
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 06 October 2022
Sec. Cancer Immunity and Immunotherapy
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1008751
This article is part of the Research Topic CAR T-cells: novel therapeutic approaches in the new era of cancer immunotherapy View all 6 articles
 Sun Il Choi1,2
Sun Il Choi1,2 Jinlong Yin1,2*
Jinlong Yin1,2*Glioblastoma (GBM) is the most common malignant brain tumor. The poor clinical outcome and overall ineffectiveness of current standard treatments, including surgery, chemotherapy, and radiation, highlight the urgent need for alternative tumor-specific therapies for GBM. Chimeric antigen receptor (CAR) T cell therapy is a revolutionary therapeutic strategy for hematological malignancies, but the optimal potency of CAR T cell therapy for solid tumors, especially GBM, has not been achieved. Although CAR T cell therapeutic strategies for GBM have been assessed in clinical trials, the current antitumor activity of CAR T cells remains insufficient. In this review, we present our perspective on genetically modifying CAR constructs, overcoming T cell dysfunctions, and developing additional treatments that can improve CAR T cell effectiveness, such as functionality, persistence, and infiltration into tumor sites. Effectively improved CAR T cells may offer patients with GBM new treatment opportunities, and this review is intended to provide a comprehensive overview for researchers to develop potent CAR T cells using genetic engineering or combinatorial preparations.
Chimeric antigen receptor (CAR) T cell therapy has emerged due to the development of genetically engineered T cell receptors (TCRs) to achieve specific target cancer-associated antigens (Figure 1). Adoptive transfer of autologous CAR T cells prolongs patient survival times and promotes remission, even in some patients who do not respond to standard treatment (1). Since the first CD19-CAR T cell therapy for patients with acute lymphoblastic leukemia (ALL) was approved by the FDA in 2017, investigators have concentrated on extending the therapeutic effects of CAR T cells not only for B cell malignancies but also for several other cancers, including solid tumors.
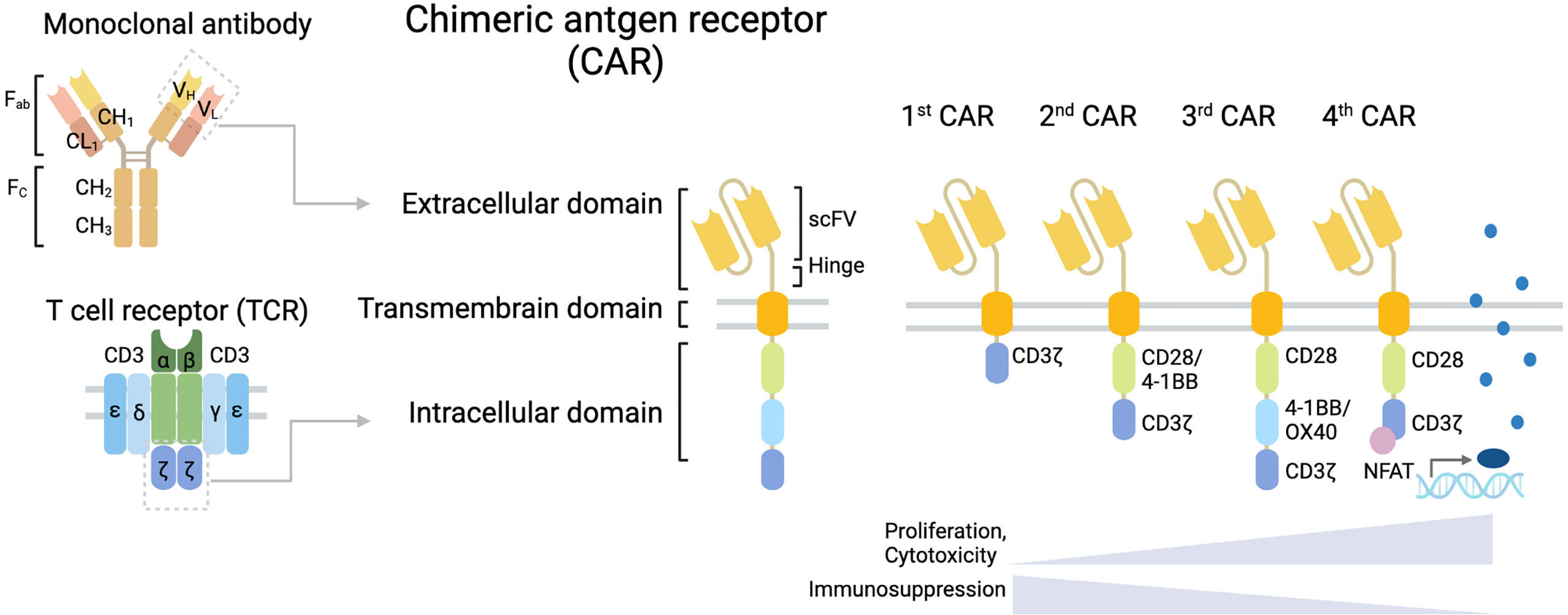
Figure 1 Structure of CAR T cell. CAR T cells have a structural design with three major domains: an extracellular domain including a hinge (space), a transmembrane domain, and an intracellular signaling domain. First-generation CARs include a single-chain variable fragment (scFv) targeting tumor-associated antigen (TAA) linked to a space in an extracellular domain, a transmembrane domain, and the CD3ζ chain of the T cell receptor (TCR) as an intracellular signaling domain. Second-generation CARs are enhanced CAR T cell activity by adding a costimulatory domain, such as CD28 or 4-1BB to support the expansion and persistence of engineered T cells. Third-generation CARs contain several costimulatory domains.
Glioblastoma (GBM), the most common and highly aggressive malignant brain tumor, has a poor prognosis with a median overall survival time of <16 months (2, 3). Currently, the approved GBM treatments typically include surgery followed by radiotherapy or chemotherapy, with a 5-year survival rate below 10%. Several immunotherapeutic approaches have been examined in clinical trials for patients with GBM. However, the impact of CAR T cells on early clinical outcomes was limited as the cells did not generate sufficient antitumor activity, and there is still no FDA-approved CAR T therapy for GBM despite safe and promising results (4–7). The development and research of CAR T cell therapy against GBM remains challenging to date due to complications, including antigen heterogeneity, loss of cells rendering CAR obsolete (T cell exhaustion), immunosuppressive molecules and cells in the tumor microenvironment (TME), and insufficient CAR T cells penetration and trafficking to tumor sites (Figure 2) (8–11).
