
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 08 December 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.797550
This article is part of the Research TopicHost-pathogen metabolic interactions determine susceptibility to bacterial infectionView all 4 articles
 Kara R. Eichelberger1*
Kara R. Eichelberger1* James E. Cassat1,2,3,4,5*
James E. Cassat1,2,3,4,5*Successful pathogens require metabolic flexibility to adapt to diverse host niches. The presence of co-infecting or commensal microorganisms at a given infection site can further influence the metabolic processes required for a pathogen to cause disease. The Gram-positive bacterium Staphylococcus aureus and the polymorphic fungus Candida albicans are microorganisms that asymptomatically colonize healthy individuals but can also cause superficial infections or severe invasive disease. Due to many shared host niches, S. aureus and C. albicans are frequently co-isolated from mixed fungal-bacterial infections. S. aureus and C. albicans co-infection alters microbial metabolism relative to infection with either organism alone. Metabolic changes during co-infection regulate virulence, such as enhancing toxin production in S. aureus or contributing to morphogenesis and cell wall remodeling in C. albicans. C. albicans and S. aureus also form polymicrobial biofilms, which have greater biomass and reduced susceptibility to antimicrobials relative to mono-microbial biofilms. The S. aureus and C. albicans metabolic programs induced during co-infection impact interactions with host immune cells, resulting in greater microbial survival and immune evasion. Conversely, innate immune cell sensing of S. aureus and C. albicans triggers metabolic changes in the host cells that result in an altered immune response to secondary infections. In this review article, we discuss the metabolic programs that govern host-pathogen interactions during S. aureus and C. albicans co-infection. Understanding C. albicans-S. aureus interactions may highlight more general principles of how polymicrobial interactions, particularly fungal-bacterial interactions, shape the outcome of infectious disease. We focus on how co-infection alters microbial metabolism to enhance virulence and how infection-induced changes to host cell metabolism can impact a secondary infection.
Microorganisms co-exist as polymicrobial communities in the human body, sharing colonization niches and competing for resources. Interactions among these commensal microbial communities facilitate persistence of microorganisms that promote health. However, in the context of a polymicrobial infection, interactions among co-infecting pathogens may exacerbate disease. Polymicrobial infections can develop when infection by one organism creates a favorable host niche for colonization by additional organisms, when two or more organisms are simultaneously introduced into the body, or when host immune defenses are weakened (1). For example, polymicrobial infections frequently develop in individuals with compromised host immune responses or disrupted biological barrier function, in which there may exist a more favorable environment for co-infection (2–4). Co-infecting pathogens can behave antagonistically, additively, or synergistically to alter disease outcome relative to mono-microbial infections (1). However, many polymicrobial infections are associated with more severe disease relative to mono-microbial infections (5–7). Because polymicrobial infections can alter the outcome of disease, it is critical to understand how the presence of co-infecting pathogens affect microbial physiology and subsequent host responses during infection.
Bacteria and fungi are frequently co-isolated from commensal microbial communities, and inter-species cross-talk facilitates stable and asymptomatic colonization of these organisms (8). However, cross-talk between bacteria and fungi can also contribute to disease progression in the context of a polymicrobial infection. The opportunistic fungal pathogen Candida albicans and the Gram-positive bacterium Staphylococcus aureus are two of the most commonly co-isolated pathogens from mixed fungal-bacterial infections in the bloodstream as well as from biofilm-associated diseases such as cystic fibrosis, periodontitis, and catheter-associated infections (4, 8–10). The frequency of mixed C. albicans-S. aureus infections is in part due to shared colonization sites in the human body as well as a propensity for these microorganisms to interact and form polymicrobial biofilms (8, 10, 11). C. albicans and S. aureus both asymptomatically colonize the skin or gastrointestinal tract, yet under certain circumstances they can also cause severe invasive disease (12, 13). A key feature of the virulence of both C. albicans and S. aureus is the ability to utilize a variety of nutrient sources to establish infection in diverse host niches (14, 15). C. albicans virulence is mediated in large part by a morphologic switch from growth as budding yeast to filamentous hyphae. C. albicans yeast colonize skin and the gastrointestinal mucosa (12). During invasive infection, C. albicans hyphal growth penetrates epithelial layers and causes tissue damage, while C. albicans yeast disseminate through the bloodstream and colonize other organs (12). C. albicans mutants that are genetically locked in either morphology are attenuated during infection, highlighting the importance of the ability to switch between the yeast and the hyphal forms to cause disease (12, 16). However, recent studies have challenged this dogma, demonstrating that certain C. albicans yeast-locked strains retain virulence in disseminated infection due to the metabolic advantages of yeast growth over hyphal growth (17). S. aureus virulence is driven by a variety of mechanisms to combat host responses and adapt to host environments. This includes production of toxins and immunomodulatory proteins that evade host immune responses (18–21).
C. albicans-S. aureus co-infection worsens invasive disease relative to infection with either organism alone. Some of the earliest studies investigating C. albicans-S. aureus co-infection identified that this polymicrobial interaction is associated with greater mortality when both organisms are inoculated into the peritoneal cavity of mice simultaneously (22–24). The enhanced virulence of C. albicans-S. aureus co-infection is due in part to physical and chemical interactions between the two microorganisms that influence microbial metabolism, virulence, and physiology. Host immune responses also play an important role in contributing to the enhanced virulence of C. albicans-S. aureus co-infection. Co-infection skews the balance of pro-inflammatory and anti-inflammatory cytokine production towards greater inflammation. Altered host responses can occur through direct interactions of host cells with the microbes that facilitate microbial dissemination, as well as through increased pro-inflammatory cytokine production by host cells as a response to co-infection (25, 26). Paradoxically, outcomes of invasive S. aureus infections are improved when S. aureus is inoculated after a C. albicans infection, suggesting cross-species protection in the context of secondary infection (27). In this review, we discuss advances towards understanding the factors that contribute to the changes in disease progression during C. albicans-S. aureus co-infection, with a focus on interactions that worsen disease outcomes. We highlight the mechanisms by which virulence is altered during acute polymicrobial infection, as well as how interactions during polymicrobial biofilm growth influence virulence and antimicrobial resistance. Finally, we discuss how polymicrobial infection alters host cell responses during co-infection and how sequential infection is protective rather than deleterious.
Co-inoculation of C. albicans and S. aureus into the peritoneal cavity of mice results in 100% mortality, while inoculation of the same dose of either organism alone does not result in lethal disease (25). Recent work identified that C. albicans influences S. aureus quorum sensing to augment its virulence, which contributes to the lethality of polymicrobial intra-peritoneal infection (Figure 1). Co-culture of C. albicans with S. aureus enhances the S. aureus quorum-sensing system Agr, or accessory gene regulator (28). The Agr system regulates a variety of toxins that are important for S. aureus pathogenesis, including α-toxin (21, 29). Todd et al. determined that co-culture of C. albicans with S. aureus enhances α-toxin levels in vitro, and co-inoculation of C. albicans and S. aureus into the peritoneum also resulted in greater α-toxin levels in the peritoneal lavage fluid relative to S. aureus mono-infection (28). While antibody treatment that neutralizes α-toxin in mice partially alleviated the mortality in a C. albicans-S. aureus co-infection, injection of purified α-toxin combined with live C. albicans was not sufficient to induce enhanced mortality (28). Thus, lethal synergism in this model may require either live organisms or involve additional S. aureus virulence factors.
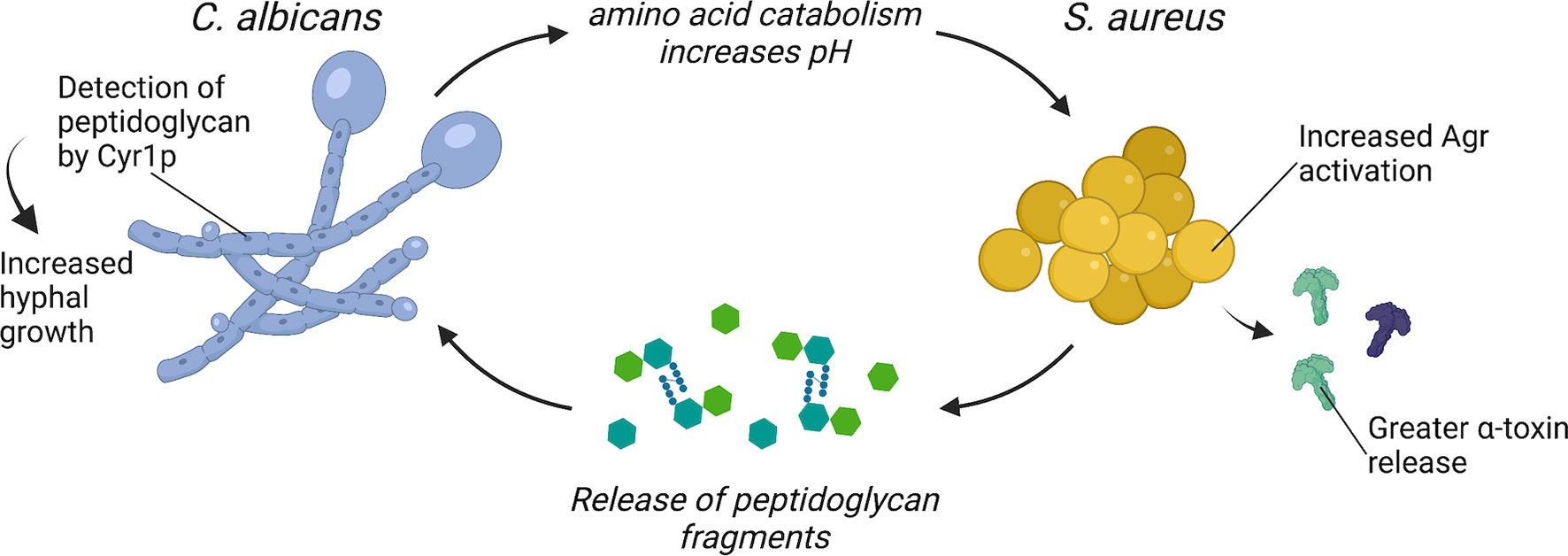
Figure 1 C. albicans and S. aureus interact to enhance virulence. C. albicans enhances S. aureus Agr activation through amino acid metabolism-driven pH modification and morphogenesis signaling. S. aureus may enhance C. albicans virulence through the release of peptidoglycan fragments that are sensed by Cyr1p and trigger hyphal morphogenesis.
One mechanism proposed for enhanced activation of S. aureus Agr system during co-culture with C. albicans requires C. albicans amino acid metabolism. During infection, C. albicans can catabolize amino acids and utilize these nutrients as a carbon source (14). Amino acids are imported via amino acid permeases, which are regulated by the transcription factors Stp1p and Stp2p. As C. albicans catabolizes amino acids, it exports ammonia, which alkalinizes the extracellular media and promotes C. albicans hyphal formation (30). C. albicans stp2Δ/Δ is defective in the alkalinization of the extracellular environment and is more efficiently killed during macrophage infection (30). The Agr system is sensitive to pH and optimal activity occurs at neutral pH (31–33). Therefore, it was hypothesized that C. albicans alkalinization of the extracellular media as a byproduct of amino acid metabolism provides an optimal pH for activation of the S. aureus Agr system during co-culture (34). Indeed, S. aureus co-cultured with C. albicans stp2Δ/Δ produces less α-toxin relative to S. aureus co-cultured with wild-type C. albicans (34). However, the role of C. albicans amino acid catabolism in promoting enhanced virulence during co-infection with S. aureus in vivo remains to be determined.
