 Bu-Fan Xiao
Bu-Fan Xiao Jing-Tao Zhang
Jing-Tao Zhang Yu-Ge Zhu
Yu-Ge Zhu Xin-Run Cui
Xin-Run Cui Zhe-Ming Lu
Zhe-Ming Lu Ben-Tong Yu2*
Ben-Tong Yu2* Nan Wu
Nan Wu- 1Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education), Department of Thoracic Surgery II, Peking University Cancer Hospital & Institute, Beijing, China
- 2Department of Thoracic Surgery, The First Affiliated Hospital of Nanchang University, Nanchang, China
- 3Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Laboratory of Biochemistry and Molecular Biology, Peking University Cancer Hospital & Institute, Beijing, China
Chimeric antigen receptor T (CAR-T) cell therapy has exhibited a substantial clinical response in hematological malignancies, including B-cell leukemia, lymphoma, and multiple myeloma. Therefore, the feasibility of using CAR-T cells to treat solid tumors is actively evaluated. Currently, multiple basic research projects and clinical trials are being conducted to treat lung cancer with CAR-T cell therapy. Although numerous advances in CAR-T cell therapy have been made in hematological tumors, the technology still entails considerable challenges in treating lung cancer, such as on−target, of−tumor toxicity, paucity of tumor-specific antigen targets, T cell exhaustion in the tumor microenvironment, and low infiltration level of immune cells into solid tumor niches, which are even more complicated than their application in hematological tumors. Thus, progress in the scientific understanding of tumor immunology and improvements in the manufacture of cell products are advancing the clinical translation of these important cellular immunotherapies. This review focused on the latest research progress of CAR-T cell therapy in lung cancer treatment and for the first time, demonstrated the underlying challenges and future engineering strategies for the clinical application of CAR-T cell therapy against lung cancer.
1 Introduction
Lung cancer is one of the most frequently occurring malignant tumors worldwide and is characterized by a substantially high malignancy and poor prognosis (1). According to the latest global cancer statistics, lung cancer remains the leading cause of cancer-related deaths worldwide (2). Lung cancer can be histologically classified into two main subtypes: small-cell lung carcinoma (SCLC) and non-small-cell lung carcinoma (NSCLC) (3). NSCLC accounts for approximately 85% of diagnosed lung cancer cases and can be further divided into adenocarcinoma, squamous cell carcinoma, and large cell carcinoma (4, 5).
The present therapeutic measures for NSCLC primarily include surgical resection, chemoradiation, molecular-targeted therapy, and immunotherapy (6). The surgical resection procedure was based on the TNM stage of NSCLC patients. Conventional or stereotactic radiotherapy is applicable to patients with surgically unresectable NSCLC (7). Platinum-based double-agent combination chemotherapy is generally accepted as the standard chemotherapy regimen for NSCLC (8). Neoadjuvant chemotherapy is applied preoperatively to downgrade the cancer stage, whereas adjuvant chemotherapy is administered postoperatively, primarily involving cisplatin-based combination regimens (7). The primary molecular-targeted therapies include epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs), anti-EGFR monoclonal antibodies, fusion gene ALK and ROS1 inhibitors, and anti-vascular endothelial growth factor receptor monoclonal antibodies (9–12). Combined therapy with multiple immune checkpoint inhibitors, such as a combination of nivolumab and ipilimumab, has been shown to achieve better response rates than monotherapy (13, 14).
Non-surgical treatment involving systemic chemotherapy plus radiotherapy is the mainstream procedure for SCLC patients because metastases occur when SCLC is newly diagnosed. Etoposide-platinum and topotecan are the standard first-line and second-line regimens for SCLC patients, respectively (15, 16). Although SCLC is very sensitive to chemotherapy, many SCLC patients relapse due to the clinical development of chemoresistance. Moreover, nivolumab was the first FDA-approved immunotherapy agent for SCLC treatment (17). Several small molecular inhibitors, including PARP inhibitors, have also been demonstrated to exert anti-tumor activity in SCLC in clinical trials (18, 19). However, due to the heterogeneity of tumors, it is imperative to explore effective novel therapies.
Chimeric antigen receptors (CARs) are engineered receptors that can enable modified T cells to recognize and kill tumor cells expressing a tumor-specific antigen (20). CAR-T cells contain two sections: autologous T cells separated from the peripheral blood of patients and integration of CARs into T cells through genetic engineering in the laboratory. Patient’s T cells are extracted, isolated, and genetically engineered to express a CAR on their surface, targeting tumor-specific antigens of cancer cells. The modified CAR-T cells are amplified in vitro and then infused back into the patients (Figure 1) (21). Subsequently, CARs can identify and bind to specific antigens expressed on cancer cells and consequently eliminate and kill cancer cells (22, 23).
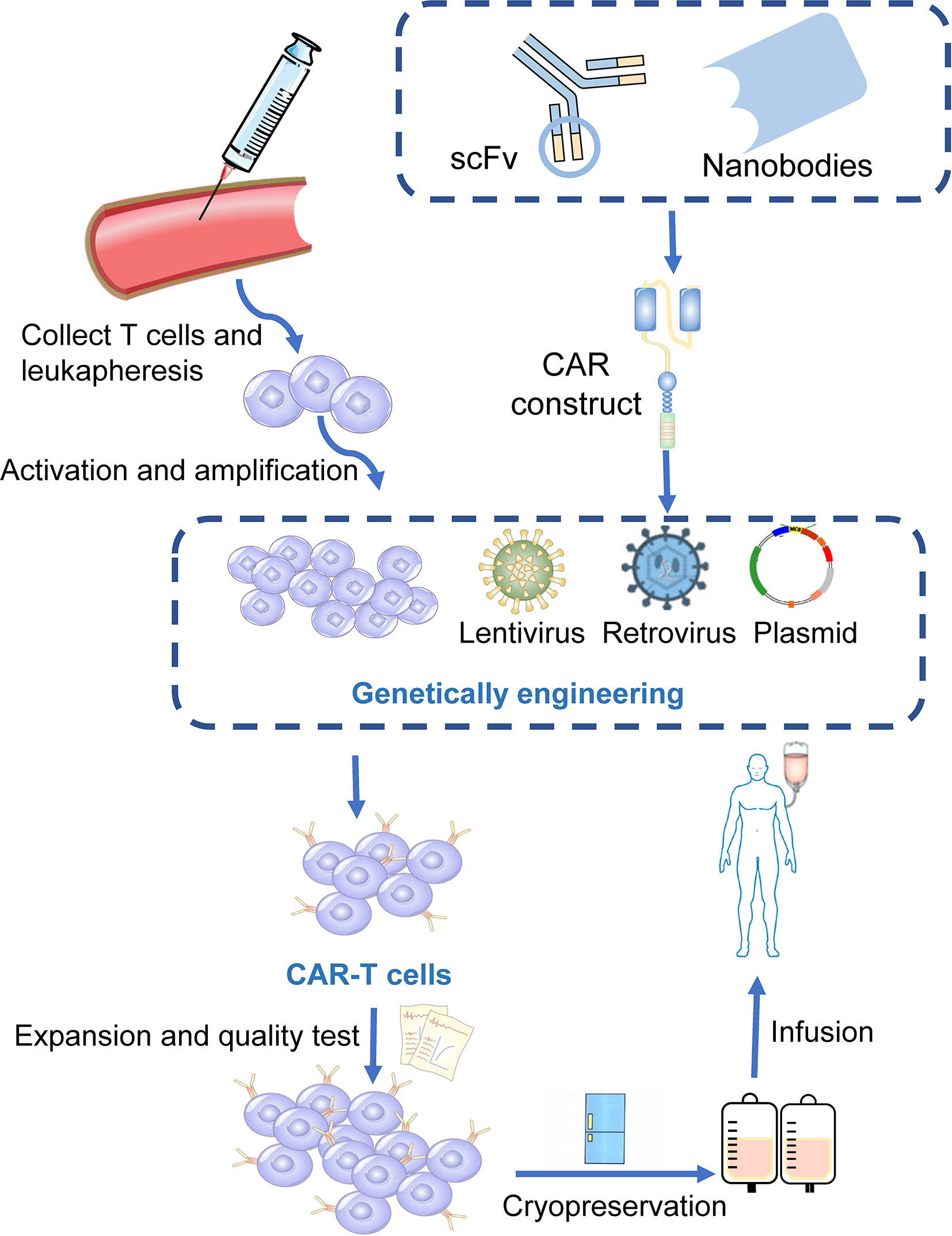
Figure 1 Manufacturing procedures of CAR-T cells. T cells are firstly collected from the peripheral blood of the patients. The activated and amplified T cells are genetically engineered with CAR structure via retroviral, lentivirus or other vectors. CAR-T cells are then expanded ex vivo and a quality control procedure is applied. Finally, those modified T cells were infused back into the patients.
CAR-T cell therapy is an emerging method against hematological malignancies and has demonstrated satisfactory curative effects, which is a substantial breakthrough in adoptive cell therapy (24, 25). CAR-T cells targeting CD19 have become a leading engineered T-cell therapy strategy against relapsed or refractory acute lymphocytic leukemia and B-cell non-Hodgkin lymphoma (26, 27). Yescarta (axicabtagene ciloleucel) and Kymriah (tisagenlecleucel) are currently approved to treat B-cell-derived malignancies, with response rates greater than 80% (28, 29). Recently, Tecartus (brexucabtagene autoleucel) has also been approved for the treatment of adult mantle cell lymphoma (30, 31). However, only targeting CD19 did not show considerable efficacy in most refractory multiple myeloma (MM) patients, partly due to the lower expression of CD19 on the cell surface of myeloma, and there is no FDA-approved CAR-T cell therapy against it (22, 32, 33). Clinical trials have indicated that CD269 (B cell maturation antigen, BCMA) and CD138 (also known as syndecan 1) molecules, which are mostly expressed in mature B cells or plasma cell surfaces, could exert substantial anti-MM activity (34–36). The unprecedented achievements of CAR-T cell therapy in hematological malignancies have also improved the use of CAR-T cells in various solid tumors.
1.1 The Design and Development of CAR Structure
CARs are artificial fusion proteins that comprise four major parts: extracellular antigen recognition and binding domains, spacer/hinge domains, transmembrane domains, and intracellular signaling domains (37, 38). Every component of the CAR structure has unique properties and has evolved to optimize the CAR function (39). The extracellular domains are responsible for recognizing and binding the targeted tumor-specific antigens, whereas intracellular signal domains primarily induce T-cell proliferation and corresponding signal transduction (Figure 2) (40). Recently, armored CAR-T cells have been engineered to overcome immunosuppressive tumor microenvironment (TME) (41). Engineered CAR-T cells can secrete various cytokines such as IL-12, chemokines, or co-expressing immunomodulatory ligands to alter the inhibitory microenvironment in the TME and support CAR-T cell function (20).
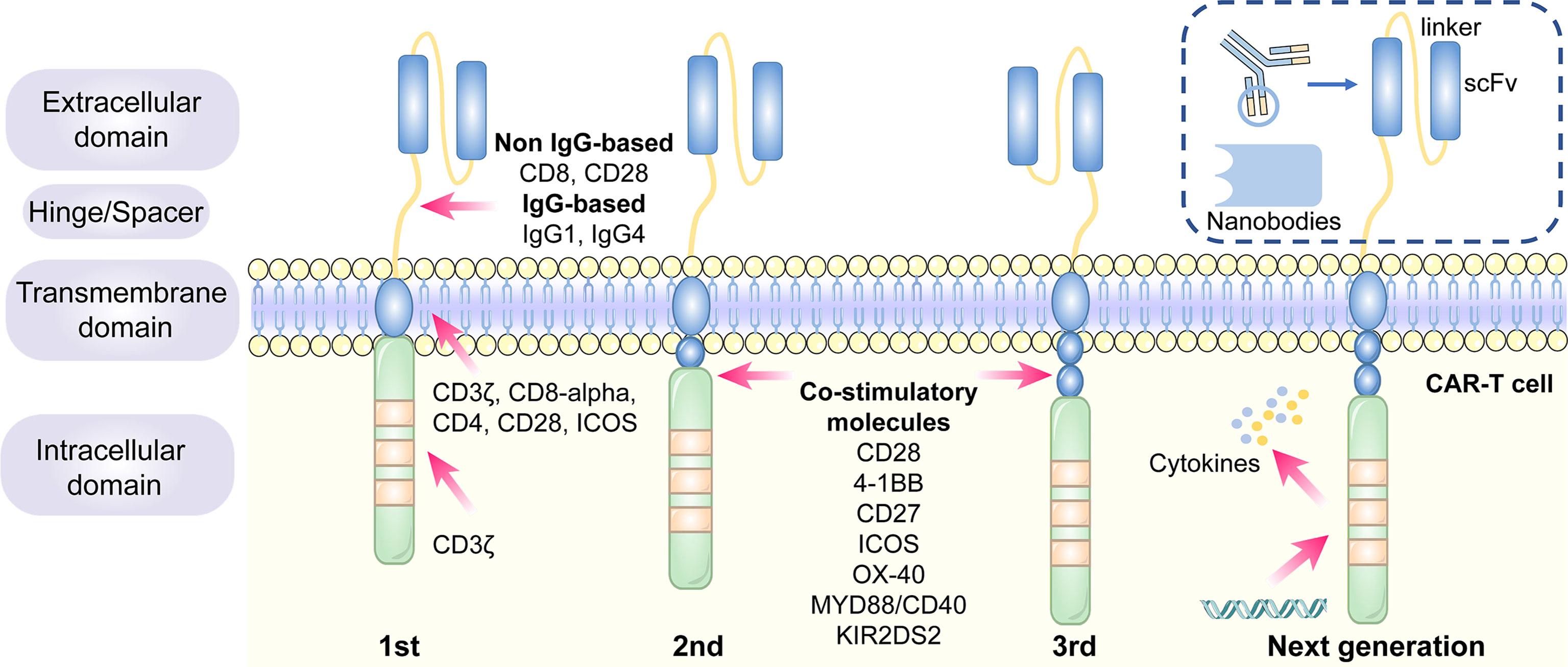
Figure 2 The structure and evolution of CAR-T cells from the first generation to the fourth generation. The CAR-T cells are consisted of extracellular tumor antigen binding domains (scFv, nanobodies), hinge regions, transmembrane regions and intracellular signaling domains. Different generations of CAR structures are primarily characterized by distinct intracellular signaling domains. The first generation of CAR-T cells only contain a CD3ζ intracellular signaling domain, with less persistence and efficacy in clinical practice. The second or third generation of CAR-T cells include one or more costimulatory molecules, and the next generation of CAR-T cells are engineered to express cytokines, which greatly improve their competence to eliminate the tumor cells.
