 Ondřej Vymazal1,2†
Ondřej Vymazal1,2† Kamila Bendíčková1†
Kamila Bendíčková1† Marco De Zuani1
Marco De Zuani1 Marcela Vlková3,4
Marcela Vlková3,4 Marcela Hortová-Kohoutková1
Marcela Hortová-Kohoutková1 Jan Frič1,5*
Jan Frič1,5*- 1International Clinical Research Center, St. Anne’s University Hospital, Brno, Czechia
- 2Department of Biology, Faculty of Medicine, Masaryk University, Brno, Czechia
- 3Department of Clinical Immunology and Allergology, Faculty of Medicine, Masaryk University, Brno, Czechia
- 4Department of Clinical Immunology and Allergology, St. Anne´s University Hospital, Brno, Czechia
- 5Department of Modern Immunotherapy, Institute of Hematology and Blood Transfusion, Prague, Czechia
Neutrophils are innate immune cells with important roles in antimicrobial defense. However, impaired or dysregulated neutrophil function can result in host tissue damage, loss of homeostasis, hyperinflammation or pathological immunosuppression. A central link between neutrophil activation and immune outcomes is emerging to be the calcineurin-nuclear factor of activated T cells (NFAT) signaling pathway, which is activated by neutrophil detection of a microbial threat via pattern recognition receptors and results in inflammatory cytokine production. This potent pro-inflammatory pathway is also the target of several immunosuppressive drugs used for the treatment of autoimmune disorders, during solid organ and hematopoietic cell transplantations, and as a part of anti-cancer therapy: but what effects these drugs have on neutrophil function, and their broader consequences for immune homeostasis and microbial defense are not yet known. Here, we bring together the emerging literature describing pathology- and drug- induced neutrophil impairment, with particular focus on their effects on calcineurin-NFAT signaling in the innate immune compartment.
Introduction
Neutrophils are the most abundant cells of the innate immune system and play a key role in antimicrobial and antifungal defenses (1). At the site of infection, pathogens are detected via pattern recognition receptors (PRRs) whose ligation leads to the activation of a complex network of signaling cascades that together orchestrate the neutrophil’s ability to kill microbes via the generation of reactive oxygen species (ROS), degranulation of effector molecules, and the release of neutrophil extracellular traps (NETs) (2, 3). Moreover, these signaling pathways stimulate the production and release of chemokines and cytokines by neutrophils that act both locally, recruiting other immune cells to the site, and systemically, to regulate the wider anti-microbial immune response (4). An emerging player is the calcineurin (CN)- nuclear factor of activated T-cell (NFAT) pathway, which is activated by Ca2+ influx, leading to NFAT translocation to nucleus and modulation of gene transcription (5), and has critical roles both in myeloid cell function and immune homeostasis (6).
Neutrophils occupy a unique and critical functional niche within effective immunity, therefore it is perhaps unsurprising that impaired neutrophil function or regulation can have profound consequences: when neutrophils are unable to respond effectively this leads to increased susceptibility to infection, and negatively impacts the inflammatory and healing process (7–11), while their exaggerated activation can result in tissue damage (10, 12, 13). Neutrophil function may be impaired by another pathology, as in the case of sepsis (14), but the picture becomes even more complicated when the cause of neutrophil dysfunction is medication being used to treat an existing illness. Recent studies have shown that several types of therapy including corticosteroids (15), cytotoxic drugs (16), and cancer chemotherapy (17, 18) can affect neutrophil functions and that this process is linked with unfavorable treatment outcomes. The pro-inflammatory pathways operating in neutrophils may also be directly affected: calcineurin inhibitors (CNI) suppress activation of the CN-NFAT pathway and are now widely used in transplantation medicine to prevent graft versus host disease and also to treat autoimmune disorders as psoriasis, severe atopic dermatitis, and rheumatoid arthritis (19, 20). While these strategies may be effective in treating the primary condition, the downstream consequences of neutrophil impairment are only just starting to be understood. What appears to unify these diverse conditions is the emergence of neutrophil subsets with profoundly immunosuppressive properties, which are only just beginning to be understood. Here, we will review the latest findings on neutrophils and their functions during CNI-based immunosuppressive therapy and in the immunosuppressive milieu of sepsis, and ask what we can learn from these two situations that might advance our knowledge of neutrophil function/regulation and enable better clinical management of affected patients.
Neutrophil Diversity in Health and Disease
For a long time, neutrophils were considered as a homogeneous population of innate myeloid cells, however, evidence has now accumulated that the population in fact exhibits considerable phenotypic and functional heterogeneity (21, 22), that is further expanded during certain pathologies or under specific conditions such as immunosuppression.
Neutrophil Subsets: A Primer
Neutrophils produced in the bone marrow are dynamically released into circulation and within a few hours migrate to tissue (23). Despite the short life in circulation, neutrophils undergo morphological and phenotypical changes referred to as aging (24, 25). In this context, so-called fresh neutrophils are released from bone marrow, and they leave blood circulation as aged neutrophils (24). Dynamic of neutrophil release and clearance changes during day and levels of fresh and aged neutrophils follows natural circadian patterns (24). Several specific functional differences of the aged neutrophils have been described to date, including lower levels of L-selectin (CD62L), increased expression of β2-integrins, enhanced ROS production, and a higher tendency to undergo NETosis (25, 26). Moreover, aging primes neutrophils for a more aggressive inflammatory response in vivo (25, 26).
Normal density neutrophils (NDNs) traditionally separate in high density fraction after density gradient centrifugation. However, low density neutrophils (LDNs) are a suggested neutrophil subpopulation with a similar density to peripheral blood mononuclear cells (PBMCs) associated with a number of pathological conditions including systemic lupus erythematosus (27), sepsis (28), tuberculosis (29, 30), HIV (31), severe fever with thrombocytopenia syndrome (32), asthma (33), cancer (34–36), and it is associated with postoperative surgical stress in patients with gastrointestinal malignancies (37). Nevertheless, the impact of LDN in these pathologies has not been fully characterized. Recently, LDNs of healthy individuals show identical properties compared to NDNs (38). LDNs have a comparable ability of oxidative flare-ups, apoptosis, a similar effect on T-cell proliferation or IFN-γ production. Compared to NDNs, only a reduced ability of NETosis was found in LDNs in healthy donors (38). Interestingly, neutrophil stimulating agents (e.g. TNF, LPS, or fMLF) induced generation of LDNs from NDNs in vitro (38) suggesting that neutrophil density alone is insufficient to distinguish individual neutrophil subsets and functional or phenotypic differences need to be discovered.
While neutrophils have been considered as simply pathogen-killing cells for a long time, it is only more recently that their complex role in health and disease, in particular, in immunosuppression, have begun to be uncovered.
