 Yan Zhou1†Yingyi Zhang2†Rui Zhao3†Zhounan Cheng1Minzhu Tang2Anni Qiu1Yang Dong1
Yan Zhou1†Yingyi Zhang2†Rui Zhao3†Zhounan Cheng1Minzhu Tang2Anni Qiu1Yang Dong1 Yihua Lu1Yulong Lian1Xun Zhuang1
Yihua Lu1Yulong Lian1Xun Zhuang1 Tian Tian1Wei Wang4*
Tian Tian1Wei Wang4* Minjie Chu1*
Minjie Chu1*- 1Department of Epidemiology, School of Public Health, Nantong University, Nantong, China
- 2Department of Occupational Disease, The Eighth People’s Hospital of Wuxi, Wuxi, China
- 3Department of Respiratory, The Eighth People’s Hospital of Wuxi, Wuxi, China
- 4Department of Occupational Health, Center for Disease Control and Prevention of Wuxi, Wuxi, China
Objective: To evaluate the association between single-nucleotide polymorphisms (SNPs) in RNA-seq identified mRNAs and silicosis susceptibility.
Methods: A comprehensive RNA-seq was performed to screen for differently expressed mRNAs in the peripheral blood lymphocytes of eight subjects exposed to silica dust (four silicosis cases and four healthy controls). Following this, the SNPs located on the shortlisted mRNAs, which may affect silicosis susceptibility, were screened through silicosis-related genome-wide association studies (GWAS) (155 silicosis cases and 141 healthy controls), whereas functional expression quantitative trait locus (eQTL)-SNPs were identified using the GTEx database. Finally, the association between functional eQTL-SNPs and silicosis susceptibility (194 silicosis cases and 235 healthy controls) was validated.
Results: A total of 70 differentially expressed mRNAs (fold change > 2 or fold change < 0.5, P < 0.05) was obtained using RNA-seq. Furthermore, 476 SNPs located on the shortlisted mRNAs, which may affect silicosis susceptibility (P < 0.05) were obtained using GWAS, whereas subsequent six functional eQTL-SNPs were identified. The mutant A allele of rs9273410 in HLA-DQB1 indicated a potential increase in silicosis susceptibility in the validation stage (additive model: odds ratio (OR)= 1.31, 95% confidence interval (CI) = 0.99–1.74, P = 0.061), whereas the combination of GWAS and the validation results indicated that the mutant A allele of rs9273410 was associated with increased silicosis susceptibility (additive model: OR = 1.35, 95% CI =1.09–1.68, P = 0.006).
Conclusion: The mutant A allele of rs9273410 was associated with increased silicosis susceptibility by modulating the expression of HLA-DQB1.
Introduction
Silicosis is a chronic progressive fibrotic lung disease, which may lead to respiratory failure or death (1, 2). The Global Silicosis Elimination Plan has established the goal of reducing the impact of silicosis by 2030; however, silicosis remains a vital issue threatening the health of individuals in various occupations in both developing and developed countries. For instance, China records the largest incidence of silicosis in the world, with more than 873,000 silicosis cases being recorded by the end of 2018 (3). According to the systematic analysis of the Global Burden of Disease Study in 2016, East Asia ranks first in the world for the highest silicosis fatality rate followed by Western Europe (4). In addition, some Australian coal mining communities are facing a severe epidemic of accelerated silicosis due to artificial stone exposure (5). Therefore, silicosis remains a major global health concern (6).
Occupational exposure to siliceous dust is widely known to be the main cause of silicosis (7). Currently, no specific therapy exists for silicosis, apart from avoiding contact with environmental silica (8). Despite the escape from the harmful workplace, persistent lung function damage and disease progression are unavoidable owing to the difficulty in removing siliceous particles from the body (9). Another cause for concern is that individuals exposed to the same working environment might present different health conditions: some people suffer from silicosis after working in an exposed environment for several years, whereas others remain healthy (10). This indicates the possibility of potentially non-modifiable risk factors related to silicosis, such as genetic variants (mainly single-nucleotide polymorphisms, SNPs) (11).
Previous genome wide association studies (GWASs) have identified numerous pulmonary fibrosis-related SNPs, which were mainly idiopathic pulmonary fibrosis (IPF)-susceptible SNP-harbouring genes such as MUC5B, AKAP13, DSP, FAM13A, DEPTOR, KIF15, and MAD1L1 (12–15). In addition, a three-stage GWAS had identified and replicated a genome-wide significant (P < 5.0 × 10-8) signal (named as variant rs73329476) that is associated with coal workers’ pneumoconiosis susceptibility and altered CPM expression (10). GWASs have proven to be successful in the discovery of SNPs associated with complex diseases; however, most SNPs identified using GWAS are in the gene desert regions, and their biological functions remain unclear. Moreover, developing post-GWAS methods for characterising the function of complex disease-associated SNPs is required. A recent report proposed that the integrative analysis of RNA sequencing (RNA-seq) data and GWAS mapping may be promising for illustrating the functional genetic architecture of underlying complex diseases (16), and the evidence that differentially expressed genes identified using RNA-seq were more likely to overlap with GWAS obtained loci have highlighted this strategy (17).
Recently, RNA-seq has become an indispensable and a powerful tool for transcriptome-wide analysis of differential gene expression, which quickly and comprehensively acquires approximately all transcriptional sequence information of a species at a certain stage (18). Studies suggest that RNA-seq can be employed to analyse the synergistic or antagonistic effects of various mRNAs associated with fibrotic lung diseases (19). A previous study identified 873 differentially expressed genes using RNA-seq from eight IPF lung samples and seven healthy controls (20). Another study using 3’ messenger RNA sequencing (mRNA-seq) and pathway enrichment analysis revealed the upregulation of pathways related to immune response and inflammatory signaling in patients with IPF compared with controls (19). A recent study on silicosis identified 600 upregulated genes and 537 downregulated genes in the silica inhalation-induced silicosis rat model group using RNA-seq and bioinformatic analyses (21). However, in silicosis human samples, RNA-seq was rarely used for the determination of specific differentially expressed genes.
In this study, we addressed the integration of RNA-seq data and GWAS mapping to effectively identify the potentially functional SNPs associated with the susceptibility of silicosis. First, RNA-Seq technology was used to identify mRNAs with distinct expression patterns among four pairs of silicosis cases and silica-exposed healthy controls. Second, candidate SNPs located on the RNA-seq-identified genes were selected, and the association between those SNPs and silicosis risk was evaluated using GWAS (155 silicosis cases and 141 healthy controls), while the potential functional SNPs with the expression quantitative trait locus (eQTL) regulation function were selected. Third, the association between the selected eQTL-SNPs and silicosis risk was validated using an additional case–control study (194 silicosis cases and 235 healthy controls).
