 Timothy Lok-Hin Chiu1†
Timothy Lok-Hin Chiu1† Daniel Leung1†
Daniel Leung1† Koon-Wing Chan1
Koon-Wing Chan1 Hok Man Yeung1
Hok Man Yeung1 Chung-Yin Wong1
Chung-Yin Wong1 Huawei Mao2
Huawei Mao2 Jianxin He3
Jianxin He3 Pandiarajan Vignesh4Weiling Liang5
Pandiarajan Vignesh4Weiling Liang5 Woei Kang Liew6Li-Ping Jiang7
Woei Kang Liew6Li-Ping Jiang7 Tong-Xin Chen8Xiang-Yuan Chen9Yin-Bo Tao9Yong-Bin Xu10
Tong-Xin Chen8Xiang-Yuan Chen9Yin-Bo Tao9Yong-Bin Xu10 Hsin-Hui Yu11Alta Terblanche12David Christopher Lung13Cheng-Rong Li14
Hsin-Hui Yu11Alta Terblanche12David Christopher Lung13Cheng-Rong Li14 Jing Chen15Man Tian16
Jing Chen15Man Tian16 Brian Eley17Xingtian Yang1Jing Yang1Wen Chin Chiang6
Brian Eley17Xingtian Yang1Jing Yang1Wen Chin Chiang6 Bee Wah Lee18,19
Bee Wah Lee18,19 Deepti Suri4
Deepti Suri4 Amit Rawat4
Amit Rawat4 Anju Gupta4
Anju Gupta4 Surjit Singh4
Surjit Singh4 Wilfred Hing Sang Wong1
Wilfred Hing Sang Wong1 Gilbert T. Chua1
Gilbert T. Chua1 Jaime Sou Da Rosa Duque1Kai-Ning Cheong20Patrick Chun-Yin Chong21
Jaime Sou Da Rosa Duque1Kai-Ning Cheong20Patrick Chun-Yin Chong21 Marco Hok-Kung Ho21Tsz-Leung Lee20
Marco Hok-Kung Ho21Tsz-Leung Lee20 Wanling Yang1
Wanling Yang1 Pamela P. Lee1*
Pamela P. Lee1* Yu Lung Lau1*
Yu Lung Lau1*- 1Department of Paediatrics and Adolescent Medicine, The University of Hong Kong, Queen Mary Hospital, Hong Kong, Hong Kong SAR, China
- 2Department of Immunology, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing, China
- 3Department of Respiratory Medicine, Beijing Children’s Hospital, Capital Medical University, National Center for Children’s Health, Beijing, China
- 4Allergy & Immunology Unit, Department of Paediatrics, Advanced Pediatrics Centre, Postgraduate Institute of Medical Education and Research, Chandigarh, India
- 5Department of Paediatrics and Adolescent Medicine, The University of Hong Kong-Shenzhen Hospital, Shenzhen, China
- 6Paediatric Immunology Service, KK Hospital, Singapore, Singapore
- 7Children’s Hospital of Chongqing Medical University, Chongqing, China
- 8Shanghai Children’s Medical Center, School of Medicine, Shanghai Jiao Tong University, Shanghai, China
- 9Department of Allergy, Immunology and Rheumatology, Guangzhou Children’s Hospital, Guangdong, China
- 10Guangzhou Women and Children’s Medical Center, Guangzhou, China
- 11Department of Paediatrics, National Taiwan University Children’s Hospital, Taipei, Taiwan
- 12Paediatric Gastroenterology and Hepatology Unit, University of Pretoria, Pretoria, South Africa
- 13Department of Pathology, Queen Elizabeth Hospital/Hong Kong Children’s Hospital, Hong Kong, Hong Kong SAR, China
- 14Department of Nephrology, Shenzhen Children’s Hospital, Shenzhen, China
- 15Department of Hematology/Oncology, Key Laboratory of Pediatric Hematology & Oncology Ministry of Health, Shanghai Children’s Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 16Department of Tuberculosis, Nanjing Chest Hospital, Nanjing, China
- 17Department of Paediatrics and Child Health, University of Cape Town and Red Cross War Memorial Children’s Hospital, Cape Town, South Africa
- 18Department of Paediatrics, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
- 19Khoo Teck Puat-National University Children’s Medical Institute, National University Health System, Singapore, Singapore
- 20Hong Kong Children’s Hospital, Hong Kong, Hong Kong SAR, China
- 21Virtus Medical, Hong Kong, Hong Kong SAR, China
Background: Chronic granulomatous disease (CGD) is an inborn error of immunity (IEI), characterised by recurrent bacterial and fungal infections. It is inherited either in an X-linked (XL) or autosomal recessive (AR) mode. Phenome refers to the entire set of phenotypes expressed, and its study allows us to generate new knowledge of the disease. The objective of the study is to reveal the phenomic differences between XL and AR-CGD by using Human Phenotype Ontology (HPO) terms.
Methods: We collected data on 117 patients with genetically diagnosed CGD from Asia and Africa referred to the Asian Primary Immunodeficiency Network (APID network). Only 90 patients with sufficient clinical information were included for phenomic analysis. We used HPO terms to describe all phenotypes manifested in the patients.
Results: XL-CGD patients had a lower age of onset, referral, clinical diagnosis, and genetic diagnosis compared with AR-CGD patients. The integument and central nervous system were more frequently affected in XL-CGD patients. Regarding HPO terms, perianal abscess, cutaneous abscess, and elevated hepatic transaminase were correlated with XL-CGD. A higher percentage of XL-CGD patients presented with BCGitis/BCGosis as their first manifestation. Among our CGD patients, lung was the most frequently infected organ, with gastrointestinal system and skin ranking second and third, respectively. Aspergillus species, Mycobacterium bovis, and Mycobacteirum tuberculosis were the most frequent pathogens to be found.
Conclusion: Phenomic analysis confirmed that XL-CGD patients have more recurrent and aggressive infections compared with AR-CGD patients. Various phenotypic differences listed out can be used as clinical handles to distinguish XL or AR-CGD based on clinical features.
Introduction
Chronic granulomatous disease (CGD) is an inborn error of immunity (IEI) that is characterised by recurrent infections caused by catalase-positive bacteria and fungi, such as Staphylococcus aureus and Aspergillus species (1). It is estimated that the prevalence of CGD is 1 in 250,000 live births among Europeans and Americans (2, 3). CGD arises from the loss of function of one of the proteins forming the NADPH oxidase complex, which generates reactive oxygen species, i.e., superoxide radicals and hydrogen peroxide for intracellular bacteria and fungi killing in phagocytes (4). Currently, there is one X-linked (XL) and five autosomal recessive (AR) forms of CGD. The gene responsible for XL-CGD is CYBB, and the other five genes responsible for AR-CGD are CYBA, NCF1, NCF2, NCF4, and CYBC1. Frequently affected organs and systems include the lung, skin, lymph node, and liver (3). Patients may suffer from pneumonia and deep and superficial abscesses. CGD patients usually present with lymphadenopathy and hepatosplenomegaly on physical examination (5).
In our study, we focus on the phenomic analysis of CGD. Phenomics stands for the acquisition of high-dimensional phenotypic data on an organism scale. Study of phenomics is usually incorporated with the study of genomics and environment so that we can know various factors which might possibly influence the complex traits displayed. Compared with genomics, phenomics is much more sophisticated and is much more difficult to be characterised (6). For this study, we use Human Phenotype Ontology (HPO) terms to describe the phenomic abnormalities. The HPO project was publicised in 2008 and provides an ontology of annotations (7), i.e., HPO terms, to describe phenotypic abnormalities encountered by clinicians. The HPO currently contains over 13,000 terms arranged in a simple class-subclass relationship such that various specific terms belong to a subclass of a parent term. The aim of the HPO system is to offer a computational bridge between genome biology and clinical medicine, as well as enabling the integration of phenotypic information across various scientific tools for clinical diagnosis and research purposes (7). Due to advancement in technology, there are more external tools available for genomic discovery project and diagnostic research. For genomic projects, HPO terms are used to filter out the list of candidate genes to be tested from whole genome sequencing using Exomiser, Phevor, or PhenIX (8, 9). External algorithms such as Phenomizer and Phenolyzer can compute clinical phenotype data written in HPO terms to give out possible differential diagnoses. Phenomizer is an external tool which utilises HPO terms to report phenotypic abnormalities. It yields a list of differential diagnoses of the patient based on the HPO data inputted by a clinician (10). However, for most IEI which are included in the genotypic classification of the International Union of the Immunological Societies, a full HPO phenomic data is still lacking currently which requires contributions from different clinical immunologists (11, 12). Therefore, it is paramount to generate a phenome of IEI for a reference for the differential diagnosis tool. This can provide sufficient information for them to diagnose future patients by importing HPO terms identified by medical practitioners.
