 Ping-Heng Tan
Ping-Heng Tan Jasmine Ji2,3
Jasmine Ji2,3 Chun-Chang Yeh
Chun-Chang Yeh Ru-Rong Ji
Ru-Rong Ji- 1Department of Anesthesiology, Chi Mei Medical Center, Tainan City, Taiwan
- 2Neuroscience Department, Wellesley College, Wellesley, Massachusetts, MA, United States
- 3Center for Translational Pain Medicine, Department of Anesthesiology, Duke University Medical Center, Durham, NC, United States
- 4Department of Anesthesiology of Tri-Service General Hospital & National Defense Medical Center, Taipei City, Taiwan
- 5Department of Neurobiology, Duke University Medical Center, Durham, NC, United States
- 6Department of Cell Biology, Duke University Medical Center, Durham, NC, United States
Interferons (IFNs) are cytokines that possess antiviral, antiproliferative, and immunomodulatory actions. IFN-α and IFN-β are two major family members of type-I IFNs and are used to treat diseases, including hepatitis and multiple sclerosis. Emerging evidence suggests that type-I IFN receptors (IFNARs) are also expressed by microglia, astrocytes, and neurons in the central and peripheral nervous systems. Apart from canonical transcriptional regulations, IFN-α and IFN-β can rapidly suppress neuronal activity and synaptic transmission via non-genomic regulation, leading to potent analgesia. IFN-γ is the only member of the type-II IFN family and induces central sensitization and microglia activation in persistent pain. We discuss how type-I and type-II IFNs regulate pain and infection via neuro-immune modulations, with special focus on neuroinflammation and neuro-glial interactions. We also highlight distinct roles of type-I IFNs in the peripheral and central nervous system. Insights into IFN signaling in nociceptors and their distinct actions in physiological vs. pathological and acute vs. chronic conditions will improve our treatments of pain after surgeries, traumas, and infections.
Introduction
The name of interferons is a reference to their ability to “interfere” with viruses (1). Interferons are a family of cytokines that play important roles in the immune, endocrine, and nervous system, including both the peripheral and central nervous systems (PNS, CNS). Interferons can be divided into three classes: type-I, type-II, and type-III interferons (IFNs). First discovered in 1957, the type-I IFN family consists of IFN-α (IFN-α1-8, 10, 13, 14, 16, 17, and 21, for a total of 13 subtypes), IFN-β, IFN-δ, IFN-ϵ, IFN-κ, IFN-τ and IFN-ω1–3. The type-II IFN family has only one member, IFN-γ (1–4). The pronociceptive and pro-inflammatory role of IFN-γ is well documented. Intrathecal injection of IFN-γ facilitates the spinal nociceptive flexor reflex in rats (5) and induces biting behavior in mice (6). Following nerve injury, IFN-γ contributes to the pathogenesis of murine neuropathic pain through activation of microglia in the spinal cord (7). Type III IFNs are lambda-IFNs, including IFN-λ1, IFN-λ2, and IFN-λ3. Type-I IFNs produce multiple biological and cellular responses, such as suppression of viral infections, inhibition of cell proliferation, promotion of antitumor activities, and modulation of immunity and pain (2, 4, 8, 9).
It is well-established that the sensing of pathogen-associated molecular patterns (PAMPs) during bacterial or viral infections, or danger-associated molecular patterns (DAMPs) following tissue injury by pattern recognition receptors results in the production of many cytokines and chemokines, including type-I IFNs, leading to innate immunity (10, 11). Toll-like receptors (TLRs) are the best-known pattern recognition receptors and are localized either on the cell surface (e.g. TLR2, TLR5, TLR4) or on intracellular compartments such as endosomes (TLR3, TLR7, TLR8, and TLR9) of immune cells and glial cells (12, 13). TLR3 and TLR7 are activated by double-stranded RNAs and singe-stranded RNAs, respectively, whereas TLR4 is activated by the bacterial product, lipopolysaccharide (LPS) (10). Following the recognition of PAMPs and DAMPs, TLRs will activate their specific signaling pathways to produce type-I IFNs. Notably, type-I IFNs can be induced by the activation of TLR3, TLR4, and TLR7, TLR8, TLR9 via distinct intracellular signaling molecules, such as TRIF for TLR3 and TLR4, and Myd88/TRAF3/6 for TLR7-9 (14) (Figure 1A).
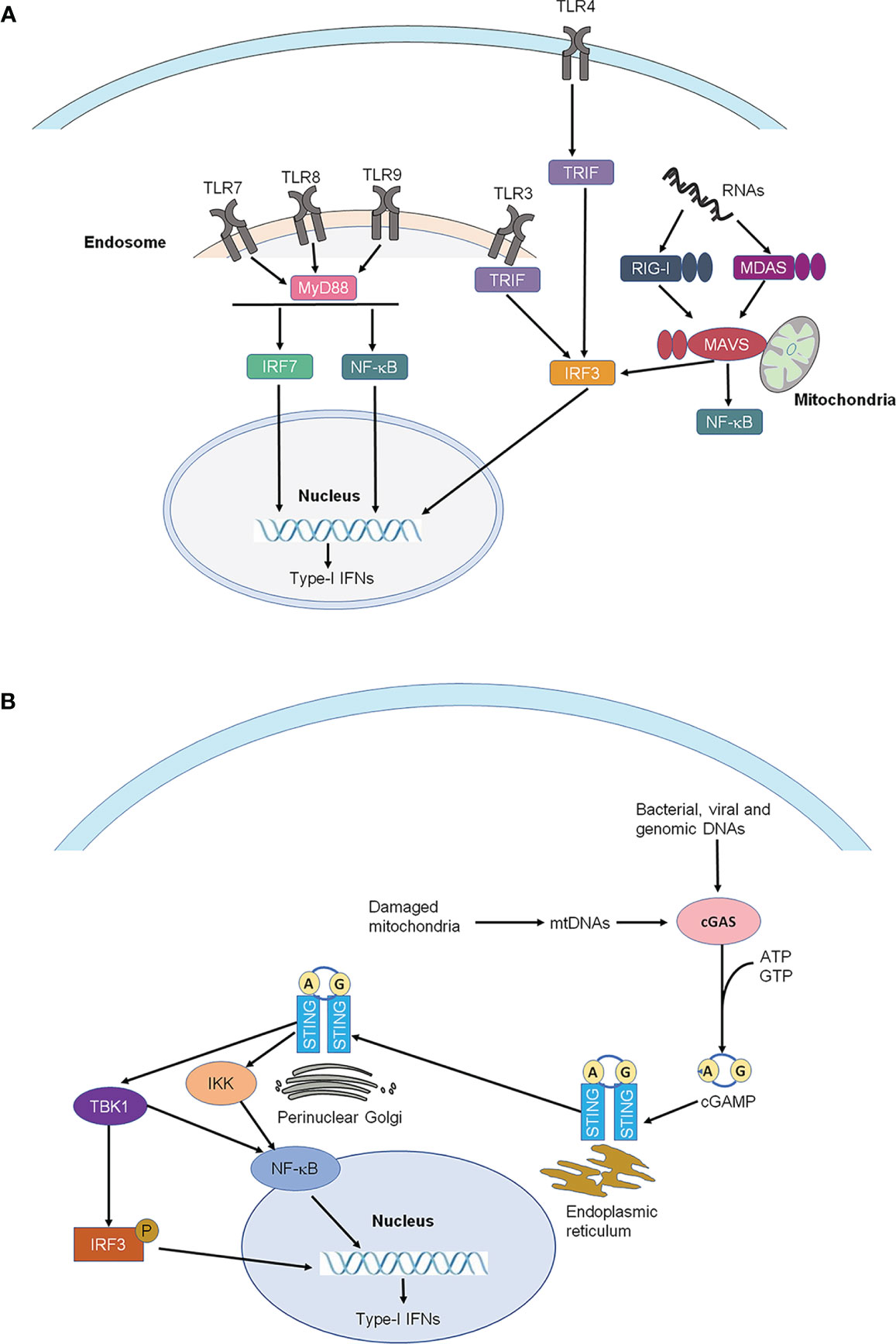
Figure 1 Production of type-I IFNs. (A) Production of type-I IFNs by activation of Toll-like receptors (TLRs) and RIG-I-like receptors (RLRs). Type-I IFNs can be induced by the activation of TLR3, TLR4, and TLR7, TLR8, TLR9 via distinct intracellular signaling molecules, such as TRIF for TLR3 and TLR4, and MYDd88 for TLR7-9. TLRs are the best-known pattern recognition receptors and are localized either on the cell surface or on intracellular compartments such as endosomes (TLR3, TLR7-9) of immune cells and glial cells. (B) Production of type-I IFNs by activation of STING. STING is an endoplasmic reticulum adaptor. Activated by viruses and intracellular DNAs, STING activates TBK1, which subsequently activates the transcription factors NFκB and interferon regulatory factor 3 (IRF3), leading to the production of type-I IFNs. IRF3, interferon regulatory factor 3; IRF7, interferon regulatory factor 7; MAVS, mitochondrial antiviral signaling proteins; MDA5, melanoma differentiation-associated protein 5; MYD88, myeloid differentiation primary response 88; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; RIG-1, retinoic acid-inducible gene; STING, stimulator of interferon genes; TBK1, TANK-binding kinase 1; TRIF, TIR-domain-containing adapter-inducing IFN-β.
In addition to TLRs, pattern recognition receptors also include retinoic acid-inducible gene I (RIG)-I-like receptors (RLRs) and stimulator of interferon genes (STING), which sense intracellular RNAs and DNAs, respectively (2 Ivashkiv and Donlin, 2014). There are three receptors in the RLRs family, including RIG-I, melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2) (14, 15). STING is an endoplasmic reticulum adaptor and regulates intracellular DNA-mediated, type-I interferon-dependent innate immunity (16, 17) (Figure 1B). Activated by select viruses and intracellular DNAs, STING activates TANK-binding kinase 1 (TBK1), which subsequently activates the transcription factors NFκB and interferon regulatory factor 3 (IRF3), leading to the production of type-I IFNs and promoting the eradication of pathogens mediated by immune cells and neoplastic cells (18). The plasmacytoid dendritic cells (pDC) are the major type-I IFN-producing cells in humans and mice following virus infection or direct stimulation with DNAs/RNAs (19–21). IFN-α and IFN-β are produced and secreted by different cell types, including natural killer (NK) cells, B cells, T cells, macrophages, fibroblasts, endothelial cells, and osteoblasts. Furthermore, IFN-α and IFN-β can stimulate macrophages and NK cells to elicit anti-viral and anti-tumor responses.
Type-I Interferon Receptor Signaling in Immune Cells
Both IFN-α and IFN-β bind to a cell surface receptor complex known as the IFN-α receptor (IFNAR) including IFNAR1 and IFNAR2 chains (4, 22, 23). IFNAR is expressed in most cell types including immune cells, neurons and glia in the spinal cord (4, 22, 23). IFNAR signaling requires the Janus activated kinases (JAKs), which consist of 4 family members: JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2). IFNAR1 is associated with TYK2, whereas IFNAR2 is associated with JAK1 (Figure 2A). Following the ligand-dependent rearrangement and dimerization of receptor subunits, these receptor-associated JAKs auto- phosphorylate for their own activation, leading to subsequent activation of the transcription factor STAT (signal transducer and activator of transcription) (4). The activation of the JAK-STAT signaling pathways appears to mediate the many biological effects of type-I IFNs via distinct gene transcription. In contrast, type-II IFN (IFN-γ) binds to IFN-γ receptor (IFNGR), including IFNGR1 and IFNGR2, which are associated with JAK1 and JAK2, respectively (4). Unlike IFN-α and IFN-β, IFN-γ is a prominent pro-inflammatory cytokine and positively regulates neuroinflammation and pathological pain (7). Thus, type-I and type-II IFNs play different roles in inflammation by coupling their target receptors with different intracellular tyrosine kinases.