Methods and Materials
Study Design
In the initial RNA-seq stage, differentially expressed mRNAs in the peripheral blood lymphocytes (PBLs) from four silicosis cases and four healthy controls with similar years of silica dust exposure and age were selected. All SNPs located on RNA-seq identified differentially expressed mRNAs with minor allele frequency (MAF) > 0.05 in the Han Chinese population were obtained using the 1000 Genome Project database. The silicosis-related GWAS (155 silicosis cases and 141 healthy controls with similar years of silica dust exposure) was used to screen for candidate positive SNPs. eQTL analysis in both the lung and whole blood was used to further screen for candidate functional eQTL-SNPs using the GTEx database. A linkage disequilibrium (LD) analysis (r2 < 0.8) was performed among these SNPs using the Ensemble database. A validation study was designed (194 silicosis cases and 235 healthy controls with similar years of silica dust exposure) to validate the association between functional eQTL-SNPs and silicosis susceptibility (Figure 1).

Figure 1 Schematic representation of the study design. RNA-seq, RNA sequencing; FC, fold change; MAF, minor allele frequency; eQTL, expression quantitative trait locus.
Study Population
To obtain the differently expressed silicosis-related mRNAs, a 1:1 individual matched case–control study was performed based on years of silica dust exposure and age. In principle, a case was generally matched to an exposed control with the most similar years of silica dust exposure (± 5) and age (± 5). Four patients with silicosis as the case group and four matched healthy individuals as the control group was selected in September 2017 from the Wuxi Institute of Occupational Diseases in Jiangsu Province.
The cases were diagnosed as stage I, stage II, or stage III silicosis according to the size, profusion, and distribution range of opacities on chest X-ray by three national certified occupational physicians based on the China National Diagnostic Criteria for Pneumoconiosis (GBZ 70-2015). The years of silica dust exposure for each individual were recorded in historical occupational health records.
The case group included in the GWAS screening stage was recruited in 2012–2016 from the Occupational Disease Institute of Wuxi. The control group of 141 healthy individuals was randomly selected from a pool of >2000 occupational dust exposed individuals who participated in the routine health surveillance of 2017 at Wuxi and were matched to the case group on the years of silica dust exposure.
In the improved Multiligase Detection Reaction (iMLDR) validation stage, 194 silicosis cases were recruited in 2017 from the Datun Mining Business Group Co. Ltd. of Xuzhou; 235 healthy controls were randomly selected from a pool of >1000 individuals with occupational silica dust exposure who participated in the routine health surveillance of 2017 in Xuzhou. These controls were matched to the cases based on the years of silica dust exposure.
Each participant filled out a structured questionnaire that included all demographic characteristics required for the study, with the help of administrative staff who are well-trained in conducting face-to-face interviews. All participants provided written informed consent, and the study protocol was approved by the Ethics Committee of Nantong University (Approval No. 2020-002).
RNA-Seq Screening
From the PBLs of four patients with silicosis and four healthy controls, the total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA) and a mRNeasy mini kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. The RNA samples were outsourced to Gminix Biotechnology Co., Ltd. (Shanghai, China) for RNA-seq using the Illumina HiSeq 2500 sequencing platform that has an average of 15 G reads. Among the identified mRNAs, the differently expressed mRNAs between the four patients with silicosis and four healthy controls were selected with P < 0.05 and fold change (FC) > 2 (cases/controls: >2-fold up-regulated or <0.5-fold down-regulated).