Currently, numerous case series about CGD patients have already been published but none of them has used HPO terms to represent their phenome. The phenomic data on CGD patients stored in Phenomizer database may not be accurate as well due to insufficient phenomic analysis. As a result, the main aim of this project is to observe and create a phenome for CGD patients in our case series. We also attempted to identify the main differences of the phenotypic data between XL-CGD and AR-CGD patients. The phenome of the respective XL or AR-CGD patients may also be sent to various external tools which use HPO terms for analysis such as Phenomizer and Exomiser so as to provide a reference for diagnosis and genetic analysis of suspected CGD patients. Significant differences are listed out to help clinicians to differentiate between them clinically.
Materials and Methods
Patient Source and Selection
The APID network is an IEI referral network established in 2009 by The University of Hong Kong, which acts as a platform to offer e-consultation and free genetic testing for IEI. There are over 100 member centres across Asia and Africa. From 2003 to 2017, 117 CGD patients referred from 18 centres were successfully genetically diagnosed and were included in this case series.
Data Collection
Clinical records and laboratory results of CGD patients, provided by their referring doctors at the time of request for genetic testing, are archived in the APID network. Patient data, including demographics, family history, age of clinical milestones, infection, and genetic results were recorded.
Phenomic Data
HPO (October 2020 version) terms, which describe individual phenotypic abnormalities in a hierarchical framework of organs, were applied for performing the phenomic analysis. Two researchers first reviewed the laboratory reports and clinical notes and then suitable HPO terms were selected from the HPO browser http://www.human-phenotype-ontology.org to describe the phenotypic abnormalities displayed. In the end, a HPO phenotypic profile for each CGD patient was generated. The HPO phenotypic profile consisting of all HPO terms which can be manifested from the clinical records was selected. No negated HPO terms, i.e., no specific phenotype was manifested in the clinical record, were used in our study. Discrepancies for the final HPO phenotypic profile were discussed and modified. Only the highest class of HPO terms in the hierarchial framework, i.e., systems affected and the most specific HPO terms were computed for detection of any significant correlation between individual phenotypic abnormality and mode of inheritance.
Genetics Data
Genetic analysis was performed using genomic DNA extracted from peripheral blood. Genomic DNA was sent to our research laboratory and the candidate genes, i.e., CYBB, CYBA, NCF1, NCF2, and NCF4, were tested by using Sanger sequencing on the basis of clinical likelihood in the research laboratory of the Department of Paediatrics and Adolescent Medicine, The University of Hong Kong. Pathogenicity of the targeted gene mutation is re-evaluated in accordance with the diagnostic interpretation guidelines published by the American Academy of Allergy, Asthma & Immunology (AAAAI) PID working group in 2020 (13).
APID Network Questionnaire
A questionnaire was also distributed online to APID network member centres across Asia and Africa. Questions regarding the availability of care for CGD patients in APID network member centres, i.e., diagnosis, laboratory tests, and treatment of CGD patients were asked. A total of 20 responses were recorded.
Ethics Approval
This research project is approved by The University of Hong Kong/Hospital Authority Hong Kong West Cluster Institutional Review Board.
Statistical Analysis
For descriptive statistics, all ages of clinical milestones were expressed in median and range (year). Univariate analysis was performed by independent-samples Mann-Whitney U test to evaluate the difference between XL-CGD and AR-CGD. First manifestation, HPO terms, and system affected are presented in the form of heat map and expressed in percentages. Fisher’s exact test was used in analysis to determine the correlation between categorical phenotypes with the genotype.
Results
Demographics Data
The demographics data of our CGD case series was displayed in the Sankey diagram in Figure 1. Of the 117 patients, 104 (88.9%) were male and 13 (11.1%) were female. XL-CGD was seen in 87 (74.4%) patients, while the remaining belonged to AR-CGD group. Out of the 30 AR CGD patients, 9 (30.0%) of them were found to have mutations in CYBA by Sanger Sequencing, 13 (43.3%) of them were found to have GT deletion in NCF1 by GeneScan® and 9 (30.0%) of them were found to have mutations in NCF2 by Sanger Sequencing, with one of them have concurrent CYBA and NCF2 mutations diagnosed. In our case series, more than half of the patients (56.7%) came from mainland China, with India (17.3%) and Hong Kong (17.3%) ranking second. Other patients either came from South-east Asia or South Africa. Details about consanguinity and family history were only available for 92 out of 117 patients, with 90 (97.8%) having no family consanguinity and 2 (2.2%) with family consanguinity. Moreover, out of the 92 patients, 69 (75.0%) have no suggestive family history and 23 (25.0%) have suggestive family history. Only 1 of them has an elder brother with known CGD.
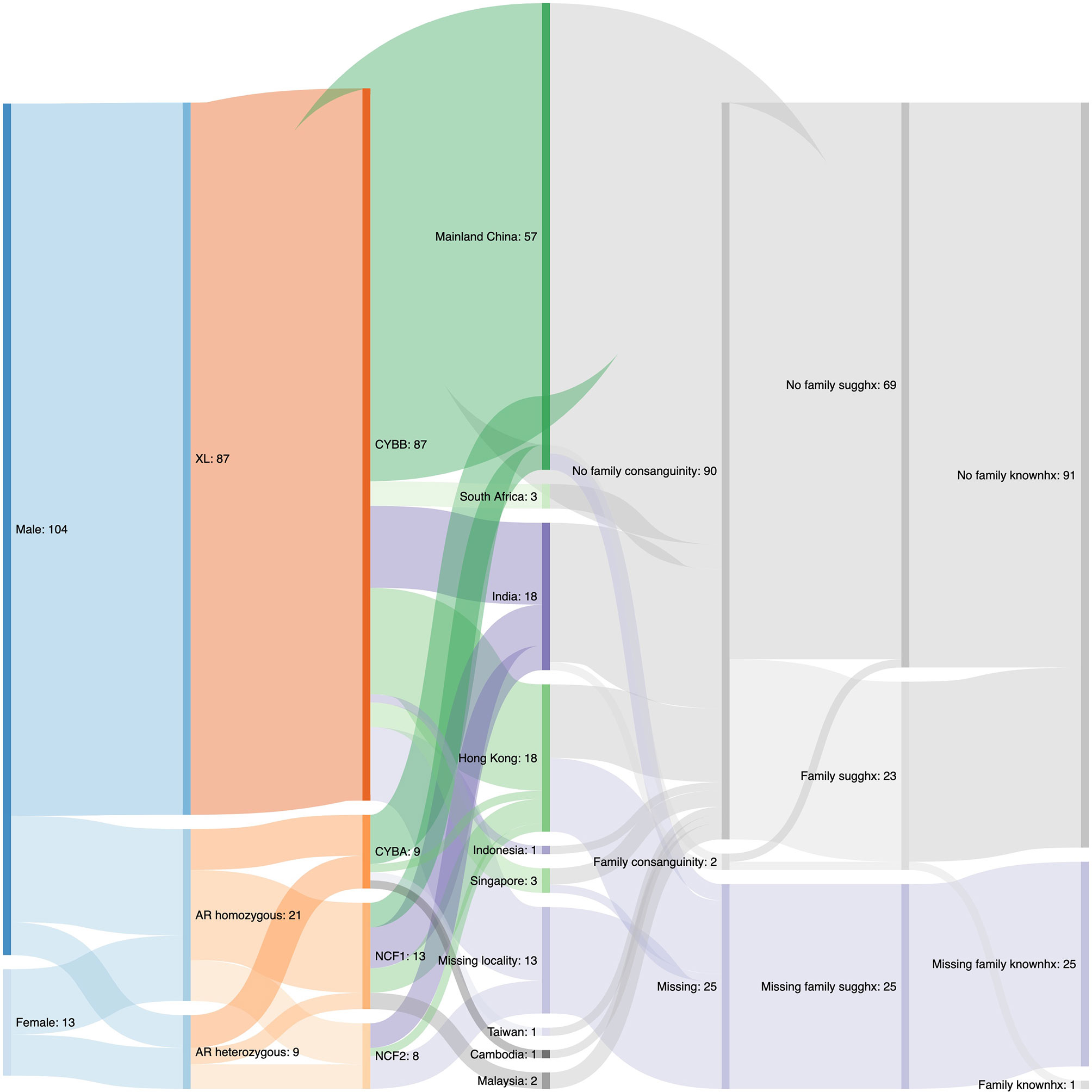
Figure 1 A Sankey diagram describing demographics data of the CGD case series. XL, X-linked; AR, autosomal recessive; sugghx, suggested history; knownhx, known history.
