 Julia Campe1,2
Julia Campe1,2 Evelyn Ullrich1,2,3,4*
Evelyn Ullrich1,2,3,4*- 1Experimental Immunology, Children’s University Hospital, Goethe University Frankfurt, Frankfurt am Main, Germany
- 2Children’s University Hospital, Goethe University Frankfurt, Frankfurt am Main, Germany
- 3Frankfurt Cancer Institute, Goethe University Frankfurt, Frankfurt am Main, Germany
- 4German Cancer Consortium (Deutsches Konsortium für Translationale Krebsforschung (DKTK)), Partner Site Frankfurt/Mainz, Frankfurt am Main, Germany
Allogenic hematopoietic stem cell transplantation (allo-HSCT) represents a potent and potentially curative treatment for many hematopoietic malignancies and hematologic disorders in adults and children. The donor-derived immunity, elicited by the stem cell transplant, can prevent disease relapse but is also responsible for the induction of graft-versus-host disease (GVHD). The pathophysiology of acute GVHD is not completely understood yet. In general, acute GVHD is driven by the inflammatory and cytotoxic effect of alloreactive donor T cells. Since several experimental approaches indicate that CD4 T cells play an important role in initiation and progression of acute GVHD, the contribution of the different CD4 T helper (Th) cell subtypes in the pathomechanism and regulation of the disease is a central point of current research. Th lineages derive from naïve CD4 T cell progenitors and lineage commitment is initiated by the surrounding cytokine milieu and subsequent changes in the transcription factor (TF) profile. Each T cell subtype has its own effector characteristics, immunologic function, and lineage specific cytokine profile, leading to the association with different immune responses and diseases. Acute GVHD is thought to be mainly driven by the Th1/Th17 axis, whereas Treg cells are attributed to attenuate GVHD effects. As the differentiation of each Th subset highly depends on the specific composition of activating and repressing TFs, these present a potent target to alter the Th cell landscape towards a GVHD-ameliorating direction, e.g. by inhibiting Th1 and Th17 differentiation. The finding, that targeting of Th1 and Th17 differentiation appears more effective for GVHD-prevention than a strategy to inhibit Th1 and Th17 cytokines supports this concept. In this review, we shed light on the current advances of potent TF inhibitors to alter Th cell differentiation and consecutively attenuate GVHD. We will focus especially on preclinical studies and outcomes of TF inhibition in murine GVHD models. Finally, we will point out the possible impact of a Th cell subset-specific immune modulation in context of GVHD.
Introduction
Allogenic hematopoietic stem cell transplantation (allo-HSCT) represents a potent and potentially curative treatment for many hematopoietic malignancies and hematologic disorders in adults and children. Its success is based on a complete replacement of the patients’ immune system by a myeloablative conditioning regimen and reconstitution from a healthy donor stem cell graft. The donor derived immunity can prevent disease relapse but is also responsible for the main complication of allo-HSCT, the graft-versus-host disease (GVHD).
Acute GVHD pathophysiology is not completely understood yet. In general, acute GVHD is driven by the inflammatory effect of donor T cells upon antigen-recognition of allo-antigens presented by host antigen-presenting cells (APCs). The subsequent alloreactive cytotoxicity of activated T cells effects the GVHD target organs (gastrointestinal tract, skin, and liver) and leads to an amplification loop of inflammation there.
Since several experimental approaches indicate that CD4 T cells play a key role in initiation and progression of acute GVHD, the contribution of the different CD4 T helper (Th) cell subtypes in the pathomechanism and regulation of the disease is a central point of current research. Acute GVHD is thought to be driven by a Th1/Th17/Th22 axis whereas Treg cells are attributed to attenuate GVHD effects. As the differentiation of each Th subset highly depends on the specific composition of activating and repressing transcription factors (TFs), these present a potent target to alter the Th cell landscape towards a GVHD-ameliorating direction by the inhibition of Th1 and Th17 differentiation. In this review, we discuss the current advances of potent of potent TF inhibitors in order to alter Th cell differentiation and attenuate GVHD in murine models.
T Helper Cell Subsets and Differentiation
To date, eight different T helper cell types are known: Th1, Th2, Th9, Th10, Th17, Th22, follicular T helper cells (Tfh) and regulatory T cells (Treg). Th cell lineages derive from naïve CD4 T cell progenitors and lineage commitment is initiated by the surrounding cytokine milieu and subsequent changes in the TF profile. Each T cell subtype has its own effector characteristics, immunologic function, and lineage specific cytokine profile, leading to the association with different immune responses (Figure 1). In this review we will focus on the Th1, Th2, Th17 and Treg subsets, the involved TFs in their differentiation as well as their impact on GVHD.
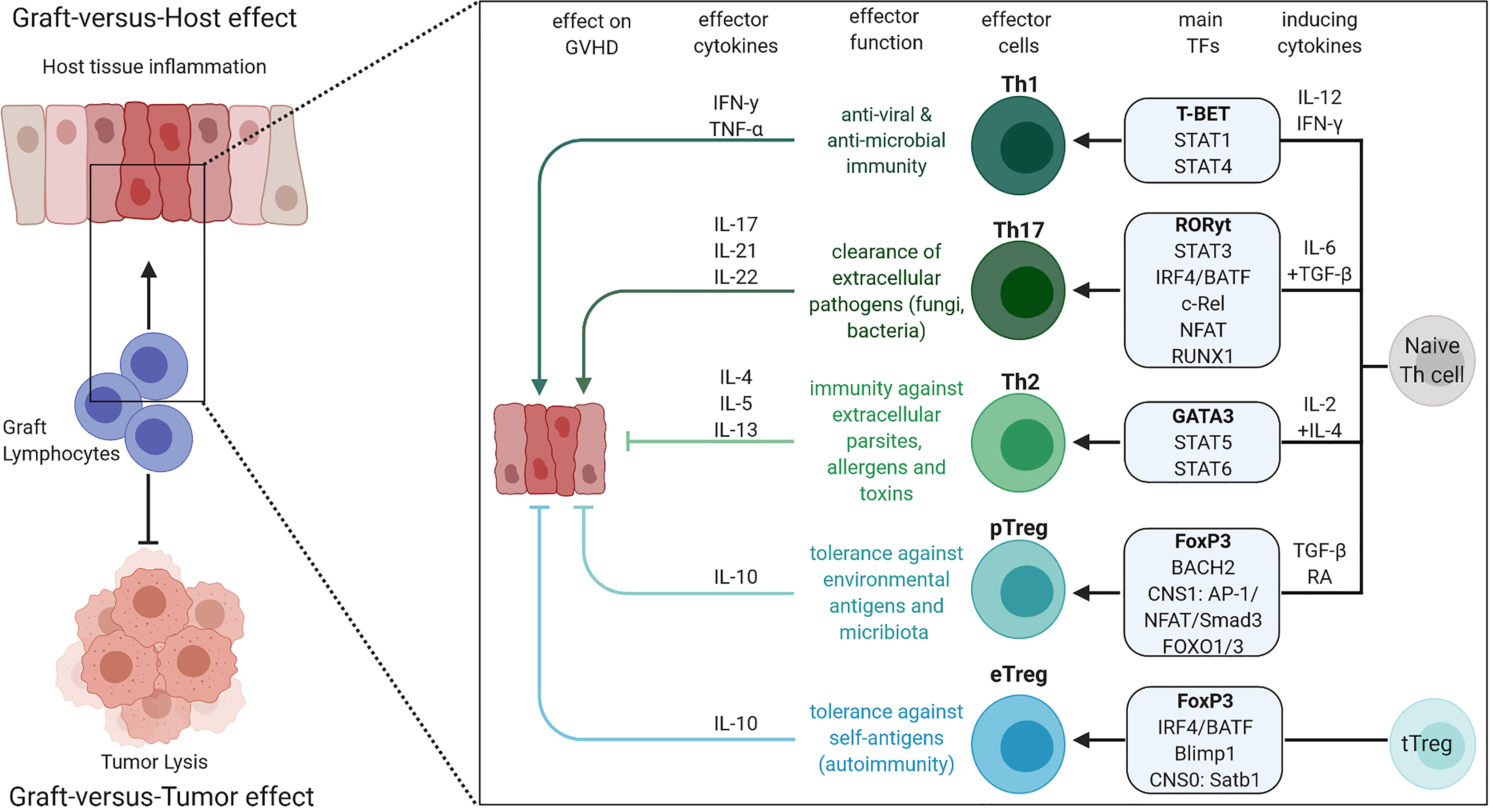
Figure 1 Overview of Th1, Th2, Th17, eTreg and pTreg differentiation, their effector cytokines, roles in the immune system and impact on GVHD. The figure was created with BioRender.com.
Th1 and Th2 Cells
In 1986, Mosmann and colleagues identified two distinct classes of CD4 helper T cells, which exhibited a different cytokine profile. The differentiation in these two classes, later called Th1 and Th2, was found to be stable and deterministic (1). Th1 cells differentiate in the presence of interferon (IFN)-γ and interleukin (IL)-12 to produce their effector cytokine IFN-γ, which has high relevance for anti-viral and anti-microbial immunity (1–3). In contrast, Th2 cells differentiate in the presence of IL-2 and IL-4 and produce the effector cytokines IL-4, IL-5 and IL-13, which play an important role in the immune response against extracellular parasites, bacteria, allergens, and toxins (1, 4–7).
In the early 2000s, Szabo et al. discovered that the underlying mechanisms of the Th1/Th2 paradigm was the initiation or repression of distinct genetic programs upon activation, directed by Th lineage specific master transcription factors (8). With this regard, T-bet was described as a master regulator of Th1 cells, which induces IFN-γ production by activating Th1 genetic programs while repressing Th2 responses (8–12). A few years earlier, GATA3 was characterized as a master transcriptional regulator for Th2 cell differentiation (13, 14).
Further studies on the mechanisms, how T-bet and GATA3 mediate Th1 and Th2 differentiation respectively, revealed the mutual inhibition of the two master TFs and the involvement of many more interacting molecules and relevant signaling cascades (15–17). T−bet was found to be induced by Signal Transducers and Activators of Transcription Protein 1 (STAT1) and IFN-γ during T cell activation and to induce STAT1 dependent processes as the induction of Interleukin-12 receptor subunit beta-2 (IL-12Rβ2) (12). Additionally, STAT4 which is activated by IL-12, and the downstream acting TF c-Rel were identified as crucial transcriptional regulators for Th1 differentiation (15, 18–22). In contrast, Th2 differentiation was associated with IL-2 dependent STAT5 signaling and IL-4 dependent STAT6 signaling pathways, which induce the expression of GATA3, IL-2 receptor (R)a and IL-4Ra as well as IL-2 and IL-4 effector cytokines (23–25).
Th17 Cells
Th17 cells were first described as an independent and distinct Th subset from Th1 and Th2 cells, producing IL-17a, IL-17f, IL-22 and IL-21 as effector cytokines in the early 2000s (26, 27). First thought that IL-23 was the inducing cytokine for Th17 cells, three groups simultaneously discovered that TGF-β and IL-6 induced Th17 differentiation (28–30), while IL-1β and tumor necrosis factor (TNF)-α can potentiate Th17 differentiation in presence of IL-6 and transforming growth factor beta TGF-β (31–33). The leading role of Th17 cells is the clearance of extracellular pathogens as fungi and bacteria but dysregulation of Th17 effects is associated with various autoimmune diseases like inflammatory bowel disease, rheumatic arthritis, experimental autoimmune encephalomyelitis (EAE), and multiple sclerosis [reviewed by Tesmer at al. (34)].
In 2006, the transcription factor retinoid acid-related orphan receptor (ROR)γt (Rorc) was identified to be uniquely expressed in mouse Th17 cells and necessary for Th17 differentiation (35). Besides, RORγt as master transcription factor, several other TFs were described to be crucial for Th17 differentiation and function. STAT3 was found to drive the transcription of Th17 specific genes like Il17a, Il17f and Il23r (36, 37) and to suppress TGF−β-induced forkhead box protein 3 (FoxP3) expression and hence regulatory T cell differentiation (28). Interferon Regulatory Factor 4 (IRF4) and Basic Leucin Zipper ATF-Like Transcription Factor (BATF) also play a significant role in Th17 differentiation by initiating the transcription of Th17 defining genes as Il17, Il21, Il23r and Rorc (38–40). IRF4 was also shown to physically interact with RORγt (38) and STAT3 (36). The transcriptional regulators c-Rel, p65, nuclear factor of activated T cells (NFAT)c2 and Runt-related transcription factor 1 (RUNX1) were found to directly regulate RORγt by binding to the Rorc promotor (41–43). Additionally, RUNX1 and hypoxia-inducible factor 1-alpha (HIF1α) physically interact with RORγt to potentiate or co-activate IL-17a expression (44, 45). Importantly, T-bet and GATA3 can inhibit RUNX1 expression or binding to DNA respectively which inhibits Th17 differentiation.
Regulatory T Cells
In contrast to the immune effector function of Th1, Th2 and Th17 cells, regulatory T cells (Tregs) are characterized by their immunosuppressive capacity and are essential mediators of self-tolerance. Already in the 1960’s it was found that a thymus-derived cell population was mediating immunologic tolerance. Later on, Sakaguchi and colleagues characterized these cells further as CD4 T cells expressing the IL-2 receptor alpha chain (CD25) (46). However, it was unclear if Tregs represent a distinct cell line until the Treg master transcription factor FoxP3 was discovered (47, 48). The importance of FoxP3 for Treg differentiation is well displayed by scurfy mice which lack FoxP3 expression and suffer from inflammatory autoimmune syndrome (47, 49). Additionally, the maintenance of FoxP3 expression after differentiation is essential for Treg immunosuppressive function (50, 51). Besides the expression of FoxP3, the development, maintenance, and function of Tregs also highly depends on TGF-β (52–55).
In contrast to other effector T helper cells, regulatory T cells differentiate in the thymus [thymus-derived Tregs (tTregs)], dependent on high affinity interaction with complexes of MHC-II and tissue-restricted self-antigens and IL-2 receptor signaling (56). However, Tregs can also differentiate from naïve T cells in the periphery (pTregs), sometimes also referred to as induced Tregs (iTregs). These cells are induced by non-self-antigens and are most likely mediating immunologic tolerance of environmental antigens and commensal microbiota [reviewed by Lee et al. (57)].
pTreg and tTreg differentiation are implemented on a transcriptional level by different involvement of regulatory elements, four conserved non-coding sequences (CNSs) of the Foxp3 locus (58). CNS1, regulated by the transcription factors Activator protein 1 (AP-1), NFAT, Small mothers against decapentaplegic homolog 3 (Smad3) and Forkhead box O (FOXO) (57, 59–62), was found to be necessary for pTreg but not for tTreg development, while CNS0, regulated by special AT-rich sequence-binding protein-1 (Satb1) is essential for tTreg generation (63). CNS2, which is regulated by the TF Protein C-ets-1 (Ets-1), cAMP response element-binding protein (CREB), RUNX, STAT5, NFAT and c-Rel is important for stable FoxP3 expression during differentiation and functionality of Tregs (58, 64–69). In contrast, CNS3 which is regulated by c-Rel and FOXO TFs influences Treg cell numbers (57, 58, 62). Additionally, gaining the full suppressor function of tTregs as effector Tregs (eTregs) depends on the transcription factors IRF4 and B lymphocyte-induced maturation protein-1 (Blimp-1), which drive the expression of the immunosuppressive cytokine IL-10 (70), while BACH2, a transcriptional repressor, inhibits the genomic binding of IRF4, and mediates pTreg differentiation and maintenance (71).
