domain. Figure 1D
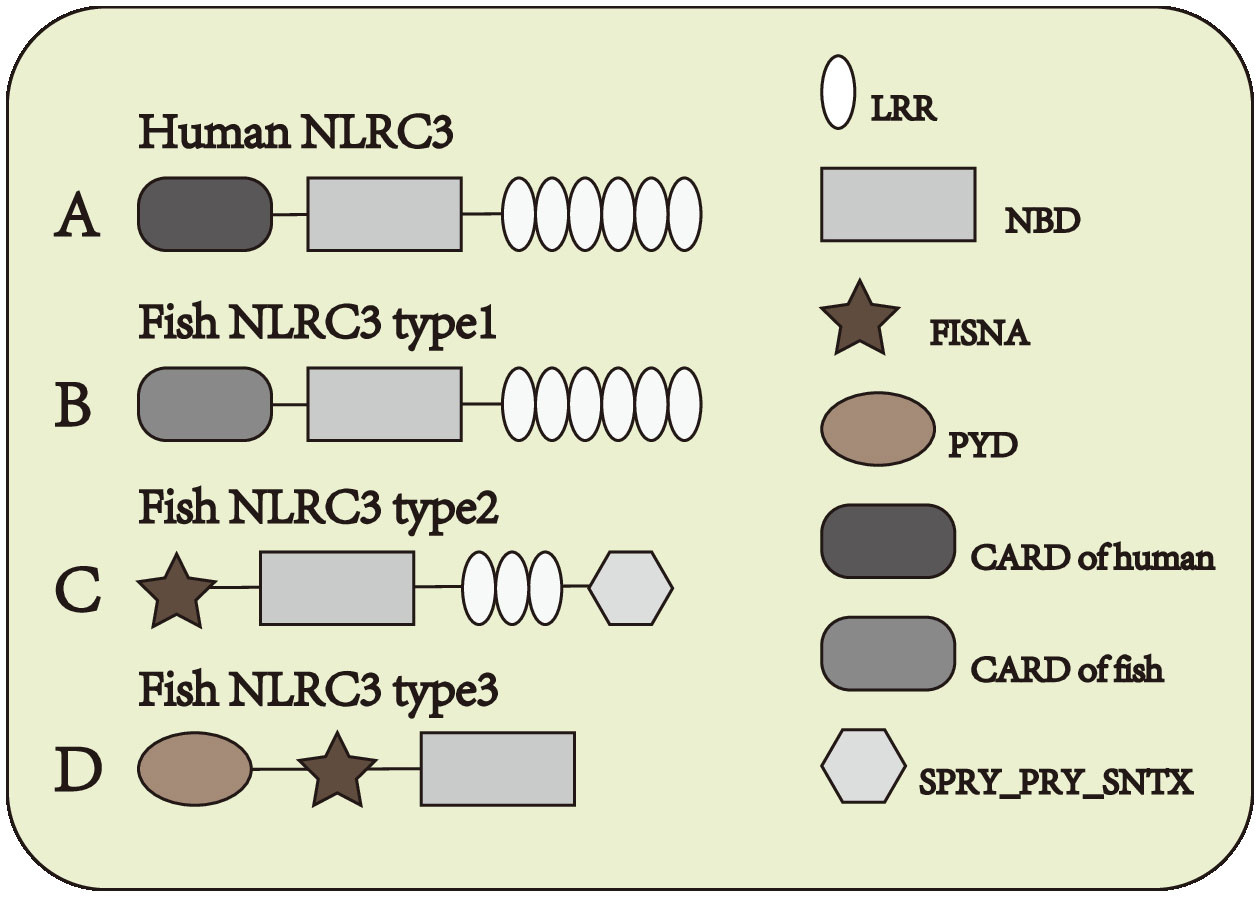
Figure 1 Structure of NLRC3 in different species. (A) From N-terminal to C-terminal, human NLRC3 is composed of CARD domain, NBD domain and LRR domain. (B–D) Three types of structures of fish NLRC3, from N-terminal to C-terminal, the composition and order of the domain are (B) CARD domain, NBD domain and LRR domain (C) FISNA domain, NBD domain, LRR domain and SPRY_PRY_SNTX domain (D) PYD domain, FISNA domain and NBD domain.
The NF-κB signaling pathway is one of the most important inflammatory signaling pathways both in innate immunity and adaptive immunity (14). While macrophages were stimulated by LPS, the NF-κB signaling pathway was activated and a variety of proinflammatory cytokines such as IL-1, IL-6, and TNF-α release. If NLRC3 was overexpressed, the activation of the NF-κB signaling pathway was inhibited. The result of co-immunoprecipitation indicated that NLRC3 interacted with TRAF6, decreased the K63-linked ubiquitination of TRAF6, and remained the K48-linked ubiquitination on it. As a result, NLRC3 not only inhibited the activation of TRAF6, but also promoted the degradation of TRAF6 through the proteasome, further reduced the abundance of TRAF6 in cytoplasm (14). Moreover, NLRC3 also negatively regulates the NF-κB signaling pathway by regulating the abundance of IRAK1 (15). NLRC3 promotes the K48-linked ubiquitination on interleukin-1 receptor-associated kinase1 (IRAK1), which decrease the abundance of IRAK1 through the proteosome (15) (Figure 2A).
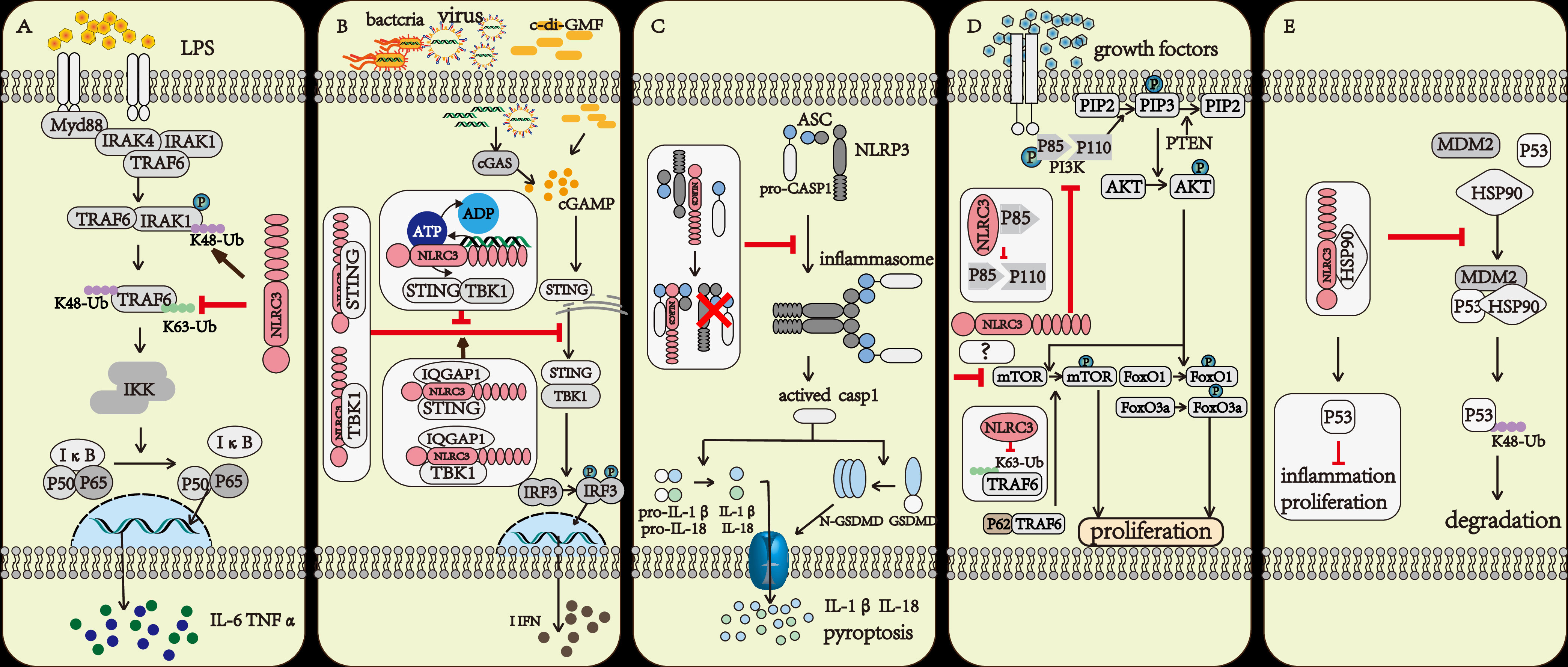
Figure 2 The regulatory mechanism of NLRC3. (A) The TRAF6 and IRAK1 activation upon LPS stimulation further activate the NF-κB signaling pathway. NLRC3 prevents the K63-linked polyubiquitination of TRAF6 and promotes the degradation of TRAF6 through proteasome, or NLRC3 promotes the K48-linked polyubiquitination of IRAK1 and promotes the degradation of IRAK1 through proteasome. (B) Upon the Bacteria, Virus infection and c-di-GMF stimulation, the DNA binding to LRR domain of NLRC3 increased the activation of ATPase on the NBD domain of NLRC3, prevented the association of NLRC3 and STING and inhibited the STING/TBK1 signaling pathway. IQGAP1 interacted with NLRC3, promoted NLRC3 to bind with STING and TBK1, and further enhanced the inhibitory effect of NLRC3 on STING/TBK1 signaling pathway. All further attenuates the production of type I interferon (I-IFN), preventing the inflammatory response of macrophages. (C) NLRC3 interacts with ASC and pro-CASP1 through the NBD domain, then prevents the formation of the inflammasome, and makes cells stay away from pyroptosis. (D) NLRC3 interacts with the p85 subunit of PI3K, then inhibits the interaction of p85 submit and catalytic submit p110α, preventing the phosphorylation and activation of the p110α submit, which blocks the transmission of downstream signals, including PI3K-AKT-mTOR and PI3K-AKT-FoxO3a/FoxO1 signaling pathways; P62 interacting with TRAF6 induced the activation of mTOR, NLRC3 prevents the K63-linked polyubiquitination of TRAF6, and negatively regulates the mTOR signaling pathway and prevents cell proliferation. (E) NLRC3 interacts with HSP90 to inhibit HSP90-mediated ubiquitination of P53 and inhibit the degradation of P53 in lysosomes.
