 Shuang Liu
Shuang Liu Wei Chong
Wei Chong- Department of Emergency, The First Affiliated Hospital of China Medical University, Shenyang, China
Sepsis is an abnormal systemic inflammatory response of the host immune system to infection and can lead to fatal multiorgan dysfunction syndrome. Epidemiological studies have shown that approximately 10-70% of sepsis cases can lead to septic cardiomyopathy. Since the pathogenesis of septic cardiomyopathy is not clear, it is difficult for medical doctors to treat the disease. Therefore, finding effective interventions to prevent and reduce myocardial damage in septic cardiomyopathy is clinically significant. Epigenetics is the study of stable genetic phenotype inheritance that does not involve changing gene sequences. Epigenetic inheritance is affected by both gene and environmental regulation. Epigenetic studies focus on the modification and influence of chromatin structure, mainly including chromatin remodelling, DNA methylation, histone modification and noncoding RNA (ncRNA)-related mechanisms. Recently, long ncRNA (lncRNA)-related mechanisms have been the focus of epigenetic studies. LncRNAs are expected to become important targets to prevent, diagnose and treat human diseases. As the energy metabolism centre of cells, mitochondria are important targets in septic cardiomyopathy. Intervention measures to prevent and treat mitochondrial damage are of great significance for improving the prognosis of septic cardiomyopathy. LncRNAs play important roles in life activities. Recently, studies have focused on the involvement of lncRNAs in regulating mitochondrial dysfunction. However, few studies have revealed the involvement of lncRNAs in regulating mitochondrial dysfunction in septic cardiomyopathy. In this article, we briefly review recent research in this area.
Sepsis and Septic Cardiomyopathy
Sepsis is an abnormal systemic inflammatory response of the host immune system to infection and can lead to fatal multiorgan dysfunction syndrome (1, 2). In severe cases, sepsis is considered a cause of death (3). Millions of human beings suffer from sepsis every year, and more than one-quarter of them lose their lives (4). According to statistics, the hospitalization rate and mortality of patients with severe sepsis increase by 8.2% and 5.6%, respectively, every year (5). Parker et al. first proposed in a 1984 study that sepsis-induced cardiac dysfunction is reversible (6). Since then, research on septic cardiomyopathy has attracted increasing attention. Epidemiological studies have shown that 10-70% of sepsis cases can lead to septic cardiomyopathy (7, 8). The mortality of patients with septic cardiomyopathy is 70%-90%, which is 2-3-fold higher than that of patients with sepsis that does not affect the heart (9, 10). Currently, there is no formal definition of septic cardiomyopathy. It is generally recognized that septic cardiomyopathy is transient cardiac dysfunction caused by sepsis and that it manifests as heart enlargement, ventricular systolic dysfunction, hypoperfusion without ventricular systolic dysfunction, poor response to fluid resuscitation and catecholamines, and so on (11–14).
It has been revealed that the specific septic cardiomyopathy pathogenesis may include an imbalance of pro- and anti-inflammatory cytokine expression, abnormal expression of Toll-like receptors and related downstream pathways, release of nitric oxide (NO) and reactive oxygen species (ROS), complement activation, abnormal calcium processing, downregulation of the adrenergic pathway, cardiomyocyte apoptosis, autonomic nervous system dysfunction, coronary microvascular disturbance, mitochondrial dysfunction, and downregulation of sarcomere and mitochondrial proteins (15–18) (Figures 1, 2).
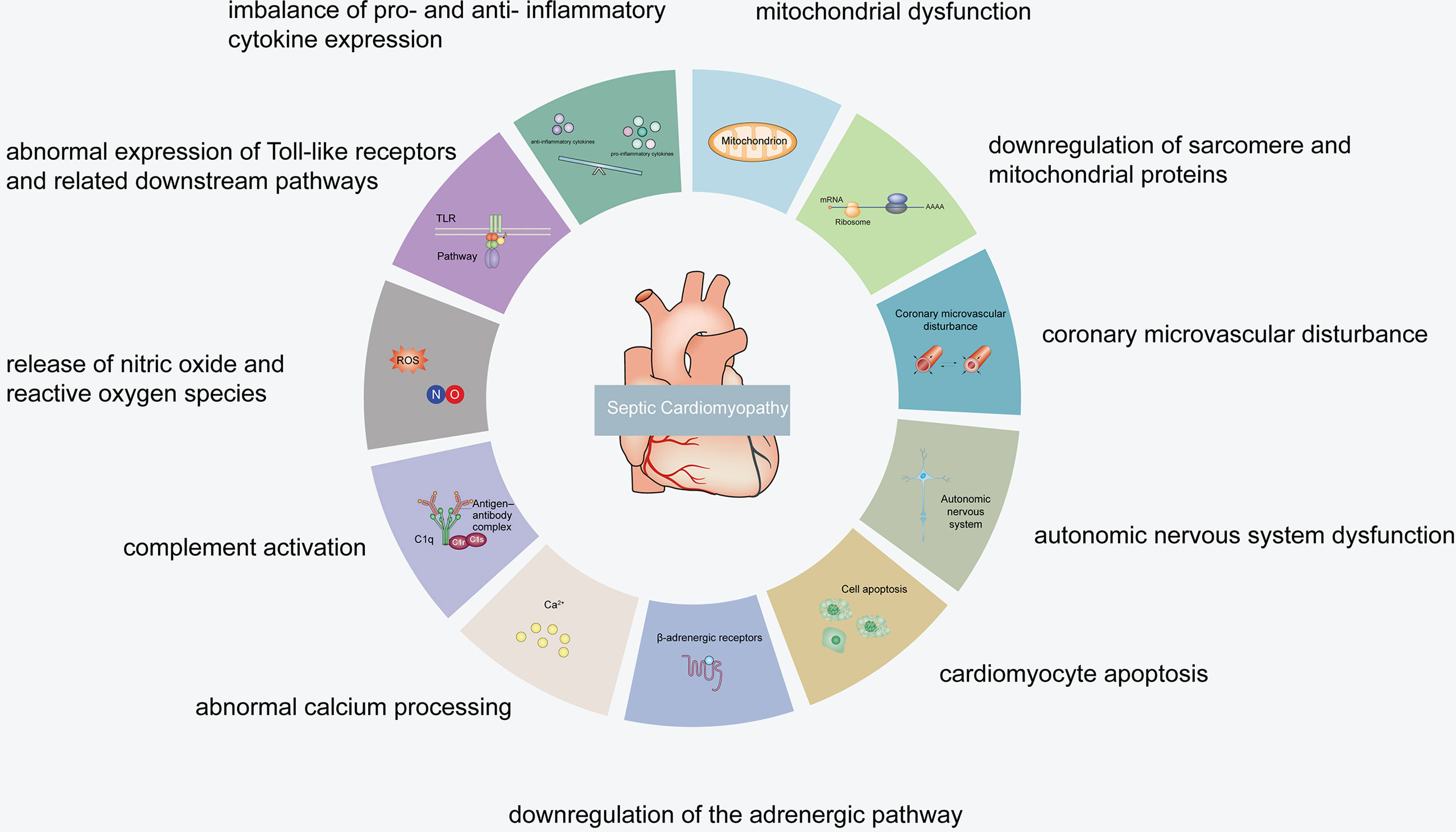
Figure 1 Septic cardiomyopathy pathogenesis.
