 Caroline Dix1,2†
Caroline Dix1,2† Johannes Zeller3,4†Hannah Stevens1,2,3
Johannes Zeller3,4†Hannah Stevens1,2,3 Steffen U. Eisenhardt4Karen S. Cheung Tung Shing5,6Tracy L. Nero5,6
Steffen U. Eisenhardt4Karen S. Cheung Tung Shing5,6Tracy L. Nero5,6 Craig J. Morton5,6,7
Craig J. Morton5,6,7 Michael W. Parker5,6,8
Michael W. Parker5,6,8 Karlheinz Peter3,6,9‡
Karlheinz Peter3,6,9‡ James D. McFadyen1,2,3,6*‡
James D. McFadyen1,2,3,6*‡- 1Department of Haematology, Alfred Hospital, Melbourne, VIC, Australia
- 2Australian Centre for Blood Diseases, Monash University, Melbourne, VIC, Australia
- 3Atherothrombosis and Vascular Biology Program, Baker Heart and Diabetes Institute, Melbourne, VIC, Australia
- 4Department of Plastic and Hand Surgery, University of Freiburg Medical Centre, Medical Faculty of the University of Freiburg, Freiburg, Germany
- 5Department of Biochemistry and Pharmacology, Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, Parkville, VIC, Australia
- 6Baker Department of Cardiometabolic Health, The University of Melbourne, Parkville, VIC, Australia
- 7Commonwealth Scientific and Industrial Research Organisation (CSIRO) Biomedical Manufacturing Program, Clayton, VIC, Australia
- 8Structural Biology Unit, St. Vincent’s Institute of Medical Research, Fitzroy, VIC, Australia
- 9Department of Cardiology, Alfred Hospital, Melbourne, VIC, Australia
C-reactive protein (CRP) is a member of the highly conserved pentraxin superfamily of proteins and is often used in clinical practice as a marker of infection and inflammation. There is now increasing evidence that CRP is not only a marker of inflammation, but also that destabilized isoforms of CRP possess pro-inflammatory and pro-thrombotic properties. CRP circulates as a functionally inert pentameric form (pCRP), which relaxes its conformation to pCRP* after binding to phosphocholine-enriched membranes and then dissociates to monomeric CRP (mCRP). with the latter two being destabilized isoforms possessing highly pro-inflammatory features. pCRP* and mCRP have significant biological effects in regulating many of the aspects central to pathogenesis of atherothrombosis and venous thromboembolism (VTE), by directly activating platelets and triggering the classical complement pathway. Importantly, it is now well appreciated that VTE is a consequence of thromboinflammation. Accordingly, acute VTE is known to be associated with classical inflammatory responses and elevations of CRP, and indeed VTE risk is elevated in conditions associated with inflammation, such as inflammatory bowel disease, COVID-19 and sepsis. Although the clinical data regarding the utility of CRP as a biomarker in predicting VTE remains modest, and in some cases conflicting, the clinical utility of CRP appears to be improved in subsets of the population such as in predicting VTE recurrence, in cancer-associated thrombosis and in those with COVID-19. Therefore, given the known biological function of CRP in amplifying inflammation and tissue damage, this raises the prospect that CRP may play a role in promoting VTE formation in the context of concurrent inflammation. However, further investigation is required to unravel whether CRP plays a direct role in the pathogenesis of VTE, the utility of which will be in developing novel prophylactic or therapeutic strategies to target thromboinflammation.
Introduction
C-reactive protein (CRP) is an acute phase reactant widely used in clinical practice as a marker of infection and/or inflammation owing to the fact that its synthesis rapidly and dramatically (up to 10,000-fold) increases after tissue injury or infection (1). However, a significant body of work exists demonstrating that destabilized isoforms of CRP possess important pro-inflammatory and pro-thrombotic properties and can therefore amplify pathological conditions associated with ‘immunothrombosis’ (2, 3). It is now appreciated that venous thromboembolism (VTE) represents a prototypical immunothrombotic response. Accordingly, VTE risk is significantly elevated in a number of conditions associated with inflammation and elevations of plasma CRP levels, particularly surgery, obesity, sepsis, cancer, inflammatory bowel disease (IBD) and COVID-19. As will be discussed below, despite evidence highlighting a direct role of CRP in amplifying thromboinflammatory responses in the setting of ischemia/reperfusion injury and transplant rejection, the direct causal role of CRP in VTE pathogenesis has not yet been thoroughly investigated. However, a significant body of research has focused on whether CRP is associated with VTE in a variety of clinical contexts. This review will discuss potential mechanisms by which CRP may play a direct role in mediating VTE pathogenesis, the clinical evidence for the role of CRP in predicting first episode VTE, recurrent VTE and in specific subsets of patients such as those with COVID-19 and cancer-associated thrombosis (CAT).
CRP structure and function
CRP is a member of the highly evolutionary conserved pentraxin superfamily of proteins and is composed of five identical protomers arranged in a pentameric configuration, termed pentameric CRP (pCRP) (4). Importantly, each CRP protomer contains a phosphocholine (PC) binding site which allows the binding of pCRP to PC exposed on inflamed, apoptotic cells and bacterial cell walls. The PC binding pockets of pCRP, contained on the ‘binding’ face of the CRP pentamer, include two critical residues for mediating CRP-PC interactions. These residues, Phe-66 and Glu-81, mediate the interactions between the methyl group and the positively charged nitrogen of PC, respectively (5). The opposite face of the CRP pentamer is the ‘effector’ face, and it has the ability to bind to, and interact with, the globular head of the complement C1q subunit and Fcγ receptors, thus endowing CRP with the ability to regulate innate immune functions (6). Intriguingly, pCRP is now understood to be a circulating precursor form of the ‘CRP system’, and it is not a pro-inflammatory molecule. In support, it has been demonstrated that the infusion of purified pCRP in healthy human volunteers does not induce pro-inflammatory effects (7).
The biological function of CRP appears significantly dependent upon conformational changes that can occur in response to a range of stimuli including binding to PC-rich membranes, exposure to an acidic microenvironment, heat or urea, or binding to misfolded proteins such as beta amyloid (3, 8). Indeed, our group recently demonstrated that upon binding of pCRP to PC-enriched membranes with curvature similar to that observed for platelet, endothelial and leukocyte derived microvesicles, pCRP relaxes its overall pentameric conformation to form pCRP* (2, 9). This structural destabilization initiates the exposure of neoepitopes at the interface of adjacent CRP protomers, thus allowing the binding of C1q to the central pore of pCRP* with subsequent activation of the classical complement pathway (Figure 1) (3, 4).
