 Lingtong Huang
Lingtong Huang Wei Wu2†
Wei Wu2†
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Immunol. , 13 January 2022
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.810677
This article is part of the Research Topic Deciphering the Landscape of Immunohematology: Enhancing our Understanding and Management of Hematological Disorders Through Advances in Immunology and Genetics View all 15 articles
Hemophagocytic lymphocytosis (HLH) is a rare disease caused by inborn errors of immunity (IEI), secondary to infection, lymphoma or autoimmune disorders, but we often overlook the fact that HLH can be secondary to inborn errors of metabolism (IEM). Here, we describe a patient who was diagnosed with glutaric aciduria type IIC complicated by features suggestive of possible HLH. The diagnosis of glutaric aciduria type IIC, a IEM, was confirmed by whole exome sequencing. The patient was treated with coenzyme Q10 and riboflavin which effectively improved her liver function. During treatment, the patient developed severe anemia and thrombocytopenia. Persistent fever, splenomegaly, cytopenias, increased ferritin, hypertriglyceridemia, hypofibrinogenemia, and hemophagocytosis in the bone marrow pointed to the diagnosis of HLH; however, the patient eventually died of gastrointestinal bleeding. After other potential causes were ruled out, the patient was diagnosed with glutaric aciduria type IIC complicated by features suggestive of possible HLH. When cytopenias occurs in IEM patients, HLH is a possible complication that cannot be ignored. This case suggests a possible relationship between IEM and risk for immune dysregulation.
Hemophagocytic lymphocytosis (HLH) is a rare fatal disease with extremely high mortality rates. It often results from genetic defects in immune system function or due to infections (such as Epstein-Barr virus, Cytomegalovirus, Parvovirus B19), tumors, and autoimmune disorders. HLH may also be caused by inborn errors of metabolism (IEM), a trigger which may often be overlooked (1). Here, we describe an adult with glutaric aciduria type IIC, a IEM, who developed features suggestive of HLH during the diagnosis and treatment of their underlying disease.
Glutaric aciduria is a systemic disease caused by errors in fatty acid oxidation and function of several mitochondrial dehydrogenase enzymes (2). In most cases, this condition has a childhood onset; however, some cases of adulthood onset disease have been reported, possibly due to late-onset multiple acyl-CoA dehydrogenase deficiency (3). Most patients develop neurological symptoms at the onset of illness (4), accompanied by repeated hypoglycemia (5), hyperlactic acidemia, and hyperlipidemia.
To our knowledge, this is the first case of glutaric aciduria type IIC complicated by HLH. Moreover, this case underscores the importance of considering HLH in patient with IEM and signs as well as symptoms of immune dysregulation. This case also provides evidence for the potential link between IEM and immune dysregulation.
The genomic DNA was randomly broken into fragments with a length of 180-280 bp by a Covaris breaker. After end repair and A-tailing, the two ends of the fragment were ligated with adapters to prepare a DNA library. The library with a specific index was pooled with up to 500,000 biotin-labeled probes for liquid phase hybridization, and then the n exons of n genes were captured by magnetic beads with streptomycin, and linearly amplified by PCR. After the increase, the library quality inspection was carried out, and the sequencing could be carried out if it was qualified. After the library was constructed, Qubit 2.0 was used for preliminary quantification, and then Agilent 2100 was used to detect the insert size of the library. After the insert size meet expectations, qPCR was used to accurately quantify the effective concentration (3nM) of the library to ensure the library quality. The library was qualified, and the Illumina platform was used for sequencing according to the effective concentration of the library and the data output requirements. AfterQC was used to evaluate the sequencing quality of the off-machine original sequencing data, and removed low-quality and contaminated reads. The filtered data was sequenced with the human hg19 reference genome using BWA software (Burrows Wheeler Aligner), and then the capture effect was evaluated. GATK software (Genome Analysis Toolkit) was used to analyze SNV (single nucletide variant) and Inde (linsertion and deletion) in the genome. Then the population database 1000 Genomes (1000 human genome dataset), Genome AD (Genome Aggregation Database dataset) 2.1.1 and ExAC (The Exome Aggregation Consortium dataset) was used to filter the analyzed SNV and Indel. The dbNSFP database was used to predict the pathogenicity of missense mutations and splicing mutations. Human Mendelian Inheritance Database (OMIM), Human Gene Mutation Database (HGMD) and Clinvar Database was used to screen for reported mutations. Finally, Sanger sequencing was used to verify all possible pathogenic sites.
A literature search was conducted on PubMed, using the keywords “glutaric aciduria” for case reports and case series written before December 2021 to assess whether this is the first case report of glutaric aciduria type IIC complicated by HLH. Another literature search was conducted on PubMed, using (Inborn errors of metabolism) AND (Hemophagocytic) for case reports and case series written before December 2021 to summarize the cases of IEM complicated by HLH. It should be noted that we did not conduct meta-analysis and systematic reviews, but only reviewed the literature that was queried.
A 27-year-old woman had persistent weakness in her upper and lower limbs for 10 years. The weakness of her upper and lower limbs did not affect her work and life. She was misdiagnosed with seronegative polymyositis for which she received 2.5 milligrams prednisone per day one year prior to admission. In the month prior to admission, she gradually became unable to take care of herself. She had no other previous medical history, nor had she traveled abroad. She was not pregnant. The patient’s parents were healthy as was her younger brother and son. There was no genetic disease in her family. Her physical examination was normal except for weakness of the upper and lower limbs.
Following admission, she developed repeated episodes of hypoglycemia, hyper lactic acidemia, and hyperlipidemia (Table 1). On the fifth day, she was transferred to critical care unit due to respiratory failure, anuria, and liver failure. Both lungs showed large patchy lesions, and the density of the liver was quantitatively measured by CT image as -40 Hu, which was lower than the density of water (0 Hu) and was similar to the density of fat (-40 Hu) (Figure 1A). Since the patient showed persistent fever, next-generation sequencing for infectious pathogens and culture of bronchoalveolar lavage fluid were performed to rule out infectious pathogens such as Cytomegalovirus, Herpes simplex virus, Epstein-Barr virus and Pneumocystis in the respiratory tract. The method of mNGS was the same as discribed before (6).
