
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 02 November 2022
Sec. Antigen Presenting Cell Biology
Volume 13 - 2022 | https://doi.org/10.3389/fimmu.2022.1007070
This article is part of the Research Topic Women in Antigen Presenting Cell Biology View all 5 articles
 Ismail Cem Yilmaz1,2†
Ismail Cem Yilmaz1,2† Emre Dunuroglu2†
Emre Dunuroglu2† Ihsan Cihan Ayanoglu2†
Ihsan Cihan Ayanoglu2† Emre Mert Ipekoglu2
Emre Mert Ipekoglu2 Muzaffer Yildirim3
Muzaffer Yildirim3 Nogay Girginkardesler4
Nogay Girginkardesler4 Yusuf Ozbel5
Yusuf Ozbel5 Seray Toz5Ahmet Ozbilgin4
Seray Toz5Ahmet Ozbilgin4 Gamze Aykut3Ihsan Gursel1,3
Gamze Aykut3Ihsan Gursel1,3 Mayda Gursel1,2*
Mayda Gursel1,2*Leishmania parasites harbor a unique network of circular DNA known as kinetoplast DNA (kDNA). The role of kDNA in leishmania infections is poorly understood. Herein, we show that kDNA delivery to the cytosol of Leishmania major infected THP-1 macrophages provoked increased parasite loads when compared to untreated cells, hinting at the involvement of cytosolic DNA sensors in facilitating parasite evasion from the immune system. Parasite proliferation was significantly hindered in cGAS- STING- and TBK-1 knockout THP-1 macrophages when compared to wild type cells. Nanostring nCounter gene expression analysis on L. major infected wild type versus knockout cells revealed that some of the most upregulated genes including, Granulysin (GNLY), Chitotriosidase-1 (CHIT1), Sialomucin core protein 24 (CD164), SLAM Family Member 7 (SLAMF7), insulin-like growth factor receptor 2 (IGF2R) and apolipoprotein E (APOE) were identical in infected cGAS and TBK1 knockout cells, implying their involvement in parasite control. Amlexanox treatment (a TBK1 inhibitor) of L. major infected wild type cells inhibited both the percentage and the parasite load of infected THP-1 cells and delayed footpad swelling in parasite infected mice. Collectively, these results suggest that leishmania parasites might hijack the cGAS-STING-TBK1 signaling pathway to their own advantage and the TBK1 inhibitor amlexanox could be of interest as a candidate drug in treatment of cutaneous leishmaniasis.
Leishmaniasis is a vector-borne neglected disease caused by protozoan parasites belonging to the Trypanosomatidae family, Kinetoplastida order, and Leishmania genus (1–6). The disease is considered to cause the ninth largest disease burden among infectious diseases (7–9) and has been listed as the second most common parasitic infection worldwide (10). Over 20 different leishmania species can be transmitted to humans during a blood meal of infected female phlebotomine sandflies (7, 11). Within the insect midgut, the parasites undergo several developmental changes and differentiate into infectious metacyclic promastigotes which are then disseminated into the mammalian host during a blood meal and are rapidly phagocytosed by mono and polymorphonuclear (PMN) phagocytes. Depending on the infecting Leishmania species and the host’s immune status, the disease manifests as cutaneous, mucocutaneous or visceral leishmaniasis (9, 12–17).
Leishmania species share a digenetic life cycle, inhabiting an insect vector and a vertebrate host. The parasites grow as motile, flagellated extracellular promastigotes in the sandfly midgut, ultimately differentiating into infectious metacyclic promastigotes (18–21). Following entry into the mammalian host, parasites rapidly localize and invade the phagocytes. Within macrophages, the parasites reside in phagolysosomal parasitophorous vacuoles, transform into non-motile, non-flagellated amastigotes and multiply by binary fission (22, 23). Macrophages are equipped with multiple germline encoded pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) originating from pathogens. In this context, the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS), its downstream adaptor stimulator of interferon genes (STING) and the kinase TBK1 constitute a fundamental pathway critical for the outcome of infections (24–26).
Emerging evidence suggests that cGAS-STING cytosolic DNA sensing pathway contributes to pathogenesis of certain parasitic infections (26–30). With respect to leishmania, whether the cGAS/STING/TBK1 signaling plays a protective or a pathological role is unknown.
Trypanosoma and leishmania parasites harbor one of the most unusual DNA structures in nature. The mitochondrial DNA of these parasites (the kinetoplast DNA (kDNA)) consist of a single network of interlocked circular DNAs composed of 25–50 maxicircles and 5,000–10,000 of minicircles (31–34). Approximately 30% of parasite DNA is of kinetoplast origin (35). Interestingly, kDNA persists in tissues of Chagas patients and presence of kDNA correlates with inflammatory cell infiltration (36). Based on this observation, it is possible that kDNA-induced immune recognition might alter the immune response during an infection.
Herein, we assessed the effect of kDNA on parasite loads in in vitro infection models and evaluated the role of cGAS-STING-TBK1 DNA-sensing pathway in the context of L. major infection. Our findings showed that stimulation of immune cells with kDNA increased parasite loads in vitro. We further showed that absence of cGAS, STING or TBK1 proteins significantly diminished parasite loads of differentiated THP-1 macrophages, implicating a role of this DNA sensing pathway in pathogenesis of leishmania infections.
L. major parasites (whom/tr/2014/cbu33) used in this study were isolated from a cutaneous lesion of a patient. The species of the isolated parasite was identified and validated (37). The isolated parasites were subjected to in vivo passaging in BALB/c mice to maintain their virulence.
L. major promastigotes were maintained and axenically cultured in plug sealed tissue culture flasks (5-6 x 106 cells/ml) in RPMI 1640 growth medium (20 mM HEPES, 100 units/ml penicillin, 100 µg/ml streptomycin, 20% (v/v) heat inactivated FBS) at 26°C inside a standard incubator without CO2. Parasites were sub-cultured at late-logarithmic phase (≈25x106 parasites/ml) by diluting the culture medium with fresh growth medium. Axenic promastigotes were passaged to a maximum of 12 passages (≈36 days) to prevent loss of virulence.
Kinetoplast DNA (kDNA) from L. major were isolated as previously described (38) with modifications. 109 parasites were sedimented at 3,000g for 10 minutes at 4°C. The pellet was resuspended in 6-8 ml NET buffer (100 mM NaCl, 10 mM EDTA (pH:8.0),10 mM Tris HCl (pH:8.0)) and mixed vigorously. The suspension was centrifuged once more. Pellet was resuspended in NET buffer. Subsequently, Proteinase K and Sarcosyl were added into the solution at final concentrations of 100 μg/ml and 1% (v/v), respectively. The mixture was incubated overnight for completion of cell lysis.
Following incubation, the lysate was centrifuged at 24,000g for 2 hours at 4°C to sediment the kDNA networks. Pellet was resuspended in NET buffer and kDNA suspension was treated with Plasmid-Safe™ ATP-Dependent DNase and RNase A. Next, one volume of phenol pH:7.9 was added onto the solution and mixed by inversions until an emulsion formed. The emulsion was centrifuged at 14,000g for 90 seconds at RT. Following centrifugation, aqueous phase was transferred to a new tube. One volume of chloroform was added onto the separated aqueous phase and the abovementioned mixing and centrifugation steps were repeated. Then, 1/3 volume of 5 M ammonium acetate solution was added onto the collected aqueous phase, followed by addition of 2-3 volumes of molecular biology grade absolute ethanol and mixing. Samples were incubated at -20°C overnight. After incubation, samples were centrifuged at maximum speed of a standard minicentrifuge and supernatants were decanted. Precipitate was washed once more with 70% ethanol, air-dried and dissolved in DNAse-free ultra-pure water. DNA concentrations of isolates were measured by using NanoDrop Microvolume Spectrophotometer.
kDNA isolates were diluted in DNA attachment buffer, pH:8.0 (40 mM HEPES-Cl,10 mM MgCl2) at a final concentration of 50 μg/ml. Mica discs were immobilized on metal discs. Approximately 50 μl of kDNA in the DNA attachment buffer was added onto freshly cleaved mica disc surface and incubated at RT for 5 minutes. Then, the excess buffer was rinsed with ultra-pure water and the sample was air-dried. Air-dried samples were scanned on an Ambient AFM (Nanomagnetics Instruments, UK) following the manufacturer’s instructions. Briefly, laser and detector positions were arranged to obtain a total force of 3.0 V with no lateral and nominal forces. Root mean square (RMS) value was set around 2-2.2. Oscillation amplitude was set to half of RMS value and samples were scanned at non-contact mode by using the NMI SPM (v2.0.37) software. Scanned images were plane-corrected and scaled by using the NMI Viewer (v1.4) software.
