 Xiaoling Li
Xiaoling Li Jie Gao
Jie Gao Jinhui Tao
Jinhui Tao- Department of Rheumatology and Immunology, The First Affiliated Hospital of University of Science and Technology of China (USTC), Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China
Gout flares require monosodium urate (MSU) to activate the NLRP3 inflammasome and secrete sufficient IL-1β. However, MSU alone is not sufficient to cause a flare. This is supported by the evidence that most patients with hyperuricemia do not develop gout throughout their lives. Recent studies have shown that, besides MSU, various purine metabolites, including adenosine triphosphate, adenosine diphosphate, and adenosine bind to different purine receptors for regulating IL-1β secretion implicated in the pathogenesis of gout flares. Purine metabolites such as adenosine triphosphate mainly activate the NLRP3 inflammasome through P2X ion channel receptors, which stimulates IL-1β secretion and induces gout flares, while some purine metabolites such as adenosine diphosphate and adenosine mainly act on the G protein-coupled receptors exerting pro-inflammatory or anti-inflammatory effects to regulate the onset and resolution of a gout flare. Given that the purine signaling pathway exerts different regulatory effects on inflammation and that, during the inflammatory process of a gout flare, an altered expression of purine metabolites and their receptors was observed in response to the changes in the internal environment. Thus, the purine signaling pathway is involved in regulating gout flare and resolution. This study was conducted to review and elucidate the role of various purine metabolites and purinergic receptors during the process.
Introduction
Gout is an inflammatory disease that manifests clinically as redness, swelling, and pain in the joints. Research involving multiple ethnicities has reported that the prevalence of gout in adults ranges between 0.68% and 3.90% (1). With the development of the economy, the incidence of gout is increasing every year, while the social danger of the disease is becoming a growing concern among scholars. Hyperuricemia underlies the pathogenesis of gout, with supersaturated uric acid being deposited in the joint cavity to form monosodium urate (MSU). The MSU stimulates abnormal IL-1β secretion to cause gout flares, which is achieved through recognition by Toll-like receptors (TLRs) or NOD-like receptors (NLRs) to activate the innate immune system (1). The activation of NLRP3 inflammasome releases large amounts of IL-1β is a central process of MSU-mediated gout flares (2). However, clinical studies have shown that most patients with hyperuricemia do not experience gout flares (3). Additionally, the presence of triggering factors, including alcohol abuse, overeating, and exertion, is often required for a gout flare, which suggests that MSU alone is not sufficient to induce gout flares, and there is a need for other causative signals to act in vivo for gout flares.
The activation of NLRP3 inflammasome and subsequent induction of the release of IL-1β exerts a central role in the initiation of gout flares. Besides MSU, extracellular DAMPs and PAMPs, including bacteria, viruses, and adenosine triphosphate (ATP), are known to be responsible for the activation of the NLRP3 inflammasome. Among them, ATP-mediated activation of the P2X7R-NLRP3 signaling pathway has gradually been recognized in the pathogenesis of gout (4, 5). Following tissue necrosis caused by MSU deposition, local ATP arising from the cellular release is increased, which can activate the P2X7 receptor leading to intra-and extracellular ion flow, activation of the NLRP3 inflammasome, and secretion of IL-1β, ultimately triggering a gout flare. Furthermore, gene polymorphisms in the P2X7 receptor have been shown to regulate IL-1β secretion in vivo. Several studies have demonstrated that a gain-in-function of the P2X7 receptor is associated with an elevated rate of gout flares (6–8). Moreover, colchicine, a classical drug for treating acute gout flares, was found to inhibit the activation of ATP-induced P2X7 receptor (9), which could be the mechanism of action against gout. This indirectly confirmed the role of ATP and P2X7 receptor in gout resolution. Therefore, as an important messenger of the purinergic pathway, ATP is considered a second pathogenic signal for gout.
Besides ATP, other purinergic metabolites, such as adenosine diphosphate (ADP) and adenosine, can bind to the corresponding purinergic receptors affecting IL-1β secretion in gout. However, the purine signaling pathway exerts different regulatory effects on inflammation. For example, in contrast to ATP and ADP, adenosine exerts an inhibitory effect on inflammation in vivo (10). In addition, the expression of purine metabolites and their receptors varies in response to changes in the inflammatory internal environment during a gout flare. MSU was found to regulate the expression of the P2X7 receptor (11). Thus, the interaction between purinergic metabolites and purinergic receptors mediates the intracellular signaling communication, which jointly regulates the onset and resolution of gout flares. In this article, we have reviewed the possible regulatory role of the purinergic signaling pathway in gout flares to understand and combat gout in a more precise manner.
Classification and Function of Purinergic Receptors
The existence of purinergic receptors in the body was first proposed by Burnstock (12) in 1972. Since then, an increasing number of purinergic receptors have been identified, and researchers have gradually noticed the role of purinergic signaling pathways in the biological effects of diseases. The purinergic receptors, also known as P receptors, are classified as P1 and P2 receptors based on their ligands; the former binds to adenosine while the latter binds to ATP, ADP, uridine triphosphate (UTP), and uridine diphosphate (UDP). The P1 receptors are G protein-coupled receptors, which can be classified into four subtypes (A1, A2A, A2B, and A3) depending on the structure of the G proteins and intracellular conductance. The A2A and A2B receptors preferentially couple to Gs and exert their effects via the AC-cAMP-PKA pathway while the A1 and A3 subtypes couple to Gi and inhibit the AC activity (13). The P2 receptors, on the other hand, are divided into two major groups, P2X and P2Y receptors, depending on the signal transduction pathway. The P2X receptors are ligand-gated ion channel receptors that recognize ATP. Upon activation, the pore channels open and allow the flow of extracellular Na+, Ca2+, K+, and the entry of macromolecules. Seven P2X receptors have been identified so far (P2X1–7 receptors). The P2Y receptors are G protein-coupled receptors that primarily recognize ATP, UTP, and its metabolites. They can be further classified into P2Y1-like receptors (coupled to Gq, including P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11 receptors) and P2Y12-like receptors (coupled to Gi, including P2Y12, P2Y13, and P2Y14 receptors) based on the intracellular signaling proteins coupled to the G proteins (14). The former receptors exert their effect via the PLC-IP3/DAG-PKC pathway while the latter inhibits the activity of AC, thus, producing different biological effects (Supplementary Table 1).
The purinergic receptors are distributed in almost all cell types, and upon recognition of the corresponding ligands, they mediate a variety of biological effects and enable the development of many diseases, including cardiovascular (15, 16), metabolic (17, 18), neurological diseases (19), and cancer (20, 21). In inflammatory diseases, purinergic receptors play an important regulatory role and participate in immune- (22), infectious- (23), and neurotransmission (24)-mediated inflammatory responses to maintain homeostasis in the organism. In the regulation of inflammation, the role of different purine signals varies. For example, the activation of P2X7 receptors promotes the inflammatory response, whereas the activation of P1 receptors inhibits it. Therefore, the P receptors can coordinate the initiation, persistence, and resolution of the inflammatory response (25). Gout is an inflammatory disease caused by abnormal purine metabolism. The purine metabolites exert complex regulatory effects on the inflammatory response, influencing the onset and resolution of gout to varying degrees.
Effect of Purinergic Signaling Pathway on Gout Flares
Gout flares are a result of a complex acute inflammatory response, where the macrophages recognize MSU deposited on the joint surface, leading to the secretion of IL-1β. This is thought to be the central process in the initiation of gout flares (26). Immediately after the release of IL-1β, the inflammatory cascade response induces neutrophil accumulation in the joints, thus, exacerbating gout flare (27). Therefore, overproduction of IL-1β is a central link in the MSU-induced gouty joint inflammation. It is essential to explore the mechanism of activation of IL-1β for investigating the pathogenesis of gout flares, and it is found that the purinergic signaling pathway is involved in the regulation of IL-1β secretion in gout.