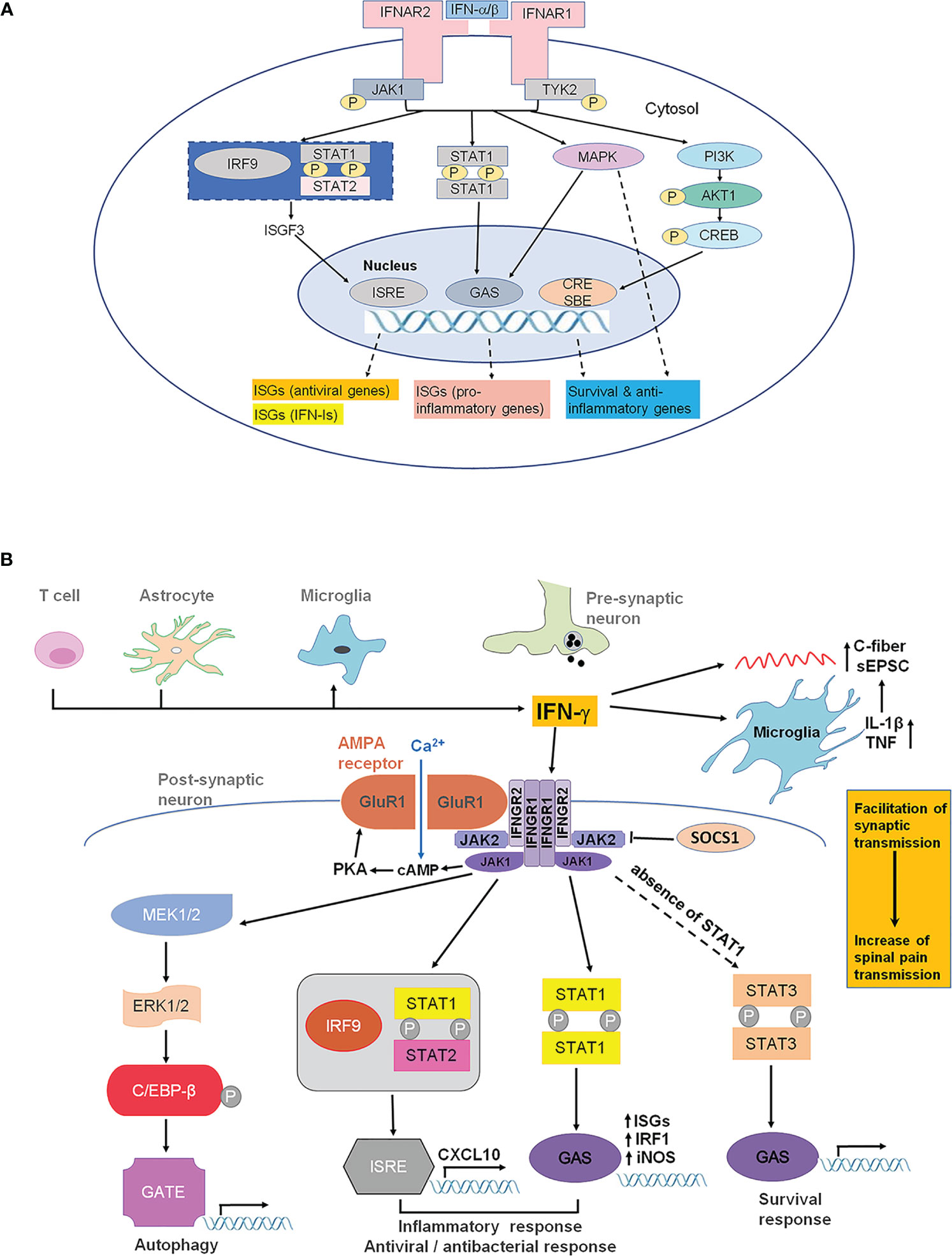
Figure 2 IFNAR and IFNGR signaling. (A) IFNAR signaling and type-I IFN-induced gene expression. IFN-α and IFN-β transmit signals through IFNAR1 and IFNAR2 (the main ligand-binding subunit). IFNAR1 and IFNAR2 are distinctly associated with TYK2 and JAK1, respectively. Activation of TYK2 and JAK1 results in activation of STAT1/2, as well as PI3K and MAPK, leading to the transcription of ISGs, including antiviral genes, type-I IFN genes, pro-inflammatory genes, as well as survival and anti-inflammatory genes, through ISRE, GAS, and CRE/SBE. Additionally, TYK2 may be associated with membrane proteins, such as ion channels, causing rapid modulation of cellular function. CRE, cyclic AMP response element; GAS, IFN-γ-activated sites; ISG, interferon-stimulated genes; ISRE, IFN-stimulated response elements; JAK1, Janus kinase 1; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; SBE, smad binding elements; STAT, signal transducer and activator of transcription; TYK2, tyrosine kinase 2. (B) IFN-γ and IFNGR signaling in the spinal cord for the pathogenesis of pain. IFN-γ can be released by T lymphocytes, astrocytes and microglial cells under pathological pain conditions (e.g., nerve injury). IFN-γ can directly and indirectly alter synaptic activity in spinal dorsal horn neurons and thereby contributes to hyperactivity (central sensitization). IFN-γ facilitates Ca2+-permeable AMPA receptors (AMPAR) coupled with IFNGR. These AMPAR lack GluR2 and induce neurotoxicity. IFN-γ can directly activate microglia and increase the release of glial mediators, such as IL-γ and TNF that modulate synaptic strength and facilitated monosynaptic C-fiber-evoked excitatory postsynaptic currents. IFNGR composes of IFNGR1 and IFNGR2 subunits, which are associated with the kinases JAK1 and JAK2. In canonical IFN-γ signaling, phosphorylation of JAK1 and JAK2 results in the phosphorylation of STAT1. A STAT1 homodimer translocates to the nucleus and binds to GAS in the promoters of IFN-γ-regulated genes (IGS) such as NOS-2 and IRF1. In cells that do not express STAT1, STAT3 can be phosphorylated by JAK1/JAK2, resulting in translocation of the STAT3 dimer to the GAS sites.
STATs are activated by IFN receptors and form either homodimers or heterodimers, causing rapid nuclear translocation and gene transcription through binding to appropriate promoter sites on IFN-stimulated genes (Figure 2A). Although the JAK-STAT pathway is the most significant pathway for IFN actions, STAT-independent signaling has also been documented (24–26). When binding to specific promoter sites, type-I IFNs can activate the ISG factor 3 (ISGF3) complex, a transcriptional factor that consists of the activated form of STAT1 and STAT2, in addition to IRF9. ISGF3 binds IFN-stimulated response elements (ISREs) to induce the expression of interferon-stimulated genes (ISGs), including antiviral genes and type-I IFNs themselves (Figure 2A). Global gene expression profiling studies have demonstrated that type-I IFN administration could induce the expression of hundreds to thousands of genes (27). Besides this canonical signaling pathway, activation of IFNAR1/2 can also result in non-canonical signaling, including the activation of PI3 kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways (2) (Figure 2A).
Type-I IFNs play a crucial role in the functioning of the immune system, mediating and coordinating different gene products to regulate defensive responses against virus infections. Type-I IFNs can induce antiviral gene products to deploy T cells against viruses (3). IFN-β can also activate the suppressor of cytokine signaling (SOCS) protein, which binds to phosphorylated tyrosine residues on JAKs and cytokine receptor chains to inhibit cytokine signaling (25). These cytokines are crucial to the production of both innate and adaptive immune responses. SOCS-1 also inactivates JAK2, decreasing the duration of IFN-γ signaling. This enables the helpful immunological actions of IFN-γ and inhibits the harmful actions of unregulated IFN-γ responses. SOCS-1 can also prevent the expression of the class II major histocompatibility complex (MHC) molecules and CD40, both of which are needed by antigen-presenting cells to activate T cells (28). Administration of type-I IFNs has been shown to be effective in treating several different types of viral infections, including hepatitis C and hepatitis B. Type-I IFNs have also been used to treat autoimmune diseases, such as multiple sclerosis (29), as well as cancers, such as lymphoma and sarcoma (28). Type-I IFNs were proposed as anti-inflammatory mediators by inducing anti-inflammatory cytokines (IL-10) and other immunosuppressive molecules (18), as well as blocking the expression of pro-inflammatory mediators, such as matrix metalloprotease 9 (MMP-9), MHC class II, ICAM-1, VCAM-1, TNF, CXCL8, and IL-12 (30, 31).
However, against acute viral infections, type-I IFNs can cause immunopathology, contributing to various systemic autoimmune diseases, including systemic lupus erythematosus, Aicardi-Goutieres syndrome, rheumatoid arthritis, systemic sclerosis, and Sjogren’s syndrome (28). Furthermore, long-term viral infections can result in high concentrations of type-I IFNs that can inhibit B cell activity or result in the production of immunosuppressive molecules (e.g. IL-10) (18). In the presence of type-I IFNs, activation of TLR4 by LPS can cause IL-10 production (10). Therefore, the pleiotropic effects of type-I IFNs are a double-edged sword, mobilizing immune cells to destroy viruses, bacteria, and other foreign bodies on the one hand, and inducing damaging neuroinflammation on the other hand. The next section will explore both the positive and negative effects of type-I IFNs in the nervous system in more detail. In contrast to type-I IFNs, type-II IFN differentially regulates gene expression and pain (Figure 2B).
IFN-α, IFN-β, and IFN-γ Regulate Pain via Neuro-Glial and Neuro-Immune Interactions
Early reports have demonstrated an antinociceptive action of IFN-α in the CNS (Table 1). In 1981, Blalock and Smith reported human leukocyte interferon, but not fibroblast interferons, might bind to opioid receptor (32, 43). Furthermore, intracerebral injection of IFN-α caused antinociception and suppression of spontaneous locomotion, which could be reversed by the opioid receptor antagonist naloxone. Unilateral microinjection of IFN-α (4, 8, 16 pmol) into the nucleus submedius, dose-dependently increased the hindpaw withdrawal latency from the noxious heat stimulus in rats, and this effect was specifically mediated by μ-opioid receptor. IFN-α also showed similar pharmacological properties as the opioid peptide β-endorphin (33, 44, 45). However, this early hypothesis of IFN-α binding to opioid receptor remains to be validated.