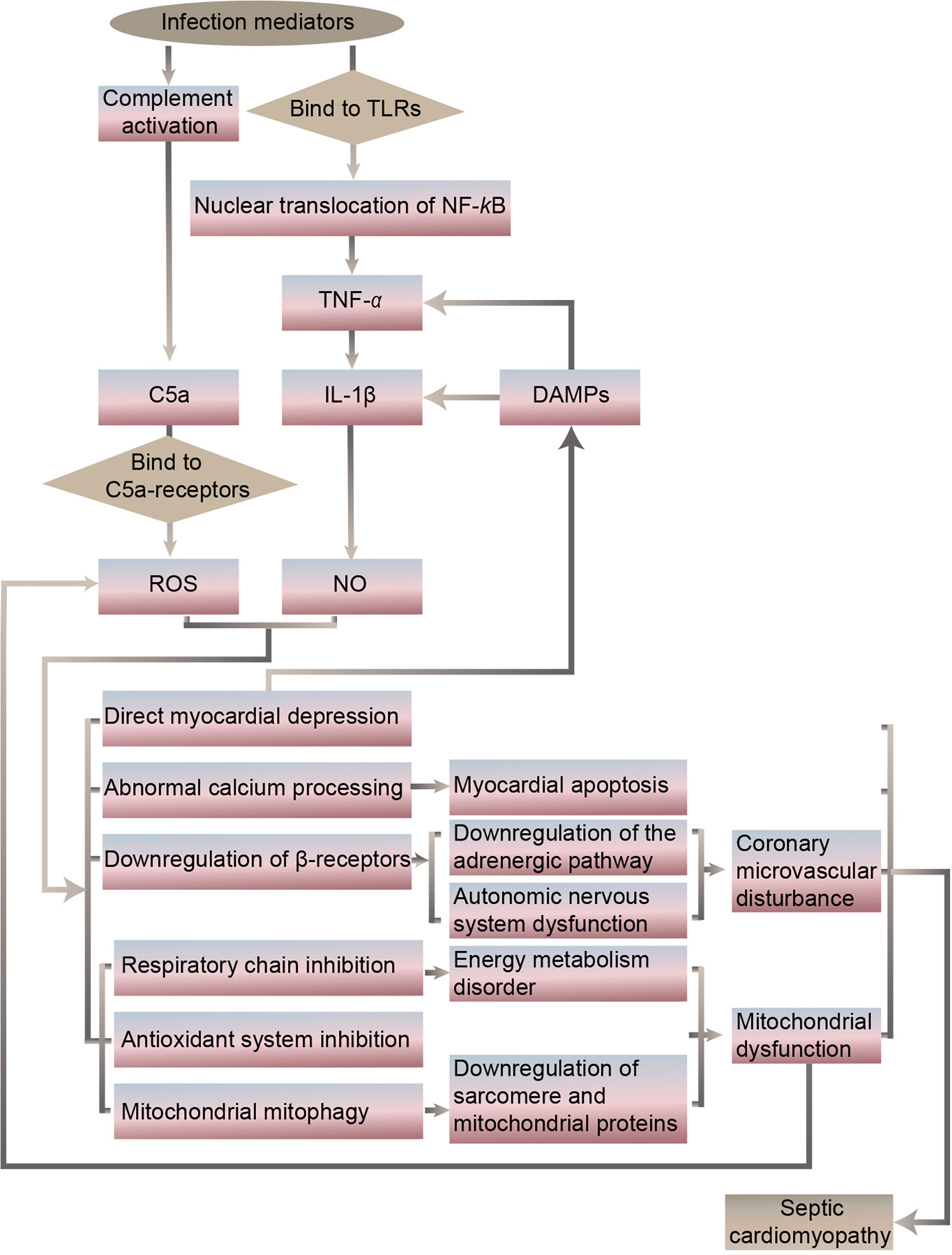
Figure 2 Potential targets of protective intervention in septic cardiomyopathy.
Mechanisms of Mitochondrial Dysfunction in Septic Cardiomyopathy
Recently, researchers have focused on preventing and reducing myocardial damage in septic cardiomyopathy. Among the septic cardiomyopathy pathogenesis, mitochondrial dysfunction deserves to be a focus and further studied (19, 20) (Figure 3). Cardiomyocytes are rich in mitochondria, especially in the areas between sarcomeres and the subsarcolemma (21). As the energy metabolism centres of cells (22), mitochondria function to generate energy through oxidative phosphorylation (OXPHOS) (23). Of the important mechanisms of septic cardiomyopathy, the specific mechanism of mitochondrial dysfunction is under debate. Studies have shown that in the pathogenesis of septic cardiomyopathy, mitochondria undergo relevant changes that lead not only to mitochondrial dysfunction but also to the mitochondrial adaptive response (24, 25).
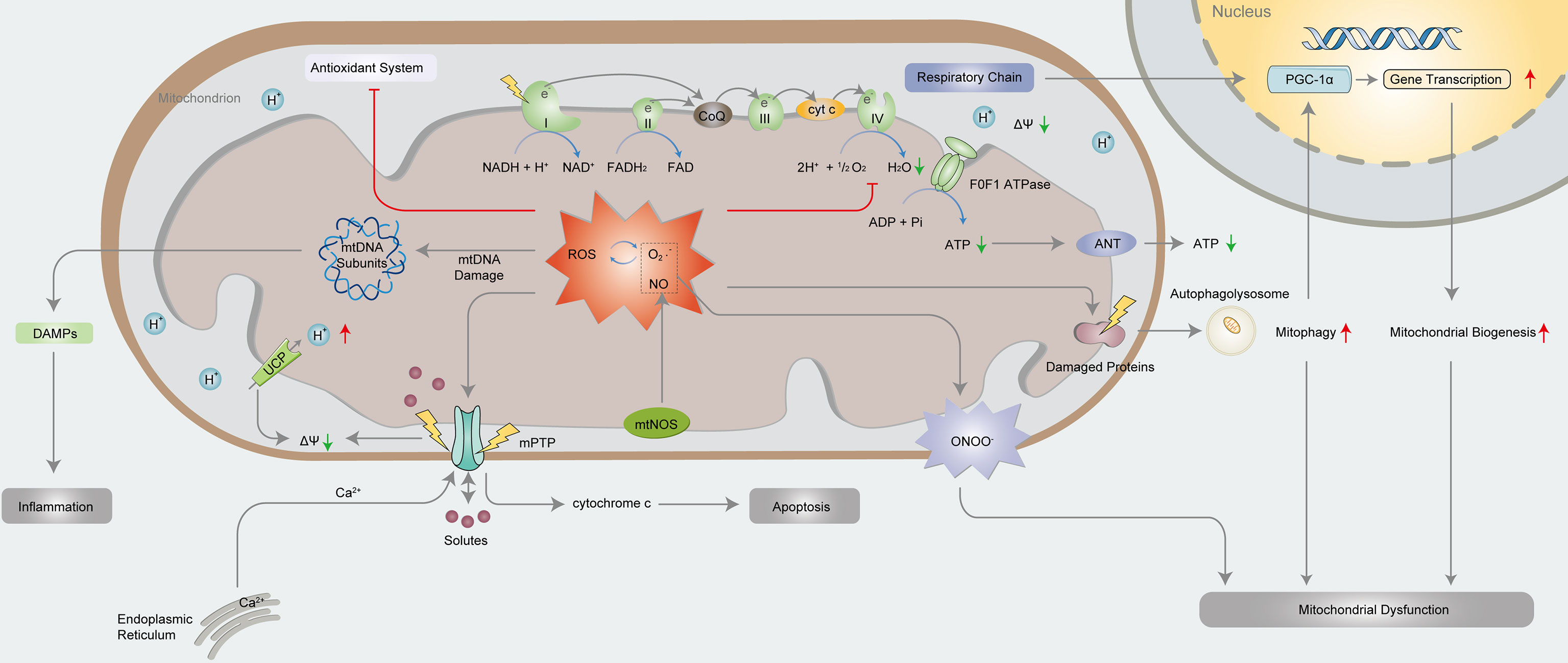
Figure 3 Mechanisms of mitochondrial dysfunction in septic cardiomyopathy.
