 Anh Duy Do
Anh Duy Do Chiu-Hsian Su1,2
Chiu-Hsian Su1,2 Yuan-Man Hsu
Yuan-Man Hsu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 14 January 2022
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.796177
This article is part of the Research Topic Role of Lipid Rafts in Anti-Microbial Immune Response Volume II View all 6 articles
Helicobacter pylori is a Gram-negative pathogen that can increase the risk of stomach cancer in infected patients. H. pylori exploits lipid rafts to infect host cells. Infection triggers clustering of Lewis x antigen (Lex) and integrins in lipid rafts to facilitate H. pylori adherence to the gastric epithelium. H. pylori infection can be treated with probiotics containing lactic acid bacteria that offer numerous benefits to the host while lacking the side effects associated with antibiotic therapy. Previously, we showed that the cell-free supernatant (CFS) derived from Lactobacillus rhamnosus JB3 (LR-JB3) at a multiplicity of infection (MOI) of 25 attenuated the pathogenicity of H. pylori. In this study, we established a mucin model to simulate the gastric environment and to further understand the influence of mucin on the pathogenesis of H. pylori. Porcine stomach mucin dramatically upregulated H. pylori virulence gene expression, including that of babA, sabA, fucT, vacA, hp0499, cagA, and cagL, as well as the adhesion and invasion ability of H. pylori and induced increased levels of IL-8 in infected-AGS cells. The CFS derived from LR-JB3 at a MOI of 25 reduced the expression of H. pylori sabA, fucT, and hp0499 in mucin, as well as that of the Lex antigen and the α5β1 integrin in AGS cells during co-cultivation. These inhibitory effects of LR-JB3 also suppressed lipid raft clustering and attenuated Lewis antigen-dependent adherence, type IV secretion system-mediated cell contact, and lipid raft-mediated entry of VacA to host cells. In conclusion, LR-JB3 could affect H. pylori infection through mediating lipid raft formation of the host cells. The currently unknown cues secreted from LR-JB3 are valuable not only for treating H. pylori infection, but also for treating diseases that are also mediated by lipid raft signaling, such as cancer and aging-associated and neurodegenerative conditions.
Helicobacter pylori is a Gram-negative pathogen that colonizes the gastric mucosa and increases the risk of stomach cancer in infected patients (1, 2). The treatment of H. pylori infection mainly relies on antibiotics, which may affect the balance of gut microflora and also facilitate the development of antibiotic-resistant strains (3, 4). Lactic acid bacteria are known to offer numerous benefits to the host, including protection against H. pylori infection (5). Previously, we indicated that oral administration of Lactobacillus rhamnosus JB3 (LR-JB3) to mice could eliminate H. pylori infection and attenuate inflammatory responses by suppressing vacA gene expression (6). LR-JB3 also induced superoxide dismutase and catalase activity, and the serum levels of beneficial amino acids, which are impaired by H. pylori infection (7). These results indicated that LR-JB3 has potential in eliminating H. pylori infection. Therefore, the mechanism that underlies LR-JB3 activity against H. pylori infection should be further investigated to support potential clinical applications.
Previously, we used a cell model to explore part of the molecular mechanism of LR-JB3 that is involved in modulating H. pylori infection. LR-JB3 at a multiplicity of infection (MOI) of 25 suppressed expression of sabA and fucT in H. pylori, translocation of CagA, and expression of the Lewis x (Lex) antigen and toll-like receptor 4 (TLR 4) in H. pylori-infected-AGS cells (8). However, it has been indicated that the Lex antigen and TLR 4 are clustered in lipid rafts to facilitate the adherence of H. pylori to the gastric epithelial cells (9–12), and disrupting lipid rafts may prevent H. pylori-associated gastric cancer (13). Lipid rafts consist of phospholipids, sphingolipids, and cholesterol and act as signaling platforms and gateways for microorganisms to adhere to host cells (10, 14–17). Furthermore, H. pylori expresses cholesteryl α-D-glucopyranoside acyltransferase (CGAT), which is encoded by the hp0499 gene, to enhance lipid raft clustering on host cell membranes (9). Thus, H. pylori can manipulate the formation of lipid rafts to promote infection. Therefore, lipid raft-mediated pathways may be involved in the mechanism of LR-JB3 against H. pylori infection.
The stomach is covered by a thick layer of mucus that physically protects epithelial cells from the gastric juices and interaction with pathogens (18); therefore, H. pylori needs to migrate through the mucus layer to colonize the epithelial cell surface (19). We used mucin to help simulate the conditions under which H. pylori encounters LR-JB3 in the stomach and then explored the mechanisms whereby LR-JB3 modulates H. pylori infection through lipid raft-mediated pathways.
H. pylori reference strain 26695 (American Type Culture Collection [ATCC] 700392) and AGS human gastric adenocarcinoma cell line (ATCC CRL-1739) were purchased from the ATCC (Manassas, VA, USA). H. pylori was grown on tryptic soy agar (TSA) supplemented with 5% sheep blood (Dr. Plate Biotech, Taipei, Taiwan) under microaerophilic conditions obtained using an anaerobic BD GasPak (BD Biosciences, Franklin Lakes, NJ, USA) at 37°C for 48–72 h. LR-JB3 isolated from a dairy product (6) was cultured in De Man, Rogosa, and Sharpe (MRS) broth (BD Biosciences, Franklin Lakes, NJ, USA) at 37°C for 12–18 h. AGS cells were cultured in Roswell Park Memorial Institute 1640 (RPMI) medium (Gibco-Life Technologies, Gaithersburg, MD, USA) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO, USA), 100 μg/mL of penicillin, and 100 μg/mL of streptomycin (Thermo Scientific, Waltham, MA, USA) at 37°C and 5% of CO2. During infection experiments, AGS cells were washed three times using phosphate buffer solution (PBS), and the culture media were replaced with antibiotic-free RPMI media containing 10% FBS for further incubation.
Porcine stomach mucin-type II (Sigma-Aldrich, St. Louis, MO, USA) in PBS at pH 7 was sterilized by autoclaving (20). H. pylori harvested from TSA agar plates was resuspended at 5 × 106 colony forming units (CFU) per mL with 50 mg/mL of mucin in PBS and incubated at 37°C and 5% CO2 for 2 h. H. pylori was then collected for mRNA isolation. Mucin-treated H. pylori was also used to infect AGS cells in the following experiments. The autoclaved mucin in PBS was centrifuged at 10,000 × g. Mucins were collected and resuspended in equal volumes of RPMI; 50 mg/mL of mucin in RPMI medium was then added into the culture medium for AGS cells in the control group.
Total mRNA was isolated using the total RNA Miniprep Purification Kit (GMBiolab, Taichung, Taiwan), and reverse transcription was performed using the MMLV Reverse Transcription Kit (Protech Technology Enterprise, Taipei, Taiwan) according to the manufacturers’ instructions. All oligonucleotide primers used in this study were synthesized by Mission Biotech, Taipei, Taiwan. The RNA sample was incubated with random primers at 65°C for 5 min and then chilled on ice to open the secondary structure of the GC-rich RNA templates. Each sample was then incubated with a mixture of 5× Reaction Buffer, dNTP pre-mix, and MMLV reverse transcriptase. RT-qPCR was performed at the following conditions: 5 min at 65°C, 10 min at 25°C, 60 min at 42°C, and a final 10 min at 72°C. qPCR was performed in 96-well optical plates (Applied Biosystems, Waltham, MA, USA). Targeted cDNA dilutions were mixed with appropriate primers (Table 1) and SYBR Green Master Mix (Thermo Fisher Scientific, Waltham, MA, USA). The PCR program was: initial denaturation at 95°C for 10 min, followed by amplification for 40 cycles with denaturation at 95°C for 10 sec; annealing at 50°C for 20 sec; and extension at 72°C for 20 sec. The melting curve analysis was: 95°C for 5 sec, 65°C for 1 min, and then an increase to 95°C at 0.08°C/sec ramp rate. Each assay was run on a qPCR QuantStudio 3 (Applied Biosystems, Waltham, MA, USA) and the fold-changes in expression were derived using the comparative ΔΔCt method (21). The 16S rRNA of H. pylori served as an internal control for sample loading and mRNA integrity (22). Results were calculated using the mean of triplicate readings.

Table 1 Primers used in this study.
