
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 10 January 2023
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1009484
This article is part of the Research TopicAdoptive Cellular Therapies in Immunoregulation and CancerView all 31 articles
 Thi-Anh-Thuy Tran1,2
Thi-Anh-Thuy Tran1,2 Young-Hee Kim1Thi-Hoang-Oanh Duong1
Young-Hee Kim1Thi-Hoang-Oanh Duong1 JayaLakshmi Thangaraj3
JayaLakshmi Thangaraj3 Tan-Huy Chu3Shin Jung1,4In-Young Kim1,4Kyung-Sub Moon1,4
Tan-Huy Chu3Shin Jung1,4In-Young Kim1,4Kyung-Sub Moon1,4 Young-Jin Kim1,4Tae-Kyu Lee1,4
Young-Jin Kim1,4Tae-Kyu Lee1,4 Chul Won Lee5
Chul Won Lee5 Hyosuk Yun5Je-Jung Lee3,6Hyun-Ju Lee3
Hyosuk Yun5Je-Jung Lee3,6Hyun-Ju Lee3 Kyung-Hwa Lee7*
Kyung-Hwa Lee7* Tae-Young Jung1,2,4*
Tae-Young Jung1,2,4*Various combination treatments have been considered to attain the effective therapy threshold by combining independent antitumor mechanisms against the heterogeneous characteristics of tumor cells in malignant brain tumors. In this study, the natural killer (NK) cells associated with bevacizumab (Bev) plus irinotecan (Iri) against glioblastoma multiforme (GBM) were investigated. For the experimental design, NK cells were expanded and activated by K562 cells expressing the OX40 ligand and membrane-bound IL-18 and IL-21. The effects of Bev and Iri on the proliferation and NK ligand expression of GBM cells were evaluated through MTT assay and flow cytometry. The cytotoxic effects of NK cells against Bev plus Iri-treated GBM cells were also predicted via the LDH assay in vitro. The therapeutic effect of different injected NK cell routes and numbers combined with the different doses of Bev and Iri was confirmed according to tumor size and survival in the subcutaneous (s.c) and intracranial (i.c) U87 xenograft NOD/SCID IL-12Rγnull mouse model. The presence of injected-NK cells in tumors was detected using flow cytometry and immunohistochemistry ex vivo. As a result, Iri was found to affect the proliferation and NK ligand expression of GBM cells, while Bev did not cause differences in these cellular processes. However, the administration of Bev modulated Iri efficacy in the i.c U87 mouse model. NK cells significantly enhanced the cytotoxic effects against Bev plus Iri-treated GBM cells in vitro. Although the intravenous (IV) injection of NK cells in combination with Bev plus Iri significantly reduced the tumor volume in the s.c U87 mouse model, only the direct intratumorally (IT) injection of NK cells in combination with Bev plus Iri elicited delayed tumor growth in the i.c U87 mouse model. Tumor-infiltrating NK cells were detected after IV injection of NK cells in both s.c and i.c U87 mouse models. In conclusion, the potential therapeutic effect of NK cells combined with Bev plus Iri against GBM cells was limited in this study. Accordingly, further research is required to improve the accessibility and strength of NK cell function in this combination treatment.
Glioblastoma multiforme (GBM) is the most frequent and aggressive type of cancer in the central nervous system (CNS). It is associated with poor prognostic factors and low survival. It accounts for 14.3% of all primary brain and other CNS tumors and 49.1% of primary malignant brain tumors. Only 6.8% of patients survive after 5 years of diagnosis (1, 2). Although advances in GBM treatments, e.g., maximal surgical resection with radiation and chemotherapy, have been applied, the mean 5-year overall survival (OS) of patients with GBM remains low (approximately 9.8%) (3, 4). Therefore, new strategies must be developed to improve the therapeutic effects of this disease.
GBM is a highly vascularized tumor that secretes a large amount of vascular endothelial growth factor (VEGF) (5). VEGF is a key mediator of tumor neovascularization (endothelial proliferation and vascular permeability) that suppresses most immune responses in tumors (6–9). As a standard treatment for GBM, bevacizumab (Bev) is a humanized monoclonal antibody approved by the FDA. It targets the VEGF receptor and consequently reduces blood flow and tumor volume (10). Furthermore, irinotecan (Iri)—a chemotherapeutic drug—is found to be effective with tumor cells by blocking a topoisomerase 1 inhibitor involved in the induction of DNA damage and subsequent apoptosis. It can cross the blood-brain barrier and exhibits unique antitumor activity against GBM as observed in preclinical and clinical investigations (11–13).
Recent clinical trials using Bev in combination with Iri and/or temozolomide in recurrent patients with GBM have demonstrated its moderate effectiveness and tolerance (14, 15). Bev combined with Iri is effective for patients with recurrent malignant gliomas and some children with recurrent low-grade gliomas. This combination is safe and has excellent activity even in the relapsed and heavily pretreated population of patients with malignant glioma, most of whom were not candidates for clinical trials (16, 17). The efficacy of Bev plus Iri can be attributed to the antitumor stem cell effect of Bev, the anti-differentiated glioma tumor cell effect of Iri, and the normalization of tumor vasculature; consequently, interstitial pressure decreases, hypoxia is minimized, and the delivery of Iri to tumors is enhanced (18, 19).
Although natural killer (NK) cells in patients with cancer are often functionally compromised owing to the immunosuppressive activity of tumors, the initial success of adoptive NK cell transfer in the treatment of hematological cancers has motivated clinical efforts devoted to using this strategy against solid cancers. NK cells not only elicit cytotoxic effects against a wide range of tumor cells of solid cancer types but also exhibit antitumor activities in preclinical xenograft mouse models of GBM. The safety of NK cell-based therapy has been evaluated in autologous and allogeneic haploidentical settings. However, the clinical efficacy of this strategy is limited (20–22). Therefore, the combination of NK cells with other treatment methods in current preclinical efforts enhances the efficacy of NK cell-based therapy. Furthermore, NK cells expanded and activated by K562-OX40L-mb-IL-18/IL-21 feeder cells have shown cytotoxic activity against multiple myeloma in in vitro and xenograft mouse models (23–25). Therefore, NK cells using K562-OX40L-mb-IL-18/IL-21 cells as feeder cells were also used to confirm therapeutic effects against GBM in this study.
Tumor cell heterogeneity is a crucial characteristic that contributes to therapeutic resistance and the recurrence of malignant brain tumors (26). Individual treatments may have a limited effect; however, combined treatments may achieve the threshold for effective therapy. Although the therapeutic effects of Bev plus Iri have been demonstrated, the role of the combination of Bev and Iri in NK therapy has not been investigated. NK cells, Bev, and Iri are known to have different working mechanisms toward target tumors (11, 27, 28). Nevertheless, more complicated possible interactions between NK cells, Bev, and Iri should be confirmed for improving their anti-GBM effects. In this study, NK cells were used in combination with Bev and Iri to further understand the preclinical success and aid in designing rational and effective combinatorial therapies for GBM in clinical settings.
Blood and tissue samples were collected from healthy donors (HDs) and GBM patients with the approval of the institutional ethical committee at Chonnam National University Hwasun Hospital. All surgical tissues and blood from GBM patients were collected at the Neurosurgery Department at Chonnam National University Hwasun Hospital.
Six-to-eight-week-old female NOD/SCID IL-12Rγnull (NSG) mice (Jackson Laboratory, Bar Harbor, MA, USA) were raised under specific pathogen-free conditions. The mice were anesthetized by the intraperitoneal injection (IP) of a 2:1 mixture of Zoletil® (Virbac Laboratories, Carros, France)/Rompun® (Bayer Korea, Anshan, Korea) at a dose of 1.5 mL/kg. All animal care procedures, experiments, and euthanasia were performed after obtaining approval from the Chonnam National University Animal Research Committee.
Human GBM cell lines, namely, U87, U118, and U343, were obtained from the Korean Cell Line Bank (Seoul, Korea) and the Brain Tumor Research Center, University of California, San Francisco. All cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, US) and 1% penicillin–streptomycin in a humidified 5% CO2 incubator at 37°C.
Human K562 (human immortalized myelogenous leukemia cell line) was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Modified K562-OX40L-mb-IL-18/IL-21 cells were used to expand and activate NK cells (25). Cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% heat-inactivated FBS (Gibco, US) and 1% penicillin–streptomycin in a humidified 5% CO2 incubator at 37°C.
Bev (Avastin®) and Iri-HCl (Campto®) were purchased from Roche (Basel, Switzerland) and Inno.N (Seoul, Republic of Korea) laboratories, respectively. Dose treatments were set at 10 mg/kg Bev plus 125 mg/m2 Iri-HCl (a high dose of Bev and Iri, Bev plus Irihigh) and 5 mg/kg Bev plus 60 mg/m2 Iri (a low dose of Bev and Iri, Bev plus Irilow) according to the previously reported clinical trial dose (29–32). The dose was converted from a human dose into a mouse dose for treatment (33). The abovementioned chemical agents were administered through IP.
The expression levels of NK ligands on GBM cells, NK markers on NK cells, and NK purity were confirmed through flow cytometry. The cells were stained with antibodies listed in Table 1. The mean fluorescence intensity (MFI) ratio was calculated by dividing the MFI of the stained cell population by that of the unstained cell population. Fluorescence minus one (FMO) stain was used in each sample in each experiment to determine positive and negative populations. Further, gates were applied from FMOs to samples. The FMO of each experiment has been described in the corresponding figure legends.
Table 1 List of antibodies used in flow cytometry.
The effects of Bev and Iri on the proliferation and survival of GBM cells (U87, U118, and U343 cells) were predicted via the MTT assay. Briefly, GBM cells (3×103 cells/well) were seeded in 96-well plates (SPL, Gyeonggi-do, Korea) and cultured with DMEM supplemented with 10% FBS and 1% P/S at 37°C in a 5% CO2 atmosphere. They were stained after 24 h and 48 h incubation with MTT (Sigma). For staining, the plates were washed with PBS, and MTT (0.5 mg/mL) was added to each well. After 4 h of incubation, the MTT solution was removed from each well. MTT formazan was then solubilized using isopropanol (Merck, Darmstadt, Germany), and optical density was read at 570 nm.