Mitochondrial Ultrastructural Damage and Decreased ATP Production
In 1994, morphological damage of myocardial mitochondria in septic cardiomyopathy was first described in an animal model (26). Studies have shown that the ultra-microstructural abnormalities of myocardial mitochondria in septic cardiomyopathy include swelling, ridge loss, matrix clearance, rupture of internal vesicles, and damage to internal and external membranes (27, 28), which are closely related to mitochondrial dysfunction (29). Specifically, ultra-microstructural abnormalities lead to the destruction of the OXPHOS process and further reduce adenosine triphosphate (ATP) production. Mitochondria are critical for synthesizing more than 90% of the ATP required by the body (30). The role of the respiratory chain represents the basic function of mitochondria. The respiratory chain is mainly composed of complexes I, II, III and IV (31), and F0F1 ATPase (32). Fatty acid β-oxidation supplies nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2), which respectively transport electrons for OXPHOS through complexes I and II. Subsequently, electrons are transported to complex III, and then to complex IV, leading to the reduction of O2 to H2O. Finally, ATP is generated under the effection of F0F1 ATPase in the mitochondrial inner membrane (33–35).
NO Production and Oxidative Stress
Sepsis is accompanied by the excessive production of NO, ROS and inflammatory cytokines (36), leading to mitochondrial dysfunction (37). Mitochondria produce NO through mitochondria NOS (mtNOS), which inhibits cytochrome c oxidase to regulate mitochondrial respiration (38). NO and O2·− produce ONOO− through diffusion-controlled reactions (39). ONOO− is a strong oxidant that can lead to direct oxidation or nitrosation damage, inhibit the OXPHOS complex and reduce O2 consumption (40, 41). Studies have shown that knockout of inducible NOS (iNOS) can attenuate injury induced by oxidative stress, impaired OXPHOS or reduced ATP synthesis, revealing the vital role of ONOO− in regulating mitochondrial dysfunction in septic cardiomyopathy (42). The increase in ROS production, especially O2·−, leads to excessive endogenous antioxidant capacity in the body (43). In turn, the excessive production of O2·− leads to further production of ROS in mitochondria, creating a vicious cycle of oxidative stress (44, 45). Excessive ROS induce protein denaturation and directly cause oxidative damage to DNA (46), which is particularly serious because mitochondrial DNA is related to the electron transport chain (ETC) (47). Furthermore, metalloproteinases and other proteases are activated, causing further functional deterioration of a variety of proteins, including antioxidant enzymes (48).
Calcium Overload and Changes in Mitochondrial Membrane Permeability
Cytoplasmic calcium homeostasis is impaired in cardiomyocytes in septic cardiomyopathy, and Ca2+ enters mitochondria through unidirectional transporters (49). In addition, the rapid oscillation of Ca2+ between mitochondria and endoplasmic reticulum also leads to mitochondrial Ca2+ overload, which further initiates the opening of mitochondrial permeability transition pore (mPTP) (50). The outer mitochondrial membrane is highly permeable, substances with molecular weights less than 1500 kDa can pass through it, while the inner mitochondrial membrane allows only substances with molecular weights less than 1.5 kDa to pass through it (51). Proton pumps in the inner mitochondrial membrane pump protons from mitochondrial matrix to outer chamber, forming a potential difference between inside and outside mitochondria, which is called the mitochondrial membrane potential (ΔΨm) (52). The mPTP opens intermittently in physiological state, and protons or positive ions in the outer chamber enter the inner chamber because of the potential difference, preventing the excessive accumulation of positive ions in the outer chamber (53). With Ca2+ overload, persistent oxidative stress, adenosine deficiency, increased phosphate concentration and mitochondrial depolarization occur, and then, the mPTP is in a mostly irreversibly opened state (54). The ΔΨM decreases rapidly, leading to ion imbalance, mitochondrial swelling and ATP depletion (55). Moreover, mPTP opening leads to the release of cytochrome c into the cytoplasm, which participates in forming apoptotic bodies with APAF-1 and the precursors of caspase-9. Apoptotic bodies activate caspase-9 facilitated by deoxy-ATP (dATP), and caspase-9 then enzymatically cleaves caspase-3 to activate it, which starts the caspase-induced apoptosis cascade of reactions that ultimately leads to cell apoptosis (56, 57). In addition, electrons produced by the mitochondrial ETC can no longer be transported to oxygen molecules, resulting in the termination of OXPHOS and the inhibition of ATP synthesis (58). To maintain the ΔΨm, mitochondria then negatively regulate F0F1 ATP synthase, leading to hydrolysis of the remaining ATP (59).
Mitochondrial Biogenesis and Mitophagy
The levels of NO, ROS and the ratio of adenosine monophosphate (AMP)/ATP increase during septic cardiomyopathy. These changes trigger mitochondrial biogenesis (60). The main mechanism of mitochondrial biogenesis is the activation of the PGC family, especially PGC-1 α. PGC-1 α is synergistically activated, and its expression leads to the increasing expression of transcription factors, mediating the expression of nuclear proteins required for the transcription and replication of nucleus- and mitochondria-encoded OXPHOS subunits and mitochondrial DNA, transcription of OXPHOS assembly factor and mitochondrial protein import components (61). Mitochondrial biogenesis stands for the growth and division of mitochondria (62). The recovery of cardiac function in septic cardiomyopathy depends partly on mitochondrial biogenesis (63). The mechanism of mitochondrial biogenesis is debated. Some studies have shown that the clearance of damaged mitochondria in sepsis can be compensated by mitochondrial biogenesis rate, producing new mitochondria. However, other studies have shown that mitochondrial biogenesis, even as a compensatory mechanism of mitochondrial dysfunction, may lead to greater mitochondrial dysfunction by disrupting the complicated processes of gene transcription and mitochondrial dynamics. In any case, mitochondrial biogenesis in septic cardiomyopathy is insufficient to compensate for mitochondrial dysfunction (64, 65). The process opposing mitochondrial biogenesis is mitochondrial autophagy (66). Mitochondrial autophagy is a mechanism by which mitochondria eliminate dysfunctional mitochondria (67). However, it is unclear whether mitochondria clear dysfunctional mitochondria only through autophagic mechanisms and/or whether autophagy is involved in programmed cell death in septic cardiomyopathy. Recent research has not clarified the relationship between mitochondrial biogenesis and mitochondrial autophagy.
The recovery of mitochondrial function is closely related to the reversal of cardiac pump function; therefore, an increasing number of in-depth targeted intervention studies are needed to prevent or even reverse mitochondrial dysfunction. Guidelines for systematic evaluation of sepsis can improve prognosis and reduce mortality. However, there is no specific treatment for sepsis complicated with damage to some organs, including the heart. Further studies on the mechanisms of mitochondrial dysfunction in septic cardiomyopathy may supply a novel strategy to supplement the treatment options.