Metacyclic parasites were resuspended in DPBS at a final concentration of 5x106 cells/ml. Carboxyfluorescein Succinimidyl Ester (CFSE) was added onto parasite suspension at a final concentration of 5 μM and incubated in dark for 20 minutes at RT. Following washing in DPBS once, resulting pellet was resuspended in Leishmania Infection Medium (RPMI 1640 supplemented with 2%(v/v) FBS, 20mM HEPES, 100 units/ml penicillin, 100µg/ml streptomycin).
Phorbol myristate acetate (PMA) differentiated THP-1 cells (105 cells/well) were infected with CFSE-labeled metacyclic parasites (106 parasites/well) in Leishmania Infection Medium to achieve a multiplicity of infection (MOI) of 1:10 (THP-1:parasites). kDNA complexed with Lipofectamine 2000 were added onto corresponding leishmania-THP-1 mixtures to reach final DNA concentrations of 0.25, 1 and 4 μg/ml. Samples were incubated at 37°C with 5% CO2 for 24 hours. Cells were then washed with pre-warmed null RPMI 1640 medium thrice, incubated with 50 μl 1X Trypsin-EDTA solution for 10-15 minutes and detached cells were collected and transferred to new microcentrifuge tubes. 500 μl 10% Regular RPMI 1640 medium was added onto cells to inactivate trypsin. Samples were centrifuged at 1,500g for 5 minutes and pellets were resuspended in 10% Regular RPMI 1640 Medium. Resuspended samples were acquired on Accuri C6 cytometer (Becton, Dickonson, U.S.A.). PMA-differentiated THP-1 cells were gated with respect to FSC-SSC parameters. Infection rates and MFI values were determined based on CFSE emission.
PMA-differentiated THP-1 cells were infected with unlabeled metacyclic parasites and stimulated with kDNA. Following 24h incubation, media were aspirated from wells. DPBS containing 1 μM SYTO® 16 Green fluorescent nucleic acid stain (permeable) and 5 μM SYTOX® Orange nucleic acid stain (impermeable) were added into the wells. Cells were incubated at 37°C for 20 minutes with staining solution. Then, staining solution was aspirated and cells were washed by PBS once. 10% Regular RPMI 1640 Medium was added onto cells and infected cells were observed on green and red channels of FLoid Cell Imaging Station and merged images from each channel were taken from different regions of the wells.
Plasmid pLEXSY-neo2.1 (Jena Biosciences, Germany) was custom-designed to encode codon optimized EGFP-LUCIFERASE (EGFP-LUC) fusion sequence. For generating transgenic parasites, 10 μg of pLEXSY EGFP-LUC vector was digested with 10 units of SwaI at RT for 2 hours. Samples were run on 0.8% (w/v) agarose gel for confirmation of successful digestion. Parasites at log phase (~15x106 parasites/ml) were centrifuged at 1,500g for 10 minutes at RT. Pellet was resuspended in DPBS, washed once and 4x107 were re-suspended in 400 μl of ice-cold electroporation buffer. 10 μg linearized expression plasmid was transferred onto parasites in the electroporation buffer and the mixture was kept on ice. DNA-parasite mixture was transferred to Gene Pulser electroporation cuvettes with 0.2 cm gap (Biorad, U.S.A.). Samples were pulsed for 30 seconds on the Gene Pulser® II Electroporation System (Biorad, U.S.A.) using the following parameters: 750 V (3750V/cm), 25 μF and 200 Ω. Pulsing was repeated twice using identical parameters and with 30 second breaks between pulses. Electroporated samples were incubated on ice for 10 minutes. After incubation, parasites were transferred to a T25 plug seal tissue culture flask filled with pre-warmed (25°C) 4 ml Leishmania growth medium. Parasites were allowed to rest for 24 hours. 24 hours after the electroporation, 50 μg/ml of Neomycin was used as a selection marker to enrich EGFP-LUC positive parasites.
THP1-Dual™ WT, THP1-Dual™ KO-STING, THP1-Dual™ KO-cGAS, THP1-Dual™ KO-TBK1 cell lines were cultured in 10% (v/v) FBS supplemented RPMI 1640 medium containing 100 μg/ml zeocin and 10 μg/ml blasticidin (In vivogen, France). For macrophage differentiation of THP-1 Dual cells, PMA was used at high concentration (50ng/ml) overnight or low concentration (5ng/ml) for 48 hours. After PMA treatment, to equalize the initial macrophage number for all cell lines, cells were detached by using accutase solution (Biolegend, U.S.A.), counted on Novocyte 2060R flow cytometer (ACEA Biosciences, U.S.A.) and distributed to 96-well plates at a density of 1x105 cells/well in Leishmania Infection Medium (RPMI 1640 supplemented with 2% FBS, 20mM HEPES, 100 units/ml penicillin, 100µg/ml streptomycin). Multiplicity of infection (MOI) was optimized as 1:10 (THP-1:stationary phase L. major). After 6h of infection, wells were washed with DPBS to remove excess parasites and fresh 10% (v/v) FBS supplemented RPMI 1640 medium was added on to the infected THP-1 cells. Percent infection rates and parasite loads (MFI) were based on the eGFP signal and were quantitated on a Novocyte 2060R flow cytometer (ACEA Biosciences, U.S.A.). In certain experiments, Amlexanox (In vivogen, France) was added into the THP-1 growth medium 1 hour prior to infection at indicated concentrations or 24 hours post-infection.
THP1-Dual™ WT, THP1-Dual™ KO-cGAS and THP1-Dual™ KO-TBK1 cells were differentiated overnight in RPMI 1640 media supplemented with 10% (v/v) FBS and 50 ng/ml PMA in 6-well tissue culture plates at a density of 2.7x106 cells/well. In vitro infection was performed by co-incubating stationary phase (2-3 days after reaching late-logarithmic phase) eGFP expressing L. major parasites with THP-1 cell lines at a MOI of 1:10 (macrophage:parasite) for 6 hours in Leishmania Infection Medium (RPMI 1640 supplemented with 2% FBS, 20mM HEPES, 100 units/ml penicillin, 100µg/ml streptomycin). The incubation period was followed by three DPBS washes to remove extracellular L. major parasites and replenishment of media. At 48 hours post-infection, supernatants were aspirated, and 1 ml of RiboEx™ was added onto each well. After cellular material was collected, total RNA isolation was performed following the manufacturers’ instructions and the RNA Clean & Concentrator™-25 (Zymo Research, U.S.A.) kit.
Following quantification of RNA samples and assessment of their purity (A260/A280 and A260/A230) using NanoDrop™ spectrophotometer, 50 ng of RNA sample for each group was processed with nCounter PanCancer Immune Profiling CS Kit (no MK) (NanoString Technologies, Inc., U.S.A.) according to the manufacturer’s instructions. Obtained raw data was analyzed using nSolver Analysis software (v4, NanoString Technologies, Inc., U.S.A.) and nCounter® Advanced Analysis Software (v2, NanoString Technologies, Inc., U.S.A.).
All protocols carried out in this study were approved by animal ethics committee of Bilkent University (Protocol No: BILAEC-2019/44). A total of seven mice were injected with 50 μl of 9x106 metacyclic L. major parasites in DPBS in their left footpads using a 1 ml syringe with a 26G needle. 41 days post-infection, 50 μl of 10 mg/ml amlexanox (n=4) or 50 μl of DMSO control (n=3) was injected into mice intraperitoneally for 12 consecutive days. A digital caliper was used for lesion surveillance by measuring both the depth and width of the footpads.
Statistical tests were performed using Graphpad Prism (v9.) unless specified otherwise Specific statistical tests used for each comparison were stated in figure legends. Asterisks represent significant p-values as detailed below.
Although cGAS binds dsDNA in a sequence-independent manner, bent DNA was shown to favor dimerization and facilitate nucleation of cGAS-DNA ladder formation (39). Interestingly, bent DNA helices were first described in the kinetoplast DNA minicircles (40). Furthermore, kDNA persistence in tissues of infected hosts correlates with inflammatory cell infiltrates (36), suggesting a role for this unusual DNA structure in pathogenesis of infections caused by protozoan parasites of the order Kinetoplastida.