NK cells were cultured and expanded using the established K562-OX40L-mb-IL-18/IL-21 feeder cells in previously described methods (25). Briefly, peripheral blood mononuclear cells isolated from HDs and GBM patients were co-cultured with gamma-irradiated (100 Gy) K562-OX40L-mb-IL-18/IL-21 feeder cells in RPMI-1640 medium containing 10% FBS, 1% penicillin/streptomycin, and 4 mM L-glutamine. NK cells were cultured in the presence of recombinant human interleukin IL-2 and IL-15 (PeproTech, Rocky Hill, NJ, USA). The recombinant IL-2 (10 U/mL) was added to the cell culture medium until day 7. From day 7, recombinant IL-2 (100 U/mL) and recombinant IL-15 (5ng/mL) were added to the cell culture medium. The cell culture medium containing cytokines was refreshed every 2–3 days. On day 14, NK cells with >90% purity were used for in vitro and in vivo experiments. For in vivo experiments, NK cells were expanded from isolated peripheral blood mononuclear cells of HDs (NK-HD cells) used for all experiments. The mice were treated with intravenously injected NK-HD cells at a low dose (1×107 NK cells/ injection, NK-IVlow) or a high dose (2×107 NK cells/ injection, NK-IVhigh), and intratumorally injected NK-HD cells (2×106 NK cells/ injection, NK-IT), respectively.
CytoTox 96 nonradioactive cytotoxicity assay (CytoTox 96, Promega, Madison, WI, USA) was performed to analyze the cytotoxic effects of NK-HD and NK-GBM cells against target GBM cells, namely, U87, U118, U343, and primary GBM (pGBM) cells, according to the manufacturer’s instructions. Briefly, GBM cells (4×104 cells/ well) with or without treatment with Bev and Iri were used as the target. The NK cells were co-cultured with the target cells at 1:1 and 1:3 (target: effector) ratio in Costar 96-well plates (SPL, Gyeonggi-do, Korea) under a 5% CO2 atmosphere at 37°C for 5 h. Then, supernatants were collected to determine the LDH concentration. The mean percentage of specific lysis was calculated as follows:
The GBM cells U87, U118, U343, and pGBM (4 × 104 cells/wells) with or without treatment with Bev and Iri were used as target cells. The NK-HD and NK-GBM cells were co-cultured with target cells at a 1:3 (target:effector) ratio in 96-well plates (SPL, Gyeonggi-do, Korea) under a 5% CO2 atmosphere at 37°C for 5 h. Subsequently, the supernatants were collected to determine the IFN-γ concentration by using the OptEIA ELISA kit (BD Bioscience) as per the manufacturer’s instructions. NK alone and NK treated with dimethyl sulfoxide (DMSO) were used as the negative control and NK treated with PMA/Ionomycin (Biolegend, 423302) was used as the positive control for released IFN-γ.
For the s.c mouse model, 5×106 U87 cells in 100 µl of PBS were mixed with 100 µl of Matrigel™ Matrix (Corning #354248) and injected into the right s.c of NSG mice. The mice were randomly allocated to the treatment arms. NK-HD cells were injected intravenously (IV). For the treatment, the mice were divided into four treatment groups: (1) control, (2) NK-IVlow, (3) Bev plus Irihigh, and (4) NK-IVlow combined with Bev plus Irihigh. The treatment was initiated when the tumor size reached approximately 100 mm3. Overall survival was quantified, and the tumor volume was calculated using the standard formula for the volume of an ellipsoid: V=4/3π [(length × width × height)/8]. Once the total tumor volume reached 1000 mm3 per animal, the mice were euthanized.
For the i.c mouse model, 5×105 U87 cells in 5 μl of PBS were stereotactically injected into the right striatum of the mice at a rate of 1 μl/min. Injection sites were estimated using the following coordinates: 1 mm posterior, 2 mm lateral from the bregma, and 4 mm deep from the cortical surface. The mice were randomly allocated to the treatment arms. NK-HD cells were injected IV or intratumorally (IT). For the treatment, the mice were divided into eight and four different treatment groups. For the eight groups, the mice were treated according to (1) control, (2) NK-IVlow, (3) NK-IT, (4) Bev plus Irilow, (5) NK-IVhigh combined with Bev plus Irilow, (6) Bev plus Irihigh, (7) NK-IVlow combined with Bev plus Irihigh, and (8) NK-IT combined with Bev plus Irihigh. For the four groups, the mice were treated according to (1) control, (2) Irihigh, (3) Bev plus Irihigh, and (4) NK-IVhigh combined with Bev plus Irihigh. The treatments were initiated when the tumor size reached approximately 5 mm3. The overall survival was quantified. Tumor size was confirmed through magnetic resonance imaging (MRI) and analyzed using the RadiAnt DICOM Viewer 2021.2 software. Tumor volume was calculated by the summation of all tumor areas in each slide and multiplication by the slide thickness.
Single tumor cells were collected from GBM patients and U87-bearing NSG mice were collected for further experiment. Single tumor cells from GBM patients were used for confirming NK ligands expression and target cells of cytotoxicity assay, while those from the U87-bearing NSG mice were used for confirming the presence of NK cells in the tumor and NK ligand expression on single tumor cells before and after treatment with NK cells, Bev, and Iri. The single tumor cells were collected according to the following protocol. First, the tumor was collected, washed with RPMI media supplemented with 10% FBS and 1% P/S, and minced into 3 mm to 4 mm pieces by using a sterile scalpel. Then, the tumor was dissociated by using a tumor dissociation kit (Miltenyi Biotech, Germany) for obtaining the single tumor cells from the s.c tumor and a brain tumor dissociation kit (Miltenyi Biotech, Germany) for obtaining the single tumor cells from the i.c tumor. Afterward, the cells were filtered using 70 or 40 μm cell strainers (Falcon), and single tumor cells were collected. Erythrocytes were removed using red blood cell lysis buffer (Miltenyi Biotech, Germany).
Tumors from the s.c and i.c mouse models were collected and fixed in 5% paraformaldehyde to confirm the presence of NK cells at tumor sites. They were then embedded in paraffin, sectioned into 4 μm coronal sections by using a microtome, and prepared for immunohistochemistry (IHC) staining. The antibodies used for the IHC experiment were anti-human CD56 (1:50 dilution; Code No. M7304; clone 123C3; DAKO; Denmark) and anti-human CD45RO (LCA; 1:100 dilution; Code No. M0701; clone 2B11+PD7/26; DAKO; Denmark). Tumor slides were scanned using the Aperio Scan Scope System (Aperio, Technology, Vista, CA, USA).
Data were statistically analyzed using SPSS 23.0 for Windows (SPSS Inc., Chicago, IL, USA). One-way or two-way ANOVA was performed for analyses across multiple groups. A log-rank test was performed on survival data with Bonferroni correction applied for comparations, and an independent sample t-test was used to compare significant differences between the two groups. Data with p<0.05 were considered statistically significant.
The viability percentages and IC50 of GBM cells (U87, U118, and U343 cells) after the treatment with Bev and Iri separately or in combination with a gradual increase in dose concentrations (0.5, 1, 2, and 4 mg/mL for Bev and 0.15, 0.3, 0.6, and 1.2 mg/mL for Iri) were determined at 24 and 48 h via the MTT assay to investigate the effects of Bev and Iri on the proliferation of GBM cells (Figures 1A–I).