Genetics Data
The genetic mutations of 117 CGD patients, with 3 patients reporting 2 unphased variants, are shown in Table 1. The commonest gene implicated was CYBB, with no diagnosed NCF4 CGD patients. In total, 87 of them had mutations identified in CYBB gene. For the remaining 30 AR-CGD patients, 20 of them had homozygous mutations while only 1 of them was confirmed with compound heterozygous. Four of them had 2 unphased heterozygous mutations found in a recessive gene and five of them had 1 heterozygous mutation found in a recessive gene.
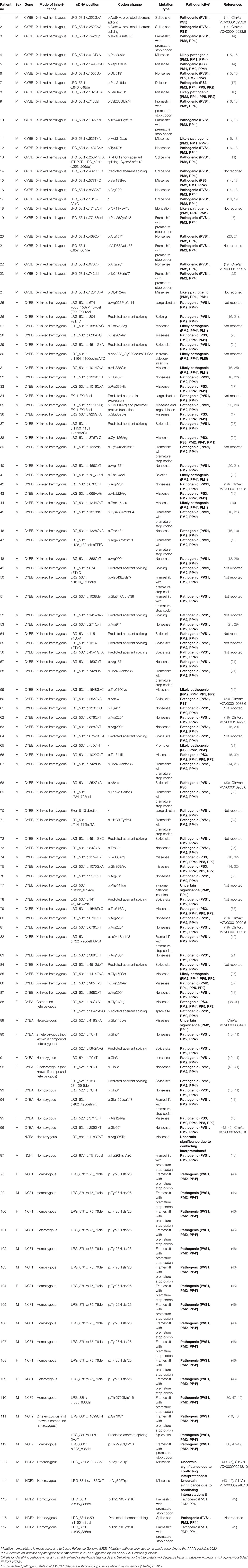
Table 1 Genetics data of CGD patients.
All AR-CGD patients with NCF1 mutations have documented GT deletion. Four patients among our CGD case series had a large deletion mutation in CYBB. There were 24 novel mutations identified in our patients, including 23 mutations in CYBB and 1 mutation in NCF2. Pathogenicity of these unreported mutations was determined by using the AAAAI guidelines in 2020 (13). Among 117 CGD patients, 99 of the CGD patients have pathogenic variants, 13 of them have likely pathogenic variants, and 5 of them have variants with uncertain significance.
Clinical Characteristics of XL and AR-CGD
The ages of clinical milestones of our CGD patients are displayed in Table 2. Ages of onset, referral, clinical diagnosis, and genetic diagnosis correlated with the mode of inheritance. Median age of onset correlated with the mode of inheritance (p = 0.01), with XL-CGD (0.2 years) lower than that of AR-CGD (0.4 years). Median age of referral to an immunology unit of XL-CGD (0.8 years) is also significantly lower than that of AR-CGD (3.5 years) and was shown to be significantly related with the mode of inheritance (p = 0.009). Median age at clinical diagnosis of XL-CGD (1.4 years) is younger than that of AR-CGD (4.8 years) with a strong correlation between XL or AR (p = 0.017). The same result was also demonstrated for the age of genetic diagnosis (p = 0.004) with XL-CGD patients showing a lower median age (2.2 years) compared with AR-CGD patients (4.8 years).

Table 2 Comparison between clinical characteristics between XL-CGD and AR-CGD using Fisher’s exact test.
First manifestations of XL and AR-CGD patients in our case series were displayed in Figure 2. Only the 5 most common first manifestations were included in the heat map, namely fever, BCGitis/BCGosis, pneumonia, cough, and lymphadenopathy, where generalized lymphadenopathy and cervical lymphadenopathy or no specific location regarding the lymphadenopathy were all categorized here. As shown in the figure, there is no significant association between the mode of inheritance and the respective first manifestation. However, it could be seen that a higher percentage of XL-CGD (18%) has BCGitis/BCGosis as their first manifestation compared with AR-CGD (4%) while a higher proportion of AR CGD (22%) has lymphadenopathy as their first manifestation compared with XL CGD (10%).
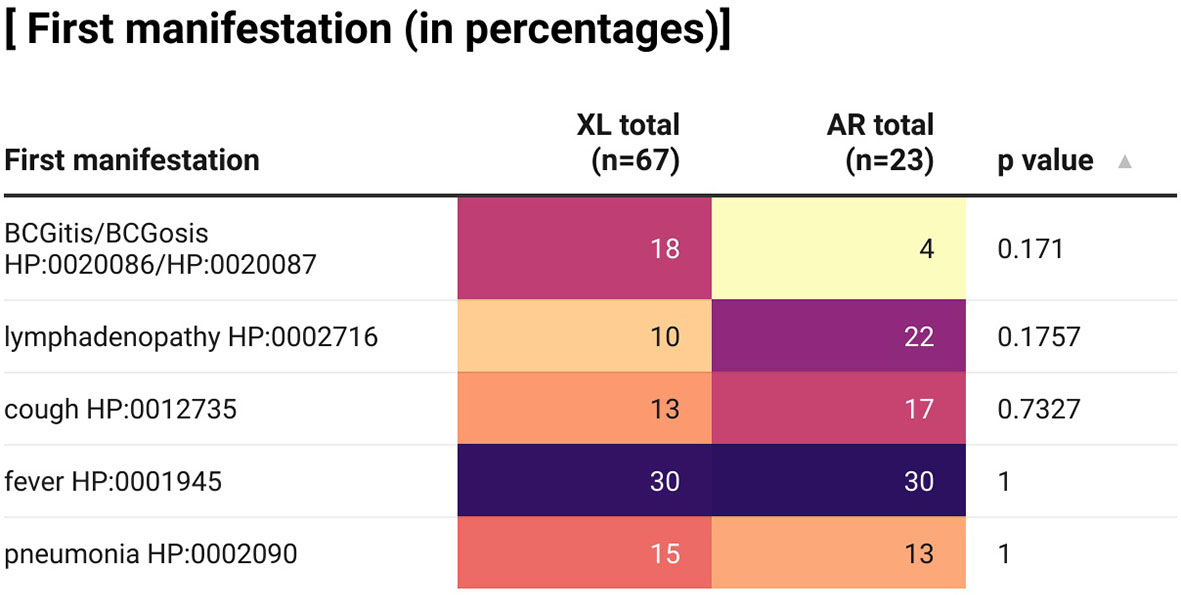
Figure 2 A heat map describing percentages of CGD patients in our case series with certain first manifestations according to their clinical records. XL, X-linked; AR, autosomal recessive; BCG, Bacillus Calmette–Guérin. Fisher’s exact test is used, p-value <0.05 is significant. This graph is created by using the app Datawrapper.
Infection Profile
Results for microbiological testing ordered based on clinical suspicion of infections were tallied, and cultured microorganisms from various infections and the 3 top locations where they were cultured are reported in Figure 3. However, the infectious etiology may not be established in every CGD case as the culture of the pathogen may not be performed or the culture reports were negative due to the use of antibiotics or antifungals prior to the culture. As shown above, a significant portion of infection was caused by uncertain bacteria or fungi. In general, the most common pathogens isolated are the Mycobacteirum bovis (n = 40), Mycobacteirum tuberculosis (n = 31), and Aspergillus species (n = 23). M. bovis was mostly isolated in the lungs, lymph nodes and the skin. M. tuberculosis was isolated in multiple systems or organs including the lungs, gastrointestinal system, skin, or disseminated infection while Aspergillus species were cultured mostly in the lungs. The most frequent locations of infections are the lungs (n = 77), gastrointestinal system (n = 27), and the skin (n = 19). Other locations of infection include the heart (n = 1), upper respiratory tract (n = 1), central nervous system (n = 1), ear (n = 2), neck (n = 2), spleen (n = 6), liver (n = 3), lymph node (n = 14), bone (n = 7), and disseminated infection (n = 3). Among the 77 episodes of infection affecting the lungs, Aspergillus species (n = 18) and M. tuberculosis (n = 15) were most often isolated, causing mostly pneumonia and bronchitis. Among the 23 episodes of infection affecting the gastrointestinal system, Rotavirus (n = 5) was most often isolated from the alimentary canal, causing mostly diarrhoea, vomiting, and enterocolitis. Other infectious etiologies causing hepato-splenomegaly and hepatic and splenic abscesses were not included here. Among the 19 episodes of infections affecting the skin, M. bovis (n = 7) is the most commonly isolated, causing cutaneous abscesses most of the time.