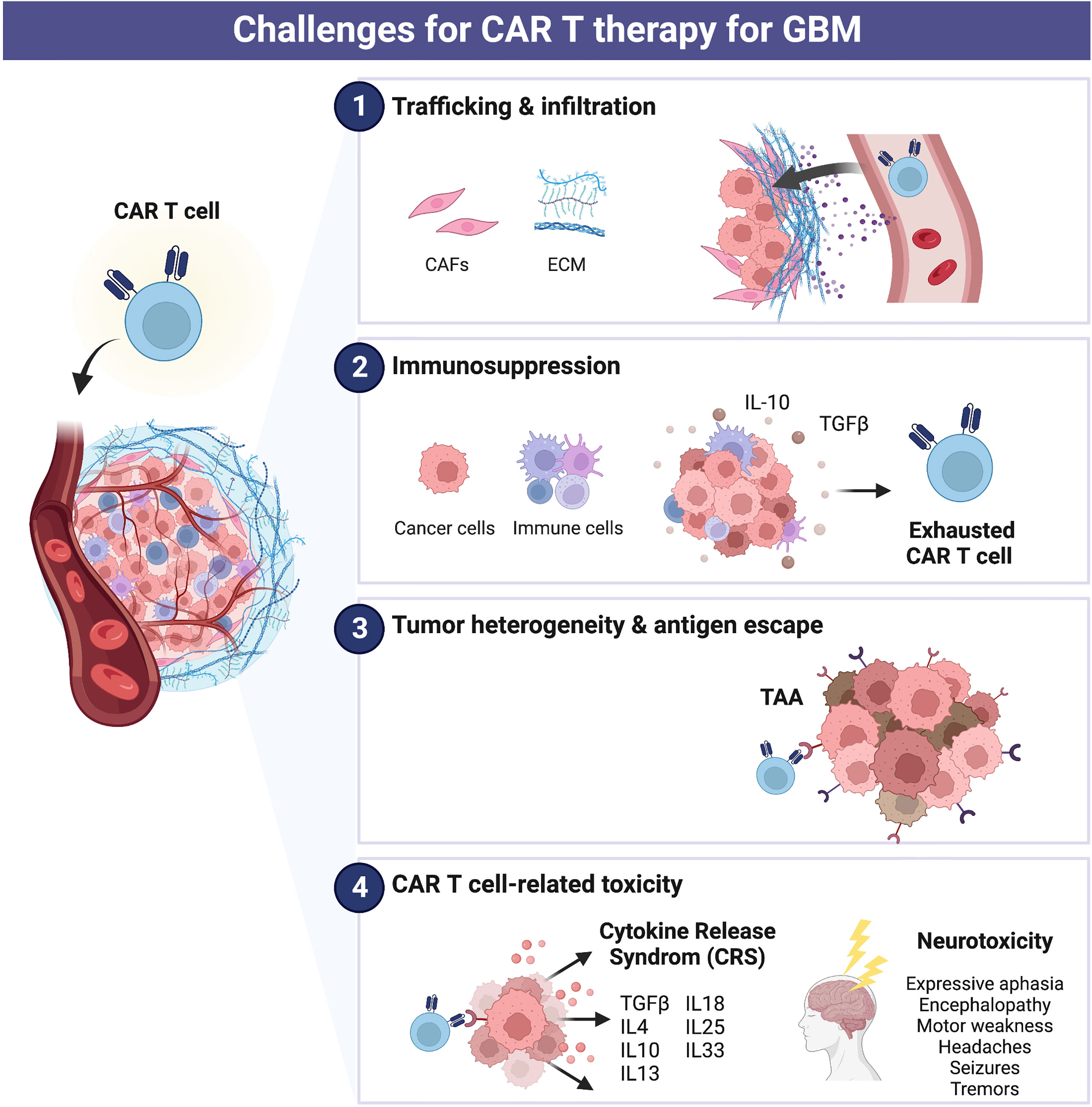
Figure 2 Challenges for CAR T therapy for glioblastoma (GBM). Current major limitations of CAR cell therapy include (1) trafficking and infiltration, (2) immunosuppression, (3) tumor heterogeneity and antigen escape, and (4) CAR T cell-related toxicity.
Here, we review current and emerging strategies in the field of CAR T cell therapy with a focus on the following three aspects (1): modification of the CAR construct, (2) overcoming T cell dysfunctions, and (3) the addition of chemical and external treatments (Figure 3). We discuss the individual strength of these technologies and their applications for GBM therapy. Although potential barriers to CAR T cell therapy still exist, effectively improved CAR T cell therapy could offer patients with GBM new treatment opportunities. This review provides a comprehensive summary of individual strengths for those aiming to develop more advanced CAR T cells using genetically modified constructs or combinatorial preparations.