1.1.1 Antigen Recognition and Binding Domains
The single-chain variable fragment (scFv) is derived from the variable heavy and variable light chains of a monoclonal antibody connected by a flexible linker (42). It is the major component of the extracellular antigen recognition and binding moieties, which can effectively recognize tumor antigen targets in a major histocompatibility complex (MHC)-independent manner and trigger CAR downstream signaling and CAR-T cells (43). The scFv sequences determine the specificity and binding affinity of the targeted antigens of the CAR (44). The high affinity of scFv has been reported to result in on-target, off-tumor toxicity, and severe cytokine release syndrome (45). Moreover, scFv can be designed to bind to soluble ligands, such as transforming growth factor-beta (TGF-β), contributing to the conversion of the immunosuppressive role of TGF-β (46). Single-domain antibodies (known as nanobodies or VHHs), whose variable regions only contain heavy chains instead of light chains, are stable camelid-derived single-domain antibodies (47). They are smaller in size and have a similar affinity to traditional scFv; however, they avoid the shortcomings of traditional scFv, such as low folding efficiency and tendency to aggregate (48, 49). In addition, cytokines (50), ligands (51–54) and antigen recognition peptides (adnectins and designed ankyrin repeat proteins) could be applied as an option for antigen recognition and binding regions of CARs (55, 56).
1.1.2 Hinge Domains
The length of the hinge regions can be adjusted to optimize the distance between CAR-T cells and targeted tumor cells, ensuring the folding efficiency of CAR scFv and providing a flexible and persistent connection for CAR signal transduction (57). In addition, the domains also augment the binding affinity of CAR-T cells and targeted cells (38). Hinge domains play a crucial role in regulating the expression and transport efficiency of CAR and the definition of the CAR signaling threshold (57). The spacer domains enable the CAR to access target epitopes that are otherwise sterically inaccessible (58). They can also be used to modulate synaptic cleft distances, as distal membrane antigen epitopes commonly require shorter spacers, whereas proximal membrane antigen epitopes require longer spacers (58, 59). Non-IgG-based spacers, including CD8 and CD28, and IgG-based spacers, such as IgG1 or IgG4, have been proven to be equally effective and are utilized in the construction of CAR hinge domains (58, 60). The spacers containing Fc domains must be changed after recognizing the targeted antigens, in case of in vivo interactions with cells expressing Fc gamma receptors that result in off-target activation of CAR-modified T cells or impaired antitumor efficacy (61).
1.1.3 Transmembrane Domains
The transmembrane domains serve as anchors to connect the extracellular antigen-binding domain to the cell membrane and transduce extracellular antigen-recognition signals to the intracellular domains (38, 58). They primarily originate from type I transmembrane proteins, including CD3ζ, CD8-alpha, CD4, or CD28 (20, 62). The stability and function of CARs are associated with transmembrane domains (38). Bridgeman et al. reported that CARs containing the CD3ζ transmembrane domain can form a complex with endogenous T cell receptor (TCR), and subsequently, may induce T cell activation (63). In vivo studies indicated that CD8-alpha resulted in lower levels of inflammatory cytokines and T-cell activation-induced death than CD28 (64). CD28 is currently the most stable transmembrane domain (39). Third-generation CAR T cells carry a B7-family inducible costimulator (ICOS) transmembrane domain (65). The persistence and anti-tumor activity of CAR-T cells is substantially promoted when the ICOS transmembrane domain is connected to an ICOS intracellular domain (62).
1.1.4 Intracellular Signaling Domains
The endodomains normally comprise a CD3ζ transducer, and one or more co-stimulatory signaling molecules such as CD28, 4-1BB (CD137), CD27, ICOS, OX-40, MYD88/CD40, and KIR2DS2 (66). This design pattern further prolongs the survival time and promotes the proliferation and antitumor activities of CAR-T cells (38, 67, 68). CD28 and 4-1BB, fused to the intracellular CD3ζ domain, are the most extensively studied and intensively applied co-stimulatory molecules (69). However, their clinical efficacy is far from each other. CAR-T cell therapy based on 4-1BB costimulatory domain is generally admitted to have more superior clinical efficacy, because 4-1BB costimulatory domain could ameliorate the exhaustion mediated by CAR signaling (70, 71). CAR-T cell product based on CD28 costimulatory domain initiates faster antitumor property, while compared with 4–1BB costimulatory domain, it is less persistent since fewer central memory T cells are formed (72) (Table 1). Additionally, CAR-T cells, incorporated two costimulatory molecules, such as ICOS and 4-1BB, have showed tremendous efficacy in preclinical mouse models (62, 73).The other co-stimulatory signaling molecules, including CD27 (74, 75), OX-40 (76, 77), MYD88/CD40 (78) and KIR2DS2 (79) have demonstrated promising efficacy in preclinical models but have not been tested in clinical trials.

Table 1 Comparison of properties of different costimulation 4-1BB versus CD28 in CAR-T cell.
1.2 The Generation of CAR−T Cells
Different generations of CAR structures, characterized by distinct intracellular signaling domains, have been designed to improve the safety and efficacy of CAR-T cell therapy against various cancers (80). First-generation CAR-T cells only contain one intracellular signaling domain, CD3ζ, with less impressive clinical efficacy for the lack of persistence and proliferative activity (38). Inclusion of the costimulatory molecules equipped with second-generation CAR-T cells with the necessary signals for activation considerably prolonged the survival time of CAR-T cells and improved clinical outcomes in cancer patients (81). Third-generation CAR-T cells aggrandize a costimulatory molecule compared with second-generation CAR-T cells, consisting of CD3ζ and two costimulatory molecules (CD27, CD28, 41BB, ICOS, OX-40, etc.), further augmenting and enhancing their competence to clear tumor cells (82, 83). In particular, the fourth generation of CAR-T cells known as T cells redirected for universal cytokine-mediated killing (TRUCK), which can recruit nuclear factor of activated T cells (NFAT) to induce the release of cytokines IL-12 IL-15 and granulocyte–macrophage colony-stimulating factor (84). The anti-tumor activity of the fourth generation of CAR-T cells is enhanced by overcoming the immunosuppressive effect of the TME (Figure 2). The fifth-generation CAR-T cells, which is proposed to remove the TCR alpha and beta chains through gene editing technology, avert the risk of graft-vs.-host disease, and manufacture “off the shelf” products, are still under investigation (85).
Although the structure of CARs is constantly evolving to promote efficacy and diminish the cytotoxic effects of CAR-T cell therapy, second-generation CAR-T cells still remain the mainstay of clinical application (86).
1.3 NSCLC and SCLC−Associated Antigens for CAR−T Cell Therapy in Preclinical Studies
CAR-T cell therapy has emerged as a novel approach to adoptive cell immunotherapy in recent decades. In solid cancers, it is more complex to construct CAR-T cells because it is difficult to identify tumor-specific antigens to be targeted. Several surface antigens have already been evaluated in preclinical studies as potential CAR-T cell therapy targets. Thereafter, we provide detailed descriptions of several novel targets.
1.3.1 Mesothelin (MSLN)
MSLN, a tumor differentiation antigen with the low expression on normal mesothelial cells, is overexpressed in a wide range of solid cancers, including lung cancer, mesothelioma, and pancreatic carcinoma; therefore, it could be used as a potential target (87, 88). High expression of MSLN is commonly correlated with negative clinical outcomes in NSCLC (67). In ex vivo experiments, MSLN-targeted CAR-T cells exerted substantial inhibitory effects on cancer cell proliferation and invasion (89). The efficiency of MSLN-targeted CAR-T cell therapy has been assessed in subcutaneous mouse lung cancer models (90). A slower growth rate of tumor size was observed in the tail vein injection of MSLN-targeted CAR-T cells (89). In summary, MSLN-targeted CAR-T cells could be feasible for MSLN-positive cancers, such as NSCLC.
1.3.2 EGFR
EGFR belongs to the HER/ErbB family of receptor tyrosine kinases that transduces extracellular growth signaling into the cells (91). More than 60% of NSCLC patients harbor activating EGFR mutations, contributing to the overexpression of EGFR, making it possible to target EGFR as a treatment for CAR-T cell therapy against NSCLC (91). EGFR-CAR T cells were found to exhibit greater cytotoxic activity in vitro (92). In nude mouse subcutaneous xenografts, EGFR-CAR T cells dramatically decreased tumor size and volume (93). The above results indicate that EGFR-targeted CAR-T cell therapy could be applied to NSCLC patients in the future (94).
1.3.3 Receptor Tyrosine Kinase-Like Orphan Receptor 1 (ROR1)
ROR1 is a crucial oncofetal glycoprotein that can sustain pro-survival and pro-apoptotic signaling in lung adenocarcinomas (95, 96). It has been proposed as a targeted antigen in CAR-T cell therapy as the overexpression of ROR1 protein has been observed in various malignancies, including lung cancer (97, 98). ROR1-CAR T cells maintained their anti-tumor activity, cytokine secretion, and proliferation in NSCLC models in vitro and in vivo (97, 99). Carolina et al. demonstrated the safety and function of second-generation ROR1 CAR-T cells in macaques (100).
1.3.4 Mucin-1 (MUC1) and Prostate Stem Cell Antigen (PSCA)
Aberrant high expression of MUC1 regulates the expression of programmed death-ligand 1 (PD-L1) in cancer cells, which could prevent cancer cells from being cleared by the immune system (101, 102). PSCA, a glycosylphosphatidylinositol (GPI)-anchored cell surface protein, belongs to the Thy-1/Ly-6 family (103). MUC-CAR T cells and PSCA-CAR T cells identify and eliminate PSCA+ or MUC1+ NSCLC cells, respectively, in vitro (104). PDX mouse subcutaneous models generated from NSCLC patients whose tumors only express PSCA or both PSCA and MUC1 were applied to explore the efficacy of PSCA and MUC1 CAR-T cells against NSCLC. Tumor growth was substantially inhibited in CAR-PSCA T cells. Thereafter, a combination of PSCA and MUC1 CAR-T cells exerted a synergistic effect on tumor survival (104). Therefore, MUC1 and PSCA could be promising CAR-T cell therapy targets for the treatment of NSCLC.
1.3.5 Human Epidermal Growth Factor Receptor 2 (HER2)
HER2 belongs to the HER/ErbB family of receptor tyrosine kinases involved in cell proliferation and angiogenesis (105). The anti-tumor effect of HER2 CAR-T cells against two NSCLC cell lines, A549 and H1650, was observed in a 96-h co-culture assay (106). Moreover, in orthotopic or subcutaneous A549 NSCLC mouse xenograft models, HER2 CAR-T cell therapy decreased tumor growth and could not completely eliminate tumors (106, 107).
1.3.6 Carcinoembryonic Antigen (CEA)
CEA is an oncofetal glycoprotein generally expressed during fetal development; however, its expression declines after birth (108). CEA levels increase rapidly in the tumorigenesis and development of lung cancer (109). Therefore, preclinical studies of CAR-T cell therapy targeting CEA have been conducted. CEA-targeted CAR-T cells have been found to eradicate advanced lung carcinomas (110).
1.3.7 PD-L1
Immunotherapy targeting programmed death-1(PD-1)/PD-L1 signaling has achieved substantial progress in NSCLC treatment. Accumulating evidence shows that PD-L1, both in tumor cells and in the TME, suppresses T cell proliferation and mediates anti-tumor immunity (111). PD-L1-targeted CAR-T cells exhibited robust cytotoxic effects against NSCLC cells in vitro and in vivo (112, 113). Therefore, PD-L1-targeted CAR-T cells could be a novel curative approach for PD-L1-positive NSCLC patients.
1.3.8 Fibroblast Activation Protein (FAP)
FAP is a marker expressed on cancer- associated fibroblasts (CAFs) in a majority of human malignancies (114). FAP molecule itself and FAP-positive cells in TME could contribute to cancer cell proliferation, invasion, angiogenesis and extracellular matrix (ECM) remodeling (115).
FAP targeted CAR-T cells inhibited the proliferation of TC1 and A549 lung cancer cells by eliminating FAP-positive stromal cells in mice models (114, 116). In contrast, another study claimed that FAP targeted CAR-T cell achieved limited antitumor efficacy and severe side effects for bone marrow stromal cells (BMSCs) were also being killed (117). Therefore, the feasibility of targeting FAP as a specific antigen in CAR-T therapy remains to be verified.
1.3.9 Other Targeted Antigens
Several tumor antigens, such as lung-specific X (LUNX), variant domain 6 of CD44 gene, melanoma-associated antigen-A1 (MAGE-A1), erythropoietin-producing hepatocellular carcinoma A2 (EphA2), and glypican-3 (GPC3), are under active investigation for application as targeted antigens of CAR-T cell therapy against NSCLC (118–122). For SCLC, CD56-and Delta-like ligand 3 (DLL-3)-targeted CAR-T cells are being explored (123, 124). Bivalent tandem CAR-T cells are equipped with two targeted antigens. CD70, B7-H3, MUC1, PSCA, PD-L1, and CD80/CD86, have exhibited enhanced antitumor efficacy in lung cancer (104, 125). B7-H3 is one of inhibitory ligands, which belongs to B7 immunoglobulin family. Although its corresponding immune checkpoint receptors remain undetermined, the inhibitory role of B7-H3 has been confirmed in preclinical studies (126). The expression of B7-H3 is aberrantly augmented in a wide range of solid tumor tissues, compared with normal tissues, which supports the possibility of targeting B7-H3 in CAR-T cell therapy against lung cancer (125, 127). CD80/CD86 are immune checkpoint ligands shared by inhibitory CTLA-4 and costimulatory CD28. CD80/CD86-targeted CAR-T cells have been generated to reverse the inhibitory CTLA4-CD86/CD86 signals and prevent the survival of B cell malignancies and other tumors including NSCLC (128). The efficacy of CAR-T cell therapy, which targets both tumor cells and tumor-associated macrophages in the TME, has also been validated in NSCLC (129) (Figure 3).