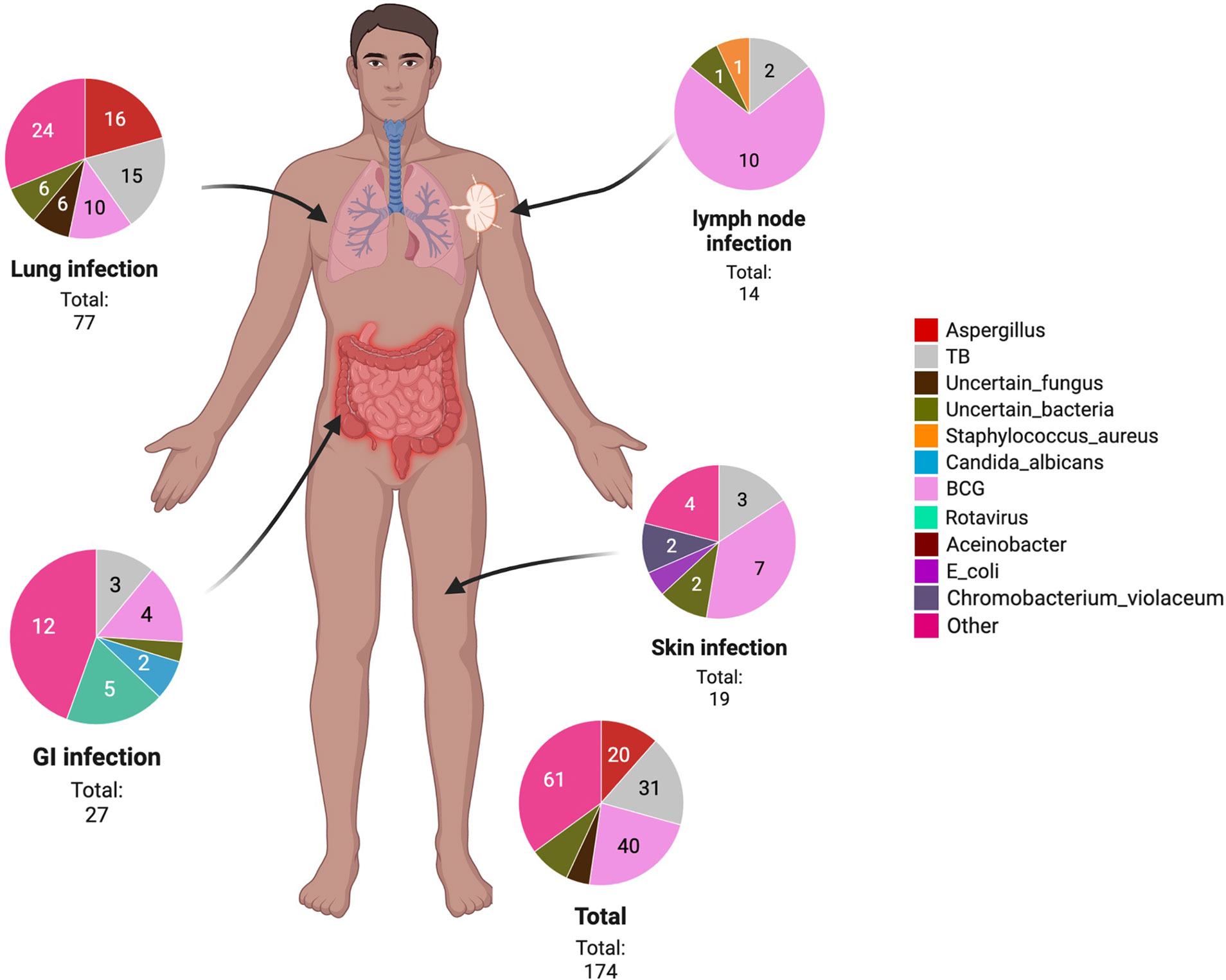
Figure 3 A body map showing the infection location of pathogens in our CGD case series. GI, gastrointestinal; TB, Mycobacterium tuberculosis; E. coli, Escherichia coli; BCG, Bacillus Calmette–Guérin.
Phenomic Analysis Between XL and AR-CGD
Regarding the phenomic analysis, we only included 90 out of 117 patients as only these 90 patients had sufficient clinical information provided. The affected systems of XL and AR-CGD patients are displayed in Figure 4 in the form of a heat map. In general, more XL-CGD patients are affected compared with AR-CGD patients in terms of the systems. Immune system was not shown in the heat map because all CGD patients had their immune system affected. As shown above, the most frequently affected systems of both XL and AR-CGD patients were the respiratory system, homeostasis system, and digestive system, respectively. A univariate analysis was performed to determine the correlation between various systems and their respective genotypes by using Fisher’s exact test. The integumentary system is significantly associated between the mode of inheritance (p = 0.0153), with more XL-CGD patients (57%) affected compared with AR-CGD patients (26%). In addition, more XL-CGD patients (13%) have their nervous system affected compared with no AR-CGD patients showing such manifestation. Although it is not statistically significant, it is still an interesting phenomenon to be reported.

Figure 4 A heat map describing percentages of CGD patients in our case series in which their organ systems are affected. HPO, Human Phenotype Ontology. **p < 0.01. This graph is created by using the app Datawrapper.
Phenotypic abnormalities of CGD patients are displayed in Figure 5 in the form of a heat map as shown above. More than 200 HPO terms describing phenotypic abnormalities were recorded in our CGD case series but only HPO terms which were manifested by more than 10% of patients would be included in the heat map. In general, more HPO terms were displayed in XL-CGD patients compared with AR-CGD patients. Among all the HPO terms recorded, both recurrent fever and pneumonia are the most frequent HPO terms identified with more than 70% of XL-CGD patients and 50% of AR-CGD patients showing this phenotypic abnormality. Other more common phenotypic abnormalities include hepatosplenomegaly, cutaneous abscess, and anaemia.
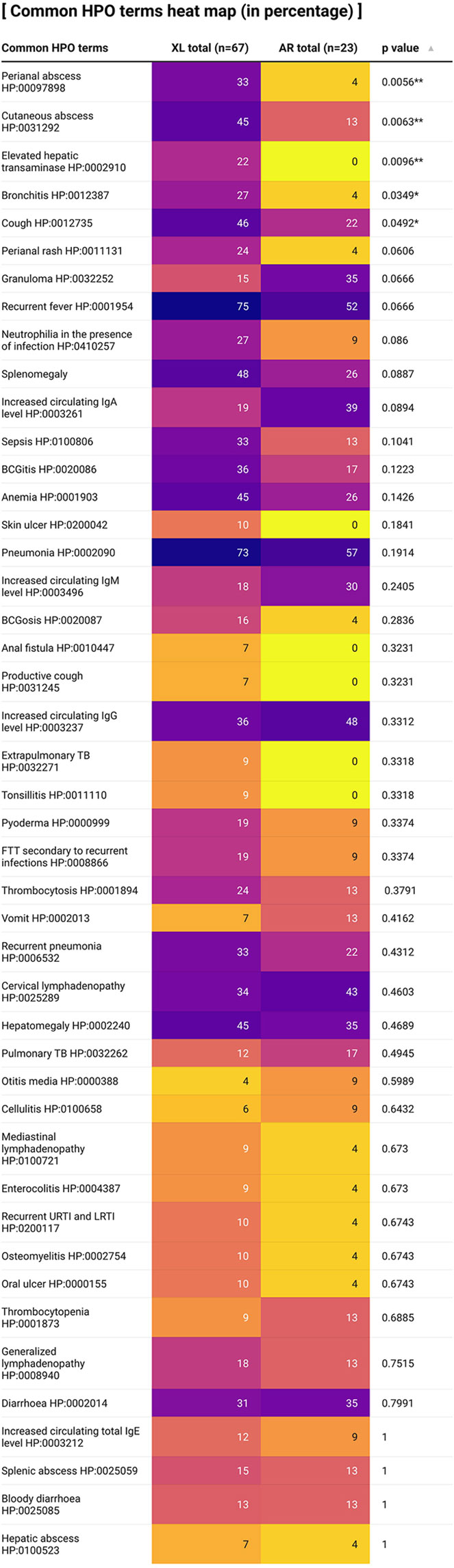
Figure 5 A heat map describing percentages of CGD patients in our case series where their clinical records displayed certain phenotypic abnormalities. Fisher’s exact test is used for statistical analysis (Only more than 5% of patients describing certain HPO term is recorded). HPO, Human Phenotype Ontology. Note: **p < 0.01; *p < 0.05. This graph is created by using the app Datawrapper.