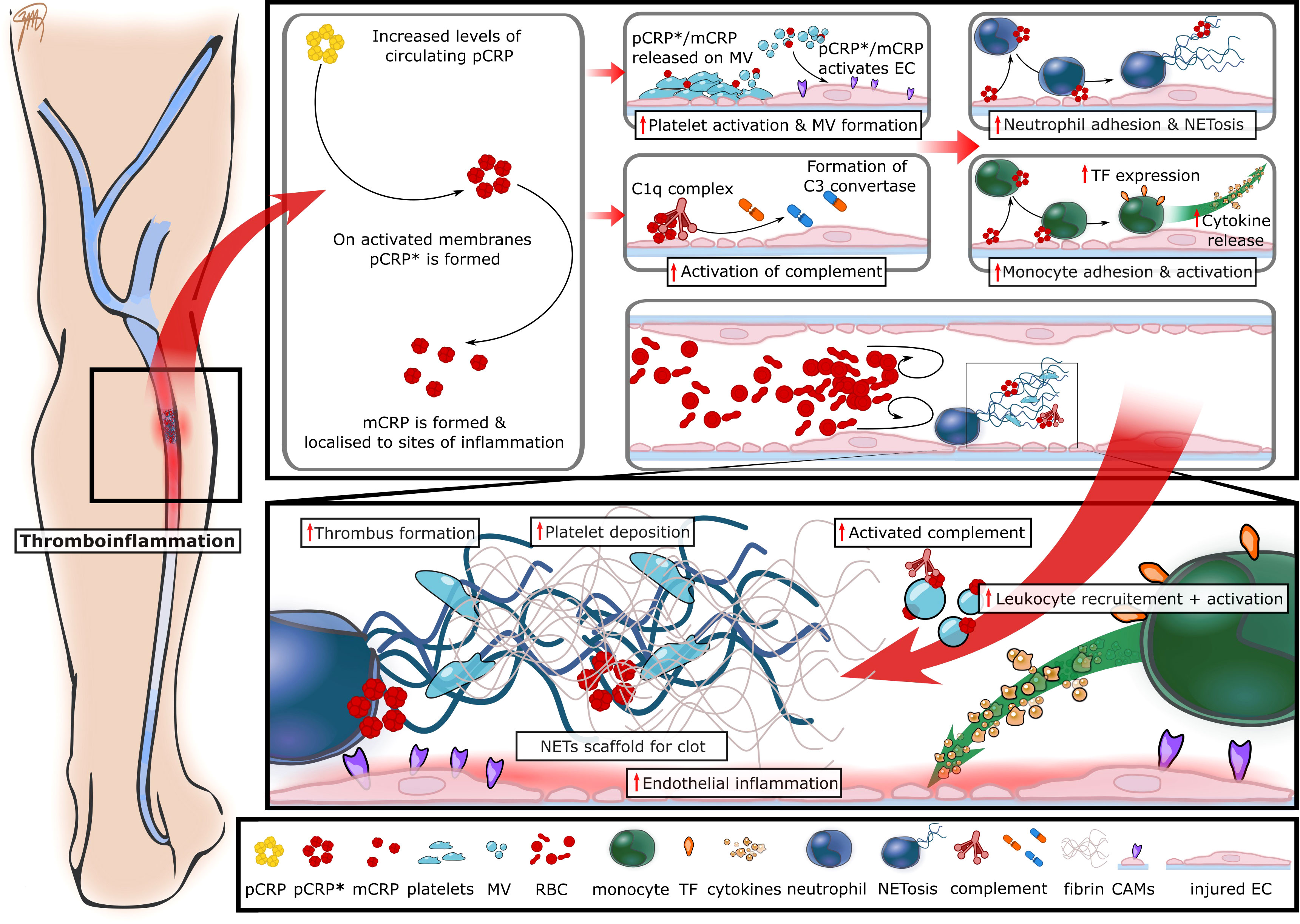
Figure 1 The proposed role of CRP in immunothrombosis and VTE pathogenesis. The inciting events leading to VTE formation are endothelial inflammation with the subsequent recruitment of platelets and leukocytes. Leukocytes provide a source of tissue factor (TF) and highly activated neutrophils undergo NETosis, thereby resulting in thrombin formation and the generation of fibrin. Circulating CRP levels rapidly increase in the setting of inflammation and pCRP binds to disturbed cell surfaces, such as inflamed endothelial cells (EC) and activated platelets where it dissociates into the pro-inflammatory and pro-thrombotic isoforms pCRP* and mCRP. These pro-inflammatory isoforms amplify further activation of the endothelium, platelets and bind to complement C1q, thereby initiating complement activation. These changes result in further recruitment and activation of leukocytes ultimately leading to increased tissue TF and NET formation, thus resulting in enhanced thrombus formation. CAMs, cell adhesion molecules; EC, endothelial cells; MV, microvesicles; RBC, red blood cells; CRP, C-reactive protein; pCRP, pentameric CRP; pCRP*, neoepitope expressing pCRP; mCRP, monomeric CRP; NETs, neutrophil extracellular traps; TF, tissue factor.
Although yet to be precisely delineated, it is currently thought that pCRP* is an ‘intermediate’ conformation that precedes dissociation of the CRP protomers and the formation of monomeric CRP (mCRP) (Figure 1). The formation of mCRP exposes a range of interprotomer neoepitopes which are thought to account for its potent pro-inflammatory and pro-thrombotic functions (2, 10). Interestingly, the dissociation of pCRP to mCRP is associated with a shift in solubility such that mCRP is relatively insoluble and therefore it is likely that this isoform does not circulate as an unbound protein in blood but rather remains anchored on activated cellular membranes, e.g. on activated platelets or extracellular vesicles (2, 3, 9, 11). These observations have resulted in the concept that pCRP and pCRP* may represent the circulating forms of CRP, whilst mCRP is likely to be generated at sites of inflammation and is predominantly tissue bound.
The concept of immunothrombosis and the pro-thrombotic and pro-inflammatory properties of pCRP* and mCRP
It is now well appreciated that venous thrombosis is a consequence of thromboinflammation. Indeed, seminal work by von Bruhl et al. demonstrate that the initial stages of VTE pathogenesis are incited by endothelial inflammation which facilitates the subsequent recruitment of platelet and leukocytes – predominantly neutrophils and monocytes (Figure 1) (12). These events culminate in the activation of recruited cells and ultimately conspire in the formation of neutrophil extracellular traps (NETs) through the process of NETosis (13–15). The formation of NETs triggers fibrin formation via activation of the contact pathway of coagulation, whilst monocytes bearing tissue factor (TF) initiate the extrinsic coagulation pathway (12). These findings demonstrate the inextricable link between the immune and coagulation systems, and led to the concept of “immunothrombosis”, an innate immune response induced by intravascular thrombus formation leading to the recognition and destruction of pathogens (16). Significantly, the pro-inflammatory and pro-thrombotic isoforms of CRP, i.e. pCRP* and mCRP, have demonstrated significant biological effects in regulating many of the aspects central to pathogenesis of atherothrombosis and VTE (17).
Indeed, pCRP* and mCRP have been demonstrated to induce endothelial inflammation and facilitate leukocyte adhesion and transmigration in vivo (18, 19). In vitro, the application of mCRP to endothelial cells inhibits nitric oxide production and increases the production of the pro-inflammatory chemokines interleukin-8 (IL-8) and monocyte chemoattractant protein-1 (MCP-1) in an NF-κB dependent manner (20). Moreover, the expression of the key adhesion molecules, vascular cell adhesion protein-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), are upregulated in response to mCRP stimulation, both of which serve to facilitate leukocyte adhesion (18, 19). Importantly, the pro-inflammatory isoforms of CRP not only promote leukocyte adhesion but also serve to enhance leukocyte activation. mCRP can directly stimulate monocytes and neutrophils resulting in activation of the leukocyte integrin αMβ2 (Mac-1), which is a critical binding partner for glycoprotein Ib (GP-Ib) on platelets (21, 22). As such, mCRP acts to enhance leukocyte recruitment and adhesion to the vasculature, consolidates leukocyte-platelet aggregation, and amplifies the local inflammatory responses. Critically in the context of VTE, CRP has been demonstrated to upregulate monocyte TF expression in addition to inducing NET formation (12, 23). These steps are critical in VTE pathogenesis since, genetic deletion, or pharmacological inhibition of these processes afford significant protection from VTE formation in animal models (12).
In addition to its effects on endothelial cells and leukocytes, mCRP can contribute to thrombosis by way of its ability to activate platelets. Although the receptor responsible for mediating mCRP-induced platelet activation remains to be delineated, a role for the platelet scavenger receptor, CD36, has been suggested (24). mCRP stimulation of platelets leads to activation of the major platelet adhesion receptor, GPIIb/IIIa, and results in alpha granule exocytosis (24). These functional responses promote thrombogenesis by enhancing platelet aggregation and leukocyte recruitment. Interestingly, platelet thrombi can also act as a scaffold for CRP dissociation which in turn acts to enhance thrombus growth (24, 25). Moreover, microvesicles enriched in lysophosphatidylcholine, and derived from activated platelets and monocytes, can bind to and destabilize pCRP to produce the pro-inflammatory CRP isoforms and induce inflammatory responses in endothelial cells (2, 9). Furthermore, activated platelets can interact directly with neutrophils and cause NET formation (26).