Genotyping Platform of the Two-Stage Case-Control Study
Genomic DNA was extracted from peripheral blood samples using a DNA extraction kit (Qiagen, Valencia, CA). A high throughput genotyping chip designed for the Asian population (Illumina Asian screening array chip) comprising 746,113 SNPs was used for genome-wide genotyping. After quality control, 155 cases and 141 controls with 544,414 autosomal SNPs were retained for subsequent analyses. And P-values from GWAS are described in Manhattan and Quantile-quantile plots (Figures 2A, B). Genotyping of the validation stage was performed using the Genesky proprietary iMLDR multiplex SNP genotyping system, which employs a multiplex PCR-ligase detection reaction method. For each SNP, the alleles were distinguished by different fluorescent labels of allele-specific oligonucleotide probe pairs. And then, different SNPs were further distinguished according to different extended lengths at the 3’end.
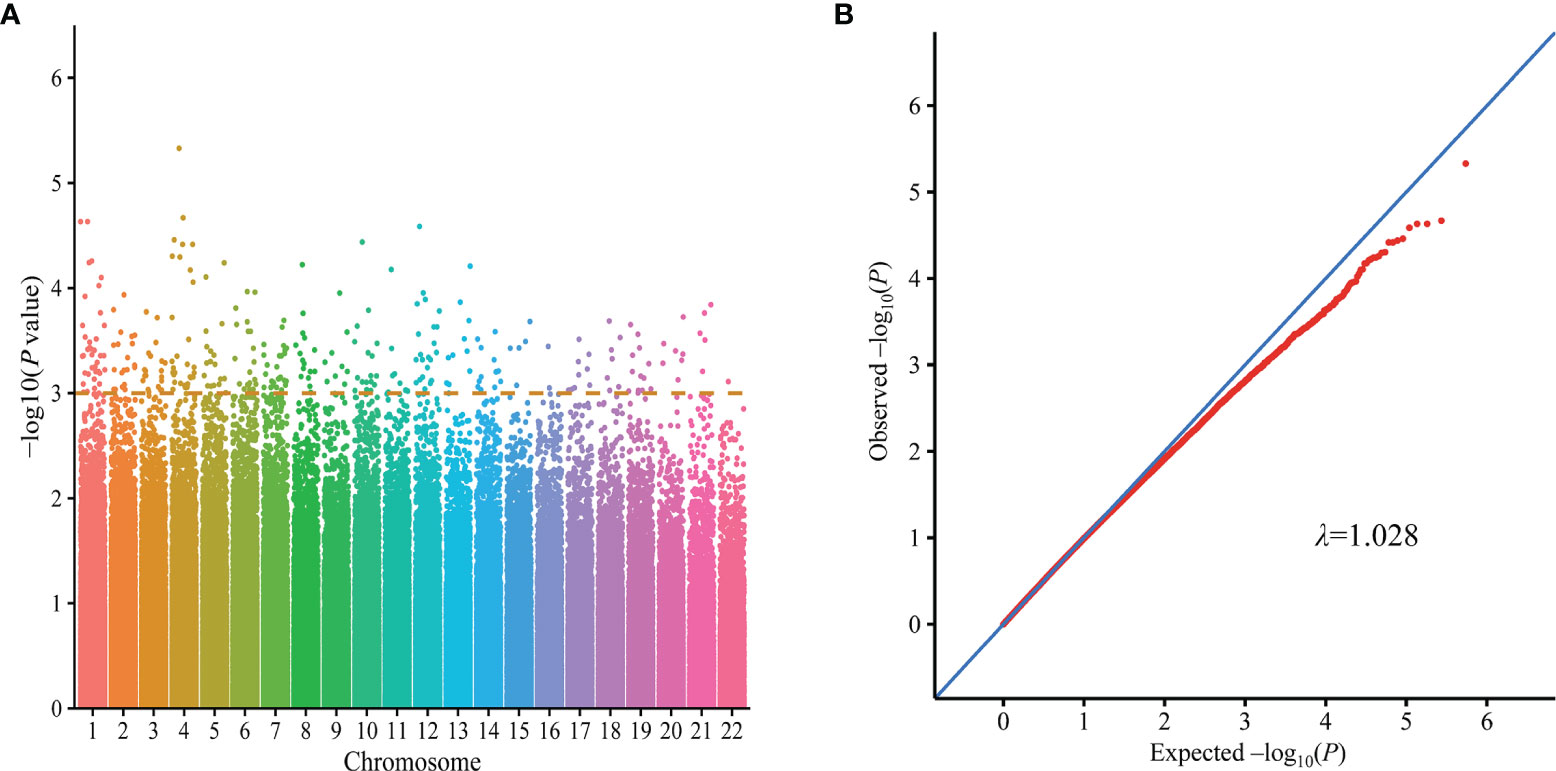
Figure 2 GWAS results on silicosis in Han Chinese population (155 cases and 141 controls). Manhattan plots represent -log10 (p-value) for SNPs distributed across all chromosomes (A), and quantile-quantile (Q-Q) plot described the distribution of the observed (y-axis) and expected (x-axis) p-values of each SNPs (B). Q-Q, quantile-quantile; GWAS, Genome-wide association study.
eQTL Analysis
Using the GTEx database (http://www.gtexportal.org/home/), eQTL analysis was performed for the lung and whole blood tissues separately. Based on the overlapping SNPs between the two tissues, candidate SNPs with eQTL functions in both the lung and whole blood tissues were obtained.
Statistical Analyses
Differences in the distribution of demographic characteristics and selected variables between the case and control groups were calculated using the two-sided chi-square tests and Student’s t-tests. Logistic regression analysis was performed to determine the association between candidate SNPs and silicosis risk based on the adjusted odds ratios (ORs) and 95% confidence intervals (CIs), adjusting for sex, age, smoking status, and silica dust exposure years. Generally, for each SNP, we assigned those carrying no minor allele to the wild-type homozygote (code 0), carrying one minor allele to the heterozygote (code 1), and carrying two minor alleles to the variant homozygote (code 2), and then we assessed the association in different genetic models: heterozygote (co-dominant) model (1 versus 0), homozygote (co-dominant) model (2 versus 0), dominant model (1 + 2 versus 0), recessive model (2 versus 1 + 0). For additive model, testing is designed specifically to reveal associations that depend additively on the minor allele. That is, subjects carrying two minor alleles (as compared with those carrying no minor allele) are twice as likely to affect the outcome in a certain direction as subjects carrying one minor allele (as compared with those carrying no minor allele). All analyses were conducted using the following software: SPSS version 20.0, STATA version 12.0 and R version 3.6.2.
Results
Characteristics of the Study Subjects
The characteristics of the subjects in the control and case groups are shown in Table 1. Generally, the sex ratios and silica dust exposure years between the case and control groups were comparable (P > 0.05). The mean age of the case group was higher than that of the control group, and the case group had higher smoking rates than those of the control group (P < 0.05). In addition, smoking was more severe in the case group than in the control group (P < 0.05).
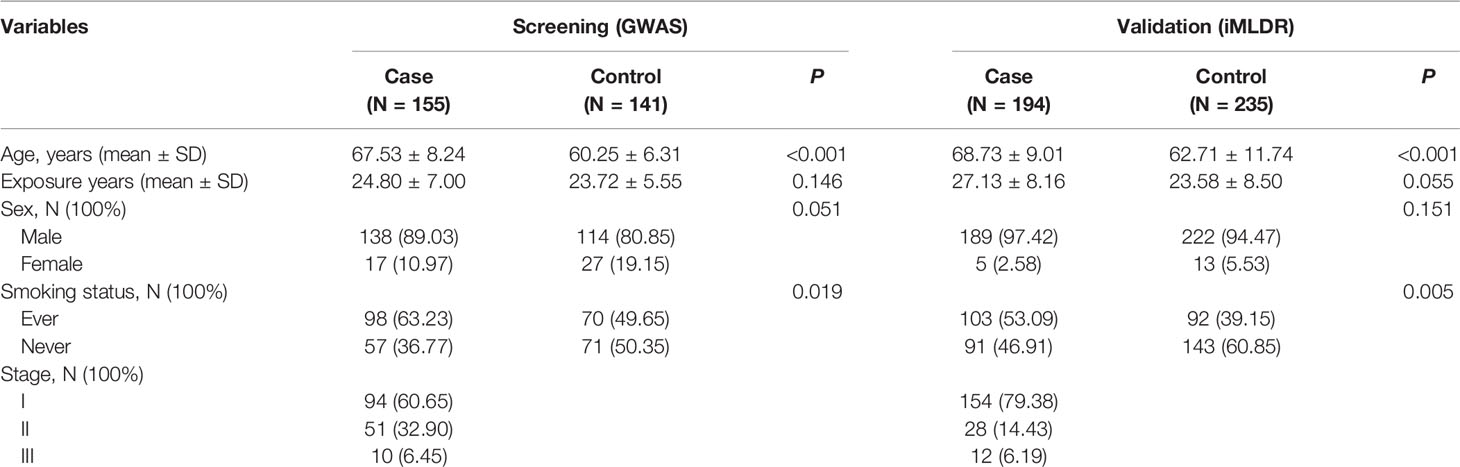
Table 1 Characteristics of the subjects enrolled in this study.
RNA-Seq Screening
RNA-seq detected 760 differently expressed mRNAs (P < 0.05) in the PBLs of four patients with silicosis and four healthy controls. A total of 70 differently expressed mRNAs with FC > 2 or FC < 0.5 were obtained from above 760 mRNAs. Among the 70 differentially expressed mRNAs, 23 had high expression and 47 had low expression (Figure 3).
