 Kanako Mitsui-Sekinaka
Kanako Mitsui-Sekinaka Yujin Sekinaka
Yujin Sekinaka Akifumi Endo
Akifumi Endo Kohsuke Imai
Kohsuke Imai Shigeaki Nonoyama
Shigeaki Nonoyama- 1Department of Pediatrics, National Defense Medical College, Saitama, Japan
- 2Department of Pediatrics and Clinical Research Center, Tokyo Medical and Dental University, Tokyo, Japan
- 3Department of Community Pediatrics, Perinatal and Maternal Medicine, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo, Japan
The Primary Immunodeficiency Database in Japan (PIDJ) is a registry of primary immunodeficiency diseases (PIDs) that was established in 2007. The database is a joint research project with research groups associated with the Ministry of Health, Labor and Welfare; the RIKEN Research Center for Allergy and Immunology (RCAI); and the Kazusa DNA Research Institute (KDRI). The PIDJ contains patient details, including the age, sex, clinical and laboratory findings, types of infections, genetic analysis results, and treatments administered. In addition, web-based case consultation is also provided. The PIDJ serves as a database for patients with PIDs and as a patient consultation service connecting general physicians with PID specialists and specialized hospitals. Thus, the database contributes to investigations related to disease pathogenesis and the early diagnosis and treatment of patients with PIDs. In the 9 years since the launch of PIDJ, 4,481 patients have been enrolled, of whom 64% have been subjected to genetic analysis. In 2017, the Japanese Society for Immunodeficiency and Autoinflammatory Diseases (JSIAD) was established to advance the diagnosis, treatment, and research in the field of PIDs and autoinflammatory diseases (AIDs). JSIAD promotes the analysis of the pathogenesis of PIDs and AIDs, enabling improved patient care and networking via the expansion of the database and construction of a biobank obtained from the PIDJ. The PIDJ was upgraded to “PIDJ ver.2” in 2019 by JSIAD. Currently, PIDJ ver.2 is used as a platform for epidemiological studies, genetic analysis, and pathogenesis evaluation for PIDs and AIDs.
Introduction
Primary immunodeficiency diseases (PIDs) are rare and genetically heterogeneous disorders that impair the immune system. Recent studies have indicated the prevalence of >400 causative genes, and genetic analysis plays an important role in confirming the PID diagnosis and the selection of treatment options, which include the use of hematopoietic stem cell transplantations, gene therapy, and biological agents. Because PIDs are rare diseases, the compilation of patient details, including genetic analysis and clinical information, can contribute significantly toward evaluating the pathogenesis involved and the establishment of optimal treatment methods. Given this background, a registry of patients with primary immunodeficiency (PIDJ: Primary Immunodeficiency Database in Japan) was established in 2007, enabling an overview of Japanese patients with PIDs.
Before the PIDJ Project
Before the launch of the PIDJ project, Japan had a retrospective, paper-based registration system. The first nationwide survey related to PIDs in Japan was performed in 1979, which was supported by the Japan Ministry of Health, in which 497 patients were registered (1). During registration, the survey included the numbers of each type of PIDs, patient age at the time of diagnosis, patient status at the time of registration, familial incidence of PIDs, and any associated complications. However, molecular analysis and sample stocking were not performed in that survey. When the causative gene associated with each patient with PIDs was identified, it was registered in each institution’s private database, as a nationwide database had not been established.
The PIDJ Project (2007~2017)
PIDs are rare diseases, with low numbers of patients but highly variable symptoms, severity, and complications, making diagnosis by non-specialists difficult. The disease can be fatal if the diagnosis is delayed or if no appropriate therapeutic intervention is performed; therefore, consultations with PID expert doctors are required. Therefore, in 2007, we launched a website for PIDJ at the RIKEN Research Center for Allergy and Immunology (RCAI) to facilitate consultations for PID patients with local physicians or expert doctors from 13 universities across Japan. In the PIDJ network, general physicians evaluating potential PID patients consult expert doctors with experience in PID diagnosis and register for PIDJ with informed consent from the patients. The clinical information of the patients is added via the internet, and PID experts advise general physicians consulting with the patient. Where further analyses are required, patients’ samples are sent to RCAI or PID expert doctors to perform immunological analysis, including FACS and genetic and functional evaluations, and patient samples are preserved for future use. Genetic analysis is performed at the Kazusa DNA Research Institute (KDRI). The important goals are to enable accurate diagnosis, recommend appropriate treatments, and provide support to connect PID patients to specialized medical institutions for prompt and appropriate medical care. Thus, consultation, registration, sample stocking, molecular diagnosis, functional analysis, and advice from PID expert doctors can easily be achieved through the PIDJ network.
