
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 14 January 2022
Sec. T Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.778916
 Niels C. Lory1,2†
Niels C. Lory1,2† Mikolaj Nawrocki2,3†
Mikolaj Nawrocki2,3† Martina Corazza2,3Joanna Schmid1Valéa Schumacher1Tanja Bedke2,3Stephan Menzel1,4Friedrich Koch-Nolte1
Martina Corazza2,3Joanna Schmid1Valéa Schumacher1Tanja Bedke2,3Stephan Menzel1,4Friedrich Koch-Nolte1 Andreas H. Guse5
Andreas H. Guse5 Samuel Huber2,3‡
Samuel Huber2,3‡ Hans-Willi Mittrücker1,2*‡
Hans-Willi Mittrücker1,2*‡Antigen recognition by the T-cell receptor induces a cytosolic Ca2+ signal that is crucial for T-cell function. The Ca2+ channel TRPM2 (transient receptor potential cation channel subfamily M member 2) has been shown to facilitate influx of extracellular Ca2+ through the plasma membrane of T cells. Therefore, it was suggested that TRPM2 is involved in T-cell activation and differentiation. However, these results are largely derived from in vitro studies using T-cell lines and non-physiologic means of TRPM2 activation. Thus, the relevance of TRPM2-mediated Ca2+ signaling in T cells remains unclear. Here, we use TRPM2-deficient mice to investigate the function of TRPM2 in T-cell activation and differentiation. In response to TCR stimulation in vitro, Trpm2-/- and WT CD4+ and CD8+ T cells similarly upregulated the early activation markers NUR77, IRF4, and CD69. We also observed regular proliferation of Trpm2-/- CD8+ T cells and unimpaired differentiation of CD4+ T cells into Th1, Th17, and Treg cells under specific polarizing conditions. In vivo, Trpm2-/- and WT CD8+ T cells showed equal specific responses to Listeria monocytogenes after infection of WT and Trpm2-/- mice and after transfer of WT and Trpm2-/- CD8+ T cells into infected recipients. CD4+ T-cell responses were investigated in the model of anti-CD3 mAb-induced intestinal inflammation, which allows analysis of Th1, Th17, Treg, and Tr1-cell differentiation. Here again, we detected similar responses of WT and Trpm2-/- CD4+ T cells. In conclusion, our results argue against a major function of TRPM2 in T-cell activation and differentiation.
T-cell receptor (TCR) stimulation causes a rapid increase of the free cytoplasmic Ca2+ concentration. The intensity of this Ca2+ signal correlates with the strength of the TCR signal and has strong impact on the activation and differentiation processes of T cells (1–4). Triggering of the TCR induces the rapid formation of the 2nd messengers nicotinic acid adenine dinucleotide phosphate (NAADP), cyclic adenosine 5′-diphosphate-ribose (cADPR), and D-myo-inositol 1,4,5-trisphosphate (IP3). NAADP targets ryanodine receptor 1 (RYR1) in the ER membrane and possibly two pore channels in the lysosomal membranes resulting in the formation of Ca2+ microdomains within milliseconds. Subsequent activation of IP3 receptors and RYR in the ER membrane by IP3 and cADPR further enhances Ca2+ release from the ER. Ca2+ depletion of the ER is sensed by stromal interaction module-1 (STIM1) which activates entry of extracellular Ca2+ via ORAI/CRAC channels in the plasma membrane and thereby causes the extended and global cytoplasmic Ca2+ signal required for effective T-cell activation (5–8).
Transient receptor potential cation channel subfamily M member 2 (TRPM2) was also identified as Ca2+ channel in T cells, but its function in T cells and particularly in TCR signaling is still unclear. TRPM2 is a Ca2+-permeable non-selective cation channel in the plasma membrane. The channel is expressed in cells of the central nervous system but is also found in leukocytes, particularly in cells of the myeloid lineage, e.g., neutrophils, macrophages, and dendritic cells (DCs). In these cells, TRPM2 has been linked to diverse functions, such as oxidative stress response, phagosome maturation, and migratory processes (9–15). Trpm2 mRNA and TRPM2 protein have been detected in T cells (16–19); however, compared to cells of the myeloid lineage, mRNA expression is relatively low in all analyzed T-cell subsets [Supplementary Figure 1 and www.immgen.org (20)].
TRPM2 is activated by adenosine 5′-diphosphate-ribose (ADPR), and recently, high-resolution structures of TRPM2 with bound ADPR were reported (18, 21–23). Using ADPR microinjection and uncaging of ADPR derivatives, activation of TRPM2 by ADPR was demonstrated in T cells (16–18, 24). Endogenous ADPR is also detected in T cells, and strong stimulation of T cells causes an increase in the ADPR concentration (17). NAADP and cADPR have been described as further agonists for TRPM2, either alone or in synergy with ADPR; however, the activity of these nucleotides on TRPM2 is controversial (5, 16, 25). TRPM2 can also sense reactive oxygen species, although the relevance of this function for T cells is unclear (19, 26). Recently, 2′-deoxyadenosine 5′-diphosphoribose (2dADPR) was identified as a further TRPM2 agonist in T cells. 2dADPR can be isolated from T cells and was more potent than ADPR in stimulation of TRPM2 currents (27).
Currently, hydrolysis of NAD by the NAD-glycohydrolase CD38 is considered to be the main source of ADPR in T cells and CD38 might also be required for 2dADPR formation (11, 25). CD38 is a type II transmembrane protein with its enzymatic activity in the extracellular part. However, a fraction of CD38 is found with an inverted orientation and thus is able to catalyze ADPR formation in the cytoplasm (28). In addition, ADPR may be cleaved from poly- or mono-ADP-ribosylated proteins. In conclusion, these data demonstrate an ADPR/2dADPR–TRPM2 pathway in T cells that can facilitate a Ca2+ influx into the cytoplasm (11, 25). However, these results are largely derived from cell lines, e.g., Jurkat cells, and rely on rather artificial activation protocols. Therefore and in light of the relatively low expression of Trpm2 in primary T cells, it is unclear to which extent they represent a relevant function of TRPM2 in primary T cells.
TRPM2-deficient (Trpm2-/-) mice are viable and fertile (29, 30). Interestingly, TRPM2 deficiency profoundly affects the immune system of these mice. Trpm2-/- mice are highly susceptible in several bacterial infection models (14, 30–33). On the other hand, mice show milder disease in inflammation and autoimmune models (19, 29, 34, 35). In most of these studies, altered susceptibility to disease can be linked to an impaired function of granulocytes, macrophages, or dendritic cells. So far, only two studies specifically analyzed T cells from Trpm2-/- mice. Wolf and colleagues found similar early Ca2+ responses following TCR stimulation of CD4+ T cells from WT and Trpm2-/- mice (7). In contrast, Melzer et al. observed reduced proliferation and cytokine production of total spleen cells and of purified CD4+ T cells from Trpm2-/- mice (19). To our knowledge, the function of TRPM2 in T cells has not been specifically addressed in mouse models in vivo.
Here, we analyze the response of CD4+ and CD8+ T cells from Trpm2-/- mice in vitro and in infection and inflammation models in vivo. We show that Trpm2-/- T cells are not impaired in the expression of early activation markers and in proliferation and differentiation to effector T-cell subsets in vitro. In vivo, Trpm2-/- CD8+ T cells show regular responses in the Listeria monocytogenes infection model both after infection of Trpm2-/- mice and after transfer of deficient T cells into infected recipients. Following anti-CD3 mAb-induced intestinal inflammation, Trpm2-/- and WT mice develop a similar disease and equally accumulate defined Th-cell subsets in the intestinal mucosa. A largely regular response of Trpm2-/- CD4+ T cells to anti-CD3 mAb treatment is also observed after T cell transfer into WT recipients. In conclusion, our results so far suggest that TRPM2 is not required for CD4+ and CD8+ T-cell activation and differentiation.
Trpm2-/- mice (Trpm2tm1Yamo) (29), Rag1-/- mice (B6.129S7−Rag1tm1Mom/J) (36), OT-1 mice [Tg(TcraTcrb)1100Mjb] (37), and CD90.1 congenic mice (B6.PL-Thy1a/CyJ) were on the C57BL/6 background. All other mice used in this study were derived from intercrosses of these mouse strains. Mice were housed under specific pathogen-free conditions at the University Medical Center Hamburg-Eppendorf. The housing was done under standard conditions with food and water ad libitum in individually ventilated cages. Mice were monitored on a daily basis. Animal experiments were approved by the local committee for animal experiments of the City of Hamburg (registration numbers: N033/2018, N067/2020). Age- and sex-matched mice were used.
Mice were i.p. infected with 104 CFU of a Listeria monocytogenes strain recombinant for ovalbumin (LmOVA) (38). Inocula were controlled by plating on TSB agar plates. From day 2 on, mice were treated with 2 mg/ml ampicillin in the drinking water. Endogenous T-cell responses were analyzed on day 8 postinfection. For T-cell co-transfer studies, CD90.1 congenic mice or Rag1-/- mice were infected with LmOVA. On the same day, infected mice received a mix of WT and Trpm2-/- OT-1 CD8+ T cells. Spleen cells from WT CD90.1+CD90.2+ OT-1 cells and Trpm2-/- CD90.1-CD90.2+ OT-1 cells were purified and mixed to reach a 1:1 ratio of CD8+ T cells. Recipient mice intravenously received a total of approx. 10,000 CD8+ T cells. Responses in CD90.1 congenic and Rag1-/- recipients were analyzed after 5 days and after 8 weeks, respectively. For analysis of endogenous T-cell response and for 8-week transfer experiments, 2 µg per mouse anti-CD45 mAb (30-F11, AF700) mAb was injected i.v., 3 min before sacrificing to label intravascular cells.