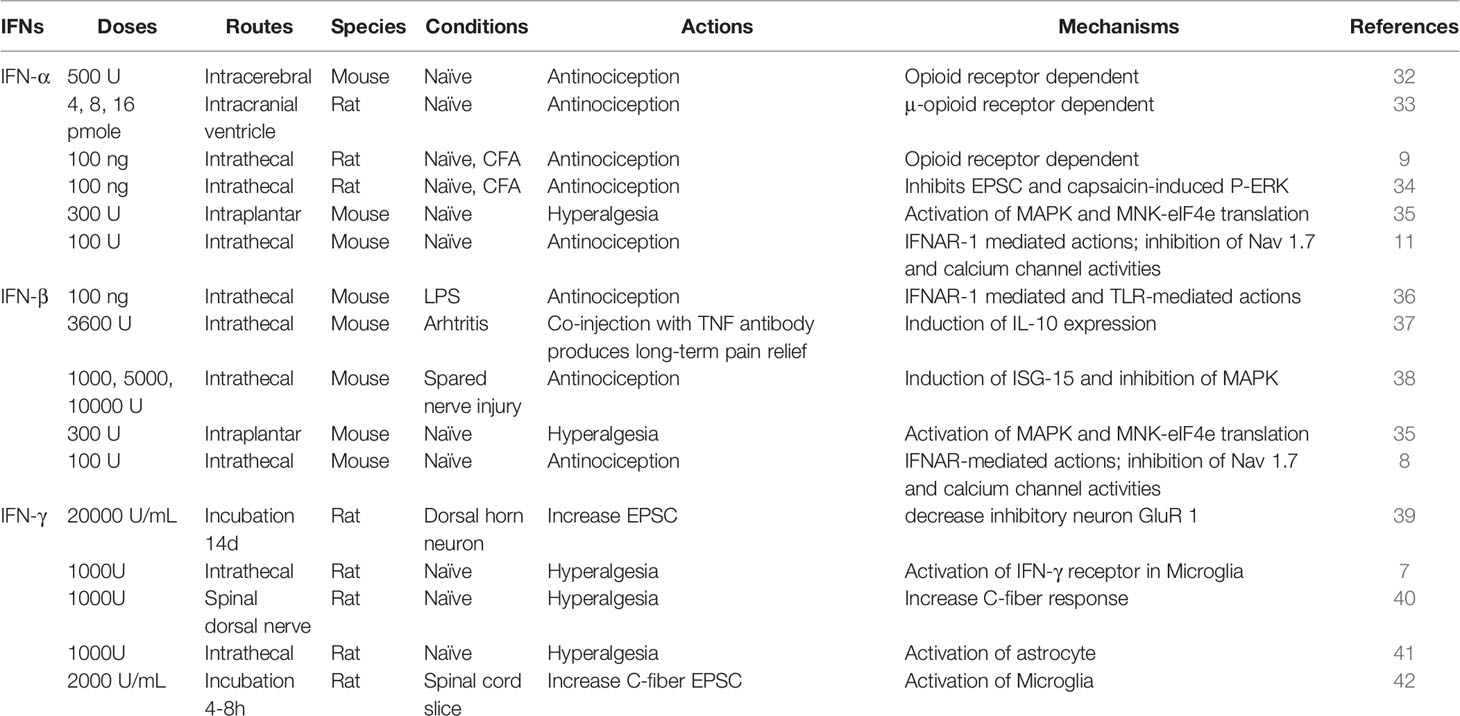
Table 1 Role of IFN-α, IFN-β, and IFN-γ in pain and the underlying mechanisms.
In 2012, we reported that intrathecal injection of short-interfering RNAs (siRNAs), at high doses (10 or 20 μg), could produce IFN-α-mediated analgesia, in a rat model of persistent inflammatory pain (9). This finding was very surprising, as the non-targeting siRNAs were used as controls that do not target specific pain genes. This is the first report demonstrating the analgesic effects of non-targeting double-stranded RNAs in the spinal cord, suggesting that caution must be taken when designing siRNAs for target validation in pain research. Notably, short double-stranded RNAs and short hairpin RNAs are able to induce type-I IFN responses in mammalian cells (46). We also found that IFN-α was markedly upregulated in the spinal cord after intrathecal administration of short (<21 bp) double-stranded RNA at a high dose (10 μg) (9). Importantly, the analgesic effect of non-targeting siRNAs can be reversed by intrathecal administration of IFN-α neutralizing antibody, supporting a critical involvement of IFN-α. Furthermore, intrathecal administration of IFN-α (30 and 100 ng) is sufficient to increase paw withdrawal latency in both naïve and inflamed rats in the radiant heat testing, in further support of an antinociceptive action of IFN-α (9).
Increasing evidence suggests that Type-I IFNs regulate pain via neuro-immune and neuro-glial interactions. TLR3 can sense double-stranded RNAs to trigger type-I IFN responses via TRIF signaling (Figure 1A) and regulates pain and itch (13, 46, 47). TLR3 is typically expressed by immune and glial cells (13). Intriguingly, TLR3 is also expressed by nociceptive sensory neurons and modulates nociceptive synaptic transmission in the spinal cord (11, 48, 49). It remains to be tested whether TLR3 is involved in the antinociception induced by the double-stranded RNAs. Von Frey testing revealed that intrathecal IFN-α (100 and 300 U) also increased paw withdrawal threshold in naïve mice (8).
Yaksh and co-workers have also demonstrated an analgesic action of IFN-β: intrathecal injection of IFN-β (100 ng) could relieve tactile allodynia induced by intrathecal TLR2 or TLR4 ligands (36). In a murine model of arthritis, intrathecal IFN-β elicited transient pain relief; however, co-administration of IFN-β and anti-TNF-α antibody produced pain relief for several weeks. This long-term pain relief may result from IFN-β-induced IL-10 expression in the spinal cord of male mice (37). Furthermore, Song and co-workers showed that a single intrathecal IFN-β administration (1000 and 5000 U) could attenuate the nerve injury-induced mechanical allodynia for several days in mice, without affecting open-field activity (38). Mechanistically, this long-lasting effect of IFN-β could be mediated by inhibition of MAPK activation, a key molecular mechanism underlying the pathogenesis of pain (50), and induction of interferon-stimulated gene 15 (ISG-15) after spared nerve injury in mice (38). Intrathecal IFN-β (100 and 300 U) also increased paw withdrawal threshold in naïve mice, in further support of an antinociceptive action of IFN-β (8).
In situ hybridization and single-cell RNAseq analyses revealed broad expression of Ifnar1 mRNA in various types of primary sensory neurons of dorsal root ganglion (DRG) of mice (8, 35, 49, 51). Furthermore, immunohistochemistry revealed the expression of IFN-α/β receptor (IFNAR) has limited expression in large fiber which expressed NF200, a cellular marker for large-myelinated neurons (Figure 3A), but expresses extensively in mouse DRG C-fiber nociceptive neurons that co-express the neuropeptide CGRP (34). Notably, LPS-induced tactile allodynia is prolonged in mice lacking Ifnar1 and this LPS-induced mechanical pain did not show any sign of recovery at 21 days of LPS in Ifnar1 null mice (36). Strikingly, selective deletion of Ifnar1 in Nav1.8-expressing nociceptors led to pain hypersensitivity and increased excitability of nociceptor neurons of naïve mice, suggesting an essential role of type-I IFN signaling in the control of nociception under the homeostasis condition (8). Conversely, incubation of dissociated wild-type DRG neurons with type-I IFN (30 U/ml) suppressed nociceptor excitability by inhibition of Nav1.7-mediated sodium currents and calcium currents (Figure 3B) (8).
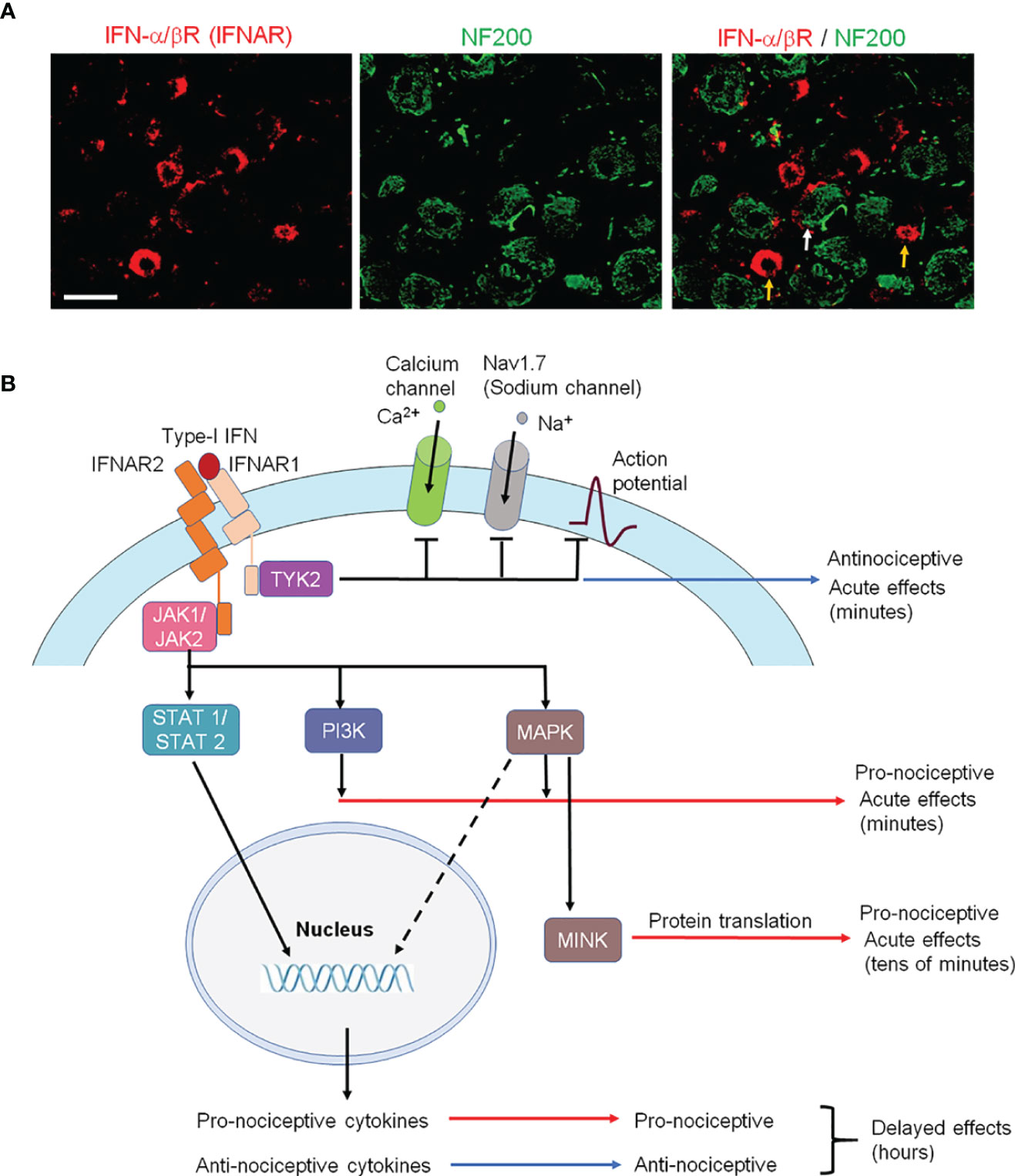
Figure 3 IFNAR signaling in nociceptive sensory neurons and its involvement in pronociceptive and antinociceptive effects. (A) Double staining of IFNα/βR (IFNAR) and NF200 in L3-L5 DRG neurons of naïve rats. Note that IFNAR is mainly expressed in NF200-negative nociceptive neurons (orange arrows) and some NF200-postive neurons (white arrow). Scale, 50 μm. Reproduced from Ref 95 (50) with permission. (B) Type-I IFN activates IFNAR1, resulting in subsequent TYK2 activation and rapid antinociception via inhibition of Na+ and Ca2+ channels and suppressing of action potential firing in nociceptors at central nervous system. IFNAR-mediated activation of JAK also results in activation of PI3K and MAPK pathways, as well as MINK/eIF4E-mediated translational pathway, leading to nociceptor sensitization at peripheral nervous system. Furthermore, IFNAR-mediated STAT activation causes delayed effects via induction of pro-inflammatory and anti-inflammatory cytokines, leading to pro-nociceptive and anti-nociceptive actions in the late-phase at central nervous system.
