 Dayene de Assis Fernandes Caldeira1
Dayene de Assis Fernandes Caldeira1 Daniel J. Weiss2
Daniel J. Weiss2 Patricia Rieken Macêdo Rocco1,3,4
Patricia Rieken Macêdo Rocco1,3,4 Pedro Leme Silva1,3,4†
Pedro Leme Silva1,3,4† Fernanda Ferreira Cruz1,3,4*†
Fernanda Ferreira Cruz1,3,4*†- 1Laboratory of Pulmonary Investigation, Institute of Biophysics Carlos Chagas Filho, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil
- 2Department of Medicine, College of Medicine, University of Vermont, Burlington, VT, United States
- 3National Institute of Science and Technology for Regenerative Medicine, Rio de Janeiro, Brazil
- 4Rio de Janeiro Innovation Network in Nanosystems for Health-NanoSAÚDE/FAPERJ, Rio de Janeiro, Brazil
Mitochondria are essential organelles for cell metabolism, growth, and function. Mitochondria in lung cells have important roles in regulating surfactant production, mucociliary function, mucus secretion, senescence, immunologic defense, and regeneration. Disruption in mitochondrial physiology can be the central point in several pathophysiologic pathways of chronic lung diseases such as chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis, and asthma. In this review, we summarize how mitochondria morphology, dynamics, redox signaling, mitophagy, and interaction with the endoplasmic reticulum are involved in chronic lung diseases and highlight strategies focused on mitochondrial therapy (mito-therapy) that could be tested as a potential therapeutic target for lung diseases.
Introduction
Mitochondria are organelles present in all eukaryotic organisms with the classic role of generating most of the cellular energy. Mitochondria are responsible for the synthesis of adenosine triphosphate (ATP) via oxidative phosphorylation (OX-PHOS) through the breakdown of sugars and fatty acids in the citric acid cycle (1). In addition to energy production, depending on the cell type, mitochondria can also be involved in other metabolic processes, such as calcium and apoptosis signaling (2–5). Considering their accepted endosymbiotic origin, mitochondria have their own transcriptional machinery, proteome, and DNA (mitochondrial DNA [mtDNA]), which makes them semi-autonomous organelles regulating their homeostasis by autophagy, fusion, and fission (6).
During homeostasis, mitochondria have important roles in lung function. Mitochondria number and intracellular organization can vary in an energy-dependent form for distinct types of airway epithelial cells (7). Mitochondria can regulate surfactant production, cellular senescence, mucociliary function, and mucus secretion (7). Mitochondria are also crucial in pulmonary immunometabolism and immune cell response, such as in alveolar macrophages (8). Appreciation for mitochondrial noncanonical functions has increased our knowledge of their role in pathophysiologic processes, including chronic lung diseases such as chronic obstructive pulmonary disease (COPD) (Figure 1), idiopathic pulmonary fibrosis (IPF) (Figure 2), and asthma (Figure 3) (9–12).
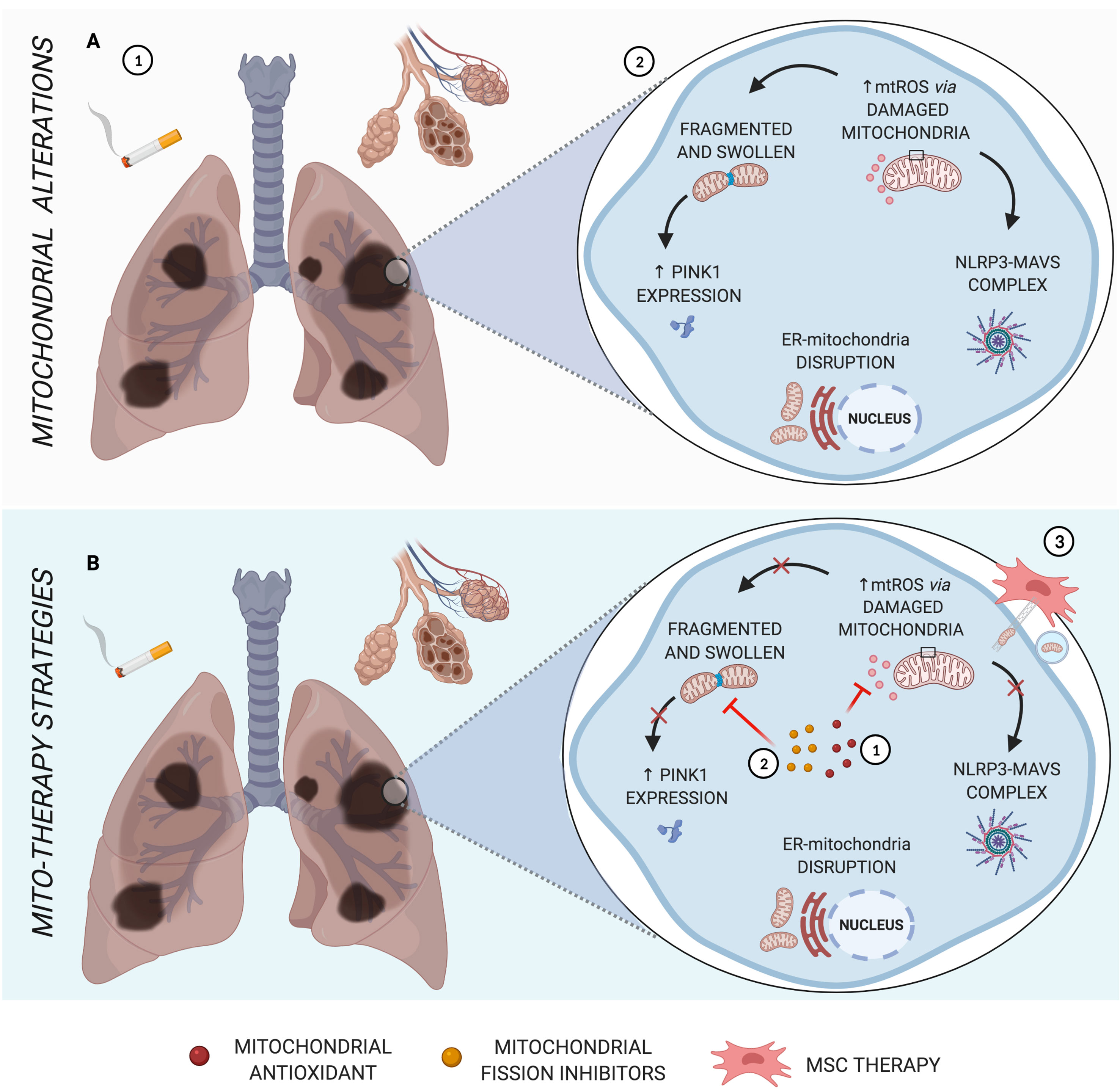
Figure 1 Main mitochondrial alterations in COPD. (A) The pathogenesis of COPD, triggered by cigarette smoke, is characterized by alveolar destruction and enlargement, as well as airway inflammation and remodeling (1). As the major source of oxidative stress, mitochondrial dysfunction leads to abnormal morphology, formation of NLRP3-MAVS complex, increased PINK1 mitophagy factor, and disruption of ER-mitochondria crosstalk (2). (B) Schematic representation of mito-therapy strategies for COPD. Mitochondrial antioxidants and fission inhibitors have a positive impact on pulmonary cells and murine models of COPD, acting in mtROS (1) and mitochondrial morphology dysfunction (2), respectively, and can act indirectly in NLRP3-MAVS complex formation, innate immune signaling for which mtROS is one activation signal. Otherwise, cell rescue from induced cigarette smoke or oxidative stress occurs via iPSC-MSC-mediated mitochondrial transfer in COPD models (3). Created with BioRender.com.
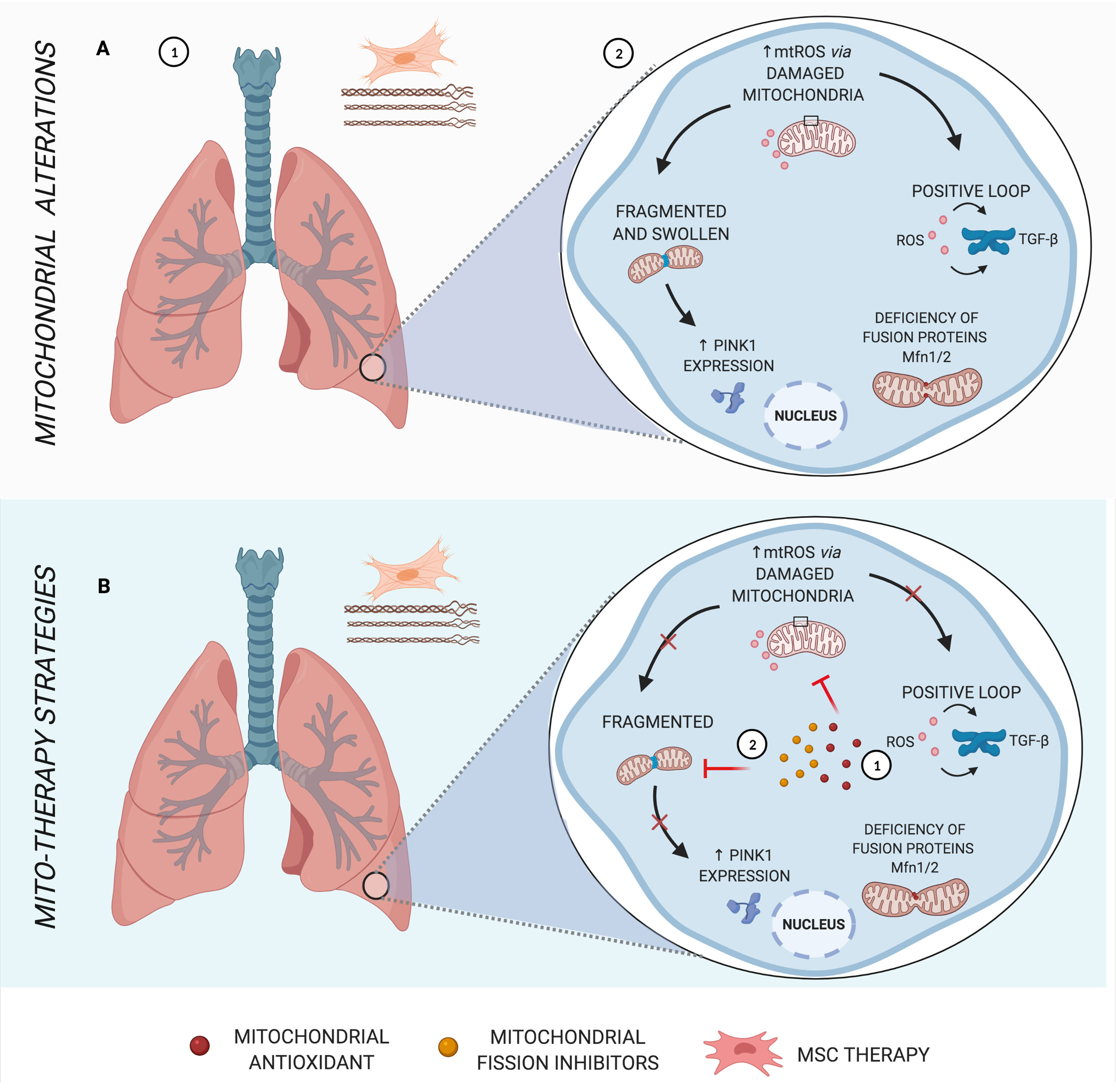
Figure 2 Main mitochondrial alterations in IPF. (A) Idiopathic pulmonary fibrosis (IPF), a parenchymal lung disease, is characterized by clusters of fibroblasts/myofibroblasts and excessive deposition of disorganized collagen and extracellular matrix, causing heterogeneous fibrosis (1). Increased mtROS in the pathogenesis of IPF induces transforming growth factor β (TGF-β), stimulating fibrogenesis in a positive loop, in addition to mitochondrial morphology alterations and deficiency in fusion proteins (2). (B) Schematic representation of mito-therapy strategies for IPF. Mitochondrial antioxidants lead to control of mitochondrial oxidative stress and, consequently, reduce TGF-β expression and activity (1), whereas mitochondrial fission inhibitors are capable of protecting pulmonary fibrosis models from mitochondrial fragmentation and posterior mitophagy factors (2). Created with BioRender.com.
