 Xiao Man Wu
Xiao Man Wu Hong Fang1,2
Hong Fang1,2 Jie Zhang
Jie Zhang Ming Xian Chang
Ming Xian Chang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 18 November 2021
Sec. Comparative Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.771277
Histone H2A is a nuclear molecule tightly associated in the form of the nucleosome. Our previous studies have demonstrated the antibacterial property of piscine H2A variants against gram-negative bacteria Edwardsiella piscicida and Gram-positive bacteria Streptococcus agalactiae. In this study, we show the function and mechanism of piscine H2A in the negative regulation of RLR signaling pathway and host innate immune response against spring viremia of carp virus (SVCV) infection. SVCV infection significantly inhibits the expression of histone H2A during an early stage of infection, but induces the expression of histone H2A during the late stage of infection such as at 48 and 72 hpi. Under normal physiological conditions, histone H2A is nuclear-localized. However, SVCV infection promotes the migration of histone H2A from the nucleus to the cytoplasm. The in vivo studies revealed that histone H2A overexpression led to the increased expression of SVCV gene and decreased survival rate. The overexpression of histone H2A also significantly impaired the expression levels of those genes involved in RLR antiviral signaling pathway. Furthermore, histone H2A targeted TBK1 and IRF3 to promote their protein degradation via the lysosomal pathway and impair the formation of TBK1-IRF3 functional complex. Importantly, histone H2A completely abolished TBK1-mediated antiviral activity and enormously impaired the protein expression of IRF3, especially nuclear IRF3. Further analysis demonstrated that the inhibition of histone H2A nuclear/cytoplasmic trafficking could relieve the protein degradation of TBK1 and IRF3, and blocked the negative regulation of histone H2A on the SVCV infection. Collectively, our results suggest that histone H2A nuclear/cytoplasmic trafficking is essential for negative regulation of RLR signaling pathway and antiviral immune response in response to SVCV infection.
1. SVCV infection promotes histone H2A nuclear/cytoplasmic trafficking.
2. Histone H2A facilitates SVCV replication.
3. Histone H2A interacts with TBK1 and IRF3 to impair the formation of TBK1-IRF3 complex.
4. Histone H2A promotes the protein degradations of TBK1 and IRF3 via the lysosomal pathway.
Pathogen-associated molecular patterns (PAMPs) are recognized by evolutionarily conserved host sensors known as pattern recognition receptors (PRRs), which include peptidoglycan recognition proteins (PGRPs), Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), C-type lectin receptors (CLRs) and DNA receptors (1–3). Among them, RLRs consisting of RIG-I (retinoic acid-inducible gene I), MDA5 (melanoma differentiation associated factor 5) and LGP2 (laboratory of genetics and physiology 2) are key sensors for virus infection, which mediate the transcriptional induction of type I interferons (IFNs) and interferon-stimulated genes (ISGs) to establish an antiviral immune response (4). It is now well known that the activated RLR signaling by interacting with the adapter protein MAVS leads to initiation of a signaling cascade that includes activation of TBK1 and IKKϵ protein kinases, which ultimately phosphorylate and activate IRF3 and NF-κB transcription factors. Following this activation, IRF3 and NF-κB translocate from the cytoplasm to the nucleus and induce transcription of IFNs and ISGs (5). RLR signaling is found conservatively present in teleost fish and mammals (6, 7).
Besides PAMPs, PRRs are also activated by damage-associated molecular patterns (DAMPs), which are host nuclear or cytoplasmic non-pathogen molecules and utilize the same signal transduction pathways to activate the immune system (8). DAMPs, sometimes termed alarmins or danger signals, are released from cells in response to exogenous and endogenous stressors such as injury or cellular death. DAMPs can also be secreted following PAMP stimulation (9). Protein sources of DAMPs include high mobility group box 1 (HMGB1), heat shock proteins (HSPs), extracellular cold-inducible RNA-binding protein (eCIRP), S100, serum amyloid A (SAA) and histones, whereas non-protein sources of DAMPs include ATP, uric acid, heparin sulfate, genomic and mitochondrial DNA, and extracellular RNAs (9, 10). Among them, histones are located mainly in the nucleus, and have been suggested as important DAMPs by binding to PRRs once they are released to the extracellular space or translocate to the cytoplasm following infection, sterile inflammation and various types of cell death (9–12). In response to stress such as hypoxia, chemical agents and infection, histones H1, H2A, H2B, H3 and H4 are frequently detected at the cell surface or cytoplasm. Extracellular histones could bind to TLRs receptors and induce NALP3 inflammation activation (13).
In teleost fish, the antibacterial properties of piscine H2A variants against gram-negative bacteria Edwardsiella piscicida and Gram-positive bacteria Streptococcus agalactiae have been investigated in zebrafish and/or grass carp (14–16). Significantly, S. agalactiae infection could induce the release of histone H2A. In the case of S. agalactiae infection, histone H2A was colocalized with NLR NOD1 both in the cytoplasm and cell nucleus. Histone H2A variant zfH2A-6 also could interact with NOD1 and cooperate with NOD1 to inhibit the proliferation of S. agalactiae (16). However, the function of piscine H2A in viral infection is still unclear.
In the present study, we firstly describe the functional characterization of piscine H2A in response to viral infection. Similar to S. agalactiae infection, SVCV infection induced the migration of histone H2A from the nucleus to the cytoplasm. Different from the antibacterial roles of histone H2A in bacterial infection, histone H2A negatively regulated antiviral immune response against SVCV infection. Functionally, the cytoplasmic histone H2A degraded TBK1 and IRF3 via the lysosomal pathway to impair the formation of TBK1-IRF3 functional complex and the expression level of nuclear IRF3 protein. All these findings shed light on a novel strategy that histone H2A is utilized by SVCV to inhibit host immune response.
All animal experiments were conducted in accordance with the Guiding Principles for the Care and Use of Laboratory Animals and were approved by the Institute of Hydrobiology, Chinese Academy of Sciences (Approval ID: IHB 2013724).
Epithelioma papulosum cyprini (EPC) and zebrafish embryonic fibroblast (ZF4) cells were maintained at 28°C in 5% CO2 in medium 199 (Hyclone) and DMEM/F12 (1:1) medium (Invitrogen) supplemented with 10% FBS, 100 U/mL penicillin and 100 mg/mL streptomycin, respectively. Spring viremia of carp virus (SVCV) was propagated in EPC cells. MG132 (S2619) and 3-MA (S2767) were purchased from Selleck. Chloroquine (C6628) and poly I:C (P9582) were purchased from Sigma-Aldrich. Leptomycin B (9676S) was purchased from Cell Signaling Technology.
The expression plasmids including pcDNA3.1-MDA5, pcDNA3.1-RIG-I, p3×FLAG-MAVS, p3×FLAG-TBK1, pTurbo-TBK1-GFP, p3×FLAG-IRF3, pTurbo-IRF3-GFP and p3×FLAG-H2A were described previously (14, 17–20). Zebrafish histone H2A-GFP plasmid was generated using the primer pairs H2AF/H2AR, which were the same primer pairs for constructing p3×FLAG-H2A plasmid (14), and inserted into the pTurboGFP-N plasmid (Everogen). These plasmids were transfected into EPC cells by Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Expression plasmids including p3×FLAG, p3×FLAG-TBK1 and/or p3×FLAG-H2A were transfected into ZF4 cells by using the Amaxa Nucleofector II transfection system (Lonza) under program T20 according to the manufacturer’s instructions.
To visualize the subcellular localization of zebrafish histone H2A, ZF4 cells were seeded onto coverslips in 24-well plates. After 24 h, the cells were infected with SVCV at a multiplicity of infection (MOI) of 1 or left untreated. At 6, 12, 24 and 48 hours post-infection (hpi), the cells were fixed with 4% formaldehyde. To investigate whether zebrafish histone H2A colocalizes with TBK1 and IRF3, EPC cells seeded in 24-well plates were transfected with 250 ng H2A-FLAG and 250 ng TBK1-GFP or IRF3-GFP. After 24 h post-transfection, the cells were infected with SVCV at the MOI of 1. At 24 hpi, the cells were fixed with 4% formaldehyde. For fluorescence microscopy analysis, the cells were permeabilized with 0.2% Triton X-100 for 15 min, and then blocked with 5% BSA for 1 h. Next, the cells were incubated in the primary antibody anti-H2A (1:200; 12349S, CST) overnight at 4°C, the secondary antibody goat anti-rabbit IgG conjugated to fluorophore Alexa Fluor 488 (1:500; A-11034, Invitrogen) for 1 h at room temperature, and finally stained with DAPI (1 μg/mL) for 15 min. The coverslips were observed with a confocal microscope (SP8; Lecia).
To investigate the effects of different multiplicity of infection on zebrafish histone H2A subcellular localization, 1×106 ZF4 cells were cultured in 6-well plates overnight and then infected with SVCV at an MOI of 0.1, 1, 5 and 10. At 24 hpi, the cells were harvested and used for preparation of nuclear and cytoplasmic extracts using a Subcellular Protein Fractionation Kit (Thermo Fisher Scientific).
To investigate the effects of different stressors on zebrafish histone H2A subcellular localization, 1×106 ZF4 cells or EPC cells passed in 6-well plates overnight were infected with SVCV at an MOI of 1 or stimulated with poly I:C (10 μg/mL) or left untreated. At 6, 24 and 48 hpi, the cells were harvested and used for preparation of nuclear and cytoplasmic extracts.
To investigate the effects of histone H2A on the protein degradations of cytoplasmic and/or nuclear IRF3, 1×106 EPC cells seeded in 6-well plates overnight were transfected with various plasmids with the indicated combination. After 24 h post-transfection, the cells were infected with SVCV at an MOI of 1. At 24 hpi, the cells were harvested and used for preparation of nuclear and cytoplasmic extracts.
To investigate the effects of inhibition of histone H2A nuclear/cytoplasmic trafficking on the protein degradations of TBK1 and IRF3, 1×106 EPC cells seeded in 6-well plates overnight were transfected with TBK1-FLAG or IRF3-FLAG (1500 ng/well) and H2A-GFP (800 ng/well). After 36 h post-transfection, the cells were treated with LMB at the concentration of 60 nm, 80 nm, 100 nm or left untreated. After 12 h, the cells were harvested and used for preparation of nuclear and cytoplasmic extracts.
Purity of the fractions was determined by Western blotting using polyclonal anti-tubulin (1:5000; ab6046; Abcam) and anti-HDAC1 (1:5000; ab41407; Abcam) antibodies (Abs). The protein expression of histone H2A was examined using monoclonal anti-H2A (1:1000; 12349S, CST) Ab.