Neutrophil Subsets Associated With Immunosuppression
The blood of healthy individuals contains both LDN and NDN (39). Recently, a population of “suppressive NDN” that is composed of mature activated cells have been defined (40). Further, in the condition of systemic inflammation, a subset of NDNs characterized by CD62low CD11bhi CD11chi that suppress T-cell proliferation through direct contact with the integrin Mac-1 was identified (41). Concurrently, recent studies revealed significant increases in the relative proportion of LDNs in patients with sepsis (28), infectious and autoimmune diseases (42), and cancer (34). Interestingly, a higher frequency of LDNs has also been found in immune-compromised patients, such as those infected with HIV or affected by common variable immunodeficiency (43). LDNs from these patients may also show profound functional alterations, for example in systemic lupus erythematosus where LDN exhibit enhanced spontaneous NETosis and relatively higher mitochondrial ROS production when compared to NDN ex vivo (44). In patients with cancer, circulating LDNs display marked immunosuppressive and pro-tumorigenic activities, similar to those documented for tumor-associated neutrophils (45–47).
These findings caused researchers to further dissect the LDN population, leading to the discovery of multiple neutrophil subsets that differ in morphology, abundance, and maturation and activation status depending on the underlying disease process (39, 48). However, the expanded LDN population in thrombocytopenia syndrome exhibiting severe fever (32), as well as in patients with malaria (49), or asthma (33) wasn´t found to be suppressive (48), and a recent study in patients with severe COVID-19 uncovered a population of CD16int LDNs with a markedly inflammatory gene expression signature whose frequency correlated with disease severity (50). Thus, it appears that the heterogeneity within the population of LDN that is expanded in these disease states can accommodate the emergence of dominant immunosuppressive, non-suppressive and pro-inflammatory features. The question arises: are there any specific cell types within the LDN population that are strongly and specifically associated with immunosuppression?
Recent studies looking at immunosuppressive neutrophils have uncovered a potential candidate LDN subpopulation but have yet to agree on a unified terminology: these cells are variously termed “granulocytic-” or “polymorphonuclear- myeloid-derived suppressor cells” (G-MDSCs or PMN-MDSCs) (47, 48, 51). Although studies of these cells are in their infancy, we know that PMN-MDSCs often contain immature neutrophils (48, 52, 53) and are observed in patients with sepsis or fungal infections caused by Aspergillus fumigatus (A. fumigatus) or Candida albicans (C. albicans) (54, 55). Ex vivo, PMN-MDSCs from these patients inhibit T-cell proliferation and IFN-γ production via ROS or arginase-1 production (48, 54, 55). Identifying PMN-MDSCs among LDNs has been challenging as there is a lack of a defined, consensus surface molecule expression panel that can distinguish between the two (38, 47). However, it is possible to confirm MDSCs’ identity functionally, through their ability to suppress immune responses (e.g. inhibition of T cell proliferation or IFN-γ production), or by their expression of biochemical and molecular markers (such as the genes encoding ARG1, NOS2, NOX2, and TGF-β) (56). It is hoped that future studies, perhaps utilizing scRNA-seq, will be able to define a unique phenotype allowing reliable identification and isolation of these cells for further study: one of the most pressing questions is which molecular pathways are involved in normal neutrophil functions, and how are they altered in immunosuppressive neutrophil subsets.
NFAT Signaling in Myeloid Immune Cells: Is This the Key Pathway Mediating Neutrophil Functions?
The CN-NFAT pathway, prototypically recognized in T cells (57, 58), has in recent years been identified as an important signaling cascade linking PRR activation, anti-microbial immunity, and myeloid cell functions (6, 59–61). Early work demonstrated NFAT activation in murine monocytes and DCs following the engagement of dectin-1 by zymosan or live C. albicans (62), with subsequent studies showing that this pathway can be stimulated by multiple pathogen-associated molecules, with varied outcomes. For example, bacterial LPS stimulation of murine DCs induced CD14-dependent extracellular Ca2+ influx, leading to CN-NFAT activation (63). Recently, these findings were enriched by a study that showed the importance of inositol triphosphate (IP3) receptor 3 (IP3R3) and IP3 kinase B (ITPKB) in LPS-induced NFAT activation in mouse and human DCs (64), signifying an atypical mechanism of Ca2+ mobilization leading to CN-NFAT activation in these cells.
CN-NFAT activation also occurs in human monocytes exposed to the ligand from Saccharomyces cerevisiae (S. cerevisiae) cell wall - zymosan (65), and similarly in murine macrophages and DCs after dectin-1 ligation by zymosan or live C. albicans (62). However, in macrophages exposed to A. fumigatus distinct pathway of TLR9- and phagocytosis-dependent NFAT activation mediated by Bruton’s tyrosine kinase (BTK), but independent on MyD88 has been reported (66).
Soon after the discovery that LPS stimulation induced CN-NFAT activation in murine DCs, similar findings were made in neutrophils, including induced expression of the genes involved in inflammation modulation including IL-10, Cox2, Erg1, and Erg2 (67, 68). Furthermore, the engagement of dectin-1 by either zymosan or live C. albicans leading to CN-NFAT activation has also been confirmed in murine and human neutrophils (Table 1) (67, 68).
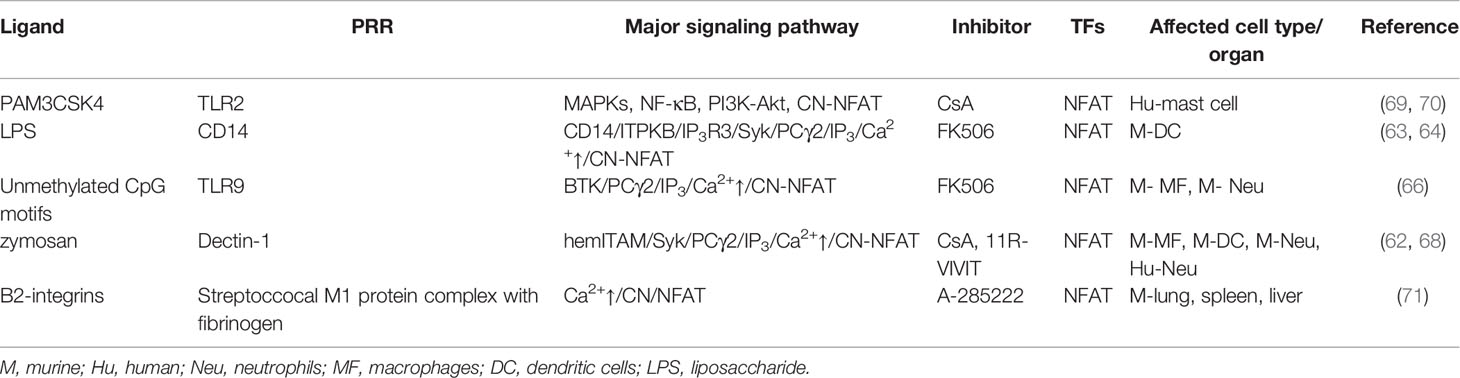
Table 1 PRR expressed by neutrophils and their signaling affected by calcineurin inhibitors demonstrated in neutrophils or derived from myeloid cells.