The adhesion and invasion abilities were defined as the numbers of H. pylori adhering and invading into the AGS cells, respectively. AGS cells were seeded into 24-well plates at 5 × 104 cells/well in antibiotic-free RPMI medium with 10% FBS at 37°C and 5% CO2 for 18 h. H. pylori treated with mucin as previously described or PBS for 2 h was then co-incubated with mucin-treated AGS cells at a MOI of 50 at 37°C and 5% CO2 for 6 h. Cell culture supernatants were removed by centrifugation at 1,500 × g for 5 min at room temperature. The cells were then washed with PBS twice, and osmotic lysis performed to calculate the total quantity of bacteria remaining, which included adhering and invading H. pylori. For this purpose, sterile water was added to the infected cells following washing and the resulting cell lysates were then resuspended in PBS and plated using serial dilutions on TSA supplemented with 5% sheep blood. These plates were then cultured with 100 μL from each dilution at 37°C for 48 h. Bacterial cell numbers were determined by manual colony counting. To determine the number of viable intracellular bacteria, the standard gentamicin assay was applied (23). The same batch of infected cells was washed three times in PBS and incubated with 100 μg/mL of the membrane-impermeable antibiotic gentamicin (Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) at 37°C and 5% CO2 for 1.5 h to remove extracellular bacteria (23). Bacterial cell numbers were then determined by manual colony counting. The adherent bacteria number was calculated by deducting the invading bacteria number from the total sum. The adhesion and invasion activities of H. pylori was determined as the mean of triplicate readings at each treatment. AGS cells incubated with 50 mg/mL of mucin in RPMI medium at 37°C and 5% CO2 for 6 h were assigned as the control group, whereas AGS cells infected by PBS-pretreated and mucin-pretreated H. pylori were assigned as the non-mucin group and the mucin treatment group, respectively. The non-mucin group was used to establish 100% adhesion and invasion. The cell-free supernatants (CFSs) were used in the detection of interleukin (IL)-8.
Detection of IL-8 in the supernatants of H. pylori-infected human AGS cells was conducted using human IL-8 ELISA Ready- SET-Go!R ® kits (eBioscience, Inc., San Diego, CA, USA) according to manuals’ instruction. Each sample was analyzed individually. Results were calculated as the mean of triplicate readings.
CFSs were collected from the cultures grown with 5 × 104 cell/mL of AGS cells, 5 × 106 CFU/mL of H. pylori, 1.25 × 106 CFU/mL of LR-JB3, and 5 × 106 CFU/mL of LR-JB3 for 2 h and were referred to as AGS, HP-100, JB3-25, and JB3-100, respectively. CFSs were mixed with 100 mg/mL of porcine stomach mucin at a 1:1 ratio for the pretreatment study. For the H. pylori pretreatment group, CFSs with mucin were co-incubated with H. pylori at 37°C for 2 h. The pretreated H. pylori was collected by 10,000 × g for 1 min at room temperature and then resuspended in RPMI medium. These H. pylori at a MOI of 50 were used to infect AGS cells in an antibiotic-free RPMI medium supplemented with 10% FBS at 37°C in 5% of CO2. For the AGS cell pretreatment group, these cells were co-incubated using CFSs with mucin at 37°C and 5% of CO2 for 2 h and then infected by mucin-pretreated H. pylori at a MOI of 50. After 6 h of incubation, the culture supernatants of both pretreatment groups were then used for the detection of IL-8 levels via ELISA. H. pylori in co-culturing conditions were assessed via adhesion and invasion assays. The total mRNA was isolated for the virulence gene expression in H. pylori. AGS cells co-incubated with 50 mg/mL of mucin in RPMI medium at 37°C and 5% of CO2 for 6 h were assigned as control group. AGS cells infected by mucin-pretreated H. pylori were referred to as the infection group. Results were calculated as the mean of triplicate readings.
Cells or bacteria were lysed with ice-cold RIPA buffer (Genestar Biotechnology, Kaohsiung, Taiwan). Protein concentration was determined using the Bradford method (Bio-Rad, Hercules, CA, USA). Protein sample (30–40 µg) was separated via SDS-PAGE using a Hoefer mini VE system (Amersham Biosciences, Piscataway, NJ, USA). Proteins were transferred to Immobilon-E polyvinylidene fluoride membrane (Merck Millipore, Carrigtwohill, County Cork, Ireland) according to the manufacturer’s instructions. Following the transfer, the membrane was washed with Tris-Buffered Saline (TBS) and blocked for 1 h at 37°C with 5% fat-free milk in TBS and 0.1% of Tween-20 (TBST). Diluted primary antibodies were added and co-incubated with the membrane at 4°C overnight. Blots were then incubated with peroxidase-conjugated secondary antibodies horseradish peroxidase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG. Following removal of the secondary antibody, blots were washed with TBST and developed using the ECL-western blotting system (Advansta, San Jose, CA, USA). Immunoblot densities were quantified using the Hansor Luminescence Imaging System LIS02 (Han-Shuo Life Technology, Taichung, Taiwan).
Twelve-millimeter diameter glasses (Paul-Marienfeld GmbH & Co. KG, Am Wöllerspfad, Lauda-Königshofen, Germany) were coated with 300 µL collagen purified from calfskin (Sigma-Aldrich, St. Louis, MO, USA) and placed on the bottom of a 24-well plate for 2 h. H. pylori and AGS cells were co-cultivated as described in the previous section. After 6 h of cultivation, the supernatant was removed and AGS cells were washed three times with PBS. AGS cells were then fixed with 4% paraformaldehyde for 20 min. Triton X-100 (1%) (Thermo Fisher Scientific, Waltham, MA, USA) was added and incubated for 20 min. The cells were then washed three times with PBS and co-incubated with Alexa Fluor 594-conjugated cholera toxin subunit β for GM1 (lipid rafts marker) staining for 1 h, or with the primary antibody for integrin α5 (1:1,000), β1 (1:500), or sialyl Lex antibody (1:50) at 4°C for 16 h. Visualization of integrins and Lewis antigens was applied using fluorescently tagged secondary antibodies (Alexa Fluor 488, green). AGS cells were then washed three times with PBS, and 4,6-diamidino-2-phenylindole (DAPI, Southern Biotech, Birmingham, USA) was added and the mixture incubated for 5 min at room temperature. Samples were imaged at a magnification of 40× using a Revole Immunofluorescence microscopy (Echo, San Diego, CA, USA).
The primary antibodies used for Western blot analysis were as follows: rat anti-integrin β1 clone AIIB2 (Merck, MABT409), rabbit anti-integrin α5 clone 5E18 ZooMAb (Sigma-Aldrich, ZRB1122-25UL), mouse anti-CD15s (sialyl Lewisx) (Santa Cruz, sc-32243), mouse anti-CagA (Santa Cruz, sc-28368), mouse anti-CagA phospho (Santa Cruz, sc-508), mouse anti-VacA (Santa Cruz, sc-32746), rabbit anti-p38 MAPK (Agrigo, ARG55258), rabbit anti-p38 MAPK phospho (Agrigo, ARG51850) rabbit anti-Cox-2 (Agrigo, ARG56421), goat anti-ATF-2 (Arigo, ARG63122), rabbit anti-ATF2 phospho (Agrigo, ARG42620), mouse anti-GAPDH (Agrigo, ARG10112). Furthermore, the secondary antibodies were as follows: donkey anti-goat IgG antibody (Arigo, ARG65352), goat anti-rabbit IgG (pre-adsorbed to 10 nm gold) (Abcam, ab27234), goat anti-rabbit IgG H&L (HRP) (Merck Millipore, AP132P), goat anti-mouse IgG H&L cross-adsorbed secondary antibody (Alexa Fluor 488) (Thermo Fisher, A-11001), goat anti-mouse IgG (HRP) (Biosciences, 554002) Goat anti-Rat IgG (HRP) (Thermo Fisher, 31470), goat anti-rabbit (Alexa Fluor 488) (Invitrogen A11034), goat anti-rat IgG H&L cross-adsorbed secondary antibody (Alexa Fluor 488) (Thermo Fisher, A-11006), and goat anti-mouse IgM CFL488 (Santa cruz, sc-395784).
Data were analyzed using SAS 9.4 software (SAS, Inc., Cary, NC, USA). Statistical significance was assessed between two groups using Student’s t test and Dunnett. Results were presented as the mean ± standard error of the mean. p<0.05 was considered to indicate statistical significance.
After entering the lumen of the stomach, H. pylori moves through the mucus layer to increase the accessibility of adhering to gastric epithelial cells (24). Therefore, we investigated the effects of mucin on H. pylori virulence gene expression in this study. The mRNA expression of sabA and babA increased nearly 50-fold compared with that of the non-mucin group (Figure 1A) while the expression of cagL, cagA, vacA, and fucT was upregulated from 4.0- to 17.7-fold. Furthermore, mucin-pretreated H. pylori showed stronger adherence and invasion abilities to AGS cells than the untreated bacteria (Figure 1B). These results indicated that mucin enhanced the virulence of H. pylori.