Cells from spleen were isolated by pressing the organ successive through 70- and 40-µm cell strainers. Cells from the kidney, lung, and liver were digested for 40 min at 37°C with 10 U/ml DNase I (Sigma-Aldrich, St. Louis, MO) and 400 µg/ml Collagenase D (Roche, Mannheim, Germany). Leukocytes were enriched by density gradient centrifugation (37.5% Easycoll, Merck Millipore, Darmstadt, Germany) and then filtered through a 30-µm strainer. Erythrocytes were depleted with lysis buffer (155 mM NH4Cl, 10 mM KHCO3, 10 µM EDTA, pH 7.2). After removal of Peyer’s patches, the small intestine was opened longitudinally and washed in PBS 1% FCS. Then, the small intestine was cut into small pieces of approx. 0.5-cm length and incubated in the presence of 5 mM EDTA in complete medium at 37°C for 30 min while shaking. Intraepithelial lymphocytes (IEL) in the supernatant were collected and enriched by centrifugation. For isolation of lamina propria lymphocytes (LPL), the remaining tissue was digested with collagenase IV (100 U/ml, Roche Diagnostics GmbH, Mannheim) and DNase I (10 U/ml Sigma-Aldrich) in complete medium at 37°C for 45 min while shaking. The digested intestinal tissue was further homogenized by passing through a metal strainer, and pooled IEL and LPL fractions were enriched by a 40%/67% Percoll gradient centrifugation. Lymphocytes were collected from the interphase.
For induction of cytokines, cells were incubated for 4 h in IMDM medium supplemented with fetal calf serum, glutamine, gentamicin, and 2-mercaptoethanol. Cells were simulated for 4 h, with phorbol 12-myristate 13-acetate (PMA, 50 ng/ml, Sigma-Aldrich) and ionomycin (1 µM, Sigma Aldrich) or with the ovalbumin257-264 peptide (10-6 M, SIINFEKL) (JPT, Berlin, Germany). Brefeldin A (10 µg/ml, Sigma Aldrich) was added to the cultures to prevent cytokine secretion. In controls, medium only contained brefeldin A.
For induction of CD69, IRF4, and NUR77, spleen cells were cultured for 4, 24, or 48 h in 96W plates coated with anti-CD3ϵ mAb (2 µg/ml, clone: 145-2C11). Anti-CD28 mAb (1 µg/ml, clone: 37.51) was added to the culture. In some of the assays, cells were CFSE labeled prior to stimulation. A division index was calculated with the FlowJo software (Tree Star, Ashland, OR, USA). For long-term culture, cells were stimulated with anti-CD3 mAb, antiCD28, mAb and IL-2 (100 U/ml IL-2, Proleukin S, Novartis, Nürnberg, Germany). After 3 days, cells were washed and further cultured with IL-7 (10 ng/ml mIL-7, PeproTech, Hamburg, Germany).
Lymphocytes were isolated from spleen and lymph nodes of WT and Trpm2-/- mice. Naive CD4+ CD25- CD44- T cells were enriched by depletion of CD25+ and CD44+ cells followed by enrichment of CD4+ T cells using MACS according to the manufacturer’s instruction (Miltenyi Biotec, Bergisch-Gladbach, Germany). The purity of CD4+ T cells obtained was about 80% as determined by flow cytometry. For each differentiation condition, the cells were cultured in a 96-well plate at 2 × 105 cells per well in 200 µl of full Click’s medium (Irvine Scientific, Santa Ana, USA) supplemented with cytokines and antibodies. For differentiation of Th1 cells, naive CD4+ T cells were cultured in the presence of 100 U/ml mIL-2, 10 ng/ml mIL-12, 10 µg/ml anti-IL-4 mAb (clone: 11B11), and 2 µg/ml anti-CD28 mAb (clone: 37.51) in plates coated with 10 µg/ml anti-CD3ϵ mAb (clone: 145 2C1). For the differentiation of Th17 cells, naive CD4+ T cells were cultured in the presence of 10 ng/ml mIL-6 and 0.25 ng/ml hTGF-β1, 10 µg/ml anti-IL-4 mAb, 10 µg/ml anti-IFN−γ mAb (clone: XMG1.2), and 2 µg/ml anti-CD28 mAb in plates coated with 10 µg/ml anti-CD3ϵ mAb. For differentiation of Treg cells, naive CD4+ T cells were cultured in the presence of 50 U/ml mIL-2 and 2 ng/ml hTGF-β1 and 2 µg/ml anti-CD28 mAb in plates coated with 2 µg/ml anti-CD3ϵ mAb. For differentiation of Tr1 cells, naive CD4+ T cells were cultured in the presence of 30 ng/ml mIL-27 and 0.25 ng/ml hTGF-β1 and 2 µg/ml anti-CD28 mAb in plates coated with 10 µg/ml anti-CD3 mAb. Cytokines and antibodies were purchased from BioLegend (San Diego, CA) and Miltenyi Biotec. After 3 days, T cells were restimulated with PMA (50 ng/ml, Sigma-Aldrich) and ionomycin (1 µM, Sigma-Aldrich) for 4 h in the presence of monensin (2 µM, BioLegend) and expression of cytokines and Foxp3 was determined by intracellular antibody staining.
After isolation from tissue or after cell culture, cells were incubated in PBS with 1% rat serum and 10 µg/ml anti-Fc-receptor mAb (clone 2.4G2, Bio X Cell, West Lebanon, NH). For extracellular staining, fluorochrome-conjugated antibodies and a fixable dead cell stain (AF750 life/dead staining or Pacific Orange succinimidyl ester, Life Technologies, Carlsbad, CA) were added. Cells were incubated for 15 min on ice. Intracellular antibody staining was conducted with the Foxp3/Transcription Factor Staining Buffer Set (eBioscience, Carlsbad, CA) according to the manufacturer’s protocol. Cells were washed with PBS 1% FCS and incubated with antibodies for intracellular staining in PBS 1% FCS for 20 min at RT.
Fluorochrome-conjugated antibodies against murine CD3 (clone 17A2, BV421), CD4 (clone RM4-5, FITC/AF700/BV605/BV785), CD8α (clone 53-6.7, PerCP/BV650), CD38 (clone 90, PE-Cy7), CD40L (clone MR1, PerCP-Cy5.5), CD44 (clone IM7, APC/BV785), CD45 (clone 30-F11, APC-Cy7/AF700/BV510/BV785), CD62L (clone MEL-14, APC/PerCP), CD69 (clone H1.2F3, PE-Cy7/V450/BV785), CD90.1 (clone HIS51, FITC/eFlour 450), CD90.2 (clone 53-2.1, PerCP), CD127 (clone A7R34, BV421), CX3CR1 (clone SA011F11, PE), Ly6C (clone AL-21, FITC), Ly6G (clone 1A8, PerCP-Cy5.5), CTLA4 (clone UC10-4F10-11, PE), PD1 (clone 29F.1A12, BV421), LAG3 (clone C9B7W, APC), IFN-γ (clone XMG1.2, APC-Cy7/BV785), IL-10 (clone JES5-16E3/APC/PE), PE), IL-17A (clone TC11-18H10.1, BV785/AF488/APC), TNF-α (clone MP6-XT22; V450), Foxp3 (clones NRRF-30, PE and FJK-16s, APC), IRF4 (clone 3E4, PE-Cy7), KI-67 (clone SolA15, PE), KLRG1 (clone 2F1, BV605), and NUR77 (clone 12.14, PE) were obtained from BioLegend, eBioscience, Thermo Fisher (Darmstadt, Germany) or BD Bioscience (Heidelberg, Germany). The anti-mouse TRPM2 mAb (clone A128) was developed by DNA immunization of rats with a plasmid expressing the full-length cDNA of mouse TRPM2. For analysis of TRPM2 expression, spleen cells were stained intracellularly with unconjugated anti-TRPM2 mAb and then stained with anti-rat-IgG antibodies (R-PE, polyclonal, Dianova, Hamburg, Germany). APC-conjugated H-2Kb/SIINFEKL dextramers were obtained from Immudex (Copenhagen, Denmark).
Cells were analyzed using Canto II, Celesta or Fortessa flow cytometers (BD Biosciences, Franklin Lakes, NJ, USA) and FlowJo software (Tree Star, Ashland, OR, USA).
Mice were i.p. injected with 15 µg anti-CD3 mAb (clone: 145 2C11) on days 0, 2, and 4. Mice were analyzed 4 h after the last injection. For the T-cell transfer approach, cells were isolated from spleen and peripheral lymph nodes of WT and Trpm2-/- mice. Total CD4+ cells were enriched with MACS according to the manufacturer’s instructions. Recipient Rag1-/- mice received 2 × 106 CD4+ cells i.v. After 4 weeks, recipients were treated i.p. with 2 µg anti-CD3 mAb on days 0, 2, and 4 and were analyzed 4 h after the last injection.
CD4+ and CD8+ T cells were isolated from spleens of WT and Trpm2-/- mice and purified by MACS (Miltenyi Biotec, Bergisch Gladbach, Germany). Total RNA from pooled CD4+ and CD8+ T cells was isolated using TRIzol® reagent (Invitrogen) according to the manufacturer’s protocol. RNA was reverse transcribed with the high-capacity cDNA synthesis Kit (Thermo Fisher, Darmstadt, Germany). cDNA concentrations for Ryr1, Tpcn1, Tpcn2, Orai1, and the control Hprt were determined with TaqMan PCR using primer and probes from Thermo Fisher: Hprt Mm03024075_m1, Ryr1 Mm01175211_m1, Tpcn1 Mm00455326_m1, Tpcn2 Mm00628260_m1, and Orai1 Mm00774349_m1.