Epigenetics and LncRNAs
Epigenetics is the study of stable genetic phenotype inheritance that does not intervene the gene sequence (68). Epigenetic modifications regulate many biological processes, including development and cell differentiation and proliferation (69). Currently, epigenetic mechanisms include the modification of DNA and proteins closely related to DNA. That is, epigenetic studies focus on the modification and influence of chromatin structure, mainly including chromatin remodelling (including advanced folding of chromatin and connections with the nuclear matrix), DNA methylation, histone modification and noncoding RNA-related mechanisms (70, 71). The reversibility of epigenetic regulation provides a targeted treatment strategy for epigenetically modified components and new ideas for innovative clinical treatment methods.
LncRNAs are endogenous RNAs with transcript lengths of more than 200 nucleotides, which do not possess the function of encoding protein. NcRNAs account for 98% of the human genome, and lncRNAs account for 80-90% of all ncRNAs (72, 73). LncRNAs are currently considered to be key epigenetic regulators (74). With increasing and in-depth research on whole-genome sequencing and function, the structure and function of lncRNAs have been found to be particularly complex (75). Although there is no consensus on the functional classification of lncRNAs, four main types are currently recognized: signals, decoys, guides and scaffolds (76). As signals or decoys, lncRNAs participate in the activation or inhibition of gene. As guides, they enlist chromatin-modifying enzymes to regulate gene expression in a cis/trans manner. As scaffolds, they enlist a variety of proteins to synthesize ribonucleoprotein complexes that regulate chromatin or histones (77). According to the classification of gene structure, lncRNAs are mainly divided into sense lncRNAs, antisense lncRNAs, intronic lncRNAs, long intergenic lncRNAs (or lincRNAs), enhancer RNAs (or erRNAs), and circular RNAs (or circRNAs) (78). LncRNAs interact with various molecules to form RNA-RNA, RNA-DNA and RNA-protein complexes, which play important roles in chromatin modification (79).
LncRNAs and Cardiovascular Diseases
Mutation or abnormal expression of lncRNAs is closely relevant to cardiovascular diseases (80, 81). Published research results mainly refer to MIAT, ANRIL, LIPCAR, and Braveheart. As early as 2006, scholars explored the relationship between MIAT and myocardial infarction. MIAT single-nucleotide polymorphisms can cause changes in the expression of myocardial infarction-related proteins (82). Overexpression of ANRIL can change sites of chromosome 9p21 that are closely relevant to the pathogenesis of coronary atherosclerosis (83). Further studies showed that ANRIL expression was positively related to the severity of coronary atherosclerosis (84). It was discovered that LIPCAR expression was upregulated during the early stage of heart failure and downregulated during the late stage, and therefore, changes in LIPCAR expression can be used to predict the risk of late cardiovascular events (85). It has been confirmed that Braveheart is closely relevant to the differentiation of mouse cardiomyocytes. Studies have shown that PRC2 can inhibit the genes necessary for the differentiation and development of cardiac cells, such as the MesP1 gene, and Braveheart can interact with SUZ12 in the PRC2 complex to further control the expression of MesP1. When the expression of Braveheart is lower than normal, mouse embryonic stem cells did not differentiate into normal cardiomyocytes, which limited heart development (86).
Effects of Regulated LncRNA Expression on Mitochondrial Function
Mitochondria are important multifunctional organelles participating in various basic biological processes (87). The integrality of mitochondrial structure and function is significant to maintain the stability of the intracellular environment. Currently, it is generally believed that the stability of the intracellular environment depends on various mitochondrial pathways regulating energy conversion and ATP production, involving ETC and tricarboxylic acid cycle (TCA) (88). Mitochondria have genetic system independent of the nucleus, and the mitochondrial genome has a complete expression mechanism (89). However, the scale of the mitochondrial genome is small (90). The biological function of mitochondria does not solely depend on the mitochondrial genome; it also depends on nucleus-encoded proteins, which are synthesized in the cytoplasm and transported into mitochondria through specific mechanisms. In other words, mitochondrial energy metabolism and intracellular environment stability depend on the simultaneously coordinated regulation and expression of the nuclear genome and mitochondrial genome (91). Increasing evidence has shown that lncRNAs can act as messengers between nucleus and mitochondria, and participate in regulating of diverse pathways (92). However, the potential regulatory mechanisms may be very complex, and relevant research is ongoing.
LncRNAs can regulate mitochondrial function and dynamics at different levels (93). Abnormal regulation of lncRNAs leads to abnormal synthesis of ATP and ROS, thus contributing to the pathological development of many diseases. Currently, research on lncRNA regulation of mitochondrial function mainly focuses on cardiovascular diseases, neurodegenerative diseases and tumour diseases (94–96). As mentioned above, cardiomyocytes are enriched with many mitochondria, and mitochondrial dysfunction is closely relevant to the pathogenesis of cardiovascular diseases.
Effects of LncRNA Regulation on Mitochondrial Dysfunction in Septic Cardiomyopathy
As previously mentioned, various mechanisms of mitochondrial dysfunction in septic cardiomyopathy have been reported. According to the literature, recent research on lncRNAs participating in the regulation of mitochondrial dysfunction in septic cardiomyopathy has mainly focused on decreases in ATP production, mitochondrial NO production and oxidative stress. Additionally, studies have shown that lipopolysaccharide (LPS) can induce an increase in ROS, a decrease in ΔΨm, the release of cytochrome c, and the upregulation of caspase-9 and caspase-3 in the cytoplasm, ultimately leading to cardiomyocyte apoptosis (97).
Cheng Xing Peng et al. explored the regulatory role of MIAT in septic myocardial injury. They found that MIAT knockdown significantly inhibited the production of mitochondrial ROS in LPS-treated HL-1 cells. In addition, the ratio of reduced glutathione to oxidized glutathione (GSH/GSSH) decreased with increasing malondialdehyde (MDA) content. This result suggested that MIAT aggravated myocardial damage by promoting oxidative stress. It was confirmed that MIAT acted on miR-330-5p directly to upregulate the TRAF6/NF-κB pathway, promoting inflammation and oxidative stress in LPS-induced cardiomyopathy (98).
RMRP inhibits the posttranscriptional regulatory effect of miR-1-5p on HSPA4 in LPS-induced mitochondrial damage. Overexpression of RMRP can significantly inhibit the decline in ΔΨm, the level of intracellular ROS, and the expression of cytoplasmic cytochrome c, caspase-9 and caspase-3, thereby inhibiting cardiomyocyte apoptosis (99). Bin Shan et al. discussed H19 regulation in septic cardiomyopathy. H19 can reduce mitochondrial inner membrane damage by regulating mitochondrial membrane potential by regulating miR-93-5p/SORBS2 pathway, thereby inhibiting mitochondrial apoptosis. Inflammatory factors, involving TNF-α, IL-1β and IL-6, were markedly downregulated in LPS-induced cardiomyocytes overexpressing H19. The expression of cytochrome c in mitochondria was upregulated, while that in cytoplasm was downregulated. This result indicated that the overexpression of H19 alleviated inflammation and mitochondrial apoptosis in LPS-induced cardiomyocytes (100). Studies have also pointed out that knocking down SOX2OT can significantly enhance cardiac function, inhibit the decline in ΔΨm, and reduce the production of mitochondrial ROS in mice with septic cardiomyopathy, while upregulating SOX2OT can reverse all of these effects. Through further research on the regulatory mechanism, it was ultimately concluded that SOX2OT aggravated mitochondrial dysfunction by downregulating the expression of SOX2, thereby affecting the prognosis of septic cardiomyopathy (101).
