 Zhipeng Zheng
Zhipeng Zheng Baohong Wang
Baohong Wang- State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Diseases, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, The First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Diverse liver diseases undergo a similar pathophysiological process in which liver regeneration follows a liver injury. Given the important role of the gut-liver axis in health and diseases, the role of gut microbiota-derived signals in liver injury and regeneration has attracted much attention. It has been observed that the composition of gut microbiota dynamically changes in the process of liver regeneration after partial hepatectomy, and gut microbiota modulation by antibiotics or probiotics affects both liver injury and regeneration. Mechanically, through the portal vein, the liver is constantly exposed to gut microbial components and metabolites, which have immense effects on the immunity and metabolism of the host. Emerging data demonstrate that gut-derived lipopolysaccharide, gut microbiota-associated bile acids, and other bacterial metabolites, such as short-chain fatty acids and tryptophan metabolites, may play multifaceted roles in liver injury and regeneration. In this perspective, we provide an overview of the possible molecular mechanisms by which gut microbiota-derived signals modulate liver injury and regeneration, highlighting the potential roles of gut microbiota in the development of gut microbiota-based therapies to alleviate liver injury and promote liver regeneration.
Introduction
The liver has an outstanding regenerative capacity (1). Liver regeneration is a well-orchestrated biological process that depends on a large series of signals. Following different types of damage, the remnant liver initiates different types of reprogramming events, which activate different progenitor cells to replace injured cells (2). The regenerating liver undergoes numerous adaptive responses in gene expression, growth factor production, and morphological structure, which have been extensively described (1, 3). The essential gene expressions required for liver regeneration cover cytokine, growth factor, and metabolic, which interact with each other and fine-tune regenerative responses to maintain hepatic homeostasis according to body demands (4). The classical mechanisms of liver regeneration focus on signaling pathways within the liver. However, recent studies have evidenced that commensal gut microbiota plays local and systematic roles in tissue repair and regeneration (5). Therefore, it is extremely important to describe the interaction between gut microbiota and liver in the regulation of liver injury and regeneration.
Through the portal vein, the liver is constantly exposed to bacterial components and gut microbial metabolites. Lipopolysaccharide (LPS) is a cell wall component of gram-negative bacteria, and a mild release of LPS from the gut can stimulate liver regeneration and tissue repair (6–8). Besides, gut microbial metabolites, such as bile acids, short-chain fatty acids (SCFAs), and tryptophan metabolites, affect host metabolism and immune system (9, 10), which may indirectly influence liver injury and regeneration. Herein, the review aims to elucidate the potential molecular mechanisms that gut microbiota interacts with the liver from the perspective of injury and regeneration, which may provide valuable clues to develop gut microbiota-based therapies for liver diseases.
Gut Microbiota and Liver Diseases
Gut microbiota dysbiosis has been found in liver diseases with distinct etiologies, including acute liver injury, viral hepatitis, non-alcoholic fatty liver disease (NAFLD), alcohol-related liver disease, autoimmune hepatitis (AIH), primary biliary cholangitis (PBC), and primary sclerosing cholangitis (PSC) (11, 12). (Table 1) Diverse liver diseases undergo a similar pathophysiological process, in which the damaged liver needs repair and gut microbiota may play an important role.
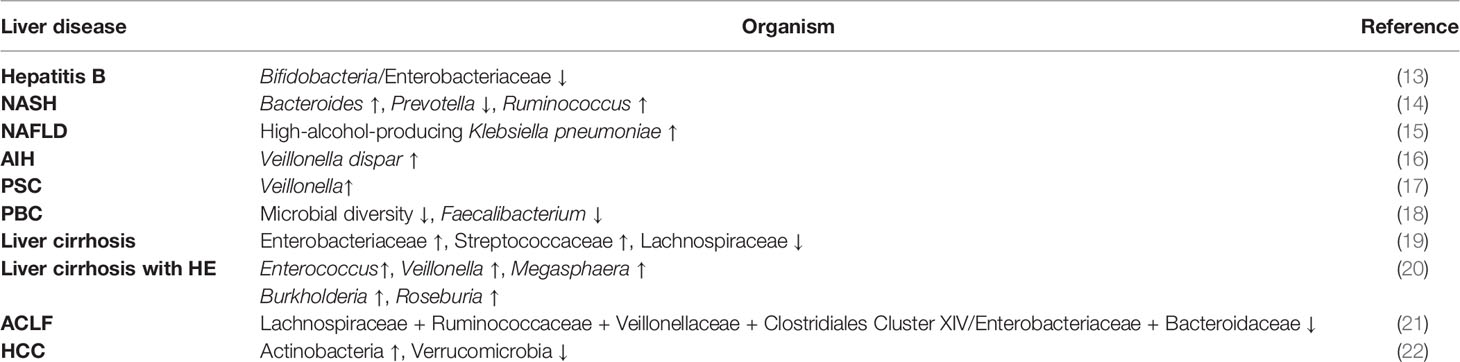
Table 1 Gut dysbiosis in liver diseases.
In pre-cirrhotic liver disease, the gut microbiota has changed. The ratio of Bifidobacteria/Enterobacteriaceae is gradually reduced in healthy individuals, hepatitis B virus carriers, patients with chronic hepatitis B, and patients with decompensated cirrhosis (13), suggesting that the alteration of gut microbiota is associated with disease progression in hepatitis B patients. In NASH patients, the relative abundance of Bacteroides significantly increases and Prevotella decreases, whereas the relative abundance of Ruminococcus is higher in F ≥ 2 fibrosis patients (14). A recent study finds that high-alcohol-producing Klebsiella pneumoniae is associated with almost 60% of NAFLD, and high-alcohol-producing Klebsiella pneumoniae supplement by oral gavage induces NAFLD in mice (15), suggesting that endogenous alcohol production by gut microbiota drives NAFLD in some cases. In addition, a gut microbiota-based metagenomic signature can be used to distinguish mild and moderate NAFLD from advanced fibrosis (23). For AIH, increased abundance of Veillonella dispar is linked to disease severity, and the combination of Veillonella, Lactobacillus, Oscillospira, and Clostridiales discriminates AIH from controls (16). The relative abundance of Veillonella is also enriched in PSC compared with healthy controls and ulcerative colitis patients without liver diseases (17). Microbial diversity is significantly reduced in PBC patients, and Faecalibacterium is further decreased in gp210-positive than gp210-negative PBC patients (18). These correlations between pre-cirrhotic liver diseases and gut microbiota indicate that the primary injury of liver diseases shape, and are shaped by, changes in gut microbiota composition.
Gut dysbiosis is more obvious in liver cirrhosis, the pathological end-stage of chronic liver disease, and can cause complications. The enrichment of potentially pathogenic bacteria Enterobacteriaceae and Streptococcaceae and the reduction of beneficial bacteria Lachnospiraceae in patients with liver cirrhosis have a positive and negative correlation with the Child-Turcotte-Pugh score, which is used to assess the severity of cirrhosis based on five clinical parameters, respectively (19). Quantitative metagenomics reveals that a combination of 15 optimal microbiota-targeted gene markers (NLF009_gene_80134, H16_gene_75905, et al.) discriminates liver cirrhosis patients from healthy individuals with a training AUC value of 0.918 and a validating AUC value of 0.836 (24). Decompensated liver cirrhosis could be accompanied by a severe central nervous system, namely hepatic encephalopathy (HE). There is no difference in stool microbiota between HE and no-HE patients, but mucosal microbiota obtained by sigmoidoscopy is different with increased abundance of Enterococcus, Veillonella, Megasphaera, and Burkholderia and decreased abundance of Roseburia in HE patients (20). The further development of decompensated liver cirrhosis towards acute-on-chronic liver failure (ACLF). The cirrhosis dysbiosis ratio (Lachnospiraceae + Ruminococcaceae + Veillonellaceae + Clostridiales Cluster XIV/Enterobacteriaceae + Bacteroidaceae) is lower in patients who progress to ACLF and associated with an elevated risk of extra-hepatic failure and death (21). Besides, most hepatocellular carcinoma (HCC) develops in the setting of advanced liver cirrhosis. Dysbiosis of gut microbiota is associated with increased inflammation, impaired intestinal barrier, and immune system disorders, which are involved in HCC development (22). The complexity and role of gut microbiota in end-stage chronic liver disease suggest that gut microbiota modulation may be a way to deal with difficulties in liver injury and regeneration.
Consequently, these studies suggest that gut microbiota may have a significant impact on the pathophysiology of liver diseases, in which abnormal ductular responses, excessive fibrosis, and impaired innate immunity can inhibit normal regeneration and lead to liver failure or tumors. How to restore the regenerative ability of the failed liver is an essential problem to be solved in clinical scenarios. A comprehensive understanding of the underlying mechanisms may enable appropriate targets of gut microbiota-based therapies to reduce the factors that inhibit liver regeneration or directly stimulate liver regeneration.
Gut Microbiota Dynamically Changes During Liver Regeneration
The relationship between gut microbiota and liver regeneration has been studied in the animal partial hepatectomy models (Figure 1). Dynamic changes of gut microbiota are observed in mice from 0 hours to 9 days after partial hepatectomy (25). Partial hepatectomy leads to a distinct change in the composition of gut microbiota, early at 1 hour after partial hepatectomy, with steadily increased Bacteroidetes and decreased Firmicutes, which account for the most abundant phyla. At the family level, increased S24-7 and Rikenellaceae make up the most abundant taxa within Bacteroidetes phylum, while decreased Clostridiaceae, Lachnospiraceae and Ruminococcaceae are the most abundant representatives in Firmicutes phylum. Moreover, alteration of S24-7, Lachnospiraceae, and Ruminococcaceae is closely associated with hepatic metabolism and proliferation. The shifts of bacterial populations persist for 9 days in mice after partial hepatectomy, which almost covers all of the priming phase, proliferative phase, and termination phase of regenerating liver (25).