As described above, neoepitope-expressing pCRP* can bind the C1q subunit and trigger activation of the classical complement pathway. Well recognized as a fundamental component of the innate immune response, there is increasing evidence highlighting an important role for complement in thrombosis (27, 28). Indeed, complement and the coagulation pathways have likely evolved from a common ancestral pathway and share many similarities since both involve the activation of inactive zymogens into potent serine proteases. Recent data has highlighted how C1q can bind to, and activate platelets, in addition to interacting with von Willebrand Factor (vWF), to enhance platelet adhesion (29). As well as the direct effects of C1q on platelet responses, downstream products of C1q activation, namely the anaphylatoxins C3a and C5a, have been shown to promote thrombosis by activating endothelial cells and neutrophils and upregulating TF expression (27). Accordingly, population-based studies have linked elevated levels of C3 and complement activation pathways with an increased risk of VTE (30).
Understanding the role of CRP in venous thromboembolism
VTE includes deep vein thrombosis (DVT) and pulmonary embolism and is associated with substantial mortality which, in contrast to arterial thromboembolic disease, has not improved in recent years (31). The historical understanding of venous thrombus formation is centred upon the mechanisms now well described as Virchow’s triad, which include vessel wall damage, venous stasis and hypercoagulability. However, mounting evidence demonstrates the critical importance of immunothrombosis and suggests that venous and arterial thrombotic disorders may be part of a continuous spectrum of disease. Indeed, it is now well appreciated that inflammation plays an important role in the pathogenesis of cardiovascular disease (32). In this regard, baseline circulating CRP levels in asymptomatic individuals constitute an independent risk factor for coronary (33) and peripheral vascular disease (34). The Women’s Health Study (35) and Physicians’ Health Study (36) both demonstrated that the circulating level of CRP is a predictor of future cardiovascular events, independent of other risk factors including hypertension, obesity and hypercholesterolemia. Additionally, short-term mortality post-acute myocardial infarct is associated with CRP responses (37). Possible explanations for these phenomena include the fact that circulating CRP levels reflect inflammation related to the atherosclerotic process and/or the extent of myocardial necrosis, and that CRP potentially directly interacts with atherosclerotic vessels (38). Indeed, preclinical studies have consistently demonstrated a convincing role for CRP, potentially via its activating isoforms, in directly amplifying tissue damage in rodent models of myocardial infarction (39). This highlights new therapeutic implications for cardiovascular and thromboinflammatory diseases (40).
Following the findings that circulating CRP levels predict cardiovascular events, and that statins lower the level of CRP in addition to cholesterol, the JUPITER study was devised to assess if rosuvastatin (compared to placebo) could reduce the risk of cardiovascular events in older individuals with normal cholesterol but high CRP (> 2 mg/L) (41, 42). The trial was stopped early due to a significant reduction in major cardiovascular events in the rosuvastatin arm (0.77 vs 1.36 per 100 person-years, hazard ratio (HR) 0.56) (41). Importantly, rosuvastatin also significantly reduced the occurrence of symptomatic VTE (0.18 vs 0.32 per 100 person-years of follow up, HR of 0.57) compared to placebo (43), as well as significantly reducing circulating CRP levels at 12 months follow up (2.2 vs 3.5 mg/L) (41, 43). As a result of these findings, there has been increasing interest in the value of circulating CRP levels in predicting VTE.
In addition to considering plasma levels of CRP, several studies have evaluated the interaction between genetic polymorphisms in the CRP gene and thrombotic risk. To date, studies pertaining to single nucleotide polymorphisms in the CRP gene and VTE do not suggest a significant association between genetically elevated CRP and VTE risk, which is in contrast to data regarding cardiovascular disease and CRP polymorphisms (44–46). Importantly, the evaluated polymorphisms in the CRP gene are not uniform across studies for arterial and venous thrombotic disease, and the utility of these genetic tests in predicting thrombotic risk is yet to be fully elucidated.
CRP in acute VTE
Acute DVT is typically associated with a classic inflammatory response (redness, heat, swelling, pain, loss of function) which can manifest clinically at the location of the DVT in most patients (47). As such, circulating CRP levels have been measured to understand the utility of CRP as a biomarker in the clinical diagnosis of VTE, and to gain insight into the potential causal relationship between inflammation and thrombosis. In this acute setting, individuals with an acute DVT have higher circulating CRP levels compared to individuals who do not have DVT (48, 49), and a higher median CRP is associated with a significantly higher odds of DVT diagnosis (48). These findings appear to be particularly prominent in patients with proximal or extensive VTE, who are more likely to demonstrate a higher circulating CRP level compared with distal or smaller thromboses (47). In addition to these acute elevations at the time of DVT diagnosis, circulating CRP levels have been shown to be elevated in the 90-days prior to a diagnosed VTE episode, which highlights inflammation as a trigger for VTE (50). However, studies have also demonstrated that circulating CRP levels typically fall in the days following acute thrombosis diagnosis and anticoagulation initiation, which could indicate that inflammation and the rise in circulating CRP levels are the result of the thrombotic event rather than the cause (49). Further research is required to fully elucidate this complex interaction. Whilst these data do not demonstrate causality, they serve to further highlight the intimate link between inflammation and thrombosis in the context of VTE.
CRP as a predictor of incident VTE
Although it is evident that acute VTE is associated with an increase in circulating CRP levels, whether long-term, low-grade CRP elevations predicts future VTE events, as it does with cardiovascular disease, remains controversial. Indeed, the results from a number of large, population-based studies examining this question have been conflicting, and associations found have typically been modest. These studies are summarized in Table 1. The differences may be partly explained by intra-individual variation in inflammatory markers, which is able to mask a true but small impact of low-grade inflammation on VTE risk, as well as significant variability in patient population by ethnicity, gender, and age (60). Additionally, timing of circulating CRP measurement in relation to the VTE episode was likely to play a role in the varied results, as well as the variation in the type of CRP assays used and their sensitivity. Moreover, given the growing awareness of the different biological activities of mCRP and pCRP*, it is important to highlight that no study to date has measured how different isoforms of CRP change in acute VTE.

Table 1 Summary of population-based studies assessing the relationship between circulating CRP levels and VTE.
Long-term population-based studies assessing baseline circulating CRP levels and future risk of VTE have shown a modest association between CRP levels and VTE (Table 1). A Danish study of over 10,000 participants found those with a baseline level of high sensitivity CRP (hsCRP) > 3 mg/L had a 2.3 times increased risk of VTE compared to those with levels < 1 mg/L, after adjustment for age, sex and use of statins over 16 years of follow up (44). Additionally, there was a stepwise increase in VTE risk as circulating CRP levels rose. The ARIC study of over 10,000 participants with an 8 year follow up period also found a higher baseline hsCRP level to be associated with an increased risk of VTE. The authors report a HR 1.6-2 times higher for values above versus below the 90th percentile of CRP (> 8.55 mg/L), after exclusion of those with prior VTE and those with very elevated circulating CRP levels (> 20 mg/L) (54). Quist-Paulsen et al. similarly found a positive association between baseline hsCRP levels and subsequent VTE, with a 1.6-fold increased risk of VTE in those with the highest quintile versus the lowest quintile of CRP. Interestingly, they found the strongest association in individuals who experienced venous thrombosis within a year of blood sampling, suggesting direct stimulation of the coagulation system by short-term inflammatory processes (56). A meta-analysis of nine population-based studies, with 81,625 participants, found an adjusted risk estimate for VTE of 1.14 (95 % CI 1.08-1.19) per standard deviation (SD) increase in baseline CRP (59).