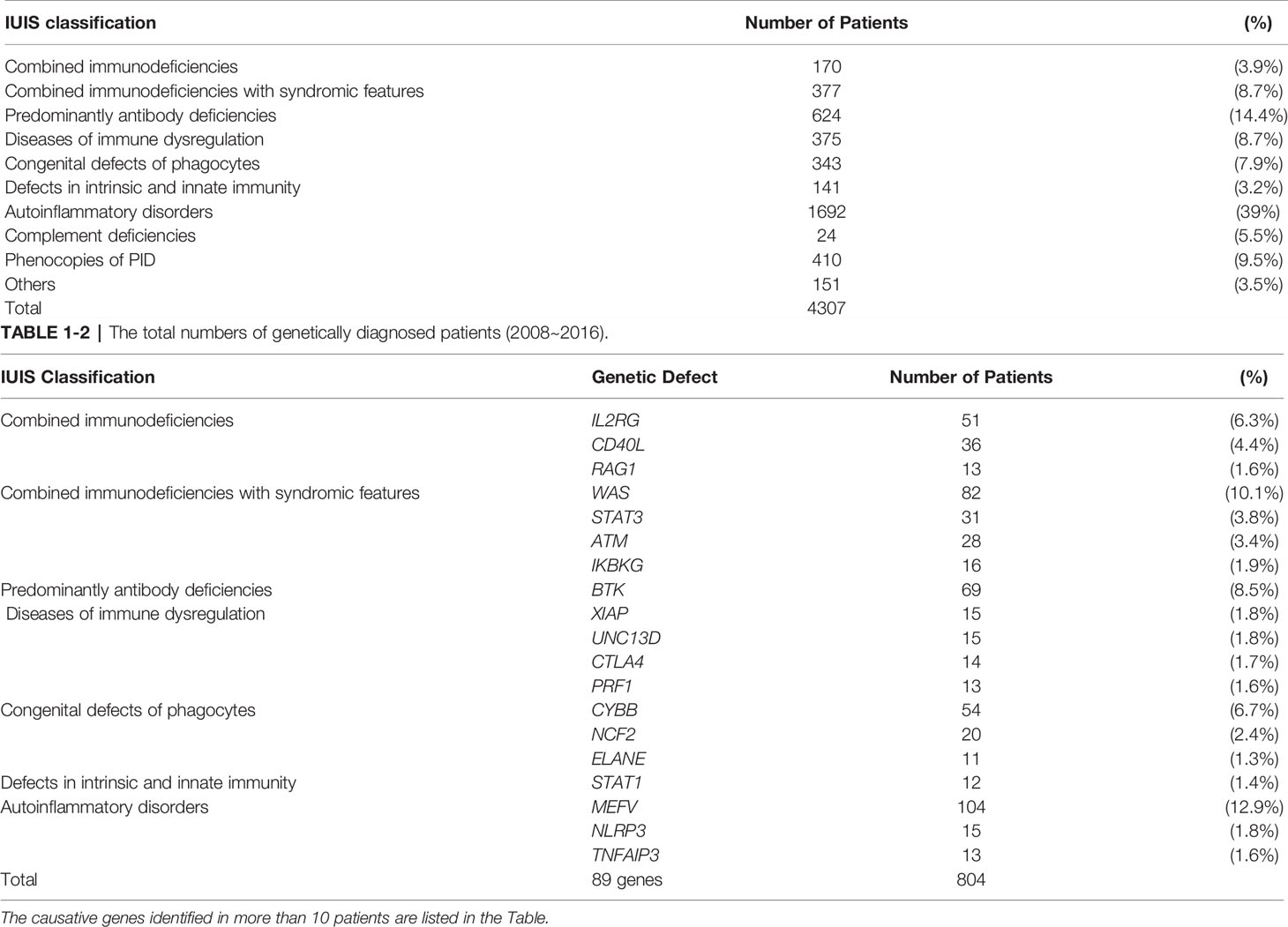
In the 9 years (from 2007 to 2017) since the launch of PIDJ, 4,481 patients have been registered. Genetic analysis has been performed in 2,869 patients (64% of all registered patients), and the causative gene was identified in 804 cases. The most common diagnosis was autoinflammatory disorders (39%), followed by predominantly antibody deficiencies (14.4%), combined immunodeficiencies with syndromic features (8.7%), diseases of immune dysregulation (8.7%), congenital defects of phagocytes (7.9%), and combined immunodeficiencies (3.9%) (Table 1).