Studies on the involvement of lncRNAs regulating mitochondrial energy metabolism in septic cardiomyopathy are also ongoing. Dongshi Liang et al. found that the increased expression of Xist is related to the decreased level of both PGC-1α and ATP, which suggested that inhibiting the expression of Xist enhanced the production of ATP, reducing sepsis-induced myocardial injury (102).
Although the aforementioned lncRNAs have been confirmed to participate in septic cardiomyopathy by regulating mitochondrial function and apoptosis, it is still unclear whether other lncRNAs are involved in regulating mitochondrial functions, and the specific regulatory mechanisms of participating lncRNAs are also unknown. To date, using gene chip hybridization technology, researchers at Zhejiang University identified 471 upregulated lncRNAs and 804 downregulated lncRNAs in myocardial tissues of septic mice. Ultimately, this group found that partial lncRNAs are mainly enriched in inflammation, immunity, energy metabolism and cell death, and predicted that certain lncRNAs may participate in mitochondrial dysfunction (103). All these results provide strong theoretical support for the continuing study of the involvement of lncRNAs in mitochondrial dysfunction in septic cardiomyopathy.
Conclusion and Perspective
LncRNAs will increasingly become targets for the intervention and treatment of septic cardiomyopathy, and the mechanism to target is closely related to lncRNA involvement in mitochondrial dysfunction. Finding intervention measures to prevent and treat mitochondrial damage is significant to improved treatment and prognosis of patients with septic cardiomyopathy. Although research on biomarkers for use in assessing the severity and prognosis of septic cardiomyopathy is ongoing, no clear markers with both sufficient sensitivity and specificity have been identified to date. Recent research has found that CitH3 may be recognized as a reliable blood biomarker for diagnosis and prognosis of sepsis (104). LncRNAs may be potential biomarkers for evaluating the severity and prognosis of septic cardiomyopathy, and they will also be the focus of the next phase of our research.
Author Contributions
WC and SL conceived the review. SL wrote the manuscript. WC revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by the project of scientific-technology plan in Shenyang, China (Grant number 19-112-4-068).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Fernando SM, Rochwerg B, Seely A. Clinical Implications of the Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). CMAJ (2018) 190(36):E1058–9. doi: 10.1503/cmaj.170149
2. Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving Sepsis Campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med (2017) 43(3):304–77. doi: 10.1007/s00134-017-4683-6
3. Morgan RW, Fitzgerald JC, Weiss SL, Nadkarni VM, Sutton RM, Berg RA. Sepsis-Associated in-Hospital Cardiac Arrest: Epidemiology, Pathophysiology, and Potential Therapies. J Crit Care (2017) 40:128–35. doi: 10.1016/j.jcrc.2017.03.023
4. Lu F, Hong Y, Liu L, Wei N, Lin Y, He J, et al. Long Noncoding Rnas: A Potential Target in Sepsis-Induced Cellular Disorder. Exp Cell Res (2021) 406(2):112756. doi: 10.1016/j.yexcr.2021.112756
5. Dombrovskiy VY, Martin AA, Sunderram J, Paz HL. Rapid Increase in Hospitalization and Mortality Rates for Severe Sepsis in the United States: A Trend Analysis From 1993 to 2003. Crit Care Med (2007) 35(5):1244–50. doi: 10.1097/01.CCM.0000261890.41311.E9
6. Parker MM, Shelhamer JH, Bacharach SL, Green MV, Natanson C, Frederick TM, et al. Profound But Reversible Myocardial Depression in Patients With Septic Shock. Ann Intern Med (1984) 100(4):483–90. doi: 10.7326/0003-4819-100-4-483
7. Beesley SJ, Weber G, Sarge T, Nikravan S, Grissom CK, Lanspa MJ, et al. Septic Cardiomyopathy. Crit Care Med (2018) 46(4):625–34. doi: 10.1097/CCM.0000000000002851
8. Zanotti-Cavazzoni SL, Hollenberg SM. Cardiac Dysfunction in Severe Sepsis and Septic Shock. Curr Opin Crit Care (2009) 15(5):392–7. doi: 10.1097/MCC.0b013e3283307a4e
9. Ehrman RR, Sullivan AN, Favot MJ, Sherwin RL, Reynolds CA, Abidov A, et al. Pathophysiology, Echocardiographic Evaluation, Biomarker Findings, and Prognostic Implications of Septic Cardiomyopathy: A Review of the Literature. Crit Care (2018) 22(1):112. doi: 10.1186/s13054-018-2043-8
10. Kakihana Y, Ito T, Nakahara M, Yamaguchi K, Yasuda T. Sepsis-Induced Myocardial Dysfunction: Pathophysiology and Management. J Intensive Care (2016) 4:22. doi: 10.1186/s40560-016-0148-1
11. Heureux ML, Sternberg M, Brath L, Turlington J, Kashiouris MG. Sepsis-Induced Cardiomyopathy: A Comprehensive Review. Curr Cardiol Rep (2020) 22(5):35. doi: 10.1007/s11886-020-01277-2
12. Flynn A, Chokkalingam Mani B, Mather PJ. Sepsis-Induced Cardiomyopathy: A Review of Pathophysiologic Mechanisms. Heart Fail Rev (2010) 15(6):605–11. doi: 10.1007/s10741-010-9176-4
13. Ravikumar N, Sayed MA, Poonsuph CJ, Sehgal R, Shirke MM, Harky A. Septic Cardiomyopathy: From Basics to Management Choices. Curr Probl Cardiol (2021) 46(4):100767. doi: 10.1016/j.cpcardiol.2020.100767
14. Wang R, Xu Y, Fang Y, Wang C, Xue Y, Wang F, et al. Pathogenetic Mechanisms of Septic Cardiomyopathy. J Cell Physiol (2021) 2021:1–10. doi: 10.1002/jcp.30527
15. Hollenberg SM, Singer M. Pathophysiology of Sepsis-Induced Cardiomyopathy. Nat Rev Cardiol (2021) 18(6):424–34. doi: 10.1038/s41569-020-00492-2
16. Martin L, Derwall M, Al Zoubi S, Zechendorf E, Reuter DA, Thiemermann C, et al. The Septic Heart: Current Understanding of Molecular Mechanisms and Clinical Implications. Chest (2019) 155(2):427–37. doi: 10.1016/j.chest.2018.08.1037
17. Tsolaki V, Makris D, Mantzarlis K, Zakynthinos E. Sepsis-Induced Cardiomyopathy: Oxidative Implications in the Initiation and Resolution of the Damage. Oxid Med Cell Longev (2017) 2017:7393525. doi: 10.1155/2017/7393525
18. Lin H, Wang W, Lee M, Meng Q, Ren H. Current Status of Septic Cardiomyopathy: Basic Science and Clinical Progress. Front Pharmacol (2020) 11:210. doi: 10.3389/fphar.2020.00210
19. Cimolai MC, Alvarez S, Bode C, Bugger H. Mitochondrial Mechanisms in Septic Cardiomyopathy. Int J Mol Sci (2015) 16(8):17763–78. doi: 10.3390/ijms160817763
20. Harrois A, Huet O, Duranteau J. Alterations of Mitochondrial Function in Sepsis and Critical Illness. Curr Opin Anaesthesiol (2009) 22(2):143–9. doi: 10.1097/ACO.0b013e328328d1cc
21. Barth E, Stämmler G, Speiser B, Schaper J. Ultrastructural Quantitation of Mitochondria and Myofilaments in Cardiac Muscle From 10 Different Animal Species Including Man. J Mol Cell Cardiol (1992) 24(7):669–81. doi: 10.1016/0022-2828(92)93381-S
22. Mokranjac D, Neupert W. Energetics of Protein Translocation Into Mitochondria. Biochim Biophys Acta (2008) 1777(7-8):758–62. doi: 10.1016/j.bbabio.2008.04.009
23. Preau S, Vodovar D, Jung B, Lancel S, Zafrani L, Flatres A, et al. Energetic Dysfunction in Sepsis: A Narrative Review. Ann Intensive Care (2021) 11(1):104. doi: 10.1186/s13613-021-00893-7
24. Schwabbauer ML. Use of the Latent Image Technique to Develop and Evaluate Problem-Solving Skills. Am J Med Technol (1975) 41(12):457–62.
25. Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox Homeostasis and Mitochondrial Dynamics. Cell Metab (2015) 22(2):207–18. doi: 10.1016/j.cmet.2015.06.006
26. Solomon MA, Correa R, Alexander HR, Koev LA, Cobb JP, Kim DK, et al. Myocardial Energy Metabolism and Morphology in a Canine Model of Sepsis. Am J Physiol (1994) 266(2 Pt 2):H757–68. doi: 10.1152/ajpheart.1994.266.2.H757
27. Song JH, Yoon SY, Park TY, Heo EY, Kim DK, Chung HS, et al. The Clinical Impact of Drug-Induced Hepatotoxicity on Anti-Tuberculosis Therapy: A Case Control Study. Respir Res (2019) 20(1):283. doi: 10.1186/s12931-019-1256-y
28. Vanasco V, Saez T, Magnani ND, Pereyra L, Marchini T, Corach A, et al. Cardiac Mitochondrial Biogenesis in Endotoxemia Is Not Accompanied by Mitochondrial Function Recovery. Free Radic Biol Med (2014) 77:1–9. doi: 10.1016/j.freeradbiomed.2014.08.009
29. Capetanaki Y. Desmin Cytoskeleton: A Potential Regulator of Muscle Mitochondrial Behavior and Function. Trends Cardiovasc Med (2002) 12(8):339–48. doi: 10.1016/S1050-1738(02)00184-6
30. Durand A, Duburcq T, Dekeyser T, Neviere R, Howsam M, Favory R, et al. Involvement of Mitochondrial Disorders in Septic Cardiomyopathy. Oxid Med Cell Longev (2017) 2017:4076348. doi: 10.1155/2017/4076348
31. Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The Role of Mitochondria in Metabolism and Cell Death. Biochem Biophys Res Commun (2017) 482(3):426–31. doi: 10.1016/j.bbrc.2016.11.088
32. Fernie AR, Carrari F, Sweetlove LJ. Respiratory Metabolism: Glycolysis, the TCA Cycle and Mitochondrial Electron Transport. Curr Opin Plant Biol (2004) 7(3):254–61. doi: 10.1016/j.pbi.2004.03.007
33. Jonckheere AI, Smeitink JA, Rodenburg RJ. Mitochondrial ATP Synthase: Architecture, Function and Pathology. J Inherit Metab Dis (2012) 35(2):211–25. doi: 10.1007/s10545-011-9382-9
34. Singer M. The Role of Mitochondrial Dysfunction in Sepsis-Induced Multi-Organ Failure. Virulence (2014) 5(1):66–72. doi: 10.4161/viru.26907
35. Szabo I, Zoratti M. Mitochondrial Channels: Ion Fluxes and More. Physiol Rev (2014) 94(2):519–608. doi: 10.1152/physrev.00021.2013
36. Prauchner CA. Oxidative Stress in Sepsis: Pathophysiological Implications Justifying Antioxidant Co-Therapy. Burns (2017) 43(3):471–85. doi: 10.1016/j.burns.2016.09.023
37. Balija TM, Lowry SF. Lipopolysaccharide and Sepsis-Associated Myocardial Dysfunction. Curr Opin Infect Dis (2011) 24(3):248–53. doi: 10.1097/QCO.0b013e32834536ce
38. Kanai AJ, Pearce LL, Clemens PR, Birder LA, VanBibber MM, Choi SY, et al. Identification of a Neuronal Nitric Oxide Synthase in Isolated Cardiac Mitochondria Using Electrochemical Detection. Proc Natl Acad Sci USA (2001) 98(24):14126–31. doi: 10.1073/pnas.241380298
39. Poli G. Introduction–Serial Review: Reactive Oxygen and Nitrogen in Inflammation(1,2). Free Radic Biol Med (2002) 33(3):301–2. doi: 10.1016/S0891-5849(02)00964-4
40. Hauser B, Bracht H, Matejovic M, Radermacher P, Venkatesh B. Nitric Oxide Synthase Inhibition in Sepsis? Lessons Learned From Large-Animal Studies. Anesth Analg (2005) 101(2):488–98. doi: 10.1213/01.ANE.0000177117.80058.4D
41. Chen X, Chen H, Deng R, Shen J. Pros and Cons of Current Approaches for Detecting Peroxynitrite and Their Applications. BioMed J (2014) 37(3):120–6. doi: 10.4103/2319-4170.134084
42. Escames G, López LC, Ortiz F, López A, García JA, Ros E, et al. Attenuation of Cardiac Mitochondrial Dysfunction by Melatonin in Septic Mice. FEBS J (2007) 274(8):2135–47. doi: 10.1111/j.1742-4658.2007.05755.x
43. Neri M, Riezzo I, Pomara C, Schiavone S, Turillazzi E. Oxidative-Nitrosative Stress and Myocardial Dysfunctions in Sepsis: Evidence From the Literature and Postmortem Observations. Mediators Inflamm (2016) 2016:3423450. doi: 10.1155/2016/3423450
44. Chen YR, Zweier JL. Cardiac Mitochondria and Reactive Oxygen Species Generation. Circ Res (2014) 114(3):524–37. doi: 10.1161/CIRCRESAHA.114.300559
45. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol Rev (2014) 94(3):909–50. doi: 10.1152/physrev.00026.2013
46. Lee HC, Wei YH. Mitochondrial Biogenesis and Mitochondrial DNA Maintenance of Mammalian Cells Under Oxidative Stress. Int J Biochem Cell Biol (2005) 37(4):822–34. doi: 10.1016/j.biocel.2004.09.010
47. Gonzalez AS, Elguero ME, Finocchietto P, Holod S, Romorini L, Miriuka SG, et al. Abnormal Mitochondrial Fusion-Fission Balance Contributes to the Progression of Experimental Sepsis. Free Radic Res (2014) 48(7):769–83. doi: 10.3109/10715762.2014.906592
48. von Dessauer B, Bongain J, Molina V, Quilodrán J, Castillo R, Rodrigo R. Oxidative Stress as a Novel Target in Pediatric Sepsis Management. J Crit Care (2011) 26(1):103.e1–7. doi: 10.1016/j.jcrc.2010.05.001
49. Joseph LC, Kokkinaki D, Valenti MC, Kim GJ, Barca E, Tomar D, et al. Inhibition of NADPH Oxidase 2 (NOX2) Prevents Sepsis-Induced Cardiomyopathy by Improving Calcium Handling and Mitochondrial Function. JCI Insight (2017) 2(17):e94248. doi: 10.1172/jci.insight.94248
50. Halestrap AP, McStay GP, Clarke SJ. The Permeability Transition Pore Complex: Another View. Biochimie (2002) 84(2-3):153–66. doi: 10.1016/S0300-9084(02)01375-5
51. Kwong JQ, Molkentin JD. Physiological and Pathological Roles of the Mitochondrial Permeability Transition Pore in the Heart. Cell Metab (2015) 21(2):206–14. doi: 10.1016/j.cmet.2014.12.001