An additional mechanism by which C. albicans may enhance S. aureus virulence during co-infection requires C. albicans morphogenesis signaling. Mice survive intraperitoneal co-infection of S. aureus and the C. albicans morphogenesis mutant efg1Δ/Δ, while mice inoculated with S. aureus and wild-type C. albicans all succumb to disease (35). Efg1p is a transcriptional regulator that induces hyphal gene expression and filamentous growth, and an EFG1 mutant, which can only grow as yeast, is highly attenuated in vivo (36, 37). To test if enhanced mortality during intraperitoneal co-infection requires a specific C. albicans morphology, Nash et al. inoculated mice with S. aureus and either a C. albicans yeast-locked strain or a C. albicans hyphal-locked strain (38). However, S. aureus co-inoculated with either yeast-locked C. albicans or hyphal-locked C. albicans is as lethal as co-infection with wild-type C. albicans in this model (38). Considering that morphology-locked strains are typically attenuated in vivo, it is surprising that morphology-locked C. albicans strains can still enhance S. aureus virulence during intraperitoneal co-infection (16, 36). There may be additional morphology-independent processes regulated by Efg1p that contribute to lethal synergism during polymicrobial intra-abdominal infection. In addition to inducing filamentous growth in C. albicans, Efg1p is a master regulator of metabolic genes, as well as genes involved in adhesion (39). For example, C. albicans efg1Δ/Δ has significantly reduced transcript levels for almost all glycolytic genes and several tricarboxylic acid cycle genes (40). Thus, C. albicans metabolism, as regulated by Efg1p, may play an additional role in enhancing S. aureus virulence during intra-abdominal polymicrobial infection.
The mechanisms by which S. aureus directly promotes C. albicans virulence are less clear. However, one bacterial-derived molecule with potent effects on C. albicans morphology and metabolism is peptidoglycan. Serum is a potent inducer of C. albicans hyphal formation, and an analysis of the hyphal-stimulating fractions of human and bovine serum found structures resembling bacterial peptidoglycan fragments (37, 41, 42). Partial hydrolysis of peptidoglycan purified from the Gram-positive bacterium S. aureus or the Gram-negative bacterium Escherichia coli potently induces hyphal formation (41). These peptidoglycan fragments are imported by C. albicans and sensed by the adenylyl cyclase protein Cyr1, which activates the Protein Kinase A (PKA) pathway and induces hyphal gene expression (41). One hypothesis is that the source of peptidoglycan fragments present in serum is from turnover of the bacteria that comprise the intestinal microbiome (42). Tan et al. confirmed that culturing C. albicans adjacent to both Gram-positive and Gram-negative bacteria induced hyphal formation, and this effect could be further augmented by treatment with β-lactam antibiotics (43). β-lactams disrupt crosslinking of new peptidoglycan fragments, freeing peptidoglycan molecules from the bacterial cell wall during the process of killing the bacteria (44). Mice that were treated with β-lactam antibiotics before and after C. albicans oral inoculation developed greater C. albicans hyphal formation in the gut (43). The β-lactam-treated mice also had higher C. albicans burdens in the kidneys, indicating increased dissemination to peripheral organs following β-lactam treatment (43). However, signaling among the various bacterial organisms that comprise the intestinal microbiota and C. albicans likely also influence C. albicans morphology and pathogenesis in the gastrointestinal tract.
N-Acetylglucosamine (GlcNAc) is also a component of peptidoglycan that can influence C. albicans metabolism and virulence. During infection, C. albicans can metabolize alternative carbon sources, such as GlcNAc, to facilitate host adaptation and survival (14). GlcNAc is also found as a component of chitin in fungal cell walls and as a part of the extracellular matrix in animals (45). However, bacteria release GlcNAc during cell wall remodeling and peptidoglycan turnover and may provide a significant source of GlcNAc to C. albicans cells during mixed fungal-bacterial infections (45, 46). GlcNAc is imported into the fungal cell via Ngt1p, is phosphorylated, and either enters an anabolic pathway for the synthesis of chitin or is catabolized for use in glycolysis (45, 47, 48). Interestingly, C. albicans mutants that cannot phosphorylate and metabolize imported GlcNAc still form hyphal filaments following GlcNAc exposure (49). This indicates a role for GlcNAc as a signaling molecule in C. albicans that is independent of its role as a nutrient and carbon source. However, metabolism of GlcNAc can also induce hyphal formation, as it releases ammonia and alkalinizes the extracellular media, which triggers hyphal formation (50). GlcNAc metabolism is important for alkalinization of the macrophage phagosome following phagocytosis of C. albicans yeast, which promotes C. albicans hyphal growth and survival within the macrophage (51). These studies collectively demonstrate that components of bacterial peptidoglycan can have potent effects on C. albicans metabolism, morphogenesis, and subsequent virulence.
During co-infection, S. aureus grows in close association with C. albicans hyphae (24). Since S. aureus releases peptidoglycan fragments during growth (52), it is feasible that peptidoglycan recycling and remodeling plays a role in altering C. albicans metabolism and morphogenesis through a ready supply of released peptidoglycan fragments (Figure 1). Peptidoglycan detection by C. albicans could be an additional mechanism contributing to the lethal synergism exhibited by C. albicans and S. aureus co-infections. Whether the ability of C. albicans to sense and respond to peptidoglycan released by neighboring bacteria is also important for C. albicans existence as a commensal within the gastrointestinal tract remains to be determined.
Biofilms pose a major clinical problem as they display tolerance to standard antimicrobial treatments and can become a niche for persistent infections. In addition to their ability to form robust biofilms during mono-infection, C. albicans and S. aureus have a propensity to form robust polymicrobial biofilms during co-infection (4, 53, 54). These are often associated with indwelling medical devices, such as catheters (55). However, certain disease conditions, such as within the cystic fibrosis lung, are also a niche for polymicrobial biofilm growth of C. albicans and S. aureus (5). In this section, we will review metabolic adaptations of both organisms during polymicrobial biofilm growth. We discuss how polymicrobial biofilm growth contributes to the altered virulence observed during a C. albicans-S. aureus co-infection relative to infection with either organism alone (Figure 2).
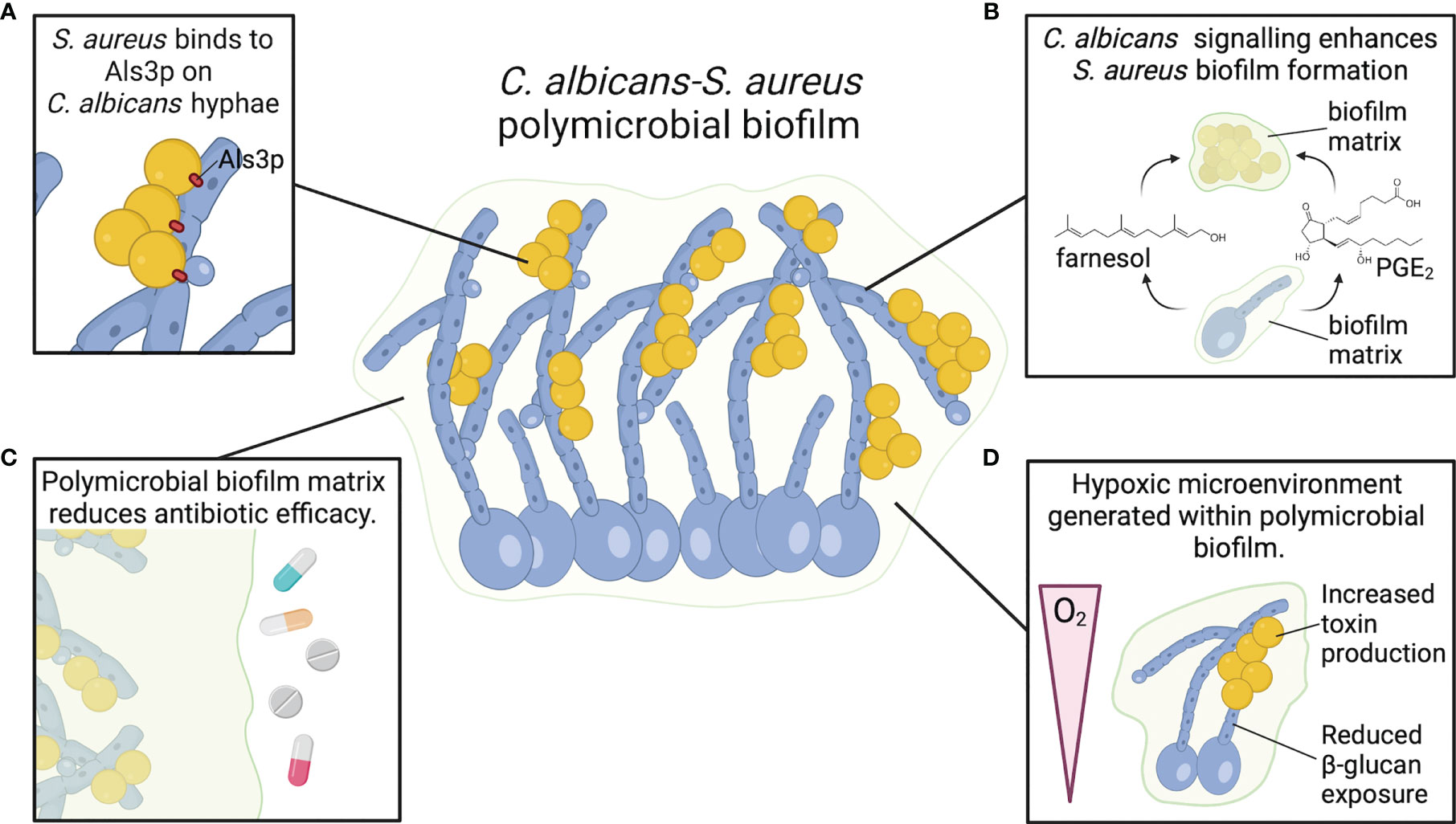
Figure 2 C. albicans and S. aureus form robust polymicrobial biofilms. (A) S. aureus adheres to Als3 protein on C. albicans hyphae, promoting strong attachment and enhancing biofilm growth. (B) C. albicans chemical signals farnesol and PGE2 enhance S. aureus biofilm growth and biomass. (C) Biofilm matrix components, particularly β-glucan, act as a physical barrier to resist antimicrobials and promote S. aureus survival. (D) C. albicans biofilms generate a hypoxic microenvironment that may enhance S. aureus toxin production.
C. albicans and S. aureus are tightly associated in polymicrobial biofilms. S. aureus forms microcolonies within the C. albicans biofilm matrix, and the bacteria primarily attach to the hyphal form of C. albicans (Figure 2A) (56–58). Binding to C. albicans hyphae is predominantly mediated by the C. albicans hyphal-specific protein Als3, which functions as an adhesin (59). However, in the presence of serum, S. aureus adheres to C. albicans als3Δ/Δ filaments to the same degree as wild-type filaments, indicating that other factors may play a role in adherence of S. aureus to C. albicans hyphae under different conditions (60). In addition to strong adherence to hyphal filaments, S. aureus colony forming units (CFU) are also significantly higher when grown with C. albicans in a polymicrobial biofilm relative to mono-microbial biofilm (56, 61, 62). However, C. albicans CFU remain the same whether grown in biofilms alone or with S. aureus (56). Biofilm growth is reduced when S. aureus is co-cultured with efg1Δ/Δ cph1Δ/Δ and bcr1Δ/Δ C. albicans mutants, but these mutants are also unable to form robust biofilms during C. albicans mono-microbial growth (60). Interestingly, S. aureus biofilm growth is enhanced when co-cultured with amphotericin-B killed, but not formalin killed, C. albicans biofilm (60). Amphotericin-B disrupts fungal cell membranes to kill C. albicans, while formalin kills cells by cross-linking proteins. Therefore, enhanced S. aureus biofilm growth during co-culture with C. albicans is not dependent on a process mediated by live C. albicans cells, but rather a surface protein that can be neutralized by formalin. Fungal glucans are an additional C. albicans cell wall component present in the extracellular matrix material of C. albicans biofilms, and glucans enhance S. aureus biofilm formation when supplied exogenously (63).