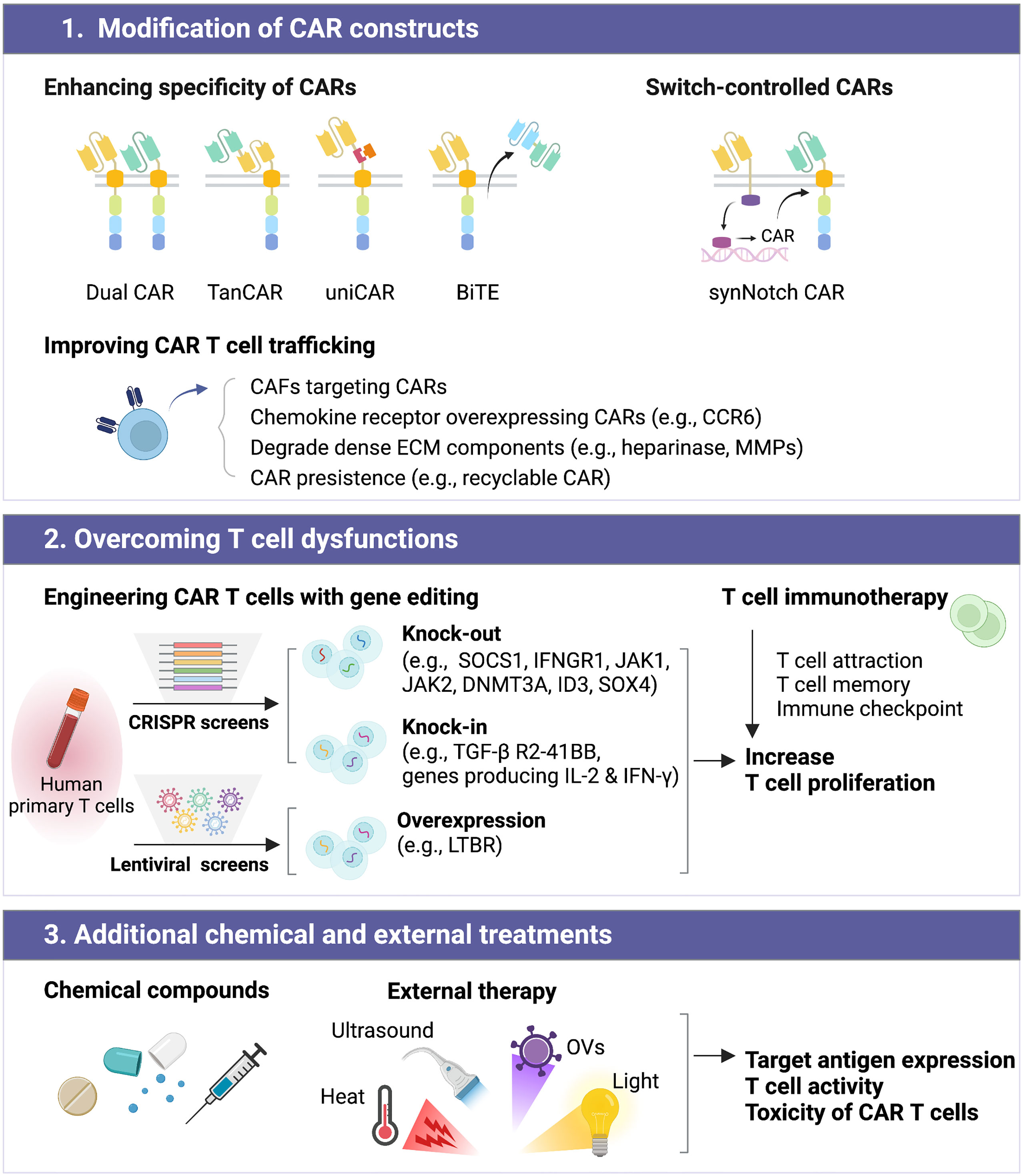
Figure 3 Improving the efficacy of CAR T cell therapy can be classified into distinct subgroups, including (1) modification of CAR constructs, (2) overcoming T cell dysfunctions, and (3) additional chemical and external treatments.
CAR T cell structure comprises three major domains: an extracellular domain, a transmembrane domain, and an intracellular signaling domain (Figure 1). Each domain has a unique function and requires an optimal design for CAR efficacy. Additionally, CAR designs have been adjusted to improve clinical effectiveness, and new design approaches are emerging. Indeed, the CAR T cells currently under development have been designed by genetically modifying the CAR construct to compensate for the weakness of existing CAR T cells, such as poor activity against GBM or severe toxicity caused by CAR T cell overactivation.
One of the reasons for the poor activity of GBM CAR T cells is the low target specificity of CARs. Although IL-13Rα2, EGFRvIII, and HER2 are the most frequently targeted antigens in preclinical and clinical studies on CAR T therapy for GBM, these antigens have important limitations (4–7). Therefore, several approaches have been investigated to overcome the antigen heterogeneity and immune escape leading to loss of antigenicity and/or immunogenicity when a single antigen is targeted in GBM, and some of these approaches reflect the strategies used for hematological malignancies.
Researchers have designed CAR T cells to have two CARs, each targeted to a different type of tumor-related antigens (DualCARs) or assembled two or more scFvs linearly (tandem CARs; TanCARs), similar to the bispecific antibody platform with dual affinity retargeting (12). However, currently, CD19/CD22- (13) and CD19/CD20-targeting approaches such as these CARs have limited the clinical experience for leukemia therapy (7, 14). Another approach is to design CARs that split from CAR molecules to the targeting domain, which is capable of binding multiple undefined antigens (universal CAR; uniCAR), and as a typical example, the T1E peptide binding to EGFR family receptors is the most relevant example for solid tumors (15). Moreover, bispecific T cell engagers (BiTEs) platforms have recently been developed capable to target multiple antigens with flexibility for combinatorial and sequential approaches and redirecting T cells activity toward tumor cells. Furthermore, other strategies are to design T cells capable of recognizing tumor cells by inducing bystander T cell activation, including the transgenic expression of cytokines (e. g., IL-18 or IL-36γ) or ligands (e.g., CD40L) (16). Similarly, one study revealed that engineered T cells to secrete the dendritic cell (DC) growth factor Fms-like tyrosine kinase 3 ligand (Flt3L) induces host T cell activation (17). In addition, in mouse GBM models, anti-Fn14×CD3 BiTE given intratumorally showed antitumor effects whereas xenografts treated with Fn14 CAR T cells recurred (18). Furthermore, to address antigenic heterogeneity in GBM, investigators have developed gene modifying EGFRvIII CAR T cells to deliver bispecific EGFR antibodies (EGFRvIII CAR T BiTEs) (19).
Most antigens targeted by CAR T cells are tumor-associated rather than tumor-specific, which means highly expressed on the tumor and have low abundance in normal tissues, capable of leading to providing on-target/off-tumor toxicities. Much research has been conducted on switch-controlled CARs, which can solve these problems by controlling activation to kill tumors while providing inhibitory mechanisms such as a CAR T cell suicide activation and methods to regulate the costimulatory pathway.
One of the best switch-controlled CAR T cells is using an antigen-specific synthetic notch (synNotch) system, conferring the expression of a second CAR upon activation (20). Numerous investigators have reported SynNotch gated CAR-T cells, such as SynNotch-GD2-B7H3, which recognize GD2 as the gate and B7H3 as the target (21), and SynNotch-HER2 (22), which controls the killing effects. The low-affinity synNotch receptor performs as a filter, limiting transcriptional induction until T cells encounter high antigen-expressing target cells. Once passed through this initial filter, the induced high-affinity CAR can perform potent T cell proliferation and killing activities. This synNotch CAR low-high circuit design allows CAR-T cells to express CAR molecules only in the presence of tumor cells with high antigen density, thereby killing tumor cells but not healthy cells with low antigen density.
In addition, two synNotch CAR researchers recently used a different approach in which stable levels of activity were maintained throughout the anticancer process, eliminating the problem of T cell depletion that has hampered traditional CAR T therapy (23, 24). They pursued two such multiantigen targeting strategies for GBM, EGFRvIII-SynNotch CAR and EphA2 or IL13Ra2 CARs (23), and approaches for mesothelioma and ovarian cancer involving alkaline phosphatase placental-like 2 (ALPPL2)-synNotch CAR and MCAM, MSLN or HER2 CAR (24). Traditional CAR T cells are designed to consistently express a kill switch; however, in these two SynNotch CAR T cells can save energy by going into standby mode when not in direct contact with the tumor.