In the rat spinal cord, IFN-α is mainly expressed by astrocytes that express glial fibrillary acidic protein (GFAP, Figure 4A) (9, 34). IFN-α is also found in vesicle-like structures of astrocytic processes in primary cultures (Figure 4B), suggesting a vesicle-mediated release from astrocytes. Interestingly, IFN-α/β receptor (IFNAR) was found to be expressed in spinal cord axonal terminals co-expressing CGRP (Figure 4C). The result indicated a neuronal expression of IFNAR in presynaptic terminals (34), although we could not exclude IFNAR expression in other cell types. A direct modulation of synaptic transmission in the spinal cord nociceptive circuit by IFN-α has been demonstrated by ex vivo electrophysiology in spinal cord slices (Figures 4D–F) (52). Perfusion of spinal cord slices with IFN-α (50 U/ml, 3 min) resulted in a rapid suppression of excitatory synaptic transmission by reducing the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) in outer lamina II (IIo) excitatory interneurons expressing somatostatin (53). IFN-α also blocked the capsaicin-induced central sensitization (52), as revealed by 1) internalization of neurokinin-1 (NK-1) receptor and 2) phosphorylation of extracellular signal-regulated kinase (ERK) in superficial dorsal horn neurons (Figures 4G, H). When naive rats or mice were intrathecally injected with a neutralizing antibody to remove endogenous IFN-α or IFN-β, it resulted in hyperalgesia (8, 9). Activation of TLR3 by Poly (I:C) upregulates IFN-β in primary cultures of microglia and astrocytes (36). Additionally, Ifnar1 null mice exhibited increased EPSCs in the spinal cord pain circuit, compared with wild-type littermates (8). Thus, the respective expression of IFN-α and IFNAR in glial cells and neurons has provided a cellular basis for spinal cord neuro-glial interactions in regulating type-I IFN-mediated nociception (Figures 4I, J) (52).
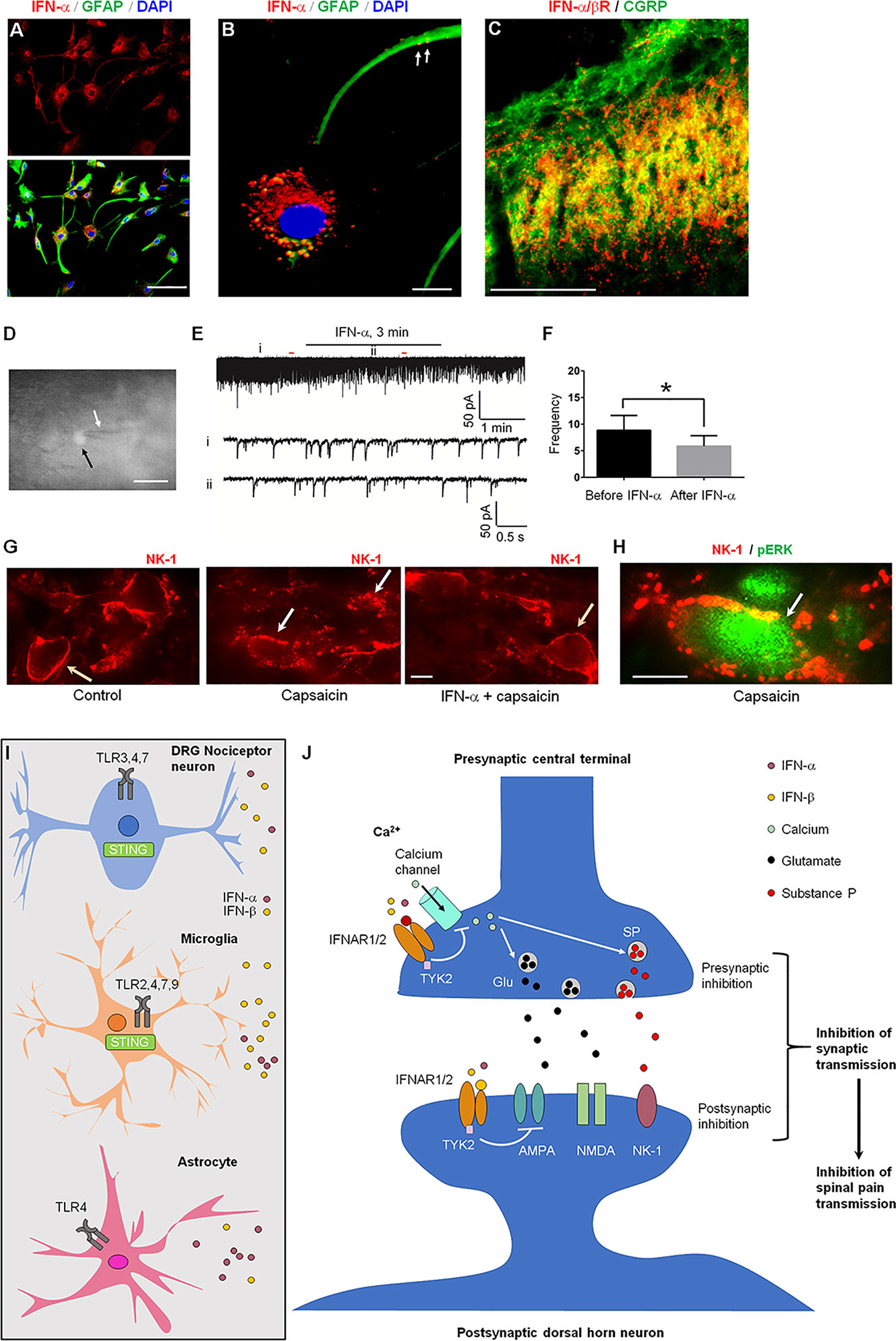
Figure 4 IFNAR signaling in the spinal cord for the inhibition of nociception. (A) IFN-α expression in cultured astrocytes. (B) Triple staining of IFN-α, GFAP, and nuclei marker DAPI in cultured astrocytes prepared from cerebral cortexes of neonatal Sprague-Dawley rats. Scales, 50 μm in A and 10 μm in B. Arrows indicate IFN-α-labeled vesicles in remote astrocyte processes. (C) Double staining of IFNAR (IFN-α/βR) and CGRP in the superficial dorsal horn. Scale, 100 μm. (D–F) IFN-α inhibits synaptic transmission in spinal cord slices. (D) The recorded neuron (somatostatin-positive, shown by white arrow) with an electrode (black arrow). (E) Patch clamp recording reveals an inhibition of spontaneous excitatory postsynaptic currents (sEPSCs) in lamina II neurons by IFN-α (25 ng/ml, 2 min). i and ii indicate traces before and after the treatment. (F) Frequency (Hz) of sEPSCs. 7 out of 9 recorded neurons respond to IFN-α. *P<0.05, n = 7 neurons. (G, H) IFN-α suppresses spinal nociceptive transmission. (G) Intraplantar capsaicin-evoked NK-1 internalization in superficial dorsal horn neurons is suppressed by intrathecal IFN-α (100 ng). The slices were stimulated with capsaicin (3 mM, 5 min) or pretreated with IFN-α 10 min before capsaicin stimulation. Orange and white arrows indicate surface-expressed and internalized NK-1 receptors, respectively. (H) Double staining showing co-localization of NK-1 (with internalization) and ERK phosphorylation (pERK) in a lamina I neuron (arrow) following capsaicin stimulation. Scales, 10 μm. A-H are reproduced from (50) with permission. (I, J) Type-I IFN and IFNAR signaling mediates neuro-glial interactions in the spinal cord for pain modulation. (I) IFN-α and IFN-β are produced by astrocytes, microglia, and nociceptive neurons following stimulation of TLRs and STING. (J) Activation of IFNAR inhibits nociception via both pre-synaptic and post-synaptic mechanisms. At the presynaptic sites, IFNAR activation inhibits calcium channels and neurotransmitter release. At the post-synaptic sites, IFNAR activation inhibits the activity of the glutamate AMPA receptors.
Recently, Donnelly et al. (8) reported STING-mediated antinociception in mice and nonhuman primates, which is governed by type-I IFNs (8). Strikingly, type-I IFN can rapidly suppress the excitability of mouse, monkey, and human nociceptors (8). Intrathecal injections of STING agonists produce sustained anti-nociception (24-48 h) in naïve mice, as well as mice with neuropathic pain and cancer pain, without showing signs of motor impairment. This antinociception by STING agonists is associated with increased expression of IFN-α and IFN-β in DRG and spinal cord tissues and abolished in Ifnar1 knockout mice. Notably, Sting knockout mice, as well as conditional knockout mice with selective deletion of Sting in nociceptor neurons, can recapitulate the phenotypes of Ifnar1 knockout mice, showing drastically increased pain sensitivity and neuronal hyperexcitability (8). Sting mRNA is expressed by both nociceptor neurons of DRG and microglia of spinal cord, suggesting that type-I IFNs can be produced by both neurons and glia (8) (Figure 4I, J).
There were also recent reports showing pronociceptive actions of type-I IFNs (35, 54, 55) (Table 1). Barragán-Iglesias, P. et al. found that intraplantar administration of either IFN-α or IFN-β (300 U in 25 μl), but not vehicle (saline), produced a persistent mechanical hypersensitivity to von Frey filament stimulation in mice, lasting for days. Furthermore, this persistent hyperalgesia depends on type-I IFN-induced MAP kinase activation and MNK/eIF4E-mediated translation in DRG neurons (35). Moreover, exposure of mouse DRG neurons to IFN-α (300 U/ml) increased nociceptor excitability (35). It is unclear if these pronociceptive actions of type-I IFN are mediated by IFNARs. Interestingly, mechanical hypersensitivity induced by high-dose intraplantar IFN-α can be blocked by intrathecal IFN-α (8), suggesting distinct actions of peripheral vs. central IFN-α. IFNAR1 and IFNAR2 are also widely expressed by vagal sensory neurons, including 70% of the TRPV1-positive neurons (presumably nociceptors). Calcium imaging revealed an acute activation of bronchopulmonary vagal nociceptors by IFN-α and IFN-β at high concentrations (1000 and 10,000 U/ml) (55). Another study showed that formalin-evoked nociceptive behavior is potentiated by systemic IFN-α administration for 8 days at very high doses (8000 IU/gram body weight/day) (54). It is important to note that peripheral IFN-β normally does not directly penetrate BBB to the CNS (56). Thus, the pronociceptive effects induced by intraplantar or systemic administration of type-I IFN is a result of peripheral and/or local actions. Hence, it is conceivable that the discrepancy of antinociceptive vs. pronociceptive effects of type-I IFNs may result from 1) location difference (peripheral vs. central) or 2) concentration difference (low vs. high) of type-I IFNs. Interestingly, TYK2 was implicated in the antinociceptive actions of type-I IFNs, and intrathecal TYK2 inhibitor blocked the STING agonist-induced analgesia and further induced hyperalgesia in naïve mice (8). It is also likely that distinct activation of downstream signaling pathways (e.g., TYK2 vs. PI3K/MAPK pathway) may lead to different regulations of neuronal excitability, as the activation of the PI3K/MAPK pathway is known to cause nociceptor sensitization (57, 58) (Figure 3B).