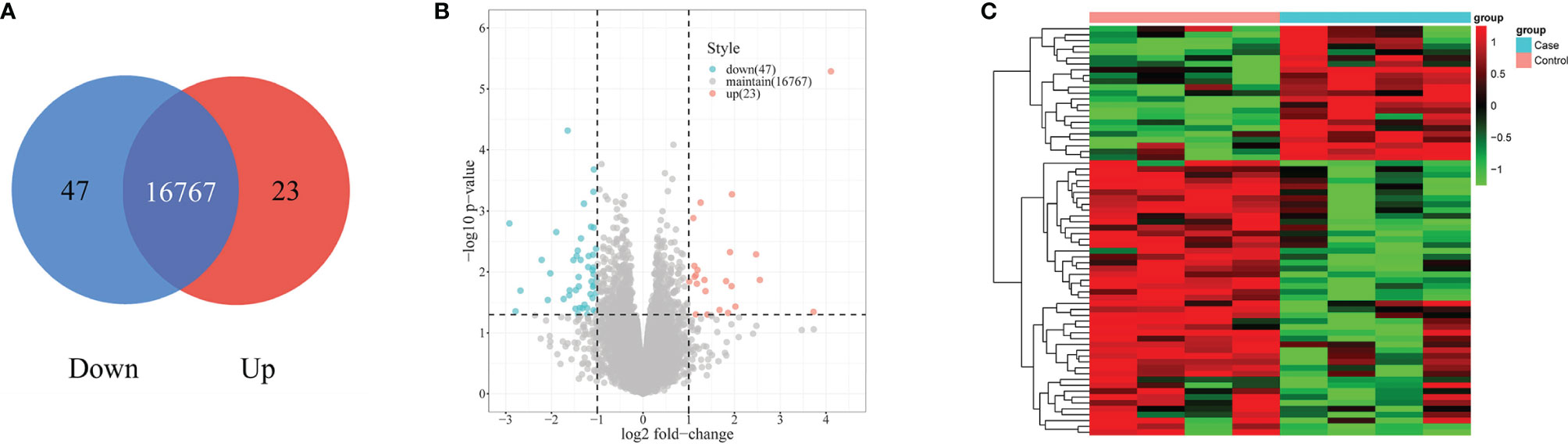
Figure 3 Differential expression of mRNAs in the case and control groups. (A) The Venn diagram shows 23 upregulated mRNAs and 47 downregulated mRNAs in the case group, and 16,767 mRNAs are maintained. (B) The differentially expressed mRNAs between the case and control groups are described using volcano plots. The red points represent the differentially expressed mRNAs on the statistical criteria of fold change >2-fold upregulated (log2 scaled) and P < 0.05 (−log10 scaled); the green points represent the differentially expressed mRNAs on the statistical criteria of fold change <1/2 downregulated (log2 scaled) and P < 0.05 (−log10 scaled). (C) Hierarchical clustering analysis of the 23 upregulated and 47 downregulated mRNAs.
Selection of Promising SNPs in Candidate mRNAs
Using the 1000 Genome Project, 69,893 SNPs located on the 70 mRNAs obtained were screened out. A total of 5,927 SNPs were selected with a minor allele frequency (MAF) value > 0.05 in the Han Chinese population.
Subsequently, the silicosis related GWAS dataset was used to evaluate the association between the shortlisted 5,927 SNPs and silicosis susceptibility. The result revealed 476 SNPs to be significantly associated with silicosis risk (P < 0.05).
To evaluate the regulatory relationship between SNPs and corresponding mRNAs, further eQTL screening with the SNPs associated with silicosis risk was carried out. Among the 476 positive SNPs, 41 and 21 SNPs with different gene expressions were found in the lung tissue and whole blood tissues, respectively. Six overlapping SNPs in the lung and whole blood were eventually selected. LD analysis of the six SNPs showed no significant LD, and these SNPs were selected for use in the validation stage.
Among the six SNPs (Table 2), three (rs2291236, rs2962375, rs7736442) were located on BASP1; rs9273410, HLA-DQB1; rs2965298, R3HDM4; and rs924150, TSHZ3. As shown in Figure 4, the gene expression levels of HLA-DQB1, TSHZ3, and BASP1 were significantly higher in the case group than the healthy control group (P < 0.05), while R3HDM4 was significantly lower in the case group than in the control group (P = 0.006).
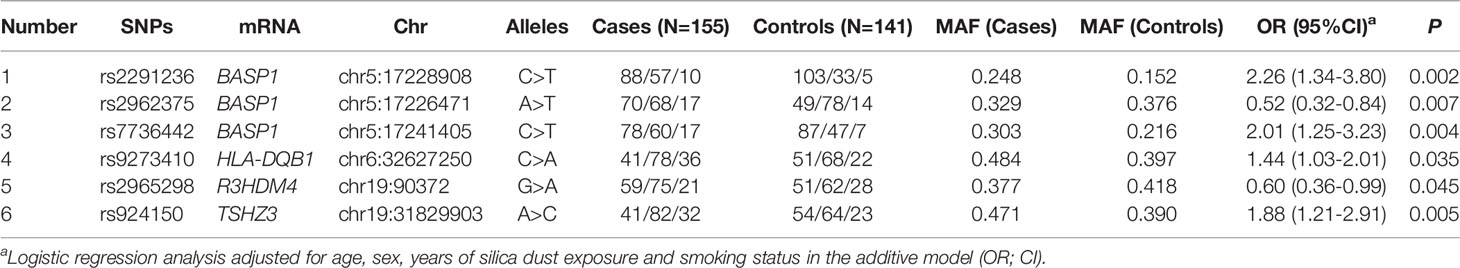
Table 2 Results of the six SNPs in the GWAS database.