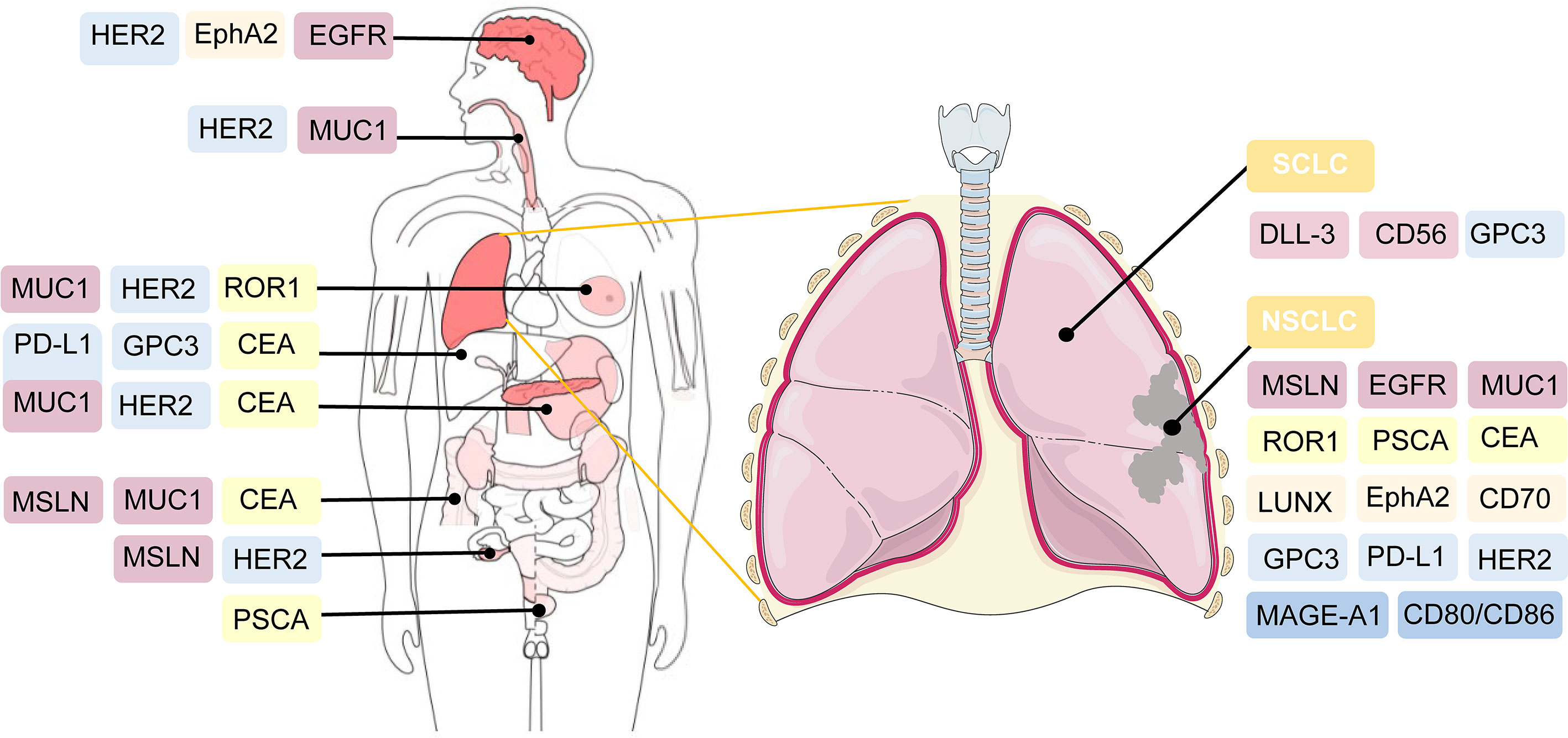
Figure 3 Potential targeted antigens for CAR-T cell therapy in preclinical and clinical trials. In the right, antigen targets are listed against SCLC and NSCLC. As shown in the left of the figure, these antigens are also broadly applied in CAR-T cell therapy against other solid tumors.
1.4 NSCLC and SCLC−Associated Antigens for CAR−T Cell Therapy in Clinical Trials
CAR-T cell treatment has achieved substantial success against several hematological malignancies. At present, the primary task is to broaden the applications of CAR-T cell therapy from merely hematologic tumors to multiple solid tumors. Thus, its safety and efficacy in solid cancers are under intensive investigation. The feasibility of CAR-T therapy against solid tumors is currently being evaluated in approximately one-third of CAR-T clinical trials (130). Among them, the majority are on CAR-T therapy for the treatment of lung cancer. The extraordinary progress of CAR-T therapy for lung cancer is promising; however, many challenges and hurdles exist. Therefore, the clinical application of CAR-T in NSCLC and SCLC treatment is still under intensive exploration. The optimal target for CAR-T cell therapy is specifically expressed or generally overexpressed in tumor cells, whereas it is expressed at very low or limited levels in normal peripheral cells or tissues (131). Current clinical trials of CAR-T therapy against NSCLC and SCLC primarily focus on MSLN, MUC1, GPC3, PSCA, EGFR, CEA, HER2, PD-L1, ROR1, and other promising targets (Table 2).
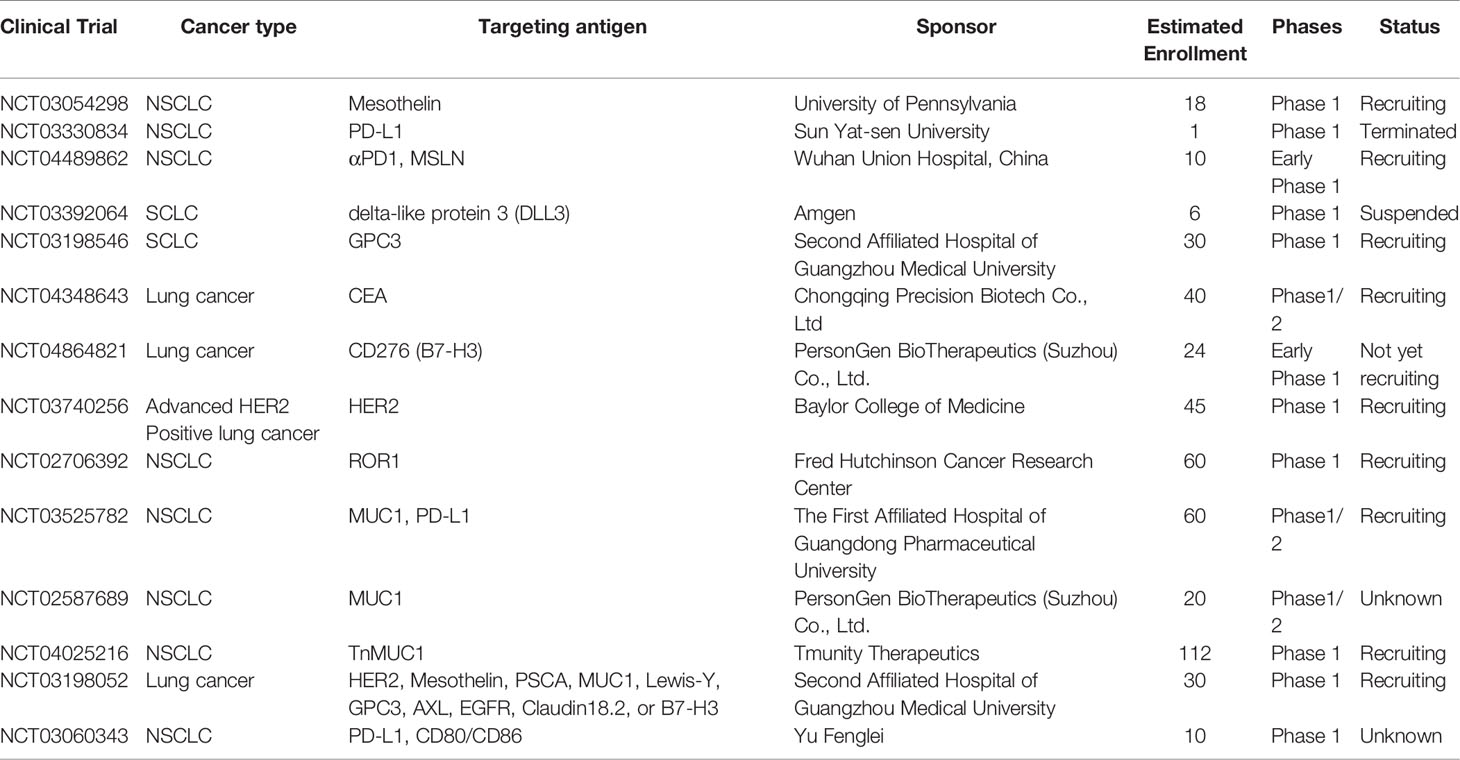
Table 2 Underlying targeting antigens of NSCLC and SCLC for CAR-T cell therapy in clinical trials.
2 Challenges and Engineering Strategies
Over the past few years, there has been a rapid increase in the use of CAR-T cell therapy to treat hematological malignancies and solid tumors. Many clinical trials have made substantial achievements; however, severe therapeutic responses to CAR-T cell therapy and unsatisfactory treatment efficacy hinder rapid development. In 2010, a patient with multiple metastases of colon cancer died after administering CAR-T cells targeting ERBB2. The patient experienced respiratory distress within 15 min after CAR-T cell transfusion and died five days after the treatment (132). Compared with hematological malignancies, solid tumors face a unique set of challenges, including issues confusing hematological malignancies, more severe and complicated related toxicities, the lack of a strongly expressed tumor-associated antigen target, low infiltration of T cells in tumor tissue, CAR-T cell exhaustion, and a highly immunosuppressive and metabolically challenging TME, which limit the safety and effectiveness of treatment (133–135). Future studies to develop practical engineering strategies to enhance the efficacy of CAR-T cell therapy and minimize adverse reactions should be conducted.
2.1 Overcoming Treatment-Related Toxicities
CAR-T cell therapy can result in a range of toxicity events. The major treatment-related toxicities include cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity (ICANS), which particularly peak in the first or second week of CAR-T cell administration, respectively (133). Patients with CRS mostly have common manifestations such as fever, tachycardia, hypoxia, dyspnea, hypertension, coagulopathy, and elevated serum cytokines, including interleukin-6 (IL-6) (136, 137). ICANS is characterized by tremor, encephalopathy, cerebellar alteration, or seizures (138). Both CRS and ICANS are caused by the activation of CAR-T cells and cytokines secreted by the associated immune cells. CAR-T cells can release pro-inflammatory cytokines, including IL-2, IL-6, and IFN-γ, and then activate more immune cells to secrete IL−1RA, IL−10, IL−6, IL−8, IFNα, and other cytokines, which eventually could lead to massive cytokine release (139). Hemophagocytic lymphohistiocytosis/macrophage activation syndrome has also been reported following CAR-T cell therapy. It is characterized by hyperinflammatory syndrome and multiple organ dysfunction (140). IL−6/IL−6R antagonists and corticosteroid usage can interrupt the inflammatory process and play a substantial role in symptom remission (141). It is critical to detect these treatment-related toxicities early and provide appropriate treatment based on the toxicity grade as soon as possible.
Selecting co-stimulatory signaling molecules and transmembrane domains could have an impact on cytokine production and CAR-T cell function. Compared with CD28/CD3ζ CAR T cells, 4–1BB/CD3ζ CAR T cells amplified more slowly, persisted for a longer time, and secreted less cytokines (142). CAR-T cells with CD8-alpha transmembrane domains have been shown to release less cytokines than those with CD28 domains (64). In addition, the inclusion of inducible caspase-9 safety switches to CARs has been verified to control the expansion of CAR-T cells and the load of cytokines (143). In summary, genetic modification of CAR designs might help reduce the generation of cytokines and the incidence of treatment-related toxicities (Figure 4).
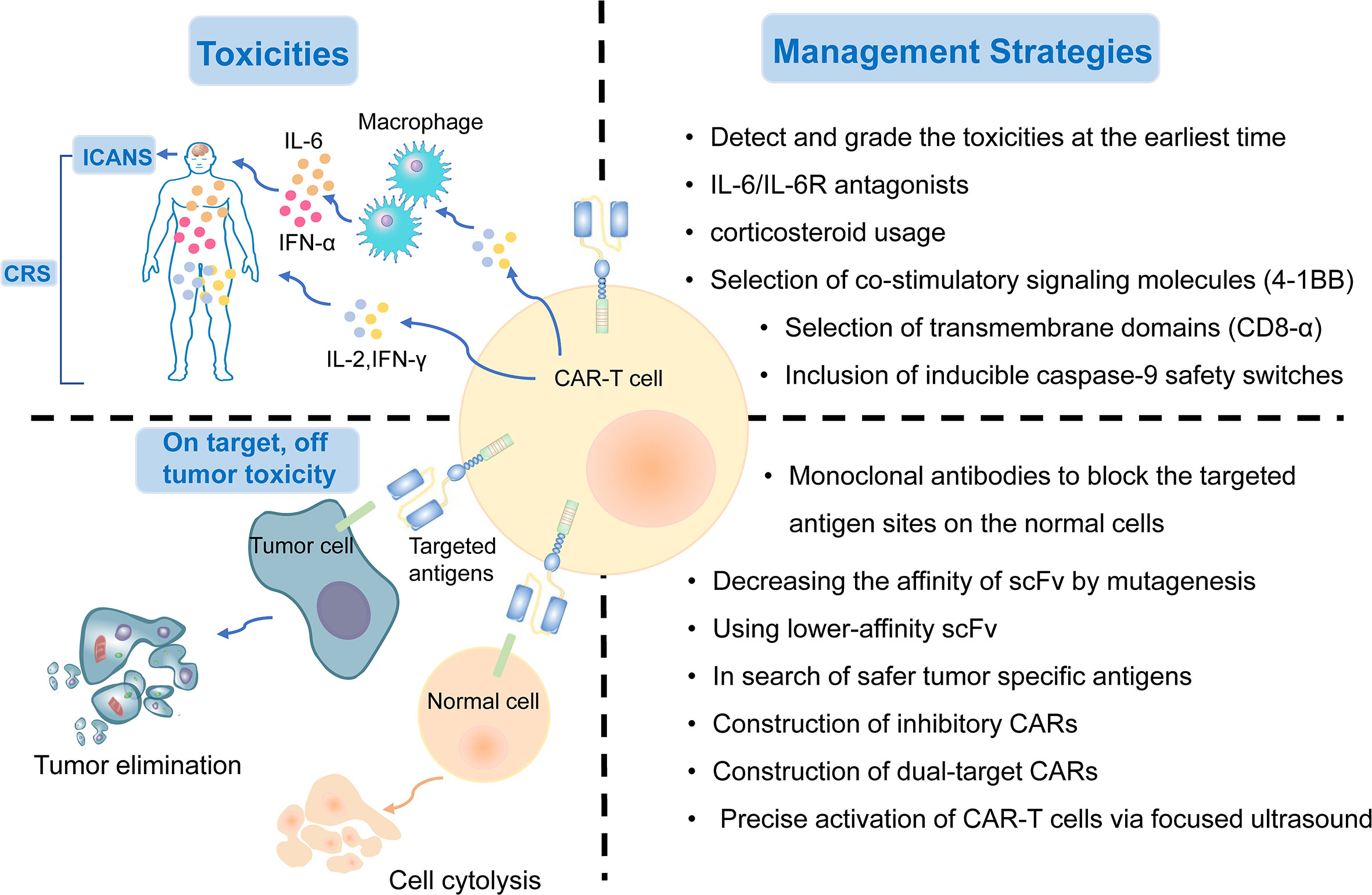
Figure 4 The treatment-related and on-target, off-tumor toxicities and corresponding management strategies of CAR-T cell therapy.