It is also noteworthy that high doses of type-I IFN treatment may generate autoantibodies, as anti-IFN-β antibodies did appear in a portion of MS patients (3-45%), especially in patients receiving subcutaneous formulation (59). IgG autoantibodies and the corresponding antigens can form the IgG-IC complex that can directly bind to the neuronal Fc-gamma receptor (FcγR) to activate nociceptors and promote pain (60). Neuromyelitis optica is an antibody-mediated autoimmune inflammatory disease of the CNS, caused by the loss of aquaporin-4 and GFAP that are mainly expressed by astrocytes. Type-I IFN contributes to the disease progression, as loss of aquaporin-4 and GFAP following intracerebral injection of IgG from a neuromyelitis optica patient was alleviated in mice lacking IFNAR (61). Patients with neuromyelitis optica also suffer from severe pain due to dysregulation of astrocytes and generation of aquaporin-4 autoantibodies (52, 62).
As the only Type-II IFN, IFN-γ was firstly reported to be able to inhibit viral replication (63). IFN-γ is released by subsets of NK cells, glial cells and activated T lymphocytes during infection and inflammation in the nervous system (64). IFN-γ not only has antiviral activity but also is involved in the polarization of the immune response and the regulation of macrophage plasticity by producing “M1” phenotype and release of pro-inflammatory cytokines such as IL-1β, IL-12, IL-23, and TNF-α (65). In general, IFN-γ signaling is short-lived to elicit functional recovery of homeostasis, including tissue repair and reestablishment of tissue physiology. Different from type 1 IFN binding to IFNAR1 and IFNAR2, IFN-γ signaling is mediated by a distinct receptor composed of the two subunits IFNGR1 and IFN-rR2 in a four-chain assembly (66) (Figure 2B), whose signal transduction cascade involves the activation of JAK1, JAK2 and the phosphorylation of STAT1(pSTAT1), STAT3, STAT5 and the indirect activation of the NF-kB module (67). pSTAT1 binds with high affinity to DNA sequences termed the γ-interferon-activated site (GAS) to initiate transcription of interferon-stimulated genes (ISG), iNOS and IRF1. Phosphorylation of C/EBPβ induced by IFN-stimulated proteolytic processing of ERK1 and ERK2 is necessary to control autophagy of infectious agents. Prolonged activation of IFN-γ resulted in the formation of the heterotrimeric transcription factor complex of STAT1 and IFN-regulatory factor 9 (Irf9), leading to altered expression of CXCL10. Non-canonical pathways could be activated in the absence of STAT1. For example, IFN-γ can activate STAT3 and subsequently activate GAS-regulated genes through JAK-STAT-dependent pathway. STAT-independent IFN-γ signaling can also occur via activation of MAPK signaling pathways. For example, IFN-γ can activate ERK1/2 to trigger binding of CCAAT/enhancer-binding protein-β (C/EBPβ) to a novel IFN-γ response element (GATE)(Figure 2B) In the CNS, IFNGR1/2 is expressed on neurons and glial cells in the spinal cord dorsal horn (6, 7, 68–70). IFN-γ is secreted by glial cells, neurons and infiltrating monocytes and is involved in promoting neuroinflammation but also neuroprotective processes such as neurogenesis and brain repair (64). Upregulation of IFN-γ was observed in the lumbar spinal dorsal horn after nerve injury (71–73). Intrathecal application of IFN-γ could induce persistent pain behavior in rodents (6, 7, 41, 72, 74), increase spinal nociceptive reflexes (75), evoked immediate biting behavior in rats (6). IFN-γ increased phospho-STAT1 levels in the spinal dorsal horn within minutes (7). Loss of IFNGR results in attenuation of tactile allodynia in mice after nerve injury (7, 71).
IFNGR has been reported to be located predominantly in postsynaptic sites on dendrites in lamina I and II and in axon terminals in the lateral spinal nucleus (68). Treatment of cultured dorsal horn neurons with IFN-γ resulted in enhanced spontaneous postsynaptic excitatory current (sEPSC), as well as reduction of AMPAR clustering on inhibitory interneurons, leading to disinhibition (39). Interestingly, disinhibition can also be induced by the pro-inflammatory cytokine IL-1β (76). Furthermore, prolonged but not acute treatment with IFN-γ facilitated monosynaptic C-fiber-evoked excitatory postsynaptic currents and this effect could be blocked by the application of minocycline, an inhibitor of microglial activation, suggesting an essential involvement of microglia (42) (Figure 2B). Stimulation of IFNGR in spinal microglia under normal conditions is sufficient to activate microglia and induce mechanical allodynia. By contrast, loss of IFNGR impaired nerve injury-induced microglia activation (7). However, another study found activation of spinal microglia after nerve injury was not completely eliminated in IFNGR-deficient mice, suggesting the involvement of IFNGR-independent mechanisms for the activation of microglia (77). Interestingly, local upregulation of IFN-γ in nucleus pulposus following disc herniation was associated with acute lumbar redicular pain (40). In this study, a significant increase in C-fiber response following application of NP and administration of IFN-γ on the dorsal nerve roots was observed. Interestingly, the single nucleotide polymorphisms (SNPs) rs2069705 in the promoter and rs2069718 in intron 3 may cause excessive expression of IFN-γ in systemic lupus erythematosus (SLE) patients (78, 79). A significant increase in Oswestry Disability Index (ODI) score, assessing physical function related to low back pain, was observed in patients with the IFN-γ rs2069705 and rs2069718 genotype (40). The potential mechanisms of disc herniation induced-neuropathic pain are central sensitization and via IFNG-GluR1 complex-GluR1 complex activated by IFN-γ (39, 80) (Figure 2B, Table 1).
Roles of Type-I IFNs in Infectious Diseases and COVID-19
Type-I IFNs play a major role in controlling encephalitis. During sterile neuroinflammation, microglia take on a pathological role; however, in viral encephalitis, their role remains unclear. Viruses can enter the CNS through the olfactory bulb (81). After intranasal vesicular stomatitis virus instillation, one study found increasing accumulation of microglia and monocytes in the olfactory bulb. Microglia depletion during encephalitis enhances virus spread, leading to increased lethality (81). Another study reported that type-I IFN signaling in astrocytes confers protection against viral encephalomyelitis and mediates IFN-γ-dependent responses (82). Furthermore, the type-I IFN receptor signaling on neurons has an important role in the activation of myeloid cells. In the infected CNS, communication between neurons, astrocytes, and microglia is essential to prevent encephalitis from becoming lethal (81). There is increased secretion of IFN-α and IFN-β in viral encephalitis (83) and secretion of IFN-α by microglia and astrocytes in human immunodeficiency virus-1 (HIV-1)-associated encephalitis was also reported (84). Aicardi-Goutières syndrome (AGS) is a genetic inflammatory disorder that affects the brain (encephalopathy), characterized by an increase in IFN-α levels in the brain. Interestingly, astrocytes were found to be the major source of IFN-α in AGS (85). Additionally, neurons differentiated either from the human NTera-2 cell line or isolated from mice infected in vivo with Theiler’s virus or La Crosse virus, can produce IFN-β (86). Human NTera-2 cell line contains pure neurons that progressively develop an extensive network of neurites and did not give rise to any glial cells, as tested by RT-PCR. Notably, neurons can produce IFN-α and IFN-β, in addition to macrophages/microglial cells and ependymal cells in mice infected by Theiler’s virus or La Crosse virus (83). However, fewer than 3% of infected neurons expressed type-I IFNs, suggesting limited production of type-I IFNs by neurons (83). It is noteworthy that IFN receptors are ubiquitously expressed in various cell types, such as macrophages, monocytes, T lymphocytes, glia, and neurons (1, 87). Following viral infections, a compliment-microglial axis results in synapse loss and memory impairment (88, 89), likewise the IL-1 and IL-1R axis inhibits adult neurogenesis and causes memory dysfunction in mice (90). Double stranded RNA is generated during viral replication and has been shown to drive anti-viral innate immune responses, sickness behavior and cognitive dysfunction, which is dependent on IFNAR1 expression and age (91). Interestingly, increased IFN-α levels were found in the CSF of HIV patients, which may contribute to HIV-induced dementia (84).
Type-I IFNs are also being examined for their role in the ongoing coronavirus disease 2019 (COVID-19) pandemic, resulting from the serve acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Type-I IFNs appear to be protective in the early stages of COVID-19. Park et al. proposes that human coronaviruses can produce viral proteins (e.g. a highly basic nucleocapsid protein (92) that inhibit IFNAR signaling, resulting in a diminished IFN response to inflammation, contributing to the pathogenesis of the disease. Type-I IFN autoantibodies may also contribute to severe COVID-19 (93, 94). Conversely, patients with severe COVID-19 showed elevated levels of IFN-α that correlated with disease severity and persisted even after disease resolution (95). IFN-β treatment has been proposed to treat patients in early stages of COVID-19, especially if applied at the site of infection, while immunomodulatory drugs may help reduce inflammation in patients in late stages of COVID-19 (96). Furthermore, type-I IFN response could exacerbate inflammation in patients with severe COVID-19, pointing to the coexistence of type-I IFN and TNF/IL-1β-driven inflammation in patients with severe COVID-19 (97). Delirium is a serious disturbance in mental abilities that is associated with confused thinking. It is noteworthy that delirium developed in 30% COVID-19 patients and was associated with increased mortality (98, 99). Cancer patients with neurologic sequelae of COVID-19 express leptomeningeal inflammatory cytokines in the absence of viral neuroinvasion, with elevated levels of IFN-γ in plasma and IFN-β in CSF (100).
Viral infections can either produce pain or inhibit pain in a context-dependent manner. Accumulating evidence suggests that nociceptors express pattern recognition receptors such as TLR3, TLR4, TLR5, and TLR7/TLR8 that can directly sense the presence of viruses, and viral infections are frequently associated with acute pain (11, 101, 102). Several types of viruses, such as herpes simplex virus (HSV) and varicella zoster virus can effectively infect sensory neurons, and in a latent phase, these viruses could be reactivated and produce intense and persistent pain through inducing skin inflammation and generating pro-inflammatory mediators (11, 103). It is also important to point out that infections can both produce pain and inhibit pain (11). HSV has been used as a gene therapy vector to deliver anti-inflammatory genes encoding IL-10 and IL-4 for sustained pain relief (104, 105). Intraplantar injection of HSV-1 increased mechanical sensory thresholds in rats (106). Furthermore, in early studies infection of rodent DRG neurons with HSV-1 or HSV-2 was shown to suppress sensory neuron excitability and sodium currents (107, 108). Intriguingly, patients not only feel pain but also experience paresthesia (numbness, tingling) after reactivation of HSV and subsequent ulceration. Thus, infections may inhibit or potentiate pain depending on the stage of the infection and distinct actions of viruses on immune cells and neurons (11, 102). Notably, many COVID survivors will also end up with chronic pain (109). The specific role of Type-I IFNs in pain sensitivity changes associated with viral infections remain to be investigated.