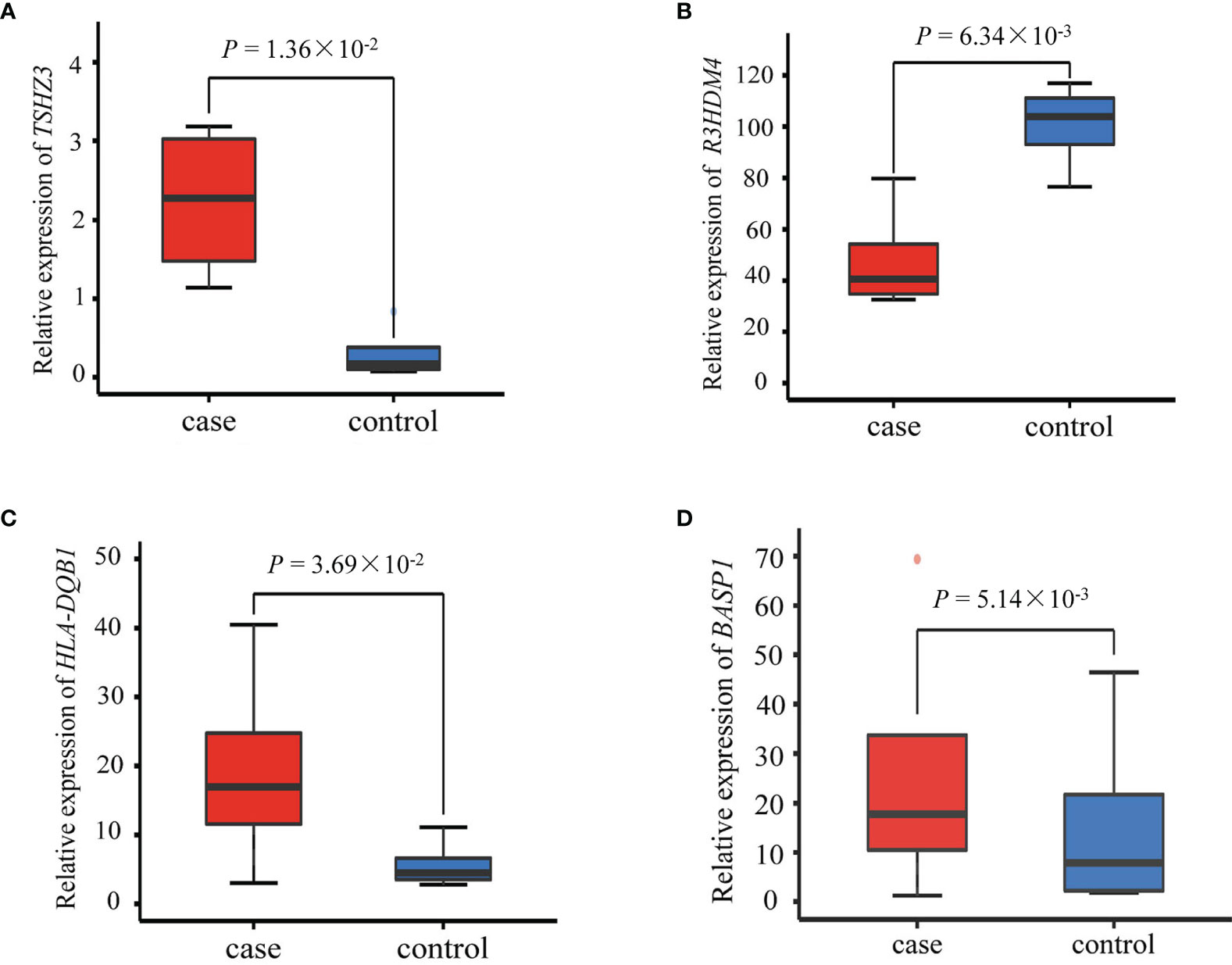
Figure 4 Differential expression of (A) TSHZ3, (B) R3HDM4, (C) HLA-DQB1 and (D) BASP1 between the four patients with silicosis and four healthy controls. P-value was calculated using the Limma R package.
Validation Between eQTL-SNPs and Silicosis Based on iMLDR Genotyping Platform
To further validate the relationship between the six eQTL-SNPs and silicosis susceptibility, the iMLDR technique was used for genotyping. The result suggested that the mutant A allele of rs9273410 may increase silicosis susceptibility (additive model: OR= 1.31, 95% CI = 0.99-1.74, P = 0.061).
The GWAS screening data were combined with the iMLDR validation data to define the association between rs9273410 and silicosis susceptibility. The two-stage case-control study revealed that the mutant A allele of rs9273410 was associated with increased silicosis susceptibility (additive model: OR= 1.35, 95% CI =1.09-1.68, P = 0.006) (Table 3).
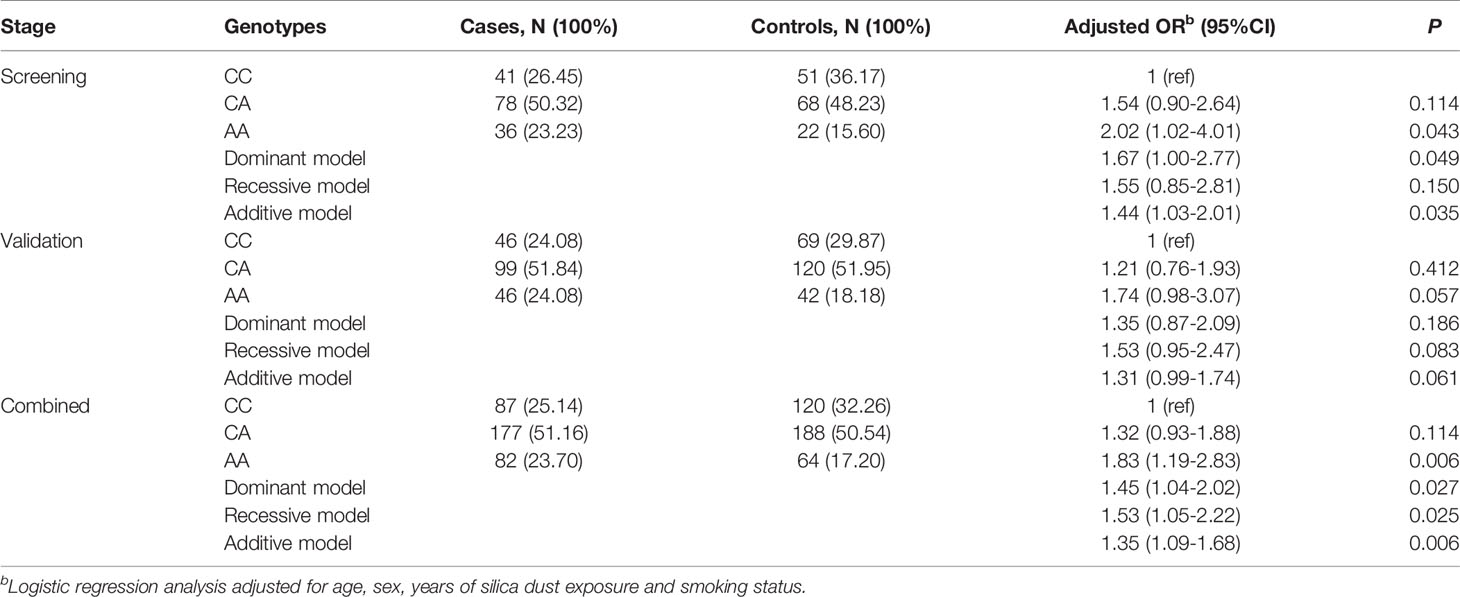
Table 3 Specific information of rs9273410 in the different stages.
eQTL Analysis
To assess the functional relevance of rs9273410 on HLA-DQB1 expression, eQTL analysis was performed using the GTEx database. As illustrated in Figure 5, significant associations were observed between HLA-DQB1 expression and rs9273410 genotypes in both the lung (P = 4.0 × 10-55) and whole blood tissue samples (P = 2.9 × 10-124). HLA-DQB1 expression level of rs9273410 was significantly lower in participants with the CA (or AA) genotype than in those with the CC genotype.
