 Angeliki Datsi
Angeliki Datsi Rüdiger V. Sorg
Rüdiger V. Sorg
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 02 November 2021
Sec. Antigen Presenting Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.770390
This article is part of the Research Topic The Therapeutic Potential of Antigen Presenting Cells View all 13 articles
Glioblastomas (GBM) are the most frequent and aggressive malignant primary brain tumor and remains a therapeutic challenge: even after multimodal therapy, median survival of patients is only 15 months. Dendritic cell vaccination (DCV) is an active immunotherapy that aims at inducing an antitumoral immune response. Numerous DCV trials have been performed, vaccinating hundreds of GBM patients and confirming feasibility and safety. Many of these studies reported induction of an antitumoral immune response and indicated improved survival after DCV. However, two controlled randomized trials failed to detect a survival benefit. This raises the question of whether the promising concept of DCV may not hold true or whether we are not yet realizing the full potential of this therapeutic approach. Here, we discuss the results of recent vaccination trials, relevant parameters of the vaccines themselves and of their application, and possible synergies between DCV and other therapeutic approaches targeting the immunosuppressive microenvironment of GBM.
Glioblastomas (GBM) are highly invasive, malignant tumors of the central nervous system. According to the 2021 World Health Organization classification, GBM are grade 4 tumors, that belong to the group of adult diffuse gliomas (1). They lack mutations in the isocitrate-dehydrogenase (IDH) gene, which now discriminates GBM from IDH-mutated grade 4 astrocytomas, which have been regarded as secondary GBM before. Based on gene expression signatures, GBM can be further subdivided into mesenchymal, proneural, neural and classical subtypes (2).
Although representing the most frequent malignant primary brain tumor (~30%–40%), GBM are rare; the yearly incidence is three to four per 100,000 adults (3). Nevertheless, they are a highly fatal tumor, responsible for 2% of cancer-related deaths, with a yearly death rate of four to five per 100,000 adults. The established therapeutic standard of care in the first-line therapy for GBM combines maximal safe resection, fractionated radiotherapy with concomitant alkylating temozolomide (TMZ) chemotherapy, followed by adjuvant TMZ treatment. This multimodal approach has improved survival of patients significantly. Nevertheless, prognosis of newly diagnosed GBM patients is dismal. Median overall survival (mOS) is only 14.6 months, and the 2-year survival rate is 27.2% (4, 5). In GBM patients with unmethylated O6-methylguanine-DNA-methyltransferase (MGMT) promoter, producing the DNA-repair enzyme, prognosis is even worse [methylated vs. unmethylated mOS is 21.7 vs. 12.7 months; (6)]. Disease recurrence is universal, there is no effective therapy for recurrent disease, and median survival after relapse is 6.2 months. Therapeutic alternatives include lomustine, carmustine, tumor-treating field (TTF) therapy, and the angiogenesis inhibitor bevacizumab (7–10). Thus, there is a clear need for novel therapeutic modalities in GBM.
Dendritic cells (DC) are professional antigen-presenting cells, which are key to the development of T-cell responses (11, 12). As immature (resting) cells, they reside in most tissues, where they sample antigens. When activated by pathological changes in the tissue, they migrate to the draining lymph nodes and present there as mature (activated) DC peptides processed from the antigenic material taken up in the tissue on human leukocyte antigen (HLA) class I and II molecules, in an immunostimulatory context of co-stimulatory and accessory molecules. Antigen-specific cytotoxic T lymphocyte (CTL) and helper T cells (TH) recognizing these peptides get activated, proliferate, and differentiate to effector cells, which execute the various actions of cellular adaptive immune responses, including the killing of target cells. DC vaccination (DCV) is an active immunotherapy seeking to exploit this pivotal role of DC therapeutically: patients are vaccinated with tumor-associated antigens (TAA)-loaded DC, with the concept that they migrate to local lymph nodes, present TAA-derived peptides on HLA molecules, and initiate an antitumoral T-cell response, which selectively kills the tumor cells and prevents tumor recurrence, due to immunological memory (13, 14).
DCV was first evaluated in 1996 in a clinical trial for B-cell lymphoma (15). In 1999, Dhodapkar et al. reported the induction of antigenic target-directed T-cell responses by DCV (16). The authors vaccinated nine healthy individuals with mature DC loaded with an influenza matrix peptide, keyhole limpet hemocyanin, or tetanus toxoid and showed induction of target-specific T-cell immunity after a single application of the vaccine. The clinical efficacy of DCV was documented in 2006 in a phase III trial in patients with hormone refractory prostate cancer (17). Patients were treated with either placebo (leukocytes) or DC loaded with a fusion protein of prostatic acid phosphatase and granulocyte-macrophage colony-stimulating factor (GM-CSF) (Provenge/sipuleucel-T). mOS of vaccinated patients was significantly improved compared with that in the placebo-treated control group (25.9 vs. 21.4 months), results that were confirmed in a second trial (18); and Provenge/sipuleucel-T was the first (and so far only) DC vaccine approved by the U.S. Food and Drug Administration in 2010 (19). In GBM, hundreds of patients have been vaccinated, mainly in smaller uncontrolled trials. Although the results are promising, few studies can provide robust evidence, and overall the efficacy of DCV in GBM is variable, ranging from little or no clinical response to significant response. Therefore, we address the questions of which parameters could possibly effect efficacy of DCV and whether and how it may be possible to improve it.
Already in 1999, Liau and colleagues documented in the 9L rat glioma model that vaccination with DC pulsed with acid-eluted peptides derived from 9L glioma cells can prolong survival of glioma-bearing animals (20). In addition, vaccination was associated with infiltration of tumors with CD8+ and, to a lesser extent, CD4+ T cells and the development of glioma 9L cell-specific CTL responses. In 2000, Heimberger et al., who had vaccinated mice with DC pulsed with lysates derived from the spontaneously arising 560 glioma cell line, which had been transfected with the murine homolog of the mutated epidermal growth factor receptor variant III (EGFRvIII) (21), reported that vaccination could protect mice from subsequent intracranial tumor challenge. Survival of vaccinated animals was significantly prolonged compared with that in control animals receiving unpulsed DC. The surviving animals showed antitumoral memory and were healthy, were neurologically normal, and showed no signs of autoimmune encephalitis. Again, vaccination was associated with the development of glioma-cell-specific CTL responses, but interestingly, those were not directed against EGFRvIII, but against other unknown TAA.
Meanwhile, numerous animal studies have been performed in prophylactic (22–24) and curative DCV settings (25–31). Overall, there is clear evidence from these initial animal studies that DCV reduces tumor growth, can prolong survival, induces tumor-specific IFNγ and CTL responses, is associated with T-cell infiltration of tumors, particularly by CD8+ T cells, and results in long-lasting antitumoral memory that provides protection from tumor re-challenge. At the same time, vaccination appears to be safe and not to be associated with the development of autoimmunity. Thus, animal studies provided a proof-of-principle for DCV of GBM. Moreover, they continue to contribute to the development of vaccination strategies to increase efficacy (32).
Active immunotherapy with DCV has been pioneered by Liau et al., who described the vaccination of a GBM patient with recurrent disease with DC pulsed with eluted peptides of an HLA class I-matched GBM cell culture in 2000 (33). Although induction of an anti-peptide immune response was observed, the patient progressed and died 3 months later. Since then, numerous studies have been published (Table 1), including six controlled, randomized trials (60, 62, 76, 79, 81, 83), and several more are underway (88). Patients included mainly adults, but also children and adolescents (34, 38, 39, 41, 48, 52, 61, 64, 65, 69, 78), and the age of vaccinated patients varied between 1 and 80 years.
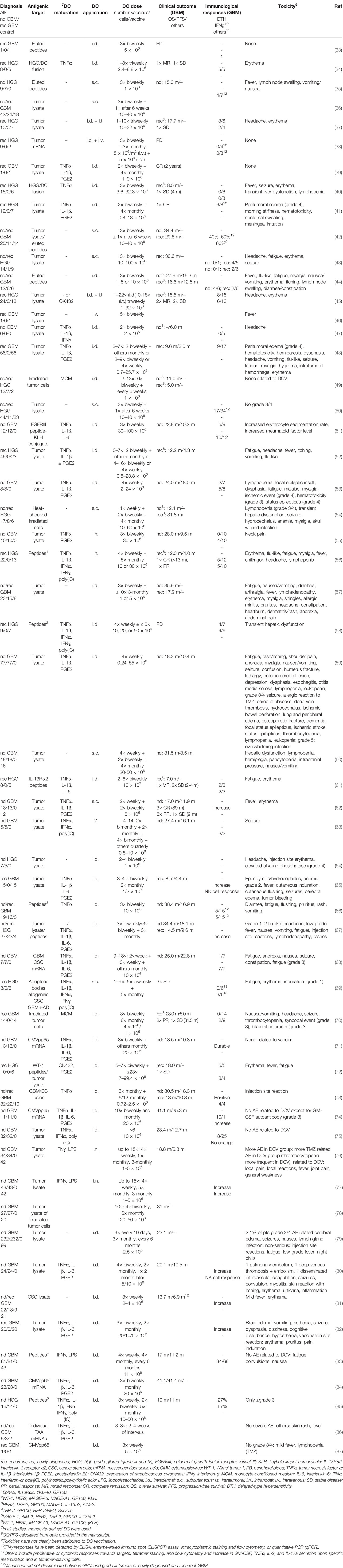
Table 1 Concluded clinical trials and case reports on dendritic cell vaccination of glioblastoma patients.
Most patients underwent cytoreductive surgery prior to DCV, but patients who were biopsied only or had no surgery at all underwent treatment as well. An association of survival and the extent of resection, which by itself is predictive for better survival (89), has also been reported for DCV (78), and a state of minimal residual disease has been indicated to be beneficial for vaccination therapy (44, 48, 65, 78). This may be due to a reduction of local immunosuppression, which correlates with the tumor size (90, 91), and the sheer number of fast growing tumor cells, which otherwise would have to be eliminated by the CTL. However, in another study, the extent of resection was not associated with survival (76), so a more detailed comparison of the absolute residual tumor volume and, in particular, the composition of the tumor [e.g., the contribution of an immunosuppressive tumor microenvironment (TME)] is required.
Therapies concomitant to DCV included radio- and chemotherapy, mainly with TMZ (4); but in several trials, DCV has been used as the sole treatment. There may be advantages but also disadvantages for vaccination in the context of TMZ chemotherapy (see also the section Dendritic Cell Dose, Vaccination Schedule, and Route of Application).
In the previous trials, mainly patients with newly diagnosed or recurrent GBM have been treated with DCV, but in several studies, grade III tumors were also included (Table 1). It is unknown whether there is a difference in the responsiveness of grade III and grade IV tumors to DCV; however, a trend for a higher immunological response rate in newly diagnosed patients has been reported (50), which may be due to the less heavy pretreatment of those patients.
Overall, DCV was well tolerated (Table 1). Severe side effects (≥grade 3) attributable to vaccination have not been observed except for one patient with gross residual tumor post-surgery, who suffered from peritumoral edema, which was controllable by glucocorticoids (41, 48). Other severe side effects have been consistent with either the respective concomitant therapies or disease progression. Frequently observed mild and easily controllable toxicities (≤grade 2), which may be attributable to DCV, are injection site reactions with itching, pain, erythema, induration, and lymph node swelling as well as flu-like symptoms, fever, fatigue, myalgia, headache, edema, and meningeal irritation, which, however, can also be observed in the course of other concomitant therapies or be due to the disease. Thus, overall toxicity of DCV therapy is limited. However, it has to be noted that Mitchel et al., who vaccinated a GBM patient with DC transfected with cytomegalovirus (CMV) phosphoprotein 65 (pp65) mRNA and applied the vaccines intradermally together with GM-CSF, reported induction of a type I hypersensitivity-like reaction with IgE, but also IgM and IgG antibodies against the GM-CSF (92), which however resolved when vaccination was continued without GM-CSF. On the one hand, these results document the potency of DCV to induce immune responses. On the other hand, however, they urge caution in using any type of protein supplement at higher doses injected together with the DC.
Induction of antigenic target-directed immune responses have been observed in the course of DCV (Table 1), with detection of IFNγ responses being most informative (50, 71, 77, 82, 83, 85), but antitumoral cytotoxic responses (35, 42–44) and an increase in tetramer positive cytotoxic T cells (43, 56, 61, 67, 72–74) have been reported as well.
Several studies identified immunological responders based on antigenic-target directed delayed-type hypersensitivity (DTH) reactions, IFNγ responses, or cytotoxic responses, which increased in the course of vaccination and reported longer survival times for responders (44, 45, 50, 55, 66, 77, 81, 82). Indeed, in the study by Wheeler et al., immunological responders (IFNγ qPCR) had a significantly longer OS (599 vs. 401 days) and time to progression (260 vs. 146 days), and the 2-year OS rates compared favorably (56% vs. 8%) for the responders compared with the non-responders (50). Moreover, long-term survivors with durable IFNγ responses have been identified (71). However, although there are reports of an association of detectable antitumoral immune responses and better clinical outcome, this is not true for all studies (67, 75). Indeed, there appears to be no strong correlation between detection of systemic antitumoral immune responses and clinical outcome, which may be due either to the parameters tested or to a failure of the systemically detectable response in reaching the brain and effectively killing the tumor cells.
Survival of vaccinated patients compared favorably with matched or historic controls (35, 44, 45, 51, 54, 57, 68, 71, 85, 86). For newly diagnosed patients, mOS ranged from 15 to 41.4 months, and the progression-free survival (PFS) ranged from 6 to 25.3 months (Table 1).
Meanwhile, several controlled studies have been published, six of them randomized (60, 62, 76, 79, 81, 83), whereas Batich et al. summarized the data of three previous similar trials they conducted (84) (for details, see Table 1).
For a phase III trial on 331 newly diagnosed GBM patients (232 in the DCV group and 99 in the control group), Liau et al. described long-term survivors and a mOS of all patients of 23.1 months. Unfortunately, however, they did not yet report conclusive data on the outcome of the study.
In two randomized phase II trials with 34 (60) and 25 (62) newly diagnosed GBM patients, mOS (31.9 and 17 months) of the vaccination groups was significantly improved compared with that of the respective control groups (15 and 10.5 months).
In another randomized phase II trial with 41 newly diagnosed and recurrent GBM patients (81), Yao et al. reported that DCV significantly prolonged mOS (13.7 vs. 10.7 months).
In two randomized phase II trials with 76 (76) and 124 newly diagnosed GBM patients (83), no significant differences in mOS (18.8 vs. 18.9 months and 17 vs. 15 months, respectively) between patients in the DCV and control groups were observed, although Wen et al. reported a significantly improved PFS for the vaccinated patients (11.2 vs. 9 months).
Batich et al. (84) merged their data on DCV with CMVpp65 mRNA transfected DC of newly diagnosed GBM patients (71, 74), either admixed with GM-CSF (11 patients) or tetanus-diphtheria toxoid (Td; six patients) conditioning of the vaccination site. They reported a mOS of vaccinated patients of 41.1 (GM-CSF) and 41.4 months (Td) compared with 18.5 months for control patients receiving unpulsed DC (six patients). Moreover, they describe long-term survival rates at 5 years of 36.4% and 33.3%.
Thus, even from these controlled trials, which revealed mixed results, it is still difficult to draw a conclusion as to the efficacy of DCV in GBM. Since markers of immunosuppression such as the PD-1+:CD8+ ratio (78), the presence of regulatory T cells (Treg) or CTLA-4 expression pre- and post-vaccination (67, 93), and an immunocapability score (77) are associated with survival after DCV, combining DCV with tools interfering with immunosuppression seems to be called for to improve efficacy. Moreover, to improve DCV efficacy may also require further optimization of the DC vaccine in respect to target antigen selection, preparation of the cells, the integration of DCV into other treatment regimens, and dosing and scheduling of the vaccination(s).
Efficacy of DCV depends on the presence of TAA or so-called neoantigens in the individual tumor, which allow specific recognition and killing of the tumor cells. The overall mutational load—the frequency of neoantigens—of this tumor entity is low, and the majority of GBM (>85%) contain only up to 10 mutations/1.4 Mb, except for patients with recurrent tumor after TMZ chemotherapy, which increases the mutational load [(94); for review, see (95)]. Nevertheless, multiple TAA have been identified for GBM (96–99).
In previous DCV studies, tumor lysates, apoptotic bodies of tumor cells, irradiated tumor cells, tumor mRNA, and fusions of tumor cells and DC as well as peptides eluted from the surface of tumor cells have been used as whole-tumor cell sources of TAA (Table 1). They are produced from the patient’s own tumor obtained from surgery and can also be derived from subsets such as cancer stem cells (68, 81), which could provide a therapeutic advantage (28, 29, 100). Whole-tumor cell sources of TAA most likely will contain multiple TAA, which are present in the individual tumor of a patient (including the various cellular subsets within the tumor), i.e., the patient’s full antigenic repertoire, ensuring antigenic diversity, thereby reducing the risk of escape of TAA-loss variants (101). Since the respective proteins are endogenously processed in the DC, in contrast to e.g., synthetic HLA class I restricted peptides, presentation on HLA class I and II molecules is possible and independent of the HLA type of the patient, thereby allowing induction of CTL as well as TH responses at the same time, which is a prerequisite for the development of an efficient CTL response. At least for tumor lysates, presentation/cross-presentation via HLA class I and II molecules of lysate-derived peptides has been shown (102). The observation of CTL responses after DCV is further evidence that the material taken up by the DC is indeed cross-presented via HLA class I (Table 1). Furthermore, whole-tumor cell sources of TAA, which have not been processed or cultured extensively, may provide the so far unknown necessary signals, allowing the DC to guide effector T cells to the brain (103).