To examine the effect of histone H2A on the promoter activities of IFNs after poly I:C stimulation, EPC cells seeded overnight in 24-well plates at 3×105 cells per well were cotransfected with the FLAG and/or p3×FLAG-H2A with the indicated concentration, 250 ng DrIFN1pro-luc or DrIFN3pro-luc reporter plasmid and 25 ng Renilla luciferase internal control reporter vector (Promega). After 24 h transfection, the cells were stimulation with poly I:C at a final concentration of 2 μg/mL or left untreated. Another 24 h later, the cells were lysed and used for luciferase activity assay by Dual-Luciferase Reporter Assay System (Promega). To examine the effect of histone H2A on the promoter activities of IFNs mediated by the normal form of RIG-I, MDA5, MAVS, TBK1 and IRF3, EPC cells seeded overnight were transfected with 200 ng various plasmids with the indicated combination. At 48 h post-transfection, the cells were washed with PBS, lysed with 100 μL of lysis buffer and measured luciferase activity. All the experiments were performed in triplicate.
In order to investigate the inducible expression of zebrafish histone H2A in response to SVCV infection, zebrafish larvae at 4 dpf (days post-fertilization) were exposed to 2×106 PFU/ml SVCV in a total volume of 5 mL water according to the methods previously reported (21) or left untreated. After immersion in the SVCV suspension for 24 h or left untreated, zebrafish larvae were maintained in 70 mm sterile disposable petri dishes with the supplemented 20 mL water. 30~40 larvae per group collected at 6, 12, 24, 48 and 72 hpi were used for RNA extraction and quantivative RT-PCR (qRT-PCR).
To determine the role of zebrafish histone H2A in SVCV infection, 100 ng/μL p3×FLAG empty plasmid or p3×FLAG-H2A was microinjected into zebrafish embryos at the one-cell stage. The typical injected volume was 2 nL. At 4 dpf, the zebrafish larvae were divided into 2 groups and used for SVCV infection (2×106 PFU/mL) or left untreated. The numbers of surviving larvae were counted daily for 6 dpi. GraphPad Prism 7 was used to generate survival curves, and the log-rank test was used to test differences in survival with WT zebrafish microinjected with p3×FLAG as the control group. To examine the expression of SVCV-N gene and confirm the overexpression of histone H2A in zebrafish larvae, 20 larvae per group were collected at 24 and 48 hpi, and used for RNA extraction and qRT-PCR. To determine the effect of histone H2A overexpression on the transcriptional regulation of antiviral genes involved in RLR signaling, 20~30 zebrafish embryos or larvae microinjected with p3×FLAG or H2A-FLAG were collected at 1, 3 and 7 dpf for RNA extraction and qRT-PCR.
To determine the regulatory effect of zebrafish histone H2A in TBK1-mediated antiviral activity and TBK1-mediated induction of IFNs and ISGs, 1×106 ZF4 cells were transfected with 2 μg p3×FLAG, TBK1-FLAG and/or H2A-FLAG alone or in combination of any two. At 36 h post-transfection, these cells were used for SVCV infection at an MOI of 1. At 48 hpi, the supernatants of transfected cells were collected for the determination of virus titers by a standard TCID50 method. The cell pellets of transfected cells were collected and used for RNA extraction. The overexpression of histone H2A in ZF4 cells and the expression of SVCV genes, IFNs and ISGs were examined by qRT-PCR.
The transfection efficiency of ZF4 cells was rather low, especially in the case of transfection of multiple plasmids. Therefore, EPC cells with higher transfection efficiency were used for overexpression experiments. Since examining the effect of histone H2A on the protein degradations of TBK1 and IRF3 was performed in EPC cells, we used EPC cells to further investigate the effects of inhibition of histone H2A nuclear/cytoplasmic trafficking on the histone H2A-mediated negative regulation of antiviral immune response in response to SVCV infection. 3×105 EPC cells seeded in 24-well plates were transfected with p3×FLAG or H2A-FLAG for 36 h, then treated with LMB at the concentration of 100 nm for 12 h or left untreated. At 48 h posttransfection, these cells were infected with SVCV at an MOI of 1. Another 24 h later, the supernatants of infected cells were collected for the determination of virus titers by a standard TCID50 method. The cell pellets of infected cells were collected for RNA extraction. The expressions of SVCV genes including SVCV-N, SVCV-P and SVCV-G were examined by qRT-PCR.
Total RNA was extracted from ZF4 cells, EPC cells or zebrafish larvae using TRIzol (Invitrogen). First-strand cDNAs were synthesized using the RevertAid First-Strand cDNA Synthesis Kit (Thermo Scientific). qRT-PCR was performed using Fast SYBR Green PCR Master mix (Bio-Rad) on a CFX96 Touch Real-Time PCR Detection System (Bio-Rad) in 96-well plates according to the procedure as follows: preincubation at 95°C for 5 min, then 45 cycles at 95°C for 15 s, 56°C for 20 s, and 72°C for 20 s. Each sample was tested in triplicate. All primers are described by previous studies (14, 22–25). The relative mRNA expression was calculated by normalizing the Ct values of target genes against the reference housekeeping gene GAPDH. The data were analyzed using the 2-△△Ct method.
To determine whether histone H2A interacted with TBK1 and IRF3 to impair the formation of TBK1-IRF3, EPC cells seeded in T25 culture bottles overnight were cotransfected with 5000 ng various plasmid with the indicated combinations. After 48 h post-transfection, the cells were lysed in IP lysis buffer containing Protease Inhibitor Cocktail. The immune complexes were captured using FLAG-tagged Protein Immunoprecipitation Kit according to the manufacturer’s protocol, washed six times with ice-cold washing buffer and examined by Western Blotting using anti-pTurboGFP (1:5000; AB513; Evrogen) and anti-FLAG (1:5000; F3165; Sigma-Aldrich) Abs. Input proteins were analyzed using anti-GAPDH (1:5000; Proteintech), anti-FLAG and anti-pTurboGFP Abs.
To examine the effect of histone H2A on the protein degradations of TBK1 and IRF3, 1×106 EPC cells seeded in 6-well plates overnight were transfected with the indicated plasmids and concentration. To determine the exact mechanism of histone H2A on the protein degradations of TBK1 and IRF3, 1×106 EPC cells seeded in 6-well plates overnight were transfected with 1500 ng TBK1-FLAG or IRF3-FLAG and 800 ng H2A-GFP. After 48 h transfection, the cells were treated with MG132, 3-MA, NH4CL and Chloroquine (CQ) at the indicated concentration for another 6 h or left untreated. The cells were lysed in RIPA lysis buffer containing Protease Inhibitor Cocktail and analyzed by Western blotting with anti-GAPDH, anti-FLAG, and anti-pTurboGFP Abs. Western blotting results were quantified using Quantity One software.
Data of qRT-PCR were expressed as means ± SD of at least three independent experiments. The significance of results was analyzed by a two-tailed Student’s T-test or a one-way ANOVA (*p < 0.05, **p < 0.01).
The inducible expression of zebrafish histone H2A (GenBank accession number MK800012) in response to bacterial infection has been characterized in our previous study (14). To determine the effect of SVCV infection on the expression regulation of zebrafish histone H2A, the hatched larvae at 4 dpf were infected with 2×106 PFU/ml SVCV. The expression of SVCV-N gene was the highest at 48 hpi. The increased 317.76-fold, 6208.65-fold, 114440.31-fold, 141146.24-fold and 73062.24 for SVCV-N were observed at 6, 12, 24, 48 and 72 hpi, respectively. Compared with the control larvae without infection, the expression of histone H2A in the larvae infected with SVCV was significantly down-regulated at 6 and 12 hpi, no significant change at 24 hpi, and significantly up-regulated at 48 and 72 hpi (Figure 1A).
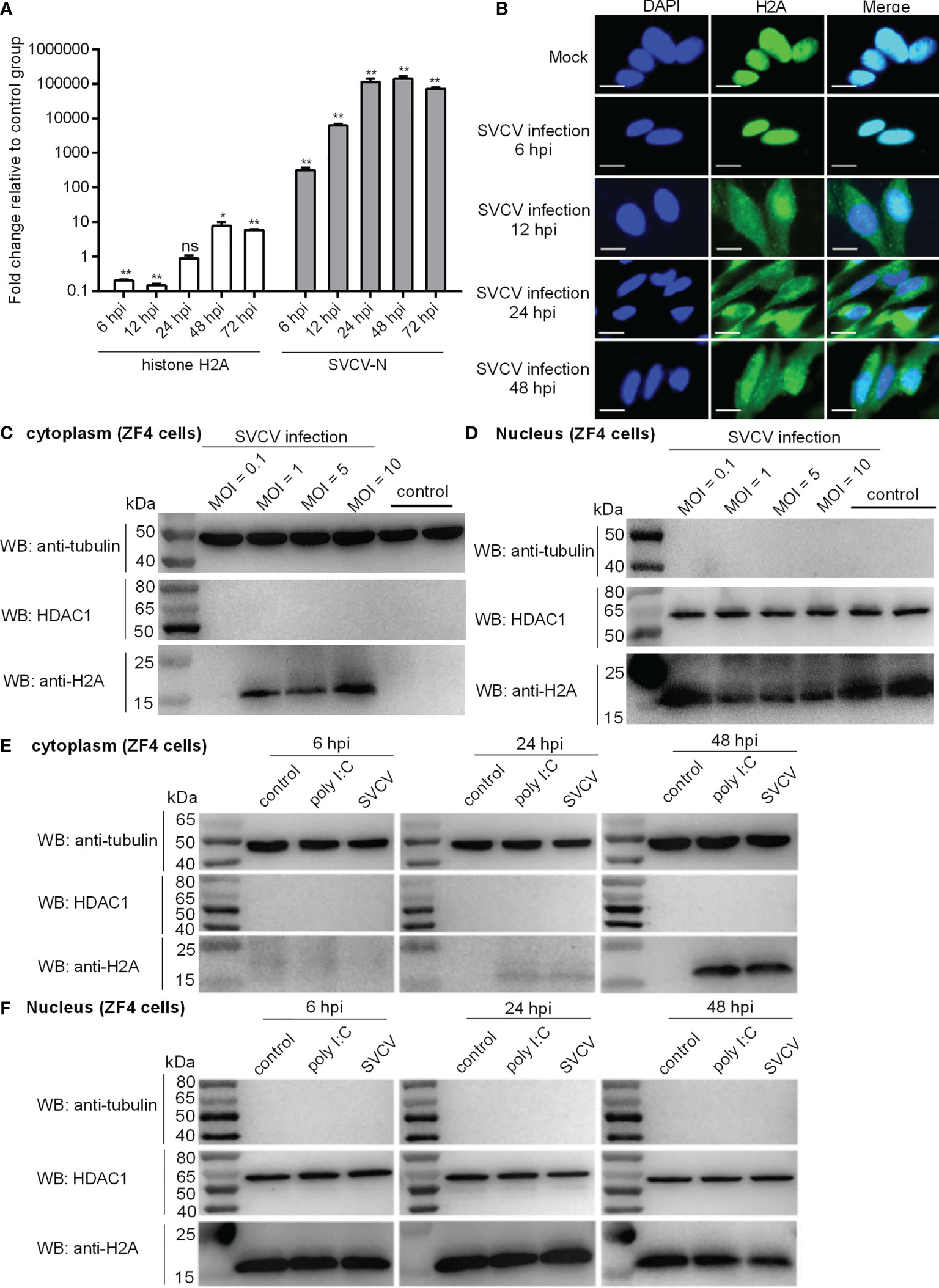
Figure 1 Expression and subcellular localization of zebrafish histone H2A. (A) The expressions of histone H2A and SVCV-N in zebrafish larvae in response to SVCV infection. Total RNA was extracted from zebrafish larvae collected at 6, 12, 24, 48 and 72 hpi. Data represented means ± SEM (n = 3), and were tested for statistical significance. *p < 0.05; **p < 0.01; ns, not significant. (B) Subcellular localization of zebrafish histone H2A by immunofluorescence analysis in the mock-infected and SVCV-infected ZF4 cells. Scale bar: 10 μm. (C) Cytoplasmic fractions from the mock-infected and SVCV-infected ZF4 cells. (D) Nuclear fractions from the mock-infected and SVCV-infected ZF4 cells. For (C, D), ZF4 cells cultured in 6-well plates overnight were infected with SVCV at an MOI of 0.1, 1, 5 and 10. At 24 hpi, the cells were harvested and used for preparation of nuclear and cytoplasmic extracts. (E) Cytoplasmic fractions from SVCV-infected, poly I:C-stimulated and untreated ZF4 cells. (F) Nuclear fractions from SVCV-infected, poly I:C-stimulated and untreated ZF4 cells. For (E, F), ZF4 cells passed in 6-well plates overnight were infected with SVCV at an MOI of 1 or stimulated with poly I:C (10 μg/mL) or left untreated. At 6, 24 and 48 hpi, the cells were harvested and used for preparation of nuclear and cytoplasmic extracts.