To assess whether Leishmania kDNA contributes to disease progression, we first isolated kDNA from the parasites. For this, kDNA was purified from late-log phase parasites and treated with Plasmid-Safe™ ATP-Dependent DNase and RNase A to eliminate residual genomic DNA and RNA contaminants in the kDNA fraction, respectively. Following purification, isolated kDNA networks were analyzed using atomic force microscopy (AFM). 5x5 and 2.5x2.5 µm scans demonstrated the typical kDNA network structure composed of interlocked circular DNAs (Figure 1A), confirming the success of the isolation protocol.
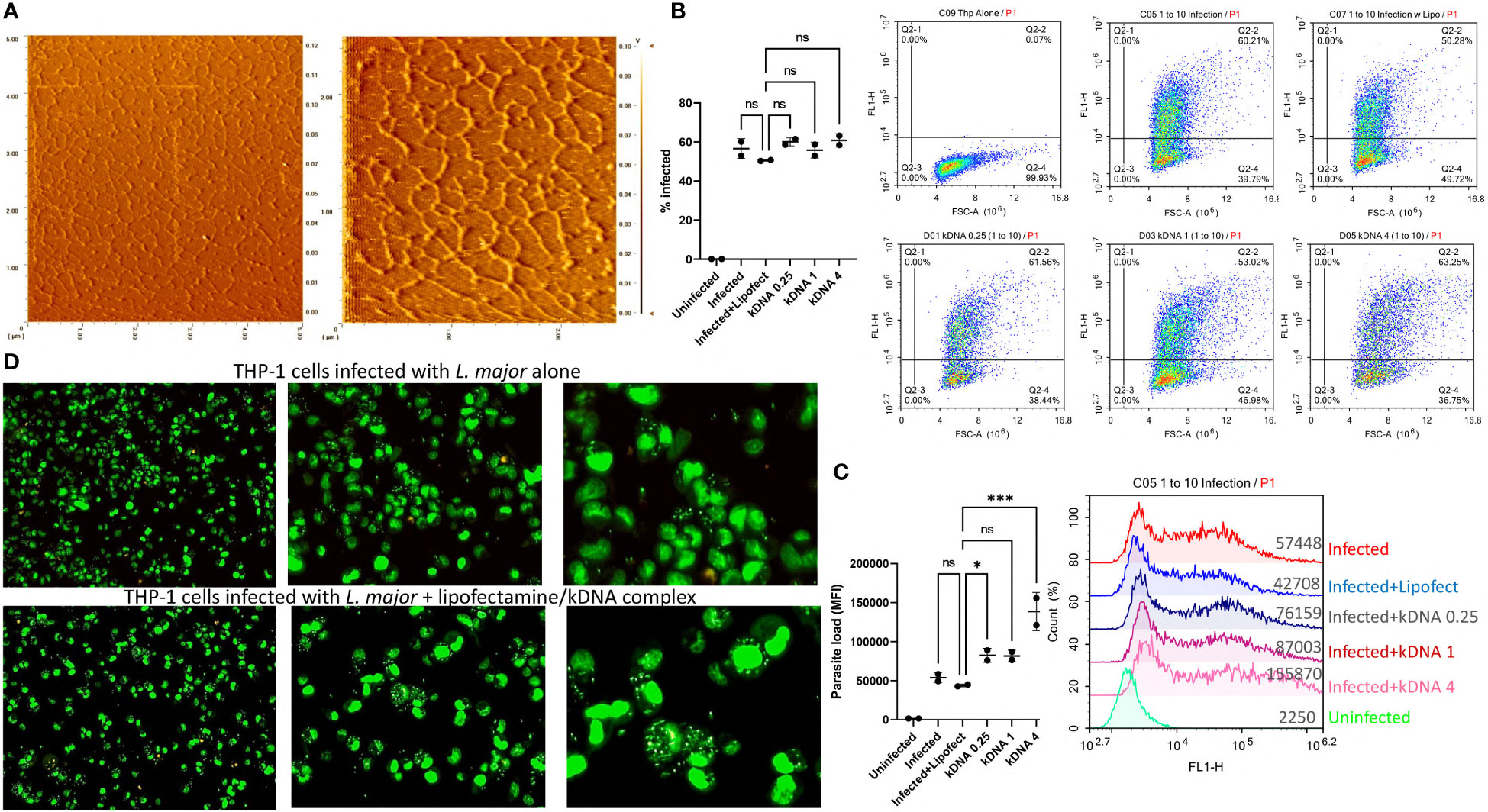
Figure 1 Effect of Cytosolic kDNA delivery on in vitro Leishmania infection. (A) AFM images of the purified kDNA networks with scanning areas of 5x5 μm (left) and 2.5x2.5 μm (right). PMA-differentiated THP-1 cells (5x105 cells/ml) were incubated with CFSE labeled parasites (5x106 parasites/ml) for 24 hours in the presence or absence of kDNA. Percentage of infected cells (B) and total parasite loads (C) were evaluated by flow cytometry. Numbers on x-axis labels indicate concentration of kDNA (μg/ml). Data represent the average of two independent experiments ± SD. All groups were compared statistically by one-way ANOVA followed by Dunnet`s multiple comparison test. (D) Fluorescence microscopy images of infected, SYTO Green stained THP-1 cells (from left to right, 10X 20X and 40X) untreated (upper panels) or treated with 1 μg/ml kDNA. Images are representatives of two independent experiments. ns=not significant, *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001
Next, PMA differentiated THP-1 macrophages were infected with CFSE labeled parasites in the presence or absence of different concentrations of kDNA and percentage of infected cells and total parasite loads (based on MFI) were evaluated by flow cytometry (Figures 1B, C). Percentage of infected THP-1 cells did not change significantly in the presence of kDNA (Figure 1B) but the parasite load was significantly increased in the presence of the highest dose of kDNA (Figure 1C). Of note, use of lipofectamine had no adverse effect on infected cell percentage or parasite loads when compared to cells infected in the absence of lipofectamine (Figures 1B, C), indicating that the kDNA-induced increased parasite load was not an artifact caused by the inclusion of the transfection reagent. Furthermore, treated THP-1 cells were responsive to kDNA delivery as evidenced by type I interferon production (Supplementary Figure 1), suggestive of activation of a cytosolic DNA sensing pathway. Of interest, kDNA was a more potent stimulator of type I interferon production when compared to stimulation with an equal concentration of linear Leishmania genomic DNA (Supplementary Figure 1).
THP-1 macrophages infected with unlabeled L. major in the absence or presence of kDNA (1 μg/ml) were also stained with the membrane permeable dye SytoGreen 16 and imaged under a fluorescence microscope. kDNA transfected cells showed higher number of parasites (Figure 1D, lower panel) per cell when compared to the untreated group (Figure 1D, upper panel).
These results suggested that cytosolic delivery of kDNA might contribute to parasitaemia in infected macrophages, indicating a role for cytosolic DNA sensors in modulating the response to leishmania infection.
To investigate the role of cytosolic DNA sensing pathway on leishmania infection rates, three different THP-1 cell lines (cGAS, STING and TBK1 knockout) were infected with eGFP expressing transgenic parasites in parallel with wild type (WT) THP-1 cells. Since PMA concentration used for macrophage differentiation may impact the maturation level of THP-1 cells and therefore the initial phagocytic activity of the macrophages, infection experiments were conducted using cells differentiated with a low (5 ng/ml) and a high (50 ng/ml) dose of PMA. When low-dose PMA differentiated cells were used, percent infected cells (Supplementary Figure 2 and Figure 2B) and parasite loads (Figures 2A, B) of WT cells were consistently higher than the knockout cells throughout the 3-day infection period. However, for the STING and TBK1 knockout cells, the initial phagocytic capacities (at 6h) were significantly lower than the WT cells (Supplementary Figure 2; Figures 2A, B) and hence the observed lower infection rates might have stemmed from fewer infecting L. major at the beginning. However, when THP-1 cells were differentiated with a higher PMA concentration, the initial phagocytic capacities of WT and cGAS-KO cells were comparable and yet by 48 and 72 h, parasite loads in the cGAS knockout cells were significantly lower than the WT cells (Figures 2C, D). Furthermore, even though the initial percentage of infected cells and parasite loads in the TBK1-KO cells exceeded that of WT cells (6h, Figures 2C, D), absence of TBK1 significantly hindered parasite proliferation at later time points (48 and 72 h). STING knockout cells internalized very little parasites by 6 h and resisted infection throughout all time points.