In contrast, a number of studies have found no effect of circulating CRP levels on VTE risk (Table 1). A study of over 6,000 individuals followed for 12.5 years found no increased risk of VTE per 1 SD increase in baseline hsCRP or across quintiles of CRP (57). This held true after adjusting for body-mass-index (BMI), smoking, diabetes or whether the VTE episode was provoked or unprovoked (57). They excluded those with prior VTE and those with hsCRP greater than 10 mg/L to remove any affect related to acute inflammation (57). The longitudinal Physicians Health Study (36) and Longitudinal Investigation of Thromboembolism Etiology (LITE) Study (52) also found CRP levels were not predictive of future VTE. The LITE study looked at 20,000 healthy participants without a history of VTE with a follow up time of 7.8 years (52), and found a linear association with factor VIII and vWF levels, but no association with CRP.
Considering the inconsistent and, at best, modest association of circulating CRP levels and VTE in all-comers, several studies have investigated different subgroups of patients with VTE to determine if there may be a stronger association. In this regard, obesity is a known risk factor for VTE and is associated with double the risk of VTE compared to people of normal weight (61). Several studies have now assessed the role of a range of biomarkers, including circulating CRP levels, in explaining the increased risk of VTE in this population. To date, these results have been conflicting (58, 62), but appear to suggest an association between elevated CRP levels and obesity in older individuals and females (36, 59, 60).
Patients undergoing major surgery are another group who are considered to be at high risk of developing VTE and circulating CRP levels may aid in identifying individuals at particularly high risk. A small study of 32 patients demonstrated those that developed VTE had a significantly higher increase in CRP levels on post-operative day 1 compared to their pre-operative baseline (63). The circulating CRP level was significantly correlated with platelet-monocyte aggregates, supporting the interplay between CRP and inflammation (63). CRP has also been shown to be associated with a higher risk of pre-operative DVT in individuals with closed femur fractures (64). After lower-risk surgery (such as laparoscopic cholecystectomy), CRP levels typically rise on post-operative day 7-10 but then normalize by day 30 and are not associated with VTE risk (65).
Overall, the measurement of plasma CRP does not appear to be a sensitive biomarker in predicting VTE risk in unselected patient groups. However, it may have utility in certain populations, including obese individuals, females and those undergoing major surgery. The lack of consistent association in unselected patient groups is likely related to normal intra-individual variation in circulating CRP levels, timing of CRP measurement as well as measurement differences. Lastly, the potential role of different isoforms of CRP as predictors of incident VTEs remains to be investigated.
CRP as a predictor of VTE recurrence
A biomarker that would identify individuals at high risk of recurrence following the first unprovoked VTE would be extremely beneficial, as it would improve individualization of the duration of anticoagulation, based on predicted recurrence risk. Currently we rely on clinical prediction scores for risk of recurrent VTE, such as Vienna, DASH and HERDOO2. However, all three have deficiencies such as an inability to stratify males, and complexities meaning they are difficult to use in routine clinical practice (66–68). In this regard, there are several clinical situations where patients are recommended to continue anticoagulation indefinitely due to the high risk of recurrent VTE, such as in CAT or antiphospholipid syndrome. However, in many other situations the decision for continued anticoagulation is less clear and relies on discussion of population-based predicted recurrence risks balanced against bleeding risks and patient wishes (69). In these situations of clinical equipoise, a novel biomarker to predict recurrent VTE to allow the use of anticoagulation in only truly high-risk patients would represent a significant advance.
Several studies have shown that circulating CRP levels are higher in those with a history of VTE compared to those without VTE, and in particular that CRP is higher in those with unprovoked compared to provoked VTE (53, 55). The Leiden Thrombophilia study found patients with a prior VTE had higher CRP levels than healthy controls, when circulating CRP was measured between 6 and 18 months following acute VTE (51). However, much of this difference was driven by the fact that 11 % of VTE patients had CRP levels above the 95th percentile of the control value (9.75 mg/L), and when these were excluded the effect disappeared (51). Regardless, this does suggest that ongoing systemic inflammation is more prominent among VTE patients (51). While these studies did not link the results to risk of VTE recurrence, a sub-study of the Leiden Thrombophilia Study followed those with a prior DVT and assessed circulating CRP levels at least 3 months after discontinuation of oral anticoagulation (70). They found those with an elevated D-dimer and/or CRP level had a higher risk of recurrence compared to those with low CRP and D-dimer levels with a HR of 1.9. Another prospective study assessing risk factors for recurrence after unprovoked VTE involving 229 patients similarly found elevated hsCRP levels, as well as iron deficiency, predicted VTE recurrence over the subsequent 24 months (71). These studies highlight the potential for using CRP in combination with other biomarkers or clinical prediction model to improve risk the risk stratification of this patient cohort.
CRP as a predictor of cancer-associated VTE
CAT is a major health problem and is the leading cause of non-cancer death in cancer patients (72). Importantly, the prevention and management of VTE in this cohort is challenging due to the increased risk of both bleeding and thrombotic complications. In this regard, the identification of a novel biomarker to improve VTE risk stratification is required (72). The identification of patients at high risk of CAT is currently based on clinical risk scores such as the Khorana score (73). While the Khorana score is well-validated, a significant number (5-7 %) of VTE events still occur in those classified as low- or intermediate-risk (74). Furthermore, those with lung cancer or hematologic malignancies classified as high risk by the Khorana score have a much lower estimated risk of VTE than other cancer types and as such is less informative in these groups of patients (74). This is evidenced by the fact that using clinical risk scores alone (Khorana), VTE prophylaxis using rivaroxaban in ambulatory cancer patients did not result in a significantly lower incidence of VTE or death over 6 months of follow up, but did result in a reduction of VTE on longer follow-up (75). The studies show that there are clearly deficiencies in using clinical risk scores alone, and other ways to stratify VTE risk in patients are needed.
CRP can be produced by cells within the tumor microenvironment including stromal cells and mesenchymal-like tumor cells (76, 77). Interestingly, activated platelets, known to destabilize pCRP to the pro-inflammatory pCRP*/mCRP isoforms, have been identified as an early marker of primary tumors and metastases (78), indicating that CRP activation at the site of cancerous tissue contributes to a pro-inflammatory state in cancer patients. Accordingly, serum levels of CRP correlate with the tumor mass in patients, with high levels of CRP reflecting advanced disease and poor prognosis (76). Regarding CAT, several biomarkers have been assessed for prediction of primary VTE risk, but CRP has not been studied as extensively as other biomarkers such as D-dimer, leukocyte and platelet count. A small study of 35 patients found that the combination of hsCRP, D-dimer and the clinical Wells score could predict the incidence of acute DVT in cancer patients (79). A study of 507 cancer patients in Germany, of which 60 developed a new VTE, found that those with VTE had higher circulating CRP levels in the 30 days prior to the VTE episode than those without any VTE in the follow up period (80). In particular, an elevated CRP level > 5 mg/L was linked with the occurrence of VTE (80). The Vienna Cancer and Thrombosis Study (CATS) followed 705 cancer patients for two years after an initial blood sample (81). They found the cumulative probability of developing VTE after 12 months was 11.7 % in those with a higher circulating CRP level (above the 75th percentile), compared to 4.9 % for those with lower CRP levels (81). After adjusting for age, gender, chemotherapy, surgery, radiotherapy and BMI, the HR of VTE per doubling of CRP was 1.1. In support, the authors report that circulating CRP level was clearly associated with a worse survival probability (81).