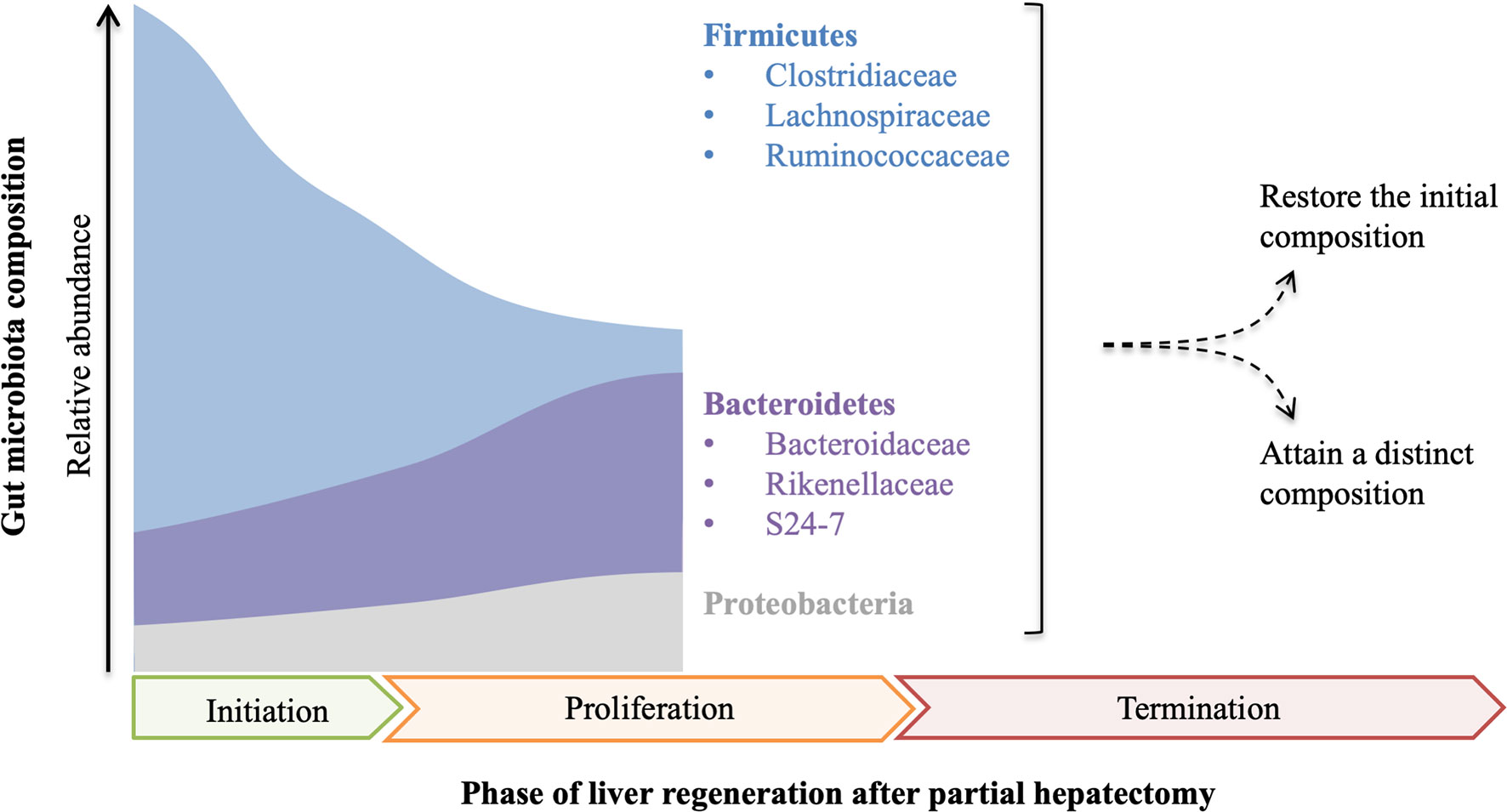
Figure 1 The composition of gut microbiota is fluctuant in the course of liver regeneration. After partial hepatectomy, the relative abundance of Firmicutes decreases while Bacteroidetes and Proteobacteria increase, which probably continues until the middle of the proliferation phase of liver regeneration. As proliferation and termination of liver regeneration progress, the final composition of gut microbiota remains controversial and needs further study.
In the other study, fluctuating alterations of gut microbiota are observed in rats after partial hepatectomy (26). In this study, the abundance of Bacteroidetes rapidly decreases at 12 hours after partial hepatectomy, but steadily increases to the initial level at 48 hours, and then decreases to a low level again at 72 hours and lasts to the endpoint. Compared with Bacteroidetes, the alteration of Firmicutes shows a different trend. The ratio of Firmicutes to Bacteroidetes (F/B) is fluctuant throughout the process of liver regeneration. Notably, the abundance of Proteobacteria has a remarkable elevation at 48 hours after partial hepatectomy, but it almost decreases to the initial level before the endpoint. At the family level, Lachnospiraceae and Ruminococcaceae increase in 12-24 hours and 3-14 days after partial hepatectomy, but they are decreased in 30-48 hours. Furthermore, cluster analysis indicates that the composition of gut microbiota is different along with the process of liver regeneration (26).
Although the changing trends of gut microbiota are not identical in the only two published studies, which may be the consequence of different experimental designs, increased Bacteroidetes and decreased Firmicutes are recognized in the priming phase and the partial proliferative phase of liver regeneration. As the proliferation and termination of liver regeneration progress, whether the composition of gut microbiota is finally restored or attains a new state remains unclear and needs further study. These data indicate that gut microbiota has a potential influence on the regenerating liver or vice versa.
Gut Microbiota Manipulations Affect Liver Injury and Regeneration
Gut microbiota is depleted in germ-free model and can be modulated by antibiotics, probiotics, prebiotics, such as dietary fibers (27), fecal microbiota transplant (FMT), and colon resection. All of these gut microbiota manipulations influence liver injury and regeneration (Figure 2).
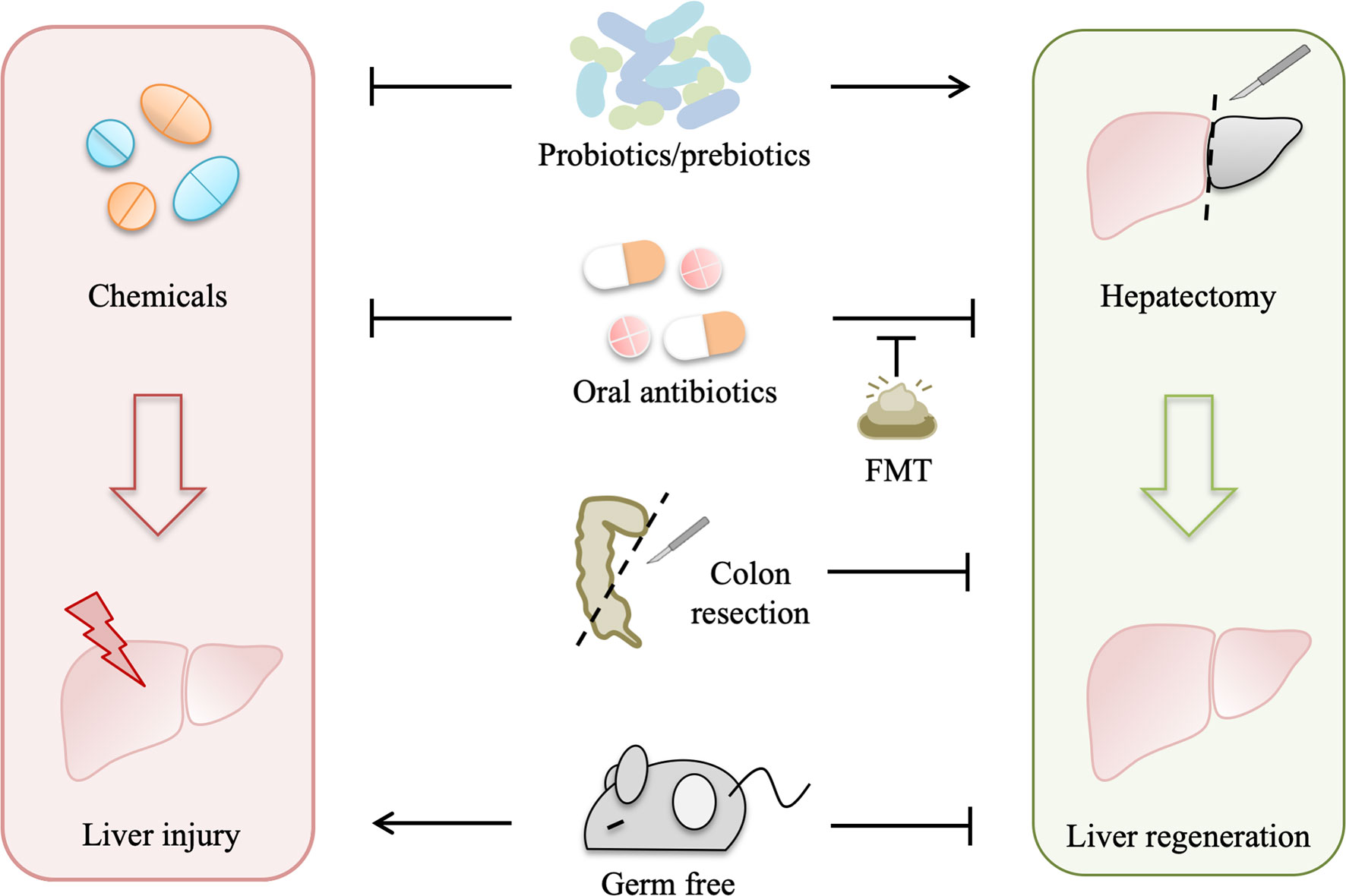
Figure 2 Gut microbiota manipulations affect liver injury and regeneration. Gut microbiota depletion by several approaches, including germ-free, antibiotics, and colon resection, suppresses liver regeneration to varying degrees, while fecal microbiota transplant (FMT) can normalize liver regeneration in the antibiotic-treated model, and probiotics/prebiotics can promote liver regeneration. In addition, both probiotics and oral antibiotics alleviate drug-induced acute liver injury, which is aggravated in germ-free rodents.
Gut bacterial depletion using oral non-absorbable antibiotics depresses liver regeneration in rats after partial hepatectomy, and liver regeneration is also impaired in germ-free mice with partial hepatectomy (7, 8). Likewise, liver regeneration is suppressed in rats with simultaneous liver and colon partial resection (28). These studies indicate that gut microbiota is required for normal liver regeneration. Interestingly, gut microbiota participates in liver injury as well as liver regeneration. Oral antibiotics prevent liver injury induced by hepatotoxic agents, such as CCl4, acetaminophen, D-Gal, and alcohol (29), whereas a complete absence of gut microbiota as in germ-free rodents can exacerbate the acute liver injury (30), suggesting that gut microbiota also contributes to the pathophysiology of drug-induced liver injury.
Moreover, FMT normalizes impaired liver regeneration in rats with gut decontamination by antibiotics (26), which further manifests that normal gut microbiota plays a driving role in liver regeneration. Probiotics improve the outcome of partial hepatectomy, not only in animal experiments but also in some clinical trials (31–33). Probiotic supplement improves mitosis in the liver of rats with simultaneous 70% partial hepatectomy and colon anastomosis probably by preventing bacteria translocation (31). In hepatocellular carcinoma (HCC) patients receiving hepatic resection, preoperative and postoperative probiotics improve liver function and reduce complications (32). In another pilot study with 19 patients subjected to right hepatectomy, symbiotics can improve liver function after liver resection in the uncomplicated subgroup (33). Likewise, emerging evidence demonstrates that probiotics alleviate drug-induced liver injury in animal experiments. Lactobacillus rhamnosus improves liver function and ameliorates alcohol-induced liver injury in mice (34, 35). Bifidobacterium adolescentis, Bacillus cereus, and Lactobacillus helveticus pretreatments can modify the gut microbiota and alleviate liver injury in D-Gal-treated rats (36–38). Akkermansia muciniphila protects mice from immune-mediated liver injury (39).
Dynamic changes of gut microbiota during liver regeneration and the benefits of FMT and probiotics on liver injury and regeneration indicate that the crosstalk between the liver and gut microbiota is important for liver regeneration, which is probably mediated by gut microbiota-derived components and metabolites.
Gut-Derived LPS Have Multiple Effects on Liver Injury and Regeneration
Gut-derived LPS, produced by enteric gram-negative bacteria, are continually presented to the liver (6), and low-grade portal venous LPS can be cleared by the liver (40). Under normal conditions, gut-derived LPS can be phagocytized and detoxified by Kupffer cells in the liver reticuloendothelial system (RES) (41). When the liver suffers from an extended injury, homeostasis between the formation and removal of gut-derived LPS is broken, which may be due to the following reasons. First, sensitivity to gut-derived LPS is increased after the initial liver damage. Second, RES injury leads to hampered detoxification and clearance of gut-derived LPS. Third, gut barrier dysfunction allows more translocation of LPS. Last, bacteria overgrowth and delay of gastrointestinal motility increase production and spillover of gut-derived LPS.