Cross Regulation of T Helper Cell Differentiation
In general, Th differentiation fates are tightly connected and regulated. For example, Th1 and Th2 cells inhibit the development of each other by their lineage specific transcription factors (72, 73) and by the cytokines IFN-γ and IL-4 (74, 75). The differentiation of Th17 cells can also be inhibited by these cytokines and by the expression of the TF T-bet (26, 72, 76). However, fully differentiated Th17 cells are resistant to IFN-γ and IL-4 inhibiting effects in vitro (27).
The T cell fate of Th17 and Tregs is connected especially tightly, as many factors were shown to have a reciprocal role in Th17 and Treg development. One reason for that is the response of both cell types to TGF-β signaling. However, IL-6 regulates the TGF-β response between both subsets, since it is necessary for Th17 induction, while it inhibits TGF-β induced Treg differentiation (77, 78). On the contrary, Tregs can lose their FoxP3 expression and reprogram to IL-17 secreting cells in the absence of TGF-β (79). Many more regulatory pathways also show that contradictive role in Th17 and Treg development. The activation of mammalian target of rapamycin (mTOR) via HIF1α promotes Th17 differentiation, whereas the lack of HIF1α and mTOR drives Treg development (80). As another example, inhibiting protein kinase CK2 was shown to block Th17 development and promotes Treg cell differentiation in mice due to a defect in STAT3 phosphorylation (81). FoxP3 itself, can also associate with RORγt and inhibit RORγt activity (82). GATA3 was shown to play a vital role in Treg differentiation as it binds to CNS2 elements and represses the development of a Th17 phenotype (83). A similar effect was reported on IL-2 which promotes Treg development and inhibits Th17 differentiation dependent on STAT5 (84). In general the opposing regulation of genes like Il17 through STAT3 and STAT5 seems to be a crucial mediator of reciprocal Th17/Treg differentiation (85).
The Impact of Th Cells in GVHD
The role of different Th-subsets in GVHD-induction and progression has been investigated with various approaches and GVHD-mouse models for quite a long time. First focusing on Th-subset specific cytokines, these studies mostly provided paradoxical results regarding the role of Th1, Th2 and Th17 cells in GVHD. However, following experiments with Th-defining TF knockout T cells improved the understanding of Th-subset involvement in GVHD. Overall, Th2 and Tregs are subsets with a protective effect on GVHD while Th1 and Th17 cells promote GVHD induction and progression. The following paragraph will give more detailed information on the various approaches revealing the role of the different Th subsets in aGVHD.
Protective T Helper Cell Subsets in GVHD
First studies examined the effect of Th2-associated cytokines in GVHD in the 1990’s. Injection of the Th2 inducing cytokines IL-2 and IL-4 led to Th2 polarization and protected recipient mice from GVHD-associated mortality (86, 87). Comparable results were observed after the administration of Granulocyte-macrophage colony-stimulating factor (GM-CSF) to recipient mice, which induced IL-4 production and inhibited GVHD-development (88). Another study confirmed the GVHD-attenuating effect of IL-4 produced by Th2 cells, also having a skewing effect on Th2 cytokines (89). On the contrary, other studies showed that the absence or neutralization of IL-4 ameliorated GVHD, implying a detrimental role of Th2 cells (90, 91). However, these contradicting results regarding the role of IL-4 in GVHD may be based on different mouse models and experimental settings (92). Despite the overall protective role of IL-4 secreting Th2 cells in GVHD, the location of these cells might define their pathogenic relevance, as they were associated with pathophysiological changes in the lung, but not in colon, liver, and skin during GVHD (93). IL-13, another Th2 effector cytokine, was also shown to have an ameliorating effect on GVHD. Although one study correlated IL-13 levels with GVHD severity in patients (94), transplantation experiments of IL-13-/- cells in an established mouse GVHD model resulted in increased mortality and decreased Th2 cytokine levels but elevated serum levels of TNF-α, a critical mediator of GVHD, in these mice (95). Further studies showed the counteracting role of IL-13 to TNF-α production and its augmenting role in IL-4 and IL-5 secretion following allo-bone marrow transplantation (96), supporting the notion that IL-13 has a protective function in GVHD. In general, the transplantation of Th2 cells to recipient mice showed beneficial effects on GVHD-survival (97, 98) and an alteration of the Th1/Th2 balance towards the Th2 cells leading to increased IL-4 levels and attenuated GVHD (98–100). Ultimately, a study investigating IL-4, IL-5, IL-9, and IL-13 quadruple cytokine-deficient T cells in a well-established mouse model demonstrated that combined Th2 cytokine deficiency resulted in enhanced T cell proliferation, higher TNF-α, IL-2, IFN-γ and IL-17a serum levels and overall aggravated GVHD (101).
A few further experiments on Th2 defining TFs gained similar results in GVHD models. Atorvastatin (AT) treatment was shown to modulate Th1/Th2 differentiation by inhibiting the production of the isoprenoid derivates farnesly-pyrophosphate (PP) and geranylgranyl-PP, of the mevalonate pathway. Inhibition of these isoprenoid derivates combined by AT or individually by a farnesyltransferase inhibitor (FTI) or a geranylgeranyltransferase inhibitor (GGTI) respectively, resulted in an upregulation of GATA3, and in case of AT and FTI treatment also an downregulation of T-bet expression in antigen-primed T cells (102). GGTI and FTIs were also shown to have ameliorating CD4 T cell specific effects on GVHD while sparing CD8 T cells in their capacity to mediate GVL and protect from viral infections (103). AT treatment also induced Th2 polarization and cytokine secretion and inhibited GVHD development by partially acting through STAT6, a transcription factor essential for Th2 differentiation in response to IL-4 and IL-13 (23, 104, 105). STAT6 was shown to be required for Th2 involved NKT-cell mediated GVHD prophylaxis (106). Additionally, transplanted STAT6-/- T cells, unable to differentiate to Th2 cells skewed towards Th1 cells and mediated aGVHD with major involvement of the colon. On the contrary, STAT4-/- T cells, which predominantly differentiated to Th2 cells, showed less severe signs of GVHD but later involvement in skin pathology (107). STAT5, another critical TF in Th2 differentiation, was found to have a dual role in Th2 and Treg differentiation in GVHD, as overexpression of STAT5 led to increased Treg numbers and attenuated GVHD, while in the absence of Tregs, anti-inflammatory Th2-cytokines increased (108).
Tregs are the second CD4 T cell subset which play a protective role in GVHD. In general, responsible for immune homeostasis and balanced immune responses, Tregs have an outstanding role in controlling GVHD development. First experiments on CD4+ CD25+ Tregs in GVHD showed that depletion of these cells aggravated GVHD, while supplementation with Tregs had the contrary effect (109, 110). The capacity of Tregs to attenuate GVHD was associated with their expansion-inhibiting effect on allogeneic T cells in the early phase of GVHD (111). The beneficial effect of Tregs in GVHD prevention was demonstrated in fully allogeneic, haploidentical and xenograft mouse models (111–116). FoxP3 expression was additionally found to negatively correlate with GVHD severity in patients (117). Importantly, murine, and human Tregs attenuate allogeneic T cell reactions, without impeding the graft-versus-leukemia (GVL) effect (111, 118–121). The use of in vitro induced Tregs (iTregs) as a GVHD therapeutic revealed effective protection in the early phase after transplantation but unstable FoxP3 expression over time led to aggravation of GVHD, making this approach less promising as initially thought (122, 123). However, additional combinatory induction with IL-2 and rapamycin was shown to stabilize FoxP3 expression in these cells (113, 124), which enabled the first successful application of iTregs as GVHD-prophylactic therapy to humans (125).
Despite this broad outline of Treg research in GVHD, many recent publications have already summarized the role of Tregs in GVHD in a detailed way (126–128), for which reason we will not go into further details at this point.
Overall, Th2 and Tregs were shown to have an attenuating and protective role in GVHD. While Th2 cells can still mediate local GVHD-associated pathophysiological changes in the lung, Tregs are an overall protective cell population in GVHD having crucial homeostatic functions, which are tightly regulated in balance with other Th-subsets.
Detrimental Th Subsets in GVHD
Contradicting first studies on Th1 cytokines in the 1990’s led to unconclusive results regarding the role of Th1 cells in GVHD. The main Th1-inducing and -associated cytokines IL-2, IFN-γ and IL-12 were found to ameliorate GVHD in several early studies which indicated a protective function of Th1 in GVHD (129–132). However, other groups showed, that increased IFN-γ levels in serum correlated with GVHD severity (133, 134) and that IFN-γ was critical for tissue pathology during GVHD (97). Besides the beneficial role of IFN-γ in the induction of GVHD-associated effects in the lung (135), it was shown to have adverse effect in acute GVHD pathology in the GI tract (93, 136–138). Additionally, the effect of IFN-γ in GVHD was found to be dependent on the irradiation regimen used (139). Overall, the reciprocal effect of IFN-γ in GVHD seems to be highly dependent on conditioning, location, timing, and the stage of allo-immune response [reviewed by Lu and Waller, (140)].
Similar to IFN-γ, contradicting findings were made, when Th17-associated cytokines were assessed in GVHD mouse models. One study suggested a protective role of IL-17a in GVHD, as IL-17-/- T cells accelerated GVHD while the systemic administration of IL-17a and the neutralization of IFN-γ prevented this effect (141). Other studies reported improved transplantation outcomes when IL-17a-/- T cells were used (142) and severe GVHD induction when in vitro generated IL-17+ cells were infused (143). Altogether, these studies indicated that, similar to IFN-γ, the role of IL-17 in GVHD is dependent on timing and conditioning regimen. IL-17 probably contributes to early development of GVHD but is dispensable for overall GVHD induction (142). Neutralization of the IL-17 inducing cytokine TGF-β was shown to increase aGVHD severity indicating an ameliorating effect of Th17 cells in GVHD (144). However, TGF-β is also relevant for the differentiation of Tregs which are GVHD protective, and its absence resulted in enhanced Th1 cell proliferation indicating Th17-independent mechanisms that lead to enhanced GVHD (144). IL-6, which induces TGF-β dependent differentiation of Th17 but not Treg cells, was found to play a relevant role in GVHD induction, as blocking of the IL-6R led to reduced GVHD pathology and Th1/Th17 cells in GVHD target organs, while absolute numbers of Tregs increased (145). However, another study showed that short-term administration of IL-6 could not confirm these beneficial effects (146). Differences between the design of these two studies indicate that the effect of IL-6 on GVHD development is dependent on conditioning, the used model, and the duration of therapy.
TNF-α, another Th1-associated cytokine, which also promotes Th17 differentiation, was shown to drive GVHD pathophysiology on several stages. For example, TNF-α is responsible for early intestinal GVHD-related toxicity (147) and TNF-receptor 1 (TNFR1) levels strongly correlate with GVHD severity (148). Additionally, the attenuating effect of TNF-blocking therapy in GVHD underlines the detrimental role of TNF-α in GVHD (149). Similarly, inhibiting the Th17 effector cytokines IL-21 and IL-23 decreased GVHD severity in various mouse models (150–152).
However, cytokines can derive from different cell types and do not necessarily represent the involvement of respective Th cell subsets. Hence, experiments examining subset defining TF knock-out CD4 T cells shed more light on the relevance of different Th cell subsets in GVHD and identified Th1 and Th17 cells as the relevant subsets promoting GVHD.
First TF-knock-out experiments to investigate the influence of Th1 differentiation on GVHD were performed with STAT6-/- and STAT4-/- T cells. STAT6-/- T cells are unable to differentiate to Th2 cells but instead show enhanced Th1 responses (23, 104, 153). In contrast, lack of STAT4 in T cells leads to impaired Th1 differentiation (154). Nikolic and colleagues investigated STAT6-/- and STAT4-/- T cells in a GVHD mouse model and found that STAT6-/- T cell recipients showed an earlier and more severe course of GVHD with severe inflammation in the GI tract in comparison to STAT4-/- T cell recipients, while only the latter group displayed severe skin disorders (107). These results indicate the detrimental role of Th1 cells in GVHD mainly affecting the GI tract but not liver and skin. Recipients of T cells with STAT1 KO, another critical STAT TF for Th1 development, also resulted in the attenuation of GVHD and increased Treg expansion (155). Comparable results were obtained in GVHD experiments with c-Rel KO T cells, which showed a dramatically reduced ability to induce GVHD in various mouse models, defects in Th1 and Th17 differentiation, enhanced Treg differentiation and a preserved Graft-versus-leukemia (GvL) effect (156).
Experiments with T cells deficient for T-bet and RORγt, the master TFs of Th1 and Th17 cells respectively also confirmed that these subsets are the most involved Th cells in GVHD induction and development. Transplanted T-bet deficient T cells screwed to Th2, Th17 and Treg subsets and led to attenuated GVHD, especially in the gut (157). The absence of RORγt in T cells only had little impact and RORγt was dispensable to induce GVHD development in two independent studies (157, 158), while one study reported an attenuated effect on GVHD if both isoforms, RORγ and RORγt were absent in CD4 transplanted T cells due to KO of the entire Rorc locus (159). However, T-bet and RORγt double KO T cells, which showed a defective differentiation of Th1 and Th17, and increased Th2 and Treg cells, induced less GVHD than T-bet KO T cells alone. This finding suggests a synergistic effect of RORγt-induced Th17 cells on Th1-mediated GVHD induction (157).
In addition, TFs linked to the reciprocal differentiation to Th17 versus Treg cells were also found to play a crucial role in attenuation of GVHD. For example, recipients of T cells with a STAT3 deficiency, a TF crucial for Th17 development, showed attenuated GVHD development and increased numbers of pTregs (160).
Summarized, Th1 and Th17 cells synergistically are the main Th subsets driving GVHD, especially with detrimental pathological effects on the GI tract. Blocking of Th1 and Th17- transcription factors was found to be a more effective strategy to prevent GVHD, than blocking Th1 and Th17-involved cytokines. Hence, the use of specific TF-blocking agents is a promising strategy to treat GVHD in the future. The following paragraph will give deeper insights in recent literature reporting the effect of a variety of Th-subset specific TF blocking agents in murine GVHD models.
Potential Strategies to Target Transcription Factors of T Helper Cell Development in GVHD
As described earlier, experiment with various Th-differentiation associated TF knock-out T cells revealed efficient attenuation of GVHD in different transplantation models. Hence, inhibition of these TFs by target-specific inhibitory agents offers a potent strategy for GVHD prophylaxis and therapy.