Biomarkers such as CRP may also further enable identification of those at lower risk of recurrence after a first CAT – potentially allowing the cessation of anticoagulation and thus mitigating the accompanying bleeding risk. Current recommendations for CAT are to instigate anticoagulation which continues indefinitely while cancer is active, or the individual is receiving anti-cancer therapy unless there are contraindications (82). To determine if circulating CRP levels, in addition to D-dimer, could predict VTE risk after cessation of anticoagulation after CAT, Jara-Palomares et al. assessed these parameters 21 days after anticoagulation cessation (83). They found a hsCRP level > 4.5 mg/L could predict VTE recurrence and would allow for 66 % of patients to stop anticoagulation based on the hsCRP level at this time point (83). The 6-month VTE recurrence rate was 8.8 %, which decreased to 1.5 % if the 21-day hsCRP level was < 4.5 mg/L (83). CRP was also evaluated as part of the CATCH trial, which compared tinzaparin with warfarin in the treatment of 900 patients with acute CAT – they found a mean circulating CRP level of 50.3 mg/L at the time of acute VTE, and that the uppermost quartile of CRP (> 75 mg/L) was associated with recurrent VTE in univariate analysis (RR 2.3) which was maintained after adjustment for competing risks such as history of VTE or metastatic disease (84).
Overall, this evidence suggests that individuals with cancer and a higher circulating CRP level have an increased risk of developing a first thrombosis, and a higher risk of recurrence following treatment for CAT if the CRP level remains high after cessation of anticoagulation.
CRP as a predictor of VTE in COVID-19
From an early stage in the coronavirus disease 2019 (COVID-19) pandemic, VTE was seen as a frequent ‘immunothrombotic’ complication in affected individuals (85, 86). Indeed, thrombotic complications have been reported to affect up to half of critically ill COVID-19 patients admitted to the intensive care unit and 7.9 % of hospitalized COVID-19 patients, significantly higher than rates seen in other respiratory illnesses (87, 88).
Elevation in circulating CRP levels in the context of COVID-19 has been shown to be associated with VTE. A case-control study of 256 non-critically ill and 144 critically ill COVID-19 patients in the USA found a circulating CRP level of > 100 mg/L at presentation to be associated with thrombotic complications with an adjusted OR of 3.04 (89). A number of subsequent retrospective studies have found higher CRP levels in those with VTE compared to those without VTE, with the positive predictive value of CRP enhanced when combined with D-dimer (90, 91). A prospective study confirmed circulating CRP levels on admission to be an independent predictor of incident pulmonary embolism during hospitalization with COVID-19 (92). Therefore, it appears CRP is useful in predicting VTE in the context of COVID-19, with D-dimer further improving risk assessment, however larger prospective studies are required to confirm the association.
Considering elevations in CRP levels have been associated with acute thrombotic episodes in the general population, it follows that it would also be associated with VTE in COVID-19. Furthermore, our current understanding of the pathophysiology of thrombosis in COVID-19 highlights important roles of platelet activation, endothelial inflammation and NETosis in the setting of COVID-19 infection (85). Recent clinical trials have suggested anti-thrombotic therapies may improve outcomes in patients with COVID-19, but at the expense of more bleeding (93). Therefore, novel approaches to target the deleterious thromboinflammatory response associated with COVID-19 infection remains an urgent unmet clinical need with the ongoing global COVID-19 pandemic.
Summary and perspectives
The data for circulating CRP levels as a predictor for VTE in the unselected general population remains mixed. However, in patients with a prior history of VTE, obesity, cancer and COVID-19 the cumulative data supports that elevations of CRP may aid in the prediction of VTE risk. Although these studies do not imply causality, further investigations are required to unravel whether circulating CRP plays a direct role in the pathogenesis of VTE. The potential pathogenic role of CRP in cardiovascular disease has spurred much debate over nearly two decades; however, it is clear that CRP binding to damaged or inflamed tissue can amplify tissue damage and inflammation, and is now implicated in the pathogenesis of a number of thromboinflammatory diseases such as ischemia/reperfusion injury. In contrast, the infusion of purified pCRP into healthy adults elicits no such inflammatory response (7). Therefore, it is tempting to speculate that CRP’s role in VTE is similar. That is, in the context of concurrent inflammation, as seen in obesity, cancer and COVID-19, a fertile environment is present for the ‘activation’ of CRP, which can then exacerbate the thromboinflammatory processes involved in VTE formation. In this regard, future studies are required to understand whether circulating CRP plays a direct role mediating VTE. Moreover, the assessment of CRP isoforms in human venous thrombosis will provide further insights regarding the potential pathogenic role of CRP in VTE. A range of therapeutic strategies directed at inhibiting the pro-inflammatory effects of CRP isoforms have been developed that display profound anti-inflammatory effects in cardiac and renal ischemia/reperfusion injury, and transplant rejection. Therefore, addressing the role of CRP, particularly its dissociation in pro-inflammatory/pro-aggregatory isoforms, in VTE is fundamental given the ongoing interest in developing novel therapeutic/preventative strategies to target thromboinflammation. Such strategies may hold promise for a range of diseases including VTE prevention/therapy and the treatment of COVID-19.
Author contributions
CD, JZ, HS, KP, JM conceived and wrote the manuscript. SE, TN, CM, MP co wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
HS is supported by a Monash University RTP Scholarship and Wheaton Family Scholarship. KP is supported by a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellowship. JM is supported by a NHMRC and Heart Foundation Early Career Fellowship. Funding from the Victorian Government Operational Infrastructure Support Scheme to St Vincent’s Institute is acknowledged. MP is a National Health and Medical Research Council of Australia Leadership Fellow.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BMI, Body-mass-index; CAMs, Cell adhesion molecules; CAT, Cancer-associated thrombosis; CI, Confidence interval; COVID-19, Coronavirus disease 19; CRP, C-reactive protein; hsCRP, High-sensitivity CRP; mCRP, Monomeric CRP; pCRP, Pentameric CRP; pCRP*, Neoepitope expressing pCRP; DVT, Deep vein thrombosis; EC, Endothelial cells; HR, Hazard ratio; HRT, Hormone replacement therapy; IBD, Inflammatory bowel disease; NETs, Neutrophil extracellular traps; MV, Microvesicles; OR, Odds ratio; PC, Phosphocholine; RBC, Red blood cells; RR, Relative risk; TF, Tissue factor; VTE, Venous thromboembolism; vWF, von Willebrand Factor.