Gut-Derived LPS Aggravate the Liver Injury
Gut-derived LPS as a cofactor plays a universal role in acute liver injury, which has been demonstrated in acute liver injury models induced by different hepatotoxic agents, including CCl4, acetaminophen, alcohol, and D-Gal (29). Under primary liver damage, gut-derived LPS can activate Kupffer cells to release pro-inflammatory mediators, such as TNF-α, interleukins (IL-1 and IL-10), lysosomal enzymes (protease and phosphatase), and superoxide, which aggravate inflammatory responses and necrosis (29). Induction of LPS tolerance protects rats from CCl4-induced liver necrosis, and LPS-binding protein also has a protective effect on acute liver injury (29).
In addition to acute liver injury, gut-derived LPS plays a critical role in chronic liver injury. Elevated plasma endotoxin is observed in patients with alcoholic liver disease (ALD) and experimental models of alcoholic liver injury, which can be attenuated by oral antibiotics (42, 43). However, LPS alone fails to mimic ethanol-induced steatosis, but together with ethanol, which is metabolized to acetaldehyde by gut bacteria and intestinal mucosa, leads to hepatocyte steatosis (42). Besides, fibrogenesis usually occurs in the advanced ALD and other chronic liver diseases, such as nonalcoholic fatty liver disease (NAFLD) and chronic hepatitis B (CHB), which can develop into cirrhosis (44). Chronic liver diseases are accompanied by dysbiosis of gut microbiota, which contributes to intestinal dysmotility, inflammation, and mucosal leakage, leading to continuous and excessive liver exposure to gut-derived LPS (45). Toll-like receptor-4 (TLR4) mutation and gut sterilization prevent hepatic fibrosis in mice, revealing that gut-derived LPS contribute to hepatic fibrosis (46). On one hand, when the liver is exposed to increased gut-derived LPS, TLR4 activation of hepatic stellate cells upregulates the production of chemokine (CCL2) and induces chemotaxis of Kupffer cells (47). On the other hand, LPS binding to TLR4 on hepatic stellate cells downregulates transforming growth factor β (TGF-β) pseudoreceptor BAMBI through Myd88- NF-κB-dependent signals, which sensitizes hepatic stellate cells to TGF-β released by Kupffer cells, promoting transdifferentiation of quiescent hepatic stellate cells to activated scar-forming myofibroblasts (46), and myofibroblasts generate extracellular matrix (ECM) materials, including collagen, laminin, and fibronectin. Excessive deposition of ECM will cause aberrant scar formation and fibrosis, which diminishes liver regeneration (48).
In addition, chronic liver diseases are driven by vicious cycles of liver injury, inflammation, repair, and regeneration, which make an opportunity for hepatocellular carcinoma (HCC) development (49). Previous TLR4-deficient, gut-sterilized, germ-free, and LPS-treated animal experiments have evidenced that gut-derived LPS contribute to hepatocarcinogenesis (50, 51). Mechanistically, the LPS-TLR4 pathway contributes to liver tumor promotion by increasing proliferation and preventing apoptosis of non-bone marrow-derived resident liver cells, and in the early phases of HCC, TLR4-dependent secretion of hepatomitogen epiregulin by hepatic stellate cells mediates HCC promotion (51).
The broad roles of gut-derived LPS in liver injury are well established and accepted, but applicating this knowledge to develop an effective treatment remains challenging, which needs further study.
Gut-Derived LPS Promote Liver Regeneration
Gut-derived LPS also plays an important role in liver regeneration. Restriction of gut-derived LPS by gut bacterial depletion, endotoxin neutralization, and induction of endotoxin tolerance significantly impairs liver regeneration in rats, which is reversed by exogenous LPS supplements (7). In addition, impaired liver regeneration is also observed in germ-free mice receiving partial hepatectomy and in rats simultaneously receiving partial hepatectomy and colon bowel resection (8, 28).
When the liver is subjected to an experimental physical or a chemical injury, gut-derived LPS will pass through the compromised liver and spill into the general circulation, leading to low-grade systemic endotoxemia, which elicits hepatotrophic factors production, such as insulin, glucagon, epidermal growth factor (EGF), vasopressin and triiodothyronine (T3) (6, 52). In addition, gut-derived LPS activates Kupffer cells by binding to TLR-4 for activation of NF-κB and subsequently stimulates the production of TNF-α, which in return activates Kupffer cells to secrete interleukin-6 (IL-6) (53). IL-6 trans-signaling through the soluble IL-6/IL-6R complex induces hepatic stellate cells to produce hepatocyte growth factor (HGF) (54). HGF cooperates with other extrahepatic factors, such as T3, insulin, and EGF, allowing the remnant hepatocytes to overcome cell-cycle checkpoint control to proliferate, which is essential for the priming phase of liver regeneration after partial hepatectomy (53, 54). Finally, when the liver regenerates sufficiently and the phagocytosis function of the Kupffer cell is restored, gut-derived LPS in the portal blood can be detoxified again (6).
Therefore, gut-derived LPS are important for liver regeneration as well as liver injury, liver fibrosis, and liver tumors, which may depend on the degree and duration of exposure (Figure 3). However, it is difficult to determine the beneficial level of gut-derived LPS for the liver in different scenarios to avoid the deleterious effects of excessive TLR-4 activation. One promising strategy is modulating gut microbiota by probiotics, prebiotics, and perhaps appropriate antibiotics, such as rifaximin, to control gut-derived LPS.
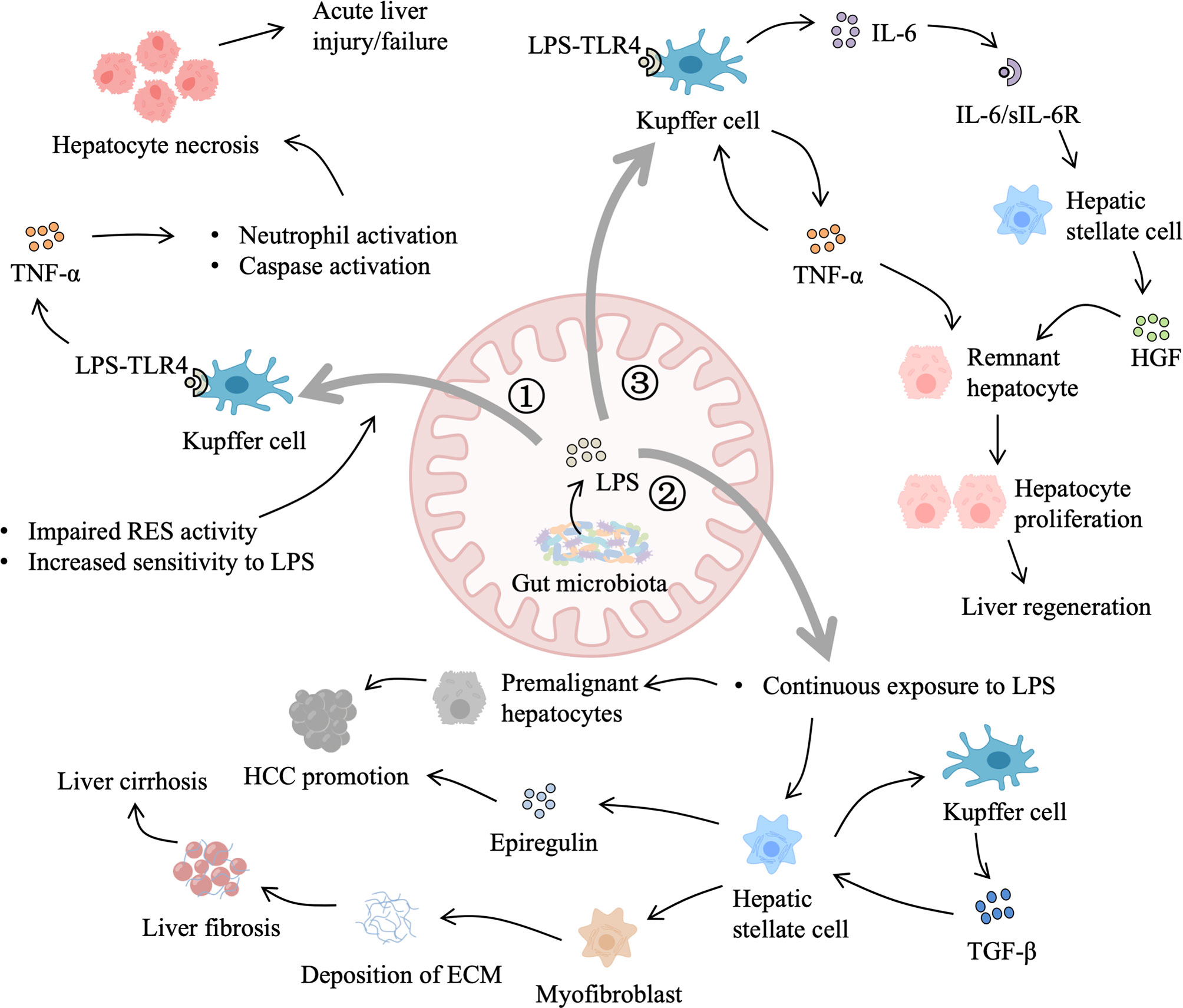
Figure 3 Role of gut-derived LPS in liver injury and regeneration. 1) When the liver suffers from an acute injury, primary damage reduces the activity of the reticuloendothelial system (RES) and increases liver sensitivity to LPS. LPS binding to Toll-like receptor 4 (TLR4) on Kupffer cells triggers the production of tumor necrosis factor-α (TNF-α), which leads to hepatocyte necrosis and aggravates the primary damage. 2) In addition, gut-derived LPS also contributes to chronic liver injury. Dysbiosis of gut microbiota leads to elevated LPS and impaired gut barrier function. Continuous LPS exposure sensitizes hepatic stellate cells to transforming growth factor-β (TGF-β) and promotes the transdifferentiation of hepatic stellate cells into myofibroblasts, resulting in the generation of extracellular matrix (ECM) materials. Excessive deposition of ECM interferes with normal regeneration, leading to liver cirrhosis and hepatocellular carcinoma (HCC) promotion. Besides, LPS accelerates the development of premalignant hepatocytes and stimulates hepatic stellate cells to secret epiregulin, which facilitates HCC promotion. 3) Moreover, after partial hepatectomy, gut-derived LPS activate Kupffer cells to secrete TNF-α, which in return stimulates Kupffer cells to produce interleukin-6 (IL-6). IL-6/IL-6 receptor complex induces the production of hepatocyte growth factor (HGF) by hepatic stellate cells. In addition, gut-derived LPS that escape Kupffer cells spilling into the general circulation elicit systemic hepatotrophic factors production. HGF, TNF-α, and other hepatotrophic factors allow remnant hepatocytes to overcome cell-cycle checkpoint and support liver regeneration.
Gut Microbiota-Associated Bile Acid Metabolism Is Involved in Liver Injury and Regeneration
Bile acids (BAs), produced from cholesterol, are assembled as primary conjugated BAs in the liver and actively transported into the biliary system. A small fraction of BAs circulates from cholangiocytes to the liver through the cholangio-hepatic shunt, while most of them are stored in the gallbladder and released into the duodenum after food intake. Approximately 95% of BAs are reabsorbed via the apical sodium-dependent bile acid transporter (ASBT) in the terminal small intestine and return to the liver through the portal vein. Conjugated primary BAs can also be deconjugated by the gut microbiota and escape reabsorption, further dehydroxylated by microbial bioconversion to secondary BAs. A part of secondary BAs is passively absorbed by colonic cells. Spillover of BAs into systemic circulation can be cleared via urinary excretion. Bile acids that are lost in urinary and fecal excretion are replenished by hepatic synthesis (55, 56).