52. Kim JE, He Q, Chen Y, Shi C, Yu K. mTOR-Targeted Therapy: Differential Perturbation to Mitochondrial Membrane Potential and Permeability Transition Pore Plays a Role in Therapeutic Response. Biochem Biophys Res Commun (2014) 447(1):184–91. doi: 10.1016/j.bbrc.2014.03.124
53. Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol (2016) 26(9):655–67. doi: 10.1016/j.tcb.2016.04.006
54. Teixeira J, Basit F, Swarts HG, Forkink M, Oliveira PJ, Willems PHGM, et al. Extracellular Acidification Induces ROS- and Mptp-Mediated Death in HEK293 Cells. Redox Biol (2018) 15:394–404. doi: 10.1016/j.redox.2017.12.018
55. Bernardi P, Di Lisa F. The Mitochondrial Permeability Transition Pore: Molecular Nature and Role as a Target in Cardioprotection. J Mol Cell Cardiol (2015) 78:100–6. doi: 10.1016/j.yjmcc.2014.09.023
56. Pérez MJ, Quintanilla RA. Development or Disease: Duality of the Mitochondrial Permeability Transition Pore. Dev Biol (2017) 426(1):1–7. doi: 10.1016/j.ydbio.2017.04.018
57. Elrod JW, Molkentin JD. Physiologic Functions of Cyclophilin D and the Mitochondrial Permeability Transition Pore. Circ J (2013) 77(5):1111–22. doi: 10.1253/circj.CJ-13-0321
58. He J, Carroll J, Ding S, Fearnley IM, Walker JE. Permeability Transition in Human Mitochondria Persists in the Absence of Peripheral Stalk Subunits of ATP Synthase. Proc Natl Acad Sci USA (2017) 114(34):9086–91. doi: 10.1073/pnas.1711201114
59. Di Lisa F, Canton M, Menabò R, Kaludercic N, Bernardi P. Mitochondria and Cardioprotection. Heart Fail Rev (2007) 12(3-4):249–60. doi: 10.1007/s10741-007-9028-z
60. Wenz T. Regulation of Mitochondrial Biogenesis and PGC-1α Under Cellular Stress. Mitochondrion (2013) 13(2):134–42. doi: 10.1016/j.mito.2013.01.006
61. Kelly DP, Scarpulla RC. Transcriptional Regulatory Circuits Controlling Mitochondrial Biogenesis and Function. Genes Dev (2004) 18(4):357–68. doi: 10.1101/gad.1177604
62. Jornayvaz FR, Shulman GI. Regulation of Mitochondrial Biogenesis. Essays Biochem (2010) 47:69–84. doi: 10.1042/bse0470069
63. Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, et al. Cardiac-Specific Induction of the Transcriptional Coactivator Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1alpha Promotes Mitochondrial Biogenesis and Reversible Cardiomyopathy in a Developmental Stage-Dependent Manner. Circ Res (2004) 94(4):525–33. doi: 10.1161/01.RES.0000117088.36577.EB
64. Reynolds CM, Suliman HB, Hollingsworth JW, Welty-Wolf KE, Carraway MS, Piantadosi CA. Nitric Oxide Synthase-2 Induction Optimizes Cardiac Mitochondrial Biogenesis After Endotoxemia. Free Radic Biol Med (2009) 46(5):564–72. doi: 10.1016/j.freeradbiomed.2008.11.007
65. Li Y, Feng YF, Liu XT, Li YC, Zhu HM, Sun MR, et al. Songorine Promotes Cardiac Mitochondrial Biogenesis via Nrf2 Induction During Sepsis. Redox Biol (2021) 38:101771. doi: 10.1016/j.redox.2020.101771
66. Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and Death Partners: Apoptosis, Autophagy and the Cross-Talk Between Them. Cell Death Differ (2009) 16(7):966–75. doi: 10.1038/cdd.2009.33
67. Lo Verso F, Carnio S, Vainshtein A, Sandri M. Autophagy Is Not Required to Sustain Exercise and PRKAA1/AMPK Activity But Is Important to Prevent Mitochondrial Damage During Physical Activity. Autophagy (2014) 10(11):1883–94. doi: 10.4161/auto.32154
68. Voelter-Mahlknecht S. Epigenetic Associations in Relation to Cardiovascular Prevention and Therapeutics. Clin Epigenet (2016) 8:4. doi: 10.1186/s13148-016-0170-0
69. Pagiatakis C, Di Mauro V. The Emerging Role of Epigenetics in Therapeutic Targeting of Cardiomyopathies. Int J Mol Sci (2021) 22(16):8721. doi: 10.3390/ijms22168721
70. Wang KC, Chang HY. Epigenomics: Technologies and Applications. Circ Res (2018) 122(9):1191–9. doi: 10.1161/CIRCRESAHA.118.310998
71. Matouk CC, Marsden PA. Epigenetic Regulation of Vascular Endothelial Gene Expression. Circ Res (2008) 102(8):873–87. doi: 10.1161/CIRCRESAHA.107.171025
72. Li J, Xuan Z, Liu C. Long Non-Coding RNAs and Complex Human Diseases. Int J Mol Sci (2013) 14(9):18790–808. doi: 10.3390/ijms140918790
73. Piccoli MT, Gupta SK, Thum T. Noncoding RNAs as Regulators of Cardiomyocyte Proliferation and Death. J Mol Cell Cardiol (2015) 89(Pt A):59–67. doi: 10.1016/j.yjmcc.2015.02.002
74. Caley DP, Pink RC, Trujillano D, Carter DR. Long Noncoding RNAs, Chromatin, and Development. ScientificWorldJournal (2010) 10:90–102. doi: 10.1100/tsw.2010.7
75. Ulitsky I, Bartel DP. LincRNAs: Genomics, Evolution, and Mechanisms. Cell (2013) 154(1):26–46. doi: 10.1016/j.cell.2013.06.020
76. Wang KC, Chang HY. Molecular Mechanisms of Long Noncoding RNAs. Mol Cell (2011) 43(6):904–14. doi: 10.1016/j.molcel.2011.08.018
77. Chen Z, Li S, Subramaniam S, Shyy JY, Chien S. Epigenetic Regulation: A New Frontier for Biomedical Engineers. Annu Rev BioMed Eng (2017) 19:195–219. doi: 10.1146/annurev-bioeng-071516-044720
78. Ma L, Bajic VB, Zhang Z. On the Classification of Long Non-Coding RNAs. RNA Biol (2013) 10(6):925–33. doi: 10.4161/rna.24604
79. Chang CP, Han P. Epigenetic and Lncrna Regulation of Cardiac Pathophysiology. Biochim Biophys Acta (2016) 1863(7 Pt B):1767–71. doi: 10.1016/j.bbamcr.2016.03.005