The stimulator of interferon gene (STING) is an intracellular DNA sensor that regulates the production of type I interferon (I-IFN) by interacting with TANK-binding kinase (TBK1) (16). When stimulated by cytosolic DNA, cyclic di-GMP (c-di-GMP), and DNA viruses, the immune response was up-regulated in NLRC3 deficient mouse embryonic fibroblasts (MEFs) and bone-marrow-derived macrophages (BMDMs). The result of co-immunoprecipitation indicated that NLRC3 prevented the interaction between STING and TBK1 via associating with both STING and TBK1 through its NBD domain. Furthermore, NLRC3 also prevented the trafficking of STING to the perinuclear and punctate region, which is important for regulating the production of I-IFN (16). During viral infection, TLR9 and STING activated the NF-κB signaling pathway, which attenuated the expression of NLRC3. This regulatory effect can be considered as a positive feedback loop that amplifies the immune response after infection (16, 17) (Figure 2B).
When regarding to the regulation of NLRC3, there has also been some progress in recent years (13, 18). Viral DNA associated with the LRR domain of NLRC3, and increased the ATPase activity of the NBD domain of NLRC3, further promoted the release of STING from NLRC3, and in the end, activated the STING/TBK1 signaling pathway (13). IQGAP1 is a famous actin- and tubulin-binding protein (18). In macrophages, IQGAP1 interacts with the actin nucleating protein, diaphanous-related formin (Dia1), to take part in phagocytosis and phagocytic cup formation during infection. The rearrangement of the cytoskeleton is important for the phagocytosis activity of macrophages (19, 20). Tocker et al. (18) demonstrated the interaction between the RGCT domain of IQGAP1 and the NBD domain of NLRC3 using yeast two-hybrid screening and coimmunoprecipitation experiments in human cell lines. And then they found in the following experiment that the deficiency of IQGAP1 promoted the activation of the I-IFN signaling pathway. Thus, together with previous evidence (16), the authors speculated that IQGAP1 interacts with NLRC3 to inhibit the STING-TBK1 signaling pathway, which in turn attenuates the production of I-IFN (18) (Figure 2B).
The inflammasome-forming subfamily of NLRs (such as NLRP3, NLRP1, NLRC4) recognize PAMP or DAMP or the changes in the intracellular environment, then recruit homologous NLRs, the apoptosis-associated speck-like protein (ASC) and pro-caspase1 to form the inflammasome, further activate CASP1, lead to pyroptosis (21). Pyroptosis, a form of programmed necrosis, amplifies inflammation during the immune response. NLRC3 could inhibit cell pyroptosis by preventing the formation of the NLRP3- and NLRC4-inflammasomes (22).Through interacting with ASC and pro-caspase-1 via the CARD domain, NLRC3 prevented the interaction of ASC and pro-caspase-1, and disrupted the formation of the ASC speck, further inhibited cell pyroptosis (22) (Figure 2C).
NLRC3 negatively regulated the PI3K-AKT-mTOR signaling pathway in mouse bone-marrow-derived macrophages during stimulated with insulin-like growth factors(IGF-1) or LPS (23). mTOR plays multiple roles during the life process of a cell such as regulating the survival, proliferation, metabolism, autophagy, and immunity of cells (24, 25). NLRC3 interacted with the p85 subunit of PI3K to prevent the interaction between the p85 submit and the p110α submit of PI3K, further inhibited the phosphorylation and activation of p110α PI3K and then blocked the transmission of downstream signals (23) (Figure 2D).
NLRC3 has also been reported to interact with TRAF6 and directly associate with mTOR further prevented the activity of mTOR (26). The signal adaptor P62 protein interacts with TRAF6 and promotes the translocation of mTOR to the lysosome and its subsequent activation (27). NLRC3 interacted with TRAF6 to promote its degradation via the proteasome (14). In 239T cells with NLRC3, TRAF6 and mTOR overexpressed, researchers observed that NLRC3 interacted with both TRAF6 and mTOR (26). Thus, we can speculate that NLRC3 interacts with TRAF6 and promotes the degradation of TRAF6 to prevent TRAF6 interaction with p62 and further prevents the activation of mTOR. NLRC3 could also directly associate with mTOR to prevent its activity, and the specific mechanisms underlying this inhibition requires to be further studied (Figure 2D).
The interaction between NLRC3 and the p85 submit of PI3K not only has effect on the PI3K/AKT/mTOR signal axis, but also on PI3K/AKT/FoxO3a or FoxO1 signaling pathways (26). FoxO3a and FoxO1 contribute to regulate cell proliferation, apoptosis, metabolism, and survival (28). AKT activates and prevents ubiquitin-mediated degradation of FoxO3a and FoxO1 (29). In colonic tissue of NLRC3 ablated mice, the activation of AKT and phosphorylation of FoxO3a and FoxO1 are higher than in WT mice. NLRC3 ablated colonic epithelial cells exhibit an increase in cellular proliferation (26) (Figure 2D).
Tumor suppressor protein P53 inhibits tumor growth in various ways and plays an important role in maintaining genomic stability and preventing abnormal cell proliferation. Ubiquitination of P53 is an important way to regulate this protein (30). NLRC3 associates with HSP90(Heat Shock Protein 90), preventing the interaction between HSP90, MDM2 and P53, further inhibiting the ubiquitin and degradation of P53 (31) (Figure 2E).
The family members of the mitogen-activated protein (MAP) kinases mediate a wide variety of cellular behaviors in response to extracellular stimuli. MAPKs are divided into four main signal pathways, P38 signal pathway is one of them, which is mainly related to the production of inflammatory cytokines and cell apoptosis (32). In DCs isolated from NLRC3 knockout mice, researchers observed that the phosphorylation of P38 and the activation of its downstream signaling pathway were up-regulated than WT (8). However, the specific mechanisms through which NLRC3 regulates the activation of the MAPK signaling pathway are still unclear and further study is needed (8).
