 Haixia Li
Haixia Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 01 November 2021
Sec. Multiple Sclerosis and Neuroimmunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.777606
This article is part of the Research Topic Demyelinating Neurological Syndromes: The Role of Autoimmunity View all 8 articles
Toll-like receptors (TLRs) are a class of proteins playing a key role in innate and adaptive immune responses. TLRs are involved in the development and progression of neuroimmune diseases via initiating inflammatory responses. Thus, targeting TLRs signaling pathway may be considered as a potential therapy for neuroimmune diseases. However, the role of TLRs is elusive and complex in neuroimmune diseases. In addition to the inadequate immune response of TLRs inhibitors in the experiments, the recent studies also demonstrated that partial activation of TLRs is conducive to the production of anti-inflammatory factors and nervous system repair. Exploring the mechanism of TLRs in neuroimmune diseases and combining with developing the emerging drug may conquer neuroimmune diseases in the future. Herein, we provide an overview of the role of TLRs in several neuroimmune diseases, including multiple sclerosis, neuromyelitis optica spectrum disorder, Guillain-Barré syndrome and myasthenia gravis. Emerging difficulties and potential solutions in clinical application of TLRs inhibitors will also be discussed.
Toll-like receptors (TLRs), as type 1 transmembrane protein receptors, recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) and then initiate immune responses (1, 2). TLRs can be activated following the recognition of lipopolysaccharides (LPS), lipoproteins, flagellin, viral and bacterial nucleic acids, leading to a combination of protein complexes and activating chromatin remodeling and transcription factors (3). Due to a variety of regulatory functions, TLRs are actively involved in the secretion of inflammatory mediators, cellular proliferation and survival (3, 4). TLRs can also bind to extracellular domains and undergo conformational changes following dimerization to recruit intercellular downstream signaling adaptors. As each TLR paralogue perform distinct functions, various heterodimers may play different roles (3). The activated TLRs triggers a cascade of cytokine and chemokine productions, contributing to the initiation and progression of cancer (5), rheumatic diseases (6), atherosclerosis (7), neurodegenerative disease (8) and autoimmune disease (9). TLRs are necessary for protecting against diseases by accelerating the healing process to restore immune homeostasis. However, excessive TLRs activity might lead to chronic and unrestricted inflammatory responses, which could aggravate diseases.
Neuroimmune diseases can be divided into the central and peripheral nervous disorders, such as multiple sclerosis (MS), neuromyelitis optica spectrum disorder (NMOSD), Guillain-Barré syndrome (GBS), and others, such as myasthenia gravis (MG). These conditions pose a threat to the human health all over the world. It has been determined that dendritic cells, circulating monocytes, Natural Killer (NK) cells, microglia/macrophages, T and B lymphocytes are involved in the pathogenesis of neuroimmune diseases (10–12). However, the precise pathogenesis of neuroimmune diseases remain largely unknown. Although immunomodulatory drugs have achieved some success in clinical practice, most of them are non-selective immunosuppressive or cytotoxic. Limited clinical efficacy and significant side effects were found in some patients (13, 14).
Accumulating evidence has revealed that TLRs play vital roles in the pathogenesis of neuroimmune diseases, and relieved clinical symptoms were observed in preclinical models by regulating TLRs (15, 16). However, clinical evidence is still insufficient (17). How to specifically target cellular type-specific function of TLRs signaling pathway in neuroimmune disease remains unclear. Here, we updated knowledge about the role of TLRs in neuroimmune diseases, and proposed several approaches to overcome obstacles for the application of TLRs inhibitors in clinical treatment.
The molecular weights of TLRs range from 90 to 150 kDa (9). The structure of TLRs can be divided into three parts: extracellular region, transmembrane region and intracellular region. Canonically, TLRs are an important pattern-recognition receptor (PRR), which consist of extracellular leucine-rich repeats (LRRs) to recognize PAMPs and DAMPs (18, 19). PAMPs are highly conserved structural components derived from microorganisms that consist of LPS, peptidoglycan, flagellin, lipoproteins and microbial nucleic acids (20, 21). Most DAMPs are endogenous molecules released from dying cells upon cellular stress or tissue damage (22, 23). The transmembrane TLRs are well-known PRRs, which function through extracellular ligand recognition. Due to its homology with the signaling domains of interleukin (IL)-1R family members, the intracellular domain structure of TLRs is known as the N-terminal cytoplasmic Toll/IL-1 receptor (TIR) mediating homotypic interactions and facilitating downstream signaling (18, 24, 25).
To date, 13 active members of the TLRs family have been identified in mammals, including 10 in humans (TLR1-10) and 12 in mice (TLR1-9 and TLR11-13) (26). TLRs are largely categorized into two subfamilies based on their localization, such as transmembrane and intracellular regions (21). Specifically, TLR1, TLR2, TLR5, TLR6 and TLR10 are located on the cell surface, while TLR3, TLR7, TLR8 and TLR9 are mainly located on the intracellular endosome membrane. The locations of TLR4, TLR11, TLR12 and TLR13 are controversial, with being expressed on the cell membrane or intracellular endosome membrane according to different cell types. They may also be expressed in both cell membrane or intracellular endosome membrane (18, 21, 27, 28). The extracellular domain of each TLR corresponds to a specific PAMP, and the collective TLR family can specially recognize different pathogens. For example, TLR4 recognizes LPS and TLR10 can sense influenza A virus infection (29).
TLRs recruit five cytosolic TIR domain-containing adaptors, including MyD88, TIR domain-containing adaptor-inducing IFN-β (TRIF), TIR domain-containing adaptor protein (TIRAP, also known as MAL), TRIF-related adaptor molecule (TRAM) and sterile α- and armadillo-motif-containing protein (SARM) (30, 31). TLRs signaling molecules induce the expression of proinflammatory cytokines via two main pathways: MyD88-dependent signaling pathway and MyD88-independent TRIF pathway (32). TLRs, with the exception of TLR3, utilize the MyD88-dependent signaling pathway. TLR4 is the only TLR that divides into MyD88-dependent and MyD88-independent (TRIF-dependent) signaling pathways (1, 2) (Figure 1).
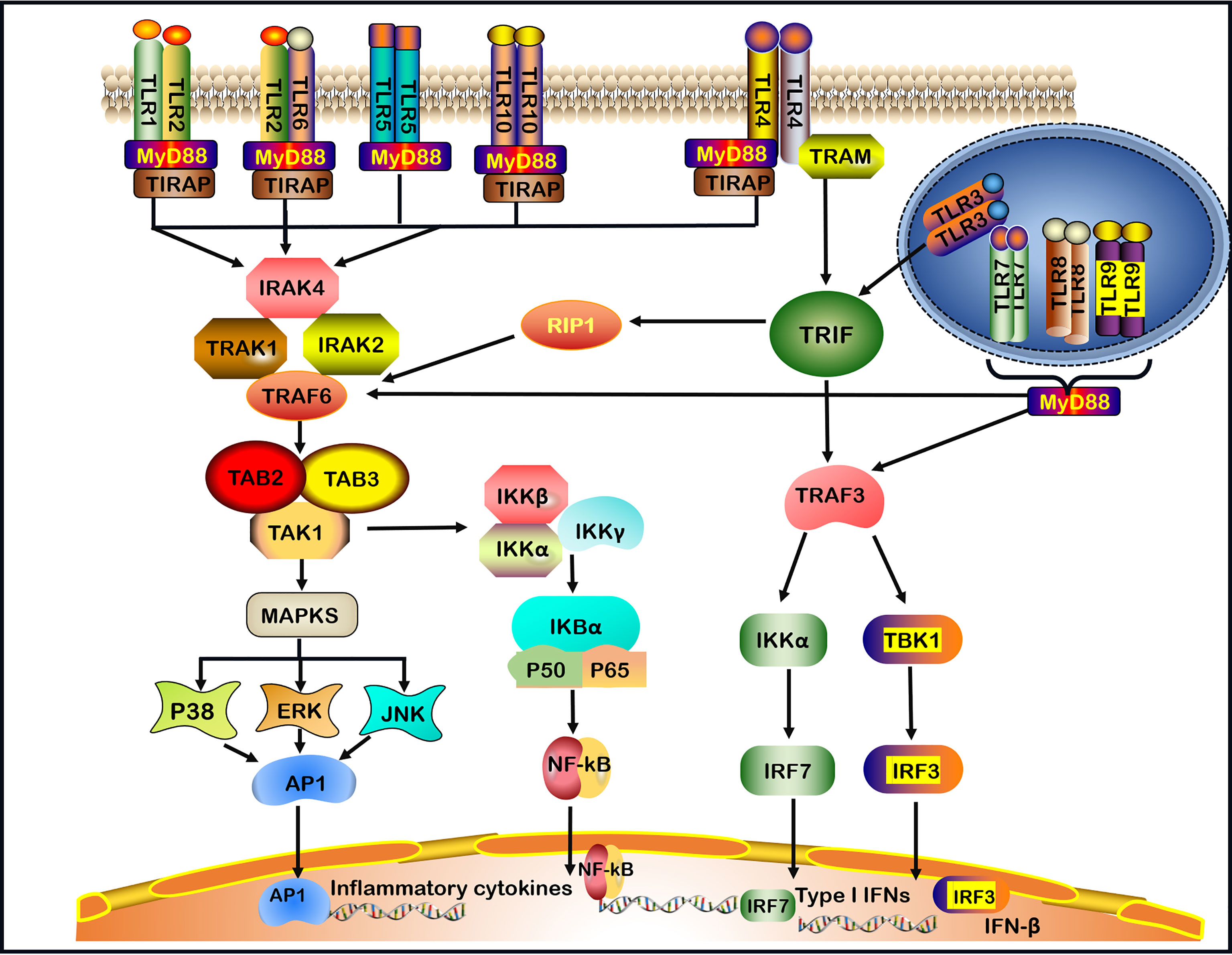
Figure 1 TLRs signaling pathways. TLRs (Toll-like receptors) recognize invading microbes and activate signaling pathways, which regulate immune and inflammatory responses. TLR1, TLR2, TLR5, TLR6, and TLR10 are located on the cell surface.TLR3, TLR7, TLR8, and TLR9 are located on the intracellular endosome membranes. All TLRs, with the exception of TLR3, by the (MyD88)- dependent signaling pathway. In addition, TLR4 signaling takes place in both MyD88-dependent and the MyD88-independent signaling pathway. In MyD88-dependent signaling pathway, the leucine-rich repeats (LRR) region of TLR binds to ligands resulting in the formation of TLRs heterodimer, such as TLR2-TLR1/TLR2-TLR6/TLR7-TLR8 heterodimer or TLR4/TLR9 homodimer, which induces the recruitment of the TIR domain-containing adaptor protein (TIRAP)/MyD88/interleukin-1 receptor-associated kinase-1 (IRAK-1)/IRAK2/IRAK-4 complex. After that, the complex continues to activate tumor necrosis factor receptor-associated factor 6 (TRAF6) and subsequent transforming growth factor-beta-activated kinase 1 (TAK1), TAK1-Binding Protein-1(TAB1) and TAK1-Binding Protein-2(TAB2), leading to the activation of mitogen-activated protein kinases (MAPKs, including subsequent activation of P38, ERK, and JNK) and nuclear factor-kappa B (NF-κB) signaling pathway, and promoting the production of pro-inflammatory cytokines. In the MyD88-independent signaling pathway, the activation of TLR3 or TLR4 can recruit TRIF. In particular, TLR4 requires a TRIF-related adaptor molecule (TRAM) for the activation of TRIF. Then, TRIF activates receptor-interacting protein 1(RIP1) and interacts with TRAF6 to promote subsequent inflammation signaling pathways. In addition, TRIF activates TRAF3, which in turn induces the activation of IRF3 and IRF7 to produce IFN-β and Type I IFNs respectively. Finally, TLR7, TLR8, TLR9 activate TRAF3 or TRAF6 and subsequent signaling pathways through the MyD88-dependent pathway.
When the LRR region of TLR bounds a ligand in the MyD88-dependent pathway, the TIR of the TLR receptor becomes allosteric and recruits MyD88 by interacting with the TIR domain-containing adaptor protein (TIRAP), also known as the MyD88 adaptor-like (Mal) protein (33). The binding of the C-terminal region of MyD88 results in a structural deformation of the N-terminal region of MyD88, which in turn activates the IL-1 receptor-associated kinase-4 (IRAK-4) participating in the recruitment, phosphorylation and degradation of IL-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2. Then, IRAKs and MyD88 combine the tumor necrosis factor receptor-associated factor 6 (TRAF6) (1, 34, 35). TRAF6 is needed to generate the Lys63-linked ubiquitin (K63-Ub) chains, which activates the complex comprising TGF-β activated kinase 1 (TAK1), TAK1‐binding protein 1 (TAB1) and TAB2 (1, 9, 35). One pathway leads to the activation of the activator protein 1 (AP-1) through activating mitogen-activated protein kinase (MAPK). MAPK comprises three subfamilies: extracellular signal-regulated kinase (ERK) 1 and 2, c-Jun N-terminal kinase (JNK) 1 and 2, as well as p38 (36). The second pathway leads to the activation of the inhibitor of kappa B kinase (IKK) complex (IKK-α, IKK-β, and IKK-γ), causing the phosphorylation of the inhibitor of the nuclear factor κappa B (IκB) protein (1) (Figure 1). Finally, the nuclear factor κappa B (NF-κB) is translocated into the nucleus, where it initiates the transcription of inflammatory cytokines and molecules, such as IL-1β, IL-6, IL-8, IL-12, IL-17, tumor necrosis factor (TNF)-α, interferon (IFN)-γ, inducible nitric oxide synthase (iNOS) and intercellular adhesion molecule-1 (ICAM-1) (9, 37).
In the MyD88-independent pathway, the TIR domain of TLR3 can directly interact with TRIF. However, TLR4 requires TRAM for the activation of TRIF (38). Subsequently, receptor-interacting protein 1 (RIP1) and TRAF6 interact with TRIF to activate NF‐κB to induce proinflammatory cytokines production. In addition, TRAF family members-associated NF-κB activator (TANK) binding kinase 1 (TBK1) alongside TRAF3 phosphorylates interferon regulatory factor-3 (IRF-3) and interferon regulatory factor-7 (IRF-7) to induce the expression of IFN-β and Type I IFNs (1, 38). TLR7, TLR8 and TLR9 activate TRAF3 or TRAF6 and the subsequent signaling pathways through the MyD88-dependent pathway, induce the production of Type I IFNs and inflammatory cytokines (39) (Figure 1).
Neuroimmune diseases are characterized by inflammation associated with neuron or axonal damage, loss of myelin sheath, and damage of neuromuscular junctions. Inappropriate or excessive activation of TLR signals may lead to neuroimmune diseases. Recent findings on the role of TLR in MS, NMOSD, GBS and MG will be discussed.
MS is a progressive autoimmune disease of the central nervous system (CNS), characterized by various clinical manifestations including motor and sensory deficits, visual disturbances and autonomic dysfunction (40). Experimental autoimmune encephalomyelitis (EAE) is a widely used animal model of MS. Pathological hallmarks of MS/EAE are composed of monocytes, CD4+ and CD8+ T cells, and B cells surrounding the venules and mediating myelin disintegration, axon loss and neuronal damage (41, 42).
TLRs play a crucial role in the pathogenesis of MS. Malika and colleagues (43) demonstrated that the expression of TLR3 and TLR4 was significantly increased in the areas surrounding inflammatory vessels and the center of MS lesions (43). TLR2 and TLR4 are actively related to the pathogenesis of MS. For example, the expression of TLR2, TLR4, and TLR9 on CD4+ and CD8+ T cells was significantly higher in patients with relapsing remitting MS (RRMS) than healthy individuals (44). The proportion of TLR+ (including TLR2, TLR4, and TLR9) Th17 cells and Tc-17 cells producing IFN-γ or IL-6 were positively correlated with the number of active brain lesions and neurological dysfunction by evaluating with expanded disability status scale (EDSS) and the number of active brain lesions by magnetic resonance imaging (MRI) scan (44) (Figure 2). Interestingly, the ligand of TLR2 (Pam3Csk4) induces more proinflammatory cytokines including IL-6, IFN-γ, IL-17 and GM-CSF than the ligand of TLR4 (LPS) and TLR9 [oligodeoxynucleotide (ODN)] from CD4+ and CD8+ T cells of MS (Figure 2). CD4+ T cells activated by Pam3Csk4 are more closely related to MS disease activity (44). Stimulation of TLR2 agonist promoted the differentiation, proliferation of Th17 cells in vitro, while inhibiting the expression of TLR2 on CD4+ T cells dramatically relieved the symptoms of EAE (45). The ligands lipopeptides and LPS bind to the TLR2 and TLR4 respectively, which adversely affect MS by increasing the production of IL-1β, IL-6 and IL-23 in antigen presenting cells (APCs) (46) (Figure 2). The expression of TLR2 in Treg cells from MS patients is significantly increased, shifting the Treg/Th17 balance towards a proinflammatory state and then promoting the progression of MS (47) (Figure 2). Furthermore, TLR2 expression is also upregulated in oligodendrocytes of MS, which inhibits the maturation of oligodendrocyte precursor cell (OPC) and remyelination through the activation of the TLR2-MyD88 signaling pathway (48) (Figure 2). Interestingly, systemic TLR2 tolerance induced by injecting low-dose Pam2CSK4 significantly enhanced remyelination in a preclinical model of MS, which resulted in the transformation of microglia from pro-inflammatory iNOS+ phenotype to non-inflammatory/pro-repair Arg1+ phenotype (49) (Figure 2). TLR2 in peripheral blood mononuclear cells (PBMCs) and CD14+ monocytes had a strong responsiveness to Pam2CSK4 stimulation in MS, suggesting that a high activity of TLR2 in MS may contribute to the pathogenesis of MS (50). The increased frequency of Th17-like cells expressing TLRs are involved in the pathogenesis of RRMS (46). The expressions of TLR2 and TLR4 on Th17 (IL-17+CD4+ T cells)/Tc-17 (IL-17+CD8+ T cells) were significantly upregulated in MS with major depressive disorder (MDD) compared with MS (46). Importantly, the selective serotonin reuptake inhibitors (SSRIs) effectively suppressed the expression and immune responsiveness of TLR2 and TLR4 on Th17/Tc-17-like cells (46). In summary, TLR2 plays a crucial role in promoting MS and inhibiting TLR2 can exert therapeutic effects on the animal model of MS.