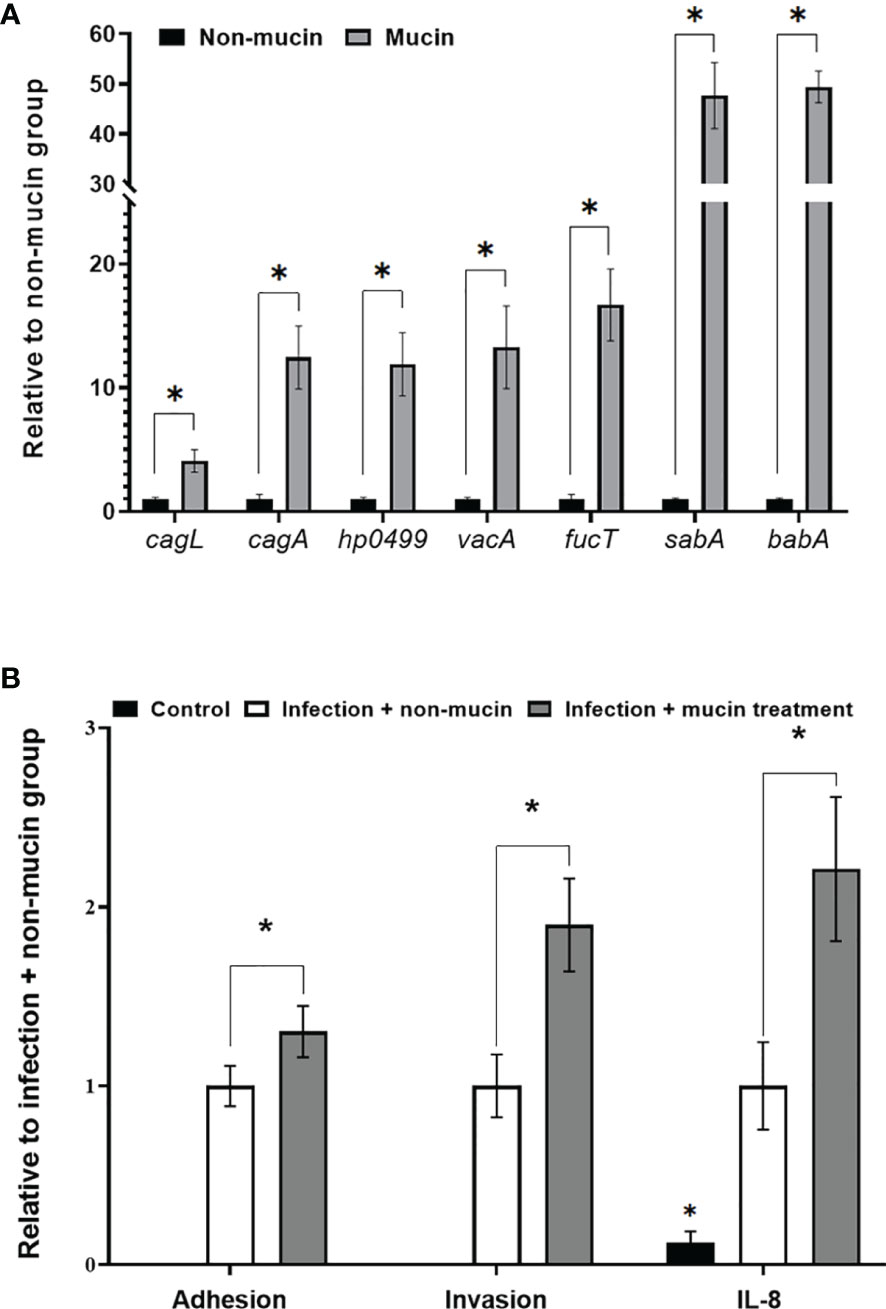
Figure 1 Effects of porcine stomach mucin on (A) virulence gene expression of H. pylori, and (B) the adhesion and invasion abilities of H. pylori to AGS cells and IL-8 levels of H. pylori-infected-AGS cells. H. pylori (5 × 106 CFU/mL) was treated with PBS (non-mucin group) or in 50 mg/mL mucin (pH 7) at 37°C and 5% CO2 for 2 h (mucin group). After cultivation, total mRNAs were isolated and assessed via RT-qPCR to measure the H. pylori virulence gene expression. PBS- or mucin-pretreated H. pylori at a MOI of 50 were co-cultivated with AGS cells at 37°C and 5% CO2 for 6 h AGS cells incubated with 50 mg/mL of mucin in RPMI medium for 6 h were assigned as the control group. The number of H. pylori adhering and invading into AGS cells was calculated by colony counting, and the IL-8 levels in the supernatant were measured by ELISA. *Indicates significant differences compared with the non-mucin group, p < 0.05.
The inhibitory effect LR-JB3-derived CFSs at a MOI of 25 (JB3-25) or 100 (JB3-100) on H. pylori infecting AGS cells through the secretion of unknown cues has been previously demonstrated, including suppressing the expression of sabA, vacA, and fucT in H. pylori, and the H. pylori-induced Lex antigen levels in AGS cells (8). Therefore, to study the effects of mucin on this phenomenon, H. pylori or AGS cells were treated using different CFSs with mucin before co-cultivation. Pretreating H. pylori JB3-25 with mucin suppressed expression of sabA, vacA, and fucT mRNAs (Figure 2A), while expression of vacA mRNA was further decreased by pretreatment with JB3-100 with mucin. The adhesion and invasion abilities of H. pylori to AGS cells as well as H. pylori-induced IL-8 levels were also attenuated by pretreating H. pylori or AGS cells using JB3-25 with mucin (Figures 2B, D). Furthermore, the Lex antigen expression in H. pylori infected-AGS cells was decreased in both pretreatment groups using JB3-25 with mucin (Figures 2C, E). This indicated that the inhibitory effects of JB3-25 remained in the presence of mucin which significantly increased the virulence of H. pylori. Interestingly, pretreating H. pylori using JB3-25 with mucin was able to affect the Lex antigen expression in AGS cells.
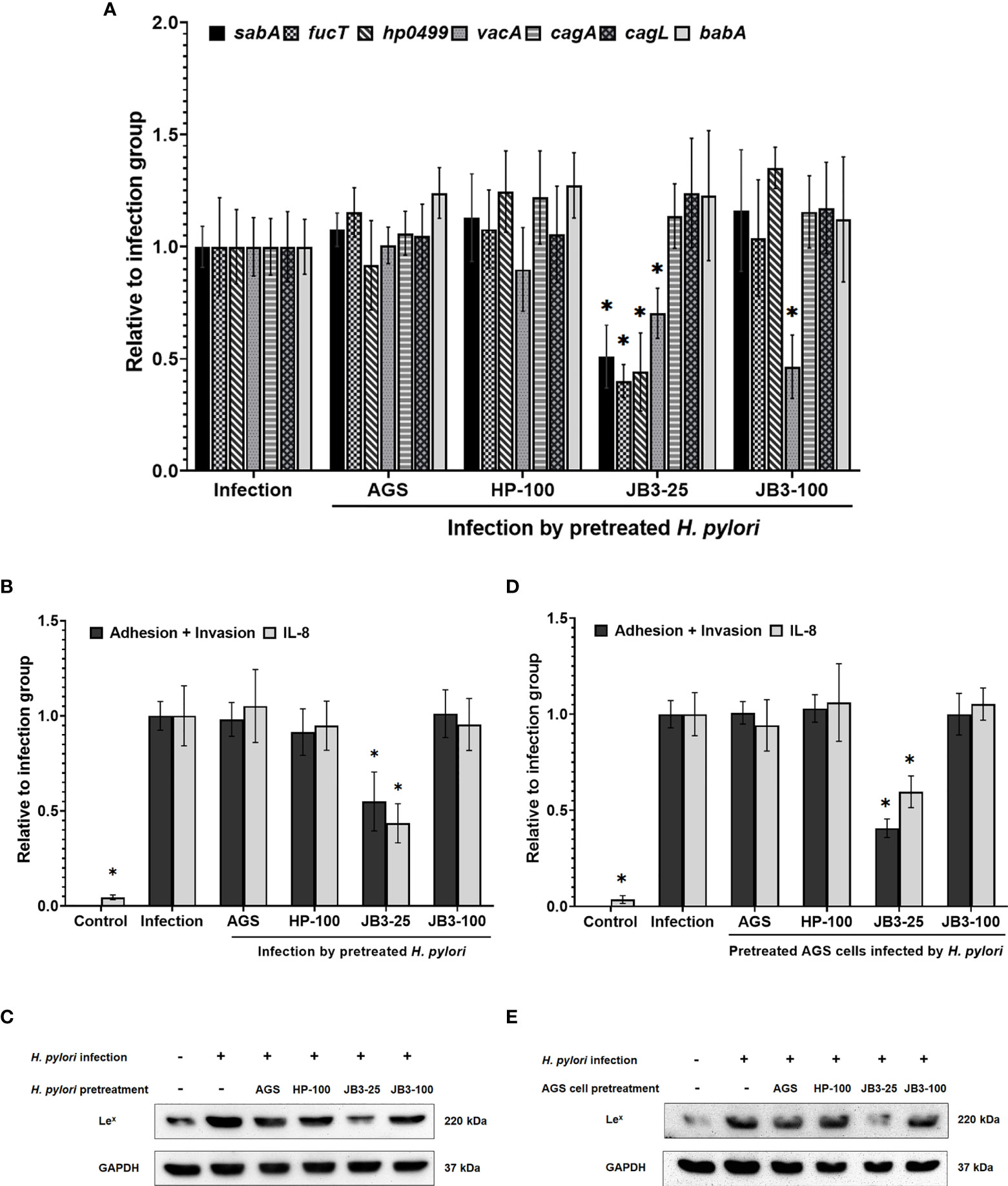
Figure 2 The effects of H. pylori (A–C) or AGS cells (D, E) pretreated using CFSs with mucin on (A) the mRNA expression of sabA, fucT, hp0499, vacA, cagA, cagL, and babA of H. pylori and (B, D) the adhesion and invasion abilities of H. pylori, IL-8 levels and (C, E) Lex antigen of H. pylori-infected-AGS cells after 6 h co-cultivation. CFSs were collected from AGS cells at 5 × 104 cell/mL, H. pylori at 5 × 106 CFU/mL, LR-JB3 at 1.25 × 106 CFU/mL, and LR-JB3 at 5 × 106 CFU/mL growing in RPMI medium were referred to as AGS, HP-100, JB3-25, and JB3-100 respectively. CFSs were mixed with 100 mg/mL of porcine stomach mucin at 1:1 ratio for pretreating H. pylori or AGS cells. After pretreatment, H. pylori and AGS cells were co-cultivated at 37°C and 5% CO2 for 6 h. The AGS cells incubated with 50 mg/mL of mucin in RPMI medium for 6 h were assigned as the control group. The AGS cells infected by mucin-pretreated H. pylori were assigned to the infection group. The number of H. pylori adhering and invading into AGS cells was calculated by colony counting, and the IL-8 levels in the supernatant were measured by ELISA. Total proteins isolated from cell lysis were used for Western blot analysis. * indicates statistically significant differences compared with the infection + non-mucin group, p < 0.05.