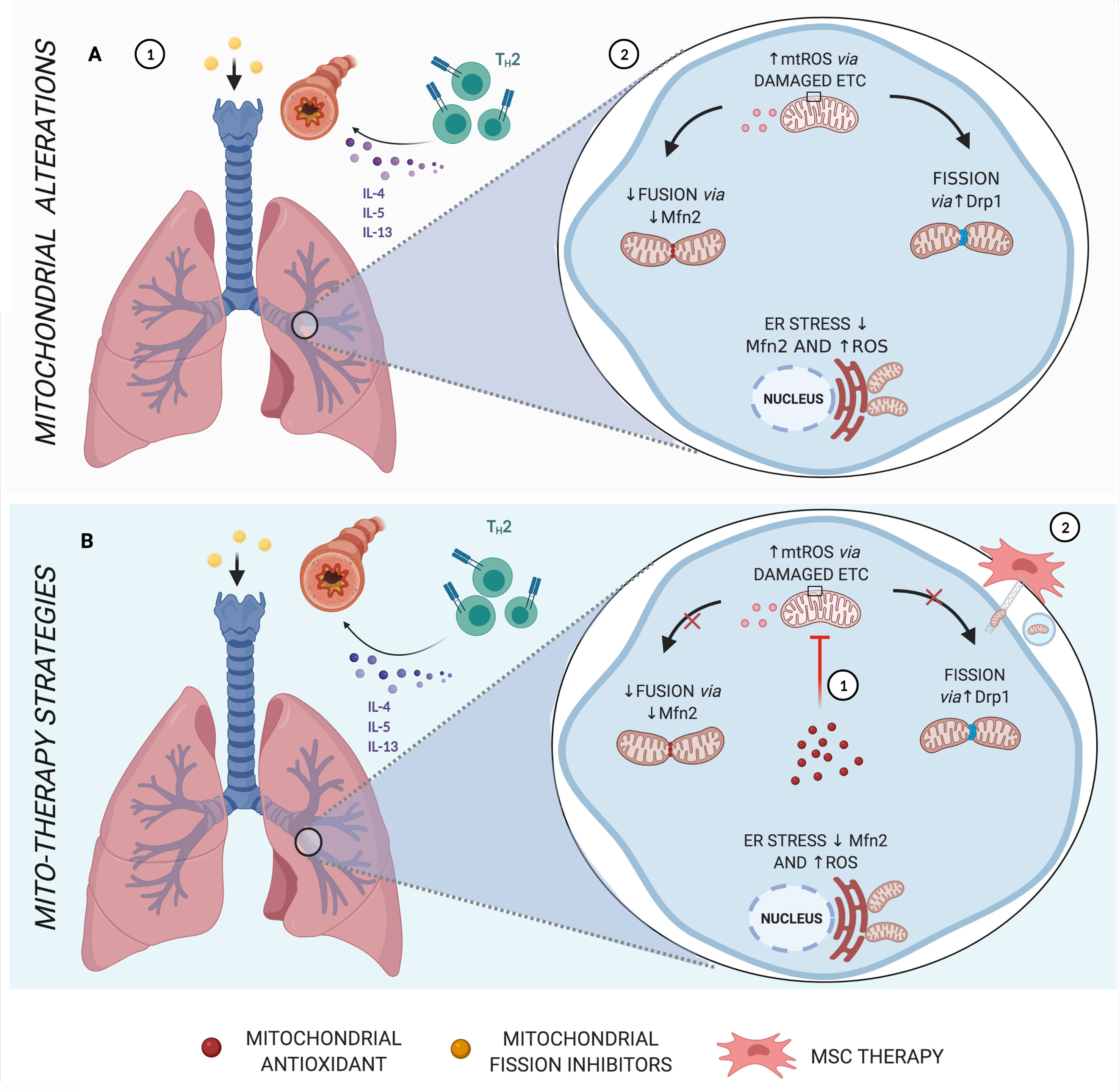
Figure 3 Main mitochondrial alterations in Asthma. (A) Asthma, which in most cases is strongly linked to allergen sensitization, is characterized by a Th2 inflammatory response via cytokines IL-4, IL-5, and IL-13, leading to bronchial hyperresponsiveness and remodeling (1). Features of asthma have also been associated with increased mtROS, endoplasmic reticulum (ER) stress, reduced fusion proteins, and increased fission dynamics (2). (B) Schematic representation of mito-therapy strategies for asthma. Mitochondrial target and localized antioxidants attenuate asthmatic pathophysiologic characteristics, especially controlling mtROS levels (1). On the other hand, mesenchymal stromal cells (MSCs) actively transfer mitochondria directly via gap junctions or through mechanisms of nanotubes and extracellular vesicles and are associated with beneficial effects in asthma models of airway injury and inflammation (2). Created with BioRender.com.
The centrality of mitochondrial dysfunction in the context of chronic respiratory diseases has been studied extensively in diseases that affect the trachea, bronchi, bronchioles (13, 14), alveoli (15, 16), and interstitium (17, 18). Mitochondrial dysfunction alters cell bioenergetics and hinders lung recovery after an insult (19). Thus, highlighting the importance and challenges of looking at mitochondria as a new target for therapeutic strategies in chronic lung diseases is essential (20–22). This review summarizes some mitochondrial physiologic and pathologic processes, as well as therapeutic strategies for chronic lung diseases considering mitochondria as the central point.
Mitochondrial Morphology
Structurally, mitochondria differ from other organelles in that their internal compartment (matrix) is kept apart from the cell cytoplasm through inner and outer membranes, separated by an intermembrane space, and all of these components play a fundamental role in their biochemical reactions (23). The conserved inner membrane ultrastructure contains invaginations or cristae where OX-PHOS enzymes, which are fundamental to mitochondrial functionality, are located (1, 24). Moreover, due to their lipid composition, small size, and partial transcriptional independence from the nucleus, mitochondria are extremely dynamic organelles (25). In response to pathophysiologic changes, mitochondria can change structurally and numerically through modifications in the behavior of their protein machinery (26).
In primary type II alveolar epithelial cells (AECII) in patients with COPD, abnormalities in the mitochondrial morphology have been reported, including loss of cristae and swollen and fragmented mitochondria (16, 27, 28). In vitro long-term exposure to cigarette smoke extract (CSE) in cultured human airway epithelial cells resulted in similar mitochondrial abnormalities (16, 27). In addition, reduced levels of prohibitin proteins (PHB1 and PHB2), present in the mitochondrial inner membrane, have been observed in lung tissue in patients with COPD and in non-COPD smokers (29). The homologous proteins PHB1 and PHB2 are essential components of fusion machinery and have been found to have a critical role in mitochondrial stability and morphogenesis and more recently in combating oxidative stress (29, 30). Collectively, these data indicate that cigarette smoke (CS) alters mitochondrial structure and functions and downregulates PHB1/PHB2 complexes, leading to increased cellular levels of reactive oxygen species (ROS) and cellular damage (29).
However, mitochondrial changes in COPD are not restricted exclusively to lung parenchyma or airway cells; they also extend to other cell types, such as skeletal muscle cells. Vastus lateralis muscle cells of patients with COPD present reduced mitochondrial fractional area, number, and enzyme activities, resulting in loss of oxidative capacity, which can help to explain the peripheral muscle dysfunction that is a hallmark of COPD (31). In addition, higher rates of apoptosis of T lymphocytes observed among patients with COPD can be partially explained by mitochondrial cytochrome c release, generally associated with abnormalities in mitochondrial morphology (32, 33). Cytochrome c can be released from swollen mitochondria via both permeability transition pore-dependent and independent mechanisms, and this liberation is associated with apoptotic cell death (34). T cell apoptosis may be also associated with the high frequency of infections and exacerbations observed among these patients, due to the resulting defective immune response (34).
Mitochondrial structure changes, such as a less dense matrix, loss of cristae, and mitochondrial cavity appearance have also been observed in experimental models of asthma induced by ovalbumin (35, 36). Allergic asthma appears to induce mitochondrial structure changes in an IL-4-dependent form, indicating the potential involvement of inflammatory cells like T lymphocytes, activated mast cells, and basophils in mitochondrial morphology changes in asthma pathogenesis (36). However, the molecular mechanism involved remains undetermined. Intense mitochondrial biogenesis was also demonstrated by a higher expression of activated mitochondria in bronchial smooth muscle (BSM) cells of asthmatic when compared to COPD and control patients (37). This indicates that although both asthma and COPD are characterized by BSM remodeling, a specific mitochondria-dependent pathway is required for BSM proliferation only in asthma (37).
Similarly, experimental models and human samples of pulmonary fibrosis demonstrated an increased number of mitochondria with either a swollen appearance or disorganized cristae in pulmonary epithelial cells (38, 39). Changes in mitochondrial morphology also occur in IPF lung fibroblasts, with disrupted membranes and altered cristae compared with normal subjects (40). Mitochondria modulate cellular senescence, a multifaceted cell phenotype that contributes directly to IPF, partially by increasing mitochondrial biogenesis, many of which become dysfunctional (41). Collectively, these results demonstrated that altered mitochondria morphology is a key pathologic feature of lung fibrosis. Comparatively, less information is currently available regarding modifications in mitochondrial morphology in the physiopathology of asthma and fibrosis compared to COPD.
Mitochondrial Dynamics
The mechanisms involved in mitochondrial membrane remodeling, termed mito-dynamics, include fusion and fission, which are tightly associated with homeostasis adjustment (42). The fusion of mitochondrial membranes, normally stimulated by energy demand and stress, restores the damaged mitochondria by diffusion and sharing of molecular components between organelles (43). The main GTPases involved in mitochondria fusion are mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2), which are integrated into the outer membrane and have domains exposed to the cell cytoplasm, together with optic atrophy 1 (Opa1), a mitochondrial dynamin associated with the inner membrane (44). On the other hand, fission divides and creates new mitochondria, so it is essential to cell cycle progression through growth and division. The presence of fission machinery is a hallmark and is mandatory for mitosis phases when mitochondria appear to be more fragmented (45, 46). Mitochondrial fission is mediated by a dynamin 1-like (Drp1) and mitochondrial fission 1 (Fis1) protein, but the mechanism has not been fully elucidated yet (47). Alterations in mito-dynamics are observed in many chronic lung diseases and contribute differently to each of them (48).
Mitochondrial dynamics dysfunction has been proposed to participate in the development of COPD (27, 49). CSE directly affects fibroblast, alveolar and small airway epithelial cells, causing substantial mitochondrial morphological defects observed after exposure to non-toxic doses through mechanisms that involve mitochondrial elongation (50, 51). Low doses of CSE also cause increased MFN expression in mouse alveolar epithelial cells (50). In contrast, mitochondrial fragmentation induced via Drp-1 recruitment is observed in human bronchial epithelial cells (HBEC) after exposure to more toxic doses of CSE (27, 52). Even more, long-term exposure to CSE causes more complex changes in mitochondrial morphology, reflecting the coexistence of different mitochondrial phenotypes, both elongated and fragmented, to different levels of chronic cigarette smoke in COPD (16, 27).
CS is also known to enhance respiratory disorders such as bronchitis and asthma, characterized by inflammatory changes, hyperresponsiveness, and increased cell proliferation of airway smooth muscle (ASM) (53). ASM cells isolated from moderate asthmatic patients appear to be more sensitive to CSE than non-asthmatic patient samples, with associated decreased expression and function of Mfn2, whereas increased Drp1-mediated mitochondrial fragmentation (49). This mitochondrial fission/fusion imbalance alters ROS dynamics and can lead to a cycle with more fragmented mitochondrial networks, elevated ROS production, and cell proliferation (49). There is still few current information on mitochondrial fission/fusion dynamics in ASM, and its importance in asthma.
Accelerated senescence is observed in lung epithelial cells in IPF, and aging. Alveolar epithelial cells derived from aged mice demonstrated accelerated lung fibrosis with enlarged mitochondria and augmented expression of OPA1 and MFN1/2 (18). This data indicates that mitochondria fusion is predominant in IPF lung epithelial cells (18). In contrast, the absence of mitochondrial fusion proteins Mfn1/2 in murine AECII is strongly associated with less production of surfactant lipids and subsequent spontaneous fibrotic remodeling in the lung, leading to higher morbidity and mortality in these animals (54). Therefore, deficiency in mitochondrial fusion may be linked to disruption in lipid metabolism, AECII injury, and further fibrosis (54). Similarly, the main protein involved in fission, Drp1, has been shown to have critical involvement in the development of pulmonary fibrosis, and when inhibited, it prevents mitochondrial fragmentation and pulmonary fibrosis in a bleomycin-induced model (55, 56). These data suggest that the relationship between mitochondrial dynamics and cell survival/death programming is complicated and may vary between individual cell types and disease conditions (49).