Taken together, these data indicate likely parallels between the pathways and effects of CN-NFAT activation in monocytes and neutrophils, further solidifying NFAT as a central player in myeloid cell immunity. However, while little direct research has been done into the role of CN-NFAT in driving neutrophil functions or in different neutrophil subsets, studies with patients undergoing immunosuppressive therapy with CNI, or with disease-associated immunosuppression, have generated intriguing insights into this topic.
Impact of Immunosuppression on Neutrophil Function And its Clinical Consequences
Immunosuppression is a concerning state for the host, leaving it open to opportunistic infection with an unresponsive defense system. This situation can result directly from pathogen infection, but may also be therapeutically induced in the case of frank autoimmunity or the need for dampened reactivity following organ transplant. Here we will explore neutrophils’ involvement in two distinct but partially-overlapping scenarios: during CNI immunosuppressive therapy and in post-sepsis immunosuppression.
Impact of Calcineurin Inhibitors on Neutrophils
Drugs inhibiting CN-NFAT signaling, such as tacrolimus (FK506) and cyclosporine A (CsA), are widely used to suppress T‐cell responses and prevent allograft rejection in transplantation medicine (20, 72). These clinically successful CNI were also recently repurposed to treat autoimmune disorders [reviewed in (59)] and as cancer chemotherapy considering that several cancer types have constitutively activated and overexpressed NFAT (73, 74). However, counterbalancing their potentially beneficial therapeutic role, a growing body of evidence indicates the potential impact of these compounds on myeloid cell function, leading to increased susceptibility to infections (59, 65, 66).
Different experimental models and clinical reports suggest that increased susceptibility to fungal and bacterial infections in patients treated with CNI is not a generic effect of inhibition of adaptive immune responses, but rather due to direct impairment of NFAT signaling in myeloid leukocytes, including neutrophils (Table 2) (6, 15, 59, 66, 68, 76, 78, 79). In vitro, treatment with CsA significantly inhibited the expression of NOD1, an intracellular receptor for bacterial peptidoglycan, in murine neutrophils, macrophages, and DCs (76). Similarly, in vivo, CsA-treatment of mice decreased the renal resistance to uropathogenic Escherichia coli infection associated with decreased expression of neutrophil-attracting chemokines CXCL2 and CXCL1, and myeloperoxidase (MPO) in mouse kidneys (76). Similarly, administration of CNI affected the production of CCL2, CCL7, CCL12, and delayed pathogen clearance in mice infected by S. cerevisiae (80). CsA treatment also negatively altered the phagocytic ability of neutrophils in the blood of human transplant recipients, which can be explained by the downregulation of Nod1 mRNA both in vitro and in vivo (76).
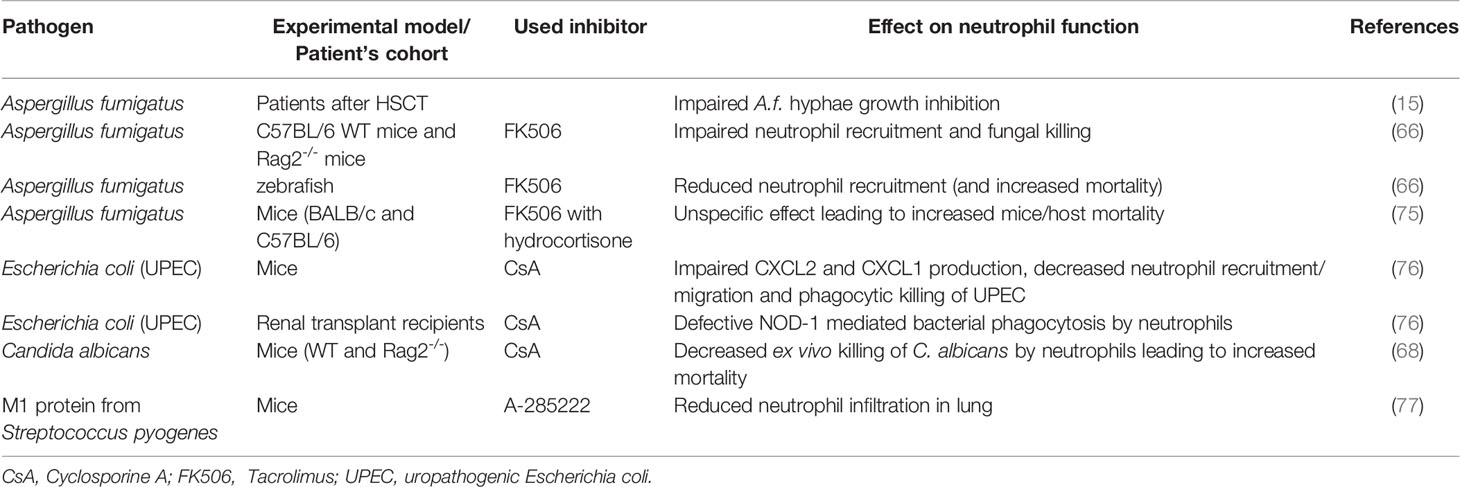
Table 2 Impact of impaired calcineurin-NFAT signaling on neutrophil functions during infection.
These molecular effects of CNI upon neutrophils can have profound consequences, most clearly demonstrated in the case of infection with the normally-harmless opportunistic pathogen, A. fumigatus. Neutrophils are the main innate immune cell type interreacting with germinating A. fumigatus conidia and hyphae to prevent their growth (81, 82); however, the ability of neutrophils to interfere with A. fumigatus is significantly impaired in CNI-treated patients after hematopoietic stem cell transplant (15) – an effect that has been mechanistically linked with increased mortality in CNI-treated A. fumigatus-infected mice (83). The impact of impaired CN-NFAT signaling on patients’ susceptibility to A. fumigatus is even more marked during GvHD or solid organ transplantation (75), partially explaining the severe and frequently fatal complication of A. fumigatus infection in patients suffering from these conditions.
Such observations led to several studies in vitro and in animal models aiming to understand how CNI affected neutrophil responses to fungal infection. FK506 treatment of mice with pulmonary aspergillosis led to reduced cytokine/chemokine responses (TNF-α; IL-6; CXCL-1 and CCL-3), delayed neutrophil recruitment, decreased neutrophil influx, and reduced fungal killing in the lung (66). The lowered ability of CNI-exposed neutrophils to kill fungal pathogens seems also to be mechanistically linked to the lack of CN-NFAT activation: neutrophil degranulation is a Ca2+-dependent process (84), which is impaired in the presence of a range of different CNI (78, 85–87). Similarly, NETosis requires the mobilization of both extracellular and intracellular calcium pools and is reduced by treatment with CsA and the macrolide immunosuppressant ascomycin (88). Recent data have also indicated a link between CNI treatment and the development and accumulation of PMN-MDSCs (89), raising the possibility that CN-NFAT signaling is also required for normal neutrophil subpopulation homeostasis.
These findings demonstrate a novel, incompletely characterized, connection between CN-NFAT signaling in neutrophils and increased susceptibility to infections, operating both at the population (PMN-MDSC emergence) and individual cell level (inhibition of cytokine production, degranulation, and NETs generation). Moreover, such data also clearly illustrate the crucial role of CN-NFAT signaling in maintaining these processes in the disease-free host.
