 Qianyan He1†
Qianyan He1† Yinzhong Ma2†
Yinzhong Ma2† Jie Liu1
Jie Liu1 Dianhui Zhang1
Dianhui Zhang1 Jiaxin Ren1Ruoyu Zhao1
Jiaxin Ren1Ruoyu Zhao1 JunLei Chang2Zhen-Ni Guo1*Yi Yang1*
JunLei Chang2Zhen-Ni Guo1*Yi Yang1*- 1Department of Neurology, The First Hospital of Jilin University, Changchun, China
- 2Shenzhen Key Laboratory of Biomimetic Materials and Cellular Immunomodulation, Institute of Biomedicine and Biotechnology, Shenzhen Institute of Advanced Technology, Chinese Academy of Sciences, Shenzhen, China
Ischemic stroke is caused by insufficient cerebrovascular blood and oxygen supply. It is a major contributor to death or disability worldwide and has become a heavy societal and clinical burden. To date, effective treatments for ischemic stroke are limited, and innovative therapeutic methods are urgently needed. Hypoxia inducible factor-1α (HIF-1α) is a sensitive regulator of oxygen homeostasis, and its expression is rapidly induced after hypoxia/ischemia. It plays an extensive role in the pathophysiology of stroke, including neuronal survival, neuroinflammation, angiogenesis, glucose metabolism, and blood brain barrier regulation. In addition, the spatiotemporal expression profile of HIF-1α in the brain shifts with the progression of ischemic stroke; this has led to contradictory findings regarding its function in previous studies. Therefore, unveiling the Janus face of HIF-1α and its target genes in different type of cells and exploring the role of HIF-1α in inflammatory responses after ischemia is of great importance for revealing the pathogenesis and identifying new therapeutic targets for ischemic stroke. Herein, we provide a succinct overview of the current approaches targeting HIF-1α and summarize novel findings concerning HIF-1α regulation in different types of cells within neurovascular units, including neurons, endothelial cells, astrocytes, and microglia, during the different stages of ischemic stroke. The current representative translational approaches focused on neuroprotection by targeting HIF-1α are also discussed.
Introduction
As the most vulnerable organ in the body, the brain requires an adequate and timely supply of oxygen and energy. Insufficient blood supply triggers a series of pathological events, leading to extensive death of neural cells in the ischemic region and neurological deficits within several hours, ultimately causing ischemic stroke. Bioenergetic failure, mitochondrial dysfunction, neuroexcitatory toxicity, inflammation, neuronal apoptosis, and protein folding errors may occur after cerebral ischemia, causing ischemic brain tissue to rapidly develop into the penumbra until irreversible infarction occurs (1). Management focuses on immediate revascularization by endovascular thrombectomy and intravenous thrombosis administration, which can improve the neurological deficits; however, time is a critical factor in both approaches. The pathological mechanisms underlying ischemic stroke have been extensively studied, which has resulted in a series of strategies and advanced methods for ischemic stroke treatment, such as anti-oxidative stress, neuroinflammation management, and neuroprotection (2, 3), with certain therapeutic effects in preclinical animal stroke models. Unfortunately, most of these strategies have failed in clinical trials; thus, there is an urgent need for innovative strategies that target the initial pathological changes, because timely treatment is critical for the prognosis of stroke patients (4, 5).
Hypoxia inducible factor-1α (HIF-1α) is considered a pivotal regulator of oxygen homeostasis and strictly regulated by oxygen levels (6, 7). HIF-1α mediates an endogenous adaptive program by regulating multiple signaling pathways and targeting downstream genes under hypoxic conditions. These genes have been found to be related to the pathophysiology of numerous neurological diseases, particularly ischemic stroke. Moreover, HIF-1α is involved not only in hypoxia but also in acute inflammatory responses, and it has recently been considered an important signaling regulator of inflammation (8, 9). A recent clinical study showed increased serum concentrations of HIF-1α at 48 h after acute ischemic stroke, and this result was positively correlated with the infarct size (P < 0.001). Although further verification is needed, HIF-1α will hopefully serve as a predictor of stroke prognosis (10). Studies have shown that HIF-1α has neuroprotective effects on cerebral ischemia, while others have reported negative effects such as interruption of the blood-brain barrier (BBB) integrity after stroke (11–14). Such divergent effects of HIF-1α in ischemic stroke may be attributed to the diversity of downstream targets, which function in different cell types within the neurovascular unit (NVU). Previous studies have found that HIF-1α is expressed in not only neurons but also astrocytes, endothelial cells, and microglia, and that it exerts cell type-specific actions (12, 15, 16). The function of neuronal HIF-1α has long attracted the interest of researchers, but the role of HIF-1α in other components of the NVU has received less attention.
In the present review, we elaborate on the regulatory mechanisms of HIF-1α and its various roles in ischemic stroke, specifically in four main types of cells in the NVU (i.e., neurons, astrocytes, endothelial cells, and microglial cells). Recent representative translational studies that propose novel therapeutic options targeting HIF-1α for ischemic stroke are also discussed.
Structure of HIF-1α and Expression After Ischemic Stroke
Initially identified as a transcription factor, HIF-1, the prime regulator of oxygen homeostasis is a stable component of cells that can be activated and induced by decreased oxygen tension (17, 18). Once HIF-1 is activated, the transcription of genes related to adaptation and survival can be promoted. As a basic-helix-loop-helix-PAS heterodimer, HIF-1 comprises α and β subunits. HIF-1β heterodimerizes with other basic-helix-loop-helix-PAS proteins and is overexpressed in cells; thus, HIF-1α protein levels predominantly govern the transcriptional activity of HIF-1 (19). HIF-1α has an N-terminal transactivation domain, an oxygen-dependent degradation domain, a C-terminal transactivation domain, and an inhibitory domain (Figure 1). Under normoxic conditions, the key residues of proline (Pro-402 and Pro-564) in the HIF-1α subunit can be hydroxylated by prolyl hydroxylase (PHD) in response to 2-oxoglutrate and iron. After hydroxylation, the HIF-1α subunit is subsequently ubiquitinated by E3 ubiquitin ligase complex, which contains a Von Hippel–Lindau protein, a negative regulator of HIF-1α transcriptional activity, and is degraded by the 26S protein (20–23). Asparagine (Asn-803) is hydroxylated by asparagine hydroxylase, which inhibits its trans-activation ability and prevents HIF-1α from binding with CBP/p300 (a transcriptional coactivator) (22). When the cellular oxygen level decreases and PHD activity is inhibited, the asparagine and prolyl in the HIF-1α protein cannot be hydroxylated, resulting in the accumulation rather than ubiquitination of HIF-1α in the cytoplasm (24, 25). The stable HIF-1α forms a heterodimer with the HIF-1β subunit after translocating into the nucleus and binds to the hypoxia response elements on the promoter of HIF-1 target genes. This stimulates and enhances the transcription of the HIF-1 target gene, triggering the in vivo steady-state response of cells to oxygen and glucose deprivation (Figure 2) (21, 26). In addition to protein hydroxylation, the activity of HIF-1α is regulated by other post-translational modifications, including phosphorylation, acetylation, S-nitrosation, and small-ubiquitin-like modifier acylation (27).
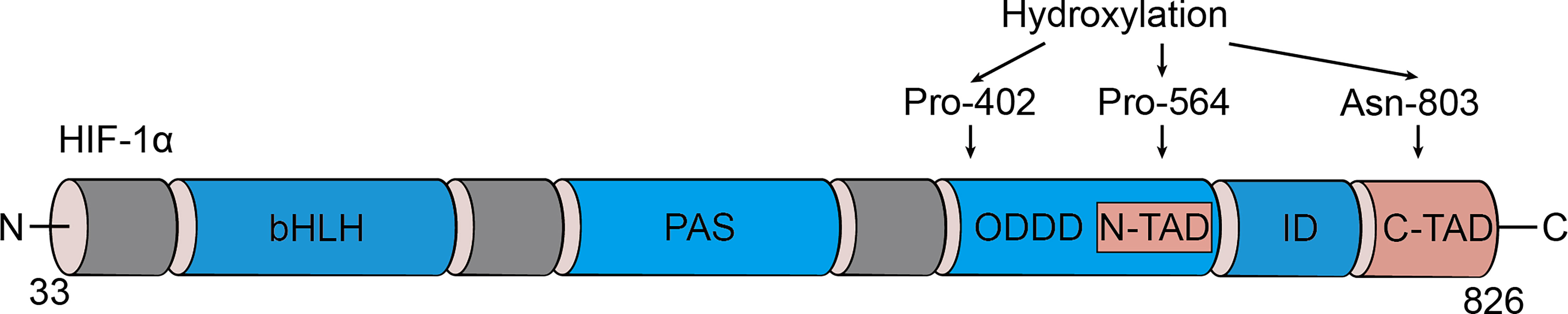
Figure 1 Domain structure of HIF-1α. HIF-1α is comprised of several conserved domains including a DNA binding (basic helix-loop-helix, bHLH) domain, protein/protein interactions and dimerization (PAS) domain, C-terminal trans-activation domain (C-TAD), N-terminal trans-activation domain (N-TAD), oxygen-dependent degradation domain (ODDD), and inhibitory domain (ID).
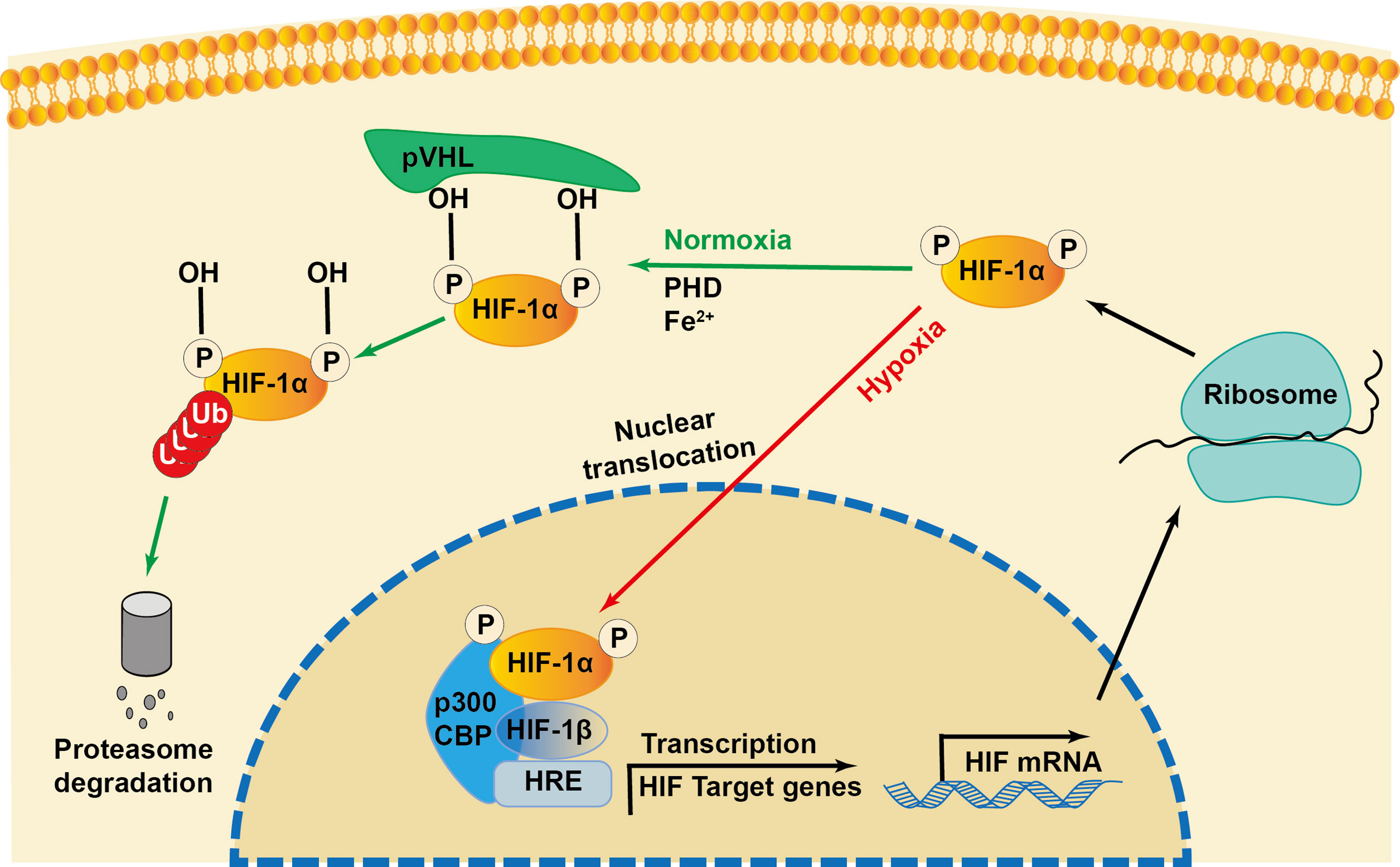
Figure 2 Regulation of HIF-1α during normoxia and hypoxia. Under normoxia, HIF-1α hydroxylase (PHD) hydroxylates the proline in the presence of iron (Fe2+), which recruits the von Hippel-Lindau protein (pVHL) to bind and initiate the proteolysis of HIF-1α by acting as a recognition component of ubiquitin ligase complex, leading to proteasomal degradation. Under hypoxia conditions, the interaction of transcriptional coactivators p300/CBP (CREB-binding protein) is activated. HIF-1α is stabilized and translocated into the nucleus and heterodimerizes with constitutively expressed HIF-1β, binds to the hypoxia responsive elements (HRE) and enhances the transcription of HIF-1α target genes.