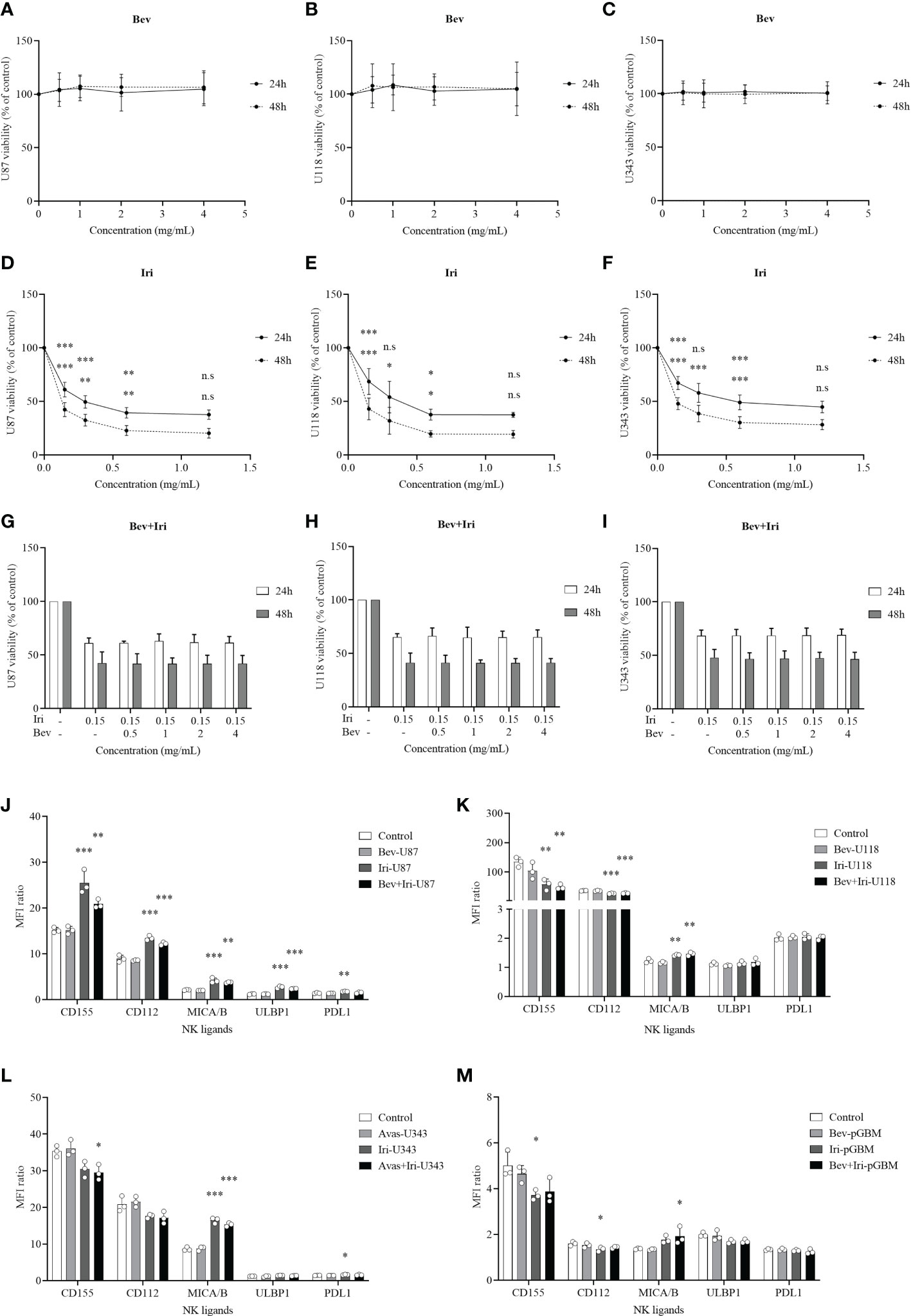
Figure 1 Effects of Bev and Iri on the proliferation of GBM cells in vitro. GBM cells (U87, U118, and U343 cells) were treated with Bev and Iri separately or in combination of according to the increased concentration of these two drugs (0.5, 1, 2, and 4 mg/mL for Bev and 0.15, 0.3, 0.6, and 1.2 mg/mL for Iri). After 24 and 48 h, cell viability was determined by MTT assay and IC50 was calculated to determine the concentration of drugs that exhibited 50% cell viability. The viability of untreated cell cultures was set at 100% (A–I). Moreover, the expression of NK ligands on GBM cells such as U87, U118, U343, and pGBM and the effects of Bev (0.5 mg/mL) and Iri (0.15 mg/mL) on their expression was clarified using flow cytometry. (J–M) Data were presented as MFI ratio calculated by dividing the MFI of the positive cells (stained cell population) by that of the negative cells (live cell population). FMO: live cells. U87, U118, and U343: human GBM cell lines; pGBM: primary GBM cells; Bev: bevacizumab; Iri: irinotecan. All data are shown as mean ± SD. p< 0.05 (*), p< 0.001 (**), p< 0.0001 (***). n.s, no significant difference.
Bev did not affect the viability of GBM cells, whereas Iri significantly decreased the viability of all GBM cells according to the increased dose statuses (0.15, 0.3, 0.6, and 1.2 mg/mL) at 24 and 48 h. In particular, 0.15 mg/mL Iri reduced 37% (p=0.000, 24 h) and 55% (p=0.000, 48 h) of U87 cell viability, 31% (p=0.000, 24 h) and 57% (p=0.000, 48 h) of U118 cell viability, and 33% (p=0.000, 24 h) and 52% (p=0.000, 48 h) of U343 cell viability compared with those in the control group. This trend was similar to 0.3 mg/mL and 0.6 mg/mL Iri. However, after 24 and 48 h, GBM cell viability did not vary between 0.6 mg/mL and 1.2 mg/mL Iri. Thus, the examined GBM cells might exhibit resistance at an Iri concentration of 1.2 mg/mL.
Iri exhibited 50% cell viability (IC50) at 0.33 mg/mL and 0.08 mg/mL for U87 cells, 0.41 mg/mL and 0.09 mg/mL for U118 cells, and 0.63 mg/mL and 0.11 mg/mL for U343 cells at 24 h and 48 h of incubation, respectively. Because Bev did not affect GBM cell viability, the minimal screening Iri dose of 0.15 mg/mL that significantly affected GBM cell proliferation but had not reached resistance on GBM cells was combined with different doses of Bev (0.5, 1, 2, and 4 mg/mL) to confirm GBM cell viability. Results of the analysis revealed that the GBM cell viability did not significantly differ among these combinations. Therefore, the screened dose of 0.5 mg/mL Bev and 0.15 mg/mL Iri were used for further in vitro analysis.
The expression levels of NK ligands (CD155, CD112, MICA/B, ULBP1, and PDL1) on GBM cells (U87, U118, and U343) and primary GBM cells (pGBM) before and after the treatments with Bev (0.5 mg/mL) and Iri (0.15 mg/mL) at 24 h were elucidated via flow cytometry (Figures 1J–M; S1). Although Iri affected the NK ligands on GBM cells, Bev did not influence their expression. In particular, the MICA/B expression was enhanced under Iri or Bev plus Iri treatment in U87 cells (p=0.000 and p=0.001), U118 cells (p=0.007 and p=0.003), and U343 cells (p=0.000 and p=0.000) compared control group. However, only Bev plus Iri increased MICA/B expression in pGBM cells (p = 0.05) compared to the control group. CD155 and CD112 expression levels increased in U87 cells with Iri (p = 0.000 and p = 0.000) and Bev plus Iri group (p = 0.007 and p = 0.000), while it decreased in U118 cells with Iri (p = 0.006 and p = 0.000) and Bev plus Iri group (p = 0.003 and p = 0.000). Furthermore, while Bev plus Iri reduced CD155 expression on U343 cells (p = 0.03), Iri caused a reduction in CD155 and CD122 in pGBM cells (p = 0.033 and p = 0.034, respectively). Moreover, only U87 cells exhibited an increase in ULBP1 expression under Iri and Bev plus Iri treatment (p = 0.000 and p = 0.000, respectively). Therefore, the synergistic effects of Iri and NK cells could be related to the MICA/B expression; however, further studies should be performed to verify this finding.
The schematic of NK cell expansion from peripheral blood mononuclear cells (PBMCs) of HDs (NK-HD) and GBM patients (NK-GBM) co-cultured with 100 Gy gamma-irradiated K562-OX40L-mb-IL-18/IL-21 feeder cells in the presence of IL-2 (from day 0 to day 6) and IL-2/IL-15 (from day 7 to day 28) is shown in Figure S2A. The NK-HD and NK-GBM cells were characterized by NK purity, expansion fold, and NK marker expression. In particular, the NK-HD and NK-GBM cells were confirmed in terms of NK purity, expansion fold, and NK marker expression every 7 days (on days 0, 7, 14, 21, and 28) (Figures S2B–D). There was no difference between the observed NK-HD and NK-GBM cells.
NK-HD and NK-GBM cell purity did not vary on days 14, 21, and 28. Although the expansion fold of NK-HD cells was higher than that of NK-GBM cells (around 4000-fold expansion for NK-HD and 3000-fold expansion for NK- GBM), it did not significantly differ between NK-HD and NK-GBM cells for 28 days. Moreover, the expression of NK markers from NK-HD and NK-GBM cells was estimated every 7 days (on days 0, 7, 14, 21, and 28). The activated and inhibited NK marker expression (NKG2D, NKp30, FASL, TRAIL, DNAM1, PD1, and NKG2A) did not differ among days 0, 7, 14, 21, and 28; similarly, the NK marker expression did not vary between NK-HD and NK-GBM cells.
The cytotoxic effects of NK-HD and NK-GBM cells against GBM cell lines (U87, U118, and U343) and pGBM cells were estimated in Figure 2. Generally, NK-HD and NK-GBM cells exhibited cytotoxic effects against all GBM cells (U87, U118, U343, and pGBM) at 1:1 and 1:3 (target:effector) ratio. Although both NK-HDs and NK-GBM enhanced the cytotoxic effects against GBM cells treated with Bev plus Iri compared to the untreated group, no difference was observed in the cytotoxicity of NK-HDs and NK-GBM against Iri-treated GBM cells and Bev plus Iri-treated GBM cells. Thus, the main effects that enhanced NK cytotoxicity against GBM cells in vitro could be related to Iri rather than Bev. This trend was observed clearly at a 1:3 (target:effector) ratio. Particularly, the cytotoxicity of NK-HD cells against GBM cells treated with Bev plus Iri was enhanced by 22% (p = 0.000) in U87 cells, 17% (p = 0.001) in U118 cells, 14% (p = 0.000) in U343 cells, and 11% (p = 0.022) in pGBM cells compared to those in the untreated group. Similarly, the cytotoxicity of NK-GBM cells against GBM cells treated with Bev plus Iri was enhanced by 22% (p = 0.02) in U87 cells, 17% (p = 0.000) in U118 cells, and 9% (p = 0.024) in U343 cells compared to those in the untreated group. However, the cytotoxicity of NK cells against GBM cells treated with Iri alone and Bev plus Iri showed no significant difference between NK-HD and NK-GBM cells, except in U343 cells.