The PI3K signaling pathways play an important part in the regulation of cell proliferation. NLRC3 prevented the interaction between P85 and P110α submits of PI3K, further inhibited the activity of mTOR and FoxO1/FoxO3a, which act as regulators of cell proliferation (23). NLRC3 also disrupted the function of mTOR through preventing the interaction between TRAF6 and P62, and NLRC3 directly interacted with mTOR to inhibit the activation of mTOR (26). Given the amount of research data and the strong research evidence, the role of NLRC3 in regulating cell proliferation is beyond all doubt. Figure 3A
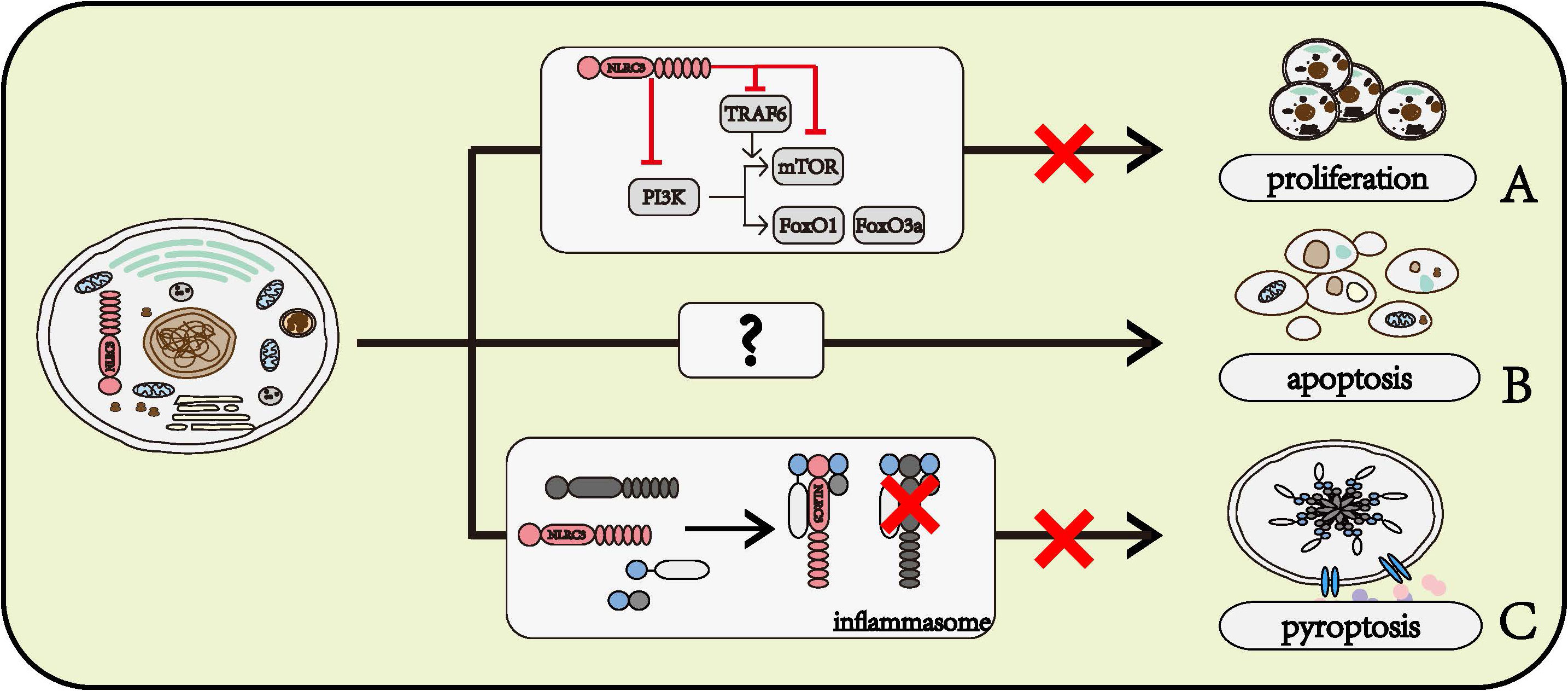
Figure 3 Roles of NLRC3 on cell fates. (A) NLRC3 inhibits cell proliferation. NLRC3 interferes the normal function of PI3K or inhibits the activation of mTOR through directly regulating itself and interfering TRAF6, thereby preventing its downstream molecules mTOR, FoxO1 and FoxO3a, and further regulate the cell proliferation. (B) NLRC3 might promote the cell apoptosis. (C) NLRC3 prevents the interaction with ASC and pro-CASP1, further inhibit the formation of inflammasome and cell pyroptosis.
In the AOM-DSS model of colorectal tumorigenesis, the activation of CASP8, CASP3, and CASP7 was lower in the colon of NLRC3 knockout mice than in WT mice (26). Flow cytometry phenotyping and TUNEL assay findings indicated the apoptosis rate of NLRC3 siRNA-treated HuH‐7 cells, a human liver cancer cell line, was significantly lower than in the scramble-siRNA-treated group after IL‐6 stimulation (33). Based on these data, we can speculate that NLRC3 has a role of promoting cell apoptosis, however, the mechanism by which NLRC3 promotes cell apoptosis remains to be determined (Figure 3B)
The pyroptosis of THP-1 has been observed to be inhibited by NLRC3 (22). NLRC3 interacts with pro-caspase1 and ASC through CARD domain, prevents the formation of inflammasome, further inhibits cell pyroptosis (22) (Figure 3C).
Macrophage is one of the most potent cells of the innate immune system. Based on existing research, it can be concluded that NLRC3 prevents the activation of macrophage. NLRC3 negatively regulated the NF-κB signaling pathway (14, 15), STING/TBK1 signaling (16), and the formation of the inflammasome (22), further prevented macrophages from producing inflammatory factors, thereby inhibited the macrophage immune response (Figure 4)
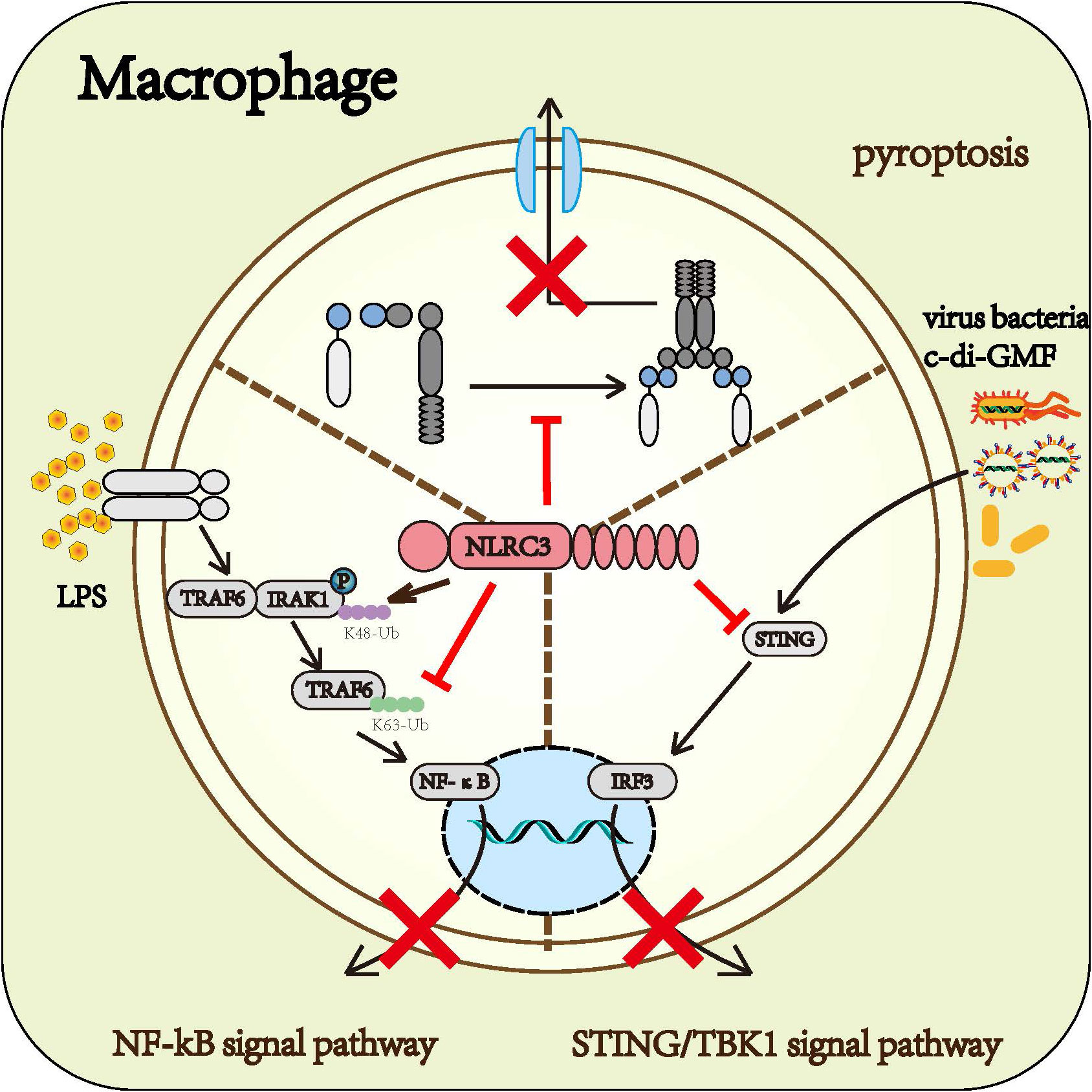
Figure 4 Roles of NLRC3 on macrophage: Upon the infection of gram-negative bacteria, NLRC3 decreases the activation of the NF-κB signal pathway and the release of multiple inflammatory cytokines such as TNF-α and IL-6 by TRAF6-dependent manner. Upon the infection of the virus, NLRC3 prevents the activation of STING/TBK1 signal pathway and the following release of I-IFN through inhibiting STING and TBK1 from functioning normally. Upon the PAMPs stimulation, NLRC3 competitively binding ASC and pro-CASP1 with NLRP3 prevents the formation of inflammasome and pyroptosis.