Activation of the P2X Receptor Signaling Pathway Leads to the Promotion of Gout Flares
The P2X receptors are ATP-gated ion channel receptors, and of these P2X receptors, the P2X7 receptor, initially defined as the P2Z receptor, possesses a unique sequence and rich function despite being the latest identified (28). Activation of the P2X7 receptor by ATP opens ion channels causing Ca2+ influx and K+ outflux to activate the NLRP3 inflammasome in the innate immune system, where Ca2+ may represent the second messenger of inflammasome activation (29). Also, the P2X7 receptor has a unique structure and function, which does not only form ion channels but also forms the pore channels, allowing the passage of up to 900 Da molecules upon continuous activation by high concentrations of ATP. The formation of such pores appears to be required to activate the NLRP3 inflammasome (30, 31). Upon activation, the NLRP3 proteins interact with apoptosis-associated speck-like protein containing a CARD (ASC), which then recruits the pro-caspase-1 forming the NLRP3 inflammasome, which subsequently undergoes cleavage to form active caspase-1. This is further processed, leading to the secretion of mature IL-1β and IL-18 that exert inflammatory effects (32). Following pharmacological inhibition of the P2X7 receptor, the level of ATP-induced IL-1β release is reduced in inflammatory cells (33). A similar reduction was observed in the levels of the P2X7 receptor, ASC, or NLRP3-deficient mouse macrophages. These findings suggested that the activation of the ATP-mediated P2X7 receptor plays a crucial role in promoting IL-1β secretion in inflammatory cells (34). Currently, it is recognized that the P2X7 receptor, among the family of P2XRs, is the most relevant in the inflammatory response.
Gout is an acute inflammatory disease based on hyperuricemia. Predisposing factors, including alcohol abuse, overeating, cold, and late nights, are required to initiate a gout flare. We evaluated the pathogenic signals embedded in these factors associated with gout flares and found that under these conditions, ATP fluctuations were observed in vivo. This, for example, included mechanical stimulation during strenuous exercise promotes ATP release (35), and the adaptive febrile response of the body during cold increases ATP (36). Dramatic changes in ATP levels can activate the P2X7R-NLRP3 signaling pathway and promote the IL-1β secretion associated with gout flares. Thus, ATP is considered to be involved in gout flares. A study by Tao et al. observed no difference in the IL-1β concentrations in peripheral blood leukocyte cultures from gout and hyperuricemia patients when stimulated with MSU alone, but upon stimulation with MSU and ATP together, IL-1β concentrations were found to be significantly higher in gout patients than that of the hyperuricemia patients (6). Thus, it is suggested that ATP is the second pathogenic signal for gout besides MSU. Importantly, the ability of the P2X7 receptor to promote IL-1β secretion upon activation may be a major determinant of gout flares, which is influenced by the gene polymorphisms of the P2X7 receptor. Studies have confirmed that gout patients possess single nucleotide polymorphism in functionally enhanced P2X7 receptor compared to hyperuricemia patients (37), thus establishing an essential role of P2X7 receptor in gout flares.
In hyperuricemic patients, synergistic action between MSU and ATP is required to fully activate the NLRP3 inflammasome, which causes sufficient secretion of IL-1β, leading to the onset of gout flares. Thus, it can be argued that as a receptor for ATP, the P2X7 receptor is an important regulator in determining gout flares, explaining why most hyperuricemic patients do not develop gouty arthritis throughout their lives.
Besides the P2X7 receptor, the P2X4 receptor can also be activated by ATP, causing an inflammatory response, which is functionally similar to the P2X7 receptor. Upon activation, it forms large-conductance pores in the cell membrane facilitating the ion flow, which subsequently activates the NLRP3 inflammasome. This further enhances the body’s inflammatory response through its synergy with the P2X7 receptor (38).
Different Roles of Activated P2Y Receptor Signaling Pathway in Gout Flares
P2Y receptors belong to the G protein-coupled receptor family and are further divided into P2Y1-like receptors (coupled to Gq) and P2Y12-like receptors (coupled to Gi), which exert regulatory effects on inflammation by affecting the activity of PLC and AC, respectively (39). Upon recognizing the corresponding nucleotide, P2Y1-like receptors coupled to Gq activate the PLC-IP3/DAG-PKC pathway, promoting Ca2+ mobilization and inflammatory response. Also, the P2Y1-like receptors can activate AC; for example, the P2Y11 receptor promotes the activation of the AC-cAMP-PKA pathway to initiate the inflammatory response (40). The P2Y12-like receptors coupled to Gi can exert an inhibitory effect on AC, further reducing the conversion of dephosphorylation of ATP to cAMP, thus, inhibiting the cAMP-PKA pathway-mediated inflammatory response.
The P2Y1-like receptors, including the P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11 receptors, can promote inflammation either through the PLC-IP3/DAG-PKC pathway or through the activation of AC. Among these P2Y1-like receptors, the P2Y2 receptor may be closely associated with gout flares. Besides its inflammatory function, the P2Y2 receptor can activate the NLRP3 inflammasome by stimulating the action of ATP release to further activate the P2X7 receptor (41).
The P2Y2 receptors are expressed on various cells, including macrophages, lymphocytes, neutrophils, and eosinophils, and are activated by multiple nucleotides such as ATP and UTP to participate in immune regulation and inflammatory responses. The activated P2Y2 receptors exhibit upregulated expression of inflammatory cytokines and chemokines, while the release of inflammatory factors is blocked by the treatment used to inhibit the P2Y2 receptors (42, 43). Furthermore, the activated P2Y2 receptors open pannexin-1 channels by inducing PLC activation and Ca2+ mobilization, which leads to further release of ATP promoting IL-1β maturation and secretion through the P2X7R-NLRP3 pathway (41). Thus, the binding of the P2Y2 receptors to ligands promoted the release of inflammatory factors and initiated the inflammatory response in multiple ways. The absence of the P2Y2 receptor is observed to reverse the increase of the nucleotide-induced IL-1β release (44).
The current study suggested that the P2Y6 receptor activated by UDP and UTP is involved in the pathogenesis of gout. Uratsuji et al. found that the P2Y6 receptors were involved in the MSU-induced inflammatory responses. The activation of the P2Y6 receptors in a variety of cells was observed to contribute to the production of inflammatory cytokines; particularly, the inhibition of the P2Y6 receptor in THP-1 cells could reduce MSU-induced IL-1β production (45). Additionally, the P2Y6 receptor affected the MSU-induced neutrophil function associated closely with gout. Sil et al. found that the inhibition of the P2Y6 receptors reduced neutrophil migration and IL-8 production, which is responsible for recruiting neutrophils to the joint cavity during a gout flare, thereby amplifying the effects of gout (46). In addition, treatment with the P2Y6 receptor antagonist MRS2578 inhibited the formation of MSU-induced neutrophil extracellular traps (NETs) in gout (46). Therefore, activation of the P2Y6 receptor may contribute to the persistence of gout flares.