CAR T cells for hematological malignancies and their corresponding target malignancies share a hematopoietic origin and therefore tend to migrate to similar areas, such as the bone marrow or lymph nodes. In comparison, most solid tumors, including GBM, do not readily attract T cells and are known as cold tumors due to their poor immunogenic status. Therefore, CAR T cells for GBM should be engineered to increase CAR T cell trafficking, allowing them to penetrate the extremely dense stroma and be attracted to tumors.
Stromal cells, such as cancer-associated fibroblasts (CAFs), express high levels of fibroblast activation protein (FAP) (25) and SLAMF7 (26) and induce remodeling of the aberrant extracellular matrix (ECM), which limits T cell motility and trafficking. Some groups have reported that combination treatment with FAP-targeting CAR T cells and tumor-targeting CAR T cells or cancer vaccines showed enhanced antitumor immunity, but there are conflicting results regarding FAP-targeting CAR T cells, which had on-target, off-tumor toxicity against bone marrow stromal cells (8). Thus, targeting stromal cells expressing FAP requires further investigation of the efficacy and toxicity profile of FAP-targeted CAR T cells (10). Meanwhile, dual-targeting CAR T cells for both malignant plasma cells and CAFs showed a potential ability to reverse TME-induced CAR T cell suppression (26).
Moreover, chemokine receptor-overexpressing CAR T cells can potentially be attracted to tumors that highly express chemokine receptors, such as CCR6, leading to effective tumor clearance (27, 28). Additionally, in parallel, technologies to enable in vivo real-time imaging of CAR T cells are being generated and assessed in preclinical and early phase clinical trials (29, 30). Such a strategy would enable the noninvasive and rapid examination to improve CAR T cell trafficking to tumor cells and antitumor function.
Furthermore, another approach is to efficiently degrade dense ECM components to promote CAR T cell infiltration to the tumor and its antitumor activity. Researchers have attempted to engineer CAR T cells with enzymes that degrade dense ECM with developed stiffness and crosslinking, such as heparinase (which degrades heparan sulfate proteoglycan) and matrix metalloproteinases (MMPs; which proteolysis all ECM components in a Zn-dependent manner) (8, 31).
In addition, CAR persistence is also important for CAR T cell trafficking. For example, CAR ubiquitination is triggered during CAR encounters with tumor antigens. Thereby, ubiquitination-blocked recyclable CAR, mutating the intracellular ubiquitination site (CARKR), could elevate endosomal CAR signaling and be recyclable back to the cell surface, leading to redirecting CAR trafficking (32).
Most patients, except for B lymphoblastic leukemia patients, do not show a sustained response to CAR T cells and develop resistance mainly causing T cell dysfunction (33, 34). Substantial efforts have been made to identify genes and pathways contributing to T cell dysfunction. In this section, we explain the modulation of CAR T cell function by structural engineering, leading to premature CAR T cell differentiation and the prevention of exhaustion.
The utility of CRISPR-Cas9 genome engineering, in other words, loss-of-function screening, has developed it possible to easily knock-out any gene in the genome with extension and customization. Deleting or inhibiting some genes enhances CAR T cell activity (35, 36). Several investigators have identified a gene signature that defines CAR and TCR dysregulation and transcription factors as key regulators of CAR T cell exhaustion using CRISPR-Cas9 screens. In CD19 CARs in CD4+ or CD8+ T cells with deletion of suppressor of cytokine signaling 1 (SOCS1), a major nonredundant checkpoint that inhibits CD4+ T cell proliferation by blocking multiple downstream signaling molecules, both IL-2 and IFN-γ, results in a markedly improved antitumor effect (37). Additionally, loss of genes in the IFN-γR signaling pathway (IFNGR1, JAK1, or JAK2) reduces overall CAR T cell binding duration and avidity in GBM, with other solid tumors being more resistant to CAR T cell-mediated death (38). The deletion of the de novo DNA methyltransferase 3 alpha (DNMT3A) in first- or second-generation CAR T cells universally preserved the cell proliferation and antitumor response during prolonged tumor exposure (39). The exhaustion-resistant DNMT3A knock-out CAR T cells have been coupled with the upregulation of IL-10, and genome-wide DNA methylation profiling revealed an atlas of genes targeted for epigenetic silencing (39). Moreover, the deletion of ID3 or SOX4 may attenuate cytotoxicity, relative to a CD8+ T to-NK-like T cell transition (NK-like CAR T cells), suggesting a potential strategy to enhance the efficacy of CAR T cell therapy against solid tumors (33). Furthermore, knocking out transforming growth factor-β receptor II (TGF-βRII) in CAR via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors (40). Recently, the results of the phase I trials have revealed that prostate-specific membrane antigen (PSMA) CAR T optimally engineered with TGF-βRII to block TGF-β signaling to demonstrate improved T cell expansion in blood, tumor trafficking, and enhanced antitumor immunity in metastatic castration-resistant prostate cancer (41). These atlases enhance CAR T cell efficacy and provide a molecular definition of CAR T cell exhaustion.
Advances in CRISPR technology have also allowed for the investigation of T cell function beyond loss-of-function screens, improving our understanding of the regulators of T cell activation as a result of gain-of-function gene perturbations and providing further insight into disease pathways. One research group utilized a widely adaptable technology for barcoding and tracking targeted integrations of large-scale nonviral DNA templates for pooled knock-in screens in primary human T cells and pooled knock-in sequencing (PoKI-seq), combining single-cell transcriptome analysis in vitro and in vivo (42). Through this platform, they engineered transforming growth factor-β (TGF-β) R2-41BB chimeric receptor to promote solid tumor clearance (42). Another group reported using genome-wide CRISPR activation (CRISPRa) and interference (CRISPRi) screens to construct gene networks regulating IL-2 and IFN-γ production in primary human T cells (43). Alterations in the cytokine response were confirmed through key hits by arrayed CRISPRa and multiplexed secretome characterization. Combining CRISPRa screening with single-cell RNA sequencing for deep molecular characterization of screened hits is possible, demonstrating how perturbations modulate T cell activation and promote cellular states characterized by notable cytokine expression profiles. The genes involved in reprogramming critical immune cell functions identified through these screens might be applied in the design of immunotherapies.