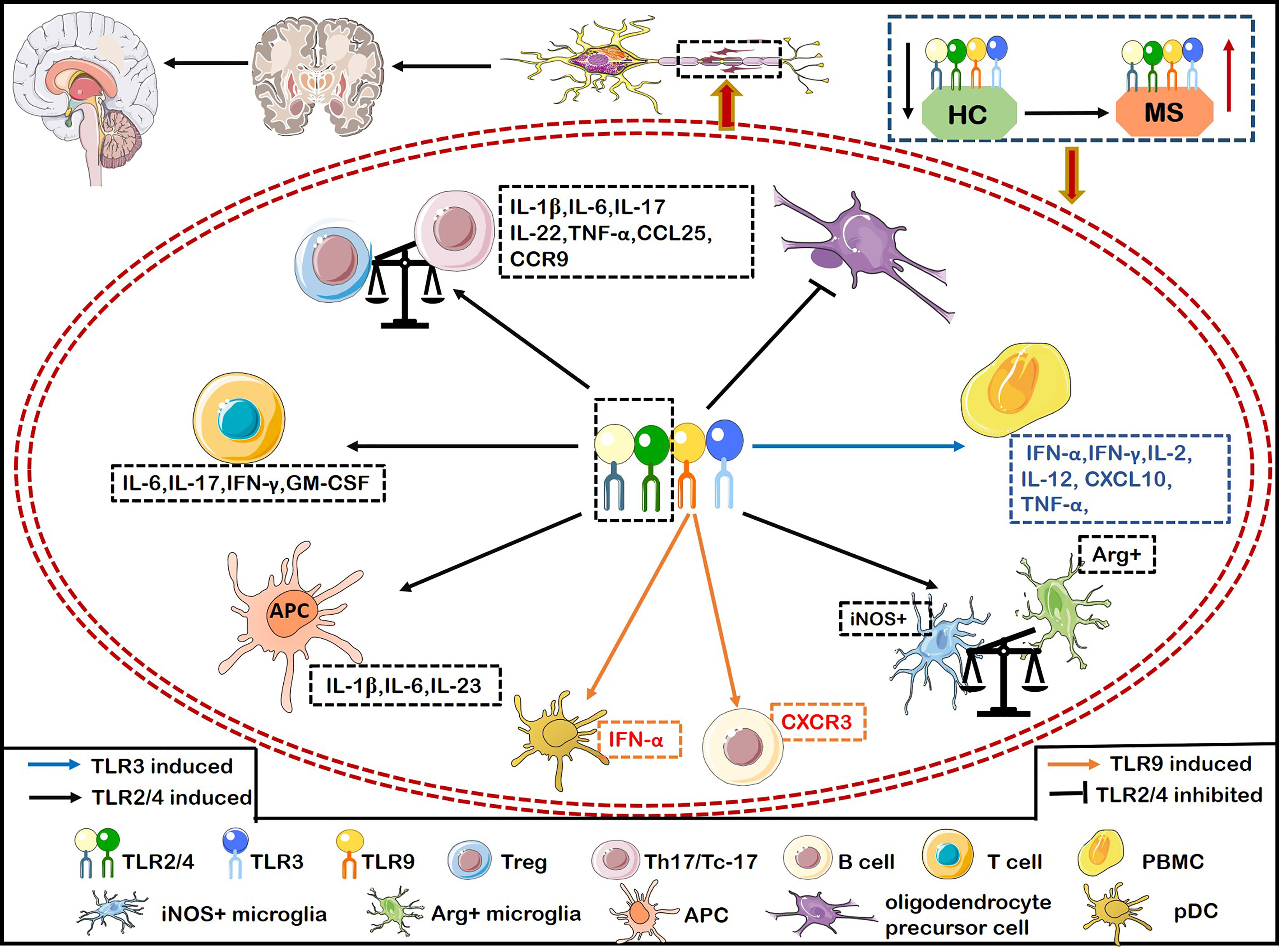
Figure 2 The inflammatory role of TLRs causes demyelination and hinders myelin regeneration in MS. Activation of TLR2/4 on T cells from MS induces the secretion of IFN-γ and IL-6, and activation of TLR2 alone also induces the secretion of IL-6, IFN-γ, IL-17, and granulocyte-macrophage colony-stimulating factor (GM-CSF). Activation of TLR2/4 on APCs induces more productions of IL-1β, IL-6, and IL-23. The expression of TLR2 on Treg breaks the balance of Treg/Th17 to exacerbate MS by transforming into a pro-inflammatory Th17-like phenotype. TLR2 is also up-regulated in oligodendrocytes of MS patients, which inhibits the maturation of oligodendrocyte precursor cell (OPC), resulting in the failure of remyelination in MS. The up-regulated expression of TLR2 on microglia increases the iNOS+ phenotype and decreases the Arg+ phenotype. The increased expression of TLR2/4 on Th17/Tc-17 cells promotes the up-regulation of IL-1β, IL-6, IL-17, IL-22, and TNF-α. Meanwhile, TLR4 expression on Th17 promotes the secretion of IL-17, IFN-r, CCL25, and CCR9, thereby aggravating inflammation and infiltration into CNS. Activation of TLR3 on PBMCs upregulates the production of IFN-α, IFNγ, IL-2, IL-12, TNF-α and CXCL10 to aggravate MS. Activation of TLR9 on B cells significantly increases the expression of T-bet to enhance the pathogenicity of B cells, and up-regulates the expression of CXCR3, thus promoting the inflammatory response of peripheral blood B cells and infiltrating into the CNS in MS.
TLR4 expression was significantly increased in bone marrow mesenchymal stem cells (MSCs) and peripheral monocytes of MS, activating its downstream molecules STAT-1, NF-κB, P38, JNK and CREB, increasing the production of CXCL10 and promoting inflammatory responses (51). Pertussis toxin (PTX), an adjuvant in inducing EAE, depended on TLR4 signaling molecules to facilitate T cell infiltration into the CNS (52, 53). Stimulating TLR4 on DCs drives the pathogenic function of T cells and assists the establishment of EAE model (54). Mice having TLR4-/-CD4+ T cells showed inadequate EAE induction, mild clinical symptoms and few demyelinating lesions (55). The effects may be attributed to inhibiting immune effects of Th17 and Th1 cells and reducing the secretion of IL-17 and IFN-γ (55). Furthermore, the inhibition of TLR4 also downregulated the expression of CCL25/CCR9 on Th17 cells, then reducing the migration and infiltration of Th17 cells into the CNS (56). A recent study showed that simultaneous activation of TLR3 with TLR2 or TLR4 in microglia caused severe neural network dysfunction by disrupting reactive oxygen and nitrogen species (oxidant-producing enzymes, inducible NO synthase and NADPH oxidase) (57). Accumulating data showed that a significantly higher abundance of both pDCs and conventional DCs (cDCs) was detected in MS. More importantly, the altered pDCs and (cDCs) are paramount to pro-inflammatory T cell response (58). Kristof et al. found that MS-derived pDCs and cDCs stimulated by TLR4 ligand, LPS, combined with IFN-γ significantly upregulated the secretion of IL-12p70 in vitro, which is important for the commitment to polarize Th1 cells (59, 60). However, no difference in TLR7 ligand, IQ-induced IFN-α secretion from pDCs and cDCs were observed between MS and healthy controls (60). In addition, the expression of CD86 on cDCs was significantly up-regulated after TLRs stimulation in MS, while no difference existed in CD80 and CD86 expression of pDCs, indicating that cDCs were in a more activated state in MS (60). The amount of investigations into the role of TLRs expressed in cDCs has escalated in recent years (61, 62), however, studies focusing on the role of TLRs expressed in cDCs in MS/EAE are sparse. Both animal and clinical studies are warranted in the future to explore the detailed mechanism which could provide an insight to a better therapeutic approach.
The role of TLR4 is not limited to promote the occurrence and development of MS. Jamie and colleagues found that TLR4 activation enhanced phagocytic activity of macrophages (promoting the clearance of myelin debris) following TLR4 agonist E6020 stimulation both in vivo and in vitro (63). TLR4 blocking inhibited microglial ability to phagocytose axon debris, which was harmful for axon outgrowth (64). Yoichiro et al. confirmed that there is a synergistical co-stimulation between TLR4 and CD40 on B cells, which increases the secretion of IL-10 during the relapse phase of MS (65). The cause of these paradoxical effects of TLR4 remains unclear. We propose that it may be related to different cellular sources of TLR4, different immune microenvironments in different stages of MS, or the cross-talk between different costimulatory molecules [CD40 and DC-specific intercellular adhesion molecule-3-grabbing nonintegrin (66)] and TLR4.
TLR3, TLR7, TLR8 and TLR9 also play an irreplaceable role in the pathogenesis of MS. Elie et al. detected TLR3 polymorphism in NK cells from MS patients, and they found that the rs3775291 allele was significantly different between MS and healthy controls (HC), and the rs3775291 (C/T or T/T) increased the incidence of MS by 71% compared with the homozygous genotype (C/C). They proposed that TLR3 mutations in NK cells are associated with MS susceptibility (67), whether the same conditions can be observed in humans in other regions or countries remains to be studied. Polyinosinic-polycytidylic acid [Poly(I:C)], a TLR3 agonist, can suppress demyelination in EAE by inducing endogenous IFN-β and peripheral CC chemokine CCL2 production (68). TLR3/MyD88 independent pathway promotes the secretion of IL-27 to suppress the mature of Th17 cells (69). The lack of Cathepsin H damages TLR3-mediated IRF3 activation, inhibits IFN-β secretion from DCs and promotes Th1 cell differentiation (70). Activation of TLR3 in astrocytes induces the expression of neuroprotective mediators, including anti-inflammatory cytokines IL-9, IL-10, and IL-11, and down-regulates the secretion of pro-inflammatory cytokines IL-12 and IL-23, and inhibits gliosis and promotes neuronal survival, angiogenesis, and myelin regeneration (71, 72).
Most previous studies suggested that TLR3 mediated the neuroprotective response during inflammatory process. However, a recent study reported that TLR3 activated by polyinosinic-polycytidylic acid on proinflammatory cytokine-pretreated astrocytes significantly promoted the production of fibronectin aggregation and led to remyelination failure in MS (73). One potential mechanism is that cytokine-induced an increase in relative mRNA of EIIIApos-Fn over EIIIBpos-Fn and a Poly(I:C)-mediated decreased in integrin affinity, which destroyed fibronectin fibrillogenesis on the cell surface (73). A similar role of TLR3 in PBMCs to promote the development of MS was recorded (74). TLR3 of PBMCs from secondary progressive MS (PMS) and benign MS (BMS) was stimulated by Poly(I:C). The mRNA expression levels of IFN-α, IFN-γ, IL-2, IL-12, TNF-α and CXCL10 in PBMCs from PMS were significantly increased (74) (Figure 2). Remarkably, the activation of TLR3 on PBMCs from BMS patients increased the expression of TNF-α and CXCL10, while the activated TLR3 also significantly upregulated the expression of the specific chimera SARM-1 (a negative regulator of TLR3-mediated immune response, inhibits TRIF, NF-κB, and IRF) that down-regulating pro-inflammatory cytokines (74). These results indicated that TLR3 in PBMCs plays different or even opposite roles in different MS disease stages. Although most of the early studies supported TLR3 activation exerting neuroprotective effects in the pathogenic courses of MS/EAE, we can see that TLR3 also appears to promote the pathogenesis of MS, which may be related to the activation of different cell types, ligands, different disease stages and different signaling pathways. Still, the priming conditions of activation need to be explored.
Functional defects of TLR7 on pDCs may inhibit the secretion of IFN-α by pDCs in MS, disrupting proper control of pDCs in the T-cell mediated autoimmunity (75). The expression of TLR7 mRNA in PBMCs and monocytes of MS was damaged (TLR9 signal damage was not observed), which inhibited the secretion of IL-6 and B cell activating factor (BAFF) (76, 77) that regulates the survival, differentiation and class switching of B cells. It is critical for the maintenance of peripheral B cell pool and the initiation of B-cell responses (78). However, these effects could be rescued following IFN-β treatment (79). From this perspective, IFN-β therapy and TLR7 re-activation are not conducive to suppress the immune response of MS. Yet, it has also been speculated that this mechanism may reduce virus-triggered relapses in MS (80). The activation of TLR7 on B cells in MS has an immunosuppressive effect (81). The inadequate expression of IFNAR1/2 and TLR7 genes in B cells are involved in decreased endogenous IFN-β secretion in RRMS. IFN-β therapy in combination with TLR7 or TLR9 agonist (Loxorubin/CpG) induced a high endogenous IFN-β expression in B cells and increased IL-10, TGF-β and IL-27 secretion in RRMS (81). TLR7-driven B cells have an abnormal immune function in MS, however, thymosin-α1 (Tα1) improved this dysregulation by reducing the production of IL-6, IL-8 and IL-1β and increasing IL-10 and IL-35 secretion (82). Furthermore, Tα1 promoted the differentiation of regulatory B cells, thereby dampening autoimmune inflammation in MS (82). In summary, TLR7 plays a double-sided immunoregulatory role in MS depending on the specific cell type.
It has been reported that damaged TLR8 signaling pathway in MS could impair the production of IL-12, suggesting that TLR8 deficiency in MS may contribute to autoimmunity (83). TLR8 deficiency leading to autoimmunity was also noted in mice (84). TLR8 participates in axonal injury by increasing the infiltration of neutrophils and leukocytes in EAE, with the signaling pathway being remained active even after focal inflammatory infiltration disappeared (85). 1,25-Dihydroxyvitamin D3 (1,25(OH)2D3) plays an anti-inflammatory role in EAE by inhibiting TLR8 in monocytes and downstream cascade signals such as MyD88, IRF-4, IRF-7 and NF-κB (86).
Dominguez et al. (87) suggested that the gene polymorphism of TLR9 (rs352162 and rs187084) is be involved in different clinical stages of MS. TLR9 on pDCs was predominantly expressed in the leptomeninges and demyelinating lesions of MS patients and enhanced the secretion of type I IFNs and IFN-α to exacerbate MS (88) (Figure 2). The pDCs activated with TLR9 agonists promoted both Th1 and Th17 responses and further induced EAE (89). IFN-β therapy inhibited the expression of TLR9 on pDCs, which reduced the secretion of IL-6, TNF-α and IFN-α and decreasing the pDCs activated by pathogens (88). Besides, TLR9 activation increased the expression of TLR-1, -2, -4, -5 and -8 in PBMCs from MS (90). This reflects the cross-regulation among members of the TLR family. TLR9−/− mice have an incremented IL-6 production by splenocytes (91) and then decreased EAE severity (92). In fact, APCs have the ability to maintain immune tolerance when facing foreign antigen stimulation, avoiding potential autoimmune reaction (93). The activation of TLR9 on APCs can break the immune tolerance state of APCs and promotes the differentiation of T lymphocytes to Th1 cells, thus inducing the occurrence of EAE (89, 94). The expression of T-bet was significantly increased when B cells were stimulated by IFN-γ and TLR9 agonist in MS, which increased the pathogenicity of B cells. The expression of CXCR3 receptor was also obviously upregulated, which promoted the infiltration of peripheral blood B cells across the BBB to the CNS (95) (Figure 2).
It seems that TLR9 plays an immune-boosting role in MS/EAE. However, recent studies displayed its immunosuppressive role in MS/EAE. Decreased expression of TLR9 on memory B cells derived from MS significantly declined the production of TLR9-mediated IL-10 by B cells (96). Activation of TLR9 up-regulates the expression of downstream molecules MyD88, TRAF6 and IRF8 promoting the development and expansion of Breg/B10 cells and the secretion of IL-10 (97). Peripheral circulation CD45 cells were recruited into the CNS when TLR9 agonist CpG oligonucleotide being intrathecal injected into EAE mice, which produced a large amount of IFN-β with immunomodulatory effect to alleviate EAE (98). Equivalently, intrathecal injection of MIS416 (a TLR9 and NOD2 bispecific innate ligand) into EAE mice up-regulated type I and II IFN, IL-10, Arg-1, CCL-2 and CXCL-10, increased the proportion of myeloid and NK cells, and reduced inflammatory T cells, which alleviated the demyelination of EAE (99).
We summarized the “pro-inflammatory” role of TLRs on various immune cells of MS, which was presented in Figure 2. Collectively, the dual role of TLRs in MS has been gradually explored. Firstly, the cellular type of expressing TLRs is a critical factor and it would be meaningful to focus on more accurate cell subtypes to explore their roles in the future. Secondly, it may be related to disease stages and activation of different signaling pathways caused by diverse TLRs ligands. Thirdly, the cross-talk between different co-stimulatory molecules on the same cell or different cells may also determine the immunomodulatory role of TLRs. It should be admitted that the contradictory effects of TLRs are the key to hinder the application of TLRs in clinic, therefore, it is necessary to clarify the causes and conditions of the contradictory mechanism.