Concluding Remarks, Clinical Relevance, and Future Directions
Type-I IFNs, including IFN-α and IFN-β, are cytokines produced by almost every type of cell in the CNS and PNS. Type-I IFNs are essential for protecting the host against infection and have been used to treat different types of viral infections, autoimmune diseases, and cancers (28, 29). For example, recombinant IFN-β is currently used as treatment for relapsing-remitting multiple sclerosis (110, 111) and recombinant IFN-α is used to treat hepatitis (112). Systemic IFN-α therapy decreased bone metastases and increased metastasis-free survival in a murine model of breast cancer metastasis (113). Although type-I IFNs are involved in CNS inflammation and neurological disease, they can produce either protective or detrimental effects depending on the disease conditions. Inappropriate or chronic production of type-I IFNs by cells in the CNS can induce a number of diseases, including autoimmune diseases and chronic and congenital infections (114, 115), generally called “cerebral interferonopathies” (116). Prolonged exposure to type-I IFNs was reported to induce sickness behavior, including depression, anxiety, cognitive impairment, and even delirium in both mice and humans (56, 117).
IFNs are the best-known cytokines that are induced by virus infection to fight against infection, and a recent study shows that type-I IFN production is controlled by STING in the pain pathway and the STING/type-I IFN axis is present in the normal DRGs and controls physiological pain under the homeostasis condition. The role of type-I IFNs in pain is still controversial, as both pronociceptive actions (35, 55) and antinociceptive actions (8, 9, 33, 34, 36–38) of IFN-α and IFN-β have been reported, depending on the locations and doses (Table 1). This discrepancy may also depend on conditions (physiological vs. pathological) and phases (acute vs. chronic).
In summary, IFNs play important physiological roles in the nervous system, regulating various brain functions, including nociception. In the spinal cord, IFN-γ promotes pain while IFN-α and IFN-β inhibit nociception. IFN-α and IFN-β have been used to treat infectious disease, cancer, and MS. However, chronic production of IFNs contributes to chronic neuroinflammation, which is associated with aging, cognitive decline, and neurological diseases. Accumulating evidence suggests that IFN signaling is also critical for neuro-glial interactions in physiological and pathological conditions. New insights into neuro-glial modulations by IFNs may lead to new therapeutics for the control of pain and neurological diseases.
Author Contributions
P-HT contributed to the second part (pain modulation), the last part (clinical relevance), Figures 2–4, and Table 1 of this review. He also obtained the permission of reproducing Figure 3A and Figure 4. JJ contributed to the first part (introduction) and second part (IFNAR signaling) and prepared Figures 1 and 2A of the review. C-CY edited the manuscript. R-RJ edited all the sections and figures/tables. All authors contributed to the article and approved the submitted version.
Funding
The manuscript was partly supported by grants from the Taiwan Ministry of Science and Technology (MOST 108-2314-B-384 -008 -MY3), Chi Mei Hospital Grants (CMFHR 10902, CMNDMC11012).
Conflict of Interest
R-RJ is a consultant of Boston Scientific and received a research grant from the company. This activity is not related to this study.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Dr. Anthony Filiano of Duke University for critical reading of this manuscript. This study was supported in part by Duke University Anesthesiology Research Funds.
References
1. Pestka S, Langer JA, Zoon KC, Samuel CE. Interferons and Their Actions. Annu Rev Biochem (1987) 56:727–77. doi: 10.1146/annurev.bi.56.070187.003455
2. Ivashkiv LB, Donlin LT. Regulation of Type I Interferon Responses. Nat Rev Immunol (2014) 14:36–49. doi: 10.1038/nri3581
3. Pestka S. The Human Interferon-Alpha Species and Hybrid Proteins. Semin Oncol (1997) 24:S9–4-S9-17.
4. Platanias LC. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat RevImmunol (2005) 5:375–86. doi: 10.1038/nri1604
5. Xu XJ, Hao JX, Olsson T, Kristensson K, van der Meide PH, Wiesenfeld-Hallin Z. Intrathecal Interferon-Gamma Facilitates the Spinal Nociceptive Flexor Reflex in the Rat. Neurosci Lett (1994) 182:263–. doi: 10.1016/0304-3940(94)90812-5
6. Robertson B, Xu XJ, Hao JX, Wiesenfeld-Hallin Z, Mhlanga J, Grant G, et al. Interferon-Gamma Receptors in Nociceptive Pathways: Role in Neuropathic Pain-Related Behaviour. Neuroreport (1997) 8:1311–6. doi: 10.1097/00001756-199703240-00050
7. Tsuda M, Masuda T, Kitano J, Shimoyama H, Tozaki-Saitoh H, Inoue K. IFN-Gamma Receptor Signaling Mediates Spinal Microglia Activation Driving Neuropathic Pain. Proc Natl Acad Sci USA (2009) 106:8032–7. doi: 10.1073/pnas.0810420106
8. Donnelly CR, Jiang C, Andriessen AS, Wang K, Wang Z, Ding H, et al. STING Controls Nociception via Type I Interferon Signalling in Sensory Neurons. Nature (2021) 591(7849):275–80. doi: 10.1038/s41586-020-03151-1
9. Tan PH, Gao YJ, Berta T, Xu ZZ, Ji RR. Short Small-Interfering RNAs Produce Interferon-Alpha-Mediated Analgesia. Br J Anaesth (2012) 108:662–9. doi: 10.1093/bja/aer492
10. Akira S, Uematsu S, Takeuchi O. Pathogen Recognition and Innate Immunity. Cell (2006) 124:783–801. doi: 10.1016/j.cell.2006.02.015
11. Donnelly CR, Chen O, Ji RR. How Do Sensory Neurons Sense Danger Signals? Trends Neurosci (2020) 43:822–38. doi: 10.1016/j.tins.2020.07.008
12. Fitzgerald KA, Kagan JC. Toll-Like Receptors and the Control of Immunity. Cell (2020) 180:1044–66. doi: 10.1016/j.cell.2020.02.041
13. Liu T, Gao YJ, Ji RR. Emerging Role of Toll-Like Receptors in the Control of Pain and Itch. Neurosci Bull (2012) 28:131–44. doi: 10.1007/s12264-012-1219-5
14. Makris S, Paulsen M, Johansson C. Type I Interferons as Regulators of Lung Inflammation. Front Immunol (2017) 8:259. doi: 10.3389/fimmu.2017.00259 eCollection 2017.
15. Goubau D, Deddouche S, Reis e Sousa C. Cytosolic Sensing of Viruses. Immunity (2013) 38:855–69. doi: 10.1016/j.immuni.2013.05.007
16. Ishikawa H, Barber GN. STING is an Endoplasmic Reticulum Adaptor That Facilitates Innate Immune Signalling. Nature (2008) 455:674–8. doi: 10.1038/nature07317
17. Ishikawa H, Ma Z, Barber GN. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nature (2009) 461:788–92. doi: 10.1038/nature08476
18. McNab F, Mayer-Barber K, Sher A, Wack A, O’Garra A. Type I Interferons in Infectious Disease. Nat Rev Immunol (2015) 15:87–103. doi: 10.1038/nri3787
19. Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, et al. Mouse Type I IFN-Producing Cells Are Immature APCs With Plasmacytoid Morphology. Nat Immunol (2001) 2:1144–50. doi: 10.1038/ni736
20. Barchet W, Cella M, Odermatt B, Asselin-Paturel C, Colonna M, Kalinke U. Virus-Induced Interferon Alpha Production by a Dendritic Cell Subset in the Absence of Feedback Signaling In Vivo. J Exp Med (2002) 195:507–16. doi: 10.1084/jem.20011666
21. Biron CA. Interferons Alpha and Beta as Immune Regulators-a New Look. Immunity (2001) 14:661–4. doi: 10.1016/S1074-7613(01)00154-6
22. Pestka S, Krause CD, Walter MR. Interferons, Interferon-Like Cytokines, and Their Receptors. Immunol Rev (2004) 202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x
23. Taniguchi T, Takaoka A. A Weak Signal for Strong Responses: Interferon-Alpha/Beta Revisited. Nat Rev Mol Cell Biol (2001) 2:378–86. doi: 10.1038/35073080
24. Fu XY, Schindler C, Improta T, Aebersold R, Darnell JE Jr. The Proteins of ISGF-3, the Interferon Alpha-Induced Transcriptional Activator, Define a Gene Family Involved in Signal Transduction. Proc Natl Acad Sci USA (1992) 89:7840–3. doi: 10.1073/pnas.89.16.7840
25. Schindler C, Shuai K, Prezioso VR, Darnell JE Jr. Interferon-Dependent Tyrosine Phosphorylation of a Latent Cytoplasmic Transcription Factor. Science (1992) 257:809–13. doi: 10.1126/science.1496401
26. Shuai K, Schindler C, Prezioso VR, Darnell JE Jr. Activation of Transcription by IFN-Gamma: Tyrosine Phosphorylation of a 91-kD DNA Binding Protein. Science (1992) 258:1808–12. doi: 10.1126/science.1281555
27. Hertzog P, Forster S, Samarajiwa S. Systems Biology of Interferon Responses. J Interferon Cytokine Res (2011) 31:5–11. doi: 10.1089/jir.2010.0126
28. Wang H, Wang J, Xia Y. Defective Suppressor of Cytokine Signaling 1 Signaling Contributes to the Pathogenesis of Systemic Lupus Erythematosus. Front Immunol (2017) . 8:1292. doi: 10.3389/fimmu.2017.01292
29. Dumitrescu L, Constantinescu CS, Tanasescu R. Recent Developments in Interferon-Based Therapies for Multiple Sclerosis. Expert Opin Biol Ther (2018) 18(6):665–80. doi: 10.1080/14712598.2018.1462793
30. Benveniste EN, Qin H. Type I Interferons as Anti-Inflammatory Mediators. Sci STKE (2007) 2007(416):pe70. doi: 10.1126/stke.4162007pe70
31. Billiau A. Anti-Inflammatory Properties of Type I Interferons. Antiviral Res (2006) 71:108–16. doi: 10.1016/j.antiviral.2006.03.006
32. Blalock JE, Smith EM. Human Leukocyte Interferon: Structural and Biological Relatedness to Adrenocorticotropic Hormone and Endorphins. Proc Natl Acad Sci USA (1980) 77:5972–4. doi: 10.1073/pnas.77.10.5972
33. Jiang CL, Son LX, Lu CL, You ZD, Wang YX, Sun LY, et al. Analgesic Effect of Interferon-Alpha via Mu Opioid Receptor in the Rat. Neurochem Int (2000) 36:193–6. doi: 10.1016/S0197-0186(99)00124-2
34. Liu CC, Gao YJ, Luo H, Berta T, Xu ZZ, Ji RR, et al. Interferon Alpha Inhibits Spinal Cord Synaptic and Nociceptive Transmission via Neuronal-Glial Interactions. Sci Rep (2016) 6:34356. doi: 10.1038/srep34356
35. Barragan-Iglesias P, Franco-Enzastiga U, Jeevakumar V, Shiers S, Wangzhou A, Granados-Soto V, et al. Type I Interferons Act Directly on Nociceptors to Produce Pain Sensitization: Implications for Viral Infection-Induced Pain. J Neurosci (2020) 40(18):3517–32. doi: 10.1523/JNEUROSCI.3055-19.2020
36. Stokes JA, Corr M, Yaksh TL. Spinal Toll-Like Receptor Signaling and Nociceptive Processing: Regulatory Balance Between TIRAP and TRIF Cascades Mediated by TNF and IFNbeta. Pain (2013) 154:733–42. doi: 10.1016/j.pain.2013.01.012
37. Woller SA, Ocheltree C, Wong SY, Bui A, Fujita Y, Goncalves Dos Santos G, et al. Neuraxial TNF and IFN-Beta Co-Modulate Persistent Allodynia in Arthritic Mice. Brain Behav Immun (2019) 76:151–8. doi: 10.1016/j.bbi.2018.11.014
38. Liu S, Karaganis S, Mo RF, Li XX, Wen RX, Song XJ. IFNbeta Treatment Inhibits Nerve Injury-Induced Mechanical Allodynia and MAPK Signaling By Activating ISG15 in Mouse Spinal Cord. J Pain (2020) 21:836–47. doi: 10.1016/j.jpain.2019.11.010
39. Vikman KS, Hill RH, Backstrom E, Robertson B, Kristensson K. Interferon-Gamma Induces Characteristics of Central Sensitization in Spinal Dorsal Horn Neurons In Vitro. Pain (2003) 106(3):241–51. doi: 10.1016/S0304-3959(03)00262-8
40. Moen G-H, Moen A, Schistad EI, Gjerstad J. Local Up-Regulation of Interferon-γ (IFN-γ) Following Disc Herniation Is Involved in the Inflammatory Response Underlying Acute Lumbar Radicular Pain. Cytokine (2017) 97:181–6. doi: 10.1016/j.cyto.2017.06.005
41. Zhou YL, Zhou SZ, Li HL, Hu ML, Li H, Guo QH, et al. Bidirectional Modulation Between Infiltrating CD3þ T-Lymphocytes and Astrocytes in the Spinal Cord Drives the Development of Allodynia in Monoarthritic Rats. Sci Rep (2018) 8:51. doi: 10.1038/s41598-017-18357-z
42. Reischer G, Heinke B, Sandkühler J. Interferon-γ Facilitates the Synaptic Transmission Between Primary Afferent C-Fibres and Lamina I Neurons in the Rat Spinal Dorsal Horn via Microglia Activation. Mol Pain (2020) 16:1744806920917249. doi: 10.1177/1744806920917249
43. Menzies RA, Patel R, Hall NR, O’Grady MP, Rier SE. Human Recombinant Interferon Alpha Inhibits Naloxone Binding to Rat Brain Membranes. Life Sci (1992) 50:PL227–32. doi: 10.1016/0024-3205(92)90555-4
44. Wang JY, Zeng XY, Fan GX, Yuan YK, Tang JS. Mu- But Not Delta- and Kappa-Opioid Receptor Mediates the Nucleus Submedius Interferon-Alpha-Evoked Antinociception in the Rat. Neurosci Lett (2006) 397:254–8. doi: 10.1016/j.neulet.2005.12.046
45. Wang YX, Jiang CL, Lu CL, Song LX, You ZD, Shao XY, et al. Distinct Domains of IFNalpha Mediate Immune and Analgesic Effects Respectively. J Neuroimmunol (2000) 108:64–7. doi: 10.1016/S0165-5728(00)00271-X
46. Bridge AJ, Pebernard S, Ducraux A, Nicoulaz AL, Iggo R. Induction of an Interferon Response by RNAi Vectors in Mammalian Cells. Nat Genet (2003) 34:263–4. doi: 10.1038/ng1173
47. Szollosi AG, McDonald I, Szabo IL, Meng J, van den Bogaard E, Steinhoff M. TLR3 in Chronic Human Itch: A Keratinocyte-Associated Mechanism of Peripheral Itch Sensitization. J Invest Dermatol (2019) 139:2393–2396 e2396.