The expression level of HIF-1α induced by stroke is related to the ischemic duration. In 1996, researchers first reported that both the HIF-1α and HIF-1β subunits were increased in rodents exposed to hypoxia for 60 min (28). Subsequently, the mRNA levels of HIF-1α and its downstream genes were found to be upregulated in the peri-infarct region at 7.5 h; they peaked at 24 h and declined at 72 h after the onset of focal cerebral ischemia in rats (29, 30). In another ischemia-reperfusion rat model, HIF-1α protein was induced at 4 h, peaked at 8 h, and declined at 24 h after reperfusion (31). The function of HIF-1α in ischemic stroke is bidirectional (Table 1). HIF-1α mediates the expression of a variety of genes that are involved in neurogenesis, angiogenesis, cell proliferation, erythropoiesis, and cellular metabolism. HIF-1α also participates in cell death, adaptive response, and cell regeneration, increasing its adaptation to ischemic stress, and consequently showing a neuroprotective role (60, 61). Conversely, several studies have reported the pernicious roles of HIF-1α in cerebral ischemic injury, including a severe inflammatory response, enhanced apoptosis, and loss of BBB integrity after ischemic stroke; this indicates that HIF-1α is likely a mediator of neuroinflammation or a factor determining the BBB permeability (62–64). Moreover, according to mounting data, HIF-1α appears to play a controversial role in cell autophagy (65–67). Taken together, HIF-1α increases after cerebral ischemia, and its expression level is dependent on ischemic duration, which may partially explain the seemingly contradictory effects on the pathological process of stroke.
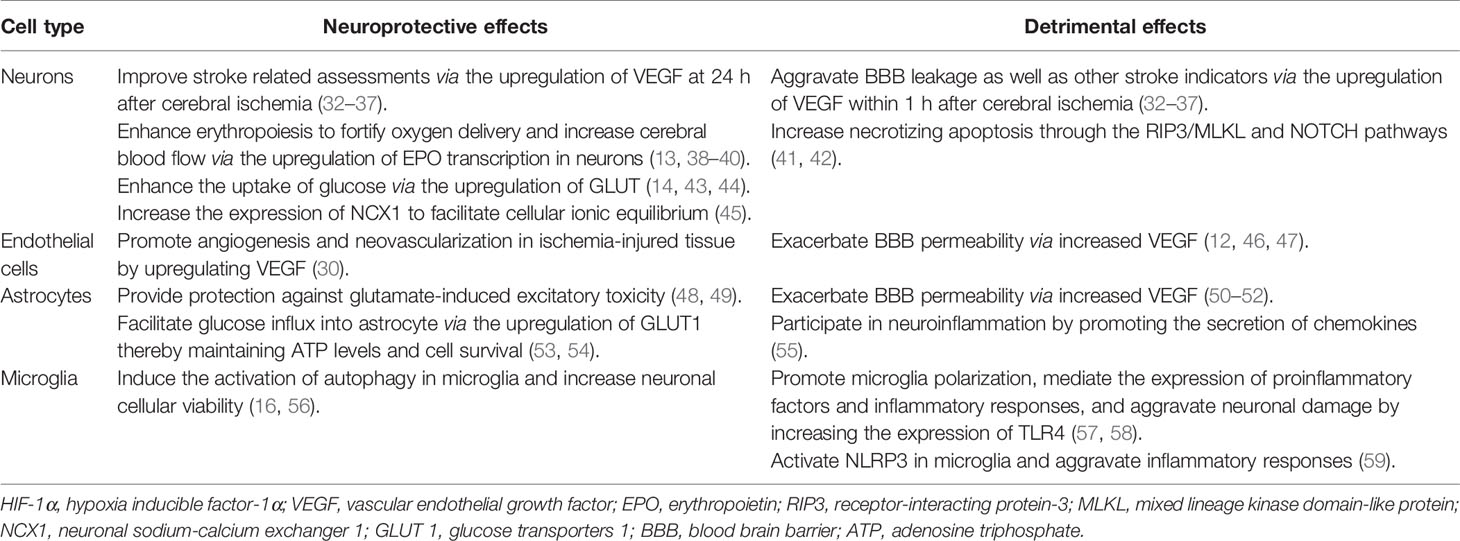
Table 1 Bidirectional roles of HIF-1α in different cells.
Functions of HIF-1α in Different NVU Cells After Ischemic Stroke
The definition of the NVU emphasizes the complexity of interactions between all perivascular components, including the basal membrane, neurons, astrocytic foot processes, pericytes, macrophages, and other leukocytes, and it integrates the contemporary concepts of the BBB (68). The central nervous system (CNS) relies on the co-operation of various types of cells and the ability of the BBB to function properly (69). During ischemic stroke, the BBB can be damaged both functionally and structurally by massive surrounding free radicals, calcium overload, ion imbalance, and autophagy, leading to brain edema or hemorrhagic transformation (70, 71). HIF-1α is widely expressed in the NVU and exhibits distinct functions, such as neuroprotection, BBB permeability regulation, angiogenesis, and inflammatory regulation (18, 61, 72). Till date, research has found that HIF-1α mediates the transcriptional expression of more than 100 genes under hypoxic conditions, and this number continues to increase (73). These HIF-1α target genes are involved in numerous processes, including cell metabolism, proliferation, survival, and death; cytoskeletal structure formation; cell adhesion; and movement. Because HIF-1α has various physiological functions in different types of cells, its phenotype becomes more complex under the context of ischemic stroke. In this article, we introduce the functions of HIF-1α and its downstream target genes in different cells in the NVU during stroke (Figure 3).
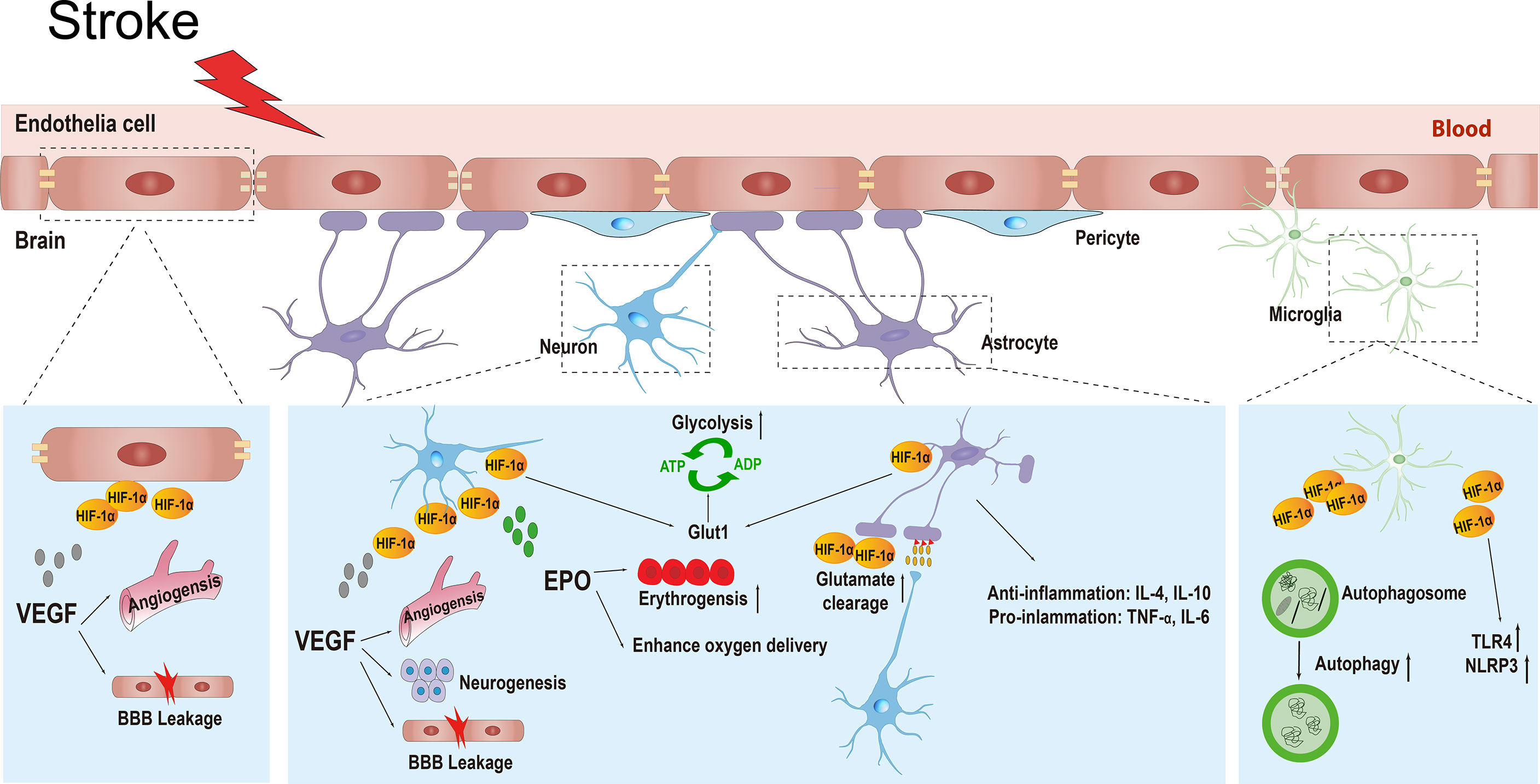
Figure 3 The various physiological functions of HIF-1α and its target genes in different types of cells in the neurovascular unit including neurons, endothelial cells, astrocytes, and microglial cells during ischemic stroke. HIF-1α, hypoxia inducible factor-1α; VEGF, vascular endothelial growth factor; BBB, blood brain barrier; EPO, erythropoietin; GLUT1, glucose transporter 1; ATP, adenosine-triphosphate; ADP, adenosine-diphosphate; IL-4, interleukin-4; IL-10, interleikin-10; TNF-α, tumor necrosis factor-α; IL-6, interleukin-6; TLR4, toll-like receptor 4; NLRP3, recombinant NLR Family, pyrin domain containing protein 3.