The P2Y12-like receptors coupled to Gi, including P2Y12, P2Y13, and P2Y14 receptors, inhibit the cAMP-PKA pathway-mediated inflammatory response and theoretically inhibit gout pathogenesis. However, due to the complexity of the mechanisms of action in different internal environments, the regulation of inflammation by P2Y12-like receptors is also diverse. For example, cAMP has long been considered an inducer of inflammation. Activation of the cAMP-PKA-ERK signaling pathway promotes IL-1β secretion (47). Additionally, the AC/cAMP/PKA upregulates IL-1β-induced IL-6 production by enhancing the JAK2/STAT3 pathway (48). However, cAMP has also been recently pointed out as an important factor in regulating inflammation relief (49). The cAMP inhibits NF-κB transcription by activating downstream PKA to reduce the production of pro-inflammatory factors. Also, it promotes the phosphorylation of CENP to increase the production of anti-inflammatory factors and stimulate macrophage polarization. Moreover, cAMP has been found to inhibit the assembly of the NLRP3 inflammasome by directly binding to NLRP3 proteins, and promotes their ubiquitination and degradation (50, 51), thus acting as a negative regulator of the NLRP3 inflammasome reducing the secretion of IL-1β. Therefore, the P2Y12-like receptors can not only exert inhibitory inflammatory effects but can also often exhibit pro-inflammatory effects through the regulation of cAMP. In microglia, extracellular ADP was observed to act on P2Y12 receptors that activated NF-κB and NLRP3 inflammasomes while the inhibition of the P2Y12 receptors reduced the IL-1β levels (52). Similarly, the P2Y14 receptors promoted the secretion of inflammatory factors IL-1α, IL-8, and IL-6 in response to MSU stimulation (53).
Complex regulatory effects of inflammation are manifested in the P2Y14 receptor. In a gout study, Li et al. found that the P2Y14 receptors on macrophages negatively regulated cAMP, which enhanced the MSU-induced activation of the NLRP3 signaling pathway. The knockdown of the P2Y14 receptor then limited the activation of the NLRP3 inflammasome, which in turn attenuated the MSU-induced inflammatory infiltration of synovial tissue (54). Thus, in acute gouty arthritis, the activation of the P2Y14 receptor can exert a pro-inflammatory effect through the cAMP/NLRP3 signaling pathway.
The P2Y receptors in gout often play conflicting roles both in promoting remission of inflammation and maintaining the persistence of inflammation. As mentioned earlier, the P2Y12-like receptor signaling pathway that inhibits inflammation can have pro-inflammatory effects through activation of NLRP3 inflammasome. Similarly, the P2Y1-like receptor signaling pathway that promotes inflammation may exhibit an inhibitory effect on inflammation. For example, in a gout mouse model, the activation of the P2Y6 receptors reduces 1-palmitoyl-2-linoleyl-3-acetyl-rac-glycerol-induced neutrophil infiltration, alleviating joint symptoms (55). Thus, the P2Y receptors play a complex role in inflammatory diseases, either as a friend or foe. Besides modulating the pro-inflammatory factors in the regulation of inflammatory responses, P2Y receptors can also influence the expression of anti-inflammatory cytokines. TGF-β and IL-10 are the major cytokines responsible for gout resolution (56). The P2Y1 receptors were found to enhance the effects of TGF-β1 and IL-10 (57, 58), while the P2Y11 receptor, although reported to inhibit TGF-β1 production through activation of cAMP, was found to upregulate the IL-10 expression (57, 58). Overall, the data suggested that the P2Y receptor signaling pathway was involved in the pathogenesis of gout in different ways.
Activation of the P1 Receptor Signaling Pathway Inhibits Inflammation in Gout Flares
The P1 receptors belong to the family of G protein-coupled receptors and include A1, A2A, A2B, and A3 receptors. These are widely expressed on immune cells and exhibit anti-inflammatory effects. The ligand of the P1 receptor, adenosine, is the main product of sequential hydrolysis of extracellular ATP, which is catalyzed by CD39 and CD73. Under physiological conditions, the adenosine concentration is maintained at low levels, but under certain conditions such as inflammation, hypoxia, and cellular injury, the concentration of adenosine is increased by degrading extracellular ATP and ADP. Adenosine is then rapidly metabolized to AMP and inosine by the action of adenosine kinase or adenosine deaminase. The contribution of adenosine in alleviating inflammation and maintaining homeostasis in the organism has been recognized widely (10).
The P1 receptor signaling exerts anti-inflammatory effects through a variety of pathways. The adenosine-activated P1 receptors (A1, A3) increase the inhibition of neutrophil adhesion, reducing the production of superoxide and secretion of inflammatory cytokines (59–61). Furthermore, the interaction of adenosine with A1, A2A, and A3 receptors inhibits the release of IL-1β from activated human peripheral blood mononuclear lymphocytes (62). Also, the interaction of adenosine with A3 receptors was found to reduce TNF-α production in the macrophage cell lines (63), while the interaction of adenosine with A1 receptors was found to reduce parasite-stimulated production of reactive oxygen species (ROS) and IL-8 from neutrophils (64). Besides its inhibitory effect on the secretion of inflammatory factors, the activation of the P1 receptor signaling pathway also promotes the secretion of anti-inflammatory cytokines, including TGF-β1 and IL-10, by the immune cells (57, 60).
The targeting of adenosine receptors has been efficient in the treatment of several inflammatory diseases. The activation of adenosine-mediated A2A receptor contributed to block the activity of NLRP3 inflammasome, thereby reducing the production of pro-inflammatory cytokines while increasing the production of anti-inflammatory cytokines (65, 66). In post-traumatic brain injury events, agonizing the A3 adenosine receptor could improve the neurocognitive function by inhibiting the activation of the NLRP3 inflammasome (67). Similarly, in arthritic mice, activating the A3 adenosine receptors could reduce the production of TNF-α and alleviate arthritic symptoms (68). Thus, increased adenosine levels following an inflammatory episode could activate the P1 receptors, exerting anti-inflammatory effects through modulation of the NLRP3 inflammasome, thus, contributing to the resolution of gout flares.
Currently, only limited evidence is available to directly demonstrate the involvement of P1 receptors in suppressing gout inflammation or promoting gout resolution. However, based on the anti-inflammatory effect of the P1 receptor signaling pathway, we can see an inevitable impact on the pathogenesis of gout through changes in adenosine concentrations during the different stages of gout flares.
Alterations in Purine Signaling Pathway During Gout Flares
As previously mentioned, the purine signaling pathway plays a different role in the inflammatory regulation of gout, with ATP playing a central role in its initiation. The type and concentration of purinergic metabolites during a gout flare are altered by various mediators in the inflammatory environment, which can influence the onset and resolution of gout.
Conversion of Purinergic Metabolites During Gout Flares
MSU alone is not sufficient for initiating a gout flare; the synergistic effect of high levels of extracellular ATP is also required. After the initiation of gout, an increased expression of nucleoside triphosphate dihydrolase 1 (CD39) and 5’-nucleotidase (CD73) can be seen in an inflammatory environment. CD39 dephosphorylates extracellular ATP to ADP and adenosine monophosphate (AMP), which is further converted to adenosine by CD73 (69, 70). Thus, the changes in the expression of CD39 and CD73 during a gout flare can determine the level of nucleotides and the state of inflammation that affects the course of gout disease.
The CD39 and CD73 are extracellular nucleotidases expressed on monocytes, macrophages, B cells, T cells, natural killer cells, dendritic cells, and neutrophils. They mainly maintain a balance between the extracellular ATP and the concentration of adenosine. This homeostasis is important for the persistence and extent of the inflammatory state (71). Changes in the levels of nucleotide enzymes can occur during gout flares. Inflammatory cytokines, oxidative stress, and a hypoxic environment may increase the activity of CD39 and CD73 (72), thereby reducing ATP levels and increasing adenosine concentration. Further, tissue hypoxia can also reduce the expression of nucleoside transporters, increasing the adenosine levels. CD39 is the main enzyme that metabolizes ATP. In vitro studies in macrophages have found that activated P2X7 receptor could upregulate the expression of cytosolic CD39 by triggering a lipid raft-dependent mechanism (73). In contrast, the elevated CD39 could limit the P2X7R-mediated pro-inflammatory response. The knockdown of CD39 results in excessive IL-1β release (74, 75). This implies that in addition to catabolizing ATP and promoting adenosine production, the interaction between CD39 and the P2X7 receptor can modulate the upregulated inflammatory state of the organism in time to restore cellular homeostasis.