Given the profound effects of common immunosuppressive drugs targeting CN (CsA, FK506) on neutrophils, the potential benefits of their use need to be carefully considered in the context of the risk of increased susceptibility to serious opportunistic infections.
Neutrophil Role in Sepsis-Induced Immunosuppression
Sepsis is a life-threatening condition caused by the dysregulated host response to infection and subsequent organ dysfunction (90). The host immune response during sepsis is complex, involving simultaneous activation of both excessive pro-inflammatory and anti-inflammatory processes (14), which ultimately results in disturbed homeostasis and profound post-sepsis immunosuppression: the predominant driving force for late morbidity and mortality in patients with sepsis (91, 92). Neutrophils are strongly affected by sepsis and are thought to play a critical role in determining the clinical outcome (93). Multiple neutrophil effector functions become impaired during the course of the disease (14, 94, 95), leading to functional paralysis of these cells in severe sepsis. One contributing factor is thought to be TLR-induced upregulation of G protein-coupled receptor kinase 2 (GRK2) expression, which ultimately desensitizes neutrophils to CXCL2 coming from the site of infection, thereby inhibiting several functions as well as their migration (93). Alongside, sepsis is associated with impaired neutrophil phagocytosis: in part, this can be linked with the release of immature neutrophils with limited phagocytic capacity from the bone marrow into the blood during sepsis and septic shock (96); but is also associated with decreased expression of the NOD1 receptor in patients with sepsis (97). These pathways are critically important, as impaired neutrophil phagocytic activity and reduced cell-surface CD64 (Fc gamma receptor) expression have been strongly correlated with poor outcomes in patients with sepsis (98). While there is some evidence from the CNI setting that NOD1-receptor-mediated neutrophil phagocytosis is CN-NFAT-dependent (76), this has yet to be assessed in cells from patients with sepsis.
In many of these studies, neutrophils are considered as a bulk population, but more recent work has begun to look at the possible involvement of the PMN-MDSCs in sepsis. Thought to arise as a result of the persistent immune activation and inflammatory environment, PMN-MDSCs have been detected in patients affected by sepsis (54, 99, 100) where they exhibit strong immunosuppressive activity (54, 101–103). A recent study showed that the frequency of PMN-MDSCs remain elevated also in sepsis survivors long after their recovery from sepsis, suggesting that these cells might contribute to the long-term adverse effects observed in sepsis survivors (100). These findings suggest a possible role of PMN-MDSCs in sepsis-induced immunosuppression and thus future research should be directed to elucidate their function and the underlying mechanisms involved.
Future Perspectives
It is now clear that neutrophils play a decisive role in different pathologies and during immunosuppressive therapies. As effector cells of innate immunity, they are crucial for pathogen clearance, but they are also involved in host homeostasis: on the flip side, neutrophil dysregulation is implicated in the development/maintenance of the immunosuppressive microenvironment found in cancer and after sepsis, but also in the inflammatory response underlying autoimmunity. This dual role of neutrophils in opposing pathological processes reflects their plasticity and heterogeneity, as more recently evidenced by the identification of functionally and phenotypically distinct neutrophil subsets.
The specific activation or immunosuppressive status of neutrophils is tightly regulated through neutrophil signaling pathways: nevertheless, the details of these signaling pathways and specifically how they might be explored as potential targets of therapeutic interventions are not sufficiently described. One such knowledge gap that is beginning to be filled concerns the calcineurin/NFAT pathway which, although its presence and activity is sufficiently demonstrated in neutrophils, has yet to reveal the consequences of its signaling in these cells under different conditions. This is likely to prove key moving forward because a detailed understanding of the immunosuppressive context of downstream PRR signaling in neutrophils is needed in order to define the mechanistic aspects which may have potential medical implications. Some progress is already being made in this regard with the development of next-generation CNI, such as the VIVIT peptide, which selectively inhibits the calcineurin/NFAT interaction, but does not compromise calcineurin’s phosphatase activity (104). A recent study showed the potential of this peptide, conjugated to nanoparticles designed to target phagocytic cells, to dampen inflammation in a murine model of arthritis (64). Not only do such approaches hold direct therapeutic potential, but they may also prove invaluable as tools with which to discriminate the roles of CN-NFAT activation in specific cell types, including neutrophils.
Overall, there is emerging evidence pointing to the importance of the CN-NFAT axis in the myeloid cell/neutrophil response to pathogens and also the effects of its impairment on myeloid cell/neutrophil function during different disorders. Although CNI-mediated immunosuppression is applied in multiple situations, the broader adverse effect on the immune system needs to be carefully evaluated: the development of more specific therapeutic strategies targeting individual cell types or over-activated pathways should be considered a priority. Our knowledge of the biology of NFAT signaling in neutrophils is in its infancy and highlights the need for expanding our understanding of the molecular mechanisms modulating neutrophil functions in health and disease.
Author Contributions
OV prepared the table, and wrote the manuscript. KB conceptualized, wrote and critically reviewed the manuscript. MZ, MH-K, and MV wrote and critically reviewed the manuscript. JF conceptualized, wrote and critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the European Social Fund and European Regional Development Fund—Proj- ect MAGNET (grant no. CZ.02.1.01/0.0/0.0/15_003/0000492) and ENOCH (CZ.02.1.01/0.0/0.0/16_019/0000868); and the Ministry of Health of the Czech Republic (grant nr. NU21-06-00408, all rights reserved) and DRO (Institute of Hematology and Blood Transfusion – UHKT, 00023736).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank Dr. Lucy Robinson from Insight Editing London for editing and critical review of the manuscript.
References
1. Zhang X, Kluger Y, Nakayama Y, Poddar R, Whitney C, DeTora A, et al. Gene Expression in Mature Neutrophils: Early Responses to Inflammatory Stimuli. J Leukoc Biol (2004) 75:358–72. doi: 10.1189/jlb.0903412
2. Mayadas TN, Cullere X, Lowell CA. The Multifaceted Functions of Neutrophils. Annu Rev Pathol (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
3. Burn GL, Foti A, Marsman G, Patel DF, Zychlinsky A. The Neutrophil. Immunity (2021) 54:1377–91. doi: 10.1016/j.immuni.2021.06.006
4. Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA. The Neutrophil as a Cellular Source of Chemokines. Immunol Rev (2000) 177:195–203. doi: 10.1034/j.1600-065x.2000.17706.x
5. Im S-H, Rao A. Activation and Deactivation of Gene Expression by Ca2+/calcineurin-NFAT-Mediated Signaling. Mol Cells (2004) 18:1–9.