48. Liu T, Berta T, Xu ZZ, Park CK, Zhang L, Lu N, et al. TLR3 Deficiency Impairs Spinal Cord Synaptic Transmission, Central Sensitization, and Pruritus in Mice. J Clin Invest (2012) 122:2195–207. doi: 10.1172/JCI45414
49. Zheng Y, Liu P, Bai L, Trimmer JS, Bean BP, Ginty DD. Deep Sequencing of Somatosensory Neurons Reveals Molecular Determinants of Intrinsic Physiological Properties. Neuron (2019) 103(4):598–616.e7. doi: 10.1016/j.neuron.2019.05.039
50. Ji RR, Gereau RW, Malcangio M, Strichartz GR. MAP Kinase and Pain. Brain Res Rev (2009) 60:135–48. doi: 10.1016/j.brainresrev.2008.12.011
51. Renthal W, Tochitsky I, Yang L, Cheng YC, Li E, Kawaguchi R, et al. Transcriptional Reprogramming of Distinct Peripheral Sensory Neuron Subtypes After Axonal Injury. Neuron (2020) 108:128–144 e129. doi: 10.1016/j.neuron.2020.07.026
52. Ji RR, Donnelly CR, Nedergaard M. Astrocytes in Chronic Pain and Itch. Nat Rev Neurosci (2019) 20(11):667–85. doi: 10.1038/s41583-019-0218-1
53. Duan B, Cheng L, Ma Q. Spinal Circuits Transmitting Mechanical Pain and Itch. Neurosci Bull (2018) 34:186–93. doi: 10.1007/s12264-017-0136-z
54. Fitzgibbon M, Kerr DM, Henry RJ, Finn DP, Roche M. Endocannabinoid Modulation of Inflammatory Hyperalgesia in the IFN-Alpha Mouse Model of Depression. Brain Behav Immun (2019) 82:372–81. doi: 10.1016/j.bbi.2019.09.006
55. Patil MJ, Ru F, Sun H, Wang J, Kolbeck RR, Dong X, et al. Acute Activation of Bronchopulmonary Vagal Nociceptors by Type I Interferons. J Physiol (2020) 598:5541–54. doi: 10.1113/JP280276
56. Blank T, Prinz M. Type I Interferon Pathway in CNS Homeostasis and Neurological Disorders. Glia (2017) 65:1397–406. doi: 10.1002/glia.23154
57. Khoutorsky A, Price TJ. Translational Control Mechanisms in Persistent Pain. Trends Neurosci (2018) 41:100–14. doi: 10.1016/j.tins.2017.11.006
58. Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-Kinase Activates ERK in Primary Sensory Neurons and Mediates Inflammatory Heat Hyperalgesia Through TRPV1 Sensitization. J Neurosci (2004) 24:8300–9. doi: 10.1523/JNEUROSCI.2893-04.2004
59. Durelli L, Ricci A. Anti-Interferon Antibodies in Multiple Sclerosis. Molecular Basis and Their Impact on Clinical Efficacy. Front Biosci (2004) 9:2192–204. doi: 10.2741/1329
60. Wang L, Jiang X, Zheng Q, Jeon SM, Chen T, Liu Y, et al. Neuronal FcgammaRI Mediates Acute and Chronic Joint Pain. J Clin Invest (2019) 129:3754–69. doi: 10.1172/JCI128010
61. Khorooshi R, Wlodarczyk A, Asgari N, Owens T. Neuromyelitis Optica-Like Pathology Is Dependent on Type I Interferon Response. Exp Neurol (2013) 247:744–7. doi: 10.1016/j.expneurol.2013.02.005
62. Bradl M, Kanamori Y, Nakashima I, Misu T, Fujihara K, Lassmann H, et al. Pain in Neuromyelitis Optica-Prevalence, Pathogenesis and Therapy. Nat Rev Neurol (2014) 10:529–36. doi: 10.1038/nrneurol.2014.129
63. Wheelock EF. Interferon-Like Virus-Inhibitor Induced in Human Leukocytes by Phytohemagglutinin. Science (1965) 149:310–1. doi: 10.1126/science.149.3681.310
64. Wong G, Goldshmit Y, Turnley AM. Interferongamma But Not TNF Alpha Promotes Neuronal Differentiation and Neurite Outgrowth of Murine Adult Neural Stem Cells. Exp Neurol (2004) 187:171–7. doi: 10.1016/j.expneurol.2004.01.009
65. Sica A, Mantovani A. Macrophage Plasticity and Polarization: In Vivo Veritas. J Clin Invest (2012) 122:787–95. doi: 10.1172/JCI59643
66. Marsters SA, Pennica D, Bach E, Schreiber RD, Ashkenazi A. Interferon Gamma Signals via a High-Affinity Multisubunit Receptor Complex That Contains Two Types of Polypeptide Chain. Proc Natl Acad Sci USA (1995) 92:5401–5. doi: 10.1073/pnas.92.12.5401
67. Green DS, Young HA, Valencia JC. Current Prospects of Type II Interferon G Signaling and Autoimmunity. J Biol Chem (2017) 292:13925–33. doi: 10.1074/jbc.R116.774745
68. Vikman KS, Robertson B, Grant G, Liljeborg A, Kristensson K. Interferon-R Receptors Are Expressed at Synapses in the Rat Superficial Dorsal Horn and Lateral Spinal Nucleus. J Neurocytol (1998) 27:749–59. doi: 10.1023/A:1006903002044
69. Rubio N, de FC. Demonstration of the Presence of a Specific Interferon-R Receptor on Murine Astrocyte Cell Surface. J Neuroimmunol (1991) 35:111–7. doi: 10.1016/0165-5728(91)90166-5
70. Torres C, Ara´nguez I, Rubio N. Expression of Interferon-R Receptors on Murine Oligodendrocytes and Its Regulation by Cytokines and Mitogens. Immunology (1995) 86:250–5.