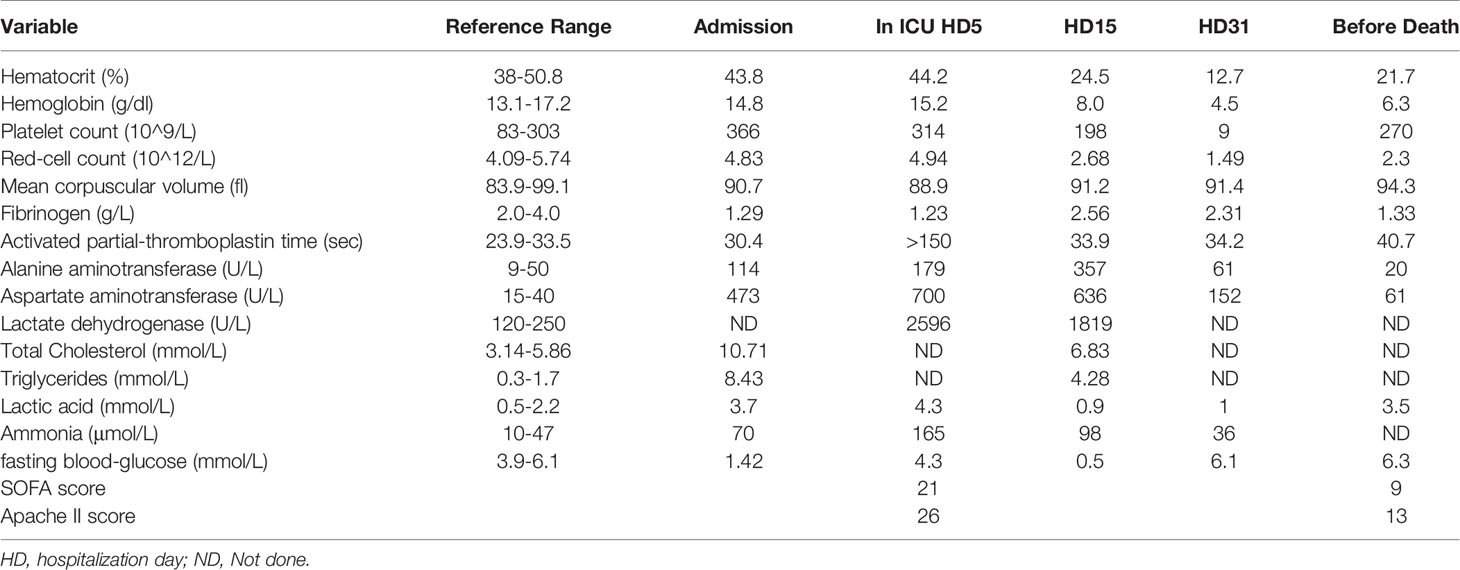
Table 1 Laboratory Data.
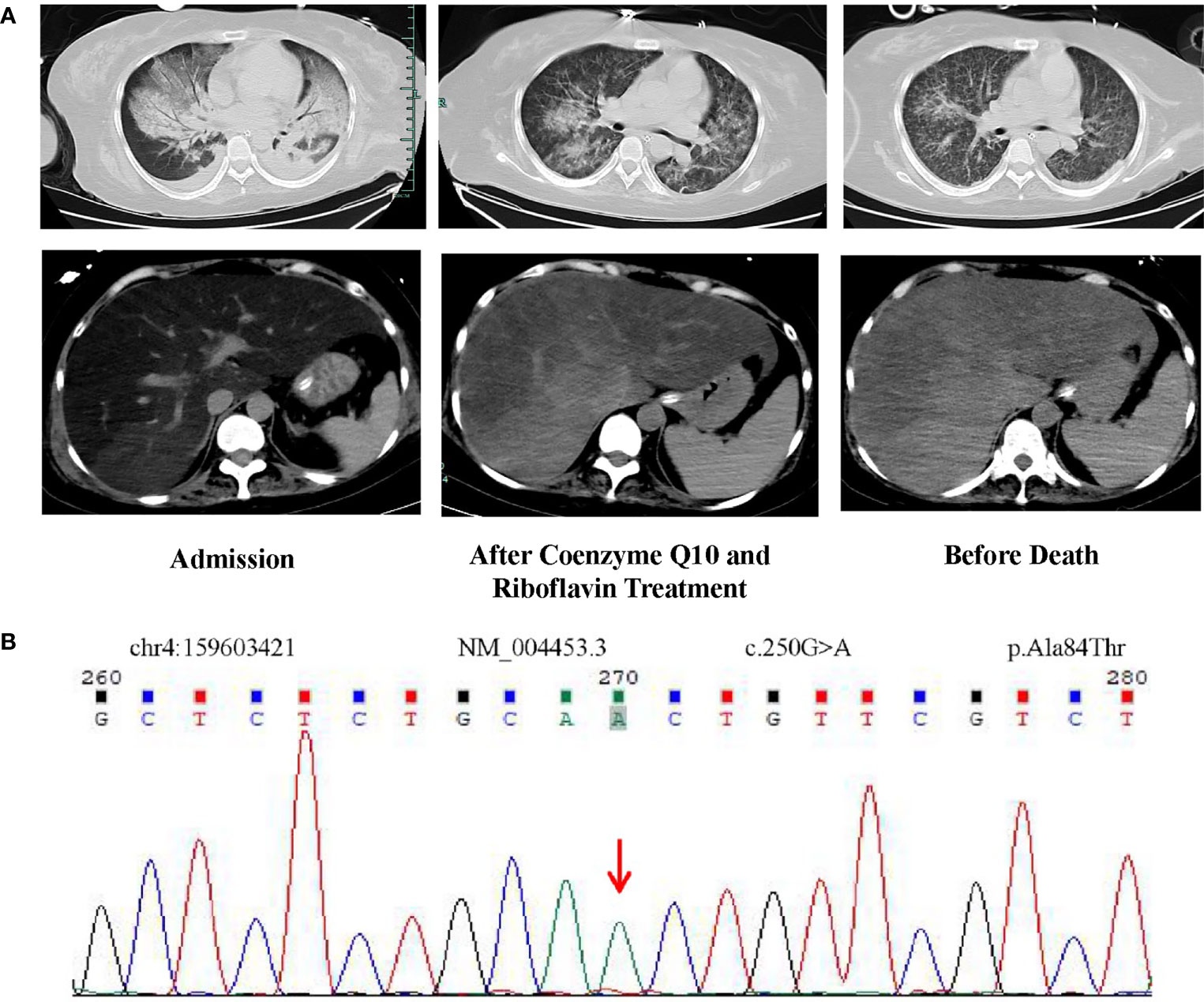
Figure 1 (A) CT images of the lungs and abdomen on admission, after coenzyme Q10 and riboflavin treatment, and before death. After treatment with riboflavin and coenzyme Q10, the CT value of the patient’s liver gradually increased from -40 Hu at the beginning to nearly normal. (B) Homozygous mutation of ETFDH gene (c.250G>A) identified by whole exome sequencing.