In normal conditions, BAs almost recycle within the enterohepatic circulation, which has important physiological roles in nutrient absorption and biliary secretion of lipids and toxic metabolites, and only traces of BAs escape to the systemic circulation. After partial hepatectomy, BAs that are reabsorbed from the intestine suddenly become too high for the remnant liver, which leads to an abrupt and massive spillover of BAs to the systemic circulation (57, 58). Bile acids overload beyond a certain threshold is deleterious (59). However, BAs are also necessary for normal liver regeneration. Depletion of BAs by cholestyramine (a BA-sequestering resin) leads to suppression of liver regeneration (60). In rats after partial hepatectomy and mice with CCl4-induced injury, elevated BAs accelerate liver regeneration, while low levels of BAs impair liver regeneration (60, 61). The similar phenomenon is observed in clinical scenarios, following a major hepatectomy, patients without external biliary drainage have better liver regeneration than those with external biliary drainage (62). Gut microbiota-dependent BA metabolism is likely to participate in liver injury and regeneration by modifying the quality (conjugated vs deconjugated and primary vs secondary BAs) and quantity of the BA pool (Figure 4).
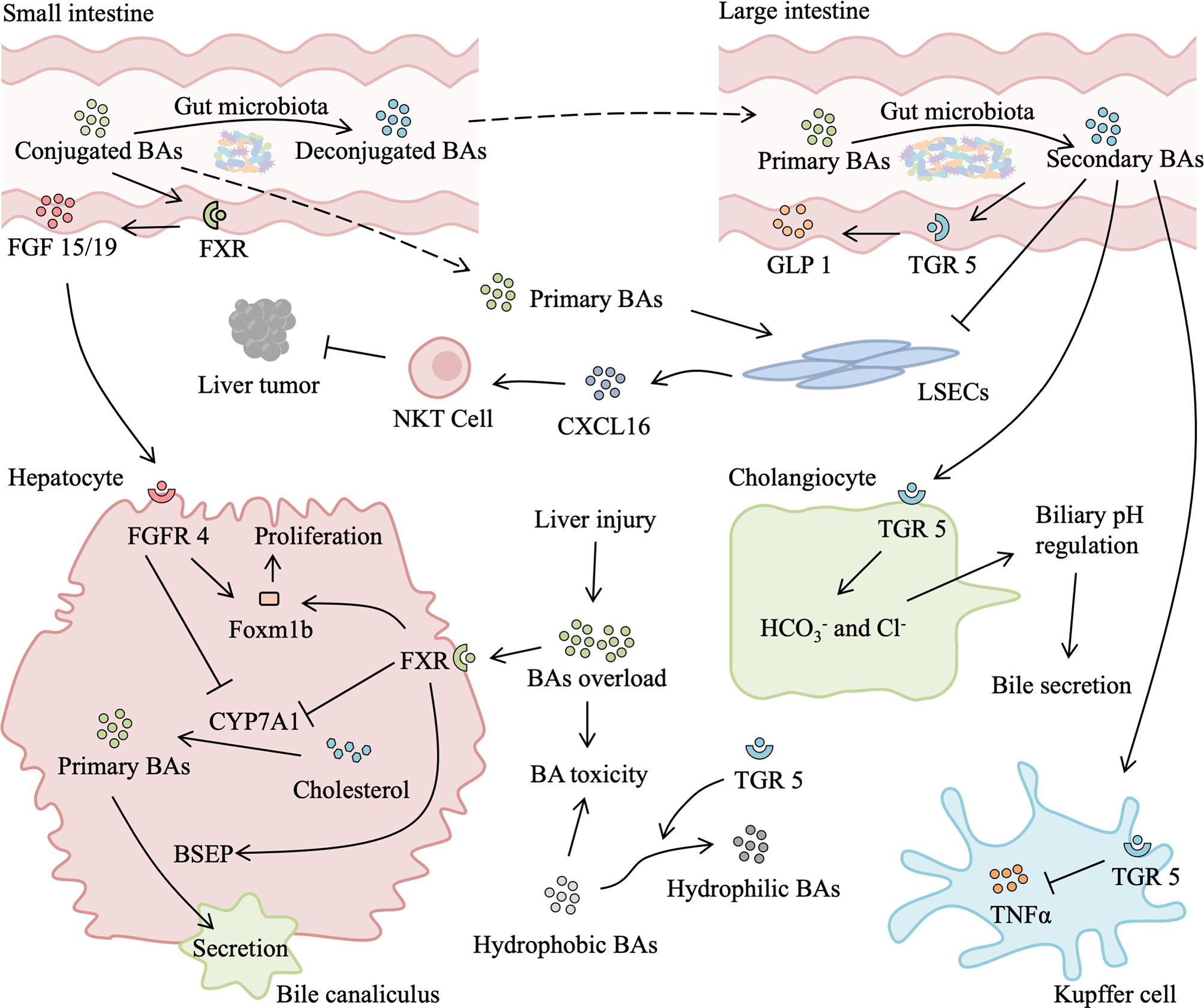
Figure 4 Role of gut microbiota-associated bile acids metabolism in liver injury and regeneration. After a liver injury or partial hepatectomy, bile acids (BAs) absorbed from the intestine suddenly become too high for the remnant liver, leading to secondary liver injury. Excessive BAs binding to hepatic farnesoid X receptor (FXR) inhibits transcription of cytochrome P450 family 7 subfamily A member 1 (CYP7A1), which reduces the production of primary BAs. In addition, activated FXR can promote the secretion of primary BAs into bile canaliculus and upregulate expression of Foxm1b, thus relieving BAs overload and facilitating liver regeneration. Meanwhile, in the small intestine, BAs activate intestinal FXR to secret fibroblast growth factor (FGF) 15/19, which binds to fibroblast growth factor receptor 4 (FGFR4). FGFR4/agonist also induces expression of Foxm1b and improves proliferation. Gut microbiota is responsible for secondary BAs production in the large intestine. Secondary BAs binding to intestinal transforming growth factor 5 (TGR5) elicits secretion of glucagon-like peptide 1 (GLP1), which activates the insulin signaling pathway. Absorbed secondary BAs binding to hepatic TGR5 has an anti-inflammatory effect by suppressing the release of TNF-α from the Kupffer cell. TGR5 activation also promotes BAs secretion by secreting and Cl- from cholangiocyte and increasing transformation of hydrophilic BAs from hydrophobic BAs, which reduce BAs load and BAs toxicity-induced liver injury, promoting liver regeneration. Moreover, primary BAs stimulate and secondary BAs suppress the expression of chemokine (C-X-C motif) ligand 16 (CXCL16) by liver sinusoidal endothelial cells (LSECs). CXCL16 is a chemokine that recruits natural killer T (NKT) cells, which suppress liver tumors.
Gut Microbiota-Mediated Deconjugation of BAs Participates in Liver Injury and Regeneration
After partial hepatectomy, to protect the remnant liver and biliary tree from excess BAs, the basolateral uptake and BAs production are decreased and the basolateral efflux and biliary excretion are increased. Deconjugation (removal of the glycine or taurine) by gut microbiota with bile salt hydrolase (BSH) prevents reabsorption of BAs in the small intestine (63). BSH is enriched in the human gut microbiota and mediates bile tolerance (64). Conjugated BAs are transported by sodium taurocholate cotransporting polypeptide (NTCP) and organic anion-transporting polypeptide (OATP) isoform, which are the major uptake transports of BAs in the liver. The mRNA levels of NTCP, OATP1, and OATP2 are decreased with the most prominent decrease of NTCP, while the protein level of NTCP is markedly decreased during the initial phase of liver regeneration (65). Downregulated NTCP relieves basolateral over uptake of BAs in the liver, but OATP expressing hepatocytes could ensure ongoing basolateral uptake of BAs after partial hepatectomy. However, one study demonstrates that the mRNA and protein levels of NTCP are unchanged after partial hepatectomy (66).
Despite the conflicting effects of partial hepatectomy on NTCP expression, high levels of BAs in the remnant liver are generally accepted. BAs overload inhibits the synthesis of BAs by negative feedback regulation through the nuclear receptor farnesoid X receptor (FXR), which is highly expressed in the liver and ileum (63). Chenodeoxycholic acid (CDCA) is the most potent efficacious ligand of FXR, followed by lithocholic acid (LCA), deoxycholic acid (DCA), and cholic acid (CA) (67). LCA and DCA are secondary BAs transformed from primary BAs by microbial 7α-dehydroxylation, which is a characteristic of Clostridium and Eubacaterium (68, 69). In the liver, FXR activation induces expression of small heterodimer partner 1 (SHP-1), which can inhibit expression of CYP7A1, the rate-limiting enzyme of BAs synthesis, by reducing the activity of liver receptor homolog 1 (LRH-1) (70). Therefore, BAs overload leads to decreased BAs production by repressing the transcription of the rate-limiting enzyme in BAs synthesis. In addition, increased basolateral efflux and biliary excretion of BAs reduce BAs concentrations in hepatocytes, which protects the liver from BA-induced liver injury. Conjugated BAs are primarily secreted into bile via the canalicular bile salt export pump (BSEP) (55). The expression of BSEP is increased from days 1 to 3 after partial hepatectomy, which depends on the activation of FXR (60). Thus, the remnant liver copes with the BAs overload via FXR to maintain normal BAs levels, including both repressions of synthesis and induction of export.
Besides, hepatic FXR plays a critical role in the expression of Foxm1b, a key regulator of the hepatic cell cycle, promoting liver regeneration after either partial hepatectomy or CCl4-induced liver injury (71). Moreover, compared with liver-specific FXR knock-out (KO) mice, conventional FXR KO mice show significantly decreased liver regeneration response at 36 h and 72 h after partial hepatectomy, suggesting that FXR activation in other tissues also contribute to liver regeneration (71).
Intestinal FXR may participate in the promotion of liver regeneration through secreting fibroblast growth factor (FGF) 15 in mice or FGF19 in humans. Trans-enterocytic BAs flux from the intestinal lumen to the basolateral side drives FXR-dependent FGF15 synthesis in the ileum (72). FGF15 reaches the liver and regulates BA homeostasis by an FGFR4-dependent activation of c-Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) pathways, leading to transcriptional inhibition of CYP7A1 in hepatocytes (72). Therefore, BAs-mediated intestinal FXR-dependent FGF15 production appears as a necessary gut-derived signal for liver protection after partial hepatectomy by maintaining BAs homeostasis. Besides, FGF15 may directly contribute to liver regeneration by stimulating the proliferation of hepatocytes and cholangiocytes (73). Mechanistically, the FGF15-FGFR4-STAT3 signaling pathway, which is required for Foxm1 transcription and cell cycle progression, controls hepatocyte proliferation in the regenerating liver (74). Furthermore, it should be noted that FXR is expressed in the kidney at high levels and in the thymus, spleen, ovary, testes, heart, and eyes at lower levels (63). Increased systemic BAs after partial hepatectomy may also act on these tissues and organs in an FXR-dependent manner and indirectly affect liver regeneration, which needs further exploration.
Taken together, gut microbiota-mediated deconjugation improves the reabsorption of BAs and the abundance of colonic primary conjugated BAs, which may induce protective as well as proliferative cascades in the damaged liver by initiating FXR-dependent responses.