To observe the formation of lipid rafts on the H. pylori-infected-AGS cells, GM1 was used as a marker to locate lipid rafts (25). Lex antigen clustered in the lipid raft after H. pylori infection (Figures 3A, B). In both pretreatment groups, the lipid raft clustering as well as Lex antigen co-localization were reduced by JB3-25 with mucin. Mucin treatment also increased the levels of hp0499 mRNA expression by 11.9-fold compared with those treated in the non-mucin group (Figure 1A), whereas pretreatment using JB3-25 with mucin suppressed hp0499 mRNA expression to the same degree as that of fucT and sabA (Figure 2A).
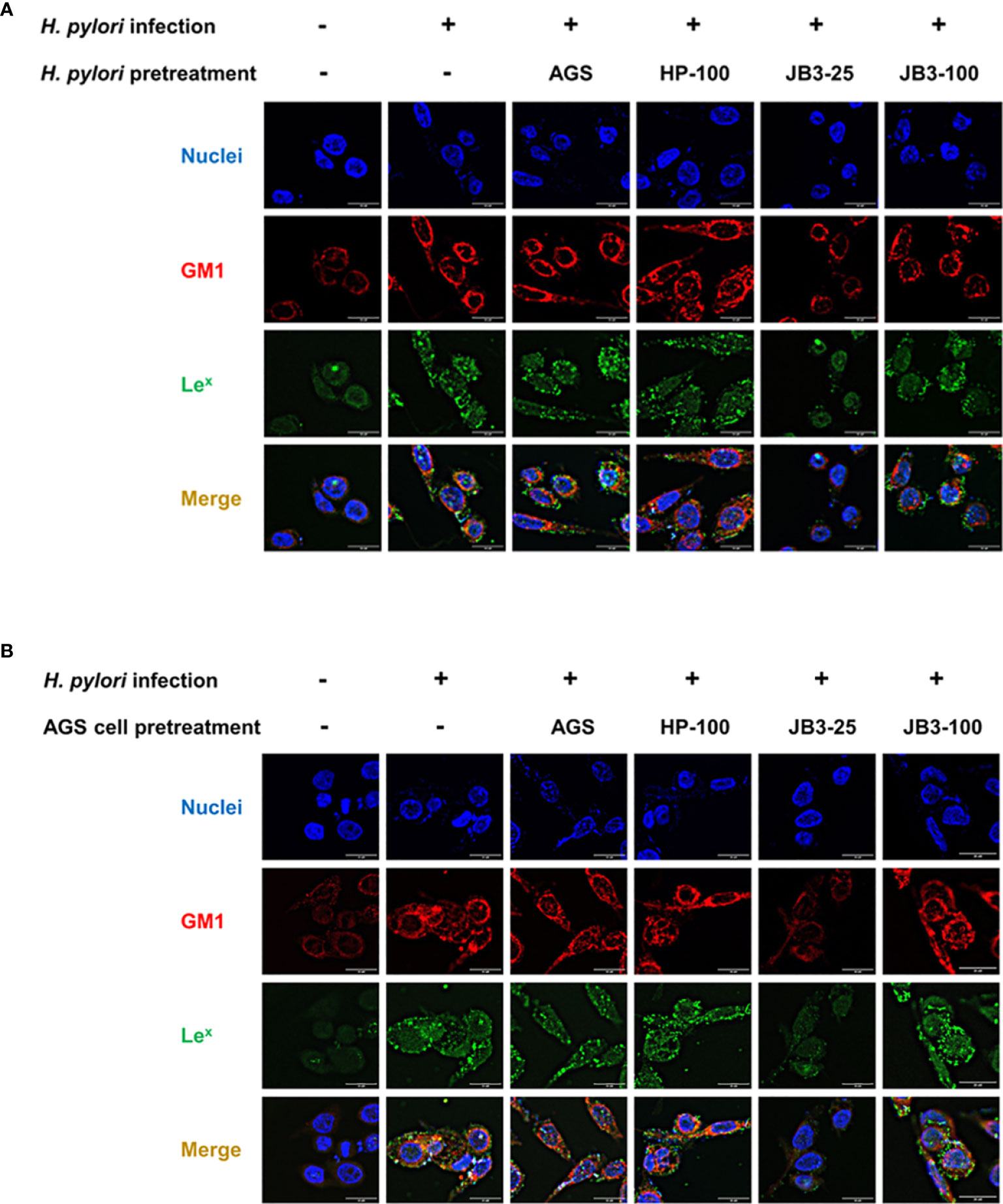
Figure 3 Representative fluorescence microscopy images demonstrated the effect of (A) H. pylori or (B) AGS cells pretreated using CFSs with mucin on the lipid rafts clustering (red) and Lex antigen (green) gathering in the rafts domain in AGS cells after 6 h co-cultivation. CFSs were collected from AGS cells at 5 × 104 cell/mL, H. pylori at 5 × 106 CFU/mL, LR-JB3 at 1.25 × 106 CFU/mL, and LR-JB3 at 5 × 106 CFU/mL growing in RPMI medium were referred to as AGS, HP-100, JB3-25, and JB3-100 respectively. CFSs were mixed with 100 mg/mL of porcine stomach mucin at 1:1 ratio for pretreating H. pylori or AGS cells. After pretreatment, H. pylori and AGS cells were co-cultivated at 37°C and 5% CO2 for 6 h The AGS cells incubated with 50 mg/mL of mucin in RPMI medium for 6 h were assigned as the control group. The AGS cells infected by mucin-pretreated H. pylori were assigned to the infection group. Samples were imaged at a magnification of 40×.
CagA is a major virulence factor of H. pylori that is delivered into host cells via the type IV secretion system (T4SS). CagL is a pilus component of T4SS and interacts with α5β1 integrin to trigger the translocation of CagA into host cells (10, 11). Translocated CagA is then phosphorylated and activates nuclear factor-kappa B (NF-κB) leading to the expression of proinflammatory cytokine IL-8 (26, 27). The expression of both cagA and cagL in H. pylori were induced by mucin but were not affected by JB3-25 with mucin (Figures 1A, 2A). However, the level of H. pylori-induced-IL-8 in AGS cells was suppressed by mucin pretreated with JB3-25 in both pretreatment groups (Figure 2B). Therefore, we then studied the association of α5β1 integrin with lipid rafts. After pretreating of H. pylori or AGS cells by JB3-25 with mucin, both lipid raft clustering and α5β1 integrin co-localization were suppressed (Figures 4A, B, D, E). The expression of α5β1 integrin (Figures 4C, F) along with the amounts of translocated CagA and phosphorylated CagA (Figures 4C, F) were all reduced in both pretreatment groups.