Although CRISPR screens offer complexity and diversity, lentiviral screens have readily been applied for the T cell phenotype and are well established, suggesting opportunities for clinical translation. For instance, one research group achieved a genome-wide gain-of-function screen in primary human CD8+ and CD4+ T cells by applying barcoded human open reading frames (ORFs) and identified the top-ranked genes and key cytokines (44). From these results, the lymphotoxin-β receptor (LTBR), which is the high-ranked ORF, causes profound transcriptional and epigenomic remodeling, activating the NF-κB pathway in overexpressed T cells, thereby increasing T cell effector functions in the chronic stimulatory environment and inducing depletion, i.e., the antigen-specific responses of CAR T cells were improved.
Several approaches to T cell immunity have been published recently. After chemotherapy with cisplatin, producing CCL20 and IL-1β at the tumor site induces to recruiting and activating innate lymphoid cells (ILC3s) in tumors. Then, activated ILC3s generate CXCL10, which attracts CD4+ and CD8+ T cells to tumors, enhancing antitumor immunity (45). Moreover, the approved pharmacologic inhibitors of cyclin-dependent kinases 4 and 6 (CDK4/6) promote the phenotypic and functional acquisition of immunologic T cell memory (46). This applies to the design of clinical trial protocols to prevent CAR T cell exhaustion during therapy. Indeed, one research group identified protein tyrosine phosphatase 1B (PTP1B) as an intracellular checkpoint that is upregulated in T cells in tumors; additionally, they revealed that deletion or inhibition of PTP1B improves the efficacy of adoptively transferred CAR T cells against solid tumors (47). Their hypothesis rationalized the relationship between cytokine-induced JAK/STAT signaling, which is well established to be fundamentally important in all aspects of immunity, particularly in T cell activation and homeostasis, and PTP1B, which can attenuate this signaling. Such successful immunotherapy cases, theories, and evidence can be actively implemented for the development of CAR T cell treatments.
Overall, transient suspension of CAR signaling molecules and receptor redesign have been proposed to improve antitumor activity. Alternatively, signaling levels might be reduced, allowing for more controlled and dynamic regulation of CARs, similar to the natural T cell receptor.
Chemical compounds can increase target antigen expression or T cell activities and have a synergetic effect on CAR T cells. The enhancer of zeste 2 polycomb repressive complex 2 (Ezh2) subunit inhibitor has upregulated GD2 expression and enhanced GD2 CAR T efficacy in Ewing sarcoma (48). Sunitinib, a multi-tyrosine kinase inhibitor, has upregulated carbonic anhydrase IX (CAIX) expression and improved the efficacy of anti-CAIX CAR T cells in renal cancer (49). Bryostatin1, a protein kinase C modulator, and its analogs were shown to upregulate CD22 to enhance CD22-CAR T cell efficacy (50, 51). The pharmacologically selective inhibition of PTP1B using MSI-1436 induced the T cell-mediated tumor suppression and enhanced PD-1 blockade response (47). Moreover, a combination of anti-PD-1-based immunotherapy with IFN-α resulted in encouraging anticancer activity in unresectable HCC patients (52). In infiltrating CD8+ T cells, IFN-α downregulates the ability to consume glucose and consequentially establishes a high glucose microenvironment promoting the transcription of CD27, which is the T cell costimulatory molecule.
By reversibly binding to CAR T cells, small compounds also play a safety switch role to address the toxicity of CAR T cell therapy by reversibly binding to CAR T cells. In lymphoma, the clinically approved drug dasatinib, which inhibits the phosphorylation of CD3, LCK, and ZAP70, results in restricted induction of NFAT and induces CD8+ and CD4+ CAR T cells to be a function-off state (53). Lenalidomide intercedes the proteasomal degradation of several target molecules through modulation of CRL4CRBN E3 ubiquitin ligase and a C2H2 zinc finger degron motif (54) that functions as a switch molecule, has been shown to be a safe induction agent for genetic vehicles designed for therapeutic uses. Duong’s research group developed dual-switch CAR T cells in which one is a potent activation switch based on rimiducid-inducible MyD88 and CD40 (iMC)-signaling elements to improve CAR T cell efficacy, and the other is an orthogonally regulated, rapamycin-induced, caspase-9-based safety switch (iRC9) to neutralize potential toxicity by this reinforced CAR (55). Similarly, Yang’s research group constructed two types of CAR T cells with the stilbenoid natural product resveratrol (3,4’,5-trihy-droxystilbene) as a switch molecule, containing gene circuits that can control the activation (on) and inactivation (off) of CAR T cells (56). These tunable dual-switch systems provide a safety switch to reduce toxicity, thus supporting greater CAR T cell expansion and long-term persistence in a drug-dependent manner.
Small molecule compounds such as those mentioned above are likely sufficiently safe; preferably, such compounds have low immunogenicity and clear pharmacodynamic and pharmacokinetic properties. Small molecule compounds should also be selectively reached to target tumor tissues via drug-directed delivery techniques, further mitigating off-target toxicity. Meanwhile, several compounds with CAR T cells have already been demonstrated to exhibit synergistic effects in clinical trials. For example, B-cell lymphoma patients achieved complete remission following a combination of anti-CD19 CAR T cells and decitabine (57). A case study revealed that CD19-CAR T cell therapy with dasatinib induced complete remission in lymphoid blast phase chronic myeloid leukemia harboring the T315I mutation in the BCR-ABL fusion gene (58). Moreover, combination CAR T cell therapy following ibrutinib in patients with relapsed or refractory CLL demonstrated the probability of one-year progression-free survival and lower CRS-associated cytokines in serum (59). Recently, the high-performance drug-regulatable system termed signal neutralization by an inhibitable protease (SNIP) using an FDA-approved small molecule with favorable pharmacokinetics in humans has been reported to validate no side effects and to outperform constitutive CARs in multiple solid tumor models (60). These SNIP CAR T cells serve as a predictable safety switch that can stop when lethal toxicity begins during drug administration, achieving more functional, less exhausted, and reliable levels of CAR T cells. As well as the reduced drug administration controls SNIP CAR T cells to be within the therapeutic range, eliminating tumor cells with high antigen expression, while preserving healthy tissue with lower antigen expression in an on-target off-tumor toxicity mouse model (60).
Moreover, especially in GBM patients, the CRS cytotoxic response is even more critical, as further elevated intracranial pressure in patients associated with swollen mass due to the effect of tumor can be lethal. Dexamethasone is routinely used to treat cerebral edema in patients with CNS tumors. Some clinical studies suggest that corticosteroids have an immunosuppressive effect on the desired antitumor effects of CAR T cells, but clinical evidence remains scarce (61). Dexamethasone upregulates cytotoxic T lymphocyte-associated antigen 4 (CTLA4), an immune checkpoint receptor expressed on T cells, and blocks the CD28 costimulatory pathway, which reduces T cell proliferation and differentiation (62). Conversely, low-dose dexamethasone did not reduce CAR T cell antitumor effects in orthotopic xenograft GBM models (63). By introducing a mathematical model, it was suggested that a critical threshold of CAR T cell death concerning the proliferation rate could guide the dose and timing of CAR T cell delivery in patients administered dexamethasone (64). Future clinical studies are required to determine the effects of dexamethasone on CAR T therapy.