As the patient had hypoglycemia, diagnosis of glycogen storage disease was considered. Due to the patient’s persistently abnormal coagulation parameters (Table 1), a liver biopsy was not performed. A muscle biopsy did not demonstrate obvious lipid deposits, but magnetic resonance imaging (MRI) of the lower limbs revealed a large amount of fat accumulation between the muscles. The patient remained aurinc due to renal failure, so the urine organic acids were not performed. Results of whole exome sequencing revealed a homozygous mutation of ETFDH gene (c.250G>A) as shown in Figure 1B. The patient was diagnosed with glutaric aciduria type IIC and was treated with 150mg riboflavin per day and 40mg coenzyme Q10 per day. The patient was alos given a high-sugar and low-fat diet. CT imaging suggested that the patient’s liver was improved significantly which was confirmed by laboratory tests (Figure 3). Despite this improvement, the patient developed severe cytopenias (45g/L of hemoglobin and 9×109/L of platelets).
Severe cytopenias are not typically seen in patients with glutaric aciduria type IIC. Other complicating diagnoses which could explain the findings of cytopenias in glutaric aciduria type IIC were considered. The patient developed acute renal failure with anuria shortly after admission suggesting consideration of thrombotic microangiopathy. Peripheral blood smear findings of thrombotic microangiopathy including mechanical haemolytic anaemia were not found, and pathogenic mutations in genes such as CD46, CFI, CFB, C3, THBD and CFH were absent (7). Acute fatty liver of pregnancy (AFLP) or hemolysis, elevated liver enzymes, and low platelets (HELLP) were also considered which may occur in pregnant women (8), and in pregnant women with IEM (8, 9); however, the possibility of pregnancy was excluded. NGS and cultures were also performed on samples of bronchoalveolar lavage fluid, peripheral blood, and peritoneal fluid, however, no pathogen was isolated which suggested the presence of aseptic inflammation. The persistent and severe hyperlipidemia suggested oxidative stress induced hemolysis; however, peripheral blood smear findings of oxidative hemolysis such as G6PD deficiency were absent. Similarly, whole exome sequencing did not document any pathogenic of G6PD enzyme deficiency-related gene mutations.
Finally, hemophagocytosis was observed on a bone marrow biopsy and aspiration. There are eight diagnostic criteria for hemophagocytic lymphocytosis (1), and the patient met six of them, including persistent fever, splenomegaly (Figure 1A), cytopenias (Figure 3), increased ferritin (1000 ng/mL, reference range 7-323 ng/mL), hypertriglyceridemia and hypofibrinogenemia (Table 1 and Figure 3), and hemophagocytosis in the bone marrow (Figure 2). Of note, serum soluble IL-2R and NK cell activity were not tested in this case. Combined with these laboratory tests, the diagnosis of HLH was suggested. Results from whole exome sequencing showed no gene mutation such as PRF1, UNC13D, STXBP2, STX1, RAB27A, LYST, AP3B1, SH2D1A or XIAP which implied that there was no primary HLH. Besides, no evidence of malignancy, infections, or autoimmune disorders were found. Therefore, we attributed the cause of HLH features to glutaric aciduria type IIC. Considering that this patient had a clear trigger, other treatments (e.g. etoposide, steroids, cyclosporine) were not administered. Supportive care including infusion of red blood cells was performed. Her hemoglobin was maintained at 60g/L, and her platelets gradually increased from 9×10^9/L to normal after the day 32 of hospitalization as liver function continued to improve (Figure 3). Unfortunately, she eventually died of gastrointestinal bleeding despite remission of the features of HLH after being hospitalized for a month and a half. The patient’s family declined an autopsy.