Statistical analyses were performed with Prism software (GraphPad Software Inc., La Jolla, CA). Results were analyzed with the tests indicated in the figure legends. A p-value of <0.05 was considered significant (p < 0.05) and is indicated with *.
Ca2+ signaling is central for T-cell activation and differentiation, and Ca2+ influx facilitated by TRPM2 could enhance or modulate these processes. In a first set of experiments, the expression of TRPM2 was determined in CD8+ T cells (Figure 1A and Supplementary Figure 2A). Compared to Ly6C+ and Ly6G+ myeloid cells (inflammatory monocytes and neutrophils), we detected only a low level of TRPM2 expression in CD8+ T cells from WT mice which was markedly reduced in CD8+ T cells from Trpm2-/- mice. Spleen cells from WT and Trpm2-/- mice were stimulated with anti-CD3ϵ and anti-CD28 mAb, and the expression of CD69 and of the transcription factors IRF4 and NUR77 (NR4A1) was determined by flow cytometry. Expression of these proteins is induced within a few hours of TCR stimulation and can be used to determine the quality of the TCR signal. After 4 h, we observed strong upregulation of all three proteins and expression of IRF4 was further increased after 24 and 48 h of culture (Figures 1B, C). CD8+ T cells from WT and Trpm2-/- mice showed similar upregulation of the proteins. Then, spleen cells were labeled with CFSE and proliferation was determined by loss of the CFSE label. Cells were stimulated with anti-CD3ϵ and anti-CD28 mAb, and after 1, 2, 3, and 4 days, CFSE staining was determined (Figures 1D, E and Supplementary Figure 2B). T-cell activation resulted in cumulative loss of CFSE staining in CD8+ T cells; however, there was no difference in proliferation between WT and Trpm2-/- cells. TRPM2 could also modulate the T-cell response at later time points. Therefore, CD8+ T cells from WT and Trpm2-/- mice were mixed and activated with anti-CD3ϵ and anti-CD28 mAb. After 3 days, activated cells were cultured in medium containing IL-7 (Figure 1F). After 21 days, CD8+ T cells expressed only low levels of CD69 and of the proliferation marker Ki-67 but had upregulated CD38. Importantly, the ratio of WT to Trpm2-/- CD8+ T cells was similar to the ratio at the start of the culture, indicating that the lack of TRPM2 did not alter the T-cell response.
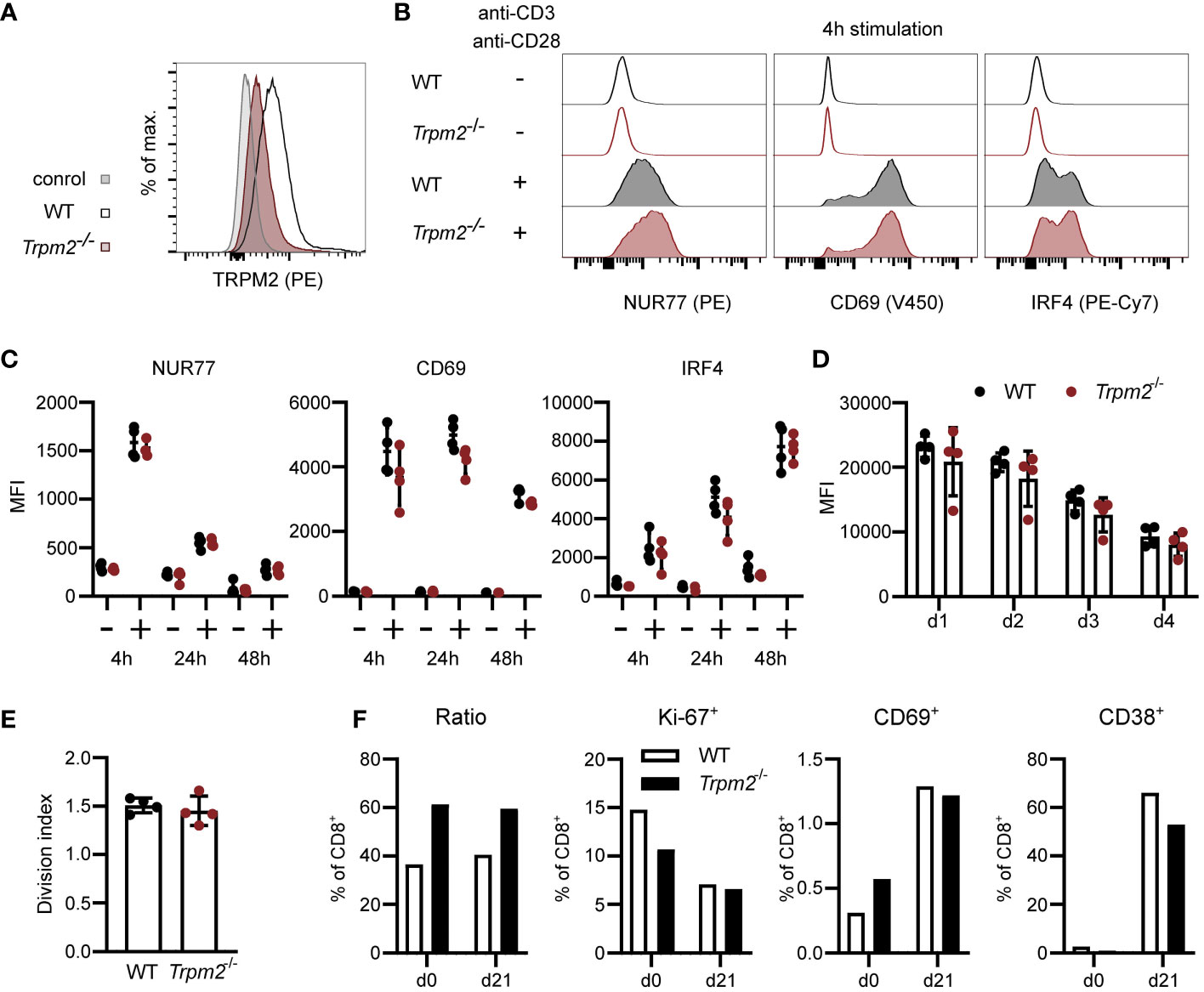
Figure 1 TRPM2 deficiency does not impair CD8+ T-cell activation in vitro. (A) CD8+ T cells were intracellularly stained with anti-TRPM2 mAb and PE-conjugated anti-rat IgG antibody (control: WT cells stained only with the secondary antibody). (B, C) Spleen cells from WT and Trpm2-/- mice were stimulated for 4, 24, or 48 h with plate-coated anti-CD3ϵ mAb and soluble anti-CD28 mAb. Expressions of CD69, NUR77, and IRF4 were determined by extra- and intracellular antibody staining. Representative staining (B) and results (C) for CD8+ T cells are shown. (D, E) Spleen cells were CFSE-labeled and stimulated with anti-CD3ϵ mAb and anti-CD28 mAb. Representative results for the mean fluorescence intensity (MFI) of CFSE of CD8+ T cells on days 1–4 (D) and the division index for day 4 (E) are given. (F) WT (CD90.1+) and Trpm2-/- (CD90.2+) spleen cells were mixed and stimulated with anti-CD3ϵ and anti-CD28 for 3 days. Then, cells were washed and incubated with IL-7. On day 21, cells were analyzed by extra- and intracellular antibody staining. Representative results for the ratio of CD90.1+ and CD90.2+ CD8+ T cells and for the expression of the proliferation markers Ki-67 and of CD69 and CD38 are shown. Bars and scatters in (C–E) give the mean ± SEM and were analyzed with the unpaired t-test. All experiments were conducted at least twice.
Similar to WT CD8+ T cells, WT CD4+ T cells expressed only low levels of TRPM2 and staining was reduced in Trpm2-/- CD4+ T cells (Figure 2A). The early response following TCR stimulation was also determined in CD4+ T cells (Figures 2B, C). CD4+ T cells showed rapid upregulation of CD69, NUR77, and IRF4, but there was no difference between WT and Trpm2-/- CD4+ T cells. We also determined the induction of the inhibitory receptors CTLA4, PD1, and LAG3 (Supplementary Figure 3). We observed an equal expression of these receptors on WT and Trpm2-/- CD4+ T cells. Differentiation of CD4+ Th cells is regulated by cytokine signals from the environment but also by the strength of the TCR stimulus and the quality of the TCR-induced Ca2+ signal. Thus, modulation of the Ca2+ signal by TRPM2 could influence the fate of Th-cell differentiation. Purified CD4+ T cells from WT and Trpm2-/- mice were stimulated under defined conditions to induce IFN-γ+ Th1 cells, IL-17A+ Th17 cells, Foxp3+ regulatory T cells (Treg cells), and Foxp3- IL-10+ regulatory 1 T cells (Tr1 cells). After 3 days, T cells were activated with PMA and ionomycin, and expression of Foxp3 and cytokines was determined by intracellular staining and flow cytometry (Figure 2D). Consistent with the conditions of differentiation, we detected upregulation of IFN-γ, IL-17A, IL-10, and Foxp3; however, WT and Trpm2-/- CD4+ T cells did not significantly differ in the generation of IFN-γ+ Th1 cells, IL-17A+ Th17 cells, and Foxp3+ Treg cells. In the experiment shown, there was a small reduction of Trpm2-/- Tr1 cells; however, this reduction was not consistent in other experiments.