Several commonly used GVHD therapeutics also rely on the modulation of TF expression or activity. Calcineurin inhibitors (CNIs) like Cyclosporine A (CyA) of tacrolimus (FK506) for example block TCR-proximal signaling by inhibition of NFAT. Even though CNIs remain standard of care for GVHD prevention, they also interfere with the valuable GVL-effect by impairing donor immunity and disruption of Treg function and survival (161–165). Combinatorial therapy with mTor inhibitors like Rapamycin (Sirolimus) and/or low-dose IL-2 administration have already shown to improve Treg reconstitution after allo-hematopoietic cell transplantation (164, 166–170).
The following section will provide more detailed information on various new therapeutic agents, divided by substance classes, which have been successfully evaluated in GVHD alone or in combination with standard of care therapeutics in the recent years (Tables 1, 2). Importantly, if not indicated by the respective clinical trial number or mentioned explicitly, this paragraph mostly summarizes results from pre-clinical GVHD mouse models and not from studies in patients. Most of them rely on the strategy of targeting TFs that mediate the reciprocal effect between Th17/Th1 and Treg differentiation, hence inducing a homeostatic effect by skewing CD4 T cell differentiation towards Tregs while preserving the GVL effect.
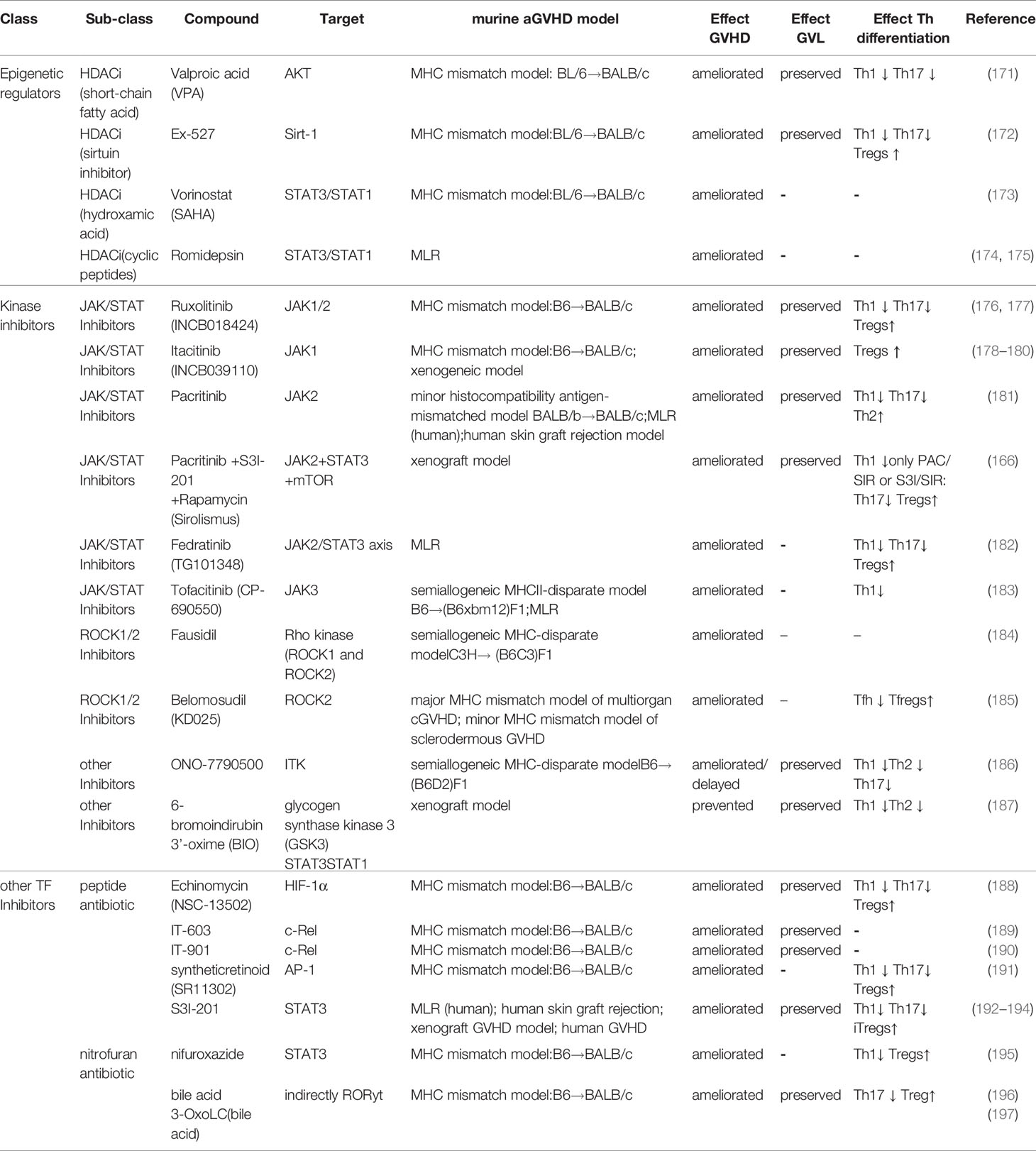
Table 1 Summary of pre-clinical studies on Th-differentiation targeting TF inhibitors.
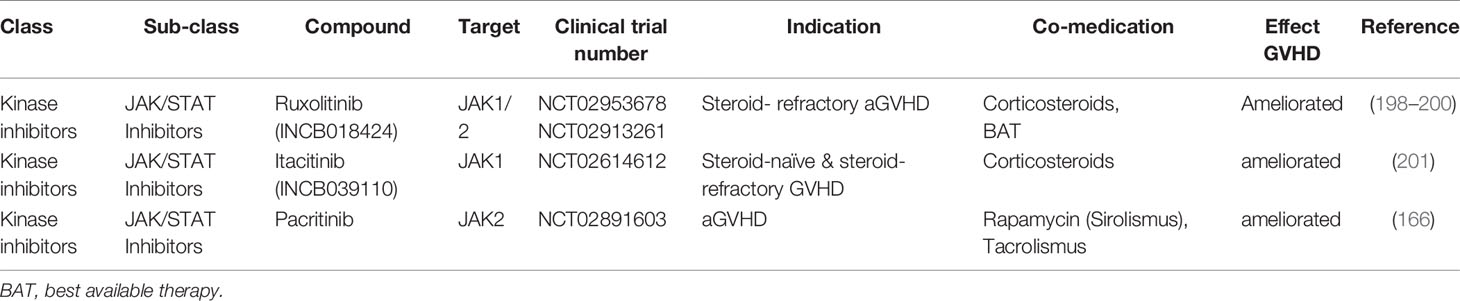
Table 2 Summary of clinical trials on Th defining TF inhibitors.
Epigenetic Modulators
Epigenetic modulation of transcription is a promising approach to indirectly inhibit TF expression. The acetylation of histones, regulated by histone acetyl transferases (HATs) and histone deacetylases (HDACs), is an epigenetic mark, which influences chromatin structure and ultimately gene expression. The use of HDACs and HDAC-inhibitors (HDACi) can modulate this balance and subsequently alter gene expression.
Valproic acid (VPA), a HDACi of the short-chain fatty acid category, was shown to indirectly decrease STAT5 phosphorylation and dampen T-bet expression in NK cells (202). In a mouse model, the administration of VPA attenuated aGVHD by downregulation of Th1 and Th17 cells (171). This effect was associated with a direct inhibition of Akt (171), a kinase which promotes Th1, Th17 and Tfh but inhibits Treg development by activation of mTOR which in turn induces T-bet, RORγt and HIF1α and inhibits FOXO1-dependent FoxP3 transcription (203–205). Importantly, the GVL-effect was preserved during VAP therapy.
Another HDACi, which showed promising effects on GVHD in preclinical models is Ex-527, a Sirtuin-1 (Sirt-1) inhibitor. Sirt-1 represses AP-1, Smad3 and FOXO-transcription factors which regulate pTreg differentiation via the CNS1 regulatory element (206–209) and was identified as a direct negative regulator of FoxP3 (210). Pre-clinical experiments in a murine GVHD mode showed that Sirt1-/- T cells were impaired in inducing aGVHD and showed an enhanced pTreg differentiation in which FoxP3 stability was increased. Ex-527 administration induced comparable effects while preserving GVL effects (172). Stabilization of FoxP3 expression by Ex-527 had already been reported earlier and associated to increased Treg suppressive function (210, 211). Another Sirt-1 inhibitor, Sirtinol, was found to decrease RORγt and IL-17A expression in CD4 T cells in vitro and to screw Th17/Treg differentiation towards Tregs, leading to a prolonging allograft survival in a mouse transplantation model (212). However, the effects of Sirnotol in GVHD were not reported yet.
Givinostat (ITF2357), a HDACi of the hydroxamic acid category, was also reported to suppress Th17 polarization and enhance FoxP3 expression and hence Treg differentiation via decreased STAT3 phosphorylation and RORγt expression downstream of IL-6R signaling. Administration of Givinostat inhibited experimental colitis development by skewing the Th17/Treg balance in the lamina propria (213) and reduced release of inflammatory IFN-γ and TNF-α in systemic inflammation (214). Virinostat (SAHA), another hydroxamic acid HDACi, inhibits STAT3 and also STAT1 phosphorylation, and was shown to attenuate GVHD and inhibit proinflammatory cytokine production during the initiation phase of GVHD (173). Additionally, blocking of STAT3 by both Givinostat and Virinostat, was shown to enhance indoleamine 2,3-dioxygenase (IDO) expression in APCs which suppresses APC allo-stimulatory functions and reduced GVHD in a murine allogeneic BM-transplantation model (215). Hence, Givinostat and Virinostat attenuate GVHD via multiple mechanisms, targeting inflammatory cytokine release, antigen presentation and T cell differentiation (216).
Romidepsin (Istodax), a cyclic peptide class HDACi, was shown to effectively suppress allo-responses in a mixed lymphocyte reaction (MLR) (174). Recently, Romidepsin was also shown to inhibit the activation of STAT1 and STAT3 via induction of suppressor of cytokine signaling 1 (SOCS1) (175). However, its effect in GVHD has not been fully assessed yet as a first study in patients was terminated due to slow accrual (clinical trail.gov, NCT02203578).
Overall, epigenetic modulators like the describes HDACi, were shown to efficiently inhibit GVHD by altering Th polarization via TF modulation. The success of HDACi in preventing GVHD is also displayed by multiple clinical studies validating their beneficial effect often in combinatory therapy in patients [reviewed by Xu et al., (217)].
Kinase Inhibitors
Another indirect way to target TFs is the inhibition of Kinases, which catalyze the transfer of activating phosphate from ATP to substrates withing signaling pathways. Hence, kinase inhibitors can indirectly block activation of the respective kinase substate like TFs and hence modulate transcription.
The most prominent examples in GVHD therapy are Janus-Kinase (JAK) inhibitors. These inhibitors block JAK/STAT signaling pathways, which have a crucial function of transmitting cytokine-receptor signals intracellularly. Early expression profiling studies and the detection of activated STAT1 and STAT3 in GHVD target organs and alloreactive donor T cells already indicated a link between GVHD and cytokine signaling through the JAK/STAT pathway (218–220).
Subsequent experiments, disrupting JAK/STAT1 signaling by the use of T cells lacking STAT1, a Th1 specific TF responding to IFN-γ Receptor (IFNγR) signaling, reported ameliorated GVHD outcomes in a minor antigen-mismatched and fully-MHC mismatched GVHD model (155). Shortly after, Ruxolitinib (INCB018424), a bioavailable JAK1/2 inhibitor, was reported to have similar mitigating effects on GVHD as IFNγR-/- T cells while the GvL effect was preserved (176, 177, 221, 222). Further mechanistical analyses revealed, that Ruxolitinib ameliorates GVHD by disrupting Th1 and Th17 differentiation but promoting Treg differentiation via indirect STAT1 and STAT3 inhibition (223). Overall, these pre-clinical data suggested Ruxolitinib as a promising candidate for GVHD treatment, which indeed has shown remarkable results in the application for steroid refractory GVHD in various clinical studies (224).
Besides Ruxolitinib inhibiting JAK1 and JAK2 simultaneously, selective JAK1, JAK2 and JAK3 inhibitors have also been investigated as potent treatment options in GVHD. The JAK3 inhibitor Tofacitinib (CP-690550) was reported to ameliorate GVHD in vivo and in vitro by selectively inhibiting Th1 differentiation but not Th17 polarization or CD4 T cell proliferation (183). Itacitinib (INCB039110), a selective JAK1 inhibitor, disrupts the JAK1/STAT3 signaling pathway and was shown to improve GVHD outcomes and survival in various mouse models, partially by reduction of CD4 and CD8 T cell numbers in the inflamed colon tissue, indicating a loss of Th17 phenotype (178–180). Itacitinib also showed promising efficiencies in the treatment of steroid-naïve and steroid-refractory GVHD in a first clinical study (201). Selective inhibition of the JAK2/STAT3 axis, an IFN-γ, IL-6 and IL-23 receptor signaling response element, by Pacritinib (SB1518) was also shown to significantly reduce GVHD in murine models (181, 225). Similar to the effects of the JAK/STAT3 inhibitor Fedratinib in early MLR experiments; Pacritinib, led to impaired expansion of Th1 and Th17 cells while Treg and Th2 responses were sustained (181, 182). A recent study also reported a successful combinatory therapy of acute GVHD with Pacritinib the STAT3 inhibitor S31-201 and the mTOR inhibitor Rapamycin in a xenogeneic mouse model and with Rapamycin and the calcineurin inhibitor Tacrolismus in patients (166).
Despite the advanced clinical validation of JAK/STAT inhibitors in GVHD [reviewed by Assal and Mapara, (224)], few other agents of the Kinase-inhibitor group have also shown beneficial effect on GVHD in pre-clinical studies. Inhibition of the glycogen synthase kinase 3 (GSK3) by the small molecule 6-bromoindirubin 3’-oxime (BIO), prevented mice from lethal GVHD in a xenogeneic model by STAT1/3 suppression and subsequent decrease of Th1 effector cytokines (187). Recent studies suggested the IL-2 inducible kinase (ITK) inhibitor ONO-7790500 as another potent therapeutic in GVHD, as administration inhibited Th1, Th2 and Th17 differentiation, inflammatory cytokine production and alloreactive T cell proliferation and significantly delayed GVHD onset and mortality (186). An earlier study with ITK-/- donor T cells in an allo-HSCT mouse model has already reported comparable beneficial effects on GVHD and observed reduced expression of IRF4, JAK1, JAK2, and STAT3 as well as phosphorylated forms of JAK1, JAK2 and STAT3 if ITK was absent in T cells, which might explain impaired differentiation capacities observed in the ITK inhibitor study (226). Rho-Kinase (ROCK) inhibitors represent a further group evaluated in pre-clinical GVHD settings. While Fausidil, a small molecule inhibiting ROCK1 and ROCK2 only had moderate ameliorating effects on GVHD-associated colitis (184), the ROCK2 inhibitor Belomosudil (KD025), which shifts the Th17/Treg balance towards homeostasis via an STAT3/STAT5-dependent mechanism, efficiently ameliorated chronic GVHD in multiple models and first clinical studies (185, 227). However, the effect of Belomosudil on aGVHD remains to be determined.