The dendritic cell (DC) is the most effective antigen-presenting cell among the immune system (34). Once stimulated by antigen, activated DCs increase the expression of surface co-stimulatory molecules and a series of cytokine to activate naïve T cells and promote their differentiation. Different cytokines secreted by DC induce different differentiation directions of naïve T cells. IL-12 promotes the differentiation towards Th1 cells, IL-4 promotes the differentiation towards Th2 cells (35); and IL-6, IL-23, and TGF-β promote the differentiation towards Th17 cells (8, 35, 36). NLRC3 can regulate the antigen-presenting function and the activity to activate and polarize CD4+ T cell Th1 and Th17 cells of DC (8). When NLRC3 was deficient in DCs, the researchers observed the production of IL-12, IL-6, and IL-23 were up-regulated. In the in vivo model, NLRC3 deficiency also led to the same result (8). Enhanced phosphorylation of P38 and the activation of downstream signaling pathway in DCs were observed in NLRC3 knockdown mice compared to DCs from wild-type (WT) mice. Thus, we can conclude that the regulatory role of NLRC3 is dependent on the P38 signaling pathway (Figure 5A). However, the specific mechanisms through which NLRC3 regulates the activation of P38 are still unclear and further study is needed (8).
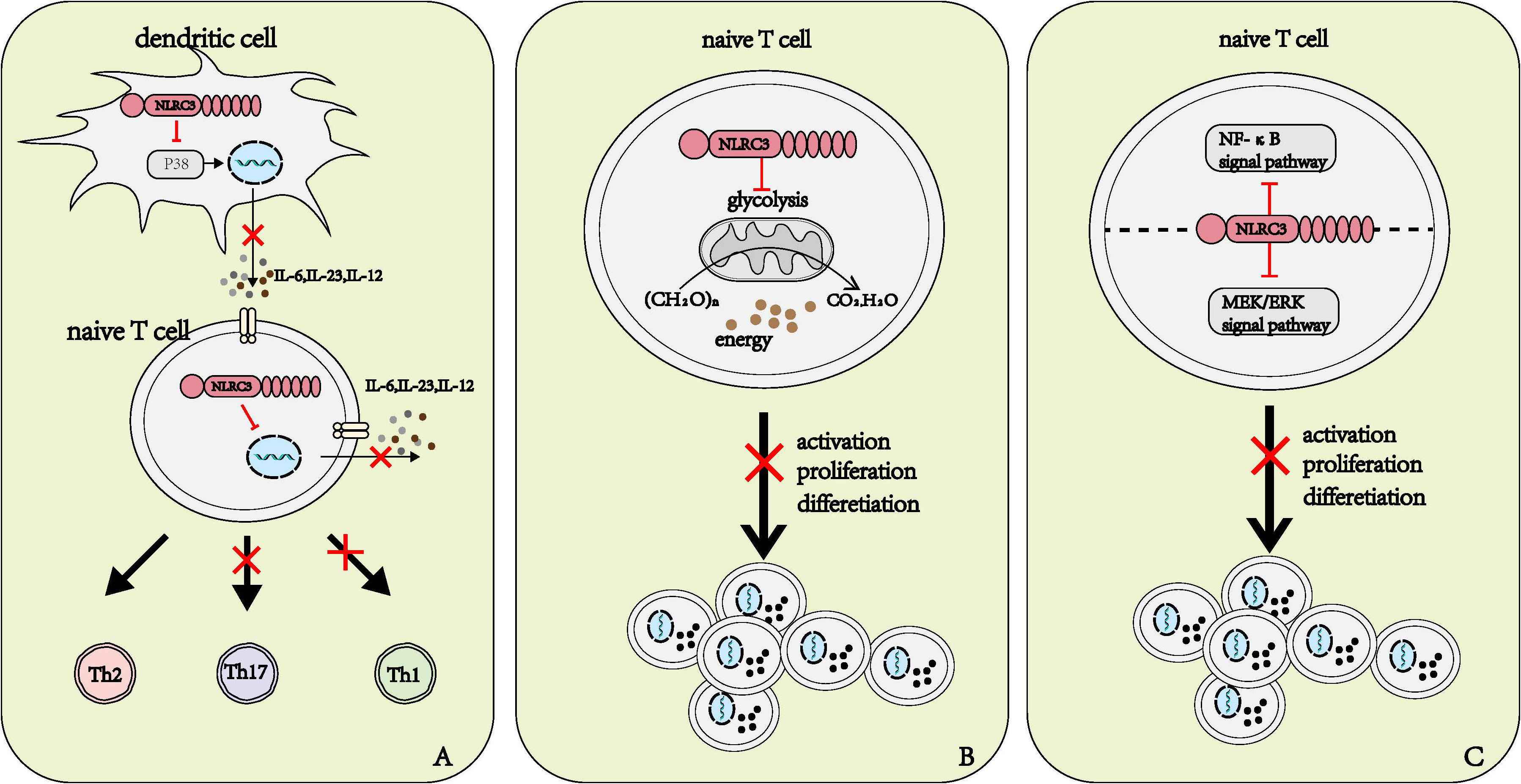
Figure 5 Roles of NLRC3 on T cells. (A) NLRC3 affects the differentiation direction of naïve T cells. NLRC3 prevents the production and release of IL-6, IL-23 and IL-12 in dendritic cells and naïve T cells, in DCs, NLRC3 executes regulatory roles by affecting P38 signal pathway. IL-12 promotes the differentiation of naïve T cells into Th1 cell, and IL-23, IL-6 promote the differentiation of naïve T cells into Th17 cell. (B, C) The potential several manners of NLRC3 affects the activation, proliferation and differentiation of T cells. (B) Sufficient energy is critical for the activation, proliferation and activation of naïve T cells, NLRC3 regulates the activation, proliferation and differentiation through negatively regulating mitochondrial function and the production of energy in naïve T cells (C) NLRC3 inhibits the activation of both NF-κB and MEK/ERK signal pathways and further inhibit the activation, proliferation and differentiation of T cells.
T cells are the most important members of the adaptive immune system. NLRC3 was first detected in T cells, and it is in T cells, thymus and lymph nodes where the expression of NLRC3 is the most abundance (11). Recently, NLRC3 was reported to negatively regulate the activation, differentiation, and proliferation of CD4+T cells in response to viruses (37), bacteria (38), and auto-antigen (37).
NLRC3 influences the polarization of T cells partly by regulating the production of cytokine. As mentioned above, NLRC3 influenced the cytokine produced by DCs to promote naïve T cell to polarize to Th1 and Th17 subsets (8). Recently, when researchers stimulated WT and NLRC3-deficient CD4+ naïve T cells with anti-CD3 and anti-CD28, they observed that some characteristic genes of Th1, Th17 and activated T cells up-regulated from the resulting heatmap of the RNA-seq expression analysis (37). We can speculate that NLRC3 affect the differentiation of naïve T cells not only by the influence on DCs, but via the direct regulatory effect on T cells. Further studies are needed to clarify the specific mechanisms of how NLRC3 regulates the differentiation of naïve T cells in a T-cell intrinsic manner (Figure 5A).
NLRC3 prevents the activation of T cells partly by influencing the metabolism of T cells. Metabolic alterations are highly correlated with T cell activation and differentiation (39). For instance, aerobic glycolysis is associated with the activation and the polarization to Th1 of T cells (40). Activated NLRC3-defincient CD4+T cells exhibited higher glycolytic capacity, glycolytic reserve, and maximal mitochondrial respiration than WT CD4+T cells (37). Such metabolic alterations provided the greater metabolic capacity to cope with sudden energy demands, in the other word, it provided energy for cell proliferation and cytokine production (37) (Figure 5B).
NLRC3 prevents the activation of T cells partly by regulating the NF-κB and MEK-ERK signal pathways (37, 38). Similar to macrophages, NLRC3 can negatively regulate the NF-κB signaling pathway by preventing the K63-linked polyubiquitin of TRAF6 in T cells (37). NLRC3 also inhibits the phosphorylation of ERK, but the specific manners by which NLRC3 influences the MEK-ERK signal pathway need further exploration (38). However, when using the inhibitors toward NF-κB or MEK-ERK signal pathways alone fail to prevent the activation of CD4+T cells with NLRC3 ablation. In a word, NLRC3 prevents the activation, proliferation and differentiation of T cells by inhibiting both NF-κB and MEK/ERK signal pathways (38) (Figure 5C).
Inflammation plays important roles in the occurrence and development of cancer. Uncontrolled inflammatory response can be detected during several stages of tumor progression, include initiation, promotion, malignant conversion, invasion, and metastasis (41, 42). Abnormal cell proliferation is the hallmark of cancer; for many years, researchers have been working on the discovery and development of drugs that inhibit the abnormal cell proliferation in cancers (43, 44).
NLRC3 has been shown to inhibit cell proliferation, promote cell apoptosis, and prevent inflammation (8, 16, 23, 26, 37, 45). Thus, we can broadly speculate that NLRC3 may be a therapeutic target for tumors or can be used as an index to diagnosis or evaluate the prognosis of tumors. Recently, a large body of literatures have confirmed the beneficial role of NLRC3 in cancer treatment.