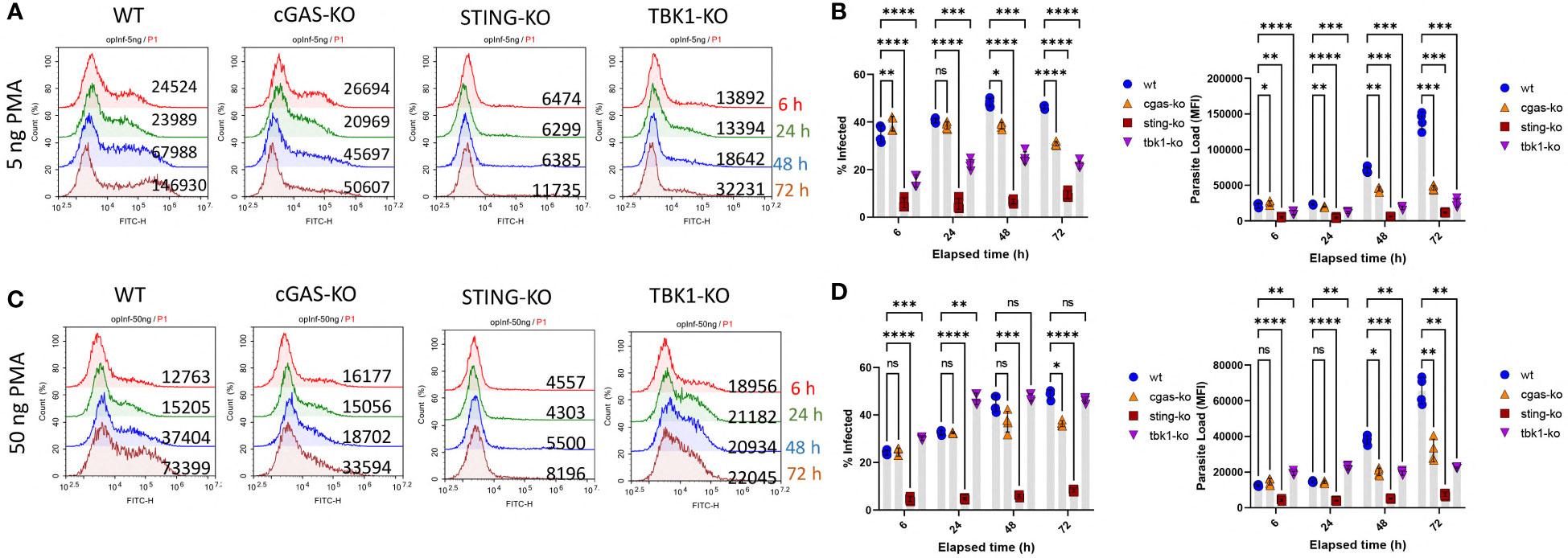
Figure 2 Kinetics of Leishmania infection in WT versus cGAS-, STING- or TBK1-knockout THP-1 cells. Low dose [(A) and (B) or high dose (C) and (D)] PMA differentiated THP-1 cells were infected with eGFP expressing L. major parasites at a MOI of 1:10 (macrophage:parasite). At indicated time points (6,24,48 and 72 h), infection percentages and parasite loads were quantified using a flow cytometer. Data presented in graphs represent the average of two independent experiments with technical replicates ± SD. Statistical comparisons to WT cells were based on two-way ANOVA followed by Tukey’s multiple comparison test. ns, not significant, *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
To gain further insight into cGAS/STING related signaling and potentiation of infection, PMA differentiated WT cells were pre-treated either with the STING inhibitor H151 or the STING activator 2’3-cGAMP followed by infection with eGFP expressing transgenic parasites. Compared to untreated WT cells, percentage of infected cells and parasite loads were significantly lower in H151 treated samples (Figure 3A and Supplementary Figure 3A). In contrast, the STING agonist 2’3-cGAMP dose dependently enhanced both the % infected cells and the parasite loads compared to untreated but infected WT controls (Figure 3B and Supplementary Figure 3B). Of note, cGAMP (20 µg/ml) failed to potentiate infection in the presence of the inhibitor H151 (Figure 3B and Supplementary Figure 3B). Comparison of infected versus infected/cGAMP-treated WT, cGAS- and STING-knockout cells demonstrated that the STING agonist enhanced infected cell percent in WT cells and parasite loads in WT and cGAS- but not in STING-knockout cells (Figures 3C–E and Supplementary Figures 3C–E). Furthermore, exogenous recombinant IFNβ supplementation also enhanced infection rate and parasite loads (Figures 3C–E and Supplementary Figures 3C–E). Collectively, these results suggest that leishmania parasites might hijack the cGAS-STING-TBK1 signaling pathway to their own advantage.
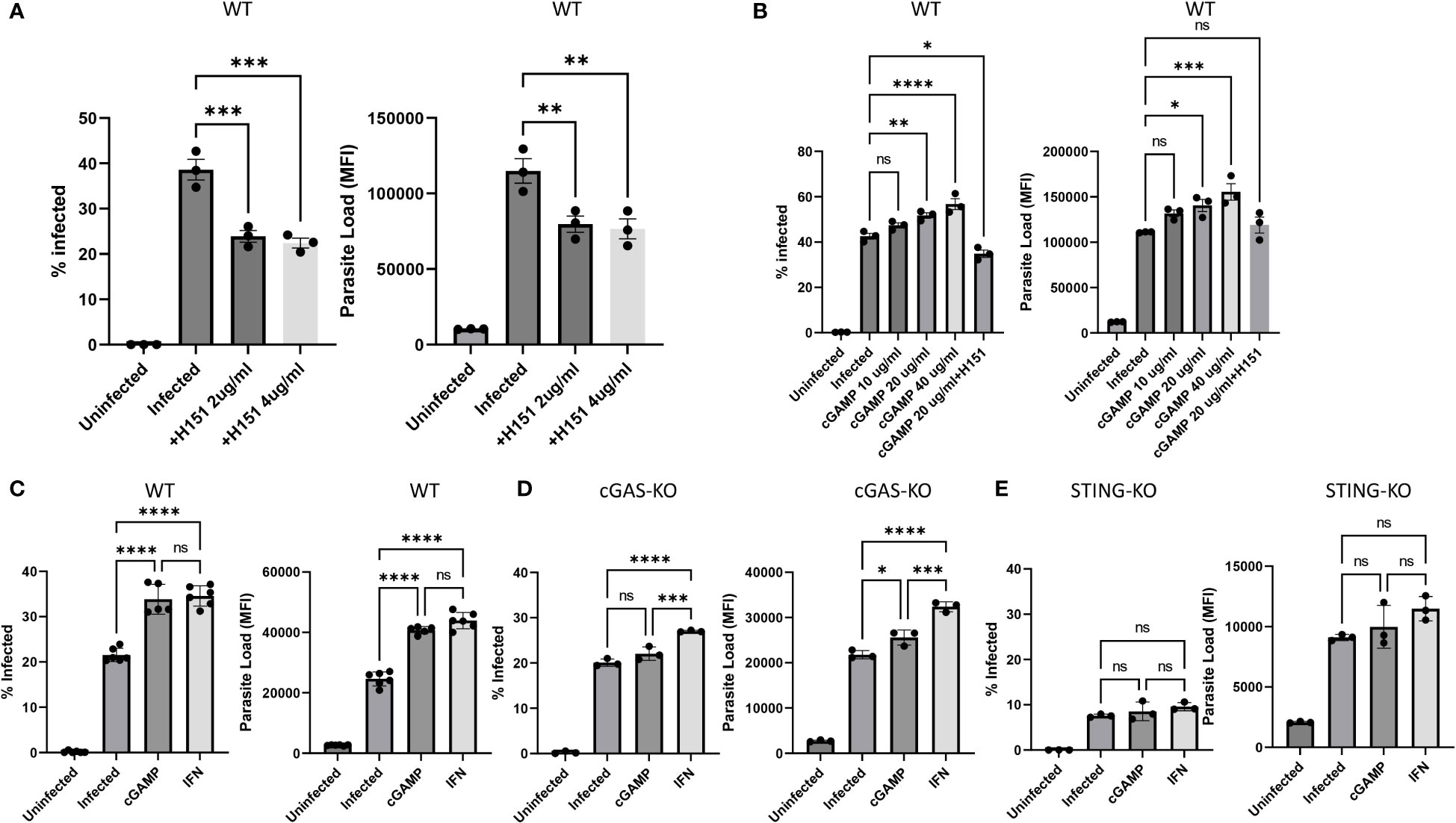
Figure 3 Effect of the STING agonist 2’3’-cGAMP, the STING antagonist H151 and recombinant IFN-β on in vitro Leishmania infection. Differentiated WT [(A), (B) and (C)], cGAS- [(D)] or STING- (E) knockout THP-1 cells were infected with eGFP expressing L. major parasites at a MOI of 1:10 (macrophage:parasite). Infection percentages and parasite loads were quantified using a flow cytometer 24 h later. H151 [2 or 4 µg/ml; (A) and (B)], cGAMP [10, 20 or 40 µg/ml; (B), (C), (D) and (E)] or recIFN-β [50 ng/ml; (C), (D) and (E)] were added 8, 2 and 8 hours prior to infection, respectively. Data represent average ± SD. All groups were compared statistically by one-way ANOVA followed by Dunnet`s multiple comparison test. ns, not significant, *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
To gain further insight into the intrinsic resistance of knockout cell lines to leishmania infection, we analyzed the gene expression profiles of WT, cGAS and TBK1 knockout cells using the NanoString nCounter PanCancer Immune Profiling CS Kit. For this, RNAs were extracted from cells 48 h-post-infection. Volcano plots comparing the fold changes in expression (Log2) with the corresponding adjusted p-values (−log10) in infected knockout cells in comparison to infected WT cells are shown in Figure 4A.