NMOSD is an antibody -mediated autoimmune diseases in the CNS characterized by inflammation, demyelination and axonal damage (100, 101). Because of the discovery of aquaporin-4 immunoglobulin G antibodies (AQP4-IgG), clinical and laboratory-based investigations have indicated that B cells are one of the fundamental roles in NMOSD immunopathology (102, 103). Th17 cells and their related cytokines, such as IL-17 and IL-21 are also involved in the development of NMOSD by accelerating the breakdown of the BBB, facilitating inflammatory cells infiltration into CNS lesions, and collaborating with B cells to release AQP4-IgG (104). Like other neuroimmune diseases, the pathogenesis of NMOSD is associated with a number of environmental and hereditary susceptibility. Growing evidence indicated that viral and bacterial infections were associated with NMOSD (105, 106). These pathogens that caused NMOSD were recognized by TLRs and triggered the secretion of proinflammatory factors. TLRs are typically expressed in human CD4+ T cells. Barrosa et al. (107) found that the expression of TLR2, TLR4 and TLR9 on non-activated CD4+ T cells of NMOSD patients was significantly increased compared with the healthy individuals. Remarkably, the co-expression of IL-17 and IL-6 were significantly enhanced in the high expression of TLR2, TLR4, and TLR9 of Th17 cells from NMOSD, and it was positively correlated with the EDSS score. The high expression of TLRs in Th17 cells aggravated the neurological dysfunction of NMOSD through the increased secretion of pro-inflammatory cytokines (107). Furthermore, IL-17 expression was significantly increased in the high expression of TLR2 and TLR9 from activated CD4+ T cells, thus promoting inflammatory responses in NMOSD (107).
Intriguingly, the high expression of IL-10 in TLR2+ Treg cells (non-classical IL-10+IL-17+Treg cells) was found only in patients with mild neurological dysfunction of NMOSD, while a significantly lower proportion was found in patients with severe neurological dysfunction (107). This supports that function of Treg cells in NMOSD is damaged (insufficient IL-10 secretion), thus leading to sustained neuroinflammation (107–110). Interestingly, the later study found that agonists of TLR9 induced IL-10 secretion from NMOSD-derived Treg cells, while TLR2 agonist did not (111). Paradoxically, TLR2 knockout animal model of psoriasis significantly weakened the mRNA expression of foxp-3 and IL-10, inhibited the proliferation of Treg cells and exacerbated psoriasiform skin inflammation. Correspondingly, the use of TLR2 agonists promoted the production of Treg cells and the secretion of IL-10 (112). Brittney et al. also reported that the nucleic acids released by bacteriolysis triggered IL-10 secretion mostly dependent on TLR2 activation (113). The contradictory role of TLR2 in Treg cells may be related to different diseases, however, there is still lack of studies on the role of TLRs in NMOSD.
Dias et al. (111) evaluated the direct effects of different TLR ligands on CD4+ T cells form NMOSD and healthy individuals, their results suggested that the agonists of TLR2 (Pam3C), TLR4 (lipopolysaccharide) (LPS) and TLR5 (FLA), but not TLR9 (ODN), elevated CD4+ T cells expansion in NMOSD patients. As expected, Pam3C, LPS, FLA and ODN did not show the obvious CD4+ T cells proliferative activities in the healthy individuals (111). Besides, they found that all TLRs agonists induced the release of IL-6, IL-17 and IL-21 by CD4+ T cells without extra stimuli, and with TLR2 and TLR4 agonists being the most effective. Of note, this still only occurred at NMOSD-derived CD4+ T cells (111). This indicates that the TLRs pathway on CD4+ T cells has altered in NMOSD patients, at least in terms of its activity. Additionally, the agonists of TLR2 and TLR4 increased the production of Tfh cells and promoted the secretion of IL-21, which was positively correlated with neurological dysfunction of NMOSD (111).
AQP4-IgG-mediated complement-dependent astrocyte injury is recognized as the core of the pathogenesis of NMOSD (114). Kazuya et al. found that damaged astrocytes caused by AQP4-IgG can release large amounts of mitochondrial DNA (mtDNA), which promotes the release of IL-1β from mononuclear cells through the activation of TLR9 and NLRP3 inflammasome-dependent manner. Subsequently, it leads to activating leukocytes, destroying BBB and promoting the migration of monocytes into the CNS (115). A later study showed that released mtDNA from damaged astrocytes by AQP4-IgG can further induce the generation of CCL2 from astrocytes (116). mtDNA acts as a molecular bridge of innate immunity and then activates monocytes by activating TLR9, and CCL2 induces monocytes to migrate into the CNS (116).
Studies on the role of TLRs in NMOSD are not as extensive as those in MS. The roles of TLRs in B cells, DCs, and microglia/macrophages of NMOSD patients still remain unknown. TLRs are actively involved in the pathogenesis of NMOSD, and further research is required to clarify cellular and molecular mechanisms and it may shed light on novel therapeutic approaches for NMOSD.
GBS is an immune-mediated demyelinating disorder of the peripheral nervous system (PNS). Clinical symptoms are characterized by tingling, limb weakness, autonomic dysfunction and numbness (117). Experimental autoimmune neuritis (EAN) is an animal model of GBS. The pathogenesis of GBS/EAN involve a variety of immune cells (such as T cells, B cells and macrophages) and a complex network of cytokines (118).
The TLR4 gene polymorphism (Asp299Gly) is closely related to an increased susceptibility to GBS (119). Apart from Asp299Gly, the Thr399Ile polymorphism is also associated with the incidence of acute motor axonal neuropathy (AMAN), a subtype of GBS (119). Anti-TLR4 antibodies interdicted the processes of demyelination of the PNS by inhibiting monocyte chemoattractant protein-1 production from Schwann cells (120). Du et al. detected mRNA levels of TLR2, TLR4, MyD88 and NF κB in PBMCs from patients with GBS, and found that they were significantly higher than healthy controls. Moreover, PBMCs from GBS produced more TNF−α and IL−1β after stimulation with TLR2 and TLR4 agonists (PGN and LPS), indicating that TLR2 and TLR4 expression on PBMCs is involved in the pathogenesis of GBS (121). In addition to TLR2 and TLR4 in GBS, TLR9 expression was also increased in PBMCs, which promoted the secretion of IFN-γ and positively correlated with the degree of disability of GBS (122). Paradoxically, Gries et al. evaluated TLR9 mRNA in CD4+ T cells and found no difference between GBS patients and healthy controls. The discrepancy may be caused by different time points of collecting PBMCs and in inconsistent disease stages of GBS. Furthermore, TLR9 may be highly expressed in PBMCs except for CD4+ T cells (123). It was noted that tlr9 mRNA was upregulated in the spleen, sciatic nerve, PBMCs and lymph nodes throughout the course of EAN, suggesting that it may be involved in the pathogenesis of EAN in different disease phages (124). TLR9 can also promote the expression of IL-12 to induce the differentiation of Th1 cells, playing a role in the pathogenesis of GBS/EAN (89).
The expression of TLR2, TLR6 and TLR11 on CD4+ T cells, and TLR2, TLR4, and TLR6 on the major histocompatibility complex class II positive (MHCII+) APCs were significantly upregulated in the acute phase of GBS and EAN, while the expression of TLR1 was decreased and the secretion of IL-17A was enhanced (123). Significant upregulation of TLR2 was also observed in sciatic nerves of EAN rats, which correlated with the disease severity (125). Peripheral TLR2 signaling pathway promotes the upregulation of IFN-γ, IL-6, and IL-17 secretion, which work coordinately to increase peripheral nerve inflammation and damage the myelin sheath and axons (118, 126). The TLRs signaling can also induce the activation of self-reactive T or B cells and activate APCs through the MyD88-dependent or -independent pathways to trigger the adaptive immunity (127). Darabi et al. (128) demonstrated that APCs activated through the TLRs signal pathway, especially TLR4 and TLR9, can induce T cells differentiate into Th1 cells and result in tissue destruction. Th1 is not sufficient to induce autoimmune pathology without induction by TLR9-activated APCs. Cross-reactivity between immune cells triggered by microbial infection is critical for autoimmune response. TLRs connects microbes and immune cells, especially under the cytokine storm, microorganisms activates APCs by activating TLRs to activate the third signal, further cross-reacting with T cells to induce and activate autoimmune T cells, triggering an autoimmune response (128).
GBS is triggered by a variety of infectious or noninfectious agents (129). The Gram-negative Campylobacter jejuni (C. jejuni) is now recognized as the primary trigger of GBS. Most infections are acquired from eating raw or undercooked poultry, unpasteurized milk and contaminated water (130). It produces a variety of glycoconjugates, including human ganglioside analogs and multiple activators of TLRs, and targets MyD88, TRIF, macrophage galactose-type lectin (MGL), etc., which induces autoimmune diseases (131). Molecular mimicry between sialylated lipooligosaccharide (LOS) structures of C. jejuni and ganglioside epitopes on the human nerves that generate cross-reactive immune response results in an autoimmune attack on the myelin or axon of peripheral nerves in GBS (130). C. jejuni is resistant to proteinase digestion, inducing the activation of neutrophils and macrophages and activating NF-κB through TLR2 and TLR4 (132, 133). The initiation of GBS by C. jejuni was strongly dependent on its pathogenic LOS structure, which triggers the innate immune system through TLR4 signaling (133). Zeb et al. developed a vaccine against C. jejuni infection by genome-wide screening. The vaccine interacts with TLR4 to trigger the release of primary and secondary immune factors to enforce humoral immune response against C. jejuni, thereby preventing GBS (134).
Zika virus (ZIKV) has emerged as a public health threat due to its teratogenic nature and associated with the occurrence of GBS (135, 136). The mechanism of ZIKV infection causing GBS is unclear. TLRs, autophagy, apoptosis and unfolded protein response (UPR) pathways are considered as a potential mechanism (137). TLR3 could be activated by ZIKV by sensing the replication intermediate of viral RNA and was upregulated in human organoids and mouse neurospheres after ZIKV infection (138). Activated TLR3 triggered the production of proinflammatory cytokines during ZIKV infection, which upregulated the STAT3 pathway and reduced the STAT1 phosphorylation in a suppressor of cytokine signaling (SOCS)-3 dependent manner, thereby inhibiting interferon response triggered by RIG-I-like receptors (RLR) and reducing the antiviral effect (139). However, the antiviral cytokine response was enhanced following the inhibition of TLR3, while the production of proinflammatory cytokines was decreased. The cross-talk between the antiviral (RLR) and inflammatory (TLR) responses may further induces GBS (139). In addition, the TLR7/8 agonist R848 blocked the ZIKV replication in monocytes (140). Whether other TLRs can be activated by ZIKV have not yet been established. We speculate that ZIKV may be involved in the pathogenesis of GBS through TLRs. In addition, there is no vaccine against ZIKV so far. In the future, the relationship between ZIKV and TLR3 may be conducive to the development of an effective anti-ZIKV vaccine to prevent GBS.
Macrophage migration inhibitory factor (MIF) is critically involved in the pathogenesis of GBS/EAN (141). It promotes the recruitment of macrophages to the PNS and the expression of proinflammatory cytokines, including TNF-α, IL-6, IL-8, and IL-12 to damage myelin and axonal (142). However, the role of MIF begins with upregulating the expression and activation of TLR4 then promotes the translocation of NF-κBp65 into the nucleus through the MyD88-dependent/independent pathway (142, 143). Altogether, these results indicate that TLRs signaling contribute to the pathogenesis of GBS.
MG is an acquired autoimmune disease characterized by neuromuscular junction transmission dysfunction, with main manifestations being fluctuating skeletal muscle fatigue, post-activity worsening symptoms associated with a reduction of acetylcholine receptor (AChR) clustering (144) and thymic hyperplasia featured with ectopic germinal center (145). Similar to other neuroimmune diseases, MG is associated with both humoral and cellular immunity (146). The exact mechanisms remain obscure and may be related to genetic and environmental factors. Evidence with chronic inflammation, TLRs activation, and persistent viral infection in MG is accumulating (147).
Wang et al. (148) detected the mRNA expression of whole TLRs in the PBMCs of both MG patients and healthy controls. They observed that all TLRs expression, except for TLR7, in MG patients were significantly different from those in the healthy controls. The levels of TLR1, TLR6 and TLR10 were considerably lower, whereas TLR2, TLR3, TLR4, TLR5, TLR8 and TLR9 mRNA were significantly upregulated in PBMCs from MG as compared to healthy controls. It is worth mentioning that the expression level of TLR9 mRNA has an evident positive relation with the clinical severity of MG. Thymic stromal cells such as thymic epithelial cells (TECs) and myoid cells, express all TLRs (149). TLR3 expression was higher in TECs cultures derived from the thymus of MG patients than healthy controls (16). Additionally, poly(I:C), the well-known agonist of TLR3, triggers the overexpression of α-AChR in TECs by releasing of IFN-β and anti-AChR antibodies (149). TLR4 overexpression and activation in MG TECs altered effector T cells (Teff)/regulatory T cells (Treg) balance, induced the production of Th17-related cytokines and drove DCs recruitment via overexpression of CCL17 and CCL22 to regulate immune cell trafficking in inflamed organs (150). TLR7 and TLR9 in MG thymus are capable of leading to abnormal B cells/plasma cells proliferation, maturation, and survival, as well as the provision of additional costimulatory signals. B cells escape from regulatory cell tolerance checkpoints depending on MyD88-depending way in MG patients once combined with the ligand like Epstein–Barr virus (EBV), which assists in the production of type I IFN and long-term inflammation (151, 152).
MiR-146a is a key modulator of innate immunity that orchestrates inflammatory signaling (153). As an inhibitor of the TLR pathway, miR-146a targets to inhibit TRAF6, IRAK1 and NF-κB to prevent inflammatory stimulation mediated by TLR overactivation (154). In addition, miR-146a deficiency promotes the activation of c-REL, which accelerates B cells proliferation, differentiation and germinal center (GC) development (155, 156). Nevertheless, miR-146a expression is significantly downregulated in hyperplastic thymus of MG and the levels of IRAK1, TRAF6 and c-REL were upregulated and negatively correlated with the level of miR-146a. Interestingly, miR-146a in the thymus of MG patients was upregulated after corticosteroid therapy, and insufficient expression of miR-146a in MG may lead to sustained TLR activation, impaired inflammatory resolution ability and thymic hyperplasia (156).
Experimental autoimmune myasthenia gravis (EAMG) is a classical experimental model for MG, which is induced by a purified antigen of AChR solubilized in complete Freund’s adjuvant (CFA) containing heat-inactivated mycobacterium tuberculosis (MTB). LPS, the TLR4 activator, has been confirmed to be efficient to replace MTB (16). TLR3’s agonist Poly(I:C) can induce transient phenomenon that express α-AChR, IFN-β and chemokines such as CXCL13 and CCL21, leading to the B-cell recruitment in EAMG (157). Although the EAMG is associated with muscle weakness due to anti-AChR antibody attack, it does not fully reproduce MG disease, because the thymus does not present ectopic GC development (158) [a classic pathological feature of MG: abnormal thymus with features of tertiary lymphoid organs, includes new angiogenesis processes, overexpression of inflammatory cytokines and chemokines, and invasion of B cells leading to ectopic GC development (159, 160)]. Fortunately, this was significantly reversed when TLR3 combined with TLR4 agonist was used in EAMG induction, however, the agonists of TLR7 and TLR9 do not have this effect. The agonist of TLR3 combined with TLR4 not only formed GC, but also prolonged symptom duration and increased CXCL13 secretion (157). Another contradictory research showed that inhibition of the TLR9 pathway by the oligodeoxynucleotide (ODN) H154 in EAMG decreased Tfh cells and B cells of GC, abated anti-AchR antibody production and terminally alleviated clinical symptoms (161). We propose that the agonists and inhibitors of TLR9 may not maintain completely opposite results. Although TLR9 does not contribute to the formation of ectopic GC, it acts on the inside of the Tfh and the B cells after the formation of GC. Overall findings indicated that inappropriate TLRs signaling activation may participate in the pathogenesis of MG/EAMG and more experimental studies are needed to confirm it.
The current treatments of neuroimmune diseases mainly include plasma exchange (PE), intravenous immunoglobulin (IVIg), application of glucocorticoid and immunosuppressive agents. Natalizumab prevents the infiltration of leukocytes into the CNS by inhibiting α4 integrin, thereby alleviating the autoimmune response of MS (162). However, the strong immunosuppression declined the ability of the immune system to monitor, potentially increasing the risk of progressive multifocal encephalopathy (PML) (163). Surprisingly, TLR3 agonists could re-establish CNS immune surveillance in EAE when α4 integrin was inhibited and reduced the risk of PML, suggesting that natalizumab therapy in combination with TLR3 agonists may restore a more appropriate immune balance in MS (163). Fingolimod (FTY720) is the first modulator of sphingosine 1-phosphate receptors (S1PR) to receive regulatory approval for relapsing-remitting MS. S1P1 is critical for the regulation of lymphocyte transport. However, Fingolimod reduces the outflow of lymphocytes from lymph nodes mediated by S1P, resulting in lymphocyte circulation depletion (164). TLR4 activation enhances the expression of chemokines through the transactivation of S1P-S1PR. FTY720 inhibited the synthesis and released proinflammatory chemokine CXCL5, CXCL10, and CCL2 that induced by TLR4 activation on astrocytes and microglia (165). Mycophenolate mofetil (MMF) is an inhibitor of inosine-5’-monophosphate dehydrogenase, which is a prodrug of mycophenolic acid (MPA). MMF inhibits the proliferation of T and B lymphocytes by depleting their guanosine nucleotides, weakening the immune response and antibody formation (166). A recent study has shown that MPA can dose-dependently downregulate the expression of CD80 and CD86 (markers of DC maturation) activated by TLR7 and TLR9 on mDCs and reduce IL-12 secretion in systemic lupus erythematosus (SLE). In pDCs, MPA inhibited IRF7 nuclear translocation and IFN-α secretion by strongly inhibiting AKT activity (167).