References
1. Gewurz H, Mold C, Siegel J, Fiedel B. C-reactive protein and the acute phase response. Adv Intern Med (1982) 27:345–72. doi: 10.1080/21548331.1982.11702332
2. Braig D, Nero TL, Koch HG, Kaiser B, Wang X, Thiele JR, et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat Commun (2017) 8:14188. doi: 10.1038/ncomms14188
3. McFadyen JD, Kiefer J, Braig D, Loseff-Silver J, Potempa LA, Eisenhardt SU, et al. Dissociation of c-reactive protein localizes and amplifies inflammation: Evidence for a direct biological role of c-reactive protein and its conformational changes. Front Immunol (2018) 9:1351. doi: 10.3389/fimmu.2018.01351
4. McFadyen JD, Zeller J, Potempa LA, Pietersz GA, Eisenhardt SU, Peter K. C-reactive protein and its structural isoforms: An evolutionary conserved marker and central player in inflammatory diseases and beyond. Subcell Biochem (2020) 94:499–520. doi: 10.1007/978-3-030-41769-7_20
5. Thompson D, Pepys MB, Wood SP. The physiological structure of human c-reactive protein and its complex with phosphocholine. Structure (1999) 7(2):169–77. doi: 10.1016/S0969-2126(99)80023-9
6. Gaboriaud C, Juanhuix J, Gruez A, Lacroix M, Darnault C, Pignol D, et al. The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J Biol Chem (2003) 278(47):46974–82. doi: 10.1074/jbc.M307764200
7. Lane T, Wassef N, Poole S, Mistry Y, Lachmann HJ, Gillmore JD, et al. Infusion of pharmaceutical-grade natural human c-reactive protein is not proinflammatory in healthy adult human volunteers. Circ Res (2014) 114(4):672–6. doi: 10.1161/CIRCRESAHA.114.302770
8. Strang F, Scheichl A, Chen YC, Wang X, Htun NM, Bassler N, et al. Amyloid plaques dissociate pentameric to monomeric c-reactive protein: A novel pathomechanism driving cortical inflammation in alzheimer's disease? Brain Pathol (2012) 22(3):337–46. doi: 10.1111/j.1750-3639.2011.00539.x
9. Habersberger J, Strang F, Scheichl A, Htun N, Bassler N, Merivirta RM, et al. Circulating microparticles generate and transport monomeric c-reactive protein in patients with myocardial infarction. Cardiovasc Res (2012) 96(1):64–72. doi: 10.1093/cvr/cvs237
10. Thiele JR, Habersberger J, Braig D, Schmidt Y, Goerendt K, Maurer V, et al. Dissociation of pentameric to monomeric c-reactive protein localizes and aggravates inflammation: In vivo proof of a powerful proinflammatory mechanism and a new anti-inflammatory strategy. Circulation (2014) 130(1):35–50. doi: 10.1161/CIRCULATIONAHA.113.007124
11. Eisenhardt SU, Habersberger J, Murphy A, Chen YC, Woollard KJ, Bassler N, et al. Dissociation of pentameric to monomeric c-reactive protein on activated platelets localizes inflammation to atherosclerotic plaques. Circ Res (2009) 105(2):128–37. doi: 10.1161/CIRCRESAHA.108.190611
12. von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med (2012) 209(4):819–35. doi: 10.1084/jem.20112322
13. Colling ME, Tourdot BE, Kanthi Y. Inflammation, infection and venous thromboembolism. Circ Res (2021) 128(12):2017–36. doi: 10.1161/CIRCRESAHA.121.318225
14. Moon MJ, McFadyen JD, Peter K. Caught at the scene of the crime: Platelets and neutrophils are conspirators in thrombosis. Arteriosclerosis Thrombosis Vasc Biol (2022) 42(1):63–6. doi: 10.1161/ATVBAHA.121.317187
15. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
16. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol (2013) 13(1):34–45. doi: 10.1038/nri3345
17. Eisenhardt SU, Habersberger J, Peter K. Monomeric c-reactive protein generation on activated platelets: The missing link between inflammation and atherothrombotic risk. Trends Cardiovasc Med (2009) 19(7):232–7. doi: 10.1016/j.tcm.2010.02.002
18. Ji SR, Ma L, Bai CJ, Shi JM, Li HY, Potempa LA, et al. Monomeric c-reactive protein activates endothelial cells via interaction with lipid raft microdomains. FASEB J (2009) 23(6):1806–16. doi: 10.1096/fj.08-116962
19. Khreiss T, Jozsef L, Potempa LA, Filep JG. Conformational rearrangement in c-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation (2004) 109(16):2016–22. doi: 10.1161/01.CIR.0000125527.41598.68
20. Li HY, Wang J, Wu YX, Zhang L, Liu ZP, Filep JG, et al. Topological localization of monomeric c-reactive protein determines proinflammatory endothelial cell responses. J Biol Chem (2014) 289(20):14283–90. doi: 10.1074/jbc.M114.555318
21. Zouki C, Haas B, Chan JS, Potempa LA, Filep JG. Loss of pentameric symmetry of c-reactive protein is associated with promotion of neutrophil-endothelial cell adhesion. J Immunol (2001) 167(9):5355–61. doi: 10.4049/jimmunol.167.9.5355
22. Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, et al. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin mac-1 (CD11b/CD18). J Exp Med (2000) 192(2):193–204. doi: 10.1084/jem.192.2.193
23. Cermak J, Key NS, Bach RR, Balla J, Jacob HS, Vercellotti GM. C-reactive protein induces human peripheral blood monocytes to synthesize tissue factor. Blood (1993) 82(2):513–20. doi: 10.1182/blood.V82.2.513.513
24. Molins B, Pena E, de la Torre R, Badimon L. Monomeric c-reactive protein is prothrombotic and dissociates from circulating pentameric c-reactive protein on adhered activated platelets under flow. Cardiovasc Res (2011) 92(2):328–37. doi: 10.1093/cvr/cvr226
25. de la Torre R, Pena E, Vilahur G, Slevin M, Badimon L. Monomerization of c-reactive protein requires glycoprotein IIb-IIIa activation: Pentraxins and platelet deposition. J Thromb Haemost. (2013) 11(11):2048–58. doi: 10.1111/jth.12415
26. Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, et al. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest (2012) 122(7):2661–71. doi: 10.1172/JCI61303
27. Subramaniam S, Jurk K, Hobohm L, Jackel S, Saffarzadeh M, Schwierczek K, et al. Distinct contributions of complement factors to platelet activation and fibrin formation in venous thrombus development. Blood (2017) 129(16):2291–302. doi: 10.1182/blood-2016-11-749879
28. Foley JH, Conway EM. Cross talk pathways between coagulation and inflammation. Circ Res (2016) 118(9):1392–408. doi: 10.1161/CIRCRESAHA.116.306853
29. Donat C, Kolm R, Csorba K, Tuncer E, Tsakiris DA, Trendelenburg M. Complement C1q enhances primary hemostasis. Front Immunol (2020) 11:1522. doi: 10.3389/fimmu.2020.01522
30. Norgaard I, Nielsen SF, Nordestgaard BG. Complement C3 and high risk of venous thromboembolism: 80517 individuals from the Copenhagen general population study. Clin Chem (2016) 62(3):525–34. doi: 10.1373/clinchem.2015.251314
31. Arshad N, Isaksen T, Hansen JB, Braekkan SK. Time trends in incidence rates of venous thromboembolism in a large cohort recruited from the general population. Eur J Epidemiol. (2017) 32(4):299–305. doi: 10.1007/s10654-017-0238-y
32. Libby P. Molecular bases of the acute coronary syndromes. Circulation (1995) 91(11):2844–50. doi: 10.1161/01.CIR.91.11.2844
33. Haverkate F, Thompson SG, Pyke SD, Gallimore JR, Pepys MB. Production of c-reactive protein and risk of coronary events in stable and unstable angina. European concerted action on thrombosis and disabilities angina pectoris study group. Lancet (9050) 1997:462–6:349. doi: 10.1016/s0140-6736(96)07591-5
34. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Plasma concentration of c-reactive protein and risk of developing peripheral vascular disease. Circulation (1998) 97(5):425–8. doi: 10.1161/01.CIR.97.5.425
35. Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH. Prospective study of c-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation (1998) 98(8):731–3. doi: 10.1161/01.CIR.98.8.731
36. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med (1997) 336(14):973–9. doi: 10.1056/NEJM199704033361401
37. Pietila KO, Harmoinen AP, Jokiniitty J, Pasternack AI. Serum c-reactive protein concentration in acute myocardial infarction and its relationship to mortality during 24 months of follow-up in patients under thrombolytic treatment. Eur Heart J (1996) 17(9):1345–9. doi: 10.1093/oxfordjournals.eurheartj.a015068