Neurons
Neurons have been compared to pacemakers in the NVU and have a central role in the physiology of the CNS (74). Because of their high energy requirements, neurons are extremely sensitive to glucose and oxygen deficiencies (75), consequently, they are the most susceptible to ischemic injuries and die in large numbers within hours of hypoxia/ischemia. The pathophysiological function of HIF-1α in neurons varies, with contradictory effects on ischemic stroke.
Roles of Vascular Endothelial Growth Factor (VEGF) Regulated by HIF-1α in Neurons
VEGF, a well-known downstream target gene of HIF-1α, is extensively involved in the pathological processes of cerebral ischemia. The family of VEGFs is characterized by their strong angiogenic properties. In the ischemic brain, VEGFs are also important regulators of neuroprotection and neurogenesis (76). The neuroprotective effect of VEGF is thought to be mediated by the regulation of neurogenesis and angiogenic actions in ischemic brains at 12 h, but not immediately (i.e., 1 h), after stroke. In middle cerebral artery occlusion (MCAO) rat models, intracerebroventricular or intravenous administration of VEGF at 1 day or 2 days, respectively, after MCAO could reduce the infarction volume, improve neurological deficits, enhance the delayed survival of newborn neurons, and stimulate angiogenesis in the ischemic penumbra (32). Conversely, administration of VEGF 1 h after ischemia significantly exacerbates the neurological deficits (32, 33). In addition to angiogenesis and neurogenesis, the impact of VEGF is mediated through the control of apoptotic factors in ischemic brain tissues. Administration of HIF-1α-siRNA significantly reduces the content of p53 and caspase-3 and is accompanied by decreased VEGF expression, thus alleviating stroke-related behavior disorders and cerebral infarction at 1 h after MCAO (34). In another experimental rodent model of MCAO, 2-methoxyestradiol (2ME2; a HIF-1α inhibitor) therapy at 30 min after MCAO significantly decreased infarction size and the number of apoptotic neurons; however, 2ME2 delivered at 8 h after MCAO did not have a protective effect. Consistent with in vivo results, 2ME2 or HIF-1α-siRNA robustly reduced the expression of VEGF with aggravated apoptosis in neurons at 12 h after OGD treatment (35). Pretreatment with HIF-1α siRNA or treatment with HIF-1α inhibitor at 10 min after MCAO was found to alleviate BBB damage by reducing the VEGF mRNA level, alleviating occludin degradation, and preventing the excretion of matrix metalloproteinase (MMP)-2 in neurons (36). In general, activation of HIF-1α is deleterious immediately (i.e., 1 h) after acute ischemic stroke; however, after 12 h, it can show neuroprotective effects via regulation of VEGF-mediated angiogenesis and neurogenesis.
Roles of Erythropoietin (EPO) Regulated by HIF-1α in Neurons
EPO, another well-known target gene of HIF-1α, has been shown to protect cells from hypoxia injury by enhancing erythropoiesis to fortify oxygen delivery and increase cerebral blood flow. Administration of deferrioxamine (an iron chelator), both in vivo and in vitro, has been found to promote the expression of HIF-1α and EPO, thus improving the tolerance against cerebral ischemia, increasing neuronal viability, and exerting protective effects on OGD-cultured neurons and MCAO rats (38). A similar study found that activation of the HIF-1α/EPO signaling pathway in HT-22 cell lines facilitated pyruvate-mediated cytoprotection (39). Hypoxic preconditioning for 180 or 300 min can lead to the production of reactive oxygen species (ROS) and induce the production threshold of HIF-1α and EPO under ischemia, thus having a neuroprotective role through the NF-κ B/JAK2-tgf5 pathway (40). In a study conducted by Li et al., rats treated with Ad-HIF-1α 1 h after transient MCAO showed reduced neuronal apoptosis on day 7, which was attributed in part to the upregulation of EPO and the suppression of active caspase-3 (13). Therefore, the increased expression of EPO regulated by HIF-1α could provide a neuronal protective effect by increasing erythropoiesis to improve oxygen delivery during ischemic brain injury.
Regulation of Energy Metabolism by HIF-1α in Neurons
Energy metabolism is regulated by HIF-1α in neurons. The brain is a vulnerable organ that requires optimal delivery of oxygen and nutrients derived from blood. To meet the metabolic needs, glucose transporters (GLUT 1) are expressed in both endothelial cells and neurons to maintain the transportation of glucose. After ischemia, the enhanced nutrient demand by neural cells in the penumbra increases the need of nutrient transport across the endothelium. The activation of HIF-1α is conducive to cell survival by promoting glucose transport activity and glycolysis and maintaining redox equilibrium (43). In OGD-cultured SH-SY5Y cells, knockdown of HIF-1α hindered important components of the cellular redox equilibrium and critical enzymes of the pentose phosphate pathway and glucose metabolism, including glucose transporters such as GLUT1 (14). Research supports that hypoxia preconditioning can alleviate the loss of neurons in the rat cortex with traumatic brain injury (44); this mechanism is primarily associated with the upregulation of HIF-1α, which induces the expression of GLUT1 and GLUT3 and ultimately increases the uptake of glucose in the neurons.
Other Target Genes and Signaling Pathways Related to HIF-1α in Neurons
Other hypoxia-mediated signaling pathways related to HIF-1α have been identified to have a neuroprotective role after cerebral ischemia. PHD is a crucial mediator for the degradation process of HIF-1α, and inhibition of PHD activity could prevent the degradation of HIF-1α. Abundant experimental evidence has confirmed that the application of PHD inhibitors or PHD knockout may stimulate a protective cellular response in ischemic brain injury through the stabilization and activation of HIF-1α (77–79) (Table 2). In addition, the β-catenin signaling pathway has been shown to be related to HIF-1α. In a rat model of MCAO, a 42-d treatment with delayed hyperbaric oxygen was found to accelerate neurogenesis and improve neurological impairments, and those benefits could be counteracted by inhibiting HIF-1α and ROS (90). In response to hypoxia, upregulated HIF-1α could facilitate the level of neuronal sodium-calcium exchanger 1 (NCX1), the vital mediator maintaining the balance between sodium and calcium to sustain the stability of the intracellular environment. Valsecchi et al., revealed that HIF-1α can alleviate cerebral infarction by the upregulation of NCX1 in MCAO rats subjected to ischemia preconditioning and in OGD-cultured neurons, indicating that NCX1 may be a new HIF-1α target gene (45).

Table 2 Therapeutic drugs that promote neuroprotection by regulating HIF-1α.
In contrast to its neuroprotective effects, HIF-1α has also been shown to be involved in some pro-necrotizing apoptosis signals and interact with the Notch signaling pathway under hypoxic conditions. The Notch intracellular domain (NICD) and HIF-1α are colocalized in the neuronal nucleus in brain tissue of MCAO mice. Overexpression of NICD and HIF-1α could aggravate cell death in OGD neuronal cell lines, where administration of 2ME2 reverses the cell death process. Furthermore, depletion of endogenous NICD and HIF-1α mediated by RNA interference were found to reduce cell death under hypoxic and ischemic conditions. Administration of HIF-1α inhibitors and γ-secretase in mice subjected to MCAO ameliorates neurological outcomes, and combination therapy was found to be superior to monotherapy (41). More recently, a study found that the simultaneous elevation of HIF-1α, mixed lineage kinase domain-like protein (MLKL), and receptor-interacting protein 3 (RIP3) in OGD HT-22 neuron cells are thought to participate in the formation and activation of the necrosome and have a significant role in necroptotic signaling. Therefore, for the first time, researchers proposed that HIF-1α is related to the occurrence of necrotizing apoptosis and may participate in ischemic brain injury and neuronal apoptosis by activating the RIP3/MLKL pathway (42).
Endothelial Cells (ECs)
Brain ECs are the main components of the BBB. They form the wall of blood vessels and are recognized as the interface of blood–CNS exchange. The properties of the BBB are largely manifested within the ECs, whose stability is essential for maintaining the integrity of the BBB (91). The proliferation and migration of endothelial cells are largely dependent on VEGF. Beyond that, VEGF also increases vascular permeability, initiating the angiogenesis through the above properties (69). After stroke, the formation of new vessels is vital, as newly formed blood networks compensate the occluded vessel and thereby rescue the penumbra. However, the degradation of the vascular basement membrane during the activation phase in angiogenesis may lead to brain edema or hemorrhagic transformation in the acute phase of stroke.
Role of VEGF Regulated by HIF-1α in ECs
Compelling evidence has suggested that VEGF is robustly induced via the activation of HIF-1α after cerebral ischemic injury (92), and the functions of VEGF in endothelial cells are also bifurcated. As one of the most effective cytokines promoting the growth of vascular endothelial, VEGF stimulates the proliferation of ECs and directly participates in angiogenesis and neovascularization in ischemia-injured tissue. In a permanent MCAO mouse model, a substantial increase of newly formed vessels was observed at the ischemia penumbra 48–72 h after the occlusion, which was related to the robust increase of VEGF and VEGF receptors (VEGFR) mediated by HIF-1 and HIF-2, suggesting the regulatory function of HIFs in the VEGF/VEGFR system during the acute phase of stroke (30). Inevitably, in the acute phase of ischemic stroke, VEGF secreted from a variety of cell types increases capillary permeability to undergo endothelium-mesenchymal transformation, which causes BBB disruption (93).
In a rat cerebral endothelial cell hypoxia-reoxygenation transwell BBB model, the accumulation of VEGF and HIF-1α robustly increased the BBB permeability, in which the effect can be antagonized by HIF-1α inhibitors (94, 95). Consistent with the aforementioned study, hypoxic exposure stabilized HIF-1α rapidly, which is concomitant with VEGF secretion. As a consequence, tight junction proteins undergo increased tyrosine-phosphorylation and delocalization, ultimately leading to the loss of BBB integrity. Moreover, accumulation or stabilization of HIF-1α can also be achieved by using deferrioxamine, which further emphasizes the interaction of HIF-1α in BBB dysfunction, especially via tight junction alterations in hypoxic conditions (12). Administration of HIF-1α inhibitor or siRNA prior to MCAO surgery significantly suppressed the degradation of tight junction proteins and the upregulation of VEGF, ultimately alleviating BBB damage (36). In addition to VEGF, MMPs are known for their ability to regulate the BBB permeability. In a stroke rat model, administration of 2ME2 was found to reduce the expression of VEGF and the elevation of active MMPs 2 and 9, thereby attenuating hemorrhagic transformation and ameliorating neurological deficits in acute hyperglycemia-induced hemorrhagic transformation (46).
BBB impairment is particularly relevant in stroke patients with diabetes mellitus. Diabetes mellitus is an established risk factor for stroke that increases the absolute risk of stroke by about 1.5 to 3 fold (96–98), and diabetic patients with stroke are predisposed to worse outcomes (99, 100). Diabetes increases the risk of cerebrovascular accidents by altering the structure and function of arteries, arterioles, and capillaries. Over time, blood vessels become increasingly stiff, tortuous, and narrowed because of diabetes. The pathological processes of atherosclerosis and thrombogenesis are accelerated, which compromises the BBB permeability (101). Studies have shown that hyperglycemia can significantly upregulate the expression of HIF-1α and its target gene VEGF in the brain microvasculature after MCAO; this further aggravates BBB disruption after ischemic brain injury. Endothelial-specific HIF-1α knockout diabetic mice showed reduced BBB disruption and decreased brain infarction. In addition, diabetic mice treated with long-term insulin showed similar HIF-1α levels in the contralateral and ipsilateral hemispheres of the brain in stroke rats (47). Therefore, the accumulation of HIF-1α caused by hyperglycemia substantially aggravates BBB damage in diabetic stroke, and therapeutic treatments targeting HIF-1α may provide new treatment options for preventing hemorrhagic transformation in these patients.