Despite above evidence, there is still a poor prognosis or serious sequelae after therapy for neuroimmune disorders. Exploring new therapeutic methods is still an urgent need. The receptors, adapter molecules and key kinases in TLRs signaling pathway can be considered as therapeutic targets. Development of TLRs antagonists is a promising direction in therapy neuroimmune diseases, which has been summarized in Table 1.
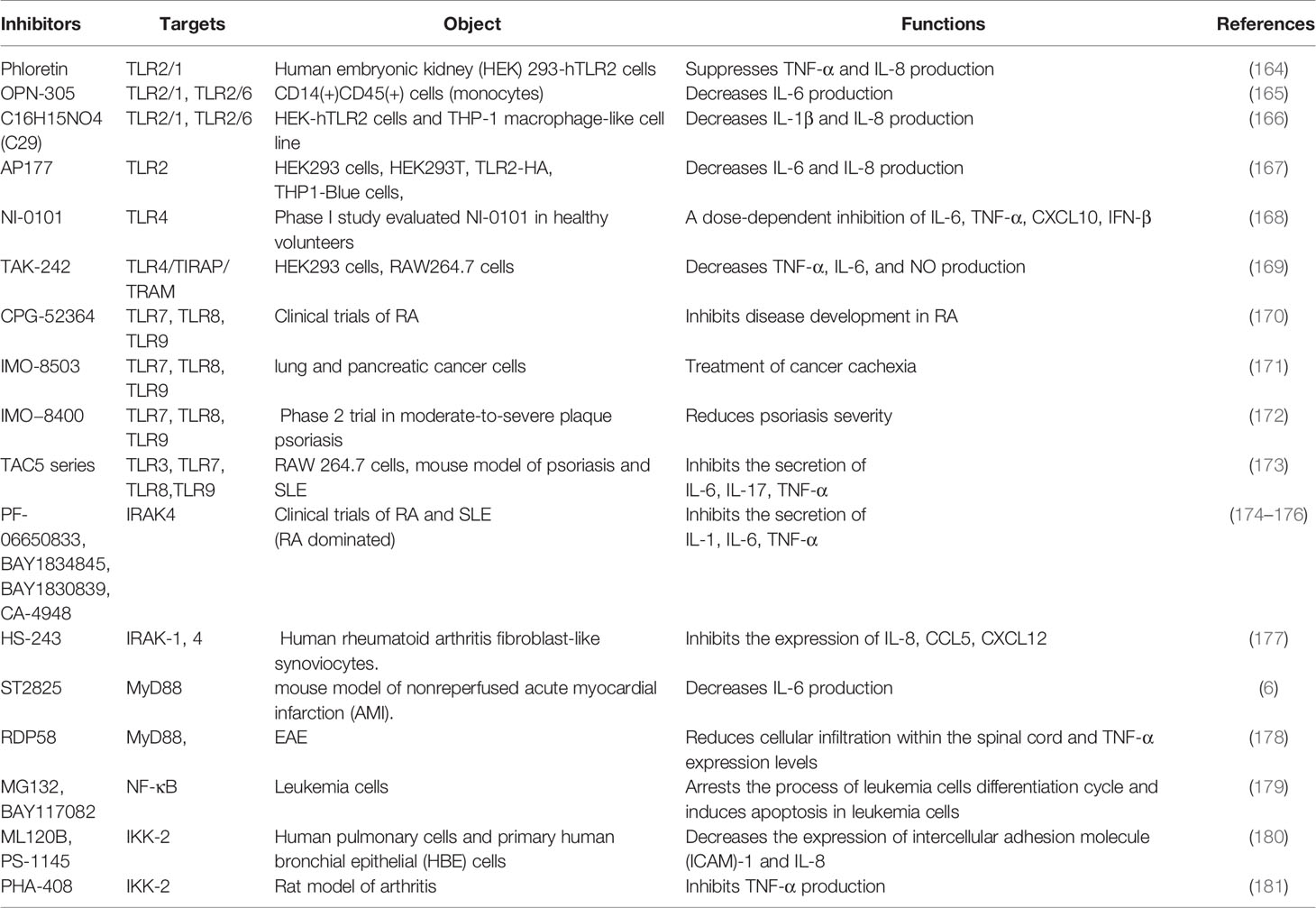
Table 1 Inhibitors of TLRs signaling pathway.
Phloretin is an inhibitor of TLR2/1 heterodimerization and can suppress the secretion of TNF-α by blocking the TLR2 signaling pathways. It is a naturally occurring dietary flavonoid that is abundant in fruits (168). OPN-305 is the first humanized IgG4 monoclonal antibody against TLR2 by targeting the ligand-binding site, preventing heterodimerization of the receptor with TLR1 or TLR6 and decreasing the production of IL-6 (169). Pragnesh et al. (182) found that the compound C16H15NO4 and a derivative ortho-vanillin inhibited the TLR2/1 and TLR2/6 signaling pathway induced by synthetic and bacterial TLR2 agonists in human HEK-TLR2 and THP-1 cells (182). AP177, an antagonist of TLR2, manifests a therapeutic potential in disordered TLR2 immune responses conditions by significantly inhibiting NF-κB activity and decreasing the secretion of cytokines IL-6 and IL-8 (183).
TLR4 is a promising therapeutic target for the treatment of neuroimmune diseases. NI 0101, an anti-TLR4 antibody, can potentially block any TLR4 ligands without obvious safety concerns. The phase I study of healthy volunteers confirmed that it inhibited the production of IL-6, TNF-α, CXCL10 and IFN-β in a dose-dependent manner with well-tolerated (170). TAK-242, a small molecule specific inhibitor of TLR4, disrupts the interactions of TLR4 with its adaptor molecules TIRAP and TRAM (171). However, the application of TAK-242 in phase III clinical trials for the treatment of severe sepsis was terminated, because it did not significantly decrease the cytokine levels in patients with severe sepsis and septic shock (69). Of note, Chaperonin 10 (Cpn10) was applied in a phase II trial in patients with MS. The expression levels of proinflammatory cytokines and new gadolinium-enhancing lesions were not significantly different between two groups with 5 mg or 10 mg weekly, respectively (172, 173).
CPG-52364, a derivative of chemical compound quinazoline with a small molecular weight, inhibits the activation of TLR7, TLR8, and TLR9 and is well tolerated in clinical trials of psoriasis and rheumatism (RA) (174). IMO-8503, another TLR7, 8, and 9 antagonist, can act as a potential treatment for cancer cachexia (175). IMO−8400, an oligonucleotide-based antagonist of TLRs 7, 8, and 9, can reduce psoriasis severity with severe adverse events (176). IMO−3100 (inhibits TLR7 and TLR9) and IRS 661 (inhibits TLR7) had been considered to be potential therapeutic compounds (18, 69). A new TAC5 series of compounds with small molecule (TAC5 and its derivatives TAC5-A, TAC5-C, TAC5-D and TAC5-E) were possible candidates for the treatment of autoimmune diseases, which effectively inhibited TLR3, TLR7, TLR8 and TLR9 signaling pathways, and significantly also inhibited the activation of NF-κB, decreased the phosphorylation of MAPK and declined the secretion of TNF-α and IL-6 (184). These findings indicate their enormous potential to treat neuroimmune diseases.
TLRs signaling pathways has been identified as a potential therapeutic target for neuroimmune diseases. The effects of molecular inhibitors and antibodies ameliorating the malfunction of innate immune caused by abnormal TLRs signaling pathway were explored (21). Inhibitors of IRAK4, such as PF-06650833, BAY1834845, BAY1830839, CA-4948 and MBS-986126, have been tested in autoimmune diseases (6, 177). Importantly, PF-06650833, BAY1834845, BAY1830839 and CA-4948 are now used in clinical trials of RA and SLE with therapeutic effects and a favorable safety (6, 177, 178). MBS-986126 reduced inflammation in preclinical models of autoimmune diseases, achieving remarkable results (179). HS-243, a highly effective IRAK inhibitor, selectively inhibits IRAK-1/4 (185). These data support a continued evaluation in clinical trials of TLRs inhibitors for the treatment of autoimmune diseases. ST2825 could inhibit MyD88 dimerization, interfering with the recruitment of IRAK1 and IRAK4 (180). RDP58 treatment decreased cellular infiltration in the spinal cord of EAE (181). The proteasome inhibitor peptide MG132 has been attributed to inhibiting NF-κB through the inhibition of IκB degradation. BAY117082 is an NF-κB inhibitor via inhibiting the IκB phosphorylation (186, 187). ML120B, PS-1145 and PHA-408 are the inhibitors of IKK-β which is a protein subunit of IκB kinase (188, 189). Taken together, many inhibitors targeting TLRs signaling pathways have been developed (Figure 3). However, few blockers have been used in clinical application for treatment in neuroimmune diseases. However, the blockers with minor side effects and high efficiency need to be developed.
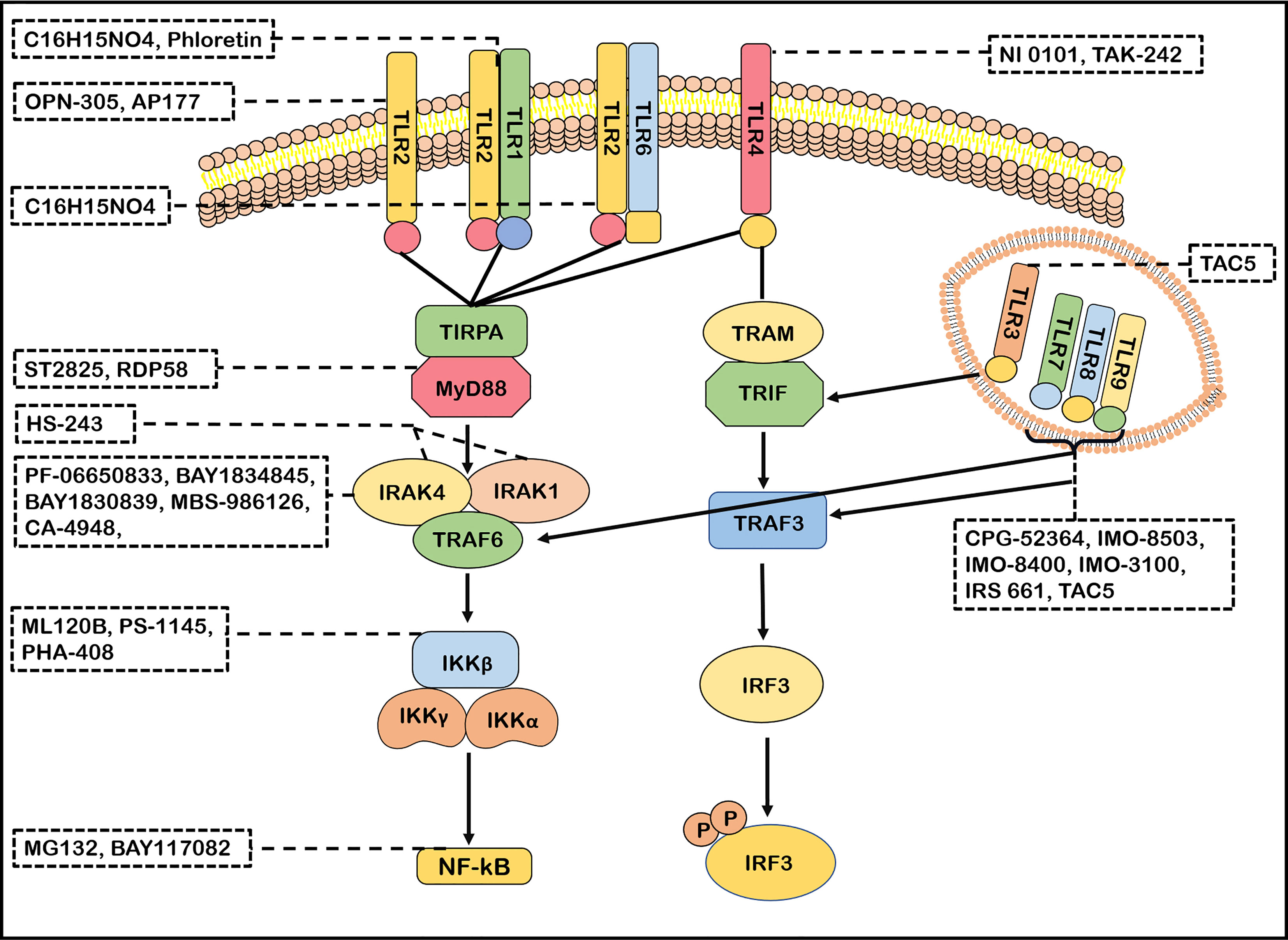
Figure 3 Antagonists of TLRs signaling pathways. Many antagonists have been developed against TLRs. Phloretin is an inhibitor of TLR2/1, OPN-305 and AP177 block TLR2 signaling. C16H15NO4 inhibits the TLR2/1 and TLR2/6 signaling pathway. TAK-242 and NI 0101 disrupt the interactions of TLR4 with its adaptor molecules TIR domain-containing adaptor protein(TIRAP). IMO-8503, CPG-52364, IMO-8503, IMO−8400, IMO−3100 and IRS 661 are the antagonists of TLR7, 8, 9. TAC5 effectively inhibits TLR3, TLR7, TLR8 and TLR9 signaling pathways to prevent the production of inflammatory factors. ST2825 and RDP58 inhibit myeloid differentiation primary response 88(MyD88)dimerization, interfering with recruitment downstream molecules. PF-06650833, BAY1834845, BAY1830839, CA-4948 and MBS-986126 are antagonists of IRAK4 and effectively block its follow-up effect. HS-243 exquisite selectively inhibits IRAK-1/4. ML120B, PS-1145 and PHA-408 are the inhibitors of IKK-β. MG132 and BAY117082 inhibit nuclear factor-kappa B (NF-κB) so that inflammatory response cannot be activated.
There are still certain problems in the application of TLRs inhibitors for clinical treatments. First, the contradictory role of TLRs in immune cells becomes a significant obstacle. As mentioned above, the conflicting role of TLR3, TLR4, TLR7, TLR8 and TLR9 in different immune cells implied that total suppression of a certain TLR signal molecules may not only inhibit immune regulatory effects of TLR, but also damage the regeneration and repair of axons, myelin sheaths and even neurons. In addition, the cross-talk between the same TLR on different cells or different TLRs on the same cell fails to precisely suppress excessive immune responses TLRs caused. Second, for the same TLR on a cell, different TLR ligands may lead to the activation of different signaling pathways, resulting in conflicting effects (71). For example, Theiler’s murine encephalomyelitis virus (TMEV) infection activated TLR3 signaling pathway, which promoted the production of CCL2, CXCL8 and proinflammatory cytokines through the activation of NF-κB and IRF-3 (190). However, the activated TLR3 by poly I:C did not show the same results in vitro. This was attributed to the cytoplasmic dsRNA-activated kinase PKR by TMEV infection, but this cannot be achieved by poly I:C stimulation. These results highlight the difference between the TLR3 response induced by direct nonlytic virus infection and by extracellular poly I:C stimulation (191). There are unknown molecules that regulate different signaling pathways after the activation of the same TLR in a complex microenvironment in vivo. The sweeping inhibition of TLR is not only one-sided but may bring harmful side effects. Therefore, finding new regulatory targets on the TLR pathway with a targeted drug delivery is crucial for the application of TLRs into clinical practice.
Epigenetic modification is an important mechanism leading to gene expression changes. The relationship between the microbiota-gut-brain axis and CNS diseases is a classic example. Human microbiota and its metabolites adjust immune cells and cytokines partial by epigenomic modifications to participate in the occurrence and development of CNS diseases (192). TLR, as PRRs expressed by a variety of cells in the gastrointestinal tract, directly binds to the microbiome to maintain intestinal homeostasis or induce gut dysbiosis resulting in inflammatory responses (193). Emerging evidences support that TLRs are involved in the pathogenesis of neurodegenerative diseases through the gut-brain axis possibly, including MS. The imbalance of the intestinal microbiome not only affects the expression level of TLRs in APCs, but also leads to the imbalance of Th17/Treg cells (192, 194). Therefore, regulating TLRs through modulating microbiota may be a safe and effective method for the treatment of neuroimmune diseases in the future.
In recent years, several studies have identified diet as one of the major factors shaping the composition of the gut microbiome, thereby affecting systemic immune systems (195). High-calorie diets and lacking physical exercise can upregulate cellular metabolism toward biosynthetic pathways and lead to the gut dysbiosis, altered intestinal immunity state and low-level systemic inflammation. Instead, low-calorie diets act on nuclear receptors and enzymes that upregulate oxidative metabolism, downregulate synthesis of proinflammatory molecules, and restore or maintain a healthy gut symbiotic microbiota (196). Modulation of the intestinal microbiota provides an opportunity for the treatment of neuroinflammatory diseases (195). Nutrients may play a role in inflammation by regulating the expression of TLRs, proinflammatory and anti-inflammatory cytokines, thus interfering with the crosstalk and signal transduction of immune cells (197). For instance, retinoid supplementation has been shown to decrease inflammatory responses by downregulating TLRs expression and secretion of proinflammatory cytokines TNF-α and IL-6 during macrophage phagocytosis (198). Vitamin B2 inhibits the activation of TLR4 and TLR6 on macrophages stimulated by LPS and zymosan, and decreased the production of TNF-α and iNOS, while increased the secretion of IL-10 (199). Vitamin D3 inhibits the expression of TLR2 and TLR4 proteins and downregulates the production of TNF-α in monocytes in a time- and dose-dependent manner (200). Moreover, regulating the microbiome by dietary amino acids affects the TLRs regulation of macrophages and DCs, and impacts the gut-microbiome-immune (201).