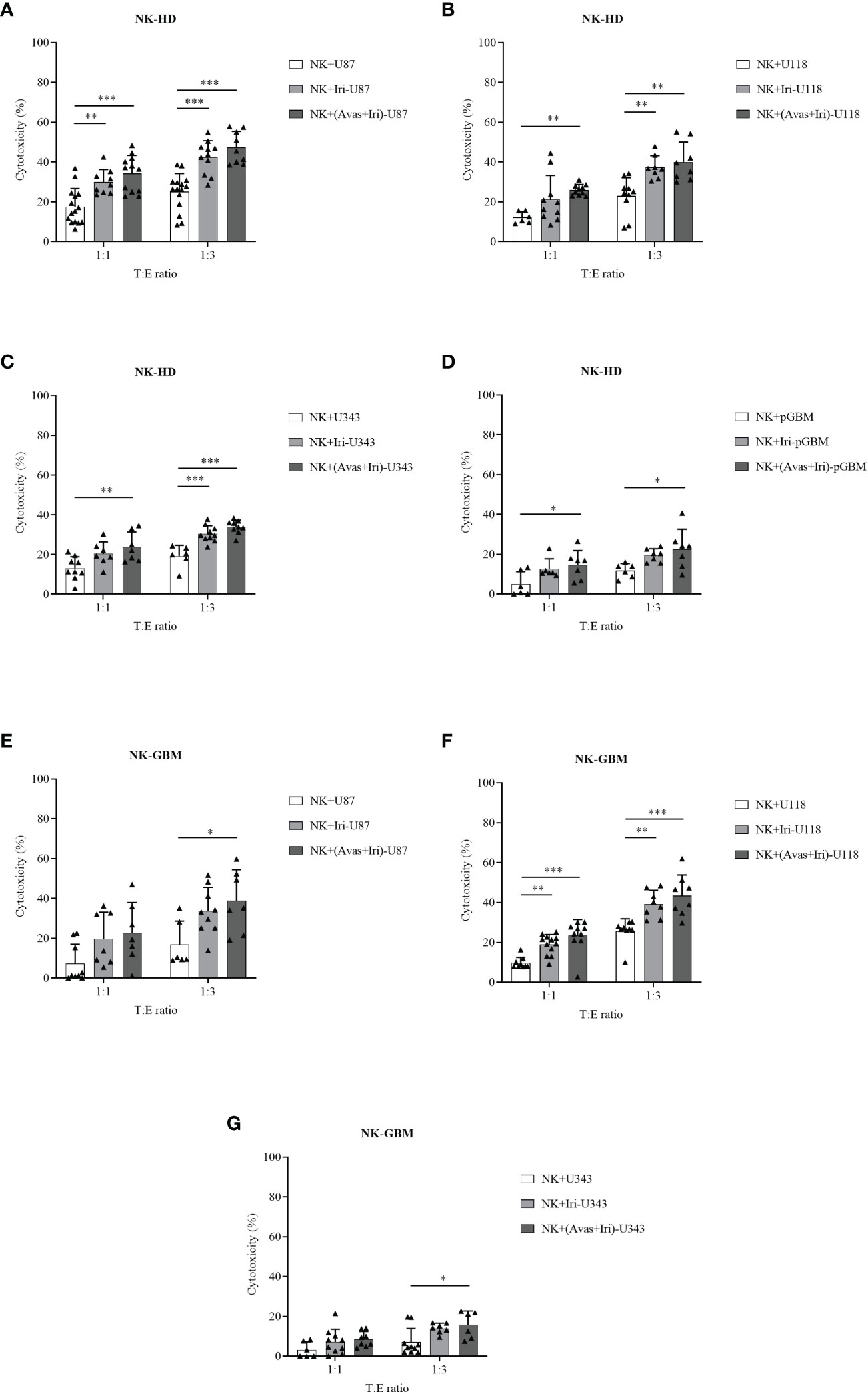
Figure 2 Cytotoxic effects of expanded NK cells from HDs (NK-HD) and GBM patients (NK-GBM) against GBM cells, Iri-treated GBM, and Bev plus Iri-treated GBM cells. The cytotoxicity effects of NK-HD (A–D) and NK-GBM cells (E–G) against GBM cells such as U87, U118, U343, pGBM at 1:1 and 1:3 (target:effector) ratio were confirmed through the LDH assay. U87, U118, and U343: human GBM cell lines; pGBM: primary GBM cells; Bev: bevacizumab (0.5 mg/mL); Iri: irinotecan (0.15 mg/mL). All data are shown as mean ± SD. p< 0.05 (*), p< 0.001 (**), p< 0.0001 (***).
The IFN-γ levels before and after co-culturing with untreated GBM cells, Iri-treated GBM cells, and Bev plus Iri-treated GBM cells can be confirmed from Figure S3. In particular, the IFN-γ levels of NK-HDs and NK-GBM cells were enhanced after co-culturing with GBM target cells. Although the IFN-γ levels of NK-HDs and NK-GBM against Iri- and Bev plus Iri-treated GBM target cells showed a slight enhancement compared to untreated GBM target cells, no difference was observed in the IFN-γ levels between Iri- and Bev plus Iri-treated GBM group. Further, the expression of NK cell markers (NKG2D, NKp30, FASL, TRAIL, DNAM1, NKG2A, and PD1) from NK-GBM cells before and after co-culturing with the untreated (U87, U118, and U343 cells), Iri-treated, and Bev plus Iri-treated GBM cells are illustrated in Figure S4. No significant differences were observed in their expression levels.
When the tumor reached around 100 mm3, U87-bearing mice were treated with NK-HD cells, Bev, and Iri according to the scheduled treatment shown in Figure 3A. Days were estimated from post-treatment. Further, mouse weight was estimated; as shown in Figure 3B, there was no difference in mouse weight between treatment groups such as NK-IVlow, Bev plus Irihigh, and NK-IVlow combined with Bev plus Irihigh group compared to the control group. The therapeutic effects of NK-HD cells combined with Bev plus Iri were confirmed from tumor volume and survival.
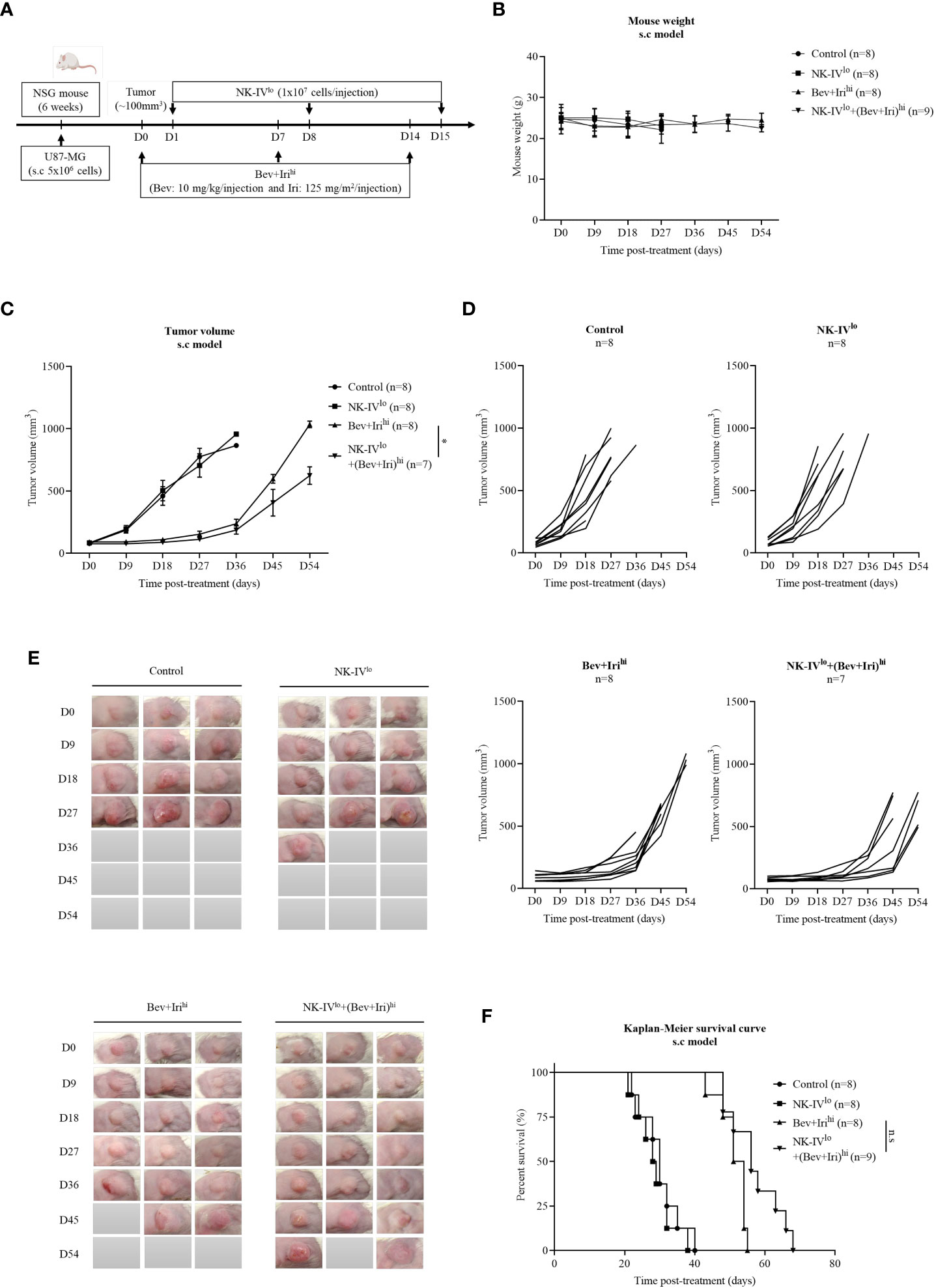
Figure 3 Experimental treatment schedules and therapeutic effects of NK-HD cells in combination with Bev plus Iri estimated in the s.c U87 mouse model. At a tumor size of 100 mm3, the mice were treated with Bev and Iri every 7 days for 14 days. NK-HD cells were injected after 1 day of Bev and Iri treatment (A). The mouse weight before and after the treatment with NK-HD cells in combination with Bev and Iri was clarified every 9 days (B). The tumor size before and after the treatment with NK-HD cells in combination with Bev and Iri was clarified every 9 days and the tumor volume was calculated as follows: V = 4/3π (length × width × height/8) (C–E). The Kaplan–Meier survival of U87-bearing mice of control, NK-IVlo, Bev plus Irihi, and NK-IVlo in combination with Bev plus Irihi group was estimated. The statistical significance of survival was determined using a log-rank test with Bonferroni correction applied for comparisons (F). NK-IVlo: intravenously injected NK-HD cells (1 × 107 NK cells); Bev+Irihi: intraperitoneally injected bevacizumab (10 mg/kg) plus irinotecan (125 mg/m2); U87: human GBM cell line. All data are shown as mean ± SD. p< 0.05 (*). n.s, no significant difference.