In order to examine the subcellular localization of histone H2A during viral infection, SVCV-infected cells were fixed at different time points post-infection and processed for analysis by confocal microscopy. At 6 hpi for early time point post-infection, histone H2A was distributed only in the nucleus of infected cells. At 12, 24 and 48 hpi, histone H2A was distributed in both the cytoplasm and nucleus of infected cells (Figure 1B).
In order to further determine the effect of SVCV infection on the histone H2A nuclear/cytoplasmic trafficking, SVCV-infected cells with the different MOIs were collected and used for subcellular fractionation. The histone H2A nuclear/cytoplasmic trafficking was obvious only at an MOI of 1, 5 or 10, but not observed at an MOI of 0.1 (Figures 1C, D).
To further investigate and confirm histone H2A nuclear/cytoplasmic trafficking in response to different stressors, ZF4 cells were stimulated by poly I:C or infected with SVCV, or left untreated. Compared with the untreated control, the protein expression of histone H2A in the cytoplasm was induced by poly I:C stimulation and SVCV infection weakly at 24 hpi and strongly at 48 hpi (Figure 1E). Compared with the untreated control, the protein expression of histone H2A in the nucleus of infected cells was decreased by SVCV infection at 48 hpi (Figure 1F). Histone H2A nuclear/cytoplasmic trafficking in response to poly I:C stimulation and SVCV infection was also confirmed in EPC cells (Figure S1 in Supplementary Material).
To investigate the possible role of histone H2A in RLR signaling, the IFN1 and IFN3 promoter activities with or without poly I:C stimulation were firstly investigated in EPC cells by Dual-Luciferase Reporter Assay. The overexpression of histone H2A inhibited the activation of zebrafish IFN1 promoter with the poly I:C stimulation (Figure 2A). In the absence of poly I:C stimulation, histone H2A also inhibited the activation of zebrafish IFN1 promoter. The induced activation of zebrafish IFN1 promoter by RIG-I, MDA5, MAVS, TBK1 and IRF3 was significantly inhibited by histone H2A (Figure 2B). Similar studies were observed for zebrafish IFN3 promoter, except for no effect of histone H2A on the IRF3-mediated promoter activity (Figures 2C, D).
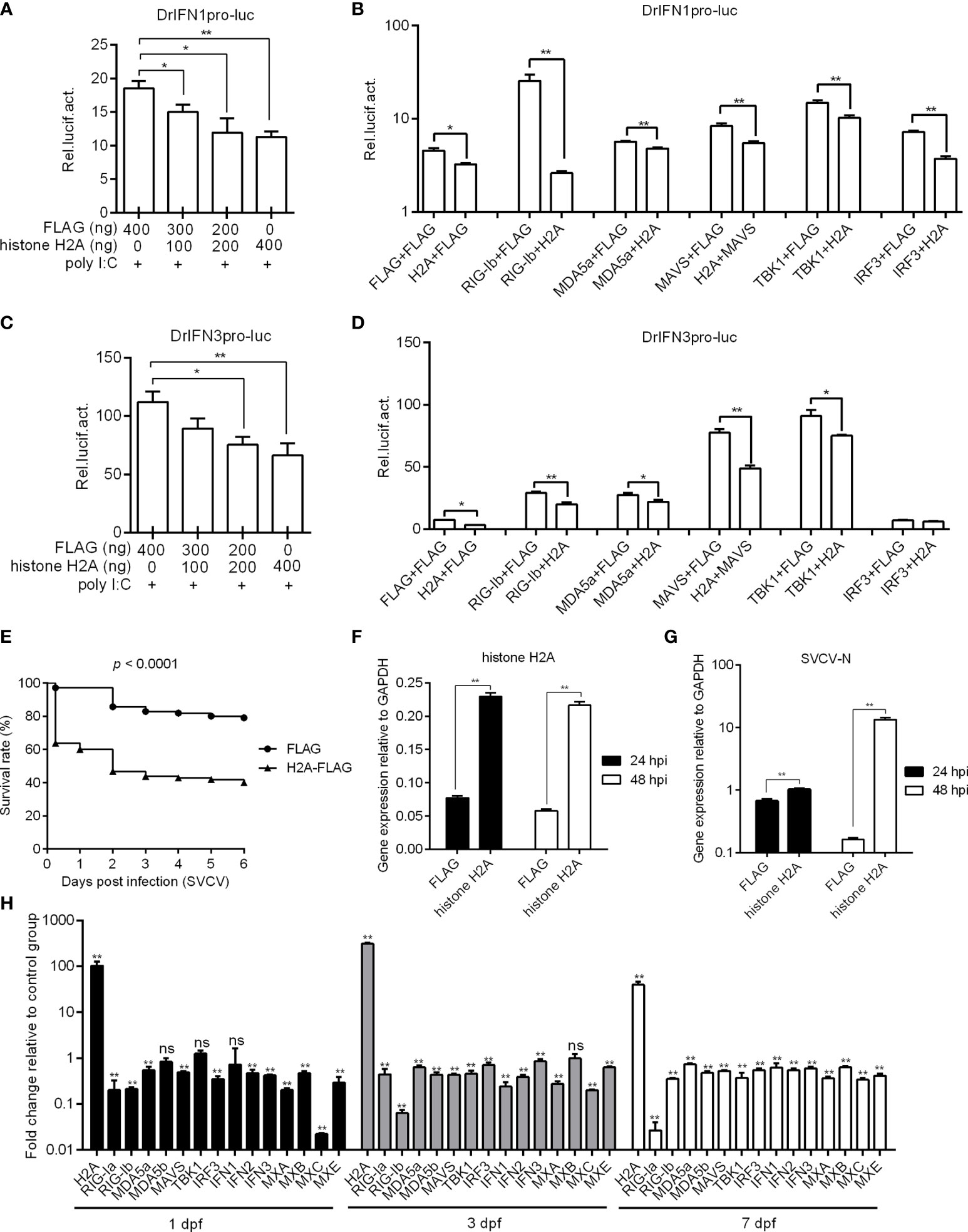
Figure 2 Zebrafish histone H2A negatively regulates RLR signaling. (A) Effect of zebrafish histone H2A on the IFN1 promoter activity mediated by poly I:C stimulation. (B) Effect of zebrafish histone H2A on the IFN1 promoter activity mediated by RLR signaling pathway. (C) Effect of zebrafish histone H2A on the IFN3 promoter activity mediated by poly I:C stimulation. (D) Effect of zebrafish histone H2A on the IFN3 promoter activity mediated by RLR signaling pathway. For (A, C), EPC cells transfected with various indicated plasmids and DNA concentration were stimulation with poly I:C. Another 24 h later, the cells were lysed and used for luciferase activity assay. For (B, D), EPC cells were transfected with the indicated combination. At 48 h post-transfection, the cells were used for luciferase activity assay. (E) Effect of zebrafish histone H2A on the larvae survival. (F) The confirmation of zebrafish histone H2A expression in the histone H2A-overexpressed larvae with SVCV infection. (G) Effect of zebrafish histone H2A on the expression of SVCV-N gene. For (E–G), zebrafish larvae microinjected with p3×FLAG or H2A-FLAG were infected with 2×106 PFU/mL SVCV for 24 h. For (E), larvae were monitored for 6 days. For (F, G), larvae were collected at 24 and 48 hpi, and used for qRT-PCR. (H) Effect of zebrafish histone H2A on the expression of antiviral genes involved in RLR signaling pathway. Zebrafish larvae microinjected with p3×FLAG or H2A-FLAG were collected at 1, 3 and 7 dpf, and used for qRT-PCR. For (A–D, F–H), data represented means ± SEM (n = 3), and were tested for statistical significance. *p < 0.05; **p < 0.01; ns, not significant.
The role of histone H2A overexpression in SVCV infection was further investigated. Compared with zebrafish larvae microinjected with FLAG empty plasmid, SVCV-infected larvae microinjected with histone H2A exhibited reduced survival rate about 40% (Figure 2E). The overexpression of histone H2A in zebrafish larvae was confirmed by qRT-PCR (Figure 2F), which significantly promoted the expression of SVCV-N both at 24 hpi and 48 hpi (Figure 2G). The function of histone H2A in the regulation of these genes involved in RLR signaling was investigated by qRT-PCR. The overexpression of histone H2A in zebrafish larvae significantly inhibited the expression of RIG-Ia, RIG-Ib, MDA5a, MDA5b, MAVS, IRF3, IFN1, IFN2, IFN3, MXA, MXB, MXC and MXE at 1, 3 and/or 7 dpf (Figure 2H). Together these results suggest a negative role for histone H2A in antiviral response and RLR signaling.