The dynamics of intracellular organization and localization of mitochondria are likely to influence several aspects of cellular physiology. A bidirectional coupling between mitochondrial morphology/dynamics and motility has been proposed as an interconnected signaling pathway involved in cellular function (57). The subcellular distribution of mitochondria can be actively modified in response to energy demand and stress. Organelle dislocation is mediated by cytoskeleton and motor proteins (Miro1/2, actin, microtubules kinesin, and dynein), which can interact with mito-dynamic proteins and can interfere with endoplasmic reticulum (ER) communication (26, 58–61). Mitochondrial intracellular movement is directly linked to calcium signals, which at different concentrations can induce mitochondrial translocation or provide a mechanism to retain mitochondria at Ca2+ signaling sites, regulating local power supply (62, 63). This can be particularly important for epithelial cells in chronic lung diseases, such as asthma and COPD, which have a high cell turnover rate and increased energy requirements (20). Other biochemical signals have been involved in the distribution of mitochondria, such as hypoxia, which has been reported to cause mitochondrial translocation to the perinuclear region (64). However, intentional subcellular mitochondrial positioning in chronic lung disease cells and what these mechanisms tell us about mitochondrial function is still a poorly studied topic.
Mitochondrial Redox Signaling
Together with NADPH oxidases, mitochondria are the major source of ROS, a mitochondrial subproduct generated during the electron transport chain (ETC) flux (65). About 3% of the electrons leak in this process, reacting with oxygen on the mitochondrial matrix to form superoxide (O2-), which is converted into hydrogen peroxide (H2O2) by superoxide dismutase 2 (SOD2) (65, 66). Although ROS are essential for physiologic functions, oxidative/antioxidative imbalance can be detrimental, particularly in organs continuously exposed to oxygen and, consequently, highly susceptible to injury mediated by ROS such as the lungs (67–70).
Mitochondrial ROS (mtROS) can act as a second messenger, promoting physiologic signals of cellular stress and leading to mitochondrial translocation (64, 67). Particularly in the lungs, due to their anatomy and continuous exposure to the environment, mitochondrial respiration is an important endogenous source of oxidative stress (71, 72). When in excess, mtROS leads to uncoupled ETC, calcium imbalance, impaired communication between the ER and mitochondria, and damaged mtDNA, and acts as an inflammatory signal (73).
Mitochondrial damage has an important role in the pathogenesis of COPD, in which ROS levels exceed the antioxidant defenses (74). The abundant ROS production can be explained by CS and CSE lipophilic components were capable of disturbing mitochondrial function and increasing the mtROS in ASM cells from patients with COPD (75). Oxidative stress causes lipid peroxidation, resulting in protein carbonylation, commonly referred to as “carbonyl stress,” that is predominantly associated with chronic diseases (76). In this cycle, carbonyl stress can damage mitochondrial proteins and drive further endogenous production of ROS (69).
Increased mtROS has been demonstrated in a number of fibrotic disorders, including pulmonary fibrosis. Oxidants have a direct impact on the production of the most potent fibrogenic cytokine, transforming growth factor β (TGF-β), inducing its gene expression. The overexpression of this central mediator of fibrogenesis increases the production of mtROS by blocking complex III activity and suppressing the antioxidant system in a reciprocal upregulation (positive loop) (77–79). mtROS also causes oxidation of lipids and proteins identified in bleomycin-induced mouse models of pulmonary fibrosis and in patients with IPF (80, 81). Similarly, exposure to asbestos fibers both in vitro and in vivo leads to increased mtROS production, which regulates lung epithelial cell apoptosis and fibrosis (82, 83).
Oxidative stress also plays an important role in allergic airway disorders. Airway remodeling and the immune response in asthma pathogenesis have been associated with mitochondrial metabolism, including the redox state (84). The most prominent stimuli of asthma, environmental factors, can lead to damage to specific chain-complex proteins, sustaining ROS generation, and can further lead to airway hyperresponsiveness (AHR) (85, 86). The cellular redox imbalance results in inflammatory infiltration and cell damage and can lead to severe asthma and reduction of the corticosteroid response (87–89). The more severe symptoms in allergic disorders have been associated with mitochondrial defects around complexes I and III, which are responsible for the majority of mtROS production due to electron leakage (85). Several markers of oxidative activity are present in people with asthma. These patients have increased production of ROS by inflammatory cells, such as macrophages, eosinophils, and neutrophils, which lead to an increased concentration of exhaled hydrogen peroxide and secretion of myeloperoxidase and eosinophil peroxidase (87–91).
Mitophagy
Mitophagy is a selective form of apoptosis for dysfunctional mitochondria, classically through phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) degradation (92). Permeabilization of the outer mitochondrial membrane via apoptosis regulator Bcl-2 associated X (BAX) and/or Bcl-2 homologous antagonist/killer (BAK), or the opening of the mitochondrial permeability transition pore (mPTP) in the inner mitochondrial membrane leading to the release of intrinsic apoptosis-induced factors, such as cytochrome c, is described to initiate the mitochondrial apoptotic pathway (93, 94). Permeabilization of the outer membrane (MOMP) and activation of fusion and fission mechanisms are necessary to release cytochrome c from cristae junctions (95, 96). Excessive levels of mtROS can induce mitophagy, which in turn removes and recycles toxic or damaged mitochondria, reducing mtROS, to maintain the intercellular balance between oxidants/antioxidants, triggering a negative feedback loop mechanism (97, 98).
Intriguingly, both enhanced and impaired mitophagy have been implicated in the pathogenesis of COPD. Pink1-deficient mice showed protection against the main characteristics of COPD, such as airspace enlargement, mucociliary clearance, and mitochondrial dysfunction (99). Accordingly, increased expression of PINK1 in lung epithelial cells of patients with COPD has also been observed, along with increased necroptosis markers, impaired alveolar macrophage autophagy (100), mitochondrial dysfunction, and morphology alteration in skeletal muscle (101). On the other hand, insufficient mitophagy and reduced expression levels of PARK2 (parkin RBR E3 ubiquitin-protein ligase) can accelerate senescence and are part of the pathogenesis of COPD (52). The PINK1-PARK2 pathway has been proposed as a crucial mechanism implicated in mitophagic degradation (102). Mitochondria with depolarized membrane stabilize PINK1, resulting in recruitment of PARK2 to mitochondria, which leads to mitochondrial substrates ubiquitination (102). Concomitant accumulation of ubiquitinated proteins is recognized as at least partly reflecting insufficient mitophagy (103).
PINK1, LC3-I/II, and other mitophagy factors, which are responsible for normalizing mitochondrial morphologic and functional integrity, play a protective role in the pathogenesis of COPD (104). The exposure of pulmonary fibroblasts to CSE led to damaged mitophagy, an increase in cell senescence, mtDNA damage, decreased mitochondrial membrane potential, and ATP levels, later restored by a specific mitochondrial antioxidant (51). These data demonstrate the important role of mitophagy in the pathogenesis of COPD, leading to senescence or programmed cell death depending on the level of damage (52).
In addition, TGF-β can also lead to mitophagy, stabilizing the mitophagy initiating protein PINK1 and inducing mtROS (38). TGF-β is known to stimulate ROS production, and oxidative stress can activate latent TGF-β, setting up a bidirectional signaling and profibrogenic cycle (78, 105). Mechanisms that activate TGF-β-mediated pro-fibrotic events and the PI3K/Akt signaling cascade are important pathways involved in the progression of pulmonary fibrosis (106, 107). In this context, berberine was capable of inhibiting PI3K/Akt/mTOR cascade activation, enhancing autophagy, and mitigating fibrotic markers in a bleomycin-induced rodent model of pulmonary fibrosis (107). PINK1 deficiency was recently correlated with pulmonary fibrosis, and its impaired expression led to an accumulation of damaged mitochondria in lung epithelial cells from patients with IPF (18). Pink1-deficient mice are more susceptible to developing pulmonary fibrosis in a bleomycin model, suggesting PINK1 may be necessary to limit fibrogenesis (38). These data together suggest that downregulation of autophagy or mitophagy is deleterious, whereas its upregulation is protective in IPF (108).
Environmental factors and allergens are the main factors involved in the development of allergic airway inflammation and asthma, leading to oxidative stress, mitochondrial dysfunction, and cellular senescence (109–112). Environmental pollutants can induce mitophagy, ROS, and mitochondrial damage, which activate the PINK/Parkin pathway (113, 114). The Ca2+/calmodulin-dependent protein kinase II (CaMKII) has been shown to be an important mediator in allergic inflammation, ROS production, and correlated with the severity of asthma (115, 116). Oxidized CaMKII stimulates transcriptional activators of TGF-β and can lead to a profibrotic phenotype, a key factor in the development of asthma airway remodeling (115, 117, 118). Recently, the allergen-ROS-ox-CaMKII-mitophagy axis was demonstrated to play an important role in the development of allergic airway inflammation, indicating that CaMKII may be a therapeutic target for asthma (119). However, studies on mitophagy and asthma are still limited.
ER Interaction and Calcium Regulation
The ER is responsible for intracellular Ca2+ storage; protein synthesis, transport, and folding; lipid and steroid synthesis; and carbohydrate metabolism (120). This organelle interacts with mitochondria through membrane ER contact sites, which involve portions of membrane known as mitochondrial associated membranes (MAMs), which play role in structural and functional linkage for intracellular functions (121, 122). At the MAMs, Ca2+ is transferred and can interfere in mitochondrial metabolism, stimulating ETC complexes and regulating ATP production (123).
ER-mitochondria interaction provides a platform for the regulation of mitochondrial dynamics and is related to different pathophysiologic contexts, such as immune response and cell death (124). ER stress, normally caused by unfolded proteins and mitochondrial dysfunction, leads to an increase in ROS production, which, in a vicious cycle, leads to further ER stress (125). However, which mechanism triggers the processes – endoplasmic/sarcoplasmic reticulum stress (ER/SR stress) or mitochondrial dysfunction – is still unclear.
ER-mitochondria crosstalk is disrupted in COPD by stress, such as inhaled tobacco products and pollutants (126). Previous studies have shown increased expression of proteins related to ER stress (chaperones, GRP78, CHOP) in lung cells from mice exposed to CS, bronchoalveolar lavage fluid, and tissue samples from chronic cigarette smokers (127–129). Similarly, AECII injury associated with ER stress markers is a well‐accepted theory in the pathogenesis of IPF (130). Increased mitochondrial content in AECIIs and mitochondrial dysfunction associated with ER stress were found in highly fibrotic areas in IPF lungs (18, 131, 132). Findings in bleomycin-treated mice and AECII of IPF lungs have shown that a disruption in the crosstalk between ER and mitochondria occurs, probably involving mitochondrial homeostasis-control mechanisms, ER stress induced by PINK1, and integrated stress response transcription factors 3 and 4 (ATF3 and ATF4) (130, 133).
ER stress-induced by TNFα and ROS has also been shown to reduce the proteins involved in the connection between ER and mitochondria through MAM, such as Mfn2, in human airway smooth muscle (hASM) cells (134). The exposure of hASM cells to TNFα, a proinflammatory cytokine that mediates the inflammatory response in asthma, led to the activation of ER stress pathways, disrupted mitochondrial proximity to the ER, and decreased Mfn2 protein expression, impairing mitochondrial mobility (134, 135). This creates the possibility of a vicious cycle with reduced Mfn2 expression and altered mitochondrial function (125).
Some aspects involving mitochondrial dynamics and ER interaction via MAMs remain enigmatic. However, the mitochondria-ER contact sites role mediating immune responses through facilitation of the NOD-like receptor protein 3 (NLRP3)-inflammasome assembly are well known, including second messenger mechanisms such as mitochondrial ROS (136, 137). These multiprotein complexes are composed of the sensor protein NOD-like receptor family, an adaptor protein with caspase domain (ASC), and a pro-caspase 1 protein. When oligomerized, these complexes sense microbial and damage signals (DAMPs and PAMPs), inducing the active form of IL-1β or IL-18 when activated (138).