The intake of probiotics might improve intestinal dysbacteriosis and reduce gut leaky, further lowing the production of inflammatory mediators, decreasing the activation of macrophages and DCs and mitigating inflammatory reactions of MS (202). Another study reported that probiotics downregulated the TLRs/MyD88/NF-κB signaling pathway, promoting the secretion of M2 polarization factors (IL-10 and IL-4) and inhibiting M1 polarization factors (TNF-α, IFN-γ, IL-1β, iNOS, COX-2, and IL-6) in the type 2 diabetes (203).
TLR2 plays a crucial role in linking the microbiome to MS. Recently, Nicholas proposed the hygiene hypothesis that reduces neuroinflammation and improves myelin repair by injecting sufficient microbial-derived molecules Lipid 654 [L654: produced by bacteroidetes and present in the serum of healthy people and MS patients, but at lower levels in MS patients (204)] into the circulation of MS patients to repeatedly stimulate TLR2 for a long time (205). Although many studies have been conducted on the role of TLRs in neuroimmune diseases, satisfactory results have not been achieved. Previous experimental methods resulted in the expression of TLRs in an ‘all-or-none’ state (205). Either failure to suppress TLR, or complete suppression of TLR leads to loss of protective immune response in the face of infection. We need to find ways to regulate TLRs expression and its immune effects at an appropriate range. In future studies, we should try to find some easily regulated ‘control buttons’ between TLRs and neuroimmune diseases, such as the microbiome.
Although many TLR inhibitors have been developed, they have not been used for clinical treatments mainly due to its inadequate response (206), especially with traditional delivery methods: insufficient stability, poor water solubility, injection site aggregation, not lasting effect, systemic toxicity, and nonspecific immune cell suppression (207–209). Developing accurate targeted drugs and effective delivery method are the most important issue. With the rapid development of material chemistry research in recent years, the application of innovative biomaterials and drug delivery devices may address these problems. Nanocarriers have often been used for drug delivery, with common nanocarriers being inorganic carriers (metal nanocrystals or carbon nanomaterials) and organic carriers (polymer nanocarriers or liposomes) (210). Nanoparticles-submicron-sized drug carriers have been actively investigated for the delivery of antibiotics, nucleic acids, peptides/proteins and chemotherapeutics (211). Polymer nanoparticles can mediate passive or active targeted drug transport, improve the drug concentration of lesions, and the stability of loading drugs. By changing the size of the polymer nanoparticles, the clearance of small drug molecules from the kidney or liver can be reduced, thereby increasing the drug cycle time (210, 212). By controlling drug release ability, the systemic side effects induced by drugs can be reduced significantly (210, 212). Polymeric nanoparticles fabricated with poly (d,l-lactide-co-glycolide, PLGA) is a biocompatible polymer, which was stable in saline and small enough to be administered by subcutaneous or intramuscular injections. It had been used as an efficient delivery platform for TLR7/8 agonists, enhancing DCs uptake and facilitating lymphatic drainage (211). We believe that the use of ‘nano-TLRs inhibitors’ in neuroimmune diseases may achieve more ideal therapeutic effects, which may greatly reduce global immunosuppressive effects in the circulation, and precisely reach the lesion center, improve absorption rate and reduce adverse drug reactions. Nanocarriers can realize targeted drug delivery for a specific cell type. For example, TLRs agonists are delivered to macrophages by nanocarriers in tumors, which promoted macrophages from M2 to M1 phenotype with an anti-tumor effect (213). Furthermore, 1-1000 nm nanoparticles were preferentially phagocytosed by APCs (214). Therefore, TLRs agonists are accurately delivered to DCs through nanoparticles to promote its maturation, playing a role of stronger antigen presentation and promoting immune responses (215, 216). The combination of TLRs inhibitors and nanocarriers would be a promising approach, which may improve therapeutic effects and reduce potential side effects.
TLRs inhibitors, as a potential immunotherapy, have been widely studied in a variety of autoimmune diseases (217, 218), but not in neuroimmune diseases such as NMOSD, GBS, MG and MS. Of note, the previous studies regarding TLRs in neuroimmune diseases mainly focused on cellular experiments in vitro and TLR expression levels in immune cells of patients. TLRs agonists have achieved promising results in tumors. Thus, we are confident that TLRs signaling pathways will also be used as therapeutic targets in neuroimmune diseases in the future, especially combining the application of next-generation sequencing and novel material chemistry technology.
HL and SL drafted the manuscript. JH, SXL, XG, and MW edited and revised the manuscript. JZ and TJ designed the framework and revised the manuscript. All authors read and approved the final manuscript.
This work was supported by grants from the General Program of the National Natural Science Foundation of China (No. 81671177), Natural Science Foundation of Jilin Province Science and Technology Development Plan Project (20190201043JC), Key Research and Development Project of Social Development Division of Jilin Science and Technology Department (20200403109SF), Special Project for Health Professionals of Jilin Provincial Finance Department (JLSWSRCZX2020-0056), as well as the grants from the Swedish Research Council(No. 2015-03005) and grants from the First hospital, Jilin University of China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mukherjee S, Huda S, Sinha Babu SP. Toll-Like Receptor Polymorphism in Host Immune Response to Infectious Diseases: A Review. Scand J Immunol (2019) 90(1):e12771. doi: 10.1111/sji.12771
2. Heinz LX, Lee J, Kapoor U. TASL is the SLC15A4-Associated Adaptor for IRF5 Activation by TLR7-9. Nature (2020) 581(7808):316–22. doi: 10.1038/s41586-020-2282-0
3. Anthoney N, Foldi I. Toll and Toll-Like Receptor Signalling in Development. Development (2018) 145(9):dev156018. doi: 10.1242/dev.156018
4. Li X, Jiang S, Tapping RI. Toll-Like Receptor Signaling in Cell Proliferation and Survival. Cytokine (2010) 49(1):1–9. doi: 10.1016/j.cyto.2009.08.010
5. Medvedev AE. Toll-Like Receptor Polymorphisms, Inflammatory and Infectious Diseases, Allergies, and Cancer. J Interferon Cytokine Res Off J Int Soc Interferon Cytokine Res (2013) 33(9):467–84. doi: 10.1089/jir.2012.0140
6. Fillatreau S, Manfroi B, Dörner T. Toll-Like Receptor Signalling in B Cells During Systemic Lupus Erythematosus. Nat Rev Rheumatol (2021) 17(2):98–108. doi: 10.1038/s41584-020-00544-4
7. Nelson CP, Erridge C. Are Toll-Like Receptors Potential Drug Targets for Atherosclerosis? Evidence From Genetic Studies to Date. Immunogenetics (2019) 71(1):1–11. doi: 10.1007/s00251-018-1092-0
8. Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 Trafficking and its Influence on LPS-Induced Pro-Inflammatory Signaling. Cell Mol Life Sci CMLS (2021) 78(4):1233–61. doi: 10.1007/s00018-020-03656-y
9. Liu Y, Yin H, Zhao M, Lu Q. TLR2 and TLR4 in Autoimmune Diseases: A Comprehensive Review. Clin Rev Allergy Immunol (2014) 47(2):136–47. doi: 10.1007/s12016-013-8402-y
10. Elemam NM, Hachim MY. Differentially Expressed Genes of Natural Killer Cells Can Distinguish Rheumatoid Arthritis Patients From Healthy Controls. Genes (Basel) (2020) 11(5):492. doi: 10.3390/genes11050492
11. Danikowski KM, Jayaraman S, Prabhakar BS. Regulatory T Cells in Multiple Sclerosis and Myasthenia Gravis. J Neuroinflamm (2017) 14(1):117. doi: 10.1186/s12974-017-0892-8
12. Heming M, Schulte-Mecklenbeck A, Brix T, Wolbert J, Ruland T, Klotz L, et al. Immune Cell Profiling of the Cerebrospinal Fluid Provides Pathogenetic Insights Into Inflammatory Neuropathies. Front Immunol (2019) 10:515. doi: 10.3389/fimmu.2019.00515
13. Lee DSW, Rojas OL. B Cell Depletion Therapies in Autoimmune Disease: Advances and Mechanistic Insights. Nat Rev Drug Discov (2021) 20(3):179–99. doi: 10.1038/s41573-020-00092-2
14. Patterson H, Nibbs R, McInnes I, Siebert S. Protein Kinase Inhibitors in the Treatment of Inflammatory and Autoimmune Diseases. Clin Exp Immunol (2014) 176(1):1–10. doi: 10.1111/cei.12248
15. Zheng C, Chen J, Chu F, Zhu J, Jin T. Inflammatory Role of TLR-MyD88 Signaling in Multiple Sclerosis. Front Mol Neurosci (2019) 12:314. doi: 10.3389/fnmol.2019.00314
16. Robinet M, Maillard S, Cron MA, Berrih-Aknin S, Le Panse R. Review on Toll-Like Receptor Activation in Myasthenia Gravis: Application to the Development of New Experimental Models. Clin Rev Allergy Immunol (2017) 52(1):133–47. doi: 10.1007/s12016-016-8549-4
17. Li X, Liu R, Su X, Pan Y, Han X, Shao C. Harnessing Tumor-Associated Macrophages as Aids for Cancer Immunotherapy. Mol Cancer (2019) 18(1):177. doi: 10.1186/s12943-019-1102-3
18. Joosten LA, Abdollahi-Roodsaz S, Dinarello CA, O’Neill L, Netea MG. Toll-Like Receptors and Chronic Inflammation in Rheumatic Diseases: New Developments. Nat Rev Rheumatol (2016) 12(6):344–57. doi: 10.1038/nrrheum.2016.61
19. Matsushima N, Tanaka T, Enkhbayar P, Mikami T, Taga M, Yamada K, et al. Comparative Sequence Analysis of Leucine-Rich Repeats (LRRs) Within Vertebrate Toll-Like Receptors. BMC Genomics (2007) 8:124. doi: 10.1186/1471-2164-8-124
20. Ryan KA, Smith MF Jr., Sanders MK, Ernst PB. Reactive Oxygen and Nitrogen Species Differentially Regulate Toll-Like Receptor 4-Mediated Activation of NF-Kappa B and Interleukin-8 Expression. Infect Immun (2004) 72(4):2123–30. doi: 10.1128/iai.72.4.2123-2130.2004
21. Li L, Acioglu C, Heary RF, Elkabes S. Role of Astroglial Toll-Like Receptors (TLRs) in Central Nervous System Infections, Injury and Neurodegenerative Diseases. Brain Behav Immun (2021) 91:740–55. doi: 10.1016/j.bbi.2020.10.007
22. Dajon M, Iribarren K, Cremer I. Toll-Like Receptor Stimulation in Cancer: A Pro- and Anti-Tumor Double-Edged Sword. Immunobiology (2017) 222(1):89–100. doi: 10.1016/j.imbio.2016.06.009
23. Chang SF, Chen CN, Lin JC, Wang HE, Mori S, Li JJ, et al. Truncated Pneumolysin From Streptococcus Pneumoniae as a TLR4-Antagonizing New Drug for Chronic Inflammatory Conditions. Cells (2020) 9(5):1183. doi: 10.3390/cells9051183
24. Li Y, Deng SL. Roles of Toll-Like Receptors in Nitroxidative Stress in Mammals. Cells (2019) 8(6):576. doi: 10.3390/cells8060576
25. Ji J, Liao Z, Rao Y, Li W, Yang C, Yuan G, et al. Thoroughly Remold the Localization and Signaling Pathway of TLR22. Front Immunol (2019) 10:3003. doi: 10.3389/fimmu.2019.03003
26. Ahmed H, Khan MA. Role of Adaptor Protein Myeloid Differentiation 88 (MyD88) in Post-Subarachnoid Hemorrhage Inflammation: A Systematic Review. Int J Mol Sci (2021) 22(8):4185. doi: 10.3390/ijms22084185
27. Xiang W, Chao ZY, Feng DY. Role of Toll-Like Receptor/MYD88 Signaling in Neurodegenerative Diseases. Rev Neurosci (2015) 26(4):407–14. doi: 10.1515/revneuro-2014-0067
28. Kawai T, Akira S. The Role of Pattern-Recognition Receptors in Innate Immunity: Update on Toll-Like Receptors. Nat Immunol (2010) 11(5):373–84. doi: 10.1038/ni.1863
29. Kawasaki T, Kawai T. Toll-Like Receptor Signaling Pathways. Front Immunol (2014) 5:461. doi: 10.3389/fimmu.2014.00461
30. Loiarro M, Volpe E, Ruggiero V, Gallo G, Furlan R, Maiorino C, et al. Mutational Analysis Identifies Residues Crucial for Homodimerization of Myeloid Differentiation Factor 88 (MyD88) and for its Function in Immune Cells. J Biol Chem (2013) 288(42):30210–22. doi: 10.1074/jbc.M113.490946
31. O’Neill LA, Bowie AG. The Family of Five: TIR-Domain-Containing Adaptors in Toll-Like Receptor Signalling. Nat Rev Immunol (2007) 7(5):353–64. doi: 10.1038/nri2079
32. O’Neill LA. When Signaling Pathways Collide: Positive and Negative Regulation of Toll-Like Receptor Signal Transduction. Immunity (2008) 29(1):12–20. doi: 10.1016/j.immuni.2008.06.004
33. Vallance TM, Zeuner MT. Toll-Like Receptor 4 Signalling and Its Impact on Platelet Function, Thrombosis, and Haemostasis. Mediators Inflamm (2017) 2017:9605894. doi: 10.1155/2017/9605894
34. Nunes KP, de Oliveira AA, Mowry FE, Biancardi VC. Targeting Toll-Like Receptor 4 Signalling Pathways: Can Therapeutics Pay the Toll for Hypertension? Br J Pharmacol (2019) 176(12):1864–79. doi: 10.1111/bph.14438
35. Strickson S, Emmerich CH, Goh ETH, Zhang J, Kelsall IR. Roles of the TRAF6 and Pellino E3 Ligases in MyD88 and RANKL Signaling. Proc Natl Acad Sci USA (2017) 114(17):E3481–e9. doi: 10.1073/pnas.1702367114
36. Küper C, Beck FX, Neuhofer W. Toll-Like Receptor 4 Activates NF-κb and MAP Kinase Pathways to Regulate Expression of Proinflammatory COX-2 in Renal Medullary Collecting Duct Cells. Am J Physiol Renal Physiol (2012) 302(1):F38–46. doi: 10.1152/ajprenal.00590.2010
37. Picard C, Casanova JL, Puel A. Infectious Diseases in Patients With IRAK-4, MyD88, NEMO, or Iκbα Deficiency. Clin Microbiol Rev (2011) 24(3):490–7. doi: 10.1128/cmr.00001-11
38. De Nardo D. Toll-Like Receptors: Activation, Signalling and Transcriptional Modulation. Cytokine (2015) 74(2):181–9. doi: 10.1016/j.cyto.2015.02.025
39. Li M, Liu J, Bi Y, Chen J, Zhao L. Potential Medications or Compounds Acting on Toll-Like Receptors in Cerebral Ischemia. Curr Neuropharmacol (2018) 16(2):160–75. doi: 10.2174/1570159x15666170601125139
40. Sato W, Yamamura T. Multiple Sclerosis: Possibility of a Gut Environment-Induced Disease. Neurochemistry Int (2019) 130:104475. doi: 10.1016/j.neuint.2019.104475
41. Mozafari S, Starost L. Multiple Sclerosis iPS-Derived Oligodendroglia Conserve Their Properties to Functionally Interact With Axons and Glia In Vivo. Sci Adv (2020) 6(49):eabc6983. doi: 10.1126/sciadv.abc6983