The tumor volume was also measured with a digital caliper; the final data are shown in Figures 3C–E; S5. As revealed by the aforementioned figures, there was no difference in tumor volume between NK-IVlow and control groups. However, the tumor volume of the mice treated with Bev plus Irihigh and NK-IVlow combined with Bev plus Irihigh showed a delay in tumor growth compared to the control and NK-IVlow groups. Bev plus Irihigh and NK-IVlow combined with Bev plus Irihigh reduced the tumor volume compared with that of the control group (p = 0.000 and p = 0.000, respectively) and NK-IVlow (p = 0.000 and p = 0.000, respectively). Moreover, NK-IVlow combined with Bev plus Irihigh also showed suppressed tumor growth compared with the Bev plus Irihigh group (p = 0.037).
Similarly, the survival of U87-bearing mice did not differ between NK-IVlow (28.8 ± 1.7 days) and the control group (30 ± 2.1 days). However, the survival of the mice treated with Bev plus Irihigh and NK-IVlow combined with Bev plus Irihigh significantly differed from that of the control and NK-IVlow groups. Bev plus Irihigh (51.3 ± 1.4 days) and NK-IVlow combined with Bev plus Irihigh (57.1 ± 2.5 days) prolonged the survival of the treated mice compared with that of the control group (p = 0.000 and p = 0.000, respectively) and the mice treated with NK-IVlow (p = 0.000 and p = 0.000, respectively). Although there was a difference in mouse survival between Bev plus Irihigh and NK-IVlow combined with Bev plus Irihigh, as revealed by the log-rank test (p = 0.018), there was no difference with the Bonferroni correction applied (Figure 3F).
When the tumor reached around 5 mm3, U87-bearing mice were treated with NK-HD cells, Bev, and Iri according to the scheduled treatment in Figure 4A. The therapeutic effects of NK-HD cells combined with Bev plus Iri were confirmed in terms of tumor volume and survival. Days were estimated from post-treatment.
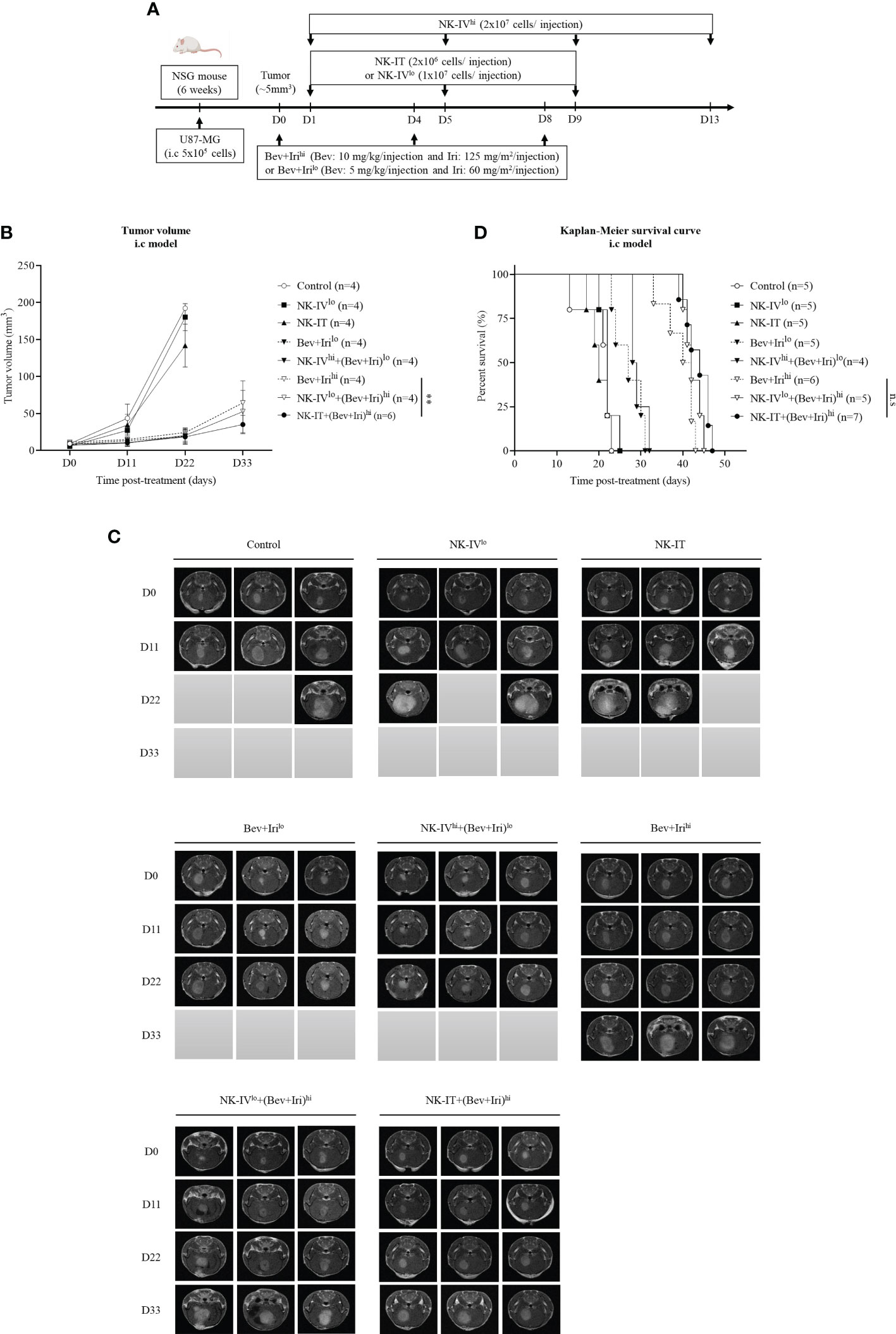
Figure 4 Experimental treatment schedule and therapeutic effects of NK-HD cells in combination with Bev plus Iri in the i.c U87 mouse model. At a tumor size of 5 mm3, the mice were treated with Bev and Iri every 4 days for 8 days. NK-HD cells were injected after 1 day of Bev and Iri treatment (A). The tumor size before and after the treatment with NK-HD cells in combination with Bev plus Iri was clarified every 6 days using MRI. The final tumor volume was analyzed using the RadiAnt DICOM Viewer 2021.2.2 software and calculated as the summation of all tumor areas in each slide and multiplication by the slide thickness (B, C). The Kaplan–Meier survival of U87-bearing mice after treatment with NK-HD cells in combination with Bev plus Iri was estimated. The statistical significance of survival was determined using a log-rank test with Bonferroni correction applied for comparisons (D). NK-IVlo: intravenously injected NK-HD cells (1 × 107 NK cells); NK-IVhi: intravenously injected NK-HD cells (2 × 107 NK cells); Irihi: intraperitoneally injected Iri (125 mg/m2); Bev+Irilo: intraperitoneally injected Bev (5 mg/kg) plus Iri (60 mg/m2); Bev+Irihi: intraperitoneally injected Bev (10 mg/kg) plus Iri (125 mg/m2); NK-IT: intratumorally injected NK-HD cells (2 × 106 NK cells); U87: human GBM cell line. All data are shown as mean ± SD. p< 0.001 (**). n.s, no significant difference.
Tumor volume was clarified via MRI and mouse weight was also estimated (Figures 4B, C; S6, S7). The tumor volume of U87-bearing mice did not differ between NK-IVlow and NK-IT compared with that of the control group. However, the tumor volumes of the mice treated with Bev plus Irilow, NK-IVhigh combined with Bev plus Irilow, Bev plus Irihigh, NK-IVlow combined with Bev plus Irihigh, and NK-IT combined with Bev plus Irihigh significantly differed from those of the control (p = 0.000, p = 0.000, p = 0.001, p = 0.000, and p = 0.000, respectively), NK-IVlow (p = 0.000, p = 0.000, p = 0.000, p = 0.000, and p = 0.000, respectively), and NK-IT (p = 0.000, p = 0.000, p = 0.001, p = 0.000, and p = 0.000, respectively) groups. Only NK-IT combined with Bev plus Irihigh delayed tumor growth compared with the Bev plus Irihigh group (p = 0.006). Moreover, Bev plus Irihigh also showed delayed tumor volume compared with Irihigh (p = 0.043). These results show the role of the combination of bevacizumab and irinotecan in the i.c U87 mouse model. The combination of NK-IVhigh with Bev plus Irihigh could not suppress the tumor size compared to Bev plus Irihigh.
A similar trend can be observed in Figure 4D, wherein the survival of U87-bearing mice did not differ between NK-IT and NK-IVlow compared with that of the control group (20.2 ± 1.8 days). However, Bev plus Irilow (27.0 ± 1.6 days) and NK-IVhigh combined with Bev plus Irilow (29.3 ± 0.9 days) prolonged the survival of the treated mice compared with those of the control (p = 0.03 and p = 0.002, respectively), NK-IVlow (n.s and p = 0.038, respectively), and NK-IT (p = 0.03 and p = 0.002, respectively). Moreover, the survival of U87-bearing mice in Bev plus Irihigh (39.5 ± 1.6 days), NK-IVlow combined with Bev plus Irihigh (42.4 ± 0.9 days), and NK-IT combined with Bev plus Irihigh (43.6 ± 1.1 days) was also prolonged compared with that of the mice treated with Bev plus Irilow (p = 0.000, p = 0.000, and p = 0.000, respectively) and NK-IVhigh combined with Bev plus Irilow (p = 0.000, p = 0.000, and p = 0.000, respectively). However, no difference in survival was observed between Bev plus Irihigh, NK-IVlow combined with Bev plus Irihigh, and NK-IT combined with Bev plus Irihigh. The combination of NK-IVhigh with Bev plus Irihigh affected mouse survival during treatment. However, the remaining mice recovered after 7 days post-treatment, and alive mice in NK-IVhigh with Bev plus Irihigh group died owing to increased tumor volume.