Increased mRNA levels of NLRP3 inflammasomes in bronchial tissues and systemically, as well higher levels of IL-18 and IL-1β were found in patients with acute exacerbation of COPD than in smokers (139). Similarly, higher expressions of NLRP3 and IL-1β were found in isolated macrophages and BALF of patients with different phenotypes of asthma, and in animal models of this disease (140–142). Additionally, knockdown Drp1 favors NLRP3 activation in mouse bone marrow-derived macrophages, and Mfn2 protein was required for NLRP3 activation after RNA virus infection to form NLRP3-Mfn2-MAVS complex (143, 144). MAVS (mitochondrial antiviral signaling), also has an important role in the pathophysiology of lung fibrosis in a bleomycin-induced model, through their main expression in pulmonary macrophages, amplifying multiple DAMPs signaling (145). MAVS aggregation is observed in lung tissues from human patients with IPF (146). MAVS is well known to induce antiviral genes, acts as a second adapter to the optimal activity of the NLRP3 inflammasome, contributing directly to IL-1β production without inducing IFN-β expression (146). Considering the close relationship between mitochondria and the ER, and their significant contribution to inflammasome activation and chronic lung diseases, MAMs and NRLP3 may be a potential new therapeutic target.
Discussion
As highlighted in this review, the contribution of mitochondrial dysfunction in the development of the main chronic lung diseases is unquestionable. All evidence suggests the urgent need and the great potential of therapeutic approaches considering mitochondria as a target.
Mitochondria as a Target and Localized Antioxidants
Restoration of the cellular antioxidant/oxidant level is a good proposal to protect cells and tissue from oxidative stress-mediated disorders (147, 148). Murine models of ovalbumin (OVA)-induced airway inflammation and hyperresponsiveness have shown attenuated asthmatic lung pathophysiologic characteristics when the redox state was restored using a small-molecular-weight thiol antioxidant compound, N-acetylcysteine amide (AD4) (87, 88). Recently, mitochondrial target antioxidants began to be widely studied as therapeutic approaches to diseases in which oxidative stress appears to be critical (149).
MitoTEMPO was reported as a SOD mimetic antioxidant that inhibits mtROS. It is combined with the lipophilic cation triphenylphosphonium (TPP+), a membrane-permeant cation that permits the accumulation of antioxidants inside mitochondria by the membrane potential generated (150). Treatment with mitochondrial-targeted antioxidant MitoTEMPO reduces significant features of asthma in cultured cells and in OVA-challenged mice, suggesting that controlling mtROS levels may reduce TGF-β expression and activity (118). MitoTEMPO also contributes to decelerating fibroblast senescence in patients with IPF (151). However, MitoTEMPO failed to inhibit airway inflammation and bronchial responsiveness in an acute ozone-induced murine model of airway inflammation and bronchial hyperresponsiveness (152). Another mitochondrial-specific antioxidant linked to TPP+, the mitoquinone (MitoQ), is a derivative of coenzyme Q and was capable of reversing mitochondrial dysfunction, inflammation, and AHR after mice exposure to ozone (69, 153). Both MitoQ and Tiron, a mitochondrial localized antioxidant, were effective in inhibiting TGF-β-induced proliferation and CXCL8 release in ASM cells from patients with COPD (69). In addition, SS-31, a mitochondrial-targeted agent, is beneficial in other respiratory conditions such as mechanical ventilation-induced diaphragm weakness and pulmonary arterial hypertension in vivo (154, 155). These data reinforce the role of mitochondrial ROS and its potential as a therapeutic approach in lung chronic disease. However, faced with so many types of mitochondrial involvement in pathological processes viewed in this review, the real contribution of mtROS is a question that remains open.
However, antioxidant therapy is still highly questioned for its disappointing results in clinical trials (156). The beneficial effects have been determined only by the pharmacologic properties, ignoring bioavailability and pharmacokinetics, by examining effects in concentrations that are often impossible to achieve in vivo (157). Now, antioxidants targeting mitochondria make this therapy more selective and effective, but before it becomes an actual therapy for patients, it is still necessary to carefully establish bioavailability and safety profiles of using more selective agents to achieve clinically relevant effects.
Mitochondrial Dynamics as a Target
Although mitochondria are very dynamic organelles and changes in fusion and fission are constantly observed in chronic lung diseases, this aspect is often overlooked when considering new therapeutic approaches. Mito-dynamics seems to be an important target because evidence indicates that when disrupted, mitochondrial function is affected negatively (158–160).
Mitochondrial division inhibitor 1 (Mdivi-1) reduces Drp1, Fis1 genes, and consequently excessive mitochondrial fission while enhancing Opa1, Mfn1, Mfn2 genes, and mitochondrial fusion activity (161). Furthermore, Mdivi-1 induced increased levels of complex I, II, and IV enzymatic activities (161). This mitochondrial division/mitophagy inhibitor was capable of reducing CS-induced cell death and mitochondrial dysfunction in vitro and protected mice from bleomycin-induced mitochondrial fragmentation and pulmonary fibrosis (56, 99). P110, which is a selective inhibitor of Drp1 enzyme activity and blocks Drp1/Fis1 interaction, was demonstrated to be neuroprotective and improve mitochondrial function and integrity (162). These data suggest that inhibitors of Drp1 might be useful for the treatment of diseases in which excessive mitochondrial fission occurs. Elucidation of mitochondrial dynamics involvement in different cellular processes is promising but still superficial, along with the impact of altered mitochondrial dynamics in chronic lung diseases physiopathology.
Cell Therapy
Interest in the therapeutic potential of cell therapy in lung biology and diseases has increased (163, 164). This research area is expanding rapidly, and several studies have demonstrated the potential of immunomodulation and regenerative effects of adult mesenchymal stromal (stem) cells (MSCs), in animal models of chronic lung diseases such as asthma, COPD, and fibrotic injuries (165–169). Promising results in animal studies and incipient clinical trials have made MSC therapy further increasingly recognizing the potential contribution of mitochondrial transfer from the MSCs as a potential mechanism of action (170, 171).
Intercellular mitochondrial transfer occurs via mechanisms including tunneling nanotube formation between two spatially separated cells, secretion of extracellular vesicles containing mitochondria, gap junctions, and cell fusion where cells will share organelles and cytosolic compounds (172). MSCs can transfer mitochondria to other cells in response to stress signals such as the release of damaged mitochondria, mtDNA, and mitochondrial products along with increased levels of ROS (173). MSC-mediated mitochondrial transfer can have an impact on inflammatory responses and cell viability and is emerging as a therapeutic strategy partially by acting as bioenergetics supplementation (174, 175).
Active mitochondrial transfer from adult stem cells to cells pretreated with ethidium bromide, with defective or deleted mtDNA by mutation, was capable of rescuing aerobic respiration of these nonfunctional mitochondria (175). BMSCs exerted protective effects on the alveolar epithelium, restoring the alveolar metabolism in an acute lung injury (ALI) model. These cells transferred mitochondria to epithelial cells via connexin-43 gap junctions, directly or through underlying mechanisms of nanotubes and microvesicles, increasing alveolar ATP production and reducing the hallmarks of ALI induced by lipopolysaccharide (176). Intercellular mitochondrial transport is regulated by Miro1, a calcium-sensitive adaptor protein that helps the mitochondria to move along microtubules inside the cells and when overexpressed, increases their mitochondrial transfer capacity and beneficial effects in asthma models (171). In addition, mitochondrial transfer from human induced pluripotent stem cell (iPSC)-derived MSCs to airway epithelial and ASM cells was also identified, rescuing cells from lung damage induced by CS or oxidative stress (177, 178). iPSC MSCs also attenuate asthma inflammation, protection attributed to mitochondrial transfer via connexin 43 (CX43)-mediated tunneling nanotube (TNT) formation (179). Taken together, these data demonstrate that BMSCs can transfer mitochondria and rescue lung damage in different contexts. However, how much of the positive effects of cell therapy in chronic lung diseases are exerted solely by mitochondrial transfer is still unknown.
Mitochondrial Therapy
Given the observed results with MSC mitochondrial transfer in experimental model systems described above, multiple strategies have been further explored, including local and systemic administration of healthy isolated exogenous mitochondria, also called mitochondrial transplantation or mitoception.
Promising outcomes have been demonstrated in in vitro and in vivo models. Preclinical studies using New Zealand White rabbits demonstrated cardioprotection in a cardiac ischemia-reperfusion injury after autologous mitochondria transplantation from biopsy samples of the pectoralis major (180). In situ mitochondrial injection was capable of enhancing post-infarct cardiac function; mitochondria were internalized by cardiomyocytes 2–8 h after transplantation (180). However, less than 10% of the transplanted mitochondria were integrated into cardiomyocytes (180). Using a similar strategy, systemic intravenously injected mitochondria isolated from cultured human hepatoma cells (HepG2) were used in mice fatty liver models, reducing lipid accumulation and restoring hepatocyte function by less well-known mechanisms (181). Mitochondrial therapy, using isolated mitochondria from C57BL/6J gastrocnemius muscle, has also shown efficacy in a murine model of lung ischemia-reperfusion injury, attenuating lung tissue injury, and mechanical parameters via vascular delivery or nebulization (182). More recently, systemic mito-therapy using a mitochondria-rich fraction isolated from BMSCs was capable of decreasing lung, liver, and kidney injury and increased the survival rate in cases of cecal ligation and puncture-induced sepsis (183).
An ongoing trial is testing arterial or tissue injection of autologous mitochondrial transplantation from skeletal muscle of the chest wall into the ischemic myocardium of patients with heart ischemia/reperfusion injury, to decrease morbidity and mortality in patients requiring extracorporeal membrane oxygenation (ECMO) (NCT#02851758). However, it is not yet fully understood if and how mitochondria present in the extracellular space exert effects on cells, and how the internalization of healthy extracellular mitochondria occurs after focal or systemic administration. Remains open in the literature the comparison between the role of MSCs paracrine secretion and mitochondrial transfer.
Conclusion
Mitochondria-targeted therapy may be a new therapeutic for restoring cellular bioenergetics and function in several airway diseases. Some mechanisms have been acknowledged, demonstrating the complex role of mitochondria in chronic lung diseases. Recent studies have challenged the initial thinking about the central role of mitochondrial oxidative stress, bringing new data about how differently mitochondrial responses can be, acquiring diverse phenotypes in morphology, dynamics, and during mitophagy in distinct diseases. In addition, mitochondria play an essential role in inflammatory signaling, via mitochondria-ER communication through MAMs activating NLRP3/MAVS complexes. Therefore, mitochondrial dysfunction was unquestionably a factor in chronic lung disease development and progression. Despite that, innovative and attractive therapy as mitochondrial antioxidants, cell therapy, and mitochondrial transfer remains with important open questions which impact directly their clinical consideration. New insights into these mechanisms may hold the key for mitochondrial target treatment, which has remained elusive.
Author Contributions
FC, PS, and PR designed this review. All authors contributed equally to literature revision and manuscript writing. All authors contributed to the article and approved the submitted version.