42. Afshar B, Khalifehzadeh-Esfahani Z, Seyfizadeh N, Rezaei Danbaran G, Hemmatzadeh M, Mohammadi H. The Role of Immune Regulatory Molecules in Multiple Sclerosis. J Neuroimmunol (2019) 337:577061. doi: 10.1016/j.jneuroim.2019.577061
43. Bsibsi M, Ravid R, Gveric D, van Noort JM. Broad Expression of Toll-Like Receptors in the Human Central Nervous System. J Neuropathol Exp Neurol (2002) 61(11):1013–21. doi: 10.1093/jnen/61.11.1013
44. Ferreira TB, Hygino J, Wing AC, Kasahara TM, Sacramento PM, Camargo S, et al. Different Interleukin-17-Secreting Toll-Like Receptor(+) T-Cell Subsets are Associated With Disease Activity in Multiple Sclerosis. Immunology (2018) 154(2):239–52. doi: 10.1111/imm.12872
45. Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, et al. Toll-Like Receptor 2 Signaling in CD4(+) T Lymphocytes Promotes T Helper 17 Responses and Regulates the Pathogenesis of Autoimmune Disease. Immunity (2010) 32(5):692–702. doi: 10.1016/j.immuni.2010.04.010
46. Sales MC, Kasahara TM. Selective Serotonin Reuptake Inhibitor Attenuates the Hyperresponsiveness of TLR2(+) and TLR4(+) Th17/Tc17-Like Cells in Multiple Sclerosis Patients With Major Depression. Immunology (2021) 162(3):290–305. doi: 10.1111/imm.13281
47. Nyirenda MH, Morandi E, Vinkemeier U, Constantin-Teodosiu D, Drinkwater S, Mee M, et al. TLR2 Stimulation Regulates the Balance Between Regulatory T Cell and Th17 Function: A Novel Mechanism of Reduced Regulatory T Cell Function in Multiple Sclerosis. J Immunol (2015) 194(12):5761–74. doi: 10.4049/jimmunol.1400472
48. Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan Blocks Oligodendrocyte Progenitor Maturation and Remyelination Through TLR2. Proc Natl Acad Sci USA (2010) 107(25):11555–60. doi: 10.1073/pnas.1006496107
49. Wasko NJ, Kulak MH, Paul D, Nicaise AM, Yeung ST, Nichols FC, et al. Systemic TLR2 Tolerance Enhances Central Nervous System Remyelination. Neuroinflamm (2019) 16(1):158. doi: 10.1186/s12974-019-1540-2
50. Fujiwara M, Anstadt EJ, Flynn B, Morse K, Ng C, Paczkowski P, et al. Enhanced TLR2 Responses in Multiple Sclerosis. Clin Exp Immunol (2018) 193(3):313–26. doi: 10.1111/cei.13150
51. Bonechi E, Aldinucci A, Mazzanti B, di Gioia M, Repice AM, Manuelli C, et al. Increased CXCL10 Expression in MS MSCs and Monocytes is Unaffected by AHSCT. Ann Clin Trans Neurol (2014) 1(9):650–8. doi: 10.1002/acn3.92
52. Kerfoot SM, Long EM, Hickey MJ, Andonegui G, Lapointe BM, Zanardo RC, et al. TLR4 Contributes to Disease-Inducing Mechanisms Resulting in Central Nervous System Autoimmune Disease. J Immunol (2004) 173(11):7070–7. doi: 10.4049/jimmunol.173.11.7070
53. Racke MK, Hu W, Lovett-Racke AE. PTX Cruiser: Driving Autoimmunity via TLR4. Trends Immunol (2005) 26(6):289–91. doi: 10.1016/j.it.2005.03.012
54. Mellanby RJ, Cambrook H, Turner DG, O’Connor RA, Leech MD, Kurschus FC, et al. TLR-4 Ligation of Dendritic Cells is Sufficient to Drive Pathogenic T Cell Function in Experimental Autoimmune Encephalomyelitis. J Neuroinflamm (2012) 9:248. doi: 10.1186/1742-2094-9-248
55. Reynolds JM, Martinez GJ, Chung Y, Dong C. Toll-Like Receptor 4 Signaling in T Cells Promotes Autoimmune Inflammation. Proc Natl Acad Sci USA (2012) 109(32):13064–9. doi: 10.1073/pnas.1120585109
56. Zhang Y, Han J, Wu M, Xu L, Wang Y, Yuan W, et al. Toll-Like Receptor 4 Promotes Th17 Lymphocyte Infiltration Via CCL25/CCR9 in Pathogenesis of Experimental Autoimmune Encephalomyelitis. J Neuroimmune Pharmacol (2019) 14(3):493–502. doi: 10.1007/s11481-019-09854-1
57. Schilling S, Chausse B, Dikmen HO, Almouhanna F, Hollnagel JO, Lewen A, et al. TLR2- and TLR3-Activated Microglia Induce Different Levels of Neuronal Network Dysfunction in a Context-Dependent Manner. Brain Behav Immun (2021) 96:80–91. doi: 10.1016/j.bbi.2021.05.013
58. Karni A, Abraham M, Monsonego A, Cai G, Freeman GJ, Hafler D, et al. Innate Immunity in Multiple Sclerosis: Myeloid Dendritic Cells in Secondary Progressive Multiple Sclerosis are Activated and Drive a Proinflammatory Immune Response. J Immunol (Baltimore Md 1950) (2006) 177(6):4196–202. doi: 10.4049/jimmunol.177.6.4196
59. Proietto AI, Mittag D, Roberts AW, Sprigg N, Wu L. The Equivalents of Human Blood and Spleen Dendritic Cell Subtypes can be Generated In Vitro From Human CD34(+) Stem Cells in the Presence of Fms-Like Tyrosine Kinase 3 Ligand and Thrombopoietin. Cell Mol Immunol (2012) 9(6):446–54. doi: 10.1038/cmi.2012.48
60. Thewissen K, Nuyts AH, Deckx N, Van Wijmeersch B, Nagels G, D’Hooghe M, et al. Circulating Dendritic Cells of Multiple Sclerosis Patients are Proinflammatory and Their Frequency is Correlated With MS-Associated Genetic Risk Factors. Multiple Sclerosis (Houndmills Basingstoke England) (2014) 20(5):548–57. doi: 10.1177/1352458513505352
61. Pérez-Cabezas B, Naranjo-Gómez M, Ruiz-Riol M, Bastos-Amador P, Fernández MA, Carmona F, et al. TLR-Activated Conventional DCs Promote γ-Secretase-Mediated Conditioning of Plasmacytoid DCs. J Leukocyte Biol (2012) 92(1):133–43. doi: 10.1189/jlb.0911452
62. Xu J, Zoltick PW, Gamero AM, Gallucci S. TLR Ligands Up-Regulate Trex1 Expression in Murine Conventional Dendritic Cells Through Type I Interferon and NF-κb-Dependent Signaling Pathways. J Leukocyte Biol (2014) 96(1):93–103. doi: 10.1189/jlb.2A0713-393RR
63. Church JS, Milich LM, Lerch JK, Popovich PG, McTigue DM. E6020, a Synthetic TLR4 Agonist, Accelerates Myelin Debris Clearance, Schwann Cell Infiltration, and Remyelination in the Rat Spinal Cord. Glia (2017) 65(6):883–99. doi: 10.1002/glia.23132
64. Rajbhandari L, Tegenge MA, Shrestha S, Ganesh Kumar N, Malik A, Mithal A, et al. Toll-Like Receptor 4 Deficiency Impairs Microglial Phagocytosis of Degenerating Axons. Glia (2014) 62(12):1982–91. doi: 10.1002/glia.22719
65. Okada Y, Ochi H, Fujii C, Hashi Y, Hamatani M, Ashida S, et al. Signaling via Toll-Like Receptor 4 and CD40 in B Cells Plays a Regulatory Role in the Pathogenesis of Multiple Sclerosis Through Interleukin-10 Production. J Autoimmun (2018) 88:103–13. doi: 10.1016/j.jaut.2017.10.011
66. Asadzadeh Manjili F, Yousefi-Ahmadipour A, Kazemi Arababadi M. The Roles Played by TLR4 in the Pathogenesis of Multiple Sclerosis; A Systematic Review Article. Immunol Lett (2020) 220:63–70. doi: 10.1016/j.imlet.2020.02.004
67. Deeba E, Koptides D, Lambrianides A, Pantzaris M, Krashias G, Christodoulou C. Complete Sequence Analysis of Human Toll-Like Receptor 3 Gene in Natural Killer Cells of Multiple Sclerosis Patients. Multiple Sclerosis Related Disord (2019) 33:100–6. doi: 10.1016/j.msard.2019.05.027
68. Touil T, Fitzgerald D, Zhang GX, Rostami A, Gran B. Cutting Edge: TLR3 Stimulation Suppresses Experimental Autoimmune Encephalomyelitis by Inducing Endogenous IFN-Beta. J Immunol (2006) 177(11):7505–9. doi: 10.4049/jimmunol.177.11.7505
69. Gooshe M, Abdolghaffari AH, Gambuzza ME, Rezaei N. The Role of Toll-Like Receptors in Multiple Sclerosis and Possible Targeting for Therapeutic Purposes. Rev Neurosci (2014) 25(5):713–39. doi: 10.1515/revneuro-2014-0026
70. Okada R, Zhang X, Harada Y, Wu Z, Nakanishi H. Cathepsin H Deficiency in Mice Induces Excess Th1 Cell Activation and Early-Onset of EAE Though Impairment of Toll-Like Receptor 3 Cascade. Inflamm Res (2018) 67(5):371–4. doi: 10.1007/s00011-018-1136-9
71. Bsibsi M, Persoon-Deen C, Verwer RW, Meeuwsen S, Ravid R, Van Noort JM. Toll-Like Receptor 3 on Adult Human Astrocytes Triggers Production of Neuroprotective Mediators. Glia (2006) 53(7):688–95. doi: 10.1002/glia.20328
72. Bsibsi M, Bajramovic JJ, Vogt MH, van Duijvenvoorden E, Baghat A, Persoon-Deen C, et al. The Microtubule Regulator Stathmin is an Endogenous Protein Agonist for TLR3. J Immunol (Baltimore Md 1950) (2010) 184(12):6929–37. doi: 10.4049/jimmunol.0902419
73. Werkman I, Sikkema AH, Versluijs JB, Qin J, de Boer P, Baron W. TLR3 Agonists Induce Fibronectin Aggregation by Activated Astrocytes: A Role of Pro-Inflammatory Cytokines and Fibronectin Splice Variants. Sci Rep (2020) 10: (1):532. doi: 10.1038/s41598-019-57069-4
74. Saresella M, Gatti A, Tortorella P, Marventano I, Piancone F, La Rosa F, et al. Toll-Like Receptor 3 Differently Modulates Inflammation in Progressive or Benign Multiple Sclerosis. Clin Immunol (Orlando Fla) (2014) 150(1):109–20. doi: 10.1016/j.clim.2013.10.012
75. Mycko MP, Cwiklinska H, Cichalewska M, Matysiak M, Lewkowicz P, Sliwinska B, et al. Plasmocytoid Dendritic Cell Deficit of Early Response to Toll-Like Receptor 7 Agonist Stimulation in Multiple Sclerosis Patients. Clin Immunol (Orlando Fla) (2014) 153(1):211–9. doi: 10.1016/j.clim.2014.04.016
76. Jego G, Bataille R, Pellat-Deceunynck C. Interleukin-6 is a Growth Factor for Nonmalignant Human Plasmablasts. Blood (2001) 97(6):1817–22. doi: 10.1182/blood.v97.6.1817
77. Roldán E, García-Pardo A, Brieva JA. VLA-4-Fibronectin Interaction is Required for the Terminal Differentiation of Human Bone Marrow Cells Capable of Spontaneous and High Rate Immunoglobulin Secretion. J Exp Med (1992) 175(6):1739–47. doi: 10.1084/jem.175.6.1739
78. Mackay F, Silveira PA, Brink R. B Cells and the BAFF/APRIL Axis: Fast-Forward on Autoimmunity and Signaling. Curr Opin Immunol (2007) 19(3):327–36. doi: 10.1016/j.coi.2007.04.008
79. Giacomini E, Severa M, Rizzo F, Mechelli R, Annibali V, Ristori G, et al. IFN-β Therapy Modulates B-Cell and Monocyte Crosstalk via TLR7 in Multiple Sclerosis Patients. Eur J Immunol (2013) 43(7):1963–72. doi: 10.1002/eji.201243212
80. Derkow K, Bauer JM, Hecker M, Paap BK, Thamilarasan M, Koczan D, et al. Multiple Sclerosis: Modulation of Toll-Like Receptor (TLR) Expression by Interferon-β Includes Upregulation of TLR7 in Plasmacytoid Dendritic Cells. PloS One (2013) 8(8):e70626. doi: 10.1371/journal.pone.0070626
81. Tao Y, Zhang X, Markovic-Plese S. Toll-Like Receptor (TLR)7 and TLR9 Agonists Enhance Interferon (IFN) Beta-1a’s Immunoregulatory Effects on B Cells in Patients With Relapsing-Remitting Multiple Sclerosis (RRMS). J Neuroimmunol (2016) 298:181–8. doi: 10.1016/j.jneuroim.2016.07.019
82. Giacomini E, Rizzo F, Etna MP, Cruciani M, Mechelli R, Buscarinu MC, et al. Thymosin-α1 Expands Deficient IL-10-Producing Regulatory B Cell Subsets in Relapsing-Remitting Multiple Sclerosis Patients. Multiple Sclerosis (Houndmills Basingstoke England) (2018) 24(2):127–39. doi: 10.1177/1352458517695892
83. Johnson TP, Tyagi R, Patel K, Schiess N, Calabresi PA, Nath A. Impaired Toll-Like Receptor 8 Signaling in Multiple Sclerosis. J Neuroinflamm (2013) 10:74. doi: 10.1186/1742-2094-10-74
84. Demaria O, Pagni PP, Traub S, de Gassart A, Branzk N, Murphy AJ, et al. TLR8 Deficiency Leads to Autoimmunity in Mice. J Clin Invest (2010) 120(10):3651–62. doi: 10.1172/jci42081
85. Soulika AM, Lee E, McCauley E, Miers L, Bannerman P, Pleasure D. Initiation and Progression of Axonopathy in Experimental Autoimmune Encephalomyelitis. J Neurosci Off J Soc Neurosci (2009) 29(47):14965–79. doi: 10.1523/jneurosci.3794-09.2009
86. Li B, Baylink DJ, Deb C, Zannetti C, Rajaallah F, Xing W, et al. 1,25-Dihydroxyvitamin D3 Suppresses TLR8 Expression and TLR8-Mediated Inflammatory Responses in Monocytes In Vitro and Experimental Autoimmune Encephalomyelitis In Vivo. PloS One (2013) 8(3):e58808. doi: 10.1371/journal.pone.0058808
87. Dominguez-Mozo MI, Garcia-Montojo M, López-Cavanillas M, De Las Heras V, Garcia-Martinez A, Arias-Leal AM, et al. Toll-Like Receptor-9 in Spanish Multiple Sclerosis Patients: An Association With the Gender. Eur J Neurol (2014) 21(3):537–40. doi: 10.1111/ene.12209
88. Balashov KE, Aung LL, Vaknin-Dembinsky A, Dhib-Jalbut S, Weiner HL. Interferon-β Inhibits Toll-Like Receptor 9 Processing in Multiple Sclerosis. Ann Neurol (2010) 68(6):899–906. doi: 10.1002/ana.22136
89. Ichikawa HT, Williams LP, Segal BM. Activation of APCs Through CD40 or Toll-Like Receptor 9 Overcomes Tolerance and Precipitates Autoimmune Disease. J Immunol (2002) 169(5):2781–7. doi: 10.4049/jimmunol.169.5.2781
90. Bayas A, Stasiolek M, Kruse N, Toyka KV, Selmaj K, Gold R. Altered Innate Immune Response of Plasmacytoid Dendritic Cells in Multiple Sclerosis. Clin Exp Immunol (2009) 157(3):332–42. doi: 10.1111/j.1365-2249.2009.03964.x
91. Marta M, Andersson A, Isaksson M, Kämpe O, Lobell A. Unexpected Regulatory Roles of TLR4 and TLR9 in Experimental Autoimmune Encephalomyelitis. Eur J Immunol (2008) 38(2):565–75. doi: 10.1002/eji.200737187
92. Prinz M, Garbe F, Schmidt H, Mildner A, Gutcher I, Wolter K, et al. Innate Immunity Mediated by TLR9 Modulates Pathogenicity in an Animal Model of Multiple Sclerosis. J Clin Invest (2006) 116(2):456–64. doi: 10.1172/jci26078
93. Garza KM, Chan SM, Suri R, Nguyen LT, Odermatt B, Schoenberger SP, et al. Role of Antigen-Presenting Cells in Mediating Tolerance and Autoimmunity. J Exp Med (2000) 191(11):2021–7. doi: 10.1084/jem.191.11.2021
94. Waldner H, Collins M, Kuchroo VK. Activation of Antigen-Presenting Cells by Microbial Products Breaks Self Tolerance and Induces Autoimmune Disease. J Clin Invest (2004) 113(7):990–7. doi: 10.1172/jci19388
95. van Langelaar J, Rijvers L, Janssen M, Wierenga-Wolf AF, Melief MJ, Siepman TA, et al. Induction of Brain-Infiltrating T-Bet-Expressing B Cells in Multiple Sclerosis. Ann Neurol (2019) 86(2):264–78. doi: 10.1002/ana.25508
96. Hirotani M, Niino M, Fukazawa T, Kikuchi S, Yabe I, Hamada S, et al. Decreased IL-10 Production Mediated by Toll-Like Receptor 9 in B Cells in Multiple Sclerosis. J Neuroimmunol (2010) 221(1-2):95–100. doi: 10.1016/j.jneuroim.2010.02.012
97. Hong J, Fang J, Lan R, Tan Q, Tian Y, Zhang M, et al. TLR9 Mediated Regulatory B10 Cell Amplification Following Sub-Total Body Irradiation: Implications in Attenuating EAE. Mol Immunol (2017) 83:52–61. doi: 10.1016/j.molimm.2017.01.011