6. Fric J, Zelante T, Wong AYW, Mertes A, Yu H-B, Ricciardi-Castagnoli P. NFAT Control of Innate Immunity. Blood (2012) 120:1380–9. doi: 10.1182/blood-2012-02-404475
7. Nathan C. Neutrophils and Immunity: Challenges and Opportunities. Nat Rev Immunol (2006) 6:173–82. doi: 10.1038/nri1785
8. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil Function: From Mechanisms to Disease. Annu Rev Immunol (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
9. Kovtun A, Messerer DAC, Scharffetter-Kochanek K, Huber-Lang M, Ignatius A. Neutrophils in Tissue Trauma of the Skin, Bone, and Lung: Two Sides of the Same Coin. J Immunol Res (2018) 2018. doi: 10.1155/2018/8173983
10. Wang J. Neutrophils in Tissue Injury and Repair. Cell Tissue Res (2018) 371:531–9. doi: 10.1007/s00441-017-2785-7
11. Peiseler M, Kubes P. More Friend Than Foe: The Emerging Role of Neutrophils in Tissue Repair. J Clin Invest (2019) 129:2629–39. doi: 10.1172/JCI124616
12. Grommes J, Soehnlein O. Contribution of Neutrophils to Acute Lung Injury. Mol Med (2011) 17:293–307. doi: 10.2119/molmed.2010.00138
13. Soehnlein O, Steffens S, Hidalgo A, Weber C. Neutrophils as Protagonists and Targets in Chronic Inflammation. Nat Rev Immunol (2017) 17:248–61. doi: 10.1038/nri.2017.10
14. Venet F, Monneret G. Advances in the Understanding and Treatment of Sepsis-Induced Immunosuppression. Nat Rev Nephrol (2018) 14:121–37. doi: 10.1038/nrneph.2017.165
15. Imbert S, Bresler P, Boissonnas A, Gauthier L, Souchet L, Uzunov M, et al. Calcineurin Inhibitors Impair Neutrophil Activity Against Aspergillus Fumigatus in Allogeneic Hematopoietic Stem Cell Transplant Recipients. J Allergy Clin Immunol (2016) 138:860–8. doi: 10.1016/j.jaci.2016.02.026
16. Allen E, Bakke A, Purtzer M, Deodhar A. Neutrophil CD64 Expression: Distinguishing Acute Inflammatory Autoimmune Disease From Systemic Infections. Ann Rheum Dis (2002) 61:522–5. doi: 10.1136/ard.61.6.522
17. Németh T, Sperandio M, Mócsai A. Neutrophils as Emerging Therapeutic Targets. Nat Rev Drug Discov (2020) 19:253–75. doi: 10.1038/s41573-019-0054-z
18. Hartmann P, Herholz K, Salzberger B, Petereit HF. Unusual and Severe Symptomatic Impairment of Neutrophil Function After One Cycle of Temozolomide in Patients With Malignant Glioma. Ann Hematol (2004) 83:212–7. doi: 10.1007/s00277-003-0802-2
19. Erdmann F, Weiwad M. Calcineurin Inhibitors: Status Quo and Perspectives. Biomol Concepts (2011) 2:65–78. doi: 10.1515/bmc.2011.011
20. Azzi JR, Sayegh MH, Mallat SG. Calcineurin Inhibitors: 40 Years Later, Can’t Live Without. J Immunol (2013) 191:5785–91. doi: 10.4049/jimmunol.1390055
21. Deniset JF, Kubes P. Neutrophil Heterogeneity: Bona Fide Subsets or Polarization States? J Leukoc Biol (2018) 103:829–38. doi: 10.1002/JLB.3RI0917-361R
22. Silvestre-Roig C, Fridlender ZG, Glogauer M, Scapini P. Neutrophil Diversity in Health and Disease. Trends Immunol (2019) 40:565–83. doi: 10.1016/j.it.2019.04.012
23. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil Kinetics in Health and Disease. Trends Immunol (2010) 31:318–24. doi: 10.1016/j.it.2010.05.006
24. Casanova-Acebes M, Pitaval C, Weiss LA, Nombela-Arrieta C, Chèvre R, A-González N, et al. Rhythmic Modulation of the Hematopoietic Niche Through Neutrophil Clearance. Cell (2013) 153:1025–35. doi: 10.1016/j.cell.2013.04.040
25. Adrover JM, Nicolás-Ávila JA, Hidalgo A. Aging: A Temporal Dimension for Neutrophils. Trends Immunol (2016) 37:334–45. doi: 10.1016/j.it.2016.03.005
26. Zhang D, Chen G, Manwani D, Mortha A, Xu C, Faith JJ, et al. Neutrophil Ageing Is Regulated by the Microbiome. Nature (2015) 525:528–32. doi: 10.1038/nature15367
27. Tay SH, Celhar T, Fairhurst A-M. Low-Density Neutrophils in Systemic Lupus Erythematosus. Arthritis Rheumatol (2020) 72:1587–95. doi: 10.1002/art.41395
28. Morisaki T, Goya T, Ishimitsu T, Torisu M. The Increase of Low Density Subpopulations and CD10 (CALLA) Negative Neutrophils in Severely Infected Patients. Surg Today (1992) 22:322–7. doi: 10.1007/BF00308740
29. La Manna MP, Orlando V, Paraboschi EM, Tamburini B, Di Carlo P, Cascio A, et al. Mycobacterium Tuberculosis Drives Expansion of Low-Density Neutrophils Equipped With Regulatory Activities. Front Immunol (2019) 10:2761. doi: 10.3389/fimmu.2019.02761
30. Deng Y, Ye J, Luo Q, Huang Z, Peng Y, Xiong G, et al. Low-Density Granulocytes Are Elevated in Mycobacterial Infection and Associated With the Severity of Tuberculosis. PloS One (2016) 11:e0153567. doi: 10.1371/journal.pone.0153567
31. Cloke T, Munder M, Taylor G, Müller I, Kropf P. Characterization of a Novel Population of Low-Density Granulocytes Associated With Disease Severity in HIV-1 Infection. PloS One (2012) 7:e48939. doi: 10.1371/journal.pone.0048939
32. Li Y, Li H, Wang H, Pan H, Zhao H, Jin H, et al. The Proportion, Origin and Pro-Inflammation Roles of Low Density Neutrophils in SFTS Disease. BMC Infect Dis (2019) 19:109. doi: 10.1186/s12879-019-3701-4
33. Fu J, Tobin MC, Thomas LL. Neutrophil-Like Low-Density Granulocytes Are Elevated in Patients With Moderate to Severe Persistent Asthma. Ann Allergy Asthma Immunol (2014) 113:635–40.e2. doi: 10.1016/j.anai.2014.08.024
34. Sagiv JY, Michaeli J, Assi S, Mishalian I, Kisos H, Levy L, et al. Phenotypic Diversity and Plasticity in Circulating Neutrophil Subpopulations in Cancer. Cell Rep (2015) 10:562–73. doi: 10.1016/j.celrep.2014.12.039