TAA represent only a small fraction of proteins; therefore, a low tumor content of the tumor sample used for preparation of the TAA would even further reduce the TAA concentration. While this may not pose a higher risk to the patients, it may compromise efficacy of the vaccine. Therefore, a high tumor cell content of the sample has to be ensured, a task that will benefit from fluorescence-guided surgery, which allows intraoperative identification of the “solid” part of the tumor (63). Nevertheless, an estimate of the tumor cell content should always be obtained and reported when publishing DCV data, to establish values relevant for efficacy, which are currently unknown. When using protein solutions, such as lysates, the protein concentration used to load the DC should also be determined and reported. Despite the abundant presence of normal self-antigens in whole-tumor cell sources of TAA, induction of autoimmunity or other severe side effects attributable to its use for vaccine production have not been reported (Table 1). However, when using whole-tumor cell sources of TAA, it has to be kept in mind that they may inhibit vaccine production, i.e., DC differentiation and maturation, or modify the function of the resulting DC, because of the presence of immunosuppressive factors produced by the tumor cells (104). Moreover, efficacy appears not to be based alone on the source of TAA, but also on how it is processed. Gark et al. reported that induction of immunogenic cell death prior to DC-loading increases survival of animals substantially and shifts responses in the brain towards TH1/CTL/TH17 (105).
As an alternative to whole-tumor cell sources of TAA, molecularly defined TAA such as specific peptides, proteins, and DC transfected with the respective target antigen mRNA have been used for DCV of GBM (Table 1). Molecularly defined TAA represent a more standardized, consistent, and reproducible source of TAA and offer the advantage of higher available target antigen concentrations and lower background, and target-specific responses can be easily monitored. They can even be produced personalized (86). Nevertheless, multiple molecularly defined TAA should be used to reduce the risk of TAA-loss variants escaping immune control (101).
It is currently unknown whether whole-tumor cell sources of TAA or molecularly defined TAA (and which ones) are superior in inducing antitumoral immune responses and more beneficial clinically in GBM. Irrespective of the source of TAA, induction of antitumoral T-cell responses by DCV has been reported in previous studies (Table 1). The controlled studies, which documented a clinical benefit, used tumor lysates, thus a whole-tumor cell source of TAA (60, 62, 81), as well as CMVpp65 transfected DC, thus a molecularly defined TAA (84). Similarly, in the controlled studies that did not report a clinical benefit, either tumor lysates (76) or a set of six defined peptides (83) was used.
When peptide and tumor lysate-loaded DC were compared in animal models, superior efficacy has been reported for lysate-loaded DC (106). Moreover, Neller et al. concluded from the analysis of 173 published immunotherapy trials on various tumor entities, including melanoma, renal cell and hepatocellular carcinomas, and lung, prostate, breast, colorectal, cervical, pancreatic, and ovarian cancers a higher objective response rate (8.1% vs. 3.6%) when whole-tumor cell TAA were used compared with molecularly defined TAA (107). Thus, there appears to be an advantage of whole-tumor cell sources of TAA. However, particularly results from DCV against CMVpp65, a target that may be present in the majority of GBM patients and appears not to be expressed in normal brain cells (108, 109), generated convincing results for the efficacy of a molecularly defined TAA (71, 74, 84).
In 1994, Sallusto et al. reported the generation of immature DC with high antigen uptake activity from blood monocytes using the cytokines GM-CSF and IL-4, which then could be matured by an additional culture period with TNFα, resulting in cells with potent T-cell stimulatory activity (110). These so-called monocyte-derived DC are potent stimulators of naïve CD4+ TH cells and can polarize TH-cell responses towards TH1, cross-present antigens, and activate CD8+ CTL [for review, see (14)]. Yet their activity depends on the maturation or activating stimulus.
Although there are now techniques to enrich the various rare blood DC populations, which may also be promising candidates for DCV (111), monocyte-derived DC have been used in all DCV trials in GBM to date (Table 1). Monocytes were enriched from either peripheral blood or leukapheresis products by adherence, immunomagnetic selection of CD14+ cells, immunomagnetic depletion of B-cells and T cells, or elutriation. Depending on the enrichment procedure, monocyte purity varied, with CD14+ selection yielding the highest purity. Higher monocyte purity may result in more stable culture conditions, reduces modulating effects of contaminating cells, and yields higher-purity DC preparations. Overall, production of the vaccine is more reproducible and results in a more homogenous cell population.
Typically, monocytes were cultured with GM-CSF and IL-4 in a first culture phase, generating immature DC within ~6 days. The antigen-uptake activity of immature DC is well developed. Therefore, they have been used for antigen loading, when tumor lysates, apoptotic bodies of tumor cells, irradiated tumor cells, fusions of tumor cells and DC, proteins, and mRNA transfection were used as source of TAA, whereas peptide pulsing was performed mainly with mature DC.
Immature DC are poor stimulators of T cells and can even induce tolerance. Only when they are activated, e.g., by proinflammatory signals, do they develop into mature DC (14, 111). Originally, TNFα has been used as maturation factor (110). In 1997, Jonuleit et al. described a more potent maturation stimulus, a cytokine cocktail containing IL-1β, IL-6, and TNFα together with PGE2 (112). This cocktail and other combinations of these factors with or without factors such as type I and II interferons, lipopolysaccharide, and toll-like receptor ligands [e.g., polyinosinic:polycytidylic acid (poly(IC))] have been used in the clinical studies summarized here (for details, see Table 1).
The optimal maturation stimulus is still a matter of debate and since immature DC sense, integrate, and translate environmental changes into signals to the T cells, differences in medium, cell density, the frequency of dead cells in cultures, etc., may result in different outcomes in regard to target cell function. Moreover, the same factors can have beneficial as well as adverse effects. For example, the combination of lipopolysaccharide and IFNγ induces semi-mature DC, which produce IL-12 and induce CTL responses (113), but it also appears to initiate an immunosuppressive program with the induction of indoleamine-2,3-dioxygenase [IDO; (114)]. Similarly, PGE2, which is part of a potent cytokine cocktail inducing DC maturation (112) and improves the migratory response of DC (115), can also induce IDO (116). In addition, the effects of the maturation factors can be further modulated by the source and processing of the TAA (104, 105). Thus, outcome of DC maturation is difficult to generalize. Therefore, the functional properties of the cells have to be determined for the respective conditions used in each manufacturing process.
Mature DC can be distinguished from immature DC by their expression of the surface molecules CD83 (117) and CD25 (118), whereas other markers such as CD40, CD80, CD86, and HLA-DR may differ only in expression density (118). Indeed, high-density expression of CD80 appears to be of utmost importance because it interacts in-cis with PD-L1, thereby blocking PD-L1 binding to PD-1 on T cells and inhibition of T-cell activation (119). However, identification of “mature” DC may be even more complex. Previously, a population of mregDC (mature DC enriched in immunoregulatory molecules) has been identified, which co-expresses maturation markers including CD83, CD40, CD80, CD86, and RelB together with immunoregulatory genes such as PD-L1, Pdcd1lg2, CD200, Fas, Socs1, Socs2, and Aldh1a2 (120).
Overall, mature DC for immunotherapy should 1) have potent TH-cell and CTL stimulatory activity; 2) polarize responses towards TH1, which is required for efficient induction of effector CTL (121, 122); 3) have to imprint effector T cells for brain tumor homing, which may require induction of VLA-4 (α4/β1 integrin), which appears to be the main integrin for lymphocyte trafficking to the brain (123); 4) express CCR7, which is required for lymph node homing (124, 125); 5) be phenotypically stable upon withdrawal of cytokines (126) to prevent re-differentiation towards immature (and possibly tolerogenic) DC after administration; 6) be resistant to immunosuppressive cytokines like TGF-β (127); and 7) not induce tolerance. Particularly, the induction of target antigen-specific tolerance, which has been reported for immature and semi-mature DC, but not fully mature DC would be detrimental to the intended induction of antitumoral immune responses (128). Indeed, de Vries and colleagues showed that TAA-loaded mature DC, but not immature DC, induce immunological responses in melanoma patients (129), and even more important, Dhodapkar et al. documented a decline of the influenza matrix peptide-specific T-cell response in healthy individuals after vaccination with matrix peptide-loaded immature DC (130).
In GBM, several clinical trials used immature DC as vaccines (Table 1). Somewhat unexpectedly, immunological responses as well as beneficial effects on survival have been reported, although Yamanaka et al., who used mature as well as immature DC in their study, reported a trend towards a better outcome in GBM patients vaccinated with the mature DC (45). Two of four controlled trials reporting a clinical benefit used immature DC (60, 81), whereas in the remaining two trials, DC were matured with TNFα, IL-1β, and PGE2 (62) or TNFα, IL-1β, IL-6, and PGE2 (84). Interestingly, both controlled trials that did not document clinical efficacy (76, 83) used lipopolysaccharide + IFNγ for maturation, thus a factor combination that also results in the induction of IDO (114). In conclusion, currently, the most potent vaccine for DCV of GBM has not been identified, and even the use of immature DC cannot be excluded, although there are strong arguments for the use of mature DC.
The minimum DC dose reported to elicit T-cell responses in healthy individuals is 2 × 106 DC/vaccine (16). A wide range of DC doses have been used in GBM-DCV trials (0.25–100 × 106 DC/individual vaccine, Table 1). In four controlled studies, which reported a significant survival benefit for vaccinated patients, doses of 2–4 × 106 (81), 6 × 106 (62), 20 × 106 (84), and 20–50 × 106 (60) DC/vaccine were used, while doses of 1–5 × 106 (76) and 11 × 106 DC/vaccine (83), i.e., in a comparable range, resulted in no clinical benefit in two other controlled trials. In several studies, immunological responders were identified based on an increase in IFNγ (qPCR, ELISPOT) after DCV, and immunological responsiveness was positively associated with survival. DC doses ranged from 1 to 50 × 106 DC (50, 71, 77, 85). No correlation with DC dose has been described for either clinical outcome or immunological responsiveness, and a dose–response relationship with an optimal dose cannot yet be defined. Because dose-limiting toxicity has not been reached in previous studies, doses tend to be maximized based on the number of cells available from the production process and the vaccination scheme. In a dose-escalating study by Prins et al. using 1, 5, and 10 × 106 DC/vaccine, no association was found between increasing DC dose and toxicity or immunologic response, but longer survival (though not statistically significant) was observed in those patients receiving the lowest DC dose (57). Although only a fraction of the injected DC reaches the lymph nodes (131, 132), this may still be far more than, e.g., in the case of infections and could actually be too much. However, Mitchel et al., who used fairly high DC doses/vaccine (20 × 106), reported an improved survival of patients, when the efficiency of migration of DC to the lymph nodes was enhanced (71). Moreover, intranodal application, which may allow to directly deliver even higher DC numbers to the lymph nodes, resulted in increased IFNγ responses after vaccination (55, 56, 77), although Buchroithner et al. could not document a survival benefit in a controlled trial using this approach (76). However, it has to be kept in mind that DC vaccines are likely to differ in potency, e.g., immature as well as mature DC have been used, and the vaccination schedule and route of application, as well as many other parameters, also influence efficacy, so an optimal DC dose is difficult to define and may need to be determined on a case-by-case basis for each vaccination strategy, yet it has the potential to improve efficacy.
Multiple vaccinations were given per patient, mainly 3–10, but also up to 23 (Table 1). Thus, many studies used a prime and boost vaccination approach. Currently, it is not clear whether multiple vaccinations improve the outcome. Jouanneau et al. showed in a GL26 orthotopic tumor model that multiple injections of TAA-loaded DC did not further improve the outcome, whereas a lysate boost resulted in a significantly prolonged survival and was associated with an increased CTL response and antibody formation, contributing to the therapeutic effect (133). Indeed, de Vleeschouwer et al. described a trend of prolonged PFS with a DCV strategy with lysate boosting, although the contribution of the lysate boosts remains unknown (48). In contrast, Okada et al. reported that DC boost vaccination further enhanced IFNγ responses (56) and Buchroithner et al. described a trend towards better survival in patients receiving more vaccines (76). Thus, the number of vaccinations and the use of DC vaccines or lysates (or any other target antigen such as e.g., peptides) for boost vaccination are parameters that influence efficacy and remain to be optimized.
Vaccines have been administered weekly, biweekly, or monthly, or in combinations thereof, frequently integrated into established treatment regimens of radiotherapy and chemotherapy. The standard of care for newly diagnosed GBM consisting of resection, and radiotherapy with concomitant and subsequent adjuvant TMZ chemotherapy (4) appears to offer several windows of opportunity for vaccination, allowing to retain the standard of care with proven efficacy while integrating DCV and potentially exploiting synergies between the two therapeutic approaches.
Immunosuppression, which is prominent in GBM (see below), correlates with tumor size, and surgical cytoreduction can at least partially restore immunological responsiveness (90, 91). Fluorescence-guided surgery (FGS) allows to increase the extent of resection safely, and radiologically complete resections could be performed in 65% of patients compared with 35% in a control group undergoing standard surgery (89) that was associated with improved survival. Thus, integrating FGS into a DCV approach (63, 75) may not only minimize residual tumor mass and thereby also immunosuppression, both of which are beneficial for immunotherapy (44, 48), but because of the extended PFS, it may also prolong the time period available for T-cell responses to clear residual tumor cells before the tumor mass becomes too large again. Moreover, FGS ensures high tumor cell frequency in the tumor samples, because the vital “solid” part of the tumor can be identified intraoperatively (63), an advantage when whole tumor cell sources of TAA such as tumor lysates are used. The substantial increase in the extent of resection by FGS may also improve safety of DCV, because in a patient with gross residual tumor after standard surgery, a grade 4 peritumoral edema has been reported, which was considered to be associated with DCV (41, 48). In addition, a maximal resection generally allows to wean glucocorticoids faster, which are perioperatively applied in GBM patients to reduce swelling in the brain—a possible advantage for DCV due to the immunosuppressive activities of the steroids. However, although glucocorticoids are potent inhibitors of T-cell immunity (134) and Keskin et al. reported induction of polyfunctional T cells after peptide vaccination only in those GBM patients that did not receive dexamethasone (135), its role during DCV is not entirely clear. Therefore, it should be used judiciously or excluded, until more data on dosing and timing of glucocorticoid use during DCV of GBM patients become available (136). Indeed, in four of the controlled trials (79, 81, 83, 84), the use of glucocorticoids was either excluded entirely or only minimal doses of 2–4 mg/day of dexamethasone were allowed.
Vaccination is either performed in the time period between radiochemotherapy and adjuvant TMZ or in the course of the adjuvant TMZ cycles around day 21 (137), because there appears to be a rationale to combine DCV with TMZ chemotherapy: 1) TMZ can improve immunological responsiveness (138–140), probably by reducing Treg (see below) (139, 141) and interfering with their recruitment to the tumor (142). 2) Although it frequently causes lymphopenia, the recovering lymphocyte compartment after chemotherapy has been shown to allow for efficient induction of antitumoral responses (143–145). 3) Dying tumor cells after radiochemotherapy or chemotherapy lead to a release of tumor antigens, which could enhance homing of tumor-specific effector CTL to the brain tumor after luminal presentation of the target peptides on HLA class I molecules on the cerebral endothelium (146). However, effects appear to depend on TMZ dose; e.g., lower but not higher TMZ doses were shown to deplete Treg (141), whereas myeloablative but not non-myeloablative doses enhanced responses to a peptide vaccine (147). In addition, results from Pellegatta et al. indicate that adjuvant TMZ may deplete CD8+ T cells previously expanded by DCV, because in contrast to NK cells, they fail to express the multidrug resistance transporter protein ABCC3 (80). A decline in responding cells after adjuvant TMZ has also been described by Batich et al. (84). Moreover, is has been shown that DCV only in the absence of TMZ, although with additional conditioning of the injection site with tetanus toxoid, results in the generation of T effector memory cells producing IFNγ, which is positively associated with survival (82). These results would argue against combining DCV and TMZ chemotherapy. Because all controlled DCV trials in GBM used TMZ in both arms, currently, it cannot be determined whether or not it affects efficacy (60, 62, 76, 81, 83, 84).
Effective induction of antitumoral T-cell immunity requires the DC to reach the T-cell areas of lymph nodes. Although the cervical nodes (148–151) or the nasopharynx-associated lymphoid tissue (152) serve as lymph node stations of brain immune responses, imprinting the brain homing phenotype of effector T cells is a function of the DC rather than that of a distinct lymph node. Therefore, effective responses targeting antigens in the brain can also be initiated in other lymph nodes besides the cervical nodes or the nasopharynx-associated lymphoid tissue (103).
Depending on the route of application, DC can be detected in different organs. Intravenous application results in a rapid enrichment in the liver, lungs, and kidneys but is the highest in the spleen, whereas after subcutaneous application, there is a marked accumulation of DC in the draining lymph nodes, with a preferential paracortical localization in the T-cell areas (153). When intradermal application is used instead, even more DC reach the T-cell areas of the lymph nodes in mice (154) as well as in humans (155), with only mature but not immature DC efficiently migrating to the lymph nodes (156). This is probably due to the expression of the CCR7 chemokine receptor on the mature DC (118, 124, 125) and the responsiveness of the cells to the chemokines CCL19 and CCL21, which are expressed constitutively by peripheral lymphatic endothelial cells and lymph node stromal cells (157).
DC can already be detected in the lymph nodes 30 min after injection, there is a maximum after 48 h, and they appear to persist for up to 14 days (158–160). Only ~5% of injected DC may reach the lymph nodes, which appears to be sufficient for effective induction of antitumoral immune responses (131, 132), although substantially higher values have been reported as well (160). A large fraction of DC remains at the injection site, rapidly becomes apoptotic, and is cleared by CD163+ macrophages (131). However, it is possible to augment DC migration to the lymph nodes by preconditioning the application site with a potent recall antigen such as tetanus/diphtheria toxoid, associated with improved survival of patients (71).
The life span of DC in the lymph nodes is limited to a few days (161, 162), and they may be removed by apoptosis (163, 164) and phagocytic clearance by macrophages (165). However, endogenous DC in skin and lymph nodes may prolong antigen presentation beyond the life span of the injected DC (166).
Overall, there appears to be an advantage of intradermal application of vaccines, and indeed, most studies have used it (Table 1). Whether the higher DC numbers delivered directly to the lymph nodes by intranodal application (55, 56, 76) are even more effective remains to be determined.
There is also the possibility of intratumoral application of DC. Pellegatta et al. have documented in an orthotopic GL261 glioma model that the efficacy of intratumoral application of GL261 lysate-loaded DC is lower than that of subcutaneous application, but that the combination of both procedures significantly improves survival (167). Since intratumorally administered DC remain in the brain parenchyma and were not detected in the cervical lymph nodes, a different mechanism than for the subcutaneously administered DC appears to be responsible for improved survival. Whether they contribute to the final maturation and shaping of the effector T-cell response (168) by acting as tissue inflammatory DC or reduce tumor cell growth in this model because of their production of TNFα (167) remains to be determined. Intratumoral application has not yet been studied in clinical trials in GBM.