HIF-1α and VEGF in Plaque Angiogenesis
Thromboembolism is the main cause of atherosclerotic stroke and is most frequently the result of plaque rupture, which is related to angiogenesis in the carotid plaque. Studies have shown that HIF-1α is relevant to plaque angiogenesis and can lead to plaque progression, bleeding, and ulceration (102, 103). In 2019, a clinical trial enrolled 54 patients who underwent carotid endarterectomy for severe internal carotid artery stenosis, of whom 20 patients were symptomatic and 34 were asymptomatic. The results of that study indicated that the serum mRNA levels of HIF-1α in carotid plaque endothelial and smooth muscle cells in symptomatic patients were 1.5-fold higher than those in asymptomatic patients, although no significant difference was observed between the two groups. Notably, the mRNA levels of VEGF were seven times higher in symptomatic patients than in asymptomatic patients (P = 0.017), indicating a correlation between VEGF and neurological deficits symptoms in patients with symptomatic carotid artery stenosis (104)
Astrocytes
Astrocytes are critical components of the BBB, as they provide support and separate neural cells by stretching their cell bodies. As important active components in the NVU, astrocytes participate in two-way communication with other cells and have multiple roles during hypoxia and ischemia, including the release of cytokines, neurotrophic factors and nutrients to support neuronal survival (105–108). Notably, astrocytes mediate dual immunoregulation in ischemic stroke via the secretion of both proinflammatory and anti-inflammatory cytokines in response to changes in the cellular microenvironment, and the activation of HIF-1α may have a crucial role in regulating inflammatory responses (109, 110).
Roles of VEGF Regulated by HIF-1α in Astrocytes
As the core components of the BBB in the NVU, astrocytes have also been found to express HIF-1α, which may affect the integrity of the BBB. In OGD-cultured primary astrocytes, HIF-1α was found to be partly involved in the VEGF-mediated astrocyte response in chronic ischemic brain injury, ultimately leading to the destruction of BBB integrity. Preventing the action of HIF-1α downregulates the expression of VEGF, which not only attenuates the proliferation of primary astrocytes but also improves the integrity of the BBB (50). Notably, sirtuin 3 (SIRT3; known as the regulator of metabolism and aging) regulates VEGF expression by inhibiting HIF-1α signaling after permanent MCAO in rats or OGD in cultured primary astrocytes. In addition, SIRT3 knockout mice reportedly show more severe BBB disruption and inflammatory responses than do wild type mice in the acute phase, suggesting an underlying regulatory mechanism of HIF-1α/VEGF signaling in astrocytes and harmful effects in the acute phase of ischemic stroke (51). In general, reversal of HIF-1α expression in the acute phase may be a worthy target for ischemic stroke therapy. Edaravone has been shown to prevent the HIF-1α from binding to the VEGF promoter in hypoxic astrocytes, thereby maintaining the permeability of capillaries and venules after ischemic stroke. The regulatory effect on HIF-1α/VEGF signaling by Edaravone may partially contribute to its edema-resolving effects in acute ischemic stroke patients (52).
HIF-1α Regulates Glucose Flux From Hypoxia in Astrocytes
Astrocytes have been shown to transport glucose from the blood to provide energy to the brain parenchyma and maintain neuronal activity, a process in which HIF-1α is a key mediator (111, 112). As a vital link between oxygen stress and nutrient cellular adaptation, HIF-1α can enhance glycolytic flux by upregulating the expression of pivotal glycolytic genes, including GLUT-1, phosphofructokinase 1, phosphoglycerate kinase-1, lactate dehydrogenase, and glucose-6-phosphate isomerase (113, 114). GLUT-1 is expressed both in neurons and astrocytes primarily at the plasma membrane and is responsible for the entry of glucose into cells without consuming energy (115). HIF-1α is activated under hypoxic conditions, resulting in the transformation of GLUTs from the organelle membrane to the cytoplasmic membrane and the activation of glycolytic enzymes (53), promoting the uptake of glucose into cells and maintaining adenosine triphosphate (ATP) levels and cell survival. HIF-1α regulates glycometabolism through the glycolytic pathway to reduce ROS production in hypoxic environments. Research has revealed that activation of astrocyte mtCB1 receptors can lead to a reduction of ROS and impact the glycolytic production of lactate through the HIF-1 pathway, leading to neuronal redox stress and reduced cell viability in mouse astrocytes (54). Embryo fibroblasts in HIF-1α-knockout mice lose the ability of glycolytic metabolism during hypoxic conditions, resulting in cell death due to ROS overload (116). These findings demonstrated that HIF-1α regulates glucose flux to prevent increased ROS production under hypoxic conditions.
HIF-1α Protects Against Glutamate From Hypoxia in Astrocytes
It is known that astrocytes can rapidly eliminate neurotransmitters released in the synaptic cleft, such as glutamate (48). The release of glutamate and the influx of calcium ions are currently considered to be among the major pathological mechanisms in ischemic stroke. Further, HIF-1α protects against glutamate from hypoxia in astrocytes. One study showed that the elevation of HIF-1α improves cell viability in primary rat astrocytes under hypoxia and high concentrations of glutamate (0.1 and 1 mM for 3 h). Pretreatment with HIF-1α inhibitor was found to reduce the survival of astrocytes under treatment with 1 mM glutamate for 3 h, suggesting that HIF-1α provides protection against glutamate-induced excitatory toxicity in hypoxic astrocytes. Moreover, a correlation between increased glutathione and HIF-1α stabilization was observed in astrocytes (49).
HIF-1α and Neuroinflammation in Astrocytes
In addition to microglial cells, astrocytes have been shown to participate in both innate and subsequent adaptive immune responses (117). Moreover, they have been found to release chemokines, including CCL2, CCL20, and CXCL10, to attract neutrophils and monocytes into the hypoxic/ischemic brain, thus increasing neuroinflammation. HIF-1α was also found to mediate the transcriptional regulation of chemokines monocyte chemoattractant proteins 1 and 5 in hypoxic astrocytes, which aggravates the neuroinflammation injury after ischemic stroke (55). Interestingly, in the initial stages of ischemic stroke, astrocytes have been found to act as negative regulators of neuroinflammation by secreting transforming growth factor-β, interleukin (IL)-4, and IL-10. As ischemic injury progresses and the neuroinflammation becomes more severe, astrocytes release proinflammatory cytokines such as IL-1β, IL-6, and tumor necrosis factor (TNF)-α, which are HIF-1-dependent cytokines (118). Collectively, these findings indicate that HIF-1α plays a vital role in astrocyte-mediated immune response after stroke.
Other Signaling Pathways Mediated by HIF-1α in Astrocytes
In addition to mediation of the previously mentioned pathophysiological functions, HIF-1α may be involved in other signaling pathways related to neuroprotection. For instance, the STAT 3/HIF-1α/VEGF signaling pathway can be activated by the inhibition of Calpain-1 activity and consequent promotion of astrocytic neurogenesis, which improves stroke prognosis (119). Ischemic preconditioning, defined as transient exposure to subsequent prolonged cerebral ischemia, represents a fundamental adaptive response to environmental stress. A study found that hypoxia preconditioning induces the expression of P450 2C11, an arachidonic acid epoxygenase expressed in astrocytes, which was found to confer protection against ischemia-reperfusion damage through increased HIF-1α and to enhance the tolerance of astrocytes to OGD-induced hypoxic-ischemic injury via the P450 cyclooxygenase pathway (120). More recently, studies have focused on the formation of proteasome and their involvement in human disease (121, 122). Low molecular mass peptide 2 (LMP2), a subunit of the immunoproteasome (which is a proteasome subtype), has been found to participate in the inflammatory pathophysiological mechanisms of ischemic stroke in both experimental and clinical studies. Inhibition of LMP2 with shRNA has been reported to alleviate infarction and attenuate the expression of TNF-α and IL-1β (121, 123). In the OGD model of primary cultured astrocytes and the MCAO model, inhibition of LMP2 led to numerous HIF-1α neuroprotective effects, suggesting that HIF-1α may have a key role in the immunoproteasome-mediated inflammatory response (124). The above findings also imply that immunoproteasome inhibitors may constitute a promising strategy for stroke treatment.
Microglia
Microglia are recognized as the resident phagocytes in the CNS and are major components of the innate immune response. As the final defense for brain parenchyma, microglia can identify subtle changes in the brain and immediately react to pathophysiological stimuli (125). As a result, activated microglia are recruited to the inflammatory site, where they eliminate toxins by phagocytosis, resolve inflammation, and repair tissue by producing anti-inflammatory factors such as IL-10 and transforming growth factor-β (TGF-β) to effectively maintain CNS homeostasis (126–128). However, microglia also promote inflammation after ischemia by secreting pro-inflammatory factors (ROS, TNF, and IL-1β). Once microglia are over-activated, uncontrolled excessive inflammation can cause host tissue damage, which is considered an essential factor leading to injury in the subacute phase of ischemic stroke.
Roles of HIF-1α in Microglia-Mediated Inflammation
The mRNA and protein levels of HIF-1a increase significantly in microglia after stimulation with pro-inflammatory cytokines TNF-α, IL-1, and IFN-γ, with the maximal induction occurring between 3 and 6 h (129). After ischemic stroke, HIF-1a facilitates the processes of microglia chemotaxis, phagocytosis, and ROS and TNF-α production by promoting the expression of the phagocytic genes cd36 and mfg-e8, which have been shown to inhibit neurogenesis and lead to neuronal injury (130). Toll-like receptor 4 (TLR4) is a Type I transmembrane protein expressed in the surface of various immune cells, monocytes, macrophages, regulatory T cells, etc. Similarly, TLR4 has also been related to microglia-activation-mediated inflammatory responses in cerebral ischemia injury processes. Activation of TLR4 was found to promote microglia polarization and increase expression of proinflammatory factors, including TNF-α and IL-1β, ultimately aggravating neuronal damage (57). Under hypoxia, HIF-1α modulates the enhanced expression of TLR4 via increased phosphorylation of NF-κB in primary BV2 cell lines, thus exacerbating the inflammatory response. Inhibition of TLR4 with siRNA or antibodies was reported to reduce the expression of phospho-NF-κB and inflammatory factors TNF-α, IL-1β, and inducible nitric oxide synthase (iNOS) (58). The phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway has been recognized as one of the upstream signaling pathways regulating the expression of HIF-1α (131). Salidroside, the bioactive component of Rhodiola rosea has shown neuroprotective effects, namely increasing the level of neuronal nuclear protein and reducing CD11b (a marker of microglia) through upregulation of the expression of HIF-1α and inhibition of the inflammatory response via the PI3K/AKT signaling pathway after cerebral ischemia in rats (132). In general, as the essential signaling regulator of inflammation, HIF-1α may be regulated by the PI3K/AKT pathway in brain tissue and participate in neuroinflammatory responses.
Therapeutic approaches targeting HIF-1α in microglia have shown neuroprotective effects. T-cell immunoglobulin and mucin domain protein-3 (TIM-3) are inhibitory molecules expressed on the T-cell surface. Research has indicated that glial TIM-3 is upregulated distinctively in activated microglia and astrocytes dependent on HIF-1 activation. Inhibition of TIM-3 has been found to substantially alleviate cerebral infarction, edema, neuronal cell death, and neutrophil infiltration in a mouse cerebral hypoxia-ischemia model, suggesting that TIM-3 is a modulator that links inflammation and subsequent brain damage after ischemia. Therefore, TIM-3 may serve as a downstream mediator of HIF-1α in the inflammatory response to hypoxic-ischemic damage (133). Arginine may protect against ischemic neuronal death after rat ischemia and reperfusion injury and inhibit inflammatory response by suppressing HIF-1α in OGD-cultured microglia and rat ischemic brain tissue (134). Glycine reduces neuroinflammation in rats with cerebral ischemia by downregulating NF-κB p65, thereby decreasing the expression of HIF-1α and ameliorating microglial polarization (135). Nucleotide-binding oligomerization domain, leucine-rich repeat and pyrin domain-containing 3 (NLRP3), one of the most studied inflammasomes (complexes of cytosolic proteins that identify pathogen signals), are predominantly activated by microglia under ischemic conditions (136). Once activated, inflammasomes produce IL-18 and IL-1β which are activated by caspase 1 and initiate the inflammatory response (137). Researchers have demonstrated that the NLRP3-dependent pyroptosis of cerebral cells is restrained by HIF-1α inhibition after 24 h of reperfusion following MCAO, indicating that HIF-1α may have a role in activating NLRP3 in microglia and mediating inflammatory responses (59).