A total of 5 HPO terms were shown to be correlated with XL or AR inheritance by Fisher’s exact test with more XL-CGD patients showing that specific phenotypic abnormality. Perianal abscess, cutaneous abscess and elevated hepatic transaminase are strongly correlated with the mode of inheritance (p < 0.001). Bronchitis and cough are correlated with the mode of inheritance as well (p < 0.05).
APID Network Questionnaire Regarding the Care of CGD Patients
The results of the questionnaire which was delivered to 20 APID network members are shown in Figure 6. As displayed, there were 16 centres who had diagnosed CGD in their clinics and most of them diagnosed their first CGD patients in the 1990s to 2010s. Only 9 APID network members have performed nitroblue tetrazolium test (NBT) after their establishment. APID network members diagnosed CGD by using NBT test initially with all of the 9 clinics performing the first NBT test before 2010s. However, starting from 1990s, APID network members started to use dihydrorhodamine (DHR) cytometry assay as well. In total, 11 APID network members have performed DHR cytometry assay in their clinics during the 1990s to 2010s.
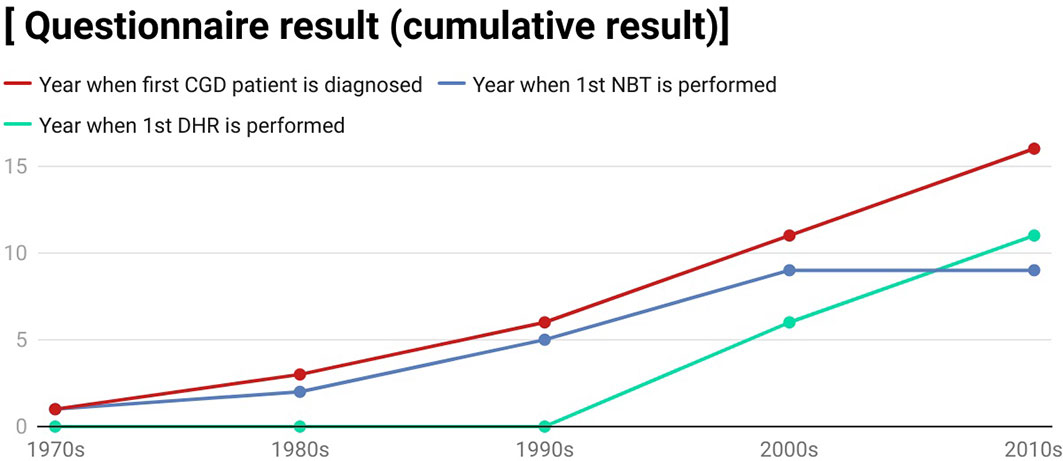
Figure 6 A line graph showing the questionnaire result which we deliver to various immunology centres. This graph is created by using the app Datawrapper.
Discussion
Our study revealed that XL-CGD and AR-CGD patients had some phenotypic differences through phenomic analysis. XL-CGD patients had their integument and the central nervous system more frequently affected. XL-CGD patients were shown to have perianal abscess, cutaneous abscess, and elevated hepatic transaminase more often as well. More XL-CGD patients presented with BCGitis/BCGosis as their first manifestation.
In our study, the most significant finding is that the integument system is more frequently affected among XL-CGD patients than AR-CGD patients. The reason behind such finding is due to more frequent perianal abscess, perianal rash, and cutaneous abscess reported among XL-CGD patients in the phenomic analysis, as it was observed in previous publications (3, 50–52). However, from reports in India and China, there is no statistical difference between XL and AR-CGD patients with episodes of superficial abscess (30, 41). Another interesting observation reported in our case series is that 13% of XL-CGD patients, but none of the AR-CGD patients had a central nervous system (CNS) abnormality. Common abnormalities under CNS include upper motor neuron dysfunction, headache, spinal cord compression, choroid plexus cyst, and unusual CNS infection, including CNS aspergillosis. The frequency of CNS aspergillosis only accounts for less than 5% in overall infections, and it has been shown that there is no significant association between the genotype and CNS aspergillosis in previous literature (53, 54). Further investigations need to be done to see whether these CNS and integumentary abnormalities are primary defects or complications of CGD or unrelated with CGD. Nevertheless, these new findings might be useful clinical handles for clinical immunologist to distinguish between XL and AR-CGD. Whenever clinicians observe some redflags, i.e., more frequent cutaneous or perianal abscesses or CNS abnormalities among CGD patients, they should suspect XL-CGD and perform targeted gene Sanger sequencing and DHR as soon as possible to confirm the diagnosis.
In addition to more frequent perianal and cutaneous abscesses seen in XL-CGD patients, a higher frequency of elevated hepatic transaminase was noted among XL-CGD patients in phenomic analysis. Previous literature has shown that abnormal liver enzymes level is common among CGD patients, occurring with at least one episode among 73% of the patients (55). However, no study has been done to correlate with the mode of inheritance and elevated hepatic transaminase. It has been hypothesised that XL-CGD patients had hepatosplenomegaly, liver abscesses, and BCGosis more often, which is a common cause of abnormal liver enzymes shown in previous literature (55). Therefore, clinicians can use the frequency of elevated hepatic transaminase to help differentiate between XL-CGD and AR-CGD.
Another notable finding in this case series is that more XL-CGD patients presented with BCGitis/BCGosis as first manifestation compared with AR-CGD patients. It has been documented that CGD patients are more prone to disseminated or local BCG infection due to defective intracellular mycobacterial killing mechanism (18, 56). BCG-related disease has been documented as a common first sign of CGD but in-depth study between genotypes of CGD has not been done. BCGitis is seen more commonly in countries where BCG vaccination is included in universal vaccination programme like mainland China, Iran, and Latin America as shown in Table 3 (67). It has been hypothesised that patients with XL-CGD had poorer control of BCG as compared with AR-CGD and hence physicians can recognise the BCG-related disease more often as their first manifestation.
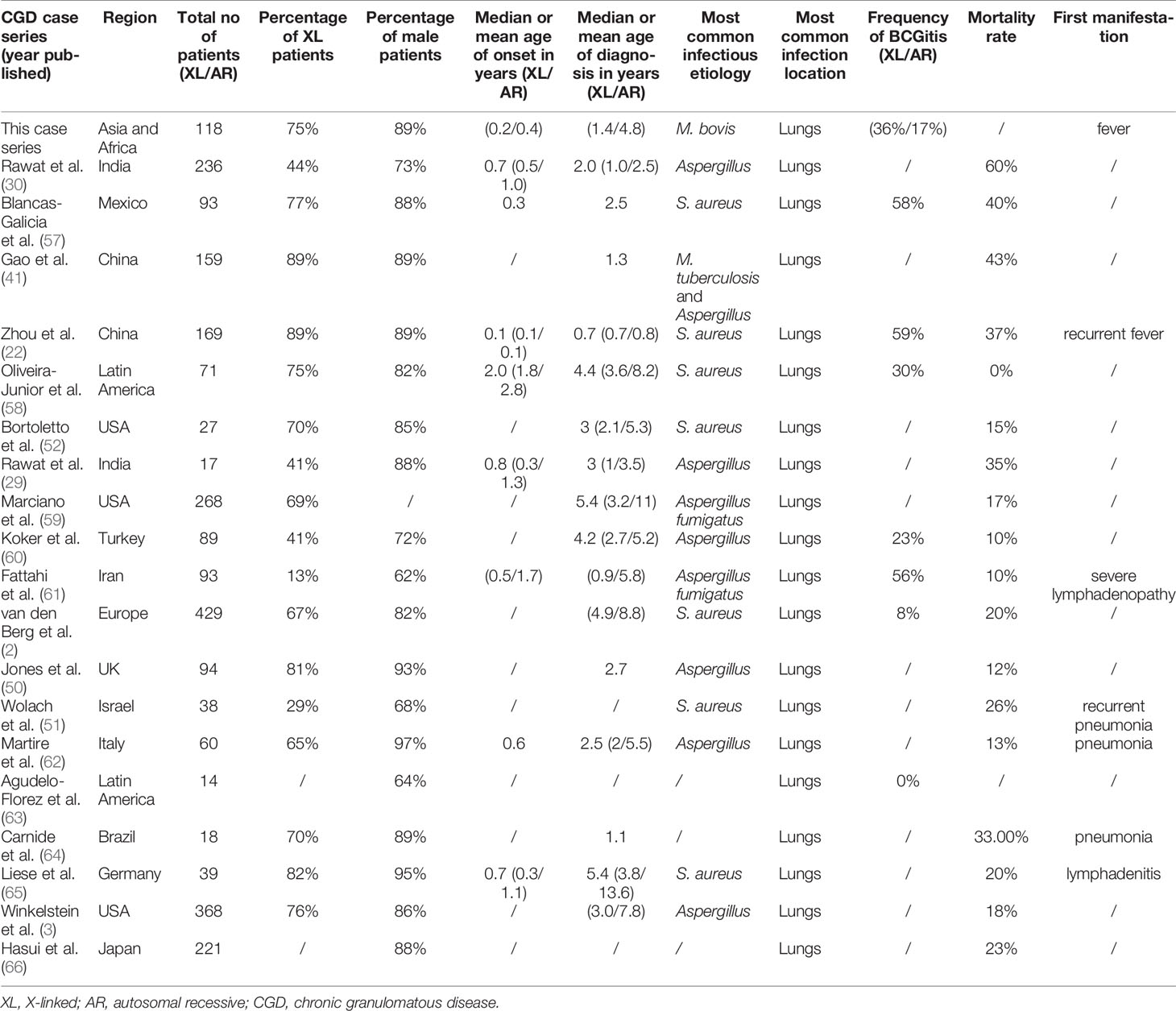
Table 3 A summary of findings regarding the differences between previous case series and this case series.