2.2 On-Target, Off-Tumor Toxicity
Although the targeted tumor-associated antigens are carefully screened, many normal cells still suffer from the attack of T cells because they express the same or similar antigens. On-target, off-tumor toxicity, manifesting multiple organ injury and failure, is an issue impeding the development of CAR-T cell treatment. Thus, there is an urgent need to explore safer targeted tumor-associated antigens for lung cancer treatment. To date, MSLN, EGFR, ROR1, MUC1, PSCA, and HER2, as described previously, are the most targeted antigens in CAR-T cell therapy for NSCLC. Several other tumor antigens, including LUNX and B7-H3, also exhibit great potential as targeted antigens in CAR-T cell therapy because they are aberrantly expressed in lung cancer tissues, with a relatively low expression in normal tissues (118, 144).
The on-target toxicity is antigen-oriented, and shielding of a CAR-targeted antigen expressed on normal tissues could minimize toxicity and optimize the efficacy of CAR-T cell therapy. Some renal cell carcinoma patients developed hepatic enzyme disorders that required discontinuation of therapy after receiving anti-carbonic anhydrase IX (CAIX) CAR-T cell therapy. This on-target toxicity can be overcome by pre-administration of parental anti-CAIX monoclonal antibodies to block the CAIX antigen sites in the liver (145). In addition, decreasing the affinity of scFv by mutagenesis or using lower-affinity scFv as a replacement could also substantially reduce on-target, off-tumor reactivity without affecting the antitumor activity (45). Other attempts include the construction of inhibitory CARs, which could protect the normal cells from being attacked by targeted CAR-T cells, and dual-target CAR-T cells, which require two signals to be full activated (146). Recently, an inducible CAR-T cell, was developed to be activated via focused ultrasound within specific tumor sites, which could dramatically mitigate the on-target, off-tumor toxicity, in comparison to conventional CAR-T cells (147) (Figure 4).
2.3 Evasion of Antitumor Immune Responses
A common mechanism for tumor cells to evade immune surveillance in CAR-T cell therapy is the downregulation or even loss of targeted antigens, whose expression level could exert a direct impact on the therapeutic efficacy (148). Targeting CD19/CD20 CAR-T cell therapies have led to promising achievements in treating B-cell malignancies in recent years (149). Tumor-associated antigens in hematologic malignancies are highly expressed and easier to target, whereas antigens in solid tumors have greater heterogeneity and lower expression levels, making it difficult to eliminate solid tumor cells (150). Intratumor heterogeneity might be a key factor contributing to the evasion of antitumor immune responses (151). In lung cancer, common targets such as MSLN, MUC1, PSCA, and epithelial cell adhesion molecule, have intratumoral heterogeneity, leading to an unsatisfactory outcome of CAT-T cell therapy in lung cancer (21). Many clinical studies have shown that when tumors relapse after treatment, tumors are found to undergo antigen loss or become antigen-negative (50, 152). This phenomenon may be mediated by the selective pressure applied by CAR-T cells to tumor cells, leading to the progressive selection of antigen-negative cells (82).
To overcome the evasion of antitumor immune responses, one approach is to engineer CARs with dual-specificity (i.e., simultaneously targeting two antigens) (153). Bispecific T cell-engagers (BiTEs), consisted of two scFvs, are produced by genetically engineered CAR-T cells to redirect both T cells and CAR-T cells against specific tumor cells (154, 155). EGFRvIII-specific CAR-T cells secreting BiTE have shown to circumvent antigen escape in glioblastoma, and its effect on lung cancers remains to be further investigated (154). Tandem CAR-T cells can mitigate antigen escape and translate into superior antitumor activity (156, 157) (Figure 5). Armored CAR-T cells secreting pro-inflammatory cytokines, such as IL-18, have also been shown to elicit an enhanced antitumor immune response in preclinical models (158).
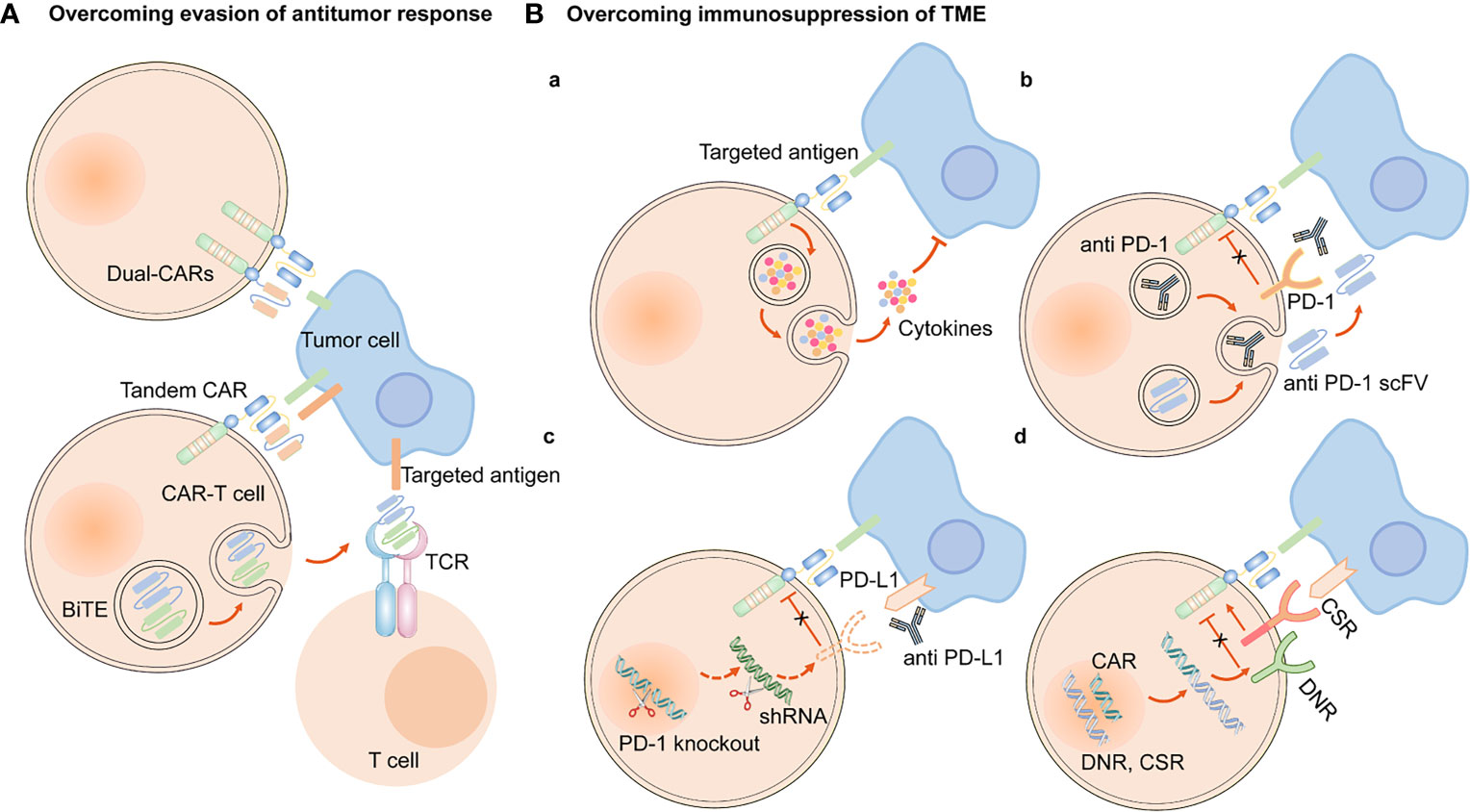
Figure 5 Engineering strategies to overcome evasion of antitumor response and immunosuppression of TME. (A) CAR-T cells are engineered to simultaneously target two antigens (dual CAR-T cells), and secret BiTE to redirect both T cells and CAR-T cells against specific tumor cells and circumvent antigen escape. Tandem CAR-T cells have bispecific receptors, which could target two different antigens. (B) (a) Armored CAR-T cells expressed immunostimulatory cytokines. Approaches to overcoming the immunosuppression of immune checkpoints in TME are as follows, (b) CAR-anti-PD-1/PD-L1 antibodies or scFv, (c) PD-1 gene knockout or downregulation of PD-1 expression by shRNA, (d) express a PD-1 DNR or a PD-1 CSR.
2.4 Physical Barriers
Cancer-associated fibroblasts (CAFs) and fibrotic environment contribute to the formation of physical barrier, preventing the CAR-T cells from being trafficked into tumor sites. Less infiltration of CAR-T cells into tumor tissues is another reason why the efficacy of CAR-T cell therapy in NSCLC is not as ideal as that in hematological malignancies.
2.4.1 CAFs
CAFs are the predominant component of stromal cells in the TME and cannot be cleared by apoptosis (159). Owing to the heterogeneity of CAFs, they could play a dual role in pro-tumorigenicity and anti-tumorigenicity (160). They could regulate the growth, invasion, and angiogenesis of tumor cells by reshaping the ECM and secreting soluble growth factors (160). Moreover, growth factors, cytokines and chemokines, including fibroblast growth factor (FGF), TGF-β, C-X-C motif chemokine ligand 12 (CXCL12), and IL-6, are also secreted by CAFs to mediate immunosuppressive responses (161). Hence, they can be applied as potential targets for anticancer treatment. However, many challenges still prevail in modulating CAFs as an ideal target for CAR-T cell therapy. As previously mentioned, FAP-targeted CAR-T cell therapy induced lethal adverse effects because CAR-T cells attacked FAP-positive BMSCs (117). In addition, CAFs have been shown to contribute to the development of therapeutic resistance because the ECM produced by CAFs could serve as a thick barrier to block the penetration of drugs (162). Accordingly, we hypothesized that the physical barrier formed by CAFs could also hinder the delivery of CAR-T cells into tumor tissues, thus diminishing the effectiveness and efficacy of CAR-T cell therapy (Figure 6).
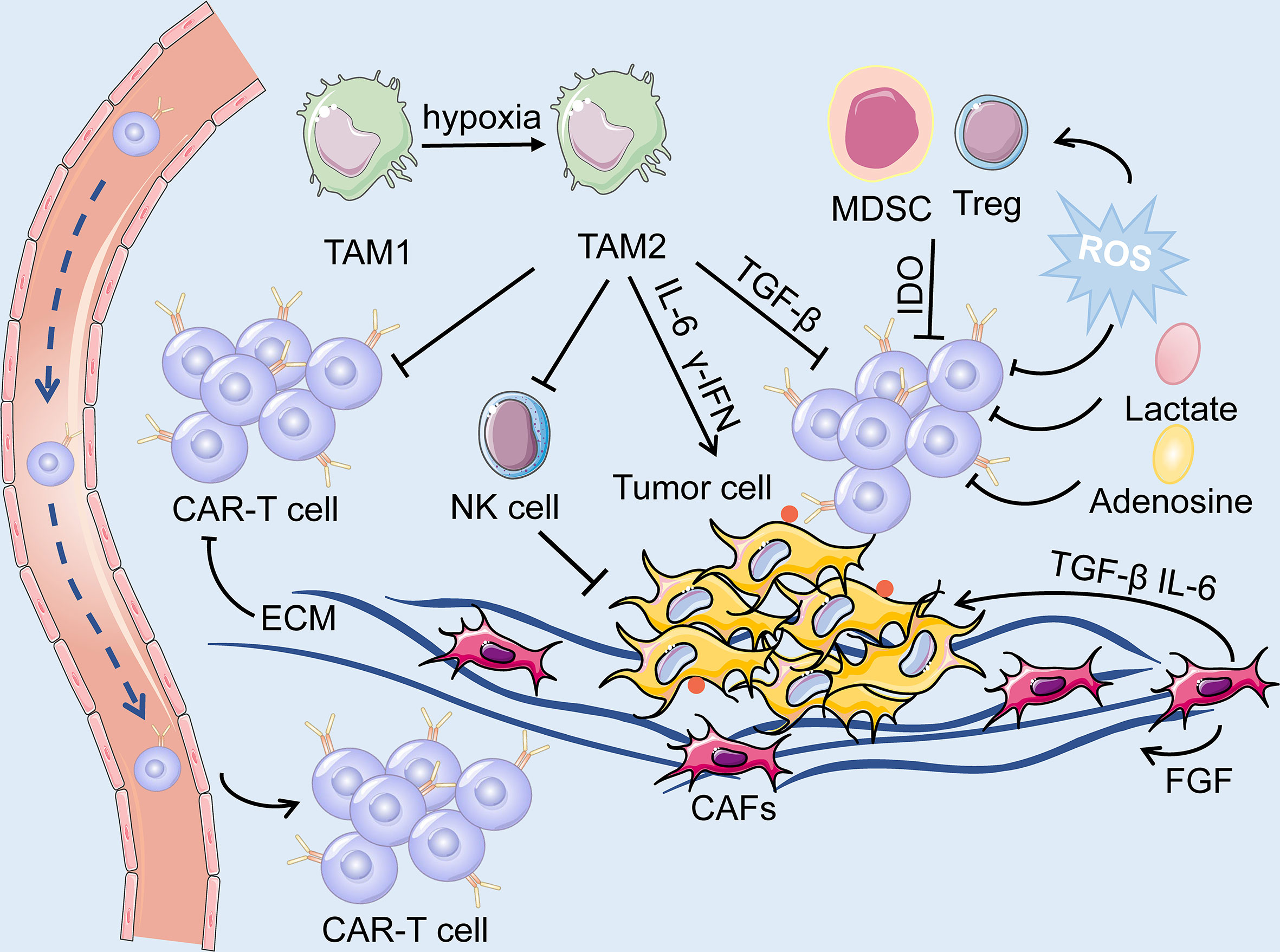
Figure 6 The TME is primarily composed of tumor cells, immune cells, immunosuppressive cells (including TAM, MDSC and Treg) and cytokines, CAFs, ECM and dysregulated tumor vasculatures. On the one hand, ECM produced by CAFs forms a physical barrier, impairing the infiltration of the CAR-T cells. On the other hand, the soluble cytokines secreted by the CAFs mediate immuno-suppressive responses, and consequently, facilitate the survival of tumor cells. The hypoxic and acidic environment directly deteriorate the metabolism of T cells while activating suppressive Tregs, leading to immunosuppression of CAR-T cells.
Several studies have been made to deplete or remodel the CAFs in the TME. One potential strategy is to apply FAP-redirected synthetic Notch CAR T cells or heparanase-modified CAR-T cells to deliver CAF remodeling molecules to suppress the expression profile of CAFs (163).
2.4.2 Fibrotic Environment
In contrast to hematological tumors, the infiltrative ability of CAR-T cells in lung cancer tissues is greatly restrained by the presence of a physical barrier. CAF activation, abnormal dense collagen, and ECM deposition contribute to developing a dense and fibrotic environment, altering the localization and migration of effector immune cells in NSCLC, which hinders immune cell infiltration and influences the efficacy of immunotherapy (164, 165). In addition, the extensive fibrotic environment mostly lacks blood vessels, which creates a hypoxic TME and further impairs immune function (166).