The differences in C. albicans and S. aureus behavior and physiology within a polymicrobial biofilm may be partly mediated by metabolic changes. Proteomic analysis of C. albicans-S. aureus polymicrobial biofilm growth identified an increased abundance of proteins primarily involved in metabolism and stress response for both organisms relative to mono-microbial biofilms (57). Metabolic exchange within the polymicrobial biofilm could also influence biofilm formation and biomass. For example, glucose is the most abundant monosaccharide present in the extracellular matrix of a C. albicans mono-culture biofilm (64). Because glucose enhances S. aureus biofilm production, greater availability of glucose provided by the C. albicans biofilm matrix may also enhance S. aureus growth and biofilm production, contributing to greater biomass in the polymicrobial biofilms (65). Altered metabolism in C. albicans-S. aureus biofilm may also contribute to enhanced persistence of the organisms in vivo. S. aureus biofilms produce lactate that modulates host macrophages and myeloid-derived suppressor cells (MDSC) to produce more IL-10, which has an anti-inflammatory effect that enables persistence of S. aureus biofilms in vivo (66). S. aureus-produced lactate may also have effects on C. albicans cells within the polymicrobial biofilm. Lactate induces reorganization of the fungal cell wall to reduce, or mask, exposure of β-glucan on C. albicans hyphae (67). Because β-glucan is a potent immunostimulatory factor, the response to lactate reduces immune detection, allowing greater C. albicans persistence and survival during infection. While most studies have focused on synergistic metabolic interactions occurring within polymicrobial biofilms, broad metabolomic analyses to characterize both synergistic and antagonistic interactions between C. albicans and S. aureus are needed.
Several chemicals produced by both S. aureus and C. albicans can influence polymicrobial biofilm formation through cross-species signaling. One of the best-studied chemical signals that can alter both C. albicans and S. aureus physiology is farnesol (68). Farnesol is a quorum-sensing molecule derived from glycolytic products via the sterol pathway in C. albicans (68). Farnesol blocks the C. albicans morphological transition from yeast to hyphae under high cell density (69). As C. albicans hyphal growth is required for the development of a biofilm, farnesol inhibits C. albicans biofilm formation (70). Depending on the concentration, farnesol exerts both synergistic and antagonistic effects on S. aureus physiology (4). High concentrations of farnesol were first determined to inhibit S. aureus biofilm growth, induce cell death, and increase sensitivity of a methicillin-sensitive S. aureus strain towards a variety of different classes of antibiotics (71). Farnesol can also inhibit S. aureus lipases, which hydrolyze host lipids to free fatty acids that promote colonization (72). However, low levels of farnesol promote biofilm formation in S. aureus (62). Additionally, Kong et al. determined that low concentrations of farnesol could increase S. aureus resistance to antibiotics via a cellular stress response (73). RNAseq analysis of S. aureus exposed to farnesol revealed an increase in expression of genes involved in stress response as well as protection against oxidative stress (74). Accordingly, farnesol treatment induces greater S. aureus resistance to killing by H2O2 (74). The ultimate effects of farnesol on S. aureus during infection may vary spatially or temporally within the polymicrobial biofilm based on the changes in C. albicans production of farnesol during development of the polymicrobial biofilm.
An additional chemical signaling molecule involved in C. albicans biofilm formation is prostaglandin E2 (PGE2). C. albicans, in addition to other pathogenic fungi, can synthesize prostaglandins de novo or following supplementation with arachidonic acid, which is metabolized to produce prostaglandins via cyclooxygenase (COX) enzymes (75). The prostaglandins synthesized by C. albicans stimulate germ tube formation, which is the precursory step to hyphal growth (75). Furthermore, C. albicans biofilm formation is significantly reduced following the use of COX inhibitor drugs to block prostaglandin biosynthesis (76). PGE2 added to S. aureus mono-culture stimulated greater biofilm growth relative to untreated S. aureus biofilms, mimicking the enhanced biomass following co-culture with C. albicans (62). These data suggest that PGE2 may be another fungal-derived molecule that influences biofilm growth of both organisms in a polymicrobial biofilm (Figure 2B).
The effects of S. aureus chemical signals on C. albicans biofilm formation and physiology are less understood. When S. aureus and C. albicans are added together to form a polymicrobial biofilm, S. aureus increases attachment of C. albicans to plastic surfaces (77). Additionally, conditioned media from a mature S. aureus biofilm significantly enhances C. albicans biofilm formation, suggesting S. aureus secreted factors contribute to biofilm formation (77). The S. aureus factor(s) that enhances C. albicans biofilm remains unknown. As discussed earlier, peptidoglycan fragments induce C. albicans hyphal formation (41, 43). Because hyphal formation is a requisite step in biofilm formation, it is possible that peptidoglycan fragments present in the S. aureus conditioned media are enhancing C. albicans biofilm. S. aureus can also negatively regulate biofilm formation through the production of staphylokinase (Sak), which reduces biomass and metabolic activity of C. albicans-S. aureus polymicrobial biofilm by causing detachment of cells from the biofilm (78). Sak also reduced C. albicans gene expression of morphogenesis regulators (78). However, it is unclear if Sak-mediated changes in C. albicans gene expression are directly mediated by Sak or if it is an indirect effect resulting from detachment of C. albicans and S. aureus cells from the polymicrobial biofilm. The effects of S. aureus chemicals and other secreted factors on polymicrobial biofilm formation during infection require further investigation.
Biofilm growth reduces antibiotic efficacy towards multiple organisms, including S. aureus and C. albicans, presenting a major barrier for treating infection. In addition to enhanced growth and biomass of a polymicrobial biofilm, C. albicans-S. aureus biofilms exhibit greater resistance to killing by antimicrobials relative to mono-microbial biofilms (Figure 2C) (56). Vancomycin is less effective at killing S. aureus when S. aureus is grown in a biofilm with C. albicans, but there are no changes in amphotericin-B fungicidal activity towards C. albicans (56). Decreased antibiotic efficacy was also observed when mice were surgically implanted with small catheters coated in S. aureus and C. albicans (79). While S. aureus is less susceptible to tigecycline in a mixed biofilm rather than a mono-culture biofilm, there is no effect on C. albicans susceptibility to anidulafungin in mixed or mono-culture biofilms (79). Treatment with a combination of anidulafungin and tigecycline improves S. aureus killing in a mixed biofilm infection (79). Anidulafungin reduces S. aureus poly-β-(1,3)-N-acetylglucosamine (PNAG) production, which is a component of S. aureus biofilm matrix for select strains (79). Although the mechanism by which anidulafungin reduces PNAG production remains to be determined, the authors suggest that it may inhibit activity of PNAG synthesis protein IcaA (79). However, the anidulafungin-mediated reduction of C. albicans biofilm growth may also contribute to enhanced killing of S. aureus in biofilm, as reducing C. albicans burdens in a polymicrobial biofilm consequently reduces the protective effect of C. albicans on S. aureus antibiotic resistance (56, 80). One proposed mechanism of reduced antibiotic efficacy towards S. aureus in a C. albicans biofilm relative to a mono-microbial biofilm is through a physical barrier of the biofilm preventing appropriate diffusion of the antibiotics. To this end, Kong et al. demonstrated that β-glucans present in the ECM of a C. albicans biofilm coat the surface of S. aureus, preventing penetration of antibiotics (63). However, there may also be a role for chemical signaling in inducing a more antibiotic-tolerant phenotype in S. aureus within the polymicrobial biofilm matrix. Exposure of S. aureus to physiologic levels of farnesol, the C. albicans quorum-sensing molecule that regulates biofilm formation, increases S. aureus survival during vancomycin treatment. The decreased vancomycin efficacy may be mediated by a global S. aureus stress response following exposure of the bacteria to farnesol (73, 74).
There is an urgent need to develop additional therapeutics to better target microorganisms growing as biofilms. Reducing biofilm biomass, specifically C. albicans burdens, improves efficacy of antibiotic treatment (56, 79). Therefore, treatments that aim to reduce biofilm biomass may improve or synergize with additional antimicrobial treatments to improve the outcomes of polymicrobial biofilm infection. Recent work demonstrated that biosurfactants limit formation of staphylococcal and C. albicans biofilm growth on plastic and metal surfaces (81, 82). Additionally, extracellular DNA contributes to biofilm biomass, and DNase treatment of C. albicans biofilms reduces biomass (83, 84). The use of biosurfactants to prevent biofilm formation and DNase treatment to reduce biofilm biomass may improve antibiotic efficacy against C. albicans-S. aureus biofilms.
Sites of infections, particularly invasive infections, are often hypoxic. Hypoxia influences C. albicans and S. aureus metabolism and subsequent interactions with host cells. Both C. albicans and S. aureus biofilm growth provide a localized hypoxic environment within the deepest sections of the biofilm (85, 86). Under hypoxic conditions, C. albicans induces transcription of genes for iron metabolism, ergosterol and heme biosynthesis, fatty acid metabolism, and cell wall biosynthesis, while reducing transcription of genes involved in mitochondrial respiration and the tricarboxylic acid cycle (87). In addition to regulating C. albicans morphogenesis, Efg1p is also a key regulator of biofilm-specific gene expression under hypoxic conditions (88). During invasive C. albicans infection, neutrophils are rapidly recruited to the site of infection and contribute to the hypoxic environment (89). In response to hypoxia, C. albicans masks β-glucan on its cell surface via a mechanism that requires C. albicans reactive oxygen species signaling within mitochondria and the cAMP-PKA pathway (90). Increased lactate produced by the recruited neutrophils under hypoxic conditions may also contribute to masking of β-glucan under hypoxic conditions, which leads to enhanced C. albicans survival following interactions with neutrophils (89). Hypoxia also impairs neutrophil responses towards S. aureus, contributes to S. aureus abscess formation, and enhances localized tissue destruction (91, 92). S. aureus toxin production is also increased under hypoxic conditions (93). Therefore, it is possible that the hypoxic microenvironment created within a polymicrobial biofilm with C. albicans could enhance S. aureus toxin production relative to S. aureus grown as a mono-culture biofilm (Figure 2D). Determining how C. albicans-S. aureus biofilm formation alters the distribution or availability of oxygen relative to mono-microbial biofilms will be important to define the consequences of hypoxia on microbial metabolism during polymicrobial biofilm growth.