Moreover, PIDJ has contributed to the identification of novel causative genes associated with PIDs. For patients in whom no mutations in known PID-causing genes are detected, cases with similar clinical symptoms and laboratory findings are selected for detailed analysis to identify novel causative genes. We have identified several causative genes of PIDs by using PIDJ database (2–13).
PIDJ ver.2, Now and Beyond
In 2017, the Japanese Society for Immunodeficiency and Autoinflammatory Diseases (JSIAD) was established to advance the diagnosis, treatment, and research in the field of PIDs and autoinflammatory diseases (AIDs). The PIDJ was upgraded to “PIDJ ver.2” in 2019 by JSIAD. With the expansion of the database and construction of a biobank, PIDJ ver.2 is being used as a platform for epidemiological studies, genetic analysis, and pathogenesis evaluation for PIDs and AIDs. Consultation, registration, sample stocking, molecular diagnosis, functional analysis, and advice from PID expert doctors are being provided using PIDJ ver.2. The PIDJ committees are organized in JSIAD to respond to consultations from general physicians. Genetic analysis (by NGS) and sample stocking are performed at the KDRI. The JSIAD collaborating facilities (81 facilities as of June 2020) participate in PIDJ ver.2 as joint research facilities and play a role in the diagnosis and treatment of PIDs and AIDs, as well as research and education about the topic. Further, on the basis of the IUIS classification (14, 15), JSIAD has developed a panel of genes recommended for genetic testing for each PID and AID related to clinical usefulness and validity (Table 2).

Table 2 Target genes responsible for PIDs and AIDs.
Discussion
PIDs include over 400 diseases caused by mutations in single genes, which makes them difficult to diagnose by general physicians. Because PIDs are rare diseases, patient registration is important for the diagnosis, genetic and functional analysis, and improved patient care. PIDJ was initially associated with 13 medical colleges, the RCAI, and KDRI, back in 2007. PIDJ is a nationwide network that includes patient consultations, registration, sample stocking, genetic diagnosis, molecular analysis, and advice by PID expert doctors.
A total of 4,481 patients have been registered in PIDJ, and genetic analysis has been performed in 2,869 patients (64% of all registered patients), and the causative gene was identified in 804 cases. Comparing our results with reports from registries in other countries (e.g., Europe, the United States, the Middle East, and Asia), PIDJ is characterized by a large number of registered AID patients (especially with MEFV mutations) (16–28). On the other hand, with regard to PIDs, adult cases that had already been diagnosed and cases that have been followed only with γ-globulin administration may not have been registered in PIDJ. This is the limitation of this registration, and future efforts should be made to ensure that all PID patients in Japan will be registered in PIDJ. We are conducting a survey of all PIDs patients in Japan to identify missing cases in the registration and are working on registering all the PIDs patients for PIDJ (29, 30).
PIDJ was upgraded to PIDJ ver.2 with the establishment of JSIAD in 2017 and has been expanded further, with additional functions to date. The PIDJ network will help doctors and researchers perform various analyses, improve disease prognosis, and advance understanding of the human immune system.
Author Contributions
KM-S did the conception and design. KM-S, KI, and AE analyzed the data. KM-S and KI wrote the manuscript. SN, YS, and KI provided critical discussion, supervised the study, and edited the manuscript. All authors reviewed the paper. All authors contributed to the article and approved the submitted version.
Funding
This work was supported in part by the Ministry of Health, Labour and Welfare, Japan (20316700, 20317089), Japan Society for the Promotion of Science (JSPS) KAKENHI (20K16942, 20K22916), the Ministry of Education, Culture, Sports, Technology, Japan (MEXT) KAKENHI (20288564), the Practical Research for Rare/Intractable Diseases from AMED (20314514, 20314940), and the Project for Baby and Infant Research of Health and Development to Adolescent and Young Adult from AMED (19188706).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all the patients and their guardians for participating for PIDJ. We also thank all physicians and researchers involved in PID patients’ care.