35. Mishalian I, Granot Z, Fridlender ZG. The Diversity of Circulating Neutrophils in Cancer. Immunobiology (2017) 222:82–8. doi: 10.1016/j.imbio.2016.02.001
36. Brandau S, Trellakis S, Bruderek K, Schmaltz D, Steller G, Elian M, et al. Myeloid-Derived Suppressor Cells in the Peripheral Blood of Cancer Patients Contain a Subset of Immature Neutrophils With Impaired Migratory Properties. J Leukoc Biol (2011) 89:311–7. doi: 10.1189/jlb.0310162
37. Kumagai Y, Ohzawa H, Miyato H, Horie H, Hosoya Y, Lefor AK, et al. Surgical Stress Increases Circulating Low-Density Neutrophils Which May Promote Tumor Recurrence. J Surg Res (2020) 246:52–61. doi: 10.1016/j.jss.2019.08.022
38. Hardisty GR, Llanwarne F, Minns D, Gillan JL, Davidson DJ, Gwyer Findlay E, et al. High Purity Isolation of Low Density Neutrophils Casts Doubt on Their Exceptionality in Health and Disease. Front Immunol (2021) 12:625922. doi: 10.3389/fimmu.2021.625922
39. Blanco-Camarillo C, Alemán OR, Rosales C. Low-Density Neutrophils in Healthy Individuals Display a Mature Primed Phenotype. Front Immunol (2021) 12:672520. doi: 10.3389/fimmu.2021.672520
40. Aarts CEM, Hiemstra IH, Béguin EP, Hoogendijk AJ, Bouchmal S, van Houdt M, et al. Activated Neutrophils Exert Myeloid-Derived Suppressor Cell Activity Damaging T Cells Beyond Repair. Blood Adv (2019) 3:3562–74. doi: 10.1182/bloodadvances.2019031609
41. Pillay J, Kamp VM, van Hoffen E, Visser T, Tak T, Lammers J-W, et al. A Subset of Neutrophils in Human Systemic Inflammation Inhibits T Cell Responses Through Mac-1. J Clin Invest (2012) 122:327–36. doi: 10.1172/JCI57990
42. Carmona-Rivera C, Kaplan MJ. Low Density Granulocytes: A Distinct Class of Neutrophils in Systemic Autoimmunity. Semin Immunopathol (2013) 35:455–63. doi: 10.1007/s00281-013-0375-7
43. Vlkova M, Chovancova Z, Nechvatalova J, Connelly AN, Davis MD, Slanina P, et al. Neutrophil and Granulocytic Myeloid-Derived Suppressor Cell-Mediated T Cell Suppression Significantly Contributes to Immune Dysregulation in Common Variable Immunodeficiency Disorders. J Immunol (2019) 202:93–104. doi: 10.4049/jimmunol.1800102
44. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA Are Interferogenic and Contribute to Lupus-Like Disease. Nat Med (2016) 22:146–53. doi: 10.1038/nm.4027
45. Sagiv JY, Voels S, Granot Z. Isolation and Characterization of Low- vs. High-Density Neutrophils in Cancer. Methods Mol Biol (2016) 1458:179–93. doi: 10.1007/978-1-4939-3801-8_13
46. Grecian R, Whyte MKB, Walmsley SR. The Role of Neutrophils in Cancer. Br Med Bull (2018) 128:5–14. doi: 10.1093/bmb/ldy029
47. McKenna E, Mhaonaigh AU, Wubben R, Dwivedi A, Hurley T, Kelly LA, et al. Neutrophils: Need for Standardized Nomenclature. Front Immunol (2021) 12:602963. doi: 10.3389/fimmu.2021.602963
48. Scapini P, Marini O, Tecchio C, Cassatella MA. Human Neutrophils in the Saga of Cellular Heterogeneity: Insights and Open Questions. Immunol Rev (2016) 273:48–60. doi: 10.1111/imr.12448
49. Rocha BC, Marques PE, Leoratti FM de S, Junqueira C, Pereira DB, Antonelli LR do V, et al. Type I Interferon Transcriptional Signature in Neutrophils and Low-Density Granulocytes Are Associated With Tissue Damage in Malaria. Cell Rep (2015) 13:2829–41. doi: 10.1016/j.celrep.2015.11.055
50. Morrissey SM, Geller AE, Hu X, Tieri D, Ding C, Klaes CK, et al. A Specific Low-Density Neutrophil Population Correlates With Hypercoagulation and Disease Severity in Hospitalized COVID-19 Patients. JCI Insight (2021) 6:148435. doi: 10.1172/jci.insight.148435
51. Cassetta L, Baekkevold ES, Brandau S, Bujko A, Cassatella MA, Dorhoi A, et al. Deciphering Myeloid-Derived Suppressor Cells: Isolation and Markers in Humans, Mice and Non-Human Primates. Cancer Immunol Immunother (2019) 68:687–97. doi: 10.1007/s00262-019-02302-2
52. Veglia F, Perego M, Gabrilovich D. Myeloid-Derived Suppressor Cells Coming of Age. Nat Immunol (2018) 19:108–19. doi: 10.1038/s41590-017-0022-x
53. Moses K, Brandau S. Human Neutrophils: Their Role in Cancer and Relation to Myeloid-Derived Suppressor Cells. Semin Immunol (2016) 28:187–96. doi: 10.1016/j.smim.2016.03.018
54. Uhel F, Azzaoui I, Grégoire M, Pangault C, Dulong J, Tadié J-M, et al. Early Expansion of Circulating Granulocytic Myeloid-Derived Suppressor Cells Predicts Development of Nosocomial Infections in Patients With Sepsis. Am J Respir Crit Care Med (2017) 196:315–27. doi: 10.1164/rccm.201606-1143OC
55. Rieber N, Singh A, Öz H, Carevic M, Bouzani M, Amich J, et al. Pathogenic Fungi Regulate Immunity by Inducing Neutrophilic Myeloid-Derived Suppressor Cells. Cell Host Microbe (2015) 17:507–14. doi: 10.1016/j.chom.2015.02.007
56. Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for Myeloid-Derived Suppressor Cell Nomenclature and Characterization Standards. Nat Commun (2016) 7:12150. doi: 10.1038/ncomms12150
57. Durand DB, Shaw JP, Bush MR, Replogle RE, Belagaje R, Crabtree GR. Characterization of Antigen Receptor Response Elements Within the Interleukin-2 Enhancer. Mol Cell Biol (1988) 8:1715–24. doi: 10.1128/MCB.8.4.1715
58. Shaw JP, Utz PJ, Durand DB, Toole JJ, Emmel EA, Crabtree GR. Identification of a Putative Regulator of Early T Cell Activation Genes. Science (1988) 241:202–5. doi: 10.1126/science.3260404
59. Bendickova K, Tidu F, Fric J. Calcineurin-NFAT Signalling in Myeloid Leucocytes: New Prospects and Pitfalls in Immunosuppressive Therapy. EMBO Mol Med (2017) 9:990–9. doi: 10.15252/emmm.201707698