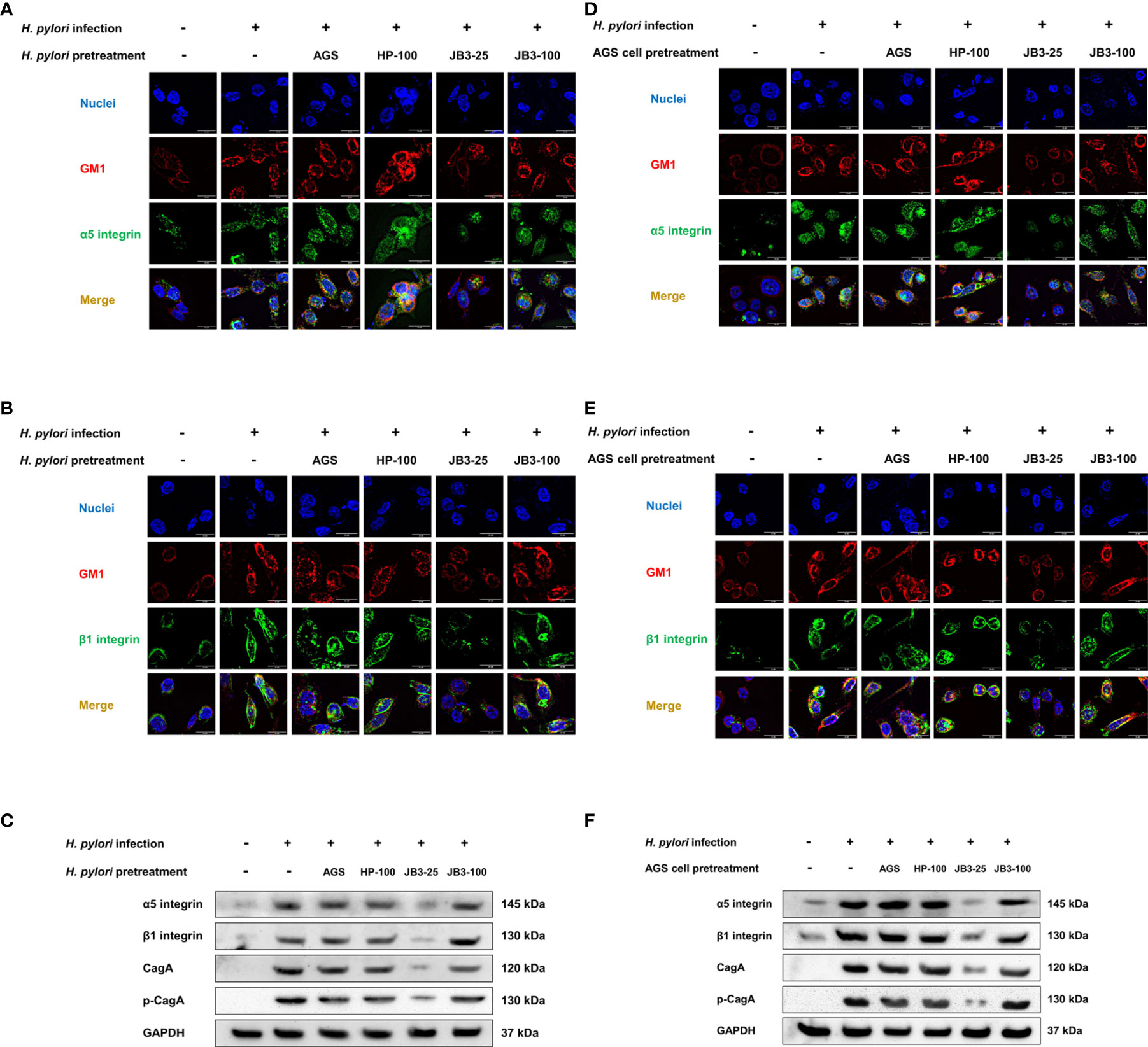
Figure 4 Representative fluorescence microscopy images and Western blot analysis of the effects of H. pylori (A–C) or AGS cells pretreated (D–F) using CFSs with mucin on the lipid rafts clustering (red), (A, C) α5 integrin (green), and (B, D) β1 integrin (green) gathering in the rafts domain, and (C, F) the expression of α5 integrin, β1 integrin, translocated CagA, and phosphorylated CagA in AGS cells after 6 h co-cultivation. CFSs were collected from AGS cells at 5 × 104 cell/mL, H. pylori at 5 × 106 CFU/mL, LR-JB3 at 1.25 × 106 CFU/mL, and LR-JB3 at 5 × 106 CFU/mL growing in RPMI medium were referred to as AGS, HP-100, JB3-25, and JB3-100 respectively. CFSs were mixed with 100 mg/mL of porcine stomach mucin at 1:1 ratio for pretreating H. pylori or AGS cells. After pretreatment, H. pylori and AGS cells were co-cultivated at 37°C and 5% CO2 for 6 h. The AGS cells incubated with 50 mg/mL of mucin in RPMI medium for 6 h were assigned as the control group. The AGS cells infected by mucin-pretreated H. pylori were assigned to the infection group. Samples were imaged at a magnification of 40×.
VacA is a multifunctional toxin that causes vacuolation, apoptosis, and autophagy and that also regulates cell adhesion and inflammatory responses (28). VacA has been demonstrated to be internalized into host cells through an interaction with fatty acid chains of phospholipids, glycosphingolipids, and sphingolipids enriched in lipid raft domains (29–31), which triggers the p38/ATF-2-mediated signaling pathway and induces inflammatory responses (32). Here, VacA was co-localized within the lipid raft after H. pylori infection (Figures 5A, C). In the H. pylori pretreatment group, both pretreatment using either JB3-25 or JB3-100 with mucin reduced the amount of VacA localizing in the lipid raft domain, whereas pretreatment using JB3-100 with mucin did not affect the clustering of lipid rafts (Figure 5A). The levels of p-p38, p-ATF2, and Cox-2 triggered by translocated VacA in H. pylori infected-AGS cells were also decreased by the same pattern (Figure 5B). However, pretreated AGS cells using JB3-100 with mucin did not suppress VacA delivery into the AGS cells, and the levels of p-p38, p-ATF2 and Cox-2 proteins were therefore maintained compared with those in the infection group (Figures 5C, D).
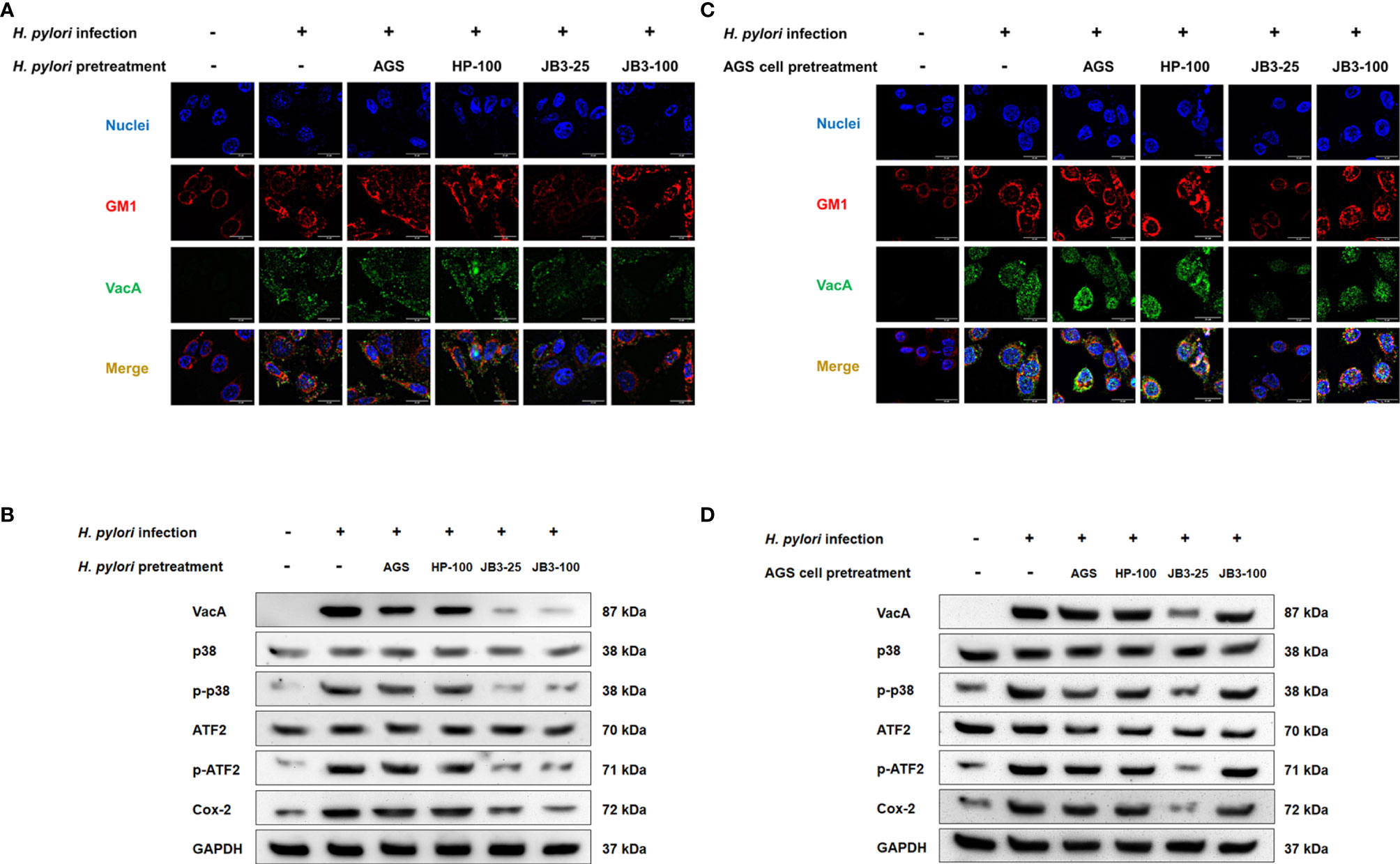
Figure 5 Representative fluorescence microscopy images and Western blot analysis demonstrated the effects of H. pylori (A, B) or AGS cells pretreated (C, D) using CFSs with mucin on the lipid rafts clustering (red), (A, C) intracellular VacA (green) gathering in the rafts domain, and (B, D) the expression of p38, phosphorylated p38, ATF-2, phosphorylated ATF-2, and Cox-2 in AGS cells after 6 h co-cultivation. CFSs were collected from AGS cells at 5 × 104 cell/mL, H. pylori at 5 × 106 CFU/mL, LR-JB3 at 1.25 × 106 CFU/mL, and LR-JB3 at 5 × 106 CFU/mL growing in RPMI medium were referred to as AGS, HP-100, JB3-25, and JB3-100 respectively. CFSs were mixed with 100 mg/mL of porcine stomach mucin at 1:1 ratio for pretreating H. pylori or AGS cells. After pretreatment, H. pylori and AGS cells were co-cultivated at 37°C and 5% CO2 for 6 h The AGS cells incubated with 50 mg/mL of mucin in RPMI medium for 6 h were assigned as the control group. The AGS cells infected by mucin-pretreated H. pylori were assigned to the infection group. Samples were imaged at a magnification of 40×.