Since the TBK1-IRF3 axis is critical for the antiviral immune response, we investigated if histone H2A was able to interact with TBK1 and IRF3. Immunoprecipitation and immunoblot assays indicated that histone H2A and TBK1 or IRF3 associated together (Figure 3A). Immunofluorescence analysis demonstrated that histone H2A colocalized with TBK1 and IRF3 (Figure 3B). Next, we investigated whether the interaction between histone H2A and TBK1 or IRF3 might influence the formation of TBK1-IRF3 functional complex. Anti-FLAG conjugated agarose beads were used to precipitate TBK1-FLAG-containing protein complex, and found IRF3-GFP (using anti-GFP antibody for IP product in lanes 5 and 7 in Figure 3C) or histone H2A-GFP (using anti-GFP antibody for IP product in lanes 6 and 7 in Figure 3C) in this complex. Compared with IRF3-GFP band for the sample without the addition of histone H2A, the addition of histone H2A significantly decreased TBK1-IRF3 interaction (using anti-GFP antibody for IP product in lane 7 in Figure 3C). Collectively, these data suggest that histone H2A interacts with TBK1 and IRF3 to impair the formation of TBK1-IRF3 complex when overexpressed in EPC cells.
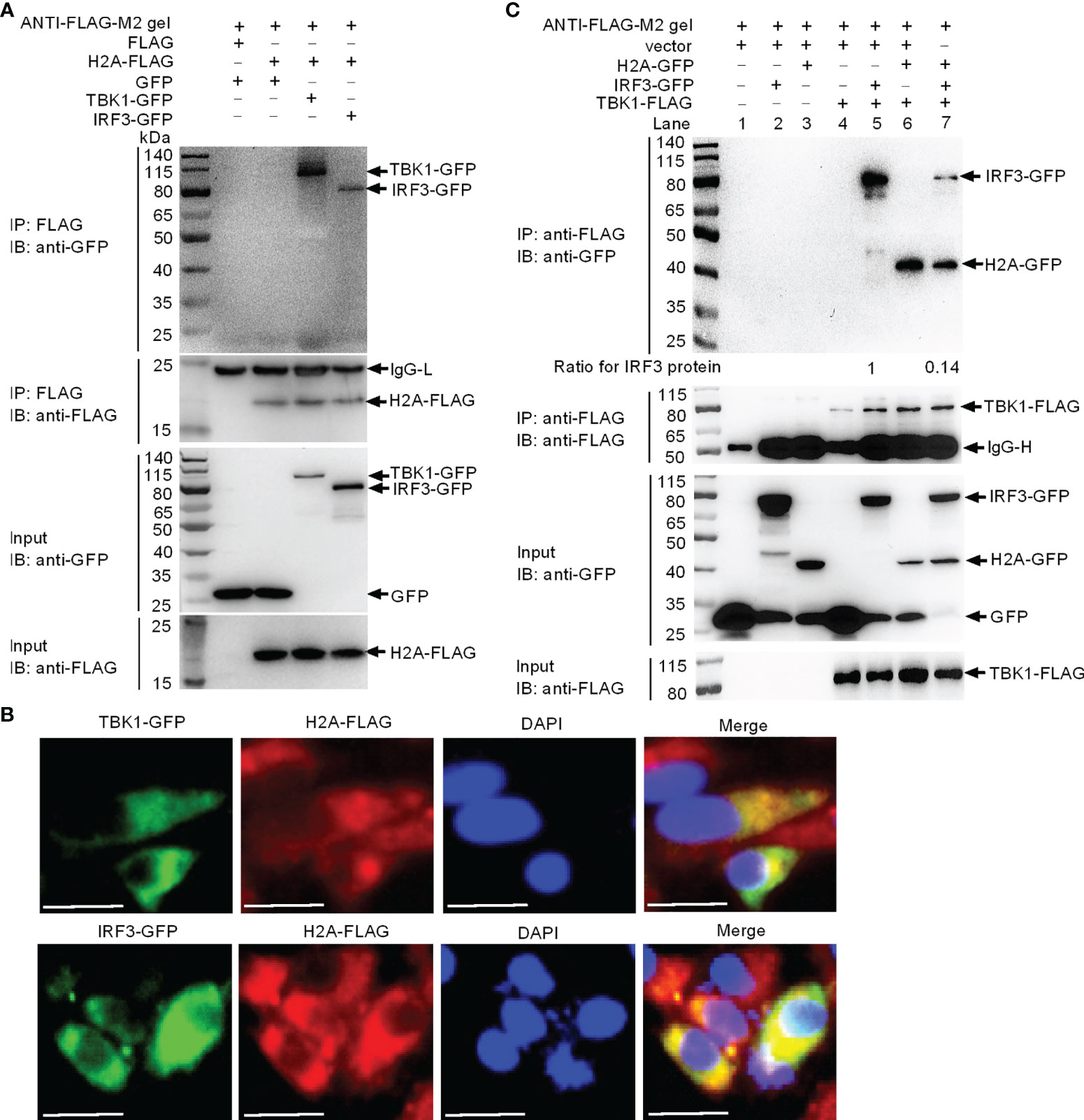
Figure 3 Zebrafish histone H2A interacts with TBK1 and IRF3 to disrupt the formation of TBK1-IRF3 complex. (A) Zebrafish histone H2A interacted with TBK1 and IRF3. (B) Zebrafish histone H2A colocalized with TBK1 and IRF3. (C) Zebrafish histone H2A interacted with TBK1 to inhibit the formation of TBK1-IRF3 complex. For (A, C), co-IP was performed with anti-FLAG-conjugated agarose beads in EPC cells. The cell lysates and bound proteins were analyzed by immunoblotting with the indicated Abs. All experiments were repeated for at least three times with similar results. The expression ratio for IRF3 protein was quantified by Quantity One. For (B), immunofluorescence analysis was performed in EPC cells transfected with H2A-FLAG and TBK1-GFP or IRF3-GFP. Scale bar: 25 μm.
Since histone H2A impaired the formation of TBK1-IRF3 complex, we next examined whether histone H2A might influence the protein levels of TBK1 and IRF3. Western blotting analysis showed that the amount of TBK1 and IRF3 proteins was significantly decreased by the overexpression of histone H2A (Figures 4A, B). To further reveal the mechanisms involved in histone H2A degrading with TBK1 and IRF3 expression during the posttranscriptional process, the effects of MG132 (inhibitor for the proteasome), early autophagy inhibitor 3-methyladenine (3-MA) and late autophagy inhibitor chloroquine (CQ) or ammonium chloride (NH4Cl) on histone H2A-mediated degradations of TBK1 and IRF3 were investigated in EPC cells. The degradations of TBK1 and IRF3 were not inhibited by MG132 (Figures 4C, D). However, the degradations of TBK1 and IRF3 were partly inhibited by 3-MA and CQ, which were dependent of the dose variation (Figures 4E–H). In addition, NH4Cl treatment only partly inhibited histone H2A-mediated degradation of IRF3, but not for TBK1 (Figures 4I, J). These data demonstrate that histone H2A degrades TBK1 and IRF3 via autophagosome-lysosome pathway.
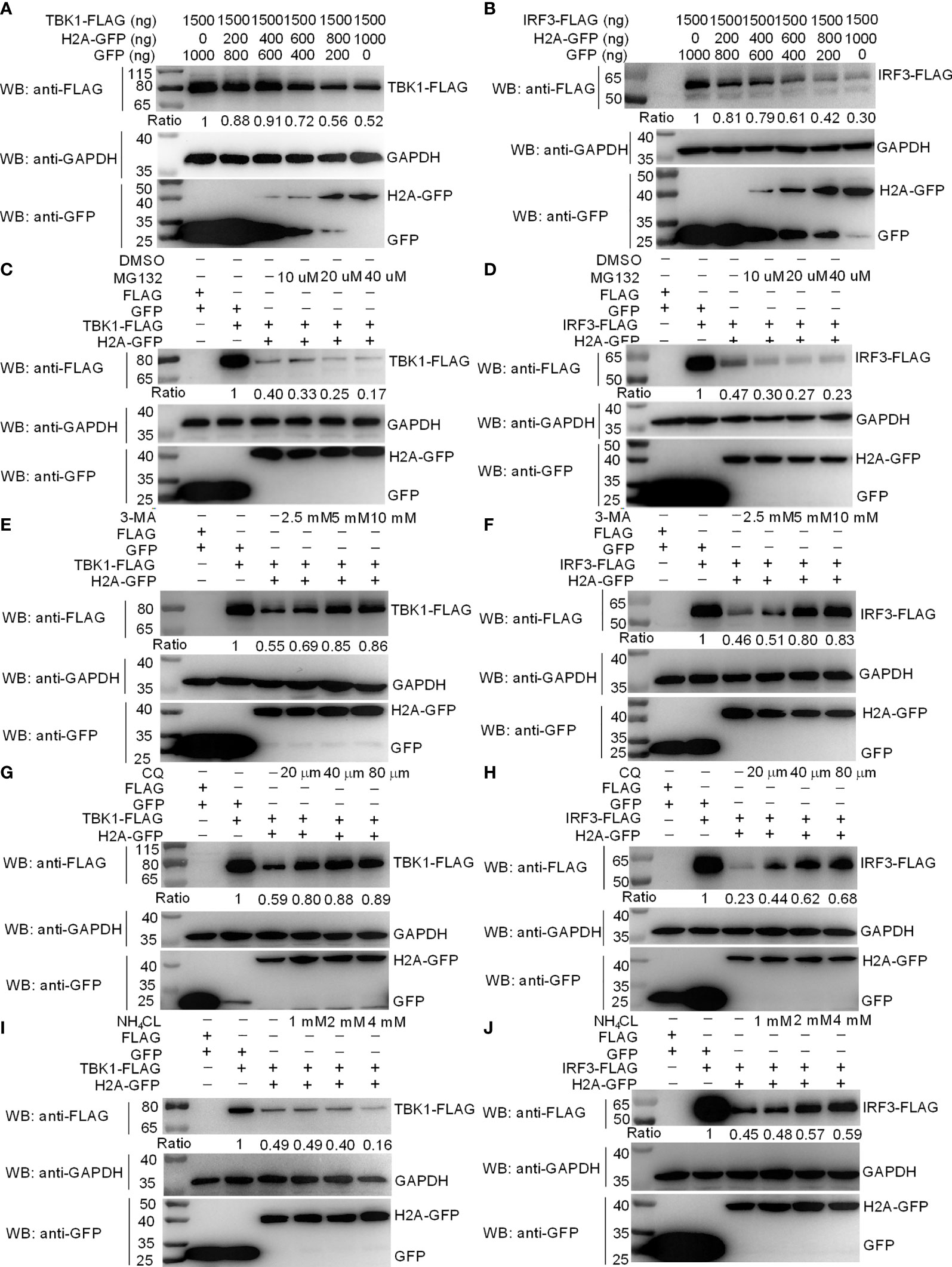
Figure 4 Zebrafish histone H2A degrades TBK1 and IRF3 via the lysosomal pathway. (A) Effect of zebrafish histone H2A on the protein expression of TBK1. (B) Effect of zebrafish histone H2A on the protein expression of IRF3. For (A, B), EPC cells seeded in six-well plates were transfected with various indicated plasmids and DNA concentration. After 48 h post-transfection, cell lysates were analyzed by Western blotting using the indicated Abs. (C) Effect of MG132 on the histone H2A-mediated protein degradation of TBK1. (D) Effect of MG132 on the histone H2A-mediated protein degradation of IRF3. (E) Effect of 3-MA on the histone H2A-mediated protein degradation of TBK1. (F) Effect of 3-MA on the histone H2A-mediated protein degradation of IRF3. (G) Effect of CQ on the histone H2A-mediated protein degradation of TBK1. (H) Effect of CQ on the histone H2A-mediated protein degradation of IRF3. (I) Effect of NH4Cl on the histone H2A-mediated protein degradation of TBK1. (J) Effect of NH4Cl on the histone H2A-mediated protein degradation of IRF3. For (C–J), EPC cells seeded in six-well plates were transfected with various indicated plasmids and DNA concentration. After 48 h post-transfection, cells were treated with DMSO, MG132, 3-MA, CQ or NH4Cl with indicated concentration for 6 h. Following this, cell lysates were analyzed by Western blotting using the indicated Abs. The expression ratio for TBK1 or IRF3 protein was quantified by Quantity One. All experiments were repeated for at least three times with similar results.