Gut Microbiota-Dependent Production of Secondary BAs Facilitates Liver Regeneration
Gut microbiota dehydrogenates primary BAs to secondary BAs mainly in the colon. Secondary BAs are the most efficient agonists of Taketa G-protein-coupled receptor 5 (TGR5) (75). TGR5 is ubiquitously expressed in many tissues, including the liver, gallbladder, intestine, brown and white adipose tissue, skeletal muscle, and so on (63). In the liver, TGR5 is highly expressed in cholangiocytes, Kupffer cells, and endothelial cells but weakly or not expressed in hepatocytes (76). Therefore, TGR5-dependent protective effects against BAs overload are likely due to other mechanisms rather than regulation of BAs synthesis. TGR5 regulates BA size and composition by reducing hydrophobicity and increasing secretion. A shift towards a more hydrophobic BAs pool is associated with inhibition of liver regeneration (77). TGR5-KO mice have a more hydrophobic BAs composition and hydrophobic BAs accumulation in the liver leads to toxic injury, which is alleviated by a BA resin enriched diet (58). In addition, TGR5 promotes cystic fibrosis transmembrane conductance regulator (CFTR)-dependent Cl- secretion and BAs uptake into biliary epithelia and reduces biliary bile acid concentrations (78). TGR5-dependent increased output of biliary and Cl- after partial hepatectomy also enhances bile secretion, which prevents the remnant liver from BAs-induced toxicity (58). Furthermore, the production and release of the cytokine after partial hepatectomy are crucial for normal liver regeneration. It has been demonstrated that the immunosuppressive effect of secondary BAs on macrophage is mediated by TGR5 (79), which inhibits LPS-induced expression of cytokines and reduces liver injury (80), by suppressing NF-κB transcription activity and its target gene expression (81). Therefore, gut microbiota-controlled activation of TGR5 may contribute to liver regeneration by regulating hepatic inflammatory response and limiting hepatocyte necrosis after partial hepatectomy. Moreover, TGR5 in skeletal muscle and brown adipose tissue promotes energy expenditure through iodothyronine deiodinase 2 enzyme (DIO2), which converts inactive thyroxine into active thyroid hormone (63). TGR5 in colonic L cells mediates synthesis and secretion of intestinal glucagon-like peptide-1 (GLP-1), which could stimulate insulin secretion (82). Thyroid hormone and insulin cooperated with other growth factors allow the hepatocyte to overcome cell-cycle checkpoint control, initiating and regulating liver regeneration (53).
Gut microbiota-dependent conversion of primary BAs to secondary BAs is also involved in the development of liver cancer (83). Primary BAs stimulate, whereas secondary BAs suppress, the expression of chemokine (C-X-C motif) ligand 16 (CXCL16) by liver sinusoidal endothelial cells, which induces accumulation of hepatic CXCR6+ natural killer T (NKT) cells and production of interferon-γ, inhibiting both primary and metastatic liver tumors.
In general, gut microbiota-dependent production of secondary BAs may reduce the inflammatory response and promote liver regeneration by TGR5-dependent regulation of the BA pool and production of T3 and GLP-1. Elucidating ways to fine-tune gut microbiota-BAs-host interaction is a promising strategy to assist normal liver regeneration.
Gut Microbial Metabolites Short-Chain Fatty Acids and Indoles May Assist Liver Regeneration in a Similar Way
Short Chain Fatty Acids May Assist Liver Regeneration
Short-chain fatty acids (SCFAs) produced by gut microbial fermentation have multiple physiological functions (84). The most abundant SCFAs in the gut are acetate, propionate, and butyrate (85, 86). Bacteroidetes produce acetate and propionate, and Firmicutes are the primary butyrate producers (87–90).
The intestinal tract is the major site of SCFAs production and the biological concentration gradient falls from the gut to the peripheral tissues. Butyrate is largely metabolized in the intestinal epithelium, and the rest is degraded in the liver (91, 92). Most propionate is degraded in the liver, and a substantial portion of acetate passes into the systemic circulation (92, 93). SCFAs function as extracellular agonists for G-protein-coupled receptor (GPR) 41 and GPR43 (94). Butyrate, but not acetate or propionate, also activates the GPR109A (95). GPR41, GPR43, and GPR109A are expressed by intestinal epithelial cells and immune cells (84). Stimulation of GPRs by SCFAs activates ERK1/2, c-JNK, and p38/mitogen-activated protein kinases (MAPKs) (96). In addition, butyrate broadly affects transcription by activating histone acetyltransferases (HATs) and suppressing nuclear class I histone (HDACs) (84). Butyrate can also act as an extracellular agonist activating peroxisome proliferator-activated receptor γ (PPAR-γ), which plays anti-inflammatory effects (97, 98).
SCFAs are important substrates for the integrity of the epithelial barrier, which limits pro-inflammatory load to the liver. Butyrate enhances gut barrier function by increasing the expression of claudin-1 and zonula occludens-1 (ZO-1), decreasing LPS translocation and inhibiting downstream inflammatory responses (99, 100). Therefore, SCFAs may indirectly affect liver injury and regeneration through maintaining gut barrier function.
In addition, SCFAs play potential roles in metabolism homeostasis during liver regeneration. Acetate, propionate, and butyrate regulate hepatic glucose and lipid homeostasis in a PPAR-γ dependent manner (101). Acetate and propionate inhibit adipocyte lipolysis via GPR43, which reduces free fatty acid (FFA) flux to the liver and ameliorates the deterioration of glucose homeostasis caused by fatty liver (102, 103). Propionate and butyrate have metabolic benefits through activating gluconeogenesis gene expression (104). Moreover, SCFAs stimulate gut hormone production of GLP-1 and peptide YY (PYY), improving the metabolic phenotype (105). Butyrate inhibits NF-κB activation in lamina propria macrophages and reduces the responsiveness of lamina propria macrophages to commensal bacteria (106, 107). Butyrate alleviates ischemia-reperfusion liver injury by preventing NF-κB activation and reducing inflammatory factors production (108). Propionate and butyrate have potential roles in the production and function of regulatory T cells via inhibiting histone deacetylase (HDAc) (109, 110).
These studies highlight the interaction between gut microbiota-derived SCFAs and the immune and metabolic homeostasis of the host, which may play an important role in liver injury and regeneration (Figure 5).
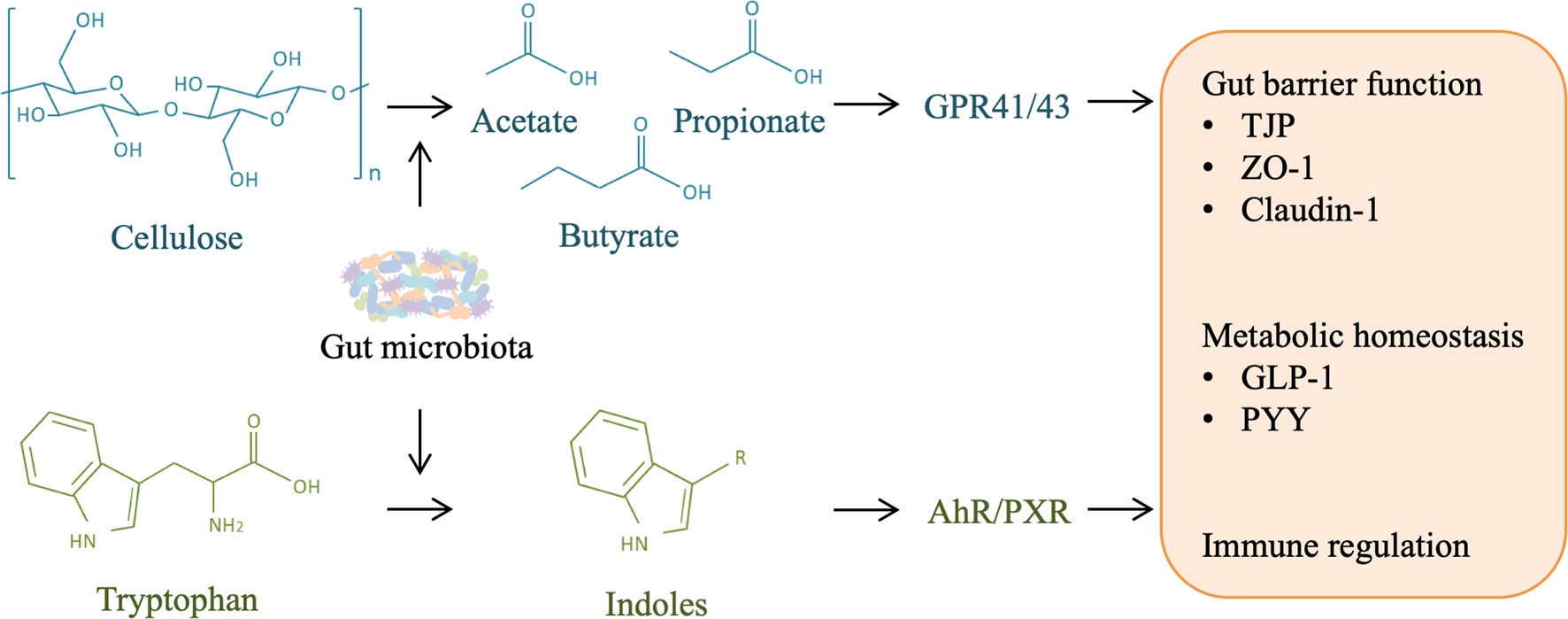
Figure 5 Role of other microbial metabolites in liver injury and regeneration. Gut microbiota is responsible for the production of short-chain fatty acids (SCFAs) and indoles from cellulose and tryptophan in food-intake, respectively. SCFAs can activate host G-protein-coupled receptor (GPR) 41/43, and indoles can activate aryl hydrocarbon receptor (AHR)/pregnane X receptor (PXR), contributing to gut barrier protection, metabolism homeostasis, and immune regulation, which may facilitate liver regeneration by alleviating liver injury.
Indoles May Assist Liver Regeneration
Tryptophan is one of the essential amino acids and must be supplied by dietary uptake. The majority of tryptophan is absorbed in the small intestine, and a significant fraction may also reach the colon. Intestinal tryptophan metabolism follows three major pathways: (1) the kynurenine pathway in epithelial and immune cells through indoleamine 2,3-dioxygenase 1 (IDO1); (2) the serotonin pathway in enterochromaffin cells through tryptophan hydroxylase 1 (TpH1); and (3) direct transformation by commensal bacteria into indole and indole derivatives (111). Numerous bacterial species can metabolize tryptophan, which has been described in a previous review (112). Indole is the most abundant metabolite of gut microbial tryptophan, which is followed by indol-3-acetic acid (IAA) and indole-3-propionic acid (IPA), in adults (112). Indole and many indole derivatives, such as IAA, IPA, indole-3-aldehyde (IAld), tryptamine (TA), 3-methylindole (skatole), and indoxyl-3-sulfate (I3S), are endogenous ligands of aryl hydrocarbon receptor (AhR) (113). AhR is initially well-known for its major role in the metabolism and elimination of environmental toxicants, but it also serves as a key transcription factor controlling many critical cellular functions and organ homeostasis (114).