Other Direct and Indirect Transcription Factor Inhibitors
Besides epigenetic regulators and kinase inhibitors other small molecules targeting TFs in a direct or indirect manner have been assessed in pre-clinical GVHD models in the last decade.
As already implicated by the successful use of JAK/STAT inhibitors, the repression or STAT3, an important activator of RORγt during Th17 differentiation, was investigated as potent strategy to prevent severe GVHD. Betts at al. reported, that the small molecule S3I-201 efficiently inhibits STAT3 expression, leading to suppressed proliferation of allo-sensitized T cells and impaired Th17 differentiation while iTreg polarization was enhanced. Mechanistically, the group uncovered that S3I-201 polarized the phosphorylation of STAT5 over STAT3 and led to activation of FoxP3 in iTregs (192). Hence, S3I-201 shifts the Th17/Treg balance towards regulatory T cells, as already reported for other STAT3 inhibitors in this review earlier. A later study of the group connected increased pSTAT3 and RORγt levels with severe aGVHD. They found that RORγt suppression was enhanced by combinatory treatment with Rapamycin and S3I-201, which abrogated the proliferation of Rapamycin-resistant T cells upon allo-sensitization in a MLR model (193). Additionally, they reported successful prevention of acute GVHD in a xenogeneic mouse model, using a combinatory treatment with S31-201, the JAK2 inhibitor Pacritinib and Rapamycin in a recently published study, as referred to earlier (166). Moreover, S3I-201 treated iTregs were found to efficiently reduce skin graft rejection and GVHD in a xenograft mouse model by reducing Th1- and Th17-mediated allorectivity, while preserving the GVL effect (194). Similarly, the STAT3 inhibitor nifuroxazide also attenuated GVHD symptoms in skin, liver and GI-tract and efficiently delayed aGVHD-associated lethality (195). Blocking of the TF AP-1 by the synthetic retinoid SR11302 also inhibited Th1/Th17 proliferation and enhanced Treg expansion by indirectly pSTAT3 blockage and STAT5 dependent FoxP3 expression, leading to diminished GVHD-associated pathology and lethality (191). Another study, which investigated the effect of GRIM19 overexpressing donor BM and T cells in GVHD, also found decreased disease-severity, Th17 polarization, and alloreactive activation due to diminished STAT3 expression. Comparable to the effect of other STAT3 inhibitors, GRIM19 overexpression also led to enhanced STAT5 expression and Treg differentiation suggesting GRIM19 induction as another potent strategy for STAT3 inhibition in the future (228).
Alongside STAT3, the inhibition of other Th1 and Th17-differentiation inducing TFs was shown to efficiently ameliorate GVHD. Inhibition of HIF1α, a key TF in Th17/Treg reciprocal differentiation, by Echinomycin (NSC-13502) was shown to efficiently attenuate GVHD and preserve anti-leukemic activity by inducing Treg expansion while diminishing Th17 responses (229). The TF c-Rel plays a role in differentiation of Th1, Th17 and Treg cells. Studies on the c-Rel inhibitor IT-603 showed ameliorating effects on GVHD, mediated through reduced alloreactivity, defective gut homing and impaired negative feedback on IL-2 production by effector T cells leading to an expansion of regulatory T cells. The attenuating effects on GVHD were additionally accompanied by a preserved graft-versus-tumor (GVT) effect and promising effects against lymphomas (189, 190). Bile acid synthesized form cholesterol, called 3-oxoLC was discovered as an inhibitory ligand of the RORγt. It efficiently altered Th17/Treg polarization towards regulatory T cells in the lamina propria suggesting a beneficial effect of bile acid metabolites in controlling intestinal-microbiome tolerance but also immune responses in GI-associated GVHD (197). Indeed, a shortly later published study reported, that the bile acid pool was reduced in patients with GVHD, and that application of bile acids reduced GVHD in several transplantation mouse models but was rather associated to alterations in antigen presentation that in Th17 differentiation (196). However, these studies suggest bile acids as potent immune modulators in the GI-tract during GVHD, partially acting through Th-subset determining TF inhibition.
Conclusion
Summarized, these data show that specific targeting of Th cell-differentiation involved transcription factors might represent a potent therapeutic strategy to prevent or ameliorate GVHD in addition to standard of care medication. However, most of the presented therapeutics have only been assessed in pre-clinical models yet and beneficial effects for patients remain to be proven. In addition, the immune modulatory effect of the presented therapeutic strategies may lead to a higher susceptibility for infections. This includes the re-activation of latent viral infections [e.g. cytomegalovirus (CMV)] but also the predisposition for newly acquired infections due to major immune suppression of especially Th1 T cells but also other immune cell populations required for viral clearance. First clinical trials with the HDACi Vorinostat and Panobinostat in GVHD patients did not show an augmentation for risk of infections while Romidepsin treated patients with T cell lymphoma more often experiences infections (230–232). Studies on the JAK1/JAK2 inhibitor Ruxolitinib also reported an increased susceptibility for viral re-activation of Hepatitis-B and varicella zoster virus in treated patients with myeloproliferative neoplasm and polycythemia vera, but also a modestly higher incidence of infection and reactivated CMV infection in patients with steroid-refractory GVHD (198, 233, 234). However, first line and second line therapies in GVHD also harbor the risk of viral re-activation and overall significant improvement in efficacy outcomes by more target specific TF inhibitors probably weights more than a moderate elevated risk for infection (198, 235). Additionally, the above-mentioned examples from clinical trials show that the risk of an enhanced susceptibility towards infections under TF inhibitor treatment is highly dependent on the drug target and specificity so that these more specific TF inhibitors might exhibit superior protection from infections that other commonly used therapeutics.
Together, given the promising results of some TF-modulators in clinical studies, we expect a fundamental contribution of TF-inhibitors to improve GVHD therapy in the future.
Author Contributions
JC and EU wrote the manuscript and approved the submitted version.
Funding
The laboratory of EU has been supported by the Frankfurt Cancer Institute FCI/DKTK (to EU), by the German Research Foundation DFG (CRC 1292, UL316/5-1), by the German Cancer Aid, the “Alfred & Angelika Gutermuth-Stiftung” and by “Menschen für Kinder e.V.”
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two Types of Murine Helper T Cell Clone. I. Definition According to Profiles of Lymphokine Activities and Secreted Proteins. J Immunol (1986) 136:2348–57.
2. Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O’Garra A, Murphy KM. Development of TH1 CD4+ T Cells Through IL-12 Produced by Listeria-Induced Macrophages. Science (1993) 260:547–9. doi: 10.1126/science.8097338
3. Lighvani AA, Frucht DM, Jankovic D, Yamane H, Aliberti J, Hissong BD, et al. T-Bet is Rapidly Induced by Interferon-Gamma in Lymphoid and Myeloid Cells. Proc Natl Acad Sci USA (2001) 98:15137–42. doi: 10.1073/pnas.261570598
4. Le Gros G, Ben-Sasson SZ, Seder R, Finkelman FD, Paul WE. Generation of Interleukin 4 (IL-4)-Producing Cells In Vivo and In Vitro: IL-2 and IL-4 are Required for In Vitro Generation of IL-4-Producing Cells. J Exp Med (1990) 172:921–9. doi: 10.1084/jem.172.3.921
5. Swain SL, Weinberg AD, English M, Huston G. IL-4 Directs the Development of Th2-Like Helper Effectors. J Immunol (1990) 145:3796–806.
6. Seder RA, Paul WE, Davis MM, Fazekas de St Groth B. The Presence of Interleukin 4 During In Vitro Priming Determines the Lymphokine-Producing Potential of CD4+ T Cells From T Cell Receptor Transgenic Mice. J Exp Med (1992) 176:1091–8. doi: 10.1084/jem.176.4.1091
7. Hsieh CS, Heimberger AB, Gold JS, O’Garra A, Murphy KM. Differential Regulation of T Helper Phenotype Development by Interleukins 4 and 10 in an Alpha Beta T-Cell-Receptor Transgenic System. Proc Natl Acad Sci USA (1992) 89:6065–9. doi: 10.1073/pnas.89.13.6065
8. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG. Glimcher LH. A Novel Transcription Factor, T-Bet, Directs Th1 Lineage Commitment. Cell (2000) 100:655–69. doi: 10.1016/s0092-8674(00)80702-3
9. Mullen AC, High FA, Hutchins AS, Lee HW, Villarino AV, Livingston DM, et al. Role of T-Bet in Commitment of TH1 Cells Before IL-12-Dependent Selection. Science (2001) 292:1907–10. doi: 10.1126/science.1059835
10. Mullen AC, Hutchins AS, High FA, Lee HW, Sykes KJ, Chodosh LA, et al. Hlx is Induced by and Genetically Interacts With T-Bet to Promote Heritable T(H)1 Gene Induction. Nat Immunol (2002) 3:652–8. doi: 10.1038/ni807
11. Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct Effects of T-Bet in TH1 Lineage Commitment and IFN-Gamma Production in CD4 and CD8 T Cells. Science (2002) 295:338–42. doi: 10.1126/science.1065543
12. Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, et al. T-Bet is a STAT1-Induced Regulator of IL-12R Expression in Naïve CD4+ T Cells. Nat Immunol (2002) 3:549–57. doi: 10.1038/ni794
13. Zheng W-P, Flavell RA. The Transcription Factor GATA-3 Is Necessary and Sufficient for Th2 Cytokine Gene Expression in CD4 T Cells. Cell (1997) 89:587–96. doi: 10.1016/s0092-8674(00)80240-8
14. Zhang DH, Cohn L, Ray P, Bottomly K, Ray A. Transcription Factor GATA-3 is Differentially Expressed in Murine Th1 and Th2 Cells and Controls Th2-Specific Expression of the Interleukin-5 Gene. J Biol Chem (1997) 272:21597–603. doi: 10.1074/jbc.272.34.21597
15. Usui T, Nishikomori R, Kitani A, Strober W. GATA-3 Suppresses Th1 Development by Downregulation of Stat4 and Not Through Effects on IL-12rβ2 Chain or T-Bet. Immunity (2003) 18:415–28. doi: 10.1016/s1074-7613(03)00057-8
16. Usui T, Preiss JC, Kanno Y, Yao ZJ, Bream JH, O’Shea JJ, et al. T-Bet Regulates Th1 Responses Through Essential Effects on GATA-3 Function Rather Than on IFNG Gene Acetylation and Transcription. J Exp Med (2006) 203:755–66. doi: 10.1084/jem.20052165
17. Suto A, Wurster AL, Reiner SL, Grusby MJ. IL-21 Inhibits IFN-Gamma Production in Developing Th1 Cells Through the Repression of Eomesodermin Expression. J Immunol (2006) 177:3721–7. doi: 10.4049/jimmunol.177.6.3721
18. Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 Responses and Enhanced Development of Th2 Cells in Stat4-Deficient Mice. Nature (1996) 382:174–7. doi: 10.1038/382174a0
19. Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, et al. Requirement for Stat4 in Interleukin-12-Mediated Responses of Natural Killer and T Cells. Nature (1996) 382:171–4. doi: 10.1038/382171a0
20. Cai G, Radzanowski T, Villegas EN, Kastelein R, Hunter CA. Identification of STAT4-Dependent and Independent Mechanisms of Resistance to Toxoplasma Gondii. J Immunol (2000) 165:2619–27. doi: 10.4049/jimmunol.165.5.2619
21. Pai S-Y, Ho I-C. C-Rel Delivers a One-Two Punch in Th1 Cell Differentiation. J Clin Invest (2002) 110:741–2. doi: 10.1172/JCI16552
22. Hilliard BA, Mason N, Xu L, Sun J, Lamhamedi-Cherradi S-E, Liou H-C, et al. Critical Roles of C-Rel in Autoimmune Inflammation and Helper T Cell Differentiation. J Clin Invest (2002) 110:843–50. doi: 10.1172/JCI15254
23. Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 Is Required for Mediating Responses to IL-4 and for the Development of Th2 Cells. Immunity (1996) 4:313–9. doi: 10.1016/s1074-7613(00)80439-2
24. Gilmour KC, Pine R, Reich NC. Interleukin 2 Activates STAT5 Transcription Factor (Mammary Gland Factor) and Specific Gene Expression in T Lymphocytes. Proc Natl Acad Sci USA (1995) 92:10772–6. doi: 10.1073/pnas.92.23.10772
25. Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 Activation Plays a Critical Role in Th2 Differentiation. Immunity (2003) 19:739–48. doi: 10.1016/S1074-7613(03)00292-9
26. Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, et al. Interleukin 17-Producing CD4+ Effector T Cells Develop via a Lineage Distinct From the T Helper Type 1 and 2 Lineages. Nat Immunol (2005) 6:1123–32. doi: 10.1038/ni1254
27. Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang Y-H, et al. A Distinct Lineage of CD4 T Cells Regulates Tissue Inflammation by Producing Interleukin 17. Nat Immunol (2005) 6:1133–41. doi: 10.1038/ni1261
28. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal Developmental Pathways for the Generation of Pathogenic Effector TH17 and Regulatory T Cells. Nature (2006) 441:235–8. doi: 10.1038/nature04753
29. Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming Growth Factor-Beta Induces Development of the T(H)17 Lineage. Nature (2006) 441:231–4. doi: 10.1038/nature04754
30. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the Context of an Inflammatory Cytokine Milieu Supports De Novo Differentiation of IL-17-Producing T Cells. Immunity (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
31. Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A Crucial Role for Interleukin (IL)-1 in the Induction of IL-17-Producing T Cells That Mediate Autoimmune Encephalomyelitis. J Exp Med (2006) 203:1685–91. doi: 10.1084/jem.20060285
32. Nakae S, Iwakura Y, Suto H, Galli SJ. Phenotypic Differences Between Th1 and Th17 Cells and Negative Regulation of Th1 Cell Differentiation by IL-17. J Leukoc Biol (2007) 81:1258–68. doi: 10.1189/jlb.1006610
33. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical Regulation of Early Th17 Cell Differentiation by Interleukin-1 Signaling. Immunity (2009) 30:576–87. doi: 10.1016/j.immuni.2009.02.007
34. Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 Cells in Human Disease. Immunol Rev (2008) 223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x
35. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The Orphan Nuclear Receptor RORgammat Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
36. Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, et al. Diverse Targets of the Transcription Factor STAT3 Contribute to T Cell Pathogenicity and Homeostasis. Immunity (2010) 32:605–15. doi: 10.1016/j.immuni.2010.05.003
37. Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu B-M, Tato C, et al. Selective Regulatory Function of Socs3 in the Formation of IL-17-Secreting T Cells. Proc Natl Acad Sci U.S.A. (2006) 103:8137–42. doi: 10.1073/pnas.0600666103
38. Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, et al. A Validated Regulatory Network for Th17 Cell Specification. Cell (2012) 151:289–303. doi: 10.1016/j.cell.2012.09.016
39. Glasmacher E, Agrawal S, Chang AB, Murphy TL, Zeng W, Vander Lugt B, et al. A Genomic Regulatory Element That Directs Assembly and Function of Immune-Specific AP-1-IRF Complexes. Science (2012) 338:975–80. doi: 10.1126/science.1228309
40. Schraml BU, Hildner K, Ise W, Lee W-L, Smith WA-E, Solomon B, et al. The AP-1 Transcription Factor Batf Controls T(H)17 Differentiation. Nature (2009) 460:405–9. doi: 10.1038/nature08114
41. Yahia-Cherbal H, Rybczynska M, Lovecchio D, Stephen T, Lescale C, Placek K, et al. NFAT Primes the Human RORC Locus for Rorγt Expression in CD4+ T Cells. Nat Commun (2019) 10:4698. doi: 10.1038/s41467-019-12680-x
42. Ruan Q, Kameswaran V, Zhang Y, Zheng S, Sun J, Wang J, et al. The Th17 Immune Response is Controlled by the Rel-Rorγ-Rorγ T Transcriptional Axis. J Exp Med (2011) 208:2321–33. doi: 10.1084/jem.20110462
43. Liu H-P, Cao AT, Feng T, Li Q, Zhang W, Yao S, et al. TGF-β Converts Th1 Cells Into Th17 Cells Through Stimulation of Runx1 Expression. Eur J Immunol (2015) 45:1010–8. doi: 10.1002/eji.201444726
44. Zhang F, Meng G, Strober W. Interactions Among the Transcription Factors Runx1, RORgammat and Foxp3 Regulate the Differentiation of Interleukin 17-Producing T Cells. Nat Immunol (2008) 9:1297–306. doi: 10.1038/ni.1663
45. Dang EV, Barbi J, Yang H-Y, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) Balance by Hypoxia-Inducible Factor 1. Cell (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
46. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic Self-Tolerance Maintained by Activated T Cells Expressing IL-2 Receptor Alpha-Chains (CD25). Breakdown of a Single Mechanism of Self-Tolerance Causes Various Autoimmune Diseases. J Immunol (1995) 155:1151–64.
47. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of CD4+CD25+ Regulatory T Cells. Nat Immunol (2003) 4:330–6. doi: 10.1038/ni904
48. Hori S, Nomura T, Sakaguchi S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science (2003) 299:1057–61. doi: 10.1126/science.1079490
49. Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T Cell Lineage Specification by the Forkhead Transcription Factor Foxp3. Immunity (2005) 22:329–41. doi: 10.1016/j.immuni.2005.01.016
50. Williams LM, Rudensky AY. Maintenance of the Foxp3-Dependent Developmental Program in Mature Regulatory T Cells Requires Continued Expression of Foxp3. Nat Immunol (2007) 8:277–84. doi: 10.1038/ni1437
51. Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, et al. Foxp3-Dependent Programme of Regulatory T-Cell Differentiation. Nature (2007) 445:771–5. doi: 10.1038/nature05543
52. Chen W, Jin W, Hardegen N, Lei K-J, Li L, Marinos N, et al. Conversion of Peripheral CD4+CD25- Naive T Cells to CD4+CD25+ Regulatory T Cells by TGF-Beta Induction of Transcription Factor Foxp3. J Exp Med (2003) 198:1875–86. doi: 10.1084/jem.20030152
53. Li MO, Wan YY. Flavell RA. T Cell-Produced Transforming Growth Factor-Beta1 Controls T Cell Tolerance and Regulates Th1- and Th17-Cell Differentiation. Immunity (2007) 26:579–91. doi: 10.1016/j.immuni.2007.03.014
54. Li MO, Sanjabi S, Flavell RA. Transforming Growth Factor-Beta Controls Development, Homeostasis, and Tolerance of T Cells by Regulatory T Cell-Dependent and -Independent Mechanisms. Immunity (2006) 25:455–71. doi: 10.1016/j.immuni.2006.07.011
55. Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen W. A Critical Function for TGF-Beta Signaling in the Development of Natural CD4+CD25+Foxp3+ Regulatory T Cells. Nat Immunol (2008) 9:632–40. doi: 10.1038/ni.1607
56. Hsieh C-S, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An Intersection Between the Self-Reactive Regulatory and Nonregulatory T Cell Receptor Repertoires. Nat Immunol (2006) 7:401–10. doi: 10.1038/ni1318
57. Lee W, Lee GR. Transcriptional Regulation and Development of Regulatory T Cells. Exp Mol Med (2018) 50:e456. doi: 10.1038/emm.2017.313
58. Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY. Role of Conserved non-Coding DNA Elements in the Foxp3 Gene in Regulatory T-Cell Fate. Nature (2010) 463:808–12. doi: 10.1038/nature08750
59. Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W. Positive and Negative Transcriptional Regulation of the Foxp3 Gene is Mediated by Access and Binding of the Smad3 Protein to Enhancer I. Immunity (2010) 33:313–25. doi: 10.1016/j.immuni.2010.09.001
60. Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT Cooperate to Induce Foxp3 Expression Through its Enhancer. Nat Immunol (2008) 9:194–202. doi: 10.1038/ni1549
61. Harada Y, Harada Y, Elly C, Ying G, Paik J-H, DePinho RA, et al. Transcription Factors Foxo3a and Foxo1 Couple the E3 Ligase Cbl-B to the Induction of Foxp3 Expression in Induced Regulatory T Cells. J Exp Med (2010) 207:1381–91. doi: 10.1084/jem.20100004
62. Ouyang W, Beckett O, Ma Q, Paik J-H, DePinho RA, Li MO. Foxo Proteins Cooperatively Control the Differentiation of Foxp3+ Regulatory T Cells. Nat Immunol (2010) 11:618–27. doi: 10.1038/ni.1884
63. Kitagawa Y, Ohkura N, Kidani Y, Vandenbon A, Hirota K, Kawakami R, et al. Guidance of Regulatory T Cell Development by Satb1-Dependent Super-Enhancer Establishment. Nat Immunol (2017) 18:173–83. doi: 10.1038/ni.3646
64. Polansky JK, Schreiber L, Thelemann C, Ludwig L, Krüger M, Baumgrass R, et al. Methylation Matters: Binding of Ets-1 to the Demethylated Foxp3 Gene Contributes to the Stabilization of Foxp3 Expression in Regulatory T Cells. J Mol Med (Berl) (2010) 88:1029–40. doi: 10.1007/s00109-010-0642-1
65. Mouly E, Chemin K, Nguyen HV, Chopin M, Mesnard L, Leite-de-Moraes M, et al. The Ets-1 Transcription Factor Controls the Development and Function of Natural Regulatory T Cells. J Exp Med (2010) 207:2113–25. doi: 10.1084/jem.20092153
66. Kim H-P, Leonard WJ. CREB/ATF-Dependent T Cell Receptor-Induced FoxP3 Gene Expression: A Role for DNA Methylation. J Exp Med (2007) 204:1543–51. doi: 10.1084/jem.20070109
67. Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY. Control of the Inheritance of Regulatory T Cell Identity by a Cis Element in the Foxp3 Locus. Cell (2014) 158:749–63. doi: 10.1016/j.cell.2014.07.031
68. Li X, Liang Y, LeBlanc M, Benner C, Zheng Y. Function of a Foxp3 Cis-Element in Protecting Regulatory T Cell Identity. Cell (2014) 158:734–48. doi: 10.1016/j.cell.2014.07.030
69. Long M, Park S-G, Strickland I, Hayden MS, Ghosh S. Nuclear factor-kappaB Modulates Regulatory T Cell Development by Directly Regulating Expression of Foxp3 Transcription Factor. Immunity (2009) 31:921–31. doi: 10.1016/j.immuni.2009.09.022
70. Cretney E, Xin A, Shi W, Minnich M, Masson F, Miasari M, et al. The Transcription Factors Blimp-1 and IRF4 Jointly Control the Differentiation and Function of Effector Regulatory T Cells. Nat Immunol (2011) 12:304–11. doi: 10.1038/ni.2006
71. Sidwell T, Liao Y, Garnham AL, Vasanthakumar A, Gloury R, Blume J, et al. Attenuation of TCR-Induced Transcription by Bach2 Controls Regulatory T Cell Differentiation and Homeostasis. Nat Commun (2020) 11:252. doi: 10.1038/s41467-019-14112-2
72. Lazarevic V, Chen X, Shim J-H, Hwang E-S, Jang E, Bolm AN, et al. T-Bet Represses T(H)17 Differentiation by Preventing Runx1-Mediated Activation of the Gene Encoding Rorγt. Nat Immunol (2011) 12:96–104. doi: 10.1038/ni.1969
73. Oestreich KJ, Huang AC, Weinmann AS. The Lineage-Defining Factors T-Bet and Bcl-6 Collaborate to Regulate Th1 Gene Expression Patterns. J Exp Med (2011) 208:1001–13. doi: 10.1084/jem.20102144
74. O’Garra A. Cytokines Induce the Development of Functionally Heterogeneous T Helper Cell Subsets. Immunity (1998) 8:275–83. doi: 10.1016/s1074-7613(00)80533-6
75. Murphy KM, Reiner SL. The Lineage Decisions of Helper T Cells. Nat Rev Immunol (2002) 2:933–44. doi: 10.1038/nri954
76. Yeh W-I, McWilliams IL, Harrington LE. Ifnγ Inhibits Th17 Differentiation and Function via Tbet-Dependent and Tbet-Independent Mechanisms. J Neuroimmunol (2014) 267:20–7. doi: 10.1016/j.jneuroim.2013.12.001
77. Kimura A, Kishimoto T. IL-6: Regulator of Treg/Th17 Balance. Eur J Immunol (2010) 40:1830–5. doi: 10.1002/eji.201040391
78. Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, et al. IL-6 Programs T(H)-17 Cell Differentiation by Promoting Sequential Engagement of the IL-21 and IL-23 Pathways. Nat Immunol (2007) 8:967–74. doi: 10.1038/ni1488
79. Xu L, Kitani A, Fuss I, Strober W. Cutting Edge: Regulatory T Cells Induce CD4+CD25-Foxp3- T Cells or are Self-Induced to Become Th17 Cells in the Absence of Exogenous TGF-Beta. J Immunol (2007) 178:6725–9. doi: 10.4049/jimmunol.178.11.6725
80. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-Dependent Glycolytic Pathway Orchestrates a Metabolic Checkpoint for the Differentiation of TH17 and Treg Cells. J Exp Med (2011) 208:1367–76. doi: 10.1084/jem.20110278
81. Ulges A, Witsch EJ, Pramanik G, Klein M, Birkner K, Bühler U, et al. Protein Kinase CK2 Governs the Molecular Decision Between Encephalitogenic TH17 Cell and Treg Cell Development. Proc Natl Acad Sci USA (2016) 113:10145–50. doi: 10.1073/pnas.1523869113
82. Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, et al. TGF-Beta-Induced Foxp3 Inhibits T(H)17 Cell Differentiation by Antagonizing RORgammat Function. Nature (2008) 453:236–40. doi: 10.1038/nature06878
83. Wang Y, Su MA, Wan YY. An Essential Role of the Transcription Factor GATA-3 for the Function of Regulatory T Cells. Immunity (2011) 35:337–48. doi: 10.1016/j.immuni.2011.08.012
84. Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, et al. Interleukin-2 Signaling via STAT5 Constrains T Helper 17 Cell Generation. Immunity (2007) 26:371–81. doi: 10.1016/j.immuni.2007.02.009
85. Yang X-P, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing Regulation of the Locus Encoding IL-17 Through Direct, Reciprocal Actions of STAT3 and STAT5. Nat Immunol (2011) 12:247–54. doi: 10.1038/ni.1995
86. Fowler DH, Kurasawa K, Husebekk A, Cohen PA, Gress RE. Cells of Th2 Cytokine Phenotype Prevent LPS-Induced Lethality During Murine Graft-Versus-Host Reaction. Regulation of Cytokines and CD8+ Lymphoid Engraftment. J Immunol (1994) 152:1004–13.
87. Fowler DH, Kurasawa K, Smith R, Eckhaus MA, Gress RE. Donor CD4-Enriched Cells of Th2 Cytokine Phenotype Regulate Graft-Versus-Host Disease Without Impairing Allogeneic Engraftment in Sublethally Irradiated Mice. Blood (1994) 84:3540–9. doi: 10.1182/blood.V84.10.3540.3540
88. Pan L, Delmonte J, Jalonen CK, Ferrara JL. Pretreatment of Donor Mice With Granulocyte Colony-Stimulating Factor Polarizes Donor T Lymphocytes Toward Type-2 Cytokine Production and Reduces Severity of Experimental Graft-Versus-Host Disease. Blood (1995) 86:4422–9. doi: 10.1182/blood.V86.12.4422.bloodjournal86124422
89. Foley JE, Jung U, Miera A, Borenstein T, Mariotti J, Eckhaus M, et al. Ex Vivo Rapamycin Generates Donor Th2 Cells That Potently Inhibit Graft-Versus-Host Disease and Graft-Versus-Tumor Effects via an IL-4-Dependent Mechanism. J Immunol (2005) 175:5732–43. doi: 10.4049/jimmunol.175.9.5732
90. Ushiyama C, Hirano T, Miyajima H, Okumura K, Ovary Z, Hashimoto H. Anti-IL-4 Antibody Prevents Graft-Versus-Host Disease in Mice After Bone Marrow Transplantation. The IgE Allotype is an Important Marker of Graft-Versus-Host Disease. J Immunol (1995) 154:2687–96.
91. Murphy WJ, Welniak LA, Taub DD, Wiltrout RH, Taylor PA, Vallera DA, et al. Differential Effects of the Absence of Interferon-Gamma and IL-4 in Acute Graft-Versus-Host Disease After Allogeneic Bone Marrow Transplantation in Mice. J Clin Invest (1998) 102:1742–8. doi: 10.1172/JCI3906
92. Fu J, Heinrichs J, Yu X-Z. Helper T-Cell Differentiation in Graft-Versus-Host Disease After Allogeneic Hematopoietic Stem Cell Transplantation. Arch Immunol Ther Exp (Warsz) (2014) 62:277–301. doi: 10.1007/s00005-014-0284-z
93. Yi T, Chen Y, Wang L, Du G, Huang D, Zhao D, et al. Reciprocal Differentiation and Tissue-Specific Pathogenesis of Th1, Th2, and Th17 Cells in Graft-Versus-Host Disease. Blood (2009) 114:3101–12. doi: 10.1182/blood-2009-05-219402
94. Jordan WJ, Brookes PA, Szydlo RM, Goldman JM, Lechler RI, Ritter MA. IL-13 Production by Donor T Cells is Prognostic of Acute Graft-Versus-Host Disease Following Unrelated Donor Stem Cell Transplantation. Blood (2004) 103:717–24. doi: 10.1182/blood-2003-01-0192
95. Hildebrandt GC, Choi SW, Mueller G, Olkiewicz KM, Moore BB, Cooke KR. The Absence of Donor-Derived IL-13 Exacerbates the Severity of Acute Graft-Versus-Host Disease Following Allogeneic Bone Marrow Transplantation. Pediatr Blood Cancer (2008) 50:911–4. doi: 10.1002/pbc.21228
96. Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, et al. Interleukin-13 Induces Tissue Fibrosis by Selectively Stimulating and Activating Transforming Growth Factor Beta(1). J Exp Med (2001) 194:809–21. doi: 10.1084/jem.194.6.809
97. Krenger W, Ferrara JL. Graft-Versus-Host Disease and the Th1/Th2 Paradigm. Immunol Res (1996) 15:50–73. doi: 10.1007/BF02918284
98. Krenger W, Cooke KR, Crawford JM, Sonis ST, Simmons R, Pan L, et al. Transplantation of Polarized Type 2 Donor T Cells Reduces Mortality Caused by Experimental Graft-Versus-Host Disease. Transplantation (1996) 62:1278–85. doi: 10.1097/00007890-199611150-00018
99. Krenger W, Snyder KM, Byon JC, Falzarano G, Ferrara JL. Polarized Type 2 Alloreactive CD4+ and CD8+ Donor T Cells Fail to Induce Experimental Acute Graft-Versus-Host Disease. J Immunol (1995) 155:585–93.