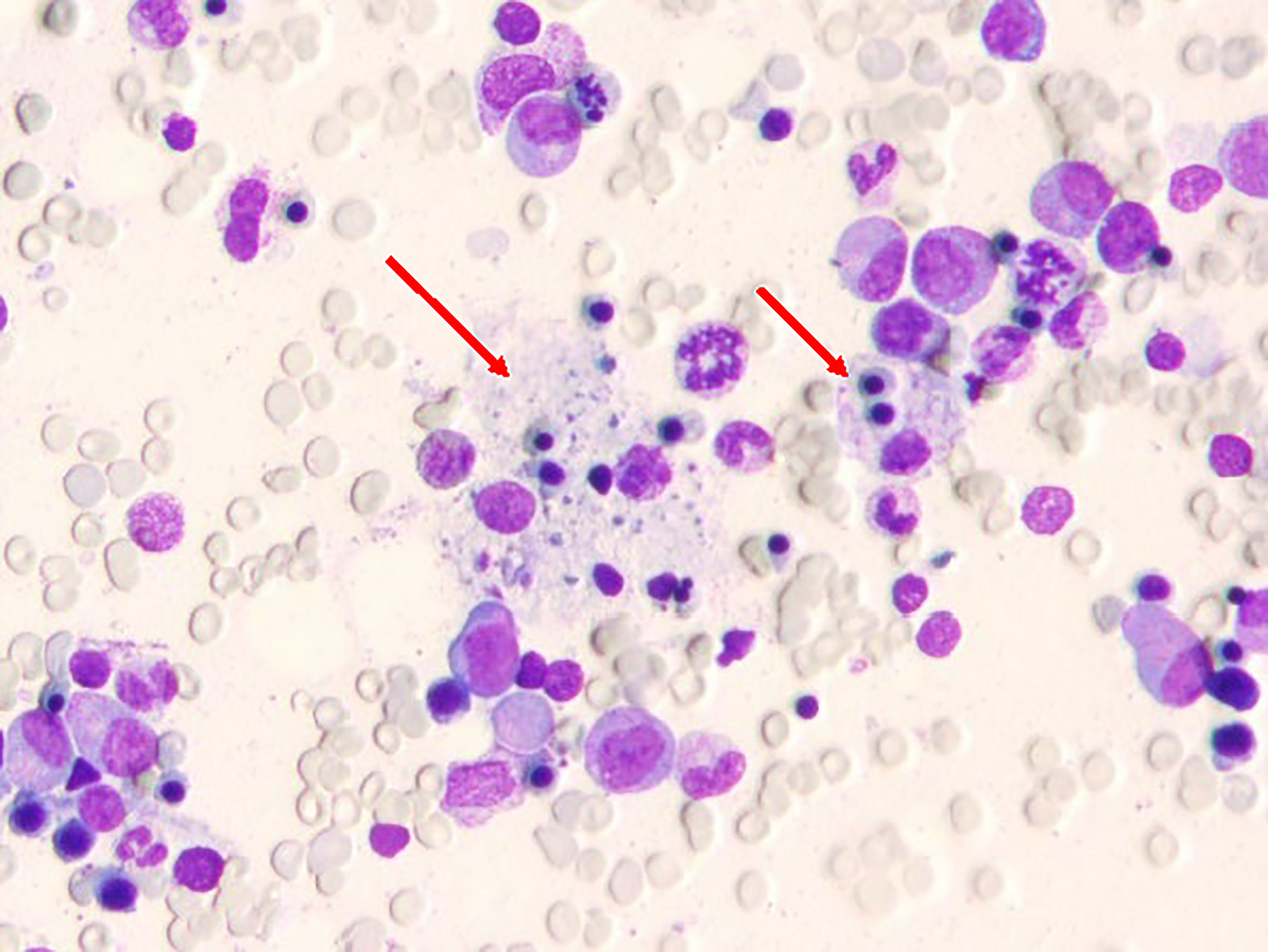
Figure 2 Bone marrow smear carried out when hemoglobin was lowest; the arrow indicates hemophagocytic cells.
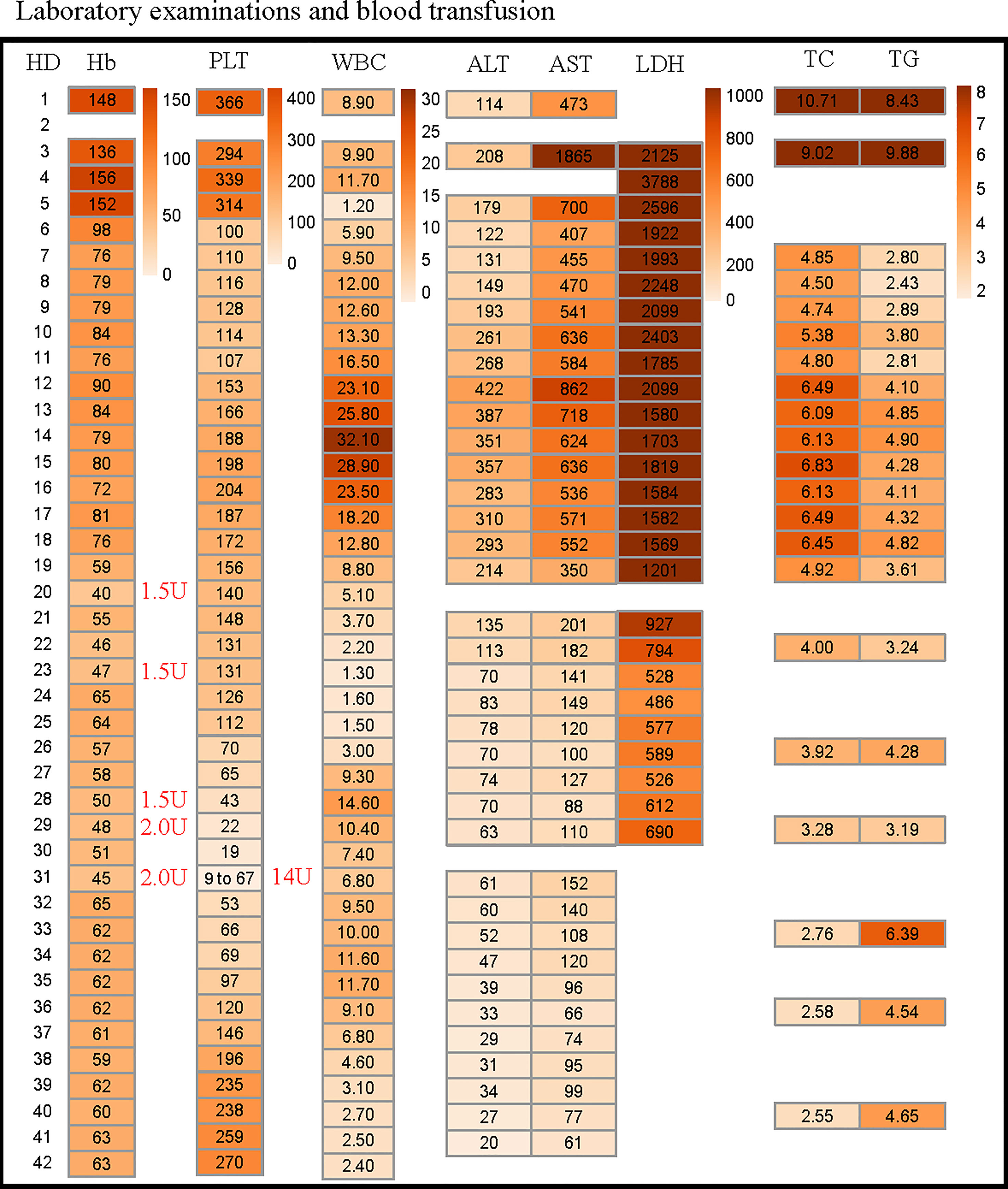
Figure 3 Changes in patient’s blood routine, liver function within 42 days after admission. The reference range for each variable are shown in Table 1. The amount of red blood cells and platelet administered to the patient are marked with red font on the right side of the corresponding time. HD, hospitalization day; Hb, hemoglobin; PLT, platelets; WBC, white blood cell; ALT, alanine aminotransferase; AST, aspartate aminotransferase; LDH, lactate dehydrogenase; TC, total cholesterol; TG, triglycerides.