Funding
Brazilian Council for Scientific and Technological Development (CNPq), Rio de Janeiro State Research Foundation (FAPERJ), Coordination for the Improvement of Higher Education Personnel (CAPES), Department of Science and Technology – Brazilian Ministry of Health (DECIT/MS), and the National Institute of Science and Technology for Regenerative Medicine/CNPq.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. McBride HM, Neuspiel M, Wasiak S. Mitochondria: More Than Just a Powerhouse. Curr Biol (2006) 16:R551–60. doi: 10.1016/j.cub.2006.06.054
2. Skulachev VP. Why Are Mitochondria Involved in Apoptosis? Permeability Transition Pores and Apoptosis as Selective Mechanisms to Eliminate Superoxide-Producing Mitochondria and Cell. FEBS Lett (1996) 397:7–10. doi: 10.1016/0014-5793(96)00989-1
3. Martinou J-C, Youle RJ. Mitochondria in Apoptosis: Bcl-2 Family Members and Mitochondrial Dynamics. Dev Cell (2011) 21:92–101. doi: 10.1016/j.devcel.2011.06.017
4. Giorgi C, Agnoletto C, Bononi A, Bonora M, De Marchi E, Marchi S, et al. Mitochondrial Calcium Homeostasis as Potential Target for Mitochondrial Medicine. Mitochondrion (2012) 12:77–85. doi: 10.1016/j.mito.2011.07.004
5. Giorgi C, Marchi S, Pinton P. The Machineries, Regulation and Cellular Functions of Mitochondrial Calcium. Nat Rev Mol Cell Biol (2018) 19:713–30. doi: 10.1038/s41580-018-0052-8
6. Emelyanov VV. Mitochondrial Connection to the Origin of the Eukaryotic Cell. Eur J Biochem (2003) 270(8):1599–618. doi: 10.1046/j.1432-1033.2003.03499.x
7. Aghapour M, Remels AHV, Pouwels SD, Bruder D, Hiemstra PS, Cloonan SM, et al. Mitochondria: At the Crossroads of Regulating Lung Epithelial Cell Function in Chronic Obstructive Pulmonary Disease. Am J Physiol Lung Cell Mol Physiol (2020) 318(1):L149–64. doi: 10.1152/ajplung.00329.2019
8. Cloonan SM, Kim K, Esteves P, Trian T, Barnes PJ. Mitochondrial Dysfunction in Lung Ageing and Disease. Eur Respir Rev (2020) 29(157):1–18. doi: 10.1183/16000617.0165-2020
9. Rodrigues GC, Rocha NN, Maia LA, Melo I, Simões AC, Antunes MA, et al. Impact of Experimental Obesity on Diaphragm Structure, Function, and Bioenergetics. J Appl Physiol (2020) 129(5):1062–74. doi: 10.1152/japplphysiol.00262.2020
10. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial Dysfunction and Oxidative Stress in Metabolic Disorders - A Step Towards Mitochondria Based Therapeutic Strategies. Biochim Biophys Acta Mol Basis Dis (2017) 1863(5):1066–77. doi: 10.1016/j.bbadis.2016.11.010
11. Zhou B, Tian R. Mitochondrial Dysfunction in Pathophysiology of Heart Failure. J Clin Invest (2018) 128:3716–26. doi: 10.1172/jci120849
12. Dornas W, Lagente V. Blocking Reactive Oxygen Species Generation Inhibits Biogenesis in Mitochondrial Dysfunction. Reactive Oxygen Species (2019) 8(22):196–201. doi: 10.20455/ros.2019.839
13. Hayashi T, Ishii A, Nakai S, Hasegawa K. Ultrastructure of Goblet-Cell Metaplasia From Clara Cell in the Allergic Asthmatic Airway Inflammation in a Mouse Model of Asthma In Vivo. Virchows Archiv (2004) 444:66–73. doi: 10.1007/s00428-003-0926-8
14. Mabalirajan U, Aich J, Leishangthem GD, Sharma SK, Dinda AK, Ghosh B. Effects of Vitamin E on Mitochondrial Dysfunction and Asthma Features in an Experimental Allergic Murine Model. J Appl Physiol (2009) 107:1285–92. doi: 10.1152/japplphysiol.00459.2009
15. Ryter SW, Rosas IO, Owen CA, Martinez FJ, Choi ME, Lee CG, et al. Mitochondrial Dysfunction as a Pathogenic Mediator of Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Ann Am Thorac Soc (2018) 15(Suppl 4):S266–72. doi: 10.1513/AnnalsATS.201808-585MG
16. Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S, et al. Prolonged Cigarette Smoke Exposure Alters Mitochondrial Structure and Function in Airway Epithelial Cells. Respir Res (2013) 14:97. doi: 10.1186/1465-9921-14-97
17. Malsin ES, Kamp DW. The Mitochondria in Lung Fibrosis: Friend or Foe? Trans Res (2018) 202:1–23. doi: 10.1016/j.trsl.2018.05.005
18. Bueno M, Lai Y-C, Romero Y, Brands J, St Croix CM, Kamga C, et al. PINK1 Deficiency Impairs Mitochondrial Homeostasis and Promotes Lung Fibrosis. J Clin Invest (2015) 125(2):521–38. doi: 10.1172/JCI74942
19. Ten VS, Ratner V. Mitochondrial Bioenergetics and Pulmonary Dysfunction: Current Progress and Future Directions. Paediatr Respir Rev (2020) 34:37–45. doi: 10.1016/j.prrv.2019.04.001
20. Prakash YS, Pabelick CM, Sieck GC. Mitochondrial Dysfunction in Airway Disease. Chest (2017) 152:618–26. doi: 10.1016/j.chest.2017.03.020
21. Demedts IK, Demoor T, Bracke KR, Joos GF, Brusselle GG. Role of Apoptosis in the Pathogenesis of COPD and Pulmonary Emphysema. Respir Res (2006) 7:1–10. doi: 10.1186/1465-9921-7-53
22. Rangarajan S, Bernard K, Thannickal VJ. Mitochondrial Dysfunction in Pulmonary Fibrosis. Ann Am Thorac Soc (2017) 14:S383–8. doi: 10.1513/annalsats.201705-370aw
23. Frey TG, Mannella CA. The Internal Structure of Mitochondria. Trends Biochem Sci (2000) 25(7):319–24. doi: 10.1016/S0968-0004(00)01609-1
24. Kühlbrandt W. Structure and Function of Mitochondrial Membrane Protein Complexes. BMC Biol (2015) 13:89. doi: 10.1186/s12915-015-0201-x
25. Soubannier V, McBride HM. Positioning Mitochondrial Plasticity Within Cellular Signaling Cascades. Biochim Biophys Acta (2009) 1793(1):154–70. doi: 10.1016/j.bbamcr.2008.07.008
26. Detmer SA, Chan DC. Functions and Dysfunctions of Mitochondrial Dynamics. Nat Rev Mol Cell Biol (2007) 8(11):870–9. doi: 10.1038/nrm2275
27. Hara H, Araya J, Ito S, Kobayashi K, Takasaka N, Yoshii Y, et al. Mitochondrial Fragmentation in Cigarette Smoke-Induced Bronchial Epithelial Cell Senescence. Am J Physiol Lung Cell Mol Physiol (2013) 305(10):L737–46. doi: 10.1152/ajplung.00146.2013
28. Kosmider B, Lin C-R, Karim L, Tomar D, Vlasenko L, Marchetti N, et al. Mitochondrial Dysfunction in Human Primary Alveolar Type II Cells in Emphysema. EBioMedicine (2019) 46:305–16. doi: 10.1016/j.ebiom.2019.07.063
29. Soulitzis N, Neofytou E, Psarrou M, Anagnostis A, Tavernarakis N, Siafakas N, et al. Downregulation of Lung Mitochondrial Prohibitin in COPD. Respir Med (2012) 106(7):954–61. doi: 10.1016/j.rmed.2012.03.019
30. Theiss AL, Sitaraman SV. The Role and Therapeutic Potential of Prohibitin in Disease. Biochim Biophys Acta (2011) 1813(6):1137–43. doi: 10.1016/j.bbamcr.2011.01.033
31. Gosker HR, Hesselink MKC, Duimel H, Ward KA, Schols AMWJ. Reduced Mitochondrial Density in the Vastus Lateralis Muscle of Patients With COPD. Eur Respir J (2007) 30(1):73–9. doi: 10.1183/09031936.00146906
32. Białas AJ, Liberski PP, Zielińska A, Kumor-Kisielewska A, Szewczyk K, Miłkowska-Dymanowska J, et al. Morphometric Analysis of Mitochondria in Lymphocytes of Patients With Exacerbations of Chronic Obstructive Pulmonary Disease - Pilot Study. Int J Chron Obstruct Pulmon Dis (2018) 13:2313–8. doi: 10.2147/COPD.S163249
33. Lim SC, Ju JY, Chi SY, Ban HJ, Kwon YS, Oh IJ, et al. Apoptosis of T Lymphocytes Isolated From Peripheral Blood of Patients With Acute Exacerbation of Chronic Obstructive Pulmonary Disease. Yonsei Med J (2011) 52(4):581–7. doi: 10.3349/ymj.2011.52.4.581
34. Kaasik A, Safiulina D, Zharkovsky A, Veksler V. Regulation of Mitochondrial Matrix Volume. Am J Physiology-Cell Physiol (2007) 292:C157–63. doi: 10.1152/ajpcell.00272.2006
35. Li M, Shang Y-X. Ultrastructural Changes in Rat Airway Epithelium in Asthmatic Airway Remodeling. Pathol Res Pract (2014) 210(12):1038–42. doi: 10.1016/j.prp.2014.03.010
36. Mabalirajan U, Dinda AK, Kumar S, Roshan R, Gupta P, Sharma SK, et al. Mitochondrial Structural Changes and Dysfunction Are Associated With Experimental Allergic Asthma. J Immunol (2008) 181(5):3540–8. doi: 10.4049/jimmunol.181.5.3540
37. Trian T, Benard G, Begueret H, Rossignol R, Girodet P-O, Ghosh D, et al. Bronchial Smooth Muscle Remodeling Involves Calcium-Dependent Enhanced Mitochondrial Biogenesis in Asthma. J Cell Biol (2007) 179:i16–6. doi: 10.1083/jcb1796oia16
38. Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, et al. Epithelial Cell Mitochondrial Dysfunction and PINK1 Are Induced by Transforming Growth Factor-Beta1 in Pulmonary Fibrosis. PloS One (2015) 10(3):e0121246. doi: 10.1371/journal.pone.0121246
39. Ganzleben I, He G-W, Günther C, Prigge E-S, Richter K, Rieker RJ, et al. PGAM5 is a Key Driver of Mitochondrial Dysfunction in Experimental Lung Fibrosis. Cell Mol Life Sci (2019) 76(23):4783–94. doi: 10.1007/s00018-019-03133-1
40. Álvarez D, Cárdenes N, Sellarés J, Bueno M, Corey C, Hanumanthu VS, et al. IPF Lung Fibroblasts Have a Senescent Phenotype. Am J Physiol Lung Cell Mol Physiol (2017) 313(6):L1164–73. doi: 10.1152/ajplung.00220.2017
41. Birch J, Barnes PJ, Passos JF. Mitochondria, Telomeres and Cell Senescence: Implications for Lung Ageing and Disease. Pharmacol Ther (2018) 183:34–49. doi: 10.1016/j.pharmthera.2017.10.005
42. Ferree A, Shirihai O. Mitochondrial Dynamics: The Intersection of Form and Function. Adv Exp Med Biol (2012) 748:13–40. doi: 10.1007/978-1-4614-3573-0_2
43. Chan DC. Mitochondrial Fusion and Fission in Mammals. Annu Rev Cell Dev Biol (2006) 22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638
44. Youle RJ, van der Bliek AM. Mitochondrial Fission, Fusion, and Stress. Sci (2012) 337(6098):1062–5. doi: 10.1126/science.1219855
45. Yu R, Lendahl U, Nistér M, Zhao J. Regulation of Mammalian Mitochondrial Dynamics: Opportunities and Challenges. Front Endocrinol (2020) 11:374. doi: 10.3389/fendo.2020.00374
46. Horbay R, Bilyy R. Mitochondrial Dynamics During Cell Cycling. Apoptosis (2016) 21(12):1327–35. doi: 10.1007/s10495-016-1295-5
47. Hu C, Huang Y, Li L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int J Mol Sci (2017) 18(1):144. doi: 10.3390/ijms18010144
48. Larson-Casey JL, He C, Carter AB. Mitochondrial Quality Control in Pulmonary Fibrosis. Redox Biol (2020) 33:101426. doi: 10.1016/j.redox.2020.101426
49. Aravamudan B, Kiel A, Freeman M, Delmotte P, Thompson M, Vassallo R, et al. Cigarette Smoke-Induced Mitochondrial Fragmentation and Dysfunction in Human Airway Smooth Muscle. Am J Physiol Lung Cell Mol Physiol (2014) 306(9):L840–54. doi: 10.1152/ajplung.00155.2013
50. Ballweg K, Mutze K, Königshoff M, Eickelberg O, Meiners S. Cigarette Smoke Extract Affects Mitochondrial Function in Alveolar Epithelial Cells. Am J Physiol Lung Cell Mol Physiol (2014) 307(11):L895–907. doi: 10.1152/ajplung.00180.2014
51. Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H, et al. Impaired Mitophagy Leads to Cigarette Smoke Stress-Induced Cellular Senescence: Implications for Chronic Obstructive Pulmonary Disease. FASEB J (2015) 29(7):2912–29. doi: 10.1096/fj.14-268276
52. Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, et al. PARK2-Mediated Mitophagy is Involved in Regulation of HBEC Senescence in COPD Pathogenesis. Autophagy (2015) 11(3):547–59. doi: 10.1080/15548627.2015.1017190
53. Walker B Jr, Stokes LD, Warren R. Environmental Factors Associated With Asthma. J Natl Med Assoc (2003) 95(2):152–66.