Several bioinformatics papers and some reports from detecting clinical specimens have demonstrated that NLRC3 is associated with the prognosis and malignancy of tumors (include colorectal cancer, Hepatocellular carcinoma, Gastric cancer, Bladder cancer and lung adenocarcinoma) (46–52).
The specific mechanism through which NLRC3 affect tumors have been reported as followed:
● PI3K-AKT-mTOR and PI3K-AKT-FoxO3a/FoxO1 signal pathways are associated with the suppressive effect of NLRC3 on colorectal cancer (23, 26).
● JAK2/STAT3 signal pathway is associated with the suppressive effect of NLRC3 on hepatocellular carcinoma (33).
● Wnt/β-catenin signal pathway is associated with the suppressive effect of NLRC3 on bladder cancer (46).
● NLRC3 is involved in CD8+T infiltration during HCC, which may be associated with the increases of CCL5 (C-C motif chemokine ligand 5) as well as CXCL9 (C-X-C motif chemokine ligand 9) (53).
The regulatory role of NLRC3 in RNA virus infection has been mainly studied in adaptive immunity, that is, the effect of NLRC3 on CD4+T cell function. First of all, NLRC3 inhibits the body’s antiviral immunity. NLRC3 knockout mice showed a higher level of immune response, higher serum cytokine content, and relatively less viral load and pathological damage to organs (37, 54). In addition, NLRC3 regulates CD4+T cell function mildly, acting more like a rheostat than a switch (37).
The regulatory effect of NLRC3 on the body’s anti-infection immunity to DNA viruses is mainly focused on innate immunity. NLRC3 inhibits the body’s immune response against DNA viruses (16). STING/TBK1 pathway plays an important role in the body’s anti-DNA virus immunity. NLRC3 interacts with STING and TBK1 to inhibit this signaling pathway (16), after recognizing viral DNA, STING interacts with TBK1 to promote IFN synthesis. In addition, viral DNA interacts with the LRR domain of NLRC3 to inhibit the binding between NLRC3 and STING, thereby activating the STING/TBK1 antiviral signaling pathway (13).
As described above, NLRC3 inhibits the activation of NF-κB signaling pathway by interacting with TRAF6 and IRAK1 (14, 15). Keratitis caused by Pseudomonas aeruginosa (PA) is extremely common and damaging. PA can injure the corneal tissue via two mechanisms: bacteria invasion and excessive local inflammation (55). NLRC3 exerts beneficial roles in PA-induced keratitis (15).The expression of NLRC3 is decreased in mouse corneal tissue and macrophages cultured in vitro after PA infection. NLRC3 prevents the activation of macrophages and attenuates the cytokines release by macrophage through negatively regulating the NF-κB signaling pathway (15)
NLRC3 mediates immune evasion of Mycobacterium tuberculosis (M. tuberculosis) after infection in vivo (38), while downregulation of NLRC3 expression has a protective effect on M. tuberculosis infection (38). M. tuberculosis is particularly devious that it can evade clearance by the host immune system. In general. macrophages engulf M. tuberculosis upon infection, but macrophages have poor bactericidal ability and carry the bacteria to the deep tissue without infection, in which forming the origin of tuberculosis granuloma. After that, chemokine binds to the chemokine receptor CCR22 of macrophages, and drives macrophages to accumulate and adhere to infected tissues, and finally form the tuberculous granuloma, with caseous necrosis of necrotic tissue and cells in the center, epithelioid cells in the periphery, and T cells and B cells in the periphery (56). Inhibiting the activation of CD4+T cells is one mechanisms of tuberculous granuloma formation (57). NLRC3 prevents the activation, proliferation, and differentiation of CD4+T cells by inhibiting the NF-κB and ERK/MEK signaling pathways, thereby attenuating the body’s anti-tuberculosis immunity, and the inhibition of CD4+T cells in turn suppresses the innate immune responses and further promotes M. tuberculosis survival (38).
Multiple sclerosis (MS) is a kind of chronic progressive inflammatory disease of the central nervous system (CNS) caused by autoimmune response. MS is characterized by the imbalance of the subsets of T cells, a large amount of Th1 and Th17 can be detected in CNS tissue, cerebrospinal fluid (CSF), and blood of patients with MS (58).
The overexpression of NLRC3 could improve the prognosis of MS (8, 37, 59–61). NLRC3 influenced the activation and polarization of the naïve T cells, and promoted the differentiation of naïve T cells into Th1 and Th17 cells (8, 37). Compared with WT mice, NLRC3 knockout mice were more likely to develop MS, and the researchers detected more infiltration of Th1 and Th17 cells in the spinal cord of NLRC3 knockout mice (37). Furthermore, data from RNA-seq studies indicated that the expression of NLRC3 is lower in CD4+T cells isolated from MS patients than in healthy volunteers (37). And when injected vaccines with NLRC3 overexpressing DCs, the MS progression in mice models was attenuated (8).
Epoxyeicosatrienoic acids (EETs) is generated during arachidonic acid metabolism and has the neuroprotective function (62). However, as soon as EET production, it is degraded by soluble epoxide hydrolase (sEH) (63). Previous research has confirmed that the sEH inhibitor TPPU has beneficial effects on neurodegenerative inflammatory diseases (64). Biliktu et al. reported that TPPU attenuated chronic MS development and the therapeutic effects of TPPU were associated with NLRC3 (61). In MS mice, TPPU increased the expression of NLRC3 in brain and spinal cord (61). Their finding was consistent with other researchers.
Alzheimer’s disease (AD) is chronic progressive neurodegenerative disease, and the formation of amyloid plaques and NFTs are the characters of AD. Amyloid plaques and NFTs activate microglia and astrocytes, lead to the release of large amounts of pro-inflammation cytokines and reactive oxygen species (ROS), and injure neurons (65).
NLRC3 has beneficial effects on AD (66).NLRC3 has been proved to be one of the most associative proteins of AD through an exome-wide rare variant analysis. The rare variants of NLRC3-coding gene had been observed in EMIF-AD (67). Overexpression of NLRC3 improved learning and memory ability in mouse, and NLRC3 inhibited the deposition of Aβ, the activation of glial cells and the degeneration of neurons in AD mouse (66). Overexpression of NLRC3 could attenuate the activation of PI3K in AD models in vivo or in vitro. And in AD mouse, researchers also detected NLRC3 in the nucleus of nerve cells (66). This is in contrast to previous reports that NLRC3 localized in cytoplasm both in physiological state and in response to LPS. The location of NLRC3 in neural nuclei may have contact with AD, which requires further investigation.
One of the most important mechanisms of cerebral ischemia-reperfusion is neuroinflammation. A large amount of pro-inflammation factors result in neuronal cell death (68). Sevoflurane was observed to play beneficial roles on cerebral ischemia-reperfusion injury, and NLRC3 enhanced the beneficial effects of sevoflurane on ischemia-reperfusion models (28). Experimental evidence suggests that the beneficial effects of NLRC3 profited from the anti-inflammatory action via preventing NF-κB signal pathway (28).
Cutaneous wound healing is an intricate progress which is composed of hemostasis, inflammation, proliferation and remodel phases. And the initial inflammatory phase and the following proliferative phase are closely related to the outcome of cutaneous wound healing (69). NLRC3 prevents the inflammation and proliferation during cutaneous wound healing, NLRC3 interacts with Hsp90, inhibits the ubiquitin of P53, further promotes the inhibition on inflammation and cell proliferation by P53 signal pathway (31) and prevents cutaneous wound healing.
Pulmonary hypertension (PH) is a chronic progressive disease, It is the prominent feature of PH that pulmonary artery smooth muscle cells (PASMCs) release inflammatory cytokines and induce pulmonary vascular remodeling (70).
A randomized controlled trial first reported the relationship between NLRC3 and PH (71). The clinical study enrolled 43 patients with PH and 20 healthy controls. The results showed that the serum concentration of NLRC3 in patients with PH was lower than that in the control group, and the concentration of serum NLRC3 was inversely proportional to the severity of PH (71).
The animal experiment evidence suggested that NLRC3 inhibited the proliferation, migration, and inflammation of PASMCs and improved the symptoms of PH. Further, in the presence of agonists of PI3K-mTOR signal pathway, the beneficial effect of NLRC3 in the PH model were reversed (72, 73). Thus, we can conclude that, NLRC3 improves the prognosis of PH by negatively regulating the PI3K-mTOR signaling pathway to suppress the proliferation, migration, and inflammation of PASMCs, NLRC3 may be the therapeutic target, diagnostic index, or prognostic index for PH.
Ventilator-induced lung injury is very common in clinical practice. Mechanical forces cause lung tissue injury, activate the immune system, and release a large number of cytokines that further destroy lung tissue, eventually lead to more serious clinical manifestations (74). When VILL induced by mechanical ventilation, the expression of NLRC3 in lung tissue of mice was downregulated (75, 76). Both dexmedetomidine and leptin have beneficial roles on the progress of VILI (75, 76). It was observed in the VILI model, the expression of NLRC3 was higher in mice treated with dexmedetomidine or leptin than in the control group (75, 76). Thus, we can speculate that, NLRC3 can alleviate the pathological damage of VILI, however the specific mechanisms involved require further investigation.
Some patients with Type 2 diabetes (T2DM) with complications such as gut flora dysbiosis, chronic gut inflammation, and increased intestinal permeability. gut flora dysbiosis induces chronic intestinal inflammation, destroys the tight junction of intestinal epithelial cells, and increases intestinal permeability (77).
NLRC3 has beneficial effect on intestinal dysfunction associated with T2DM (78). Overexpression of NLRC3 ameliorated epithelial integrity and up-regulated the expression of tight junction proteins ZO-1 and occludin in colonic epithelial cells (78). In addition, researchers also observed that Butyrate, a short-chain fatty acid (SCFA), binds to the G-protein coupled receptor 43 (GPR43) on the colonic epithelial cell to stimulate the expression of NLRC3 (78).
Based on the finding, we can conclude that NLRC3 is involved in mediating the excessive inflammatory response and uncontrolled cell proliferation. The development and progression of a large number of diseases undergo both pathological process, such as cancer, autoimmune diseases and stroke (Table 1). Thus, NLRC3 is a promising molecule and may be an important diagnostic marker and therapeutic target for many diseases. However, how NLRC3 exerts its regulatory activity on intracellular cell signaling has not been thoroughly evaluated; areas requiring further study include NLRC3-mediated mechanisms in the promotion of cell apoptosis, on the differentiation naïve T cells, and regulation of the cell cycle. In addition, the studies of NLRC3 are limited to macrophages, dendritic cells, T cells, and epithelial cells models, however NLRC3 has also been detected in other cells, such as B cells, neutrophils, and basophils, thus, further studies investigating the role of NLRC3 on these cells may be meaningful.
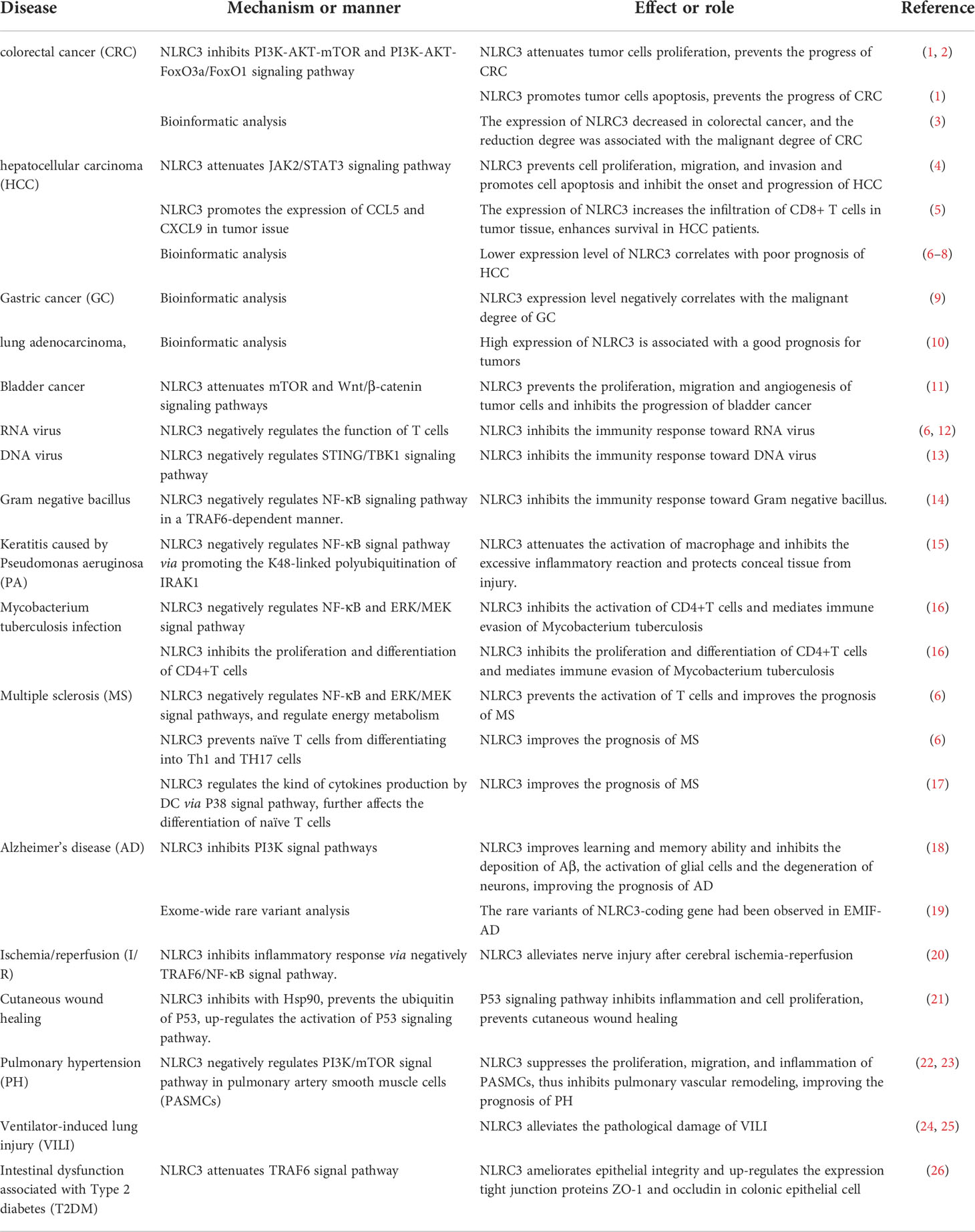
Table 1 Roles of NLRC3 in diseases.
NLRC3 as a member of the PRR family, its function in pathogen recognition has been addressed by some researchers. In contrast to many other NLRs, the LRR domain of NLRC3—which recognizes pathogens—is positively charged. This is an interesting discovery, as numerous antigens are negatively charged, which means that NLRC3 might have the ability to recognize antigens. Further research is needed to confirm this speculation.
In conclusion, the proven functions of NLRC3 are preventing the activation of the immune system, inhibiting the inflammatory response, attenuating cell proliferation, and promoting apoptosis. In addition, the roles and application of NLRC3 in specific disease processes, requires further insight and exploration on how to translate mechanism research to a direct clinical application. In addition, we speculate that NLRC3 may have the function of recognizing antigens, whether this hypothesis is true, and what type of antigens NLRC3 can recognize, warrants further research.
JX and YS designed the review. DS and JX wrote the manuscript with supervision of YS. All authors critically revised the manuscript and approved it for publication.
This study was supported by National Natural Science Foundation of China (No. 81772047, 81971818, 82002026 and 82272217) and the National Key Research and Development Project (2021YFC2500802).
Thanks for researchers who contribute to the exploration of NLRC3.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sahoo BR. Structure of fish toll-like receptors (Tlr) and nod-like receptors (Nlr). Int J Biol Macromol (2020) 161:1602–17. doi: 10.1016/j.ijbiomac.2020.07.293
2. Kawai T, Akira S. Tlr signaling. Semin Immunol (2007) 19(1):24–32. doi: 10.1016/j.smim.2006.12.004
3. Chang CL, Hsu SYT. Roles of Clr/Ramp receptor signaling in reproduction and development. Curr Protein Pept Sci (2013) 14(5):393–406. doi: 10.2174/13892037113149990056
4. Guo H, Gibson SA, Ting JPY. Gut microbiota, nlr proteins, and intestinal homeostasis. J Exp Med (2020) 217(10):e20181832. doi: 10.1084/jem.20181832
5. Ablasser A, Hur S. Regulation of cgas- and rlr-mediated immunity to nucleic acids. Nat Immunol (2020) 21(1):17–29. doi: 10.1038/s41590-019-0556-1
6. Huang J, Xie P, Dong Y, An W. Inhibition of Drp1 sumoylation by alr protects the liver from ischemia-reperfusion injury. Cell Death Differ (2021) 28(4):1174–92. doi: 10.1038/s41418-020-00641-7
7. Caruso R, Warner N, Inohara N, Núñez G. Nod1 and Nod2: Signaling, host defense, and inflammatory disease. Immunity (2014) 41(6):898–908. doi: 10.1016/j.immuni.2014.12.010
8. Fu Y, Zhan X, Wang Y, Jiang X, Liu M, Yang Y, et al. Nlrc3 expression in dendritic cells attenuates Cd4 T cell response and autoimmunity. EMBO J (2019) 38(16):e101397. doi: 10.15252/embj.2018101397
9. Nagai-Singer MA, Morrison HA, Allen IC. Nlrx1 is a multifaceted and enigmatic regulator of immune system function. Front Immunol (2019) 10:2419. doi: 10.3389/fimmu.2019.02419
10. He Y, Hara H, Núñez G. Mechanism and regulation of Nlrp3 inflammasome activation. Trends Biochem Sci (2016) 41(12):1012–21. doi: 10.1016/j.tibs.2016.09.002
11. Conti BJ, Davis BK, Zhang J, O’Connor W, Williams KL, Ting JPY. Caterpiller 16.2 (Clr16.2), a novel Nbd/Lrr family member that negatively regulates T cell function. J Biol Chem (2005) 280(18):18375–85. doi: 10.1074/jbc.M413169200
12. Gültekin Y, Eren E, Özören N. Overexpressed Nlrc3 acts as an anti-inflammatory cytosolic protein. J Innate Immun (2015) 7(1):25–36. doi: 10.1159/000363602
13. Li X, Deng M, Petrucelli AS, Zhu C, Mo J, Zhang L, et al. Viral DNA binding to Nlrc3, an inhibitory nucleic acid sensor, unleashes sting, a cyclic dinucleotide receptor that activates type I interferon. Immunity (2019) 50(3):591–9. doi: 10.1016/j.immuni.2019.02.009
14. Schneider M, Zimmermann AG, Roberts RA, Zhang L, Swanson KV, Wen H, et al. The innate immune sensor Nlrc3 attenuates toll-like receptor signaling Via modification of the signaling adaptor Traf6 and transcription factor nf-Kb. Nat Immunol (2012) 13(9):823–31. doi: 10.1038/ni.2378
15. Guo L, Kong Q, Dong Z, Dong W, Fu X, Su L, et al. Nlrc3 promotes host resistance against pseudomonas aeruginosa-induced keratitis by promoting the degradation of Irak1. Int J Mol Med (2017) 40(3):898–906. doi: 10.3892/ijmm.2017.3077
16. Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, et al. Nlrc3, a member of the nlr family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor sting. Immunity (2014) 40(3):329–41. doi: 10.1016/j.immuni.2014.01.010
17. Mangan MSJ, Latz E. Nlrc3 puts the brakes on sting. Immunity (2014) 40(3):305–6. doi: 10.1016/j.immuni.2014.02.007
18. Tocker AM, Durocher E, Jacob KD, Trieschman KE, Talento SM, Rechnitzer AA, et al. The scaffolding protein Iqgap1 interacts with Nlrc3 and inhibits type I ifn production. J Immunol (2017) 199(8):2896–909. doi: 10.4049/jimmunol.1601370
19. Okada M, Hozumi Y, Iwazaki K, Misaki K, Yanagida M, Araki Y, et al. Dgkζ is involved in lps-activated phagocytosis through Iqgap1/Rac1 pathway. Biochem Biophys Res Commun (2012) 420(2):479–84. doi: 10.1016/j.bbrc.2012.03.057
20. Brandt DT, Marion S, Griffiths G, Watanabe T, Kaibuchi K, Grosse R. Dia1 and Iqgap1 interact in cell migration and phagocytic cup formation. J Cell Biol (2007) 178(2):193–200. doi: 10.1083/jcb.200612071
21. Kesavardhana S, Kanneganti T-D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol (2017) 29(5):201–10. doi: 10.1093/intimm/dxx018
22. Eren E, Berber M, Özören N. Nlrc3 protein inhibits inflammation by disrupting Nalp3 inflammasome assembly Via competition with the adaptor protein asc for pro-Caspase-1 binding. J Biol Chem (2017) 292(30):12691–701. doi: 10.1074/jbc.M116.769695
23. Karki R, Man SM, Malireddi RKS, Kesavardhana S, Zhu Q, Burton AR, et al. Nlrc3 is an inhibitory sensor of Pi3k-mtor pathways in cancer. Nature (2016) 540(7634):583–7. doi: 10.1038/nature20597
24. Hua H, Kong Q, Zhang H, Wang J, Luo T, Jiang Y. Targeting mtor for cancer therapy. J Hematol Oncol (2019) 12(1):71. doi: 10.1186/s13045-019-0754-1
25. Saxton RA, Sabatini DM. Mtor signaling in growth, metabolism, and disease. Cell (2017) 168(6):960–76. doi: 10.1016/j.cell.2017.02.004
26. Karki R, Malireddi RKS, Zhu Q, Kanneganti T-D. Nlrc3 regulates cellular proliferation and apoptosis to attenuate the development of colorectal cancer. Cell Cycle (2017) 16(13):1243–51. doi: 10.1080/15384101.2017.1317414
27. Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. K63 polyubiquitination and activation of mtor by the P62-Traf6 complex in nutrient-activated cells. Mol Cell (2013) 51(3):283–96. doi: 10.1016/j.molcel.2013.06.020
28. Li W, Zhang Y, Hu Z, Xu Y. Overexpression of Nlrc3 enhanced inhibition effect of sevoflurane on inflammation in an ischaemia reperfusion cell model. Folia Neuropathol (2020) 58(3):213–22. doi: 10.5114/fn.2020.100064
29. Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and Foxo3a Via the proteasome. J Biol Chem (2003) 278(14):12361–6. doi: 10.1074/jbc.M213069200
30. Kwon S-K, Saindane M, Baek K-H. P53 stability is regulated by diverse deubiquitinating enzymes. Biochim Biophys Acta Rev Cancer (2017) 1868(2):404–11. doi: 10.1016/j.bbcan.2017.08.001
31. Qin Y, Wu K, Zhang Z, Pan R, Lin Z, Zhang W, et al. Nlrc3 deficiency promotes cutaneous wound healing due to the inhibition of P53 signaling. Biochim Biophys Acta Mol Basis Dis (2022) 1868(11):166518. doi: 10.1016/j.bbadis.2022.166518
32. Zarubin T, Han J. Activation and signaling of the P38 map kinase pathway. Cell Res (2005) 15(1):11–8. doi: 10.1038/sj.cr.7290257
33. Kang J-H, Li M-J, Luan P-P, Jiang D-K, Chen Y-W, Xu X, et al. Nlrc3 silencing accelerates the invasion of hepatocellular carcinoma cell Via il-6/Jak2/Stat3 pathway activation. Cell Biol Int (2020) 44(10):2053–64. doi: 10.1002/cbin.11414
34. Murphy TL, Grajales-Reyes GE, Wu X, Tussiwand R, Briseño CG, Iwata A, et al. Transcriptional control of dendritic cell development. Annu Rev Immunol (2016) 34:93–119. doi: 10.1146/annurev-immunol-032713-120204
35. Walsh KP, Mills KHG. Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol (2013) 34(11):521–30. doi: 10.1016/j.it.2013.07.006
36. Worbs T, Hammerschmidt SI, Förster R. Dendritic cell migration in health and disease. Nat Rev Immunol (2017) 17(1):30–48. doi: 10.1038/nri.2016.116
37. Uchimura T, Oyama Y, Deng M, Guo H, Wilson JE, Rampanelli E, et al. The innate immune sensor Nlrc3 acts as a rheostat that fine-tunes T cell responses in infection and autoimmunity. Immunity (2018) 49(6):1049–61. doi: 10.1016/j.immuni.2018.10.008
38. Hu S, Du X, Huang Y, Fu Y, Yang Y, Zhan X, et al. Nlrc3 negatively regulates Cd4+ T cells and impacts protective immunity during mycobacterium tuberculosis infection. PloS Pathog (2018) 14(8):e1007266. doi: 10.1371/journal.ppat.1007266
39. Pearce EL, Poffenberger MC, Chang C-H, Jones RG. Fueling immunity: Insights into metabolism and lymphocyte function. Science (2013) 342(6155):1242454. doi: 10.1126/science.1242454
40. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science (2016) 354(6311):481–4. doi: 10.1126/science.aaf6284
41. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