Macrophages are the primary sites of signaling events that regulate the onset and remission of gout. During the disease progression, macrophages show a shift from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 phenotype. Zanin et al. found (76) that M1 macrophages showed a decrease in the expression of CD39 and CD73 along with a reduction in ATP and AMP hydrolysis, whereas the M2 macrophages displayed higher CD39 and CD73 expression, as well as increased ATP and AMP hydrolysis. Also, the addition of adenosine led to an increase in the expression of the M2 macrophage gene, which in turn promoted the conversion of M1 phenotype to M2 phenotype (77). Moreover, after the initiation of inflammation, adenosine was found to decrease the production of pro-inflammatory cytokines and increase the secretion of anti-inflammatory cytokines by macrophages (78). These data suggested that purinergic signaling may be involved in gout resolution by regulating the conversion of macrophages from the M1 to M2 phenotype.
Changes of Purinergic Receptors During Gout Flares
Besides changes in the purine metabolites, the expression of purinergic receptors seems to be altered in response to the internal environment during the inflammatory process of gout flares. Among them, the changes in the expression of the P2X7 receptor have the most important impact. In addition to directly activating the NLRP3 inflammasome, MSU was found to stimulate the expression of P2X7 receptors and P2X4 receptors, secreting IL-1β and causing gout flares (11). Besides, an interesting experiment found that ethanol can upregulate the expression of P2X7 receptors to induce NLRP3 inflammasome activation (79), which partly explains why alcohol consumption predisposes to gout flares.
The expression of the P2Y receptor is also altered during a gout flare. The MSU stimulation of human keratinocytes resulted in an increased expression of P2Y6 receptors and P2Y14 receptors (45, 53). The P2Y6 receptor belongs to the P2Y1-like receptor family, which promotes the inflammatory responses and the upregulation by MSU facilitates the initiation and persistence of gout. In contrast, the P2Y14 receptor belonging to the P2Y12-like receptors inhibits cAMP and exhibits an inflammatory suppressive effect. The upregulation of P2Y14 by MSU appears to be detrimental to the development of gout. However, its upregulation can also promote gout pathogenesis through the negative effect of cAMP on the NLRP3 inflammasome (54).
The transition of macrophages from the phenotypes M1 to M2 promotes remission of gout inflammation. Upon macrophage activation, an increase was observed in the expression of P1 receptors (A1 and A3), which was accompanied by a decrease in the production of pro-inflammatory cytokines IL-6 and TNF-α and an increase in the production of anti-inflammatory factor IL-10. This promoted macrophage polarization and contributed to the suppression of inflammation (80, 81).
Modulation of Purinergic Signaling in Different Stages of Gout Flares
The previous section showed in vivo conversions of purine metabolites at different stages of a gout flare. Early in the flare, large amounts of ATP were released extracellularly by the necrotic, apoptotic, and inflammatory cells through pannexins or connexin channels (82). First, ATP was bound to the P2X7 receptors in the cytosol. Then, in concert with MSU, it stimulated the activation of the NLRP3 inflammasome, thus, prompting IL-1β secretion. Next, the expressions of CD39 and CD73 were increased in the inflammatory hypoxic environment following gout initiation, which promoted the dephosphorylation of ATP to adenosine. This inhibited the inflammatory response and promotes the self-resolution of gout flares by activating the P1 receptor signaling pathway. During the process, changes in the expression of purinergic receptor induced by the internal environment contributed to the ordered changes in purine signaling that regulated both gout flare and resolution (Figure 1). The main reasons for this seemingly contradictory mechanism are the ordered changes in the expression of purine metabolites and their receptors in the inflammatory environment, as well as the different purine signaling pathways they mediate in response to inflammation. Also, the accumulation of acidic metabolites in the inflammatory environment inhibits the cAMP/PKA signaling pathway reducing the IL-1β production (83), which indirectly counteracts the purinergic signaling-mediated cAMP/PKA pathway activity, favoring gout resolution.
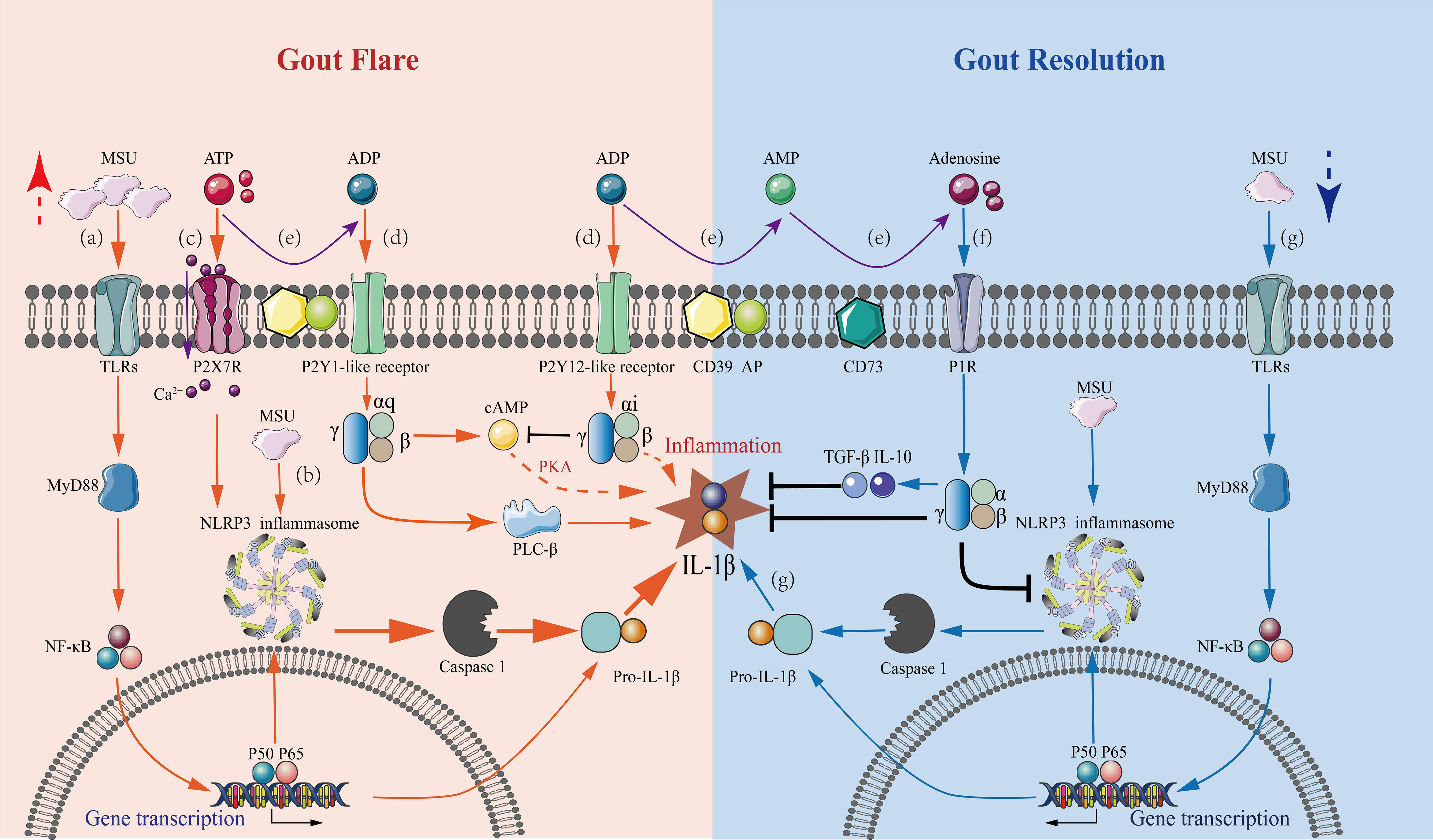