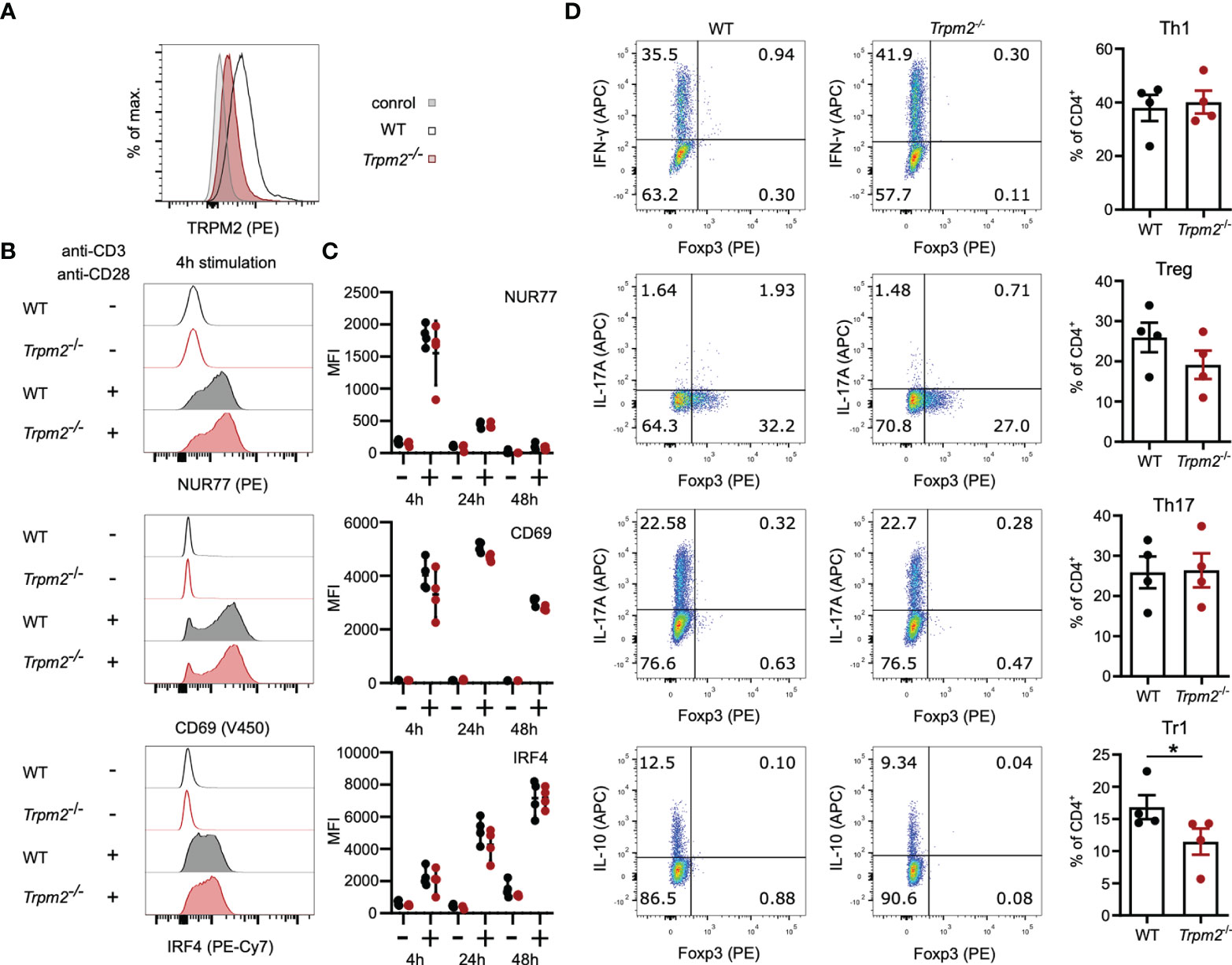
Figure 2 TRPM2 deficiency does not impair CD4+ T-cell activation and differentiation in vitro. (A) CD4+ T cells were intracellularly stained with anti-TRPM2 mAb and PE-conjugated anti-rat IgG antibody (control: WT cells stained only with the secondary antibody). (B, C) Spleen cells from WT and Trpm2-/- mice were stimulated for 4, 24, or 48 h with plate-coated anti-CD3ϵ mAb and soluble anti-CD28 mAb. Expressions of CD69, NUR77, and IRF4 were determined by extra- and intracellular antibody staining. Representative staining (B) and results (C) for CD4+ T cells are shown. (C) Purified naïve CD4+ T cells were stimulated with plate-bound anti-CD3ϵ mAb and soluble anti-CD28 mAb and cultured under Th1-, Th17-, Treg-, and Tr1-polarizing conditions. After 4 days, T cells were stimulated with PMA/ionomycin in the presence of monensin, and cytokine and Foxp3 expression was measured by flow cytometry. Shown are representative dot plots and corresponding bar graphs. Th1 cells were defined as IFN-γ+ CD4+ T cells, Th17 cells as IL-17A+ CD4+ T cells, Treg cells as Foxp3+ CD4+ T cells, and Tr1 cells as Foxp3- IL-10+ CD4+ T cells. Bars and scatters in (C, D) give the mean ± SEM and were analyzed with the unpaired t-test. *p < 0.05. All experiments were conducted at least twice.
Absence of TRPM2 could be compensated by higher expression of other Ca2+ channels. Therefore, mRNA was isolated from WT and Trpm2-/- T cells and the expression of Ryr1, Tpcn1, Tpcn2, and Orai1 coding for Ca2+ channels that might compensate for the absence in T cells was measured by RT-PCR (Supplementary Figure 4). We detected similar mRNA levels for all analyzed Ca2+ channels in WT and Trpm2-/- T cells.
In conclusion, our data so far provide no evidence for a substantial role of TRPM2 in the activation and differentiation of CD4+ and CD8+ T cells.
In order to analyze the role TRPM2 in CD8+ T cells in vivo, we used the Listeria monocytogenes infection model. WT and Trpm2-/- mice were infected with an ovalbumin-recombinant strain of L. monocytogenes (LmOVA) which induce a strong CD8+ T cell response against the OVA257-264 peptide (38). Since Trpm2-/- mice are more susceptible to L. monocytogenes (30, 31), mice were treated after 2 days with ampicillin in the drinking water which results in the rapid elimination of Listeria but does only marginally effect the T-cell response (39). Eight days postinfection, frequencies and total numbers of ovalbumin-specific CD8+ T cells were determined using OVA257-264H-2Kb dextramers (Figures 3A–C). WT and Trpm2-/- mice showed similar numbers of dextramers+ CD8+ T cells in spleen and liver, the main sites of listeria replication. Dextramer+ CD8+ T cells in both mouse strains were CD44hiCD62Llo and similar frequencies expressed CX3CR1 and KLRG1, indicative for highly activated effector T cells (Figures 3D–F). Dextramer+ CD8+ T cells did also not differ with regard to the upregulation of CD38 (Figure 3G). Spleen and liver cells were also incubated with OVA257-264 peptide, and the induction of TNF-α and IFN-γ was determined by intracellular cytokine staining (Figure 3H). Again, we observed similar frequencies of TNF-α+IFN-γ+ CD8+ T cells. There was also no difference in the production of IFN-γ and TNF-α by CD4+ T cells in response to polyclonal restimulation (Figure 3I).