54. Chung K-P, Hsu C-L, Fan L-C, Huang Z, Bhatia D, Chen Y-J, et al. Mitofusins Regulate Lipid Metabolism to Mediate the Development of Lung Fibrosis. Nat Commun (2019) 10(1):3390. doi: 10.1038/s41467-019-11327-1
55. Zhang J, Xu P, Wang Y, Wang M, Li H, Lin S, et al. Astaxanthin Prevents Pulmonary Fibrosis by Promoting Myofibroblast Apoptosis Dependent on Drp1-Mediated Mitochondrial Fission. J Cell Mol Med (2015) 19(9):2215–31. doi: 10.1111/jcmm.12609
56. Carter AB, Brent Carter A. Mitochondrial Dynamics In Lung Fibrosis. Free Radical Biol Med (2017) 112:17. doi: 10.1016/j.freeradbiomed.2017.10.370
57. Varadi A, Johnson-Cadwell LI, Cirulli V, Yoon Y, Allan VJ, Rutter GA. Cytoplasmic Dynein Regulates the Subcellular Distribution of Mitochondria by Controlling the Recruitment of the Fission Factor Dynamin-Related Protein-1. J Cell Sci (2004) 117(Pt 19):4389–400. doi: 10.1242/jcs.01299
58. Quintana A, Hoth M. Mitochondrial Dynamics and Their Impact on T Cell Function. Cell Calcium (2012) 52:57–63. doi: 10.1016/j.ceca.2012.02.005
59. Campello S, Lacalle RA, Bettella M, Mañes S, Scorrano L, Viola A. Orchestration of Lymphocyte Chemotaxis by Mitochondrial Dynamics. J Exp Med (2006) 203(13):2879–86. doi: 10.1084/jem.20061877
60. Westermann B. Mitochondrial Fusion and Fission in Cell Life and Death. Nat Rev Mol Cell Biol (2010) 11:872–84. doi: 10.1038/nrm3013
61. Bavister BD. The Mitochondrial Contribution to Stem Cell Biology. Reprod Fertil Dev (2006) 18:829. doi: 10.1071/rd06111
62. Yi M, Weaver D, Hajnóczky G. Control of Mitochondrial Motility and Distribution by the Calcium Signal. J Cell Biol (2004) 167:661–72. doi: 10.1083/jcb.200406038
63. Jeyaraju DV, Cisbani G, Pellegrini L. Calcium Regulation of Mitochondria Motility and Morphology. Biochim Biophys Acta (2009) 1787(11):1363–73. doi: 10.1016/j.bbabio.2008.12.005
64. Al-Mehdi A-B, Pastukh VM, Swiger BM, Reed DJ, Patel MR, Bardwell GC, et al. Perinuclear Mitochondrial Clustering Creates an Oxidant-Rich Nuclear Domain Required for Hypoxia-Induced Transcription. Sci Signal (2012) 5(231):ra47. doi: 10.1126/scisignal.2002712
65. Liu Y, Fiskum G, Schubert D. Generation of Reactive Oxygen Species by the Mitochondrial Electron Transport Chain. J Neurochemistry (2002) 80(5):780–7. doi: 10.1046/j.0022-3042.2002.00744.x
66. Scialò F, Fernández-Ayala DJ, Sanz A. Role of Mitochondrial Reverse Electron Transport in ROS Signaling: Potential Roles in Health and Disease. Front Physiol (2017) 8:428. doi: 10.3389/fphys.2017.00428
67. Schumacker PT, Gillespie MN, Nakahira K, Choi AMK, Crouser ED, Piantadosi CA, et al. Mitochondria in Lung Biology and Pathology: More Than Just a Powerhouse. Am J Physiol Lung Cell Mol Physiol (2014) 306(11):L962–74. doi: 10.1152/ajplung.00073.2014
68. Hockenbery DM. Mitochondria and Cell Death. New York, NY, USA: Humana Press (2016). p. 232. Available at: https://play.google.com/store/books/details?id=Q9AYDAAAQBAJ.
69. Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, et al. Oxidative Stress-Induced Mitochondrial Dysfunction Drives Inflammation and Airway Smooth Muscle Remodeling in Patients With Chronic Obstructive Pulmonary Disease. J Allergy Clin Immunol (2015) 136(3):769–80. doi: 10.1016/j.jaci.2015.01.046
70. Holguin F. Oxidative Stress in Airway Diseases. Ann Am Thorac Soc (2013) 10:S150–7. doi: 10.1513/annalsats.201305-116aw
71. van der Vliet A, Janssen-Heininger YMW, Anathy V. Oxidative Stress in Chronic Lung Disease: From Mitochondrial Dysfunction to Dysregulated Redox Signaling. Mol Aspects Med (2018) 63:59–69. doi: 10.1016/j.mam.2018.08.001
72. Caldeira D de AF, Mesquita FM, Pinheiro FG, Oliveira DF, Oliveira LFS, Nascimento JHM, et al. Acute Exposure to C60 Fullerene Damages Pulmonary Mitochondrial Function and Mechanics. Nanotoxicology (2020) 15(3):1–14. doi: 10.1080/17435390.2020.1863498
73. Liu X, Chen Z. The Pathophysiological Role of Mitochondrial Oxidative Stress in Lung Diseases. J Transl Med (2017) 15(1):207. doi: 10.1186/s12967-017-1306-5
74. Li F, Xu M, Wang M, Wang L, Wang H, Zhang H, et al. Roles of Mitochondrial ROS and NLRP3 Inflammasome in Multiple Ozone-Induced Lung Inflammation and Emphysema. Respir Res (2018) 19(1):230. doi: 10.1186/s12931-018-0931-8
75. van der Toorn M, van der Toorn M, Rezayat D, Kauffman HF, Bakker SJL, Gans ROB, et al. Lipid-Soluble Components in Cigarette Smoke Induce Mitochondrial Production of Reactive Oxygen Species in Lung Epithelial Cells. Am J Physiology-Lung Cell Mol Physiol (2009) 297:L109–14. doi: 10.1152/ajplung.90461.2008
76. Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A. Protein Carbonylation in Human Diseases. Trends Mol Med (2003) 9p:169–76. doi: 10.1016/s1471-4914(03)00031-5
77. Meng X-M, Nikolic-Paterson DJ, Lan HY. TGF-β: The Master Regulator of Fibrosis. Nat Rev Nephrol (2016) 12p:325–38. doi: 10.1038/nrneph.2016.48
78. Jain M, Rivera S, Monclus EA, Synenki L, Zirk A, Eisenbart J, et al. Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J Biol Chem (2013) 288:770–7. doi: 10.1074/jbc.m112.431973
79. Liu R-M, Desai LP. Reciprocal Regulation of TGF-β and Reactive Oxygen Species: A Perverse Cycle for Fibrosis. Redox Biol (2015) 6:565–77. doi: 10.1016/j.redox.2015.09.009
80. Bonvallot V, Baeza-Squiban A, Baulig A, Brulant S, Boland S, Muzeau F, et al. Organic Compounds From Diesel Exhaust Particles Elicit a Proinflammatory Response in Human Airway Epithelial Cells and Induce Cytochrome P450 1A1 Expression. Am J Respir Cell Mol Biol (2001) 25(4):515–21. doi: 10.1165/ajrcmb.25.4.4515
81. Gazdhar A, Lebrecht D, Roth M, Tamm M, Venhoff N, Foocharoen C, et al. Time-Dependent and Somatically Acquired Mitochondrial DNA Mutagenesis and Respiratory Chain Dysfunction in a Scleroderma Model of Lung Fibrosis. Sci Rep (2015) 4:1–7. doi: 10.1038/srep05336
82. Cheresh P, Kim S-J, Tulasiram S, Kamp DW. Oxidative Stress and Pulmonary Fibrosis. Biochim Biophys Acta (2013) 1832(7):1028–40. doi: 10.1016/j.bbadis.2012.11.021
83. Buder-Hoffmann SA, Shukla A, Barrett TF, MacPherson MB, Lounsbury KM, Mossman BT. A Protein Kinase Cdelta-Dependent Protein Kinase D Pathway Modulates ERK1/2 and JNK1/2 Phosphorylation and Bim-Associated Apoptosis by Asbestos. Am J Pathol (2009) 174(2):449–59. doi: 10.2353/ajpath.2009.080180
84. Zifa E, Daniil Z, Skoumi E, Stavrou M, Papadimitriou K, Terzenidou M, et al. Mitochondrial Genetic Background Plays a Role in Increasing Risk to Asthma. Mol Biol Rep (2012) 39:4697–708. doi: 10.1007/s11033-011-1262-8
85. Aguilera-Aguirre L, Bacsi A, Saavedra-Molina A, Kurosky A, Sur S, Boldogh I. Mitochondrial Dysfunction Increases Allergic Airway Inflammation. J Immunol (2009) 183(8):5379–87. doi: 10.4049/jimmunol.0900228
86. Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, et al. Ultrafine Particulate Pollutants Induce Oxidative Stress and Mitochondrial Damage. Environ Health Perspect (2003) 111:455–60. doi: 10.1289/ehp.6000
87. Lee KS, Kim SR, Park HS, Park SJ, Min KH, Lee KY, et al. A Novel Thiol Compound, N-Acetylcysteine Amide, Attenuates Allergic Airway Disease by Regulating Activation of NF-kappaB and Hypoxia-Inducible Factor-1alpha. Exp Mol Med (2007) 39(6):756–68. doi: 10.1038/emm.2007.82
88. Whitekus MJ, Li N, Zhang M, Wang M, Horwitz MA, Nelson SK, et al. Thiol Antioxidants Inhibit the Adjuvant Effects of Aerosolized Diesel Exhaust Particles in a Murine Model for Ovalbumin Sensitization. J Immunol (2002) 168:2560–7. doi: 10.4049/jimmunol.168.5.2560
89. Riedl MA, Nel AE. Importance of Oxidative Stress in the Pathogenesis and Treatment of Asthma. Curr Opin Allergy Clin Immunol (2008) 8(1):49–56. doi: 10.1097/ACI.0b013e3282f3d913
90. Emelyanov A, Fedoseev G, Abulimity A, Rudinski K, Fedoulov A, Karabanov A, et al. Elevated Concentrations of Exhaled Hydrogen Peroxide in Asthmatic Patients. Chest (2001) 120:1136–9. doi: 10.1378/chest.120.4.1136
91. Aldridge RE, Chan T, van Dalen CJ, Senthilmohan R, Winn M, Venge P, et al. Eosinophil Peroxidase Produces Hypobromous Acid in the Airways of Stable Asthmatics. Free Radic Biol Med (2002) 33(6):847–56. doi: 10.1016/s0891-5849(02)00976-0
92. Youle RJ, Narendra DP. Mechanisms of Mitophagy. Nat Rev Mol Cell Biol (2011) 12(1):9–14. doi: 10.1038/nrm3028
93. Urbani A, Babu M. Mitochondria in Health and in Sickness. Springer Nat (2019) 277:71–82. doi: 10.1007/978-981-13-8367-0
94. Kubli DA, Gustafsson ÅB.. Mitochondria and Mitophagy: The Yin and Yang of Cell Death Control. Circ Res (2012) 111(9):1208–21. doi: 10.1161/CIRCRESAHA.112.265819
95. Todt F, Cakir Z, Reichenbach F, Emschermann F, Lauterwasser J, Kaiser A, et al. Differential Retrotranslocation of Mitochondrial Bax and Bak. EMBO J (2015) 34(1):67–80. doi: 10.15252/embj.201488806
96. Scorrano L. Cristae Remodeling and Mitochondrial Fragmentation: A Checkpoint for Cytochrome C Release and Apoptosis? Apoptosome (2010) 1:253–70. doi: 10.1007/978-90-481-3415-1_13
97. Sena LA, Chandel NS. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol Cell (2012) 48:158–67. doi: 10.1016/j.molcel.2012.09.025