98. Dieu RS, Wais V, Sørensen MZ, Marczynska J, Dubik M, Kavan S, et al. Central Nervous System-Endogenous TLR7 and TLR9 Induce Different Immune Responses and Effects on Experimental Autoimmune Encephalomyelitis. Front Neurosci (2021) 15:685645. doi: 10.3389/fnins.2021.685645
99. Dubik M, Marczynska J, Mørch MT, Webster G, Jensen KN, Wlodarczyk A, et al. Innate Signaling in the CNS Prevents Demyelination in a Focal EAE Model. Front Neurosci (2021) 15:682451. doi: 10.3389/fnins.2021.682451
100. Bruscolini A, La Cava M, Mallone F, Marcelli M, Ralli M. Controversies in the Management of Neuromyelitis Optica Spectrum Disorder. Expert Rev Neurother (2019) 19(11):1127–33. doi: 10.1080/14737175.2019.1648210
101. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International Consensus Diagnostic Criteria for Neuromyelitis Optica Spectrum Disorders. Neurology (2015) 85(2):177–89. doi: 10.1212/wnl.0000000000001729
102. Wu Y, Zhong L, Geng J. Neuromyelitis Optica Spectrum Disorder: Pathogenesis, Treatment, and Experimental Models. Multiple Sclerosis Related Disord (2019) 27:412–8. doi: 10.1016/j.msard.2018.12.002
103. Jarius S, Wildemann B. AQP4 Antibodies in Neuromyelitis Optica: Diagnostic and Pathogenetic Relevance. Nat Rev Neurol (2010) 6(7):383–92. doi: 10.1038/nrneurol.2010.72
104. Lin J, Li X, Xia J. Th17 Cells in Neuromyelitis Optica Spectrum Disorder: A Review. Int J Neurosci (2016) 126(12):1051–60. doi: 10.3109/00207454.2016.1163550
105. Smyk DS, Koutsoumpas AL, Mytilinaiou MG, Rigopoulou EI, Sakkas LI, Bogdanos DP. Helicobacter Pylori and Autoimmune Disease: Cause or Bystander. World J Gastroenterol (2014) 20(3):613–29. doi: 10.3748/wjg.v20.i3.613
106. Masuda S, Mori M, Arai K, Uzawa A, Muto M, Uchida T, et al. Epstein-Barr Virus Persistence and Reactivation in Neuromyelitis Optica. J Neurol Neurosurg Psychiatry (2015) 86(10):1137–42. doi: 10.1136/jnnp-2014-308095
107. Barros PO, Dias ASO, Kasahara TM, Ornelas AMM, Aguiar RS, Leon SA, et al. Expansion of IL-6(+) Th17-Like Cells Expressing TLRs Correlates With Microbial Translocation and Neurological Disabilities in NMOSD Patients. J Neuroimmunol (2017) 307:82–90. doi: 10.1016/j.jneuroim.2017.04.001
108. Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 Cells Transdifferentiate Into Regulatory T Cells During Resolution of Inflammation. Nature (2015) 523(7559):221–5. doi: 10.1038/nature14452
109. Cho EB, Cho HJ, Seok JM, Min JH, Kang ES, Kim BJ. The IL-10-Producing Regulatory B Cells (B10 Cells) and Regulatory T Cell Subsets in Neuromyelitis Optica Spectrum Disorder. Neurol Sci Off J Ital Neurol Soc Ital Soc Clin Neurophysiol (2018) 39(3):543–9. doi: 10.1007/s10072-018-3248-y
110. Brill L, Lavon I, Vaknin-Dembinsky A. Foxp3+ Regulatory T Cells Expression in Neuromyelitis Optica Spectrum Disorders. Multiple Sclerosis Related Disord (2019) 30:114–8. doi: 10.1016/j.msard.2019.01.047
111. Dias ASO, Sacramento PM, Lopes LM, Sales MC, Castro C, Araújo A, et al. TLR-2 and TLR-4 Agonists Favor Expansion of CD4(+) T Cell Subsets Implicated in the Severity of Neuromyelitis Optica Spectrum Disorders. Multiple Sclerosis Related Disord (2019) 34:66–76. doi: 10.1016/j.msard.2019.06.018
112. Nakao M, Sugaya M. TLR2 Deficiency Exacerbates Imiquimod-Induced Psoriasis-Like Skin Inflammation Through Decrease in Regulatory T Cells and Impaired IL-10 Production. Int J Mol Sci (2020) 21(22):8560. doi: 10.3390/ijms21228560
113. Nguyen BN, Chávez-Arroyo A. TLR2 and Endosomal TLR-Mediated Secretion of IL-10 and Immune Suppression in Response to Phagosome-Confined Listeria Monocytogenes. PLoS Pathog (2020) 16(7):e1008622. doi: 10.1371/journal.ppat.1008622
114. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, et al. Intrathecal Pathogenic Anti-Aquaporin-4 Antibodies in Early Neuromyelitis Optica. Ann Neurol (2009) 66(5):617–29. doi: 10.1002/ana.21802
115. Yamashita K, Kinoshita M, Miyamoto K, Namba A, Shimizu M, Koda T, et al. Cerebrospinal Fluid Mitochondrial DNA in Neuromyelitis Optica Spectrum Disorder. J Neuroinflamm (2018) 15(1):125. doi: 10.1186/s12974-018-1162-0
116. Shimizu M, Okuno T, Kinoshita M, Sumi H, Fujimura H, Yamashita K, et al. Mitochondrial DNA Enhance Innate Immune Responses in Neuromyelitis Optica by Monocyte Recruitment and Activation. Sci Rep (2020) 10(1):13274. doi: 10.1038/s41598-020-70203-x
117. Rodríguez Y, Rojas M, Pacheco Y, Acosta-Ampudia Y, Ramírez-Santana C, Monsalve DM, et al. Guillain-Barré Syndrome, Transverse Myelitis and Infectious Diseases. Cell Mol Immunol (2018) 15(6):547–62. doi: 10.1038/cmi.2017.142
118. Zhang HL, Zheng XY, Zhu J. Th1/Th2/Th17/Treg Cytokines in Guillain-Barré Syndrome and Experimental Autoimmune Neuritis. Cytokine Growth Factor Rev (2013) 24(5):443–53. doi: 10.1016/j.cytogfr.2013.05.005
119. Nyati KK, Prasad KN, Verma A, Singh AK, Rizwan A, Sinha S, et al. Association of TLR4 Asp299Gly and Thr399Ile Polymorphisms With Guillain-Barré Syndrome in Northern Indian Population. J Neuroimmunol (2010) 218(1-2):116–9. doi: 10.1016/j.jneuroim.2009.10.018
120. Karanth S, Yang G, Yeh J, Richardson PM. Nature of Signals That Initiate the Immune Response During Wallerian Degeneration of Peripheral Nerves. Exp Neurol (2006) 202(1):161–6. doi: 10.1016/j.expneurol.2006.05.024
121. Du Y, Zhang G, Zhang Z, Wang Q, Ma R, Zhang L, et al. Toll-Like Receptor 2 and -4 are Involved in the Pathogenesis of the Guillain-Barré Syndrome. Mol Med Rep (2015) 12(2):3207–13. doi: 10.3892/mmr.2015.3730
122. Wang YZ, Liang QH, Ramkalawan H, Wang YL, Yang YF, Zhou WB, et al. Expression of Toll-Like Receptors 2, 4 and 9 in Patients With Guillain-Barré Syndrome. Neuroimmunomodulation (2012) 19(1):60–8. doi: 10.1159/000328200
123. Gries M, Davies L, Liu Y, Bachhuber A, Spiegel J, Dillmann U, et al. Response of Toll-Like Receptors in Experimental Guillain-Barré Syndrome: A Kinetic Analysis. Neurosci Lett (2012) 518(2):154–60. doi: 10.1016/j.neulet.2012.04.077
124. Deng YN, Zhou WB. Expression of TLR4 and TLR9 mRNA in Lewis Rats With Experimental Allergic Neuritis. Neuroimmunomodulation (2007) 14(6):337–43. doi: 10.1159/000127433
125. Zhang ZY, Zhang Z, Schluesener HJ. Toll-Like Receptor-2, CD14 and Heat-Shock Protein 70 in Inflammatory Lesions of Rat Experimental Autoimmune Neuritis. Neuroscience (2009) 159(1):136–42. doi: 10.1016/j.neuroscience.2008.12.034
126. Zimmermann C, Weber A, Mausberg AK, Kieseier BC, Hartung HP, Hofstetter HH. T Cell Activation Status Determines the Cytokine Pattern Induced by Zymosan and Bacterial DNA Both in Thymocytes and Splenocytes. Clin Exp Immunol (2013) 172(2):245–53. doi: 10.1111/cei.12037
127. Nyati KK, Prasad KN. Role of Cytokines and Toll-Like Receptors in the Immunopathogenesis of Guillain-Barré Syndrome. Mediators Inflamm (2014) 2014:758639. doi: 10.1155/2014/758639
128. Darabi K, Karulin AY, Boehm BO, Hofstetter HH, Fabry Z, LaManna JC, et al. The Third Signal in T Cell-Mediated Autoimmune Disease? J Immunol (2004) 173(1):92–9. doi: 10.4049/jimmunol.173.1.92
129. Wu X, Li C, Zhang B, Shen D, Li T, Liu K, et al. Predictors for Mechanical Ventilation and Short-Term Prognosis in Patients With Guillain-Barré Syndrome. Crit Care (London England) (2015) 19(1):310. doi: 10.1186/s13054-015-1037-z
130. Nyati KK, Nyati R. Role of Campylobacter Jejuni Infection in the Pathogenesis of Guillain-Barré Syndrome: An Update. BioMed Res Int (2013) 2013:852195. doi: 10.1155/2013/852195
131. Phongsisay V. The Immunobiology of Campylobacter Jejuni: Innate Immunity and Autoimmune Diseases. Immunobiology (2016) 221(4):535–43. doi: 10.1016/j.imbio.2015.12.005
132. Phongsisay V, Hara H, Fujimoto S. Toll-Like Receptors Recognize Distinct Proteinase-Resistant Glycoconjugates in Campylobacter Jejuni and Escherichia Coli. Mol Immunol (2015) 64(1):195–203. doi: 10.1016/j.molimm.2014.11.020
133. Mousavi S, Bereswill S, Heimesaat MM. Novel Clinical Campylobacter Jejuni Infection Models Based on Sensitization of Mice to Lipooligosaccharide, a Major Bacterial Factor Triggering Innate Immune Responses in Human Campylobacteriosis. Microorganisms (2020) 8(4):482. doi: 10.3390/microorganisms8040482
134. Zeb A, Ali SS, Azad AK, Safdar M, Anwar Z, Suleman M, et al. Genome-Wide Screening of Vaccine Targets Prioritization and Reverse Vaccinology Aided Design of Peptides Vaccine to Enforce Humoral Immune Response Against Campylobacter Jejuni. Comput Biol Med (2021) 133:104412. doi: 10.1016/j.compbiomed.2021.104412
135. Dhiman G, Abraham R, Griffin DE. Human Schwann Cells are Susceptible to Infection With Zika and Yellow Fever Viruses, But Not Dengue Virus. Sci Rep (2019) 9(1):9951. doi: 10.1038/s41598-019-46389-0
136. Haque A, Akçeşme FB. Pant AB. A Review of Zika Virus: Hurdles Toward Vaccine Development and the Way Forward. Antiviral Ther (2018) 23(4):285–93. doi: 10.3851/imp3215
137. Ojha CR, Rodriguez M, Lapierre J, Muthu Karuppan MK, Branscome H, Kashanchi F, et al. Complementary Mechanisms Potentially Involved in the Pathology of Zika Virus. Front Immunol (2018) 9:2340. doi: 10.3389/fimmu.2018.02340
138. Dang J, Tiwari SK, Lichinchi G, Qin Y, Patil VS, Eroshkin AM, et al. Zika Virus Depletes Neural Progenitors in Human Cerebral Organoids Through Activation of the Innate Immune Receptor Tlr3. Cell Stem Cell (2016) 19(2):258–65. doi: 10.1016/j.stem.2016.04.014
139. Plociennikowska A, Frankish J, Moraes T, Del Prete D, Kahnt F, Acuna C, et al. TLR3 Activation by Zika Virus Stimulates Inflammatory Cytokine Production Which Dampens the Antiviral Response Induced by RIG-I-Like Receptors. J Virol (2021) 95(10). doi: 10.1128/jvi.01050-20
140. Vanwalscappel B, Tada T, Landau NR. Toll-Like Receptor Agonist R848 Blocks Zika Virus Replication by Inducing the Antiviral Protein Viperin. Virology (2018) 522:199–208. doi: 10.1016/j.virol.2018.07.014
141. Nicoletti F, Créange A, Orlikowski D, Bolgert F, Mangano K, Metz C, et al. Macrophage Migration Inhibitory Factor (MIF) Seems Crucially Involved in Guillain-Barré Syndrome and Experimental Allergic Neuritis. J Neuroimmunol (2005) 168(1-2):168–74. doi: 10.1016/j.jneuroim.2005.07.019
142. Shen D, Lang Y, Chu F, Wu X, Wang Y, Zheng X, et al. Roles of Macrophage Migration Inhibitory Factor in Guillain-Barré Syndrome and Experimental Autoimmune Neuritis: Beneficial or Harmful? Expert Opin Ther Targets (2018) 22(7):567–77. doi: 10.1080/14728222.2018.1484109
143. Wang YZ, Tian FF, Liu H, Zhang W, Li J, Xiao B, et al. Macrophage Migration Inhibitory Factor is Necessary for the Lipo-Oligosaccharide-Induced Response by Modulation of Toll-Like Receptor 4 in Monocytes From GBS Patients. J Neuroimmunol (2013) 257(1-2):67–75. doi: 10.1016/j.jneuroim.2013.01.006
144. Evoli A. Myasthenia Gravis: New Developments in Research and Treatment. Curr Opin Neurol (2017) 30(5):464–70. doi: 10.1097/wco.0000000000000473
145. Barnett C, Katzberg HD, Keshavjee S, Bril V. Thymectomy for non-Thymomatous Myasthenia Gravis: A Propensity Score Matched Study. Orphanet J Rare Dis (2014) 9:214. doi: 10.1186/s13023-014-0214-5
147. Cavalcante P, Cufi P, Mantegazza R, Berrih-Aknin S, Bernasconi P, Le Panse R. Etiology of Myasthenia Gravis: Innate Immunity Signature in Pathological Thymus. Autoimmun Rev (2013) 12(9):863–74. doi: 10.1016/j.autrev.2013.03.010
148. Wang YZ, Yan M, Tian FF, Zhang JM, Liu Q, Yang H, et al. Possible Involvement of Toll-Like Receptors in the Pathogenesis of Myasthenia Gravis. Inflammation (2013) 36(1):121–30. doi: 10.1007/s10753-012-9526-6
149. Cufi P, Dragin N, Weiss JM, Martinez-Martinez P, De Baets MH, Roussin R, et al. Implication of Double-Stranded RNA Signaling in the Etiology of Autoimmune Myasthenia Gravis. Ann Neurol (2013) 73(2):281–93. doi: 10.1002/ana.23791
150. Cordiglieri C, Marolda R, Franzi S, Cappelletti C, Giardina C, Motta T, et al. Innate Immunity in Myasthenia Gravis Thymus: Pathogenic Effects of Toll-Like Receptor 4 Signaling on Autoimmunity. J Autoimmun (2014) 52:74–89. doi: 10.1016/j.jaut.2013.12.013
151. Cavalcante P, Barzago C, Baggi F, Antozzi C, Maggi L, Mantegazza R, et al. Toll-Like Receptors 7 and 9 in Myasthenia Gravis Thymus: Amplifiers of Autoimmunity? Ann New York Acad Sci (2018) 1413(1):11–24. doi: 10.1111/nyas.13534
152. Cavalcante P, Galbardi B, Franzi S, Marcuzzo S, Barzago C, Bonanno S, et al. Increased Expression of Toll-Like Receptors 7 and 9 in Myasthenia Gravis Thymus Characterized by Active Epstein-Barr Virus Infection. Immunobiology (2016) 221(4):516–27. doi: 10.1016/j.imbio.2015.12.007
153. Saba R, Sorensen DL, Booth SA. MicroRNA-146a: A Dominant, Negative Regulator of the Innate Immune Response. Front Immunol (2014) 5:578. doi: 10.3389/fimmu.2014.00578
154. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-Dependent Induction of microRNA miR-146, an Inhibitor Targeted to Signaling Proteins of Innate Immune Responses. Proc Natl Acad Sci USA (2006) 103(33):12481–6. doi: 10.1073/pnas.0605298103
155. Heise N, De Silva NS, Silva K, Carette A, Simonetti G, Pasparakis M, et al. Germinal Center B Cell Maintenance and Differentiation are Controlled by Distinct NF-κb Transcription Factor Subunits. J Exp Med (2014) 211(10):2103–18. doi: 10.1084/jem.20132613
156. Bortone F, Scandiffio L, Marcuzzo S, Bonanno S, Frangiamore R, Motta T, et al. miR-146a in Myasthenia Gravis Thymus Bridges Innate Immunity With Autoimmunity and Is Linked to Therapeutic Effects of Corticosteroids. Front Immunol (2020) 11:142. doi: 10.3389/fimmu.2020.00142
157. Robinet M, Villeret B, Maillard S, Cron MA, Berrih-Aknin S, Le Panse R. Use of Toll-Like Receptor Agonists to Induce Ectopic Lymphoid Structures in Myasthenia Gravis Mouse Models. Front Immunol (2017) 8:1029. doi: 10.3389/fimmu.2017.01029
158. Meinl E, Klinkert WE, Wekerle H. The Thymus in Myasthenia Gravis. Changes Typical for the Human Disease are Absent in Experimental Autoimmune Myasthenia Gravis of the Lewis Rat. Am J Pathol (1991) 139(5):995–1008.