The current gold standard in treating H. pylori infection is triple therapy combining a proton pump inhibitor and two antibiotics (33). However, antibiotic therapy has limitations including development of antibiotic-resistant strains (34) and disruption of the balance in intestinal microflora, which can lead to gastrointestinal diseases (35). Considering these side effects of antibiotic therapy, a new therapeutic strategy for H. pylori infection is urgently required. We have previously shown that LR-JB3 has potential in attenuating H. pylori infection in vivo and in vitro (6–8). Therefore, in this study, we further investigated a number of the underlying mechanisms.
Mucus plays a key role as a physical barrier that protects epithelial cells from the acidic gastric juice of the stomach and also from the entrance of pathogens (36). Studies have indicated that mucin promotes the expression of virulence genes in Pseudomonas aeruginosa and Clostridium septicum (37, 38); however, most of the cell-based models do not include mucus in the study. Furthermore, Lactobacilli has been shown to colonize in the mucus layer of the stomach where H. pylori expresses urease to reduce mucus viscosity to facilitate movement toward the gastric epithelial cells (19, 39). The mucus layer is the location where LR-JB3 and H. pylori first encounter each other in the stomach. Therefore, mucins were used in our cell model to offer an alternative view of the interaction between LR-JB3 and H. pylori.
In this study, the presence of mucin dramatically increased the expression of sabA and babA in H. pylori. These genes both encode outer membrane proteins that are involved in the adherence of H. pylori to highly glycosylated mucins via binding to the Lewis b (Leb) antigen and sialyl-Lewis x (SLex) antigen, respectively (40). FucT is an enzyme involved in the Lex biosynthesis of lipopolysaccharide (LPS) O-antigen of H. pylori (2) and fucT mRNA expression was also significantly increased by the mucin. Thus, our data suggests that mucin treatment facilitated H. pylori adhering to gastric epithelial cells in a Lewis antigen-dependent manner. Furthermore, the expression of two well-known virulent factors in H. pylori, cagA and vacA, was also increased. These virulence factors affect various host cell pathways such as cell inflammatory response, vacuolation, and apoptosis (41). Thus, passing through the mucus layer of the stomach increased the ability of H. pylori to infect gastric epithelial cells. However, LR-JB3 inhibited H. pylori colonization, which is agreement with our previous findings (8), even though the virulence of H. pylori was boosted by mucin.
Interestingly, the infection of JB3-25-pretreated H. pylori could also suppress the expression of infection-induced α5β1 integrin and Lex antigen in AGS cells. H. pylori infection is known to trigger the expression of α5β1integrin to enhance CagA translocation, thereby promoting gastric pathogenesis (42). Furthermore, H. pylori infection has been shown to stimulate the CagA-dependent expression of β3 GlcNAc T5 (β3GnT5), which is a GlcNAc transferase essential for the expression of Lewis antigens on the epithelial cell surface (43). Therefore, JB3-25 may regulate the cellular responses of AGS cells through modulating the virulence of H. pylori. The unknown cue in JB3-25 suppressed the ability of H. pylori to induce the expression of α5β1integrin in AGS cells, sequentially reduce the amount of the translocated CagA, and then downregulate the expression of Lewis antigens in AGS cells.
α5β1 integrin and Lex antigen are gathered in the raft domain to promote the adhesion of H. pylori to epithelial cells and initiate pathogenicity (9). The lipid raft plays an important role in the process of H. pylori infection. Pretreating AGS cells by JB3-25 reduced lipid raft clustering, and pretreating H. pylori also decreased its expression of CGAT, which participates in the lipid raft formation of the host cells. An inhibitor of CGAT, amiodarone, could prevent H. pylori from adhering to AGS cells and effectively suppressed the translocation of CagA into AGS cells (9). Therefore, the unknown cue in JB3-25 may contain CGAT inhibitor-like molecules to interfere with the gathering of the lipid raft and further attenuating H. pylori infection.
In our previous study, the levels of 15 amino acid levels were decreased in H. pylori-infected mice (7). Among these, phenylalanine and proline are able to reduce cellular cholesterol levels through repressing the ABCA1 gene that mediates cholesterol synthesis (44). LR-JB3 treatments could restore plasma levels of phenylalanine and proline in infected mice. A cholesterol synthesis inhibitor simvastatin could also reduce CagA translocation via disrupting lipid raft clustering (13). β-cyclodextrin has been reported as an agent that disrupts cholesterol and sphingolipid-rich microdomains to facilitate the disassembly of lipid rafts leading to reduce VacA internalization into Hela cells (45). Thus, the unknown cue in JB3-25 may disrupt the formation of lipid rafts through regulating the cholesterol levels in host cells.
In contrast to JB3-25, JB3-100 specifically reduced the expression of the vacA gene. This result is consistent with our previous finding (8). Here, mucin treatment increased vacA gene expression by 13-fold compared with that in the non-mucin group, although the inhibitory effect of LR-JB3 on vacA expression was still maintained. JB3-100 pretreatment did not affect infection-induced lipid raft formation and the expression of α5β1 integrin and Lex antigen in pretreated AGS cells. Thus, JB3-100 only acted on H. pylori but not on host cells. The major difference between JB3-25 and JB3-100 are the densities of LR-JB3 in the cultures. This finding implies that bacterial density plays an important role in producing metabolites that interact with both H. pylori and AGS cells.
A summary of the possible pathways of CFSs derived from LR-JB3 against H. pylori is shown in Figure 6. Briefly, H. pylori infection triggers the formation of lipid rafts and recruitment of the Lex antigen and α5β1 integrin to the lipid raft domains to facilitate the adherence of H. pylori to epithelial cells; CGAT is also secreted by H. pylori and is delivered to epithelial cells to enhance lipid rafts clustering. H. pylori expresses SabA and FucT to increase Lewis antigen-dependent adherence. CagL the adhesion subunit of T4SS binds to α5β1 integrin for T4SS-dependent CagA delivery causing the expression of IL-8. T4SS also enhances the adherence of H. pylori to AGS cells. Furthermore, the translocated VacA is also gathered in the lipid raft and can stimulate the p38/ATF-2-mediated signal pathway to induce inflammatory responses. The unknown cue derived from JB3-25 (blue triangle) can interfere with lipid raft formation, reducing CGAT expression in H. pylori, and suppressing the expressions of the Lex antigen and α5β1 integrin in AGS cells. Therefore, Lewis antigen-dependent adherence, T4SS-mediated cell contact, and lipid raft-mediated entry of VacA are all attenuated. Furthermore, another unknown cue (yellow triangle) from JB3-25 and JB3-100 inhibits the internalization of VacA resulting in a reduced Cox-2 level mediated by the p38/ATF-2-mediated signal pathway. The reduced intracellular levels of VacA are caused by suppressed expression in H. pylori using LR-JB3 treatment. The mechanism of unknown cues from LR-JB3 that interfere with the lipid raft clustering on H. pylori-infected-AGS cells requires further investigation.