Taken together, HIF-1a in microglia may serve as a potential therapeutic target for relieving inflammation reactions and neuronal damage after cerebral ischemia.
HIF-1α and Autophagy in Microglia
Autophagy is a process by which cytoplasmic material is degraded by lysosomes or vacuoles and then recycled to adapt to exogenous disruption (138). It is a vital process for tissue homeostasis that mediates and balances the favorable and adverse effects of inflammation. In microglia, autophagy appears to function mainly as an inhibitor of inflammatory responses (139, 140). Previous research has shown that deficiencies in autophagy caused by nutrient deprivation induce the activation and inflammation of microglia (140). More recently, researchers have found that HIF-1α may play a role in the autophagy-mediated activation of microglia (141). Under hypoxic conditions, the intensified autophagy appears to be mediated by the accumulation of HIF-1α in microglia, resulting in the inhibition of functions of autophagy-associated genes and the prevention of neuronal death in the initial stages. Moreover, genetic knockdown or pharmacological inhibition of HIF-1α was found to suppress autophagy and increase cellular viability (16). Another study confirmed that the inhibition of LC3-II (a direct indicator of autophagy) by HIF-1α induced the activation of autophagy in microglia (56). Although research has shown a correlation between HIF-1α and autophagy in microglia, the detailed mechanisms remain unclear.
Pericytes
In addition to neurons, astrocytes, endothelial cells, and microglial cells, pericytes are an important component for maintenance of the vascular integrity of the NVU. However, few studies have focused on the role of HIF-1α in pericytes in the context of ischemic stroke. One study indicated that HIF-1 deficiency reduced ischemia-induced pericyte death, alleviated BBB permeability and preserved their coverage of the CNS microvasculature translating into a more stable barrier after transient MCAO in conditional mutant mice with pericyte-targeted HIF-1 loss of function (142). Further studies may focus on unveiling the biological function of HIF-1α in pericytes under pathological and physiological conditions, thus providing more favorable evidence on potential therapeutic targets for ischemic stroke. In addition to the abovementioned CNS cells, macrophages, neutrophils, dendritic cells, and lymphocytes are involved in the inflammatory response mediated by HIF-1α (143). However, more rigorous studies are needed to elucidate the specific roles of HIF-1α in infiltrated immune cells. HIF-1α is now considered a potential novel therapeutic target for ischemic stroke.
Conclusion
In the treatment of acute ischemic stroke, intravenous thrombolysis with recombinant tissue plasminogen activator and mechanical thrombectomy have been shown to be extremely effective. These treatments, however, are still limited by their strict time frames. As a cellular sensor of oxygen level, HIF-1α is now considered as a potential novel therapeutic target for ischemic stroke and will hopefully serve as a predictor of stroke prognosis. An increasing number of new therapeutic strategies and agents focused on neuroprotection via regulation of HIF-1α are under development (Table 2), but these are yet to be tested in clinical trials. In the present review, we highlight the neuroprotective and detrimental effects of HIF-1α in in different cell types within the CNS under the context of stroke. We also summarize the representative therapeutic strategies based on the regulation of HIF-1α with the aim of providing guidance for future research on developing HIF-1α as a therapeutic target. However, differences in the functioning of HIF-1α between cell types and in different environments make it a challenging target in acute ischemic stroke. Simply activating or inhibiting the expression of HIF-1α for a therapeutic goal may lead to paradoxical results. Future studies are needed to focus on certain types of cells and bidirectional functions to gain a better understanding of HIF-1α in pre-clinical models of ischemic stroke before progressing to clinical trials. In conclusion, HIF-1α plays an important role in the aftermath of stroke and is a promising target for the treatment of ischemic stroke.
Author Contributions
QH and YM designed and wrote the manuscript. JL, DZ, JR, RZ revise the manuscript. JC, Z-NG, and YY gave constructive advice and participated in proof-reading of this paper. All authors contributed to the article and approved the submitted version.
Funding
This review was supported by the National Natural Science Foundation of China to YY (Grant No. 81771243), the Science and technology department of Jilin province (20180623052TC), and the Jilin Provincial Key Laboratory (20190901005JC) to YY.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
References
1. George PM, Steinberg GK. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron (2015) 87(2):297–309. doi: 10.1016/j.neuron.2015.05.041
2. Datta A, Sarmah D, Mounica L, Kaur H, Kesharwani R, Verma G, et al. Cell Death Pathways in Ischemic Stroke and Targeted Pharmacotherapy. Transl Stroke Res (2020) 11(6):1185–202. doi: 10.1007/s12975-020-00806-z
3. Su XT, Wang L, Ma SM, Cao Y, Yang NN, Lin LL, et al. Mechanisms of Acupuncture in the Regulation of Oxidative Stress in Treating Ischemic Stroke. Oxid Med Cell Longev (2020) 2020:7875396. doi: 10.1155/2020/7875396
4. Al-Mufti F, Amuluru K, Roth W, Nuoman R, El-Ghanem M, Meyers PM. Cerebral Ischemic Reperfusion Injury Following Recanalization of Large Vessel Occlusions. Neurosurgery (2018) 82(6):781–9. doi: 10.1093/neuros/nyx341
5. Ramagiri S, Taliyan R. Protective Effect of Remote Limb Post Conditioning via Upregulation of Heme Oxygenase-1/BDNF Pathway in Rat Model of Cerebral Ischemic Reperfusion Injury. Brain Res (2017) 1669:44–54. doi: 10.1016/j.brainres.2017.05.016
6. Salceda S, Caro J. Hypoxia-Inducible Factor 1alpha (HIF-1alpha) Protein is Rapidly Degraded by the Ubiquitin-Proteasome System Under Normoxic Conditions. Its Stabilization by Hypoxia Depends on Redox-Induced Changes. J Biol Chem (1997) 272(36):22642–7. doi: 10.1074/jbc.272.36.22642
7. Kallio PJ, Wilson WJ, O’Brien S, Makino Y, Poellinger L. Regulation of the Hypoxia-Inducible Transcription Factor 1alpha by the Ubiquitin-Proteasome Pathway. J Biol Chem (1999) 274(10):6519–25. doi: 10.1074/jbc.274.10.6519
8. Lipton P. Ischemic Cell Death in Brain Neurons. Physiol Rev (1999) 79(4):1431–568. doi: 10.1152/physrev.1999.79.4.1431
9. Eltzschig HK, Bratton DL, Colgan SP. Targeting Hypoxia Signalling for the Treatment of Ischaemic and Inflammatory Diseases. Nat Rev Drug Discov (2014) 13(11):852–69. doi: 10.1038/nrd4422
10. Xue L, Chen H, Lu K, Huang J, Duan H, Zhao Y. Clinical Significance of Changes in Serum Neuroglobin and HIF-1α Concentrations During the Early-Phase of Acute Ischemic Stroke. J Neurol Sci (2017) 375:52–7. doi: 10.1016/j.jns.2017.01.039
11. Barteczek P, Li L, Ernst AS, Böhler LI, Marti HH, Kunze R. Neuronal HIF-1α and HIF-2α Deficiency Improves Neuronal Survival and Sensorimotor Function in the Early Acute Phase After Ischemic Stroke. J Cereb Blood Flow Metab (2017) 37(1):291–306. doi: 10.1177/0271678x15624933
12. Engelhardt S, Al-Ahmad AJ, Gassmann M, Ogunshola OO. Hypoxia Selectively Disrupts Brain Microvascular Endothelial Tight Junction Complexes Through a Hypoxia-Inducible Factor-1 (HIF-1) Dependent Mechanism. J Cell Physiol (2014) 229(8):1096–105. doi: 10.1002/jcp.24544
13. Li J, Tao T, Xu J, Liu Z, Zou Z, Jin M. HIF−1α Attenuates Neuronal Apoptosis by Upregulating EPO Expression Following Cerebral Ischemia−Reperfusion Injury in a Rat MCAO Model. Int J Mol Med (2020) 45(4):1027–36. doi: 10.3892/ijmm.2020.4480
14. Guo S, Miyake M, Liu KJ, Shi H. Specific Inhibition of Hypoxia Inducible Factor 1 Exaggerates Cell Injury Induced by In Vitro Ischemia Through Deteriorating Cellular Redox Environment. J Neurochem (2009) 108(5):1309–21. doi: 10.1111/j.1471-4159.2009.05877.x
15. Vangeison G, Carr D, Federoff HJ, Rempe DA. The Good, the Bad, and the Cell Type-Specific Roles of Hypoxia Inducible Factor-1 Alpha in Neurons and Astrocytes. J Neurosci (2008) 28(8):1988–93. doi: 10.1523/jneurosci.5323-07.2008
16. Wang X, Ma J, Fu Q, Zhu L, Zhang Z, Zhang F, et al. Role of Hypoxia−Inducible Factor−1α in Autophagic Cell Death in Microglial Cells Induced by Hypoxia. Mol Med Rep (2017) 15(4):2097–105. doi: 10.3892/mmr.2017.6277
17. Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA Binding, and Transactivation Properties of Hypoxia-Inducible Factor 1. J Biol Chem (1996) 271(30):17771–8. doi: 10.1074/jbc.271.30.17771
18. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-Inducible Factor 1 is a Basic-Helix-Loop-Helix-PAS Heterodimer Regulated by Cellular O2 Tension. Proc Natl Acad Sci USA (1995) 92(12):5510–4. doi: 10.1073/pnas.92.12.5510
19. Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, et al. Hypoxia Response Elements in the Aldolase A, Enolase 1, and Lactate Dehydrogenase A Gene Promoters Contain Essential Binding Sites for Hypoxia-Inducible Factor 1. J Biol Chem (1996) 271(51):32529–37. doi: 10.1074/jbc.271.51.32529
20. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-Alpha to the Von Hippel-Lindau Ubiquitylation Complex by O2-Regulated Prolyl Hydroxylation. Science (2001) 292(5516):468–72. doi: 10.1126/science.1059796
21. Mahon PC, Hirota K, Semenza GL. FIH-1: A Novel Protein That Interacts With HIF-1alpha and VHL to Mediate Repression of HIF-1 Transcriptional Activity. Genes Dev (2001) 15(20):2675–86. doi: 10.1101/gad.924501
22. Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an Asparaginyl Hydroxylase Enzyme That Regulates the Transcriptional Activity of Hypoxia-Inducible Factor. Genes Dev (2002) 16(12):1466–71. doi: 10.1101/gad.991402
23. Kaelin WG Jr., Ratcliffe PJ. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol Cell (2008) 30(4):393–402. doi: 10.1016/j.molcel.2008.04.009
24. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol Cell (2010) 40(2):294–309. doi: 10.1016/j.molcel.2010.09.022
25. Freedman SJ, Sun ZY, Poy F, Kung AL, Livingston DM, Wagner G, et al. Structural Basis for Recruitment of CBP/p300 by Hypoxia-Inducible Factor-1 Alpha. Proc Natl Acad Sci USA (2002) 99(8):5367–72. doi: 10.1073/pnas.082117899
26. Greer SN, Metcalf JL, Wang Y, Ohh M. The Updated Biology of Hypoxia-Inducible Factor. EMBO J (2012) 31(11):2448–60. doi: 10.1038/emboj.2012.125