The binding of chemokines and their corresponding receptors can mediate the trafficking of CAR-T cells through fibrotic environment. Hence, one approach to enhance the infiltration level of CAR-T cells is to engineer them to express chemokines or transgenic chemokine receptors (167). The CAR-T cells engineered to express IL-7 and CCL19 have been validated to increase the infiltration of peripheral CAR-T cells and dendritic cells and into tumor tissues and enhance the anti-tumor immune responses (168). Another engineering strategy is to construct enzyme-modified CAR-T cells to express heparanase, which accelerates the degradation of ECM and facilitates CAR-T cell trafficking to tumor sites (169). In addition, local injection of CAR-T cells is under investigation.
2.5 Immune Suppression in the TME
The TME of lung cancer has an immunosuppressive effect, as T cell activity is suppressed due to anti-inflammatory cytokines and upregulated immune checkpoint ligands. Additionally, the immunosuppressive cells, such as myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), tumor associated macrophages, and tumor associated neutrophils are broadly present in the TME (Figure 6). CAR-T cell therapy against lung cancer is less efficient because of immune suppression of the TME and loss of CAR-T cell function.
One engineering approach to overcome the immunosuppressive role of TME is to establish armored CAR-T cells that secrete pro-inflammatory cytokines or chemokines, such as IL-12, IL-15, and IL-18 (20). These cells can recruit and activate innate immune cells such as natural killer (NK) cells and macrophages, and reprogram the immunosuppressive TME, which subsequently supports the proliferative and antitumor activity of CAR-T cells (170). In addition, based on blocking immune checkpoints, genetic knockdown of immune checkpoint receptors in CAR-T cells, such as PD-1, was demonstrated to enhance the anti-tumor effect. The clinical outcomes are being actively assessed in clinical trials on lung cancer (171). Other strategies include engineering CAR-T cells to secrete immune-checkpoint inhibitors, including anti–PD-1 scFv and anti–PD-L1 antibodies, to express PD-1 dominant-negative receptors (DNR) or PD-1 chimeric switch receptors (CSR) (113, 172, 173) (Figure 5).
2.6 Metabolic Profile of the TME
Cumulating evidence supports that metabolism plays an essential role in the immune response because it could regulate the function and activity of T cells. The inhibition of T cell metabolism may directly deteriorate the activity of T cells while activating suppressive Tregs, resulting in immuno-suppression (174). The proliferation of CAR-T cells, secretion of cytokines, and elimination of tumor cells are all energy-demanding processes. However, tumor cells mostly consume a large proportion of energy and nutrients, while generate a mass of immunosuppressive metabolites, such as adenosine, lactate, and kynurenine (135, 174). Moreover, indolamine-2,3-dioxygenase (IDO) secreted by tumor cells and MDSC could catalyze tryptophan into kynurenine, leading to the inactivation of CAR-T cells and the proliferation of Tregs (175) (Figure 6). On the other hand, the dysregulated vasculatures also result in an extremely hypoxic and acidic TME. All of the above elements contribute to the formation of the metabolically hostile TME, which further impairs the function of CAR-T cells.
Reprogramming the CAR-T cells to adjust their metabolic properties through genetic or pharmacological inhibition of adenosine receptors A1 and A2AR substantially elevated CAR T cell efficacy in breast cancer, which appears to be a promising method to enhance CAR-T cell function in the TME (176). Additionally, ROS generated by MDSC exerts a negative impact on CAR-T cells, and therefore, the reduction of ROS might be a potential strategy to overcome the metabolic profile of TME. Furthermore, CD28 and 4-1BB, the co-stimulatory domains of CAR-T cells, respectively, improved the metabolic fitness of CAR-T cells in melanoma by upregulating the intake of glucose and the expression of glycolytic enzymes, and enhancing mitochondrial biogenesis and oxidative metabolism (177, 178). However, limited data are available on the metabolic reprogramming of CAR-T cells in lung cancer.
2.7 CAR-T Cell Exhaustion
The existence of inhibitory ligands in the TME and endogenous TCRs leads to the gradual exhaustion of CAR-T cells (134). Clinical evidence has confirmed that CAR-T cell exhaustion markedly limits the efficacy of CAR-T cell therapy; therefore, it is imperative to prevent or reduce CAR-T cell exhaustion. However, it is difficult to reverse the cell exhaustion process directly by dedifferentiating T cells for exhaustion, which is a transcriptional and epigenetic forced differentiation state (179). Therefore, less differentiated T cell populations, such as naive T cells, whose proliferative activity is more robust, are selected for CAR-T cell manufacture (180). The negative regulators inducing T-cell exhaustion include PD-1, CTLA4, T-cell immunoglobulin and mucin domain 3, and lymphocyte-activation gene 3, which could restrain the activity of T cells while promoting the suppressive function of Tregs (181–183).
The above research advancements may shed light on new strategies to increase CAR-T cell persistence. Engineering strategies to inhibit these negative regulators primarily involve: (1) immune checkpoint blockades, (2) genetic knockdown of negative regulators in CAR-T cells, (3) PD-1 DNR, and (4) autocrine secretion of anti–PD-1 scFv and anti–PD-L1 antibodies from CAR T cells (20, 73, 182, 184). At present, combination therapy of CAR-T cells and immune checkpoint blockades has been utilized to overcome CAR-T cell exhaustion in clinical trials of NSCLC (185). CRISPR/Cas9-mediated knockdown of negative regulators in CAR-T cells may become a novel therapeutic approach to increase the persistence of CAR-T cells (182). CAR-T cells targeting PD-L1z, equipped with CAR-T cells with intrinsic blockade properties of PD-1, demonstrated efficacious antitumor activity in NSCLC models (113). CAR-T cells secreting anti–PD-L1 antibodies have been demonstrated to combat T cell exhaustion in a renal cell carcinoma mouse model (172) (Figure 5). In addition, transient cessation of CAR signaling, 4-1BB and CD28 costimulatory signaling, c-Jun, and transcription factors, such as nuclear receptor subfamily 4 group A, NFAT, and thymocyte selection-associated high mobility group box protein have also been shown to regulate T cell exhaustion (179, 184, 186). Further studies are required to apply these findings to enhance CAR-T cell resistance to exhaustion.
3 Future Outlook
CAR-T cell therapy has emerged as a novel and effective immunotherapy against multiple cancers, especially hematological malignancies. The same issues, such as CAR-T therapy-related toxicities, on-target, off-tumor toxicity, and evasion of antitumor responses, have plagued the treatment of hematologic malignancies; the treatment of solid tumors encounters even greater challenges. Moreover, the physical barrier impedes the infiltration of CAR-T cells to tumor sites, and the TME is immunosuppressive. In recent years, the successful improvements in the safety and efficacy of the therapy have facilitated the application of CAR-T therapy in solid tumors, including lung cancer. CAR structures persistently undergo evolution to enhance efficacy and reduce the cytotoxic effects of CAR-T cell therapy. In addition, the engineering solutions mentioned above are in their early stages and are being progressively developed towards the clinical application phase, and further investigations are expected (Figure 7). Among these engineering strategies, gene editing technology is one of powerful tools to improve the efficacy and safety of CAR-T cell therapy and is driving the application of this novel cancer therapy. The manufacture of “off the shelf” CAR-T cell products by disrupting the TCR alpha/beta chains through TALENs or CRISPR/Cas9 platform, is currently undergoing the evaluation of clinical trials (187). The inclusion of inducible caspase-9 safety switches to CARs could regulate the production of cytokines to prevent CRS (143). CRISPR/Cas9-mediated knockdown of negative immune checkpoints enables the CAR-T cells to resist the immunosuppressive TME. It is too early to appreciate the promising prospects of this novel immunotherapy approach in lung cancer treatment until more clinical trials to investigate these engineering strategies are conducted and evaluated.
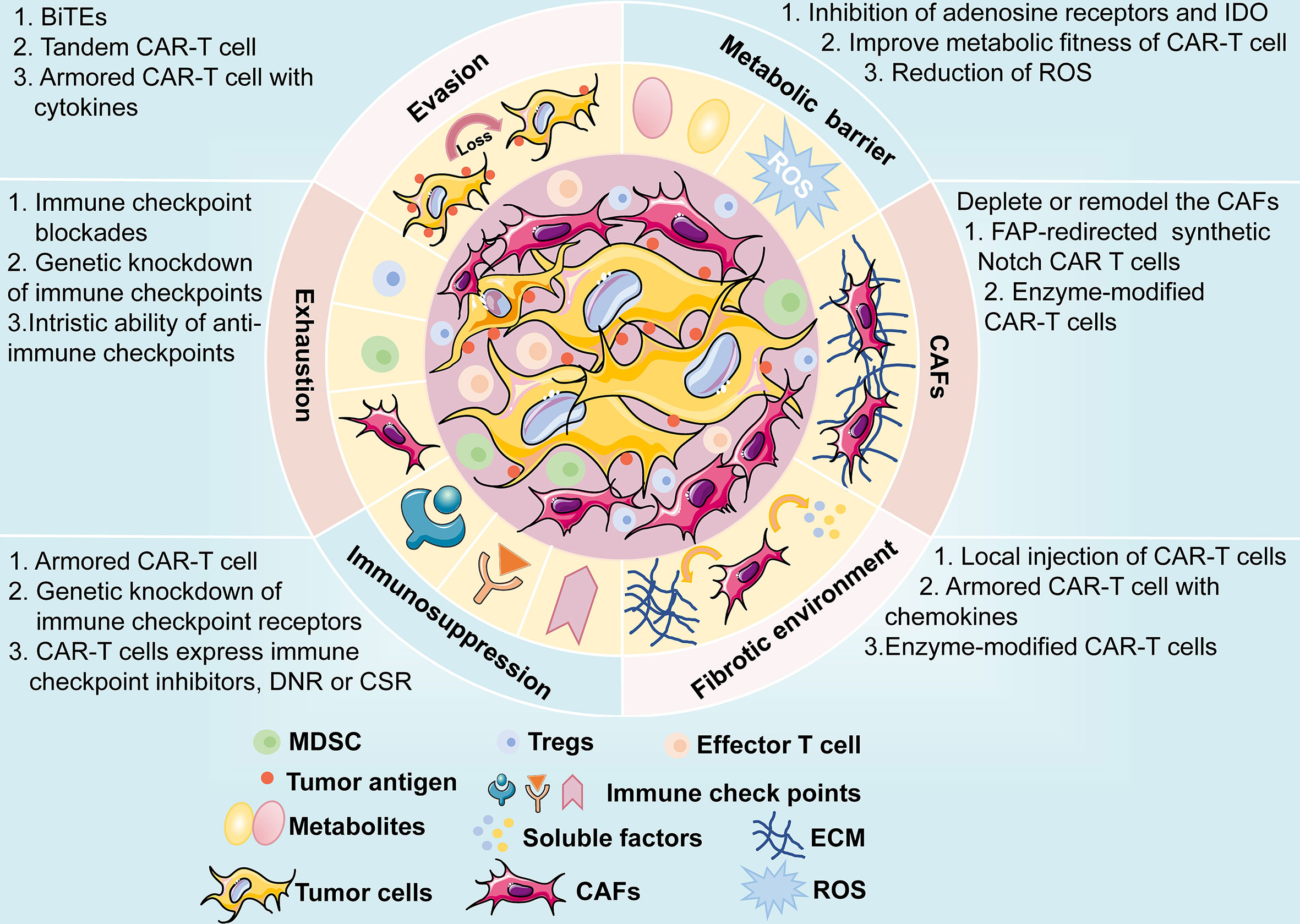
Figure 7 A brief overview of potential challenges faced by CAR-T cell therapy, including antigen evasion, metabolic barrier, CAFs, fibrotic environment, immunosuppression of TME, and exhaustion of CAR-T cells. The possible mechanisms and engineering strategies are also presented.