38. Lagrand WK, Visser CA, Hermens WT, Niessen HW, Verheugt FW, Wolbink GJ, et al. C-reactive protein as a cardiovascular risk factor: More than an epiphenomenon? Circulation (1999) 100(1):96–102. doi: 10.1161/01.CIR.100.1.96
39. Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, et al. Targeting c-reactive protein for the treatment of cardiovascular disease. Nature (2006) 440(7088):1217–21. doi: 10.1038/nature04672
40. Zeller J, Bogner B, McFadyen JD, Kiefer J, Braig D, Pietersz G, et al. Transitional changes in the structure of c-reactive protein create highly pro-inflammatory molecules: Therapeutic implications for cardiovascular diseases. Pharmacol Ther (2022) 235:108165. doi: 10.1016/j.pharmthera.2022.108165
41. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr., Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated c-reactive protein. N Engl J Med (2008) 359(21):2195–207. doi: 10.1056/NEJMoa0807646
42. Albert MA, Danielson E, Rifai N, Ridker PM, Investigators P. Effect of statin therapy on c-reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA (2001) 286(1):64–70. doi: 10.1001/jama.286.1.64
43. Glynn RJ, Danielson E, Fonseca FA, Genest J, Gotto AM Jr., Kastelein JJ, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med (2009) 360(18):1851–61. doi: 10.1056/NEJMoa0900241
44. Zacho J, Tybjaerg-Hansen A, Nordestgaard BG. C-reactive protein and risk of venous thromboembolism in the general population. Arterioscler Thromb Vasc Biol (2010) 30(8):1672–8. doi: 10.1161/ATVBAHA.109.198473
45. Zee RY, Hegener HH, Cook NR, Ridker PM. C-reactive protein gene polymorphisms and the risk of venous thromboembolism: a haplotype-based analysis. J Thromb Haemost (2004) 2(8):1240–3. doi: 10.1111/j.1538-7836.2004.00773.x
46. Lange LA, Carlson CS, Hindorff LA, Lange EM, Walston J, Durda JP, et al. Association of polymorphisms in the CRP gene with circulating c-reactive protein levels and cardiovascular events. JAMA (2006) 296(22):2703–11. doi: 10.1001/jama.296.22.2703
47. Syrjala H, Haukipuro K, Kiviniemi H. Acute phase response and deep lower limb venous thrombosis. J Clin Pathol (1990) 43(6):519–20. doi: 10.1136/jcp.43.6.519-b
48. Korte WC, Wuillemin WA, Caliezi C, Riesen WF. CRP measured by a high sensitivity assay correlates with clinical probability (Wells score) testing for deep venous thrombosis. Thromb Haemost (2004) 91(4):841–2. doi: 10.1055/s-0037-1614281
49. Roumen-Klappe EM, den Heijer M, van Uum SH, van der Ven-Jongekrijg J, van der Graaf F, Wollersheim H. Inflammatory response in the acute phase of deep vein thrombosis. J Vasc Surg (2002) 35(4):701–6. doi: 10.1067/mva.2002.121746
50. Grimnes G, Isaksen T, Tichelaar Y, Brox J, Braekkan SK, Hansen JB. C-reactive protein and risk of venous thromboembolism: results from a population-based case-crossover study. Haematologica (2018) 103(7):1245–50. doi: 10.3324/haematol.2017.186957
51. Kamphuisen PW, Eikenboom JC, Vos HL, Pablo R, Sturk A, Bertina RM, et al. Increased levels of factor VIII and fibrinogen in patients with venous thrombosis are not caused by acute phase reactions. Thromb Haemost (1999) 81(5):680–3. doi: 10.1055/s-0037-1614553
52. Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Tracy RP, Aleksic N, et al. Coagulation factors, inflammation markers, and venous thromboembolism: The longitudinal investigation of thromboembolism etiology (LITE). Am J Med (2002) 113(8):636–42. doi: 10.1016/S0002-9343(02)01345-1
53. Vormittag R, Vukovich T, Schonauer V, Lehr S, Minar E, Bialonczyk C, et al. Basal high-sensitivity-C-reactive protein levels in patients with spontaneous venous thromboembolism. Thromb Haemost (2005) 93(3):488–93. doi: 10.1160/TH04-11-0745
54. Folsom AR, Lutsey PL, Astor BC, Cushman M. C-reactive protein and venous thromboembolism. a prospective investigation in the ARIC cohort. Thromb Haemost (2009) 102(4):615–9. doi: 10.1160/TH09-04-0274
55. Luxembourg B, Schmitt J, Humpich M, Glowatzki M, Dressler D, Seifried E, et al. Cardiovascular risk factors in idiopathic compared to risk-associated venous thromboembolism: A focus on fibrinogen, factor VIII, and high-sensitivity c-reactive protein (hs-CRP). Thromb Haemost (2009) 102(4):668–75. doi: 10.1160/TH-09-02-0104
56. Quist-Paulsen P, Naess IA, Cannegieter SC, Romundstad PR, Christiansen SC, Rosendaal FR, et al. Arterial cardiovascular risk factors and venous thrombosis: results from a population-based, prospective study (the HUNT 2). Haematologica (2010) 95(1):119–25. doi: 10.3324/haematol.2009.011866
57. Hald EM, Braekkan SK, Mathiesen EB, Njolstad I, Wilsgaard T, Brox J, et al. High-sensitivity c-reactive protein is not a risk factor for venous thromboembolism: The tromso study. Haematologica (2011) 96(8):1189–94. doi: 10.3324/haematol.2010.034991
58. Olson NC, Cushman M, Lutsey PL, McClure LA, Judd S, Tracy RP, et al. Inflammation markers and incident venous thromboembolism: The REasons for geographic and racial differences in stroke (REGARDS) cohort. J Thromb Haemost (2014) 12(12):1993–2001. doi: 10.1111/jth.12742
59. Kunutsor SK, Seidu S, Blom AW, Khunti K, Laukkanen JA. Serum c-reactive protein increases the risk of venous thromboembolism: A prospective study and meta-analysis of published prospective evidence. Eur J Epidemiol. (2017) 32(8):657–67. doi: 10.1007/s10654-017-0277-4
60. Horvei LD, Grimnes G, Hindberg K, Mathiesen EB, Njolstad I, Wilsgaard T, et al. C-reactive protein, obesity, and the risk of arterial and venous thrombosis. J Thromb Haemost (2016) 14(8):1561–71. doi: 10.1111/jth.13369
61. Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Polak JF, Folsom AR. Cardiovascular risk factors and venous thromboembolism incidence: The longitudinal investigation of thromboembolism etiology. Arch Intern Med (2002) 162(10):1182–9. doi: 10.1001/archinte.162.10.1182
62. Cushman M, O'Meara ES, Heckbert SR, Zakai NA, Rosamond W, Folsom AR. Body size measures, hemostatic and inflammatory markers and risk of venous thrombosis: The longitudinal investigation of thromboembolism etiology. Thromb Res (2016) 144:127–32. doi: 10.1016/j.thromres.2016.06.012
63. Shih L, Kaplan D, Kraiss LW, Casper TC, Pendleton RC, Peters CL, et al. Platelet-monocyte aggregates and c-reactive protein are associated with VTE in older surgical patients. Sci Rep (2016) 6:27478. doi: 10.1038/srep27478
64. Zhang J, Zhao K, Li J, Meng H, Zhu Y, Zhang Y. Age over 65 years and high levels of c-reactive protein are associated with the risk of preoperative deep vein thrombosis following closed distal femur fractures: A prospective cohort study. J Orthop Surg Res (2020) 15(1):559. doi: 10.1186/s13018-020-02089-4
65. Ulrych J, Kvasnicka T, Fryba V, Komarc M, Malikova I, Burget F, et al. 28 day post-operative persisted hypercoagulability after surgery for benign diseases: A prospective cohort study. BMC Surg (2016) 16:16. doi: 10.1186/s12893-016-0128-3
66. Rodger MA, Kahn SR, Wells PS, Anderson DA, Chagnon I, Le Gal G, et al. Identifying unprovoked thromboembolism patients at low risk for recurrence who can discontinue anticoagulant therapy. CMAJ (2008) 179(5):417–26. doi: 10.1503/cmaj.080493
67. Eichinger S, Heinze G, Jandeck LM, Kyrle PA. Risk assessment of recurrence in patients with unprovoked deep vein thrombosis or pulmonary embolism: The Vienna prediction model. Circulation (2010) 121(14):1630–6. doi: 10.1161/CIRCULATIONAHA.109.925214
68. Tosetto A, Iorio A, Marcucci M, Baglin T, Cushman M, Eichinger S, et al. Predicting disease recurrence in patients with previous unprovoked venous thromboembolism: A proposed prediction score (DASH). J Thromb Haemost (2012) 10(6):1019–25. doi: 10.1111/j.1538-7836.2012.04735.x
69. Kearon C, Kahn SR. Long-term treatment of venous thromboembolism. Blood (2020) 135(5):317–25. doi: 10.1182/blood.2019002364
70. Roshani S, Vossen C, Christiansen S, Rosendaal F, Reitsma P, Cushman M, et al. Inflammation markers, d-dimer and the risk of recurrent venous thrombosis. (Leiden, The Netherlands: Leiden University) (2012).