The tumor-infiltrating NK-HD cells of U87-bearing mice were confirmed via flow cytometry with hCD45 and IHC with hCD56 and hCD45 markers; the results are presented in Figures 5A–F. The percentages of hCD45+ cells in tumors were clarified on the s.c and i.c U87 mouse models. In general, a few hCD45+ cells were detected in the tumor in the NK treatment group (NK-IVlow) and NK cells in combination with Bev plus Iri group (NK-IVlow combined Bev plus Irihigh or NK-IVhigh combined Bev plus Irilow) in both s.c and i.c U87 mouse models, which was confirmed with the presence of hCD45 and hCD56 in the tumor with IHC assay. Although NK-IVlow combined Bev plus Irihigh showed a slight enhancement compared to NK-IVlow, no difference was observed in the s.c and i.c U87 mouse models. Moreover, the presence of NK cells in NK-IVhigh combined with Bev plus Irilow was confirmed by flow cytometry and IHC in the i.c U87 mouse model.
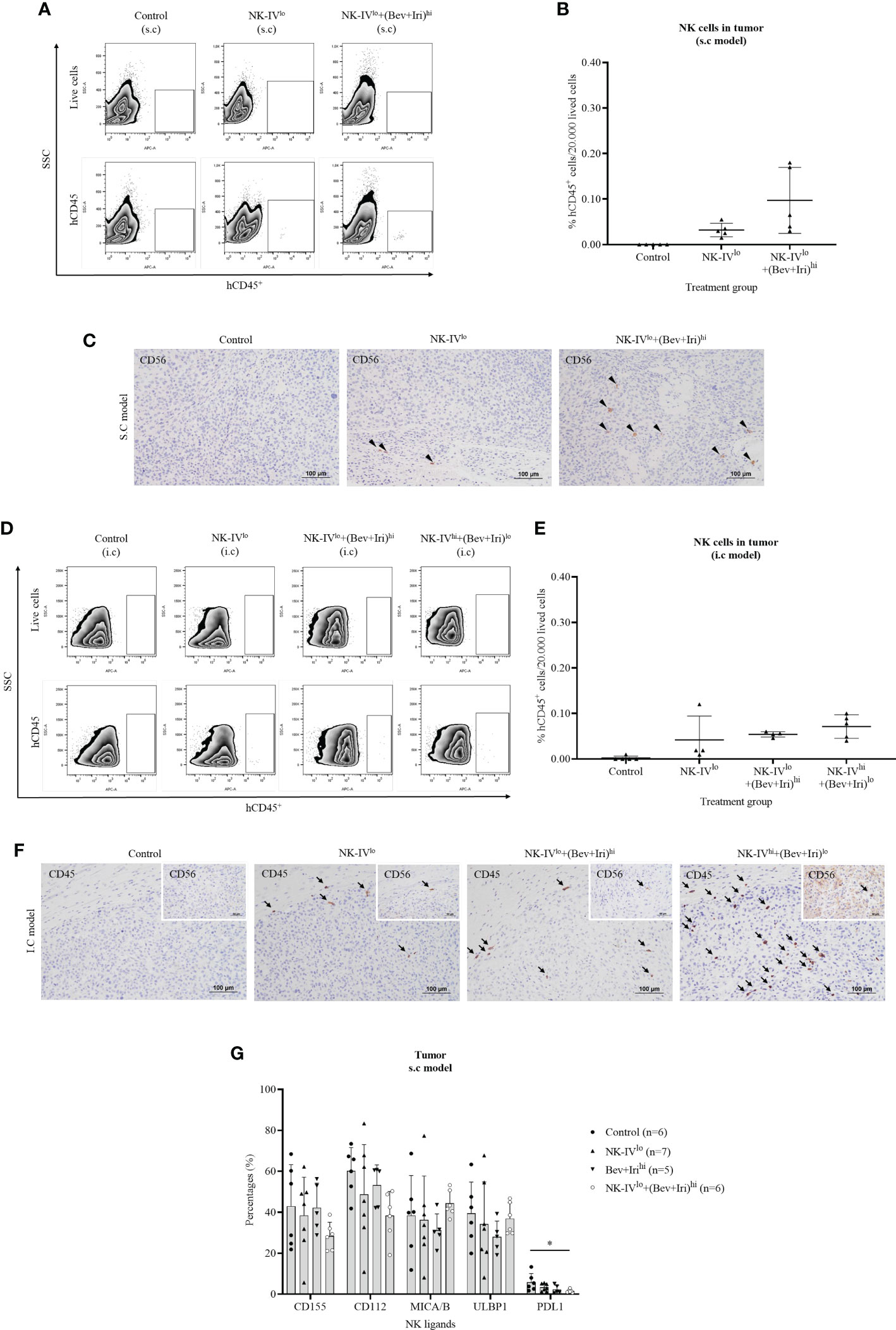
Figure 5 Presence of NK-HD cells in the tumor of s.c and i.c U87-bearing mice with or without treatment of NK-HD cells alone or NK-HD cells in combination with Bev plus Iri was confirmed through flow cytometry by hCD45 and immunohistochemistry (IHC) with hCD45 and hCD56 (A–F). The expression levels of NK ligands such as CD155, CD112, MICA/B, ULBP1, and PDL1 on single tumor cells with or without treatment of NK-HD cells alone or NK cells in combination with Bev plus Iri were also clarified using flow cytometry (G). Data are presented by percentages of positive cells. FMO: live cells without staining NK ligand markers; NK-IVlo: intravenously injected NK-HD cells (1 × 107 NK cells); NK-IVhi: intravenously injected NK-HD cells (2 × 107 NK cells); Bev+Irilo: intraperitoneally injected Bev (5 mg/kg) plus Iri (60 mg/m2); Bev+Irihi: intraperitoneally injected Bev (10 mg/kg) plus Iri (125 mg/m2); U87: human GBM cell line. All data are shown as mean ± SD. p< 0.05 (*).
The expression of NK ligands on single tumor cells in the s.c U87 mouse model is illustrated in Figures 5G; S8. The expression of NK ligands (CD155, CD112, MICA/B, and ULBP1) on single tumor cells in the s.c model did not vary among the control, NK-IVlow, Bev plus Irihigh, and NK-IVlow combined with Bev plus Irihigh groups. However, NK-IVlow combined with Bev plus Irihigh reduced the PDL1 expression compared with that of the control group in the s.c U87 mouse model (p=0.041).
Scientific reports and clinical studies have demonstrated the promising effects of NK cell therapy on cancer, particularly in GBM (34–37) (Table 2). However, NK cell function is frequently impaired in solid tumors. One of the reasons for this occurrence is that the solid tumor-associated microenvironment often becomes hypoxic and induces TGF-β, which promotes the downregulation of activated NK cell receptors in patients with cancer; consequently, their killing activity that targets tumors is impaired (38). Among various approaches, adoptive NK therapy, which involves ex vivo expanded and activated NK cells has emerged as a promising solution to overcome immunosuppression commonly observed in solid tumors by increasing the number and antitumor activity of NK cells (39). Therefore, we used NK cells that were expanded and activated according to the previously established protocol by using a feeder cell, K562 expressing the OX40 ligand and membrane-bound IL-18 and IL-21, which showed potential effects on multiple myeloma in the previous study (23); we wanted to confirm the therapeutic effects of these established NK cells in the GBM setting. Interestingly, NK-GBM showed no difference compared to NK-HDs in terms of purity, expansion fold, activated markers, and slightly lower cytotoxic effects against GBM cells with this established protocol.
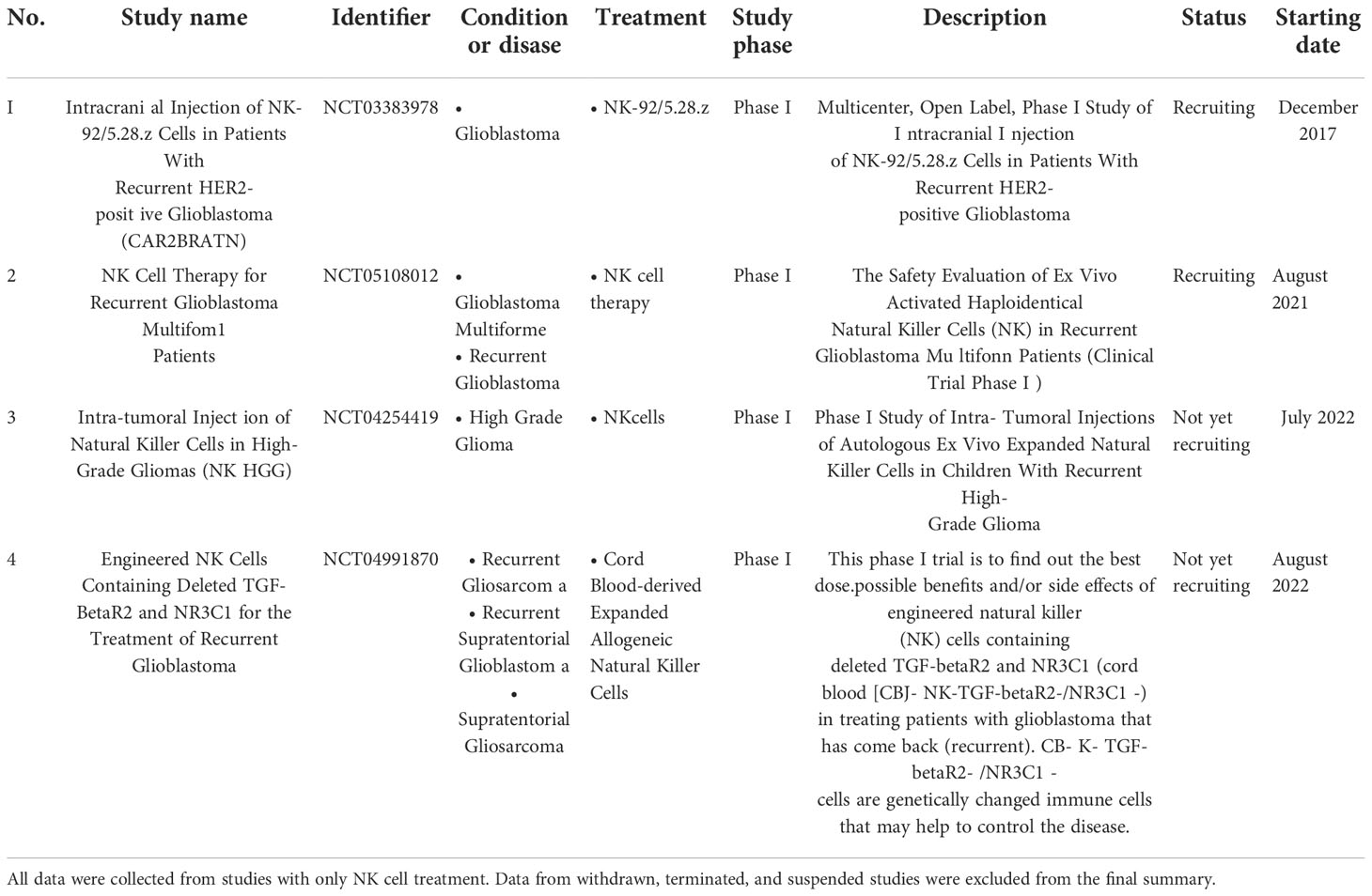
Table 2 List of completed and ongoing clinical trials of NK cell therapy on GBM was obtained from https://www.clinicaltrials.gov (keywords: glioblastoma and NK cells) until November 2022.