Author Contributions
B-TY and NW contributed significantly to fund support and the conception of the review. B-FX, J-TZ and Y-GZ contributed to wrote the manuscript. X-RC contributed to make preparations and revise the manuscript. Z-ML helped proposed some constructive suggestions. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key Research and Development Program of China (No. 2018YFC0910700), Beijing Human Resources and Social Security Bureau (Beijing Millions of Talents Project, 2018A05), Beijing Municipal Administration of Hospitals’ Youth Programme (QMS20191107), National Natural Science Foundation of China (No. 81972842), Beijing Natural Science Foundation (No. 7192036), Natural Science Foundation of Jiangxi Province (20202BABL206088).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wang X, Gong Y, Yao J, Chen Y, Li Y, Zeng Z, et al. Establishment of Criteria for Molecular Differential Diagnosis of MPLC and IPM. Front Oncol (2020) 10:614430. doi: 10.3389/fonc.2020.614430
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
3. Niemira M, Collin F, Szalkowska A, Bielska A, Chwialkowska K, Reszec J, et al. Molecular Signature of Subtypes of Non-Small-Cell Lung Cancer by Large-Scale Transcriptional Profiling: Identification of Key Modules and Genes by Weighted Gene Co-Expression Network Analysis (WGCNA). Cancers (2019) 12:37-60. doi: 10.3390/cancers12010037
4. Herbst R, Morgensztern D, Boshoff C. The Biology and Management of Non-Small Cell Lung Cancer. Nature (2018) 553:446–54. doi: 10.1038/nature25183
5. Li J, Zheng Q, Zhao X, Zhao J, An T, Wu M, et al. Nomogram Model for Predicting Cause-Specific Mortality in Patients With Stage I Small-Cell Lung Cancer: A Competing Risk Analysis. BMC Cancer (2020) 20:793. doi: 10.1186/s12885-020-07271-9
6. He S, Lin J, Xu Y, Lin L, Feng J. A Positive Feedback Loop Between ZNF205-AS1 and EGR4 Promotes Non-Small Cell Lung Cancer Growth. J Cell Mol Med (2019) 23:1495–508. doi: 10.1111/jcmm.14056
7. Duma N, Santana-Davila R, Molina JR. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin Proc (2019) 94:1623–40. doi: 10.1016/j.mayocp.2019.01.013
8. Johnson ML, Patel JD. Chemotherapy and Targeted Therapeutics as Maintenance of Response in Advanced Non-Small Cell Lung Cancer. Semin Oncol (2014) 41:93–100. doi: 10.1053/j.seminoncol.2013.12.007
9. Wu SG, Shih JY. Management of Acquired Resistance to EGFR TKI-Targeted Therapy in Advanced Non-Small Cell Lung Cancer. Mol Cancer (2018) 17:38. doi: 10.1186/s12943-018-0777-1
10. Golding B, Luu A, Jones R, Viloria-Petit AM. The Function and Therapeutic Targeting of Anaplastic Lymphoma Kinase (ALK) in Non-Small Cell Lung Cancer (NSCLC). Mol Cancer (2018) 17:52. doi: 10.1186/s12943-018-0810-4
11. Roys A, Chang X, Liu Y, Xu X, Wu Y, Zuo D. Resistance Mechanisms and Potent-Targeted Therapies of ROS1-Positive Lung Cancer. Cancer Chemother Pharmacol (2019) 84:679–88. doi: 10.1007/s00280-019-03902-6
12. Janning M, Loges S. Anti-Angiogenics: Their Value in Lung Cancer Therapy. Oncol Res Treat (2018) 41:172–80. doi: 10.1159/000488119
13. Allaeys T, Berzenji L, Van Schil PE. Surgery After Induction Targeted Therapy and Immunotherapy for Lung Cancer. Cancers (2021) 13:2603–18. doi: 10.3390/cancers13112603
14. Cascone T, William WN, Weissferdt A, Lin HY, Leung CH, Carter BW, et al. Neoadjuvant Nivolumab (N) or Nivolumab Plus Ipilimumab (NI) for Resectable Non-Small Cell Lung Cancer (NSCLC): Clinical and Correlative Results From the NEOSTAR Study. J Clin Oncol (2019) 37:8504–4. doi: 10.1200/JCO.2019.37.15_suppl.8504
15. Waqar SN, Morgensztern D. Treatment Advances in Small Cell Lung Cancer (SCLC). Pharmacol Ther (2017) 180:16–23. doi: 10.1016/j.pharmthera.2017.06.002
16. Zhao H, Ren D, Liu H, Chen J. Comparison and Discussion of the Treatment Guidelines for Small Cell Lung Cancer. Thorac Cancer (2018) 9:769–74. doi: 10.1111/1759-7714.12765
17. Yang S, Zhang Z, Wang Q. Emerging Therapies for Small Cell Lung Cancer. J Hematol Oncol (2019) 12:47. doi: 10.1186/s13045-019-0736-3
18. Saltos A, Shafique M, Chiappori A. Update on the Biology, Management, and Treatment of Small Cell Lung Cancer (SCLC). Front Oncol (2020) 10:1074. doi: 10.3389/fonc.2020.01074
19. Barayan R, Ran X, Lok BH. PARP Inhibitors for Small Cell Lung Cancer and Their Potential for Integration Into Current Treatment Approaches. J Thorac Dis (2020) 12:6240–52. doi: 10.21037/jtd.2020.03.89
20. Rafiq S, Hackett CS, Brentjens RJ. Engineering Strategies to Overcome the Current Roadblocks in CAR T Cell Therapy. Nat Rev Clin Oncol (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
21. Chen N, Li X, Chintala NK, Tano ZE, Adusumilli PS. Driving CARs on the Uneven Road of Antigen Heterogeneity in Solid Tumors. Curr Opin Immunol (2018) 51:103–10. doi: 10.1016/j.coi.2018.03.002
22. Grywalska E, Sosnowska-Pasiarska B, Smok-Kalwat J, Pasiarski M, Niedzwiedzka-Rystwej P, Rolinski J. Paving the Way Toward Successful Multiple Myeloma Treatment: Chimeric Antigen Receptor T-Cell Therapy. Cells (2020) 9:983. doi: 10.3390/cells9040983
23. Stock S, Schmitt M, Sellner L. Optimizing Manufacturing Protocols of Chimeric Antigen Receptor T Cells for Improved Anticancer Immunotherapy. Int J Mol Sci (2019) 20:6223. doi: 10.3390/ijms20246223
24. Han D, Xu Z, Zhuang Y, Ye Z, Qian Q. Current Progress in CAR-T Cell Therapy for Hematological Malignancies. J Cancer (2021) 12:326–34. doi: 10.7150/jca.48976
25. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T Cell Immunotherapy for Human Cancer. Science (2018) 359:1361–5. doi: 10.1126/science.aar6711
26. Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS. Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy in Acute Lymphocytic Leukaemia: A Systematic Review and Meta-Analysis. Lancet Haematol (2020) 7:e816–26. doi: 10.1016/S2352-3026(20)30277-5
27. Makita S, Imaizumi K, Kurosawa S, Tobinai K. Chimeric Antigen Receptor T-Cell Therapy for B-Cell Non-Hodgkin Lymphoma: Opportunities and Challenges. Drugs Context (2019) 8:212567. doi: 10.7573/dic.212567
28. King AC, Orozco JS. Axicabtagene Ciloleucel: The First FDA-Approved CAR T-Cell Therapy for Relapsed/Refractory Large B-Cell Lymphoma. J Adv Pract Oncol (2019) 10:878–82. doi: 10.6004/jadpro.2019.10.8.9
29. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med (2019) 380:45–56. doi: 10.1056/NEJMoa1804980
30. Maus MV, Alexander S, Bishop MR, Brudno JN, Callahan C, Davila ML, et al. Society for Immunotherapy of Cancer (SITC) Clinical Practice Guideline on Immune Effector Cell-Related Adverse Events. J Immunother Cancer (2020) 8:e001511. doi: 10.1136/jitc-2020-001511
31. Jain P, Nastoupil L, Westin J, Lee HJ, Navsaria L, Steiner RE, et al. Outcomes and Management of Patients With Mantle Cell Lymphoma After Progression on Brexucabtagene Autoleucel Therapy. Br J Haematol (2021) 192:e38–42. doi: 10.1111/bjh.17197
32. Garfall AL, Stadtmauer EA, Hwang WT, Lacey SF, Melenhorst JJ, Krevvata M, et al. Anti-CD19 CAR T Cells With High-Dose Melphalan and Autologous Stem Cell Transplantation for Refractory Multiple Myeloma. JCI Insight (2019) 4:e127684. doi: 10.1172/jci.insight.127684
33. Rodriguez-Lobato LG, Ganzetti M, Fernandez de Larrea C, Hudecek M, Einsele H, Danhof S. CAR T-Cells in Multiple Myeloma: State of the Art and Future Directions. Front Oncol (2020) 10:1243. doi: 10.3389/fonc.2020.01243
34. Ding L, Hu Y, Huang H. Novel Progresses of Chimeric Antigen Receptor (CAR) T Cell Therapy in Multiple Myeloma. Stem Cell Investig (2021) 8:1. doi: 10.21037/sci-2020-029
35. Chen KH, Wada M, Pinz KG, Liu H, Shuai X, Chen X, et al. A Compound Chimeric Antigen Receptor Strategy for Targeting Multiple Myeloma. Leukemia (2018) 32:402–12. doi: 10.1038/leu.2017.302
36. Lin Q, Zhao J, Song Y, Liu D. Recent Updates on CAR T Clinical Trials for Multiple Myeloma. Mol Cancer (2019) 18:154. doi: 10.1186/s12943-019-1092-1
37. Lam N, Trinklein ND, Buelow B, Patterson GH, Ojha N, Kochenderfer JN. Anti-BCMA Chimeric Antigen Receptors With Fully Human Heavy-Chain-Only Antigen Recognition Domains. Nat Commun (2020) 11:283. doi: 10.1038/s41467-019-14119-9
38. Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, et al. Recent Advances in CAR-T Cell Engineering. J Hematol Oncol (2020) 13:86. doi: 10.1186/s13045-020-00910-5
39. Zhang C, Liu J, Zhong JF, Zhang X. Engineering CAR-T Cells. Biomark Res (2017) 5:22. doi: 10.1186/s40364-017-0102-y
40. Yu S, Li A, Liu Q, Li T, Yuan X, Han X, et al. Chimeric Antigen Receptor T Cells: A Novel Therapy for Solid Tumors. J Hematol Oncol (2017) 10:78. doi: 10.1186/s13045-017-0444-9
41. Yeku OO, Purdon TJ, Koneru M, Spriggs D, Brentjens RJ. Armored CAR T Cells Enhance Antitumor Efficacy and Overcome the Tumor Microenvironment. Sci Rep (2017) 7:10541. doi: 10.1038/s41598-017-10940-8
42. Zhang H, Zhao P, Huang H. Engineering Better Chimeric Antigen Receptor T Cells. Exp Hematol Oncol (2020) 9:34. doi: 10.1186/s40164-020-00190-2
43. Singh AP, Zheng X, Lin-Schmidt X, Chen W, Carpenter TJ, Zong A, et al. Development of a Quantitative Relationship Between CAR-Affinity, Antigen Abundance, Tumor Cell Depletion and CAR-T Cell Expansion Using a Multiscale Systems PK-PD Model. MAbs (2020) 12:1688616. doi: 10.1080/19420862.2019.1688616
44. Abreu TR, Fonseca NA, Goncalves N, Moreira JN. Current Challenges and Emerging Opportunities of CAR-T Cell Therapies. J Control Release (2020) 319:246–61. doi: 10.1016/j.jconrel.2019.12.047
45. Fujiwara K, Masutani M, Tachibana M, Okada N. Impact of scFv Structure in Chimeric Antigen Receptor on Receptor Expression Efficiency and Antigen Recognition Properties. Biochem Biophys Res Commun (2020) 527:350–7. doi: 10.1016/j.bbrc.2020.03.071
46. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-Cell Responses to Soluble Factors With Chimeric Antigen Receptors. Nat Chem Biol (2018) 14:317–24. doi: 10.1038/nchembio.2565
47. Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, et al. Nanobody-Based CAR T Cells That Target the Tumor Microenvironment Inhibit the Growth of Solid Tumors in Immunocompetent Mice. Proc Natl Acad Sci USA (2019) 116:7624–31. doi: 10.1073/pnas.1817147116
48. Ingram JR, Schmidt FI, Ploegh HL. Exploiting Nanobodies' Singular Traits. Annu Rev Immunol (2018) 36:695–715. doi: 10.1146/annurev-immunol-042617-053327
49. Mo F, Duan S, Jiang X, Yang X, Hou X, Shi W, et al. Nanobody-Based Chimeric Antigen Receptor T Cells Designed by CRISPR/Cas9 Technology for Solid Tumor Immunotherapy. Signal Transduct Target Ther (2021) 6:80. doi: 10.1038/s41392-021-00462-1
50. Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma After Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med (2016) 375:2561–9. doi: 10.1056/NEJMoa1610497
51. Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients With AML/MDS and Multiple Myeloma. Cancer Immunol Res (2019) 7:100–12. doi: 10.1158/2326-6066.CIR-18-0307
52. Wang Y, Xu Y, Li S, Liu J, Xing Y, Xing H, et al. Targeting FLT3 in Acute Myeloid Leukemia Using Ligand-Based Chimeric Antigen Receptor-Engineered T Cells. J Hematol Oncol (2018) 11:60. doi: 10.1186/s13045-018-0603-7
53. Lee L, Draper B, Chaplin N, Philip B, Chin M, Galas-Filipowicz D, et al. An APRIL-Based Chimeric Antigen Receptor for Dual Targeting of BCMA and TACI in Multiple Myeloma. Blood (2018) 131:746–58. doi: 10.1182/blood-2017-05-781351
54. Nakazawa Y, Matsuda K, Kurata T, Sueki A, Tanaka M, Sakashita K, et al. Anti-Proliferative Effects of T Cells Expressing a Ligand-Based Chimeric Antigen Receptor Against CD116 on CD34(+) Cells of Juvenile Myelomonocytic Leukemia. J Hematol Oncol (2016) 9:27. doi: 10.1186/s13045-016-0256-3
55. Siegler E, Li S, Kim YJ, Wang P. Designed Ankyrin Repeat Proteins as Her2 Targeting Domains in Chimeric Antigen Receptor-Engineered T Cells. Hum Gene Ther (2017) 28:726–36. doi: 10.1089/hum.2017.021
56. Han X, Cinay GE, Zhao Y, Guo Y, Zhang X, Wang P. Adnectin-Based Design of Chimeric Antigen Receptor for T Cell Engineering. Mol Ther J Am Soc Gene Ther (2017) 25:2466–76. doi: 10.1016/j.ymthe.2017.07.009
57. Fujiwara K, Tsunei A, Kusabuka H, Ogaki E, Tachibana M, Okada N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells (2020) 9:1182. doi: 10.3390/cells9051182
58. Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T Design: Elements and Their Synergistic Function. EBioMedicine (2020) 58:102931. doi: 10.1016/j.ebiom.2020.102931
59. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin Cancer Res (2013) 19:3153–64. doi: 10.1158/1078-0432.CCR-13-0330
60. Schafer D, Henze J, Pfeifer R, Schleicher A, Brauner J, Mockel-Tenbrinck N, et al. A Novel Siglec-4 Derived Spacer Improves the Functionality of CAR T Cells Against Membrane-Proximal Epitopes. Front Immunol (2020) 11:1704. doi: 10.3389/fimmu.2020.01704
61. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol Res (2015) 3:125–35. doi: 10.1158/2326-6066.CIR-14-0127
62. Guedan S, Posey AD Jr, Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T Cell Persistence Through ICOS and 4-1BB Costimulation. JCI Insight (2018) 3:e96976. doi: 10.1172/jci.insight.96976
63. Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The Optimal Antigen Response of Chimeric Antigen Receptors Harboring the CD3zeta Transmembrane Domain Is Dependent Upon Incorporation of the Receptor Into the Endogenous TCR/CD3 Complex. J Immunol (Baltimore Md 1950) (2010) 184:6938–49. doi: 10.4049/jimmunol.0901766
64. Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A Safe and Potent Anti-CD19 CAR T Cell Therapy. Nat Med (2019) 25:947–53. doi: 10.1038/s41591-019-0421-7
65. Wan Z, Shao X, Ji X, Dong L, Wei J, Xiong Z, et al. Transmembrane Domain-Mediated Lck Association Underlies Bystander and Costimulatory ICOS Signaling. Cell Mol Immunol (2020) 17:143–52. doi: 10.1038/s41423-018-0183-z
66. Weinkove R, George P, Dasyam N, McLellan AD. Selecting Costimulatory Domains for Chimeric Antigen Receptors: Functional and Clinical Considerations. Clin Trans Immunol (2019) 8:e1049. doi: 10.1002/cti2.1049
67. Qu J, Mei Q, Chen L, Zhou J. Chimeric Antigen Receptor (CAR)-T-Cell Therapy in Non-Small-Cell Lung Cancer (NSCLC): Current Status and Future Perspectives. Cancer Immunol Immunother (2021) 70:619–31. doi: 10.1007/s00262-020-02735-0
68. Chandran SS, Klebanoff CA. T Cell Receptor-Based Cancer Immunotherapy: Emerging Efficacy and Pathways of Resistance. Immunol Rev (2019) 290:127–47. doi: 10.1111/imr.12772
69. van der Stegen SJ, Hamieh M, Sadelain M. The Pharmacology of Second-Generation Chimeric Antigen Receptors. Nat Rev Drug Discov (2015) 14:499–509. doi: 10.1038/nrd4597
70. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB Costimulation Ameliorates T Cell Exhaustion Induced by Tonic Signaling of Chimeric Antigen Receptors. Nat Med (2015) 21:581–90. doi: 10.1038/nm.3838
71. Philipson BI, O'Connor RS, May MJ, June CH, Albelda SM, Milone MC. 4-1BB Costimulation Promotes CAR T Cell Survival Through Noncanonical NF-KappaB Signaling. Sci Signal (2020) 13:e8248. doi: 10.1126/scisignal.aay8248
72. Sun C, Shou P, Du H, Hirabayashi K, Chen Y, Herring LE, et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell (2020) 37:216–225 e6. doi: 10.1016/j.ccell.2019.12.014
73. Larson RC, Maus MV. Recent Advances and Discoveries in the Mechanisms and Functions of CAR T Cells. Nat Rev Cancer (2021) 21:145–61. doi: 10.1038/s41568-020-00323-z
74. Song DG, Powell DJ. Pro-Survival Signaling via CD27 Costimulation Drives Effective CAR T-Cell Therapy. Oncoimmunology (2012) 1:547–9. doi: 10.4161/onci.19458
75. Duong CP, Westwood JA, Yong CS, Murphy A, Devaud C, John LB, et al. Engineering T Cell Function Using Chimeric Antigen Receptors Identified Using a DNA Library Approach. PloS One (2013) 8:e63037. doi: 10.1371/journal.pone.0063037
76. Hombach AA, Chmielewski M, Rappl G, Abken H. Adoptive Immunotherapy With Redirected T Cells Produces CCR7- Cells That Are Trapped in the Periphery and Benefit From Combined CD28-OX40 Costimulation. Hum Gene Ther (2013) 24:259–69. doi: 10.1089/hum.2012.247
77. Hombach AA, Heiders J, Foppe M, Chmielewski M, Abken H. OX40 Costimulation by a Chimeric Antigen Receptor Abrogates CD28 and IL-2 Induced IL-10 Secretion by Redirected CD4(+) T Cells. Oncoimmunology (2012) 1:458–66. doi: 10.4161/onci.19855
78. Mata M, Gerken C, Nguyen P, Krenciute G, Spencer DM, Gottschalk S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov (2017) 7:1306–19. doi: 10.1158/2159-8290.CD-17-0263
79. Wang E, Wang LC, Tsai CY, Bhoj V, Gershenson Z, Moon E, et al. Generation of Potent T-Cell Immunotherapy for Cancer Using DAP12-Based, Multichain, Chimeric Immunoreceptors. Cancer Immunol Res (2015) 3:815–26. doi: 10.1158/2326-6066.CIR-15-0054
80. Singh AK, McGuirk JP. CAR T Cells: Continuation in a Revolution of Immunotherapy. Lancet Oncol (2020) 21:e168–78. doi: 10.1016/S1470-2045(19)30823-X
81. van der Stegen S, Hamieh M, Sadelain M. The Pharmacology of Second-Generation Chimeric Antigen Receptors. Nat Rev Drug Discov (2015) 14:499–509. doi: 10.1038/nrd4597
82. D'Aloia MM, Zizzari IG, Sacchetti B, Pierelli L, Alimandi M. CAR-T Cells: The Long and Winding Road to Solid Tumors. Cell Death Dis (2018) 9:282. doi: 10.1038/s41419-018-0278-6
83. Guedan S, Ruella M, June CH. Emerging Cellular Therapies for Cancer. Annu Rev Immunol (2019) 37:145–71. doi: 10.1146/annurev-immunol-042718-041407
84. Chmielewski M, Abken H. TRUCKs: The Fourth Generation of CARs. Expert Opin Biol Ther (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
85. Zhao L, Cao YJ. Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol (2019) 10:2250. doi: 10.3389/fimmu.2019.02250
86. Zhong S, Cui Y, Liu Q, Chen S. CAR-T Cell Therapy for Lung Cancer: A Promising But Challenging Future. J Thorac Dis (2020) 12:4516–21. doi: 10.21037/jtd.2020.03.118
87. Klampatsa A, Dimou V, Albelda SM. Mesothelin-Targeted CAR-T Cell Therapy for Solid Tumors. Expert Opin Biol Ther (2021) 21:473–86. doi: 10.1080/14712598.2021.1843628
88. Hagemann UB, Ellingsen C, Schuhmacher J, Kristian A, Mobergslien A, Cruciani V, et al. Mesothelin-Targeted Thorium-227 Conjugate (MSLN-TTC): Preclinical Evaluation of a New Targeted Alpha Therapy for Mesothelin-Positive Cancers. Clin Cancer Res (2019) 25:4723–34. doi: 10.1158/1078-0432.CCR-18-3476
89. Ye L, Lou Y, Lu L, Fan X. Mesothelin-Targeted Second Generation CAR-T Cells Inhibit Growth of Mesothelin-Expressing Tumors In Vivo. Exp Ther Med (2019) 17:739–47. doi: 10.3892/etm.2018.7015
90. Morello A, Sadelain M, Adusumilli PS. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov (2016) 6:133–46. doi: 10.1158/2159-8290.CD-15-0583
91. da Cunha Santos G, Shepherd FA, Tsao MS. EGFR Mutations and Lung Cancer. Annu Rev Pathol (2011) 6:49–69. doi: 10.1146/annurev-pathol-011110-130206
92. Zhang Z, Jiang J, Wu X, Zhang M, Luo D, Zhang R, et al. Chimeric Antigen Receptor T Cell Targeting EGFRvIII for Metastatic Lung Cancer Therapy. Front Med (2019) 13:57–68. doi: 10.1007/s11684-019-0683-y
93. Li H, Huang Y, Jiang DQ, Cui LZ, He Z, Wang C, et al. Antitumor Activity of EGFR-Specific CAR T Cells Against Non-Small-Cell Lung Cancer Cells In Vitro and in Mice. Cell Death Dis (2018) 9:177. doi: 10.1038/s41419-017-0238-6
94. Liu D. CAR-T "the Living Drugs", Immune Checkpoint Inhibitors, and Precision Medicine: A New Era of Cancer Therapy. J Hematol Oncol (2019) 12:113. doi: 10.1186/s13045-019-0819-1
95. Yamaguchi T, Yanagisawa K, Sugiyama R, Hosono Y, Shimada Y, Arima C, et al. NKX2-1/TITF1/TTF-1-Induced ROR1 Is Required to Sustain EGFR Survival Signaling in Lung Adenocarcinoma. Cancer Cell (2012) 21:348–61. doi: 10.1016/j.ccr.2012.02.008
96. Schiavone G, Epistolio S, Martin V, Molinari F, Barizzi J, Mazzucchelli L, et al. Functional and Clinical Significance of ROR1 in Lung Adenocarcinoma. BMC Cancer (2020) 20:1085. doi: 10.1186/s12885-020-07587-6
97. Wallstabe L, Gottlich C, Nelke LC, Kuhnemundt J, Schwarz T, Nerreter T, et al. ROR1-CAR T Cells Are Effective Against Lung and Breast Cancer in Advanced Microphysiologic 3D Tumor Models. JCI Insight (2019) 4:e126345. doi: 10.1172/jci.insight.126345
98. Balakrishnan A, Goodpaster T, Randolph-Habecker J, Hoffstrom BG, Jalikis FG, Koch LK, et al. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin Cancer Res (2017) 23:3061–71. doi: 10.1158/1078-0432.CCR-16-2083
99. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al. Immunogenic Chemotherapy Enhances Recruitment of CAR-T Cells to Lung Tumors and Improves Antitumor Efficacy When Combined With Checkpoint Blockade. Cancer Cell (2021) 39:193–208.e10. doi: 10.1016/j.ccell.2020.11.005
100. Berger C, Sommermeyer D, Hudecek M, Berger M, Balakrishnan A, Paszkiewicz PJ, et al. Safety of Targeting ROR1 in Primates With Chimeric Antigen Receptor-Modified T Cells. Cancer Immunol Res (2015) 3:206–16. doi: 10.1158/2326-6066.Cir-14-0163
101. Bouillez A, Adeegbe D, Jin C, Hu X, Tagde A, Alam M, et al. MUC1-C Promotes the Suppressive Immune Microenvironment in Non-Small Cell Lung Cancer. Oncoimmunology (2017) 6:e1338998. doi: 10.1080/2162402X.2017.1338998
102. Jiang ZB, Huang JM, Xie YJ, Zhang YZ, Chang C, Lai HL, et al. Evodiamine Suppresses Non-Small Cell Lung Cancer by Elevating CD8(+) T Cells and Downregulating the MUC1-C/PD-L1 Axis. J Exp Clin Cancer Res (2020) 39:249. doi: 10.1186/s13046-020-01741-5
103. Saeki N, Gu J, Yoshida T, Wu X. Prostate Stem Cell Antigen: A Jekyll and Hyde Molecule? Clin Cancer Res (2010) 16:3533–8. doi: 10.1158/1078-0432.Ccr-09-3169
104. Wei X, Lai Y, Li J, Qin L, Xu Y, Zhao R, et al. PSCA and MUC1 in Non-Small-Cell Lung Cancer as Targets of Chimeric Antigen Receptor T Cells. Oncoimmunology (2017) 6:e1284722. doi: 10.1080/2162402x.2017.1284722
105. Soliman AM, Alqahtani AS, Ghorab M. Novel Sulphonamide Benzoquinazolinones as Dual EGFR/HER2 Inhibitors, Apoptosis Inducers and Radiosensitizers. J Enzyme Inhib Med Chem (2019) 34:1030–40. doi: 10.1080/14756366.2019.1609469
106. McKenna MK, Englisch A, Brenner B, Smith T, Hoyos V, Suzuki M, et al. Mesenchymal Stromal Cell Delivery of Oncolytic Immunotherapy Improves CAR-T Cell Antitumor Activity. Mol Ther J Am Soc Gene Ther (2021) 29:1808–20. doi: 10.1016/j.ymthe.2021.02.004
107. Gao Q, Wang S, Chen X, Cheng S, Zhang Z, Li F, et al. Cancer-Cell-Secreted CXCL11 Promoted CD8(+) T Cells Infiltration Through Docetaxel-Induced-Release of HMGB1 in NSCLC. J Immunother Cancer (2019) 7:42. doi: 10.1186/s40425-019-0511-6
108. Zeltsman M, Dozier J, McGee E, Ngai D, Adusumilli PS. CAR T-Cell Therapy for Lung Cancer and Malignant Pleural Mesothelioma. Trans Res J Lab Clin Med (2017) 187:1–10. doi: 10.1016/j.trsl.2017.04.004
109. Li X, Liu M, Zhang H, Liu H, Chen J. Clinical Study of Apatinib Combined With EGFR-TKI in the Treatment of Chronic Progression After EGFR-TKI Treatment in Non-Small Cell Lung Cancer (Chictr1800019185). Thorac Cancer (2020) 11:819–26. doi: 10.1111/1759-7714.13303
110. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors That Exhibit Augmented Activity Against Advanced Solid Tumors. Cell Rep (2017) 21:3205–19. doi: 10.1016/j.celrep.2017.11.063
111. Chen Y, Pei Y, Luo J, Huang Z, Yu J, Meng X. Looking for the Optimal PD-1/PD-L1 Inhibitor in Cancer Treatment: A Comparison in Basic Structure, Function, and Clinical Practice. Front Immunol (2020) 11:1088. doi: 10.3389/fimmu.2020.01088
112. Liu M, Wang X, Li W, Yu X, Flores-Villanueva P, Xu-Monette ZY, et al. Targeting PD-L1 in Non-Small Cell Lung Cancer Using CAR T Cells. Oncogenesis (2020) 9:72. doi: 10.1038/s41389-020-00257-z
113. Qin L, Zhao R, Chen D, Wei X, Wu Q, Long Y, et al. Chimeric Antigen Receptor T Cells Targeting PD-L1 Suppress Tumor Growth. Biomark Res (2020) 8:19. doi: 10.1186/s40364-020-00198-0
114. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor Effects of Chimeric Receptor Engineered Human T Cells Directed to Tumor Stroma. Mol Ther J Am Soc Gene Ther (2013) 21:1611–20. doi: 10.1038/mt.2013.110
115. Busek P, Mateu R, Zubal M, Kotackova L, Sedo A. Targeting Fibroblast Activation Protein in Cancer - Prospects and Caveats. Front Biosci (Landmark Ed) (2018) 23:1933–68. doi: 10.2741/4682
116. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting Fibroblast Activation Protein in Tumor Stroma With Chimeric Antigen Receptor T Cells can Inhibit Tumor Growth and Augment Host Immunity Without Severe Toxicity. Cancer Immunol Res (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
117. Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune Targeting of Fibroblast Activation Protein Triggers Recognition of Multipotent Bone Marrow Stromal Cells and Cachexia. J Exp Med (2013) 210:1125–35. doi: 10.1084/jem.20130110
118. Hu Z, Zheng X, Jiao D, Zhou Y, Sun R, Wang B, et al. LunX-CAR T Cells as a Targeted Therapy for Non-Small Cell Lung Cancer. Mol Ther Oncolytics (2020) 17:361–70. doi: 10.1016/j.omto.2020.04.008
119. Porcellini S, Asperti C, Corna S, Cicoria E, Valtolina V, Stornaiuolo A, et al. CAR T Cells Redirected to CD44v6 Control Tumor Growth in Lung and Ovary Adenocarcinoma Bearing Mice. Front Immunol (2020) 11:99. doi: 10.3389/fimmu.2020.00099
120. Mao Y, Fan W, Hu H, Zhang L, Michel J, Wu Y, et al. MAGE-A1 in Lung Adenocarcinoma as a Promising Target of Chimeric Antigen Receptor T Cells. J Hematol Oncol (2019) 12:106. doi: 10.1186/s13045-019-0793-7
121. Li N, Liu S, Sun M, Chen W, Xu X, Zeng Z, et al. Chimeric Antigen Receptor-Modified T Cells Redirected to EphA2 for the Immunotherapy of Non-Small Cell Lung Cancer. Trans Oncol (2018) 11:11–7. doi: 10.1016/j.tranon.2017.10.009
122. Shimizu Y, Suzuki T, Yoshikawa T, Endo I, Nakatsura T. Next-Generation Cancer Immunotherapy Targeting Glypican-3. Front Oncol (2019) 9:248. doi: 10.3389/fonc.2019.00248
123. Crossland DL, Denning WL, Ang S, Olivares S, Mi T, Switzer K, et al. Antitumor Activity of CD56-Chimeric Antigen Receptor T Cells in Neuroblastoma and SCLC Models. Oncogene (2018) 37:3686–97. doi: 10.1038/s41388-018-0187-2
124. Owen DH, Giffin MJ, Bailis JM, Smit MD, Carbone DP, He K. DLL3: An Emerging Target in Small Cell Lung Cancer. J Hematol Oncol (2019) 12:61. doi: 10.1186/s13045-019-0745-2