71. Potaczek DP, Jankowska EA, Wypasek E, Undas A. Iron deficiency: A novel risk factor of recurrence in patients after unprovoked venous thromboembolism. Pol Arch Med Wewn. (2016) 126(3):159–65. doi: 10.20452/pamw.3311
72. Kim AS, Khorana AA, McCrae KR. Mechanisms and biomarkers of cancer-associated thrombosis. Transl Res (2020) 225:33–53. doi: 10.1016/j.trsl.2020.06.012
73. Khorana AA, Kuderer NM, Culakova E, Lyman GH, Francis CW. Development and validation of a predictive model for chemotherapy-associated thrombosis. Blood (2008) 111(10):4902–7. doi: 10.1182/blood-2007-10-116327
74. Mulder FI, Candeloro M, Kamphuisen PW, Di Nisio M, Bossuyt PM, Guman N, et al. The khorana score for prediction of venous thromboembolism in cancer patients: A systematic review and meta-analysis. Haematologica (2019) 104(6):1277–87. doi: 10.3324/haematol.2018.209114
75. Khorana AA, Soff GA, Kakkar AK, Vadhan-Raj S, Riess H, Wun T, et al. Rivaroxaban for thromboprophylaxis in high-risk ambulatory patients with cancer. N Engl J Med (2019) 380(8):720–8. doi: 10.1056/NEJMoa1814630
76. Koukourakis MI, Kambouromiti G, Pitsiava D, Tsousou P, Tsiarkatsi M, Kartalis G. Serum c-reactive protein (CRP) levels in cancer patients are linked with tumor burden and are reduced by anti-hypertensive medication. Inflammation (2009) 32(3):169–75. doi: 10.1007/s10753-009-9116-4
77. Potempa LA, Rajab IM, Olson ME, Hart PC. C-reactive protein and cancer: Interpreting the differential bioactivities of its pentameric and monomeric, modified isoforms. Front Immunol (2021) 12:744129. doi: 10.3389/fimmu.2021.744129
78. Yap ML, McFadyen JD, Wang X, Zia NA, Hohmann JD, Ziegler M, et al. Targeting activated platelets: A unique and potentially universal approach for cancer imaging. Theranostics (2017) 7(10):2565–74. doi: 10.7150/thno.19900
79. Setiawan B, Rosalina R, Pangarsa EA, Santosa D, Suharti C. Clinical evaluation for the role of high-sensitivity c-reactive protein in combination with d-dimer and wells score probability test to predict the incidence of deep vein thrombosis among cancer patients. Int J Gen Med (2020) 13:587–94. doi: 10.2147/IJGM.S261718
80. Kroger K, Weiland D, Ose C, Neumann N, Weiss S, Hirsch C, et al. Risk factors for venous thromboembolic events in cancer patients. Ann Oncol (2006) 17(2):297–303. doi: 10.1093/annonc/mdj068
81. Kanz R, Vukovich T, Vormittag R, Dunkler D, Ay C, Thaler J, et al. Thrombosis risk and survival in cancer patients with elevated c-reactive protein. J Thromb Haemost (2011) 9(1):57–63. doi: 10.1111/j.1538-7836.2010.04069.x
82. Streiff MB, Abutalib SA, Farge D, Murphy M, Connors JM, Piazza G. Update on guidelines for the management of cancer-associated thrombosis. Oncologist (2021) 26(1):e24–40. doi: 10.1002/onco.13596
83. Jara-Palomares L, Solier-Lopez A, Elias-Hernandez T, Asensio-Cruz MI, Blasco-Esquivias I, Sanchez-Lopez V, et al. D-dimer and high-sensitivity c-reactive protein levels to predict venous thromboembolism recurrence after discontinuation of anticoagulation for cancer-associated thrombosis. Br J Cancer (2018) 119(8):915–21. doi: 10.1038/s41416-018-0269-5
84. Khorana AA, Kamphuisen PW, Meyer G, Bauersachs R, Janas MS, Jarner MF, et al. Tissue factor as a predictor of recurrent venous thromboembolism in malignancy: Biomarker analyses of the CATCH trial. J Clin Oncol (2017) 35(10):1078–85. doi: 10.1200/JCO.2016.67.4564
85. Shaw RJ, Bradbury C, Abrams ST, Wang G, Toh CH. COVID-19 and immunothrombosis: emerging understanding and clinical management. Br J Haematol (2021) 194(3):518–29. doi: 10.1111/bjh.17664
86. McFadyen JD, Stevens H, Peter K. The emerging threat of (Micro)Thrombosis in COVID-19 and its therapeutic implications. Circ Res (2020) 127(4):571–87. doi: 10.1161/CIRCRESAHA.120.317447
87. Nopp S, Moik F, Jilma B, Pabinger I, Ay C. Risk of venous thromboembolism in patients with COVID-19: A systematic review and meta-analysis. Res Pract Thromb Haemost (2020) 4(7):1178–91. doi: 10.1002/rth2.12439
88. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
89. Al-Samkari H, Karp Leaf RS, Dzik WH, Carlson JCT, Fogerty AE, Waheed A, et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood (2020) 136(4):489–500. doi: 10.1182/blood.2020006520
90. Dujardin RWG, Hilderink BN, Haksteen WE, Middeldorp S, Vlaar APJ, Thachil J, et al. Biomarkers for the prediction of venous thromboembolism in critically ill COVID-19 patients. Thromb Res (2020) 196:308–12. doi: 10.1016/j.thromres.2020.09.017
91. Cai C, Guo Y, You Y, Hu K, Cai F, Xie M, et al. Deep venous thrombosis in COVID-19 patients: A cohort analysis. Clin Appl Thromb Hemost (2020) 26:1–10. doi: 10.1177/1076029620982669
92. Garcia-Ortega A, Oscullo G, Calvillo P, Lopez-Reyes R, Mendez R, Gomez-Olivas JD, et al. Incidence, risk factors, and thrombotic load of pulmonary embolism in patients hospitalized for COVID-19 infection. J Infect (2021) 82(2):261–9. doi: 10.1016/j.jinf.2021.01.003
Keywords: C-reactive protein, venous thromboembolism, immunothrombosis, COVID-19, Thromboinflammation
Citation: Dix C, Zeller J, Stevens H, Eisenhardt SU, Shing KSCT, Nero TL, Morton CJ, Parker MW, Peter K and McFadyen JD (2022) C-reactive protein, immunothrombosis and venous thromboembolism. Front. Immunol. 13:1002652. doi: 10.3389/fimmu.2022.1002652
Received: 25 July 2022; Accepted: 22 August 2022;
Published: 13 September 2022.
Edited by:
Guirong Wang, Upstate Medical University, United StatesReviewed by:
Lawrence Albert Potempa, Roosevelt University, United StatesDaniel P Potaczek, University of Marburg, Germany
Yi Wu, Xi’an Jiaotong University, China
Copyright © 2022 Dix, Zeller, Stevens, Eisenhardt, Shing, Nero, Morton, Parker, Peter and McFadyen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James D. McFadyen, amFtZXMubWNmYWR5ZW5AbW9uYXNoLmVkdQ==
†These authors have contributed equally to this work share first authorship
‡These authors have contributed equally to this work and share senior authorship