The main limitation of our study is that the clinical data provided to the APID network might be insufficient. Some of the CGD patients from our study were genetically diagnosed 20 years ago, during which the awareness and understanding of CGD was still inadequate in many countries, leading to an underreporting of CGD patients with atypical features. The authors do not have full access to the complete medical records of the CGD patients and hence some major phenotypic data of the CGD patients in our case series may be missed. Microbiological culture tests were not performed in some cases, leading to omissions in our infection profile. Staphylococcus aureus, for example, has been reported to be the most common pathogen causing skin abscesses in previous CGD case series but was not shown in our study. In addition, the clinical data provided to us were only up to the time when the patient was clinically diagnosed with CGD, and hence no follow-up clinical data could be computed and analysed. As a result, survival and death analysis cannot be done. As NBT or DHR assays were not always available and our cases came from many centres with different testing methodologies (68), therefore the functional phenotype of residual reactive oxygen species production could not be analysed in our case series. Since there were only 23 AR-CGD cases with sufficient clinical information, there might not be enough power resulting in false-negative results in our phenomic analysis comparing between AR and XL-CGD.
In conclusion, more severe integument infections, CNS, and hepatic enzyme abnormalities were observed in XL-CGD patients compared with AR-CGD patients. A summary of key findings regarding the differences between previous case series and this case series is presented in Table 3. Whenever clinicians identify such phenomic features among our children suspected to have IEI, they should suspect a diagnosis of XL-CGD and perform DHR as soon as possible. This can help speed up the diagnostic process and hence start prophylactic treatment as well as offering targeted genetic testing.
Data Availability Statement
The datasets presented in this article are not readily available because of ethical restrictions. Requests to access the datasets should be directed to bGF1eWx1bmdAaGt1Lmhr.
Ethics Statement
The studies involving human participants were reviewed and approved by Hospital Authority Hong Kong West Cluster-University of Hong Kong Institutional Review Board. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author Contributions
YL conceptualised the study. YL and DL designed the study. K-WC and C-YW performed genetic study. TC, HY, K-WC, and DL curated mutations. TC and DL phenotyped the patients, analysed data, and penned the manuscript. Other authors referred patients and provided clinical care and clinical data. All authors critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
The work is funded by the Society for Relief of Disabled Children and Jeffrey Modell Foundation.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with the authors DL, HM, SS, PPL and YLL.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zerbe CS, Marciano B, Holland SM. Chronic Granulomatous Disease: Pathogenesis, Clinical Manifestations, and Diagnosis. UpToDate (2020).
2. van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic Granulomatous Disease: The European Experience. PloS One (2009) 4(4):e5234. doi: 10.1371/journal.pone.0005234
3. Winkelstein JA, Marino MC, Johnston RB Jr., Boyle J, Curnutte J, Gallin JI, et al. Chronic Granulomatous Disease. Report on a National Registry of 368 Patients. Medicine (2000) 79(3):155–69. doi: 10.1097/00005792-200005000-00003
4. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, Biochemical, and Clinical Features of Chronic Granulomatous Disease. Medicine (2000) 79(3):170–200. doi: 10.1097/00005792-200005000-00004
5. Esfandbod M, Kabootari M. Images in Clinical Medicine. Chronic Granulomatous Disease. N Engl J Med (2012) 367(8):753. doi: 10.1056/NEJMicm1106350
6. Houle D, Govindaraju DR, Omholt S. Phenomics: The Next Challenge. Nat Rev Genet (2010) 11(12):855–66. doi: 10.1038/nrg2897
7. Kohler S, Vasilevsky NA, Engelstad M, Foster E, McMurry J, Ayme S, et al. The Human Phenotype Ontology in 2017. Nucleic Acids Res (2017) 45(D1):D865–D76. doi: 10.1093/nar/gkw1039
8. Bowles NE, Jou CJ, Arrington CB, Kennedy BJ, Earl A, Matsunami N, et al. Exome Analysis of a Family With Wolff-Parkinson-White Syndrome Identifies a Novel Disease Locus. Am J Med Genet A (2015) 167A(12):2975–84. doi: 10.1002/ajmg.a.37297
9. Pippucci T, Parmeggiani A, Palombo F, Maresca A, Angius A, Crisponi L, et al. A Novel Null Homozygous Mutation Confirms CACNA2D2 as a Gene Mutated in Epileptic Encephalopathy. PloS One (2013) 8(12):e82154. doi: 10.1371/journal.pone.0082154
10. Kohler S, Schulz MH, Krawitz P, Bauer S, Dolken S, Ott CE, et al. Clinical Diagnostics in Human Genetics With Semantic Similarity Searches in Ontologies. Am J Hum Genet (2009) 85(4):457–64. doi: 10.1016/j.ajhg.2009.09.003
11. Kohler S, Gargano M, Matentzoglu N, Carmody LC, Lewis-Smith D, Vasilevsky NA, et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res (2021) 49(D1):D1207–D17. doi: 10.1093/nar/gkaa1043
12. Haimel M, Pazmandi J, Heredia RJ, Dmytrus J, Bal SK, Zoghi S, et al. Curation and Expansion of Human Phenotype Ontology for Defined Groups of Inborn Errors of Immunity. J Allergy Clin Immunol (2021) 149(1):369–78. doi: 10.1016/j.jaci.2021.04.033
13. Chinn IK, Chan AY, Chen K, Chou J, Dorsey MJ, Hajjar J, et al. Diagnostic Interpretation of Genetic Studies in Patients With Primary Immunodeficiency Diseases: A Working Group Report of the Primary Immunodeficiency Diseases Committee of the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol (2020) 145(1):46–69. doi: 10.1016/j.jaci.2019.09.009
14. Kannengiesser C, Gérard B, El Benna J, Henri D, Kroviarski Y, Chollet-Martin S, et al. Molecular Epidemiology of Chronic Granulomatous Disease in a Series of 80 Kindreds: Identification of 31 Novel Mutations. Hum Mutat (2008) 29(9):E132–49. doi: 10.1002/humu.20820
15. Hui Y, Chan S, Lau Y. Identification of Mutations in Seven Chinese Patients With X-Linked Chronic Granulomatous Disease. Blood (1996) 88(10):4021–8. doi: 10.1182/blood.V88.10.4021.bloodjournal88104021
16. Roos D, Kuhns DB, Maddalena A, Bustamante J, Kannengiesser C, de Boer M, et al. Hematologically Important Mutations: The Autosomal Recessive Forms of Chronic Granulomatous Disease (Second Update). Blood Cells Mol Dis (2010) 44(4):291–9. doi: 10.1016/j.bcmd.2010.01.009
17. Babior BM. NADPH Oxidase: An Update. Blood J Am Soc Hematol (1999) 93(5):1464–76. doi: 10.1182/blood.V93.5.1464