100. Foley JE, Mariotti J, Ryan K, Eckhaus M, Fowler DH. Th2 Cell Therapy of Established Acute Graft-Versus-Host Disease Requires IL-4 and IL-10 and is Abrogated by IL-2 or Host-Type Antigen-Presenting Cells. Biol Blood Marrow Transplant (2008) 14:959–72. doi: 10.1016/j.bbmt.2008.06.007
101. Tawara I, Maeda Y, Sun Y, Lowler KP, Liu C, Toubai T, et al. Combined Th2 Cytokine Deficiency in Donor T Cells Aggravates Experimental Acute Graft-vs-Host Disease. Exp Hematol (2008) 36:988–96. doi: 10.1016/j.exphem.2008.02.010
102. Dunn SE, Youssef S, Goldstein MJ, Prod’homme T, Weber MS, Zamvil SS, et al. Isoprenoids Determine Th1/Th2 Fate in Pathogenic T Cells, Providing a Mechanism of Modulation of Autoimmunity by Atorvastatin. J Exp Med (2006) 203:401–12. doi: 10.1084/jem.20051129
103. Hechinger A-K, Maas K, Dürr C, Leonhardt F, Prinz G, Marks R, et al. Inhibition of Protein Geranylgeranylation and Farnesylation Protects Against Graft-Versus-Host Disease via Effects on CD4 Effector T Cells. Haematologica (2013) 98:31–40. doi: 10.3324/haematol.2012.065789
104. Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, et al. Lack of IL-4-Induced Th2 Response and IgE Class Switching in Mice With Disrupted Stat6 Gene. Nature (1996) 380:630–3. doi: 10.1038/380630a0
105. Zeiser R, Youssef S, Baker J, Kambham N, Steinman L, Negrin RS. Preemptive HMG-CoA Reductase Inhibition Provides Graft-Versus-Host Disease Protection by Th-2 Polarization While Sparing Graft-Versus-Leukemia Activity. Blood (2007) 110:4588–98. doi: 10.1182/blood-2007-08-106005
106. Hashimoto D, Asakura S, Miyake S, Yamamura T, van Kaer L, Liu C, et al. Stimulation of Host NKT Cells by Synthetic Glycolipid Regulates Acute Graft-Versus-Host Disease by Inducing Th2 Polarization of Donor T Cells. J Immunol (2005) 174:551–6. doi: 10.4049/jimmunol.174.1.551
107. Nikolic B, Lee S, Bronson RT, Grusby MJ, Sykes M. Th1 and Th2 Mediate Acute Graft-Versus-Host Disease, Each With Distinct End-Organ Targets. J Clin Invest (2000) 105:1289–98. doi: 10.1172/JCI7894
108. Vogtenhuber C, Bucher C, Highfill SL, Koch LK, Goren E, Panoskaltsis-Mortari A, et al. Constitutively Active Stat5b in CD4+ T Cells Inhibits Graft-Versus-Host Disease Lethality Associated With Increased Regulatory T-Cell Potency and Decreased T Effector Cell Responses. Blood (2010) 116:466–74. doi: 10.1182/blood-2009-11-252825
109. Cohen JL, Trenado A, Vasey D, Klatzmann D, Salomon BL. CD4(+)CD25(+) Immunoregulatory T Cells: New Therapeutics for Graft-Versus-Host Disease. J Exp Med (2002) 196:401–6. doi: 10.1084/jem.20020090
110. Taylor PA, Lees CJ, Blazar BR. The Infusion of Ex Vivo Activated and Expanded CD4(+)CD25(+) Immune Regulatory Cells Inhibits Graft-Versus-Host Disease Lethality. Blood (2002) 99:3493–9. doi: 10.1182/blood.v99.10.3493
111. Edinger M, Hoffmann P, Ermann J, Drago K, Fathman CG, Strober S, et al. CD4+CD25+ Regulatory T Cells Preserve Graft-Versus-Tumor Activity While Inhibiting Graft-Versus-Host Disease After Bone Marrow Transplantation. Nat Med (2003) 9:1144–50. doi: 10.1038/nm915
112. Wysocki CA, Jiang Q, Panoskaltsis-Mortari A, Taylor PA, McKinnon KP, Su L, et al. Critical Role for CCR5 in the Function of Donor CD4+CD25+ Regulatory T Cells During Acute Graft-Versus-Host Disease. Blood (2005) 106:3300–7. doi: 10.1182/blood-2005-04-1632
113. Zhang P, Tey S-K, Koyama M, Kuns RD, Olver SD, Lineburg KE, et al. Induced Regulatory T Cells Promote Tolerance When Stabilized by Rapamycin and IL-2 In Vivo. J Immunol (2013) 191:5291–303. doi: 10.4049/jimmunol.1301181
114. Mutis T, van Rijn RS, Simonetti ER, Aarts-Riemens T, Emmelot ME, van Bloois L, et al. Human Regulatory T Cells Control Xenogeneic Graft-Versus-Host Disease Induced by Autologous T Cells in RAG2-/-Gammac-/- Immunodeficient Mice. Clin Cancer Res (2006) 12:5520–5. doi: 10.1158/1078-0432.CCR-06-0035
115. Hippen KL, Harker-Murray P, Porter SB, Merkel SC, Londer A, Taylor DK, et al. Umbilical Cord Blood Regulatory T-Cell Expansion and Functional Effects of Tumor Necrosis Factor Receptor Family Members OX40 and 4-1BB Expressed on Artificial Antigen-Presenting Cells. Blood (2008) 112:2847–57. doi: 10.1182/blood-2008-01-132951
116. Hippen KL, Merkel SC, Schirm DK, Sieben CM, Sumstad D, Kadidlo DM, et al. Massive Ex Vivo Expansion of Human Natural Regulatory T Cells (T(regs)) With Minimal Loss of In Vivo Functional Activity. Sci Transl Med (2011) 3:83ra41. doi: 10.1126/scitranslmed.3001809
117. Miura Y, Thoburn CJ, Bright EC, Phelps ML, Shin T, Matsui EC, et al. Association of Foxp3 Regulatory Gene Expression With Graft-Versus-Host Disease. Blood (2004) 104:2187–93. doi: 10.1182/blood-2004-03-1040
118. Jones SC, Murphy GF, Korngold R. Post-Hematopoietic Cell Transplantation Control of Graft-Versus-Host Disease by Donor CD425 T Cells to Allow an Effective Graft-Versus-Leukemia Response. Biol Blood Marrow Transplant (2003) 9:243–56. doi: 10.1053/bbmt.2003.50027
119. Trenado A, Charlotte F, Fisson S, Yagello M, Klatzmann D, Salomon BL, et al. Recipient-Type Specific CD4+CD25+ Regulatory T Cells Favor Immune Reconstitution and Control Graft-Versus-Host Disease While Maintaining Graft-Versus-Leukemia. J Clin Invest (2003) 112:1688–96. doi: 10.1172/JCI17702
120. Nguyen VH, Zeiser R, Dasilva DL, Chang DS, Beilhack A, Contag CH, et al. In Vivo Dynamics of Regulatory T-Cell Trafficking and Survival Predict Effective Strategies to Control Graft-Versus-Host Disease Following Allogeneic Transplantation. Blood (2007) 109:2649–56. doi: 10.1182/blood-2006-08-044529
121. Martelli MF, Di Ianni M, Ruggeri L, Falzetti F, Carotti A, Terenzi A, et al. HLA-Haploidentical Transplantation With Regulatory and Conventional T-Cell Adoptive Immunotherapy Prevents Acute Leukemia Relapse. Blood (2014) 124:638–44. doi: 10.1182/blood-2014-03-564401
122. Koenecke C, Czeloth N, Bubke A, Schmitz S, Kissenpfennig A, Malissen B, et al. Alloantigen-Specific De Novo-Induced Foxp3+ Treg Revert In Vivo and do Not Protect From Experimental GVHD. Eur J Immunol (2009) 39:3091–6. doi: 10.1002/eji.200939432
123. Beres A, Komorowski R, Mihara M, Drobyski WR. Instability of Foxp3 Expression Limits the Ability of Induced Regulatory T Cells to Mitigate Graft Versus Host Disease. Clin Cancer Res (2011) 17:3969–83. doi: 10.1158/1078-0432.CCR-10-3347
124. Shin H-J, Baker J, Leveson-Gower DB, Smith AT, Sega EI, Negrin RS. Rapamycin and IL-2 Reduce Lethal Acute Graft-Versus-Host Disease Associated With Increased Expansion of Donor Type CD4+CD25+Foxp3+ Regulatory T Cells. Blood (2011) 118:2342–50. doi: 10.1182/blood-2010-10-313684
125. MacMillan ML, Hippen KL, McKenna DH, Kadidlo D, Sumstad D, DeFor TE, et al. First-In-Human Phase 1 Trial of Induced Regulatory T Cells for Graft-Versus-Host Disease Prophylaxis in HLA-Matched Siblings. Blood Adv (2021) 5:1425–36. doi: 10.1182/bloodadvances.2020003219
126. Elias S, Rudensky AY. Therapeutic Use of Regulatory T Cells for Graft-Versus-Host Disease. Br J Haematol (2019) 187:25–38. doi: 10.1111/bjh.16157
127. Whangbo JS, Antin JH, Koreth J. The Role of Regulatory T Cells in Graft-Versus-Host Disease Management. Expert Rev Hematol (2020) 13:141–54. doi: 10.1080/17474086.2020.1709436
128. Guo W-W, Su X-H, Wang M-Y, Han M-Z, Feng X-M, Jiang E-L. Regulatory T Cells in GVHD Therapy. Front Immunol (2021) 12:697854. doi: 10.3389/fimmu.2021.697854
129. Sykes M, Romick ML, Sachs DH. Interleukin 2 Prevents Graft-Versus-Host Disease While Preserving the Graft-Versus-Leukemia Effect of Allogeneic T Cells. Proc Natl Acad Sci U.S.A. (1990) 87:5633–7. doi: 10.1073/pnas.87.15.5633
130. Sykes M, Szot GL, Nguyen PL, Pearson DA. Interleukin-12 Inhibits Murine Graft-Versus-Host Disease. Blood (1995) 86:2429–38. doi: 10.1182/blood.V86.6.2429.bloodjournal8662429
131. Brok HP, Heidt PJ, van der Meide PH, Zurcher C, Vossen JM. Interferon-Gamma Prevents Graft-Versus-Host Disease After Allogeneic Bone Marrow Transplantation in Mice. J Immunol (1993) 151:6451–9.
132. Yang YG, Dey BR, Sergio JJ, Pearson DA, Sykes M. Donor-Derived Interferon Gamma is Required for Inhibition of Acute Graft-Versus-Host Disease by Interleukin 12. J Clin Invest (1998) 102:2126–35. doi: 10.1172/JCI4992
133. Dickinson AM, Sviland L, Hamilton PJ, Usher P, Taylor P, Jackson G, et al. Cytokine Involvement in Predicting Clinical Graft-Versus-Host Disease in Allogeneic Bone Marrow Transplant Recipients. Bone Marrow Transplant (1994) 13:65–70.
134. Takatsuka H, Yamada S, Okamoto T, Fujimori Y, Wada H, Iwata N, et al. Predicting the Severity of Intestinal Graft-Versus-Host Disease From Leukotriene B4 Levels After Bone Marrow Transplantation. Bone Marrow Transplant (2000) 26:1313–6. doi: 10.1038/sj.bmt.1702712
135. Sun K, Hsiao H-H, Li M, Ames E, Bouchlaka M, Welniak LA, et al. IFN-γ Receptor-Deficient Donor T Cells Mediate Protection From Graft-Versus-Host Disease and Preserve Graft-Versus-Tumor Responses After Allogeneic Bone Marrow Transplantation. J Immunol (2012) 189:2033–42. doi: 10.4049/jimmunol.1102853
136. Mowat AM. Antibodies to IFN-Gamma Prevent Immunologically Mediated Intestinal Damage in Murine Graft-Versus-Host Reaction. Immunology (1989) 68:18–23.