A major obstacle to the therapeutic vaccination of GBM with DC is that the antitumoral immune response must be elicited in the context of immunosuppression. Humoral and cell contact-dependent mechanisms originating not only from the tumor cells themselves (including intrinsic mechanisms of immune evasion) (169) but also from the cells of the TME, such as Treg, tumor-associated macrophages (TAM), and myeloid-derived suppressor cells (MDSC), can inhibit antitumor immunity (Figure 1). Moreover, immune checkpoints—control mechanisms that limit and thereby prevent excessive immune responses—may be activated in the TME, also resulting in inefficient responses and T-cell dysfunction. Whether DCV by itself can tip the balance towards immunity is unclear, but efficacy may require the combination with additional therapeutic strategies (Figure 2) to overcome the adverse effects of immunosuppression and immune checkpoint regulation.
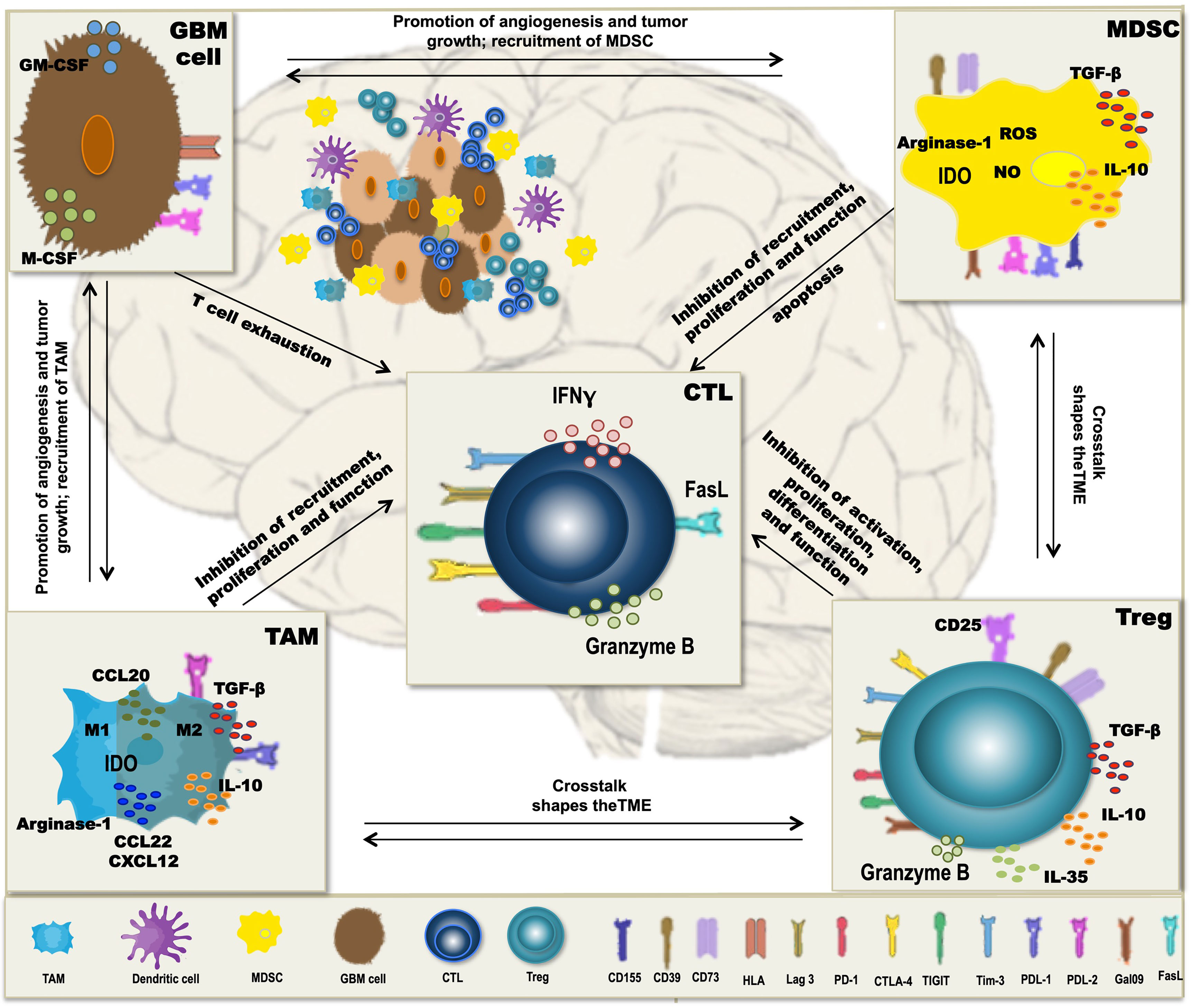
Figure 1 Mechanisms of immunosuppression in glioblastoma (GBM). In GBM, tumor-associated macrophages (TAM), myeloid-derived suppressor cells (MDSC), and regulatory T cells (Treg) form a potent immunosuppressive tumor microenvironment (TME), which inhibits antitumor immunity and thereby interferes with dendritic cell vaccination (DCV). Besides the intrinsic immune escape mechanisms of the tumor cells, immune checkpoint molecules, like PD-L1, PD-L2, Tim-3, Lag-3, CD155, and galectin-9 that normally control the extent of immune responses, are expressed on the immunosuppressive cells of the TME, contributing to T-cell dysfunction and subsequently inefficient antitumoral immune responses. Cells of the TME secrete cytokines such as TGF-β, IL-10, and IL-35 and the chemokines CCL20, CCL22, and CXCL12, which inhibit T-cell proliferation and function and contribute to a crosstalk between the different TME cell types, thereby further enhancing immunosuppression. Additional mechanisms include the activity of indoleamine-2,3-dioxigenase (IDO) and arginase-1 as well as production of reactive oxygen species (ROS) and nitric oxide (NO), all interfering with a proper differentiation, expansion, and function of effector T cells.
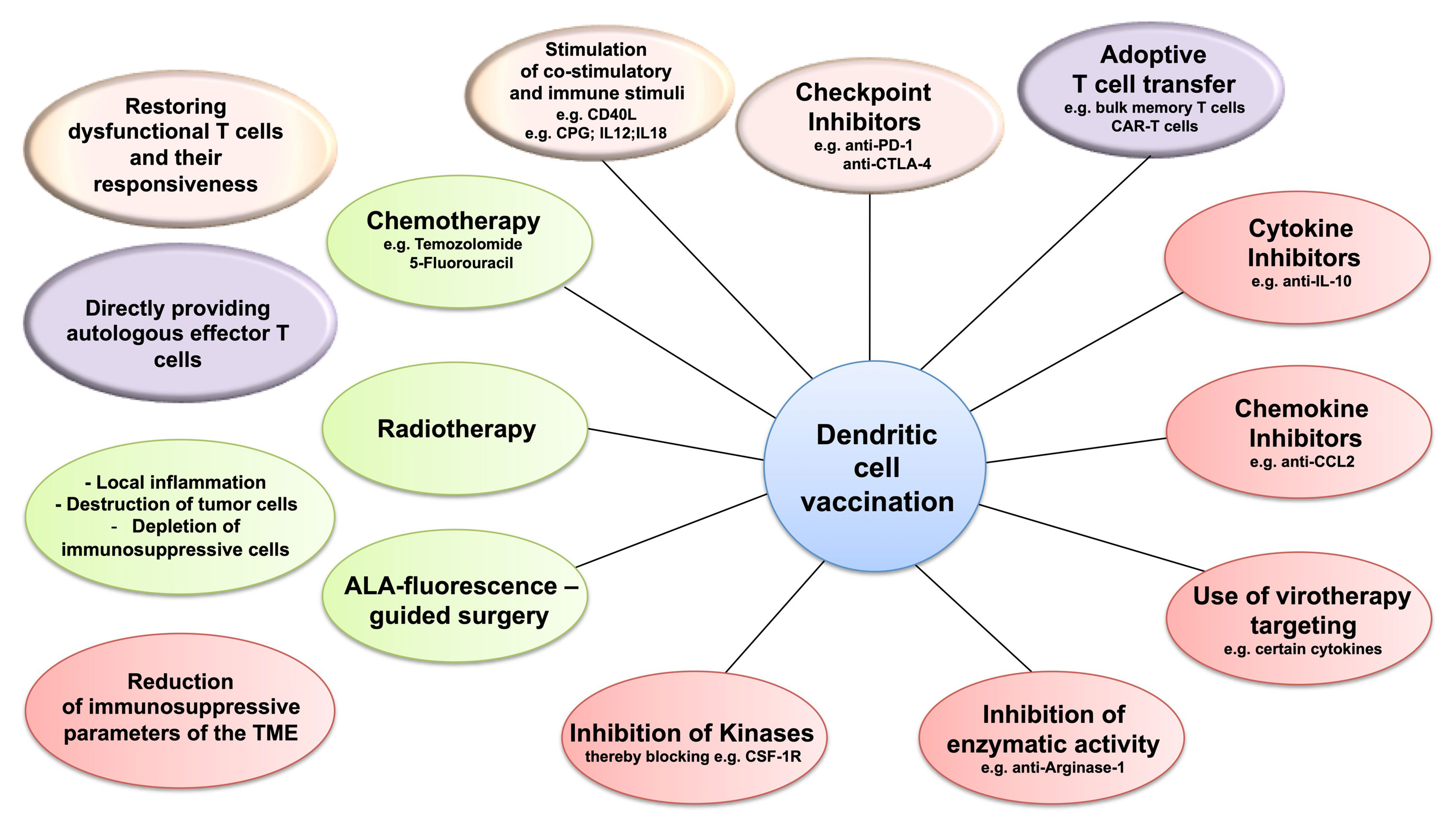
Figure 2 Dendritic cell vaccination (DCV) and targeting the immunosuppression in the tumor microenvironment (TME). Combining DCV with therapies targeting the three immunosuppressive cell populations of the TME—regulatory T cells (Treg), tumor-associated macrophages (TAM), or myeloid-derived suppressor cells (MDSC)—might improve efficacy. Potential target strategies include restoring the responsiveness of the dysfunctional T cells (pink), applying effector T cells by adoptive transfer (purple), depleting immunosuppressive cells, and modulating the inflammatory conditions in the TME (green) and blocking the mechanisms of immunosuppression (red).
Treg are a distinct immunosuppressive T-cell subpopulation, which contribute to maintaining immunological tolerance, limiting excessive immune responses, and promoting homeostasis and tissue regeneration. They either develop in the thymus (thymic (t)Treg) as a distinct T-cell lineage or differentiate in the periphery (peripheral (p)Treg) from naïve T cells [reviewed in (170, 171)]. Naïve/resting Treg reside mainly in the blood and secondary lymphoid organs. They can be identified as CD3+/CD4+/CD25low/CD127−/low T cells, which express the transcription factor FoxP3 (172–176), although FoxP3 may also be transiently expressed at low levels in the course of activation of human conventional CD4+ and CD8+ T cells (177). They further express CD45RA, CD62L, and CCR7 (CD197), whereas expression of CTLA-4 (CD152) and CD45R0 is absent on the naïve/resting Treg. Upon T-cell receptor stimulation, they differentiate into highly proliferative and suppressive effector Treg, which are characterized by a CD3+/CD4+/CD25high/CD127−/low/FoxP3high/CD45RA−/CD45R0+/CCR7−/CD62L−/CTLA-4+ immunophenotype [reviewed in (170, 178)], with delineation of cells further improved by the presence of CD15s on effector Treg, but not conventional effector T cells (179). Depending on the stimulatory context and the affinity of T-cell receptor recognition, there is induction of chemokine receptors (e.g., CCR2, CCR4, and CXCR3) and adhesion molecules (e.g., lymphocyte function-associated antigen-1 (LFA-1), integrin-α4, and integrin-β1), which guide them to their target sites, as well as of effector molecules, which mediate selective mechanisms of immunosuppression. Indeed, effector Treg can upregulate transcription factors associated with distinct TH-effector phenotypes, allowing suppression to be tailored to the respective polarized responses, and it may be further modulated by the local microenvironment at the target site (170, 178, 180–182).
Multiple mechanisms contribute to effector Treg function:
1. They secrete the immunosuppressive cytokines IL-10, TGF-β, and IL-35, which inhibit T-cell activation and proliferation either directly or by suppressing the stimulatory activity of DC, and contribute to the formation of tolerogenic DC and the generation of additional Treg (183–186). These effects are further enhanced by cytokine-mediated crosstalk between the immunosuppressive cells of the TME, e.g., TGF-β stimulates MDSC proliferation and suppressive activity (187). Moreover, TGF-β downregulates intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) on blood vessels and thus inhibits conventional T-cell infiltration into the TME (188).
2. Treg release granzyme and perforin, which induce apoptosis of effector T cells (189, 190). Moreover, CD4+ TH-effector cells expressing death receptor 5 (DR5) can also be killed by Treg expressing the corresponding ligand TNF-related apoptosis-inducing ligand (TRAIL) (191).
3. Due to the high-density expression of high-affinity IL-2 receptors (CD25/CD122/CD132) on Treg, they act as an IL-2 sink and deprive conventional T cells of IL-2, which suppresses their expansion and differentiation to effector cells and may cause effector T cells to become anergic or apoptotic (192).
4. Treg can release adenosine nucleosides. They express CD39 (ectonucleoside triphosphate diphosphohydrolase-1) and CD73 (ecto-5′-nucleotidase), which together convert adenosine triphosphate to adenosine, with extracellular adenosine inhibiting DC antigen presentation as well as proliferation and cytokine secretion of activated T cells through the A2A receptor (193–195). It further promotes differentiation and proliferation of Treg, expansion of MDSC, and polarization of M2 macrophages [reviewed in (196)]. Treg also contain high levels of cyclic adenosine monophosphate, which they can transfer into conventional T cells via gap junctions, thereby inhibiting their proliferation and IL-2 production upon activation (197).
5. Effector Treg express several surface molecules, which interfere with the activation, proliferation, differentiation, and effector function of conventional T cells. CTLA-4 is an immune checkpoint regulator. It binds to CD80/CD86 on DC with higher affinity than CD28 on conventional T cells, leading to anergy, apoptosis, or even the conversion of the activated conventional T cells into Treg, due to the absence of the co-stimulatory signal. However, CTLA-4 not only competes with CD28 for CD80/CD86 binding but also depletes them from the surface of DC by transendocytosis, thereby further preventing co-stimulation (198, 199). Moreover, it induces IDO in DC and causes their conversion to tolerogenic DC. IDO is an enzyme that degrades the essential amino acid tryptophan to kynurenine. The resulting local tryptophan depletion as well as the interaction of kynurenine with the aryl hydrocarbon receptor prevents T-cell proliferation and can induce differentiation of CD4+ T cells into Treg (200–202). Other immune checkpoint regulators including PD-1 and its ligand PD-L1, lymphocyte-activation gene 3 (Lag-3), T-cell immunoglobulin mucin-3 (Tim-3), and T-cell immunoreceptor with Ig and ITIM domains (TIGIT) can also be expressed on Treg and contribute to immunosuppression (203–208) [reviewed in (209, 210)].
In various solid tumors, there is a high frequency of tumor-infiltrating effector Treg and particularly a high Treg : CD8+ T-cell ratio, which negatively correlate with prognosis (211), indicating that there is a naturally occurring antitumoral immune response. Treg cannot be detected in normal brain, and they are rare in low-grade brain tumors, yet despite lymphopenia, Treg frequencies are increased in the TME as well as in the blood of GBM patients (212–215). Frequencies may vary by GBM subtype, with the IDHwildtype (Iso-citrate dehydrogenase) and mesenchymal subtypes having higher Treg frequencies than IDHmutated (216) and proneural and classical GBM subtypes, respectively (217). It has been reported that there is an age-dependent increase in the Treg : CD8+ ratio, with a maximal increase in the 60–69 years age group, which coincides with the median age (64 years) of GBM patients at diagnosis (218). Nevertheless, the association of Treg frequency with prognosis is moderate at the most (188, 214, 215, 219), in contrast to CD4+ and CD8+ effector T-cell infiltrates, which are positively associated with survival (188). However, association appears to require the concomitant presence of a low immunosuppressive signature (220). Indeed, a negative association of the Treg : CD8+ T cell ratio with survival has been reported (221), which could indicate that there are patients with natural antitumoral immunity and that only in these patients are Treg associated with poor prognosis.
In mouse models of GBM, a time-dependent accumulation of Treg after implantation of tumor cells has been described (222–224). Interestingly, Treg numbers increased first in blood and later, but still in the asymptomatic phase, in the tumor tissue (223, 224). Thus, Treg appear to be recruited to the tumor already in an early phase, when tumor cell numbers are still low and are not a consequence of the immune system simply being overwhelmed by the tumor mass at a later phase of tumor development. The chemokines CCL2 and CCL22, which are produced by brain cells as well as GBM cells and cells of the TME, appear to be responsible for the recruitment of the CCR4+ Treg to the TME of GBM (142, 215, 225).
Treg-mediated immunosuppression has been targeted to enhance the efficacy of natural or induced antitumoral immunity mainly in preclinical models of GBM. This includes depletion of Treg by anti-CD25 antibodies (145, 222, 226, 227), interference with their immunosuppressive activity, e.g., by blocking surface molecules such as CTLA-4 (222, 228, 229), TIGIT (230), Tim-3 (231), and PD-1 (229, 230, 232) or enzymes like ecto-5′-nucleotidase (CD73) (233) and IDO (229), or by preventing the accumulation of Treg in the TME by blocking CCR4-mediated migration (225). Irrespective of the approach, these studies could document a beneficial effect, with increased survival and reduction of tumor burden associated with restoration of antitumoral immunity, particularly when different strategies were combined.