80. Archer K, Broskova Z, Bayoumi AS, Teoh JP, Davila A, Tang Y, et al. Long Non-Coding RNAs as Master Regulators in Cardiovascular Diseases. Int J Mol Sci (2015) 16(10):23651–67. doi: 10.3390/ijms161023651
81. Uchida S, Dimmeler S. Long Noncoding Rnas in Cardiovascular Diseases. Circ Res (2015) 116(4):737–50. doi: 10.1161/CIRCRESAHA.116.302521
82. Ishii N, Ozaki K, Sato H, Mizuno H, Saito S, Takahashi A, et al. Identification of a Novel Non-Coding RNA, MIAT, That Confers Risk of Myocardial Infarction. J Hum Genet (2006) 51(12):1087–99. doi: 10.1007/s10038-006-0070-9
83. Motterle A, Pu X, Wood H, Xiao Q, Gor S, Ng FL, et al. Functional Analyses of Coronary Artery Disease Associated Variation on Chromosome 9p21 in Vascular Smooth Muscle Cells. Hum Mol Genet (2012) 21(18):4021–9. doi: 10.1093/hmg/dds224
84. Holdt LM, Teupser D. Recent Studies of the Human Chromosome 9p21 Locus, Which Is Associated With Atherosclerosis in Human Populations. Arterioscler Thromb Vasc Biol (2012) 32(2):196–206. doi: 10.1161/ATVBAHA.111.232678
85. Kumarswamy R, Bauters C, Volkmann I, Maury F, Fetisch J, Holzmann A, et al. Circulating Long Noncoding RNA, LIPCAR, Predicts Survival in Patients With Heart Failure. Circ Res (2014) 114(10):1569–75. doi: 10.1161/CIRCRESAHA.114.303915
86. Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, et al. Braveheart, a Long Noncoding RNA Required for Cardiovascular Lineage Commitment. Cell (2013) 152(3):570–83. doi: 10.1016/j.cell.2013.01.003
87. Liu X, Shan G. Mitochondria Encoded Non-Coding RNAs in Cell Physiology. Front Cell Dev Biol (2021) 9:713729. doi: 10.3389/fcell.2021.713729
88. Steenbergen C, Das S, Su J, Wong R, Murphy E. Cardioprotection and Altered Mitochondrial Adenine Nucleotide Transport. Basic Res Cardiol (2009) 104(2):149–56. doi: 10.1007/s00395-009-0002-x
89. Yasukawa T, Kang D. An Overview of Mammalian Mitochondrial DNA Replication Mechanisms. J Biochem (2018) 164(3):183–93. doi: 10.1093/jb/mvy058
90. Sharma N, Pasala MS, Prakash A. Mitochondrial DNA: Epigenetics and Environment. Environ Mol Mutagen (2019) 60(8):668–82. doi: 10.1002/em.22319
91. Kaniak-Golik A, Skoneczna A. Mitochondria-Nucleus Network for Genome Stability. Free Radic Biol Med (2015) 82:73–104. doi: 10.1016/j.freeradbiomed.2015.01.013
92. Dong Y, Yoshitomi T, Hu JF, Cui J. Long Noncoding RNAs Coordinate Functions Between Mitochondria and the Nucleus. Epigenet Chromatin (2017) 10(1):41. doi: 10.1186/s13072-017-0149-x
93. Gusic M, Prokisch H. NcRNAs: New Players in Mitochondrial Health and Disease. Front Genet (2020) 11:95. doi: 10.3389/fgene.2020.00095
94. Jusic A, Devaux Y. Mitochondrial Noncoding RNA-Regulatory Network in Cardiovascular Disease. Basic Res Cardiol (2020) 115(3):23. doi: 10.1007/s00395-020-0783-5
95. Rezaei O, Nateghinia S, Estiar MA, Taheri M, Ghafouri-Fard S. Assessment of the Role of Non-Coding RNAs in the Pathophysiology of Parkinson’s Disease. Eur J Pharmacol (2021) 896:173914. doi: 10.1016/j.ejphar.2021.173914
96. Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-Coding RNAs: A New Frontier in the Study of Human Diseases. Cancer Lett (2013) 339(2):159–66. doi: 10.1016/j.canlet.2013.06.013
97. Smeding L, Plötz FB, Groeneveld AB, Kneyber MC. Structural Changes of the Heart During Severe Sepsis or Septic Shock. Shock (2012) 37(5):449–56. doi: 10.1097/SHK.0b013e31824c3238
98. Xing PC, An P, Hu GY, Wang DL, Zhou MJ. LncRNA MIAT Promotes Inflammation and Oxidative Stress in Sepsis-Induced Cardiac Injury by Targeting MiR-330-5p/TRAF6/NF-κb Axis. Biochem Genet (2020) 58(5):783–800. doi: 10.1007/s10528-020-09976-9
99. Han Y, Cai Y, Lai X, Wang Z, Wei S, Tan K, et al. LncRNA RMRP Prevents Mitochondrial Dysfunction and Cardiomyocyte Apoptosis via the MiR-1-5p/Hsp70 Axis in LPS-Induced Sepsis Mice. Inflammation (2020) 43(2):605–18. doi: 10.1007/s10753-019-01141-8
100. Shan B, Li JY, Liu YJ, Tang XB, Zhou Z, Luo LX. LncRNA H19 Inhibits the Progression of Sepsis-Induced Myocardial Injury via Regulation of the MiR-93-5p/SORBS2 Axis. Inflammation (2021) 44(1):344–57. doi: 10.1007/s10753-020-01340-8
101. Chen M, Guan Y, Li A, Zhao YZ, Zhang L, Zhang L, et al. LncRNA SOX2OT Mediates Mitochondrial Dysfunction in Septic Cardiomyopathy. DNA Cell Biol (2019) 38(11):1197–206. doi: 10.1089/dna.2019.4839
102. Liang D, Jin Y, Lin M, Xia X, Chen X, Huang A. Down-Regulation of Xist and MiR-7a-5p Improves LPS-Induced Myocardial Injury. Int J Med Sci (2020) 17(16):2570–7. doi: 10.7150/ijms.45408
103. Shi Y, Zheng X, Zheng M, Wang L, Chen Y, Shen Y. Identification of Mitochondrial Function-Associated LncRNAs in Septic Mice Myocardium. J Cell Biochem (2021) 122(1):53–68. doi: 10.1002/jcb.29831
Keywords: lncRNA, epigenetics, sepsis, septic cardiomyopathy, mitochondrial dysfunction
Citation: Liu S and Chong W (2021) Roles of LncRNAs in Regulating Mitochondrial Dysfunction in Septic Cardiomyopathy. Front. Immunol. 12:802085. doi: 10.3389/fimmu.2021.802085
Received: 26 October 2021; Accepted: 10 November 2021;
Published: 24 November 2021.
Edited by:
Veron Ramsuran, University of KwaZulu-Natal, South AfricaReviewed by:
Peter A. Ward, University of Michigan, United StatesCopyright © 2021 Liu and Chong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Chong, d2Nob25nQGNtdS5lZHUuY24=