Figure 1 Mechanisms underlying purinergic signaling pathways in the regulation of gout flares and resolution. A series of purinergic receptor-mediated intracellular signaling events in macrophages are involved in gout flare and resolution, with the regulation of IL-1β levels being a central event. Gout flare: (A) The binding of extracellular MSU to TLR induces intracellular transcription and accumulation of the pro-IL-1β gene through activation of MyD88-NFκB signaling. (B) The uptake of MSU then activates the NLRP3 inflammasome, releasing active caspase-1, which cleaves pro-IL-1β to mature IL-1β. (C) Increased extracellular ATP binds to P2X7 receptors causing Ca2+ influx, which stimulates the NLRP3 inflammasome activation in concert with MSU, leading to massive IL-1β secretion. (D) Extracellular ATP and ADP stimulate the P2Y1-like receptors coupled to Gq, activating the PLC-IP3/DAG-PKC signaling pathway and cAMP-PKA pathway, which promotes the release of IL-1β. The stimulation of P2Y12-like receptors coupled to Gi exerts an inhibitory effect on the AC-cAMP-PKA pathway reducing the IL-1β production. However, inhibition of IL-1β production via other pathways also exists. Gout resolution: (E) In an inflammatory hypoxic environment, the activation of CD39 and CD73 during gout flares results in the progressive degradation of ATP and ADP to adenosine. (F) The resultant adenosine activates P1 receptors, decreasing the secretion of pro-inflammatory cytokine IL-1β, thus, promoting the production of anti-inflammatory cytokines TGF-β1 and IL-10. (G) Under the inflammatory conditions of gout, ATP is degraded, which affects the sustained stimulation of the P2X7R-NLRP3 signaling pathway. This results in a marked reduction of IL-1β secretion by MSU stimulation alone, which is not sufficient to sustain gout, and the condition then tends toward inflammatory remission.
Gout is a self-limiting disease with numerous causes contributing to its self-resolution. It is widely recognized that after initiating the inflammatory response in gout, neutrophils phagocytose MSU to form NETs. The aggregated NETs act by reducing the expression of pro-inflammatory cytokines (IL-1β, IL-6, IL-8), which promote the production of anti-inflammatory cytokines (TGF-β1, IL-1Ra, IL-10, IL-37), attenuating the inflammatory response (1, 84). Additionally, CD14 plays a role in the self-relief of gout. The knockdown of CD14 can reduce the activation of MSU-induced NLRP3 and release IL-1β (85). Studies have observed that a reduction of CD14 expression in gout patients may contribute to gout resolution (86). Besides neutrophils and CD14, purinergic signaling pathways play a synergistic role in gout resolution through the regulation of inflammatory responses.
Conclusion and Outlook
In summary, purinergic signaling pathways are involved in regulating the entire process of gout flare and resolution. After the initiation of the inflammatory response in gout, inflammation-induced microenvironmental changes lead to an orderly alteration in the type of purinergic metabolites in the body, gradually converting a large amount of ATP released from necrotic cells into ADP and adenosine. These purinergic metabolites stimulate the corresponding purinergic receptors and exert different regulatory effects on the inflammatory response. With the conversion of ATP to adenosine, the pro-inflammatory response mediated by the P2X and P2Y receptor signaling pathway shifts to an anti-inflammatory effect mediated mainly by the P1Y receptor signaling pathway, which then balances and restores the body’s homeostasis. These changes may therefore be an important mechanism of action for self-resolution of gout flares.
The purinergic signaling pathways can be potential targets for future interventions in gout pathogenesis. Currently, attempts are made to use targeted purinergic receptor therapy in gout. In a recent study, researchers designed and synthesized a novel P2Y14 receptor antagonist, which reduced the MSU-induced joint swelling and inflammatory infiltration in an acute gouty arthritis mouse model (87, 88), thus, improving the clinical value of targeted purinergic signaling in gout. However, the regulation of inflammation by purinergic signaling is much more complex than expected. It is not only manifested by the complexity of purinergic receptor-mediated signaling mechanisms, for example, the same receptor exhibiting opposite effects on inflammation in different settings, but it is also manifested by the diversity of their ligands. Besides activation by purinergic metabolites, receptors can be activated by additional endogenous ligands. A complex regulation network is formed between the endogenous ligands and P2Y receptors, finely tuning the nucleotide receptor signaling pathway (89). Therefore, the internal environment also has an important influence on purinergic signaling. An in-depth study of the regulatory mechanisms underlying different purinergic signals and the integration of various upstream and downstream influencing factors can accurately determine the role of purinergic signaling in gout flares, providing effective strategies for clinical intervention.