However, specific inhibition of Treg or of their functional activity may not be required. Higher doses of non-fractionated radiotherapy (234) and chemotherapy with low-dose TMZ (141) or cyclophosphamide (235) have also been reported to reduce Treg, although it is unclear whether the long-lasting lymphopenia induced by the standard concomitant radiochemotherapy of GBM patients is beneficial for antitumoral immunity (144, 145, 236). Moreover, immunogenic stimuli such as CPG oligonucleotides (237, 238), virotherapy (239, 240), or the use of agonistic antibodies specific for co-stimulatory receptors such as CD40 (241) and OX40 [CD134; (242)] may be sufficient to tip the balance towards antitumoral immunity, associated with a reduction in Treg. Another promising approach is to modulate the metabolism of Treg, which appears to be tightly linked to their survival and function in the TME [reviewed in (243)]. Whether combinations with therapeutic approaches targeting Treg or their function result in increased efficacy has not extensively been studied and requires further investigation. Curtin and colleagues reported that anti-CD25 depletion of Treg in combination with intratumoral delivery of an adenoviral vector expressing Fms-like tyrosine kinase 3 ligand and herpes simplex type 1-thymidine kinase inhibited clonal expansion of tumor antigen-specific T cells, T-cell dependent tumor regression, and long-term survival of animals (239), whereas a trend towards improved survival has been reported for the combination of radiotherapy and anti-IDO (244). Particularly, the timing of Treg depletion appears to be of utmost importance. Several of the target molecules (e.g., CD25 and CTLA-4) are not specific to Treg but are upregulated in the course of activation on conventional T cells as well. Similarly, although anti-PD1 treatment may enhance antitumoral immunity, at the same time, it may increase the suppressive activity of Treg (245). Thus, depletion has to be performed prior to the promotion of antitumoral immunity or other treatments, to avoid suppression of the antitumoral effector response (145, 239, 246). Moreover, long-term survival of glioma-bearing mice was only observed when the animals were treated by a combination of systemic and intracranial, but not by systemic anti-CD25 antibody treatment alone (247). Thus, depleting/blocking agents have either to be administered directly to the tumor in the brain or to be able to pass the blood–brain barrier efficiently. Local administration may also prevent uncontrolled inflammation and autoimmune phenomena due to the systemic elimination of Treg and thereby of an important control mechanism of immunity and tolerance.
Evidence for a prominent role of Treg for the efficacy of DCV comes from several studies: Driessen et al. identified a lower Treg frequency in cured rats compared with non-cured rats after DCV in a 9L gliosarcoma model (248). Fong et al. and Prins et al. observed a prolonged survival in patients whose Treg decreased after vaccination with peptide-pulsed DC (67, 93), with higher Treg values before vaccination having been shown by Erhart et al. to be negatively associated with survival after vaccination with lysate-loaded DC (77). Although Batich et al. described long-term survivors after anti-CMVpp6 DCV, they observed increases in Treg that were, however, paralleled by an increase of the CD8+:Treg ratio (74). Thus, the absolute numbers or frequencies of Treg by themselves may not be informative enough. Moreover, several studies reported a beneficial effect on survival when DCV was combined with anti-CD25 treatment, particularly when depletion was performed prior to vaccination (106, 227, 249). Thus, depletion of Treg or interference with their activity has the potential to improve the outcome of DCV.
MDSC are a heterogeneous population of immature myeloid cells with potent immunosuppressive activity. In the TME, they constantly interact with the infiltrating T cells, particularly CTL, and suppress their function (250, 251), thereby supporting tumor growth and progression (252). MDSC are divided into two general subsets: polymorphonuclear (PMN)-MDSC (CD11b+/CD14−/CD15+ or CD11b+/CD14−/CD66b+), similar to neutrophils, and monocytic MDSC (CD11b+/CD14+/HLA-DR−/lo//CD15−), similar to monocytes (250). However, while they are phenotypically similar to neutrophils and monocytes, they are functionally distinct (253, 254).
MDSC can be detected in cancer patients or during chronic inflammation (250, 251, 255), when persistent low-level stimulation of myelopoiesis results in the development of the immunosuppressive myeloid cells (256). They develop in the bone marrow and traffic into solid tumors, where they accumulate mediated by factors such as GM-CSF, M-CSF, G-CSF, VEGF, IFNγ, IL-6, and IL-4, which are secreted by the tumor cells themselves or other cells of the TME (257, 258).
MDSC, similar to Treg and in part overlapping (see above), use multiple mechanisms of immunosuppression [for review, see (259)], in particular including reactive oxygen species (ROS) and nitric oxide (NO)-dependent pathways. ROS and NO cause apoptosis of immune cells (260) and block entry of CTL into the tumor and responsiveness of T cells to HLA stimulation through the nitration of chemokines and T-cell receptors, respectively (261, 262). MDSC also inhibit extravasation of T cells by downregulating CD44 and CD164 (259) and lymph node re-circling through downregulation of CD62L on naïve T cells via expression of ADAM17 (263). Arginase 1 catabolizes the non-essential amino acid arginine and depletes it from the microenvironment. Similarly, there is a depletion of cysteine by consumption and sequestration and of tryptophan due to IDO activity. This lack of amino acids in the TME inhibits proliferation of activated T cells. Like Treg, MDSC can produce immunosuppressive cytokines such as IL-10 and TGF-β and extracellular adenosine through the enzymatic activity of CD39 and CD73. Moreover, they express ligands of immune checkpoint regulatory pathways such as PD-L1, PD-L2, CD155, and galectin-9, which dampen and suppress T-cell responses and may even cause T-cell apoptosis upon interaction with their receptors on T cells (259, 264). Moreover, there is extensive crosstalk between MDSC and Treg, further enhancing immunosuppression (265, 266).
In GBM, MDSC are a major immunosuppressive component of the TME (267). They are also significantly increased in the blood of patients, associated with a higher concentration of the MDSC-specific protein S100A8/9 and arginase activity in the serum (268). More recently, Alban et al. confirmed the presence of MDSC in the blood of GBM patients, while this cell population was completely absent in low-grade glioma patients and healthy individuals (269). In the tumor, MDSC can be found in close proximity to cancer stem cells, and their presence correlates negatively with OS (270). Analogous to Treg, MDSC appear to be recruited to the brain tumor in an early phase and have already been detected in premalignant lesions in a mouse model (271).
Early studies have confirmed the immunosuppressive activity of MDSC in glioma patients and shown that their depletion can restore the disturbed T-cell function (272). Depletion of MDSC with low-dose 5-fluorouracil (5-FU) resulted in prolonged survival in a glioma mouse model (270), and in a clinical study, it has been documented that metronomic capecitabine reduces MDSC, associated with an increase in T-cell infiltrates in the tumors (273). Moreover, their frequency correlates negatively with the response to immunotherapy as reviewed by Stewart and Smyth (274).
Thus, there is evidence for a prominent role of MDSC-mediated immunosuppression in GBM. It has been proposed that targeting MDSC might improve the response to other therapeutic approaches, particularly immunotherapy. The following main targeting strategies are considered: 1) depletion of MDSC, 2) blockage of their migration towards the tumor site, 3) abrogation of their immunosuppressive activity, and 4) pushing them into differentiation towards mature myeloid cells (258). Gao et al. have recently summarized all studies and agents targeting MDSC (275). Therefore, in the following section, only a few examples of the intervention with the immunosuppressive mechanisms of MDSC are presented.
The depletion of MDSC can either be achieved directly by low-dose chemotherapy with, e.g., 5-FU (270), capecitabine (273), or ibudilast (269), or indirectly by promoting their differentiation to either M1 macrophages by docetaxel (276) or towards DC with paclitaxel (277). Full maturation of MDSC can be induced by all-trans retinoic acid [ATRA; (278)]. Blocking of the CSF-1 receptor (CSF-1R) signaling via pexidartinib reduced MDSC as well as M2 macrophages (279), and STAT3 inhibitors can reduce the number of MDSC and interfere with their functional activity (271). Furthermore, because MDSC share several mechanisms of immunosuppression with Treg such as the utilization of checkpoint regulatory pathways, the same inhibiting strategies can be used (see Regulatory T Cells section).
Evidence for a role of MDSC for efficacy of DCV comes from a study on small cell lung cancer patients, who were vaccinated with p53-loaded DC together with ATRA treatment. DCV alone did not change the frequency of MDSC, which however was reduced twofold by ATRA. Moreover, the combination therapy resulted in a significant increase of specific immune responses (280). In GBM, standard radiochemotherapy has been reported to reduce MDSC in a mouse model (281), but effects have not yet been assessed in humans in detail. Nevertheless, there may be a rationale for depletion of MDSC or interference with their activity together with DCV.
Macrophages display a high plasticity in response to microenvironmental cues, allowing them to acquire distinct phenotypes and perform diverse functions. In general, macrophages are divided into classically activated M1 macrophages and alternatively activated M2 macrophages (282, 283). Differentiation of monocytes to M1 macrophages is induced by GM-CSF and proinflammatory cytokines like IFNγ and TNFα, whereas M2 macrophages differentiate in the presence of M-CSF and anti-inflammatory stimuli (284–288). M2 macrophages can be further subdivided into M2a, M2b, and M2c macrophages (289, 290), with IL-4 and IL-13, immune complexes and TLR agonists, and IL-10, TGF-β, and glucocorticoids representing the major polarizing factors, respectively. Functionally, M1 macrophages are proinflammatory, promoting immunity, wound healing, and tissue regeneration (291, 292). In contrast, M2 macrophages are rather anti-inflammatory; and involvement in wound healing, tissue repair and TH2 polarization (293–295), phagocytic and immunomodulatory activity (293, 296–299), and involvement in immunosuppression and angiogenesis (289, 300, 301) have been reported for the M2a, M2b, and M2c subtypes, respectively.
TAM are components of the TME. They frequently exhibit an M2-like phenotype and generally act pro-tumorally, and their presence is associated with poor prognosis. They originate from bone marrow-derived circulating monocytes and accumulate in the tumor due to the presence of M-CSF, GM-CSF, and CCL2 as well as other factors (302, 303). In the tumor, they differentiate into anti-inflammatory M2-like TAM by tumor and TME-derived factors like M-CSF, IL-4, IL-10, and TGF-β. Depending on the local conditions, they polarize towards the M2a, M2b, or M2c subtypes (296, 302, 304–306).
Besides other tumor promoting activities, the anti-inflammatory M2-like TAM promote immunosuppression. They express immunosuppressive cytokines such as TGF-β and IL-10, which not only inhibit T-cell proliferation and function but also contribute together with chemokines, such as CCL20, CCL22, and CXCL12, to an extensive crosstalk with Treg and MDSC, further enhancing immunosuppression (307–314). Moreover, similar to Treg and MDSC, depletion of amino acids (tryptophan and arginine) and expression of ligands of immune checkpoint regulatory pathways contribute to the immunosuppressive activity of TAM (201, 312, 315–320).
In GBM, TAM make up approximately 30%–50% of all cells in the TME (321–323). They are associated with poor prognosis and tumor progression (324–327). Besides infiltrating TAM, in GBM, there is a secondary population of brain-resident monocytic cells, the microglia, which may also be modulated in its activity by the tumor and other cells of the TME (328–331). Bone marrow-derived monocytes are recruited to the brain by cytokines and chemokines such as IL-1β and CCL2 (313, 332), where they mainly differentiate towards anti-inflammatory M2-like macrophages, particularly towards M2c (330, 333). Moreover, the polarization towards M2-like macrophages is an evolving process, which is highly dependent on a hypoxic TME. With increasing hypoxia, the cells polarize more and more towards the M2 subtype (334–336). In line with this observation, there appears to be an increase in M2-like macrophages in recurrent compared with primary tumors, especially in the mesenchymal subtype due to NF-1 deficiency (296, 337).
Due to their importance in promoting tumor progression and immunosuppression, therapeutic targeting of TAM and their function is being attempted. Choi et al. have summarized different molecular targets in preclinical and clinical investigations (338). These include blocking recruitment of TAM into the tumor by targeting chemokines such as CCL2 and CXCL12, which resulted in a reduction in tumor size. Further approaches target the functional characteristics of macrophages, by utilizing small-molecule inhibitors for PI3K, Ras/MAPK signaling, or the IDO pathway, leading, e.g., to reduced IL-10 secretion from M2 macrophages.
A different approach is the reversion of the M2 phenotype to the proinflammatory M1 phenotype by using, e.g., oncolytic virotherapy. Van den Bossche et al. cultured human M2 macrophages with cancer stem cells infected with Delta24-RGD virus and observed a transition towards the M1 phenotype of the cells. Patients treated with this viral particles showed an increase in M1 macrophages in their tumor tissue compared with untreated controls (339). Saha et al. demonstrated that the application of an oncolytic herpes simplex virus (oHSV) expressing IL-12 in combination with antibodies against CTLA-4 and PD-L1 shows a regression of almost all tumors in two GBM mouse models by inducing an effector T-cell influx and an increase in the M1 phenotype of macrophages (340). Thus, local immunostimulatory conditions in the tumor may alter the immunosuppressiveness of the TME. Indeed, DCV in a mouse model revealed a reduction of TAM and MDSC after treatment (341). Moreover, Dammeijer et al. reported that the kinase inhibitor PLX3397 (pexidartinib), targeting CSF-1R signaling, results in a reduction of TAM in a mouse model for malignant mesothelioma but did not influence survival. However, when combined with DCV, survival was increased, which was associated with a reduction of TAM and an increase in effector T-cell infiltration (342). These results suggest that combining DCV with depletion, blocking, or re-polarization of TAM may improve efficacy of the treatment of GBM.
T-cell activation by DC for antitumoral immunity requires the differentiation and expansion of antigen-experienced effector memory T cells (343), with interferon-γ (IFNγ)-producing THelper1 (TH1) cells being required for efficient induction of antitumoral effector cytotoxic T cells (121). Antigen-experienced effector memory T cells are characterized by the expression of the surface markers CD45RO and CD69 (and CD103 on tissue resident cells) in the absence of CCR7 (C-C chemokine receptor type 7) and CD62L (343–345). Effector function of these TH1 and Tc1 cells is defined by the expression of the transcription factors (TF) T-bet and Eomes, and the effector potency depends on a delicate balance of these two TFs (346). Highly potent effector T cells present with a T-bethigh/Eomeslow profile. They produce high levels of IFNγ, perforin, and granzyme B (347).
The activity of these effector T cells needs to be tightly regulated in order to prevent excessive immune reactions and uncontrolled inflammation, which may cause destruction of healthy tissue. Therefore, in the course of activation, T cells upregulate immune checkpoint receptors on their surface, including PD-1, CTLA-4, Lag-3, and Tim-3 that upon interaction with their ligands provide a negative feedback to attenuate proliferation and function of the activated T cells, thereby preventing overreactions (348, 349).
In various cancers, including GBM, these immune checkpoint mechanisms, which normally promote self-tolerance and protect against autoimmunity, contribute to tumor immune escape (348–350). Tumor cells or components of the TME such as TAM, Treg, and MDSC express ligands of immune checkpoint receptors, which upon interaction with their receptors on tumor infiltrating T cells cause partial dysfunction of the T cells, a process also referred to as “T-cell exhaustion,” because similarly dysfunctional T cells can be found in chronic infections and after repetitive T-cell stimulation (348, 349, 351). Compared with CD8+ T cells generated in response to acute infections, such as an acute CMV infection, exhausted antigen-specific CD8+ T cells generated in response to chronic infections or cancer are characterized by reduced proliferation rates, diminished cytotoxicity, and lower cytokine production. Additionally, they express non-transiently checkpoint receptors. T-cell dysfunction has been observed in an early stage of cancer, and it becomes more severe upon tumor progression (352, 353), protecting the tumor cells from the effector mechanisms of the T cells.
In GBM, PD-1/PD-L1 is the best characterized immune checkpoint mechanism. In the majority of tumors, cells of the TME (354) as well as tumor cells express PD-L1 (355), although expression may be restricted to a minor subpopulation of tumor cells only [0%–87%; median 2.8%; (356)]. The respective receptor, PD-1, is expressed on tumor-infiltrating CD4+ and CD8+ T cells (355). Expression on the tumor-infiltrating T cells is higher than on their counterparts in blood (346, 357), and they are functionally impaired (346, 358). Besides PD-1, expression of Tim-3, Lag-3, and CTLA-4 has also been observed on infiltrating T cells in GBM (359).
The dysfunctional state of the T cells may not be permanent but appears to have to be maintained by receptor–ligand interactions, in contrast to T-cell senescence, which is not reversible (353, 360). When the respective immune checkpoint receptors are blocked, e.g., by receptor or ligand-specific monoclonal antibodies, T cells can be reinvigorated: their function and proliferation are restored (359, 361). Indeed, in many tumor entities, application of monoclonal antibodies blocking immune checkpoint receptors or their ligands has resulted in a survival benefit for the patients (361).
In mouse models of GBM, blocking of CTLA-4, PD-1/PD-L1, and TIGIT has been shown to result in increased survival, frequently associated with depletion of immunosuppressive cells and an influx of effector T cells into the tumor (222, 228–231). Thus, there appears to be an intrinsic antitumoral immune response in cancer patients, which can be enhanced by interference with the immune checkpoint pathways. Although this type of immunotherapy results in durable responses in many tumors, this is only true for a fraction of patients (20%–50%, depending on the cancer type), and therapy is associated with severe immune-related adverse events (362). In contrast to many other tumors, however, in GBM, interference with immune checkpoint pathways has not been successful. No survival benefit has been reported so far in several clinical trials (363–367), except for one of three trials applying an anti-PD-1 monoclonal antibody in a neoadjuvant setting (368–370). The reason for the therapeutic failure of immune checkpoint interference in GBM is currently unknown. Whether other checkpoint regulators or possibly other immune escape mechanisms play a more prominent role in GBM than in other tumors and therefore the loss of PD-1 signaling due to blocking is irrelevant remains to be determined. However, reinvigoration of exhausted T cells may also have limitations in GBM. It depends on how terminally differentiated/exhausted T cells are. The function of exhausted T cells showing a T-betlow/Eomeshigh TF expression and a concomitant expression of multiple immune checkpoint receptors cannot be restored (346). It is further essential to distinguish between progenitor exhausted T cells, which express the TF TCF1 and the TCF1−/Tim-3+ terminally exhausted T cells, which co-express the TF TOX, vital for their persistence in the tumor environment with chronic antigen stimulation, with the later population not being re-invigoratable, thus not responding to checkpoint blockade (352, 353, 371). It has been suggested that immune checkpoint inhibition improves OS by overcoming the exhaustion state of tumor-infiltrating T cells resulting in an increased effector response, but functional proof about that is still missing up to date. Although several studies have described an increase in TCF1+ tumor-infiltrating T cells, it is unclear whether they are re-invigorated from the exhausted cells, represent new “non-exhausted” or “non-terminally differentiated” clonotypes, or are recruited from the periphery following immune checkpoint inhibition.