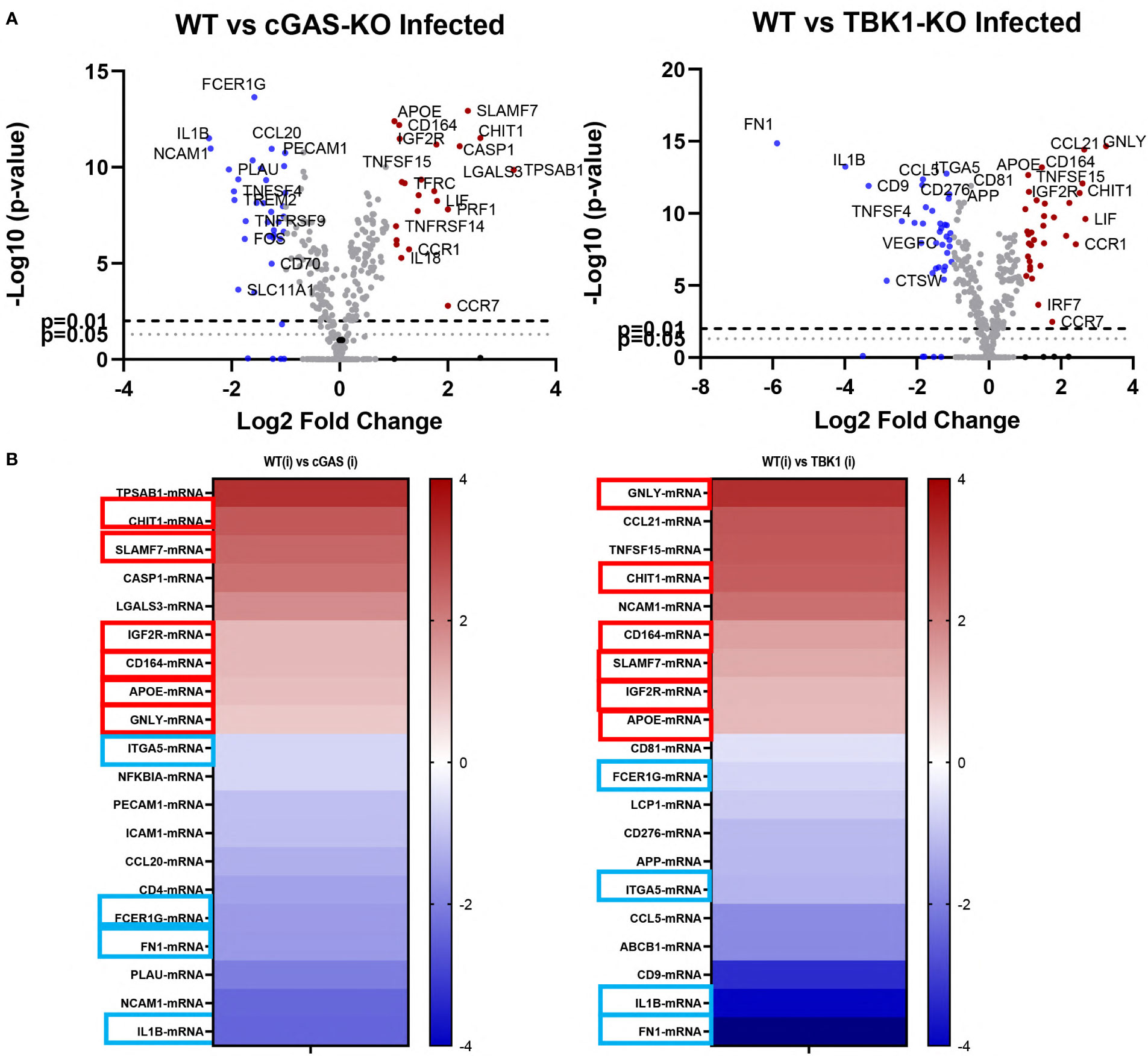
Figure 4 Differentially expressed genes in infected WT versus cGAS and TBK1 knockout cells. (A) Volcano plots of differentially expressed genes between L. major infected WT and knockout cell lines (n=3). Upregulated (fold change ≥ 2) and downregulated (fold change ≤ 2) genes are shown in red and blue, respectively. Dashed lines represent p=0.01 and dotted lines show p=0.05. The log2FC is plotted on the x-axis, and the negative log10FDR (adjusted p-value) is plotted on the y-axis. (B) Heatmaps showing the top 10 most up- or down-regulated genes in the infected knockout cells with respect to infected WT controls. Six of the shared upregulated genes and 4 of the shared downregulated genes are enclosed in boxes (red boxes, upregulated; blue boxes, downregulated). Statistical analysis for differentially expressed genes were performed using nSolver Analysis software (v4, NanoString Technologies, Inc., U.S.A.) and nCounter® Advanced Analysis Software (v2, NanoString Technologies, Inc., U.S.A.).
Of the 770 genes found in the panel, 24 and 37 genes were differentially up- and downregulated in infected cGAS knockout cells when compared to infected WT cells, respectively (Figure 4A, left plot). In the TBK1 knockout cells, 37 genes were upregulated and 39 downregulated when compared to infected WT cells (Figure 4A, right plot). Out of the top ten most up- or down-regulated genes in both knockout cells, six of the upregulated and four of the downregulated genes were identical (Figure 4B). Granulysin (GNLY), Chitotriosidase-1 (CHIT1), Sialomucin core protein 24 (CD164), SLAM Family Member 7 (SLAMF7), insulin-like growth factor receptor 2 (IGF2R) and apolipoprotein E (APOE) genes were upregulated in infected cGAS and TBK1 knockout cell lines in comparison to infected WT controls. The four shared down-regulated genes were Fc Epsilon Receptor Ig (FCER1G), Integrin Subunit Alpha 5 (ITGA5), Interleukin 1 beta (IL1B) and Fibronectin 1 (FN1). Collectively, these commonly up- and down-regulated genes might play a role in modulating the response of cells during infection with leishmania parasites.
Since TBK1 knockout cells were highly resistant to infection, we next wanted to evaluate the effect of a TBK1 inhibitor on leishmania infection in WT cells. For this, WT THP-1 cells were pre-treated with increasing concentrations of amlexanox prior to a 24-hour L. major infection assay (Supplementary Figure 4). Amlexanox treatment potently inhibited both the percentage and the parasite load of infected cells in a dose dependent manner. Similarly, another TBK1 inhibitor, BX795, also significantly reduced parasite loads in infected cells (Supplementary Figure 5), although this decrease was less pronounced when compared to amlexanox-induced infection suppression.