60. Zanoni I, Granucci F. Regulation and Dysregulation of Innate Immunity by NFAT Signaling Downstream of Pattern Recognition Receptors (PRRs). Eur J Immunol (2012) 42:1924–31. doi: 10.1002/eji.201242580
61. Santus W, Barresi S, Mingozzi F, Broggi A, Orlandi I, Stamerra G, et al. Skin Infections Are Eliminated by Cooperation of the Fibrinolytic and Innate Immune Systems. Sci Immunol (2017) 2:eaan2725. doi: 10.1126/sciimmunol.aan2725
62. Goodridge HS, Simmons RM, Underhill DM. Dectin-1 Stimulation by Candida Albicans Yeast or Zymosan Triggers NFAT Activation in Macrophages and Dendritic Cells. J Immunol (2007) 178:3107–15. doi: 10.4049/jimmunol.178.5.3107
63. Zanoni I, Ostuni R, Capuano G, Collini M, Caccia M, Ronchi AE, et al. CD14 Regulates the Dendritic Cell Life Cycle After LPS Exposure Through NFAT Activation. Nature (2009) 460:264–8. doi: 10.1038/nature08118
64. Marongiu L, Mingozzi F, Cigni C, Marzi R, Di Gioia M, Garrè M, et al. Inositol 1,4,5-Trisphosphate 3-Kinase B Promotes Ca2+ Mobilization and the Inflammatory Activity of Dendritic Cells. Sci Signal (2021) 14:eaaz2120. doi: 10.1126/scisignal.aaz2120
65. Bendíčková K, Tidu F, De Zuani M, Kohoutková MH, Andrejčinová I, Pompeiano A, et al. Calcineurin Inhibitors Reduce NFAT-Dependent Expression of Antifungal Pentraxin-3 by Human Monocytes. J Leukoc Biol (2020) 107:497–508. doi: 10.1002/JLB.4VMA0318-138R
66. Herbst S, Shah A, Mazon Moya M, Marzola V, Jensen B, Reed A, et al. Phagocytosis-Dependent Activation of a TLR9-BTK-Calcineurin-NFAT Pathway Co-Ordinates Innate Immunity to Aspergillus Fumigatus. EMBO Mol Med (2015) 7:240–58. doi: 10.15252/emmm.201404556
67. Vega Rioja A, Chacon PJ, Monteseirín Mateo J, Bekay R, Alba Jiménez G, Martín Nieto J, et al. Expression of the Transcription Factor NFAT2 in Human Neutrophils: IgE-Dependent, Ca2+- and Calcineurin-Mediated NFAT2 Activation. J Cell Sci (2007) 120(Pt 14):2328–37. doi: 10.1242/jcs.000331
68. Greenblatt MB, Aliprantis A, Hu B, Glimcher LH. Calcineurin Regulates Innate Antifungal Immunity in Neutrophils. J Exp Med (2010) 207:923–31. doi: 10.1084/jem.20092531
69. Jin M, Yu B, Zhang W, Zhang W, Xiao Z, Mao Z, et al. Toll-Like Receptor 2-Mediated MAPKs and NF-κb Activation Requires the GNAO1-Dependent Pathway in Human Mast Cells. Integr Biol (Camb) (2016) 8:968–75. doi: 10.1039/c6ib00097e
70. Zaidi AK, Thangam ERRB, Ali H. Distinct Roles of Ca2+ Mobilization and G Protein Usage on Regulation of Toll-Like Receptor Function in Human and Murine Mast Cells. Immunology (2006) 119:412–20. doi: 10.1111/j.1365-2567.2006.02450.x
71. Zhang S, Wu M, Gao H. Editorial: NFAT Signaling: No FAT as New Weapon to Fight Shock. J Leukoc Biol (2015) 97:997–9. doi: 10.1189/jlb.4CE1214-616R
72. Flanagan WM, Corthésy B, Bram RJ, Crabtree GR. Nuclear Association of a T-Cell Transcription Factor Blocked by FK-506 and Cyclosporin A. Nature (1991) 352:803–7. doi: 10.1038/352803a0
73. Müller MR, Rao A. NFAT, Immunity and Cancer: A Transcription Factor Comes of Age. Nat Rev Immunol (2010) 10:645–56. doi: 10.1038/nri2818
74. Qin J-J, Nag S, Wang W, Zhou J, Zhang W-D, Wang H, et al. NFAT as Cancer Target: Mission Possible? Biochim Biophys Acta (2014) 1846:297–311. doi: 10.1016/j.bbcan.2014.07.009
75. Herbst S, Shah A, Carby M, Chusney G, Kikkeri N, Dorling A, et al. A New and Clinically Relevant Murine Model of Solid-Organ Transplant Aspergillosis. Dis Model Mech (2013) 6:643–51. doi: 10.1242/dmm.010330
76. Tourneur E, Ben Mkaddem S, Chassin C, Bens M, Goujon J-M, Charles N, et al. Cyclosporine A Impairs Nucleotide Binding Oligomerization Domain (Nod1)-Mediated Innate Antibacterial Renal Defenses in Mice and Human Transplant Recipients. PloS Pathog (2013) 9(1):e1003152. doi: 10.1371/journal.ppat.1003152
77. Zhang S, Zhang S, Garcia-Vaz E, Herwald H, Gomez MF, Thorlacius H. Streptococcal M1 Protein Triggers Chemokine Formation, Neutrophil Infiltration, and Lung Injury in an NFAT-Dependent Manner. J Leukoc Biol (2015) 97:1003–10. doi: 10.1189/jlb.3HI0214-123RR
78. Forrest MJ, Jewell ME, Koo GC, Sigal NH. FK-506 and Cyclosporin a: Selective Inhibition of Calcium Ionophore-Induced Polymorphonuclear Leukocyte Degranulation. Biochem Pharmacol (1991) 42:1221–8. doi: 10.1016/0006-2952(91)90257-6
79. Vandewalle A, Tourneur E, Bens M, Chassin C, Werts C. Calcineurin/NFAT Signaling and Innate Host Defence: A Role for NOD1-Mediated Phagocytic Functions. Cell Commun Signal (2014) 12:8. doi: 10.1186/1478-811X-12-8
80. Busch R, Murti K, Liu J, Patra AK, Muhammad K, Knobeloch K-P, et al. NFATc1 Releases BCL6-Dependent Repression of CCR2 Agonist Expression in Peritoneal Macrophages From Saccharomyces Cerevisiae Infected Mice. Eur J Immunol (2016) 46:634–46. doi: 10.1002/eji.201545925
81. Schaffner A, Douglas H, Braude A. Selective Protection Against Conidia by Mononuclear and Against Mycelia by Polymorphonuclear Phagocytes in Resistance to Aspergillus. Observations on These Two Lines of Defense In Vivo and In Vitro With Human and Mouse Phagocytes. J Clin Invest (1982) 69:617–31. doi: 10.1172/jci110489
82. Cunha C, Kurzai O, Löffler J, Aversa F, Romani L, Carvalho A. Neutrophil Responses to Aspergillosis: New Roles for Old Players. Mycopathologia (2014) 178:387–93. doi: 10.1007/s11046-014-9796-7