Furthermore, immune checkpoint interference can only enhance but not induce antitumoral responses. The low mutational load [(94); for review, see (95)] and the low frequency [20%–30% (50, 83)] of preexisting antitumoral T-cell responses in GBM patients may therefore limit efficacy of immune checkpoint interference as well. In agreement with this, PD-1 or PD-L1 blockage combined with DCV in mouse models resulted in CD8+ T cell-dependent long-term survival, which was not observed with the respective monotherapies (372, 373). Similar results have been obtained by Wang et al., who vaccinated GBM patients with personalized TAA-pulsed DC combined with low-dose cyclophosphamide, poly(IC), imiquimod, and anti-PD-1 antibody, which induced antigen-specific CD4+ and CD8+ T-cell responses, which were associated with a favorable outcome when compared with the respective monotherapies (86). Moreover, Jan et al. and Yao et al. reported for DCV that a lower PD-1+:CD8+ ratio in TIL as well as in blood lymphocytes is associated with longer survival (78, 81), and Fong et al. described an association of decreased CTLA-4 expression with survival after DCV (93). Thus, there appears to be a rationale for combining DCV and blocking of immune checkpoint regulatory pathways to increase efficacy. DCV itself might be able to trigger the expansion of the above-described TCF1+/TOX− neoantigen-specific T cells from the periphery or even the tumor site and hence enhance tumor killing and survival.
Overall, targeting any one of the three immunosuppressive cell populations in the TME of GBM as well as the immune checkpoint regulatory pathways (Figure 2) appears to represent a promising approach by itself, but in particular in combination with DCV.
Even after more than 10 years of DCV in GBM and after more than 1,000 patients having been vaccinated, it is still difficult to draw conclusions as to the efficacy of DCV. However, there are promising results urging to further develop it as a therapeutic tool, which will require not only optimizing the DC vaccines in respect to target antigen selection, preparation of the cells, and integration of DCV into other treatment regimens but also dosing and scheduling of the vaccination(s). Future vaccination strategies will have also to take into account immunosuppression in GBM and the means to overcome it, which are now becoming increasingly available, to improve efficacy in GBM patients.
Both authors contributed equally to the article and approved the submitted version.
This study was supported by a grant from the Federal Ministry of Education and Research (BMBF; grant #01KG1242).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors are grateful to Marc Kuballa for helpful discussion and for critically reading the manuscript.
1. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 Who Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
2. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in Pdgfra, Idh1, Egfr, and Nf1. Cancer Cell (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
3. Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, et al. The Epidemiology of Glioma in Adults: A "State of the Science" Review. Neuro Oncol (2014) 16:896–913. doi: 10.1093/neuonc/nou087
4. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy Plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N Engl J Med (2005) 352:987–96. doi: 10.1056/NEJMoa043330
5. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of Radiotherapy With Concomitant and Adjuvant Temozolomide Versus Radiotherapy Alone on Survival in Glioblastoma in a Randomised Phase Iii Study: 5-Year Analysis of the Eortc-Ncic Trial. Lancet Oncol (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
6. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. Mgmt Gene Silencing and Benefit From Temozolomide in Glioblastoma. N Engl J Med (2005) 352:997–1003. doi: 10.1056/NEJMoa043331
7. Herrlinger U, Tzaridis T, Mack F, Steinbach JP, Schlegel U, Sabel M, et al. Lomustine-Temozolomide Combination Therapy Versus Standard Temozolomide Therapy in Patients With Newly Diagnosed Glioblastoma With Methylated Mgmt Promoter (Ceteg/Noa-09): A Randomised, Open-Label, Phase 3 Trial. Lancet (2019) 393:678–88. doi: 10.1016/S0140-6736(18)31791-4
8. Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA (2017) 318:2306–16. doi: 10.1001/jama.2017.18718
9. Fisher JP, Adamson DC. Current Fda-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines (2021) 9:324. doi: 10.3390/biomedicines9030324
10. Cruz Da Silva E, Mercier MC, Etienne-Selloum N, Dontenwill M, Choulier L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases Ii, Iii, Iv Clinical Trials. Cancers (Basel) (2021) 13:1795. doi: 10.3390/cancers13081795
11. Yin X, Chen S, Eisenbarth SC. Dendritic Cell Regulation of T Helper Cells. Annu Rev Immunol (2021) 39:759–90. doi: 10.1146/annurev-immunol-101819-025146
12. Cabeza-Cabrerizo M, Cardoso A, Minutti CM, Pereira da Costa M, Reis ESC. Dendritic Cells Revisited. Annu Rev Immunol (2021) 39:131–66. doi: 10.1146/annurev-immunol-061020-053707
13. Wang Y, Xiang Y, Xin VW, Wang XW, Peng XC, Liu XQ, et al. Dendritic Cell Biology and its Role in Tumor Immunotherapy. J Hematol Oncol (2020) 13:107. doi: 10.1186/s13045-020-00939-6
14. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic Cells in Cancer Immunology and Immunotherapy. Nat Rev Immunol (2020) 20:7–24. doi: 10.1038/s41577-019-0210-z
15. Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, et al. Vaccination of Patients With B-Cell Lymphoma Using Autologous Antigen-Pulsed Dendritic Cells. Nat Med (1996) 2:52–8. doi: 10.1038/nm0196-52
16. Dhodapkar MV, Steinman RM, Sapp M, Desai H, Fossella C, Krasovsky J, et al. Rapid Generation of Broad T-Cell Immunity in Humans After a Single Injection of Mature Dendritic Cells. J Clin Invest (1999) 104:173–80. doi: 10.1172/JCI6909
17. Small EJ, Schellhammer PF, Higano CS, Redfern CH, Nemunaitis JJ, Valone FH, et al. Placebo-Controlled Phase Iii Trial of Immunologic Therapy With Sipuleucel-T (Apc8015) in Patients With Metastatic, Asymptomatic Hormone Refractory Prostate Cancer. J Clin Oncol (2006) 24:3089–94. doi: 10.1200/JCO.2005.04.5252
18. Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T Immunotherapy for Castration-Resistant Prostate Cancer. N Engl J Med (2010) 363:411–22. doi: 10.1056/NEJMoa1001294
19. U.S. Food and Drug Administration. PROVENGE (Sipuleucel-T) (2021). Available at: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/provenge-sipuleucel-t (Accessed October 7th, 2021).
20. Liau LM, Black KL, Prins RM, Sykes SN, DiPatre PL, Cloughesy TF, et al. Treatment of Intracranial Gliomas With Bone Marrow-Derived Dendritic Cells Pulsed With Tumor Antigens. J Neurosurg (1999) 90:1115–24. doi: 10.3171/jns.1999.90.6.1115
21. Heimberger AB, Crotty LE, Archer GE, McLendon RE, Friedman A, Dranoff G, et al. Bone Marrow-Derived Dendritic Cells Pulsed With Tumor Homogenate Induce Immunity Against Syngeneic Intracerebral Glioma. J Neuroimmunol (2000) 103:16–25. doi: 10.1016/S0165-5728(99)00172-1
22. Insug O, Ku G, Ertl HC, Blaszczyk-Thurin M. A Dendritic Cell Vaccine Induces Protective Immunity to Intracranial Growth of Glioma. Anticancer Res (2002) 22:613–21.
23. Prins RM, Odesa SK, Liau LM. Immunotherapeutic Targeting of Shared Melanoma-Associated Antigens in a Murine Glioma Model. Cancer Res (2003) 63:8487–91.
24. Ciesielski MJ, Apfel L, Barone TA, Castro CA, Weiss TC, Fenstermaker RA. Antitumor Effects of a Xenogeneic Survivin Bone Marrow Derived Dendritic Cell Vaccine Against Murine Gl261 Gliomas. Cancer Immunol Immunother (2006) 55:1491–503. doi: 10.1007/s00262-006-0138-6
25. Aoki H, Mizuno M, Natsume A, Tsugawa T, Tsujimura K, Takahashi T, et al. Dendritic Cells Pulsed With Tumor Extract-Cationic Liposome Complex Increase the Induction of Cytotoxic T Lymphocytes in Mouse Brain Tumor. Cancer Immunol Immunother (2001) 50:463–8. doi: 10.1007/s002620100220
26. Ni HT, Spellman SR, Jean WC, Hall WA, Low WC. Immunization With Dendritic Cells Pulsed With Tumor Extract Increases Survival of Mice Bearing Intracranial Gliomas. J Neurooncol (2001) 51:1–9. doi: 10.1023/a:1006452726391
27. Zhu X, Lu C, Xiao B, Qiao J, Sun Y. An Experimental Study of Dendritic Cells-Mediated Immunotherapy Against Intracranial Gliomas in Rats. J Neurooncol (2005) 74:9–17. doi: 10.1007/s11060-004-3339-x
28. Pellegatta S, Poliani PL, Corno D, Menghi F, Ghielmetti F, Suarez-Merino B, et al. Neurospheres Enriched in Cancer Stem-Like Cells are Highly Effective in Eliciting a Dendritic Cell-Mediated Immune Response Against Malignant Gliomas. Cancer Res (2006) 66:10247–52. doi: 10.1158/0008-5472.CAN-06-2048
29. Pellegatta S, Poliani PL, Corno D, Grisoli M, Cusimano M, Ubiali F, et al. Dendritic Cells Pulsed With Glioma Lysates Induce Immunity Against Syngeneic Intracranial Gliomas and Increase Survival of Tumor-Bearing Mice. Neurol Res (2006) 28:527–31. doi: 10.1179/016164106X116809
30. Ciesielski MJ, Kozbor D, Castanaro CA, Barone TA, Fenstermaker RA. Therapeutic Effect of a T Helper Cell Supported Ctl Response Induced by a Survivin Peptide Vaccine Against Murine Cerebral Glioma. Cancer Immunol Immunother (2008) 57:1827–35. doi: 10.1007/s00262-008-0510-9
31. Fujita M, Zhu X, Ueda R, Sasaki K, Kohanbash G, Kastenhuber ER, et al. Effective Immunotherapy Against Murine Gliomas Using Type 1 Polarizing Dendritic Cells–Significant Roles of Cxcl10. Cancer Res (2009) 69:1587–95. doi: 10.1158/0008-5472.CAN-08-2915
32. Mac Keon S, Ruiz MS, Gazzaniga S, Wainstok R. Dendritic Cell-Based Vaccination in Cancer: Therapeutic Implications Emerging From Murine Models. Front Immunol (2015) 6:243. doi: 10.3389/fimmu.2015.00243
33. Liau LM, Black KL, Martin NA, Sykes SN, Bronstein JM, Jouben-Steele L, et al. Treatment of a Patient by Vaccination With Autologous Dendritic Cells Pulsed With Allogeneic Major Histocompatibility Complex Class I-Matched Tumor Peptides. Case Rep Neurosurg Focus (2000) 9:e8. doi: 10.3171/foc.2000.9.6.9
34. Kikuchi T, Akasaki Y, Irie M, Homma S, Abe T, Ohno T. Results of a Phase I Clinical Trial of Vaccination of Glioma Patients With Fusions of Dendritic and Glioma Cells. Cancer Immunol Immunother (2001) 50:337–44. doi: 10.1007/s002620100205
35. Yu JS, Wheeler CJ, Zeltzer PM, Ying H, Finger DN, Lee PK, et al. Vaccination of Malignant Glioma Patients With Peptide-Pulsed Dendritic Cells Elicits Systemic Cytotoxicity and Intracranial T-Cell Infiltration. Cancer Res (2001) 61:842–7.
36. Wheeler CJ, Black KL, Liu G, Ying H, Yu JS, Zhang W, et al. Thymic Cd8+ T Cell Production Strongly Influences Tumor Antigen Recognition and Age-Dependent Glioma Mortality. J Immunol (2003) 171:4927–33. doi: 10.4049/jimmunol.171.9.4927
37. Yamanaka R, Abe T, Yajima N, Tsuchiya N, Homma J, Kobayashi T, et al. Vaccination of Recurrent Glioma Patients With Tumour Lysate-Pulsed Dendritic Cells Elicits Immune Responses: Results of a Clinical Phase I/Ii Trial. Br J Cancer (2003) 89:1172–9. doi: 10.1038/sj.bjc.6601268
38. Caruso DA, Orme LM, Neale AM, Radcliff FJ, Amor GM, Maixner W, et al. Results of a Phase 1 Study Utilizing Monocyte-Derived Dendritic Cells Pulsed With Tumor RNA in Children and Young Adults With Brain Cancer. Neuro Oncol (2004) 6:236–46. doi: 10.1215/S1152851703000668
39. De Vleeschouwer S, Van Calenbergh F, Demaerel P, Flamen P, Rutkowski S, Kaempgen E, et al. Transient Local Response and Persistent Tumor Control in a Child With Recurrent Malignant Glioma: Treatment With Combination Therapy Including Dendritic Cell Therapy. Case Rep J Neurosurg (2004) 100:492–7. doi: 10.3171/ped.2004.100.5.0492
40. Kikuchi T, Akasaki Y, Abe T, Fukuda T, Saotome H, Ryan JL, et al. Vaccination of Glioma Patients With Fusions of Dendritic and Glioma Cells and Recombinant Human Interleukin 12. J Immunother (2004) 27:452–9. doi: 10.1097/00002371-200411000-00005
41. Rutkowski S, De Vleeschouwer S, Kaempgen E, Wolff JE, Kuhl J, Demaerel P, et al. Surgery and Adjuvant Dendritic Cell-Based Tumour Vaccination for Patients With Relapsed Malignant Glioma, a Feasibility Study. Br J Cancer (2004) 91:1656–62. doi: 10.1038/sj.bjc.6602195
42. Wheeler CJ, Das A, Liu G, Yu JS, Black KL. Clinical Responsiveness of Glioblastoma Multiforme to Chemotherapy After Vaccination. Clin Cancer Res (2004) 10:5316–26. doi: 10.1158/1078-0432.CCR-04-0497
43. Yu JS, Liu G, Ying H, Yong WH, Black KL, Wheeler CJ. Vaccination With Tumor Lysate-Pulsed Dendritic Cells Elicits Antigen-Specific, Cytotoxic T-Cells in Patients With Malignant Glioma. Cancer Res (2004) 64:4973–9. doi: 10.1158/0008-5472.CAN-03-3505
44. Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, et al. Dendritic Cell Vaccination in Glioblastoma Patients Induces Systemic and Intracranial T-Cell Responses Modulated by the Local Central Nervous System Tumor Microenvironment. Clin Cancer Res (2005) 11:5515–25. doi: 10.1158/1078-0432.CCR-05-0464
45. Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, Kobayashi T, et al. Clinical Evaluation of Dendritic Cell Vaccination for Patients With Recurrent Glioma: Results of a Clinical Phase I/Ii Trial. Clin Cancer Res (2005) 11:4160–7. doi: 10.1158/1078-0432.CCR-05-0120
46. Khan JA, Yaqin S. Dendritic Cell Therapy With Improved Outcome in Glioma Multiforme–a Case Report. J Zhejiang Univ Sci B (2006) 7:114–7. doi: 10.1631/jzus.2006.B0114
47. Okada H, Lieberman FS, Walter KA, Lunsford LD, Kondziolka DS, Bejjani GK, et al. Autologous Glioma Cell Vaccine Admixed With Interleukin-4 Gene Transfected Fibroblasts in the Treatment of Patients With Malignant Gliomas. J Transl Med (2007) 5:67. doi: 10.1186/1479-5876-5-67
48. de Vleeschouwer S, Fieuws S, Rutkowski S, Van Calenbergh F, Van Loon J, Goffin J, et al. Postoperative Adjuvant Dendritic Cell-Based Immunotherapy in Patients With Relapsed Glioblastoma Multiforme. Clin Cancer Res (2008) 14:3098–104. doi: 10.1158/1078-0432.CCR-07-4875
49. Walker DG, Laherty R, Tomlinson FH, Chuah T, Schmidt C. Results of a Phase I Dendritic Cell Vaccine Trial for Malignant Astrocytoma: Potential Interaction With Adjuvant Chemotherapy. J Clin Neurosci (2008) 15:114–21. doi: 10.1016/j.jocn.2007.08.007
50. Wheeler CJ, Black KL, Liu G, Mazer M, Zhang XX, Pepkowitz S, et al. Vaccination Elicits Correlated Immune and Clinical Responses in Glioblastoma Multiforme Patients. Cancer Res (2008) 68:5955–64. doi: 10.1158/0008-5472.CAN-07-5973
51. Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE 2nd, Lally-Goss D, et al. An Epidermal Growth Factor Receptor Variant Iii-Targeted Vaccine is Safe and Immunogenic in Patients With Glioblastoma Multiforme. Mol Cancer Ther (2009) 8:2773–9. doi: 10.1158/1535-7163.MCT-09-0124
52. Ardon H, De Vleeschouwer S, Van Calenbergh F, Claes L, Kramm CM, Rutkowski S, et al. Adjuvant Dendritic Cell-Based Tumour Vaccination for Children With Malignant Brain Tumours. Pediatr Blood Cancer (2010) 54:519–25. doi: 10.1002/pbc.22319
53. Ardon H, Van Gool S, Lopes IS, Maes W, Sciot R, Wilms G, et al. Integration of Autologous Dendritic Cell-Based Immunotherapy in the Primary Treatment for Patients With Newly Diagnosed Glioblastoma Multiforme: A Pilot Study. J Neurooncol (2010) 99:261–72. doi: 10.1007/s11060-010-0131-y
54. Chang CN, Huang YC, Yang DM, Kikuta K, Wei KJ, Kubota T, et al. A Phase I/Ii Clinical Trial Investigating the Adverse and Therapeutic Effects of a Postoperative Autologous Dendritic Cell Tumor Vaccine in Patients With Malignant Glioma. J Clin Neurosci (2011) 18:1048–54. doi: 10.1016/j.jocn.2010.11.034
55. Fadul CE, Fisher JL, Hampton TH, Lallana EC, Li Z, Gui J, et al. Immune Response in Patients With Newly Diagnosed Glioblastoma Multiforme Treated With Intranodal Autologous Tumor Lysate-Dendritic Cell Vaccination After Radiation Chemotherapy. J Immunother (2011) 34:382–9. doi: 10.1097/CJI.0b013e318215e300
56. Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of Cd8+ T-Cell Responses Against Novel Glioma-Associated Antigen Peptides and Clinical Activity by Vaccinations With {Alpha}-Type 1 Polarized Dendritic Cells and Polyinosinic-Polycytidylic Acid Stabilized by Lysine and Carboxymethylcellulose in Patients With Recurrent Malignant Glioma. J Clin Oncol (2011) 29:330–6. doi: 10.1200/JCO.2010.30.7744
57. Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene Expression Profile Correlates With T-Cell Infiltration and Relative Survival in Glioblastoma Patients Vaccinated With Dendritic Cell Immunotherapy. Clin Cancer Res (2011) 17:1603–15. doi: 10.1158/1078-0432.CCR-10-2563
58. Akiyama Y, Oshita C, Kume A, Iizuka A, Miyata H, Komiyama M, et al. Alpha-Type-1 Polarized Dendritic Cell-Based Vaccination in Recurrent High-Grade Glioma: A Phase I Clinical Trial. BMC Cancer (2012) 12:623. doi: 10.1186/1471-2407-12-623
59. Ardon H, Van Gool SW, Verschuere T, Maes W, Fieuws S, Sciot R, et al. Integration of Autologous Dendritic Cell-Based Immunotherapy in the Standard of Care Treatment for Patients With Newly Diagnosed Glioblastoma: Results of the Hgg-2006 Phase I/Ii Trial. Cancer Immunol Immunother (2012) 61:2033–44. doi: 10.1007/s00262-012-1261-1
60. Cho DY, Yang WK, Lee HC, Hsu DM, Lin HL, Lin SZ, et al. Adjuvant Immunotherapy With Whole-Cell Lysate Dendritic Cells Vaccine for Glioblastoma Multiforme: A Phase Ii Clinical Trial. World Neurosurg (2012) 77:736–44. doi: 10.1016/j.wneu.2011.08.020
61. Iwami K, Shimato S, Ohno M, Okada H, Nakahara N, Sato Y, et al. Peptide-Pulsed Dendritic Cell Vaccination Targeting Interleukin-13 Receptor Alpha2 Chain in Recurrent Malignant Glioma Patients With Hla-a*24/a*02 Allele. Cytotherapy (2012) 14:733–42. doi: 10.3109/14653249.2012.666633
62. Jie X, Hua L, Jiang W, Feng F, Feng G, Hua Z. Clinical Application of a Dendritic Cell Vaccine Raised Against Heat-Shocked Glioblastoma. Cell Biochem Biophys (2012) 62:91–9. doi: 10.1007/s12013-011-9265-6
63. Valle RD, de Cerio AL, Inoges S, Tejada S, Pastor F, Villanueva H, et al. Dendritic Cell Vaccination in Glioblastoma After Fluorescence-Guided Resection. World J Clin Oncol (2012) 3:142–9. doi: 10.5306/wjco.v3.i11.142
64. Lasky JL 3rd, Panosyan EH, Plant A, Davidson T, Yong WH, Prins RM, et al. Autologous Tumor Lysate-Pulsed Dendritic Cell Immunotherapy for Pediatric Patients With Newly Diagnosed or Recurrent High-Grade Gliomas. Anticancer Res (2013) 33:2047–56.