References
1. Hayakawa H, Iwata T, Yata J, Kobayashi N. Primary Immunodeficiency Syndrome in Japan. I. Overview of a Nationwide Survey on Primary Immunodeficiency Syndrome. J Clin Immunol (1981) 1(1):31–9. doi: 10.1007/BF00915474
2. Kato T, Crestani E, Kamae C, Honma K, Yokosuka Y, Ikegawa T, et al. RAG1 Deficiency may Present Clinically as Selective IgA Deficiency. J Clin Immunol (2015) 35(3):280–8. doi: 10.1007/s10875-015-0146-4
3. Tsujita Y, Mitsui-Sekinaka K, Imai K, Yeh TW, Mitsuiki N, Asano T, et al. Phosphatase and Tensin Homolog (PTEN) Mutation can Cause Activated Phosphatidylinositol 3-Kinase δ Syndrome-Like Immunodeficiency. J Allergy Clin Immunol (2016) 138(6):1672–80. doi: 10.1016/j.jaci.2016.03.055
4. Hoshino A, Okada S, Yoshida K, Nishida N, Okuno Y, Ueno H, et al. Abnormal Hematopoiesis and Autoimmunity in Human Subjects With Germline IKZF1 Mutations. J Allergy Clin Immunol (2017) 140(1):223–31. doi: 10.1016/j.jaci.2016.09.029
5. Sekinaka Y, Mitsuiki N, Imai K, Yabe M, Yabe H, Mitsui-Sekinaka K, et al. Common Variable Immunodeficiency Caused by FANC Mutations. J Clin Immunol (2017) 37(5):434–44. doi: 10.1007/s10875-017-0396-4
6. Kadowaki T, Ohnishi H, Kawamoto N, Hori T, Nishimura K, Kobayashi C, et al. Haploinsufficiency of A20 Causes Autoinflammatory and Autoimmune Disorders. J Allergy Clin Immunol (2018) 141(4):1485–8.e11. doi: 10.1016/j.jaci.2017.10.039
7. Tozawa Y, Abdrabou SSMA, Nogawa-Chida N, Nishiuchi R, Ishida T, Suzuki Y, et al. A Deep Intronic Mutation of C.1166-285 T > G in SLC46A1 Is Shared by Four Unrelated Japanese Patients With Hereditary Folate Malabsorption (HFM). Clin Immunol (2019) 208:108256. doi: 10.1016/j.clim.2019.108256
8. Okano T, Imai K, Naruto T, Okada S, Yamashita M, Yeh TW, et al. Whole-Exome Sequencing-Based Approach for Germline Mutations in Patients With Inborn Errors of Immunity. J Clin Immunol (2020) 40(5):729–40. doi: 10.1007/s10875-020-00798-3
9. Matsuda T, Kambe N, Ueki Y, Kanazawa N, Izawa K, Honda Y, et al. Clinical Characteristics and Treatment of 50 Cases of Blau Syndrome in Japan Confirmed by Genetic Analysis of the NOD2 Mutation. Ann Rheum Dis (2020) 79(11):1492–9. doi: 10.1136/annrheumdis-2020-217320
10. Yeh TW, Okano T, Naruto T, Yamashita M, Okamura M, Tanita K, et al. APRIL-Dependent Lifelong Plasmacyte Maintenance and Immunoglobulin Production in Humans. J Allergy Clin Immunol (2020) 146(5):1109–20.e4. doi: 10.1016/j.jaci.2020.03.025
11. Tanita K, Sakura F, Nambu R, Tsumura M, Imanaka Y, Ohnishi H, et al. Clinical and Immunological Heterogeneity in Japanese Patients With Gain-Of-Function Variants in STAT3. J Clin Immunol (2021) 41(4):780–90. doi: 10.1007/s10875-021-00975-y
12. Yamashita M, Kuehn HS, Okuyama K, Okada S, Inoue Y, Mitsuiki N, et al. A Variant in Human AIOLOS Impairs Adaptive Immunity by Interfering With IKAROS. Nat Immunol (2021) 22(7):893–903. doi: 10.1038/s41590-021-00951-z
13. Kanazawa N, Hemmi H, Kinjo N, Ohnishi H, Hamazaki J, Mishima H, et al. Heterozygous Missense Variant of the Proteasome Subunit Beta-Type 9 Causes Neonatal-Onset Autoinflammation and Immunodeficiency. Nat Commun (2021) 12(1):6819. doi: 10.1038/s41467-021-27085-y
14. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification From the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. doi: 10.1007/s10875-019-00737-x
15. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
16. CEREDIH: The French PID study group. The French National Registry of Primary Immunodeficiency Diseases. Clin Immunol (2010) 135(2):264–72. doi: 10.1016/j.clim.2010.02.021