125. Yang M, Tang X, Zhang Z, Gu L, Wei H, Zhao S, et al. Tandem CAR-T Cells Targeting CD70 and B7-H3 Exhibit Potent Preclinical Activity Against Multiple Solid Tumors. Theranostics (2020) 10:7622–34. doi: 10.7150/thno.43991
126. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
127. Kontos F, Michelakos T, Kurokawa T, Sadagopan A, Schwab JH, Ferrone CR, et al. B7-H3: An Attractive Target for Antibody-Based Immunotherapy. Clin Cancer Res (2021) 27:1227–35. doi: 10.1158/1078-0432.CCR-20-2584
128. Lin S, Cheng L, Ye W, Li S, Zheng D, Qin L, et al. Chimeric CTLA4-CD28-CD3z T Cells Potentiate Antitumor Activity Against CD80/CD86-Positive B Cell Malignancies. Front Immunol (2021) 12:642528. doi: 10.3389/fimmu.2021.642528
129. Chu W, Zhou Y, Tang Q, Wang M, Ji Y, Yan J, et al. Bi-Specific Ligand-Controlled Chimeric Antigen Receptor T-Cell Therapy for Non-Small Cell Lung Cancer. Biosci Trends (2018) 12:298–308. doi: 10.5582/bst.2018.01048
130. Springuel L, Lonez C, Alexandre B, Van Cutsem E, Machiels JH, Van Den Eynde M, et al. Chimeric Antigen Receptor-T Cells for Targeting Solid Tumors: Current Challenges and Existing Strategies. BioDrugs (2019) 33:515–37. doi: 10.1007/s40259-019-00368-z
131. Kiesgen S, Chicaybam L, Chintala NK, Adusumilli PS. Chimeric Antigen Receptor (CAR) T-Cell Therapy for Thoracic Malignancies. J Thorac Oncol (2018) 13(1):16–26. doi: 10.1016/j.jtho.2018.01.001
132. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther J Am Soc Gene Ther (2010) 18:843–51. doi: 10.1038/mt.2010.24
133. Morris EC, Neelapu SS, Giavridis T, Sadelain M. Cytokine Release Syndrome and Associated Neurotoxicity in Cancer Immunotherapy. Nat Rev Immunol (2021) 1–12. doi: 10.1038/s41577-021-00547-6
134. Kasakovski D, Xu L, Li Y. T Cell Senescence and CAR-T Cell Exhaustion in Hematological Malignancies. J Hematol Oncol (2018) 11:91. doi: 10.1186/s13045-018-0629-x
135. Hou AJ, Chen LC, Chen YY. Navigating CAR-T Cells Through the Solid-Tumour Microenvironment. Nat Rev Drug Discov (2021) 20(7):531–50. doi: 10.1038/s41573-021-00189-2
136. Frey N, Porter D. Cytokine Release Syndrome With Chimeric Antigen Receptor T Cell Therapy. Biol Blood Marrow Transplant (2019) 25:e123–7. doi: 10.1016/j.bbmt.2018.12.756
137. Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine Release Syndrome. J Immunother Cancer (2018) 6:56. doi: 10.1186/s40425-018-0343-9
138. Lee DW, Santomasso BD, Locke FL, Ghobadi A, Turtle CJ, Brudno JN, et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated With Immune Effector Cells. Biol Blood Marrow Transplant (2019) 25:625–38. doi: 10.1016/j.bbmt.2018.12.758
139. Rivera AM, May S, Lei M, Qualls S, Bushey K, Rubin DB, et al. CAR T-Cell-Associated Neurotoxicity: Current Management and Emerging Treatment Strategies. Crit Care Nurs Q (2020) 43:191–204. doi: 10.1097/CNQ.0000000000000302
140. Carter SJ, Tattersall RS, Ramanan AV. Macrophage Activation Syndrome in Adults: Recent Advances in Pathophysiology, Diagnosis and Treatment. Rheumatology (Oxford) (2019) 58:5–17. doi: 10.1093/rheumatology/key006
141. Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, et al. Chimeric Antigen Receptor T-Cell Therapy — Assessment and Management of Toxicities. Nat Rev Clin Oncol (2018) 15:47–62. doi: 10.1038/nrclinonc.2017.148
142. Salter AI, Ivey RG, Kennedy JJ, Voillet V, Rajan A, Alderman EJ, et al. Phosphoproteomic Analysis of Chimeric Antigen Receptor Signaling Reveals Kinetic and Quantitative Differences That Affect Cell Function. Sci Signal (2018) 11:eaat6753. doi: 10.1126/scisignal.aat6753
143. Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, et al. Inducible Caspase-9 Selectively Modulates the Toxicities of CD19-Specific Chimeric Antigen Receptor-Modified T Cells. Mol Ther J Am Soc Gene Ther (2017) 25:580–92. doi: 10.1016/j.ymthe.2017.01.011
144. Liu J, Yang S, Cao B, Zhou G, Zhang F, Wang Y, et al. Targeting B7-H3 via Chimeric Antigen Receptor T Cells and Bispecific Killer Cell Engagers Augments Antitumor Response of Cytotoxic Lymphocytes. J Hematol Oncol (2021) 14:21. doi: 10.1186/s13045-020-01024-8
145. Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of Metastatic Renal Cell Carcinoma With CAIX CAR-Engineered T Cells: Clinical Evaluation and Management of on-Target Toxicity. Mol Ther J Am Soc Gene Ther (2013) 21:904–12. doi: 10.1038/mt.2013.17
146. Yu S, Yi M, Qin S, Wu K. Next Generation Chimeric Antigen Receptor T Cells: Safety Strategies to Overcome Toxicity. Mol Cancer (2019) 18:125. doi: 10.1186/s12943-019-1057-4
147. Wu Y, Liu Y, Huang Z, Wang X, Jin Z, Li J, et al. Control of the Activity of CAR-T Cells Within Tumours via Focused Ultrasound. Nat Biomed Eng (2021). doi: 10.1038/s41551-021-00779-w
148. Song DG, Ye Q, Poussin M, Chacon JA, Figini M, Powell DJ Jr. Effective Adoptive Immunotherapy of Triple-Negative Breast Cancer by Folate Receptor-Alpha Redirected CAR T Cells Is Influenced by Surface Antigen Expression Level. J Hematol Oncol (2016) 9:56. doi: 10.1186/s13045-016-0285-y
149. Tong C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y, et al. Optimized Tandem CD19/CD20 CAR-Engineered T Cells in Refractory/Relapsed B-Cell Lymphoma. Blood (2020) 136:1632–44. doi: 10.1182/blood.2020005278
150. Majzner RG, Mackall CL. Tumor Antigen Escape From CAR T-Cell Therapy. Cancer Discov (2018) 8:1219–26. doi: 10.1158/2159-8290.CD-18-0442
151. Caswell DR, Swanton C. The Role of Tumour Heterogeneity and Clonal Cooperativity in Metastasis, Immune Evasion and Clinical Outcome. BMC Med (2017) 15:133. doi: 10.1186/s12916-017-0900-y
152. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients With Recurrent Glioblastoma. Sci Transl Med (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
153. Rafiq S, Brentjens RJ. Tumors Evading CARs-The Chase Is On. Nat Med (2018) 24:1492–3. doi: 10.1038/s41591-018-0212-6
154. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T Cells Secreting BiTEs Circumvent Antigen Escape Without Detectable Toxicity. Nat Biotechnol (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
155. Goebeler M-E, Bargou RC. T Cell-Engaging Therapies — BiTEs and Beyond. Nat Rev Clin Oncol (2020) 17:418–34. doi: 10.1038/s41571-020-0347-5
156. Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T Cells Targeting HER2 and IL13Rα2 Mitigate Tumor Antigen Escape. J Clin Invest (2016) 126:3036–52. doi: 10.1172/jci83416
157. Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids (2013) 2:e105. doi: 10.1038/mtna.2013.32
158. Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and Through Activation of the Endogenous Immune System. Cell Rep (2018) 23:2130–41. doi: 10.1016/j.celrep.2018.04.051
159. Nurmik M, Ullmann P, Rodriguez F, Haan S, Letellier E. In Search of Definitions: Cancer-Associated Fibroblasts and Their Markers. Int J Cancer (2020) 146:895–905. doi: 10.1002/ijc.32193
160. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat Rev Cancer (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
161. Biffi G, Tuveson DA. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol Rev (2021) 101:147–76. doi: 10.1152/physrev.00048.2019
162. Kobayashi H, Enomoto A, Woods SL, Burt AD, Takahashi M, Worthley DL. Cancer-Associated Fibroblasts in Gastrointestinal Cancer. Nat Rev Gastroenterol Hepatol (2019) 16:282–95. doi: 10.1038/s41575-019-0115-0
163. Henze J, Tacke F, Hardt O, Alves F, Al Rawashdeh W. Enhancing the Efficacy of CAR T Cells in the Tumor Microenvironment of Pancreatic Cancer. Cancers (2020) 12:1389–409. doi: 10.3390/cancers12061389
164. Valkenburg KC, de Groot AE, Pienta KJ. Targeting the Tumour Stroma to Improve Cancer Therapy. Nat Rev Clin Oncol (2018) 15:366–81. doi: 10.1038/s41571-018-0007-1
165. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix Architecture Defines the Preferential Localization and Migration of T Cells Into the Stroma of Human Lung Tumors. J Clin Invest (2012) 122:899–910. doi: 10.1172/JCI45817
166. Jiang H, Hegde S, DeNardo DG. Tumor-Associated Fibrosis as a Regulator of Tumor Immunity and Response to Immunotherapy. Cancer Immunol Immunother (2017) 66:1037–48. doi: 10.1007/s00262-017-2003-1
167. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced Tumor Trafficking of GD2 Chimeric Antigen Receptor T Cells by Expression of the Chemokine Receptor CCR2b. J Immunother (Hagerstown Md 1997) (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
168. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 Expression in CAR-T Cells Improves Immune Cell Infiltration and CAR-T Cell Survival in the Tumor. Nat Biotechnol (2018) 36:346–51. doi: 10.1038/nbt.4086
169. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes. Nat Med (2015) 21:524–9. doi: 10.1038/nm.3833
170. Dragon AC, Zimmermann K, Nerreter T, Sandfort D, Lahrberg J, Kloss S, et al. CAR-T Cells and TRUCKs That Recognize an EBNA-3C-Derived Epitope Presented on HLA-B*35 Control Epstein-Barr Virus-Associated Lymphoproliferation. J Immunother Cancer (2020) 8:e000736. doi: 10.1136/jitc-2020-000736
171. McGowan E, Lin Q, Ma G, Yin H, Chen S, Lin Y. PD-1 Disrupted CAR-T Cells in the Treatment of Solid Tumors: Promises and Challenges. BioMed Pharmacother (2020) 121:109625. doi: 10.1016/j.biopha.2019.109625
172. Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric Antigen Receptor T Cells Secreting Anti-PD-L1 Antibodies More Effectively Regress Renal Cell Carcinoma in a Humanized Mouse Model. Oncotarget (2016) 7:34341–55. doi: 10.18632/oncotarget.9114
173. Tay JC, Zha S, Wang S. Chimeric Switch Receptor: Switching for Improved Adoptive T-Cell Therapy Against Cancers. Immunotherapy (2017) 9:1339–49. doi: 10.2217/imt-2017-0103
174. Beckermann KE, Dudzinski SO, Rathmell JC. Dysfunctional T Cell Metabolism in the Tumor Microenvironment. Cytokine Growth Factor Rev (2017) 35:7–14. doi: 10.1016/j.cytogfr.2017.04.003
175. Newick K, O'Brien S, Moon E, Albelda SM. CAR T Cell Therapy for Solid Tumors. Annu Rev Med (2017) 68:139–52. doi: 10.1146/annurev-med-062315-120245
176. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the Adenosine 2A Receptor Enhances Chimeric Antigen Receptor T Cell Efficacy. J Clin Invest (2017) 127:929–41. doi: 10.1172/JCI89455
177. Pellegrino M, Del Bufalo F, De Angelis B, Quintarelli C, Caruana I, de Billy E. Manipulating the Metabolism to Improve the Efficacy of CAR T-Cell Immunotherapy. Cells (2020) 10:14-29 doi: 10.3390/cells10010014
178. Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, et al. 4-1BB Costimulation Induces T Cell Mitochondrial Function and Biogenesis Enabling Cancer Immunotherapeutic Responses. J Exp Med (2018) 215:1091–100. doi: 10.1084/jem.20171068
179. Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining 'T Cell Exhaustion'. Nat Rev Immunol (2019) 19:665–74. doi: 10.1038/s41577-019-0221-9
180. Sadelain M, Riviere I, Riddell S. Therapeutic T Cell Engineering. Nature (2017) 545:423–31. doi: 10.1038/nature22395
181. Solinas C, De Silva P, Bron D, Willard-Gallo K, Sangiolo D. Significance of TIM3 Expression in Cancer: From Biology to the Clinic. Semin Oncol (2019) 46:372–9. doi: 10.1053/j.seminoncol.2019.08.005
182. Hong M, Clubb JD, Chen YY. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell (2020) 38:473–88. doi: 10.1016/j.ccell.2020.07.005
183. Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of Response and Resistance to CD19 Chimeric Antigen Receptor (CAR) T Cell Therapy of Chronic Lymphocytic Leukemia. Nat Med (2018) 24:563–71. doi: 10.1038/s41591-018-0010-1
184. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T Cells With Cell-Intrinsic PD-1 Checkpoint Blockade Resist Tumor-Mediated Inhibition. J Clin Invest (2016) 126:3130–44. doi: 10.1172/JCI83092
185. Sanghera C, Sanghera R. Immunotherapy - Strategies for Expanding Its Role in the Treatment of All Major Tumor Sites. Cureus (2019) 11:e5938. doi: 10.7759/cureus.5938
186. Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. C-Jun Overexpression in CAR T Cells Induces Exhaustion Resistance. Nature (2019) 576:293–300. doi: 10.1038/s41586-019-1805-z
Keywords: chimeric antigen receptor, T cell, immunotherapy, lung cancer, engineering strategy
Citation: Xiao B-F, Zhang J-T, Zhu Y-G, Cui X-R, Lu Z-M, Yu B-T and Wu N (2021) Chimeric Antigen Receptor T-Cell Therapy in Lung Cancer: Potential and Challenges. Front. Immunol. 12:782775. doi: 10.3389/fimmu.2021.782775
Received: 24 September 2021; Accepted: 13 October 2021;
Published: 01 November 2021.
Edited by:
Niels Schaft, University Hospital Erlangen, GermanyReviewed by:
Jamshid Hadjati, Tehran University of Medical Sciences, IranYozo Nakazawa, Shinshu University School of Medicine, Japan
Markus Martin Chmielewski, University Hospital of Cologne, Germany
Copyright © 2021 Xiao, Zhang, Zhu, Cui, Lu, Yu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ben-Tong Yu, eXViZW50b25nQDEyNi5jb20=; Nan Wu, bmFud3VAYmptdS5lZHUuY24=
†These authors have contributed equally to this work