18. Lee PP, Chan KW, Jiang L, Chen T, Li C, Lee TL, et al. Susceptibility to Mycobacterial Infections in Children With X-Linked Chronic Granulomatous Disease: A Review of 17 Patients Living in a Region Endemic for Tuberculosis. Pediatr Infect Dis J (2008) 27(3):224–30. doi: 10.1097/INF.0b013e31815b494c
19. Rae J, Newburger PE, Dinauer MC, Noack D, Hopkins PJ, Kuruto R, et al. X-Linked Chronic Granulomatous Disease: Mutations in the CYBB Gene Encoding the Gp91-Phox Component of Respiratory-Burst Oxidase. Am J Hum Genet (1998) 62(6):1320–31. doi: 10.1086/301874
20. Ariga T, Sakiyama Y, Furuta H, Matsumoto S. Molecular Genetic Studies of Two Families With X-Linked Chronic Granulomatous Disease: Mutation Analysis and Definitive Determination of Carrier Status in Patients’ Sisters. Eur J Haematol (1994) 52(2):99–102. doi: 10.1111/j.1600-0609.1994.tb01293.x
21. Kuribayashi F, de Boer M, Leusen JH, Verhoeven AJ, Roos D. A Novel Polymorphism in the Coding Region of CYBB, the Human Gp91-Phox Gene. Hum Genet (1996) 97(5):611–3. doi: 10.1007/BF02281870
22. Zhou Q, Hui X, Ying W, Hou J, Wang W, Liu D, et al. A Cohort of 169 Chronic Granulomatous Disease Patients Exposed to BCG Vaccination: A Retrospective Study From a Single Center in Shanghai, China (2004–2017). J Clin Immunol (2018) 38(3):260–72. doi: 10.1007/s10875-018-0486-y
23. Biberstine-Kinkade KJ, DeLeo FR, Epstein RI, LeRoy BA, Nauseef WM, Dinauer MC. Heme-Ligating Histidines in Flavocytochromeb 558: IDENTIFICATION OF SPECIFIC HISTIDINES IN Gp91phox. J Biol Chem (2001) 276(33):31105–12. doi: 10.1074/jbc.M103327200
24. von Goessel H, Hossle JP, Seger R, Gungor T. Characterization of 17 New Cases of X-Linked Chronic Granulomatous Disease With Seven Novel Mutations in the CYBB Gene. Exp Hematol (2006) 34(4):528–35. doi: 10.1016/j.exphem.2006.01.005
25. Wu J, Wang W-F, Zhang Y-D, Chen T-X. Clinical Features and Genetic Analysis of 48 Patients With Chronic Granulomatous Disease in a Single Center Study From Shanghai, China (2005–2015): New Studies and a Literature Review. J Immunol Res (2017) 2017:8745254. doi: 10.1155/2017/8745254
26. Stasia MJ, Lardy B, Maturana A, Rousseau P, Martel C, Bordigoni P, et al. Molecular and Functional Characterization of a New X-Linked Chronic Granulomatous Disease Variant (X91+) Case With a Double Missense Mutation in the Cytosolic Gp91phox C-Terminal Tail. Biochim Biophys Acta (BBA) Mol Basis Dis (2002) 1586(3):316–30. doi: 10.1016/S0925-4439(01)00110-7
28. Patiño PJ, Perez JE, Lopez JA, Condino-Neto A, Grumach AS, Botero JH, et al. Molecular Analysis of Chronic Granulomatous Disease Caused by Defects in Gp91-Phox. Hum Mutat (1999) 13(1):29–37. doi: 10.1002/(SICI)1098-1004(1999)13:1<29::AID-HUMU3>3.0.CO;2-X
29. Rawat A, Singh S, Suri D, Gupta A, Saikia B, Minz RW, et al. Chronic Granulomatous Disease: Two Decades of Experience From a Tertiary Care Centre in North West India. J Clin Immunol (2014) 34(1):58–67. doi: 10.1007/s10875-013-9963-5
30. Rawat A, Vignesh P, Sudhakar M, Sharma M, Suri D, Jindal A, et al. Clinical, Immunological, and Molecular Profile of Chronic Granulomatous Disease: A Multi-Centric Study of 236 Patients From India. Front Immunol (2021) 12:625320. doi: 10.3389/fimmu.2021.625320
31. Suzuki S, Kumatori A, Haagen I-A, Fujii Y, Sadat MA, Jun HL, et al. PU. 1 as an Essential Activator for the Expression of Gp91phox Gene in Human Peripheral Neutrophils, Monocytes, and B Lymphocytes. Proc Natl Acad Sci (1998) 95(11):6085–90. doi: 10.1073/pnas.95.11.6085
32. Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH Oxidase and Survival in Chronic Granulomatous Disease. New Engl J Med (2010) 363(27):2600–10. doi: 10.1056/NEJMoa1007097
33. Xu H, Tian W, Li S-J, Zhang L-Y, Liu W, Zhao Y, et al. Clinical and Molecular Features of 38 Children With Chronic Granulomatous Disease in Mainland China. J Clin Immunol (2014) 34(6):633–41. doi: 10.1007/s10875-014-0061-0
34. Kulkarni M, Hule G, de Boer M, van Leeuwen K, Kambli P, Aluri J, et al. Approach to Molecular Diagnosis of Chronic Granulomatous Disease (CGD): An Experience From a Large Cohort of 90 Indian Patients. J Clin Immunol (2018) 38(8):898–916. doi: 10.1007/s10875-018-0567-y
35. Gérard B, El Benna J, Alcain F, Gougerot-Pocidalo MA, Grandchamp B, Chollet-Martin S. Characterization of 11 Novel Mutations in the X-Linked Chronic Granulomatous Disease (CYBB Gene). Hum Mutat (2001) 18(2):163–. doi: 10.1002/humu.1166
36. Kaneda M, Sakuraba H, Ohtake A, Nishida A, Kiryu C, Kakinuma K. Missense Mutations in the Gp91-Phox Gene Encoding Cytochrome B 558 in Patients With Cytochrome B Positive and Negative X-Linked Chronic Granulomatous Disease. Blood J Am Soc Hematol (1999) 93(6):2098–104. doi: 10.1182/blood.V93.6.2098.406k09_2098_2104
37. Barese C, Copelli S, Zandomeni R, Oleastro M, Zelazko M, Rivas EM. X-Linked Chronic Granulomatous Disease: First Report of Mutations in Patients of Argentina. J Pediatr Hematol Oncol (2004) 26(10):656–60. doi: 10.1097/01.mph.0000139455.29962.be
38. Chang Y-H, Yu H-H, Lau Y-L, Chan K-W, Chiang B-L. A New Autosomal Recessive, Heterozygous Pair of Mutations of CYBA in a Patient With Chronic Granulomatous Disease. Ann Allergy Asthma Immunol (2010) 105(2):183–5. doi: 10.1016/j.anai.2010.05.013
39. Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, et al. Hematologically Important Mutations: X-Linked Chronic Granulomatous Disease (Third Update). Blood Cells Mol Dis (2010) 45(3):246–65. doi: 10.1016/j.bcmd.2010.07.012
40. Ishibashi F, Nunoi H, Endo F, Matsuda I, Kanegasaki S. Statistical and Mutational Analysis of Chronic Granulomatous Disease in Japan With Special Reference to Gp91-Phox and P22-Phox Deficiency. Hum Genet (2000) 106(5):473–81. doi: 10.1007/s004390000288
41. Gao LW, Yin QQ, Tong YJ, Gui JG, Liu XY, Feng XL, et al. Clinical and Genetic Characteristics of Chinese Pediatric Patients With Chronic Granulomatous Disease. Pediatr Allergy Immunol (2019) 30(3):378–86. doi: 10.1111/pai.13033
42. Wang S, Wang T, Xiang Q, Xiao M, Cao Y, Xu H, et al. Clinical and Molecular Features of Chronic Granulomatous Disease in Mainland China and a XL-CGD Female Infant Patient After Prenatal Diagnosis. J Clin Immunol (2019) 39(8):762–75. doi: 10.1007/s10875-019-00680-x
43. Cross AR, Noack D, Rae J, Curnutte JT, Heyworth PG. Hematologically Important Mutations: The Autosomal Recessive Forms of Chronic Granulomatous Disease (First Update). Blood Cells Mol Dis (2000) 26(5):561–5. doi: 10.1006/bcmd.2000.0333
44. Kim-Howard X, Sun C, Molineros JE, Maiti AK, Chandru H, Adler A, et al. Allelic Heterogeneity in NCF2 Associated With Systemic Lupus Erythematosus (SLE) Susceptibility Across Four Ethnic Populations. Hum Mol Genet (2014) 23(6):1656–68. doi: 10.1093/hmg/ddt532
45. Patinño PJ, Rae J, Noack D, Erickson R, Ding J, de Olarte D, et al. Molecular Characterization of Autosomal Recessive Chronic Granulomatous Disease Caused by a Defect of the Nicotinamide Adenine Dinucleotide Phosphate (Reduced Form) Oxidase Component P67-Phox. Blood J Am Soc Hematol (1999) 94(7):2505–14. doi: 10.1182/blood.V94.7.2505.419k10_2505_2514