65. Pellegatta S, Eoli M, Frigerio S, Antozzi C, Bruzzone MG, Cantini G, et al. The Natural Killer Cell Response and Tumor Debulking are Associated With Prolonged Survival in Recurrent Glioblastoma Patients Receiving Dendritic Cells Loaded With Autologous Tumor Lysates. Oncoimmunology (2013) 2:e23401. doi: 10.4161/onci.23401
66. Phuphanich S, Wheeler CJ, Rudnick JD, Mazer M, Wang H, Nuno MA, et al. Phase I Trial of a Multi-Epitope-Pulsed Dendritic Cell Vaccine for Patients With Newly Diagnosed Glioblastoma. Cancer Immunol Immunother (2013) 62:125–35. doi: 10.1007/s00262-012-1319-0
67. Prins RM, Wang X, Soto H, Young E, Lisiero DN, Fong B, et al. Comparison of Glioma-Associated Antigen Peptide-Loaded Versus Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination in Malignant Glioma Patients. J Immunother (2013) 36:152–7. doi: 10.1097/CJI.0b013e3182811ae4
68. Vik-Mo EO, Nyakas M, Mikkelsen BV, Moe MC, Due-Tonnesen P, Suso EM, et al. Therapeutic Vaccination Against Autologous Cancer Stem Cells With Mrna-Transfected Dendritic Cells in Patients With Glioblastoma. Cancer Immunol Immunother (2013) 62:1499–509. doi: 10.1007/s00262-013-1453-3
69. Olin MR, Low W, McKenna DH, Haines SJ, Dahlheimer T, Nascene D, et al. Vaccination With Dendritic Cells Loaded With Allogeneic Brain Tumor Cells for Recurrent Malignant Brain Tumors Induces a Cd4(+)Il17(+) Response. J Immunother Cancer (2014) 2:4. doi: 10.1186/2051-1426-2-4
70. Hunn MK, Bauer E, Wood CE, Gasser O, Dzhelali M, Ancelet LR, et al. Dendritic Cell Vaccination Combined With Temozolomide Retreatment: Results of a Phase I Trial in Patients With Recurrent Glioblastoma Multiforme. J Neurooncol (2015) 121:319–29. doi: 10.1007/s11060-014-1635-7
71. Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, et al. Tetanus Toxoid and Ccl3 Improve Dendritic Cell Vaccines in Mice and Glioblastoma Patients. Nature (2015) 519:366–9. doi: 10.1038/nature14320
72. Sakai K, Shimodaira S, Maejima S, Udagawa N, Sano K, Higuchi Y, et al. Dendritic Cell-Based Immunotherapy Targeting Wilms’ Tumor 1 in Patients With Recurrent Malignant Glioma. J Neurosurg (2015) 123:989–97. doi: 10.3171/2015.1.JNS141554
73. Akasaki Y, Kikuchi T, Homma S, Koido S, Ohkusa T, Tasaki T, et al. Phase I/Ii Trial of Combination of Temozolomide Chemotherapy and Immunotherapy With Fusions of Dendritic and Glioma Cells in Patients With Glioblastoma. Cancer Immunol Immunother (2016) 65:1499–509. doi: 10.1007/s00262-016-1905-7
74. Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ, et al. Long-Term Survival in Glioblastoma With Cytomegalovirus Pp65-Targeted Vaccination. Clin Cancer Res (2017) 23:1898–909. doi: 10.1158/1078-0432.CCR-16-2057
75. Inoges S, Tejada S, de Cerio AL, Gallego Perez-Larraya J, Espinos J, Idoate MA, et al. A Phase Ii Trial of Autologous Dendritic Cell Vaccination and Radiochemotherapy Following Fluorescence-Guided Surgery in Newly Diagnosed Glioblastoma Patients. J Transl Med (2017) 15:104. doi: 10.1186/s12967-017-1202-z
76. Buchroithner J, Erhart F, Pichler J, Widhalm G, Preusser M, Stockhammer G, et al. Audencel Immunotherapy Based on Dendritic Cells has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase Ii Randomized Trial. Cancers (Basel) (2018) 10:372. doi: 10.3390/cancers10100372
77. Erhart F, Buchroithner J, Reitermaier R, Fischhuber K, Klingenbrunner S, Sloma I, et al. Immunological Analysis of Phase Ii Glioblastoma Dendritic Cell Vaccine (Audencel) Trial: Immune System Characteristics Influence Outcome and Audencel Up-Regulates Th1-Related Immunovariables. Acta Neuropathol Commun (2018) 6:135. doi: 10.1186/s40478-018-0621-2
78. Jan CI, Tsai WC, Harn HJ, Shyu WC, Liu MC, Lu HM, et al. Predictors of Response to Autologous Dendritic Cell Therapy in Glioblastoma Multiforme. Front Immunol (2018) 9:727. doi: 10.3389/fimmu.2018.00727
79. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS, et al. First Results on Survival From a Large Phase 3 Clinical Trial of an Autologous Dendritic Cell Vaccine in Newly Diagnosed Glioblastoma. J Transl Med (2018) 16:142. doi: 10.1186/s12967-018-1507-6
80. Pellegatta S, Eoli M, Cuccarini V, Anghileri E, Pollo B, Pessina S, et al. Survival Gain in Glioblastoma Patients Treated With Dendritic Cell Immunotherapy is Associated With Increased Nk But Not Cd8(+) T Cell Activation in the Presence of Adjuvant Temozolomide. Oncoimmunology (2018) 7:e1412901. doi: 10.1080/2162402X.2017.1412901
81. Yao Y, Luo F, Tang C, Chen D, Qin Z, Hua W, et al. Molecular Subgroups and B7-H4 Expression Levels Predict Responses to Dendritic Cell Vaccines in Glioblastoma: An Exploratory Randomized Phase Ii Clinical Trial. Cancer Immunol Immunother (2018) 67:1777–88. doi: 10.1007/s00262-018-2232-y
82. Eoli M, Corbetta C, Anghileri E, Di Ianni N, Milani M, Cuccarini V, et al. Expansion of Effector and Memory T Cells is Associated With Increased Survival in Recurrent Glioblastomas Treated With Dendritic Cell Immunotherapy. Neurooncol Adv (2019) 1:vdz022. doi: 10.1093/noajnl/vdz022
83. Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC, et al. A Randomized Double-Blind Placebo-Controlled Phase Ii Trial of Dendritic Cell Vaccine Ict-107 in Newly Diagnosed Patients With Glioblastoma. Clin Cancer Res (2019) 25:5799–807. doi: 10.1158/1078-0432.CCR-19-0261
84. Batich KA, Mitchell DA, Healy P, Herndon JE 2nd, Sampson JH. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin Cancer Res (2020) 26:5297–303. doi: 10.1158/1078-0432.CCR-20-1082
85. Mitsuya K, Akiyama Y, Iizuka A, Miyata H, Deguchi S, Hayashi N, et al. Alpha-Type-1 Polarized Dendritic Cell-Based Vaccination in Newly Diagnosed High-Grade Glioma: A Phase Ii Clinical Trial. Anticancer Res (2020) 40:6473–84. doi: 10.21873/anticanres.14669
86. Wang QT, Nie Y, Sun SN, Lin T, Han RJ, Jiang J, et al. Tumor-Associated Antigen-Based Personalized Dendritic Cell Vaccine in Solid Tumor Patients. Cancer Immunol Immunother (2020) 69:1375–87. doi: 10.1007/s00262-020-02496-w
87. Gumrukcu S, Nguyen TX, White RL, Howell GT, Musikanth P. Allogeneic Natural Killer and Cytomegalovirus (Cmv)-Pp65 Pulsed Dendritic Cells Induced Complete Response Through 15 Months in a Patient With Recurrent Glioblastoma: A Case Study. Am J Case Rep (2021) 22:e931030. doi: 10.12659/AJCR.931030
88. Sprooten J, Ceusters J, Coosemans A, Agostinis P, De Vleeschouwer S, Zitvogel L, et al. Trial Watch: Dendritic Cell Vaccination for Cancer Immunotherapy. Oncoimmunology (2019) 8:e1638212. doi: 10.1080/2162402X.2019.1638212
89. Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ. Fluorescence-Guided Surgery With 5-Aminolevulinic Acid for Resection of Malignant Glioma: A Randomised Controlled Multicentre Phase Iii Trial. Lancet Oncol (2006) 7:392–401. doi: 10.1016/S1470-2045(06)70665-9
90. Ruffini PA, Rivoltini L, Silvani A, Boiardi A, Parmiani G. Factors, Including Transforming Growth Factor Beta, Released in the Glioblastoma Residual Cavity, Impair Activity of Adherent Lymphokine-Activated Killer Cells. Cancer Immunol Immunother (1993) 36:409–16. doi: 10.1007/BF01742258
91. Choi SH, Stuckey DW, Pignatta S, Reinshagen C, Khalsa JK, Roozendaal N, et al. Tumor Resection Recruits Effector T Cells and Boosts Therapeutic Efficacy of Encapsulated Stem Cells Expressing Ifnbeta in Glioblastomas. Clin Cancer Res (2017) 23:7047–58. doi: 10.1158/1078-0432.CCR-17-0077
92. Mitchell DA, Sayour EJ, Reap E, Schmittling R, DeLeon G, Norberg P, et al. Severe Adverse Immunologic Reaction in a Patient With Glioblastoma Receiving Autologous Dendritic Cell Vaccines Combined With Gm-Csf and Dose-Intensified Temozolomide. Cancer Immunol Res (2015) 3:320–5. doi: 10.1158/2326-6066.CIR-14-0100
93. Fong B, Jin R, Wang X, Safaee M, Lisiero DN, Yang I, et al. Monitoring of Regulatory T Cell Frequencies and Expression of Ctla-4 on T Cells, Before and After Dc Vaccination, can Predict Survival in Gbm Patients. PloS One (2012) 7:e32614. doi: 10.1371/journal.pone.0032614
94. Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, et al. Mutational Burden, Immune Checkpoint Expression, and Mismatch Repair in Glioma: Implications for Immune Checkpoint Immunotherapy. Neuro Oncol (2017) 19:1047–57. doi: 10.1093/neuonc/nox026
95. Finocchiaro G, Langella T, Corbetta C, Pellegatta S. Hypermutations in Gliomas: A Potential Immunotherapy Target. Discov Med (2017) 23:113–20.
96. Dettling S, Stamova S, Warta R, Schnolzer M, Rapp C, Rathinasamy A, et al. Identification of Crkii, Cfl1, Cntn1, Nme2, and Tkt as Novel and Frequent T-Cell Targets in Human Idh-Mutant Glioma. Clin Cancer Res (2018) 24:2951–62. doi: 10.1158/1078-0432.CCR-17-1839
97. Dutoit V, Herold-Mende C, Hilf N, Schoor O, Beckhove P, Bucher J, et al. Exploiting the Glioblastoma Peptidome to Discover Novel Tumour-Associated Antigens for Immunotherapy. Brain (2012) 135:1042–54. doi: 10.1093/brain/aws042
98. Saikali S, Avril T, Collet B, Hamlat A, Bansard JY, Drenou B, et al. Expression of Nine Tumour Antigens in a Series of Human Glioblastoma Multiforme: Interest of Egfrviii, Il-13ralpha2, Gp100 and Trp-2 for Immunotherapy. J Neurooncol (2007) 81:139–48. doi: 10.1007/s11060-006-9220-3
99. Zhang JG, Kruse CA, Driggers L, Hoa N, Wisoff J, Allen JC, et al. Tumor Antigen Precursor Protein Profiles of Adult and Pediatric Brain Tumors Identify Potential Targets for Immunotherapy. J Neurooncol (2008) 88:65–76. doi: 10.1007/s11060-008-9534-4
100. Finocchiaro G, Pellegatta S. Immunotherapy With Dendritic Cells Loaded With Glioblastoma Stem Cells: From Preclinical to Clinical Studies. Cancer Immunol Immunother (2016) 65:101–9. doi: 10.1007/s00262-015-1754-9
101. Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic Escape After Prolonged Progression-Free Survival With Epidermal Growth Factor Receptor Variant Iii Peptide Vaccination in Patients With Newly Diagnosed Glioblastoma. J Clin Oncol (2010) 28:4722–9. doi: 10.1200/JCO.2010.28.6963
102. De Vleeschouwer S, Arredouani M, Ade M, Cadot P, Vermassen E, Ceuppens JL, et al. Uptake and Presentation of Malignant Glioma Tumor Cell Lysates by Monocyte-Derived Dendritic Cells. Cancer Immunol Immunother (2005) 54:372–82. doi: 10.1007/s00262-004-0615-8
103. Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, Aurrand-Lions M, et al. Homing Phenotypes of Tumor-Specific Cd8 T Cells are Predetermined at the Tumor Site by Crosspresenting Apcs. Immunity (2005) 22:175–84. doi: 10.1016/j.immuni.2004.12.008
104. Erhart F, Weiss T, Klingenbrunner S, Fischhuber K, Reitermaier R, Halfmann A, et al. Spheroid Glioblastoma Culture Conditions as Antigen Source for Dendritic Cell-Based Immunotherapy: Spheroid Proteins are Survival-Relevant Targets But can Impair Immunogenic Interferon Gamma Production. Cytotherapy (2019) 21:643–58. doi: 10.1016/j.jcyt.2019.03.002
105. Garg AD, Vandenberk L, Koks C, Verschuere T, Boon L, Van Gool SW, et al. Dendritic Cell Vaccines Based on Immunogenic Cell Death Elicit Danger Signals and T Cell-Driven Rejection of High-Grade Glioma. Sci Transl Med (2016) 8:328ra27. doi: 10.1126/scitranslmed.aae0105
106. Grauer OM, Sutmuller RP, van Maren W, Jacobs JF, Bennink E, Toonen LW, et al. Elimination of Regulatory T Cells is Essential for an Effective Vaccination With Tumor Lysate-Pulsed Dendritic Cells in a Murine Glioma Model. Int J Cancer (2008) 122:1794–802. doi: 10.1002/ijc.23284
107. Neller MA, Lopez JA, Schmidt CW. Antigens for Cancer Immunotherapy. Semin Immunol (2008) 20:286–95. doi: 10.1016/j.smim.2008.09.006
108. Mitchell DA, Xie W, Schmittling R, Learn C, Friedman A, McLendon RE, et al. Sensitive Detection of Human Cytomegalovirus in Tumors and Peripheral Blood of Patients Diagnosed With Glioblastoma. Neuro Oncol (2008) 10:10–8. doi: 10.1215/15228517-2007-035
109. Cobbs CS, Harkins L, Samanta M, Gillespie GY, Bharara S, King PH, et al. Human Cytomegalovirus Infection and Expression in Human Malignant Glioma. Cancer Res (2002) 62:3347–50.