Previous studies have shown that piscine TBK1 plays an important role in the antiviral immune responses against SVCV and GCRV infection (20, 26). We therefore further investigated the role of histone H2A in TBK1-mediated antiviral activity. The supernate of ZF4 cells transfected with TBK1 and/or histone H2A were used for the detection of SVCV viral loads. Compared with the control group transfected with empty plasmids, lower viral loads were observed for ZF4 cells transfected with TBK1, higher viral loads for ZF4 cells co-transfected with histone H2A and TBK1, and the highest viral loads for ZF4 cells transfected with histone H2A (Figure 5A). The cell precipitates of ZF4 cells transfected with TBK1 and/or histone H2A were used for the examination of expressions of viral genes. The overexpression of histone H2A was confirmed by qRT-PCR (Figure 5B). Compared with the control group transfected with empty plasmids, the expression of SVCV-N or SVCV-G was significantly decreased by TBK1 overexpression, but increased by the coexpression of histone H2A and TBK1 (Figures 5C, D). All these data suggest that histone H2A can completely abolish TBK1-mediated antiviral activity.
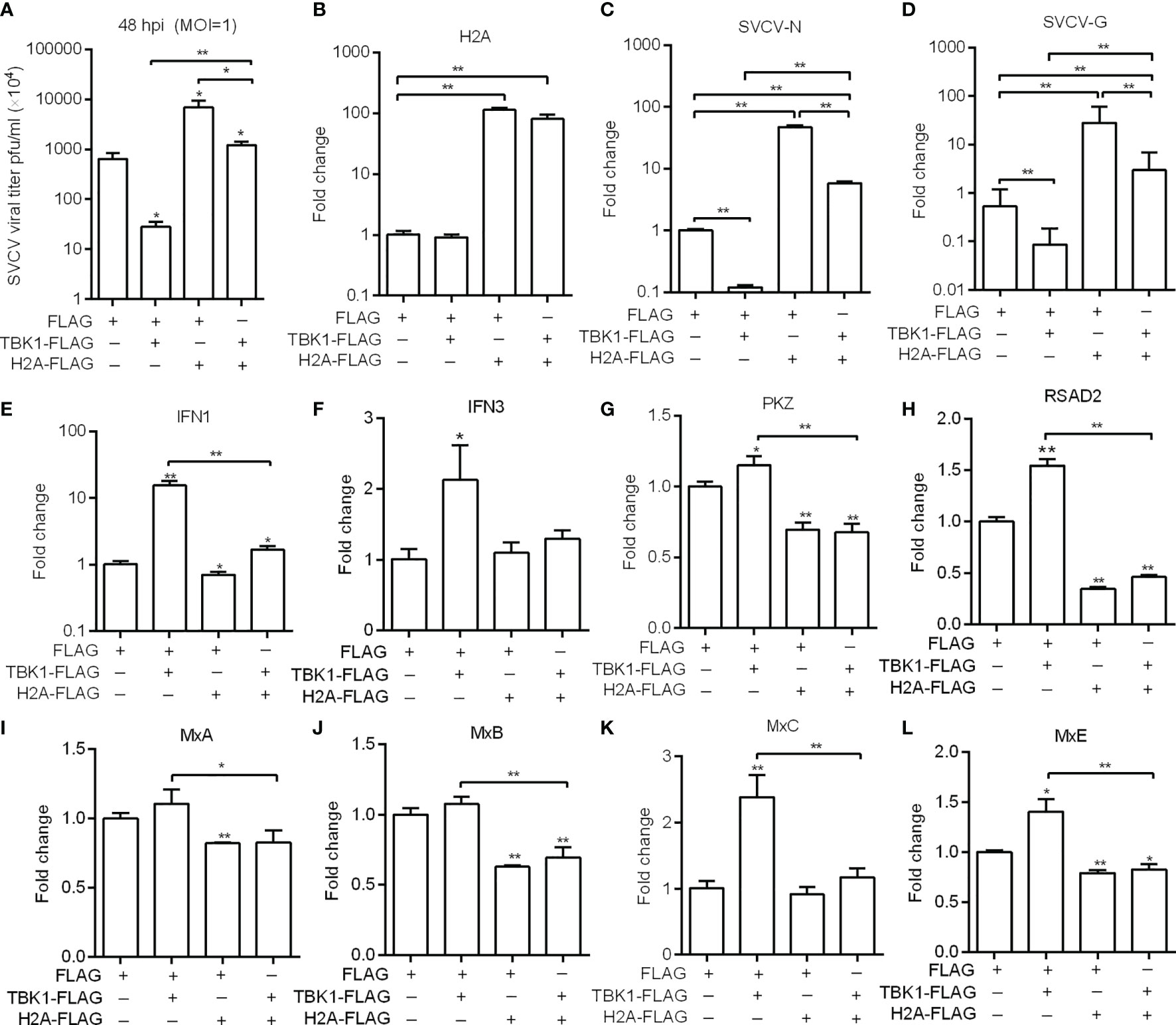
Figure 5 Zebrafish histone H2A inhibits the effects mediated by TBK1. (A) Effect of zebrafish histone H2A on the TBK1-mediated antiviral activity. (B) The confirmation of zebrafish histone H2A expression in the histone H2A-overexpressed ZF4 cells with SVCV infection. (C) Effect of zebrafish histone H2A on the expression of SVCV-N gene mediated by TBK1. (D) Effect of zebrafish histone H2A on the expression of SVCV-G gene mediated by TBK1. (E) Effect of zebrafish histone H2A on the expression of IFN1 mediated by TBK1. (F) Effect of zebrafish histone H2A on the expression of IFN3 mediated by TBK1. (G) Effect of zebrafish histone H2A on the expression of PKZ mediated by TBK1. (H) Effect of zebrafish histone H2A on the expression of RSAD2 mediated by TBK1. (I) Effect of zebrafish histone H2A on the expression of MxA mediated by TBK1. (J) Effect of zebrafish histone H2A on the expression of MxB mediated by TBK1. (K) Effect of zebrafish histone H2A on the expression of MxC mediated by TBK1. (L) Effect of zebrafish histone H2A on the expression of MxE mediated by TBK1. For (A–L) ZF4 cells were transfected with the indicated plasmids. At 36 h post-transfection, these cells were used for SVCV infection at an MOI of 1. At 48 hpi, the supernatants of transfected cells were collected for the determination of virus titers. The cell pellets of transfected cells were collected and used for qRT-PCR. Data represented means ± SEM (n = 3), and were tested for statistical significance. *p < 0.05; **p < 0.01. The asterisk above the error bars indicates statistical significance using the group microinjected with empty plasmid as the control group. The asterisk above the bracket indicates statistical significance between the two groups connected by the bracket.
TBK1 can induce a higher expression of IFNs and ISGs (20, 27). We next investigated whether histone H2A inhibited TBK1-mediated induction of antiviral effector genes in the case of SVCV infection. The overexpression of histone H2A significantly inhibited the expression of IFN1, PKZ, RSAD2, MxA, MxB and MxE. Furthermore, the overexpression of histone H2A significantly inhibited TBK1-mediated induction of IFN1 and ISGs including PKZ, RSAD2, MxC and MxE (Figures 5E–L).
IRF3 normally shuttles between nucleus and cytoplasm. To investigate the location of histone H2A-mediated IRF3 degradation, cytoplasmic and nuclear fractions were prepared from SVCV-infected EPC cells transfected with various indicated plasmids. Overexpression of histone H2A was found to degrade cytoplasmic IRF3 in a dose dependent manner (Figure 6A). Notably, histone H2A also promoted the degradation of nuclear IRF3 protein (Figure 6B).