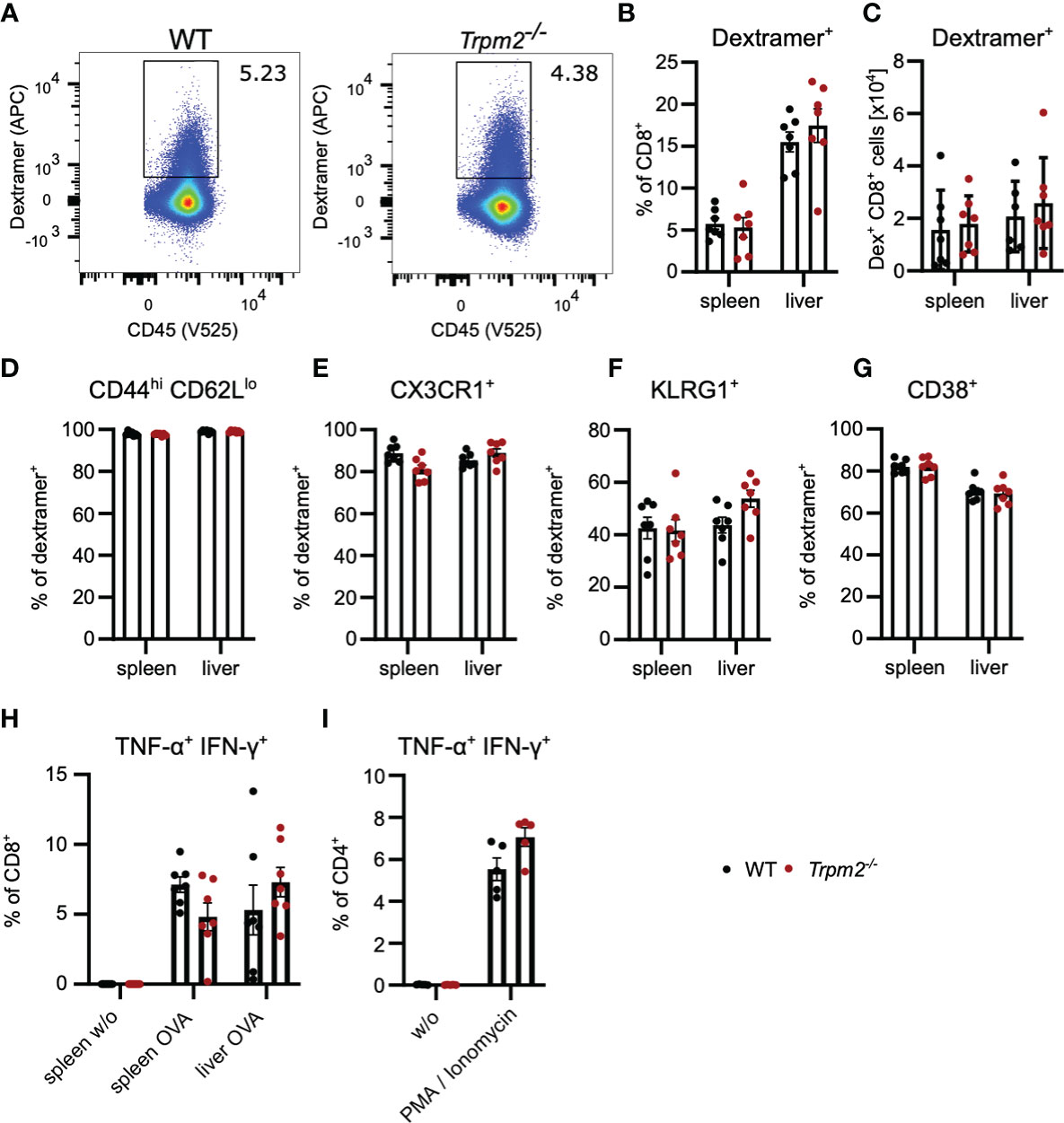
Figure 3 TRPM2 deficiency does not impair the CD8+ T-cell response to L. monocytogenes. Trpm2-/- and WT mice were infected with LmOVA. Mice were treated with ampicillin in the drinking water from day 2 on until analysis at day 8. Ovalbumin-specific CD8+ T cells were detected with H−2Kb OVA257-264-dextramers. (A) Representative dextramers staining of CD8-gated T cells. (B) Frequencies and (C) numbers of dextramers+ CD8+ T cells in spleen and liver of infected mice. (D–G) Percentage of CD44hiCD62Llo (D), CX3CR1+ (E), KLRG1+ (F), and CD38+ (G) cells among dextramer+ CD8+ T cells. (H) Spleen and liver cells were stimulated for 4 h with OVA257-264 peptide and the expression of IFN-γ and TNF-α in CD8+ T cells was determined by intracellular cytokine staining. (I) Spleen cells were stimulated for 4 h with PMA/ionomycin, and the expression of IFN-γ and TNF-α in CD4+ T cells was determined by intracellular cytokine staining. Bars in (C–I) give the mean ± SEM and were analyzed with the unpaired t-test. Data from one of three experiments are shown.
More excessive initial inflammation and altered function of TRPM2-deficient innate immune cells could mask a defect of CD8+ T cells in Trpm2-/- mice. Therefore, we used a competitive T-cell transfer assay to characterize the function of TRPM2 in CD8+ T cells. Trpm2-/- mice were crossed with OT-1 mice which are transgenic for an MHC class I-restricted OVA257-264-specific TCR (37). CD8+ T cells from WT and Trpm2-/- mice were mixed roughly at a 1:1 ratio, and 1 × 104 CD8+ T cells were transferred into recipient mice infected with LmOVA. Donor and recipient cells differed in the expression of CD90.1 and CD90.2, which allowed identification of the different cell populations. Five days post transfer and infection, CD8+ T cells derived from both donors could be detected in spleen and liver (Figures 4A, B). However, the ratio of WT to Trpm2-/- cells in both tissues was similar to that of the transferred CD8+ T-cells and both populations were similar in their expression profiles of CD44, CD62L, and KLRG1 (Figures 4C–E). In addition, after stimulation with the OVA257-264 peptide, WT and Trpm2-/- OT-1 T-cells showed similar induction of IFN-γ and TNF-α and of NURF77 (Figures 4F, G). Thus, TRPM2 deficiency restricted to CD8+ T cells did not significantly impair their response during acute infection.
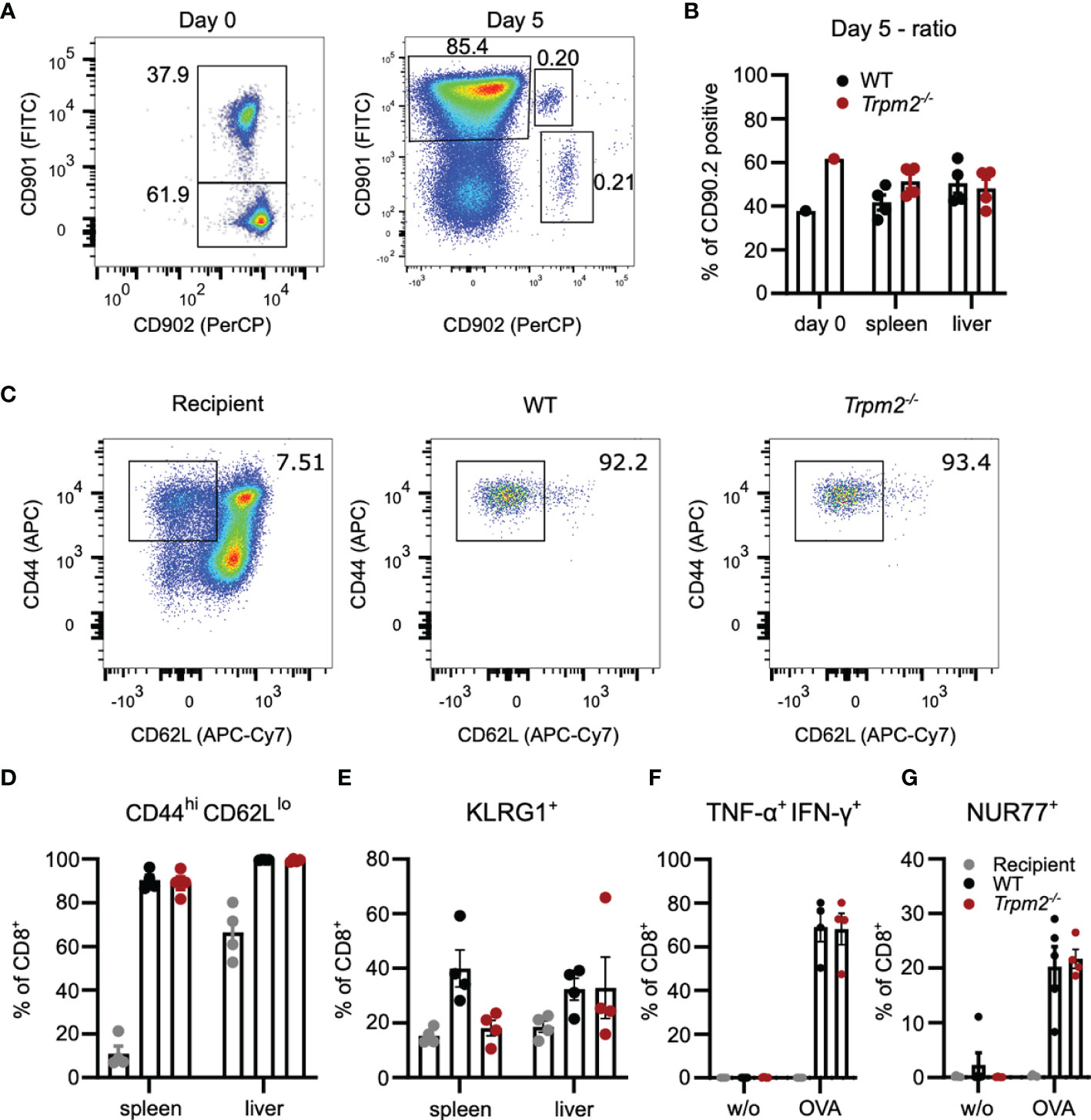
Figure 4 TRPM2 deficiency does not impair the acute CD8+ T-cell response to L. monocytogenes in a T-cell transfer model. Trpm2-/- CD8+ OT-1 T cells (CD90.1-CD90.2+) and WT CD8+ OT-1 T cells (CD90.1+CD90.2+) were mixed roughly at a ratio 1:1 and 104 CD8+ T cells were i.v. transferred to CD90.1+CD90.2- WT mice. Recipients were infected on the same day with LmOVA and analyzed 5 days later. (A) Representative dot plots for CD90.1 and CD90.2 staining of the transferred CD8+ T-cell population (Day 0) and of the CD8+ T cells from the spleen at day 7. (In the experiment shown, we transferred cells with a 2:3 ratio of WT to Trpm2-/- CD8+ T cells.) (B) Frequencies of WT and Trpm2-/- cells among CD8+ donor T cells at day 0 (mix before transfer) and day 5 postinfection. (C) Representative dot plots for CD44 and CD62L expression of donor and recipient CD8+ T cells. (D) Frequencies of CD44hiCD62Llo donor and recipient CD8+ T cells in spleen and liver. (E) Frequencies of KLRG1+ donor and recipient CD8+ T cells in spleen and liver. (F) Spleen cells were stimulated for 4 h with OVA257-264 peptide and the frequencies of IFN-γ+ TNF-α+ donor and recipient CD8+ T cells was determined by intracellular cytokine staining. (G) Frequencies of NUR77+ donor and recipient CD8+ T cells after OVA257-264 peptide stimulation of spleen cells. Bars give the mean ± SEM. Results in (D–G) and were analyzed with ANOVA with Dunnett’s multiple-comparison test. Experiments were conducted twice.