Although typically less severe than the infections caused by S. aureus, Staphylococcus epidermidis can also cause recalcitrant biofilm-related infections and can grow as a polymicrobial biofilm with C. albicans (23, 94). Early studies determined that C. albicans and S. epidermidis have greater biomass in the polymicrobial biofilm relative to mono-microbial biofilm growth on a catheter disc model (94). Fluconazole is less effective against C. albicans and vancomycin is less effective against some strains of S. epidermidis when both organisms are grown in a polymicrobial biofilm (94). S. epidermidis added to pre-formed C. albicans biofilms adhere to C. albicans, and single cell force spectroscopy was used to demonstrate that the strong attachment is primarily mediated by C. albicans hyphal proteins Als1 and Als3, as well as O-linked mannans on the C. albicans cell surface (95, 96). Surgical implantation of a S. epidermidis-C. albicans infected catheter in mice resulted in greater S. epidermidis growth on the catheter and enhanced dissemination, highlighting a role for C. albicans in enhancement of staphylococcal virulence in vivo following polymicrobial biofilm growth (83). Microarray analysis revealed 223 differentially expressed S. epidermidis genes following biofilm growth with C. albicans relative to growth in the absence of C. albicans (83). C. albicans polymicrobial biofilm growth increases expression of genes encoding the global transcriptional regulator SarA and nucleic acid metabolism pathways, while reducing genes involved in carbohydrate and amino acid metabolism pathways (83). Pammi et al. demonstrated a role for S. epidermidis extracellular DNA in contributing to the greater biofilm growth and biomass of a S. epidermidis-C. albicans polymicrobial biofilm (83). Taken together, S. epidermidis, like S. aureus, has enhanced biofilm growth when co-cultured with C. albicans. Comparing the metabolic changes that are species-dependent may reveal both shared and unique mechanisms of staphylococcal interactions with fungi during polymicrobial biofilm growth.
Host immune responses during acute C. albicans-S. aureus co-infections typically contribute to disease severity rather than promote resolution of infection. While increased virulence during C. albicans-S. aureus intraperitoneal co-infection is mediated in part by alterations to microbial physiology, host immune responses also contribute to the lethal synergism of co-infection. Intraperitoneal co-infection of C. albicans and S. aureus induces increased levels of pro-inflammatory cytokines IL-6 and G-CSF and chemokines CXCL-1, CCL2, and CCL3 as well as greater neutrophil influx to the peritoneal cavity compared to infection with either organism alone (25). The robust inflammatory response during intraperitoneal C. albicans-S. aureus infection is driven in part through host inflammatory mediator PGE2 (25). Treating mice with a NSAID, which inhibits PGE2 production, reduces neutrophil recruitment and improved survival of mice (25). The contribution of C. albicans-derived PGE2 to lethality during intraperitoneal co-infection of mice is unknown. The addition of pro-inflammatory cytokines also enhances the growth of S. aureus in liquid culture (97, 98). Whether the enhanced pro-inflammatory cytokine production during C. albicans-S. aureus directly promotes greater S. aureus growth during co-infection in the peritoneum is also unknown. Oral co-infection of C. albicans and S. aureus results in greater dissemination of S. aureus to the kidneys relative to S. aureus inoculated alone by “hijacking” of macrophages (26, 99–101). It was initially proposed that S. aureus binds Als3p on C. albicans hyphae and is physically pushed into the subepithelial layer as C. albicans hyphae grow, facilitating spread beyond the initial infection site in a “hitchhiking” mechanism (99). However, it was later determined that C. albicans hyphae attract macrophages that fail to phagocytose the large hyphal filaments but instead phagocytose S. aureus bound to the hyphae in a “bait-and-switch” mechanism (26). The engulfed S. aureus evade macrophage-mediated killing, and instead the S. aureus-loaded macrophages traffic to the draining lymph node and facilitate bacterial spread to other organs, such as the kidneys (26). Supporting a role of macrophages in enhancing S. aureus dissemination during oral co-infection, treatment of mice orally co-infected with C. albicans and S. aureus with high levels of steroids reduces circulating immune cells and decreases the incidence of S. aureus dissemination beyond the oral mucosa to the kidneys (101). There may also be a secondary role for C. albicans-induced epithelial damage in mediating S. aureus dissemination during oral co-infection. Co-inoculating mice with S. aureus and C. albicans ece1Δ/Δ, which cannot produce the candidalysin toxin, results in smaller tongue lesions and reduced dissemination to the kidneys relative to co-infection with wild-type C. albicans (101). Infection of the oral mucosa with C. albicans and S. aureus highlights a role for macrophage phagocytosis in facilitating dissemination of S. aureus beyond the initial site of infection.
C. albicans and S. aureus secreted factors produced during co-infection also negatively impact host cells by direct toxicity. Epithelial cell lines exhibit greater cell death (as measured by lactate dehydrogenase release) following treatment with conditioned media from C. albicans-S. aureus biofilms compared to treatment with conditioned media from either organism grown as a mono-microbial biofilm (102) . S. aureus mono-culture biofilm growth produces higher levels of toxins leukocidin A/B and α-toxin compared to planktonic cell growth, which inhibits macrophage phagocytosis and induces macrophage cytotoxicity (103). Because C. albicans enhances S. aureus α-toxin secretion in planktonic co-culture, C. albicans may further enhance S. aureus toxin production in a polymicrobial biofilm (28). Chemical signals produced by C. albicans-S. aureus biofilms also target host cells (104). Farnesol, the C. albicans quorum sensing molecule that impacts both C. albicans and S. aureus biofilm growth, induces apoptosis of human squamous carcinoma cells (105). Additionally, even brief exposure of pre-osteoblastic cell lines to farnesol inhibited cell spreading, suggesting that chemical signaling from polymicrobial biofilms that form on orthopedic devices may influence bone cell physiology (106). Additional work to understand the full effects of polymicrobial biofilm growth on host cell responses during infection is needed. Because host responses can be influenced by the local environment of the infection site, it will be important to analyze host responses during C. albicans-S. aureus co-infection in multiple models.
There is significant overlap in immune responses to both C. albicans and S. aureus. For example, a Th1/Th17 response is required for clearance of both S. aureus and C. albicans in mice (107). STAT3 is activated downstream of Th17 cytokine signaling and is important for clearance of C. albicans and S. aureus. STAT3 deficiencies predispose individuals to both S. aureus and C. albicans skin and mucosal infections (108). IL-17, a Th17 signature cytokine, is important for host defense against both C. albicans and S. aureus skin and mucosal infections (109). IL-17 induces epithelial cells to produce antimicrobial peptides as well as CXC chemokines, which contribute to neutrophil recruitment (109). Pro-inflammatory cytokines, antimicrobial peptides, and neutrophils are important for S. aureus clearance during skin infection, while the epithelial cell-derived antimicrobial peptides are critical for protection against C. albicans mucosal infections (110–112). Additionally, a vaccine derived from the C. albicans Als3 protein not only provides protection against C. albicans infections but also S. aureus bacteremia and skin and soft tissue infection, and the protection against both organisms is mediated via STAT3 and Th17 signaling (113–115). The dual-species protection of the Als3p-derived vaccine may result from structural similarities between Als3p and S. aureus surface adhesins, such as collagen binding protein and clumping factor (116). Interestingly, Als3p is also the main factor to which S. aureus binds on C. albicans hyphal filaments, although this interaction was not shown to involve S. aureus collagen binding protein or clumping factor (59, 99).
In addition to providing cross-protection via adaptive immune responses, C. albicans also induces protection against secondary S. aureus infections through innate immune memory, often referred to as trained immunity (Figure 3). Trained immunity is the process by which an initial infection induces epigenetic changes in innate immune cells that alter their response to a secondary infection (117, 118). A key feature of trained immunity is that the immune cell returns to a baseline after the initial infection is cleared, but the epigenetic changes induced by the initial infection persist (119). During a secondary infection, the trained immune cells generate a robust immune response that contributes to clearance of the secondary infection (119). C. albicans infection and the fungal cell wall protein β-glucan are potent inducers of trained immunity. Non-lethal C. albicans injection in mice provides protection against a secondary lethal C. albicans challenge (27, 120). Protection occurs via C. albicans β-glucan signaling through Dectin-1 on monocytes, which is associated with changes in histone methylation, indicating an epigenetic mechanism (120). Powering the trained immunity effect induced by β-glucan is a shift in monocyte metabolism from oxidative phosphorylation to aerobic glycolysis (121). Additionally, training induced by C. albicans β-glucan not only provides protection against a secondary C. albicans infection, but also secondary challenge by other microorganisms, including S. aureus (27, 121, 122). These data indicate that host exposure to C. albicans influences subsequent S. aureus infection by providing protection via innate immune memory. However, the protection generated during primary infection may depend on the degree of immune stimulation by C. albicans. A high infectious dose of C. albicans induces a tolerant phenotype in monocytes in vitro, a state in which monocytes have a diminished response to secondary challenge (118, 123). The amount of β-glucan exposure during infection may also vary based on the infection site and local environment. Typically, β-glucan comprises the inner cell wall layer of C. albicans, and C. albicans can modulate levels of β-glucan display in the cell wall during infection in response to host cues such as pH or available lactate (67, 124–126). Therefore, C. albicans regulation of β-glucan exposure in vivo likely influences the training phenotype. Other signaling mechanisms stimulated during infection may also contribute to the generation of innate immune cell memory. Treatment of mice with monophosphoryl lipid A, a Toll-like receptor 4 agonist, induces protection against a secondary infection with either C. albicans or S. aureus (127, 128). Cell signaling pathways induced by multiple PAMPs during infection, particularly polymicrobial infections, may influence the development of innate immune memory.
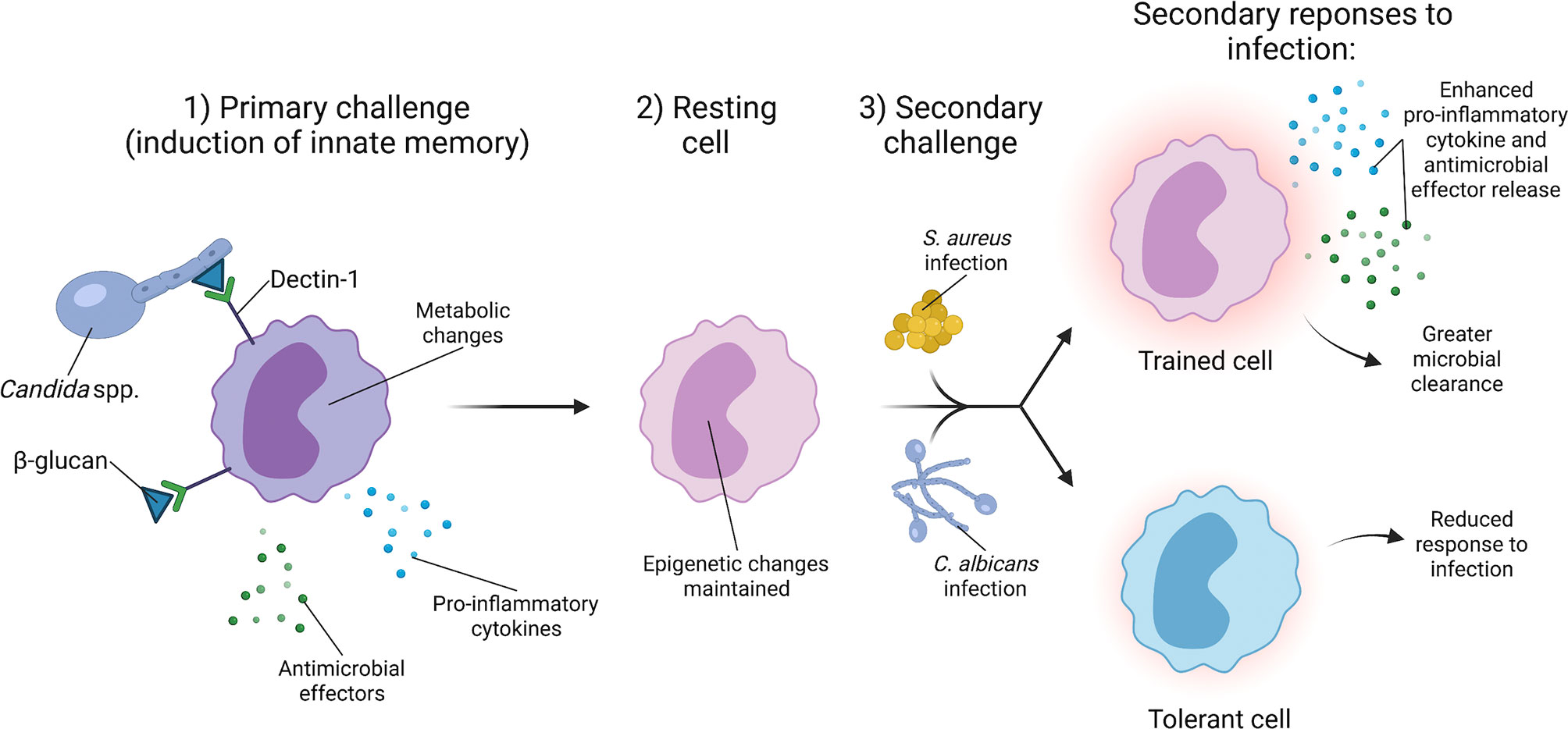
Figure 3 C. albicans induces innate immune training to provide broad protection against secondary infections. The generation of innate immune memory is dependent upon C. albicans cell wall component β-glucan signaling through Dectin-1 on monocytes. Dectin-1 signaling induces epigenetic changes and a metabolic shift from oxidative phosphorylation to aerobic glycolysis. Upon clearance of the primary C. albicans infection, monocytes enter a resting state, but retain the epigenetic changes from the primary challenge. Secondary challenge with a lethal infection of C. albicans or other organisms such as S. aureus causes the trained monocyte to produce a robust immune response that facilitates greater clearance of secondary S. aureus and C. albicans infections.