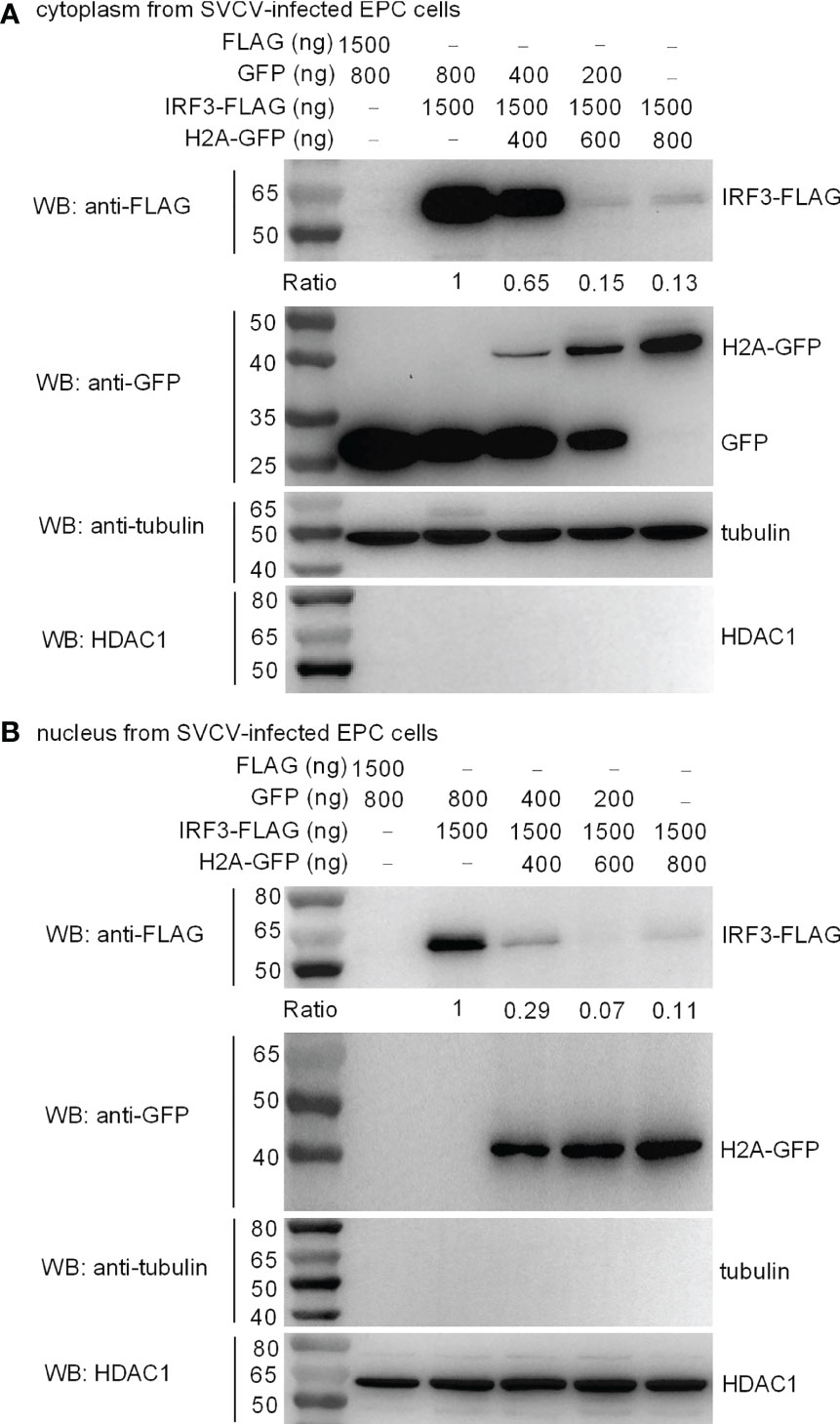
Figure 6 Zebrafish histone H2A impairs the protein expression of cytoplasmic (A) and nuclear IRF3 protein (B). For (A, B), EPC cells seeded in 6-well plates were transfected with various indicated plasmids. After 36 h post-transfection, the cells were infected with SVCV. After 12 h, the cells were harvested and used for preparation of nuclear and cytoplasmic extracts. The expression ratio for IRF3 protein was quantified by Quantity One.
Histone H2A was translocated into cytoplasm after SVCV infection. To directly confirm the nuclear/cytoplasmic trafficking of histone H2A is required for the protein degradations of TBK1 and IRF3 and the negative regulation of histone H2A in SVCV infection, nuclear export inhibitor LMB was used. LMB treatment (100 nM) largely blocked the nuclear export of histone H2A, and almost completely abolished histone H2A-induced TBK1 degradation in SVCV-infected cells (Figures 7A, B). Inhibition of histone H2A nuclear/cytoplasmic trafficking by LMB also inhibited histone H2A-induced IRF3 degradation both in the cytoplasm and the nucleus (Figures 7C, D). Furthermore, LMB treatment significantly decreased the SVCV replication mediated by histone H2A in the supernatants of infected cells (Figure 7E). Most of all, the expressions of SVCV genes including SVCV-N, SVCV-P and SVCV-G were significantly decreased by the histone H2A and LMB treatment compared with the control group transfected with empty plasmids and the group transfected with histone H2A but without LMB treatment (Figures 7F–H). Taken together, these results suggest that the cytoplasmic localization of histone H2A is required for the protein degradations of TBK1 and IRF3 and the negative regulation of histone H2A on the SVCV infection.
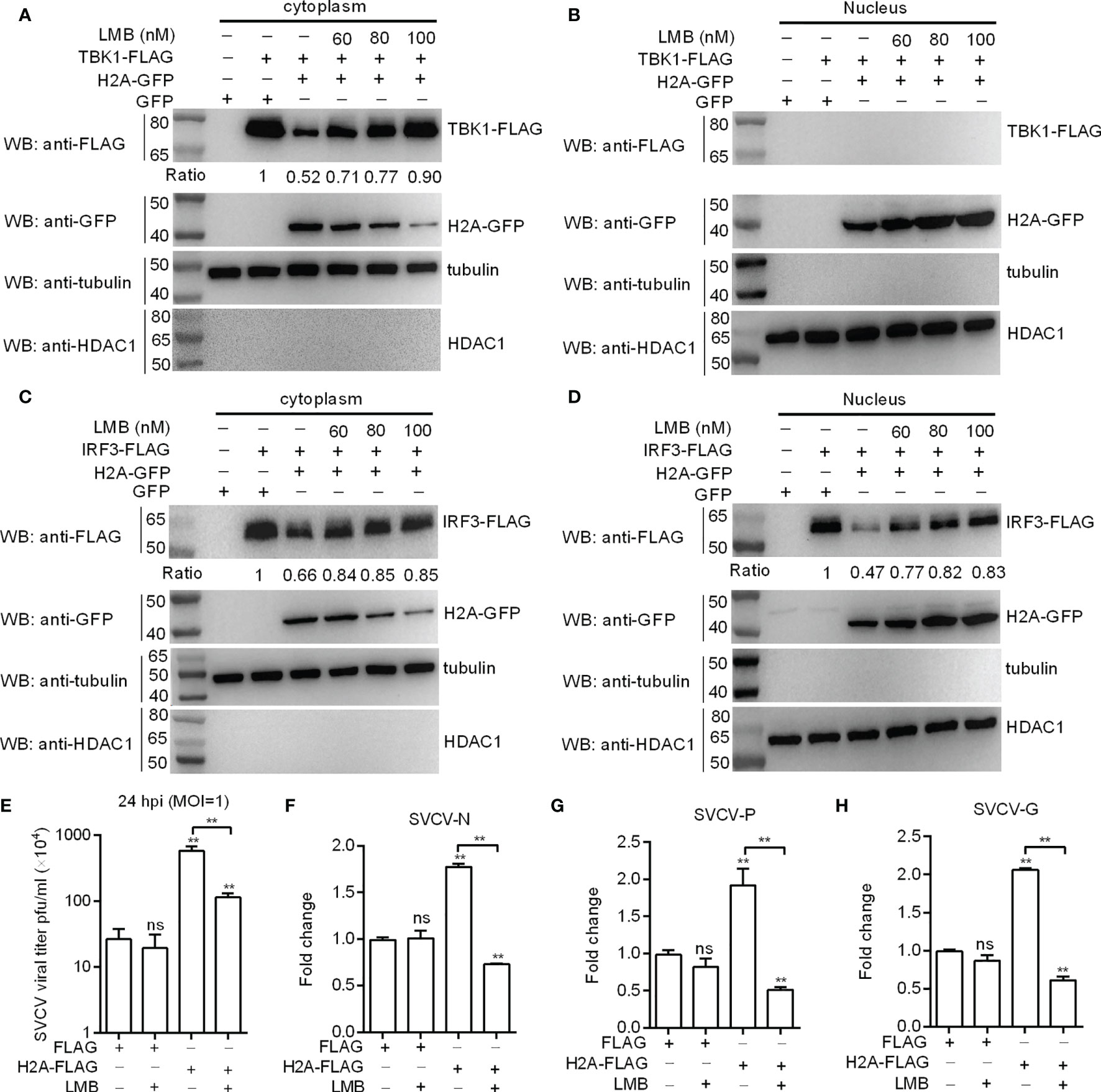
Figure 7 Histone H2A nuclear/cytoplasmic trafficking is essential for the negative regulation of histone H2A. (A) Effect of the inhibition of histone H2A nuclear/cytoplasmic trafficking on the protein degradation of cytoplasmic TBK1. (B) Effect of the inhibition of histone H2A nuclear/cytoplasmic trafficking on the protein degradation of nuclear TBK1. (C) Effect of the inhibition of histone H2A nuclear/cytoplasmic trafficking on the protein degradation of cytoplasmic IRF3. (D) Effect of the inhibition of histone H2A nuclear/cytoplasmic trafficking on the protein degradation of nuclear IRF3. For A–D), EPC cells seeded in 6-well plates were transfected with various indicated plasmids. After 36 h post-transfection, the cells were treated with LMB at the indicated concentration or left untreated. After 12 h, the cells were harvested and used for preparation of nuclear and cytoplasmic extracts. The expression ratio for TBK1 or IRF3 protein was quantified by Quantity One. (E) Effect of the inhibition of histone H2A nuclear/cytoplasmic trafficking on the histone H2A-mediated SVCV replication. (F) Effect of the inhibition of histone H2A nuclear/cytoplasmic trafficking on the expression of SVCV-N mediated by histone H2A. (G) Effect of the inhibition of histone H2A nuclear/cytoplasmic trafficking on the expression of SVCV-P mediated by histone H2A. (H) Effect of the inhibition of histone H2A nuclear/cytoplasmic trafficking on the expression of SVCV-G mediated by histone H2A. For (E–H), EPC cells seeded in 24-well plates were transfected with p3×FLAG or H2A-FLAG, then treated with LMB or left untreated. After 48 h posttransfection, the cells were infected with SVCV. Another 24 h later, the supernatants of infected cells were collected for the determination of virus titers. The cell pellets of infected cells were collected for qRT-PCR. Data represented means ± SEM (n = 3), and were tested for statistical significance. **p < 0.01; ns, not significant. The asterisk above the error bars indicates statistical significance using the group microinjected with empty plasmid as the control group. The asterisk above the bracket indicates statistical significance between the two groups connected by the bracket.
Histones are localized to the nucleus, and are fundamental structural components of chromatin. However many studies have shown the extrachromosomal functions of histones. Histones are found to translocate to the cytoplasm following infection or in response to various exogenous and endogenous stressors, and mediate functions in the cytoplasm. For example, histone H1.2 showed a time-dependent and X-ray dose-dependent increase in the cytoplasm after X-ray irradiation, and promoted the mitochondrial apoptotic pathway (28). Double-stranded DNA (dsDNA) stimulation induced the aggregation of histone H2B and the interaction between histone H2B and MAVS in the cytoplasm, which triggered antiviral innate immune responses against DNA viruses (29). This study demonstrates for the first time that SVCV, a negative ssRNA virus, induces the translocation of nuclear histone H2A to the cytoplasm. Histone H2A nuclear/cytoplasmic trafficking triggers the protein degradations of TBK1 and IRF3 via the lysosomal pathway, which leads to the impaired piscine antiviral response.