Author Contributions
XL wrote the manuscript. JG drafted the figures. JT contributed to provide the general idea and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81771774) and Anhui Provincial Key research and development plan (201904a07020103).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.785425/full#supplementary-material
References
1. Dalbeth N, Gosling AL, Gaffo A, Abhishek A. Gout. Lancet (2021) 397:1843–55. doi: 10.1016/S0140-6736(21)00569-9
2. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-Associated Uric Acid Crystals Activate the NALP3 Inflammasome. Nature (2006) 440:237–41. doi: 10.1038/nature04516
3. Bardin T, Richette P. Definition of Hyperuricemia and Gouty Conditions. Curr Opin Rheumatol (2014) 26:186–91. doi: 10.1097/BOR.0000000000000028
4. Gicquel T, Le Daré B, Boichot E, Lagente V. Purinergic Receptors: New Targets for the Treatment of Gout and Fibrosis. Fundam Clin Pharmacol (2017) 31:136–46. doi: 10.1111/fcp.12256
5. Tao JH, Zhang Y, Li XP. P2X7R: A Potential Key Regulator of Acute Gouty Arthritis. Semin Arthritis Rheumatol (2013) 43:376–80. doi: 10.1016/j.semarthrit.2013.04.007
6. Tao JH, Cheng M, Tang JP, Dai XJ, Zhang Y, Li XP, et al. Single Nucleotide Polymorphisms Associated With P2X7R Function Regulate the Onset of Gouty Arthritis. PloS One (2017) 12:e0181685. doi: 10.1371/journal
7. Ying Y, Chen Y, Li Z, Huang H, Gong Q. Investigation Into the Association Between P2RX7 Gene Polymorphisms and Susceptibility to Primary Gout and Hyperuricemia in a Chinese Han Male Population. Rheumatol Int (2017) 37:571–78. doi: 10.1007/s00296-017-3669-6
8. Gong QY, Chen Y. Correlation Between P2X7 Receptor Gene Polymorphisms and Gout. Rheumatol Int (2015) 35:1307–10. doi: 10.1007/s00296-015-3258-5
9. Marques-da-Silva C, Chaves MM, Castro NG, Coutinho-Silva R, Guimaraes MZ. Colchicine Inhibits Cationic Dye Uptake Induced by ATP in P2X2 and P2X7 Receptor-Expressing Cells: Implications for its Therapeutic Action. Br J Pharmacol (2011) 163:912–26. doi: 10.1111/j.1476-5381.2011.01254.x
10. Linden J, Cekic C. Regulation of Lymphocyte Function by Adenosine. Arterioscler Thromb Vasc Biol (2012) 32:2097–103. doi: 10.1161/ATVBAHA.111.226837
11. Gicquel T, Robert S, Loyer P, Victoni T, Bodin A, Ribault C, et al. IL-1β Production is Dependent on the Activation of Purinergic Receptors and NLRP3 Pathway in Human Macrophages. FASEB J (2015) 29:4162–73. doi: 10.1096/fj.14-267393
13. Pasquini S, Contri C, Borea PA, Vincenzi F, Varani K. Adenosine and Inflammation: Here, There and Everywhere. Int J Mol Sci (2021) 22:7685. doi: 10.3390/ijms22147685
14. Klaver D, Thurnher M. Control of Macrophage Inflammation by P2Y Purinergic Receptors. Cells (2021) 10:1098. doi: 10.3390/cells10051098
15. Godoy-Marín H, Duroux R, Jacobson KA, Soler C, Colino-Lage H, Jiménez-Sábado V, et al. Adenosine A2A Receptors Are Upregulated in Peripheral Blood Mononuclear Cells From Atrial Fibrillation Patients. Int J Mol Sci (2021) 22:3467. doi: 10.3390/ijms22073467
16. Tian G, Zhou J, Quan Y, Kong Q, Wu W, Liu X. P2Y1 Receptor Agonist Attenuates Cardiac Fibroblasts Activation Triggered by TGF-β1. Front Pharmacol (2021) 12:627773. doi: 10.3389/fphar.2021.627773
17. Jain S, Jacobson KA. Purinergic Signaling in Diabetes and Metabolism. Biochem Pharmacol (2021) 187:114393. doi: 10.1016/j.bcp.2020.114393
18. Jain S, Pydi SP, Toti KS, Robaye B, Idzko M, Gavrilova O, et al. Lack of Adipocyte Purinergic P2Y6 Receptor Greatly Improves Whole Body Glucose Homeostasis. Proc Natl Acad Sci USA (2020) 117:30763–74. doi: 10.1073/pnas.2006578117
19. D’Angelo V, Giorgi M, Paldino E, Cardarelli S, Fusco FR, Saverioni I, et al. A2A Receptor Dysregulation in Dystonia DYT1 Knock-Out Mice. Int J Mol Sci (2021) 22:2691. doi: 10.3390/ijms22052691
20. Cao Y, Wang X, Li Y, Evers M, Zhang H, Chen X. Extracellular and Macropinocytosis Internalised ATP Work Together to Induce Epithelial-Mesenchymal Transition and Other Early Metastatic Activities in Lung Cancer. Cancer Cell Int (2019) 19:254. doi: 10.1186/s12935-019-0973-0
21. Zhang Y, Ding J, Wang L. The Role of P2X7 Receptor in Prognosis and Metastasis of Colorectal Cancer. Adv Med Sci (2019) 64:388–94. doi: 10.1016/j.advms.2019.05.002
22. Shakya AK, Naik RR, Almasri IM, Kaur A. Role and Function of Adenosine and its Receptors in Inflammation, Neuroinflammation, IBS, Autoimmune Inflammatory Disorders, Rheumatoid Arthritis and Psoriasis. Curr Pharm Des (2019) 25:2875–91. doi: 10.2174/1381612825666190716145206
23. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity (2017) 47:15–31. doi: 10.1016/j.immuni.2017.06.020
24. Illes P, Rubini P, Ulrich H, Zhao Y, Tang Y. Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells (2020) 9:1108. doi: 10.3390/cells9051108
25. Di Virgilio F, Sarti AC, Coutinho-Silva R. Purinergic Signaling, DAMPs, and Inflammation. Am J Physiol Cell Physiol (2020) 318:C832–35. doi: 10.1152/ajpcell.00053.2020
26. Schett G, Dayer JM, Manger B. Interleukin-1 Function and Role in Rheumatic Disease. Nat Rev Rheumatol (2016) 12:14–24. doi: 10.1038/nrrheum.2016.166
27. Mitroulis I, Kambas K, Ritis K. Neutrophils. IL-1beta, and Gout:is There a Link? Semin Immunopathol (2013) 35:501–12. doi: 10.1007/s00281-013-0361-0
28. North RA. Molecular Physiology of P2X Receptors. Physiol Rev (2002) 82:1013–67. doi: 10.1152/physrev.00015.2002
29. Latz E, Xiao TS, Stutz A. Activation and Regulation of the Inflammasomes. Nat Rev Immunol (2013) 13:397–411. doi: 10.1038/nri3452
30. Monif M, Reid CA, Powell KL, Smart ML, Williams DA. The P2X7 Receptor Drives Microglial Activation and Proliferation: A Trophic Role for P2X7R Pore. J Neurosci (2009) 29:3781–91. doi: 10.1523/JNEUROSCI.5512-08.2009
31. Burnstock G. P2X Ion Channel Receptors and Inflammation. Purinergic Signal (2016) 12:59–67. doi: 10.1007/s11302-015-9493-0
32. Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front Immunol (2019) 10:2538. doi: 10.3389/fimmu.2019.02538
33. Faria RX, Oliveira FH, Salles JP, Oliveira AS, von Ranke NL, Bello ML, et al. 1,4-Naphthoquinones Potently Inhibiting P2X7 Receptor Activity. Eur J Med Chem (2018) 143:1361–72. doi: 10.1016/j.ejmech.2017.10.033
34. Liu YH, Chang YC, Chen LK, Su PA, Ko WC, Tsai YS, et al. The ATP-P2X7 Signaling Axis Is an Essential Sentinel for Intracellular Clostridium Difficile Pathogen-Induced Inflammasome Activation. Front Cell Infect Microbiol (2018) 8:84. doi: 10.3389/fcimb.2018.00084