110. Sallusto F, Lanzavecchia A. Efficient Presentation of Soluble Antigen by Cultured Human Dendritic Cells is Maintained by Granulocyte/Macrophage Colony-Stimulating Factor Plus Interleukin 4 and Downregulated by Tumor Necrosis Factor Alpha. J Exp Med (1994) 179:1109–18. doi: 10.1084/jem.179.4.1109
111. Bol KF, Schreibelt G, Rabold K, Wculek SK, Schwarze JK, Dzionek A, et al. The Clinical Application of Cancer Immunotherapy Based on Naturally Circulating Dendritic Cells. J Immunother Cancer (2019) 7:109. doi: 10.1186/s40425-019-0580-6
112. Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, et al. Pro-Inflammatory Cytokines and Prostaglandins Induce Maturation of Potent Immunostimulatory Dendritic Cells Under Fetal Calf Serum-Free Conditions. Eur J Immunol (1997) 27:3135–42. doi: 10.1002/eji.1830271209
113. Felzmann T, Huttner KG, Breuer SK, Wimmer D, Ressmann G, Wagner D, et al. Semi-Mature Il-12 Secreting Dendritic Cells Present Exogenous Antigen to Trigger Cytolytic Immune Responses. Cancer Immunol Immunother (2005) 54:769–80. doi: 10.1007/s00262-004-0637-2
114. Jurgens B, Hainz U, Fuchs D, Felzmann T, Heitger A. Interferon-Gamma-Triggered Indoleamine 2,3-Dioxygenase Competence in Human Monocyte-Derived Dendritic Cells Induces Regulatory Activity in Allogeneic T Cells. Blood (2009) 114:3235–43. doi: 10.1182/blood-2008-12-195073
115. Legler DF, Krause P, Scandella E, Singer E, Groettrup M. Prostaglandin E2 is Generally Required for Human Dendritic Cell Migration and Exerts its Effect via Ep2 and Ep4 Receptors. J Immunol (2006) 176:966–73. doi: 10.4049/jimmunol.176.2.966
116. Trabanelli S, Lecciso M, Salvestrini V, Cavo M, Ocadlikova D, Lemoli RM, et al. Pge2-Induced Ido1 Inhibits the Capacity of Fully Mature Dcs to Elicit an in Vitro Antileukemic Immune Response. J Immunol Res (2015) 2015:253191. doi: 10.1155/2015/253191
117. Zhou LJ, Tedder TF. Human Blood Dendritic Cells Selectively Express Cd83, a Member of the Immunoglobulin Superfamily. J Immunol (1995) 154:3821–35.
118. Sorg RV, Ozcan Z, Brefort T, Fischer J, Ackermann R, Muller M, et al. Clinical-Scale Generation of Dendritic Cells in a Closed System. J Immunother (2003) 26:374–83. doi: 10.1097/00002371-200307000-00010
119. Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, et al. Restriction of Pd-1 Function by Cis-Pd-L1/Cd80 Interactions is Required for Optimal T Cell Responses. Science (2019) 364:558–66. doi: 10.1126/science.aav7062
120. Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, et al. A Conserved Dendritic-Cell Regulatory Program Limits Antitumour Immunity. Nature (2020) 580:257–62. doi: 10.1038/s41586-020-2134-y
121. Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, Nakui M, et al. Distinct Role of Antigen-Specific T Helper Type 1 (Th1) and Th2 Cells in Tumor Eradication in Vivo. J Exp Med (1999) 190:617–27. doi: 10.1084/jem.190.5.617
122. Yamanaka R, Honma J, Tsuchiya N, Yajima N, Kobayashi T, Tanaka R. Tumor Lysate and Il-18 Loaded Dendritic Cells Elicits Th1 Response, Tumor-Specific Cd8+ Cytotoxic T Cells in Patients With Malignant Glioma. J Neurooncol (2005) 72:107–13. doi: 10.1007/s11060-004-3550-9
123. Wilson EH, Weninger W, Hunter CA. Trafficking of Immune Cells in the Central Nervous System. J Clin Invest (2010) 120:1368–79. doi: 10.1172/JCI41911
124. Sallusto F, Schaerli P, Loetscher P, Schaniel C, Lenig D, Mackay CR, et al. Rapid and Coordinated Switch in Chemokine Receptor Expression During Dendritic Cell Maturation. Eur J Immunol (1998) 28:2760–9. doi: 10.1002/(SICI)1521-4141(199809)28:09<2760::AID-IMMU2760>3.0.CO;2-N
125. Caux C, Ait-Yahia S, Chemin K, de Bouteiller O, Dieu-Nosjean MC, Homey B, et al. Dendritic Cell Biology and Regulation of Dendritic Cell Trafficking by Chemokines. Springer Semin Immunopathol (2000) 22:345–69. doi: 10.1007/s002810000053
126. Romani N, Reider D, Heuer M, Ebner S, Kampgen E, Eibl B, et al. Generation of Mature Dendritic Cells From Human Blood. An Improved Method With Special Regard to Clinical Applicability. J Immunol Methods (1996) 196:137–51. doi: 10.1016/0022-1759(96)00078-6
127. Grauer O, Poschl P, Lohmeier A, Adema GJ, Bogdahn U. Toll-Like Receptor Triggered Dendritic Cell Maturation and Il-12 Secretion are Necessary to Overcome T-Cell Inhibition by Glioma-Associated Tgf-Beta2. J Neurooncol (2007) 82:151–61. doi: 10.1007/s11060-006-9274-2
128. Dudek AM, Martin S, Garg AD, Agostinis P. Immature, Semi-Mature, and Fully Mature Dendritic Cells: Toward a Dc-Cancer Cells Interface That Augments Anticancer Immunity. Front Immunol (2013) 4:438. doi: 10.3389/fimmu.2013.00438
129. de Vries IJ, Lesterhuis WJ, Scharenborg NM, Engelen LP, Ruiter DJ, Gerritsen MJ, et al. Maturation of Dendritic Cells is a Prerequisite for Inducing Immune Responses in Advanced Melanoma Patients. Clin Cancer Res (2003) 9:5091–100.
130. Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-Specific Inhibition of Effector T Cell Function in Humans After Injection of Immature Dendritic Cells. J Exp Med (2001) 193:233–8. doi: 10.1084/jem.193.2.233
131. Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, van Rossum MM, et al. Limited Amounts of Dendritic Cells Migrate Into the T-Cell Area of Lymph Nodes But Have High Immune Activating Potential in Melanoma Patients. Clin Cancer Res (2009) 15:2531–40. doi: 10.1158/1078-0432.CCR-08-2729
132. Lesterhuis WJ, de Vries IJ, Schreibelt G, Lambeck AJ, Aarntzen EH, Jacobs JF, et al. Route of Administration Modulates the Induction of Dendritic Cell Vaccine-Induced Antigen-Specific T Cells in Advanced Melanoma Patients. Clin Cancer Res (2011) 17:5725–35. doi: 10.1158/1078-0432.CCR-11-1261
133. Jouanneau E, Poujol D, Gulia S, Le Mercier I, Blay JY, Belin MF, et al. Dendritic Cells are Essential for Priming But Inefficient for Boosting Antitumour Immune Response in an Orthotopic Murine Glioma Model. Cancer Immunol Immunother (2006) 55:254–67. doi: 10.1007/s00262-005-0040-7
134. Giles AJ, Hutchinson MND, Sonnemann HM, Jung J, Fecci PE, Ratnam NM, et al. Dexamethasone-Induced Immunosuppression: Mechanisms and Implications for Immunotherapy. J Immunother Cancer (2018) 6:51. doi: 10.1186/s40425-018-0371-5
135. Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature (2019) 565:234–9. doi: 10.1038/s41586-018-0792-9
136. Young JS, Dayani F, Morshed RA, Okada H, Aghi MK. Immunotherapy for High Grade Gliomas: A Clinical Update and Practical Considerations for Neurosurgeons. World Neurosurg (2019) 124:397–409. doi: 10.1016/j.wneu.2018.12.222
137. Heimberger AB, Sun W, Hussain SF, Dey M, Crutcher L, Aldape K, et al. Immunological Responses in a Patient With Glioblastoma Multiforme Treated With Sequential Courses of Temozolomide and Immunotherapy: Case Study. Neuro Oncol (2008) 10:98–103. doi: 10.1215/15228517-2007-046
138. Park SD, Kim CH, Kim CK, Park JA, Sohn HJ, Hong YK, et al. Cross-Priming by Temozolomide Enhances Antitumor Immunity of Dendritic Cell Vaccination in Murine Brain Tumor Model. Vaccine (2007) 25:3485–91. doi: 10.1016/j.vaccine.2006.12.060
139. Kim TG, Kim CH, Park JS, Park SD, Kim CK, Chung DS, et al. Immunological Factors Relating to the Antitumor Effect of Temozolomide Chemoimmunotherapy in a Murine Glioma Model. Clin Vaccine Immunol (2010) 17:143–53. doi: 10.1128/CVI.00292-09
140. Kim CH, Woo SJ, Park JS, Kim HS, Park MY, Park SD, et al. Enhanced Antitumour Immunity by Combined Use of Temozolomide and Tat-Survivin Pulsed Dendritic Cells in a Murine Glioma. Immunology (2007) 122:615–22. doi: 10.1111/j.1365-2567.2007.02680.x
141. Banissi C, Ghiringhelli F, Chen L, Carpentier AF. Treg Depletion With a Low-Dose Metronomic Temozolomide Regimen in a Rat Glioma Model. Cancer Immunol Immunother (2009) 58:1627–34. doi: 10.1007/s00262-009-0671-1
142. Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential Migration of Regulatory T Cells Mediated by Glioma-Secreted Chemokines can be Blocked With Chemotherapy. Cancer Immunol Immunother (2008) 57:123–31. doi: 10.1007/s00262-007-0336-x
143. Asavaroengchai W, Kotera Y, Mule JJ. Tumor Lysate-Pulsed Dendritic Cells can Elicit an Effective Antitumor Immune Response During Early Lymphoid Recovery. Proc Natl Acad Sci U S A (2002) 99:931–6. doi: 10.1073/pnas.022634999
144. Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater Chemotherapy-Induced Lymphopenia Enhances Tumor-Specific Immune Responses That Eliminate Egfrviii-Expressing Tumor Cells in Patients With Glioblastoma. Neuro Oncol (2011) 13:324–33. doi: 10.1093/neuonc/noq157
145. Mitchell DA, Cui X, Schmittling RJ, Sanchez-Perez L, Snyder DJ, Congdon KL, et al. Monoclonal Antibody Blockade of Il-2 Receptor Alpha During Lymphopenia Selectively Depletes Regulatory T Cells in Mice and Humans. Blood (2011) 118:3003–12. doi: 10.1182/blood-2011-02-334565
146. Galea I, Bernardes-Silva M, Forse PA, van Rooijen N, Liblau RS, Perry VH. An Antigen-Specific Pathway for Cd8 T Cells Across the Blood-Brain Barrier. J Exp Med (2007) 204:2023–30. doi: 10.1084/jem.20070064
147. Sanchez-Perez LA, Choi BD, Archer GE, Cui X, Flores C, Johnson LA, et al. Myeloablative Temozolomide Enhances Cd8(+) T-Cell Responses to Vaccine and is Required for Efficacy Against Brain Tumors in Mice. PloS One (2013) 8:e59082. doi: 10.1371/journal.pone.0059082
148. Karman J, Ling C, Sandor M, Fabry Z. Dendritic Cells in the Initiation of Immune Responses Against Central Nervous System-Derived Antigens. Immunol Lett (2004) 92:107–15. doi: 10.1016/j.imlet.2003.10.017
149. Hatterer E, Davoust N, Didier-Bazes M, Vuaillat C, Malcus C, Belin MF, et al. How to Drain Without Lymphatics? Dendritic Cells Migrate From the Cerebrospinal Fluid to the B-Cell Follicles of Cervical Lymph Nodes. Blood (2006) 107:806–12. doi: 10.1182/blood-2005-01-0154
150. Hatterer E, Touret M, Belin MF, Honnorat J, Nataf S. Cerebrospinal Fluid Dendritic Cells Infiltrate the Brain Parenchyma and Target the Cervical Lymph Nodes Under Neuroinflammatory Conditions. PloS One (2008) 3:e3321. doi: 10.1371/journal.pone.0003321
151. Steel CD, Hahto SM, Ciavarra RP. Peripheral Dendritic Cells are Essential for Both the Innate and Adaptive Antiviral Immune Responses in the Central Nervous System. Virology (2009) 387:117–26. doi: 10.1016/j.virol.2009.01.032
152. D’Agostino PM, Gottfried-Blackmore A, Anandasabapathy N, Bulloch K. Brain Dendritic Cells: Biology and Pathology. Acta Neuropathol (2012) 124:599–614. doi: 10.1007/s00401-012-1018-0
153. Eggert AA, Schreurs MW, Boerman OC, Oyen WJ, de Boer AJ, Punt CJ, et al. Biodistribution and Vaccine Efficiency of Murine Dendritic Cells are Dependent on the Route of Administration. Cancer Res (1999) 59:3340–5.
154. Eggert AA, van der Voort R, Torensma R, Moulin V, Boerman OC, Oyen WJ, et al. Analysis of Dendritic Cell Trafficking Using Egfp-Transgenic Mice. Immunol Lett (2003) 89:17–24. doi: 10.1016/S0165-2478(03)00105-6
155. Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK. Migration of Human Dendritic Cells After Injection in Patients With Metastatic Malignancies. Cancer Res (1999) 59:56–8.
156. De Vries IJ, Krooshoop DJ, Scharenborg NM, Lesterhuis WJ, Diepstra JH, Van Muijen GN, et al. Effective Migration of Antigen-Pulsed Dendritic Cells to Lymph Nodes in Melanoma Patients is Determined by Their Maturation State. Cancer Res (2003) 63:12–7.