98. Sureshbabu A, Bhandari V. Targeting Mitochondrial Dysfunction in Lung Diseases: Emphasis on Mitophagy. Front Physiol (2013) 4:384. doi: 10.3389/fphys.2013.00384
99. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, et al. Mitophagy-Dependent Necroptosis Contributes to the Pathogenesis of COPD. J Clin Invest (2014) 124(9):3987–4003. doi: 10.1172/JCI74985
100. Monick MM, Powers LS, Walters K, Lovan N, Zhang M, Gerke A, et al. Identification of an Autophagy Defect in Smokers’ Alveolar Macrophages. J Immunol (2010) 185(9):5425–35. doi: 10.4049/jimmunol.1001603
101. Puig-Vilanova E, Rodriguez DA, Lloreta J, Ausin P, Pascual-Guardia S, Broquetas J, et al. Oxidative Stress, Redox Signaling Pathways, and Autophagy in Cachectic Muscles of Male Patients With Advanced COPD and Lung Cancer. Free Radic Biol Med (2015) 79:91–108. doi: 10.1016/j.freeradbiomed.2014.11.006
102. Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-Mediated Mitophagy is Dependent on VDAC1 and P62/SQSTM1. Nat Cell Biol (2010) 2):119–31. doi: 10.1038/ncb2012
103. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The Selective Autophagy Substrate P62 Activates the Stress Responsive Transcription Factor Nrf2 Through Inactivation of Keap1. Nat Cell Biol (2010) 12(3):213–23. doi: 10.1038/ncb2021
104. Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, et al. Thyroid Hormone Inhibits Lung Fibrosis in Mice by Improving Epithelial Mitochondrial Function. Nat Med (2018) 24:39–49. doi: 10.1038/nm.4447
105. Liu R-M, Gaston Pravia KA. Oxidative Stress and Glutathione in TGF-Beta-Mediated Fibrogenesis. Free Radic Biol Med (2010) 48(1):1–15. doi: 10.1016/j.freeradbiomed.2009.09.026
106. Hong M, Wilkes MC, Penheiter SG, Gupta SK, Edens M, Leof EB. Non-Smad Transforming Growth Factor-β Signaling Regulated by Focal Adhesion Kinase Binding the P85 Subunit of Phosphatidylinositol 3-Kinase. J Biol Chem (2011) 286(20):17841–50. doi: 10.1074/jbc.M111.233676
107. Chitra P, Saiprasad G, Manikandan R, Sudhandiran G. Berberine Inhibits Smad and Non-Smad Signaling Cascades and Enhances Autophagy Against Pulmonary Fibrosis. J Mol Med (2015) 93(9):1015–31. doi: 10.1007/s00109-015-1283-1
108. Aggarwal S, Mannam P, Zhang J. Differential Regulation of Autophagy and Mitophagy in Pulmonary Diseases. Am J Physiol Lung Cell Mol Physiol (2016) 311(2):L433–52. doi: 10.1152/ajplung.00128.2016
109. Jung KH, Torrone D, Lovinsky-Desir S, Perzanowski M, Bautista J, Jezioro JR, et al. Short-Term Exposure to PM2.5 and Vanadium and Changes in Asthma Gene DNA Methylation and Lung Function Decrements Among Urban Children. Respir Res (2017) 18:1–11. doi: 10.1186/s12931-017-0550-9
110. Berhane K, Chang C-C, McConnell R, Gauderman WJ, Avol E, Rapapport E, et al. Association of Changes in Air Quality With Bronchitic Symptoms in Children in California, 1993-2012. JAMA (2016) 315(14):1491–501. doi: 10.1001/jama.2016.3444
111. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial Dysfunction Induces Senescence With a Distinct Secretory Phenotype. Cell Metab (2016) 23(2):303–14. doi: 10.1016/j.cmet.2015.11.011
112. Sachdeva K, Do DC, Zhang Y, Hu X, Chen J, Gao P. Environmental Exposures and Asthma Development: Autophagy, Mitophagy, and Cellular Senescence. Front Immunol (2019) 10:2787. doi: 10.3389/fimmu.2019.02787
113. Wang H-T, Lin J-H, Yang C-H, Haung C-H, Weng C-W, Maan-Yuh Lin A, et al. Acrolein Induces mtDNA Damages, Mitochondrial Fission and Mitophagy in Human Lung Cells. Oncotarget (2017) 8(41):70406–21. doi: 10.18632/oncotarget.19710
114. Qiu Y-N, Wang G-H, Zhou F, Hao J-J, Tian L, Guan L-F, et al. PM2.5 Induces Liver Fibrosis via Triggering ROS-Mediated Mitophagy. Ecotoxicology Environ Saf (2019) 167:178–87. doi: 10.1016/j.ecoenv.2018.08.050
115. Sanders PN, Koval OM, Jaffer OA, Prasad AM, Businga TR, Scott JA, et al. CaMKII is Essential for the Proasthmatic Effects of Oxidation. Sci Transl Med (2013) 5(195):195ra97. doi: 10.1126/scitranslmed.3006135
116. Sebag SC, Koval OM, Paschke JD, Winters CJ, Jaffer OA, Dworski R, et al. Mitochondrial CaMKII Inhibition in Airway Epithelium Protects Against Allergic Asthma. JCI Insight (2017) 2(3):e88297. doi: 10.1172/jci.insight.88297
117. Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, et al. Oxidized CaMKII Causes Cardiac Sinus Node Dysfunction in Mice. J Clin Invest (2011) 121(8):3277–88. doi: 10.1172/JCI57833
118. Jaffer OA, Brent Carter A, Sanders PN, Dibbern ME, Winters CJ, Murthy S, et al. Mitochondrial-Targeted Antioxidant Therapy Decreases Transforming Growth Factor-β–Mediated Collagen Production in a Murine Asthma Model. Am J Respir Cell Mol Biol (2015) 52:106–15. doi: 10.1165/rcmb.2013-0519oc
119. Zhang Y, Do DC, Hu X, Wang J, Zhao Y, Mishra S, et al. CaMKII Oxidation Regulates Cockroach Allergen–Induced Mitophagy in Asthma. J Allergy Clin Immunol (2020) 147(4):1464–77. doi: 10.1016/j.jaci.2020.08.033
120. Schwarz DS, Blower MD. The Endoplasmic Reticulum: Structure, Function and Response to Cellular Signaling. Cell Mol Life Sci (2016) 73:79–94. doi: 10.1007/s00018-015-2052-6
121. Phillips MJ, Voeltz GK. Structure and Function of ER Membrane Contact Sites With Other Organelles. Nat Rev Mol Cell Biol (2016) 17(2):69–82. doi: 10.1038/nrm.2015.8
122. Giorgi C, Missiroli S, Patergnani S, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-Associated Membranes: Composition, Molecular Mechanisms, and Physiopathological Implications. Antioxid Redox Signal (2015) 22(12):995–1019. doi: 10.1089/ars.2014.6223
123. Glancy B, Balaban RS. Role of Mitochondrial Ca2 in the Regulation of Cellular Energetics. Biochemistry (2012) 51:2959–73. doi: 10.1021/bi2018909
124. Marchi S, Patergnani S, Pinton P. The Endoplasmic Reticulum–Mitochondria Connection: One Touch, Multiple Functions. Biochim Biophys Acta (BBA) - Bioenergetics (2014) 1837:461–9. doi: 10.1016/j.bbabio.2013.10.015
125. Delmotte P, Sieck GC. Endoplasmic Reticulum Stress and Mitochondrial Function in Airway Smooth Muscle. Front Cell Dev Biol (2019) 7:374. doi: 10.3389/fcell.2019.00374
126. Manevski M, Muthumalage T, Devadoss D, Sundar IK, Wang Q, Singh KP, et al. Cellular Stress Responses and Dysfunctional Mitochondrial–cellular Senescence, and Therapeutics in Chronic Respiratory Diseases. Redox Biol (2020) 33:101443. doi: 10.1016/j.redox.2020.101443
127. Kelsen SG, Duan X, Ji R, Perez O, Liu C, Merali S. Cigarette Smoke Induces an Unfolded Protein Response in the Human Lung. Am J Respir Cell Mol Biol (2008) 38:541–50. doi: 10.1165/rcmb.2007-0221oc
128. Kenche H, Baty CJ, Vedagiri K, Shapiro SD, Blumental-Perry A. Cigarette Smoking Affects Oxidative Protein Folding in Endoplasmic Reticulum by Modifying Protein Disulfide Isomerase. FASEB J (2013) 27(3):965–77. doi: 10.1096/fj.12-216234
129. Aksoy MO, Kim V, Cornwell WD, Rogers TJ, Kosmider B, Bahmed K, et al. Secretion of the Endoplasmic Reticulum Stress Protein, GRP78, Into the BALF is Increased in Cigarette Smokers. Respir Res (2017) 18(1):78. doi: 10.1186/s12931-017-0561-6
130. Bueno M, Brands J, Voltz L, Fiedler K, Mays B, St Croix C, et al. ATF3 Represses PINK1 Gene Transcription in Lung Epithelial Cells to Control Mitochondrial Homeostasis. Aging Cell (2018) 17(2):e12720. doi: 10.1111/acel.12720
131. Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, et al. Epithelial Endoplasmic Reticulum Stress and Apoptosis in Sporadic Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med (2008) 178(8):838–46. doi: 10.1164/rccm.200802-313OC
132. Lawson WE, Cheng D-S, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, et al. Endoplasmic Reticulum Stress Enhances Fibrotic Remodeling in the Lungs. Proc Natl Acad Sci USA (2011) 108(26):10562–7. doi: 10.1073/pnas.1107559108
133. Jiang D, Cui H, Xie N, Banerjee S, Liu R-M, Dai H, et al. ATF4 Mediates Mitochondrial Unfolded Protein Response in Alveolar Epithelial Cells. Am J Respir Cell Mol Biol (2020) 63(4):478–89. doi: 10.1165/rcmb.2020-0107OC
134. Yap J, Chen X, Delmotte P, Sieck GC. Tnfα Selectively Activates the IRE1α/XBP1 Endoplasmic Reticulum Stress Pathway in Human Airway Smooth Muscle Cells. Am J Physiol Lung Cell Mol Physiol (2020) ;318(3):L483–93. doi: 10.1152/ajplung.00212.2019
135. Delmotte P, Zavaletta VA, Thompson MA, Prakash YS, Sieck GC. Tnfα Decreases Mitochondrial Movement in Human Airway Smooth Muscle. Am J Physiol Lung Cell Mol Physiol (2017) 313(1):L166–76. doi: 10.1152/ajplung.00538.2016
136. Misawa T, Takahama M, Saitoh T. Mitochondria-Endoplasmic Reticulum Contact Sites Mediate Innate Immune Responses. Adv Exp Med Biol (2017) 997:187–97. doi: 10.1007/978-981-10-4567-7_14
137. Jo E-K, Kim JK, Shin D-M, Sasakawa C. Molecular Mechanisms Regulating NLRP3 Inflammasome Activation. Cell Mol Immunol (2016) 13(2):148–59. doi: 10.1038/cmi.2015.95
138. Yokoyama A. Advances in Asthma: Pathophysiology, Diagnosis and Treatment Springer (2018). Available at: https://play.google.com/store/books/details?id=vnWADwAAQBAJ.