The competitive transfer assay was also used to determine the role of TRPM2 in CD8+ memory T-cell formation. To exclude rejection of donor cells, Rag1-/- mice were used as recipients. Eight weeks post transfer and LmOVA infection, donor cells in the spleen, liver, lung, kidney, and bone marrow were analyzed. In the spleen, liver, and bone marrow, we observed ratios of WT to Trpm2-/- cells similar to the ratio of the transferred cell population (Figure 5A). Interestingly, WT cells were slightly more prominent in lung and kidney, indicating a disadvantage of Trpm2-/- CD8+ T cells in migration into or survival within these tissues. Phenotypical characterization revealed similar expression profiles for CD44 and CD62L cells with high frequencies of CD44hiCD62Llo effector/effector memory T cells in the liver, lung, and kidney, and somewhat lower frequencies of these cells in the spleen and bone marrow (Figure 5B). Upon peptide restimulation of CD8+ T cells from the spleen of recipients, we observed similar frequencies of TNF-α+IFN-γ+ and NUR77+ T cells in both CD8+ T-cell populations (Figures 5C, D).

Figure 5 TRPM2 deficiency does not impair the long-term CD8+ T-cell response to L. monocytogenes in a T-cell transfer model. 5,000 Trpm2-/- CD8+ OT-1 T cells (CD90.1-CD90.2+) and 5,000 WT CD8+ OT-1 T cells (CD90.1+CD90.2+) were mixed and i.v. transferred to Rag1-/- mice. Recipients were infected on the same day with LmOVA and analyzed 8 weeks later. (A) Frequencies of WT and Trpm2-/- cells among CD8+ donor T cells at day 0 (mix before transfer) and 8 weeks postinfection in spleen, liver, lung, kidney, and bone marrow (BM). (B) Frequencies of CD44hiCD62Llo donor CD8+ T cells in different tissues. (C) Spleen cells were stimulated for 4 h with OVA257-264 peptide, and the frequencies of IFN-γ+ TNF-α+ donor and recipient CD8+ T cells were determined by intracellular cytokine staining. (D) Frequencies of NUR77+ donor and recipient CD8+ T cells after OVA257-264 peptide stimulation of spleen cells. Bars in give the mean ± SEM. Results in (B–D) and were analyzed with the unpaired t-test. One representative experiment of three is shown.
In order to test the response of Trpm2-/- CD4+ T cells in vivo, we used the model of anti-CD3 mAb-induced intestinal inflammation. In this model, repeated injection of anti-CD3 mAb causes systemic T-cell activation. A main hallmark of the model is the activation and accumulation of Th1 and Th17 cells in the small intestine resulting in inflammation of the intestinal mucosa, and diarrhea and weight loss as disease manifestations. As a consequence of the inflammation, Th17 cells differentiate to IL-10-secreting Tr1 cells and enhanced frequencies of both Tr1 cells and conventional Foxp3+ Treg cells are found in the small intestinal mucosa. Thus, the anti-CD3 application model allows the analyses of Th1, Th17, and Treg cell responses as well as the formation of Tr1 cells (40–43). Four days after anti-CD3 mAb treatment, mice had lost about 15% of their weight; however, weight loss was similarly extensive in WT and Trpm2-/- mice (Figure 6A). Characterization of T cells from the small intestinal mucosa revealed similar frequencies and numbers of CD4+ Th1, Th17, Treg, and Tr1 cells in WT and Trpm2-/- mice (Figure 6B). Thus, deficiency of TRPM2 did not affect the CD4+ T-cell response in this model.
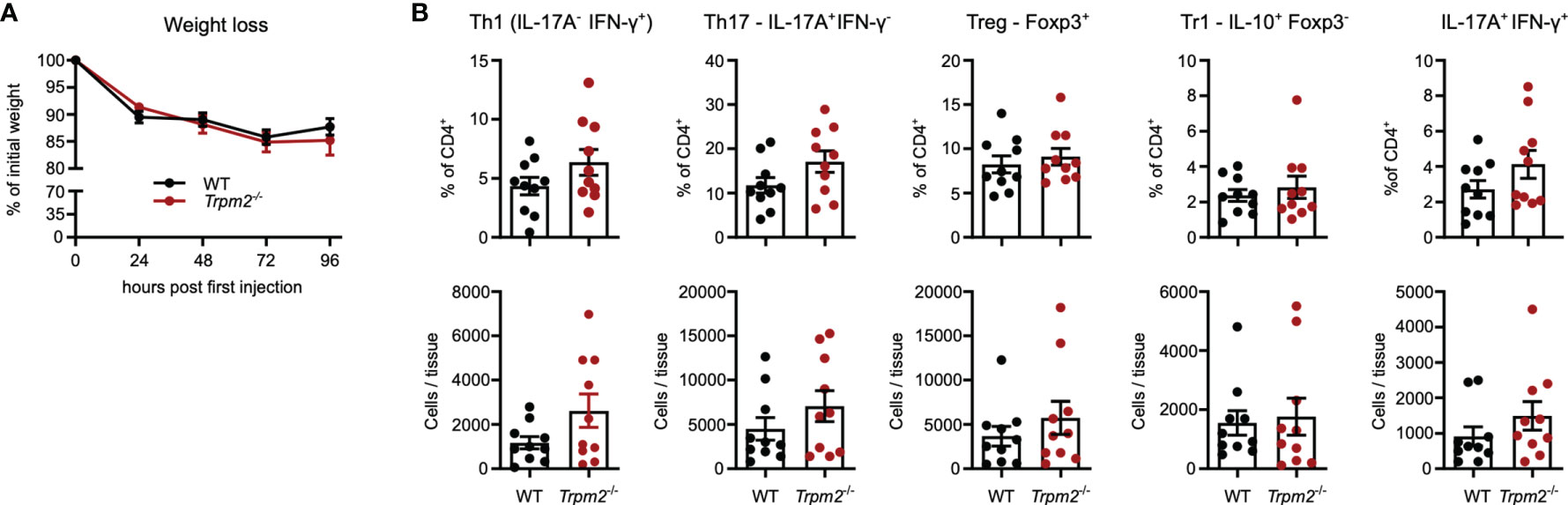
Figure 6 TRPM2 deficiency does not impair the CD4+ T-cell response in response to anti-CD3 mAb induced intestinal inflammation. Mice were i.p. injected with anti-CD3 mAb on days 0, 2, and 4 and analyzed 4 h after the last injection. (A) Percentage of initial body weight. (B) Frequencies and numbers of different CD4+ T-cell populations in the small intestinal mucosa. Bars depict mean ± SEM. Results were analyzed with the Mann–Whitney test. Data were pooled from two independent experiments.
As discussed in the context of the infection model, TRPM2 deficiency in cells other than T cells could mask an altered CD4+ T-cells response. Therefore, Rag1-/- mice were reconstituted with CD4+ T cells from either WT or Trpm2-/- mice and then treated with anti-CD3 mAb. Under these conditions, weight loss was less pronounced but similar in mice reconstituted with WT and Trpm2-/- cells (Figure 7A). Recipients did not differ in the frequencies and numbers of intestinal IFN-γ+ Th1 cells, IL-17A+ Th17 cells, and IFN-γ+IL-17A+ CD4+ T cells as well as in frequencies and numbers of Foxp3+ cells (Figure 7B).
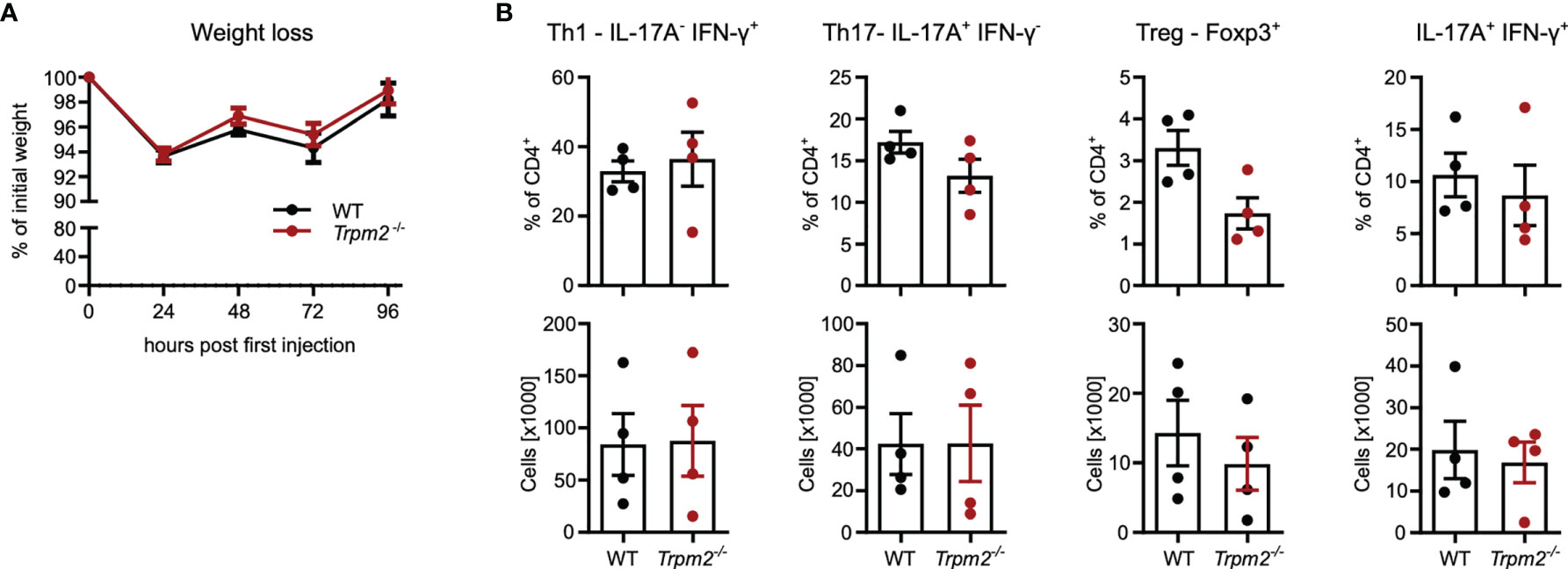
Figure 7 TRPM2 deficiency does not impair response of transferred CD4+ T cells in the model of anti-CD3 mAb induced intestinal inflammation. Total CD4+ T cells from WT and Trpm2-/- mice were transferred into Rag1-/- mice. After 4 weeks, recipients were treated with anti-CD3 mAb on days 0, 2, and 4 and were analyzed 4 h after the last injection. (A) Percentage of initial body weight. (B) Frequencies and numbers of different CD4+ T-cell populations in the small intestinal mucosa. Bars depict mean ± SEM. Results were analyzed with the Mann–Whitney test.