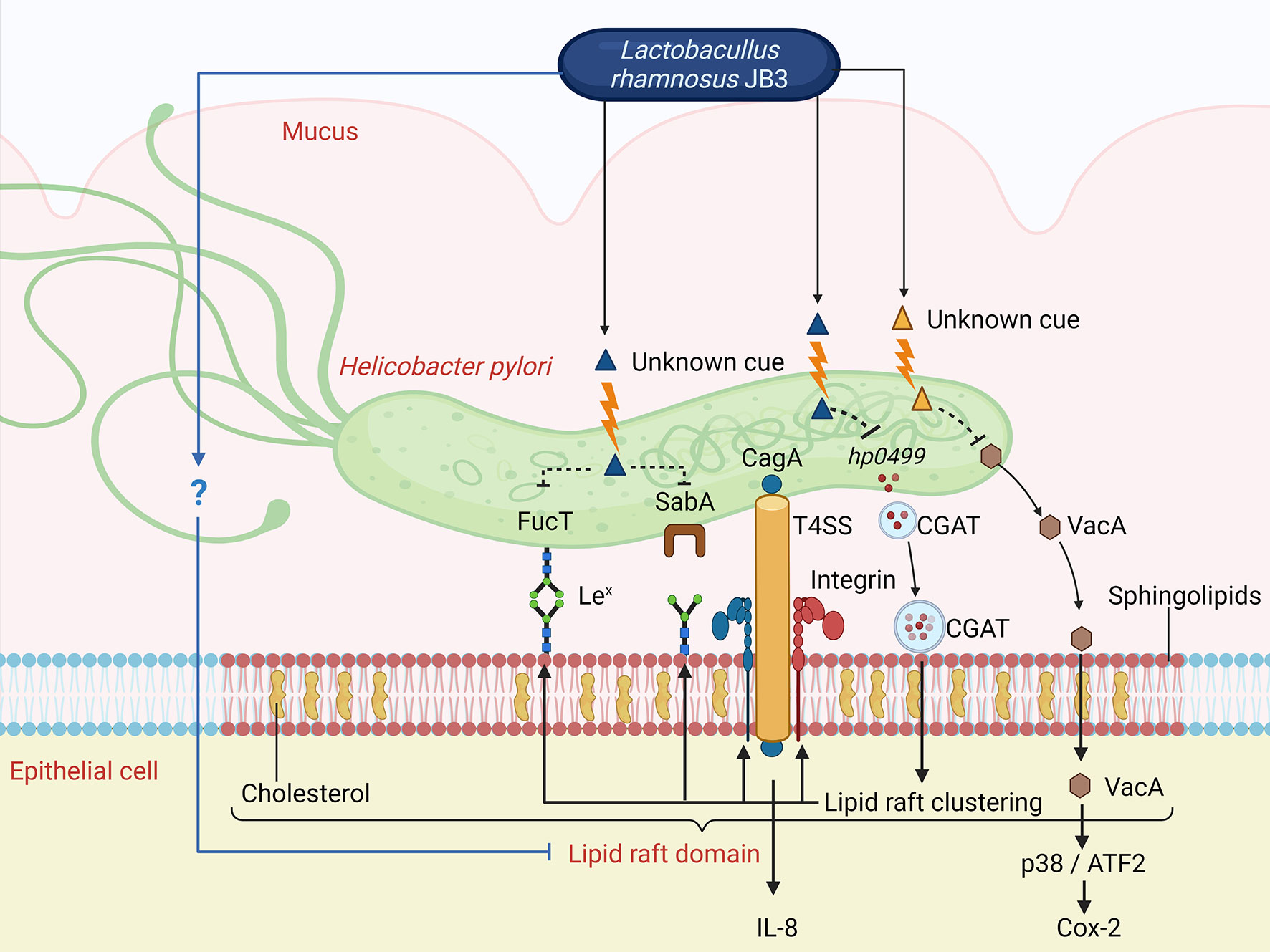
Figure 6 The possible pathways of CFSs derived from LR-JB3 against H. pylori infection (for further details see in the text). Lex, Lewis x antigen; T4SS, type IV secretion system; ATF2, Activating transcription factor 2; CGAT, Cholesteryl α-D-glucoside 6-acyltransferase. The figure was created with BioRender.com.
To the best of our knowledge, this is the first study to show that L. rhamnosus can affect H. pylori infection through mediating lipid raft formation of the host cells. The unknown cues secreted from LR-JB3 are valuable not only for treating H. pylori infection but also for treating diseases mediated by lipid raft signaling, such as cancers and neurodegenerative and aging-associated diseases (46).
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Y-MH designed this study. ADD and C-HS performed experiments; Y-MH and ADD wrote the paper. All authors approved this final manuscript. All authors have read and agreed to the published version of the manuscript.
This research was funded by Ministry of Science and Technology (Taiwan) grant number MOST109-2320-B-039-009-MY3 and China Medical University Hospital grant number DMR-108-141.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
This research was supported by grants from the Ministry of Science and Technology (Taiwan) and by China Medical University Hospital, Taiwan. We thank enago for their English language editing.
1. Wu JY, Lee YC, Graham DY. The Eradication of Helicobacter Pylori to Prevent Gastric Cancer: A Critical Appraisal. Expert Rev Gastroenterol Hepatol (2019) 13(1):17–24. doi: 10.1080/17474124.2019.1542299
2. Oleastro M, Ménard A. The Role of Helicobacter Pylori Outer Membrane Proteins in Adherence and Pathogenesis. Biology (2013) 2(3):1110–34. doi: 10.3390/biology2031110
3. Zou Y, Qian X, Liu X, Song Y, Song C, Wu S, et al. The Effect of Antibiotic Resistance on Helicobacter Pylori Eradication Efficacy: A Systematic Review and Meta-Analysis. Helicobacter (2020) 25(4):e12714. doi: 10.1111/hel.12714
4. Ramirez J, Guarner F, Bustos Fernandez L, Maruy A, Sdepanian VL, Cohen H. Antibiotics as Major Disruptors of Gut Microbiota. Front Cell Infect Microbiol (2020) 10:731. doi: 10.3389/fcimb.2020.572912
5. Kamiya S, Yonezawa H, Osaki T. Role of Probiotics in Eradication Therapy for Helicobacter Pylori Infection. Adv Exp Med Biol (2019) 1149:243–55. doi: 10.1007/5584_2019_369
6. Chen ME, Su CH, Yang JS, Lu CC, Hou YC, Wu JB, et al. Baicalin, Baicalein, and Lactobacillus Rhamnosus JB3 Alleviated Helicobacter Pylori Infections In Vitro and In Vivo. J Food Sci (2018) 83(12):3118–25. doi: 10.1111/1750-3841.14372
7. Lin C-C, Huang W-C, Su C-H, Lin W-D, Wu W-T, Yu B, et al. Effects of Multi-Strain Probiotics on Immune Responses and Metabolic Balance in Helicobacter Pylori-Infected Mice. Nutrients (2020) 12(8):2476. doi: 10.3390/nu12082476
8. Do AD, Chang CC, Su CH, Hsu YM. Lactobacillus Rhamnosus JB3 Inhibits Helicobacter Pylori Infection Through Multiple Molecular Actions. Helicobacter (2021) 26:e12806. doi: 10.1111/hel.12806
9. Jan HM, Chen YC, Yang TC, Ong LL, Chang CC, Muthusamy S, et al. Cholesteryl Alpha-D-Glucoside 6-Acyltransferase Enhances the Adhesion of Helicobacter Pylori to Gastric Epithelium. Commun Biol (2020) 3(1):120. doi: 10.1038/s42003-020-0855-y
10. Murata-Kamiya N, Kikuchi K, Hayashi T, Higashi H, Hatakeyama M. Helicobacter Pylori Exploits Host Membrane Phosphatidylserine for Delivery, Localization, and Pathophysiological Action of the CagA Oncoprotein. Cell Host Microbe (2010) 7(5):399–411. doi: 10.1016/j.chom.2010.04.005
11. Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter Exploits Integrin for Type IV Secretion and Kinase Activation. Nature (2007) 449(7164):862–6. doi: 10.1038/nature06187
12. Lu DY, Chen HC, Yang MS, Hsu YM, Lin HJ, Tang CH, et al. Ceramide and Toll-Like Receptor 4 Are Mobilized Into Membrane Rafts in Response to Helicobacter Pylori Infection in Gastric Epithelial Cells. Infect Immun (2012) 80(5):1823–33. doi: 10.1128/IAI.05856-11
13. Lin C-J, Liao W-C, Lin H-J, Hsu Y-M, Lin C-L, Chen Y-A, et al. Statins Attenuate Helicobacter Pylori CagA Translocation and Reduce Incidence of Gastric Cancer: In Vitro and Population-Based Case-Control Studies. PloS One (2016) 11(1):e0146432. doi: 10.1371/journal.pone.0146432
14. Simons K, Ikonen E. Functional Rafts in Cell Membranes. Nature (1997) 387(6633):569–72. doi: 10.1038/42408
15. Wang M, Hajishengallis G. Lipid Raft-Dependent Uptake, Signalling and Intracellular Fate of Porphyromonas Gingivalis in Mouse Macrophages. Cell Microbiol (2008) 10(10):2029–42. doi: 10.1111/j.1462-5822.2008.01185.x
16. Frisan T. Bacterial Genotoxins: The Long Journey to the Nucleus of Mammalian Cells. Biochim Biophys Acta -Biomembranes (2016) 1858(3):567–75. doi: 10.1016/j.bbamem.2015.08.016
17. Chen Y, Huang K, Chen L-K, Wu H-Y, Hsu C-Y, Tsai Y-S, et al. Membrane Cholesterol Is Crucial for Clostridium Difficile Surface Layer Protein Binding and Triggering Inflammasome Activation. Front Immunol (2020) 11. doi: 10.3389/fimmu.2020.01675
18. Hansson GC. Role of Mucus Layers in Gut Infection and Inflammation. Curr Opin Microbiol (2012) 15(1):57–62. doi: 10.1016/j.mib.2011.11.002
19. Huang Y, Wang Q-l, Cheng D-d, Xu W-t, Lu N-h. Adhesion and Invasion of Gastric Mucosa Epithelial Cells by Helicobacter Pylori. Front Cell Infect Microbiol (2016) 6:159. doi: 10.3389/fcimb.2016.00159
20. Aristoteli LP, Willcox MD. Mucin Degradation Mechanisms by Distinct Pseudomonas Aeruginosa Isolates In Vitro. Infect Immun (2003) 71(10):5565–75. doi: 10.1128/IAI.71.10.5565-5575.2003
21. Livak KJ, Schmittgen TD. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods (2001) 25(4):402–8. doi: 10.1006/meth.2001.1262
22. Li L, Kelly LK, Ayub K, Graham DY, Go MF. Genotypes of Helicobacter Pylori Obtained From Gastric Ulcer Patients Taking or Not Taking NSAIDs. Am J Gastroenterol (1999) 94(6):1502–7. doi: 10.1111/j.1572-0241.1999.01133.x