The effect of amlexanox on Leishmania infection was further analyzed in a 3-day in vitro infection model and an in vivo therapeutic treatment model (Figure 5). As expected, amlexanox addition to THP-1 cells 1 hour prior to infection, potently inhibited both the infection rate and proliferation of L. major parasites (Figures 5A-C, purple line). To evaluate whether amlexanox could exert an inhibitory effect on already infected cells, the drug was added to cells 24 h-post infection. The results showed that there was still a significant inhibitory effect of late amlexanox addition on infected cell percent and parasite load by 72 h (Figures 5A-C, red line).
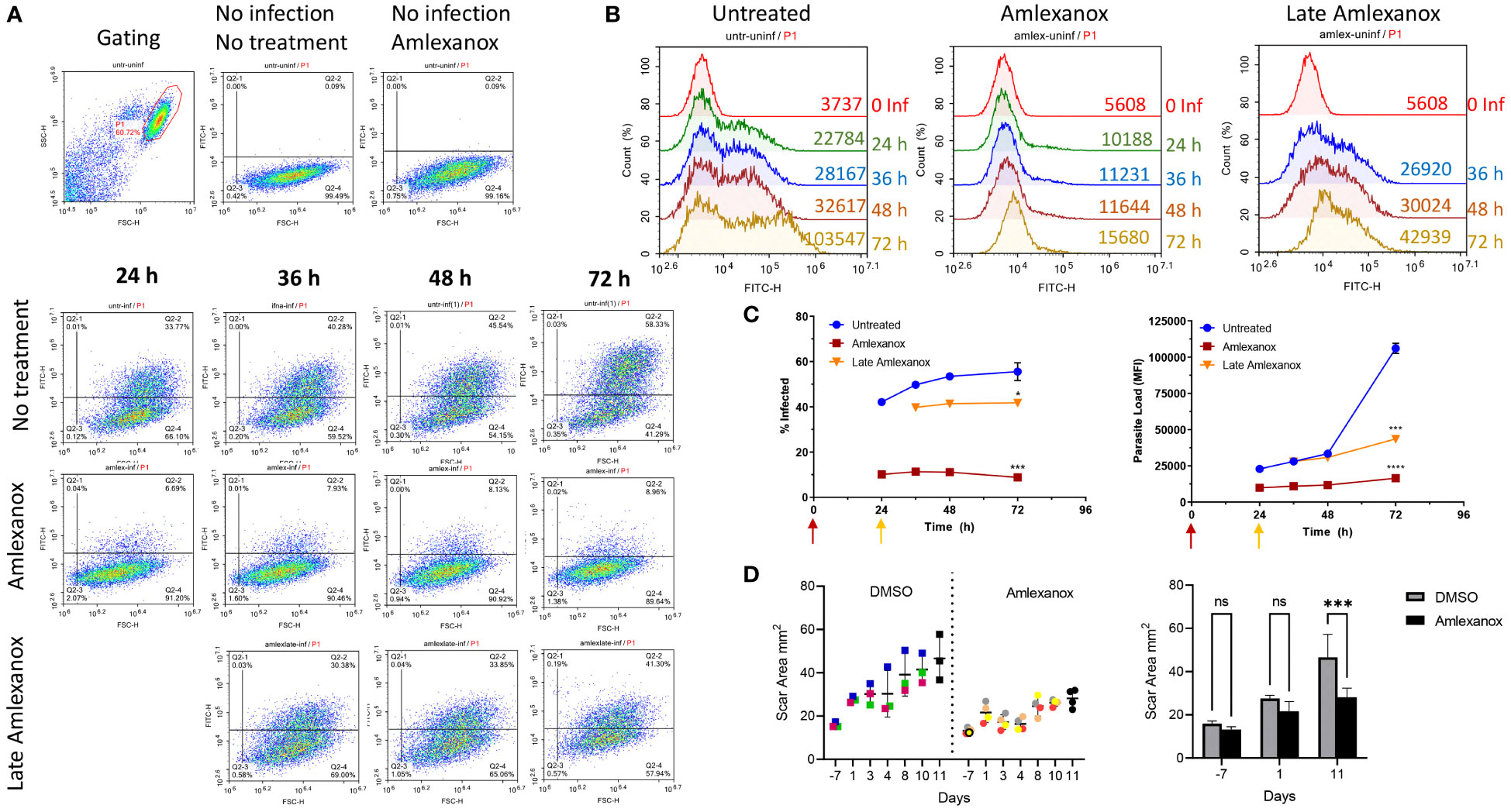
Figure 5 Amlexanox-induced inhibition of L. major infection in WT THP1 cells in a 3-day infection model. Differentiated THP-1 cells were infected with eGFP expressing L. major parasites at a MOI of 1:10 (macrophage:parasite). Infection percentages and parasite loads were quantified using a flow cytometer at the indicated time points. Amlexanox was added either 1 hour prior to infection or 24 h post infection (late amlexanox). (A) and (B) show representative flow cytometry density and histogram plots, corresponding to infected cell percent and parasite load, respectively. (C) Data presented in graphs are representative of two independent experiments with similar results Amlexanox was added at different time points in treated groups (purple, 1 h pre-infection; red, 24 h post-infection). 72 h data were statistically compared to untreated group using one-way ANOVA followed by Bonferroni’s multiple comparison test. (D) Footpad swelling in L. major infected mice treated or not with amlexanox starting on day 41 post-infection for or a duration of 12 days. Results were based on digital caliper measurement of footpad depth and width of DMSO injected controls (n=3) and amlexanox injected (n=4) treatment group. Data were statistically compared using multiple unpaired t tests. ns, not significant, *p≤0.05, ***p≤0.001, ****p≤0.0001.
Since in vitro infection experiments revealed a potent anti-parasitic effect of amlexanox treatment, we wanted to assess the drug’s anti-leishmanial activity in vivo. For this, seven mice were infected with L. major and on day 41 post-footpad challenge, the drug (n=4) or DMSO control (n=3) was injected to mice on 12 consecutive days. Throughout the therapy period footpad swelling was monitored by a digital caliper (Figure 5D). Days presented in the Figures correspond to the initiation and continuation of amlexanox therapy. Results demonstrated that amlexanox treatment delayed footpad swelling ˜2-fold (30.31% change between days 1 and 11), when compared to the DMSO control group (69.14% change between days 1 and 11) (Figure 5D). These results collectively suggest that amlexanox could be a new candidate drug for treatment of cutaneous leishmaniasis caused by L. major.
Multiple pattern recognition receptors have been demonstrated to be involved in the modulation of host response during the course of leishmania infections (41–45). Yet, the role of the cGAS-STING DNA sensing pathway in regulation of leishmania infections remains largely unexplored. In this study, we revealed a critical role of the cGAS-STING-TBK1 signaling pathway in modulating parasite loads in infected macrophages. The cGAS-STING-TBK1 pathway was previously shown to be exploited by non-viral pathogens to favor their persistence. For example, Listeria monocytogenes, Staphylococcus aureus and Neisseria gonorrhea exploit the cGAS-STING–TBK1 pathway to impair antibacterial defenses, promoting bacterial persistence (46–48). With respect to infections caused by protozoan parasites, stimulation of the cGAS-STING-TBK1 pathway can yield detrimental or protective outcomes (26). For example, one study demonstrated that hijacking of the cGAS-STING-TBK1-IRF3 pathway by Toxoplasma gondii, promoted its replication both in vitro and in vivo (30). In contrast, another study found a protective effect of cGAS, STING and type-I interferons during Toxoplasma gondii infection (49).
Evidence suggests that cGAS preferentially binds to U-turns or bent DNA helices which favor dimerization and nucleation of protein-DNA ladder formation (39). In this context, bent DNA was first discovered due to electrophoretic anomalies exhibited by the AT-rich kDNA of kinetoplastid parasites (40, 50). Therefore, it is highly likely that the affinity of cGAS towards optimally pre-structured kDNA would be much higher in magnitude when compared to linear genomic DNA. Consistent with this view, our results demonstrated that kDNA delivery to the cytosol magnified parasite burden of infected macrophages. Whether these unique DNA networks from different kinetoplastida also share a role of modulating host defenses remains to be explored.
Our data on gene expression analysis in infected cGAS and TBK1 knockout cell lines identified a number of shared up- and down-regulated genes which collectively might contribute to establishment of infection resistance. Among the shared up-regulated genes, SLAMF7 stands out as a receptor linked to superactivated macrophage phenotype (51). Similarly, granulysin and chitotriosidase expression in macrophages were associated with microbial defense (52, 53). Among the downregulated genes, ITGA5 (integrin subunit alpha 5) stands out since the protein encoded by this gene is a constituent of fibronectin receptor (integrin α5β1) which has a prominent role in promastigote internalization (54).