139. Wang H, Lv C‘er, Wang S, Ying H, Weng Y, Yu W. NLRP3 Inflammasome Involves in the Acute Exacerbation of Patients With Chronic Obstructive Pulmonary Disease. Inflammation (2018) 41(4):1321–33. doi: 10.1007/s10753-018-0780-0
140. Kim SR, Kim DI, Kim SH, Lee H, Lee KS, Cho SH, et al. NLRP3 Inflammasome Activation by Mitochondrial ROS in Bronchial Epithelial Cells is Required for Allergic Inflammation. Cell Death Dis (2014) 5:e1498. doi: 10.1038/cddis.2014.460
141. Tan H-TT, Hagner S, Ruchti F, Radzikowska U, Tan G, Altunbulakli C, et al. Tight Junction, Mucin, and Inflammasome-Related Molecules are Differentially Expressed in Eosinophilic, Mixed, and Neutrophilic Experimental Asthma in Mice. Allergy (2019) 74(2):294–307. doi: 10.1111/all.13619
142. Liu W, Liu S, Verma M, Zafar I, Good JT, Rollins D, et al. Mechanism of T2/T17-Predominant and Neutrophilic T2/T17-Low Subtypes of Asthma. J Allergy Clin Immunol (2017) 139(5):1548–58.e4. doi: 10.1016/j.jaci.2016.08.032
143. Park S, Won J-H, Hwang I, Hong S, Lee HK, Yu J-W. Defective Mitochondrial Fission Augments NLRP3 Inflammasome Activation. Sci Rep (2015) 5:15489. doi: 10.1038/srep15489
144. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial Protein Mitofusin 2 is Required for NLRP3 Inflammasome Activation After RNA Virus Infection. Proc Natl Acad Sci USA (2013) 110(44):17963–8. doi: 10.1073/pnas.1312571110
145. Kim S-H, Lee JY, Yoon CM, Shin HJ, Lee SW, Rosas I, et al. Mitochondrial Antiviral Signalling Protein is Crucial for the Development of Pulmonary Fibrosis. Eur Respir J (2020) 57(4):2000652. doi: 10.1183/13993003.00652-2020
146. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The Adaptor MAVS Promotes NLRP3 Mitochondrial Localization and Inflammasome Activation. Cell (2013) 153(2):348–61. doi: 10.1016/j.cell.2013.02.054
147. Grinberg L, Fibach E, Amer J, Atlas D. N-Acetylcysteine Amide, a Novel Cell-Permeating Thiol, Restores Cellular Glutathione and Protects Human Red Blood Cells From Oxidative Stress. Free Radic Biol Med (2005) 38(1):136–45. doi: 10.1016/j.freeradbiomed.2004.09.025
148. Boyd-Kimball D, Sultana R, Abdul HM, Butterfield DA. Gamma-Glutamylcysteine Ethyl Ester-Induced Up-Regulation of Glutathione Protects Neurons Against Abeta(1-42)-Mediated Oxidative Stress and Neurotoxicity: Implications for Alzheimer’s Disease. J Neurosci Res (2005) 79(5):700–6. doi: 10.1002/jnr.20394
149. Sheu S-S, Nauduri D, Anders MW. Targeting Antioxidants to Mitochondria: A New Therapeutic Direction. Biochim Biophys Acta (2006) 1762(2):256–65. doi: 10.1016/j.bbadis.2005.10.007
150. Trnka J, Blaikie FH, Smith RAJ, Murphy MP. A Mitochondria-Targeted Nitroxide is Reduced to Its Hydroxylamine by Ubiquinol in Mitochondria. Free Radic Biol Med (2008) 44(7):1406–19. doi: 10.1016/j.freeradbiomed.2007.12.036
151. Schuliga M, Pechkovsky DV, Read J, Waters DW, Blokland KEC, Reid AT, et al. Mitochondrial Dysfunction Contributes to the Senescent Phenotype of IPF Lung Fibroblasts. J Cell Mol Med (2018) 22(12):5847–61. doi: 10.1111/jcmm.13855
152. Xu M, Wang L, Wang M, Wang H, Zhang H, Chen Y, et al. Mitochondrial ROS and NLRP3 Inflammasome in Acute Ozone-Induced Murine Model of Airway Inflammation and Bronchial Hyperresponsiveness. Free Radic Res (2019) 53(7):780–90. doi: 10.1080/10715762.2019.1630735
153. Kelso GF, Porteous CM, Coulter CV, Hughes G, Porteous WK, Ledgerwood EC, et al. Selective Targeting of a Redox-Active Ubiquinone to Mitochondria Within Cells: Antioxidant and Antiapoptotic Properties. J Biol Chem (2001) 276(7):4588–96. doi: 10.1074/jbc.M009093200
154. Powers SK, Hudson MB, Bradley Nelson W, Talbert EE, Min K, Szeto HH, et al. Mitochondria-Targeted Antioxidants Protect Against Mechanical Ventilation-Induced Diaphragm Weakness*. Crit Care Med (2011) 39:1749–59. doi: 10.1097/ccm.0b013e3182190b62
155. Lu H-I, Huang T-H, Sung P-H, Chen Y-L, Chua S, Chai H-Y, et al. Administration of Antioxidant Peptide SS-31 Attenuates Transverse Aortic Constriction-Induced Pulmonary Arterial Hypertension in Mice. Acta Pharmacol Sin (2016) 37(5):589–603. doi: 10.1038/aps.2015.162
156. Decramer M, Rutten-van Mölken M, Dekhuijzen PNR, Troosters T, van Herwaarden C, Pellegrino R, et al. Effects of N-Acetylcysteine on Outcomes in Chronic Obstructive Pulmonary Disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): A Randomised Placebo-Controlled Trial. Lancet (2005) 365(9470):1552–60. doi: 10.1016/S0140-6736(05)66456-2
157. Foronjy R, Wallace A, D’Armiento J. The Pharmokinetic Limitations of Antioxidant Treatment for COPD. Pulm Pharmacol Ther (2008) 21(2):370–9. doi: 10.1016/j.pupt.2007.10.004
158. Ong S-B, Hausenloy DJ. Mitochondrial Dynamics as a Therapeutic Target for Treating Cardiac Diseases. Handb Exp Pharmacol (2017) 240:251–79. doi: 10.1007/164_2016_7
159. Guo C, Wang J, Jing L, Ma R, Liu X, Gao L, et al. Mitochondrial Dysfunction, Perturbations of Mitochondrial Dynamics and Biogenesis Involved in Endothelial Injury Induced by Silica Nanoparticles. Environ Pollut (2018) 236:926–36. doi: 10.1016/j.envpol.2017.10.060
160. Skuratovskaia D, Komar A, Vulf M, Litvinova L. Mitochondrial Destiny in Type 2 Diabetes: The Effects of Oxidative Stress on the Dynamics and Biogenesis of Mitochondria. PeerJ (2020) 8:e9741. doi: 10.7717/peerj.9741
161. Manczak M, Kandimalla R, Yin X, Reddy PH. Mitochondrial Division Inhibitor 1 Reduces Dynamin-Related Protein 1 and Mitochondrial Fission Activity. Hum Mol Genet (2019) 28(2):177–99. doi: 10.1093/hmg/ddy335
162. Qi X, Qvit N, Su Y-C, Mochly-Rosen D. A Novel Drp1 Inhibitor Diminishes Aberrant Mitochondrial Fission and Neurotoxicity. J Cell Sci (2013) 126(Pt 3):789–802. doi: 10.1242/jcs.114439
163. Antunes M, Lapa JR, Rocco P. Mesenchymal Stromal Cell Therapy in COPD: From Bench to Bedside. Int J Chronic Obstructive Pulmonary Dis (2017) 12:3017–27. doi: 10.2147/copd.s146671
164. Weiss DJ, Bertoncello I, Borok Z, Kim C, Panoskaltsis-Mortari A, Reynolds S, et al. Stem Cells and Cell Therapies in Lung Biology and Lung Diseases. Proc Am Thorac Soc (2011) J8(3):223–72. doi: 10.1513/pats.201012-071DW
165. Du Y-M, Zhuansun Y-X, Chen R, Lin L, Lin Y, Li J-G. Mesenchymal Stem Cell Exosomes Promote Immunosuppression of Regulatory T Cells in Asthma. Exp Cell Res (2018) 363(1):114–20. doi: 10.1016/j.yexcr.2017.12.021
166. Periera-Simon S, Xia X, Catanuto P, Coronado R, Kurtzberg J, Bellio M, et al. Anti-Fibrotic Effects of Different Sources of MSC in Bleomycin-Induced Lung Fibrosis in C57BL6 Male Mice. Respirology (2020) 26(2):161–70. doi: 10.1111/resp.13928
167. Gu W, Song L, Li X-M, Wang D, Guo X-J, Xu W-G. Mesenchymal Stem Cells Alleviate Airway Inflammation and Emphysema in COPD Through Down-Regulation of Cyclooxygenase-2 via P38 and ERK MAPK Pathways. Sci Rep (2015) 5:8733. doi: 10.1038/srep08733
168. Takeda K, Webb TL, Ning F, Shiraishi Y, Regan DP, Chow L, et al. Mesenchymal Stem Cells Recruit CCR2 Monocytes To Suppress Allergic Airway Inflammation. J Immunol (2018) 200(4):1261–9. doi: 10.4049/jimmunol.1700562
169. Cruz FF, Borg ZD, Goodwin M, Sokocevic D, Wagner DE, Coffey A, et al. Systemic Administration of Human Bone Marrow-Derived Mesenchymal Stromal Cell Extracellular Vesicles Ameliorates Aspergillus Hyphal Extract-Induced Allergic Airway Inflammation in Immunocompetent Mice. Stem Cells Transl Med (2015) 4(11):1302–16. doi: 10.5966/sctm.2014-0280
170. Li C, Cheung MKH, Han S, Zhang Z, Chen L, Chen J, et al. Mesenchymal Stem Cells and Their Mitochondrial Transfer: A Double-Edged Sword. Biosci Rep (2019) 39(5):BSR20182417. doi: 10.1042/BSR20182417
171. Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, et al. Miro1 Regulates Intercellular Mitochondrial Transport & Enhances Mesenchymal Stem Cell Rescue Efficacy. EMBO J (2014) 33(9):994–1010. doi: 10.1002/embj.201386030
172. Torralba D, Baixauli F, Sánchez-Madrid F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front Cell Dev Biol (2016) 4:org/10.3389/fcell.2016.00107. doi: 10.3389/fcell.2016.00107
173. Paliwal S, Chaudhuri R, Agrawal A, Mohanty S. Regenerative Abilities of Mesenchymal Stem Cells Through Mitochondrial Transfer. J BioMed Sci (2018) 25(1):31. doi: 10.1186/s12929-018-0429-1
174. Han D, Zheng X, Wang X, Jin T, Cui L, Chen Z. Mesenchymal Stem/Stromal Cell-Mediated Mitochondrial Transfer and the Therapeutic Potential in Treatment of Neurological Diseases. Stem Cells Int (2020) 2020:8838046. doi: 10.1155/2020/8838046
175. Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial Transfer Between Cells can Rescue Aerobic Respiration. Proc Natl Acad Sci USA (2006) 103(5):1283–8. doi: 10.1073/pnas.0510511103
176. Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, et al. Mitochondrial Transfer From Bone-Marrow-Derived Stromal Cells to Pulmonary Alveoli Protects Against Acute Lung Injury. Nat Med (2012) 18(5):759–65. doi: 10.1038/nm.2736
177. Li X, Zhang Y, Yeung SC, Liang Y, Liang X, Ding Y, et al. Mitochondrial Transfer of Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells to Airway Epithelial Cells Attenuates Cigarette Smoke-Induced Damage. Am J Respir Cell Mol Biol (2014) 51(3):455–65. doi: 10.1165/rcmb.2013-0529OC
178. Li X, Michaeloudes C, Zhang Y, Wiegman CH, Adcock IM, Lian Q, et al. Mesenchymal Stem Cells Alleviate Oxidative Stress-Induced Mitochondrial Dysfunction in the Airways. J Allergy Clin Immunol (2018) 141(5):1634–45.e5. doi: 10.1016/j.jaci.2017.08.017
179. Yao Y, Fan X-L, Jiang D, Zhang Y, Li X, Xu Z-B, et al. Connexin 43-Mediated Mitochondrial Transfer of iPSC-MSCs Alleviates Asthma Inflammation. Stem Cell Rep (2018) 11(5):1120–35. doi: 10.1016/j.stemcr.2018.09.012
180. Masuzawa A, Black KM, Pacak CA, Ericsson M, Barnett RJ, Drumm C, et al. Transplantation of Autologously Derived Mitochondria Protects the Heart From Ischemia-Reperfusion Injury. Am J Physiol Heart Circ Physiol (2013) 304(7):H966–82. doi: 10.1152/ajpheart.00883.2012
181. Fu A, Shi X, Zhang H, Fu B. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front Pharmacol (2017) 8:241. doi: 10.3389/fphar.2017.00241
182. Moskowitzova K, Orfany A, Liu K, Ramirez-Barbieri G, Thedsanamoorthy JK, Yao R, et al. Mitochondrial Transplantation Enhances Murine Lung Viability and Recovery After Ischemia-Reperfusion Injury. Am J Physiol Lung Cell Mol Physiol (2020) 318(1):L78–88. doi: 10.1152/ajplung.00221.2019
Keywords: mitochondrial dysfunction, mitochondrial dynamics, mitochondrial morphology, mitotherapy, chronic lung diseases, reactive species of oxygen (ROS)
Citation: Caldeira DdAF, Weiss DJ, Rocco PRM, Silva PL and Cruz FF (2021) Mitochondria in Focus: From Function to Therapeutic Strategies in Chronic Lung Diseases. Front. Immunol. 12:782074. doi: 10.3389/fimmu.2021.782074
Received: 23 September 2021; Accepted: 29 October 2021;
Published: 23 November 2021.
Edited by:
You-Me Kim, Korea Advanced Institute of Science and Technology, South KoreaReviewed by:
Leah Reznikov, University of Florida, United StatesKalina Atanasova, University of Florida, United States
Copyright © 2021 Caldeira, Weiss, Rocco, Silva and Cruz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fernanda Ferreira Cruz, ZmZjcnV6QGJpb2YudWZyai5icg==
†These authors share senior authorship