Abnormal blood biochemical examinations such as lactic acid, blood glucose and blood lipids in adults can often lead clinicians to consider the diagnosis of a IEM. In addition to biochemical examinations, whole exome sequencing has aided in the rapid diagnosis of IEM. In this case, the patient showed no obvious neurological symptoms except for upper and lower extremities weakness. The patient’s condition progressed to severe hypoglycemia and hyperlipidemia, which is consistent with the clinical manifestations of glutaric aciduria type II. In the east of China, homozygous mutation of ETFDH gene (c.250G>A) is the most common cause of glutaric aciduria type IIC (10, 11). This genetic mutation was found in this case (Figure 1B). Given that the CT appearance of liver (Figure 1A), lipid deposition was suspected. The patient was eventually diagnosed with glutaric aciduria type IIC. Consequently, the patient was administered a high-dose coenzyme Q10 and riboflavin—the two drugs recommended for the disease (2) and the clinical manifestations improved rapidly.
Few people would consider HLH in the differential diagnosis of cytopenias in IEM patients. She had very serious liver damage and hypertriglyceridemia. She also had multi organ failure, including anuria, respiratory failure and liver failure, which made it easy to overlook the HLH features. As a critically ill patient, all of her clinical symptoms were non-specific. After considering and excluding important diagnoses associated with acute onset of cytopenias, the diagnosis of HLH was considered. HLH is a fatal disease which is often caused by genetic defects, or it may develop secondary to malignancy, autoimmune diseases, and infections (1). However, none of these factors were found during the disease course of our patient. Therefore, we attributed the occurrence of HLH to glutaric aciduria type IIC.
In the past 30 years, cases of IEM complicated by HLH have been reported (Table 2). Almost all cases occured in children, so in the treatment of adult IEM patients, the diagnosis of HLH may be overlooked. Some reported cases were associated with Lysosomal Storage Disease (LSD), including Gaucher Disease (GD) (21, 22), Chediak-Higashi Syndrome (CHS) (29), Griscelli’s Disease (28), Hermansky-Pudlak Syndrome Type II (HPSII) (23, 24), Wolman’s Disease (a type of lysosomal acid lipase deficiency) (25–27). NK cell dysfunction could be found in some LSDs (e.g. CHS, Griscelli’s Disease, HPSII) because of lysosomal dysfunction, so it is also classified as IEI. Excluding LSD, many forms of IEM can lead to the occurrence of HLH. Disorders of lipid metabolism such as glutaric aciduria type IIC and long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency (13) or disorders of organic acid metabolism such as lysinuric protein intolerance (LPI) (19, 20, 35), methylmalonic acidemia (17), propionic acidemia (17, 18) may be complicated by HLH. There are many other rare IEMs complicated by HLH that are also reported such as biotinidase deficiency (12), hepatolenticular degeneration (33), mevalonate kinase deficiency (30, 31), pyrimidine deficiency (32), disorder of glycogen metabolism (14, 15), prolidase deficiency (16) and cobalamin C disease (34).