Having established that cGAS-STING-TBK1 axis plays an important role in regulation of parasite burden in infected macrophages, we next asked whether a TBK1 inhibitor could be of benefit in inhibiting parasitemia in vitro and in vivo. Indeed amlexanox, a specific inhibitor of IKKϵ and TBK1 proved to be highly effective in controlling parasite loads both in vitro and in vivo. This result merits further investigation on the potential of amlexanox as a surrogate treatment strategy for cutaneous leishmaniasis. In summary, our results imply that kDNA of leishmania parasites potentially engage cGAS and modify the response of host cells in a STING- and TBK1-dependent manner to promote a pro-parasitic state. Future studies investigating the role of cGAS-STING-TBK1 pathway using knockout mouse models of leishmania infection and research on inhibitors of this pathway should unravel specific mechanisms involved in leishmania-mediated immune evasion.
The data presented in the study are deposited in the Gene Expression Omnibus (GEO) repository, accession number GSE211590. Data is available through the following link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE211590.
All protocols carried out in this study were approved by animal ethics committee of Bilkent University (Protocol No: BILAEC-2019/44).
IY, ED, IA, IG and MG designed the study, analyzed the data and wrote the manuscript. EI, MY and GA assisted experiments. NG and AO provided leishmania parasites and shared culture conditions, YO and ST provided leishmania parasites and shared kinetoplast DNA isolation protocol. All authors contributed to the article and approved the submitted version.
This work is supported by TUBITAK grants 115S073 and 219S678 to MG.
We thank Dr. Deniz Kahraman of Middle East Technical University, Informatics Institute for technical help with the nCounter Nanostring assay.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2022.1007070/full#supplementary-material
1. Bañuls A-L, Hide M, Prugnolle F. Leishmania and the leishmaniases: A parasite genetic update and advances in taxonomy, epidemiology and pathogenicity in humans. Adv Parasit (2007) 64:1–458. doi: 10.1016/s0065-308x(06)64001-3
2. Lukeš J, Skalický T, Týč J, Votýpka J, Yurchenko V. Evolution of parasitism in kinetoplastid flagellates. Mol Biochem Parasit (2014) 195:115–22. doi: 10.1016/j.molbiopara.2014.05.007
3. Simpson AGB, Stevens JR, Lukeš J. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol (2006) 22:168–74. doi: 10.1016/j.pt.2006.02.006
4. Andrade-Narváez FJ, Vargas-González A, Canto-Lara SB, Damián-Centeno AG. Clinical picture of cutaneous leishmaniases due to leishmania (Leishmania) mexicana in the Yucatan peninsula, Mexico. Memórias Instituto Oswaldo Cruz (2001) 96:163–7. doi: 10.1590/s0074-02762001000200005
5. Desjeux P. Leishmaniasis: current situation and new perspectives. Comp Immunol Microbiol Infect Dis (2004) 27:305–18. doi: 10.1016/j.cimid.2004.03.004
6. Hotez PJ, Bottazzi ME, Franco-Paredes C, Ault SK, Periago MR. The neglected tropical diseases of Latin America and the Caribbean: A review of disease burden and distribution and a roadmap for control and elimination. PloS Negl Trop D (2008) 2:e300. doi: 10.1371/journal.pntd.0000300
7. Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, et al. Leishmaniasis worldwide and global estimates of its incidence. PloS One (2012) 7:e35671. doi: 10.1371/journal.pone.0035671
8. Hoyos CL, Cajal SP, Juarez M, Marco JD, D’Amato AMA, Cayo M, et al. Epidemiology of American tegumentary leishmaniasis and trypanosoma cruzi infection in the northwestern Argentina. BioMed Res Int (2016) 2016:6456031. doi: 10.1155/2016/6456031
9. Kevric I, Cappel MA, Keeling JH. New world and old world leishmania infections a practical review. Dermatol Clin (2015) 33:579–93. doi: 10.1016/j.det.2015.03.018
10. World Health Organization. WHO control of the leishmaniases. World Health Organization Technical Rep Ser, (2010) vol. xii–xiii, 1–186. Geneva, ISBN 978 92 4 120949 6.
11. Gramiccia M, Gradoni L. The current status of zoonotic leishmaniases and approaches to disease control. Int J Parasitol (2005) 35:1169–80. doi: 10.1016/j.ijpara.2005.07.001
12. Torres-Guerrero E, Quintanilla-Cedillo MR, Ruiz-Esmenjaud J, Arenas R. Leishmaniasis: a review. F1000research (2017) 6:750. doi: 10.12688/f1000research.11120.1
13. Mann S, Frasca K, Scherrer S, Henao-Martínez AF, Newman S, Ramanan P, et al. A review of leishmaniasis: Current knowledge and future directions. Curr Trop Med Rep (2021) 8:121–32. doi: 10.1007/s40475-021-00232-7
14. Bailey MS, Lockwood DNJ. Cutaneous leishmaniasis. Clin Dermatol (2007) 25:203–11. doi: 10.1016/j.clindermatol.2006.05.008
15. Davies CR, Kaye P, Croft SL, Sundar S. Leishmaniasis: new approaches to disease control. Bmj (2003) 326:377–82. doi: 10.1136/bmj.326.7385.377
16. Alvar J, Arana B. Leishmaniasis-impact and therapeutic needs. Drug Discovery (2018), 1–23 In Rivas L; Gil C (ed), United Kingdom. doi: 10.1039/9781788010177-00001
17. Lessa MM, Lessa HA, Castro TWN, Oliveira A, Scherifer A, Machado P, et al. Mucosal leishmaniasis: epidemiological and clinical aspects. Braz J Otorhinolar (2007) 73:843–7. doi: 10.1016/s1808-8694(15)31181-2
18. Gossage SM, Rogers ME, Bates PA. Two separate growth phases during the development of leishmania in sand flies: implications for understanding the life cycle. Int J Parasitol (2003) 33:1027–34. doi: 10.1016/s0020-7519(03)00142-5
19. Serafim TD, Coutinho-Abreu IV, Oliveira F, Meneses C, Kamhawi S, Valenzuela JG. Sequential blood meals promote leishmania replication and reverse metacyclogenesis augmenting vector infectivity. Nat Microbiol (2018) 3:548–55. doi: 10.1038/s41564-018-0125-7
20. Rogers ME, Chance ML, Bates PA. The role of promastigote secretory gel in the origin and transmission of the infective stage of leishmania mexicana by the sandfly lutzomyia longipalpis. Parasitology (2002) 124:495–507. doi: 10.1017/s0031182002001439
21. Rogers ME. The role of leishmania proteophosphoglycans in sand fly transmission and infection of the mammalian host. Front Microbiol (2012) 3:223. doi: 10.3389/fmicb.2012.00223
22. Mathison BA, Pritt BS. Encyclopedia of infection and immunity. (2022), 3, 136–203, Editor: Nima Rezaei, Elsevier. doi: 10.1016/b978-0-12-818731-9.00107-5
23. Duque GA, Descoteaux A. Leishmania survival in the macrophage: where the ends justify the means. Curr Opin Microbiol (2015) 26:32–40. doi: 10.1016/j.mib.2015.04.007
24. Motwani M, Pesiridis S, Fitzgerald KA. DNA Sensing by the cGAS–STING pathway in health and disease. Nat Rev Genet (2019) 20:657–74. doi: 10.1038/s41576-019-0151-1
25. Cheng Z, Dai T, He X, Zhang Z, Xie F, Wang S, et al. The interactions between cGAS-STING pathway and pathogens. Signal Transduct Target Ther (2020) 5:91. doi: 10.1038/s41392-020-0198-7
26. Sun Y, Cheng Y. STING or sting: cGAS-STING-Mediated immune response to protozoan parasites. Trends Parasitol (2020) 36:773–84. doi: 10.1016/j.pt.2020.07.001