It was reported that histones could act as regulators of TLR or NLR signaling. In mice, histone H4 directly interacts with TLR2 and TLR4, and activates TLR2 and TLR4 to induce the production of proinflammatory cytokines in a MyD88-dependent manner (30). Nucleotide binding oligomerzation domain 1 (NOD1), NOD2 and NLRP3 belong to the NLR family. Once released from the nucleosome, extracellular histones have been reported to activate NLRP3 inflammasome (31). Extracellular histone H3, whose stimulation could elevate the expression of NLRP3 and NOD2, caused pyroptosis in sepsis via NOD2 and VSIG4/NLRP3 pathways (32). In teleost fish, our previous studies revealed that histone H2A variant cooperated with NOD1 or RIP2 of NLR signaling to induce the transcription of antibacterial PRRs including NOD1, PGRP2, PGRP5 and PGRP6 (14, 16). The present study showed that the overexpression of histone H2A significantly impaired the expression of antiviral RLRs. All these results demonstrate that histones acting as alarmin signals can activate or suppress PRRs such as TLRs, NLRs, RLRs and PGRPs. Furthermore, previous studies revealed that TLR2 and TLR4 may be PRRs for recognizing histones. In mouse peritoneal macrophages (PEMs), the most effective histones for the induction of cytokine and chemokine release were H2A and H2B. However in mouse PEMs exposed to histones, the absence of TLR2 and TLR4 dramatically reduced the cytokine responses (33). Since piscine histone H2A can impair the expression of RLRs, it is interesting to further investigate the effect and mechanisms of histone H2A-mediated ubiquitination in RIG-I antiviral activity.
Histone variants allow for greater control of DNA replication, repair or transcription. The variants of histone H2A include H2A.X, H2A.Z, MacroH2A, H2A.Bbd, etc (34). Among them, H2A.Z is the most studied variant of H2A. H2A.Z has been associated with both transcription activation and transcription inhibition (35). In human cells, H2A.Z acts as a negative regulator of antiviral responses via GCN5 and BRD2 (36). In shrimp Penaeus vannamei, the white spot syndrome virus (WSSV) decreased the expression of histones H2A and H4 to promote viral replication (37). Although the lysine rich histones H2A and H2B were less potent compared to the arginine rich histones H3, H3.3 and H4, all histones inhibited infectivity of influenza A H3N2 strain Phil82 in mammals (38). Here, we provided evidence that zebrafish histone H2A negatively regulated antiviral response against SVCV infection. Interesting, our previous studies revealed that nucleotide polymorphisms of piscine histone H2A affect disease resistance of zebrafish and grass carp against bacterial infection (15, 39). Whether there is a correlation between piscine H2A nucleotide polymorphisms and viral infection needs further investigation.
TBK1-IRF3 axis plays a pivotal role in inducing type I IFN signaling. To date, identified regulators of TBK1-IRF3 signaling axis have been shown to regulate innate immune responses against various types of pathogens (7, 40). Since histones act as DAMPs, and can engage PRRs to trigger immune signaling pathways just like PAMPs, how histones regulate TBK1-IRF3 axis remains an important, but less understood question. A study showed that histone H2B interacted with IFI16 (gamma-interferon-inducible protein 16) and BRCA1 in the nucleus, and viral infections such as EBV, KSHV and HSV-1 induced the cytoplasmic distribution of H2B-IFI16 and H2B-BRCA1 complexes via Ran-GTP protein. Importantly, the H2B-IFI16-BRCA1 complex caused TBK1 and IRF3 phosphorylation and nuclear translocation of pIRF3, which resulted in IFN-β production (41). The significant finding in the present study is that several lines of evidence confirm the negative regulation of piscine histone H2A on the TBK1-IRF3 axis. First, histone H2A inhibited the IFN1 and/or IFN3 promoter activities mediated by TBK1 and IRF3. Second, histone H2A interacted with TBK1 and IRF3 to impair the formation of TBK1-IRF3 complex. Third, histone H2A degraded TBK1 and IRF3 via autophagosome-lysosome pathway. Fourth, histone H2A inhibited TBK1-mediated antiviral activity and degraded cytoplasmic and nuclear IRF3 protein. Fifth, inhibition of histone H2A nuclear/cytoplasmic trafficking by Leptomycin B inhibited the protein degradations of TBK1 and IRF3. All these findings together demonstrate a novel negative regulatory feedback loop between cytoplasmic histone H2A and TBK1-IRF3 axis that regulates RLR signaling.
In summary, we report in this work that piscine histone H2A is critically involved in the negative regulation of TBK1 and IRF3 in antiviral IFN response. Based on the experimental data, a working model is presented for explaining how histone H2A is utilized by SVCV to evade host immune response. Under normal physiological conditions, histone H2A is nuclear-localized. SVCV infection induces the translocation of nuclear histone H2A to the cytoplasm. The cytoplasmic histone H2A targets TBK1 and IRF3 for the protein degradations of TBK1 and IRF3 via the lysosomal pathway, which lead to the impaired formation of TBK1-IRF3 functional complex and the decreased expression of nuclear IRF3 protein. Moreover, cytoplasmic histone H2A also blocks TBK1-mediated antiviral activity. All these together impair the antiviral immune response in response to SVCV infection (Figure 8). Further work is needed to assess the mechanism of histone H2A nuclear/cytoplasmic trafficking following infection.
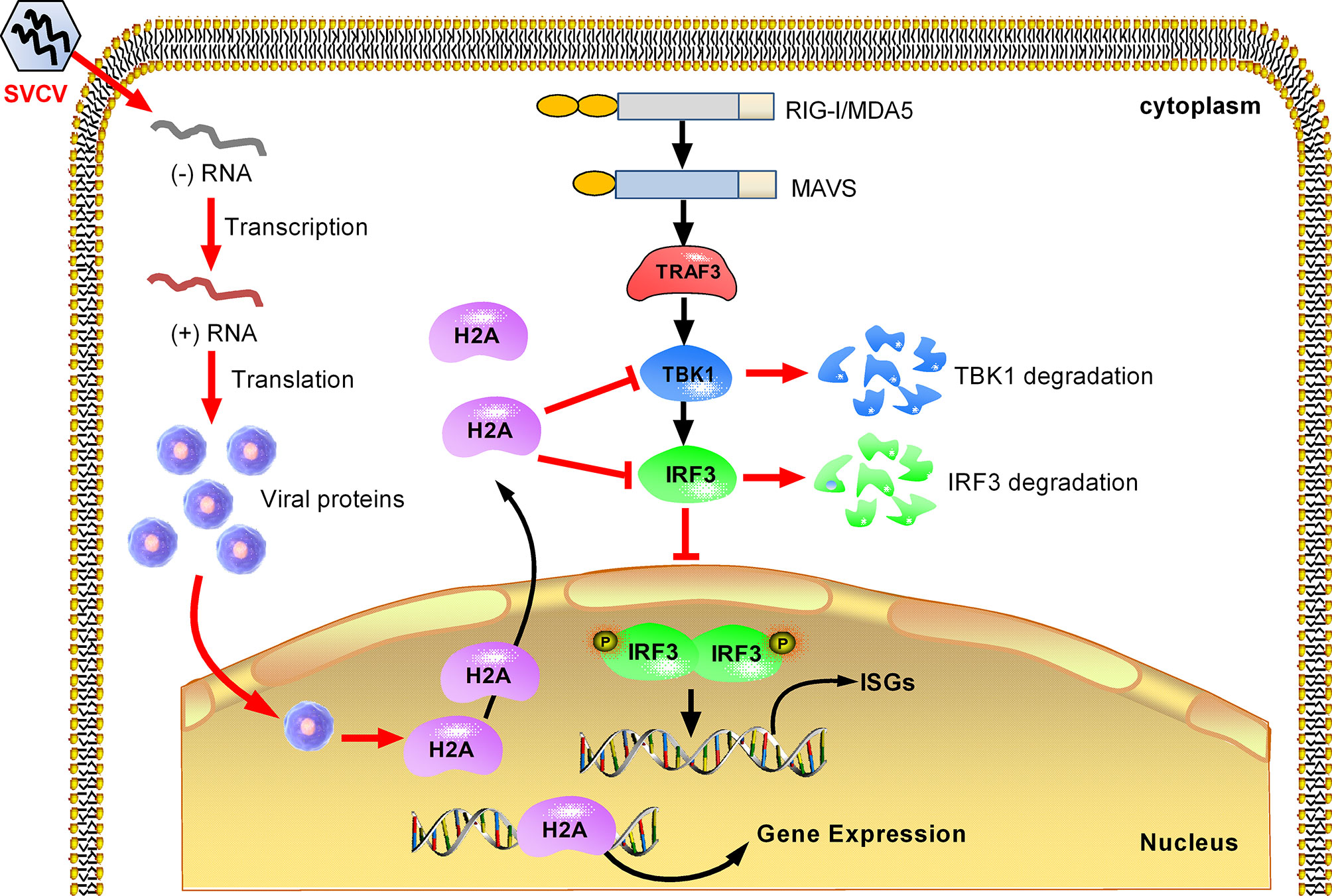
Figure 8 Proposed model illustrating how histone H2A is utilized by SVCV to evade host immune response. Histone H2A is nuclear-localized in the absence of pathogen infection. SVCV infection promotes histone H2A nuclear/cytoplasmic trafficking. In the cytoplasm, histone H2A degrades TBK1 and IRF3, which lead to the impaired formation of TBK1-IRF3 functional complex and the decreased expression of nuclear IRF3 protein. In the cell nucleus, the impaired expressions of IRF3 and/or histone H2A inhibit transcriptional regulation of immune genes, and impede the production of type I IFNs and ISGs in response to SVCV infection.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
All animal experiments were conducted in accordance with the Guiding Principles for the Care and Use of Laboratory Animals and were approved by the Institute of Hydrobiology, Chinese Academy of Sciences (Approval ID: IHB 2013724).