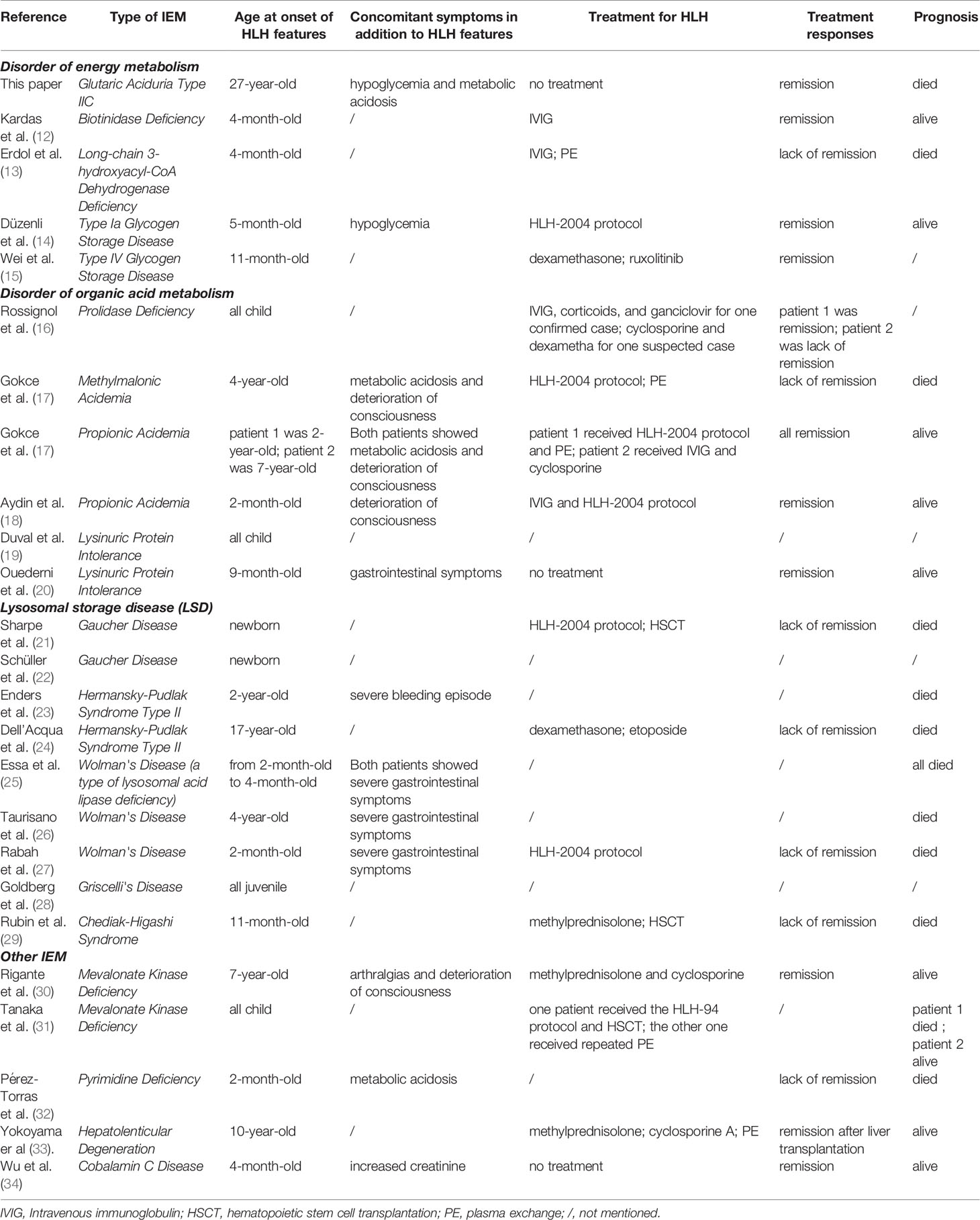
Table 2 Reported cases of IEM complicated by HLH.
These signs and symptoms of HLH occurring in the context of IEM indicate that different IEM complicated by HLH have heterogeneity. In addition to the typical symptoms of HLH, most IEM patients with HLH also have many clinical manifestations that may be related to the primary disease. Some patients may develop metabolic encephalopathy (17, 18, 30), and some patients may have severe gastrointestinal symptoms (20, 26, 27). Metabolic acidosis is also a relatively common clinical manifestation in IEM complicated by HLH (17, 32). Treatments may be variable and may include IVIG, etoposide, cyclosporine, plasma exchange and hematopoietic cell transplantation (table 2). Non-LSD IEM patients may not need targeted treatment of HLH features and signs of HLH may regress as the primary disease improves (19, 20, 34, 35). For the treatment of IEM complicated by HLH, we recommend that the patient’s HLH features be carefully monitored with respect to the response of the treatment of the underlying IEM. In this case, although the patient eventually died of gastrointestinal bleeding, the patient responded well to riboflavin and coenzyme Q10, and her HLH features showed signs of remission which suggests that in patients with IEM, the treatment of the primary disease may be crucial. However, it should be noted that the treatment of such patients still requires the cooperation of metabolic physicians, immunologists, hematologists and intensive care physicians to develop an individualized treatment plan.
However, we cannot clarify the causal relationship between glutaric aciduria type IIC and HLH and we have not explored the pathogenesis which are the limitations of this case report. The rare incidence of IEM and the rare complication of HLH limit the ability to often consider the diagnosis of HLH when faced with a patient with a IEM. Also, due to insufficient knowledge of the potential association of IEM and HLH, many patients may be misdiagnosed. Many potential links between metabolism and immunity have been discovered (36, 37). This case provides evidence for the relationship between IEM and impaired immune function. When cytopenias occur in IEM patients, HLH is a possible complication that cannot be ignored.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the ethics committee of the First Affiliated Hospital of Zhejiang University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
XF, LH, WW, YZ, HY wrote the first draft made the initial diagnosis. XF, LH, LT participate in the discussion of the diagnosis. We all and cared for the patient and reviewed the final manuscript.