Given the production site, the role of gut microbial tryptophan metabolism is preponderant in intestinal AhR activity. Activation of AhR in the gut improves intestinal barrier function by decreasing gut permeability and mucosal inflammation (83). Indole upregulates the expression of genes involved in the maintenance of epithelial-cell tight-junction resistance (115, 116). Indoleacrylic acid (IA) improves intestinal epithelial barrier function by promoting goblet cell differentiation and mucus production, which is possibly mediated by AhR activation (117). Moreover, in the context of indole, IPA also regulates intestinal barrier function by acting as a ligand for the pregnane X receptor (PXR) (118). These studies suggest that tryptophan metabolites enhance the intestinal epithelial barrier function by AhR and PXR signaling pathways, which decreases translocation of gut-derived LPS and then could participate in liver injury and regeneration. Similar to SCFAs, indole is also able to modulate the secretion of GLP-1 from colonic L cells and influence host metabolism in an AhR-dependent manner (119, 120). GLP-1 has an important role in glucose homeostasis and liver function, which is probably involved in liver regeneration.
In addition, AhR is expressed by many immune cells and plays an important role in the regulation of immune response in health and disease (113). Gut microbiota-derived IAA and TA reduce inflammatory responses in macrophages and hepatocytes (121), suggesting that gut microbial tryptophan metabolites could regulate immune responses in the liver as well, which indicates their potential roles in liver injury and regeneration. However, the exact effects of gut microbial tryptophan metabolites on liver regeneration still need further research (Figure 5).
Conclusion
Mechanisms of liver regeneration are complex. The crosstalk between gut microbiota and liver allows that gut-derived signals are orchestrated in liver injury and regeneration. Gut-derived LPS and gut microbiota-associated bile acid metabolism appear to have multiple effects on liver injury and regeneration, while SCFAs and tryptophan metabolites produced by gut microbiota have potential benefits in liver regeneration. A comprehensive understanding of these roles of gut-derived signals in liver injury and regeneration will enable the further development of rational specific therapies to either directly improve liver regeneration or prevent complications that appear in the process of liver regeneration. The role of gut-derived signals in animal models of liver regeneration has been briefly described in this review, but lessons learned from animal models still need to be confirmed in more clinical settings, such as chronic liver injury with abnormal liver architecture and advanced liver fibrosis, in which normal regeneration of compromised liver is expected. However, gut-derived signals have the promise to motivate translational and interventional studies of liver injury and regeneration.
Author Contributions
ZZ managed the literature research and wrote the first draft of the manuscript. BW critically reviewed the manuscript and provided valuable discussions and criticisms. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by National Natural Science Foundation of China [grant numbers 82170668 and 81790633], Sino-German Center for Research Promotion [grant numbers GZ1546], and CAMS Innovation Fund for Medical Sciences [grant numbers 2019-I2M-5-045].
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Michalopoulos GK. Hepatostat: Liver Regeneration and Normal Liver Tissue Maintenance. Hepatology (2017) 65(4):1384–92. doi: 10.1002/hep.28988
2. Michalopoulos GK, Khan Z. Liver Stem Cells: Experimental Findings and Implications for Human Liver Disease. Gastroenterology (2015) 149(4):876–82. doi: 10.1053/j.gastro.2015.08.004
3. Forbes SJ, Newsome PN. Liver Regeneration - Mechanisms and Models to Clinical Application. Nat Rev Gastroenterol Hepatol (2016) 13(8):473–85. doi: 10.1038/nrgastro.2016.97
4. Fausto N, Campbell JS, Riehle KJ. Liver Regeneration. Hepatology (2006) 43(2 Suppl 1):S45–53. doi: 10.1002/hep.20969
5. Shavandi A, Saeedi P, Gerard P, Jalalvandi E, Cannella D, Bekhit AE, et al. The Role of Microbiota in Tissue Repair and Regeneration. J Tissue Eng Regener Med (2020) 14(3):539–55. doi: 10.1002/term.3009
6. Cornell RP. Gut-Derived Endotoxin Elicits Hepatotrophic Factor Secretion for Liver Regeneration. Am J Physiol (1985) 249(5 Pt 2):R551–62. doi: 10.1152/ajpregu.1985.249.5.R551
7. Cornell RP. Restriction of Gut-Derived Endotoxin Impairs DNA Synthesis for Liver Regeneration. Am J Physiol (1985) 249(5 Pt 2):R563–9. doi: 10.1152/ajpregu.1985.249.5.R563
8. Cornell RP, Liljequist BL, Bartizal KF. Depressed Liver Regeneration After Partial Hepatectomy of Germ-Free, Athymic and Lipopolysaccharide-Resistant Mice. Hepatology (1990) 11(6):916–22. doi: 10.1002/hep.1840110603
9. Van Treuren W, Dodd D. Microbial Contribution to the Human Metabolome: Implications for Health and Disease. Annu Rev Pathol (2019) 24(15):345–69. doi: 10.1146/annurev-pathol-020117-043559
10. Garrett WS. Immune Recognition of Microbial Metabolites. Nat Rev Immunol (2019) 20(2):91–2. doi: 10.1038/s41577-019-0252-2
11. Bajaj JS, Khoruts A. Microbiota Changes and Intestinal Microbiota Transplantation in Liver Diseases and Cirrhosis. J Hepatol (2020) 72(5):1003–27. doi: 10.1016/j.jhep.2020.01.017
12. Tokuhara D. Role of the Gut Microbiota in Regulating Non-Alcoholic Fatty Liver Disease in Children and Adolescents. Front Nutr (2021) 8:700058. doi: 10.3389/fnut.2021.700058
13. Lu H, Wu Z, Xu W, Yang J, Chen Y, Li L, et al. Intestinal Microbiota was Assessed in Cirrhotic Patients With Hepatitis B Virus Infection. Intestinal Microbiota of HBV Cirrhotic Patients. Microb Ecol (2011) 61(3):693–703. doi: 10.1007/s00248-010-9801-8
14. Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, et al. The Severity of Nonalcoholic Fatty Liver Disease Is Associated With Gut Dysbiosis and Shift in the Metabolic Function of the Gut Microbiota. Hepatology (2016) 63(3):764–75. doi: 10.1002/hep.28356
15. Yuan J, Chen C, Cui J, Lu J, Yan C, Wei X, et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella Pneumoniae. Cell Metab (2019) 30(4):675–88.e7. doi: 10.1016/j.cmet.2019.08.018
16. Wei Y, Li Y, Yan L, Sun C, Miao Q, Wang Q, et al. Alterations of Gut Microbiome in Autoimmune Hepatitis. Gut (2020) 69(3):569–77. doi: 10.1136/gutjnl-2018-317836
17. Kummen M, Holm K, Anmarkrud JA, Nygard S, Vesterhus M, Hoivik ML, et al. The Gut Microbial Profile in Patients With Primary Sclerosing Cholangitis Is Distinct From Patients With Ulcerative Colitis Without Biliary Disease and Healthy Controls. Gut (2017) 66(4):611–9. doi: 10.1136/gutjnl-2015-310500
18. Tang R, Wei Y, Li Y, Chen W, Chen H, Wang Q, et al. Gut Microbial Profile Is Altered in Primary Biliary Cholangitis and Partially Restored After UDCA Therapy. Gut (2018) 67(3):534–41. doi: 10.1136/gutjnl-2016-313332
19. Chen Y, Yang F, Lu H, Chen W, Chen H, Wang Q, et al. Characterization of Fecal Microbial Communities in Patients With Liver Cirrhosis. Hepatology (2011) 54(2):562–72. doi: 10.1002/hep.24423
20. Bajaj JS, Hylemon PB, Ridlon JM, Heuman DM, Daita K, White MB, et al. Colonic Mucosal Microbiome Differs From Stool Microbiome in Cirrhosis and Hepatic Encephalopathy and Is Linked to Cognition and Inflammation. Am J Physiol Gastrointest Liver Physiol (2012) 303(6):G675–85. doi: 10.1152/ajpgi.00152.2012
21. Bajaj JS, Vargas HE, Reddy KR, Lai JC, O'Leary JG, Tandon P, et al. Association Between Intestinal Microbiota Collected at Hospital Admission and Outcomes of Patients With Cirrhosis. Clin Gastroenterol Hepatol (2019) 17(4):756–65.e3. doi: 10.1016/j.cgh.2018.07.022
22. Rattan P, Minacapelli CD, Rustgi V. The Microbiome and Hepatocellular Carcinoma. Liver Transpl (2020) 26(10):1316–27. doi: 10.1002/lt.25828
23. Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut Microbiome-Based Metagenomic Signature for Non-Invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab (2017) 25(5):1054–62.e5. doi: 10.1016/j.cmet.2017.04.001
24. Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, et al. Alterations of the Human Gut Microbiome in Liver Cirrhosis. Nature (2014) 513(7516):59–64. doi: 10.1038/nature13568
25. Liu HX, Rocha CS, Dandekar S, Wan YJ. Functional Analysis of the Relationship Between Intestinal Microbiota and the Expression of Hepatic Genes and Pathways During the Course of Liver Regeneration. J Hepatol (2016) 64(3):641–50. doi: 10.1016/j.jhep.2015.09.022
26. Bao Q, Yu L, Chen D, Li L. Variation in the Gut Microbial Community Is Associated With the Progression of Liver Regeneration. Hepatol Res (2019) 50(1):121–36. doi: 10.1111/hepr.13424.
27. Gasaly N, de Vos P, Hermoso MA. Impact of Bacterial Metabolites on Gut Barrier Function and Host Immunity: A Focus on Bacterial Metabolism and Its Relevance for Intestinal Inflammation. Front Immunol (2021) 12:658354. doi: 10.3389/fimmu.2021.658354
28. Miyazaki M, Kohda S, Itoh H, Kaiho T, Kimura F, Ambiru S, et al. Inhibition of Hepatic Regeneration After 70% Partial Hepatectomy by Simultaneous Resection of the Bowel in Rats. Eur Surg Res (1995) 27(6):396–405. doi: 10.1159/000129426
29. Nolan JP. The Role of Intestinal Endotoxin in Liver Injury: A Long and Evolving History. Hepatology (2010) 52(5):1829–35. doi: 10.1002/hep.23917
30. Hartmann P, Chu H, Duan Y, Schnabl B. Gut Microbiota in Liver Disease: Too Much Is Harmful, Nothing at All Is Not Helpful Either. Am J Physiol Gastrointest Liver Physiol (2019) 316(5):G563–73. doi: 10.1152/ajpgi.00370.201
31. Seehofer D, Rayes N, Schiller R, Stockmann M, Muller AR, Schirmeier A, et al. Probiotics Partly Reverse Increased Bacterial Translocation After Simultaneous Liver Resection and Colonic Anastomosis in Rats. J Surg Res (2004) 117(2):262–71. doi: 10.1016/j.jss.2003.11.021
32. Rifatbegovic Z, Mesic D, Ljuca F, Zildzic M, Avdagic M, Grbic K, et al. Effect of Probiotics on Liver Function After Surgery Resection for Malignancy in the Liver Cirrhotic. Med Arh (2010) 64(4):208–11.