137. Burman AC, Banovic T, Kuns RD, Clouston AD, Stanley AC, Morris ES, et al. IFNgamma Differentially Controls the Development of Idiopathic Pneumonia Syndrome and GVHD of the Gastrointestinal Tract. Blood (2007) 110:1064–72. doi: 10.1182/blood-2006-12-063982
138. Jasperson LK, Bucher C, Panoskaltsis-Mortari A, Mellor AL, Munn DH, Blazar BR. Inducing the Tryptophan Catabolic Pathway, Indoleamine 2,3-Dioxygenase (IDO), for Suppression of Graft-Versus-Host Disease (GVHD) Lethality. Blood (2009) 114:5062–70. doi: 10.1182/blood-2009-06-227587
139. Welniak LA, Blazar BR, Anver MR, Wiltrout RH, Murphy WJ. Opposing Roles of Interferon-Gamma on CD4+ T Cell-Mediated Graft-Versus-Host Disease: Effects of Conditioning. Biol Blood Marrow Transplant (2000) 6:604–12. doi: 10.1016/s1083-8791(00)70025-5
140. Lu Y, Waller EK. Dichotomous Role of Interferon-Gamma in Allogeneic Bone Marrow Transplant. Biol Blood Marrow Transplant (2009) 15:1347–53. doi: 10.1016/j.bbmt.2009.07.015
141. Yi T, Zhao D, Lin C-L, Zhang C, Chen Y, Todorov I, et al. Absence of Donor Th17 Leads to Augmented Th1 Differentiation and Exacerbated Acute Graft-Versus-Host Disease. Blood (2008) 112:2101–10. doi: 10.1182/blood-2007-12-126987
142. Kappel LW, Goldberg GL, King CG, Suh DY, Smith OM, Ligh C, et al. IL-17 Contributes to CD4-Mediated Graft-Versus-Host Disease. Blood (2009) 113:945–52. doi: 10.1182/blood-2008-08-172155
143. Carlson MJ, West ML, Coghill JM, Panoskaltsis-Mortari A, Blazar BR, Serody JS. In Vitro-Differentiated TH17 Cells Mediate Lethal Acute Graft-Versus-Host Disease With Severe Cutaneous and Pulmonary Pathologic Manifestations. Blood (2009) 113:1365–74. doi: 10.1182/blood-2008-06-162420
144. Banovic T, MacDonald KP, Morris ES, Rowe V, Kuns R, Don A, et al. TGF-Beta in Allogeneic Stem Cell Transplantation: Friend or Foe? Blood (2005) 106:2206–14. doi: 10.1182/blood-2005-01-0062
145. Chen X, Das R, Komorowski R, Beres A, Hessner MJ, Mihara M, et al. Blockade of Interleukin-6 Signaling Augments Regulatory T-Cell Reconstitution and Attenuates the Severity of Graft-Versus-Host Disease. Blood (2009) 114:891–900. doi: 10.1182/blood-2009-01-197178
146. Tawara I, Koyama M, Liu C, Toubai T, Thomas D, Evers R, et al. Interleukin-6 Modulates Graft-Versus-Host Responses After Experimental Allogeneic Bone Marrow Transplantation. Clin Cancer Res (2011) 17:77–88. doi: 10.1158/1078-0432.CCR-10-1198
147. Cooke KR, Hill GR, Crawford JM, Bungard D, Brinson YS, Delmonte J, et al. Tumor Necrosis Factor- Alpha Production to Lipopolysaccharide Stimulation by Donor Cells Predicts the Severity of Experimental Acute Graft-Versus-Host Disease. J Clin Invest (1998) 102:1882–91. doi: 10.1172/JCI4285
148. Levine JE. Implications of TNF-α in the Pathogenesis and Management of GVHD. Int J Hematol (2011) 93:571–7. doi: 10.1007/s12185-011-0803-1
149. Couriel D, Saliba R, Hicks K, Ippoliti C, de Lima M, Hosing C, et al. Tumor Necrosis Factor-Alpha Blockade for the Treatment of Acute GVHD. Blood (2004) 104:649–54. doi: 10.1182/blood-2003-12-4241
150. Bucher C, Koch L, Vogtenhuber C, Goren E, Munger M, Panoskaltsis-Mortari A, et al. IL-21 Blockade Reduces Graft-Versus-Host Disease Mortality by Supporting Inducible T Regulatory Cell Generation. Blood (2009) 114:5375–84. doi: 10.1182/blood-2009-05-221135
151. Hippen KL, Bucher C, Schirm DK, Bearl AM, Brender T, Mink KA, et al. Blocking IL-21 Signaling Ameliorates Xenogeneic GVHD Induced by Human Lymphocytes. Blood (2012) 119:619–28. doi: 10.1182/blood-2011-07-368027
152. Das R, Komorowski R, Hessner MJ, Subramanian H, Huettner CS, Cua D, et al. Blockade of Interleukin-23 Signaling Results in Targeted Protection of the Colon and Allows for Separation of Graft-Versus-Host and Graft-Versus-Leukemia Responses. Blood (2010) 115:5249–58. doi: 10.1182/blood-2009-11-255422
153. Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, et al. Essential Role of Stat6 in IL-4 Signalling. Nature (1996) 380:627–30. doi: 10.1038/380627a0
154. Nishikomori R, Usui T, Wu C-Y, Morinobu A, O’Shea JJ, Strober W. Activated STAT4 has an Essential Role in Th1 Differentiation and Proliferation That is Independent of its Role in the Maintenance of IL-12R Beta 2 Chain Expression and Signaling. J Immunol (2002) 169:4388–98. doi: 10.4049/jimmunol.169.8.4388
155. Ma H, Lu C, Ziegler J, Liu A, Sepulveda A, Okada H, et al. Absence of Stat1 in Donor CD4⁺ T Cells Promotes the Expansion of Tregs and Reduces Graft-Versus-Host Disease in Mice. J Clin Invest (2011) 121:2554–69. doi: 10.1172/JCI43706
156. Yu Y, Wang D, Kaosaard K, Liu C, Fu J, Haarberg K, et al. C-Rel is an Essential Transcription Factor for the Development of Acute Graft-Versus-Host Disease in Mice. Eur J Immunol (2013) 43:2327–37. doi: 10.1002/eji.201243282
157. Yu Y, Wang D, Liu C, Kaosaard K, Semple K, Anasetti C, et al. Prevention of GVHD While Sparing GVL Effect by Targeting Th1 and Th17 Transcription Factor T-Bet and Rorγt in Mice. Blood (2011) 118:5011–20. doi: 10.1182/blood-2011-03-340315
158. Iclozan C, Yu Y, Liu C, Liang Y, Yi T, Anasetti C, et al. T Helper17 Cells are Sufficient But Not Necessary to Induce Acute Graft-Versus-Host Disease. Biol Blood Marrow Transplant (2010) 16:170–8. doi: 10.1016/j.bbmt.2009.09.023
159. Fulton LM, Carlson MJ, Coghill JM, Ott LE, West ML, Panoskaltsis-Mortari A, et al. Attenuation of Acute Graft-Versus-Host Disease in the Absence of the Transcription Factor Rorγt. J Immunol (2012) 189:1765–72. doi: 10.4049/jimmunol.1200858
160. Laurence A, Amarnath S, Mariotti J, Kim YC, Foley J, Eckhaus M, et al. STAT3 Transcription Factor Promotes Instability of Ntreg Cells and Limits Generation of Itreg Cells During Acute Murine Graft-Versus-Host Disease. Immunity (2012) 37:209–22. doi: 10.1016/j.immuni.2012.05.027
161. Vaeth M, Bäuerlein CA, Pusch T, Findeis J, Chopra M, Mottok A, et al. Selective NFAT Targeting in T Cells Ameliorates GvHD While Maintaining Antitumor Activity. Proc Natl Acad Sci USA (2015) 112:1125–30. doi: 10.1073/pnas.1409290112
162. Singh K, Stempora L, Harvey RD, Kirk AD, Larsen CP, Blazar BR, et al. Superiority of Rapamycin Over Tacrolimus in Preserving Nonhuman Primate Treg Half-Life and Phenotype After Adoptive Transfer. Am J Transplant (2014) 14:2691–703. doi: 10.1111/ajt.12934
163. Zeiser R, Nguyen VH, Beilhack A, Buess M, Schulz S, Baker J, et al. Inhibition of CD4+CD25+ Regulatory T-Cell Function by Calcineurin-Dependent Interleukin-2 Production. Blood (2006) 108:390–9. doi: 10.1182/blood-2006-01-0329
164. Cutler C, Logan B, Nakamura R, Johnston L, Choi S, Porter D, et al. Tacrolimus/sirolimus vs Tacrolimus/Methotrexate as GVHD Prophylaxis After Matched, Related Donor Allogeneic HCT. Blood (2014) 124:1372–7. doi: 10.1182/blood-2014-04-567164
165. Pidala J, Kim J, Jim H, Kharfan-Dabaja MA, Nishihori T, Fernandez HF, et al. A Randomized Phase II Study to Evaluate Tacrolimus in Combination With Sirolimus or Methotrexate After Allogeneic Hematopoietic Cell Transplantation. Haematologica (2012) 97:1882–9. doi: 10.3324/haematol.2012.067140
166. Pidala J, Walton K, Elmariah H, Kim J, Mishra A, Bejanyan N, et al. Pacritinib Combined With Sirolimus and Low-Dose Tacrolimus for GVHD Prevention After Allogeneic Hematopoietic Cell Transplantation: Preclinical and Phase I Trial Results. Clin Cancer Res (2021) 27:2712–22. doi: 10.1158/1078-0432.CCR-20-4725
167. Pidala J, Kim J, Alsina M, Ayala E, Betts BC, Fernandez HF, et al. Prolonged Sirolimus Administration After Allogeneic Hematopoietic Cell Transplantation is Associated With Decreased Risk for Moderate-Severe Chronic Graft-Versus-Host Disease. Haematologica (2015) 100:970–7. doi: 10.3324/haematol.2015.123588
168. Gooptu M, Kim HT, Howard A, Choi SW, Soiffer RJ, Antin JH, et al. Effect of Sirolimus on Immune Reconstitution Following Myeloablative Allogeneic Stem Cell Transplantation: An Ancillary Analysis of a Randomized Controlled Trial Comparing Tacrolimus/Sirolimus and Tacrolimus/Methotrexate (Blood and Marrow Transplant Clinical Trials Network/BMT CTN 0402). Biol Blood Marrow Transplant (2019) 25:2143–51. doi: 10.1016/j.bbmt.2019.06.029
169. Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu M-F, Liu H, et al. Ultra Low-Dose IL-2 for GVHD Prophylaxis After Allogeneic Hematopoietic Stem Cell Transplantation Mediates Expansion of Regulatory T Cells Without Diminishing Antiviral and Antileukemic Activity. Clin Cancer Res (2014) 20:2215–25. doi: 10.1158/1078-0432.CCR-13-3205
170. Betts BC, Pidala J, Kim J, Mishra A, Nishihori T, Perez L, et al. IL-2 Promotes Early Treg Reconstitution After Allogeneic Hematopoietic Cell Transplantation. Haematologica (2017) 102:948–57. doi: 10.3324/haematol.2016.153072
171. Long J, Chang L, Shen Y, Gao W-H, Wu Y-N, Dou H-B, et al. Valproic Acid Ameliorates Graft-Versus-Host Disease by Downregulating Th1 and Th17 Cells. J Immunol (2015) 195:1849–57. doi: 10.4049/jimmunol.1500578
172. Daenthanasanmak A, Iamsawat S, Chakraborty P, Nguyen HD, Bastian D, Liu C, et al. Targeting Sirt-1 Controls GVHD by Inhibiting T-Cell Allo-Response and Promoting Treg Stability in Mice. Blood (2019) 133:266–79. doi: 10.1182/blood-2018-07-863233
173. Leng C, Li C, Ziegler J, Lokshin A, Lentzsch S, Mapara MY. Inhibition of Graft-Versus-Host Disease (GVHD) by Histone Deacetylase Inhibitor (HDAC) SAHA Leads to Downregulation of STAT1 Activation. Blood (2005) 106:1311. doi: 10.1182/blood.V106.11.1311.1311
174. Li N, Zhang C, Lin C-L, McCulloch B, Wright J, Kirschbaum M, et al. A Radiation-Free Immune and Epigenetics Based Conditioning Regimen for Allogeneic HCT: Combination of Anti-CD3 and Romidepsin. Blood (2006) 108:3204. doi: 10.1182/blood.V108.11.3204.3204
175. Cleophas MC, Crişan TO, Klück V, Hoogerbrugge N, Netea-Maier RT, Dinarello CA, et al. Romidepsin Suppresses Monosodium Urate Crystal-Induced Cytokine Production Through Upregulation of Suppressor of Cytokine Signaling 1 Expression. Arthritis Res Ther (2019) 21:50. doi: 10.1186/s13075-019-1834-x
176. Choi J, Ziga ED, Ritchey J, Collins L, Prior JL, Cooper ML, et al. Ifnγr Signaling Mediates Alloreactive T-Cell Trafficking and GVHD. Blood (2012) 120:4093–103. doi: 10.1182/blood-2012-01-403196
177. Choi J, Cooper ML, Alahmari B, Ritchey J, Collins L, Holt M, et al. Pharmacologic Blockade of JAK1/JAK2 Reduces GvHD and Preserves the Graft-Versus-Leukemia Effect. PloS One (2014) 9:e109799. doi: 10.1371/journal.pone.0109799
178. Juvekar A, Ruggeri B, Condon S, Borkowski A, Huber R, Smith P. Itacitinib, a JAK1 Selective Inhibitor Preserves Graft-Versus-Leukemia (GVL), Enhances Survival and Is Highly Efficacious in a MHC-Mismatched Mouse Model of Acute GvHD. Blood (2018) 132:4522. doi: 10.1182/blood-2018-99-112144
179. Courtois J, Ritacco C, Dubois S, Canti L, Vandenhove B, Seidel L, et al. Itacitinib Prevents Xenogeneic GVHD in Humanized Mice. Bone Marrow Transplant (2021) 56:2672–81. doi: 10.1038/s41409-021-01363-1
180. Covington M, He X, Scuron M, Li J, Collins R, Juvekar A, et al. Preclinical Characterization of Itacitinib (INCB039110), a Novel Selective Inhibitor of JAK1, for the Treatment of Inflammatory Diseases. Eur J Pharmacol (2020) 885:173505. doi: 10.1016/j.ejphar.2020.173505
181. Betts BC, Bastian D, Iamsawat S, Nguyen H, Heinrichs JL, Wu Y, et al. Targeting JAK2 Reduces GVHD and Xenograft Rejection Through Regulation of T Cell Differentiation. Proc Natl Acad Sci U.S.A. (2018) 115:1582–7. doi: 10.1073/pnas.1712452115
182. Betts BC, Abdel-Wahab O, Curran SA, St Angelo ET, Koppikar P, Heller G, et al. Janus Kinase-2 Inhibition Induces Durable Tolerance to Alloantigen by Human Dendritic Cell-Stimulated T Cells Yet Preserves Immunity to Recall Antigen. Blood (2011) 118:5330–9. doi: 10.1182/blood-2011-06-363408
183. Park H-B, Oh K, Garmaa N, Seo MW, Byoun O-J, Lee H-Y, et al. CP-690550, a Janus Kinase Inhibitor, Suppresses CD4+ T-Cell-Mediated Acute Graft-Versus-Host Disease by Inhibiting the Interferon-γ Pathway. Transplantation (2010) 90:825–35. doi: 10.1097/TP.0b013e3181f24e59
184. Iyengar S, Zhan C, Lu J, Korngold R, Schwartz DH. Treatment With a Rho Kinase Inhibitor Improves Survival From Graft-Versus-Host Disease in Mice After MHC-Haploidentical Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant (2014) 20:1104–11. doi: 10.1016/j.bbmt.2014.04.029