35. Sauer H, Hescheler J, Wartenberg M. Mechanical Strain-Induced Ca(2+) Waves are Propagated via ATP Release and Purinergic Receptor Activation. Am J Physiol Cell Physiol (2000) 279:C295–307. doi: 10.1152/ajpcell.2000.279.2.C295
36. Lowell BB, Spiegelman BM. Towards a Molecular Understanding of Adaptive Thermogenesis. Nature (2000) 404:652–60. doi: 10.1038/35007527
37. Tao JH, Tang JP, Chen M, Li XP, Liu Q, Wang YL. Study on the Association Between P2X7R Gene Rs1621388 Polymorphism and Gout Susceptibility. Chin J Dis Control Prev (2017) 21:698–701. doi: 10.16462/j.cnki.zhjbkz.2017.07.012
38. Kanellopoulos JM, Almeida-da-Silva CLC, Rüütel Boudinot S, Ojcius DM. Structural and Functional Features of the P2X4 Receptor: An Immunological Perspective. Front Immunol (2021) 12. doi: 10.3389/fimmu
39. Cattaneo M. P2Y12 Receptors: Structure and Function. J Thromb Haemost (2015) Suppl 1:S10–6. doi: 10.1111/jth.12952
40. Franke H, Krügel U, Illes P. P2 Receptors and Neuronal Injury. Pflugers Arch (2006) 452:622–44. doi: 10.1007/s00424-006-0071-8
41. Thorstenberg ML, Rangel Ferreira MV, Amorim N, Canetti C, Morrone FB, Alves Filho JC, et al. Purinergic Cooperation Between P2Y2 and P2X7 Receptors Promote Cutaneous Leishmaniasis Control: Involvement of Pannexin-1 and Leukotrienes. Front Immunol (2018) 9:1531. doi: 10.3389/fimmu.2018.01531
42. Kobayashi T, Kouzaki H, Kita H. Human Eosinophils Recognise Endogenous Danger Signal Crystalline Uric Acid and Produce Pro-Inflammatory Cytokines Mediated by Autocrine ATP. J Immunol (2010) 184:6350–8. doi: 10.4049/jimmunol.0902673
43. Kobayashi T, Soma T, Noguchi T, Nakagome K, Nakamoto H, Kita H, et al. ATP Drives Eosinophil Effector Responses Through P2 Purinergic Receptors. Allergol Int (2015) 64 Suppl(0):S30–6. doi: 10.1016/j.alit.2015.04.009
44. de la Rosa G, Gómez AI, Baños MC, Pelegrín P. Signaling Through Purinergic Receptor P2Y2 Enhances Macrophage IL-1β Production. Int J Mol Sci (2020) 21:4686. doi: 10.3390/ijms21134686
45. Uratsuji H, Tada Y, Kawashima T, Kamata M, Hau CS, Asano Y, et al. P2Y6 Receptor Signaling Pathway Mediates Inflammatory Responses Induced by Monosodium Urate Crystals. J Immunol (2012) 188:436–44. doi: 10.4049/jimmunol.1003746
46. Sil P, Hayes CP, Reaves BJ, Breen P, Quinn S, Sokolove J, et al. P2Y6 Receptor Antagonist MRS2578 Inhibits Neutrophil Activation and Aggregated Neutrophil Extracellular Trap Formation Induced by Gout-Associated Monosodium Urate Crystals. J Immunol (2017) 198:428–42. doi: 10.4049/jimmunol.1600766
47. Permpoonputtana K, Porter JE, Govitrapong P. Calcitonin Gene-Related Peptide Mediates an Inflammatory Response in Schwann Cells Via cAMP-Dependent ERK Signaling Cascade. Life Sci (2016) 144:19–25. doi: 10.1016/j.lfs.2015.11.015
48. Tanabe K, Kozawa O, Iida H. cAMP/PKA Enhances Interleukin-1β-Induced Interleukin-6 Synthesis Through STAT3 in Glial Cells. Cell Signal (2016) 28:19–24. doi: 10.1016/j.cellsig.2015.10.009
49. Tavares LP, Negreiros-Lima GL, Lima KM, E Silva PMR, Pinho V, Teixeira MM, et al. Blame the Signaling: Role of cAMP for the Resolution of Inflammation. Pharmacol Res (2020) 159:105030. doi: 10.1016/j.phrs.2020.105030
50. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The Calcium-Sensing Receptor Regulates the NLRP3 Inflammasome Through Ca2+ and cAMP. Nature (2012) 492:123–7. doi: 10.1038/nature11588
51. Sokolowska M, Chen LY, Liu Y, Martinez-Anton A, Qi HY, Logun C, et al. Prostaglandin E2 Inhibits NLRP3 Inflammasome Activation Through EP4 Receptor and Intracellular Cyclic AMP in Human Macrophages. J Immunol (2015) 194:5472–87. doi: 10.4049/jimmunol.1401343
52. Suzuki T, Kohyama K, Moriyama K, Ozaki M, Hasegawa S, Ueno T, et al. Extracellular ADP Augments Microglial Inflammasome and NF-κb Activation via the P2Y12 Receptor. Eur J Immunol (2020) 50:205–19. doi: 10.1002/eji.201848013
53. Uratsuji H, Tada Y, Hau CS, Shibata S, Kamata M, Kawashima T, et al. Monosodium Urate Crystals Induce Functional Expression of P2Y14 Receptor in Human Keratinocytes. J Invest Dermatol (2016) 136:1293–6. doi: 10.1016/j.jid.2016.01.026
54. Li H, Jiang W, Ye S, Zhou M, Liu C, Yang X, et al. P2Y14 Receptor has a Critical Role in Acute Gouty Arthritis by Regulating Pyroptosis of Macrophages. Cell Death Dis (2020) 11:394. doi: 10.1038/s41419-020-2609-7
55. Shin SH, Jeong J, Kim JH, Sohn KY, Yoon SY, Kim JW. 1-Palmitoyl-2-Linoleoyl-3-Acetyl-Rac-Glycerol (PLAG) Mitigates Monosodium Urate (MSU)-Induced Acute Gouty Inflammation in BALB/c Mice. Front Immunol (2020) 11:710. doi: 10.3389/fimmu.2020.00710
56. Scanu A, Oliviero F, Ramonda R, Frallonardo P, Dayer JM, Punzi L. Cytokine Levels in Human Synovial Fluid During the Different Stages of Acute Gout: Role of Transforming Growth Factor β1 in the Resolution Phase. Ann Rheum Dis (2012) 71:621–4. doi: 10.1136/annrheumdis-2011-200711
57. Zuccarini M, Giuliani P, Buccella S, Di Liberto V, Mudò G, Belluardo N, et al. Modulation of the TGF-β1-Induced Epithelial to Mesenchymal Transition (EMT) Mediated by P1and P2 Purine Receptors in MDCK Cells. Purinergic Signal (2017) 13:429–42. doi: 10.1007/s11302-017-9571-6
58. Seo DR, Kim SY, Kim KY, Lee HG, Moon JH, Lee JS, et al. Cross Talk Between P2 Purinergic Receptors Modulates Extracellular ATP-Mediated Interleukin-10 Production in Rat Microglial Cells. Exp Mol Med (2008) 40:19–26. doi: 10.3858/emm.2008.40.1.19
59. Barletta KE, Ley K, Mehrad B. Regulation of Neutrophil Function by Adenosine. Arterioscler Thromb Vasc Biol (2012) 32:856–64. doi: 10.1161/ATVBAHA.111.226845
60. Koscso B, Csoka B, Selmeczy Z, Himer L, Pacher P, Virag L, et al. Adenosine Augments IL-10 Production by Microglial Cells Through an A2B Adenosine Receptor-Mediated Process. J Immunol (2012) 188:445e453. doi: 10.4049/jimmunol.1101224
61. Haskó G, Pacher P. Regulation of Macrophage Function by Adenosine. Arterioscler Thromb Vasc Biol (2012) 32:865–9. doi: 10.1161/ATVBAHA.111.226852
62. Sipka S, Kovács I, Szántó S, Szegedi G, Brugós L, Bruckner G, et al. Adenosine Inhibits the Release of Interleukin-1beta in Activated Human Peripheral Mononuclear Cells. Cytokine (2005) 31:258–63. doi: 10.1016/j.cyto.2005.05.002
63. Sajjadi FG, Takabayashi K, Foster AC, Domingo RC, Firestein GS. Inhibition of TNF-Alpha Expression by Adenosine: Role of A3 Adenosine Receptors. J Immunol (1996) 156:3435–42.