33. Rayes N, Pilarski T, Stockmann M, Bengmark S, Neuhaus P, Seehofer D. Effect of Pre- and Probiotics on Liver Regeneration After Resection: A Randomised, Double-Blind Pilot Study. Benef Microbes (2012) 3(3):237–44. doi: 10.3920/BM2012.0006
34. Wang Y, Kirpich I, Liu Y, Ma Z, Barve S, McClain CJ, et al. Lactobacillus Rhamnosus GG Treatment Potentiates Intestinal Hypoxia-Inducible Factor, Promotes Intestinal Integrity and Ameliorates Alcohol-Induced Liver Injury. Am J Pathol (2011) 179(6):2866–75. doi: 10.1016/j.ajpath.2011.08.039
35. Wang Y, Liu Y, Kirpich I, Ma Z, Wang C, Zhang M, et al. Lactobacillus Rhamnosus GG Reduces Hepatic TNFalpha Production and Inflammation in Chronic Alcohol-Induced Liver Injury. J Nutr Biochem (2013) 24(9):1609–15. doi: 10.1016/j.jnutbio.2013.02.001
36. Li Y, Lv L, Ye J, Fang D, Shi D, Wu W, et al. Bifidobacterium Adolescentis CGMCC 15058 Alleviates Liver Injury, Enhances the Intestinal Barrier and Modifies the Gut Microbiota in D-Galactosamine-Treated Rats. Appl Microbiol Biotechnol (2019) 103(1):375–93. doi: 10.1007/s00253-018-9454-y
37. Li YT, Ye JZ, Lv LX, Xu H, Yang LY, Jiang XW, et al. Pretreatment With Bacillus Cereus Preserves Against D-Galactosamine-Induced Liver Injury in a Rat Model. Front Microbiol (2019) 10:1751. doi: 10.3389/fmicb.2019.01751
38. Wang Q, Lv L, Jiang H, Wang K, Yan R, Li Y, et al. Lactobacillus Helveticus R0052 Alleviates Liver Injury by Modulating Gut Microbiome and Metabolome in D-Galactosamine-Treated Rats. Appl Microbiol Biotechnol (2019) 103(23-24):9673–86. doi: 10.1007/s00253-019-10211-8
39. Wu W, Lv L, Shi D, Ye J, Fang D, Guo F, et al. Protective Effect of Akkermansia Muciniphila Against Immune-Mediated Liver Injury in a Mouse Model. Front Microbiol (2017) 8:1804. doi: 10.3389/fmicb.2017.01804
40. Prytz H, Holst-Christensen J, Korner B, Liehr H. Portal Venous and Systemic Endotoxaemia in Patients Without Liver Disease and Systemic Endotoxaemia in Patients With Cirrhosis. Scand J Gastroenterol (1976) 11(8):857–63. doi: 10.1080/00365521.1976.12097199
41. Nolan JP. Endotoxin, Reticuloendothelial Function, and Liver Injury. Hepatology (1981) 1(5):458–65. doi: 10.1002/hep.1840010516
42. Rao R. Endotoxemia and Gut Barrier Dysfunction in Alcoholic Liver Disease. Hepatology (2009) 50(2):638–44. doi: 10.1002/hep.23009
43. Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics Prevent Liver Injury in Rats Following Long-Term Exposure to Ethanol. Gastroenterology (1995) 108(1):218–24. doi: 10.1016/0016-5085(95)90027-6
44. Bataller R, Brenner DA. Liver Fibrosis. J Clin Invest (2005) 115(2):209–18. doi: 10.1172/JCI24282
45. Carotti S, Guarino MP, Vespasiani-Gentilucci U, Morini S. Starring Role of Toll-Like Receptor-4 Activation in the Gut-Liver Axis. World J Gastrointest Pathophysiol (2015) 6(4):99–109. doi: 10.4291/wjgp.v6.i4.99
46. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 Enhances TGF-Beta Signaling and Hepatic Fibrosis. Nat Med (2007) 13(11):1324–32. doi: 10.1038/nm1663
47. Krenkel O, Tacke F. Liver Macrophages in Tissue Homeostasis and Disease. Nat Rev Immunol (2017) 17(5):306–21. doi: 10.1038/nri.2017.11
48. Issa R, Zhou X, Trim N, Millward-Sadler H, Krane S, Benyon C, et al. Mutation in Collagen-1 That Confers Resistance to the Action of Collagenase Results in Failure of Recovery From CCl4-Induced Liver Fibrosis, Persistence of Activated Hepatic Stellate Cells, and Diminished Hepatocyte Regeneration. FASEB J (2003) 17(1):47–9. doi: 10.1096/fj.02-0494fje
49. Yu LX, Schwabe RF. The Gut Microbiome and Liver Cancer: Mechanisms and Clinical Translation. Nat Rev Gastroenterol Hepatol (2017) 14(9):527–39. doi: 10.1038/nrgastro.2017.72
50. Yu LX, Yan HX, Liu Q, Yang W, Wu HP, Dong W, et al. Endotoxin Accumulation Prevents Carcinogen-Induced Apoptosis and Promotes Liver Tumorigenesis in Rodents. Hepatology (2010) 52(4):1322–33. doi: 10.1002/hep.23845
51. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell (2012) 21(4):504–16. doi: 10.1016/j.ccr.2012.02.007
52. Cornell RP. Endotoxin-Induced Hyperinsulinemia and Hyperglucagonemia After Experimental Liver Injury. Am J Physiol (1981) 241(6):E428–35. doi: 10.1152/ajpendo.1981.241.6.E428
53. Taub R. Liver Regeneration: From Myth to Mechanism. Nat Rev Mol Cell Biol (2004) 5(10):836–47. doi: 10.1038/nrm1489
54. Fazel Modares N, Polz R, Haghighi F, Lamertz L, Behnke K, Zhuang Y, et al. IL-6 Trans-Signaling Controls Liver Regeneration After Partial Hepatectomy. Hepatology (2019). doi: 10.1016/S0618-8278(19)30621-8
55. Chiang JYL, Ferrell JM. Bile Acid Metabolism in Liver Pathobiology. Gene Expr (2018) 18(2):71–87. doi: 10.3727/105221618X15156018385515
56. Schneider KM, Albers S, Trautwein C. Role of Bile Acids in the Gut-Liver Axis. J Hepatol (2018) 68(5):1083–5. doi: 10.1016/j.jhep.2017.11.025
57. Doignon I, Julien B, Serriere-Lanneau V, Garcin I, Alonso G, Nicou A, et al. Immediate Neuroendocrine Signaling After Partial Hepatectomy Through Acute Portal Hyperpressure and Cholestasis. J Hepatol (2011) 54(3):481–8. doi: 10.1016/j.jhep.2010.07.012
58. Pean N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, et al. The Receptor TGR5 Protects the Liver From Bile Acid Overload During Liver Regeneration in Mice. Hepatology (2013) 58(4):1451–60. doi: 10.1002/hep.26463
59. Merlen G, Ursic-Bedoya J, Jourdainne V, et al. Bile Acids and Their Receptors During Liver Regeneration: "Dangerous Protectors". Mol Aspects Med (2017) 56:25–33. doi: 10.1016/j.mam.2017.03.002
60. Huang W, Ma K, Zhang J, Qatanani M, Cuvillier J, Liu J, et al. Nuclear Receptor-Dependent Bile Acid Signaling Is Required for Normal Liver Regeneration. Science (2006) 312(5771):233–6. doi: 10.1126/science.1121435
61. Ueda J, Chijiiwa K, Nakano K, Zhao G, Tanaka M. Lack of Intestinal Bile Results in Delayed Liver Regeneration of Normal Rat Liver After Hepatectomy Accompanied by Impaired Cyclin E-Associated Kinase Activity. Surgery (2002) 131(5):564–73. doi: 10.1067/msy.2002.123008
62. Otao R, Beppu T, Isiko T, Mima K, Okabe H, Hayashi H, et al. External Biliary Drainage and Liver Regeneration After Major Hepatectomy. Br J Surg (2012) 99(11):1569–74. doi: 10.1002/bjs.8906
63. Wahlstrom A, Sayin SI, Marschall HU, Backhed F. Intestinal Crosstalk Between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab (2016) 24(1):41–50. doi: 10.1016/j.cmet.2016.05.005
64. Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and Comparative Metagenomic Analysis of Bile Salt Hydrolase Activity in the Human Gut Microbiome. Proc Natl Acad Sci USA (2008) 105(36):13580–5. doi: 10.1073/pnas.0804437105
65. Vos TA, Ros JE, Havinga R, Moshage H, Kuipers F, Jansen PL, et al. Regulation of Hepatic Transport Systems Involved in Bile Secretion During Liver Regeneration in Rats. Hepatology (1999) 29(6):1833–9. doi: 10.1002/hep.510290638
66. Csanaky IL, Aleksunes LM, Tanaka Y, Klaassen CD. Role of Hepatic Transporters in Prevention of Bile Acid Toxicity After Partial Hepatectomy in Mice. Am J Physiol Gastrointest Liver Physiol (2009) 297(3):G419–33. doi: 10.1152/ajpgi.90728.2008
67. Chiang JY. Bile Acid Metabolism and Signaling. Compr Physiol (2013) 3(3):1191–212. doi: 10.1002/cphy.c120023
68. Ridlon JM, Kang DJ, Hylemon PB. Isolation and Characterization of a Bile Acid Inducible 7alpha-Dehydroxylating Operon in Clostridium Hylemonae TN271. Anaerobe (2010) 16(2):137–46. doi: 10.1016/j.anaerobe.2009.05.004
69. Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of Bile Salt Biotransformations by Intestinal Bacteria. Gut Microbes (2016) 7(1):22–39. doi: 10.1080/19490976.2015.1127483
70. Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A Regulatory Cascade of the Nuclear Receptors FXR, SHP-1, and LRH-1 Represses Bile Acid Biosynthesis. Mol Cell (2000) 6(3):517–26. doi: 10.1016/S1097-2765(00)00051-4
71. Zhang L, Wang YD, Chen WD, Wang X, Lou G, Liu N, et al. Promotion of Liver Regeneration/Repair by Farnesoid X Receptor in Both Liver and Intestine in Mice. Hepatology (2012) 56(6):2336–43. doi: 10.1002/hep.25905
72. Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast Growth Factor 15 Functions as an Enterohepatic Signal to Regulate Bile Acid Homeostasis. Cell Metab (2005) 2(4):217–25. doi: 10.1016/j.cmet.2005.09.001
73. Uriarte I, Fernandez-Barrena MG, Monte MJ, Latasa MU, Chang HC, Carotti S, et al. Identification of Fibroblast Growth Factor 15 as a Novel Mediator of Liver Regeneration and its Application in the Prevention of Post-Resection Liver Failure in Mice. Gut (2013) 62(6):899–910. doi: 10.1136/gutjnl-2012-302945
74. Padrissa-Altes S, Bachofner M, Bogorad RL, Pohlmeier L, Rossolini T, Bohm F, et al. Control of Hepatocyte Proliferation and Survival by Fgf Receptors Is Essential for Liver Regeneration in Mice. Gut (2015) 64(9):1444–53. doi: 10.1136/gutjnl-2014-307874
75. Martinot E, Sedes L, Baptissart M, Lobaccaro JM, Caira F, Beaudoin C, et al. Bile Acids and Their Receptors. Mol Aspects Med (2017) 56:2–9. doi: 10.1016/j.mam.2017.01.006
76. Chiang JYL. Bile Acid Metabolism and Signaling in Liver Disease and Therapy. Liver Res (2017) 1(1):3–9. doi: 10.1016/j.livres.2017.05.001
77. Garcia-Rodriguez JL, Barbier-Torres L, Fernandez-Alvarez S, Gutierrez-de Juan V, Monte MJ, Halilbasic E, et al. SIRT1 Controls Liver Regeneration by Regulating Bile Acid Metabolism Through Farnesoid X Receptor and Mammalian Target of Rapamycin Signaling. Hepatology (2014) 59(5):1972–83. doi: 10.1002/hep.26971
78. Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Haussinger D. The Membrane-Bound Bile Acid Receptor TGR5 Is Localized in the Epithelium of Human Gallbladders. Hepatology (2009) 50(3):861–70. doi: 10.1002/hep.23032
79. Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G Protein-Coupled Receptor Responsive to Bile Acids. J Biol Chem (2003) 278(11):9435–40. doi: 10.1074/jbc.M209706200
80. Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and Function of the Bile Acid Receptor TGR5 in Kupffer Cells. Biochem Biophys Res Commun (2008) 372(1):78–84. doi: 10.1016/j.bbrc.2008.04.171
81. Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-Protein-Coupled Bile Acid Receptor, Gpbar1 (TGR5), Negatively Regulates Hepatic Inflammatory Response Through Antagonizing Nuclear Factor Kappa Light-Chain Enhancer of Activated B Cells (NF-KappaB) in Mice. Hepatology (2011) 54(4):1421–32. doi: 10.1002/hep.24525
82. Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-Mediated Bile Acid Sensing Controls Glucose Homeostasis. Cell Metab (2009) 10(3):167–77. doi: 10.1016/j.cmet.2009.08.001
83. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut Microbiome-Mediated Bile Acid Metabolism Regulates Liver Cancer via NKT Cells. Science (2018) 360(6391). doi: 10.1126/science.aan5931
84. Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell (2016) 165(6):1332–45. doi: 10.1016/j.cell.2016.05.041
85. Macfarlane GT, Macfarlane S. Bacteria, Colonic Fermentation, and Gastrointestinal Health. J AOAC Int (2012) 95(1):50–60. doi: 10.5740/jaoacint.SGE_Macfarlane
86. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. Short Chain Fatty Acids in Human Large Intestine, Portal, Hepatic and Venous Blood. Gut (1987) 28(10):1221–7. doi: 10.1136/gut.28.10.1221
87. El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The Abundance and Variety of Carbohydrate-Active Enzymes in the Human Gut Microbiota. Nat Rev Microbiol (2013) 11(7):497–504. doi: 10.1038/nrmicro3050
88. Flint HJ, Duncan SH, Scott KP, Louis P. Links Between Diet, Gut Microbiota Composition and Gut Metabolism. Proc Nutr Soc (2015) 74(1):13–22. doi: 10.1017/S0029665114001463
89. Levy M, Thaiss CA, Elinav E. Metabolites: Messengers Between the Microbiota and the Immune System. Genes Dev (2016) 30(14):1589–97. doi: 10.1101/gad.284091.116
90. Rios-Covian D, Ruas-Madiedo P, Margolles A, Gueimonde M, de Los Reyes-Gavilan CG, Salazar N, et al. Intestinal Short Chain Fatty Acids and Their Link With Diet and Human Health. Front Microbiol (2016) 7:185. doi: 10.3389/fmicb.2016.00185
91. Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol Cell (2012) 48(4):612–26. doi: 10.1016/j.molcel.2012.08.033
92. Bloemen JG, Venema K, van de Poll MC, Olde Damink SW, Buurman WA, Dejong CH. Short Chain Fatty Acids Exchange Across the Gut and Liver in Humans Measured at Surgery. Clin Nutr (2009) 28(6):657–61. doi: 10.1016/j.clnu.2009.05.011
93. Morrison DJ, Preston T. Formation of Short Chain Fatty Acids by the Gut Microbiota and Their Impact on Human Metabolism. Gut Microbes (2016) 7(3):189–200. doi: 10.1080/19490976.2015.1134082
94. Brown AJ, Goldsworthy SM, Barnes AA, Eilert MM, Tcheang L, Daniels D, et al. The Orphan G Protein-Coupled Receptors GPR41 and GPR43 Are Activated by Propionate and Other Short Chain Carboxylic Acids. J Biol Chem (2003) 278(13):11312–9. doi: 10.1074/jbc.M211609200
95. Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, et al. GPR109A Is a G-Protein-Coupled Receptor for the Bacterial Fermentation Product Butyrate and Functions as a Tumor Suppressor in Colon. Cancer Res (2009) 69(7):2826–32. doi: 10.1158/0008-5472.CAN-08-4466
96. Seljeset S, Siehler S. Receptor-Specific Regulation of ERK1/2 Activation by Members of the "Free Fatty Acid Receptor" Family. J Recept Signal Transduct Res (2012) 32(4):196–201. doi: 10.3109/10799893.2012.692118
97. Alex S, Lange K, Amolo T, Grinstead JS, Haakonsson AK, Szalowska E, et al. Short-Chain Fatty Acids Stimulate Angiopoietin-Like 4 Synthesis in Human Colon Adenocarcinoma Cells by Activating Peroxisome Proliferator-Activated Receptor Gamma. Mol Cell Biol (2013) 33(7):1303–16. doi: 10.1128/MCB.00858-12
98. Croasdell A, Duffney PF, Kim N, Lacy SH, Sime PJ, Phipps RP. PPARgamma and the Innate Immune System Mediate the Resolution of Inflammation. PPAR Res (2015) 2015:549691. doi: 10.1155/2015/549691
99. Wang HB, Wang PY, Wang X, Wan YL, Liu YC. Butyrate Enhances Intestinal Epithelial Barrier Function via Up-Regulation of Tight Junction Protein Claudin-1 Transcription. Dig Dis Sci (2012) 57(12):3126–35. doi: 10.1007/s10620-012-2259-4
100. Liu B, Qian J, Wang Q, Ma Z, Qiao Y. Butyrate Protects Rat Liver Against Total Hepatic Ischemia Reperfusion Injury With Bowel Congestion. PloS One (2014) 9(8):e106184. doi: 10.1371/journal.pone.0106184
101. den Besten G, Bleeker A, Gerding A, van Eunen K, Havinga R, van Dijk TH, et al. Short-Chain Fatty Acids Protect Against High-Fat Diet-Induced Obesity via a PPARgamma-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes (2015) 64(7):2398–408. doi: 10.2337/db14-1213
102. Crouse JR, Gerson CD, DeCarli LM, Lieber CS. Role of Acetate in the Reduction of Plasma Free Fatty Acids Produced by Ethanol in Man. J Lipid Res (1968) 9(4):509–12. doi: 10.1016/S0022-2275(20)42731-2
103. Ge H, Li X, Weiszmann J, Wang P, Baribault H, Chen JL, et al. Activation of G Protein-Coupled Receptor 43 in Adipocytes Leads to Inhibition of Lipolysis and Suppression of Plasma Free Fatty Acids. Endocrinology (2008) 149(9):4519–26. doi: 10.1210/en.2008-0059
104. De Vadder F, Kovatcheva-Datchary P, Goncalves D, Vinera J, Zitoun C, Duchampt A, et al. Microbiota-Generated Metabolites Promote Metabolic Benefits via Gut-Brain Neural Circuits. Cell (2014) 156(1-2):84–96. doi: 10.1016/j.cell.2013.12.016
105. Kaji I, Karaki S, Kuwahara A. Short-Chain Fatty Acid Receptor and Its Contribution to Glucagon-Like Peptide-1 Release. Digestion (2014) 89(1):31–6. doi: 10.1159/000356211
106. Luhrs H, Gerke T, Muller JG, Melcher R, Schauber J, Boxberge F, et al. Butyrate Inhibits NF-kappaB Activation in Lamina Propria Macrophages of Patients With Ulcerative Colitis. Scand J Gastroenterol (2002) 37(4):458–66. doi: 10.1080/003655202317316105
107. Chang PV, Hao L, Offermanns S, Medzhitov R. The Microbial Metabolite Butyrate Regulates Intestinal Macrophage Function via Histone Deacetylase Inhibition. Proc Natl Acad Sci USA (2014) 111(6):2247–52. doi: 10.1073/pnas.1322269111
108. Qiao YL, Qian JM, Wang FR, Ma ZY, Wang QW. Butyrate Protects Liver Against Ischemia Reperfusion Injury by Inhibiting Nuclear Factor Kappa B Activation in Kupffer Cells. J Surg Res (2014) 187(2):653–9. doi: 10.1016/j.jss.2013.08.028
109. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature (2013) 504(7480):451–5. doi: 10.1038/nature12726
110. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature (2013) 504(7480):446–50. doi: 10.1038/nature12721
111. Agus A, Planchais J, Sokol H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe (2018) 23(6):716–24. doi: 10.1016/j.chom.2018.05.003
112. Roager HM, Licht TR. Microbial Tryptophan Catabolites in Health and Disease. Nat Commun (2018) 9(1):3294. doi: 10.1038/s41467-018-05470-4
113. Gutierrez-Vazquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity (2018) 48(1):19–33. doi: 10.1016/j.immuni.2017.12.012
114. Roman AC, Carvajal-Gonzalez JM, Merino JM, Mulero-Navarro S, Fernandez-Salguero PM. The Aryl Hydrocarbon Receptor in the Crossroad of Signalling Networks With Therapeutic Value. Pharmacol Ther (2018) 185:50–63. doi: 10.1016/j.pharmthera.2017.12.003
115. Bansal T, Alaniz RC, Wood TK, Jayaraman A. The Bacterial Signal Indole Increases Epithelial-Cell Tight-Junction Resistance and Attenuates Indicators of Inflammation. Proc Natl Acad Sci USA (2010) 107(1):228–33. doi: 10.1073/pnas.0906112107
116. Shimada Y, Kinoshita M, Harada K, Mizutani M, Masahata K, Kayama H, et al. Commensal Bacteria-Dependent Indole Production Enhances Epithelial Barrier Function in the Colon. PloS One (2013) 8(11):e80604. doi: 10.1371/journal.pone.0080604
117. Wlodarska M, Luo C, Kolde R, d'Hennezel E, Annand JW, Heim CE, et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe (2017) 22(1):25–37.e6. doi: 10.1016/j.chom.2017.06.007
118. Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, et al. Symbiotic Bacterial Metabolites Regulate Gastrointestinal Barrier Function via the Xenobiotic Sensor PXR and Toll-Like Receptor 4. Immunity (2014) 41(2):296–310. doi: 10.1016/j.immuni.2014.06.014
119. Chimerel C, Emery E, Summers DK, Keyser U, Gribble FM, Reimann F. Bacterial Metabolite Indole Modulates Incretin Secretion From Intestinal Enteroendocrine L Cells. Cell Rep (2014) 9(4):1202–8. doi: 10.1016/j.celrep.2014.10.032
120. Natividad JM, Agus A, Planchais J, Lamas B, Jarry AC, Martin R, et al. Impaired Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is a Key Factor in Metabolic Syndrome. Cell Metab (2018) 28(5):737–49.e4. doi: 10.1016/j.cmet.2018.07.001
Keywords: gut microbiota (GM), liver injury and regeneration, lipopolisaccharide (LPS), bile acid (BA), SCFA (short chain fatty acids), tryptophan metabolites, gut microbial metabolites
Citation: Zheng Z and Wang B (2021) The Gut-Liver Axis in Health and Disease: The Role of Gut Microbiota-Derived Signals in Liver Injury and Regeneration. Front. Immunol. 12:775526. doi: 10.3389/fimmu.2021.775526
Received: 14 September 2021; Accepted: 19 November 2021;
Published: 10 December 2021.
Edited by:
Alina Maria Holban, University of Bucharest, RomaniaReviewed by:
Daisuke Tokuhara, Osaka City University, JapanNaschla Gasaly, University of Chile, Chile
Marcela A. Hermoso Ramello, University of Chile, Chile, in collaboration with reviewer NG
Copyright © 2021 Zheng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Baohong Wang, d2FuZ2Jhb2hvbmd6anVAemp1LmVkdS5jbg==