In conclusion, we could not demonstrate a substantial role of TRPM2 in the in vivo response of CD4+ T cells.
Upon TCR stimulation, we observed a similar induction of IRF4, NUR77, and CD69 after 4, 24, and 48 h in both CD4+ and CD8+ T cells from WT and Trpm2-/- mice. This result is consistent with the regular early Ca2+ signal after TCR stimulation of Trpm2-/- T cells reported by Wolf and colleagues (7) and indicates that TRPM2 is not required for early T-cell activation. In line with this concept, Trpm2-/- CD8+ T cells were not impaired in their proliferation after polyclonal stimulation in vitro and in their response to L. monocytogenes infection in vivo. The absence of effect is further consistent with the relatively low expression levels of mRNA for Trpm2, Cd38, and Bst1/Cd157 (coding for a NAD-glycohydrolase closely related to CD38 (44) (Supplementary Figure 1, Immgen.org (20)) and of TRPM2 protein (Figures 1A, 2A and Supplementary Figure 2A) in naive mouse CD4+ and CD8+ T cells. Our results are in contrast to results from Melzer et al., who reported reduced in vitro proliferation and cytokine production of Trpm2-/- T cells (19). Currently, we have no valid explanation for these inconsistent results.
Differentiation of CD4+ Th cells is regulated by signals from the environment, in particular inflammatory cytokines, but also by the quality of the TCR-signal (2–4). Thus, TRPM2-facilitated Ca2+ signaling could affect Th-cell differentiation. However, under defined in vitro conditions, we observed similar Th1, Th17, Tr1, an Treg-cell differentiation of purified WT and Trpm2-/- CD4+ T cells. Consistent with the results from the in vitro assays, Th-cell differentiation was also only marginally affected in anti-CD3 mAb-induced inflammation, which is associated with intestinal accumulation of Th1, Th17, Tr1, and Treg cells (40–43). Thus, TRPM2 has, at least in our experimental models, no major function in CD4+ Th-cell differentiation. Interestingly, Trpm2-/- mice and WT mice developed comparable symptoms to anti-CD3 mAb treatment. This differs to the attenuated disease of Trpm2-/- mice in the dextran sulfate sodium (DSS) model (29). In contrast to the anti-CD3 mAb model in which disease is caused by the response of T cells, the DSS colitis model is primarily driven by innate immune mechanisms after damage and bacterial infiltration of the colon mucosa. Consistent with this notion, the milder course of DSS colitis of Trpm2-/- mice is associated with reduced neutrophil but similar T cell accumulation (29).
TRPM2 could be relevant at later stages of the T-cell response for instance due to increased expression levels of TRPM2 or of ADPR generating enzymes. mRNA expression analysis did not reveal major changes in Trpm2 levels in different T-cell subsets sorted from naïve and infected mice. In contrast, the expression of CD38 was upregulated on effector and memory T-cell subsets (Figure 1F and Supplementary Figure 1). However, our results from long-term in vitro coculture and the analysis of CD8+ T cells 8 weeks post co-transfer and infection argue against such a late function, since we detect no major changes in the ratio of WT and Trpm2-/- donor cells in cell culture and in the spleen and liver, the main organs of L. monocytogenes infection in the mouse. Interestingly, there was a slightly reduced accumulation of Trpm2-/- CD8+ T cells in the lung and kidney. Thus, TRPM2 might be required for the migration of T cells to these tissues or the survival of T cells within these tissues. The CD38–ADPR–TRPM2 axis has been linked to chemokine signaling in myeloid cells (9), and it is possible that this pathway is also active in chemokine signaling in T cells and thereby required for migration of activated T cells to peripheral tissues such as lung and kidney. However, this function would be restricted to only certain tissues and chemokine receptors since we did not observe impaired migration of CD8+ T cells to liver and bone marrow or of CD4+ T cells to the intestinal mucosa.
In summary, our results do not support a major function of TRPM2 in T-cell activation and differentiation. However, we cannot exclude a role of TRPM2 in T-cell subsets or differentiation stages not analyzed in our assays. Our analyses were entirely conducted with mouse T cells and in mouse models. Thus, we can also not exclude that TRPM2 has a more substantial role in human T cells.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by Behörde für Justiz und Verbraucherschutz der Freien und Hansestadt Hamburg.
Conceptualization: NL, MN, AHG, SH, H-WM. Data curation: NL, MN, MC, JS, VS. Methodology: NL, MN, TB, SM, FK-N, SH, H-WM. Formal analysis: NL, MN. Project administration: SH, H-WM. Writing, review, editing: all authors. All authors contributed to the article and approved the submitted version.
This work was supported in part by the Deutsche Forschungsgemeinschaft (project number 335447717, SFB1328: to SH, H-WM, FK-N, and AHG). SH has an endowed Heisenberg-Professorship awarded by the Deutsche Forschungsgemeinschaft. MN has a scholarship from the Else Kröner Stiftung (iPRIME).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Dietmar Zehn for LmOVA and Dr. Yasuo Mori for Trpm2-/- mice.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.778916/full#supplementary-material
ADPR, adenosine 5′-diphosphate-ribose; cADPR, cyclic adenosine 5′-diphosphate-ribose; 2dADPR, 2′-deoxyadenosine 5′-diphosphoribose; IP3, D-myo-inositol 1,4,5-trisphosphate; Lm, Listeria monocytogenes; LmOVA, ovalbumin recombinant Listeria monocytogenes; NAADP, nicotinic acid adenine dinucleotide phosphate; OVA, ovalbumin; PMA, phorbol 12-myristate 13-acetate; RYR, ryanodine receptor; TPC, two-pore channel; TRPM2, transient receptor potential cation channel subfamily M member 2.
1. Le Borgne M, Raju S, Zinselmeyer BH, Le VT, Li J, Wang Y, et al. Real-Time Analysis of Calcium Signals During the Early Phase of T Cell Activation Using a Genetically Encoded Calcium Biosensor. J Immunol (2016) 196(4):1471–9. doi: 10.4049/jimmunol.1502414
2. Corse E, Gottschalk RA, Allison JP. Strength of TCR-Peptide/MHC Interactions and In Vivo T Cell Responses. J Immunol (2011) 186(9):5039–45. doi: 10.4049/jimmunol.1003650
3. Guichard V, Bonilla N, Durand A, Audemard-Verger A, Guilbert T, Martin B, et al. Calcium-Mediated Shaping of Naive CD4 T-Cell Phenotype and Function. Elife (2017) 6:e27215. doi: 10.7554/eLife.27215
4. Rogers PR, Croft M. Peptide Dose, Affinity, and Time of Differentiation can Contribute to the Th1/Th2 Cytokine Balance. J Immunol (1999) 163(3):1205–13.