Innate immune training by Candida also influences the outcome of polymicrobial infection. Lilly et al. revealed that inoculation of mice intraperitoneally with other Candida species, such as C. dubliniensis, provides protection against a secondary co-infection challenge with a lethal dose of C. albicans and S. aureus (129). Mice that are rechallenged have greater neutrophil recruitment to the peritoneal cavity as well as enhanced C. albicans clearance, suggesting a role for neutrophils in providing protection against secondary infection (129). Inoculation of mice with C. dubliniensis provides protection against a lethal C. albicans-S. aureus infection for up to 60 days and was not dependent upon B and T cells (130). One hypothesis for C. dubliniensis-mediated immune protection is that following inoculation of certain Candida species, the bone marrow is transiently colonized, myelopoiesis is altered to skew cell development towards suppressor cells, and upon secondary challenge by a lethal polymicrobial infection, suppressive cells are activated that minimize the harmful pro-inflammatory response that occurs when naïve mice are co-infected (130, 131). Whether the protective effect conferred by primary C. dubliniensis inoculation is also mediated by β-glucan and Dectin-1 signaling is unknown.
While Candida and β-glucan induce a robust trained immunity response, S. aureus has a more limited ability to induce innate immune memory. Inoculating mice with S. aureus to induce a skin and soft tissue infection (SSTI) conferred protection against a secondary challenge with S. aureus after the initial infection cleared (132). Macrophages isolated from the S. aureus-infected mice were able to induce greater killing of S. aureus when infected ex vivo as compared to macrophages isolated from naïve mice (133). Enhanced macrophage-mediated killing is specific for S. aureus as the macrophages from S. aureus-infected animals did not enhance killing of S. epidermidis, Enterococcus faecalis, or E. coli (133). Additionally, protection against secondary SSTI infection is limited to the site of inoculation and is dependent upon the resident dermal macrophages present at the site of inoculation (134). Transfer of bone marrow cells from mice that received an initial S. aureus intradermal challenge failed to induce protection against secondary challenge. This differs from training with β-glucan, which induces changes in the hematopoietic progenitor cells of the bone marrow (134, 135). S. aureus may also negatively impact trained immunity during growth as small colony variants, or SCVs. SCVs can arise from mutations in metabolic genes and are associated with chronic S. aureus infections due to their long-term persistence within host cells and increased resistance to standard treatments (136). S. aureus ΔhemB is a SCV with altered metabolic activity that fails to induce protection against a secondary S. aureus challenge in a murine intradermal infection model (137). The absence of trained immunity was proposed to be the result of an enhanced use of fumarate by S. aureus ΔhemB, which depletes this metabolite from the local environment and prevents macrophages from using fumarate to drive the epigenetic changes required for training (137). Taken together, these studies demonstrate that S. aureus can produce a localized trained immunity that protects against secondary S. aureus challenge in an SSTI murine model of infection, and the protection is in part due to host metabolism as well as the activity of resident dermal macrophages.
C. albicans and S. aureus are formidable opportunistic pathogens, and their interactions during acute polymicrobial infection further worsen disease. Physical interactions mediate tight association of S. aureus with C. albicans hyphal filaments, facilitating enhanced chemical signaling and metabolite exchange between the two organisms. Adherence of S. aureus to C. albicans is particularly important for polymicrobial biofilm growth, which reduces antibiotic efficacy due to physical shielding of S. aureus from antibiotics as well as influencing S. aureus stress responses. C. albicans-S. aureus co-infection induces a severe pro-inflammatory response characterized by neutrophil influx, greater microbial burdens, and cytokine responses. Paradoxically, separating the timing of C. albicans and S. aureus inoculation induces a protective effect due to the induction of trained immunity by C. albicans that provides protection against a secondary S. aureus infection. Metabolic changes during co-infection underpin a variety of outcomes of C. albicans-S. aureus interactions, particularly during biofilm growth and when interacting with host cells. It is important to establish clinically relevant models of polymicrobial infection, particularly chronic infection models, to better characterize metabolic changes during C. albicans-S. aureus co-infection. Future studies that examine molecular interactions among C. albicans, S. aureus, and host immune cells during polymicrobial infection are needed to identify therapies that may target these challenging infections. Determining how C. albicans and S. aureus physiology changes during various co-infection models may reveal additional mechanisms of interaction that can be targeted to reduce mortality and enhance antimicrobial efficacy.
KE drafted the manuscript, which was edited by KE and JC. All authors contributed to the article and approved the submitted version.
JC was supported by R01AI132560 (NIAID), R01AI145992 (NIAID), R01AI161022 (NIAID), a Senior Research Award from the Crohn’s and Colitis Foundation, and a Career Award for Medical Scientists from the Burroughs Wellcome Fund. KE was supported by 2T32AI095202-12.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank the members of the Cassat lab for thoroughly reviewing the manuscript. Figures were created using BioRender.com.
1. Brogden KA, Guthmiller JM, Taylor CE. Human Polymicrobial Infections. Lancet (2005) 365(9455):253–5. doi: 10.1016/S0140-6736(05)70155-0
2. Dyess DL, Garrison RN, Fry DE. Candida Sepsis: Implications of Polymicrobial Blood-Borne Infection. Arch Surg-chicago (1985) 120(3):345–8. doi: 10.1001/archsurg.1985.01390270083014
3. Rolston KVI, Bodey GP, Safdar A. Polymicrobial Infection in Patients With Cancer: An Underappreciated and Underreported Entity. Clin Infect Dis (2007) 45(2):228–33. doi: 10.1086/518873
4. Carolus H, Dyck KV, Dijck PV. Candida albicans and Staphylococcus Species: A Threatening Twosome. Front Microbiol (2019) 10:2162. doi: 10.3389/fmicb.2019.02162
5. Peters BM, Jabra-Rizk MA, O’May GA, Costerton JW, Shirtliff ME. Polymicrobial Interactions: Impact on Pathogenesis and Human Disease. Clin Microbiol Rev (2012) 25(1):193–213. doi: 10.1128/CMR.00013-11
6. Short FL, Murdoch SL, Ryan RP. Polybacterial Human Disease: The Ills of Social Networking. Trends Microbiol (2014) 22(9):508–16. doi: 10.1016/j.tim.2014.05.007
7. Tay WH, Chong KKL, Kline KA. Polymicrobial–Host Interactions During Infection. J Mol Biol (2016) 428(17):3355–71. doi: 10.1016/j.jmb.2016.05.006
8. Peleg AY, Hogan DA, Mylonakis E. Medically Important Bacterial–Fungal Interactions. Nat Rev Microbiol (2010) 8(5):340–9. doi: 10.1038/nrmicro2313
9. Klotz SA, Chasin BS, Powell B, Gaur NK, Lipke PN. Polymicrobial Bloodstream Infections Involving Candida Species: Analysis of Patients and Review of the Literature. Diagn Micr Infect Dis (2007) 59(4):401–6. doi: 10.1016/j.diagmicrobio.2007.07.001
10. Shirtliff ME, Peters BM, Jabra-Rizk MA. Cross-Kingdom Interactions: Candida albicans and Bacteria. FEMS Microbiol Lett (2009) 299(1):1–8. doi: 10.1111/j.1574-6968.2009.01668.x
11. Douglas LJ. Candida Biofilms and Their Role in Infection. Trends Microbiol (2003) 11(1):30–6. doi: 10.1016/S0966-842X(02)00002-1
12. Gow NAR, van de Veerdonk FL, Brown AJP, Netea MG. Candida albicans Morphogenesis and Host Defence: Discriminating Invasion From Colonization. Nat Rev Microbiol (2012) 10(2):112–22. doi: 10.1038/nrmicro2711
13. Krismer B, Weidenmaier C, Zipperer A, Peschel A. The Commensal Lifestyle of Staphylococcus aureus and Its Interactions With the Nasal Microbiota. Nat Rev Microbiol (2017) 15(11):675–87. doi: 10.1038/nrmicro.2017.104
14. Miramón P, Lorenz MC. A Feast for Candida: Metabolic Plasticity Confers an Edge for Virulence. PLoS Pathog (2017) 13(2):e1006144. doi: 10.1371/journal.ppat.1006144
15. Horn CM, Kielian T. Crosstalk Between Staphylococcus aureus and Innate Immunity: Focus on Immunometabolism. Front Immunol (2021) 11:621750. doi: 10.3389/fimmu.2020.621750
16. Peters BM, Palmer GE, Nash AK, Lilly EA, Fidel PL, Noverr MC, et al. Fungal Morphogenetic Pathways Are Required for the Hallmark Inflammatory Response During Candida albicans Vaginitis. Infect Immun (2014) 82(2):532–43. doi: 10.1128/IAI.01417-13
17. Dunker C, Polke M, Schulze-Richter B, Schubert K, Rudolphi S, Gressler AE, et al. Rapid Proliferation Due to Better Metabolic Adaptation Results in Full Virulence of a Filament-Deficient Candida albicans Strain. Nat Commun (2021) 12(1):3899. doi: 10.1038/s41467-021-24095-8