46. Roos D, de Boer M, Köker MY, Dekker J, Singh-Gupta V, Åhlin A, et al. Chronic Granulomatous Disease Caused by Mutations Other Than the Common GT Deletion in NCF1, the Gene Encoding the P47phox Component of the Phagocyte NADPH Oxidase. Hum Mutat (2006) 27(12):1218–29. doi: 10.1002/humu.20413
47. Köker M, Sanal Ö, Van Leeuwen K, De Boer M, Metin A, Patıroğlu T, et al. Four Different NCF2 Mutations in Six Families From Turkey and an Overview of NCF2 Gene Mutations. Eur J Clin Invest (2009) 39(10):942–51. doi: 10.1111/j.1365-2362.2009.02195.x
48. Vignesh P, Rawat A, Kumar A, Suri D, Gupta A, Lau YL, et al. Chronic Granulomatous Disease Due to Neutrophil Cytosolic Factor (NCF2) Gene Mutations in Three Unrelated Families. J Clin Immunol (2017) 37(2):109–12. doi: 10.1007/s10875-016-0366-2
49. Noack D, Rae J, Cross AR, Muñoz J, Salmen S, Mendoza JA, et al. Autosomal Recessive Chronic Granulomatous Disease Caused by Novel Mutations in NCF-2, the Gene Encoding the P67-Phox Component of Phagocyte NADPH Oxidase. Hum Genet (1999) 105(5):460–7. doi: 10.1007/s004399900152
50. Jones LB, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, et al. Special Article: Chronic Granulomatous Disease in the United Kingdom and Ireland: A Comprehensive National Patient-Based Registry. Clin Exp Immunol (2008) 152(2):211–8. doi: 10.1111/j.1365-2249.2008.03644.x
51. Wolach B, Gavrieli R, de Boer M, Gottesman G, Ben-Ari J, Rottem M, et al. Chronic Granulomatous Disease in Israel: Clinical, Functional and Molecular Studies of 38 Patients. Clin Immunol (2008) 129(1):103–14. doi: 10.1016/j.clim.2008.06.012
52. Bortoletto P, Lyman K, Camacho A, Fricchione M, Khanolkar A, Katz BZ. Chronic Granulomatous Disease: A Large, Single-Center US Experience. Pediatr Infect Dis J (2015) 34(10):1110–4. doi: 10.1097/INF.0000000000000840
53. Falcone EL, Holland SM. Invasive Fungal Infection in Chronic Granulomatous Disease: Insights Into Pathogenesis and Management. Curr Opin Infect Dis (2012) 25(6):658–69. doi: 10.1097/QCO.0b013e328358b0a4
54. Blumental S, Mouy R, Mahlaoui N, Bougnoux ME, Debre M, Beaute J, et al. Invasive Mold Infections in Chronic Granulomatous Disease: A 25-Year Retrospective Survey. Clin Infect Dis (2011) 53(12):e159–69. doi: 10.1093/cid/cir731
55. Hussain N, Feld JJ, Kleiner DE, Hoofnagle JH, Garcia-Eulate R, Ahlawat S, et al. Hepatic Abnormalities in Patients With Chronic Granulomatous Disease. Hepatology (2007) 45(3):675–83. doi: 10.1002/hep.21524
56. Lau Y, Chan G, Ha S, Hui Y, Yuen K. The Role of Phagocytic Respiratory Burst in Host Defense Against Mycobacterium Tuberculosis. Clin Infect Dis (1998) 26(1):226–7. doi: 10.1086/517036
57. Blancas-Galicia L, Santos-Chavez E, Deswarte C, Mignac Q, Medina-Vera I, Leon-Lara X, et al. Genetic, Immunological, and Clinical Features of the First Mexican Cohort of Patients With Chronic Granulomatous Disease. J Clin Immunol (2020) 40(3):475–93. doi: 10.1007/s10875-020-00750-5
58. de Oliveira-Junior EB, Zurro NB, Prando C, Cabral-Marques O, Pereira PV, Schimke LF, et al. Clinical and Genotypic Spectrum of Chronic Granulomatous Disease in 71 Latin American Patients: First Report From the LASID Registry. Pediatr Blood Cancer (2015) 62(12):2101–7. doi: 10.1002/pbc.25674
59. Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, et al. Common Severe Infections in Chronic Granulomatous Disease. Clin Infect Dis (2015) 60(8):1176–83. doi: 10.1093/cid/ciu1154
60. Koker MY, Camcioglu Y, van Leeuwen K, Kilic SS, Barlan I, Yilmaz M, et al. Clinical, Functional, and Genetic Characterization of Chronic Granulomatous Disease in 89 Turkish Patients. J Allergy Clin Immunol (2013) 132(5):1156–63 e5. doi: 10.1016/j.jaci.2013.05.039
61. Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance Pattern and Clinical Aspects of 93 Iranian Patients With Chronic Granulomatous Disease. J Clin Immunol (2011) 31(5):792–801. doi: 10.1007/s10875-011-9567-x
62. Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, et al. Clinical Features, Long-Term Follow-Up and Outcome of a Large Cohort of Patients With Chronic Granulomatous Disease: An Italian Multicenter Study. Clin Immunol (2008) 126(2):155–64. doi: 10.1016/j.clim.2007.09.008
63. Agudelo-Florez P, Prando-Andrade CC, Lopez JA, Costa-Carvalho BT, Quezada A, Espinosa FJ, et al. Chronic Granulomatous Disease in Latin American Patients: Clinical Spectrum and Molecular Genetics. Pediatr Blood Cancer (2006) 46(2):243–52. doi: 10.1002/pbc.20455
64. Carnide EG, Jacob CA, Castro AM, Pastorino AC. Clinical and Laboratory Aspects of Chronic Granulomatous Disease in Description of Eighteen Patients. Pediatr Allergy Immunol (2005) 16(1):5–9. doi: 10.1111/j.1399-3038.2005.00225.x
65. Liese J, Kloos S, Jendrossek V, Petropoulou T, Wintergerst U, Notheis G, et al. Long-Term Follow-Up and Outcome of 39 Patients With Chronic Granulomatous Disease. J Pediatr (2000) 137(5):687–93. doi: 10.1067/mpd.2000.109112
66. Hasui M, THE STUDY GROUP OF PHAGOCYTE DISORDERS OF JAPAN, MASAFUMI. Chronic Granulomatous Disease in Japan: Incidence and Natural History. Pediatr Int (1999) 41(5):589–93. doi: 10.1046/j.1442-200x.1999.01129.x
67. Liang W, Li H, Liu C, Zhong G, Wu Q, Liu X, et al. Correlation Between Genotype and Pathogen in Chronic Granulomatous Disease. Chin J Appl Clin Pediatr (2021) (24):364–8. doi: 10.1046/j.1442-200x.1999.01129.x
Keywords: chronic granulomatous disease (CGD), inborn error of immunity (IEI), human phenotype ontology (HPO), phenome, genetics
Citation: Chiu TL-H, Leung D, Chan K-W, Yeung HM, Wong C-Y, Mao H, He J, Vignesh P, Liang W, Liew WK, Jiang L-P, Chen T-X, Chen X-Y, Tao Y-B, Xu Y-B, Yu H-H, Terblanche A, Lung DC, Li C-R, Chen J, Tian M, Eley B, Yang X, Yang J, Chiang WC, Lee BW, Suri D, Rawat A, Gupta A, Singh S, Wong WHS, Chua GT, Duque JSDR, Cheong K-N, Chong PC-Y, Ho MH-K, Lee T-L, Yang W, Lee PP and Lau YL (2022) Phenomic Analysis of Chronic Granulomatous Disease Reveals More Severe Integumentary Infections in X-Linked Compared With Autosomal Recessive Chronic Granulomatous Disease. Front. Immunol. 12:803763. doi: 10.3389/fimmu.2021.803763
Received: 28 October 2021; Accepted: 27 December 2021;
Published: 24 January 2022.
Edited by:
Hirokazu Kanegane, Tokyo Medical and Dental University, JapanReviewed by:
Antonio Condino-Neto, University of São Paulo, BrazilAnete S. Grumach, Faculdade de Medicina do ABC, Brazil
Copyright © 2022 Chiu, Leung, Chan, Yeung, Wong, Mao, He, Vignesh, Liang, Liew, Jiang, Chen, Chen, Tao, Xu, Yu, Terblanche, Lung, Li, Chen, Tian, Eley, Yang, Yang, Chiang, Lee, Suri, Rawat, Gupta, Singh, Wong, Chua, Duque, Cheong, Chong, Ho, Lee, Yang, Lee and Lau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pamela P. Lee, cHB3bGVlQGhrdS5oaw==; Yu Lung Lau, bGF1eWx1bmdAaGt1Lmhr
†These authors share first authorship