17. Gathmann B, Binder N, Ehl S, Kindle G, ESID Registry Working Party. The European Internet-Based Patient and Research Database for Primary Immunodeficiencies: Update 2011. Clin Exp Immunol (2012) 167(3):479–91. doi: 10.1111/j.1365-2249.2011.04542.x
18. Stray-Pedersen A, Abrahamsen T, Froland S. Primary Immunodeficiency Diseases in Norway. J Clin Immunol (2000) 20(6):477–85. doi: 10.1023/A:1026416017763
19. Llambi M, Etxagobe G, Matamoros F. The Spanish Registry of Primary Immunodeficiencies (REDIP). Allergol Immunopathol (Madr) (2001) 29(3):122–5. doi: 10.1016/S0301-0546(01)79031-3
20. El-Helou SM, Biegner AK, Bode S, Ehl SR, Heeg M, Maccari ME, et al. The German National Registry of Primary Immunodeficiencies (2012-2017). Front Immunol (2019) 10:1272. doi: 10.3389/fimmu.2019.01272
21. Lougaris V, Pession A, Baronio M, Soresina A, Rondelli R, Gazzurelli L, et al. The Italian Registry for Primary Immunodeficiencies (Italian Primary Immunodeficiency Network; IPINet): Twenty Years of Experience (1999-2019). J Clin Immunol (2020) 40(7):1026–37. doi: 10.1007/s10875-020-00844-0
22. Leiva LE, Zelazco M, Oleastro M, Carneiro-Sampaio M, Condino-Neto A, Costa-Carvalho BT, et al. Primary Immunodeficiency Diseases in Latin America: The Second Report of the LAGID Registry. J Clin Immunol (2007) 27(1):101–8. doi: 10.1007/s10875-006-9052-0
23. Boyle JM, Buckley RH. Population Prevalence of Diagnosed Primary Immunodeficiency Diseases in the United States. J Clin Immunol (2007) 27(5):497–502. doi: 10.1007/s10875-007-9103-1
24. Rezaei N, Aghamohammadi A, Moin M, Pourpak Z, Movahedi M, Gharagozlou M, et al. Frequency and Clinical Manifestations of Patients With Primary Immunodeficiency Disorders in Iran. Update From the Iranian Primary Immunodeficiency Registry. J Clin Immunol (2006) 26(6):519–32. doi: 10.1007/s10875-006-9047-x
25. Massaad MJ, Zainal M, Al-Herz W. Frequency and Manifestations of Autoimmunity Among Children Registered in the Kuwait National Primary Immunodeficiency Registry. Front Immunol (2020) 11:1119. doi: 10.3389/fimmu.2020.01119
26. Kirkpatrick P, Riminton S. Primary Immunodeficiency Diseases in Australia and New Zealand. J Clin Immunol (2007) 27(5):517–24. doi: 10.1007/s10875-007-9105-z
27. Lim D, Thong B, Ho S, Shek LPC, Lou J, Leonget KP, et al. Primary Immunodeficiency Diseases in Singapore – the Last 11 Years. Singapore Med J (2003) 44(11):579–86.
28. Lee WI, Huang JL, Jaing TH, Shyur SD, Yang KD, Chien YH, et al. Distribution, Clinical Features and Treatment in Taiwanese Patients With Symptomatic Primary Immunodeficiency Diseases (PIDs) in a Nationwide Population-Based Study During 1985-2010. Immunobiology (2011) 216(12):1286–94. doi: 10.1016/j.imbio.2011.06.002
29. Ishimura M, Takada H, Doi T, Imai K, Sasahara Y, Kanegane H, et al. Nationwide Survey of Patients With Primary Immunodeficiency Diseases in Japan. J Clin Immunol (2011) 31(6):968–76. doi: 10.1007/s10875-011-9594-7
Keywords: primary immunodeficiency, Primary Immunodeficiency Database in Japan, Japanese Society for Immunodeficiency and Autoinflammatory Diseases, consultation, genetic analysis, pathogenesis
Citation: Mitsui-Sekinaka K, Sekinaka Y, Endo A, Imai K and Nonoyama S (2022) The Primary Immunodeficiency Database in Japan. Front. Immunol. 12:805766. doi: 10.3389/fimmu.2021.805766
Received: 30 October 2021; Accepted: 15 December 2021;
Published: 10 January 2022.
Edited by:
Xiaodong Zhao, Chongqing Medical University, ChinaReviewed by:
Esther De Vries, Tilburg University, NetherlandsCopyright © 2022 Mitsui-Sekinaka, Sekinaka, Endo, Imai and Nonoyama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kohsuke Imai, a2ltYWkucGVkQHRtZC5hYy5qcA==