This work was supported by the National Natural Science Foundation of China grant 81872672 (to LT).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to acknowledge Prof. Lucio Luzzatto from Muhimbili University of Health and Allied Sciences participated in discussion and analyzed the peripheral blood smears to rule out the diagnosis of oxidative hemolysis. We deeply appreciate David Buchbinder for reviewing and editing this manuscript carefully to improve the quality of the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.810677/full#supplementary-material
1. Canna SW, Marsh RA. Pediatric Hemophagocytic Lymphohistiocytosis. Blood (2020) 135:1332–43. doi: 10.1182/blood.2019000936
2. Gempel K, Topaloglu H, Talim B, Schneiderat P, Schoser BG, Hans VH, et al. The Myopathic Form of Coenzyme Q10 Deficiency is Caused by Mutations in the Electron-Transferring-Flavoprotein Dehydrogenase (ETFDH) Gene. Brain (2007) 130:2037–44. doi: 10.1093/brain/awm054
3. Antozzi C, Garavaglia B, Mora M, Rimoldi M, Morandi L, Ursino E, et al. Late-Onset Riboflavin-Responsive Myopathy With Combined Multiple Acyl Coenzyme A Dehydrogenase and Respiratory Chain Deficiency. Neurology (1994) 44:2153–8. doi: 10.1212/WNL.44.11.2153
4. Kolker S, Koeller DM, Okun JG, Hoffmann GF. Pathomechanisms of Neurodegeneration in Glutaryl-CoA Dehydrogenase Deficiency. Ann Neurol (2004) 55:7–12. doi: 10.1002/ana.10784
5. Dusheiko G, Kew MC, Joffe BI, Lewin JR, Mantagos S, Tanaka K. Recurrent Hypoglycemia Associated With Glutaric Aciduria Type II in an Adult. N Engl J Med (1979) 301:1405–9. doi: 10.1056/NEJM197912273012601
6. Huang L, Zhang X, Fang X. Case Report: Epstein-Barr Virus Encephalitis Complicated With Brain Stem Hemorrhage in an Immune-Competent Adult. Front Immunol (2021) 12:618830. doi: 10.3389/fimmu.2021.618830
7. Fakhouri F, Fremeaux-Bacchi V. Thrombotic Microangiopathy in aHUS and Beyond: Clinical Clues From Complement Genetics. Nat Rev Nephrol (2021) 17:543–53. doi: 10.1038/s41581-021-00424-4
8. Yang Z, Yamada J, Zhao Y, Strauss AW, Ibdah JA. Prospective Screening for Pediatric Mitochondrial Trifunctional Protein Defects in Pregnancies Complicated by Liver Disease. JAMA (2002) 288:2163–6. doi: 10.1001/jama.288.17.2163
9. Wilcken B, Leung KC, Hammond J, Kamath R, Leonard JV. Pregnancy and Fetal Long-Chain 3-Hydroxyacyl Coenzyme A Dehydrogenase Deficiency. Lancet (1993) 341:407–8. doi: 10.1016/0140-6736(93)92993-4
10. Xi J, Wen B, Lin J, Zhu W, Luo S, Zhao C, et al. Clinical Features and ETFDH Mutation Spectrum in a Cohort of 90 Chinese Patients With Late-Onset Multiple Acyl-CoA Dehydrogenase Deficiency. J Inherit Metab Dis (2014) 37:399–404. doi: 10.1007/s10545-013-9671-6
11. Chokchaiwong S, Kuo YT, Hsu SP, Hsu YC, Lin SH, Zhong WB, et al. ETF-QO Mutants Uncoupled Fatty Acid Beta-Oxidation and Mitochondrial Bioenergetics Leading to Lipid Pathology. Cells (2019) 8:106. doi: 10.3390/cells8020106
12. Kardas F, Patiroglu T, Unal E, Chiang SC, Bryceson YT, Kendirci M. Hemophagocytic Syndrome in a 4-Month-Old Infant With Biotinidase Deficiency. Pediatr Blood Cancer (2012) 59:191–3. doi: 10.1002/pbc.23247
13. Erdol S, Ture M, Baytan B, Yakut T, Saglam H. An Unusual Case of LCHAD Deficiency Presenting With a Clinical Picture of Hemophagocytic Lymphohistiocytosis: Secondary HLH or Coincidence? J Pediatr Hematol Oncol (2016) 38:661–2. doi: 10.1097/MPH.0000000000000626
14. Duzenli Kar Y, Ozdemir ZC, Kiral E, Kilic Yildirim G, Dinleyici EC, Bor O. Hemophagocytic Lymphohystiocytosis Associated With Type Ia Glycogen Storage Disease. J Pediatr Hematol Oncol (2019) 41:e260–2. doi: 10.1097/MPH.0000000000001208
15. Wei A, Ma H, Li Z, Zhang L, Zhang R, Wang T. Type IV Glycogen Storage Disease Associated With Hemophagocytic Lymphohistiocytosis: A Case Report. J Pediatr Hematol Oncol (2020) 42:368–9. doi: 10.1097/MPH.0000000000001694
16. Rossignol F, Duarte Moreno MS, Benoist JF, Boehm M, Bourrat E, Cano A, et al. Quantitative Analysis of the Natural History of Prolidase Deficiency: Description of 17 Families and Systematic Review of Published Cases. Genet Med (2021) 23:1604–15. doi: 10.1038/s41436-021-01200-2
17. Gokce M, Unal O, Hismi B, Gumruk F, Coskun T, Balta G, et al. Secondary Hemophagocytosis in 3 Patients With Organic Acidemia Involving Propionate Metabolism. Pediatr Hematol Oncol (2012) 29:92–8. doi: 10.3109/08880018.2011.601402
18. Aydin Koker S, Yesilbas O, Koker A, Sevketoglu E. Propionic Acidemia: An Extremely Rare Cause of Hemophagocytic Lymphohistiocytosis in an Infant. Arch Argent Pediatr (2020) 118:e174–7. doi: 10.5546/aap.2020.eng.e174
19. Duval M, Fenneteau O, Doireau V, Faye A, Emilie D, Yotnda P, et al. Intermittent Hemophagocytic Lymphohistiocytosis is a Regular Feature of Lysinuric Protein Intolerance. J Pediatr (1999) 134:236–9. doi: 10.1016/S0022-3476(99)70423-3