The combination of different treatment methods to target tumors with different mechanisms aids in overcoming the high degree of heterogeneity in solid tumors. Specifically, combination therapy of different targeted drugs with adoptively transferred NK cells helps to improve the therapeutic effects against solid tumors (40). Among different drugs, Bev and Iri have been studied separately or in combination in clinical trials for treating GBM (11, 41) (Table 3). Although Bev did not affect the proliferation of GBM cells in vitro in a previous study (42), the combination of anti-angiogenic drugs (Bev) with NK cell therapies was found to boost NK cell infiltration into the solid tumor, thereby improving efficacy (43). A trial combining bevacizumab and allogeneic NK therapy is investigating this combination approach (NCT02857920). Moreover, Bev in combination with Iri enhances the function of irinotecan through the normalization of tumor vasculature; therefore, the delivery of irinotecan into tumors is increased. A combination of Bev and Iri showed an active regimen with acceptable toxicity in recurrent malignant gliomas (44). In addition, the combination of Bev and Iri delayed tumor growth compared to Iri alone in the i.c U87 mouse model in a previous study (45). In our study, although Bev also did not affect the proliferation and NK ligand expression of GBM cells in vitro, Bev combined with Iri delayed tumor growth compared to Iri alone. A combination of Bev and Iri also reduced tumor volume and extended survival in these s.c and i.c U87 mouse models. We also clarified the role of Iri in increasing the cytotoxicity of NK cells against GBM cells through enhancement of the MICA/B expression; however, we were unable to find the relationship between this toxicity related to released IFN-γ and NK marker expression after co-culturing with Bev plus Iri-treated GBM cells. Although this is the first study to highlight the potential for using NK cells combined with Bev and Iri, a more detailed role of NK cells, Bev, and Iri in this combination should be investigated.

Table 3 A list of completed and ongoing clinical trials of combination treatment of Bev and Iri on GBM was obtained from www.clinicaltrials.gov (keywords: glioblastoma, bevacizumab, and irinotecan) until November 2022.
U87 cells, which are implanted into immunodeficient mice such as nude, NOD/SCID, and NOD/SCID gamma (NSG), are widely employed and have proved useful in the assessment of GBM angiogenesis and anti-angiogenic therapeutic approaches (46–48). In this study, the U87 mouse model was also used to confirm the therapeutic effects of NK cells in combination with Bev and Iri. However, the lack of NK cells plus Bev resulted in weak evidence to confirm the synergistic effects of Bev in enhancing NK cell infiltration. Although U87 tumors are known to lack tumor infiltration (48), a low level of macrophages and lymphocytes are still able to infiltrate U87 human GBM xenografts in nude mice, as reported in a previous study (49). In agreement with this previous finding, in the present study, the NK cells detected in tumors were very low in numbers in both s.c and i.c models by IV injection of NK cells. However, we did not clarify that low levels of NK cells infiltrated the U87 tumor after treatment due to the low migration and infiltration of NK cell target tumor or NK cells in the tumor and their death after performing a function when they were detected. More experiments should be conducted to highlight this issue using another xenograft mouse model, which displays a highly infiltrative and invasive growth pattern like U251 tumors or the GBM murine model.
Our study highlights the potential use of adoptive transfer NK cells in combination with Bev and Iri by delaying tumor growth in both s.c and i.c U87 mouse models. We showed that Iri alone has short-term effects on U87 tumors owing to the regrowth of tumors post-treatment. However, Bev combined with Iri delayed tumor growth compared to Iri alone. Moreover, NK cells combined with Bev and Iri showed slow tumor growth compared to Bev plus Iri. Thus, NK cells have targeted resistance with Bev and Iri cells. However, this combination failed to improve mouse survival. Further, NK cells have been reported to contribute to antitumor immunity not only by directly eliminating malignant cells but also by regulating tumor-specific adaptive immune response through crosstalk with dendritic cells (50). However, the complete function of NK cell therapy alone or in combination with Bev and Iri was not examined in a complete human system.
Although the NK cells that were expanded by using K562-OX40L-mb-IL-18/IL-21 feeder cells showed cytotoxic effects against GBM cells in vitro and no therapeutic effects on single treatment with s.c and i.c U87 GBM models, the combination of NK cells with Bev and Iri enhanced cytotoxic effects against GBM cells in vitro and delayed tumor progression during early-stage treatment. However, the NK cell combination with Bev plus Iri was unable to improve survival in both s.c and i.c U87 GBM models with this setting. Therefore, other settings should be considered to highlight the synergistic effects among NK cells, Bev, and Iri.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by Institutional Review Board of Chonnam National University Hwasun Hospital. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Chonnam National University Animal Research Committee.
T-A-TT, Y-HK, K-HL, and T-YJ designed and performed the experiments. T-A-TT, Y-HK, T-H-OD, K-HL, and T-YJ analyzed and presented the data. T-A-TT and T-YJ wrote the article. JT and T-HC contributed to the preliminary experiments. SJ, I-YK, K-SM, Y-JK, T-KL, J-JL, CL, HY, K-HL, and T-YJ contributed intellectually to the research. All authors contributed to the article and approved the submitted version.
This study was supported by a grant (grant no. HCRI19011) from the Chonnam National University Hwasun Hospital Institute for Biomedical Science (Republic of Korea) and by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT, and Future Planning (2020R1I1A3073338).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1009484/full#supplementary-material
1. Ostrom QT, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the united states in 2014-2018. Neuro Oncol (2021) 23(12 Suppl 2):iii1–iii105. doi: 10.1093/neuonc/noab200
2. Miller KD, Ostrom QT, Kruchko C, Patil N, Tihan T, Cioffi G, et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J Clin (2021) 71(5):381–406. doi: 10.3322/caac.21693
3. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol (2009) 10(5):459–66. doi: 10.1016/S1470-2045(09)70025-7
4. Ostrom QT, Cote DJ, Ascha M, Kruchko C, Barnholtz-Sloan JS. Adult glioma incidence and survival by race or ethnicity in the united states from 2000 to 2014. JAMA Oncol (2018) 4(9):1254–62. doi: 10.1001/jamaoncol.2018.1789
5. Krcek R, Matschke V, Theis V, Adamietz IA, Buhler H, Theiss C. Vascular endothelial growth factor, irradiation, and axitinib have diverse effects on motility and proliferation of glioblastoma multiforme cells. Front Oncol (2017) 7:182. doi: 10.3389/fonc.2017.00182
6. Ferrara N. VEGF and intraocular neovascularization: From discovery to therapy. Transl Vis Sci Technol (2016) 5(2):10. doi: 10.1167/tvst.5.2.10
7. Mukwaya A, Jensen L, Lagali N. Relapse of pathological angiogenesis: functional role of the basement membrane and potential treatment strategies. Exp Mol Med (2021) 53(2):189–201. doi: 10.1038/s12276-021-00566-2
8. Al-Ostoot FH, Salah S, Khamees HA, Khanum SA. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat Res Commun (2021) 28:100422. doi: 10.1016/j.ctarc.2021.100422
9. Haibe Y, Kreidieh M, El Hajj H, Khalifeh I, Mukherji D, Temraz S, et al. Resistance mechanisms to anti-angiogenic therapies in cancer. Front Oncol (2020) 10:221. doi: 10.3389/fonc.2020.00221
10. Cohen MH, Shen YL, Keegan P, Pazdur R. FDA Drug approval summary: Bevacizumab (Avastin (R)) as treatment of recurrent glioblastoma multiforme. Oncologist (2009) 14(11):1131–8. doi: 10.1634/theoncologist.2009-0121
11. Vredenburgh JJ, Desjardins A, Reardon DA, Friedman HS. Experience with irinotecan for the treatment of malignant glioma. Neuro Oncol (2009) 11(1):80–91. doi: 10.1215/15228517-2008-075
12. Cloughesy TF, Filka E, Kuhn J, Nelson G, Kabbinavar F, Friedman H, et al. Two studies evaluating irinotecan treatment for recurrent malignant glioma using an every-3-week regimen. Cancer-Am Cancer Soc (2003) 97(9):2381–6. doi: 10.1002/cncr.11306
13. Cloughesy TF, Filka E, Nelson G, Kabbinavar F, Friedman H, Miller LL, et al. Irinotecan treatment for recurrent malignant glioma using an every-3-week regimen. Am J Clin Oncol-Canc (2002) 25(2):204–8. doi: 10.1097/00000421-200204000-00022