18. Balasubramanian D, Harper L, Shopsin B, Torres VJ. Staphylococcus aureus Pathogenesis in Diverse Host Environments. Pathog Dis (2017) 75:ftx005. doi: 10.1093/femspd/ftx005
19. de Jong NWM, van Kessel KPM, van Strijp JAG. Immune Evasion by Staphylococcus aureus. Microbiol Spectr (2019) 7(2):1–27. doi: 10.1128/microbiolspec.GPP3-0061-2019
20. Tam K, Torres VJ. Staphylococcus aureus Secreted Toxins and Extracellular Enzymes. Microbiol Spectr (2019) 7(2):1–34. doi: 10.1128/microbiolspec.GPP3-0039-2018
21. Butrico CE, Cassat JE. Quorum Sensing and Toxin Production in Staphylococcus aureus Osteomyelitis: Pathogenesis and Paradox. Toxins (2020) 12(8):516. doi: 10.3390/toxins12080516
22. Carlson E. Synergistic Effect of Candida albicans and Staphylococcus aureus on Mouse Mortality. Infect Immun (1982) 38(3):921–4. doi: 10.1128/iai.38.3.921-924.1982
23. Carlson E. Effect of Strain of Staphylococcus aureus on Synergism With Candida albicans Resulting in Mouse Mortality and Morbidity. Infect Immun (1983) 42(1):285–92. doi: 10.1128/iai.42.1.285-292.1983
24. Carlson E, Johnson G. Protection by Candida albicans of Staphylococcus aureus in the Establishment of Dual Infection in Mice. Infect Immun (1985) 50(3):655–9. doi: 10.1128/iai.50.3.655-659.1985
25. Peters BM, Noverr MC. Candida albicans-Staphylococcus aureus Polymicrobial Peritonitis Modulates Host Innate Immunity. Infect Immun (2013) 81(6):2178–89. doi: 10.1128/IAI.00265-13
26. Allison DL, Scheres N, Willems HME, Bode CS, Krom BP, Shirtliff ME. The Host Immune System Facilitates Disseminated Staphylococcus aureus Disease Due to Phagocytic Attraction to Candida albicans During Coinfection: A Case of Bait and Switch. Infect Immun (2019) 87(11):1–14. doi: 10.1128/IAI.00137-19
27. Bistoni F, Vecchiarelli A, Cenci E, Puccetti P, Marconi P, Cassone A. Evidence for Macrophage-Mediated Protection Against Lethal Candida albicans Infection. Infect Immun (1986) 51(2):668–74. doi: 10.1128/iai.51.2.668-674.1986
28. Todd OA, Fidel PL, Harro JM, Hilliard JJ, Tkaczyk C, Sellman BR, et al. Candida albicans Augments Staphylococcus aureus Virulence by Engaging the Staphylococcal Agr Quorum Sensing System. mBio (2019) 10(3):e00910–19. doi: 10.1128/mBio.00910-19
29. Wang B, Muir TW. Regulation of Virulence in Staphylococcus aureus: Molecular Mechanisms and Remaining Puzzles. Cell Chem Biol (2016) 23(2):214–24. doi: 10.1016/j.chembiol.2016.01.004
30. Vylkova S, Lorenz MC. Modulation of Phagosomal pH by Candida albicans Promotes Hyphal Morphogenesis and Requires Stp2p, a Regulator of Amino Acid Transport. PLoS Pathog (2014) 10(3):e1003995. doi: 10.1371/journal.ppat.1003995
31. Regassa LB, Betley MJ. Alkaline pH Decreases Expression of the Accessory Gene Regulator (Agr) in Staphylococcus aureus. J Bacteriol (1992) 174(15):5095–100. doi: 10.1128/jb.174.15.5095-5100.1992
32. Regassa LB, Novick RP, Betley MJ. Glucose and Nonmaintained pH Decrease Expression of the Accessory Gene Regulator (Agr) in Staphylococcus aureus. Infect Immun (1992) 60(8):3381–8. doi: 10.1128/iai.60.8.3381-3388.1992
33. Jenul C, Horswill AR. Regulation of Staphylococcus aureus Virulence. Microbiol Spectr (2019) 7(2):1–21. doi: 10.1128/microbiolspec.GPP3-0031-2018
34. Todd OA, Noverr MC, Peters BM. Candida albicans Impacts Staphylococcus aureus Alpha-Toxin Production via Extracellular Alkalinization. mSphere (2019) 4(6):e00780–19. doi: 10.1128/mSphere.00780-19
35. Nash EE, Peters BM, Fidel PL, Noverr MC. Morphology-Independent Virulence of Candida Species During Polymicrobial Intra-Abdominal Infections With Staphylococcus aureus. Infect Immun (2016) 84(1):90–8. doi: 10.1128/IAI.01059-15
36. Lo H-J, Köhler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, Fink GR. Nonfilamentous C. Albicans Mutants Are Avirulent. Cell (1997) 90(5):939–49. doi: 10.1016/s0092-8674(00)80358-x
37. Sudbery PE. Growth of Candida albicans Hyphae. Nat Rev Microbiol (2011) 9(10):737–48. doi: 10.1038/nrmicro2636
38. Nash EE, Peters BM, Palmer GE, Fidel PL, Noverr MC. Morphogenesis Is Not Required for Candida albicans-Staphylococcus aureus Intra-Abdominal Infection-Mediated Dissemination and Lethal Sepsis. Infect Immun (2014) 82(8):3426–35. doi: 10.1128/IAI.01746-14
39. Lassak T, Schneider E, Bussmann M, Kurtz D, Manak JR, Srikantha T, et al. Target Specificity of the Candida albicans Efg1 Regulator. Mol Microbiol (2011) 82(3):602–18. doi: 10.1111/j.1365-2958.2011.07837.x
40. Doedt T, Krishnamurthy S, Bockmühl DP, Tebarth B, Stempel C, Russell CL, et al. APSES Proteins Regulate Morphogenesis and Metabolism in Candida albicans. Mol Biol Cell (2004) 15(7):3167–80. doi: 10.1091/mbc.e03-11-0782
41. Xu X-L, Lee RTH, Fang H-M, Wang Y-M, Li R, Zou H, et al. Bacterial Peptidoglycan Triggers Candida albicans Hyphal Growth by Directly Activating the Adenylyl Cyclase Cyr1p. Cell Host Microbe (2008) 4(1):28–39. doi: 10.1016/j.chom.2008.05.014
42. Wang Y, Xu X-L. Bacterial Peptidoglycan-Derived Molecules Activate Candida albicans Hyphal Growth. Commun Integr Biol (2014) 1(2):137–9. doi: 10.4161/cib.1.2.6870
43. Tan CT, Xu X, Qiao Y, Wang Y. A Peptidoglycan Storm Caused by β-Lactam Antibiotic’s Action on Host Microbiota Drives Candida albicans Infection. Nat Commun (2021) 12(1):2560. doi: 10.1038/s41467-021-22845-2
44. Kong K, Schneper L, Mathee K. Beta-Lactam Antibiotics: From Antibiosis to Resistance and Bacteriology. Apmis (2010) 118(1):1–36. doi: 10.1111/j.1600-0463.2009.02563.x
45. Konopka JB. N-Acetylglucosamine Functions in Cell Signaling. Sci (2012) 2012:489208. doi: 10.6064/2012/489208
46. Park JT, Uehara T. How Bacteria Consume Their Own Exoskeletons (Turnover and Recycling of Cell Wall Peptidoglycan). Microbiol Mol Biol R (2008) 72(2):211–27. doi: 10.1128/MMBR.00027-07
47. Alvarez FJ, Konopka JB. Identification of an N-Acetylglucosamine Transporter That Mediates Hyphal Induction in Candida albicans. Mol Biol Cell (2007) 18(3):965–75. doi: 10.1091/mbc.e06-10-0931
48. Min K, Naseem S, Konopka JB. N-Acetylglucosamine Regulates Morphogenesis and Virulence Pathways in Fungi. J Fungi (2019) 6(1):8. doi: 10.3390/jof6010008
49. Naseem S, Gunasekera A, Araya E, Konopka JB. N-Acetylglucosamine (GlcNAc) Induction of Hyphal Morphogenesis and Transcriptional Responses in Candida albicans Are Not Dependent on Its Metabolism. J Biol Chem (2011) 286(33):28671–80. doi: 10.1074/jbc.M111.249854
50. Naseem S, Araya E, Konopka JB. Hyphal Growth in Candida albicans Does Not Require Induction of Hyphal-Specific Gene Expression. Mol Biol Cell (2015) 26(6):1174–87. doi: 10.1091/mbc.E14-08-1312
51. Vesely EM, Williams RB, Konopka JB, Lorenz MC. N-Acetylglucosamine Metabolism Promotes Survival of Candida albicans in the Phagosome. mSsphere (2017) 2(5):e00357–17. doi: 10.1128/mSphere.00357-17
52. Borisova M, Gaupp R, Duckworth A, Schneider A, Dalügge D, Mühleck M, et al. Peptidoglycan Recycling in Gram-Positive Bacteria Is Crucial for Survival in Stationary Phase. mBio (2016) 7(5):e00923–16. doi: 10.1128/mBio.00923-16
53. Finkel JS, Mitchell AP. Genetic Control of Candida albicans Biofilm Development. Nat Rev Microbiol (2011) 9(2):109–18. doi: 10.1038/nrmicro2475
54. Moormeier DE, Bayles KW. Staphylococcus aureus Biofilm: A Complex Developmental Organism. Mol Microbiol (2017) 104(3):365–76. doi: 10.1111/mmi.13634
55. Kojic EM, Darouiche RO. Candida Infections of Medical Devices. Clin Microbiol Rev (2004) 17(2):255–67. doi: 10.1128/CMR.17.2.255-267.2004
56. Harriott MM, Noverr MC. Candida albicans and Staphylococcus aureus Form Polymicrobial Biofilms: Effects on Antimicrobial Resistance. Antimicrob Agents Ch (2009) 53(9):3914–22. doi: 10.1128/AAC.00657-09
57. Peters BM, Jabra-Rizk MA, Scheper MA, Leid JG, Costerton JW, Shirtliff ME. Microbial Interactions and Differential Protein Expression in Staphylococcus aureus –Candida albicans Dual-Species Biofilms. FEMS Immunol Med Mic (2010) 59(3):493–503. doi: 10.1111/j.1574-695X.2010.00710.x
58. Zago CE, Silva S, Sanitá PV, Barbugli PA, Dias CMI, Lordello VB, et al. Dynamics of Biofilm Formation and the Interaction Between Candida albicans and Methicillin-Susceptible (MSSA) and -Resistant Staphylococcus aureus (MRSA). PLoS One (2015) 10(4):e0123206. doi: 10.1371/journal.pone.0123206
59. Peters BM, Ovchinnikova ES, Krom BP, Schlecht LM, Zhou H, Hoyer LL, et al. Staphylococcus aureus Adherence to Candida albicans Hyphae Is Mediated by the Hyphal Adhesin Als3p. Microbiology (2012) 158(12):2975–86. doi: 10.1099/mic.0.062109-0
60. Harriott MM, Noverr MC. Ability of Candida albicans Mutants To Induce Staphylococcus aureus Vancomycin Resistance During Polymicrobial Biofilm Formation. Antimicrob Agents Ch (2010) 54(9):3746–55. doi: 10.1128/AAC.00573-10
61. Peters BM, Ward RM, Rane HS, Lee SA, Noverr MC. Efficacy of Ethanol Against Candida albicans and Staphylococcus aureus Polymicrobial Biofilms. Antimicrob Agents Ch (2013) 57(1):74–82. doi: 10.1128/AAC.01599-12
62. Krause J, Geginat G, Tammer I. Prostaglandin E2 From Candida albicans Stimulates the Growth of Staphylococcus aureus in Mixed Biofilms. PLoS One (2015) 10(8):e0135404. doi: 10.1371/journal.pone.0135404