157. Forster R, Davalos-Misslitz AC, Rot A. Ccr7 and its Ligands: Balancing Immunity and Tolerance. Nat Rev Immunol (2008) 8:362–71. doi: 10.1038/nri2297
158. Macatonia SE, Knight SC, Edwards AJ, Griffiths S, Fryer P. Localization of Antigen on Lymph Node Dendritic Cells After Exposure to the Contact Sensitizer Fluorescein Isothiocyanate. Functional and Morphological Studies. J Exp Med (1987) 166:1654–67. doi: 10.1084/jem.166.6.1654
159. Ruedl C, Koebel P, Bachmann M, Hess M, Karjalainen K. Anatomical Origin of Dendritic Cells Determines Their Life Span in Peripheral Lymph Nodes. J Immunol (2000) 165:4910–6. doi: 10.4049/jimmunol.165.9.4910
160. Garg S, Oran A, Wajchman J, Sasaki S, Maris CH, Kapp JA, et al. Genetic Tagging Shows Increased Frequency and Longevity of Antigen-Presenting, Skin-Derived Dendritic Cells in Vivo. Nat Immunol (2003) 4:907–12. doi: 10.1038/ni962
161. Ingulli E, Mondino A, Khoruts A, Jenkins MK. In Vivo Detection of Dendritic Cell Antigen Presentation to Cd4(+) T Cells. J Exp Med (1997) 185:2133–41. doi: 10.1084/jem.185.12.2133
162. Kamath AT, Henri S, Battye F, Tough DF, Shortman K. Developmental Kinetics and Lifespan of Dendritic Cells in Mouse Lymphoid Organs. Blood (2002) 100:1734–41. doi: 10.1182/blood.V100.5.1734.h81702001734_1734_1741
163. Chen M, Wang YH, Wang Y, Huang L, Sandoval H, Liu YJ, et al. Dendritic Cell Apoptosis in the Maintenance of Immune Tolerance. Science (2006) 311:1160–4. doi: 10.1126/science.1122545
164. Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, et al. Elimination of Antigen-Presenting Cells and Autoreactive T Cells by Fas Contributes to Prevention of Autoimmunity. Immunity (2007) 26:629–41. doi: 10.1016/j.immuni.2007.03.016
165. Granucci F, Zanoni I. The Dendritic Cell Life Cycle. Cell Cycle (2009) 8:3816–21. doi: 10.4161/cc.8.23.9998
166. Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, et al. Migratory Dendritic Cells Transfer Antigen to a Lymph Node-Resident Dendritic Cell Population for Efficient Ctl Priming. Immunity (2006) 25:153–62. doi: 10.1016/j.immuni.2006.04.017
167. Pellegatta S, Poliani PL, Stucchi E, Corno D, Colombo CA, Orzan F, et al. Intra-Tumoral Dendritic Cells Increase Efficacy of Peripheral Vaccination by Modulation of Glioma Microenvironment. Neuro Oncol (2010) 12:377–88. doi: 10.1093/neuonc/nop024
168. Masson F, Calzascia T, Di Berardino-Besson W, de Tribolet N, Dietrich PY, Walker PR. Brain Microenvironment Promotes the Final Functional Maturation of Tumor-Specific Effector Cd8+ T Cells. J Immunol (2007) 179:845–53. doi: 10.4049/jimmunol.179.2.845
169. Pearson JRD, Cuzzubbo S, McArthur S, Durrant LG, Adhikaree J, Tinsley CJ, et al. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front Immunol (2020) 11:582106. doi: 10.3389/fimmu.2020.582106
170. Shevyrev D, Tereshchenko V. Treg Heterogeneity, Function, and Homeostasis. Front Immunol (2019) 10:3100. doi: 10.3389/fimmu.2019.03100
171. Savage PA, Klawon DEJ, Miller CH. Regulatory T Cell Development. Annu Rev Immunol (2020) 38:421–53. doi: 10.1146/annurev-immunol-100219-020937
172. Hori S, Nomura T, Sakaguchi S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science (2003) 299:1057–61. doi: 10.1126/science.1079490
173. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 Programs the Development and Function of Cd4+Cd25+ Regulatory T Cells. Nat Immunol (2003) 4:330–6. doi: 10.1038/ni904
174. Khattri R, Cox T, Yasayko SA, Ramsdell F. An Essential Role for Scurfin in Cd4+Cd25+ T Regulatory Cells. Nat Immunol (2003) 4:337–42. doi: 10.1038/ni909
175. Liu W, Putnam AL, Xu-Yu Z, Szot GL, Lee MR, Zhu S, et al. Cd127 Expression Inversely Correlates With Foxp3 and Suppressive Function of Human Cd4+ T Reg Cells. J Exp Med (2006) 203:1701–11. doi: 10.1084/jem.20060772
176. Yu N, Li X, Song W, Li D, Yu D, Zeng X, et al. Cd4(+)Cd25 (+)Cd127 (Low/-) T Cells: A More Specific Treg Population in Human Peripheral Blood. Inflammation (2012) 35:1773–80. doi: 10.1007/s10753-012-9496-8
177. Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient Expression of Foxp3 in Human Activated Nonregulatory Cd4+ T Cells. Eur J Immunol (2007) 37:129–38. doi: 10.1002/eji.200636435
178. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T Cells and Human Disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
179. Miyara M, Chader D, Sage E, Sugiyama D, Nishikawa H, Bouvry D, et al. (Cd15s) Identifies Highly Differentiated and Most Suppressive Foxp3high Regulatory T Cells in Humans. Proc Natl Acad Sci U S A (2015) 112:7225–30. doi: 10.1073/pnas.1508224112
180. Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The Transcription Factor T-Bet Controls Regulatory T Cell Homeostasis and Function During Type 1 Inflammation. Nat Immunol (2009) 10:595–602. doi: 10.1038/ni.1731
181. Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, et al. Regulatory T-Cell Suppressor Program Co-Opts Transcription Factor Irf4 to Control T(H)2 Responses. Nature (2009) 458:351–6. doi: 10.1038/nature07674
182. Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, et al. Cd4+ Regulatory T Cells Control Th17 Responses in a Stat3-Dependent Manner. Science (2009) 326:986–91. doi: 10.1126/science.1172702
183. Asseman C, Mauze S, Leach MW, Coffman RL, Powrie F. An Essential Role for Interleukin 10 in the Function of Regulatory T Cells That Inhibit Intestinal Inflammation. J Exp Med (1999) 190:995–1004. doi: 10.1084/jem.190.7.995
184. Nakamura K, Kitani A, Strober W. Cell Contact-Dependent Immunosuppression by Cd4(+)Cd25(+) Regulatory T Cells is Mediated by Cell Surface-Bound Transforming Growth Factor Beta. J Exp Med (2001) 194:629–44. doi: 10.1084/jem.194.5.629
185. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The Inhibitory Cytokine Il-35 Contributes to Regulatory T-Cell Function. Nature (2007) 450:566–9. doi: 10.1038/nature06306
186. Vignali DA, Collison LW, Workman CJ. How Regulatory T Cells Work. Nat Rev Immunol (2008) 8:523–32. doi: 10.1038/nri2343
187. Lee CR, Kwak Y, Yang T, Han JH, Park SH, Ye MB, et al. Myeloid-Derived Suppressor Cells are Controlled by Regulatory T Cells via Tgf-Beta During Murine Colitis. Cell Rep (2016) 17:3219–32. doi: 10.1016/j.celrep.2016.11.062
188. Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-Cell Infiltration Positively Impacts Survival of Glioblastoma Patients and is Impaired by Tumor-Derived Tgf-Beta. Clin Cancer Res (2011) 17:4296–308. doi: 10.1158/1078-0432.CCR-10-2557
189. Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T Regulatory Cells can Use the Perforin Pathway to Cause Autologous Target Cell Death. Immunity (2004) 21:589–601. doi: 10.1016/j.immuni.2004.09.002
190. Gondek DC, Lu LF, Quezada SA, Sakaguchi S, Noelle RJ. Cutting Edge: Contact-Mediated Suppression by Cd4+Cd25+ Regulatory Cells Involves a Granzyme B-Dependent, Perforin-Independent Mechanism. J Immunol (2005) 174:1783–6. doi: 10.4049/jimmunol.174.4.1783
191. Ren X, Ye F, Jiang Z, Chu Y, Xiong S, Wang Y. Involvement of Cellular Death in Trail/Dr5-Dependent Suppression Induced by Cd4(+)Cd25(+) Regulatory T Cells. Cell Death Differ (2007) 14:2076–84. doi: 10.1038/sj.cdd.4402220
192. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. Cd4+Cd25+Foxp3+ Regulatory T Cells Induce Cytokine Deprivation-Mediated Apoptosis of Effector Cd4+ T Cells. Nat Immunol (2007) 8:1353–62. doi: 10.1038/ni1536
193. Kobie JJ, Shah PR, Yang L, Rebhahn JA, Fowell DJ, Mosmann TR. T Regulatory and Primed Uncommitted Cd4 T Cells Express Cd73, Which Suppresses Effector Cd4 T Cells by Converting 5’-Adenosine Monophosphate to Adenosine. J Immunol (2006) 177:6780–6. doi: 10.4049/jimmunol.177.10.6780
194. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine Generation Catalyzed by Cd39 and Cd73 Expressed on Regulatory T Cells Mediates Immune Suppression. J Exp Med (2007) 204:1257–65. doi: 10.1084/jem.20062512
195. Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, et al. Expression of Ectonucleotidase Cd39 by Foxp3+ Treg Cells: Hydrolysis of Extracellular Atp and Immune Suppression. Blood (2007) 110:1225–32. doi: 10.1182/blood-2006-12-064527
196. Sek K, Molck C, Stewart GD, Kats L, Darcy PK, Beavis PA. Targeting Adenosine Receptor Signaling in Cancer Immunotherapy. Int J Mol Sci (2018) 19:3837. doi: 10.3390/ijms19123837
197. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic Adenosine Monophosphate is a Key Component of Regulatory T Cell-Mediated Suppression. J Exp Med (2007) 204:1303–10. doi: 10.1084/jem.20062129
198. Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ Natural Regulatory T Cells Preferentially Form Aggregates on Dendritic Cells in Vitro and Actively Inhibit Their Maturation. Proc Natl Acad Sci U S A (2008) 105:10113–8. doi: 10.1073/pnas.0711106105
199. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-Endocytosis of Cd80 and Cd86: A Molecular Basis for the Cell-Extrinsic Function of Ctla-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
200. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of Tryptophan Catabolism by Regulatory T Cells. Nat Immunol (2003) 4:1206–12. doi: 10.1038/ni1003
201. Mellor AL, Munn DH. Ido Expression by Dendritic Cells: Tolerance and Tryptophan Catabolism. Nat Rev Immunol (2004) 4:762–74. doi: 10.1038/nri1457
202. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An Interaction Between Kynurenine and the Aryl Hydrocarbon Receptor can Generate Regulatory T Cells. J Immunol (2010) 185:3190–8. doi: 10.4049/jimmunol.0903670
203. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of Lag-3 in Regulatory T Cells. Immunity (2004) 21:503–13. doi: 10.1016/j.immuni.2004.08.010
204. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T Cells Inhibit Dendritic Cells by Lymphocyte Activation Gene-3 Engagement of Mhc Class Ii. J Immunol (2008) 180:5916–26. doi: 10.4049/jimmunol.180.9.5916
205. Ito T, Hanabuchi S, Wang YH, Park WR, Arima K, Bover L, et al. Two Functional Subsets of Foxp3+ Regulatory T Cells in Human Thymus and Periphery. Immunity (2008) 28:870–80. doi: 10.1016/j.immuni.2008.03.018
206. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The Surface Protein Tigit Suppresses T Cell Activation by Promoting the Generation of Mature Immunoregulatory Dendritic Cells. Nat Immunol (2009) 10:48–57. doi: 10.1038/ni.1674
207. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg Cells Expressing the Coinhibitory Molecule Tigit Selectively Inhibit Proinflammatory Th1 and Th17 Cell Responses. Immunity (2014) 40:569–81. doi: 10.1016/j.immuni.2014.02.012
208. DiDomenico J, Lamano JB, Oyon D, Li Y, Veliceasa D, Kaur G, et al. The Immune Checkpoint Protein Pd-L1 Induces and Maintains Regulatory T Cells in Glioblastoma. Oncoimmunology (2018) 7:e1448329. doi: 10.1080/2162402X.2018.1448329
209. Gianchecchi E, Fierabracci A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of Pd-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front Immunol (2018) 9:2374. doi: 10.3389/fimmu.2018.02374
210. Dees S, Ganesan R, Singh S, Grewal IS. Regulatory T Cell Targeting in Cancer: Emerging Strategies in Immunotherapy. Eur J Immunol (2021) 51:280–91. doi: 10.1002/eji.202048992
211. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic Value of Tumor-Infiltrating Foxp3+ Regulatory T Cells in Cancers: A Systematic Review and Meta-Analysis. Sci Rep (2015) 5:15179. doi: 10.1038/srep15179
212. Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased Regulatory T-Cell Fraction Amidst a Diminished Cd4 Compartment Explains Cellular Immune Defects in Patients With Malignant Glioma. Cancer Res (2006) 66:3294–302. doi: 10.1158/0008-5472.CAN-05-3773
213. El Andaloussi A, Lesniak MS. An Increase in Cd4+Cd25+Foxp3+ Regulatory T Cells in Tumor-Infiltrating Lymphocytes of Human Glioblastoma Multiforme. Neuro Oncol (2006) 8:234–43. doi: 10.1215/15228517-2006-006
214. Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, et al. Incidence and Prognostic Impact of Foxp3+ Regulatory T Cells in Human Gliomas. Clin Cancer Res (2008) 14:5166–72. doi: 10.1158/1078-0432.CCR-08-0320
215. Jacobs JF, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJ, Wesseling P, et al. Prognostic Significance and Mechanism of Treg Infiltration in Human Brain Tumors. J Neuroimmunol (2010) 225:195–9. doi: 10.1016/j.jneuroim.2010.05.020
216. Richardson LG, Nieman LT, Stemmer-Rachamimov AO, Zheng XS, Stafford K, Nagashima H, et al. Idh-Mutant Gliomas Harbor Fewer Regulatory T Cells in Humans and Mice. Oncoimmunology (2020) 9:1806662. doi: 10.1080/2162402X.2020.1806662
217. Kaffes I, Szulzewsky F, Chen Z, Herting CJ, Gabanic B, Velazquez Vega JE, et al. Human Mesenchymal Glioblastomas are Characterized by an Increased Immune Cell Presence Compared to Proneural and Classical Tumors. Oncoimmunology (2019) 8:e1655360. doi: 10.1080/2162402X.2019.1655360
218. Ladomersky E, Scholtens DM, Kocherginsky M, Hibler EA, Bartom ET, Otto-Meyer S, et al. The Coincidence Between Increasing Age, Immunosuppression, and the Incidence of Patients With Glioblastoma. Front Pharmacol (2019) 10:200. doi: 10.3389/fphar.2019.00200
219. Thomas AA, Fisher JL, Rahme GJ, Hampton TH, Baron U, Olek S, et al. Regulatory T Cells are Not a Strong Predictor of Survival for Patients With Glioblastoma. Neuro Oncol (2015) 17:801–9. doi: 10.1093/neuonc/nou363
220. Pereira MB, Barros LRC, Bracco PA, Vigo A, Boroni M, Bonamino MH, et al. Transcriptional Characterization of Immunological Infiltrates and Their Relation With Glioblastoma Patients Overall Survival. Oncoimmunology (2018) 7:e1431083. doi: 10.1080/2162402X.2018.1431083
221. Sayour EJ, McLendon P, McLendon R, De Leon G, Reynolds R, Kresak J, et al. Increased Proportion of Foxp3+ Regulatory T Cells in Tumor Infiltrating Lymphocytes is Associated With Tumor Recurrence and Reduced Survival in Patients With Glioblastoma. Cancer Immunol Immunother (2015) 64:419–27. doi: 10.1007/s00262-014-1651-7
222. Grauer OM, Nierkens S, Bennink E, Toonen LW, Boon L, Wesseling P, et al. Cd4+Foxp3+ Regulatory T Cells Gradually Accumulate in Gliomas During Tumor Growth and Efficiently Suppress Antiglioma Immune Responses in Vivo. Int J Cancer (2007) 121:95–105. doi: 10.1002/ijc.22607
223. Kennedy BC, Maier LM, D’Amico R, Mandigo CE, Fontana EJ, Waziri A, et al. Dynamics of Central and Peripheral Immunomodulation in a Murine Glioma Model. BMC Immunol (2009) 10:11. doi: 10.1186/1471-2172-10-11
224. Tran Thang NN, Derouazi M, Philippin G, Arcidiaco S, Di Berardino-Besson W, Masson F, et al. Immune Infiltration of Spontaneous Mouse Astrocytomas is Dominated by Immunosuppressive Cells From Early Stages of Tumor Development. Cancer Res (2010) 70:4829–39. doi: 10.1158/0008-5472.CAN-09-3074
225. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. Ccl2 Produced by the Glioma Microenvironment is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res (2016) 76:5671–82. doi: 10.1158/0008-5472.CAN-16-0144
226. El Andaloussi A, Han Y, Lesniak MS. Prolongation of Survival Following Depletion of Cd4+Cd25+ Regulatory T Cells in Mice With Experimental Brain Tumors. J Neurosurg (2006) 105:430–7. doi: 10.3171/jns.2006.105.3.430
227. Fecci PE, Sweeney AE, Grossi PM, Nair SK, Learn CA, Mitchell DA, et al. Systemic Anti-Cd25 Monoclonal Antibody Administration Safely Enhances Immunity in Murine Glioma Without Eliminating Regulatory T Cells. Clin Cancer Res (2006) 12:4294–305. doi: 10.1158/1078-0432.CCR-06-0053
228. Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, et al. Systemic Ctla-4 Blockade Ameliorates Glioma-Induced Changes to the Cd4+ T Cell Compartment Without Affecting Regulatory T-Cell Function. Clin Cancer Res (2007) 13:2158–67. doi: 10.1158/1078-0432.CCR-06-2070
229. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable Therapeutic Efficacy Utilizing Combinatorial Blockade Against Ido, Ctla-4, and Pd-L1 in Mice With Brain Tumors. Clin Cancer Res (2014) 20:5290–301. doi: 10.1158/1078-0432.CCR-14-0514
230. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. Tigit and Pd-1 Dual Checkpoint Blockade Enhances Antitumor Immunity and Survival in Gbm. Oncoimmunology (2018) 7:e1466769. doi: 10.1080/2162402X.2018.1466769
231. Li J, Liu X, Duan Y, Liu Y, Wang H, Lian S, et al. Combined Blockade of T Cell Immunoglobulin and Mucin Domain 3 and Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 Results in Durable Therapeutic Efficacy in Mice With Intracranial Gliomas. Med Sci Monit (2017) 23:3593–602. doi: 10.12659/msm.903098
232. Galstyan A, Markman JL, Shatalova ES, Chiechi A, Korman AJ, Patil R, et al. Blood-Brain Barrier Permeable Nano Immunoconjugates Induce Local Immune Responses for Glioma Therapy. Nat Commun (2019) 10:3850. doi: 10.1038/s41467-019-11719-3
233. Azambuja JH, Schuh RS, Michels LR, Iser IC, Beckenkamp LR, Roliano GG, et al. Blockade of Cd73 Delays Glioblastoma Growth by Modulating the Immune Environment. Cancer Immunol Immunother (2020) 69:1801–12. doi: 10.1007/s00262-020-02569-w
234. Riva M, Wouters R, Nittner D, Ceuster J, Sterpin E, Giovannoni R, et al. Radiation Dose-Escalation and Dose-Fractionation Modulate the Immune Microenvironment, Cancer Stem Cells and Vasculature in Experimental High-Grade Gliomas. J Neurosurg Sci (2020). doi: 10.23736/S0390-5616.20.05060-2
235. Wu J, Waxman DJ. Metronomic Cyclophosphamide Eradicates Large Implanted Gl261 Gliomas by Activating Antitumor Cd8(+) T-Cell Responses and Immune Memory. Oncoimmunology (2015) 4:e1005521. doi: 10.1080/2162402X.2015.1005521
236. Dutoit V, Philippin G, Widmer V, Marinari E, Vuilleumier A, Migliorini D, et al. Impact of Radiochemotherapy on Immune Cell Subtypes in High-Grade Glioma Patients. Front Oncol (2020) 10:89. doi: 10.3389/fonc.2020.00089
237. El Andaloussi A, Sonabend AM, Han Y, Lesniak MS. Stimulation of Tlr9 With Cpg Odn Enhances Apoptosis of Glioma and Prolongs the Survival of Mice With Experimental Brain Tumors. Glia (2006) 54:526–35. doi: 10.1002/glia.20401
238. Grauer OM, Molling JW, Bennink E, Toonen LW, Sutmuller RP, Nierkens S, et al. Tlr Ligands in the Local Treatment of Established Intracerebral Murine Gliomas. J Immunol (2008) 181:6720–9. doi: 10.4049/jimmunol.181.10.6720
239. Curtin JF, Candolfi M, Fakhouri TM, Liu C, Alden A, Edwards M, et al. Treg Depletion Inhibits Efficacy of Cancer Immunotherapy: Implications for Clinical Trials. PloS One (2008) 3:e1983. doi: 10.1371/journal.pone.0001983
240. Qiao J, Dey M, Chang AL, Kim JW, Miska J, Ling A, et al. Intratumoral Oncolytic Adenoviral Treatment Modulates the Glioma Microenvironment and Facilitates Systemic Tumor-Antigen-Specific T Cell Therapy. Oncoimmunology (2015) 4:e1022302. doi: 10.1080/2162402X.2015.1022302
241. Kosaka A, Ohkuri T, Okada H. Combination of an Agonistic Anti-Cd40 Monoclonal Antibody and the Cox-2 Inhibitor Celecoxib Induces Anti-Glioma Effects by Promotion of Type-1 Immunity in Myeloid Cells and T-Cells. Cancer Immunol Immunother (2014) 63:847–57. doi: 10.1007/s00262-014-1561-8
242. Jahan N, Talat H, Curry WT. Agonist Ox40 Immunotherapy Improves Survival in Glioma-Bearing Mice and is Complementary With Vaccination With Irradiated Gm-Csf-Expressing Tumor Cells. Neuro Oncol (2018) 20:44–54. doi: 10.1093/neuonc/nox125
243. Atif M, Mohr A, Conti F, Scatton O, Gorochov G, Miyara M. Metabolic Optimisation of Regulatory T Cells in Transplantation. Front Immunol (2020) 11:2005. doi: 10.3389/fimmu.2020.02005
244. Kesarwani P, Prabhu A, Kant S, