
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 23 September 2022
Sec. Inflammation
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1010882
This article is part of the Research TopicCommunity Series in Translational Insights into Mechanisms and Therapy of Organ Dysfunction in Sepsis and Trauma, volume IIView all 12 articles
 Sophie Dennhardt1,2,3
Sophie Dennhardt1,2,3 Wiebke Pirschel1,2
Wiebke Pirschel1,2 Bianka Wissuwa1,2
Bianka Wissuwa1,2 Diana Imhof4
Diana Imhof4 Christoph Daniel5
Christoph Daniel5 Jan T. Kielstein6
Jan T. Kielstein6 Isabel Hennig-Pauka7
Isabel Hennig-Pauka7 Kerstin Amann5
Kerstin Amann5 Florian Gunzer8
Florian Gunzer8 Sina M. Coldewey1,2,3*
Sina M. Coldewey1,2,3*Hemolytic-uremic syndrome (HUS) can occur as a systemic complication of infections with Shiga toxin (Stx)-producing Escherichia coli and is characterized by microangiopathic hemolytic anemia and acute kidney injury. Hitherto, therapy has been limited to organ-supportive strategies. Erythropoietin (EPO) stimulates erythropoiesis and is approved for the treatment of certain forms of anemia, but not for HUS-associated hemolytic anemia. EPO and its non-hematopoietic analog pyroglutamate helix B surface peptide (pHBSP) have been shown to mediate tissue protection via an innate repair receptor (IRR) that is pharmacologically distinct from the erythropoiesis-mediating receptor (EPO-R). Here, we investigated the changes in endogenous EPO levels in patients with HUS and in piglets and mice subjected to preclinical HUS models. We found that endogenous EPO was elevated in plasma of humans, piglets, and mice with HUS, regardless of species and degree of anemia, suggesting that EPO signaling plays a role in HUS pathology. Therefore, we aimed to examine the therapeutic potential of EPO and pHBSP in mice with Stx-induced HUS. Administration of EPO or pHBSP improved 7-day survival and attenuated renal oxidative stress but did not significantly reduce renal dysfunction and injury in the employed model. pHBSP, but not EPO, attenuated renal nitrosative stress and reduced tubular dedifferentiation. In conclusion, targeting the EPO-R/IRR axis reduced mortality and renal oxidative stress in murine HUS without occurrence of thromboembolic complications or other adverse side effects. We therefore suggest that repurposing EPO for the treatment of patients with hemolytic anemia in HUS should be systematically investigated in future clinical trials.
Hemolytic-uremic syndrome (HUS) belongs to the group of thrombotic microangiopathies and includes atypical and typical HUS. The latter accounts for approximately 90% of HUS cases and is a life-threatening systemic complication of infections with certain bacterial pathogens, most commonly Shiga toxin (Stx)-producing Escherichia coli (STEC) (1).
STEC-infections can cause diarrhea or hemorrhagic colitis with bloody diarrhea. STEC-HUS typically presents with microangiopathic hemolytic anemia, thrombocytopenia, acute kidney injury (AKI) and other organ dysfunctions (1). One-third of patients with STEC-HUS also develop long-term renal and up to 4%, long-term neurological sequelae (2). Renal oxidative stress was shown to be an important factor in HUS pathogenesis in pediatric HUS (3, 4). Its pathophysiological relevance was also demonstrated in a murine model of Stx2-induced HUS (5).
To date, organ supportive therapy in the intensive care unit (ICU), including hemodialysis, fluid resuscitation as well erythrocyte transfusion if indicated, has been the standard of care in patients with STEC-HUS (6). In the absence of targeted therapeutic options, there is a medical need to further investigate molecular therapeutic approaches for the treatment of this life-threatening systemic syndrome.
Due to the low incidence – of e. g. 0.07 per 100,000 persons/year for Germany (7) up to 0.67 per 100,000 persons/year for Argentina (8) – conducting prospective randomized clinical trials has proven difficult. To provide tools for preclinical studies, we have recently introduced and characterized several animal models for STEC-HUS, including an infection model employing the Northern German outbreak strain O104:H4 of 2011 and the well-characterized outbreak strain O157:H7 86-24 in gnotobiotic piglets (9), as well as a clinically relevant mouse model (10–12) reflecting most aspects of human pathology by repeatedly exposing animals to low doses of Stx isolated from an EHEC O157:H7 86-24. All previously described small animal models of STEC-HUS mimic well the critical illness, renal failure and microangiopathy seen in humans, while being limited in their modeling of hemolytic anemia, which is oftentimes masked by hemoconcentration secondary to fluid deficiency (10, 13, 14).
Erythropoietin (EPO), a pleiotropic hormone that has been shown to exert tissue-protective effects via the innate repair receptor (IRR) independent of its hematopoietic properties via the EPO receptor (EPO-R) homodimer (15, 16), appears to be a promising candidate to be further evaluated in HUS. Preclinical trials usually use one magnitude higher doses of EPO compared with clinical trials (1000-5000 IU/kg versus 300 IU/kg) (17). This could explain why preclinical tissue protection following EPO administration could often not be translated into the clinical setting. Furthermore, beside its potential beneficial effects, the thrombogenic effect of this erythropoiesis-stimulating hormone was critically discussed in the context of clinical studies (18, 19), as tissue-protective effects require high levels of EPO and administration of high EPO doses can increase the risk of thrombosis (16) and hypertension (17). For this reason, EPO-derived non-hematopoietic small peptide activators of the IRR, such as the pyroglutamate helix B surface peptide (pHBSP), have been developed (15). Preclinically, pHBSP has been shown to convey tissue protection in numerous disease models (20), such as nephroprotection in ischemia-reperfusion models of AKI (21, 22). Furthermore, pHBSP has been successfully employed in phase II studies in patients with diabetes or sarcoidosis and neuropathic pain (23, 24).
HUS can be considered as a specific phenotype of sepsis, as it represents a combination of infection and organ dysfunction. We previously reported that EPO attenuated AKI (25) and cardiac dysfunction (26) in mouse models of endotoxemia and cecal ligation and puncture-induced sepsis. There is no clinical evidence on the beneficial or harmful effects of EPO in patients with sepsis and there is only little evidence regarding the beneficial or harmful effects of EPO in ICU patients in general (27, 28). To our knowledge, the tissue-protective effects of EPO or pHBSP in HUS have not been investigated, yet. One trial that aimed to investigate the effects of EPO on the microcirculation of patients with severe sepsis was discontinued due to lack of recruitment (NCT01087450). We did not find the results of a study that examined the immunomodulatory effects of activated protein C and/or EPO in sepsis (NCT00229034). A third study examining the effects of EPO on renal function in critically ill patients with and without multiorgan failure appears to be not completed (EudraCT number: 2008-003733-24). Altogether, there is no evidence from randomized controlled trials regarding the effects of EPO administration in sepsis and HUS. Despite this lack of evidence, it is however conceivable, that in the presence of severe hemolytic anemia in patients with HUS, the stimulation of hematopoiesis by EPO could be beneficial by reducing the need for allogenic erythrocyte transfusion, which carry non-negligible risks (29, 30).
Anemia is usually accompanied by a regulatory increase in endogenous EPO production due to hypoxia-mediated feedback mechanisms. In patients with iron deficiency or hemolytic anemia, serum EPO levels exceeding 2000 mU/ml (two orders of magnitude above normal range) have been reported (31). In contrast, renal anemia is often accompanied by insufficient EPO production due to direct damage to EPO-producing cells in the kidney (32) or inhibition of EPO production by various cytokines (33). Already in the early 1990’s, it was hypothesized that anemias associated with low serum EPO levels might respond to treatment with recombinant EPO (34).
Currently, EPO can be considered in the treatment of anemia associated with chemotherapy (35) and chronic kidney disease (36). The current national guideline for the treatment of HUS in pediatric patients states that EPO can be considered in the treatment of HUS-associated hemolytic anemia as expert consensus without citing evidence (37). So far, no adverse effects have been observed in children receiving EPO for the treatment of hemolytic anemias (38–41). However, a reduction of red blood cell transfusion could not be shown in two small studies (38, 39). Hitherto, there are no sufficiently powered clinical studies in this context. Even a neutral effect of EPO in terms of nephroprotection in the absence of side effects might positively impact the therapy of HUS-associated hemolytic anemia and save the need for red blood cell transfusions.
In view of the above, we consider it necessary and promising to assess the role of EPO in this orphan disease which comprises a specific form of septic organ failure. The first objective of this study is to further elucidate the role of endogenous EPO and its potential clinical relevance in HUS by measuring endogenous EPO levels in different species: human patients (infection with EHEC O104:H4, 2011 German outbreak), gnotobiotic piglets (subjected to EHEC O104:H4 or EHEC O157:H7 86-24) and mice (subjected to Stx2 derived from EHEC O157:H7 86-24). The second objective of this study is to examine the effect of EPO and the non-hematopoietic peptide pHBSP in a murine model of HUS by measuring surrogate parameters of kidney injury and dysfunction, intrarenal barrier integrity, microangiopathy, oxidative and nitrosative stress and metabolome. Hereby, we aim to provide preclinical evidence to assess whether treatment of HUS with EPO or pHBSP should be considered and further investigated, particularly for HUS-associated hemolytic anemia.
Plasma samples of patients with STEC-HUS (n = 27, median age 47, 24 female) were provided by the German STEC-HUS Registry (42). These samples were taken within 10 d after admission to the hospital (HUS acute stage) and at the day of the last plasmapheresis (HUS pre-discharge). We analyzed the subcohort of 7 patients of which samples were available from day 1 to day 3 after hospital admission and within 3 d before hospital discharge. Age- and sex-matched samples of healthy controls (n = 21, median age 63.5, 15 female) were provided by Jena University Hospital [ICROS study (43)]. Ethic approval was obtained by the primary investigators from the Ethics Committee of Hannover Medical School (1123-2011) and of the Friedrich Schiller University Jena (5276-09/17). Participants provided written informed consent prior to inclusion in the respective studies.
Samples from gnotobiotic piglets (9) were collected 4 to 6 days after STEC-infection or mock infection (sham). Experimental procedures in gnotobiotic piglets were approved by the local permitting authorities in the Lower Saxony State Office for Consumer Protection and Food Safety and in accordance with the requirements of the national animal welfare law (Approval Number: 33.9-42502-04-13/1149) in accordance with the German legislation following the guidelines of FELASA and ARRIVE (9).
All procedures performed in mice were approved by the regional animal welfare committee and the Thuringian State Office for Consumer Protection (registration number 02-058/14) and performed in accordance with the German legislation. HUS-like disease in mice was induced by repetitive doses of Stx2 as described previously (10). Briefly, wild-type C57BL/6J mice aged 10-16 weeks weighing 20-30 g were randomly assigned to one of four groups (sham n = 16, Stx+vehicle n = 26, Stx+EPO n = 22, or Stx+pHBSP n = 21) and received 3x25 ng/kg body weight (BW) Stx or 0.9% NaCl i.v. on days 0, 3, and 6. EPO (1000 IU/kg BW) was applied s.c. 1 h after initial Stx injection. Due to its short plasma half-life, pHBSP (30 µg/kg BW) was applied s.c. every 24 h starting 1 h after initial Stx injection. Vehicle (Ringer’s solution) was applied s.c. starting 1 h after initial Stx injection. Mice received 3x800 µl Ringer’s solution each day for volume replacement. In compliance with ethical regulations, survival was monitored for 7 days using humane endpoints (mice were euthanized when reaching a high-grade disease state, Figure 3). Disease progression was monitored by weight loss and activity-based HUS score (ranging from 1-normally active, 2-active with slight restrictions, 3-active with clear intermissions, 4-slowed, 5-lethargic, 6-moribund, to 7-dead) as described previously (10). Animals were exsanguinated in deep ketamine/xylazine anesthesia (10). Renal tissue, blood and plasma samples were collected on day 7. An additional experiment with a comparable survival rate including sham and Stx+vehicle mice was performed to compensate for the lower survival rate in the Stx+vehicle group and to increase the statistical power in the analysis of day-7-samples for plasma urea and creatinine and all histological [Schiff’s periodic acid (PAS), acid fuchsin orange G (SFOG)] and immunohistochemistry (IHC) stainings (kidney-injury molecule 1 (KIM-1), cluster of differentiation 31 (CD31), E-cadherin, glycoprotein 1b (GP1b), nitrotyrosine and NADPH oxygenase 1 (NOX-1), Figures 4–7). Plasma EPO levels (Figure 2B) and plasma metabolome (Figure 8) were measured in an independent experimental setup with comparable survival rates since availability of plasma per animal was limited.
Stx purification was performed as described previously (10). Human recombinant EPO (Epoeitin beta, Hoffmann-La Roche) was diluted to 1000 U/kg BW in Ringer’s solution (vehicle). A standard N-(9-fluorenyl)methoxycarbonyl protocol for automated solid-phase peptide synthesis was implemented for pHBSP synthesis (Supplementary Methods). pHBSP was diluted to 30 µg/kg BW in Ringer’s solution (vehicle).
Human (42), piglet (9) and murine (10) blood samples were taken as described elsewhere. Hematology and analysis of laboratory chemistry parameters in murine samples were performed as recently described (10). Briefly, blood counts were analyzed using impedance technology implemented in the pocH100iv system (Sysmex, Kobe, Japan). Laboratory parameters were analyzed by an Architect™ c16200/ci8200 automated clinical chemistry system (Abbott Diagnostics, Abbott. Park, USA). This system uses the Jaffè method for plasma creatinine measurements and the urease/nicotinamide adenine dinucleotide hydrogen method for plasma urea measurements. The following parameters were measured using commercially available ELISA kits: serum or plasma EPO and murine plasma neutrophil-gelatinase-associated lipocalin (NGAL, Supplementary Table S1).
Processing of kidney samples, histopathological analysis and IHC stainings were performed as described previously (10). SFOG and thrombocyte (GP1b) staining was performed as described recently (11). Until antigen demasking, sections for E-cadherin, NOX-1 and nitrotyrosine staining were treated similarly. The Vector M.O.M. Immunodetection kit (Vector Laboratories) was used for E-cadherin staining. Blocking of nitrotyrosine staining was performed in normal goat serum. IHC sections were washed with Tris(hydroxymethyl)aminomethan (TRIS) buffer during staining [50 mM TRIS, 300 mM NaCl, pH was adjusted to 7.6 with HCl (all Carl Roth); 0.04% Tween®20 (Sigma Aldrich)], incubated with primary antibodies overnight at 4°C in appropriate dilutions (Supplementary Table S2) and subsequently incubated for 30 min with secondary antibodies (Supplementary Table S3). NOX-1 sections were incubated in primary antibody directly after antigen retrieval. Nitrotyrosine sections were stained using the CSA kit and rabbit Link (Dako).
Quantification and scoring of histopathological and IHC stainings were performed as described previously (10). For quantification of renal E-cadherin expression, all cut and completely positively stained tubules (with entirely visible lumen, excluding those with spotty staining) were counted in 12 adjacent cortical fields [one cortical field = region of interest (ROI)] per section in 400x magnification. Nitrotyrosine staining was quantified by superimposing a 10x10 grid (area of 0.0977 mm²) over each field and counting positively stained fields in 20 adjacent cortical fields per section in 400x magnification (blinded). NOX-1 staining was analyzed using a scoring system (0 to 3; 0: < 25%, 1: 25-50%, 2: 50-75%, 3: > 75% positive staining per field, 12 fields/section, magnification 200x, blinded). Quantification of SFOG and thrombocyte staining was performed as described recently (11).
Targeted metabolomics from heparinized plasma was performed at Biocrates Life Sciences by mass spectrometry using an MxP® Quant 500 kit. Data analysis was performed using R version 3.4.4 (R Core Team) as follows: Readings below detection level were set to half of detection level for each analyte separately. Metabolome data was then log 2 transformed without any further normalization. Z scores were calculated using mean and standard deviation of all samples. Trends along the four sample groups were tested using linear regression models where sample group assignment was used as the independent variable and each analyte as the dependent variable in a separate model. Prior to linear regression, the independent variable sample group assignment was transformed to a pseudo-continuous variable where all samples of group “sham” were set to 0, samples of group “Stx+pHBSP” were set to 1/3, samples of group “Stx+EPO” were set to 2/3 and samples of group “Stx+vehicle” were set to 1. Applying this transformation, the linear regression models test whether there is an increasing or decreasing trend along the four groups in the assigned order. P-values for all models were Benjamini-Hochberg adjusted (44). Analytes with adjusted P-values below 0.05 and an absolute effect of one or bigger were considered significantly changing along the four sample groups.
Data are depicted as mean ± SD for n observations. GraphPad Prism 7.05 (GraphPad Software) was used for data analysis applying Student’s t-test and Wilcoxon signed rank test for comparisons between 2 groups. One-way ANOVA with Holm-Sidak post hoc test (parametric data) or Kruskal-Wallis test with Dunn’s post hoc test (nonparametric data) were used for comparisons between more than 2 groups. Survival was depicted as Kaplan-Meier curve and analyzed by Mantel-Cox test. Association between EPO levels in patients with STEC-HUS and hematological and laboratory parameters and piglet samples was performed with GraphPad Prism 7.05 implementing non-parametric Spearman’s rank correlation coefficients. A P-value < 0.05 was considered significant. Mean ± SD and P-values for all analyses are given in Supplementary Table S4.
In patients with STEC-HUS, EPO serum levels were elevated in the acute stage of disease compared with healthy controls (Figure 1A, upper panel, Supplementary Tables S5, S6 for individual values). Pre-discharge, EPO levels decreased compared to the acute phase, yet continued to be higher than in healthy controls. In a subgroup, in which blood samples were consequently taken within 3 days after admission and 3 days before discharge from the hospital, a reduction of serum EPO levels was observed in 6 out of 7 HUS patients (Figure 1A, lower panel). Several hematological, laboratory and clinical parameters at hospital admission are listed in Table 1 and EPO values were correlated with these parameters. Anemia was present in 6 out of 7 patients (Figures 1B, C) and there was a trend towards a negative correlation between endogenous EPO levels and hemoglobin (Hb; r = -0.5357, P = 0.2357) or hematocrit (r = -0.6071, P = 0.1667) at hospital admission. No correlation was observed between endogenous EPO levels and lactate dehydrogenase (LDH) or creatinine at hospital admission (Figures 1D, E), although intriguingly, the lowest endogenous EPO levels were observed in patients with pronounced kidney dysfunction (Figure 1E).
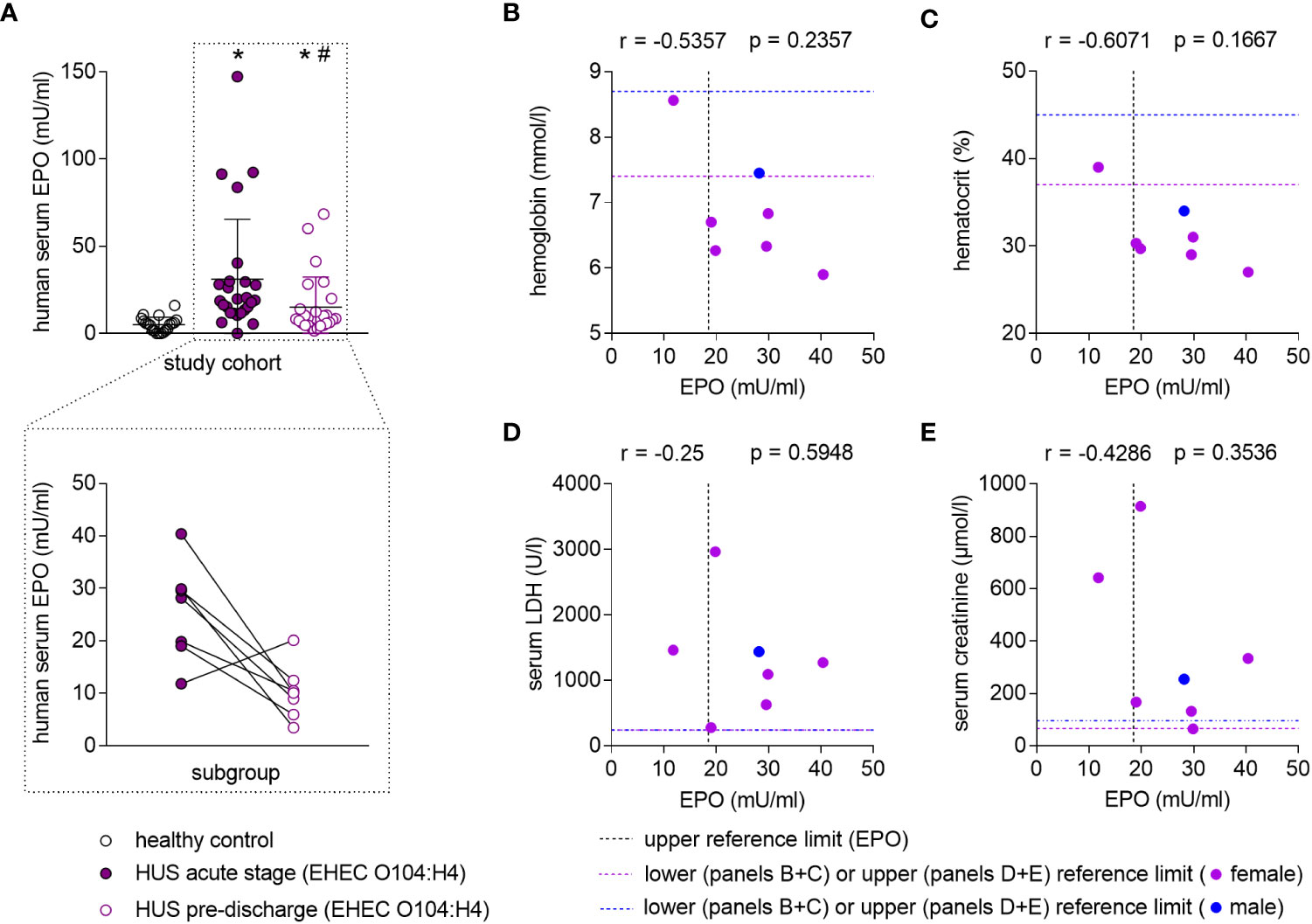
Figure 1 Endogenous EPO levels in patients with STEC-HUS and their correlation with anemia and kidney damage. (A) Upper panel: EPO serum levels in healthy controls (n = 20) and HUS patients (n = 26) within 10 d after admission to the hospital (HUS acute stage) and the last plasmapheresis (HUS pre-discharge) determined by ELISA. Data are expressed as scatter dot plot with mean ± SD. *P < 0.05 vs. healthy control (Mann-Whitney U-test), #P < 0.05 vs. HUS acute stage (Wilcoxon signed-rank test). Lower panel: Pairwise comparison of EPO serum levels of a subcohort of patients (n = 7) in which blood samples were available from day 1 to 3 after hospital admission and within 3 days before hospital discharge. Data are expressed as scatter dot plot with connecting line between respective measurements. (B-E) Correlation analysis implementing Spearman’s correlation coefficient (r) for correlations of endogenous EPO levels with (B) hemoglobin (n = 7), (C) hematocrit (n = 7), (D) lactate dehydrogenase (LDH, n = 7) and (E) creatinine (n = 7) in the subcohort. The upper reference limit for EPO in health are indicated by dashed vertical lines. Gender-specific lower or upper reference limits for the respective parameters of anemia and kidney damage are indicated by dashed horizontal lines.
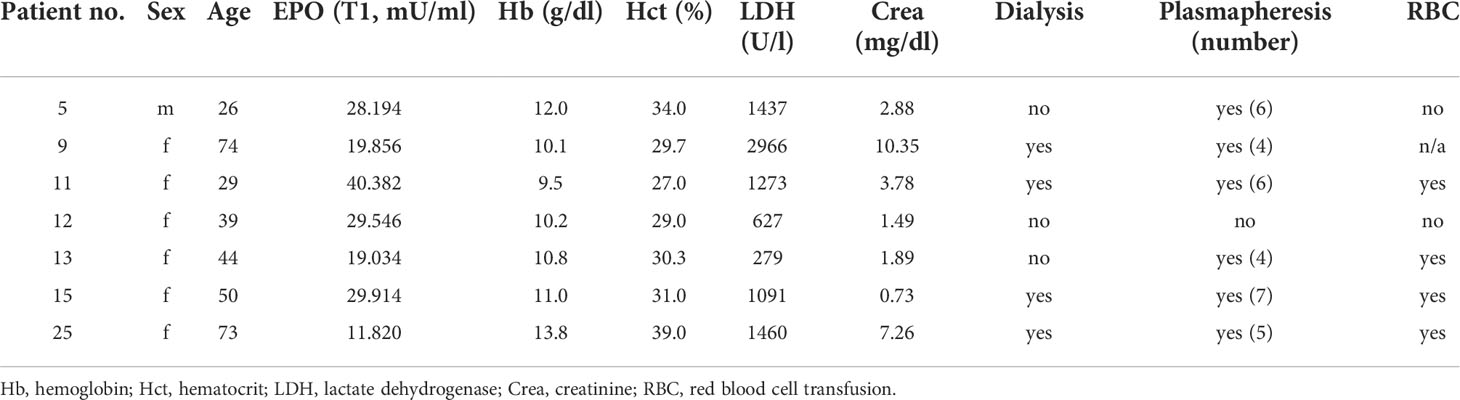
Table 1 Hematological, laboratory and renal function parameters in the subgroup of patients with STEC-HUS assessed at hospital admission.
While anemia appears to be a major driver of increased EPO secretion in patients with STEC-HUS, we wondered whether other mechanisms might also play a role. Considering that – at least in our limited subcohort-endogenous EPO was lowest in those patients with pronounced kidney dysfunction and injury, we hypothesize that these patients might profit from therapeutic EPO administration.
To further assess the role of EPO in HUS, we measured endogenous EPO levels in two well-characterized animal models of this condition. In gnotobiotic piglets with STEC-HUS, EPO levels were elevated in EHEC O104:H4- and EHEC O157:H7-infected piglets compared with sham piglets (Figure 2A). STEC-infected gnotobiotic piglets did not display clear signs of anemia or kidney injury compared with mock-infected sham animals (Table 2). No correlations were observed between EPO-levels and hematological parameters (Supplementary Table S7). In O157:H7-infected piglets, although LDH was comparably low, there was a trend towards negative correlation with EPO levels (Supplementary Table S7). In C57BL/6 mice subjected to Stx, slight hemoconcentration was observed (Supplementary Table 3, significant increase in Hb in Stx+vehicle compared with sham mice). Despite the absence of anemia in Stx+vehicle mice, EPO plasma levels were elevated compared with sham mice (Figure 2B). There was no correlation between plasma EPO levels and plasma NGAL as surrogate parameter for kidney damage in mice (Supplementary Figure S1).
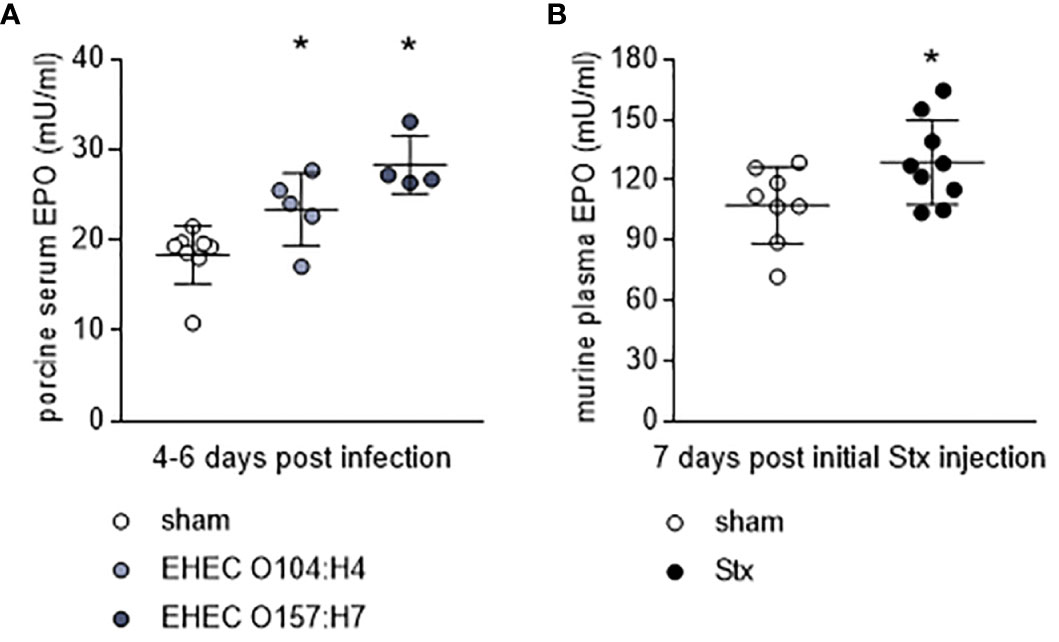
Figure 2 Endogenous EPO levels in animal models of HUS. (A) EPO serum levels in gnotobiotic piglets that were either mock-infected (sham) or infected with EHEC O157:H7 or EHEC O104:H4 determined by ELISA. Samples were taken 4-6 d after infection (sham n = 8, EHEC O104:H4 n = 5, EHEC O157:H7 n = 4). Data are expressed as scatter dot plot with mean ± SD. *P < 0.05 vs. sham (one-way ANOVA, Holm-Sidak’s multiple comparison test). (B) EPO plasma levels in mice with HUS on day 7 of experiment (sham n = 8, Stx n = 10) determined by ELISA. Data are expressed as scatter dot plot with mean ± SD, *P < 0.05 vs. sham (unpaired t-test).
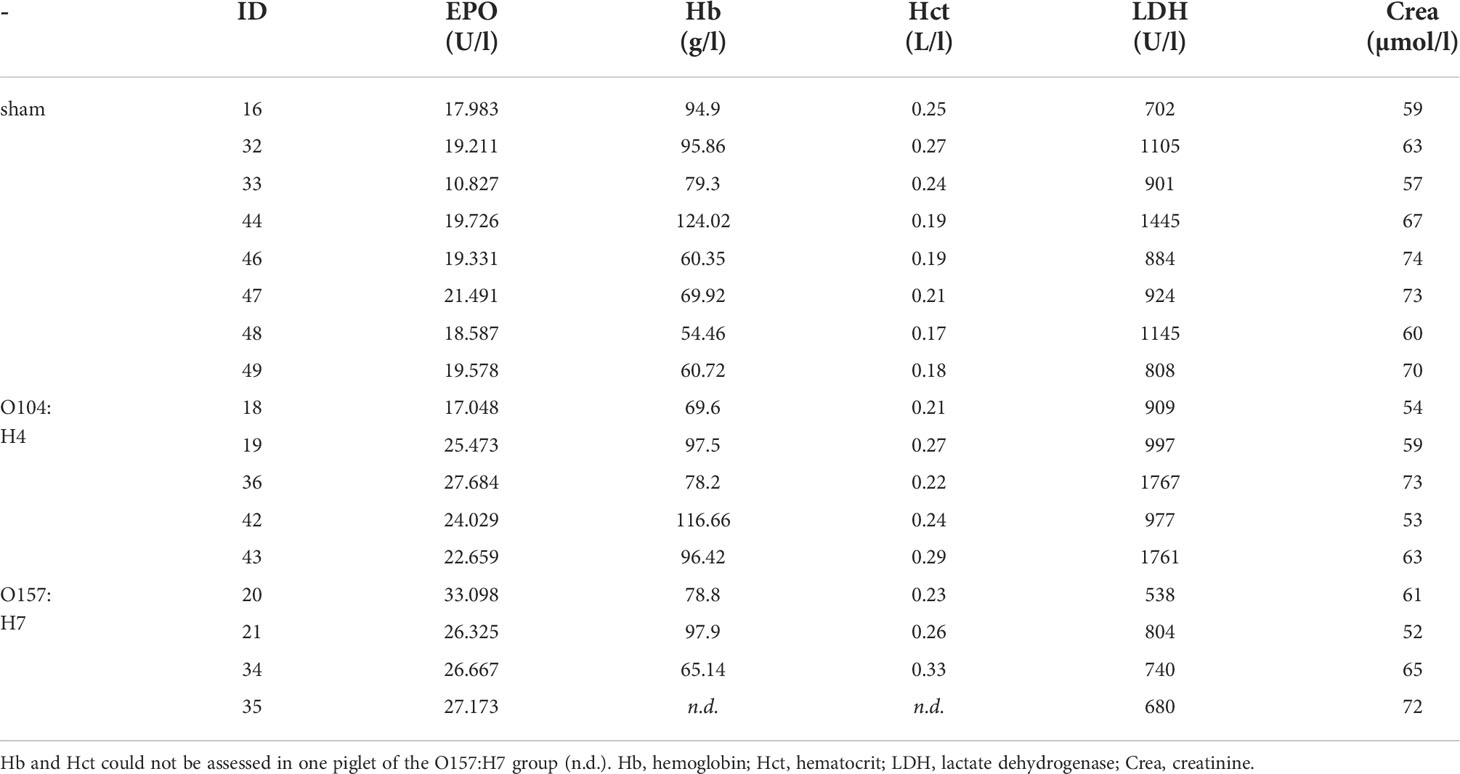
Table 2 Hematological, laboratory and renal function parameters in STEC-infected piglets and mock-infected controls (sham) assessed 4-6 days after infection.
As anemia appears not to be the sole driver of increased EPO levels during HUS and patients with pronounced kidney damage showed the lowest EPO levels, we next wanted to analyze the therapeutic potential of EPO and the non-hematopoietic EPO derivative pHBSP in a murine model of HUS.
7-day survival was significantly increased in Stx+EPO (68.2%) and Stx+pHBSP mice (76.2%) compared with Stx+vehicle mice (42.3%; Figure 3A). Consistently, disease progression was reduced on days 6 and 7 in these treatment groups (Figure 3B). All surviving Stx-challenged animals lost up to 20% weight until the end of the experiment. Statistical comparison of weight loss was performed until the first animals had to be euthanized at day 5 (Figure 3C). Weight loss in Stx+EPO mice was less pronounced compared with Stx+vehicle mice on days 4 and 5 (Figure 3C). While neurological involvement assessed by hind limb clasping reflex frequently occurred in Stx+vehicle mice (approx. 39%, P = 0.0067 vs. sham, Table 3), it was less common in Stx+EPO (approx. 27%; Table 3) and Stx+pHBSP mice (approx. 24%; Table 3). No differences were observed in plasma alanine aminotransferase (ALAT) and aspartate aminotransferase (ASAT), while Hb and/or hematocrit as indicators of hemoconcentration were increased in all Stx-challenged mice (Table 3). Compared with sham mice, Hb was elevated in Stx+EPO mice, while it was not in Stx+pHBSP mice. However, there was no significant difference between Stx+EPO mice and Stx-vehicle mice. The effects of EPO-treatment on Hb might be masked by the slight hemoconcentration observed in all Stx-challenged groups.
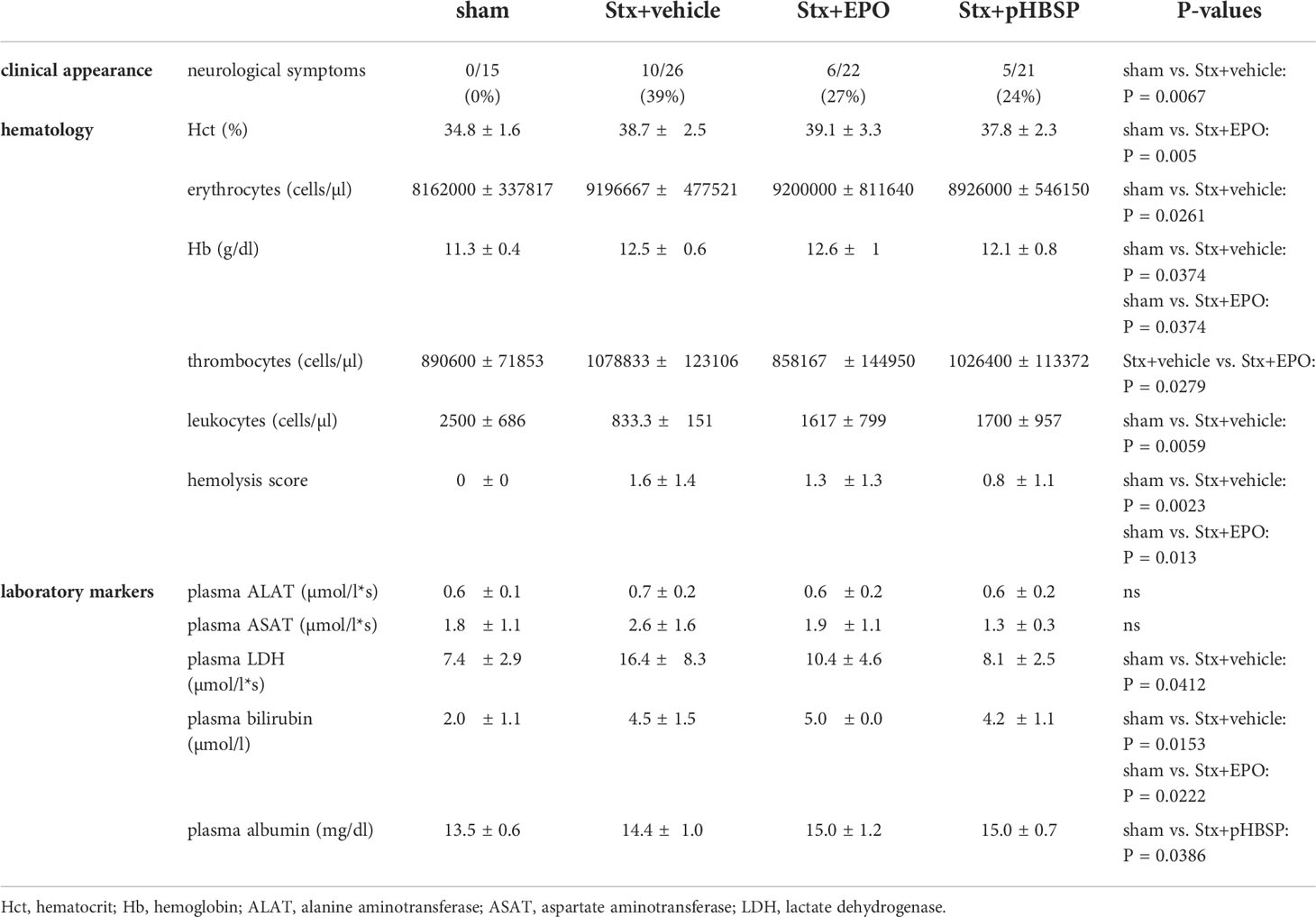
Table 3 Hematological and laboratory parameters of mice with HUS and effects of EPO and pHBSP treatment.
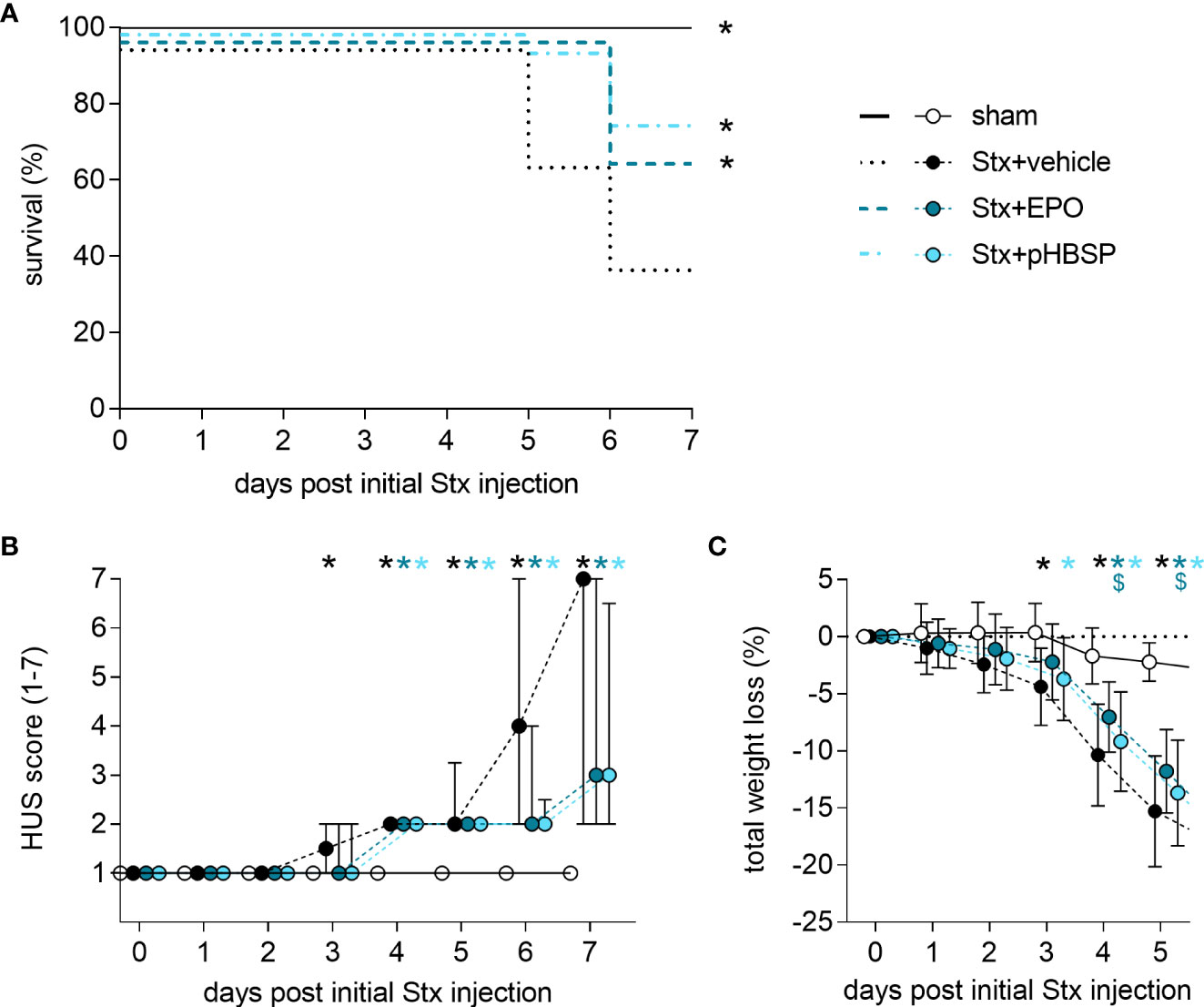
Figure 3 Effect of EPO and pHBSP treatment on survival and clinical presentation of mice with HUS. (A-C) Mice received either Stx or vehicle intravenously to induce experimental HUS. Mice with HUS were treated with vehicle (Ringer’s solution 1 h post HUS induction s.c.), EPO (1 h post HUS induction s.c., 1000 IU/kg BW once) or pHBSP (1 h post HUS induction s.c., 30 µg/kg BW every 24 h; sham n = 15, Stx+vehicle n = 26, Stx+EPO n = 22, Stx+pHBSP n = 21). (A) Kaplan-Meier 7-day survival curves of sham mice, Stx-challenged mice treated with vehicle, Stx-challenged mice treated with EPO and Stx-challenged mice treated with pHBSP with humane endpoints. *P < 0.05 vs. Stx+vehicle (log-rank Mantel-Cox test). (B) Activity-based HUS score (ranging from 1-normally active, 2-active with slight restrictions, 3-active with clear intermissions, 4-slowed, 5-lethargic, 6-moribund, to 7-dead) was assessed three times daily and is depicted every 24 h until day 7 starting with first Stx injection. Data are expressed as median ± interquartile range. *P < 0.05 vs. sham (Kruskal-Wallis test, Dunn’s multiple comparison test at every single time point). (C) Total weight loss referred to day 0 was assessed every 24 h and is depicted until day 5. Data are expressed as mean ± SD. *P < 0.05 sham vs. the respective color-coded group, $P < 0.05 vs. Stx+vehicle (two-way ANOVA, Holm-Sidak’s multiple comparison test).
After observing significantly increased survival rates in EPO- and pHBSP-treated animals, we analyzed the impact of both treatments on kidney injury and dysfunction. Stx+vehicle, Stx+EPO and Stx+pHBSP mice presented with increased plasma urea, creatinine and NGAL compared with sham mice (Figures 4A–C). PAS staining revealed renal tissue damage in all Stx-challenged groups irrespective of treatment (Figure 4D). KIM-1 expression was elevated in Stx+vehicle, Stx+EPO and Stx+pHBSP mice compared with sham mice (Figure 4E). In Stx+pHBSP compared with Stx+vehicle mice, KIM-1 expression was reduced (Figure 4E).
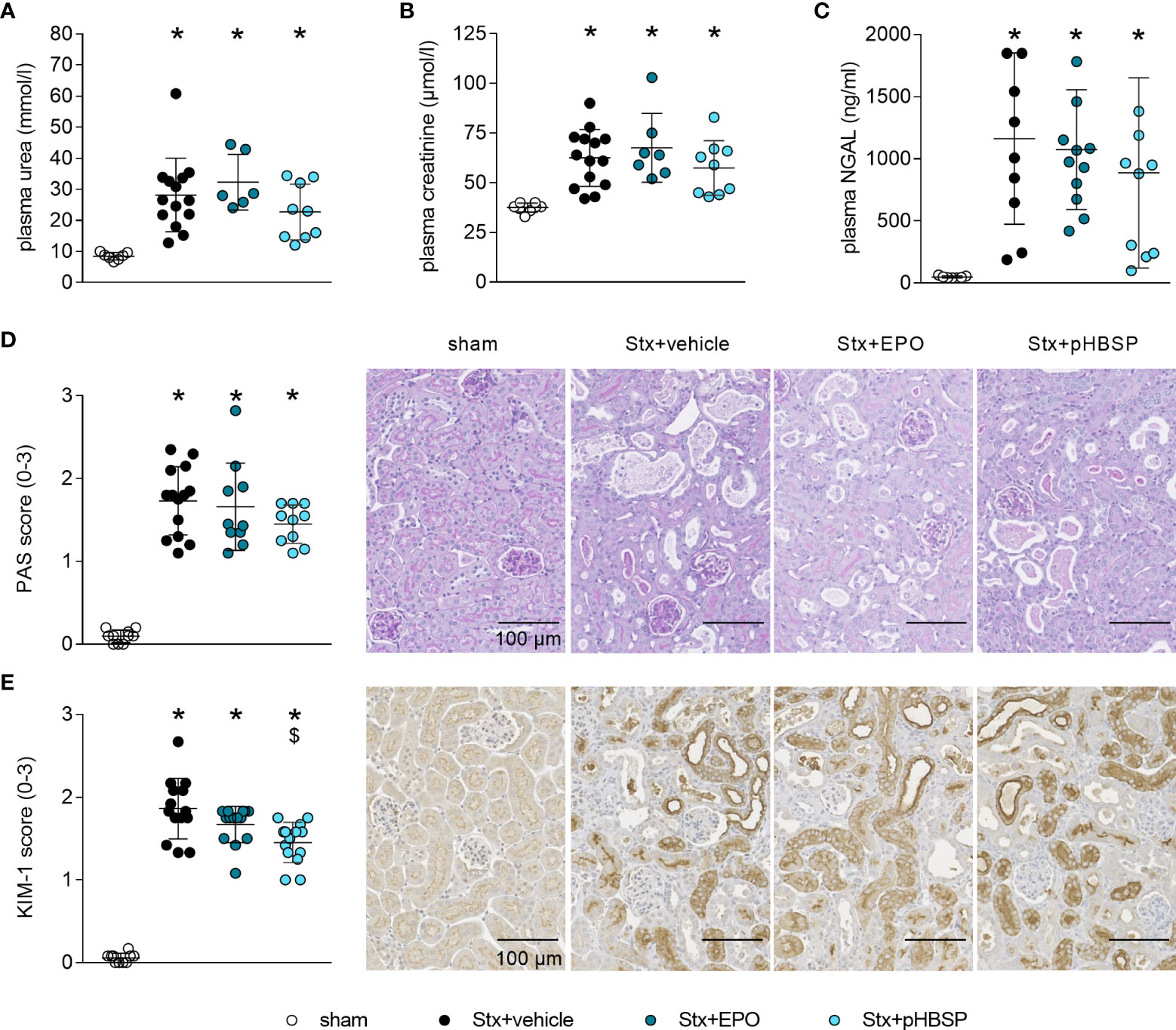
Figure 4 Effect of EPO and pHBSP treatment on kidney dysfunction and tubular injury in mice with HUS. Kidney injury and dysfunction was assessed on day 7 of HUS experiment. Plasma samples of mice with HUS and sham mice were analyzed for the kidney dysfunction parameters (A) urea and (B) creatinine (sham n = 7, Stx+vehicle n = 14, Stx+EPO n = 6, Stx+pHBSP n = 9) by an Architect™ ci16200 System (Abbott), as well as the kidney injury marker (C) NGAL (sham n = 7, Stx+vehicle n = 10, Stx+EPO n = 12, Stx+pHBSP n = 10) by ELISA. (A-C) Data are expressed as scatter dot plot with mean ± SD. *P < 0.05 vs. sham (one-way ANOVA, Holm-Sidak’s multiple comparison test). Quantification data as well as representative images (scale bar 100 µm) of (D) PAS staining (sham n = 10, Stx+vehicle n = 14, Stx+EPO n = 10, Stx+pHBSP n = 10, 10 fields per slide, score 0: no damage, 1: < 25% damaged, 2: 25–50% damaged, 3: > 50% damaged) and (E) immunohistochemical KIM-1 staining (score 0: < 25%, 1: 25–50%, 2: 50–75%, 3: > 75% strong positive staining per visual field, sham n = 10, Stx+vehicle n = 14, Stx+EPO n = 14, Stx+pHBSP n = 14, 12 fields per slide) in renal sections of sham and Stx-challenged mice. (D, E) Data are expressed as scatter dot plot with mean ± SD. *P < 0.05 vs. sham, $P < 0.05 vs. Stx+vehicle, (Kruskal-Wallis test, Dunn’s multiple comparison test).
The integrity of endothelial and epithelial barriers in the kidney is crucial for its function. It was demonstrated before that Stx can damage both endothelial as well as epithelial cells in the kidney and thereby influence the integrity of these barriers (1). We stained kidney sections for endothelial and epithelial cell markers to assess the influence of EPO and pHBSP treatment on intrarenal barriers. Loss of renal CD31-positive endothelial cells (Figure 5A) and reduced E-cadherin expression was detected in kidneys of all Stx-challenged groups compared with sham mice (Figure 5B). However, E-cadherin expression was less decreased in Stx+pHBSP compared with Stx+vehicle and Stx+EPO mice (Figure 5B).
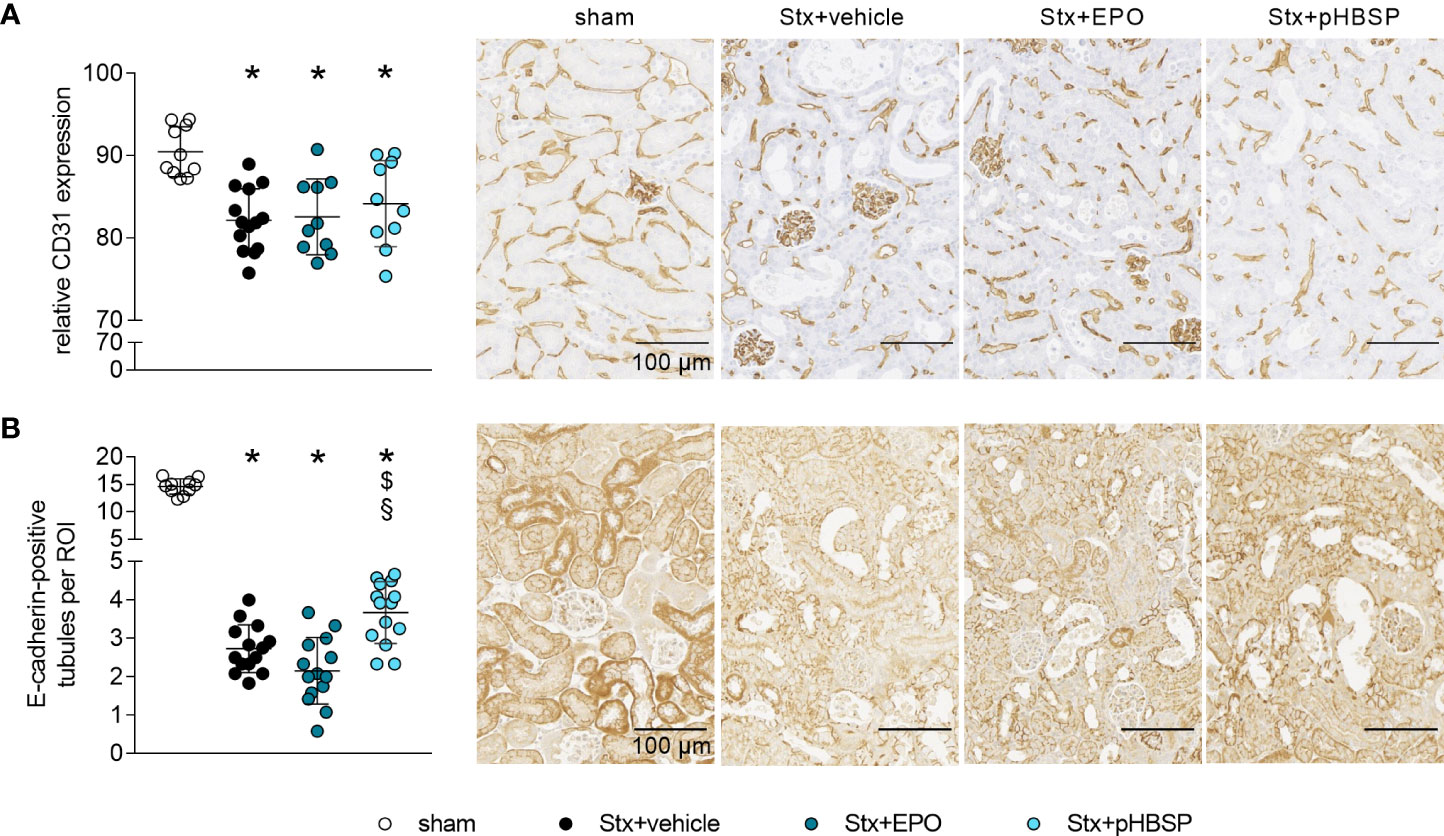
Figure 5 Effect of EPO and pHBSP treatment on intrarenal barriers in mice with HUS. Intrarenal barriers were assessed on day 7 of HUS experiment. Quantification data as well as representative images (scale bar 100 µm) of immunohistochemical (A) CD31 staining (sham n = 10, Stx+vehicle n = 14, Stx+EPO n = 10, Stx+pHBSP n = 10) and (B) E-cadherin (sham n = 10, Stx+vehicle n = 14, Stx+EPO n = 14, Stx+pHBSP n = 14), in renal sections of sham mice and mice subjected to Stx. (A, B) Data are expressed as scatter dot plot with mean ± SD. *P < 0.05 vs. sham, $P < 0.05 vs. Stx+vehicle, §P < 0.05 vs. Stx+EPO (one-way ANOVA, Holm-Sidak’s multiple comparison test).
GP1b was stained to highlight thrombocytes in the kidneys as indicators of microangiopathy (Figure 6A). An insignificant elevation of thrombocytes was observed in all Stx-challenged animals regardless of treatment, although it was weakest in the Stx+pHBSP group (Figure 6A). SFOG staining was performed to visualize fibrin deposits as indicators of microthrombi in renal sections (Figure 6B). Fibrin deposits were observed in all Stx-challenged groups compared with sham animals. There was an insignificant trend towards lower mean values of fibrin deposition in Stx+EPO and Stx+pHBSP mice compared with Stx+vehicle mice (Figure 6B).
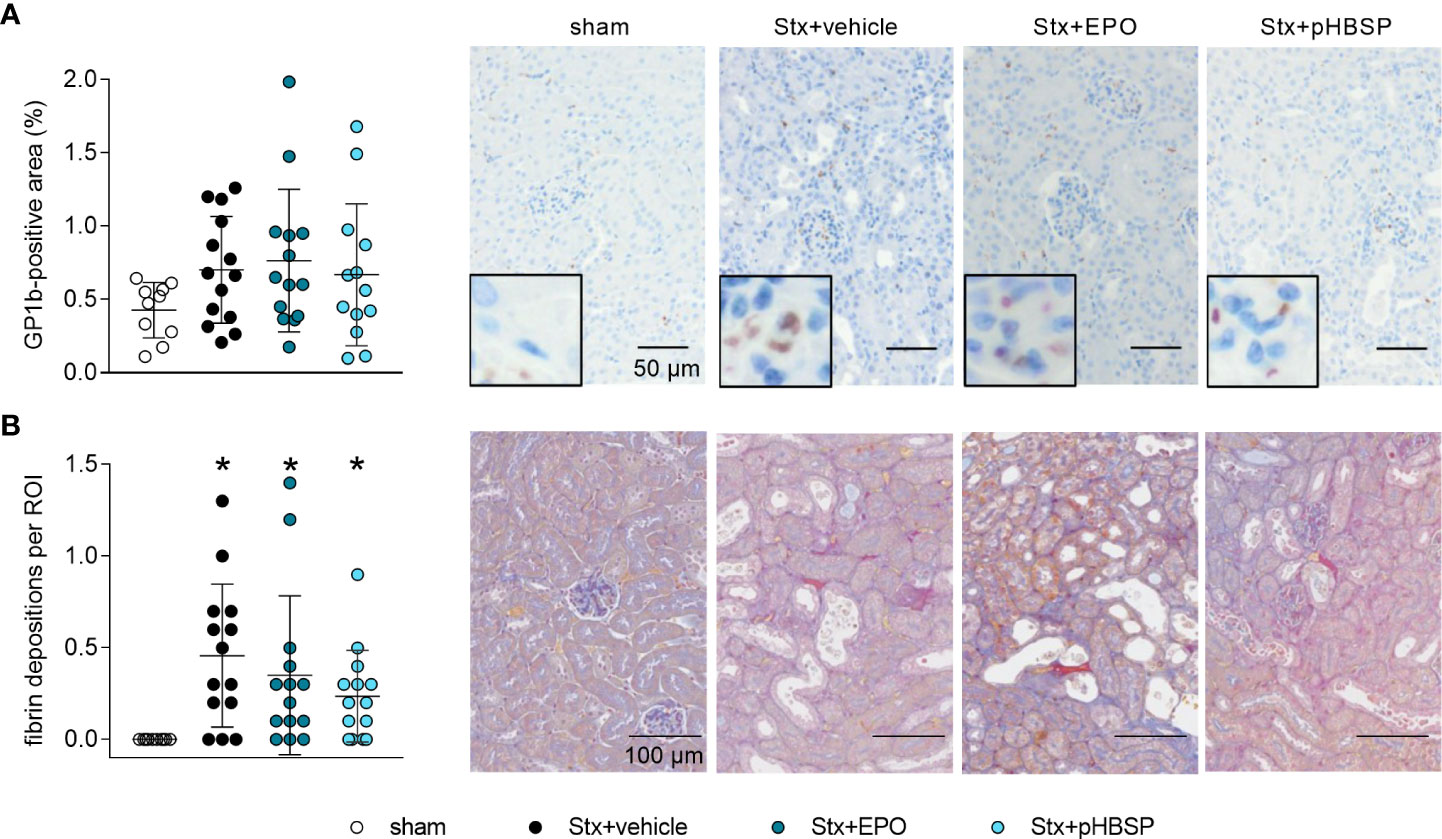
Figure 6 Effects of EPO and pHBSP treatment on microangiopathy in mice with HUS. Thrombocytes (indicated by glycoprotein 1b (GP1b) staining) and renal fibrin depositions (indicated by acid fuchsin orange G (SFOG) staining) were assessed on day 7 of HUS experiment. Quantification data and representative images of (A) immunohistochemical staining of thrombocytes (sham n = 10, Stx+vehicle n = 14, Stx+EPO n = 14, Stx+pHBSP n = 13) and (B) fibrin depositions (sham n = 10, Stx+vehicle n = 14, Stx+EPO n = 14, Stx+pHBSP n = 14) in kidney sections of sham mice and Stx-challenged mice. Quantitative data are expressed as scatter dot plot with mean ± SD. *P < 0.05 vs. sham (Kruskal-Wallis test, Dunn’s multiple comparison test), scale bar 100 µm.
To further characterize the potential protective mechanism of EPO and pHBSP in HUS, surrogate parameters of nitrosative (nitrotyrosin formation) and oxidative (NOX-1 expression) stress were analyzed. Renal nitrotyrosine formation was elevated in all Stx-challenged groups compared with sham mice (Figure 7A). Compared with Stx+vehicle mice, formation of nitrotyrosine was decreased in the kidneys of Stx+EPO and Stx+pHBSP mice (Figure 7A). Compared with Stx+EPO mice, nitrotyrosine staining was even further decreased in Stx+pHBSP mice (Figure 7A). Of note, the predominantly glomerular staining of nitrotyrosine in Stx+vehicle mice was mitigated in Stx+EPO mice and attenuated in Stx+pHBSP mice (Figure 7A, arrowheads). NOX-1 expression was increased only in Stx+vehicle compared with sham mice, whereas Stx+pHBSP mice displayed lower NOX-1 expression compared with Stx+vehicle and Stx+EPO mice (Figure 7B).
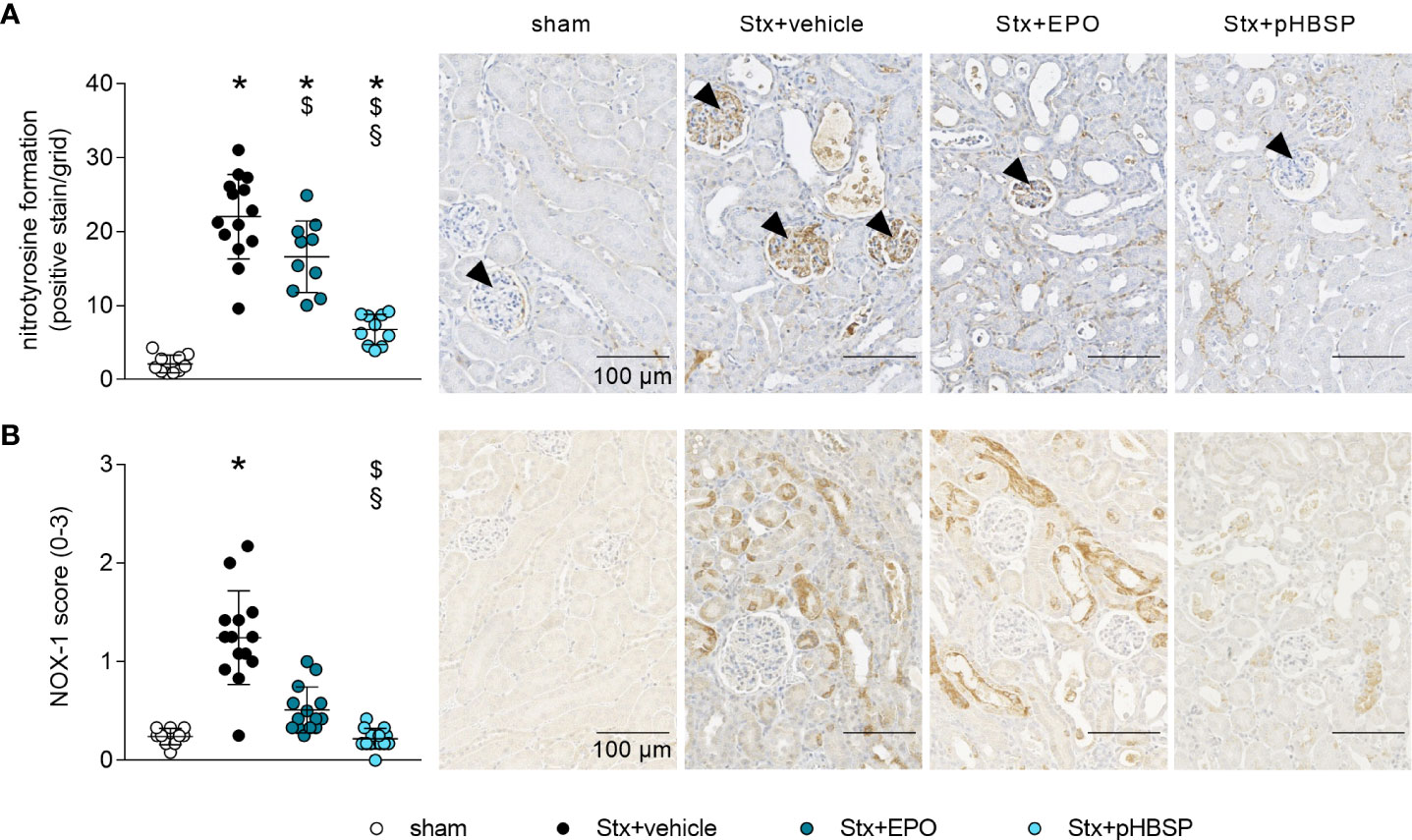
Figure 7 Effects of EPO and pHBSP treatment on nitrosative and oxidative stress in mice with HUS. Nitrosative and oxidative stress were assessed on day 7 of HUS experiment. Quantification data and representative images of immunohistochemical staining of (A) nitrotyrosine (sham n = 10, Stx+vehicle n = 14, Stx+EPO n = 10, Stx+pHBSP n = 10, arrowheads indicating glomeruli) and (B) NOX-1 (sham n = 10, Stx+vehicle n = 14, Stx+EPO n = 14, Stx+pHBSP n = 14) in kidney sections of sham mice and Stx-challenged mice. Quantitative data are expressed as scatter dot plot with mean ± SD. *P < 0.05 vs. sham, $P < 0.05 vs. Stx+vehicle, §P < 0.05 vs. Stx+EPO (A: one-way ANOVA, Holm-Sidak’s multiple comparison test, B: Kruskal-Wallis test, Dunn’s multiple comparison test), scale bar 100 µm.
Oxidative stress and metabolism are closely interrelated and studies on the metabolome have not yet been reported in animal models of HUS. Therefore, we performed targeted metabolomics in the plasma of all four groups. Of the 630 metabolites analyzed in plasma, 426 were within the limit of detection. These 426 metabolites were analyzed using a linear regression model to test whether their abundance increases or decreases in the assigned order: sham–Stx+pHBSP–Stx+EPO–Stx+vehicle. 32 metabolites fitted this hypothesis, they are highlighted in heatmaps (Figures 8A, B) and assigned to the following substance classes: amino acids and derivatives (7/32), alkaloids (1/32), aminoxides (1/32, heatmap A) and lipids (23/32, triacylglycerides in heatmap B).
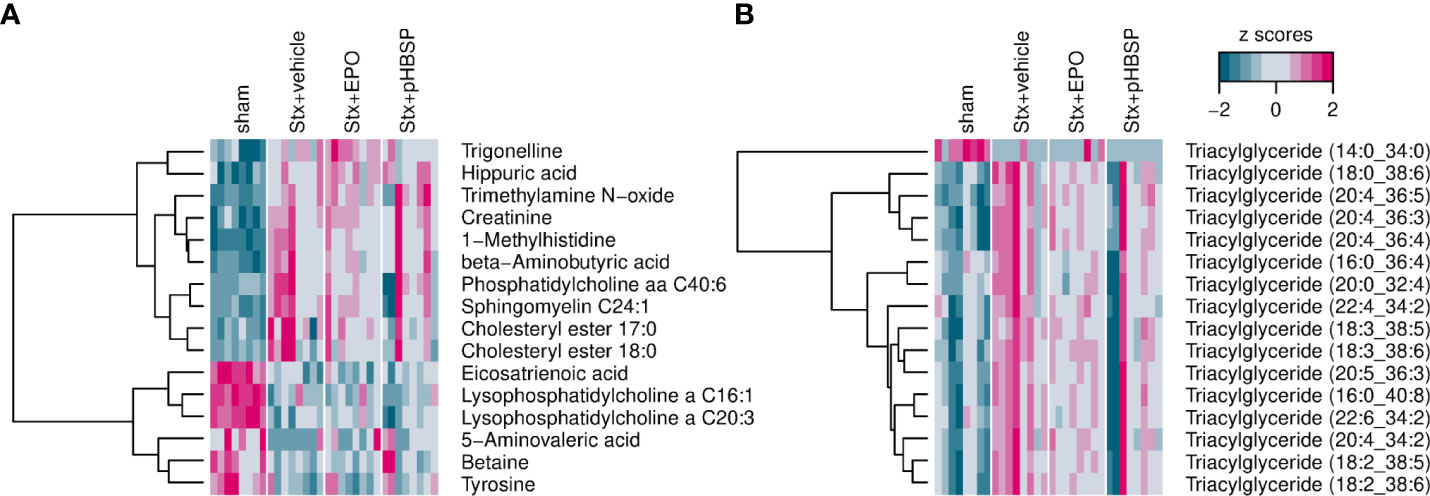
Figure 8 Effect of EPO and pHBSP treatment on plasma metabolome in mice with HUS. Metabolites in plasma of sham mice and Stx-challenged mice were assessed on day 7 of HUS experiment. Statistic modelling was performed to test the hypothesis that their abundance either increases or decreases in the following order: sham–Stx+pHBSP–Stx+EPO–Stx+vehicle (A) Small metabolites in murine plasma showing a significant trend for the four groups under investigation. (B) Triacylglyceroles in murine plasma showing a significant trend for the four groups under investigation. Data are shown as heatmaps depicting z-scores for all samples. The analytes were hierarchically clustered using Ward’s minimum variance method (45) and an euclidian distance between z scores. Dendograms provide information about distances between clusters.
To date, the role of EPO in HUS, a systemic orphan disease with occurrence of microangiopathic hemolytic anemia and AKI, has not been systematically investigated. We hypothesized that EPO treatment may be beneficial in patients with HUS-mediated hemolytic anemia and, targeting the IRR axis with EPO or non-hematopoietic EPO analogs, such as pHBSP, may convey nephroprotection in HUS.
In response to cellular hypoxia (46), a regulatory increase in renal EPO secretion in patients with anemia would be expected. To our knowledge, there are no systematic studies on changes in endogenous EPO levels in adult patients with STEC-HUS and only one brief report in pediatric patients (32). We observed increased EPO serum levels in a small cohort of adult patients with STEC-HUS compared to healthy controls already at a reduced Hb level of about 11 g/dl. Our data showed no correlation between the degree of anemia and EPO levels. This observation is consistent with the above-mentioned report in children with STEC-HUS (32) and in critically ill patients with AKI of other origins (47–50). However, we cannot exclude a gender bias, as our patient cohort was mainly female as women were more frequently affected in the 2011 EHEC outbreak (51). Therefore, our observations should be validated in a larger, gender balanced cohort.
In the kidneys, EPO is produced by fibroblast-like type I interstitial cells located between peritubular capillaries and the proximal convoluted tubule (52). Thus, EPO expression is likely to be impaired when EPO-producing cells are damaged. Consistent with these considerations, patients with very high LDH or creatinine levels as surrogate parameters for tissue damage and renal dysfunction had the lowest EPO levels in our study. However, we found no correlation between LDH or creatinine levels and EPO levels in patients with HUS. We attribute this to the small number of patients studied. Nevertheless, clinical data suggests that EPO response appears to be impaired in STEC-HUS and EPO regulation may play an important role in the pathogenesis and disease resolution. Analyzing serum EPO levels in a larger cohort of patients with HUS and acute kidney injury of other origins may help to further elucidate whether the degree of renal dysfunction and/or injury correlates with the endogenous EPO production. Here, we pursued our line of thought by further conducting studies in animals with experimental HUS.
In gnotobiotic piglets with EHEC infection (9) and in mice after Stx challenge, we found an increase in EPO expression despite an absence of anemia in these animal models. We and others reported earlier that the extent of systemic hemolysis and subsequent anemia in experimental HUS is not as pronounced as in patients with HUS (9, 10, 14, 53, 54).
Based on pathophysiological considerations and evidence, these findings could imply that EPO expression in HUS does not increase proportionally to the extent of anemia but rather depends on the degree of renal hypoxia as first mechanistically described by Semenza et al. (55, 56). Microthrombotic occlusion in the kidneys can result in local hypoxia increasing the expression of the master regulator hypoxia-induced factor 1α (HIF-1α) and thereby EPO (57). However, direct damage to the EPO-expressing cells in the kidneys of HUS patients with hemolytic anemia may, especially in cases with severe renal injury, also result in insufficient EPO production to adequately stimulate hematopoiesis. Thus, patients with severe renal injury could benefit from treatment with exogenous EPO (58, 59).
In our experimental setting, we observed a survival benefit in mice with HUS treated with either EPO or pHBSP. Tissue-protective effects of EPO and non-hematopoietic analogs have been the subject of discussion in the literature for nearly two decades (15, 16). Despite the observed beneficial effects of EPO in preclinical models of endotoxemia, sepsis, hemorrhagic shock and ischemia-reperfusion induced AKI (60–63), EPO failed to convey nephroprotection in several clinical trials that included patients following cardiac surgery (64, 65), cardiac arrest (66), kidney-transplantation (67) as well as ICU patients at risk for the development of AKI (EARLYARF trial) (68). However, in a small pilot trial with patients suffering from AKI after coronary artery bypass grafting surgery, treatment with EPO had a beneficial effect on all-cause mortality (69). Corwin et al. reported that administration of high-dose EPO (40,000 IU) to intensive care patients reduced the number of red blood cell transfusions without affecting mortality or clinical outcome (27). In a follow-up study of ICU patients, a subcohort of trauma patients treated with high-dose EPO showed no reduced need for red blood cell transfusions, however mortality was significantly reduced (18). Notably, an increase in thrombotic vascular events was noted in ICU patients who had not received thromboprophylaxis at baseline (18).
In the subset of mice surviving up to day 7 of the HUS experiments, we further investigated surrogate parameters for renal dysfunction and injury as well as barrier integrity, microangiopathy, oxidative and nitrosative stress and metabolome in plasma and/or renal tissue samples. As we have observed previously that AKI is accompanied by electrolyte imbalances, we propose that, in this model, mice die due to a severe AKI-induced hyperkalemia with subsequent cardiac arrhythmias and cardiac arrest (unpublished data). As fewer animals survived up to day 7 in the Stx+vehicle group (11/26, 42.3% survival) compared with the Stx+EPO (15/22, 68.2% survival) and Stx+pHBSP (16/21, 76.2% survival) group, the results need to be carefully interpreted in the light of a reverse survivorship bias, that might explain why we did not observe an effect of treatment with EPO or pHBSP on plasma creatinine, urea and NGAL, as surrogate parameters for AKI and renal dysfunction. However, we observed significant effects of EPO and/or pHBSP treatment on barrier integrity, oxidative and nitrosative stress and on selected metabolites in the samples studied.
Intact renal endothelial and epithelial barriers are important for the physiological function of the kidneys. We observed a pronounced Stx-induced damage of renal endothelial cells independent of treatment. Treatment with pHBSP, but not EPO led to a decrease in KIM-1 and an increase in E-cadherin expression in Stx-challenged mice in our setting. This could indicate less renal damage and enhanced epithelial barrier integrity, as tubular dedifferentiation – an important step in AKI progression – is characterised by a downregulation of E-cadherin (70). Although not resulting in amelioration of AKI in our model, this could have contributed to the increased survival observed after pHBSP treatment in Stx-challenged mice. In line with our results, the pharmaceutical upregulation of E-cadherin expression also proved nephroprotective in preclinical studies of cis-platin-induced AKI (71, 72).
In our recent study, we observed survival benefits in animals with reduced microangiopathy (11). Microangiopathy is a pathophysiological hallmark of HUS. As EPO can exert pro-thrombogenic effects, we consequently assessed the effects of EPO or pHBSP treatment on fibrin deposition and thrombocytes as surrogate parameters of microangiopathy to evaluate the potential of adverse treatment effects in HUS. Notably, we did not observe an aggravation of microthrombi formation or fibrin deposition in mice with HUS treated with either EPO or pHBSP.
Oxidative stress is involved in the pathogenesis of STEC-HUS (3–5). Nitrosative stress resembles a subtype of oxidative stress that has been shown to contribute to cell death (73). We observed an intense glomerular staining of nitrotyrosine in Stx+vehicle mice. Nitrotyrosine results from the reaction of the peroxynitrite radical – created when excess nitric oxide (NO) and superoxide radicals combine (74) – with accessible tyrosine residues (75). While basal NO plays a critical role in the regulation of the perfusion and vascular tone in glomeruli (74), it has been shown previously that inducible NO synthase (iNOS) induction and overproduction of NO had deleterious effects in ischemic AKI (76). In mice, iNOS is – among others – expressed in macrophages and glomerular mesangial cells and inducible by various pro-inflammatory stimuli (77). EPO and pHBSP have been demonstrated to reduce the expression of iNOS either on protein level in AKI of different etiologies (63, 78) or on mRNA level in lung and brain injury (79, 80). Thus, the reduction in renal nitrotyrosine staining in Stx+EPO and Stx+pHBSP mice could result from an inhibition of iNOS in the glomeruli of these mice.
Due to its critical role in the reabsorption of nutrients and maintenance of homeostasis, energy demand and metabolic activity are high in the kidneys (81). By analyzing the plasma metabolome, we observed an increase in triglycerides and cholesteryl esters in Stx-challenged mice that was consistently reported in HUS patients (82). Notably, we observed that alterations in some metabolites associated with kidney injury, e. g. trimethylamine N-oxide (83, 84) and trigonelline (85, 86) as well as lipid compounds were less distinct in mice with HUS after EPO or pHBSP treatment. In line with our results, a recent study demonstrated that EPO reduces lipidemia by stimulating lipid catabolism in peripheral adipose tissue (87). Further studies are required to examine the complex metabolic changes in animals and patients with HUS.
Taken together, we found that treatment with EPO or pHBSP positively affects several pathomechanisms of STEC-HUS which might explain the observed improvement in clinical outcome of mice with HUS. Further systematic studies in this context in animal models and even more importantly larger patient cohorts are needed to provide sufficient evidence to adjust clinical management.
We report here for the first time, that I) EPO levels in patients with STEC-HUS, STEC-infected piglets and Stx-challenged mice are elevated in a uniform manner, II) EPO or pHBSP treatment of mice with HUS improves survival and disease outcome, III) protective effects of pHBSP and EPO are associated with reduced renal oxidative stress, and IV) treatment with pHBSP in Stx-challenged mice is additionally associated with ameliorated nitrosative stress, less KIM-1 expression and tubular dedifferentiation. In the light of our results demonstrating favourable tissue-protective effects of EPO in a preclinical model, treatment of HUS-induced hemolytic anemia with EPO should be considered in patients. Further studies are needed to evaluate the effect of EPO and pHBSP treatment in clinical studies in patients with STEC-HUS.
The metabolomics dataset presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ebi.ac.uk/biostudies/BioStudies accession number S-BSST657. Original data can be requested from c2luYS5jb2xkZXdleUBtZWQudW5pLWplbmEuZGU=.
The studies involving human participants were reviewed and approved by Ethics Committee of Hannover Medical School (1123-2011) and Ethics Committee of Friedrich Schiller University Jena (5276-09/17). The patients/participants provided their written informed consent to participate in this study. The animal studies were reviewed and approved by the Lower Saxony State Office for Consumer Protection and Food Safety (Approval Number: 33.9-42502-04-13/1149) and the regional animal welfare committee and the Thuringian State Office for Consumer Protection (registration number 02-058/14).
SC designed and planned the study; planned, performed and analyzed experiments and wrote the manuscript. SD planned, performed and analyzed experiments and wrote the manuscript. WP planned and performed histology and immunohistochemistry and analyzed data. BW planned and performed animal experiments. FG provided purified Stx and gnotobiotic piglet samples and contributed important intellectual content. DI synthesized and purified pHBSP and contributed important intellectual content. CD and KA performed histology and immunohistochemistry and contributed important intellectual content to histology and immunohistochemistry. JK provided HUS patient samples. IH-P provided gnotobiotic piglet samples. All authors provided important intellectual content and revised the manuscript prior to submission.
The research leading to these results has received funding from the German Research Foundation (DFG; Research Unit FOR1738, grant no. CO912/2-1 to SC and IM97/7-2 to DI) and the Federal Ministry of Education and Research (BMBF; ZIK Septomics Research Center, Translational Septomics, grant no. 03Z22JN12 to SC and Center for Sepsis Control and Care, project TaSep, grant no. 01EO1502 to SC).
We would like to thank J. Fischer (Septomics Research Center, Jena) and Dr. H. H. Brewitz (University of Bonn) for technical assistance, Prof. Dr. J. Menne (KRH Klinikum Siloah, Hannover) for provision of HUS patient samples and D. Driesch (BioControl) for support in metabolomics data analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1010882/full#supplementary-material
1. Joseph A, Cointe A, Mariani Kurkdjian P, Rafat C, Hertig A. Shiga toxin-associated hemolytic uremic syndrome: A narrative review. Toxins (Basel) (2020) 12(2):32. doi: 10.3390/toxins12020067
2. Spinale JM, Ruebner RL, Copelovitch L, Kaplan BS. Long-term outcomes of shiga toxin hemolytic uremic syndrome. Pediatr Nephrol (2013) 28:2097–105. doi: 10.1007/s00467-012-2383-6
3. Ferraris V, Acquier A, Ferraris JR, Vallejo G, Paz C, Mendez CF. Oxidative stress status during the acute phase of haemolytic uraemic syndrome. Nephrol Dial Transplant (2011) 26:858–64. doi: 10.1093/ndt/gfq511
4. Kanzelmeyer NK, Pape L, Chobanyan-Jurgens K, Tsikas D, Hartmann H, Fuchs AJ, et al. L-arginine/NO pathway is altered in children with haemolytic-uraemic syndrome (HUS). Oxid Med Cell Longev (2014) 2014:203512. doi: 10.1155/2014/203512
5. Gomez SA, Abrey-Recalde MJ, Panek CA, Ferrarotti NF, Repetto MG, Mejias MP, et al. The oxidative stress induced in vivo by shiga toxin-2 contributes to the pathogenicity of haemolytic uraemic syndrome. Clin Exp Immunol (2013) 173:463–72. doi: 10.1111/cei.12124
6. Adamski J. Thrombotic microangiopathy and indications for therapeutic plasma exchange. Hematol Am Soc Hematol Educ Program (2014) 2014:444–9. doi: 10.1182/asheducation-2014.1.444
7. RKI. Infektionsepidemiologisches jahrbuch meldepflichtiger krankheiten für 2020. Robert Koch Institut (2020). Available at: https://www.rki.de/DE/Content/Infekt/Jahrbuch/Jahrbuch_2020.html.
8. Ministerio de Salud. Boletín integrado de vigilancia N560 SE30 2021 (2021). Available at: https://bancos.salud.gob.ar/recurso/boletin-integrado-de-vigilancia-n560-se30-2021.
9. Wöchtl B, Gunzer F, Gerner W, Gasse H, Koch M, Bago Z, et al. Comparison of clinical and immunological findings in gnotobiotic piglets infected with escherichia coli O104:H4 outbreak strain and EHEC O157:H7. Gut Pathog (2017) 9:30. doi: 10.1186/s13099-017-0179-8
10. Dennhardt S, Pirschel W, Wissuwa B, Daniel C, Gunzer F, Lindig S, et al. Modeling hemolytic-uremic syndrome: In-depth characterization of distinct murine models reflecting different features of human disease. Front Immunol (2018) 9:1459. doi: 10.3389/fimmu.2018.01459
11. Pirschel W, Mestekemper AN, Wissuwa B, Krieg N, Kroller S, Daniel C, et al. Divergent roles of haptoglobin and hemopexin deficiency for disease progression of shiga-toxin-induced hemolytic-uremic syndrome in mice. Kidney Int (2022) 101(6):1171–85. doi: 10.1016/j.kint.2021.12.024
12. Sobbe IV, Krieg N, Dennhardt S, Coldewey SM. Involvement of NF-kappaB1 and the non-canonical NF-kappaB signaling pathway in the pathogenesis of acute kidney injury in shiga-toxin-2-induced hemolytic-uremic syndrome in mice. Shock (2021) 56:573–81. doi: 10.1097/SHK.0000000000001558
13. Keepers TR, Psotka MA, Gross LK, Obrig TG. A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J Am Soc Nephrol (2006) 17:3404–14. doi: 10.1681/ASN.2006050419
14. Sauter KA, Melton-Celsa AR, Larkin K, Troxell ML, O’Brien AD, Magun BE. Mouse model of hemolytic-uremic syndrome caused by endotoxin-free shiga toxin 2 (Stx2) and protection from lethal outcome by anti-Stx2 antibody. Infect Immun (2008) 76:4469–78. doi: 10.1128/IAI.00592-08
15. Brines M, Patel NS, Villa P, Brines C, Mennini T, De Paola M, et al. Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc Natl Acad Sci United States America (2008) 105:10925–30. doi: 10.1073/pnas.0805594105
16. Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci United States America (2004) 101:14907–12. doi: 10.1073/pnas.0406491101
17. Pearl RG. Erythropoietin and organ protection: lessons from negative clinical trials. Crit Care (2014) 18:526. doi: 10.1186/s13054-014-0526-9
18. Corwin HL, Gettinger A, Fabian TC, May A, Pearl RG, Heard S, et al. Efficacy and safety of epoetin alfa in critically ill patients. N Engl J Med (2007) 357:965–76. doi: 10.1056/NEJMoa071533
19. Ehrenreich H, Weissenborn K, Prange H, Schneider D, Weimar C, Wartenberg K, et al. Recombinant human erythropoietin in the treatment of acute ischemic stroke. Stroke (2009) 40:e647–56. doi: 10.1161/STROKEAHA.109.564872
20. Collino M, Thiemermann C, Cerami A, Brines M. Flipping the molecular switch for innate protection and repair of tissues: Long-lasting effects of a non-erythropoietic small peptide engineered from erythropoietin. Pharmacol Ther (2015) 151:32–40. doi: 10.1016/j.pharmthera.2015.02.005
21. Netrebenko AS, Gureev VV, Pokrovskii MV, Gureeva AV, Tsuverkalova YM, Rozhkov IS. Assessment of the nephroprotective properties of the erythropoietin mimetic peptide and infliximab in kidney ischemia-reperfusion injury in rats. Arch Razi Inst (2021) 76:995–1004. doi: 10.22092/ari.2021.355849.1728
22. Patel NS, Kerr-Peterson HL, Brines M, Collino M, Rogazzo M, Fantozzi R, et al. Delayed administration of pyroglutamate helix b surface peptide (pHBSP), a novel nonerythropoietic analog of erythropoietin, attenuates acute kidney injury. Mol Med (2012) 18:719–27. doi: 10.2119/molmed.2012.00093
23. Brines M, Dunne AN, van Velzen M, Proto PL, Ostenson CG, Kirk RI, et al. ARA 290, a nonerythropoietic peptide engineered from erythropoietin, improves metabolic control and neuropathic symptoms in patients with type 2 diabetes. Mol Med (2015) 20:658–66. doi: 10.2119/molmed.2014.00215
24. Dahan A, Dunne A, Swartjes M, Proto PL, Heij L, Vogels O, et al. ARA 290 improves symptoms in patients with sarcoidosis-associated small nerve fiber loss and increases corneal nerve fiber density. Mol Med (2013) 19:334–45. doi: 10.2119/molmed.2013.00122
25. Coldewey SM, Khan AI, Kapoor A, Collino M, Rogazzo M, Brines M, et al. Erythropoietin attenuates acute kidney dysfunction in murine experimental sepsis by activation of the beta-common receptor. Kidney Int (2013) 84:482–90. doi: 10.1038/ki.2013.118
26. Khan AI, Coldewey SM, Patel NS, Rogazzo M, Collino M, Yaqoob MM, et al. Erythropoietin attenuates cardiac dysfunction in experimental sepsis in mice via activation of the beta-common receptor. Dis Model Mech (2013) 6:1021–30. doi: 10.1242/dmm.011908
27. Corwin HL, Gettinger A, Pearl RG, Fink MP, Levy MM, Shapiro MJ, et al. Efficacy of recombinant human erythropoietin in critically ill patients: A randomized controlled trial. JAMA (2002) 288:2827–35. doi: 10.1001/jama.288.22.2827
28. MacLaren R, Gasper J, Jung R, Vandivier RW. Use of exogenous erythropoietin in critically ill patients. J Clin Pharm Ther (2004) 29:195–208. doi: 10.1111/j.1365-2710.2004.00552.x
29. Pasricha SR, Frazer DM, Bowden DK, Anderson GJ. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with beta-thalassemia major: A longitudinal study. Blood (2013) 122:124–33. doi: 10.1182/blood-2012-12-471441
30. Rasmussen SR, Kandler K, Nielsen RV, Jakobsen PC, Ranucci M, Ravn HB. Association between transfusion of blood products and acute kidney injury following cardiac surgery. Acta Anaesthesiol Scand (2020) 64:1397–404. doi: 10.1111/aas.13664
31. Schrezenmeier H, Noe G, Raghavachar A, Rich IN, Heimpel H, Kubanek B. Serum erythropoietin and serum transferrin receptor levels in aplastic anaemia. Br J Haematol (1994) 88:286–94. doi: 10.1111/j.1365-2141.1994.tb05020.x
32. Exeni R, Donato H, Rendo P, Antonuccio M, Rapetti MC, Grimoldi I, et al. Low levels of serum erythropoietin in children with endemic hemolytic uremic syndrome. Pediatr Nephrol (1998) 12:226–30. doi: 10.1007/s004670050443
33. Faquin WC, Schneider TJ, Goldberg MA. Effect of inflammatory cytokines on hypoxia-induced erythropoietin production. Blood (1992) 79:1987–94. doi: 10.1182/blood.V79.8.1987.1987
35. Deutsche Krebsgesellschaft, AWMF. Supportive therapie bei onkologischen PatientInnen - langversion 1.0 (2016). Available at: http://leitlinienprogramm-onkologie.de/Supportive-Therapie.95.0.html.
36. K.D.I.O.K.A.W. Group. KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney Inter Supplement (2012) pp:279–335. doi: 10.1038/kisup.2012.35
37. Gesellschaft für Pädiatrische Nephrologie e.V. (GPN), AWMF. Hämolytisch-urämisches syndrom im kindesalter (2022). Available at: https://www.awmf.org/leitlinien/detail/ll/166-002.html.
38. Balestracci A, Capone MA, Meni Battaglia L, Toledo I, Martin SM, Beaudoin L, et al. Erythropoietin in children with hemolytic uremic syndrome: A pilot randomized controlled trial. Pediatr Nephrol (2022) 37(10):2383–92. doi: 10.1007/s00467-022-05474-9
39. Balestracci A, Martin SM, Toledo I, Alvarado C, Wainsztein RE. Early erythropoietin in post-diarrheal hemolytic uremic syndrome: A case-control study. Pediatr Nephrol (2015) 30:339–44. doi: 10.1007/s00467-014-2911-7
40. Pape L, Ahlenstiel T, Kreuzer M, Drube J, Froede K, Franke D, et al. Early erythropoietin reduced the need for red blood cell transfusion in childhood hemolytic uremic syndrome: a randomized prospective pilot trial. Pediatr Nephrol (2009) 24:1061–4. doi: 10.1007/s00467-008-1087-4
41. Woo da E, Lee JM, Kim YK, Park YH. Recombinant human erythropoietin therapy for a jehovah’s witness child with severe anemia due to hemolytic-uremic syndrome. Korean J Pediatr (2016) 59:100–3. doi: 10.3345/kjp.2016.59.2.100
42. Kielstein JT, Beutel G, Fleig S, Steinhoff J, Meyer TN, Hafer C, et al. Best supportive care and therapeutic plasma exchange with or without eculizumab in shiga-toxin-producing e. coli O104:H4 induced haemolytic-uraemic syndrome: an analysis of the German STEC-HUS registry. Nephrol Dial Transplant (2012) 27:3807–15. doi: 10.1093/ndt/gfs394
43. Coldewey SM, Neu C, Baumbach P, Scherag A, Goebel B, Ludewig K, et al. Identification of cardiovascular and molecular prognostic factors for the medium-term and long-term outcomes of sepsis (ICROS): protocol for a prospective monocentric cohort study. BMJ Open (2020) 10:e036527. doi: 10.1136/bmjopen-2019-036527
44. Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med (1990) 9:811–8. doi: 10.1002/sim.4780090710
45. Murtagh F, Legendre P. Ward’s hierarchical agglomerative clustering method: Which algorithms implement ward’s criterion? J Classif (2014) 31:274–95. doi: 10.1007/s00357-014-9161-z
46. Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol (2020) 21:268–83. doi: 10.1038/s41580-020-0227-y
47. Matsuura R, Doi K, Komaru Y, Miyamoto Y, Yoshida T, Isegawa T, et al. Endogenous erythropoietin and hepatic dysfunction in acute kidney injury requiring renal replacement therapy. Nephron (2019) 142:10–6. doi: 10.1159/000496618
48. Morgera S, Heering P, Szentandrasi T, Niederau C, Grabensee B. Erythropoietin in patients with acute renal failure and continuous veno-venous haemofiltration. Int Urol Nephrol (1997) 29:245–50. doi: 10.1007/BF02551350
49. Tamion F, Le Cam-Duchez V, Menard JF, Girault C, Coquerel A, Bonmarchand G. Erythropoietin and renin as biological markers in critically ill patients. Crit Care (2004) 8:R328–35. doi: 10.1186/cc2902
50. Yamashita T, Noiri E, Hamasaki Y, Matsubara T, Ishii T, Yahagi N, et al. Erythropoietin concentration in acute kidney injury is associated with insulin-like growth factor-binding protein-1. Nephrol (Carlton) (2016) 21:693–9. doi: 10.1111/nep.12656
51. Frank C, Werber D, Cramer JP, Askar M, Faber M, an der Heiden M, et al. Epidemic profile of shiga-toxin-producing escherichia coli O104:H4 outbreak in Germany. N Engl J Med (2011) 365:1771–80. doi: 10.1056/NEJMoa1106483
52. Maxwell PH, Osmond MK, Pugh CW, Heryet A, Nicholls LG, Tan CC, et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int (1993) 44:1149–62. doi: 10.1038/ki.1993.362
53. Gunzer F, Hennig-Pauka I, Waldmann KH, Sandhoff R, Grone HJ, Kreipe HH, et al. Gnotobiotic piglets develop thrombotic microangiopathy after oral infection with enterohemorrhagic escherichia coli. Am J Clin Pathol (2002) 118:364–75. doi: 10.1309/UMW9-D06Q-M94Q-JGH2
54. Walsh PR, Johnson S. Treatment and management of children with haemolytic uraemic syndrome. Arch Dis Child (2018) 103:285–91. doi: 10.1136/archdischild-2016-311377
55. Semenza GL, Koury ST, Nejfelt MK, Gearhart JD, Antonarakis SE. Cell-type-specific and hypoxia-inducible expression of the human erythropoietin gene in transgenic mice. Proc Natl Acad Sci United States America (1991) 88:8725–9. doi: 10.1073/pnas.88.19.8725
56. Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc Natl Acad Sci United States America (1991) 88:5680–4. doi: 10.1073/pnas.88.13.5680
57. Corrado C, Fontana S. Hypoxia and HIF signaling: One axis with divergent effects. Int J Mol Sci 21 (2020) 21(66):5611. doi: 10.3390/ijms21165611
58. Fishbane S, Coyne DW. How I treat renal anemia. Blood (2020) 136:783–9. doi: 10.1182/blood.2019004330
59. Mikhail A, Brown C, Williams JA, Mathrani V, Shrivastava R, Evans J, et al. Renal association clinical practice guideline on anaemia of chronic kidney disease. BMC Nephrol (2017) 18:345. doi: 10.1186/s12882-017-0688-1
60. Heitrich M, Garcia DM, Stoyanoff TR, Rodriguez JP, Todaro JS, Aguirre MV. Erythropoietin attenuates renal and pulmonary injury in polymicrobial induced-sepsis through EPO-r, VEGF and VEGF-R2 modulation. BioMed Pharmacother (2016) 82:606–13. doi: 10.1016/j.biopha.2016.05.045
61. Kwak J, Kim JH, Jang HN, Jung MH, Cho HS, Chang SH, et al. Erythropoietin ameliorates Ischemia/Reperfusion-induced acute kidney injury via inflammasome suppression in mice. Int J Mol Sci 21 (2020) 21(10):3453. doi: 10.3390/ijms21103453
62. Ranjbaran M, Kadkhodaee M, Seifi B, Adelipour M, Azarian B. Erythropoietin attenuates experimental haemorrhagic shock-induced renal damage through an iNOS- dependent mechanism in male wistar rats. Injury (2017) 48:262–9. doi: 10.1016/j.injury.2017.01.010
63. Stoyanoff TR, Rodriguez JP, Todaro JS, Colavita JPM, Torres AM, Aguirre MV. Erythropoietin attenuates LPS-induced microvascular damage in a murine model of septic acute kidney injury. BioMed Pharmacother (2018) 107:1046–55. doi: 10.1016/j.biopha.2018.08.087
64. de Seigneux S, Ponte B, Weiss L, Pugin J, Romand JA, Martin PY, et al. Epoetin administrated after cardiac surgery: effects on renal function and inflammation in a randomized controlled study. BMC Nephrol (2012) 13:132. doi: 10.1186/1471-2369-13-132
65. Kim JH, Shim JK, Song JW, Song Y, Kim HB, Kwak YL. Effect of erythropoietin on the incidence of acute kidney injury following complex valvular heart surgery: a double blind, randomized clinical trial of efficacy and safety. Crit Care (2013) 17:R254. doi: 10.1186/cc13081
66. Guillemet L, Jamme M, Bougouin W, Geri G, Deye N, Vivien B, et al. Effects of early high-dose erythropoietin on acute kidney injury following cardiac arrest: exploratory post hoc analyses from an open-label randomized trial. Clin Kidney J (2020) 13:413–20. doi: 10.1093/ckj/sfz068
67. Sureshkumar KK, Hussain SM, Ko TY, Thai NL, Marcus RJ. Effect of high-dose erythropoietin on graft function after kidney transplantation: a randomized, double-blind clinical trial. Clin J Am Soc Nephrol (2012) 7:1498–506. doi: 10.2215/CJN.01360212
68. Endre ZH, Walker RJ, Pickering JW, Shaw GM, Frampton CM, Henderson SJ, et al. Early intervention with erythropoietin does not affect the outcome of acute kidney injury (the EARLYARF trial). Kidney Int (2010) 77:1020–30. doi: 10.1038/ki.2010.25
69. Oh SW, Chin HJ, Chae DW, Na KY. Erythropoietin improves long-term outcomes in patients with acute kidney injury after coronary artery bypass grafting. J Korean Med Sci (2012) 27:506–11. doi: 10.3346/jkms.2012.27.5.506
70. Zheng G, Lyons JG, Tan TK, Wang Y, Hsu TT, Min D, et al. Disruption of e-cadherin by matrix metalloproteinase directly mediates epithelial-mesenchymal transition downstream of transforming growth factor-beta1 in renal tubular epithelial cells. Am J Pathol (2009) 175:580–91. doi: 10.2353/ajpath.2009.080983
71. Gao L, Liu MM, Zang HM, Ma QY, Yang Q, Jiang L, et al. Restoration of e-cadherin by PPBICA protects against cisplatin-induced acute kidney injury by attenuating inflammation and programmed cell death. Lab Invest (2018) 98:911–23. doi: 10.1038/s41374-018-0052-5
72. Ni J, Hou X, Wang X, Shi Y, Xu L, Zheng X, et al. 3-deazaneplanocin a protects against cisplatin-induced renal tubular cell apoptosis and acute kidney injury by restoration of e-cadherin expression. Cell Death Dis (2019) 10:355. doi: 10.1038/s41419-019-1589-y
73. Wang F, Yuan Q, Chen F, Pang J, Pan C, Xu F, et al. Fundamental mechanisms of the cell death caused by nitrosative stress. Front Cell Dev Biol (2021) 9:742483. doi: 10.3389/fcell.2021.742483
74. Goligorsky MS, Brodsky SV, Noiri E. NO bioavailability, endothelial dysfunction, and acute renal failure: new insights into pathophysiology. Semin Nephrol (2004) 24:316–23. doi: 10.1016/j.semnephrol.2004.04.003
75. Korkmaz A, Kolankaya D. Inhibiting inducible nitric oxide synthase with rutin reduces renal ischemia/reperfusion injury. Can J Surg (2013) 56:6–14. doi: 10.1503/cjs.004811
76. Noiri E, Peresleni T, Miller F, Goligorsky MS. In vivo targeting of inducible NO synthase with oligodeoxynucleotides protects rat kidney against ischemia. J Clin Invest (1996) 97:2377–83. doi: 10.1172/JCI118681
77. Sharma K, Danoff TM, DePiero A, Ziyadeh FN. Enhanced expression of inducible nitric oxide synthase in murine macrophages and glomerular mesangial cells by elevated glucose levels: possible mediation via protein kinase c. Biochem Biophys Res Commun (1995) 207:80–8. doi: 10.1006/bbrc.1995.1156
78. Yang FL, Subeq YM, Chiu YH, Lee RP, Lee CJ, Hsu BG. Recombinant human erythropoietin reduces rhabdomyolysis-induced acute renal failure in rats. Injury (2012) 43:367–73. doi: 10.1016/j.injury.2011.11.013
79. Chen H, Luo B, Yang X, Xiong J, Liu Z, Jiang M, et al. Therapeutic effects of nonerythropoietic erythropoietin analog ARA290 in experimental autoimmune encephalomyelitis rat. J Neuroimmunol (2014) 268:64–70. doi: 10.1016/j.jneuroim.2014.01.006
80. Liu Y, Lu J, Wang X, Chen L, Liu S, Zhang Z, et al. Erythropoietin-derived peptide protects against acute lung injury after rat traumatic brain injury. Cell Physiol Biochem (2017) 41:2037–44. doi: 10.1159/000475434
81. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol (2017) 13:629–46. doi: 10.1038/nrneph.2017.107
82. Kaplan BS, Gale D, Ipp T. Hyperlipidemia in the hemolytic-uremic syndrome. Pediatrics (1971) 47:776–9. doi: 10.1542/peds.47.4.776
83. Sun G, Yin Z, Liu N, Bian X, Yu R, Su X, et al. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem Biophys Res Commun (2017) 493:964–70. doi: 10.1016/j.bbrc.2017.09.108
84. Tang WH, Wang Z, Kennedy DJ, Wu Y, Buffa JA, Agatisa-Boyle B, et al. Gut microbiota-dependent trimethylamine n-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ Res (2015) 116:448–55. doi: 10.1161/CIRCRESAHA.116.305360
85. Chihanga T, Ma Q, Nicholson JD, Ruby HN, Edelmann RE, Devarajan P, et al. NMR spectroscopy and electron microscopy identification of metabolic and ultrastructural changes to the kidney following ischemia-reperfusion injury. Am J Physiol Renal Physiol (2018) 314(2)F154–66. doi: 10.1152/ajprenal.00363.2017
86. Won AJ, Kim S, Kim YG, Kim KB, Choi WS, Kacew S, et al. Discovery of urinary metabolomic biomarkers for early detection of acute kidney injury. Mol Biosyst (2016) 12:133–44. doi: 10.1039/C5MB00492F
Keywords: erythropoietin, hemolytic-uremic syndrome, shiga toxin, mice, pyroglutamate helix B surface peptide, microangiopathic hemolytic anemia, thrombotic microangiopathy
Citation: Dennhardt S, Pirschel W, Wissuwa B, Imhof D, Daniel C, Kielstein JT, Hennig-Pauka I, Amann K, Gunzer F and Coldewey SM (2022) Targeting the innate repair receptor axis via erythropoietin or pyroglutamate helix B surface peptide attenuates hemolytic-uremic syndrome in mice. Front. Immunol. 13:1010882. doi: 10.3389/fimmu.2022.1010882
Received: 03 August 2022; Accepted: 30 August 2022;
Published: 23 September 2022.
Edited by:
Manuela Mengozzi, Brighton and Sussex Medical School, United KingdomReviewed by:
Adrien Joseph, Assistance Publique Hopitaux De Paris, FranceCopyright © 2022 Dennhardt, Pirschel, Wissuwa, Imhof, Daniel, Kielstein, Hennig-Pauka, Amann, Gunzer and Coldewey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sina M. Coldewey, c2luYS5jb2xkZXdleUBtZWQudW5pLWplbmEuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.