63. Kong EF, Tsui C, Kucharíková S, Andes D, Dijck PV, Jabra-Rizk MA. Commensal Protection of Staphylococcus aureus Against Antimicrobials by Candida albicans Biofilm Matrix. mBio (2016) 7(5):e01365–16. doi: 10.1128/mBio.01365-16
64. Baillie GS, Douglas LJ. Matrix Polymers of Candida Biofilms and Their Possible Role in Biofilm Resistance to Antifungal Agents. J Antimicrob Chemoth (2000) 46(3):397–403. doi: 10.1093/jac/46.3.397
65. Boles BR, Horswill AR. Agr-Mediated Dispersal of Staphylococcus aureus Biofilms. PLoS Pathog (2008) 4(4):e1000052. doi: 10.1371/journal.ppat.1000052
66. Heim CE, Bosch ME, Yamada KJ, Aldrich AL, Chaudhari SS, Klinkebiel D, et al. Lactate Production by Staphylococcus aureus Biofilm Inhibits HDAC11 to Reprogramme the Host Immune Response During Persistent Infection. Nat Microbiol (2020) 5(10):1271–84. doi: 10.1038/s41564-020-0756-3
67. Ballou ER, Avelar GM, Childers DS, Mackie J, Bain JM, Wagener J, et al. Lactate Signalling Regulates Fungal β-Glucan Masking and Immune Evasion. Nat Microbiol (2016) 2(2):16238. doi: 10.1038/nmicrobiol.2016.238
68. Han T-L, Cannon RD, Villas-Bôas SG. The Metabolic Basis of Candida albicans Morphogenesis and Quorum Sensing. Fungal Genet Biol (2011) 48(8):747–63. doi: 10.1016/j.fgb.2011.04.002
69. Hornby JM, Jensen EC, Lisec AD, Tasto JJ, Jahnke B, Shoemaker R, et al. Quorum Sensing in the Dimorphic Fungus Candida albicans Is Mediated by Farnesol. Appl Environ Microb (2001) 67(7):2982–92. doi: 10.1128/AEM.67.7.2982-2992.2001
70. Ramage G, Saville SP, Wickes BL, López-Ribot JL. Inhibition of Candida albicans Biofilm Formation by Farnesol, a Quorum-Sensing Molecule. Appl Environ Microb (2002) 68(11):5459–63. doi: 10.1128/AEM.68.11.5459-5463.2002
71. Jabra-Rizk MA, Meiller TF, James CE, Shirtliff ME. Effect of Farnesol on Staphylococcus aureus Biofilm Formation and Antimicrobial Susceptibility. Antimicrob Agents Ch (2006) 50(4):1463–9. doi: 10.1128/AAC.50.4.1463-1469.2006
72. Kuroda M, Nagasaki S, Ito R, Ohta T. Sesquiterpene Farnesol as a Competitive Inhibitor of Lipase Activity of Staphylococcus aureus. FEMS Microbiol Lett (2007) 273(1):28–34. doi: 10.1111/j.1574-6968.2007.00772.x
73. Kong EF, Tsui C, Kucharíková S, Dijck PV, Jabra-Rizk MA. Modulation of Staphylococcus aureus Response to Antimicrobials by the Candida albicans Quorum Sensing Molecule Farnesol. Antimicrob Agents Ch (2017) 61(12):e01573–17. doi: 10.1128/AAC.01573-17
74. Vila T, Kong EF, Ibrahim A, Piepenbrink K, Shetty AC, McCracken C, et al. Candida albicans Quorum-Sensing Molecule Farnesol Modulates Staphyloxanthin Production and Activates the Thiol-Based Oxidative-Stress Response in Staphylococcus aureus. Virulence (2019) 10(1):625–42. doi: 10.1080/21505594.2019.1635418
75. Noverr MC, Phare SM, Toews GB, Coffey MJ, Huffnagle GB. Pathogenic Yeasts Cryptococcus neoformans and Candida albicans Produce Immunomodulatory Prostaglandins. Infect Immun (2001) 69(5):2957–63. doi: 10.1128/IAI.69.5.2957-2963.2001
76. Alem MAS, Douglas LJ. Effects of Aspirin and Other Nonsteroidal Anti-Inflammatory Drugs on Biofilms and Planktonic Cells of Candida albicans. Antimicrob Agents Ch (2004) 48(1):41–7. doi: 10.1128/AAC.48.1.41-47.2004
77. Lin YJ, Alsad L, Vogel F, Koppar S, Nevarez L, Auguste F, et al. Interactions Between Candida albicans and Staphylococcus aureus Within Mixed Species Biofilms. Bios (2013) 84(1):30–9. doi: 10.1893/0005-3155-84.1.30
78. Liu H, Chen H, Sun Y, Zhang X, Lu H, Li J, et al. Characterization of the Mechanism and Impact of Staphylokinase on the Formation of Candida albicans and Staphylococcus aureus Polymicrobial Biofilms. J Med Microbiol (2019) 68(3):355–67. doi: 10.1099/jmm.0.000914
79. Rogiers O, Holtappels M, Siala W, Lamkanfi M, Bambeke FV, Lagrou K, et al. Anidulafungin Increases the Antibacterial Activity of Tigecycline in Polymicrobial Candida albicans/Staphylococcus aureus Biofilms on Intraperitoneally Implanted Foreign Bodies. J Antimicrob Chemoth (2018) 73(10):2806–14. doi: 10.1093/jac/dky246
80. Luo Y, McAuley DF, Fulton CR, Pessoa JS, McMullan R, Lundy FT. Targeting Candida albicans in Dual-Species Biofilms With Antifungal Treatment Reduces Staphylococcus aureus and MRSA In Vitro. PLoS One (2021) 16(4):e0249547. doi: 10.1371/journal.pone.0249547
81. Ceresa C, Rinaldi M, Tessarolo F, Maniglio D, Fedeli E, Tambone E, et al. Inhibitory Effects of Lipopeptides and Glycolipids on C. Albicans–Staphylococcus Spp. Dual-Species Biofilms. Front Microbiol (2021) 11:545654. doi: 10.3389/fmicb.2020.545654
82. Tambone E, Marchetti A, Ceresa C, Piccoli F, Anesi A, Nollo G, et al. Counter-Acting Candida albicans-Staphylococcus aureus Mixed Biofilm on Titanium Implants Using Microbial Biosurfactants. Polymers-basel (2021) 13(15):2420. doi: 10.3390/polym13152420
83. Pammi M, Liang R, Hicks J, Mistretta T-A, Versalovic J. Biofilm Extracellular DNA Enhances Mixed Species Biofilms of Staphylococcus epidermidis and Candida albicans. BMC Microbiol (2013) 13(1):257–7. doi: 10.1186/1471-2180-13-257
84. Sapaar B, Nur A, Hirota K, Yumoto H, Murakami K, Amoh T, et al. Effects of Extracellular DNA From Candida albicans and Pneumonia-Related Pathogens on Candida Biofilm Formation and Hyphal Transformation. J Appl Microbiol (2014) 116(6):1531–42. doi: 10.1111/jam.12483
85. Rani SA, Pitts B, Beyenal H, Veluchamy RA, Lewandowski Z, Davison WM, et al. Spatial Patterns of DNA Replication, Protein Synthesis, and Oxygen Concentration Within Bacterial Biofilms Reveal Diverse Physiological States. J Bacteriol (2007) 189(11):4223–33. doi: 10.1128/JB.00107-07
86. Fox EP, Cowley ES, Nobile CJ, Hartooni N, Newman DK, Johnson AD. Anaerobic Bacteria Grow Within Candida albicans Biofilms and Induce Biofilm Formation in Suspension Cultures. Curr Biol (2014) 24(20):2411–6. doi: 10.1016/j.cub.2014.08.057
87. Chung H, Lee Y-H. Hypoxia: A Double-Edged Sword During Fungal Pathogenesis? Front Microbiol (2020) 11:1920. doi: 10.3389/fmicb.2020.01920
88. Stichternoth C, Ernst JF. Hypoxic Adaptation by Efg1 Regulates Biofilm Formation by Candida albicans. Appl Environ Microb (2009) 75(11):3663–72. doi: 10.1128/AEM.00098-09
89. Lopes JP, Stylianou M, Backman E, Holmberg S, Jass J, Claesson R, et al. Evasion of Immune Surveillance in Low Oxygen Environments Enhances Candida albicans Virulence. mBio (2018) 9(6):e02120–18. doi: 10.1128/mBio.02120-18
90. Pradhan A, Avelar GM, Bain JM, Childers DS, Larcombe DE, Netea MG, et al. Hypoxia Promotes Immune Evasion by Triggering β-Glucan Masking on the Candida albicans Cell Surface via Mitochondrial and cAMP-Protein Kinase A Signaling. mBio (2018) 9(6):e01318–18. doi: 10.1128/mBio.01318-18
91. McGovern NN, Cowburn AS, Porter L, Walmsley SR, Summers C, Thompson AAR, et al. Hypoxia Selectively Inhibits Respiratory Burst Activity and Killing of Staphylococcus aureus in Human Neutrophils. J Immunol (2011) 186(1):453–63. doi: 10.4049/jimmunol.1002213
92. Hajdamowicz NH, Hull RC, Foster SJ, Condliffe AM. The Impact of Hypoxia on the Host-Pathogen Interaction Between Neutrophils and Staphylococcus aureus. Int J Mol Sci (2019) 20(22):5561. doi: 10.3390/ijms20225561
93. Wilde AD, Snyder DJ, Putnam NE, Valentino MD, Hammer ND, Lonergan ZR, et al. Bacterial Hypoxic Responses Revealed as Critical Determinants of the Host-Pathogen Outcome by TnSeq Analysis of Staphylococcus aureus Invasive Infection. PLoS Pathog (2015) 11(12):e1005341. doi: 10.1371/journal.ppat.1005341
94. Adam B, Baillie GS, Douglas LJ. Mixed Species Biofilms of Candida albicans and Staphylococcus epidermidis. J Med Microbiol (2002) 51(4):344–9. doi: 10.1099/0022-1317-51-4-344
95. El-Azizi MA, Starks SE, Khardori N. Interactions of Candida albicans With Other Candida Spp. And Bacteria in the Biofilms. J Appl Microbiol (2004) 96(5):1067–73. doi: 10.1111/j.1365-2672.2004.02213.x
96. Beaussart A, Herman P, El-Kirat-Chatel S, Lipke PN, Kucharíková S, Dijck PV, et al. Single-Cell Force Spectroscopy of the Medically Important Staphylococcus epidermidis – Candida albicans Interaction. Nanoscale (2013) 5(22):10894–900. doi: 10.1039/c3nr03272h
97. Kanangat S, Bronze MS, Meduri GU, Postlethwaite A, Stentz F, Tolley E, et al. Enhanced Extracellular Growth of Staphylococcus aureus in the Presence of Selected Linear Peptide Fragments of Human Interleukin (IL)-1β and IL-1 Receptor Antagonist. J Infect Dis (2001) 183(1):65–9. doi: 10.1086/317645
98. Domenico EGD, Cavallo I, Bordignon V, Prignano G, Sperduti I, Gurtner A, et al. Inflammatory Cytokines and Biofilm Production Sustain Staphylococcus aureus Outgrowth and Persistence: A Pivotal Interplay in the Pathogenesis of Atopic Dermatitis. Sci Rep-uk (2018) 8(1):9573. doi: 10.1038/s41598-018-27421-1
99. Schlecht LM, Peters BM, Krom BP, Freiberg JA, Hänsch GM, Filler SG, et al. Systemic Staphylococcus aureus Infection Mediated by Candida albicans Hyphal Invasion of Mucosal Tissue. Microbiology+ (2015) 161(1):168–81. doi: 10.1099/mic.0.083485-0
100. Todd OA, Peters BM. Candida albicans and Staphylococcus aureus Pathogenicity and Polymicrobial Interactions: Lessons Beyond Koch’s Postulates. J Fungi (2019) 5(3):81. doi: 10.3390/jof5030081