5. Ernst IM, Fliegert R, Guse AH. Adenine Dinucleotide Second Messengers and T-Lymphocyte Calcium Signaling. Front Immunol (2013) 4:259. doi: 10.3389/fimmu.2013.00259
6. Trebak M, Kinet J-P. Calcium Signalling in T Cells. Nat Rev Immunol (2019) 19(3):154–69. doi: 10.1038/s41577-018-0110-7
7. Wolf IM, Diercks B-P, Gattkowski E, Czarniak F, Kempski J, Werner R, et al. Frontrunners of T Cell Activation: Initial, Localized Ca2+ Signals Mediated by NAADP and the Type 1 Ryanodine Receptor. Sci Signal (2015) 8(398):ra102–ra. doi: 10.1126/scisignal.aab0863
8. Diercks BP, Werner R, Weidemuller P, Czarniak F, Hernandez L, Lehmann C, et al. ORAI1, STIM1/2, and RYR1 Shape Subsecond Ca(2+) Microdomains Upon T Cell Activation. Sci Signal (2018) 11(561):eaat0358. doi: 10.1126/scisignal.aat0358
9. Partida-Sanchez S, Gasser A, Fliegert R, Siebrands CC, Dammermann W, Shi G, et al. Chemotaxis of Mouse Bone Marrow Neutrophils and Dendritic Cells is Controlled by Adp-Ribose, the Major Product Generated by the CD38 Enzyme Reaction. J Immunol (2007) 179(11):7827–39. doi: 10.4049/jimmunol.179.11.7827
10. Sumoza-Toledo A, Lange I, Cortado H, Bhagat H, Mori Y, Fleig A, et al. Dendritic Cell Maturation and Chemotaxis is Regulated by TRPM2-Mediated Lysosomal Ca2+ Release. FASEB J (2011) 25(10):3529–42. doi: 10.1096/fj.10-178483
11. Knowles H, Li Y, Perraud A-L. The TRPM2 Ion Channel, an Oxidative Stress and Metabolic Sensor Regulating Innate Immunity and Inflammation. Immunol Res (2013) 55(1):241–8. doi: 10.1007/s12026-012-8373-8
12. Wang G, Cao L, Liu X, Sieracki NA, Di A, Wen X, et al. Oxidant Sensing by TRPM2 Inhibits Neutrophil Migration and Mitigates Inflammation. Dev Cell (2016) 38(5):453–62. doi: 10.1016/j.devcel.2016.07.014
13. Beceiro S, Radin JN, Chatuvedi R, Piazuelo MB, Horvarth DJ, Cortado H, et al. TRPM2 Ion Channels Regulate Macrophage Polarization and Gastric Inflammation During Helicobacter Pylori Infection. Mucosal Immunol (2017) 10(2):493–507. doi: 10.1038/mi.2016.60
14. Di A, Kiya T, Gong H, Gao X, Malik AB. Role of the Phagosomal Redox-Sensitive TRP Channel TRPM2 in Regulating Bactericidal Activity of Macrophages. J Cell Sci (2017) 130(4):735–44. doi: 10.1242/jcs.196014
15. Morad H, Luqman S, Tan C-H, Swann V, McNaughton PA. TRPM2 Ion Channels Steer Neutrophils Towards a Source of Hydrogen Peroxide. Sci Rep (2021) 11(1):9339. doi: 10.1038/s41598-021-88224-5
16. Beck A, Kolisek M, Bagley LA, Fleig A, Penner R. Nicotinic Acid Adenine Dinucleotide Phosphate and Cyclic ADP-Ribose Regulate TRPM2 Channels in T Lymphocytes. FASEB J (2006) 20(7):962–4. doi: 10.1096/fj.05-5538fje
17. Gasser A, Glassmeier Gn, Fliegert R, Langhorst MF, Meinke S, Hein Dr, et al. Activation of T Cell Calcium Influx by the Second Messenger ADP-Ribose. J Biol Chem (2006) 281(5):2489–96. doi: 10.1074/jbc.M506525200
18. Sano Y, Inamura K, Miyake A, Mochizuki S, Yokoi H, Matsushime H, et al. Immunocyte Ca2+ Influx System Mediated by LTRPC2. Science (2001) 293(5533):1327–30. doi: 10.1126/science.1062473
19. Melzer N, Hicking G, Göbel K, Wiendl H. TRPM2 Cation Channels Modulate T Cell Effector Functions and Contribute to Autoimmune CNS Inflammation. PloS One (2012) 7(10):e47617. doi: 10.1371/journal.pone.0047617
20. Heng TS, Painter MW. The Immunological Genome Project: Networks of Gene Expression in Immune Cells. Nat Immunol (2008) 9(10):1091–4. doi: 10.1038/ni1008-1091
21. Perraud A-L, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, et al. ADP-Ribose Gating of the Calcium-Permeable LTRPC2 Channel Revealed by Nudix Motif Homology. Nature (2001) 411(6837):595–9. doi: 10.1038/35079100
22. Wang L, Fu T-M, Zhou Y, Xia S, Greka A, Wu H. Structures and Gating Mechanism of Human TRPM2. Science (2018) 362(6421):eaav4809. doi: 10.1126/science.aav4809
23. Huang Y, Winkler PA, Sun W, Lü W, Du J. Architecture of the TRPM2 Channel and its Activation Mechanism by ADP-Ribose and Calcium. Nature (2018) 562(7725):145–9. doi: 10.1038/s41586-018-0558-4
24. Yu P, Wang Q, Zhang L-H, Lee H-C, Zhang L, Yue J. A Cell Permeable NPE Caged ADP-Ribose for Studying TRPM2. PloS One (2012) 7(12):e51028. doi: 10.1371/journal.pone.0051028
25. Fliegert R, Riekehr WM, Guse AH. Does Cyclic ADP-Ribose (cADPR) Activate the Non-Selective Cation Channel Trpm2? Front Immunol (2020) 11:2018. doi: 10.3389/fimmu.2020.02018
26. Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-Permeable Channel Activated by Changes in Redox Status Confers Susceptibility to Cell Death. Mol Cell (2002) 9(1):163–73. doi: 10.1016/S1097-2765(01)00438-5
27. Fliegert R, Bauche A, Wolf Perez AM, Watt JM, Rozewitz MD, Winzer R, et al. 2’-Deoxyadenosine 5’-Diphosphoribose is an Endogenous TRPM2 Superagonist. Nat Chem Biol (2017) 13(9):1036–44. doi: 10.1038/nchembio.2415
28. Zhao YJ, Lam CM, Lee HC. The Membrane-Bound Enzyme CD38 Exists in Two Opposing Orientations. Sci Signal (2012) 5(241):ra67. doi: 10.1126/scisignal.2002700
29. Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, et al. TRPM2-Mediated Ca 2+ Influx Induces Chemokine Production in Monocytes That Aggravates Inflammatory Neutrophil Infiltration. Nat Med (2008) 14(7):738–47. doi: 10.1038/nm1758
30. Knowles H, Heizer JW, Li Y, Chapman K, Ogden CA, Andreasen K, et al. Transient Receptor Potential Melastatin 2 (TRPM2) Ion Channel is Required for Innate Immunity Against Listeria Monocytogenes. Proc Natl Acad Sci USA (2011) 108(28):11578–83. doi: 10.1073/pnas.1010678108
31. Robledo-Avila FH, Ruiz-Rosado J, Brockman KL, Partida-Sánchez S. The TRPM2 Ion Channel Regulates Inflammatory Functions of Neutrophils During Listeria Monocytogenes Infection. Front Immunol (2020) 11:97. doi: 10.3389/fimmu.2020.00097
32. Qian X, Zhao H, Chen X, Li J. Disruption of Transient Receptor Potential Melastatin 2 Decreases Elastase Release and Bacterial Clearance in Neutrophils. Innate Immun (2018) 24(2):122–30. doi: 10.1177/1753425918759181
33. Tripathi JK, Sharma A, Sukumaran P, Sun Y, Mishra BB, Singh BB, et al. Oxidant Sensor Cation Channel TRPM2 Regulates Neutrophil Extracellular Trap Formation and Protects Against Pneumoseptic Bacterial Infection. FASEB J (2018) 32(12):fj201800605. doi: 10.1096/fj.201800605
34. Gelderblom M, Melzer N, Schattling B, Gob E, Hicking G, Arunachalam P, et al. Transient Receptor Potential Melastatin Subfamily Member 2 Cation Channel Regulates Detrimental Immune Cell Invasion in Ischemic Stroke. Stroke (2014) 45(11):3395–402. doi: 10.1161/STROKEAHA.114.005836
35. Garcia-Rodriguez S, Rosal-Vela A, Botta D, Cumba Garcia LM, Zumaquero E, Prados-Maniviesa V, et al. CD38 Promotes Pristane-Induced Chronic Inflammation and Increases Susceptibility to Experimental Lupus by an Apoptosis-Driven and TRPM2-Dependent Mechanism. Sci Rep (2018) 8(1):3357. doi: 10.1038/s41598-018-21337-6
36. Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-Deficient Mice Have No Mature B and T Lymphocytes. Cell (1992) 68(5):869–77. doi: 10.1016/0092-8674(92)90030-G
37. Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T Cell Receptor Antagonist Peptides Induce Positive Selection. Cell (1994) 76(1):17–27. doi: 10.1016/0092-8674(94)90169-4
38. Zehn D, Lee SY, Bevan MJ. Complete But Curtailed T-Cell Response to Very Low-Affinity Antigen. Nature (2009) 458(7235):211–4. doi: 10.1038/nature07657
39. Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, Pamer EG. Early Programming of T Cell Populations Responding to Bacterial Infection. J Immunol (2000) 165(12):6833–9. doi: 10.4049/jimmunol.165.12.6833
40. Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, et al. Control of TH17 Cells Occurs in the Small Intestine. Nature (2011) 475(7357):514–8. doi: 10.1038/nature10228
41. Huber S, Gagliani N, Esplugues E, O’Connor J, Huber FJ, Chaudhry A, et al. Th17 Cells Express Interleukin-10 Receptor and are Controlled by Foxp3– and Foxp3+ Regulatory CD4+ T Cells in an Interleukin-10-Dependent Manner. Immunity (2011) 34(4):554–65. doi: 10.1016/j.immuni.2011.01.020
42. Gagliani N, Vesely MCA, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 Cells Transdifferentiate Into Regulatory T Cells During Resolution of Inflammation. Nature (2015) 523(7559):221–U5. doi: 10.1038/nature14452
43. Brockmann L, Soukou S, Steglich B, Czarnewski P, Zhao L, Wende S, et al. Molecular and Functional Heterogeneity of IL-10-Producing CD4(+) T Cells. Nat Commun (2018) 9(1):5457. doi: 10.1038/s41467-018-07581-4
Keywords: TRPM2, T cells, T-cell activation, TCR signaling, ADPR, calcium signaling
Citation: Lory NC, Nawrocki M, Corazza M, Schmid J, Schumacher V, Bedke T, Menzel S, Koch-Nolte F, Guse AH, Huber S and Mittrücker H-W (2022) TRPM2 Is Not Required for T-Cell Activation and Differentiation. Front. Immunol. 12:778916. doi: 10.3389/fimmu.2021.778916
Received: 17 September 2021; Accepted: 20 December 2021;
Published: 14 January 2022.
Edited by:
Andrew D. Wells, Children’s Hospital of Philadelphia, United StatesReviewed by:
Prabhakaran Kumar, University of Illinois at Chicago, United StatesCopyright © 2022 Lory, Nawrocki, Corazza, Schmid, Schumacher, Bedke, Menzel, Koch-Nolte, Guse, Huber and Mittrücker. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans-Willi Mittrücker, aC5taXR0cnVlY2tlckB1a2UuZGU=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.