27. Brahimi-Horn C, Mazure N, Pouysségur J. Signalling via the Hypoxia-Inducible Factor-1alpha Requires Multiple Posttranslational Modifications. Cell Signal (2005) 17(1):1–9. doi: 10.1016/j.cellsig.2004.04.010
28. Wiener CM, Booth G, Semenza GL. In Vivo Expression of mRNAs Encoding Hypoxia-Inducible Factor 1. Biochem Biophys Res Commun (1996) 225(2):485–8. doi: 10.1006/bbrc.1996.1199
29. Sharp FR, Bergeron M, Bernaudin M. Hypoxia-Inducible Factor in Brain. Adv Exp Med Biol (2001) 502:273–91. doi: 10.1007/978-1-4757-3401-0_18
30. Marti HJ, Bernaudin M, Bellail A, Schoch H, Euler M, Petit E, et al. Hypoxia-Induced Vascular Endothelial Growth Factor Expression Precedes Neovascularization After Cerebral Ischemia. Am J Pathol (2000) 156(3):965–76. doi: 10.1016/s0002-9440(10)64964-4
31. Mu D, Jiang X, Sheldon RA, Fox CK, Hamrick SE, Vexler ZS, et al. Regulation of Hypoxia-Inducible Factor 1alpha and Induction of Vascular Endothelial Growth Factor in a Rat Neonatal Stroke Model. Neurobiol Dis (2003) 14(3):524–34. doi: 10.1016/j.nbd.2003.08.020
32. Sun Y, Jin K, Xie L, Childs J, Mao XO, Logvinova A, et al. VEGF-Induced Neuroprotection, Neurogenesis, and Angiogenesis After Focal Cerebral Ischemia. J Clin Invest (2003) 111(12):1843–51. doi: 10.1172/jci17977
33. Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, et al. VEGF Enhances Angiogenesis and Promotes Blood-Brain Barrier Leakage in the Ischemic Brain. J Clin Invest (2000) 106(7):829–38. doi: 10.1172/jci9369
34. Chen C, Hu Q, Yan J, Yang X, Shi X, Lei J, et al. Early Inhibition of HIF-1alpha With Small Interfering RNA Reduces Ischemic-Reperfused Brain Injury in Rats. Neurobiol Dis (2009) 33(3):509–17. doi: 10.1016/j.nbd.2008.12.010
35. Yeh SH, Ou LC, Gean PW, Hung JJ, Chang WC. Selective Inhibition of Early–But Not Late–Expressed HIF-1α is Neuroprotective in Rats After Focal Ischemic Brain Damage. Brain Pathol (2011) 21(3):249–62. doi: 10.1111/j.1750-3639.2010.00443.x
36. Sun Y, Chen X, Zhang X, Shen X, Wang M, Wang X, et al. β2-Adrenergic Receptor-Mediated HIF-1α Upregulation Mediates Blood Brain Barrier Damage in Acute Cerebral Ischemia. Front Mol Neurosci (2017) 10:257. doi: 10.3389/fnmol.2017.00257
37. Abdel-Latif RG, Rifaai RA, Amin EF. Empagliflozin Alleviates Neuronal Apoptosis Induced by Cerebral Ischemia/Reperfusion Injury Through HIF-1α/VEGF Signaling Pathway. Arch Pharm Res (2020) 43(5):514–25. doi: 10.1007/s12272-020-01237-y
38. Li YX, Ding SJ, Xiao L, Guo W, Zhan Q. Desferoxamine Preconditioning Protects Against Cerebral Ischemia in Rats by Inducing Expressions of Hypoxia Inducible Factor 1 Alpha and Erythropoietin. Neurosci Bull (2008) 24(2):89–95. doi: 10.1007/s12264-008-0089-3
39. Ryou MG, Liu R, Ren M, Sun J, Mallet RT, Yang SH. Pyruvate Protects the Brain Against Ischemia-Reperfusion Injury by Activating the Erythropoietin Signaling Pathway. Stroke (2012) 43(4):1101–7. doi: 10.1161/strokeaha.111.620088
40. Liu J, Narasimhan P, Yu F, Chan PH. Neuroprotection by Hypoxic Preconditioning Involves Oxidative Stress-Mediated Expression of Hypoxia-Inducible Factor and Erythropoietin. Stroke (2005) 36(6):1264–9. doi: 10.1161/01.Str.0000166180.91042.02
41. Cheng YL, Park JS, Manzanero S, Choi Y, Baik SH, Okun E, et al. Evidence That Collaboration Between HIF-1α and Notch-1 Promotes Neuronal Cell Death in Ischemic Stroke. Neurobiol Dis (2014) 62:286–95. doi: 10.1016/j.nbd.2013.10.009
42. Yang XS, Yi TL, Zhang S, Xu ZW, Yu ZQ, Sun HT, et al. Hypoxia-Inducible Factor-1 Alpha is Involved in RIP-Induced Necroptosis Caused by In Vitro and In Vivo Ischemic Brain Injury. Sci Rep (2017) 7(1):5818. doi: 10.1038/s41598-017-06088-0
43. Hu CJ, Iyer S, Sataur A, Covello KL, Chodosh LA, Simon MC. Differential Regulation of the Transcriptional Activities of Hypoxia-Inducible Factor 1 Alpha (HIF-1alpha) and HIF-2alpha in Stem Cells. Mol Cell Biol (2006) 26(9):3514–26. doi: 10.1128/mcb.26.9.3514-3526.2006
44. Wu X, Wang C, Wang J, Zhu M, Yao Y, Liu J. Hypoxia Preconditioning Protects Neuronal Cells Against Traumatic Brain Injury Through Stimulation of Glucose Transport Mediated by HIF-1α/GLUTs Signaling Pathway in Rat. Neurosurg Rev (2021) 44(1):411–22. doi: 10.1007/s10143-019-01228-8
45. Valsecchi V, Pignataro G, Del Prete A, Sirabella R, Matrone C, Boscia F, et al. NCX1 is a Novel Target Gene for Hypoxia-Inducible Factor-1 in Ischemic Brain Preconditioning. Stroke (2011) 42(3):754–63. doi: 10.1161/STROKEAHA.110.597583
46. Chen C, Ostrowski RP, Zhou C, Tang J, Zhang JH. Suppression of Hypoxia-Inducible Factor-1alpha and its Downstream Genes Reduces Acute Hyperglycemia-Enhanced Hemorrhagic Transformation in a Rat Model of Cerebral Ischemia. J Neurosci Res (2010) 88(9):2046–55. doi: 10.1002/jnr.22361
47. Zhang Z, Yan J, Shi H. Role of Hypoxia Inducible Factor 1 in Hyperglycemia-Exacerbated Blood-Brain Barrier Disruption in Ischemic Stroke. Neurobiol Dis (2016) 95:82–92. doi: 10.1016/j.nbd.2016.07.012
48. Becerra-Calixto A, Cardona-Gómez GP. The Role of Astrocytes in Neuroprotection After Brain Stroke: Potential in Cell Therapy. Front Mol Neurosci (2017) 10:88. doi: 10.3389/fnmol.2017.00088
49. Badawi Y, Ramamoorthy P, Shi H. Hypoxia-Inducible Factor 1 Protects Hypoxic Astrocytes Against Glutamate Toxicity. ASN Neuro (2012) 4(4):231–41. doi: 10.1042/AN20120006
50. Schmid-Brunclik N, Burgi-Taboada C, Antoniou X, Gassmann M, Ogunshola OO. Astrocyte Responses to Injury: VEGF Simultaneously Modulates Cell Death and Proliferation. Am J Physiol Regul Integr Comp Physiol (2008) 295(3):R864–73. doi: 10.1152/ajpregu.00536.2007
51. Yang X, Zhang Y, Geng K, Yang K, Shao J, Xia W. Sirt3 Protects Against Ischemic Stroke Injury by Regulating HIF-1α/VEGF Signaling and Blood-Brain Barrier Integrity. Cell Mol Neurobiol (2020) 41(6):1203–15. doi: 10.1007/s10571-020-00889-0
52. Ishikawa A, Yoshida H, Metoki N, Toki T, Imaizumi T, Matsumiya T, et al. Edaravone Inhibits the Expression of Vascular Endothelial Growth Factor in Human Astrocytes Exposed to Hypoxia. Neurosci Res (2007) 59(4):406–12. doi: 10.1016/j.neures.2007.08.008
53. Dombrowski L, Faure R, Marette A. Sustained Activation of Insulin Receptors Internalized in GLUT4 Vesicles of Insulin-Stimulated Skeletal Muscle. Diabetes (2000) 49(11):1772–82. doi: 10.2337/diabetes.49.11.1772
54. Jimenez-Blasco D, Busquets-Garcia A, Hebert-Chatelain E, Serrat R, Vicente-Gutierrez C, Ioannidou C, et al. Glucose Metabolism Links Astroglial Mitochondria to Cannabinoid Effects. Nature (2020) 583(7817):603–8. doi: 10.1038/s41586-020-2470-y
55. Mojsilovic-Petrovic J, Callaghan D, Cui H, Dean C, Stanimirovic DB, Zhang W. Hypoxia-Inducible Factor-1 (HIF-1) is Involved in the Regulation of Hypoxia-Stimulated Expression of Monocyte Chemoattractant Protein-1 (MCP-1/CCL2) and MCP-5 (Ccl12) in Astrocytes. J Neuroinflamm (2007) 4:12. doi: 10.1186/1742-2094-4-12
56. Yang Z, Zhao TZ, Zou YJ, Zhang JH, Feng H. Hypoxia Induces Autophagic Cell Death Through Hypoxia-Inducible Factor 1alpha in Microglia. PloS One (2014) 9(5):e96509. doi: 10.1371/journal.pone.0096509
57. Tu XK, Yang WZ, Shi SS, Wang CH, Zhang GL, Ni TR, et al. Spatio-Temporal Distribution of Inflammatory Reaction and Expression of TLR2/4 Signaling Pathway in Rat Brain Following Permanent Focal Cerebral Ischemia. Neurochem Res (2010) 35(8):1147–55. doi: 10.1007/s11064-010-0167-6
58. Yao L, Kan EM, Lu J, Hao A, Dheen ST, Kaur C, et al. Toll-Like Receptor 4 Mediates Microglial Activation and Production of Inflammatory Mediators in Neonatal Rat Brain Following Hypoxia: Role of TLR4 in Hypoxic Microglia. J Neuroinflamm (2013) 10:23. doi: 10.1186/1742-2094-10-23
59. Jiang Q, Geng X, Warren J, Eugene Paul Cosky E, Kaura S, Stone C, et al. Hypoxia Inducible Factor-1α (HIF-1α) Mediates NLRP3 Inflammasome-Dependent-Pyroptotic and Apoptotic Cell Death Following Ischemic Stroke. Neuroscience (2020) 448:126–39. doi: 10.1016/j.neuroscience.2020.09.036
60. Chen WY, Chang MS. IL-20 is Regulated by Hypoxia-Inducible Factor and Up-Regulated After Experimental Ischemic Stroke. J Immunol (2009) 182(8):5003–12. doi: 10.4049/jimmunol.0803653
61. Declercq W, Vanden Berghe T, Vandenabeele P. RIP Kinases at the Crossroads of Cell Death and Survival. Cell (2009) 138(2):229–32. doi: 10.1016/j.cell.2009.07.006
62. Chakrabarti S, Rizvi M, Morin K, Garg R, Freedman JE. The Role of CD40L and VEGF in the Modulation of Angiogenesis and Inflammation. Vascul Pharmacol (2010) 53(3-4):130–7. doi: 10.1016/j.vph.2010.05.003
63. Ashina K, Tsubosaka Y, Kobayashi K, Omori K, Murata T. VEGF-Induced Blood Flow Increase Causes Vascular Hyper-Permeability In Vivo. Biochem Biophys Res Commun (2015) 464(2):590–5. doi: 10.1016/j.bbrc.2015.07.014
64. Cheng YL, Park JS, Manzanero S, Choi Y, Baik SH, Okun E, et al. Evidence That Collaboration Between HIF-1alpha and Notch-1 Promotes Neuronal Cell Death in Ischemic Stroke. Neurobiol Dis (2014) 62:286–95. doi: 10.1016/j.nbd.2013.10.009
65. Lu N, Li X, Tan R, An J, Cai Z, Hu X, et al. HIF-1α/Beclin1-Mediated Autophagy Is Involved in Neuroprotection Induced by Hypoxic Preconditioning. J Mol Neurosci (2018) 66(2):238–50. doi: 10.1007/s12031-018-1162-7
66. Niu G, Zhu D, Zhang X, Wang J, Zhao Y, Wang X. Role of Hypoxia-Inducible Factors 1α (Hif1α) in SH-SY5Y Cell Autophagy Induced by Oxygen-Glucose Deprivation. Med Sci Monit (2018) 24:2758–66. doi: 10.12659/msm.905140
67. Wang Y, Dong XX, Cao Y, Liang ZQ, Han R, Wu JC, et al. P53 Induction Contributes to Excitotoxic Neuronal Death in Rat Striatum Through Apoptotic and Autophagic Mechanisms. Eur J Neurosci (2009) 30(12):2258–70. doi: 10.1111/j.1460-9568.2009.07025.x
68. Andreone BJ, Lacoste B, Gu C. Neuronal and Vascular Interactions. Annu Rev Neurosci (2015) 38:25–46. doi: 10.1146/annurev-neuro-071714-033835
69. Obermeier B, Daneman R, Ransohoff RM. Development, Maintenance and Disruption of the Blood-Brain Barrier. Nat Med (2013) 19(12):1584–96. doi: 10.1038/nm.3407
70. Liu A, Zhu W, Sun L, Han G, Liu H, Chen Z, et al. Ginsenoside Rb1 Administration Attenuates Focal Cerebral Ischemic Reperfusion Injury Through Inhibition of HMGB1 and Inflammation Signals. Exp Ther Med (2018) 16(4):3020–6. doi: 10.3892/etm.2018.6523
71. Hu L, Chen W, Tian F, Yuan C, Wang H, Yue H. Neuroprotective Role of Fucoxanthin Against Cerebral Ischemic/Reperfusion Injury Through Activation of Nrf2/HO-1 Signaling. BioMed Pharmacother (2018) 106:1484–9. doi: 10.1016/j.biopha.2018.07.088