23. Lai C-H, Kuo C-H, Chen P-Y, Poon S-K, Chang C-S, Wang W-C. Association of Antibiotic Resistance and Higher Internalization Activity in Resistant Helicobacter Pylori Isolates. J Antimicrob Chemother (2006) 57(3):466–71. doi: 10.1093/jac/dki479
24. Celli JP, Turner BS, Afdhal NH, Keates S, Ghiran I, Kelly CP, et al. Helicobacter Pylori Moves Through Mucus by Reducing Mucin Viscoelasticity. Proc Natl Acad Sci (2009) 106(34):14321–6. doi: 10.1073/pnas.0903438106
25. Moreno-Altamirano MMB, Aguilar-Carmona I, Sánchez-García FJ. Expression of GM1, a Marker of Lipid Rafts, Defines Two Subsets of Human Monocytes With Differential Endocytic Capacity and Lipopolysaccharide Responsiveness. Immunology (2007) 120(4):536–43. doi: 10.1111/j.1365-2567.2006.02531.x
26. Backert S, Tegtmeyer N. Type IV Secretion and Signal Transduction of Helicobacter Pylori CagA Through Interactions With Host Cell Receptors. Toxins (2017) 9(4):115. doi: 10.3390/toxins9040115
27. Fazeli Z, Alebouyeh M, Tavirani MR, Azimirad M, Yadegar A. Helicobacter Pylori CagA Induced Interleukin-8 Secretion in Gastric Epithelial Cells. Gastroenterol Hepatol bed to bench (2016) 9(Suppl1):S42. doi: 10.22037/ghfbb.v0i0.1001
28. Nakayama M, Hisatsune J, Yamasaki E, Nishi Y, Wada A, Kurazono H, et al. Clustering of Helicobacter Pylori VacA in Lipid Rafts, Mediated by Its Receptor, Receptor-Like Protein Tyrosine Phosphatase β, Is Required for Intoxication in AZ-521 Cells. Infect Immun (2006) 74(12):6571–80. doi: 10.1128/IAI.00356-06
29. Molinari M, Galli C, de Bernard M, Norais N, Ruysschaert J-M, Rappuoli R, et al. The Acid Activation of Helicobacter Pylori Toxin VacA: Structural and Membrane Binding Studies. Biochem Biophys Res Commun (1998) 248(2):334–40. doi: 10.1006/bbrc.1998.8808
30. Roche N, Ilver D, Ångström J, Barone S, Telford JL, Teneberg S. Human Gastric Glycosphingolipids Recognized by Helicobacter Pylori Vacuolating Cytotoxin VacA. Microbes infection (2007) 9(5):605–14. doi: 10.1016/j.micinf.2007.01.023
31. Rolando M, Buchrieser C. A Comprehensive Review on the Manipulation of the Sphingolipid Pathway by Pathogenic Bacteria. Front Cell Dev Biol (2019) 7:168. doi: 10.3389/fcell.2019.00168
32. Nakayama M, Kimura M, Wada A, Yahiro K, Ogushi K, Niidome T, et al. Helicobacter Pylori VacA Activates the P38/Activating Transcription Factor 2-Mediated Signal Pathway in AZ-521 Cells. J Biol Chem (2004) 279(8):7024–8. doi: 10.1074/jbc.M308898200
33. Malfertheiner P, Megraud F, O'morain C, Gisbert J, Kuipers E, Axon A, et al. Management of Helicobacter Pylori Infection—the Maastricht V/Florence Consensus Report. Gut (2017) 66(1):6–30. doi: 10.1136/gutjnl-2016-312288
34. Alba C, Blanco A, Alarcón T. Antibiotic Resistance in Helicobacter Pylori. Curr Opin Infect Dis (2017) 30(5):489–97. doi: 10.1097/QCO.0000000000000396
35. Vangay P, Ward T, Gerber JS, Knights D. Antibiotics, Pediatric Dysbiosis, and Disease. Cell Host Microbe (2015) 17(5):553–64. doi: 10.1016/j.chom.2015.04.006
36. Hessey S, Spencer J, Wyatt J, Sobala G, Rathbone B, Axon A, et al. Bacterial Adhesion and Disease Activity in Helicobacter Associated Chronic Gastritis. Gut (1990) 31(2):134–8. doi: 10.1136/gut.31.2.134
37. Lory S, Jin S, Boyd JM, Rakeman JL, Bergman P. Differential Gene Expression by Pseudomonas Aeruginosa During Interaction With Respiratory Mucus. Am J Respir Crit Care Med (1996) 154(4):S183. JAjor, medicine cc. doi: 10.1164/ajrccm/154.4_Pt_2.S183
38. Macfarlane S, Hopkins MJ, Macfarlane GT. Toxin Synthesis and Mucin Breakdown are Related to Swarming Phenomenon in Clostridium Septicum. Infection Immun (2001) 69(2):1120–6. doi: 10.1128/IAI.69.2.1120-1126.2001
39. Van Tassell ML, Miller MJ. Lactobacillus Adhesion to Mucus. Nutrients (2011) 3(5):613–36. doi: 10.3390/nu3050613
40. Baj J, Forma A, Sitarz M, Portincasa P, Garruti G, Krasowska D, et al. Helicobacter Pylori Virulence Factors-Mechanisms of Bacterial Pathogenicity in the Gastric Microenvironment. Cells (2020) 10(1). doi: 10.3390/cells10010027
41. Kao CY, Sheu BS, Wu JJ. Helicobacter Pylori Infection: An Overview of Bacterial Virulence Factors and Pathogenesis. BioMed J (2016) 39(1):14–23. doi: 10.1016/j.bj.2015.06.002
42. Yeh Y-C, Cheng H-C, Yang H-B, Chang W-L, Sheu B-S. H. Pylori CagL-Y58/E59 Prime Higher Integrin α5β1 in Adverse pH Condition to Enhance Hypochlorhydria Vicious Cycle for Gastric Carcinogenesis. PloS One (2013) 8(8):e72735. doi: 10.1371/journal.pone.0072735
43. Marcos NT, Magalhães A, Ferreira B, Oliveira MJ, Carvalho AS, Mendes N, et al. Helicobacter Pylori Induces β3gnt5 in Human Gastric Cell Lines, Modulating Expression of the SabA Ligand Sialyl–Lewis X. J Clin Invest (2008) 118(6):2325–36. doi: 10.1172/JCI34324
44. Banno A, Wang J, Okada K, Mori R, Mijiti M, Nagaoka S. Identification of a Novel Cholesterol-Lowering Dipeptide, Phenylalanine-Proline (FP), and Its Down-Regulation of Intestinal ABCA1 in Hypercholesterolemic Rats and Caco-2 Cells. Sci Rep (2019) 9(1):1–12. doi: 10.1038/s41598-019-56031-8
45. Patel HK, Willhite DC, Patel RM, Ye D, Williams CL, Torres EM, et al. Plasma Membrane Cholesterol Modulates Cellular Vacuolation Induced by the Helicobacter Pylori Vacuolating Cytotoxin. Infection Immun (2002) 70(8):4112–23. doi: 10.1128/IAI.70.8.4112-4123.2002
Keywords: Helicobacter pylori, Lactobacillus rhamnosus, mucus, lipid raft, Lewis x antigen, α5β1 integrin
Citation: Do AD, Su C-H and Hsu Y-M (2022) Antagonistic Activities of Lactobacillus rhamnosus JB3 Against Helicobacter pylori Infection Through Lipid Raft Formation. Front. Immunol. 12:796177. doi: 10.3389/fimmu.2021.796177
Received: 16 October 2021; Accepted: 24 December 2021;
Published: 14 January 2022.
Edited by:
Chih-Ho Lai, Chang Gung University, TaiwanReviewed by:
Abbas Yadegar, Shahid Beheshti University of Medical Sciences, IranCopyright © 2022 Do, Su and Hsu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuan-Man Hsu, eXVhbm1oQHRodS5lZHUudHc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.