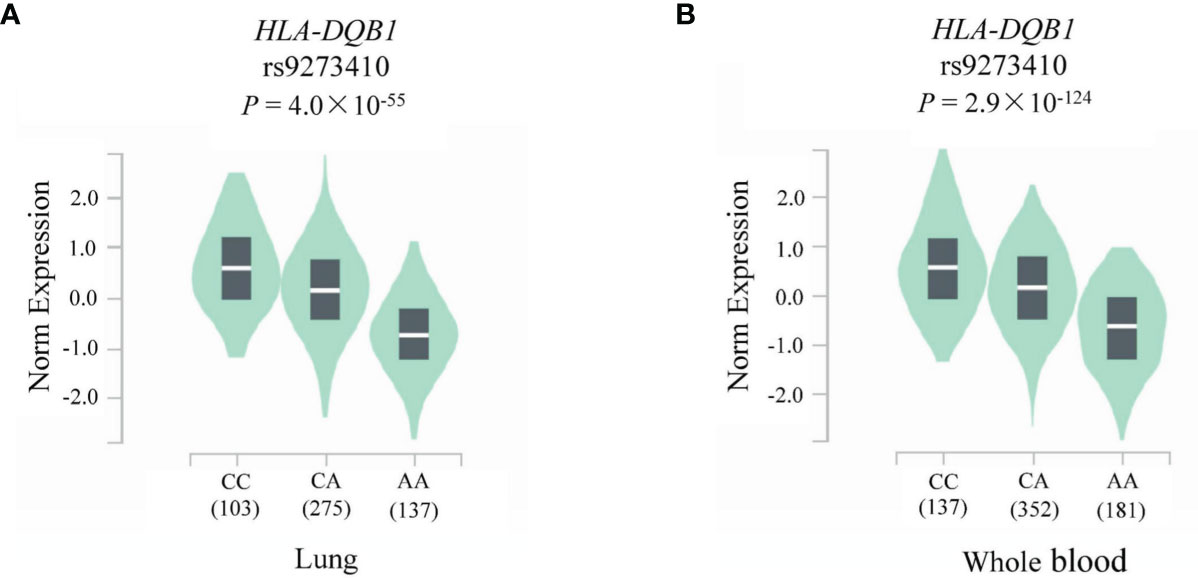
Figure 5 HLA-DQB1 relative expression levels in the (A) lung and (B) whole blood according to rs9273410 genotypes.
Discussion
In this multi-stage study, we systematically appraised the association between functional eQTL-SNPs and silicosis susceptibility by integration of RNA-seq data and GWAS mapping. The two-stage case-control study revealed that the mutant A allele of rs9273410 was potentially associated with increased silicosis susceptibility.
Increasing evidence suggests that mRNA is involved in the regulation of various fibrotic diseases. For instance, the interaction between galectin-3 and ST2 can be used to identify high systemic fibrosis in patients with acute heart failure (22). Moreover, gene-specific hydroxymethylation, which is based on CRISPR/Cas9, can affect gene expression and alleviate renal fibrosis (23). Recently, Junsuk et al. demonstrated that alternative polyadenylation, which is attributed to NUDT21 reduction, significantly increased the expression of fibrotic mediators and proteins in lung fibroblasts by shortening the 3’-untranslated regions (3’-UTRs) of mRNAs, stabilising their transcripts and eventually activating pathological signalling pathways (24). A study reported that HDAC8 expression increased in IPF lung tissue, indicating that HDAC8 expression contributes to pulmonary fibrosis and HDAC8 inhibition can treat IPF and other fibrotic lung diseases (25). In addition, Liu et al. reported that the upregulation of NRF2-mediated LOC344887 contributes to the antifibrotic potential of SFN by repressing the expression of CDH2 and other fibrotic genes (26). These studies indicate that the differential expression of mRNA is associated with the occurrence and development of fibrotic diseases. Therefore, this study aimed to fill the gap between mRNA and silicosis susceptibility.
The identified SNP rs9273410 is located in the 3’-UTR region of HLA-DQB1. Recently, an increasing number of studies have reported that HLA-DQB1 in the MHC region may promote inflammatory response, which is a characteristic of silicosis (27). HLA-DQB1 has the strongest genetic association with scleroderma because of excessive fibrosis of the internal organs (28). Severin et al. reported that two HLA-DQB1 alleles (HLA-DQB1*0303 and HLA-DQB1*0609) were significantly associated with the rapid progression of hepatic fibrosis (29). In addition, Louise et al. selected 50,008 unique samples from the UK Biobank and reported six novel genome-wide significant signals of association with the extremes of FEV1, including independent signals at two previously reported loci (NPNT and HLA-DQB1/HLA-DQA2). Their results also revealed that HLA-DQB1 was associated with chronic obstructive pulmonary disease (30). A recent study indicated that several HLA–tumour–peptide interactions were the major factors modulating lung cancer susceptibility (31). A previous study also revealed a genome-wide significant association between the HLA region and fibrotic idiopathic interstitial pneumonias (27). Our study is the first to report the susceptibility of HLA-DQB1 for silicosis. Therefore, this study indicates that HLA-DQB1 may promote lung fibrotic disease development.
The SNP rs9273410 is present in the binding site of miR-888, which is associated with the development of various diseases (32). Huang et al. reported that E-cadherin is directly targeted by miR-888, which exists in MCF-7 side population sphere cells (33). Therefore, the biological rationality indicates that SNP rs9273410 may regulate miR-888 and influence fibrotic disease development through epithelial–mesenchymal transition. In addition, miR-888 was significantly upregulated in lung adenocarcinoma, suggesting that miR-888 plays a role as an oncogene in the progression of lung adenocarcinoma and is a potential therapeutic target for patients with lung adenocarcinoma (32). Further studies investigating the potential mechanisms of rs9273410 that contribute to silicosis via miR-888 regulation are warranted.