72. Semenza GL. Targeting HIF-1 for Cancer Therapy. Nat Rev Cancer (2003) 3(10):721–32. doi: 10.1038/nrc1187
73. Powis G, Kirkpatrick L. Hypoxia Inducible Factor-1alpha as a Cancer Drug Target. Mol Cancer Ther (2004) 3(5):647–54.
74. Muoio V, Persson PB, Sendeski MM. The Neurovascular Unit - Concept Review. Acta Physiol (Oxf) (2014) 210(4):790–8. doi: 10.1111/apha.12250
75. Deleglise B, Lassus B, Soubeyre V, Doulazmi M, Brugg B, Vanhoutte P, et al. Dysregulated Neurotransmission Induces Trans-Synaptic Degeneration in Reconstructed Neuronal Networks. Sci Rep (2018) 8(1):11596. doi: 10.1038/s41598-018-29918-1
76. Rosenstein JM, Mani N, Silverman WF, Krum JM. Patterns of Brain Angiogenesis After Vascular Endothelial Growth Factor Administration In Vitro and In Vivo. Proc Natl Acad Sci USA (1998) 95(12):7086–91. doi: 10.1073/pnas.95.12.7086
77. Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, et al. Hypoxia-Inducible Factor Prolyl 4-Hydroxylase Inhibition. A Target for Neuroprotection in the Central Nervous System. J Biol Chem (2005) 280(50):41732–43. doi: 10.1074/jbc.M504963200
78. Hamrick SE, McQuillen PS, Jiang X, Mu D, Madan A, Ferriero DM. A Role for Hypoxia-Inducible Factor-1alpha in Desferoxamine Neuroprotection. Neurosci Lett (2005) 379(2):96–100. doi: 10.1016/j.neulet.2004.12.080
79. Reischl S, Li L, Walkinshaw G, Flippin LA, Marti HH, Kunze R. Inhibition of HIF Prolyl-4-Hydroxylases by FG-4497 Reduces Brain Tissue Injury and Edema Formation During Ischemic Stroke. PloS One (2014) 9(1):e84767. doi: 10.1371/journal.pone.0084767
80. Ogle ME, Gu X, Espinera AR, Wei L. Inhibition of Prolyl Hydroxylases by Dimethyloxaloylglycine After Stroke Reduces Ischemic Brain Injury and Requires Hypoxia Inducible Factor-1alpha. Neurobiol Dis (2012) 45(2):733–42. doi: 10.1016/j.nbd.2011.10.020
81. Kuan CY, Chen HR, Gao N, Kuo YM, Chen CW, Yang D, et al. Brain-Targeted Hypoxia-Inducible Factor Stabilization Reduces Neonatal Hypoxic-Ischemic Brain Injury. Neurobiol Dis (2021) 148:105200. doi: 10.1016/j.nbd.2020.105200
82. Kunze R, Zhou W, Veltkamp R, Wielockx B, Breier G, Marti HH. Neuron-Specific Prolyl-4-Hydroxylase Domain 2 Knockout Reduces Brain Injury After Transient Cerebral Ischemia. Stroke (2012) 43(10):2748–56. doi: 10.1161/STROKEAHA.112.669598
83. Huang X, Li Y, Li J, Feng Y, Xu X. Tanshinone IIA Dampens the Cell Proliferation Induced by Ischemic Insult in Rat Astrocytes via Blocking the Activation of HIF-1α/SDF-1 Signaling. Life Sci (2014) 112(1-2):59–67. doi: 10.1016/j.lfs.2014.07.020
84. Bhullar KS, Rupasinghe HP. Partridgeberry Polyphenols Protect Rat Primary Cortical Neurons From Oxygen-Glucose Deprivation-Reperfusion-Induced Injury via Suppression of Inflammatory Adipokines and Regulation of HIF-1alpha and PPARgamma. Nutr Neurosci (2016) 19(6):260–8. doi: 10.1179/1476830515Y.0000000026
85. Zhang Q, Qian Z, Pan L, Li H, Zhu H. Hypoxia-Inducible Factor 1 Mediates the Anti-Apoptosis of Berberine in Neurons During Hypoxia/Ischemia. Acta Physiol Hung (2012) 99(3):311–23. doi: 10.1556/APhysiol.99.2012.3.8
86. Chen M, Zou W, Chen M, Cao L, Ding J, Xiao W, et al. Ginkgolide K Promotes Angiogenesis in a Middle Cerebral Artery Occlusion Mouse Model via Activating JAK2/STAT3 Pathway. Eur J Pharmacol (2018) 833:221–9. doi: 10.1016/j.ejphar.2018.06.012
87. Yang F, Zhou L, Wang D, Wang Z, Huang QY. Minocycline Ameliorates Hypoxia-Induced Blood-Brain Barrier Damage by Inhibition of HIF-1alpha Through SIRT-3/PHD-2 Degradation Pathway. Neuroscience (2015) 304:250–9. doi: 10.1016/j.neuroscience.2015.07.051
88. Hu Q, Liu L, Zhou L, Lu H, Wang J, Chen X, et al. Effect of Fluoxetine on HIF-1alpha- Netrin/VEGF Cascade, Angiogenesis and Neuroprotection in a Rat Model of Transient Middle Cerebral Artery Occlusion. Exp Neurol (2020) 329:113312. doi: 10.1016/j.expneurol.2020.113312
89. Wang Z, Tsai LK, Munasinghe J, Leng Y, Fessler EB, Chibane F, et al. Chronic Valproate Treatment Enhances Postischemic Angiogenesis and Promotes Functional Recovery in a Rat Model of Ischemic Stroke. Stroke (2012) 43(9):2430–6. doi: 10.1161/STROKEAHA.112.652545
90. Hu Q, Liang X, Chen D, Chen Y, Doycheva D, Tang J, et al. Delayed Hyperbaric Oxygen Therapy Promotes Neurogenesis Through Reactive Oxygen Species/Hypoxia-Inducible Factor-1alpha/Beta-Catenin Pathway in Middle Cerebral Artery Occlusion Rats. Stroke (2014) 45(6):1807–14. doi: 10.1161/STROKEAHA.114.005116
91. Chang J, Mancuso MR, Maier C, Liang X, Yuki K, Yang L, et al. Gpr124 is Essential for Blood-Brain Barrier Integrity in Central Nervous System Disease. Nat Med (2017) 23(4):450–60. doi: 10.1038/nm.4309
92. Yan J, Zhou B, Taheri S, Shi H. Differential Effects of HIF-1 Inhibition by YC-1 on the Overall Outcome and Blood-Brain Barrier Damage in a Rat Model of Ischemic Stroke. PloS One (2011) 6(11):e27798. doi: 10.1371/journal.pone.0027798
93. Ferrara N. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocr Rev (2004) 25(4):581–611. doi: 10.1210/er.2003-0027
94. Yeh WL, Lu DY, Lin CJ, Liou HC, Fu WM. Inhibition of Hypoxia-Induced Increase of Blood-Brain Barrier Permeability by YC-1 Through the Antagonism of HIF-1alpha Accumulation and VEGF Expression. Mol Pharmacol (2007) 72(2):440–9. doi: 10.1124/mol.107.036418
95. Fischer S, Clauss M, Wiesnet M, Renz D, Schaper W, Karliczek GF. Hypoxia Induces Permeability in Brain Microvessel Endothelial Cells via VEGF and NO. Am J Physiol (1999) 276(4):C812–20. doi: 10.1152/ajpcell.1999.276.4.C812
96. Kissela BM, Khoury J, Kleindorfer D, Woo D, Schneider A, Alwell K, et al. Epidemiology of Ischemic Stroke in Patients With Diabetes: The Greater Cincinnati/Northern Kentucky Stroke Study. Diabetes Care (2005) 28(2):355–9. doi: 10.2337/diacare.28.2.355
97. Janghorbani M, Hu FB, Willett WC, Li TY, Manson JE, Logroscino G, et al. Prospective Study of Type 1 and Type 2 Diabetes and Risk of Stroke Subtypes: The Nurses’ Health Study. Diabetes Care (2007) 30(7):1730–5. doi: 10.2337/dc06-2363
98. Sarwar N, Gao P, Seshasai SR, Gobin R, Kaptoge S, Di Angelantonio E, et al. Diabetes Mellitus, Fasting Blood Glucose Concentration, and Risk of Vascular Disease: A Collaborative Meta-Analysis of 102 Prospective Studies. Lancet (2010) 375(9733):2215–22. doi: 10.1016/s0140-6736(10)60484-9
99. Lau LH, Lew J, Borschmann K, Thijs V, Ekinci EI. Prevalence of Diabetes and its Effects on Stroke Outcomes: A Meta-Analysis and Literature Review. J Diabetes Investig (2019) 10(3):780–92. doi: 10.1111/jdi.12932
100. Krinock MJ, Singhal NS. Diabetes, Stroke, and Neuroresilience: Looking Beyond Hyperglycemia. Ann N Y Acad Sci (2021) 1495(1):78–98. doi: 10.1111/nyas.14583
101. Yahagi K, Kolodgie FD, Lutter C, Mori H, Romero ME, Finn AV, et al. Pathology of Human Coronary and Carotid Artery Atherosclerosis and Vascular Calcification in Diabetes Mellitus. Arterioscler Thromb Vasc Biol (2017) 37(2):191–204. doi: 10.1161/atvbaha.116.306256
102. Luque A, Turu M, Juan-Babot O, Cardona P, Font A, Carvajal A, et al. Overexpression of Hypoxia/Inflammatory Markers in Atherosclerotic Carotid Plaques. Front Biosci (2008) 13:6483–90. doi: 10.2741/3168
103. Higashida T, Kanno H, Nakano M, Funakoshi K, Yamamoto I. Expression of Hypoxia-Inducible Angiogenic Proteins (Hypoxia-Inducible Factor-1alpha, Vascular Endothelial Growth Factor, and E26 Transformation-Specific-1) and Plaque Hemorrhage in Human Carotid Atherosclerosis. J Neurosurg (2008) 109(1):83–91. doi: 10.3171/jns/2008/109/7/0083
104. Basic J, Stojkovic S, Assadian A, Rauscher S, Duschek N, Kaun C, et al. The Relevance of Vascular Endothelial Growth Factor, Hypoxia Inducible Factor-1 Alpha, and Clusterin in Carotid Plaque Instability. J Stroke Cerebrovasc Dis (2019) 28(6):1540–5. doi: 10.1016/j.jstrokecerebrovasdis.2019.03.009
105. Lin JH, Lou N, Kang N, Takano T, Hu F, Han X, et al. A Central Role of Connexin 43 in Hypoxic Preconditioning. J Neurosci (2008) 28(3):681–95. doi: 10.1523/jneurosci.3827-07.2008
106. Takano T, Oberheim N, Cotrina ML, Nedergaard M. Astrocytes and Ischemic Injury. Stroke (2009) 40(3 Suppl):S8–12. doi: 10.1161/strokeaha.108.533166
107. Liddelow SA, Barres BA. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity (2017) 46(6):957–67. doi: 10.1016/j.immuni.2017.06.006