42. Greten FR, Grivennikov SI. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity (2019) 51(1):27–41. doi: 10.1016/j.immuni.2019.06.025
43. Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, et al. Colorectal cancer. Nat Rev Dis Primers (2015) 1:15065. doi: 10.1038/nrdp.2015.65
44. Smyth EC, Lagergren J, Fitzgerald RC, Lordick F, Shah MA, Lagergren P, et al. Oesophageal cancer. Nat Rev Dis Primers (2017) 3:17048. doi: 10.1038/nrdp.2017.48
45. Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, et al. Nlrp12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical nf-Kb signaling. Immunity (2012) 36(5):742–54. doi: 10.1016/j.immuni.2012.03.012
46. Chen Z, Yang L, Chen L, Li J, Zhang F, Xing Y, et al. Mir-190b promotes tumor growth and metastasis Via suppressing Nlrc3 in bladder carcinoma. FASEB J (2020) 34(3):4072–84. doi: 10.1096/fj.201901764R
47. Liu R, Truax AD, Chen L, Hu P, Li Z, Chen J, et al. Expression profile of innate immune receptors, nlrs and Aim2, in human colorectal cancer: Correlation with cancer stages and inflammasome components. Oncotarget (2015) 6(32):33456–69. doi: 10.18632/oncotarget.5587
48. Ma Y-Y, Zhang G-H, Li J, Wang S-B, Hu Z-M, Zhang C-W, et al. The correlation of Nlrc3 expression with the progression and prognosis of hepatocellular carcinoma. Hum Pathol (2018) 82:273–81. doi: 10.1016/j.humpath.2018.07.031
49. Pan L, Fang J, Chen M-Y, Zhai S-T, Zhang B, Jiang Z-Y, et al. Promising key genes associated with tumor microenvironments and prognosis of hepatocellular carcinoma. World J Gastroenterol (2020) 26(8):789–803. doi: 10.3748/wjg.v26.i8.789
50. Qi Z, Yan F, Chen D, Xing W, Li Q, Zeng W, et al. Identification of prognostic biomarkers and correlations with immune infiltrates among cgas-sting in hepatocellular carcinoma. Biosci Rep (2020) 40(10):BSR20202603. doi: 10.1042/BSR20202603
51. Wang H, Shen L, Li Y, Lv J. Integrated characterisation of cancer genes identifies key molecular biomarkers in stomach adenocarcinoma. J Clin Pathol (2020) 73(9):579–86. doi: 10.1136/jclinpath-2019-206400
52. Deng L, Long F, Wang T, Dai L, Chen H, Yang Y, et al. Identification of an immune classification and prognostic genes for lung adenocarcinoma based on immune cell signatures. Front Med (Lausanne) (2022) 9:855387. doi: 10.3389/fmed.2022.855387
53. Wang C, Shi J, Xu J, Fu Q, Ding Y, Yang J, et al. Nlrc3 high expression represents a novel predictor for positive overall survival correlated with Ccl5 and Cxcl9 in hcc patients. Front Oncol (2022) 12:815326. doi: 10.3389/fonc.2022.815326
54. Ma R, Zhang X, Shu J, Liu Z, Sun W, Hou S, et al. Knockout mice showed renal pathological changes after htnv infection. Front Immunol (2021) 12:692509. doi: 10.3389/fimmu.2021.692509
55. Engel LS, Callegan MC, Hobden JA, Reidy JJ, Hill JM, O’Callaghan RJ. Effectiveness of specific Antibiotic/Steroid combinations for therapy of experimental pseudomonas aeruginosa keratitis. Curr Eye Res (1995) 14(3):229–34. doi: 10.3109/02713689509033519
56. Ramakrishnan L. Revisiting the role of the granuloma in tuberculosis. Nat Rev Immunol (2012) 12(5):352–66. doi: 10.1038/nri3211
57. Goldberg MF, Saini NK, Porcelli SA. Evasion of innate and adaptive immunity by mycobacterium tuberculosis. Microbiol Spectr (2014) 2(5):MGM2-0005-2013. doi: 10.1128/microbiolspec.MGM2-0005-2013
58. Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple sclerosis: Mechanisms and immunotherapy. Neuron (2018) 97(4):742–68. doi: 10.1016/j.neuron.2018.01.021
59. Chen L, Cao S-Q, Lin Z-M, He S-J, Zuo J-P. Nod-like receptors in autoimmune diseases. Acta Pharmacol Sin (2021) 42(11):1742–56. doi: 10.1038/s41401-020-00603-2
60. Holley CL, Coll RC, Schroder K. Nlrc3 restrains responses to a T. Immunity (2018) 49(6):989–91. doi: 10.1016/j.immuni.2018.12.003
61. Biliktu M, Senol SP, Temiz-Resitoglu M, Guden DS, Horat MF, Sahan-Firat S, et al. Pharmacological inhibition of soluble epoxide hydrolase attenuates chronic experimental autoimmune encephalomyelitis by modulating inflammatory and anti-inflammatory pathways in an inflammasome-dependent and -independent manner. Inflammopharmacology (2020) 28(6):1509–24. doi: 10.1007/s10787-020-00691-w
62. Wang L, Luo G, Zhang L-F, Geng H-X. Neuroprotective effects of epoxyeicosatrienoic acids. Prostaglandins Other Lipid Mediat (2018) 138. doi: 10.1016/j.prostaglandins.2018.07.002
63. Kodani SD, Morisseau C. Role of epoxy-fatty acids and epoxide hydrolases in the pathology of neuro-inflammation. Biochimie (2019) 159:59–65. doi: 10.1016/j.biochi.2019.01.020
64. Wagner KM, McReynolds CB, Schmidt WK, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases. Pharmacol Ther (2017) 180:62–76. doi: 10.1016/j.pharmthera.2017.06.006
65. Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. Lancet (2021) 397(10284):1577–90. doi: 10.1016/S0140-6736(20)32205-4
66. Zha L, Yu Z, Fang J, Zhou L, Guo W, Zhou J. Nlrc3 delays the progression of ad in App/Ps1 mice Via inhibiting Pi3k activation. Oxid Med Cell Longev (2020) 2020:5328031. doi: 10.1155/2020/5328031
67. Neumann A, Küçükali F, Bos I, Vos SJB, Engelborghs S, De Pooter T, et al. Rare variants in Iffo1, dtnb, Nlrc3 and Slc22a10 associate with alzheimer’s disease csf profile of neuronal injury and inflammation. Mol Psychiatry (2022) 27(4):1990–9. doi: 10.1038/s41380-022-01437-6
68. Lucas S-M, Rothwell NJ, Gibson RM. The role of inflammation in cns injury and disease. Br J Pharmacol (2006) 147 Suppl 1:S232–S40. doi: 10.1038/sj.bjp.0706400
69. Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med (1999) 341(10):738–46. doi: 10.1056/NEJM199909023411006
70. Thenappan T, Ormiston ML, Ryan JJ, Archer SL. Pulmonary arterial hypertension: Pathogenesis and clinical management. BMJ (2018) 360:j5492. doi: 10.1136/bmj.j5492
71. Zha L-H, Zhou J, Li T-Z, Luo H, He J-N, Zhao L, et al. Nlrc3: A novel noninvasive biomarker for pulmonary hypertension diagnosis. Aging Dis (2018) 9(5):843–51. doi: 10.14336/AD.2017.1102
72. Zha L-H, Zhou J, Li T-Z, Luo H, Zhang M-Q, Li S, et al. Nlrc3 inhibits mct-induced pulmonary hypertension in rats via attenuating Pi3k activation. J Cell Physiol (2019) 234(9):15963–76. doi: 10.1002/jcp.28255
73. Zha L-H, Zhou J, Tan Y, Guo S, Zhang M-Q, Li S, et al. Nlrc3 inhibits pdgf-induced pasmcs proliferation Via Pi3k-mtor pathway. J Cell Physiol (2020) 235(12):9557–67. doi: 10.1002/jcp.29763
74. Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med (2013) 369(22):2126–36. doi: 10.1056/NEJMra1208707
75. Zhang B, Zhang X, Li Q, Ma F, Sun L, Wang M. Dexmedetomidine attenuates ventilator-induced lung injury in rats by up-regulating Nlrc3. Ann Palliat Med (2020) 9(5):2474–84. doi: 10.21037/apm-19-375
76. Zhang X, Qin WW, Ma FG, Sun LX, Han W. Leptin reduces ventilator-induced lung injury in rats by regulating Nlrp3, Nlrc4 and Nlrc3. J Biol Regul Homeost Agents (2021) 35(1):59–69. doi: 10.23812/20-258-A
77. Yang G, Wei J, Liu P, Zhang Q, Tian Y, Hou G, et al. Role of the gut microbiota in type 2 diabetes and related diseases. Metabolism (2021) 117:154712. doi: 10.1016/j.metabol.2021.154712
Keywords: NLRC3, NOD-like receptor, NF-κB, sting, pyroptosis, PI3K, immune response, immune-related diseases
Citation: Sun D, Xu J, Zhang W, Song C, Gao C, He Y and Shang Y (2022) Negative regulator NLRC3: Its potential role and regulatory mechanism in immune response and immune-related diseases. Front. Immunol. 13:1012459. doi: 10.3389/fimmu.2022.1012459
Received: 05 August 2022; Accepted: 04 October 2022;
Published: 20 October 2022.
Edited by:
Junji Xing, Houston Methodist Research Institute, United StatesReviewed by:
Ming Xian Chang, Institute of Hydrobiology (CAS), ChinaCopyright © 2022 Sun, Xu, Zhang, Song, Gao, He and Shang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiqian Xu, eGpxNTQ1Ni5nb29kQDE2My5jb20=; You Shang, eW91X3NoYW5naHVzdEAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.