This study has several merits. First, the combination of GWAS and RNA-seq results helped to identify more precisely the potentially functional SNPs associated with silicosis susceptibility, and the genes that were identified in both the RNA-seq and GWAS had a higher probability of revealing the molecular basis of silicosis development. Second, the functional eQTL-SNP rs9273410 was obtained through the GWAS screening (155 cases and 141 controls) and iMLDR validation (194 cases and 235 controls). This double validation adds value to our results. Third, in the eQTL analysis, we considered both the lung and whole blood tissues, increasing the coverage area of our results compared to previous results when either the lung or whole blood tissues were considered. Finally, in the RNA-seq screening stage, to match the exposure years between the case and control groups, four healthy controls based on 1:1 individual matching on silica dust exposure years and age were selected as controls, thereby enhancing the study’s statistical efficiency. However, some limitations of our study still exist. Firstly, in the GWAS screening and iMLDR validation stages, in order to retain all available cases to evaluate the association between genetic variants and the susceptibility of developing silicosis, we mainly matched the cases and controls by the years of silica dust exposure, while age of the two groups were not comparable. Although we have adjusted for age in the further logistic regression analysis, we cannot exclude the possibility that this factor may affect our results, and further studies with strict matching should be worth attention. Secondly, although this study was designed as a multi-stage study that integrates RNA-seq, GWAS and iMLDR, it was carried out mainly on population-based case-control studies and further functional verification of basic biological experiments should be included to reveal the potential mechanisms.
In conclusion, our study identified an eQTL-SNP rs9273410 located on HLA-DQB1 that might contribute to the development of silicosis. However, further evaluation of the biological mechanisms of rs9273410 and HLA-DQB1 is warranted.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://figshare.com/s/4d5a6a115f52f78d3674.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Committee of Nantong University (Approval No. 2020-002). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
Conceptualization, YaZ, ZC and YiZ. Data curation, YaZ, YD, and AQ. Formal analysis, RZ, MT and YiZ. Funding acquisition, WW and MC. Investigation, YaZ, YD, AQ, YiZ, and MC. Methodology, YiL, YuL, XZ, and TT. Resources, YiZ, WW, and MC. Software, YaZ and ZC. Supervision, YiL, YuL, XZ, and TT. Visualization, RZ, MT, and MC. Writing—original draft, YaZ, RZ, and YiZ. Writing—review and editing, YaZ and MC. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20191449, SBK2021020088). The scientific research projects of Jiangsu commission of health (Z2019004, H2019020). Jiangsu Association of Science and Technology Youth Science and Technology Talents Enrollment Project in 2019. Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX21-3126, KYCX20-2852).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hoy RF, Chambers DC. Silica-Related Diseases in the Modern World. Allergy (2020) 75:2805–17. doi: 10.1111/all.14202
2. Vacek PM, Glenn RE, Rando RJ, Parker JE, Kanne JP, Henry DA, et al. Exposure‒response Relationships for Silicosis and Its Progression in Industrial Sand Workers. Scand J Work Environ Health (2019) 45:280–8. doi: 10.5271/sjweh.3786
3. Qin X, Lin X, Liu L, Li Y, Li X, Deng Z, et al. Macrophage-Derived Exosomes Mediate Silica-Induced Pulmonary Fibrosis by Activating Fibroblast in an Endoplasmic Reticulum Stress-Dependent Manner. J Cell Mol Med (2021) 25:4466–77. doi: 10.1111/jcmm.16524
4. GBD 2016 Occupational Chronic Respiratory Risk Factors Collaborators, GBD 2016 Occupational Chronic Respiratory Risk Factors Collaborators. Global and Regional Burden of Chronic Respiratory Disease in 2016 Arising From Non-Infectious Airborne Occupational Exposures: A Systematic Analysis for the Global Burden of Disease Study 2016. Occup Environ Med (2020) 77:142–50. doi: 10.1136/oemed-2019-106013
5. Wood C, Yates D. Respiratory Surveillance in Mineral Dust-Exposed Workers. Breathe (Sheff) (2020) 16:190632. doi: 10.1183/20734735.0362-2019
6. Steenland K, Ward E. Silica: A Lung Carcinogen. CA Cancer J Clin (2014) 64:63–9. doi: 10.3322/caac.21214
7. Mandrioli D, Schlunssen V, Adam B, Cohen RA, Colosio C, Chen W, et al. WHO/ILO Work-Related Burden of Disease and Injury: Protocol for Systematic Reviews of Occupational Exposure to Dusts and/or Fibres and of the Effect of Occupational Exposure to Dusts and/or Fibres on Pneumoconiosis. Environ Int (2018) 119:174–85. doi: 10.1016/j.envint.2018.06.005
8. Riley L, Urbine D. Chronic Silicosis With Progressive Massive Fibrosis. N Engl J Med (2019) 380:2256. doi: 10.1056/NEJMicm1809675
9. Leung CC, Yu IT, Chen W. Silicosis. Lancet (2012) 379:2008–18. doi: 10.1016/S0140-6736(12)60235-9
10. Chu M, Wu S, Wang W, Yu Y, Zhang M, Sang L, et al. Functional Variant of the Carboxypeptidase M (CPM) Gene May Affect Silica-Related Pneumoconiosis Susceptibility by Its Expression: A Multistage Case-Control Study. Occup Environ Med (2019) 76:169–74. doi: 10.1136/oemed-2018-105545
11. Salum KCR, Castro MCS, Nani ASF, Kohlrausch FB. Is Individual Genetic Susceptibility a Link Between Silica Exposure and Development or Severity of Silicosis? A Systematic Review. Inhal Toxicol (2020) 32:375–87. doi: 10.1080/08958378.2020.1825569
12. Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A Common MUC5B Promoter Polymorphism and Pulmonary Fibrosis. N Engl J Med (2011) 364:1503–12. doi: 10.1056/NEJMoa1013660
13. Allen RJ, Porte J, Braybrooke R, Flores C, Fingerlin TE, Oldham JM, et al. Genetic Variants Associated With Susceptibility to Idiopathic Pulmonary Fibrosis in People of European Ancestry: A Genome-Wide Association Study. Lancet Respir Med (2017) 5:869–80. doi: 10.1016/S2213-2600(17)30387-9
14. Moore C, Blumhagen RZ, Yang IV, Walts A, Powers J, Walker T, et al. Resequencing Study Confirms That Host Defense and Cell Senescence Gene Variants Contribute to the Risk of Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2019) 200:199–208. doi: 10.1164/rccm.201810-1891OC
15. Allen RJ, Guillen-Guio B, Oldham JM, Ma SF, Dressen A, Paynton ML, et al. Genome-Wide Association Study of Susceptibility to Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2020) 201:564–74. doi: 10.1164/rccm.201905-1017OC
16. Conde L, Bracci PM, Richardson R, Montgomery SB, Skibola CF. Integrating GWAS and Expression Data for Functional Characterization of Disease-Associated SNPs: An Application to Follicular Lymphoma. Am J Hum Genet (2013) 92:126–30. doi: 10.1016/j.ajhg.2012.11.009
17. Liu Y, Morley M, Brandimarto J, Hannenhalli S, Hu Y, Ashley EA, et al. RNA-Seq Identifies Novel Myocardial Gene Expression Signatures of Heart Failure. Genomics (2015) 105:83–9. doi: 10.1016/j.ygeno.2014.12.002
18. Stark R, Grzelak M, Hadfield J. RNA Sequencing: The Teenage Years. Nat Rev Genet (2019) 20:631–56. doi: 10.1038/s41576-019-0150-2
19. Sala MA, Balderas-Martinez YI, Buendia-Roldan I, Abdala-Valencia H, Nam K, Jain M, et al. Inflammatory Pathways Are Upregulated in the Nasal Epithelium in Patients With Idiopathic Pulmonary Fibrosis. Respir Res (2018) 19:233. doi: 10.1186/s12931-018-0932-7
20. Nance T, Smith KS, Anaya V, Richardson R, Ho L, Pala M, et al. Transcriptome Analysis Reveals Differential Splicing Events in IPF Lung Tissue. PloS One (2014) 9:e97550. doi: 10.1371/journal.pone.0097550
21. Zhao H, Jiang Z, Lv R, Li X, Xing Y, Gao Y, et al. Transcriptome Profile Analysis Reveals a Silica-Induced Immune Response and Fibrosis in a Silicosis Rat Model. Toxicol Lett (2020) 333:42–8. doi: 10.1016/j.toxlet.2020.07.021
22. Dong R, Zhang M, Hu Q, Zheng S, Soh A, Zheng Y, et al. Galectin-3 as a Novel Biomarker for Disease Diagnosis and a Target for Therapy (Review). Int J Mol Med (2018) 41:599–614. doi: 10.3892/ijmm.2017.3311
23. Xu X, Tan X, Tampe B, Wilhelmi T, Hulshoff MS, Saito S, et al. High-Fidelity CRISPR/Cas9- Based Gene-Specific Hydroxymethylation Rescues Gene Expression and Attenuates Renal Fibrosis. Nat Commun (2018) 9:3509. doi: 10.1038/s41467-018-05766-5
24. Ko J, Mills T, Huang J, Chen NY, Mertens TCJ, Collum SD, et al. Transforming Growth Factor Beta1 Alters the 3’-UTR of mRNA to Promote Lung Fibrosis. J Biol Chem (2019) 294:15781–94. doi: 10.1074/jbc.RA119.009148
25. Saito S, Zhuang Y, Suzuki T, Ota Y, Bateman ME, Alkhatib AL, et al. HDAC8 Inhibition Ameliorates Pulmonary Fibrosis. Am J Physiol Lung Cell Mol Physiol (2019) 316:L175–86. doi: 10.1152/ajplung.00551.2017
26. Liu P, Luo G, Dodson M, Schmidlin CJ, Wei Y, Kerimoglu B, et al. The NRF2-LOC344887 Signaling Axis Suppresses Pulmonary Fibrosis. Redox Biol (2021) 38:101766. doi: 10.1016/j.redox.2020.101766
27. Fingerlin TE, Zhang W, Yang IV, Ainsworth HC, Russell PH, Blumhagen RZ, et al. Genome-Wide Imputation Study Identifies Novel HLA Locus for Pulmonary Fibrosis and Potential Role for Auto-Immunity in Fibrotic Idiopathic Interstitial Pneumonia. BMC Genet (2016) 17:74. doi: 10.1186/s12863-016-0377-2
28. Tsou PS, Sawalha AH. Unfolding the Pathogenesis of Scleroderma Through Genomics and Epigenomics. J Autoimmun (2017) 83:73–94. doi: 10.1016/j.jaut.2017.05.004
29. Kamdem SD, Moyou-Somo R, Brombacher F, Nono JK. Host Regulators of Liver Fibrosis During Human Schistosomiasis. Front Immunol (2018) 9:2781. doi: 10.3389/fimmu.2018.02781
30. Wain LV, Shrine N, Miller S, Jackson VE, Ntalla I, Soler Artigas M, et al. Novel Insights Into the Genetics of Smoking Behaviour, Lung Function, and Chronic Obstructive Pulmonary Disease (UK BiLEVE): A Genetic Association Study in UK Biobank. Lancet Respir Med (2015) 3:769–81. doi: 10.1016/S2213-2600(15)00283-0
31. Ferreiro-Iglesias A, Lesseur C, McKay J, Hung RJ, Han Y, Zong X, et al. Fine Mapping of MHC Region in Lung Cancer Highlights Independent Susceptibility Loci by Ethnicity. Nat Commun (2018) 9:3927. doi: 10.1038/s41467-018-05890-2
32. Cao JX. Mir888 Regulates Cancer Progression by Targeting Multiple Targets in Lung Adenocarcinoma. Oncol Rep (2019) 41:3367–76. doi: 10.3892/or.2019.7118
Keywords: silicosis, mRNA, HLA-DQB1, rs9273410, susceptibility
Citation: Zhou Y, Zhang Y, Zhao R, Cheng Z, Tang M, Qiu A, Dong Y, Lu Y, Lian Y, Zhuang X, Tian T, Wang W and Chu M (2022) Integrating RNA-Seq With GWAS Reveals a Novel SNP in Immune-Related HLA-DQB1 Gene Associated With Occupational Pulmonary Fibrosis Risk: A Multi-Stage Study. Front. Immunol. 12:796932. doi: 10.3389/fimmu.2021.796932
Received: 18 October 2021; Accepted: 20 December 2021;
Published: 17 January 2022.
Edited by:
Dong Weng, Tongji University, ChinaCopyright © 2022 Zhou, Zhang, Zhao, Cheng, Tang, Qiu, Dong, Lu, Lian, Zhuang, Tian, Wang and Chu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minjie Chu, Y2h1bWluamllQG50dS5lZHUuY24=; Wei Wang, b25ld2F5MTk4NUAxNjMuY29t
†These authors have contributed equally to this work