14. Lu G, Rao M, Zhu P, Liang B, El-Nazer RT, Fonkem E, et al. Triple-drug therapy with bevacizumab, irinotecan, and temozolomide plus tumor treating fields for recurrent glioblastoma: A retrospective study. Front Neurol (2019) 10:42. doi: 10.3389/fneur.2019.00042
15. Moller S, Grunnet K, Hansen S, Schultz H, Holmberg M, Sorensen M, et al. A phase II trial with bevacizumab and irinotecan for patients with primary brain tumors and progression after standard therapy. Acta Oncol (2012) 51(6):797–804. doi: 10.3109/0284186X.2012.681063
16. Ali SA, McHayleh WM, Ahmad A, Sehgal R, Braffet M, Rahman M, et al. Bevacizumab and irinotecan therapy in glioblastoma multiforme: a series of 13 cases. J Neurosurg (2008) 109(2):268–72. doi: 10.3171/JNS/2008/109/8/0268
17. Gururangan S, Fangusaro J, Poussaint TY, McLendon RE, Onar-Thomas A, Wu S, et al. Efficacy of bevacizumab plus irinotecan in children with recurrent low-grade gliomas–a pediatric brain tumor consortium study. Neuro Oncol (2014) 16(2):310–7. doi: 10.1093/neuonc/not154
18. Vredenburgh JJ, Desjardins A, Herndon JE, Marcello J, Reardon DA, Quinn JA, et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J Clin Oncol (2007) 25(30):4722–9. doi: 10.1200/JCO.2007.12.2440
19. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev (2011) 91:1071. doi: 10.1152/physrev.00038.2010
20. Zhang CC, Burger MC, Jennewein L, Genssler S, Schonfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. Jnci-J Natl Cancer I (2016) 108(5):dj375. doi: 10.1093/jnci/djv375
21. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front Immunol (2019) 10:3038. doi: 10.3389/fimmu.2019.03038
22. Khatua S, Cooper LJN, Sandberg DI, Ketonen L, Johnson JM, Rytting ME, et al. Phase I study of intraventricular infusions of autologous ex vivo expanded NK cells in children with recurrent medulloblastoma and ependymoma. Neuro-Oncology (2020) 22(8):1214–25. doi: 10.1093/neuonc/noaa047
23. Thangaraj JL, Ahn SY, Jung SH, Vo MC, Chu TH, Thi Phan MT, et al. Expanded natural killer cells augment the antimyeloma effect of daratumumab, bortezomib, and dexamethasone in a mouse model. Cell Mol Immunol (2021) 18(7):1652–61. doi: 10.1038/s41423-021-00686-9
24. Thangaraj JL, Jung SH, Vo MC, Chu TH, Phan MT, Lee KH, et al. Expanded natural killer cells potentiate the antimyeloma activity of daratumumab, lenalidomide, and dexamethasone in a myeloma xenograft model. Cancer Immunol Immunother (2022). doi: 10.1007/s00262-022-03322-1
25. Thangaraj JL, Phan MT, Kweon S, Kim J, Lee JM, Hwang I, et al. Expansion of cytotoxic natural killer cells in multiple myeloma patients using K562 cells expressing OX40 ligand and membrane-bound IL-18 and IL-21. Cancer Immunol Immunother (2022) 71(3):613–25. doi: 10.1007/s00262-021-02982-9
26. Qazi MA, Vora P, Venugopal C, Sidhu SS, Moffat J, Swanton C, et al. Intratumoral heterogeneity: pathways to treatment resistance and relapse in human glioblastoma. Ann Oncol (2017) 28(7):1448–56. doi: 10.1093/annonc/mdx169
27. Garcia J, Hurwitz HI, Sandler AB, Miles D, Coleman RL, Deurloo R, et al. Bevacizumab (Avastin (R)) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat Rev (2020) 86:102017. doi: 10.1016/j.ctrv.2020.102017
28. Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol (2017) 8:1124. doi: 10.3389/fimmu.2017.01124
29. Vredenburgh JJ, Desjardins A, Herndon JE 2nd, Dowell JM, Reardon DA, Quinn JA, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res (2007) 13(4):1253–9. doi: 10.1158/1078-0432.CCR-06-2309
30. Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I, et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol (2009) 27(5):740–5. doi: 10.1200/JCO.2008.16.3055
31. Kenmotsu H, Niho S, Tsuboi M, Wakabayashi M, Ishii G, Nakagawa K, et al. Randomized phase III study of irinotecan plus cisplatin versus etoposide plus cisplatin for completely resected high-grade neuroendocrine carcinoma of the lung: JCOG1205/1206. J Clin Oncol (2020) 38(36):4292–301. doi: 10.1200/JCO.20.01806
32. Gleeson JP, Keane F, Keegan NM, Mammadov E, Harrold E, Alhusaini A, et al. Similar overall survival with reduced vs. standard dose bevacizumab monotherapy in progressive glioblastoma. Cancer Med-Us (2020) 9(2):469–75. doi: 10.1002/cam4.2616
33. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm (2016) 7(2):27–31. doi: 10.4103/0976-0105.177703
34. Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol (2021) 14(1):7. doi: 10.1186/s13045-020-01014-w
35. Pan C, Zhai Y, Li G, Jiang T, Zhang W. NK cell-based immunotherapy and therapeutic perspective in gliomas. Front Oncol (2021) 11:751183. doi: 10.3389/fonc.2021.751183
36. Sedgwick AJ, Ghazanfari N, Constantinescu P, Mantamadiotis T, Barrow AD. The role of NK cells and innate lymphoid cells in brain cancer. Front Immunol (2020) 11:1549. doi: 10.3389/fimmu.2020.01549
37. Burger MC, Zhang C, Harter PN, Romanski A, Strassheimer F, Senft C, et al. CAR-engineered NK cells for the treatment of glioblastoma: Turning innate effectors into precision tools for cancer immunotherapy. Front Immunol (2019) 10:2683. doi: 10.3389/fimmu.2019.02683
38. Chambers AM, Lupo KB, Matosevic S. Tumor microenvironment-induced immunometabolic reprogramming of natural killer cells. Front Immunol (2018) 9:2517. doi: 10.3389/fimmu.2018.02517
39. Shin MH, Kim J, Lim SA, Kim J, Kim SJ, Lee KM. NK cell-based immunotherapies in cancer. Immune Netw (2020) 20(2):e14. doi: 10.4110/in.2020.20.e14
40. Li L, Li W, Wang C, Yan X, Wang Y, Niu C, et al. Adoptive transfer of natural killer cells in combination with chemotherapy improves outcomes of patients with locally advanced colon carcinoma. Cytotherapy (2018) 20(1):134–48. doi: 10.1016/j.jcyt.2017.09.009
41. Agha CA, Ibrahim S, Hassan A, Elias DA, Fathallah-Shaykh HM. Bevacizumab is active as a single agent against recurrent malignant gliomas. Anticancer Res (2010) 30(2):609–11.
42. von Baumgarten L, Brucker D, Tirniceru A, Kienast Y, Grau S, Burgold S, et al. Bevacizumab has differential and dose-dependent effects on glioma blood vessels and tumor cells. Clin Cancer Res (2011) 17(19):6192–205. doi: 10.1158/1078-0432.CCR-10-1868
43. Xu C, Liu D, Chen Z, Zhuo F, Sun H, Hu J, et al. Umbilical cord blood-derived natural killer cells combined with bevacizumab for colorectal cancer treatment. Hum Gene Ther (2019) 30(4):459–70. doi: 10.1089/hum.2018.011
44. Mesti T, Moltara ME, Boc M, Rebersek M, Ocvirk J. Bevacizumab and irinotecan in recurrent malignant glioma, a single institution experience. Radiol Oncol (2015) 49(1):80–5. doi: 10.2478/raon-2014-0021
45. Wang W, Sivakumar W, Torres S, Jhaveri N, Vaikari VP, Gong A, et al. Effects of convection-enhanced delivery of bevacizumab on survival of glioma-bearing animals. Neurosurg Focus (2015) 38(3):E8. doi: 10.3171/2015.1.FOCUS14743
46. Kirsch M, Strasser J, Allende R, Bello L, Zhang J, Black PM. Angiostatin suppresses malignant glioma growth in vivo. Cancer Res (1998) 58(20):4654–9.
47. Schmidt NO, Ziu M, Carrabba G, Giussani C, Bello L, Sun Y, et al. Antiangiogenic therapy by local intracerebral microinfusion improves treatment efficiency and survival in an orthotopic human glioblastoma model. Clin Cancer Res (2004) 10(4):1255–62. doi: 10.1158/1078-0432.CCR-03-0052
48. Haddad AF, Young JS, Amara D, Berger MS, Raleigh DR, Aghi MK, et al. Mouse models of glioblastoma for the evaluation of novel therapeutic strategies. Neurooncol Adv (2021) 3(1):vdab100. doi: 10.1093/noajnl/vdab100
49. Candolfi M, Curtin JF, Nichols WS, Muhammad AG, King GD, Pluhar GE, et al. Intracranial glioblastoma models in preclinical neuro-oncology: neuropathological characterization and tumor progression. J Neurooncol (2007) 85(2):133–48. doi: 10.1007/s11060-007-9400-9
Keywords: natural killer cells, bevacizumab, irinotecan, U87 cell line, glioblastoma
Citation: Tran T-A-T, Kim Y-H, Duong T-H-O, Thangaraj J, Chu T-H, Jung S, Kim I-Y, Moon K-S, Kim Y-J, Lee T-K, Lee CW, Yun H, Lee J-J, Lee H-J, Lee K-H and Jung T-Y (2023) Natural killer cell therapy potentially enhances the antitumor effects of bevacizumab plus irinotecan in a glioblastoma mouse model. Front. Immunol. 13:1009484. doi: 10.3389/fimmu.2022.1009484
Received: 02 August 2022; Accepted: 30 November 2022;
Published: 10 January 2023.
Edited by:
Peter Brossart, University of Bonn, GermanyReviewed by:
Adeleh Taghi Khani, Beckman Research Institute, United StatesCopyright © 2023 Tran, Kim, Duong, Thangaraj, Chu, Jung, Kim, Moon, Kim, Lee, Lee, Yun, Lee, Lee, Lee and Jung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tae-Young Jung, anVuZy10eUBjaG9ubmFtLmFjLmty; Kyung-Hwa Lee, bWRrYXlsZWVAY2hvbm5hbS5hYy5rcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.