101. Dyck KV, Viela F, Mathelié-Guinlet M, Demuyser L, Hauben E, Jabra-Rizk MA, et al. Adhesion of Staphylococcus aureus to Candida albicans During Co-Infection Promotes Bacterial Dissemination Through the Host Immune Response. Front Cell Infect Microbiol (2021) 10:624839. doi: 10.3389/fcimb.2020.624839
102. de Carvalho Dias K, Barbugli PA, de Patto F, Lordello VB, Penteado L de A, Medeiros AI, et al. Soluble Factors From Biofilm of Candida albicans and Staphylococcus aureus Promote Cell Death and Inflammatory Response. BMC Microbiol (2017) 17(1):146. doi: 10.1186/s12866-017-1031-5
103. Scherr TD, Hanke ML, Huang O, James DBA, Horswill AR, Bayles KW, et al. Staphylococcus aureus Biofilms Induce Macrophage Dysfunction Through Leukocidin AB and Alpha-Toxin. mBio (2015) 6(4):e01021–15. doi: 10.1128/mBio.01021-15
104. Bernard C, Girardot M, Imbert C. Candida albicans Interaction With Gram-Positive Bacteria Within Interkingdom Biofilms. J Mycol Médicale (2019) 30(1):100909. doi: 10.1016/j.mycmed.2019.100909
105. Scheper MA, Shirtliff ME, Meiller TF, Peters BM, Jabra-Rizk MA. Farnesol, a Fungal Quorum-Sensing Molecule Triggers Apoptosis in Human Oral Squamous Carcinoma Cells. Neoplasia (2008) 10(9):954–63. doi: 10.1593/neo.08444
106. Unnanuntana A, Bonsignore L, Shirtliff ME, Greenfield EM. The Effects of Farnesol on Staphylococcus aureus Biofilms and Osteoblasts. J Bone Jt Surg (2009) 91(11):2683–92. doi: 10.2106/JBJS.H.01699
107. Lin L, Ibrahim AS, Xu X, Farber JM, Avanesian V, Baquir B, et al. Th1-Th17 Cells Mediate Protective Adaptive Immunity Against Staphylococcus aureus and Candida albicans Infection in Mice. PLoS Pathog (2009) 5(12):e1000703. doi: 10.1371/journal.ppat.1000703
108. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-Negative Mutations in the DNA-Binding Domain of STAT3 Cause Hyper-IgE Syndrome. Nature (2007) 448(7157):1058–62. doi: 10.1038/nature06096
109. Li J, Casanova J-L, Puel A. Mucocutaneous IL-17 Immunity in Mice and Humans: Host Defense vs. Excessive Inflammation. Mucosal Immunol (2018) 11(3):581–9. doi: 10.1038/mi.2017.97
110. McLoughlin RM, Solinga RM, Rich J, Zaleski KJ, Cocchiaro JL, Risley A, et al. CD4+ T Cells and CXC Chemokines Modulate the Pathogenesis of Staphylococcus aureus Wound Infections. Proc Natl Acad Sci (2006) 103(27):10408–13. doi: 10.1073/pnas.0508961103
111. Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 Cells and IL-17 Receptor Signaling Are Essential for Mucosal Host Defense Against Oral Candidiasis. J Exp Med (2009) 206(2):299–311. doi: 10.1084/jem.20081463
112. Trautwein-Weidner K, Gladiator A, Nur S, Diethelm P, LeibundGut-Landmann S. IL-17-Mediated Antifungal Defense in the Oral Mucosa Is Independent of Neutrophils. Mucosal Immunol (2015) 8(2):221–31. doi: 10.1038/mi.2014.57
113. Spellberg BJ, Ibrahim AS, Avanesian V, Fu Y, Myers C, Phan QT, et al. Efficacy of the Anti-Candida Rals3p-N or Rals1p-N Vaccines Against Disseminated and Mucosal Candidiasis. J Infect Dis (2006) 194(2):256–60. doi: 10.1086/504691
114. Spellberg B, Ibrahim AS, Yeaman MR, Lin L, Fu Y, Avanesian V, et al. The Antifungal Vaccine Derived From the Recombinant N Terminus of Als3p Protects Mice Against the Bacterium Staphylococcus aureus. Infect Immun (2008) 76(10):4574–80. doi: 10.1128/IAI.00700-08
115. Yeaman MR, Filler SG, Chaili S, Barr K, Wang H, Kupferwasser D, et al. Mechanisms of NDV-3 Vaccine Efficacy in MRSA Skin Versus Invasive Infection. Proc Natl Acad Sci (2014) 111(51):E5555–63. doi: 10.1073/pnas.1415610111
116. Sheppard DC, Yeaman MR, Welch WH, Phan QT, Fu Y, Ibrahim AS, et al. Functional and Structural Diversity in the Als Protein Family of Candida albicans. J Biol Chem (2004) 279(29):30480–9. doi: 10.1074/jbc.M401929200
117. Netea MG, Quintin J, van der Meer JWM. Trained Immunity: A Memory for Innate Host Defense. Cell Host Microbe (2011) 9(5):355–61. doi: 10.1016/j.chom.2011.04.006
118. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining Trained Immunity and Its Role in Health and Disease. Nat Rev Immunol (2020) 20(6):375–88. doi: 10.1038/s41577-020-0285-6
119. Divangahi M, Aaby P, Khader SA, Barreiro LB, Bekkering S, Chavakis T, et al. Trained Immunity, Tolerance, Priming and Differentiation: Distinct Immunological Processes. Nat Immunol (2021) 22(1):2–6. doi: 10.1038/s41590-020-00845-6
120. Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, et al. Candida albicans Infection Affords Protection Against Reinfection via Functional Reprogramming of Monocytes. Cell Host Microbe (2012) 12(2):223–32. doi: 10.1016/j.chom.2012.06.006
121. Cheng S-C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α–Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science (2014) 345(6204):1250684–1250684. doi: 10.1126/science.1250684
122. Luzio NRD, Williams DL. Protective Effect of Glucan Against Systemic Staphylococcus aureus Septicemia in Normal and Leukemic Mice. Infect Immun (1978) 20(3):804–10. doi: 10.1128/iai.20.3.804-810.1978
123. Cheng S-C, Scicluna BP, Arts RJW, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad Defects in the Energy Metabolism of Leukocytes Underlie Immunoparalysis in Sepsis. Nat Immunol (2016) 17(4):406–13. doi: 10.1038/ni.3398
124. Wheeler RT, Kombe D, Agarwala SD, Fink GR. Dynamic, Morphotype-Specific Candida albicans β-Glucan Exposure During Infection and Drug Treatment. PLoS Pathog (2008) 4(12):e1000227. doi: 10.1371/journal.ppat.1000227
125. Cottier F, Sherrington S, Cockerill S, Toledo V del O, Kissane S, Tournu H, et al. Remasking of Candida albicans β-Glucan in Response to Environmental pH Is Regulated by Quorum Sensing. mBio (2019) 10(5):e02347–19. doi: 10.1128/mBio.02347-19
126. Garcia-Rubio R, de Oliveira HC, Rivera J, Trevijano-Contador N. The Fungal Cell Wall: Candida, Cryptococcus, and Aspergillus Species. Front Microbiol (2020) 10:2993. doi: 10.3389/fmicb.2019.02993
127. Romero CD, Varma TK, Hobbs JB, Reyes A, Driver B, Sherwood ER. The Toll-Like Receptor 4 Agonist Monophosphoryl Lipid A Augments Innate Host Resistance to Systemic Bacterial Infection. Infect Immun (2011) 79(9):3576–87. doi: 10.1128/IAI.00022-11
128. Fensterheim BA, Young JD, Luan L, Kleinbard RR, Stothers CL, Patil NK, et al. The TLR4 Agonist Monophosphoryl Lipid A Drives Broad Resistance to Infection via Dynamic Reprogramming of Macrophage Metabolism. J Immunol (2018) 200(11):ji1800085. doi: 10.4049/jimmunol.1800085
129. Lilly EA, Ikeh M, Nash EE, Fidel PL, Noverr MC. Immune Protection Against Lethal Fungal-Bacterial Intra-Abdominal Infections. mBio (2018) 9(1):e01472–17. doi: 10.1128/mBio.01472-17
130. Lilly EA, Yano J, Esher SK, Hardie E, Fidel PL, Noverr MC. Spectrum of Trained Innate Immunity Induced by Low-Virulence Candida Species Against Lethal Polymicrobial Intra-Abdominal Infection. Infect Immun (2019) 87(8):1–12. doi: 10.1128/IAI.00348-19
131. Esher SK, Fidel PL, Noverr MC. Candida/Staphylococcal Polymicrobial Intra-Abdominal Infection: Pathogenesis and Perspectives for a Novel Form of Trained Innate Immunity. J Fungi (2019) 5(2):37. doi: 10.3390/jof5020037
132. Chan LC, Chaili S, Filler SG, Miller LS, Solis NV, Wang H, et al. Innate Immune Memory Contributes to Host Defense Against Recurrent Skin and Skin Structure Infections Caused by Methicillin-Resistant Staphylococcus aureus. Infect Immun (2017) 85(2):e00876–16. doi: 10.1128/IAI.00876-16
133. Chan LC, Rossetti M, Miller LS, Filler SG, Johnson CW, Lee HK, et al. Protective Immunity in Recurrent Staphylococcus aureus Infection Reflects Localized Immune Signatures and Macrophage-Conferred Memory. Proc Natl Acad Sci (2018) 115(47):201808353. doi: 10.1073/pnas.1808353115
134. Feuerstein R, Forde AJ, Lohrmann F, Kolter J, Ramirez NJ, Zimmermann J, et al. Resident Macrophages Acquire Innate Immune Memory in Staphylococcal Skin Infection. Elife (2020) 9:e55602. doi: 10.7554/eLife.55602
135. Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, Grinenko T, et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell (2018) 172(1–2):147–61.e12. doi: 10.1016/j.cell.2017.11.034
136. Kahl BC, Becker K, Löffler B. Clinical Significance and Pathogenesis of Staphylococcal Small Colony Variants in Persistent Infections. Clin Microbiol Rev (2016) 29(2):401–27. doi: 10.1128/CMR.00069-15
Keywords: Candida albicans, Staphylococcus aureus, polymicrobial infection, polymicrobial biofilm, fungal-bacterial interactions
Citation: Eichelberger KR and Cassat JE (2021) Metabolic Adaptations During Staphylococcus aureus and Candida albicans Co-Infection. Front. Immunol. 12:797550. doi: 10.3389/fimmu.2021.797550
Received: 18 October 2021; Accepted: 19 November 2021;
Published: 08 December 2021.
Edited by:
Tammy Kielian, University of Nebraska Medical Center, United StatesReviewed by:
Brian M. Peters, University of Tennessee Health Science Center (UTHSC), United StatesCopyright © 2021 Eichelberger and Cassat. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kara R. Eichelberger, a2FyYS5laWNoZWxiZXJnZXJAdnVtYy5vcmc=; James E. Cassat, amltLmNhc3NhdEB2dW1jLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.