159. Truffault F, de Montpreville V, Eymard B, Sharshar T, Le Panse R, Berrih-Aknin S. Thymic Germinal Centers and Corticosteroids in Myasthenia Gravis: An Immunopathological Study in 1035 Cases and a Critical Review. Clin Rev Allergy Immunol (2017) 52(1):108–24. doi: 10.1007/s12016-016-8558-3
160. Mitsdoerffer M, Peters A. Tertiary Lymphoid Organs in Central Nervous System Autoimmunity. Front Immunol (2016) 7:451. doi: 10.3389/fimmu.2016.00451
161. Zhang P, Yang CL, Liu RT, Li H, Zhang M, Zhang N, et al. Toll-Like Receptor 9 Antagonist Suppresses Humoral Immunity in Experimental Autoimmune Myasthenia Gravis. Mol Immunol (2018) 94:200–8. doi: 10.1016/j.molimm.2018.01.005
162. Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A Randomized, Placebo-Controlled Trial of Natalizumab for Relapsing Multiple Sclerosis. New Engl J Med (2006) 354(9):899–910. doi: 10.1056/NEJMoa044397
163. Hussain RZ, Cravens PC, Doelger R, Dentel B, Herndon E, Loof N, et al. TLR3 Agonism Re-Establishes CNS Immune Competence During α4-Integrin Deficiency. Ann Clin Trans Neurol (2018) 5(12):1543–61. doi: 10.1002/acn3.664
164. Huwiler A, Zangemeister-Wittke U. The Sphingosine 1-Phosphate Receptor Modulator Fingolimod as a Therapeutic Agent: Recent Findings and New Perspectives. Pharmacol Ther (2018) 185:34–49. doi: 10.1016/j.pharmthera.2017.11.001
165. O’Sullivan SA, O’Sullivan C, Healy LM, Dev KK, Sheridan GK. Sphingosine 1-Phosphate Receptors Regulate TLR4-Induced CXCL5 Release From Astrocytes and Microglia. J Neurochem (2018) 144(6):736–47. doi: 10.1111/jnc.14313
166. Fulton B, Markham A. Mycophenolate Mofetil. A Review of its Pharmacodynamic and Pharmacokinetic Properties and Clinical Efficacy in Renal Transplantation. Drugs (1996) 51(2):278–98. doi: 10.2165/00003495-199651020-00007
167. Shigesaka M, Ito T. Mycophenolic Acid, the Active Form of Mycophenolate Mofetil, Interferes With IRF7 Nuclear Translocation and Type I IFN Production by Plasmacytoid Dendritic Cells. Arthritis Res Ther (2020) 22(1):264. doi: 10.1186/s13075-020-02356-z
168. Kim J, Durai P, Jeon D, Jung ID, Lee SJ, Park YM, et al. Phloretin as a Potent Natural TLR2/1 Inhibitor Suppresses TLR2-Induced Inflammation. Nutrients (2018) 10(7):868. doi: 10.3390/nu10070868
169. Reilly M, Miller RM, Thomson MH, Patris V, Ryle P, McLoughlin L, et al. Randomized, Double-Blind, Placebo-Controlled, Dose-Escalating Phase I, Healthy Subjects Study of Intravenous OPN-305, A Humanized Anti-TLR2 Antibody. Clin Pharmacol Ther (2013) 94(5):593–600. doi: 10.1038/clpt.2013.150
170. Monnet E, Lapeyre G, Poelgeest EV, Jacqmin P, Graaf K, Reijers J, et al. Evidence of NI-0101 Pharmacological Activity, an Anti-TLR4 Antibody, in a Randomized Phase I Dose Escalation Study in Healthy Volunteers Receiving LPS. Clin Pharmacol Ther (2017) 101(2):200–8. doi: 10.1002/cpt.522
171. Matsunaga N, Tsuchimori N, Matsumoto T, Ii M. TAK-242 (Resatorvid), a Small-Molecule Inhibitor of Toll-Like Receptor (TLR) 4 Signaling, Binds Selectively to TLR4 and Interferes With Interactions Between TLR4 and its Adaptor Molecules. Mol Pharmacol (2011) 79(1):34–41. doi: 10.1124/mol.110.068064
172. Broadley SA, Vanags D, Williams B, Johnson B, Feeney D, Griffiths L, et al. Results of a Phase IIa Clinical Trial of an Anti-Inflammatory Molecule, Chaperonin 10, in Multiple Sclerosis. Multiple sclerosis (Houndmills Basingstoke England) (2009) 15(3):329–36. doi: 10.1177/1352458508099141
173. Ma CJ, Ni L, Zhang Y, Zhang CL, Wu XY, Atia AN, et al. PD-1 Negatively Regulates Interleukin-12 Expression by Limiting STAT-1 Phosphorylation in Monocytes/Macrophages During Chronic Hepatitis C Virus Infection. Immunology (2011) 132(3):421–31. doi: 10.1111/j.1365-2567.2010.03382.x
174. Lai CY, Su YW, Lin KI, Hsu LC. Natural Modulators of Endosomal Toll-Like Receptor-Mediated Psoriatic Skin Inflammation. J Immunol Res (2017) 2017:7807313. doi: 10.1155/2017/7807313
175. Calore F, Londhe P, Fadda P, Nigita G, Casadei L, Marceca GP, et al. The TLR7/8/9 Antagonist IMO-8503 Inhibits Cancer-Induced Cachexia. Cancer Res (2018) 78(23):6680–90. doi: 10.1158/0008-5472.can-17-3878
176. Balak DM, van Doorn MB, Arbeit RD, Rijneveld R, Klaassen E, Sullivan T, et al. IMO-8400, a Toll-Like Receptor 7, 8, and 9 Antagonist, Demonstrates Clinical Activity in a Phase 2a, Randomized, Placebo-Controlled Trial in Patients With Moderate-to-Severe Plaque Psoriasis. Clin Immunol (Orlando Fla) (2017) 174:63–72. doi: 10.1016/j.clim.2016.09.015
177. Wiese MD, Manning-Bennett AT, Abuhelwa AY. Investigational IRAK-4 Inhibitors for the Treatment of Rheumatoid Arthritis. Expert Opin invest Drugs (2020) 29(5):475–82. doi: 10.1080/13543784.2020.1752660
178. Danto SI, Shojaee N, Singh RSP, Li C, Gilbert SA, Manukyan Z, et al. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of PF-06650833, a Selective Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) Inhibitor, in Single and Multiple Ascending Dose Randomized Phase 1 Studies in Healthy Subjects. Arthritis Res Ther (2019) 21(1):269. doi: 10.1186/s13075-019-2008-6
179. Dudhgaonkar S, Ranade S. Selective IRAK4 Inhibition Attenuates Disease in Murine Lupus Models and Demonstrates Steroid Sparing Activity. J Immuno (2017) 198: (3):1308–19. doi: 10.4049/jimmunol.1600583
180. Van Tassell BW, Seropian IM, Toldo S, Salloum FN, Smithson L, Varma A, et al. Pharmacologic Inhibition of Myeloid Differentiation Factor 88 (MyD88) Prevents Left Ventricular Dilation and Hypertrophy After Experimental Acute Myocardial Infarction in the Mouse. J Cardiovasc Pharmacol (2010) 55(4):385–90. doi: 10.1097/FJC.0b013e3181d3da24
181. DeVry CG, Valdez M, Gao L, Wang J, Kotsch K, Volk HD, et al. RDP58, a Novel Immunomodulatory Peptide, Ameliorates Clinical Signs of Disease in the Lewis Rat Model of Acute Experimental Autoimmune Encephalomyelitis. J Neuroimmunol (2004) 152(1-2):33–43. doi: 10.1016/j.jneuroim.2004.03.020
182. Mistry P, Laird MH, Schwarz RS, Greene S, Dyson T, Snyder GA, et al. Inhibition of TLR2 Signaling by Small Molecule Inhibitors Targeting a Pocket Within the TLR2 TIR Domain. Proc Natl Acad Sci USA (2015) 112(17):5455–60. doi: 10.1073/pnas.1422576112
183. Chang YC, Kao WC, Wang WY, Wang WY, Yang RB, Peck K. Identification and Characterization of Oligonucleotides That Inhibit Toll-Like Receptor 2-Associated Immune Responses. FASEB J Off Publ Fed Am Soc Exp Biol (2009) 23(9):3078–88. doi: 10.1096/fj.09-129312
184. Patra MC, Achek A, Kim GY, Panneerselvam S. A Novel Small-Molecule Inhibitor of Endosomal TLRs Reduces Inflammation and Alleviates Autoimmune Disease Symptoms in Murine Models. Cells (2020) 9(7):1648. doi: 10.3390/cells9071648
185. Scarneo SA, Hughes PF, Yang KW, Carlson DA, Gurbani D, Westover KD, et al. A Highly Selective Inhibitor of Interleukin-1 Receptor-Associated Kinases 1/4 (IRAK-1/4) Delineates the Distinct Signaling Roles of IRAK-1/4 and the TAK1 Kinase. J Biol Chem (2020) 295(6):1565–74. doi: 10.1074/jbc.RA119.011857
186. Zanotto-Filho A, Delgado-Cañedo A, Schröder R, Becker M, Klamt F, Moreira JC. The Pharmacological NFkappaB Inhibitors BAY117082 and MG132 Induce Cell Arrest and Apoptosis in Leukemia Cells Through ROS-Mitochondria Pathway Activation. Cancer Lett (2010) 288(2):192–203. doi: 10.1016/j.canlet.2009.06.038
187. Krishnan N, Bencze G, Cohen P, Tonks NK. The Anti-Inflammatory Compound BAY-11-7082 is a Potent Inhibitor of Protein Tyrosine Phosphatases. FEBS J (2013) 280(12):2830–41. doi: 10.1111/febs.12283
188. Newton R, Holden NS, Catley MC, Oyelusi W, Leigh R, Proud D, et al. Repression of Inflammatory Gene Expression in Human Pulmonary Epithelial Cells by Small-Molecule IkappaB Kinase Inhibitors. J Pharmacol Exp Ther (2007) 321(2):734–42. doi: 10.1124/jpet.106.118125
189. Li J, Wang X, Zhang F, Yin H. Toll-Like Receptors as Therapeutic Targets for Autoimmune Connective Tissue Diseases. Pharmacol Ther (2013) 138(3):441–51. doi: 10.1016/j.pharmthera.2013.03.003
190. So EY, Kang MH, Kim BS. Induction of Chemokine and Cytokine Genes in Astrocytes Following Infection With Theiler’s Murine Encephalomyelitis Virus is Mediated by the Toll-Like Receptor 3. Glia (2006) 53(8):858–67. doi: 10.1002/glia.20346
191. Carpentier PA, Williams BR, Miller SD. Distinct Roles of Protein Kinase R and Toll-Like Receptor 3 in the Activation of Astrocytes by Viral Stimuli. Glia (2007) 55(3):239–52. doi: 10.1002/glia.20450
192. Chen B, Sun L, Zhang X. Integration of Microbiome and Epigenome to Decipher the Pathogenesis of Autoimmune Diseases. J Autoimmun (2017) 83:31–42. doi: 10.1016/j.jaut.2017.03.009
193. de Kivit S, Tobin MC, Forsyth CB, Keshavarzian A, Landay AL. Regulation of Intestinal Immune Responses Through TLR Activation: Implications for Pro- and Prebiotics. Front Immunol (2014) 5:60. doi: 10.3389/fimmu.2014.00060
194. Keogh CE, Rude KM, Gareau MG. Role of Pattern Recognition Receptors and the Microbiota in Neurological Disorders. J Physiol (2021) 599(5):1379–89. doi: 10.1113/jp279771
195. Lombardi VC, De Meirleir KL, Subramanian K, Nourani SM, Dagda RK, Delaney SL, et al. Nutritional Modulation of the Intestinal Microbiota; Future Opportunities for the Prevention and Treatment of Neuroimmune and Neuroinflammatory Disease. J Nutr Biochem (2018) 61:1–16. doi: 10.1016/j.jnutbio.2018.04.004
196. Riccio P, Rossano R. Nutrition Facts in Multiple Sclerosis. ASN Neuro (2015) 7(1):1759091414568185. doi: 10.1177/1759091414568185
197. Tourkochristou E, Triantos C, Mouzaki A. The Influence of Nutritional Factors on Immunological Outcomes. Front Immunol (2021) 12:665968. doi: 10.3389/fimmu.2021.665968
198. Kim SY, Koo JE, Song MR, Lee JY. Retinol Suppresses the Activation of Toll-Like Receptors in MyD88- and STAT1-Independent Manners. Inflammation (2013) 36(2):426–33. doi: 10.1007/s10753-012-9562-2
199. Mazur-Bialy AI, Pochec E, Plytycz B. Immunomodulatory Effect of Riboflavin Deficiency and Enrichment - Reversible Pathological Response Versus Silencing of Inflammatory Activation. J Physiol Pharmacol an Off J Polish Physiol Soc (2015) 66(6):793–802.
200. Sadeghi K, Wessner B, Laggner U, Ploder M, Tamandl D, Friedl J, et al. Vitamin D3 Down-Regulates Monocyte TLR Expression and Triggers Hyporesponsiveness to Pathogen-Associated Molecular Patterns. Eur J Immunol (2006) 36(2):361–70. doi: 10.1002/eji.200425995
201. Ma N, Ma X. Dietary Amino Acids and the Gut-Microbiome-Immune Axis: Physiological Metabolism and Therapeutic Prospects. Compr Rev Food Sci Food Saf (2019) 18: (1):221–42. doi: 10.1111/1541-4337.12401
202. Morshedi M, Hashemi R, Moazzen S, Sahebkar A, Hosseinifard ES. Immunomodulatory and Anti-Inflammatory Effects of Probiotics in Multiple Sclerosis: A Systematic Review. J Neuroinflamm (2019) 16(1):231. doi: 10.1186/s12974-019-1611-4
203. Wang Y, Wu Y, Sailike J, Sun X, Abuduwaili N, Tuoliuhan H, et al. Fourteen Composite Probiotics Alleviate Type 2 Diabetes Through Modulating Gut Microbiota and Modifying M1/M2 Phenotype Macrophage in Db/Db Mice. Pharmacol Res (2020) 161:105150. doi: 10.1016/j.phrs.2020.105150
204. Farrokhi V, Nemati R, Nichols FC, Yao X, Anstadt E, Fujiwara M, et al. Bacterial Lipodipeptide, Lipid 654, is a Microbiome-Associated Biomarker for Multiple Sclerosis. Clin Trans Immunol (2013) 2(11):e8. doi: 10.1038/cti.2013.11
205. Wasko NJ, Nichols F, Clark RB. Multiple Sclerosis, the Microbiome, TLR2, and the Hygiene Hypothesis. Autoimmun Rev (2020) 19(1):102430. doi: 10.1016/j.autrev.2019.102430
206. Curtis JR, Singh JA. Use of Biologics in Rheumatoid Arthritis: Current and Emerging Paradigms of Care. Clin Ther (2011) 33(6):679–707. doi: 10.1016/j.clinthera.2011.05.044
207. Varshney D, Qiu SY, Graf TP, McHugh KJ. Employing Drug Delivery Strategies to Overcome Challenges Using TLR7/8 Agonists for Cancer Immunotherapy. AAPS J (2021) 23(4):90. doi: 10.1208/s12248-021-00620-x
208. Sooklert K, Boonwong C, Ekpo P, Rojanathanes R, Patarakul K, Chirathaworn C, et al. Effect of Gold Nanoparticles on the TLR2-Mediated Inflammatory Responses Induced by Leptospira in TLR2-Overexpressed HEK293 Cells. Nanomaterials (Basel) (2020) 10(12):208. doi: 10.3390/nano10122522
209. Feng X, Liu J, Xu W, Li G, Ding J. Tackling Autoimmunity With Nanomedicines. Nanomedicine (Lond) (2020) 15(16):1585–97. doi: 10.2217/nnm-2020-0102
210. Zeb A, Rana I, Choi HI, Lee CH, Baek SW, Lim CW, et al. Potential and Applications of Nanocarriers for Efficient Delivery of Biopharmaceuticals. Pharmaceutics (2020) 12(12):1184. doi: 10.3390/pharmaceutics12121184
211. Kim H, Griffith TS. Poly(d,l-Lactide-Co-Glycolide) Nanoparticles as Delivery Platforms for TLR7/8 Agonist-Based Cancer Vaccine. J Pharmacol Exp Ther (2019) 370(3):715–24. doi: 10.1124/jpet.118.254953
212. Bourzac K. Nanotechnology: Carrying Drugs. Nature (2012) 491(7425):S58–60. doi: 10.1038/491s58a
213. Ni K. Nanoscale Metal-Organic Framework Co-Delivers TLR-7 Agonists and Anti-CD47 Antibodies to Modulate Macrophages and Orchestrate Cancer Immunotherapy. J Am Chem Soc (2020) 142(29):12579–84. doi: 10.1021/jacs.0c05039
214. Lu L, Duong VT, Shalash AO, Skwarczynski M. Chemical Conjugation Strategies for the Development of Protein-Based Subunit Nanovaccines. Nanovaccines.Vaccines (Basel) (2021) 9(6). doi: 10.3390/vaccines9060563
215. Widmer J, Thauvin C, Mottas I, Nguyen VN, Delie F, Allémann E, et al. Polymer-Based Nanoparticles Loaded With a TLR7 Ligand to Target the Lymph Node for Immunostimulation. Int J Pharm (2018) 535(1-2):444–51. doi: 10.1016/j.ijpharm.2017.11.031
216. Zhang L, Wu S, Qin Y, Fan F, Zhang Z, Huang C, et al. Targeted Codelivery of an Antigen and Dual Agonists by Hybrid Nanoparticles for Enhanced Cancer Immunotherapy. Nano Lett (2019) 19(7):4237–49. doi: 10.1021/acs.nanolett.9b00030
217. Arleevskaya MI, Larionova RV, Brooks WH, Bettacchioli E, Renaudineau Y. Toll-Like Receptors, Infections, and Rheumatoid Arthritis. Clin Rev Allergy Immunol (2020) 58: (2):172–81. doi: 10.1007/s12016-019-08742-z
Keywords: toll-like receptors, neuroimmune diseases, inhibitors, multiple sclerosis, neuromyelitis optica spectrum disorder, Guillain-Barré syndrome, Myasthenia gravis
Citation: Li H, Liu S, Han J, Li S, Gao X, Wang M, Zhu J and Jin T (2021) Role of Toll-Like Receptors in Neuroimmune Diseases: Therapeutic Targets and Problems. Front. Immunol. 12:777606. doi: 10.3389/fimmu.2021.777606
Received: 15 September 2021; Accepted: 15 October 2021;
Published: 01 November 2021.
Edited by:
Clio Mavragani, National and Kapodistrian University of Athens, GreeceCopyright © 2021 Li, Liu, Han, Li, Gao, Wang, Zhu and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tao Jin, jin_tao@jlu.edu.cn
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.