185. Flynn R, Paz K, Du J, Reichenbach DK, Taylor PA, Panoskaltsis-Mortari A, et al. Targeted Rho-Associated Kinase 2 Inhibition Suppresses Murine and Human Chronic GVHD Through a Stat3-Dependent Mechanism. Blood (2016) 127:2144–54. doi: 10.1182/blood-2015-10-678706
186. Kondo T, Ikegawa S, Fukumi T, Sumii Y, Sugiura H, Sando Y, et al. Pretransplant Short-Term Exposure of Donor Graft Cells to ITK Selective Inhibitor Ameliorates Acute Graft-Versus-Host Disease by Inhibiting Effector T Cell Differentiation While Sparing Regulatory T Cells. Immunohorizons (2021) 5:424–37. doi: 10.4049/immunohorizons.2100042
187. Klamer G, Shen S, Song E, Rice AM, Knight R, Lindeman R, et al. GSK3 Inhibition Prevents Lethal GVHD in Mice. Exp Hematol (2013) 41:39–55.e10. doi: 10.1016/j.exphem.2012.09.005
188. Kong D, Park EJ, Stephen AG, Calvani M, Cardellina JH, Monks A, et al. Echinomycin, a Small-Molecule Inhibitor of Hypoxia-Inducible Factor-1 DNA-Binding Activity. Cancer Res (2005) 65:9047–55. doi: 10.1158/0008-5472.CAN-05-1235
189. Shono Y, Tuckett AZ, Ouk S, Liou H-C, Altan-Bonnet G, Tsai JJ, et al. A Small-Molecule C-Rel Inhibitor Reduces Alloactivation of T Cells Without Compromising Antitumor Activity. Cancer Discov (2014) 4:578–91. doi: 10.1158/2159-8290.CD-13-0585
190. Shono Y, Tuckett AZ, Liou H-C, Doubrovina E, Derenzini E, Ouk S, et al. Characterization of a C-Rel Inhibitor That Mediates Anticancer Properties in Hematologic Malignancies by Blocking NF-κb-Controlled Oxidative Stress Responses. Cancer Res (2016) 76:377–89. doi: 10.1158/0008-5472.CAN-14-2814
191. Park M-J, Moon S-J, Lee S-H, Kim E-K, Yang E-J, Min J-K, et al. Blocking Activator Protein 1 Activity in Donor Cells Reduces Severity of Acute Graft-Versus-Host Disease Through Reciprocal Regulation of IL-17-Producing T Cells/Regulatory T Cells. Biol Blood Marrow Transplant (2014) 20:1112–20. doi: 10.1016/j.bbmt.2014.04.031
192. Betts BC, Veerapathran A, Pidala J, Yu X-Z, Anasetti C. STAT5 Polarization Promotes Itregs and Suppresses Human T-Cell Alloresponses While Preserving CTL Capacity. J Leukoc Biol (2014) 95:205–13. doi: 10.1189/jlb.0313154
193. Betts BC, Sagatys EM, Veerapathran A, Lloyd MC, Beato F, Lawrence HR, et al. CD4+ T Cell STAT3 Phosphorylation Precedes Acute GVHD, and Subsequent Th17 Tissue Invasion Correlates With GVHD Severity and Therapeutic Response. J Leukoc Biol (2015) 97:807–19. doi: 10.1189/jlb.5A1114-532RR
194. Walton K, Fernandez MR, Sagatys EM, Reff J, Kim J, Lee MC, et al. Metabolic Reprogramming Augments Potency of Human Pstat3-Inhibited Itregs to Suppress Alloreactivity. JCI Insight (2020) 5(9):e136437. doi: 10.1172/jci.insight.136437
195. Jia H, Cui J, Jia X, Zhao J, Feng Y, Zhao P, et al. Therapeutic Effects of STAT3 Inhibition by Nifuroxazide on Murine Acute Graft Graft-vs.-Host Disease: Old Drug, New Use. Mol Med Rep (2017) 16:9480–6. doi: 10.3892/mmr.2017.7825
196. Haring E, Uhl FM, Andrieux G, Proietti M, Bulashevska A, Sauer B, et al. Bile Acids Regulate Intestinal Antigen Presentation and Reduce Graft-Versus-Host Disease Without Impairing the Graft-Versus-Leukemia Effect. Haematologica (2021) 106:2131–46. doi: 10.3324/haematol.2019.242990
197. Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, et al. Bile Acid Metabolites Control TH17 and Treg Cell Differentiation. Nature (2019) 576:143–8. doi: 10.1038/s41586-019-1785-z
198. Zeiser R, von Bubnoff N, Butler J, Mohty M, Niederwieser D, Or R, et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-Versus-Host Disease. N Engl J Med (2020) 382:1800–10. doi: 10.1056/NEJMoa1917635
199. Jagasia M, Perales M-A, Schroeder MA, Ali H, Shah NN, Chen Y-B, et al. Ruxolitinib for the Treatment of Steroid-Refractory Acute GVHD (REACH1): A Multicenter, Open-Label Phase 2 Trial. Blood (2020) 135:1739–49. doi: 10.1182/blood.2020004823
200. Jagasia M, Zeiser R, Arbushites M, Delaite P, Gadbaw B. Bubnoff N Von. Ruxolitinib for the Treatment of Patients With Steroid-Refractory GVHD: An Introduction to the REACH Trials. Immunotherapy (2018) 10:391–402. doi: 10.2217/imt-2017-0156
201. Schroeder MA, Khoury HJ, Jagasia M, Ali H, Schiller GJ, Staser K, et al. A Phase 1 Trial of Itacitinib, a Selective JAK1 Inhibitor, in Patients With Acute Graft-Versus-Host Disease. Blood Adv (2020) 4:1656–69. doi: 10.1182/bloodadvances.2019001043
202. Alvarez-Breckenridge CA, Yu J, Price R, Wei M, Wang Y, Nowicki MO, et al. The Histone Deacetylase Inhibitor Valproic Acid Lessens NK Cell Action Against Oncolytic Virus-Infected Glioblastoma Cells by Inhibition of STAT5/T-BET Signaling and Generation of Gamma Interferon. J Virol (2012) 86:4566–77. doi: 10.1128/JVI.05545-11
203. Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The Kinase mTOR Regulates the Differentiation of Helper T Cells Through the Selective Activation of Signaling by Mtorc1 and Mtorc2. Nat Immunol (2011) 12:295–303. doi: 10.1038/ni.2005
204. Merkenschlager M. Boehmer H Von. PI3 Kinase Signalling Blocks Foxp3 Expression by Sequestering Foxo Factors. J Exp Med (2010) 207:1347–50. doi: 10.1084/jem.20101156
205. Abdullah L, Hills LB, Winter EB, Huang YH. Diverse Roles of Akt in T Cells. Immunometabolism (2021) 3(1):e210007. doi: 10.20900/immunometab20210007
206. Zhang R, Chen H-Z, Liu J-J, Jia Y-Y, Zhang Z-Q, Yang R-F, et al. SIRT1 Suppresses Activator Protein-1 Transcriptional Activity and Cyclooxygenase-2 Expression in Macrophages. J Biol Chem (2010) 285:7097–110. doi: 10.1074/jbc.M109.038604
207. Li J, Qu X, Ricardo SD, Bertram JF, Nikolic-Paterson DJ. Resveratrol Inhibits Renal Fibrosis in the Obstructed Kidney: Potential Role in Deacetylation of Smad3. Am J Pathol (2010) 177:1065–71. doi: 10.2353/ajpath.2010.090923
208. Huang H, Tindall DJ. Dynamic FoxO Transcription Factors. J Cell Sci (2007) 120:2479–87. doi: 10.1242/jcs.001222
209. Giannakou ME, Partridge L. The Interaction Between FOXO and SIRT1: Tipping the Balance Towards Survival. Trends Cell Biol (2004) 14:408–12. doi: 10.1016/j.tcb.2004.07.006
210. Kwon H-S, Lim HW, Wu J, Schnölzer M, Verdin E, Ott M. Three Novel Acetylation Sites in the Foxp3 Transcription Factor Regulate the Suppressive Activity of Regulatory T Cells. J Immunol (2012) 188:2712–21. doi: 10.4049/jimmunol.1100903
211. Beier UH, Wang L, Han R, Akimova T, Liu Y, Hancock WW. Histone Deacetylases 6 and 9 and Sirtuin-1 Control Foxp3+ Regulatory T Cell Function Through Shared and Isoform-Specific Mechanisms. Sci Signal (2012) 5:ra45. doi: 10.1126/scisignal.2002873
212. Ye Q, Zhang M, Wang Y, Fu S, Han S, Wang L, et al. Sirtinol Regulates the Balance of Th17/Treg to Prevent Allograft Rejection. Cell Biosci (2017) 7:55. doi: 10.1186/s13578-017-0182-2
213. Glauben R, Sonnenberg E, Wetzel M, Mascagni P, Siegmund B. Histone Deacetylase Inhibitors Modulate Interleukin 6-Dependent CD4+ T Cell Polarization In Vitro and In Vivo. J Biol Chem (2014) 289:6142–51. doi: 10.1074/jbc.M113.517599
214. Leoni F, Fossati G, Lewis EC, Lee J-K, Porro G, Pagani P, et al. The Histone Deacetylase Inhibitor ITF2357 Reduces Production of Pro-Inflammatory Cytokines In Vitro and Systemic Inflammation In Vivo. Mol Med (2005) 11:1–15. doi: 10.2119/2006-00005.Dinarello
215. Reddy P, Sun Y, Toubai T, Duran-Struuck R, Clouthier SG, Weisiger E, et al. Histone Deacetylase Inhibition Modulates Indoleamine 2,3-Dioxygenase-Dependent DC Functions and Regulates Experimental Graft-Versus-Host Disease in Mice. J Clin Invest (2008) 118:2562–73. doi: 10.1172/JCI34712
216. Choi S, Reddy P. HDAC Inhibition and Graft Versus Host Disease. Mol Med (2011) 17:404–16. doi: 10.2119/molmed.2011.00007
217. Xu X, Li X, Zhao Y, Huang H. Immunomodulatory Effects of Histone Deacetylation Inhibitors in Graft-Vs.-Host Disease After Allogeneic Stem Cell Transplantation. Front Immunol (2021) 12:641910. doi: 10.3389/fimmu.2021.641910
218. Sugerman PB, Faber SB, Willis LM, Petrovic A, Murphy GF, Pappo J, et al. Kinetics of Gene Expression in Murine Cutaneous Graft-Versus-Host Disease. Am J Pathol (2004) 164:2189–202. doi: 10.1016/S0002-9440(10)63776-5
219. Leng C, Gries M, Ziegler J, Lokshin A, Mascagni P, Lentzsch S, et al. Reduction of Graft-Versus-Host Disease by Histone Deacetylase Inhibitor Suberonylanilide Hydroxamic Acid Is Associated With Modulation of Inflammatory Cytokine Milieu and Involves Inhibition of STAT1. Exp Hematol (2006) 34:776–87. doi: 10.1016/j.exphem.2006.02.014
220. Ma H-H, Ziegler J, Li C, Sepulveda A, Bedeir A, Grandis J, et al. Sequential Activation of Inflammatory Signaling Pathways During Graft-Versus-Host Disease (GVHD): Early Role for STAT1 and STAT3. Cell Immunol (2011) 268:37–46. doi: 10.1016/j.cellimm.2011.01.008
221. Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical Characterization of the Selective JAK1/2 Inhibitor INCB018424: Therapeutic Implications for the Treatment of Myeloproliferative Neoplasms. Blood (2010) 115:3109–17. doi: 10.1182/blood-2009-04-214957
222. Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and Efficacy of INCB018424, a JAK1 and JAK2 Inhibitor, in Myelofibrosis. N Engl J Med (2010) 363:1117–27. doi: 10.1056/NEJMoa1002028
223. Spoerl S, Mathew NR, Bscheider M, Schmitt-Graeff A, Chen S, Mueller T, et al. Activity of Therapeutic JAK 1/2 Blockade in Graft-Versus-Host Disease. Blood (2014) 123:3832–42. doi: 10.1182/blood-2013-12-543736
224. Assal A, Mapara MY. Janus Kinase Inhibitors and Cell Therapy. Front Immunol (2021) 12:740847. doi: 10.3389/fimmu.2021.740847
225. Magenau J, Reddy P. Next Generation Treatment of Acute Graft-Versus-Host Disease. Leukemia (2014) 28:2283–91. doi: 10.1038/leu.2014.195
226. Mammadli M, Huang W, Harris R, Sultana A, Cheng Y, Tong W, et al. Targeting Interleukin-2-Inducible T-Cell Kinase (ITK) Differentiates GVL and GVHD in Allo-HSCT. Front Immunol (2020) 11:593863. doi: 10.3389/fimmu.2020.593863
227. Jagasia M, Lazaryan A, Bachier CR, Salhotra A, Weisdorf DJ, Zoghi B, et al. ROCK2 Inhibition With Belumosudil (KD025) for the Treatment of Chronic Graft-Versus-Host Disease. J Clin Oncol (2021) 39:1888–98. doi: 10.1200/JCO.20.02754
228. Park M-J, Lee SH, Lee S-H, Kim E-K, Lee EJ, Moon Y-M, et al. GRIM19 Ameliorates Acute Graft-Versus-Host Disease (GVHD) by Modulating Th17 and Treg Cell Balance Through Down-Regulation of STAT3 and NF-AT Activation. J Transl Med (2016) 14:206. doi: 10.1186/s12967-016-0963-0
229. Yao Y, Wang L, Zhou J, Zhang X. HIF-1α Inhibitor Echinomycin Reduces Acute Graft-Versus-Host Disease and Preserves Graft-Versus-Leukemia Effect. J Transl Med (2017) 15:28. doi: 10.1186/s12967-017-1132-9
230. Choi SW, Braun T, Henig I, Gatza E, Magenau J, Parkin B, et al. Vorinostat Plus Tacrolimus/Methotrexate to Prevent GVHD After Myeloablative Conditioning, Unrelated Donor HCT. Blood (2017) 130:1760–7. doi: 10.1182/blood-2017-06-790469
231. Perez L, Fernandez H, Kharfan-Dabaja M, Khimani F, Betts B, Mishra A, et al. A Phase 2 Trial of the Histone Deacetylase Inhibitor Panobinostat for Graft-Versus-Host Disease Prevention. Blood Adv (2021) 5:2740–50. doi: 10.1182/bloodadvances.2021004225
232. Foss F, Coiffier B, Horwitz S, Pro B, Prince HM, Sokol L, et al. Tolerability to Romidepsin in Patients With Relapsed/Refractory T-Cell Lymphoma. Biomark Res (2014) 2:16. doi: 10.1186/2050-7771-2-16
233. Gauthier N, Soret-Dulphy J, Zhao L-P, Daltro De Oliveira R, Marcault C, Parquet N, et al. Ruxolitinib Treatment Is Associated With Increased Incidence of Infections and Higher Risk of HSV/Vzv Recurrence in Patients With Myeloproliferative Neoplasm (MPN) Related Myelofibrosis (Mf). Blood (2020) 136:8. doi: 10.1182/blood-2020-141260
234. Sadjadian P, Wille K, Griesshammer M. Ruxolitinib-Associated Infections in Polycythemia Vera: Review of the Literature, Clinical Significance, and Recommendations. Cancers (Basel) (2020) 12(11):3132. doi: 10.3390/cancers12113132
Keywords: aGVHD, GvL, CD4+ T cells, T helper cell differentiation, transcription factors
Citation: Campe J and Ullrich E (2022) T Helper Cell Lineage-Defining Transcription Factors: Potent Targets for Specific GVHD Therapy? Front. Immunol. 12:806529. doi: 10.3389/fimmu.2021.806529
Received: 31 October 2021; Accepted: 14 December 2021;
Published: 05 January 2022.
Edited by:
Christian Koenecke, Hannover Medical School, GermanyReviewed by:
Robert Zeiser, University of Freiburg, GermanyMauro Di Ianni, University of Studies G. d’Annunzio Chieti and Pescara, Italy
Copyright © 2022 Campe and Ullrich. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Evelyn Ullrich, RXZlbHluLlVsbHJpY2hAa2d1LmRl