27. Liang L, Shen Y, Hu Y, Liu H, Cao J. cGAS exacerbates schistosoma japonicum infection in a STING-type I IFN-dependent and independent manner. PloS Pathog (2022) 18:e1010233. doi: 10.1371/journal.ppat.1010233
28. Gallego-Marin C, Schrum JE, Andrade WA, Shaffer SA, Giraldo LF, Lasso AM, et al. Cyclic GMP–AMP synthase is the cytosolic sensor of plasmodium falciparum genomic DNA and activates type I IFN in malaria. J Immunol (2018) 200:768–74. doi: 10.4049/jimmunol.1701048
29. Majumdar T, Sharma S, Kumar M, Hussain M, Chauhan N, Kalia I, et al. Tryptophan-kynurenine pathway attenuates β-catenin-dependent pro-parasitic role of STING-TICAM2-IRF3-IDO1 signalosome in toxoplasma gondii infection. Cell Death Dis (2019) 10:161. doi: 10.1038/s41419-019-1420-9
30. Majumdar T, Chattopadhyay S, Ozhegov E, Dhar J, Goswami R, Sen GC, et al. Induction of interferon-stimulated genes by IRF3 promotes replication of toxoplasma gondii. PloS Pathog (2015) 11:e1004779. doi: 10.1371/journal.ppat.1004779
31. Laurent M, Steinert M. Electron microscopy of kinetoplastic DNA from trypanosoma mega. Proc Natl Acad Sci (1970) 66:419–24. doi: 10.1073/pnas.66.2.419
32. Simpson L, da Silva A. Isolation and characterization of kinetoplast DNA from leishmania tarentolae. J Mol Biol (1971) 56:443–73. doi: 10.1016/0022-2836(71)90394-9
33. Shapiro TA, Englund PT. The structure and replication of kinetoplast DNA. Annu Rev Microbiol (1995) 49:117–43. doi: 10.1146/annurev.mi.49.100195.001001
34. Lukesš J, Guilbride DL, Votyípka J, Ziíkovaí A, Benne R, Englund PT. Kinetoplast DNA network: Evolution of an improbable structure. Eukaryot Cell (2002) 1:495–502. doi: 10.1128/ec.1.4.495-502.2002
35. Simpson L, Neckelmann N, de la Cruz VF, Simpson AM, Feagin JE, Jasmer DP, et al. Comparison of the maxicircle (mitochondrial) genomes of leishmania tarentolae and trypanosoma brucei at the level of nucleotide sequence. J Biol Chem (1987) 262:6182–96. doi: 10.1016/s0021-9258(18)45555-x
36. Tarleton RL, Zhang L. Chagas disease etiology: Autoimmunity or parasite persistence? Parasitol Today (1999) 15:94–9. doi: 10.1016/s0169-4758(99)01398-8
37. Özbilgin A, Çulha G, Uzun S, Harman M, Topal SG, Okudan F, et al. Leishmaniasis in Turkey: first clinical isolation of leishmania major from 18 autochthonous cases of cutaneous leishmaniasis in four geographical regions. Trop Med Int Health (2016) 21:783–91. doi: 10.1111/tmi.12698
38. Akman L, Aksu HSZ, Wang RQ, Ozensoy S, Ozbel Y, Alkan Z, et al. Multi site DNA polymorphism analyses of leishmania isolates define their genotypes predicting clinical epidemiology of leishmaniasis in a specific region. J Eukaryot Microbiol (2000) 47:545–54. doi: 10.1111/j.1550-7408.2000.tb00088.x
39. Andreeva L, Hiller B, Kostrewa D, Lässig C, Mann CC de O, Drexler DJ, et al. cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein–DNA ladders. Nature (2017) 549:394–8. doi: 10.1038/nature23890
40. Marini JC, Levene SD, Crothers DM, Englund PT. Bent helical structure in kinetoplast DNA. Proc Natl Acad Sci (1982) 79:7664–8. doi: 10.1073/pnas.79.24.7664
41. Becker I, Salaiza N, Aguirre M, Delgado J, Carrillo-Carrasco N, Kobeh LG, et al. Leishmania lipophosphoglycan (LPG) activates NK cells through toll-like receptor-2. Mol Biochem Parasit (2003) 130:65–74. doi: 10.1016/s0166-6851(03)00160-9
42. Flandin J, Chano F, Descoteaux A. RNA Interference reveals a role for TLR2 and TLR3 in the recognition of leishmania donovani promastigotes by interferon–γ primed macrophages. Eur J Immunol (2006) 36:411–20. doi: 10.1002/eji.200535079
43. Liese J, Schleicher U, Bogdan C. TLR9 signaling is essential for the innate NK cell response in murine cutaneous leishmaniasis. Eur J Immunol (2007) 37:3424–34. doi: 10.1002/eji.200737182
44. Schleicher U, Liese J, Knippertz I, Kurzmann C, Hesse A, Heit A, et al. NK cell activation in visceral leishmaniasis requires TLR9, myeloid DCs, and IL-12, but is independent of plasmacytoid DCs. J Exp Med (2007) 204:893–906. doi: 10.1084/jem.20061293
45. Sacramento LA, da Costa JL, de Lima MHF, Sampaio PA, Almeida RP, Cunha FQ, et al. Toll-like receptor 2 is required for inflammatory process development during leishmania infantum infection. Front Microbiol (2017) 8:262. doi: 10.3389/fmicb.2017.00262
46. Nandakumar R, Tschismarov R, Meissner F, Prabakaran T, Krissanaprasit A, Farahani E, et al. Intracellular bacteria engage a STING-TBK1-MVB12b pathway to enable paracrine cGAS-STING signaling. Nat Microbiol (2019) 4:701–13. doi: 10.1038/s41564-019-0367-z
47. Scumpia PO, Botten GA, Norman JS, Kelly-Scumpia KM, Spreafico R, Ruccia AR, et al. Opposing roles of toll-like receptor and cytosolic DNA-STING signaling pathways for staphylococcus aureus cutaneous host defense. PloS Pathog (2017) 13:e1006496. doi: 10.1371/journal.ppat.1006496
48. Andrade WA, Firon A, Schmidt T, Hornung V, Fitzgerald KA, Kurt-Jones EA, et al. Group b streptococcus degrades cyclic-di-AMP to modulate STING-dependent type I interferon production. Cell Host Microbe (2016) 20:49–59. doi: 10.1016/j.chom.2016.06.003
49. Wang P, Li S, Zhao Y, Zhang B, Li Y, Liu S, et al. The GRA15 protein from toxoplasma gondii enhances host defense responses by activating the interferon stimulator STING. J Biol Chem (2019) 294:16494–508. doi: 10.1074/jbc.ra119.009172
50. Rettig M, Germann MW, Wang S, Wilson WD. Molecular basis for sequence dependent induced DNA bending. Chembiochem (2013) 14:323–31. doi: 10.1002/cbic.201200706
51. Simmons DP, Nguyen HN, Gomez-Rivas E, Jeong Y, Jonsson AH, Chen AF, et al. SLAMF7 engagement superactivates macrophages in acute and chronic inflammation. Sci Immunol (2022) 7:eabf2846. doi: 10.1126/sciimmunol.abf2846
52. Kida Y, Kuwano K, Zhang Y, Arai S. Acholeplasma laidlawii up regulates granulysin gene expression via transcription factor activator protein 1 in a human monocytic cell line, THP 1. Immunology (2001) 104:324–32. doi: 10.1046/j.1365-2567.2001.01310.x
53. Yap J, McCurdy S, Alcala M, Irei J, Garo J, Regan W, et al. Expression of chitotriosidase in macrophages modulates atherosclerotic plaque formation in hyperlipidemic mice. Front Physiol (2020) 11:714. doi: 10.3389/fphys.2020.00714
Keywords: leishmania, kinetoplast DNA (kDNA), cGAS, STING, TBK1, amlexanox, 2'3'-cGAMP, H151
Citation: Yilmaz IC, Dunuroglu E, Ayanoglu IC, Ipekoglu EM, Yildirim M, Girginkardesler N, Ozbel Y, Toz S, Ozbilgin A, Aykut G, Gursel I and Gursel M (2022) Leishmania kinetoplast DNA contributes to parasite burden in infected macrophages: Critical role of the cGAS-STING-TBK1 signaling pathway in macrophage parasitemia. Front. Immunol. 13:1007070. doi: 10.3389/fimmu.2022.1007070
Received: 29 July 2022; Accepted: 19 October 2022;
Published: 02 November 2022.
Edited by:
Bénédicte Manoury, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Sam Campos, University of Arizona, United StatesCopyright © 2022 Yilmaz, Dunuroglu, Ayanoglu, Ipekoglu, Yildirim, Girginkardesler, Ozbel, Toz, Ozbilgin, Aykut, Gursel and Gursel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mayda Gursel, bWF5ZGEuZ3Vyc2VsQGliZy5lZHUudHI=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.