Recently, several unique approaches to interrogate CAR-mediated antitumor immunity have been reported. Newly designed controllable CAR T cells, including ultrasound (FUS) control of induced heat-shock-protein promoter (Hsp) in CD19 CAR T cells (65) and photothermal modulating an IL-15 superagonist or a natural killer group 2D ligand (NKG2DL) BiTE (66), have also been reported. These switch-CAR T cells also reduced tumor burden, did not affect the surrounding tissue, improved survival, and mitigated antigen escape. As another example, the light-switchable CAR (LiCAR) T cells exhibit allowing spatial, temporal control, and T cell-mediated antitumor therapeutic effects through real-time photo-dependent activation (67).
Furthermore, engineered oncolytic viruses (OVs) exhibit tumor selectivity, targeted transgene delivery to tumors, and desirable immunogenic properties making a promising treatment approach for solid tumors. An engineered OV expressing a non-signaling truncated CD19 (CD19t) protein for selectively delivery to tumor enables targeting by CD19-CAR T cells. Infected tumor cells with an oncolytic vaccinia virus coding for CD19t (OV19t) produces de novo CD19 at the cell surface and subsequent virus-mediated tumor cytolysis (68). Recently, another OV study revealed that induction of the native TCR with viral or virally encoded epitopes enhances CAR T cell proliferation, activity, and efficacy in mice with intracranial GBM tumors (69).
As a breakthrough treatment for GBM, CAR T therapy is crucial, through better optimization of CAR T cells is needed. Lessons learned from T cell immunotherapies have set the new stage for the application of CAR T therapy, and it is critical to identify more selective approaches, leveraging our understanding of CAR T cell resistance and toxicity. As genetic engineering rapidly advances, we have an exciting and unique opportunity to improve outcomes for GBM patients. Further basic scientific studies of CAR T cell development involving comparisons with CAR T cells for hematological malignancies are necessary to promote the translation of immunotherapy and establish it as efficient anticancer treatment.
The conception and design of the paper were performed by SIC and JY. The initial draft was completed by SIC. Editing and further drafts were performed by JY. All authors reviewed the final version of the manuscript.
This work was supported by a grant from the National Natural Science Foundation of China (82173228).
Figures were created using BioRender.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Charrot S, Hallam S. Car-T cells: Future perspectives. Hemasphere (2019) 3(2):e188. doi: 10.1097/HS9.0000000000000188
2. McKinnon C, Nandhabalan M, Murray SA, Plaha P. Glioblastoma: Clinical presentation, diagnosis, and management. BMJ (2021) 374:n1560. doi: 10.1136/bmj.n1560
3. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 who classification of tumors of the central nervous system: A summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
4. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med (2016) 375(26):2561–9. doi: 10.1056/NEJMoa1610497
5. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. Her2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: A phase 1 dose-escalation trial. JAMA Oncol (2017) 3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184
6. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused egfrviii-directed car T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9(399):peaaa0984. doi: 10.1126/scitranslmed.aaa0984
7. Dunn GP, Cloughesy TF, Maus MV, Prins RM, Reardon DA, Sonabend AM. Emerging immunotherapies for malignant glioma: From immunogenomics to cell therapy. Neuro Oncol (2020) 22(10):1425–38. doi: 10.1093/neuonc/noaa154
8. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in car T cell therapy. Nat Rev Clin Oncol (2020) 17(3):147–67. doi: 10.1038/s41571-019-0297-y
9. Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of car T cells. Nat Rev Cancer (2021) 21(3):145–61. doi: 10.1038/s41568-020-00323-z
10. Hou AJ, Chen LC, Chen YY. Navigating car-T cells through the solid-tumour microenvironment. Nat Rev Drug Discov (2021) 20(7):531–50. doi: 10.1038/s41573-021-00189-2
11. Akbari B, Ghahri-Saremi N, Soltantoyeh T, Hadjati J, Ghassemi S, Mirzaei HR. Epigenetic strategies to boost car T cell therapy. Mol Ther (2021) 29(9):2640–59. doi: 10.1016/j.ymthe.2021.08.003
12. Wagner J, Wickman E, DeRenzo C, Gottschalk S. Car T cell therapy for solid tumors: Bright future or dark reality? Mol Ther (2020) 28(11):2320–39. doi: 10.1016/j.ymthe.2020.09.015
13. Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. Cd22-targeted car T cells induce remission in b-all that is naive or resistant to Cd19-targeted car immunotherapy. Nat Med (2018) 24(1):20–8. doi: 10.1038/nm.4441
14. Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T Cells expressing Cd19/Cd20 bispecific chimeric antigen receptors prevent antigen escape by malignant b cells. Cancer Immunol Res (2016) 4(6):498–508. doi: 10.1158/2326-6066.CIR-15-0231
15. Fu W, Lei C, Liu S, Cui Y, Wang C, Qian K, et al. Car exosomes derived from effector car-T cells have potent antitumour effects and low toxicity. Nat Commun (2019) 10(1):4355. doi: 10.1038/s41467-019-12321-3
16. Li X, Daniyan AF, Lopez AV, Purdon TJ, Brentjens RJ. Cytokine il-36gamma improves car T-cell functionality and induces endogenous antitumor response. Leukemia (2021) 35(2):506–21. doi: 10.1038/s41375-020-0874-1
17. Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3l drives epitope spreading and antitumor immunity. Nat Immunol (2020) 21(8):914–26. doi: 10.1038/s41590-020-0676-7
18. Li G, Zhang Z, Cai L, Tang X, Huang J, Yu L, et al. Fn14-targeted bite and car-T cells demonstrate potent preclinical activity against glioblastoma. Oncoimmunology (2021) 10(1):1983306. doi: 10.1080/2162402X.2021.1983306
19. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. Car-T cells secreting bites circumvent antigen escape without detectable toxicity. Nat Biotechnol (2019) 37(9):1049–58. doi: 10.1038/s41587-019-0192-1
20. Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell (2016) 167(2):419–32.e16. doi: 10.1016/j.cell.2016.09.011
21. Moghimi B, Muthugounder S, Jambon S, Tibbetts R, Hung L, Bassiri H, et al. Preclinical assessment of the efficacy and specificity of Gd2-B7h3 synnotch car-T in metastatic neuroblastoma. Nat Commun (2021) 12(1):511. doi: 10.1038/s41467-020-20785-x
22. Hernandez-Lopez RA, Yu W, Cabral KA, Creasey OA, Lopez Pazmino MDP, Tonai Y, et al. T Cell circuits that sense antigen density with an ultrasensitive threshold. Science (2021) 371(6534):1166–71. doi: 10.1126/science.abc1855
23. Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. Synnotch-car T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med (2021) 13(591):eabe7378. doi: 10.1126/scitranslmed.abe7378
24. Hyrenius-Wittsten A, Su Y, Park M, Garcia JM, Alavi J, Perry N, et al. Synnotch car circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Transl Med (2021) 13(591):eabd8836. doi: 10.1126/scitranslmed.abd8836
25. Valkenburg KC, de Groot AE, Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol (2018) 15(6):366–81. doi: 10.1038/s41571-018-0007-1
26. Sakemura R, Hefazi M, Siegler EL, Cox MJ, Larson DP, Hansen MJ, et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to cart-cell therapy in multiple myeloma. Blood (2022) 139(26):3708–21. doi: 10.1182/blood.2021012811
27. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. Cxcr1- or Cxcr2-modified car T cells Co-opt il-8 for maximal antitumor efficacy in solid tumors. Nat Commun (2019) 10(1):4016. doi: 10.1038/s41467-019-11869-4
28. Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, et al. Cxcr2-modified car-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol (2020) 50(5):712–24. doi: 10.1002/eji.201948457
29. Sakemura R, Can I, Siegler EL, Kenderian SS. In vivo cart cell imaging: Paving the way for success in cart cell therapy. Mol Ther Oncolytics (2021) 20:625–33. doi: 10.1016/j.omto.2021.03.003
30. Sakemura R, Bansal A, Siegler EL, Hefazi M, Yang N, Khadka RH, et al. Development of a clinically relevant reporter for chimeric antigen receptor T-cell expansion, trafficking, and toxicity. Cancer Immunol Res (2021) 9(9):1035–46. doi: 10.1158/2326-6066.CIR-20-0901
31. Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, Kenderian SS. Car T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol Ther Oncolytics (2022) 25:69–77. doi: 10.1016/j.omto.2022.03.009
32. Li W, Qiu S, Chen J, Jiang S, Chen W, Jiang J, et al. Chimeric antigen receptor designed to prevent ubiquitination and downregulation showed durable antitumor efficacy. Immunity (2020) 53(2):456–70.e6. doi: 10.1016/j.immuni.2020.07.011
33. Good CR, Aznar MA, Kuramitsu S, Samareh P, Agarwal S, Donahue G, et al. An nk-like car T cell transition in car T cell dysfunction. Cell (2021) 184(25):6081–100.e26. doi: 10.1016/j.cell.2021.11.016
34. Cox MJ, Lucien F, Sakemura R, Boysen JC, Kim Y, Horvei P, et al. Leukemic extracellular vesicles induce chimeric antigen receptor T cell dysfunction in chronic lymphocytic leukemia. Mol Ther (2021) 29(4):1529–40. doi: 10.1016/j.ymthe.2020.12.033
35. Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of car T-cells with crispr-Cas9 gene editing. Mol Cancer (2022) 21(1):78. doi: 10.1186/s12943-022-01559-z
36. Buquicchio FA, Satpathy AT. Interrogating immune cells and cancer with crispr-Cas9. Trends Immunol (2021) 42(5):432–46. doi: 10.1016/j.it.2021.03.003
37. Sutra Del Galy A, Menegatti S, Fuentealba J, Lucibello F, Perrin L, Helft J, et al. In vivo genome-wide crispr screens identify Socs1 as intrinsic checkpoint of Cd4(+) Th1 cell response. Sci Immunol (2021) 6(66):eabe8219. doi: 10.1126/sciimmunol.abe8219
38. Larson RC, Kann MC, Bailey SR, Haradhvala NJ, Llopis PM, Bouffard AA, et al. Car T cell killing requires the ifngammar pathway in solid but not liquid tumours. Nature (2022) 604(7906):563–70. doi: 10.1038/s41586-022-04585-5
39. Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, et al. Deleting Dnmt3a in car T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med (2021) 13(620):eabh0272. doi: 10.1126/scitranslmed.abh0272
40. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. Tgf-beta inhibition Via crispr promotes the long-term efficacy of car T cells against solid tumors. JCI Insight (2020) 5(4):e133977. doi: 10.1172/jci.insight.133977
41. Narayan V, Barber-Rotenberg JS, Jung IY, Lacey SF, Rech AJ, Davis MM, et al. Psma-targeting tgfbeta-insensitive armored car T cells in metastatic castration-resistant prostate cancer: A phase 1 trial. Nat Med (2022) 28(4):724–34. doi: 10.1038/s41591-022-01726-1
42. Roth TL, Li PJ, Blaeschke F, Nies JF, Apathy R, Mowery C, et al. Pooled knockin targeting for genome engineering of cellular immunotherapies. Cell (2020) 181(3):728–44.e21. doi: 10.1016/j.cell.2020.03.039
43. Schmidt R, Steinhart Z, Layeghi M, Freimer JW, Bueno R, Nguyen VQ, et al. Crispr activation and interference screens decode stimulation responses in primary human T cells. Science (2022) 375(6580):eabj4008. doi: 10.1126/science.abj4008
44. Legut M, Gajic Z, Guarino M, Daniloski Z, Rahman JA, Xue X, et al. A genome-scale screen for synthetic drivers of T cell proliferation. Nature (2022) 603(7902):728–35. doi: 10.1038/s41586-022-04494-7
45. Bruchard M, Geindreau M, Perrichet A, Truntzer C, Ballot E, Boidot R, et al. Recruitment and activation of type 3 innate lymphoid cells promote antitumor immune responses. Nat Immunol (2022) 23(2):262–74. doi: 10.1038/s41590-021-01120-y