64. Frasson AP, Menezes CB, Goelzer GK, Gnoatto SCB, Garcia SC, Tasca T. Adenosine Reduces Reactive Oxygen Species and Interleukin-8 Production by Trichomonas Vaginalis-Stimulated Neutrophils. Purinergic Signal (2017) 13:569–77. doi: 10.1007/s11302-017-9584-1
65. Zhou Y, Zeng X, Li G, Yang Q, Xu J, Zhang M, et al. Inactivation of Endothelial Adenosine A2A Receptors Protects Mice From Cerebral Ischaemia-Induced Brain Injury. Br J Pharmacol (2019) 176:2250–63. doi: 10.1111/bph.14673
66. Ohta A, Sitkovsky M. Role of G-Protein-Coupled Adenosine Receptors in Downregulation of Inflammation and Protection From Tissue Damage. Nature (2001) 414:916–20. doi: 10.1038/414916a
67. Farr SA, Cuzzocrea S, Esposito E, Campolo M, Niehoff ML, Doyle TM, et al. Adenosine A3 Receptor as a Novel Therapeutic Target to Reduce Secondary Events and Improve Neurocognitive Functions Following Traumatic Brain Injury. J Neuroinflammation (2020) 17:339. doi: 10.1186/s12974-020-02009-7
68. Baharav E, Bar-Yehuda S, Madi L, Silberman D, Rath-Wolfson L, Halpren M, et al. Anti-Inflammatory Effect of A3 Adenosine Receptor Agonists in Murine Autoimmune Arthritis Models. J Rheumatol (2005) 32:469–76.
69. Yegutkin GG. Nucleotide- and Nucleoside-Converting Ectoenzymes: Important Modulators of Purinergic Signaling Cascade. Biochim Biophys Acta (2008) 1783:673–94. doi: 10.1016/j.bbamcr.2008.01.024
70. Liu X, Ma L, Zhang S, Ren Y, Dirksen RT. CD73 Controls Extracellular Adenosine Generation in the Trigeminal Nociceptive Nerves. J Dent Res (2017) 96:671–77. doi: 10.1177/0022034517692953
71. Murphy PS, Wang J, Bhagwat SP, Munger JC, Janssen WJ, Wright TW, et al. CD73 Regulates Anti-Inflammatory Signaling Between Apoptotic Cells and Endotoxin-Conditioned Tissue Macrophages. Cell Death Differ (2017) 24:559–70. doi: 10.1038/cdd.2016.159
72. Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, et al. Ecto-5’-Nucleotidase (CD73) Regulation by Hypoxia-Inducible Factor-1 Mediates Permeability Changes in Intestinal Epithelia. J Clin Invest (2002) 110:993–1002. doi: 10.1172/JCI15337
73. Savio LEB, de Andrade Mello P, Santos SACS, de Sousa JC, Oliveira SDS, Minshall RD, et al. P2X7 Receptor Activation Increases Expression of Caveolin-1 and Formation of Macrophage Lipid Rafts, Thereby Boosting CD39 Activity. J Cell Sci (2020) 133:jcs237560. doi: 10.1242/jcs.237560
74. Savio LEB, de Andrade Mello P, Figliuolo VR, de Avelar Almeida TF, Santana PT, Oliveira SDS, et al. CD39 Limits P2X7 Receptor Inflammatory Signaling and Attenuates Sepsis-Induced Liver Injury. J Hepatol (2017) 67:716–26. doi: 10.1016/j.jhep.2017.05.021
75. Lévesque SA, Kukulski F, Enjyoji K, Robson SC, Sévigny J. NTPDase1 Governs P2X7-Dependent Functions in Murine Macrophages. Eur J Immunol (2010) 40:1473–85. doi: 10.1002/eji.200939741
76. Zanin RF, Braganhol E, Bergamin LS, Campesato LF, Filho AZ, Moreira JC, et al. Differential Macrophage Activation Alters the Expression Profile of NTPDase and Ecto-5’-Nucleotidase. PloS One (2012) 7:e31205. doi: 10.1371/journal.pone.0031205
77. Barberà-Cremades M, Baroja-Mazo A, Pelegrín P. Purinergic Signaling During Macrophage Differentiation Results in M2 Alternative Activated Macrophages. J Leukoc Biol (2016) 99:289–99. doi: 10.1189/jlb.1A0514-267RR
78. Haskó G, Cronstein BN. Adenosine: An Endogenous Regulator of Innate Immunity. Trends Immunol (2004) 25:33–9. doi: 10.1016/j.it.2003.11.003
79. Le Daré B, Victoni T, Bodin A, Vlach M, Vene E, Loyer P, et al. Ethanol Upregulates the P2X7 Purinergic Receptor in Human Macrophages. Fundam Clin Pharmacol (2019) 33:63–74. doi: 10.1111/fcp.12433
80. Antonioli L, Csóka B, Fornai M, Colucci R, Kókai E, Blandizzi C, et al. Adenosine and Inflammation: What’s New on the Horizon? Drug Discov Today (2014) 19:1051–68. doi: 10.1016/j.drudis.2014.02.010
81. Haskó G, Szabó C, Németh ZH, Kvetan V, Pastores SM, Vizi ES. Adenosine Receptor Agonists Differentially Regulate IL-10, TNF-Alpha, and Nitric Oxide Production in RAW 264.7 Macrophages and in Endotoxemic Mice. J Immunol (1996) 157:4634–40.
82. Eltzschig HK, Eckle T, Mager A, Küper N, Karcher C, Weissmüller T, et al. ATP Release From Activated Neutrophils Occurs via Connexin 43 and Modulates Adenosine-Dependent Endothelial Cell Function. Circ Res (2006) 99:1100–8. doi: 10.1161/01.RES.0000250174.31269.70
83. Jin Y, Sato K, Tobo A, Mogi C, Tobo M, Murata N, et al. Inhibition of Interleukin-1β Production by Extracellular Acidification Through the TDAG8/cAMP Pathway in Mouse Microglia. J Neurochem (2014) 129:683–95. doi: 10.1111/jnc.12661
84. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated Neutrophil Extracellular Traps Limit Inflammation by Degrading Cytokines and Chemokines. Nat Med (2014) 20:511–7. doi: 10.1038/nm.3547
85. Scott P, Ma H, Viriyakosol S, Terkeltaub R, Liu-Bryan R. Engagement of CD14 Mediates the Inflammatory Potential of Monosodium Urate Crystals. J Immunol (2006) 177:6370–8. doi: 10.4049/jimmunol.177.9.6370
86. Duan L, Luo J, Fu Q, Shang K, Wei Y, Wang Y, et al. Decreased Expression of CD14 in MSU-Mediated Inflammation May Be Associated With Spontaneous Remission of Acute Gout. J Immunol Res (2019) 2019:7143241. doi: 10.1155/2019/7143241
87. Lu R, Wang Y, Liu C, Zhang Z, Li B, Meng Z, et al. Design, Synthesis and Evaluation of 3-Amide-5-Aryl Benzoic Acid Derivatives as Novel P2Y14R Antagonists With Potential High Efficiency Against Acute Gouty Arthritis. Eur J Med Chem (2021) 216:113313. doi: 10.1016/j.ejmech.2021.113313
88. Wang W, Liu C, Li H, Tian S, Liu Y, Wang N, et al. Discovery of Novel and Potent P2Y14R Antagonists via Structure-Based Virtual Screening for the Treatment of Acute Gouty Arthritis. J Adv Res (2020) 23:133–42. doi: 10.1016/j.jare.2020.02.007
Keywords: purinergic signaling, gout flare, ATP, Adenosine, P2X7R, IL-1β
Citation: Li X, Gao J and Tao J (2021) Purinergic Signaling in the Regulation of Gout Flare and Resolution. Front. Immunol. 12:785425. doi: 10.3389/fimmu.2021.785425
Received: 29 September 2021; Accepted: 15 November 2021;
Published: 01 December 2021.
Edited by:
Xiaoxia Zhu, Fudan University, ChinaCopyright © 2021 Li, Gao and Tao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhui Tao, dGFvamluaHVpQHVzdGMuZWR1LmNu
†These authors have contributed equally to this work and share first authorship