108. Liu Z, Chopp M. Astrocytes, Therapeutic Targets for Neuroprotection and Neurorestoration in Ischemic Stroke. Prog Neurobiol (2016) 144:103–20. doi: 10.1016/j.pneurobio.2015.09.008
109. Lau LT, Yu AC. Astrocytes Produce and Release Interleukin-1, Interleukin-6, Tumor Necrosis Factor Alpha and Interferon-Gamma Following Traumatic and Metabolic Injury. J Neurotrauma (2001) 18(3):351–9. doi: 10.1089/08977150151071035
110. Hellwig-Bürgel T, Stiehl DP, Wagner AE, Metzen E, Jelkmann W. Review: Hypoxia-Inducible Factor-1 (HIF-1): A Novel Transcription Factor in Immune Reactions. J Interferon Cytokine Res (2005) 25(6):297–310. doi: 10.1089/jir.2005.25.297
111. Magistretti PJ, Allaman I. Lactate in the Brain: From Metabolic End-Product to Signalling Molecule. Nat Rev Neurosci (2018) 19(4):235–49. doi: 10.1038/nrn.2018.19
112. Barros LF, Weber B. CrossTalk Proposal: An Important Astrocyte-to-Neuron Lactate Shuttle Couples Neuronal Activity to Glucose Utilisation in the Brain. J Physiol (2018) 596(3):347–50. doi: 10.1113/jp274944
113. Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, et al. The Transcription Factor HIF-1alpha Plays a Critical Role in the Growth Factor-Dependent Regulation of Both Aerobic and Anaerobic Glycolysis. Genes Dev (2007) 21(9):1037–49. doi: 10.1101/gad.1529107
114. Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, et al. The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1alpha. Cell (2010) 140(2):280–93. doi: 10.1016/j.cell.2009.12.041
115. Parra-Abarca J, Rivera-Ramírez N, Villa-Maldonado LF, García-Hernández U, Aguilera P, Arias-Montaño JA. Histamine H(1) and H(3) Receptor Activation Increases the Expression of Glucose Transporter 1 (GLUT-1) in Rat Cerebro-Cortical Astrocytes in Primary Culture. Neurochem Int (2019) 131:104565. doi: 10.1016/j.neuint.2019.104565
116. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-Mediated Expression of Pyruvate Dehydrogenase Kinase: A Metabolic Switch Required for Cellular Adaptation to Hypoxia. Cell Metab (2006) 3(3):177–85. doi: 10.1016/j.cmet.2006.02.002
117. Perry VH, Nicoll JA, Holmes C. Microglia in Neurodegenerative Disease. Nat Rev Neurol (2010) 6(4):193–201. doi: 10.1038/nrneurol.2010.17
118. Dong Y, Benveniste EN. Immune Function of Astrocytes. Glia (2001) 36(2):180–90. doi: 10.1002/glia.1107
119. Wu X, Liu S, Hu Z, Zhu G, Zheng G, Wang G. Enriched Housing Promotes Post-Stroke Neurogenesis Through Calpain 1-STAT3/HIF-1alpha/VEGF Signaling. Brain Res Bull (2018) 139:133–43. doi: 10.1016/j.brainresbull.2018.02.018
120. Liu M, Alkayed NJ. Hypoxic Preconditioning and Tolerance via Hypoxia Inducible Factor (HIF) 1alpha-Linked Induction of P450 2C11 Epoxygenase in Astrocytes. J Cereb Blood Flow Metab (2005) 25(8):939–48. doi: 10.1038/sj.jcbfm.9600085
121. Chen X, Zhang X, Wang Y, Lei H, Su H, Zeng J, et al. Inhibition of Immunoproteasome Reduces Infarction Volume and Attenuates Inflammatory Reaction in a Rat Model of Ischemic Stroke. Cell Death Dis (2015) 6(1):e1626. doi: 10.1038/cddis.2014.586
122. Lü L, Wang H. Transient Focal Cerebral Ischemia Upregulates Immunoproteasomal Subunits. Cell Mol Neurobiol (2012) 32(6):965–70. doi: 10.1007/s10571-012-9854-y
123. Chen X, Wang Y, Fu M, Lei H, Cheng Q, Zhang X. Plasma Immunoproteasome Predicts Early Hemorrhagic Transformation in Acute Ischemic Stroke Patients. J Stroke Cerebrovasc Dis (2017) 26(1):49–56. doi: 10.1016/j.jstrokecerebrovasdis.2016.08.027
124. Chen X, Zhang X, Chen T, Jiang X, Wang X, Lei H, et al. Inhibition of Immunoproteasome Promotes Angiogenesis via Enhancing Hypoxia-Inducible Factor-1alpha Abundance in Rats Following Focal Cerebral Ischaemia. Brain Behav Immun (2018) 73:167–79. doi: 10.1016/j.bbi.2018.04.009
125. Rivest S. Regulation of Innate Immune Responses in the Brain. Nat Rev Immunol (2009) 9(6):429–39. doi: 10.1038/nri2565
126. Jeon SB, Yoon HJ, Park SH, Kim IH, Park EJ. Sulfatide, a Major Lipid Component of Myelin Sheath, Activates Inflammatory Responses as an Endogenous Stimulator in Brain-Resident Immune Cells. J Immunol (2008) 181(11):8077–87. doi: 10.4049/jimmunol.181.11.8077
127. Glass CK, Natoli G. Molecular Control of Activation and Priming in Macrophages. Nat Immunol (2016) 17(1):26–33. doi: 10.1038/ni.3306
128. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
129. de Lemos ML, de la Torre AV, Petrov D, Brox S, Folch J, Pallàs M, et al. Evaluation of Hypoxia Inducible Factor Expression in Inflammatory and Neurodegenerative Brain Models. Int J Biochem Cell Biol (2013) 45(7):1377–88. doi: 10.1016/j.biocel.2013.04.011
130. Bok S, Kim YE, Woo Y, Kim S, Kang SJ, Lee Y, et al. Hypoxia-Inducible Factor-1α Regulates Microglial Functions Affecting Neuronal Survival in the Acute Phase of Ischemic Stroke in Mice. Oncotarget (2017) 8(67):111508–21. doi: 10.18632/oncotarget.22851
131. Chen H, Xiong T, Qu Y, Zhao F, Ferriero D, Mu D. mTOR Activates Hypoxia-Inducible Factor-1α and Inhibits Neuronal Apoptosis in the Developing Rat Brain During the Early Phase After Hypoxia-Ischemia. Neurosci Lett (2012) 507(2):118–23. doi: 10.1016/j.neulet.2011.11.058
132. Wei Y, Hong H, Zhang X, Lai W, Wang Y, Chu K, et al. Salidroside Inhibits Inflammation Through PI3K/Akt/HIF Signaling After Focal Cerebral Ischemia in Rats. Inflammation (2017) 40(4):1297–309. doi: 10.1007/s10753-017-0573-x
133. Koh HS, Chang CY, Jeon SB, Yoon HJ, Ahn YH, Kim HS, et al. The HIF-1/Glial TIM-3 Axis Controls Inflammation-Associated Brain Damage Under Hypoxia. Nat Commun (2015) 6:6340. doi: 10.1038/ncomms7340
134. Chen SF, Pan MX, Tang JC, Cheng J, Zhao D, Zhang Y, et al. Arginine is Neuroprotective Through Suppressing HIF-1α/LDHA-Mediated Inflammatory Response After Cerebral Ischemia/Reperfusion Injury. Mol Brain (2020) 13(1):63. doi: 10.1186/s13041-020-00601-9
135. Liu R, Liao XY, Pan MX, Tang JC, Chen SF, Zhang Y, et al. Glycine Exhibits Neuroprotective Effects in Ischemic Stroke in Rats Through the Inhibition of M1 Microglial Polarization via the NF-kappaB P65/Hif-1alpha Signaling Pathway. J Immunol (2019) 202(6):1704–14. doi: 10.4049/jimmunol.1801166
136. Luo Y, Reis C, Chen S. NLRP3 Inflammasome in the Pathophysiology of Hemorrhagic Stroke: A Review. Curr Neuropharmacol (2019) 17(7):582–9. doi: 10.2174/1570159x17666181227170053
137. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in Health and Disease. Nature (2012) 481(7381):278–86. doi: 10.1038/nature10759
138. Levine B, Mizushima N, Virgin HW. Autophagy in Immunity and Inflammation. Nature (2011) 469(7330):323–35. doi: 10.1038/nature09782
139. Williamson WR, Wang D, Haberman AS, Hiesinger PR. A Dual Function of V0-ATPase A1 Provides an Endolysosomal Degradation Mechanism in Drosophila Melanogaster Photoreceptors. J Cell Biol (2010) 189(5):885–99. doi: 10.1083/jcb.201003062
140. Sumpter R Jr., Levine B. Autophagy and Innate Immunity: Triggering, Targeting and Tuning. Semin Cell Dev Biol (2010) 21(7):699–711. doi: 10.1016/j.semcdb.2010.04.003
141. Su P, Zhang J, Wang D, Zhao F, Cao Z, Aschner M, et al. The Role of Autophagy in Modulation of Neuroinflammation in Microglia. Neuroscience (2016) 319:155–67. doi: 10.1016/j.neuroscience.2016.01.035
142. Tsao CC, Baumann J, Huang SF, Kindler D, Schroeter A, Kachappilly N, et al. Pericyte Hypoxia-Inducible Factor-1 (HIF-1) Drives Blood-Brain Barrier Disruption and Impacts Acute Ischemic Stroke Outcome. Angiogenesis (2021) 24(4):823–42. doi: 10.1007/s10456-021-09796-4
Keywords: HIF-1α, ischemic stroke, hypoxia, neurovascular unit, neuroprotection, neuroinflammation
Citation: He Q, Ma Y, Liu J, Zhang D, Ren J, Zhao R, Chang J, Guo Z-N and Yang Y (2021) Biological Functions and Regulatory Mechanisms of Hypoxia-Inducible Factor-1α in Ischemic Stroke. Front. Immunol. 12:801985. doi: 10.3389/fimmu.2021.801985
Received: 26 October 2021; Accepted: 26 November 2021;
Published: 13 December 2021.
Edited by:
Yujie Chen, Army Medical University, ChinaReviewed by:
Charles Kozhikkadan Davis, University of Wisconsin-Madison, United StatesJichao Yuan, Third Military Medical University, China
Copyright © 2021 He, Ma, Liu, Zhang, Ren, Zhao, Chang, Guo and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Yang, eWFuZ195aUBqbHUuZWR1LmNu; Zhen-Ni Guo, emhlbjFuaTJAamx1LmVkdS5jbg==
†These authors have contributed equally to this work