83. Zelante T, Wong AYW, Mencarelli A, Foo S, Zolezzi F, Lee B, et al. Impaired Calcineurin Signaling in Myeloid Cells Results in Downregulation of Pentraxin-3 and Increased Susceptibility to Aspergillosis. Mucosal Immunol (2017) 10:470–80. doi: 10.1038/mi.2016.52
84. Lacy P. Mechanisms of Degranulation in Neutrophils. Allergy Asthma Clin Immunol (2006) 2:98–108. doi: 10.1186/1710-1492-2-3-98
85. Kharazmi A, Svenson M, Nielsen H, Birgens HS. Effect of Cyclosporin A on Human Neutrophil and Monocyte Function. Scand J Immunol (1985) 21:585–91. doi: 10.1111/j.1365-3083.1985.tb01848.x
86. Hörl WH, Wanner C, Riegel W, Schlosser W, Wilms H, Schollmeyer P. Reduction of Degranulation of Polymorphonuclear Leukocytes by Immunosuppression in Patients Following Cadaveric Renal Transplantation. Transplantation (1989) 47:144–8. doi: 10.1097/00007890-198901000-00032
87. Yan P, Nanamori M, Sun M, Zhou C, Cheng N, Li N, et al. The Immunosuppressant Cyclosporin A Antagonizes Human Formyl Peptide Receptor Through Inhibition of Cognate Ligand Binding. J Immunol (2006) 177:7050–8. doi: 10.4049/jimmunol.177.10.7050
88. Gupta AK, Giaglis S, Hasler P, Hahn S. Efficient Neutrophil Extracellular Trap Induction Requires Mobilization of Both Intracellular and Extracellular Calcium Pools and Is Modulated by Cyclosporine A. PloS One (2014) 9:e97088. doi: 10.1371/journal.pone.0097088
89. Wang X, Bi Y, Xue L, Liao J, Chen X, Lu Y, et al. The Calcineurin-NFAT Axis Controls Allograft Immunity in Myeloid-Derived Suppressor Cells Through Reprogramming T Cell Differentiation. Mol Cell Biol (2015) 35:598–609. doi: 10.1128/MCB.01251-14
90. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA (2016) 315:801–10. doi: 10.1001/jama.2016.0287
91. Boomer JS, To K, Chang KC, Takasu O, Osborne DF, Walton AH, et al. Immunosuppression in Patients Who Die of Sepsis and Multiple Organ Failure. JAMA (2011) 306:2594–605. doi: 10.1001/jama.2011.1829
92. Hotchkiss RS, Monneret G, Payen D. Sepsis-Induced Immunosuppression: From Cellular Dysfunctions to Immunotherapy. Nat Rev Immunol (2013) 13:862–74. doi: 10.1038/nri3552
93. Sônego F, Castanheira FV e S, Ferreira RG, Kanashiro A, Leite CAVG, Nascimento DC, et al. Paradoxical Roles of the Neutrophil in Sepsis: Protective and Deleterious. Front Immunol (2016) 7:155. doi: 10.3389/fimmu.2016.00155
94. Kovach MA, Standiford TJ. The Function of Neutrophils in Sepsis. Curr Opin Infect Dis (2012) 25:321–7. doi: 10.1097/QCO.0b013e3283528c9b
95. Shen X, Cao K, Jiang J, Guan W, Du J. Neutrophil Dysregulation During Sepsis: An Overview and Update. J Cell Mol Med (2017) 21:1687–97. doi: 10.1111/jcmm.13112
96. Demaret J, Venet F, Friggeri A, Cazalis M-A, Plassais J, Jallades L, et al. Marked Alterations of Neutrophil Functions During Sepsis-Induced Immunosuppression. J Leukoc Biol (2015) 98:1081–90. doi: 10.1189/jlb.4A0415-168RR
97. Esquerdo KF, Sharma NK, Brunialti MKC, Baggio-Zappia GL, Assunção M, Azevedo LCP, et al. Inflammasome Gene Profile Is Modulated in Septic Patients, With a Greater Magnitude in Non-Survivors. Clin Exp Immunol (2017) 189:232–40. doi: 10.1111/cei.12971
98. Danikas DD, Karakantza M, Theodorou GL, Sakellaropoulos GC, Gogos CA. Prognostic Value of Phagocytic Activity of Neutrophils and Monocytes in Sepsis. Correlation to CD64 and CD14 Antigen Expression. Clin Exp Immunol (2008) 154:87–97. doi: 10.1111/j.1365-2249.2008.03737.x
99. Darcy CJ, Minigo G, Piera KA, Davis JS, McNeil YR, Chen Y, et al. Neutrophils With Myeloid Derived Suppressor Function Deplete Arginine and Constrain T Cell Function in Septic Shock Patients. Crit Care (2014) 18:R163. doi: 10.1186/cc14003
100. De Zuani M, Hortová-Kohoutková M, Andrejčinová I, Tomášková V, Šrámek V, Helán M, et al. Human Myeloid-Derived Suppressor Cell Expansion During Sepsis Is Revealed by Unsupervised Clustering of Flow Cytometric Data. Eur J Immunol (2021) 51(7):1785–91. doi: 10.1002/eji.202049141
101. Janols H, Bergenfelz C, Allaoui R, Larsson A-M, Rydén L, Björnsson S, et al. A High Frequency of MDSCs in Sepsis Patients, With the Granulocytic Subtype Dominating in Gram-Positive Cases. J Leukoc Biol (2014) 96:685–93. doi: 10.1189/jlb.5HI0214-074R
102. Mathias B, Delmas AL, Ozrazgat-Baslanti T, Vanzant EL, Szpila BE, Mohr AM, et al. Human Myeloid-Derived Suppressor Cells Are Associated With Chronic Immune Suppression After Severe Sepsis/Septic Shock. Ann Surg (2017) 265:827–34. doi: 10.1097/SLA.0000000000001783
103. Schrijver IT, Théroude C, Roger T. Myeloid-Derived Suppressor Cells in Sepsis. Front Immunol (2019) 10:327. doi: 10.3389/fimmu.2019.00327
Keywords: immunosuppression, calcineurin inbibitors, neutrophil (PMN) function, sepsis, NFAT signaling, pattern recognition receptor (PRR)
Citation: Vymazal O, Bendíčková K, De Zuani M, Vlková M, Hortová-Kohoutková M and Frič J (2021) Immunosuppression Affects Neutrophil Functions: Does Calcineurin-NFAT Signaling Matter? Front. Immunol. 12:770515. doi: 10.3389/fimmu.2021.770515
Received: 07 September 2021; Accepted: 19 October 2021;
Published: 02 November 2021.
Edited by:
Ivan Zanoni, Harvard Medical School, United StatesReviewed by:
Valentina Poli, Boston Children’s Hospital and Harvard Medical School, United StatesLaura Marongiu, University of Milano-Bicocca, Italy
Copyright © 2021 Vymazal, Bendíčková, De Zuani, Vlková, Hortová-Kohoutková and Frič. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Frič, amFuLmZyaWNAZm51c2EuY3o=
†These authors have contributed equally to this work