20. Ouederni M, Ben Khaled M, Rekaya S, Ben Fraj I, Mellouli F, Bejaoui M. A Nine-Month-Old-Boy With Atypical Hemophagocytic Lymphohistiocytosis. Mediterr J Hematol Infect Dis (2017) 9:e2017057. doi: 10.4084/MJHID.2017.057
21. Sharpe LR, Ancliff P, Amrolia P, Gilmour KC, Vellodi A. Type II Gaucher Disease Manifesting as Haemophagocytic Lymphohistiocytosis. J Inherit Metab Dis (2009) 32 Suppl 1:S107–10. doi: 10.1007/s10545-009-1091-2
22. Schuller S, Attarbaschi A, Berger A, Hutter C, Klebermass-Schrehof K, Steiner M. Hemophagocytic Lymphohistiocytosis Triggered by Gaucher Disease in a Preterm Neonate. Pediatr Hematol Oncol (2016) 33:462–7. doi: 10.1080/08880018.2016.1234011
23. Enders A, Zieger B, Schwarz K, Yoshimi A, Speckmann C, Knoepfle EM, et al. Lethal Hemophagocytic Lymphohistiocytosis in Hermansky-Pudlak Syndrome Type II. Blood (2006) 108:81–7. doi: 10.1182/blood-2005-11-4413
24. Dell’Acqua F, Saettini F, Castelli I, Badolato R, Notarangelo LD, Rizzari C. Hermansky-Pudlak Syndrome Type II and Lethal Hemophagocytic Lymphohistiocytosis: Case Description and Review of the Literature. J Allergy Clin Immunol Pract (2019) 7:2476–8.e2475. doi: 10.1016/j.jaip.2019.04.001
25. Al Essa M, Nounou R, Sakati N, Le Quesne G, Joshi S, Archibald A, et al. Wolman’s Disease: The King Faisal Specialist Hospital and Research Centre Experience. Ann Saudi Med (1998) 18:120–4. doi: 10.5144/0256-4947.1998.120
26. Taurisano R, Maiorana A, De Benedetti F, Dionisi-Vici C, Boldrini R, Deodato F. Wolman Disease Associated With Hemophagocytic Lymphohistiocytosis: Attempts for an Explanation. Eur J Pediatr (2014) 173:1391–4. doi: 10.1007/s00431-014-2338-y
27. Rabah F, Al-Hashmi N, Beshlawi I. Wolman’s Disease With Secondary Hemophagocytic Lymphohistiocytosis. Pediatr Hematol Oncol (2014) 31:576–8. doi: 10.3109/08880018.2014.920942
28. Goldberg J, Nezelof C. Lymphohistiocytosis: A Multi-Factorial Syndrome of Macrophagic Activation Clinico-Pathological Study of 38 Cases. Hematol Oncol (1986) 4:275–89. doi: 10.1002/hon.2900040405
29. Rubin CM, Burke BA, McKenna RW, McClain KL, White JG, Nesbit ME Jr, et al. The Accelerated Phase of Chediak-Higashi Syndrome. An Expression of the Virus-Associated Hemophagocytic Syndrome? Cancer (1985) 56:524–30. doi: 10.1002/1097-0142(19850801)56:3<524::aid-cncr2820560320>3.0.co;2-z
30. Rigante D, Capoluongo E, Bertoni B, Ansuini V, Chiaretti A, Piastra M, et al. First Report of Macrophage Activation Syndrome in Hyperimmunoglobulinemia D With Periodic Fever Syndrome. Arthritis Rheum (2007) 56:658–61. doi: 10.1002/art.22409
31. Tanaka T, Yoshioka K, Nishikomori R, Sakai H, Abe J, Yamashita Y, et al. National Survey of Japanese Patients With Mevalonate Kinase Deficiency Reveals Distinctive Genetic and Clinical Characteristics. Mod Rheumatol (2019) 29:181–7. doi: 10.1080/14397595.2018.1442639
32. Perez-Torras S, Mata-Ventosa A, Drogemoller B, Tarailo-Graovac M, Meijer J, Meinsma R, et al. Deficiency of Perforin and Hcnt1, a Novel Inborn Error of Pyrimidine Metabolism, Associated With a Rapidly Developing Lethal Phenotype Due to Multi-Organ Failure. Biochim Biophys Acta Mol Basis Dis (2019) 1865:1182–91. doi: 10.1016/j.bbadis.2019.01.013
33. Yokoyama S, Kasahara M, Morioka D, Fukuda A, Arai K, Mori T, et al. Successful Living-Donor Liver Transplantation for Wilson’s Disease With Hemophagocytic Syndrome. Transplantation (2007) 84:1067–9. doi: 10.1097/01.tp.0000285993.73978.54
34. Wu S, Gonzalez-Gomez I, Coates T, Yano S. Cobalamin C Disease Presenting With Hemophagocytic Lymphohistiocytosis. Pediatr Hematol Oncol (2005) 22:717–21. doi: 10.1080/08880010500278871
35. Mauhin W, Habarou F, Gobin S, Servais A, Brassier A, Grisel C, et al. Update on Lysinuric Protein Intolerance, a Multi-Faceted Disease Retrospective Cohort Analysis From Birth to Adulthood. Orphanet J Rare Dis (2017) 12:3. doi: 10.1186/s13023-016-0550-8
36. Voss K, Hong HS, Bader JE, Sugiura A, Lyssiotis CA, Rathmell JC. A Guide to Interrogating Immunometabolism. Nat Rev Immunol (2021) 21:637–52. doi: 10.1038/s41577-021-00529-8
Keywords: glutaric aciduria, hemophagocytic lymphocytosis, hemophagocytic syndrome, cytopenia, inborn errors of metabolism, IEM
Citation: Huang L, Wu W, Zhu Y, Yu H, Tang L and Fang X (2022) Case Report: Hemophagocytic Lymphocytosis in a Patient With Glutaric Aciduria Type IIC. Front. Immunol. 12:810677. doi: 10.3389/fimmu.2021.810677
Received: 07 November 2021; Accepted: 24 December 2021;
Published: 13 January 2022.
Edited by:
Markus G. Seidel, Medical University of Graz, AustriaReviewed by:
Mihnea-Alexandru Găman, Carol Davila University of Medicine and Pharmacy, RomaniaCopyright © 2022 Huang, Wu, Zhu, Yu, Tang and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xueling Fang, eHVlbGluZ2ZhbmdAemp1LmVkdS5jbg==; Lingling Tang, bGx0YW5nQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.