46. Lelliott EJ, Kong IY, Zethoven M, Ramsbottom KM, Martelotto LG, Meyran D, et al. Cdk4/6 inhibition promotes antitumor immunity through the induction of T-cell memory. Cancer Discov (2021) 11(10):2582–601. doi: 10.1158/2159-8290.CD-20-1554
47. Wiede F, Lu KH, Du X, Zeissig MN, Xu R, Goh PK, et al. Ptp1b is an intracellular checkpoint that limits T-cell and car T-cell antitumor immunity. Cancer Discov (2022) 12(3):752–73. doi: 10.1158/2159-8290.CD-21-0694
48. Kailayangiri S, Altvater B, Lesch S, Balbach S, Gottlich C, Kuhnemundt J, et al. Ezh2 inhibition in Ewing sarcoma upregulates Gd2 expression for targeting with gene-modified T cells. Mol Ther (2019) 27(5):933–46. doi: 10.1016/j.ymthe.2019.02.014
49. Li H, Ding J, Lu M, Liu H, Miao Y, Li L, et al. Caix-specific car-T cells and sunitinib show synergistic effects against metastatic renal cancer models. J Immunother (2020) 43(1):16–28. doi: 10.1097/CJI.0000000000000301
50. Ramakrishna S, Highfill SL, Walsh Z, Nguyen SM, Lei H, Shern JF, et al. Modulation of target antigen density improves car T-cell functionality and persistence. Clin Cancer Res (2019) 25(17):5329–41. doi: 10.1158/1078-0432.CCR-18-3784
51. Hardman C, Ho S, Shimizu A, Luu-Nguyen Q, Sloane JL, Soliman MSA, et al. Synthesis and evaluation of designed pkc modulators for enhanced cancer immunotherapy. Nat Commun (2020) 11(1):1879. doi: 10.1038/s41467-020-15742-7
52. Hu B, Yu M, Ma X, Sun J, Liu C, Wang C, et al. Interferon-a potentiates anti-Pd-1 efficacy by remodeling glucose metabolism in the hepatocellular carcinoma microenvironment. Cancer Discov (2022)12(7):1718–41. doi: 10.1158/2159-8290.CD-21-1022
53. Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/Off switch for car T cells. Sci Transl Med (2019) 11(499):eaau5907. doi: 10.1126/scitranslmed.aau5907
54. Jan M, Scarfo I, Larson RC, Walker A, Schmidts A, Guirguis AA, et al. Reversible on- and off-switch chimeric antigen receptors controlled by lenalidomide. Sci Transl Med (2021) 13(575):eabb6295. doi: 10.1126/scitranslmed.abb6295
55. Duong MT, Collinson-Pautz MR, Morschl E, Lu A, Szymanski SP, Zhang M, et al. Two-dimensional regulation of car-T cell therapy with orthogonal switches. Mol Ther Oncolytics (2019) 12:124–37. doi: 10.1016/j.omto.2018.12.009
56. Yang L, Yin J, Wu J, Qiao L, Zhao EM, Cai F, et al. Engineering genetic devices for in vivo control of therapeutic T cell activity triggered by the dietary molecule resveratrol. Proc Natl Acad Sci USA (2021) 118(34):e2106612118. doi: 10.1073/pnas.2106612118
57. Li S, Xue L, Wang M, Qiang P, Xu H, Zhang X, et al. Decitabine enhances cytotoxic effect of T cells with an anti-Cd19 chimeric antigen receptor in treatment of lymphoma. Onco Targets Ther (2019) 12:5627–38. doi: 10.2147/OTT.S198567
58. Zhang H, Hu Y, Chang AH, Huang H. Successful treatment of T315i bcr-abl mutated lymphoid blast phase chronic myeloid leukemia with chimeric antigen receptor T cell therapy followed by dasatinib. Regener Ther (2020) 14:40–2. doi: 10.1016/j.reth.2019.11.003
59. Gauthier J, Hirayama AV, Purushe J, Hay KA, Lymp J, Li DH, et al. Feasibility and efficacy of Cd19-targeted car T cells with concurrent ibrutinib for cll after ibrutinib failure. Blood (2020) 135(19):1650–60. doi: 10.1182/blood.2019002936
60. Labanieh L, Majzner RG, Klysz D, Sotillo E, Fisher CJ, Vilches-Moure JG, et al. Enhanced safety and efficacy of protease-regulated car-T cell receptors. Cell (2022) 185(10):1745–63.e22. doi: 10.1016/j.cell.2022.03.041
61. Karschnia P, Jordan JT, Forst DA, Arrillaga-Romany IC, Batchelor TT, Baehring JM, et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with car T cells. Blood (2019) 133(20):2212–21. doi: 10.1182/blood-2018-12-893396
62. Giles AJ, Hutchinson MND, Sonnemann HM, Jung J, Fecci PE, Ratnam NM, et al. Dexamethasone-induced immunosuppression: Mechanisms and implications for immunotherapy. J Immunother Cancer (2018) 6(1):51. doi: 10.1186/s40425-018-0371-5
63. Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of Il13ralpha2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther (2018) 26(1):31–44. doi: 10.1016/j.ymthe.2017.10.002
64. Brummer AB, Yang X, Ma E, Gutova M, Brown CE, Rockne RC. Dose-dependent thresholds of dexamethasone destabilize car T-cell treatment efficacy. PloS Comput Biol (2022) 18(1):e1009504. doi: 10.1371/journal.pcbi.1009504
65. Wu Y, Liu Y, Huang Z, Wang X, Jin Z, Li J, et al. Control of the activity of car-T cells within tumours Via focused ultrasound. Nat BioMed Eng (2021) 5(11):1336–47. doi: 10.1038/s41551-021-00779-w
66. Miller IC, Zamat A, Sun LK, Phuengkham H, Harris AM, Gamboa L, et al. Enhanced intratumoural activity of car T cells engineered to produce immunomodulators under photothermal control. Nat BioMed Eng (2021) 5(11):1348–59. doi: 10.1038/s41551-021-00781-2
67. Nguyen NT, Huang K, Zeng H, Jing J, Wang R, Fang S, et al. Nano-optogenetic engineering of car T cells for precision immunotherapy with enhanced safety. Nat Nanotechnol (2021) 16(12):1424–34. doi: 10.1038/s41565-021-00982-5
68. Park AK, Fong Y, Kim SI, Yang J, Murad JP, Lu J, et al. Effective combination immunotherapy using oncolytic viruses to deliver car targets to solid tumors. Sci Transl Med (2020) 12(559):eaaz1863. doi: 10.1126/scitranslmed.aaz1863
Keywords: glioblastoma, cancer therapy, cell therapy, immunotherapy, CAR T
Citation: Choi SI and Yin J (2022) Prospective approaches to enhancing CAR T cell therapy for glioblastoma. Front. Immunol. 13:1008751. doi: 10.3389/fimmu.2022.1008751
Received: 01 August 2022; Accepted: 22 September 2022;
Published: 06 October 2022.
Edited by:
Costanza Maria Cristiani, Magna Græcia University of Catanzaro, ItalyReviewed by:
Haopeng Wang, ShanghaiTech University, ChinaCopyright © 2022 Choi and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinlong Yin, amx5aW5AaGVudS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.