MXC conceived and designed the experiments. XMW, HF and JZ performed the experiments and analyzed the data. MXC, XMW, JZ and YHB interpreted data for the work. MXC and XMW wrote the manuscript. MXC and YHB revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by the National Natural Science Foundation of China Grant (31873046), Wuhan Application Foundation Frontier Project (2019020701011467), and Strategic Priority Research Program of the Chinese Academy of Sciences Grant XDA24010308.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.771277/full#supplementary-material
1. Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
2. Kumar H, Kawai T, Akira S. Pathogen Recognition by the Innate Immune System. Int Rev Immunol (2011) 30:16–34. doi: 10.3109/08830185.2010.529976
3. Zhang Z, Long Q, Xie J. Roles of Peptidoglycan Recognition Protein (PGRP) in Immunity and Implications for Novel Anti-Infective Measures. Crit Rev Eukaryot Gene Expr (2012) 22:259–68. doi: 10.1615/CritRevEukarGeneExpr.v22.i3.90
4. Rehwinkel J, Gack MU. RIG-I-Like Receptors: Their Regulation and Roles in RNA Sensing. Nat Rev Immunol (2020) 20:537–51. doi: 10.1038/s41577-020-0288-3
5. Kell AM, Gale M Jr. RIG-I in RNA Virus Recognition. Virology (2015) 479-480:110–21. doi: 10.1016/j.virol.2015.02.017
6. Chen SN, Zou PF, Nie P. Retinoic Acid-Inducible Gene I (RIG-I)-Like Receptors (RLRs) in Fish: Current Knowledge and Future Perspectives. Immunology (2017) 151:16–25. doi: 10.1111/imm.12714
7. Chang MX. The Negative Regulation of Retinoic Acid-Inducible Gene I (RIG-I)-Like Receptors (RLRs) Signaling Pathway in Fish. Dev Comp Immunol (2021) 119:104038. doi: 10.1016/j.dci.2021.104038
8. Denning NL, Aziz M, Gurien SD, Wang P. DAMPs and NETs in Sepsis. Front Immunol (2019) 10:2536. doi: 10.3389/fimmu.2019.02536
9. Huang J, Xie Y, Sun X, Zeh HJ 3rd, Kang R, Lotze MT, et al. DAMPs, Ageing, and Cancer: The ‘DAMP Hypothesis’. Ageing Res Rev (2015) 24:3–16. doi: 10.1016/j.arr.2014.10.004
10. Murao A, Aziz M, Wang H, Brenner M, Wang P. Release Mechanisms of Major DAMPs. Apoptosis (2021) 26:152–62. doi: 10.1007/s10495-021-01663-3
11. Wu D, Ingram A, Lahti JH, Mazza B, Grenet J, Kapoor A, et al. Apoptotic Release of Histones From Nucleosomes. J Biol Chem (2002) 277:12001–8. doi: 10.1074/jbc.M109219200
12. Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, et al. Endogenous Histones Function as Alarmins in Sterile Inflammatory Liver Injury Through Toll-Like Receptor 9 in Mice. Hepatology (2011) 54:999–1008. doi: 10.1002/hep.24501
13. Chen R, Kang R, Fan XG, Tang D. Release and Activity of Histone in Diseases. Cell Death Dis (2014) 5:e1370. doi: 10.1038/cddis.2014.337
14. Wu XM, Cao L, Nie P, Chang MX. Histone H2A Cooperates With RIP2 to Induce the Expression of Antibacterial Genes and MHC Related Genes. Dev Comp Immunol (2019) 101:103455. doi: 10.1016/j.dci.2019.103455
15. Wu XM, Fang H, Nie P, Chang MX. Nucleotide Polymorphism and Function of Piscine Histone H2A in Edwardsiella Piscicida Infection. J Fish China (2020) 44:1–14.
16. Wu X, Xiong F, Fang H, Zhang J, Chang M. Crosstalks Between NOD1 and Histone H2A Contribute to Host Defense Against Streptococcus Agalactiae Infection in Zebrafish. Antibiotics (Basel) (2021) 10:861. doi: 10.3390/antibiotics10070861
17. Zou PF, Chang MX, Xue NN, Liu XQ, Li JH, Fu JP, et al. Melanoma Differentiation-Associated Gene 5 in Zebrafish Provoking Higher Interferon-Promoter Activity Through Signalling Enhancing of its Shorter Splicing Variant. Immunology (2014) 141:192–202. doi: 10.1111/imm.12179
18. Zou PF, Chang MX, Li Y, Zhang SH, Fu JP, Chen SN, et al. Higher Antiviral Response of RIG-I Through Enhancing RIG-I/MAVS-Mediated Signaling by Its Long Insertion Variant in Zebrafish. Fish Shellfish Immunol (2015) 43:13–24. doi: 10.1016/j.fsi.2014.12.001
19. Chen WQ, Hu YW, Zou PF, Ren SS, Nie P, Chang MX. MAVS Splicing Variants Contribute to the Induction of Interferon and Interferon-Stimulated Genes Mediated by RIG-I-Like Receptors. Dev Comp Immunol (2015) 49:19–30. doi: 10.1016/j.dci.2014.10.017
20. Hu YW, Zhang J, Wu XM, Cao L, Nie P, Chang MX. TANK-Binding Kinase 1 (TBK1) Isoforms Negatively Regulate Type I Interferon Induction by Inhibiting TBK1-IRF3 Interaction and IRF3 Phosphorylation. Front Immunol (2018) 9:84. doi: 10.3389/fimmu.2018.00084
21. Cao L, Wu XM, Nie P, Chang MX. The Negative Regulation of Piscine CD44c in Viral and Bacterial Infection. Dev Comp Immunol (2019) 96:135–43. doi: 10.1016/j.dci.2019.03.005
22. Wu XM, Zhang J, Li PW, Hu YW, Cao L, Ouyang S, et al. NOD1 Promotes Antiviral Signaling by Binding Viral RNA and Regulating the Interaction of MDA5 and MAVS. J Immunol (2020) 204:2216–31. doi: 10.4049/jimmunol.1900667
23. Levraud JP, Boudinot P, Colin I, Benmansour A, Peyrieras N, Herbomel P, et al. Identification of the Zebrafish IFN Receptor: Implications for the Origin of the Vertebrate IFN System. J Immunol (2007) 178:4385–94. doi: 10.4049/jimmunol.178.7.4385
24. Aggad D, Mazel M, Boudinot P, Mogensen KE, Hamming OJ, Hartmann R, et al. The Two Groups of Zebrafish Virus-Induced Interferons Signal via Distinct Receptors With Specific and Shared Chains. J Immunol (2009) 183:3924–31. doi: 10.4049/jimmunol.0901495
25. López-Muñoz A, Roca FJ, Meseguer J, Mulero V. New Insights Into the Evolution of IFNs: Zebrafish Group II IFNs Induce a Rapid and Transient Expression of IFN-Dependent Genes and Display Powerful Antiviral Activities. J Immunol (2009) 182:3440–9. doi: 10.4049/jimmunol.0802528
26. Yan C, Xiao J, Li J, Chen H, Liu J, Wang C, et al. TBK1 of Black Carp Plays an Important Role in Host Innate Immune Response Against SVCV and GCRV. Fish Shellfish Immunol (2017) 69:108–18. doi: 10.1016/j.fsi.2017.08.016
27. Zhang L, Chen WQ, Hu YW, Wu XM, Nie P, Chang MX. TBK1-Like Transcript Negatively Regulates the Production of IFN and IFN-Stimulated Genes Through RLRs-MAVS-TBK1 Pathway. Fish Shellfish Immunol (2016) 54:135–43. doi: 10.1016/j.fsi.2016.04.002
28. Konishi A, Shimizu S, Hirota J, Takao T, Fan Y, Matsuoka Y, et al. Involvement of Histone H1.2 in Apoptosis Induced by DNA Double-Strand Breaks. Cell (2003) 114:673–88. doi: 10.1016/S0092-8674(03)00719-0
29. Kobiyama K, Takeshita F, Jounai N, Sakaue-Sawano A, Miyawaki A, Ishii KJ, et al. Extrachromosomal Histone H2B Mediates Innate Antiviral Immune Responses Induced by Intracellular Double-Stranded DNA. J Virol (2010) 84:822–32. doi: 10.1128/JVI.01339-09
30. Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hägele H, Lichtnekert J, et al. Histones From Dying Renal Cells Aggravate Kidney Injury via TLR2 and TLR4. J Am Soc Nephrol (2012) 23:1375–88. doi: 10.1681/ASN.2011111077
31. Allam R, Darisipudi MN, Tschopp J, Anders HJ. Histones Trigger Sterile Inflammation by Activating the NLRP3 Inflammasome. Eur J Immunol (2013) 43:3336–42. doi: 10.1002/eji.201243224
32. Shi CX, Wang Y, Chen Q, Jiao FZ, Pei MH, Gong ZJ. Extracellular Histone H3 Induces Pyroptosis During Sepsis and May Act Through NOD2 and VSIG4/NLRP3 Pathways. Front Cell Infect Microbiol (2020) 10:196. doi: 10.3389/fcimb.2020.00196
33. Fattahi F, Grailer JJ, Lu H, Dick RS, Parlett M, Zetoune FS, et al. Selective Biological Responses of Phagocytes and Lungs to Purified Histones. J Innate Immun (2017) 9:300–17. doi: 10.1159/000452951
34. Bönisch C, Hake SB. Histone H2A Variants in Nucleosomes and Chromatin: More or Less Stable? Nucleic Acids Res (2012) 40:10719–41. doi: 10.1093/nar/gks865
35. Hu G, Cui K, Northrup D, Liu C, Wang C, Tang Q, et al. H2A.Z Facilitates Access of Active and Repressive Complexes to Chromatin in Embryonic Stem Cell Self-Renewal and Differentiation. Cell Stem Cell (2013) 12:180–92. doi: 10.1016/j.stem.2012.11.003
36. Au-Yeung N, Horvath CM. Histone H2A.Z Suppression of Interferon-Stimulated Transcription and Antiviral Immunity Is Modulated by GCN5 and BRD2. iScience (2018) 6:68–82. doi: 10.1016/j.isci.2018.07.013
37. Encinas-García T, Loreto-Quiroz DL, Mendoza-Cano F, Peña-Rodriguez A, Fimbres-Olivarria D, Re-Vega E, et al. White Spot Syndrome Virus Down-Regulates Expression of Histones H2A and H4 of Penaeus Vannamei to Promote Viral Replication. Dis Aquat Organ (2019) 137:73–9. doi: 10.3354/dao03428
38. Hoeksema M, Tripathi S, White M, Qi L, Taubenberger J, van Eijk M, et al. Arginine-Rich Histones Have Strong Antiviral Activity for Influenza A Viruses. Innate Immun (2015) 21:736–45. doi: 10.1177/1753425915593794
39. Chang MX, Wu XM, Cao L. Nucleic Acid Molecules, Expression Vectors and Applications, Antibacterial Drugs and Methods. Natl Invention Patent ZL (2019) 1 0460046:4.
40. Zhao W. Negative Regulation of TBK1-Mediated Antiviral Immunity. FEBS Lett (2013) 587:542–8. doi: 10.1016/j.febslet.2013.01.052
Keywords: histone H2A, nuclear/cytoplasmic trafficking, negative regulation, RLR signaling, SVCV infection
Citation: Wu XM, Fang H, Zhang J, Bi YH and Chang MX (2021) Histone H2A Nuclear/Cytoplasmic Trafficking Is Essential for Negative Regulation of Antiviral Immune Response and Lysosomal Degradation of TBK1 and IRF3. Front. Immunol. 12:771277. doi: 10.3389/fimmu.2021.771277
Received: 06 September 2021; Accepted: 29 October 2021;
Published: 18 November 2021.
Edited by:
Maria Forlenza, Wageningen University and Research, NetherlandsReviewed by:
Krzysztof Rakus, Jagiellonian University, PolandCopyright © 2021 Wu, Fang, Zhang, Bi and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming Xian Chang, bWluZ3hpYW5jaGFuZ0BpaGIuYWMuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.