71. Costigan M, Moss A, Latremoliere A, Johnston C, VermaGandhu M, Herbert TA, et al. T-Cell Infiltration and Signaling in the Adult Dorsal Spinal Cord Is a Major Contributor to Neuropathic Pain-Like Hypersensitivity. J Neurosci (2009) 29:14415–22. doi: 10.1523/JNEUROSCI.4569-09.2009
72. Chen X-M, Xu J, Song J-G, Zheng B-J, Wang X-R. Electroacupuncture Inhibits Excessive Interferon-C Evoked Up-Regulation of P2X4 Receptor in Spinal Microglia in a CCI Rat Model for Neuropathic Pain. Br J Anaesth (2015) 114:150–7. doi: 10.1093/bja/aeu199
73. Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS Role of Toll-Like Receptor 4 in Innate Neuroimmunity and Painful Neuropathy. Proc Natl Acad Sci USA (2005) 102:5856–61. doi: 10.1073/pnas.0501634102
74. Vikman KS, Siddall PJ, Duggan AW. Increased Responsiveness of Rat Dorsal Horn Neurons In Vivo Following Prolonged Intrathecal Exposure to Interferon-R. Neuroscience (2005) 135:969–77. doi: 10.1016/j.neuroscience.2005.06.059
75. Xu X-J, Hao J-X, Olsson T, Kristensson K, van der Meide PH, Wiesenfeld-Hallin Z. Intrathecal Interferon-C Facilitates the Spinal Nociceptive Flexor Reflex in the Rat. Neurosci Lett (1994) 182:263–6. doi: 10.1016/0304-3940(94)90812-5
76. Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine Mechanisms of Central Sensitization: Distinct and Overlapping Role of Interleukin-1beta, Interleukin-6, and Tumor Necrosis Factor-Alpha in Regulating Synaptic and Neuronal Activity in the Superficial Spinal Cord. J Neurosci (2008) 28:5189–94. doi: 10.1523/JNEUROSCI.3338-07.2008
77. Scholz J, Woolf CJ. The Neuropathic Pain Triad: Neurons, Immune Cells and Glia. Nat Neurosci (2007) 10:1361–8. doi: 10.1038/nn1992
78. Kim K, Cho SK, Sestak A, Namjou B, Kang C, Bae SC. Interferon-Gamma Gene Polymorphisms Associated With Susceptibility to Systemic Lupus Erythematosus. Ann Rheum Dis (2010) 69(6):1247–50. doi: 10.1136/ard.2009.117572
79. Harigai M, Kawamoto M, Hara M, Kubota T, Kamatani N, Miyasaka N. Excessive Production of IFN-Gamma in Patients With Systemic Lupus Erythematosus and Its Contribution to Induction of B Lymphocyte Stimulator/B Cell-Activating Factor/TNF Ligand Superfamily-13B. J Immunol (2008) 181(3):2211–9. doi: 10.4049/jimmunol.181.3.2211
80. Mizuno T, Zhang G, Takeuchi H, Kawanokuchi J, Wang J, Sonobe Y. Et al. Interferon-Gamma Directly Induces Neurotoxicity Through a Neuron Specific, Calcium-Permeable Complex of IFN-Gamma Receptor and AMPA GluR1 Receptor. FASEB J (2008) 22(6):1797–806. doi: 10.1096/fj.07-099499
81. Chhatbar C, Detje CN, Grabski E, Borst K, Spanier J, Ghita L, et al. Type I Interferon Receptor Signaling of Neurons and Astrocytes Regulates Microglia Activation During Viral Encephalitis. Cell Rep (2018) 25:118–29 e114. doi: 10.1016/j.celrep.2018.09.003
82. Hwang M, Bergmann CC. Alpha/Beta Interferon (IFN-Alpha/Beta) Signaling in Astrocytes Mediates Protection Against Viral Encephalomyelitis and Regulates IFN-Gamma-Dependent Responses. J Virol (2018) 92(10):e01901–17. doi: 10.1128/JVI.01901-17
83. Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, et al. Neurons Produce Type I Interferon During Viral Encephalitis. Proc Natl Acad Sci USA (2006) 103:7835–40. doi: 10.1073/pnas.0602460103
84. Rho MB, Wesselingh S, Glass JD, McArthur JC, Choi S, Griffin J, et al. A Potential Role for Interferon-Alpha in the Pathogenesis of HIV-Associated Dementia. Brain Behav Immun (1995) 9:366–77. doi: 10.1006/brbi.1995.1034
85. van Heteren JT, Rozenberg F, Aronica E, Troost D, Lebon P, Kuijpers TW. Astrocytes Produce Interferon-Alpha and CXCL10, But Not IL-6 or CXCL8, in Aicardi-Goutieres Syndrome. Glia (2008) 56:568–78. doi: 10.1002/glia.20639
86. Prehaud C, Megret F, Lafage M, Lafon M. Virus Infection Switches TLR-3-Positive Human Neurons to Become Strong Producers of Beta Interferon. J Virol (2005) 79:12893–904. doi: 10.1128/JVI.79.20.12893-12904.2005
87. Aguet M. High-Affinity Binding of 125I-Labelled Mouse Interferon to a Specific Cell Surface Receptor. Nature (1980) 284:459–61. doi: 10.1038/284459a0
88. Salter MW, Stevens B. Microglia Emerge as Central Players in Brain Disease. Nat Med (2017) 23:1018–27. doi: 10.1038/nm.4397
89. Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A Complement-Microglial Axis Drives Synapse Loss During Virus-Induced Memory Impairment. Nature (2016) 534:538–43. doi: 10.1038/nature18283
90. Garber C, Vasek MJ, Vollmer LL, Sun T, Jiang X, Klein RS. Astrocytes Decrease Adult Neurogenesis During Virus-Induced Memory Dysfunction via IL-1. Nat Immunol (2018) 19:151–61. doi: 10.1038/s41590-017-0021-y
91. McGarry N, Murray CL, Garvey S, Wilkinson A, Tortorelli L, Ryan L, et al. Double Stranded RNA Drives Anti-Viral Innate Immune Responses, Sickness Behavior and Cognitive Dysfunction Dependent on dsRNA Length, IFNAR1 Expression and Age. Brain Behav Immun (2021) 95:413–28. doi: 10.1016/j.bbi.2021.04.016
92. Lu X, Pan J, Tao J, Guo D. SARS-CoV Nucleocapsid Protein Antagonizes IFN-Beta Response by Targeting Initial Step of IFN-Beta Induction Pathway, and Its C-Terminal Region is Critical for the Antagonism. Virus Genes (2011) 42:37–45. doi: 10.1007/s11262-010-0544-x
93. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies Against Type I IFNs in Patients With Life-Threatening COVID-19. Science (2020) 370(6515):eabd4585. doi: 10.1126/science.abd4585
94. de Prost N, Bastard P, Arrestier R, Fourati S, Mahevas M, Burrel S, et al. Plasma Exchange to Rescue Patients With Autoantibodies Against Type I Interferons and Life-Threatening COVID-19 Pneumonia. J Clin Immunol (2021) 41(3):536–44. doi: 10.1007/s10875-021-00994-9
95. Park A, Iwasaki A. Type I and Type III Interferons - Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe (2020) 27:870–8. doi: 10.1016/j.chom.2020.05.008
96. Sa Ribero M, Jouvenet N, Dreux M, Nisole S. Interplay Between SARS-CoV-2 and the Type I Interferon Response. PloS Pathog (2020) 16:e1008737. doi: 10.1371/journal.ppat.1008737
97. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and Influenza Highlights the Role of Type I Interferons in Development of Severe COVID-19. Sci Immunol (2020) 5(49):eabd1554. doi: 10.1126/sciimmunol.abd1554
98. Pranata R, Huang I, Lim MA, Yonas E, Vania R, Kuswardhani RAT. Delirium and Mortality in Coronavirus Disease 2019 (COVID-19) - A Systematic Review and Meta-Analysis. Arch Gerontol Geriatr (2021) 95:104388. doi: 10.1016/j.archger.2021.104388
99. Shao SC, Lai CC, Chen YH, Chen YC, Hung MJ, Liao SC. Prevalence, Incidence and Mortality of Delirium in Patients With COVID-19: A Systematic Review and Meta-Analysis. Age Ageing (2021) 50(5):1445–53. doi: 10.1093/ageing/afab103
100. Remsik J, Wilcox JA, Babady NE, McMillen TA, Vachha BA, Halpern NA, et al. Inflammatory Leptomeningeal Cytokines Mediate COVID-19 Neurologic Symptoms in Cancer Patients. Cancer Cell (2021) 39:276–83 e273. doi: 10.1016/j.ccell.2021.01.007
101. Chiu IM. Infection, Pain, and Itch. Neurosci Bull (2018) 34:109–19. doi: 10.1007/s12264-017-0098-1
102. Ji RR, Chamessian A, Zhang YQ. Pain Regulation by Non-Neuronal Cells and Inflammation. Science (2016) 354:572–7. doi: 10.1126/science.aaf8924
103. Silva JR, Lopes AH, Talbot J, Cecilio NT, Rossato MF, Silva RL, et al. Neuroimmune-Glia Interactions in the Sensory Ganglia Account for the Development of Acute Herpetic Neuralgia. J Neurosci (2017) 37:6408–22. doi: 10.1523/JNEUROSCI.2233-16.2017
104. Hao S, Mata M, Glorioso JC, Fink DJ. HSV-Mediated Expression of Interleukin-4 in Dorsal Root Ganglion Neurons Reduces Neuropathic Pain. Mol Pain (2006) 2:6. doi: 10.1186/1744-8069-2-6
105. Zhou Z, Peng X, Hao S, Fink DJ, Mata M. HSV-Mediated Transfer of Interleukin-10 Reduces Inflammatory Pain Through Modulation of Membrane Tumor Necrosis Factor Alpha in Spinal Cord Microglia. Gene Ther (2008) 15:183–90. doi: 10.1038/sj.gt.3303054
106. Andoh T, Shiraki K, Kurokawa M, Kuraishi Y. Paresthesia Induced by Cutaneous Infection With Herpes Simplex Virus in Rats. Neurosci Lett (1995) 190(2):101–4. doi: 10.1016/0304-3940(95)11512-u
107. Fukuda J, Kurata T, Yamaguchi K. Specific Reduction in Na Currents After Infection With Herpes Simplex Virus in Cultured Mammalian Nerve Cells. Brain Res (1983) 268:367–71. doi: 10.1016/0006-8993(83)90506-1
108. Oakes SG, Petry RW, Ziegler RJ, Pozos RS. Electrophysiological Changes of HSV-1-Infected Dorsal Root Ganglia Neurons in Culture. J Neuropathol Exp Neurol (1981) 40:380–89. doi: 10.1097/00005072-198107000-00003
109. Clauw DJ, Häuser W, Cohen SP, Fitzcharles MA. Considering the Potential for an Increase in Chronic Pain After the COVID-19 Pandemic. Pain (2020) 161(8):1694–7. doi: 10.1097/j.pain.0000000000001950
110. Fogarty E, Schmitz S, Tubridy N, Walsh C, Barry M. Comparative Efficacy of Disease-Modifying Therapies for Patients With Relapsing Remitting Multiple Sclerosis: Systematic Review and Network Meta-Analysis. Mult Scler Relat Disord (2016) 9:23–30. doi: 10.1016/j.msard.2016.06.001
111. Sormani MP, Bruzzi P. Can We Measure Long-Term Treatment Effects in Multiple Sclerosis? Nat Rev Neurol (2015) 11:176–82. doi: 10.1038/nrneurol.2014.237
112. Ferri C, Sebastiani M, Giuggioli D, Colaci M, Fallahi P, Piluso A, et al. Hepatitis C Virus Syndrome: A Constellation of Organ- and Non-Organ Specific Autoimmune Disorders, B-Cell Non-Hodgkin’s Lymphoma, and Cancer. World J Hepatol (2015) 7:327–43. doi: 10.4254/wjh.v7.i3.327
113. Parker BS, Rautela J, Hertzog PJ. Antitumour Actions of Interferons: Implications for Cancer Therapy. Nat Rev Cancer (2016) 16:131–44. doi: 10.1038/nrc.2016.14
114. Crow MK, Olferiev M, Kirou KA. Targeting of Type I Interferon in Systemic Autoimmune Diseases. Transl Res (2015) 165:296–305. doi: 10.1016/j.trsl.2014.10.005
115. Meyrelles ARI, Siqueira JD, Hofer CB, Costa TP, Azevedo AP, Guimaraes BV, et al. HIV/HPV Co-Infection During Pregnancy in Southeastern Brazil: Prevalence, HPV Types, Cytological Abnormalities and Risk Factors. Gynecol Oncol (2013) 128:107–12. doi: 10.1016/j.ygyno.2012.10.003
116. McGlasson S, Jury A, Jackson A, Hunt D. Type I Interferon Dysregulation and Neurological Disease. Nat Rev Neurol (2015) 11:515–23. doi: 10.1038/nrneurol.2015.143
Keywords: infection, microglia, astrocytes, IFN-α, IFN-β, IFN-γ, spinal cord, nociceptors
Citation: Tan P-H, Ji J, Yeh C-C and Ji R-R (2021) Interferons in Pain and Infections: Emerging Roles in Neuro-Immune and Neuro-Glial Interactions. Front. Immunol. 12:783725. doi: 10.3389/fimmu.2021.783725
Received: 26 September 2021; Accepted: 19 October 2021;
Published: 05 November 2021.
Edited by:
Lilian Basso, Toulouse Institute for Infectious and Inflammatory Diseases (INSERM), FranceReviewed by:
Sebastien Talbot, Université de Montréal, CanadaLaure Garnier, Université de Genève, Switzerland
Copyright © 2021 Tan, Ji, Yeh and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping-Heng Tan, dGFucGhwaGRAeWFob28uY29tLnR3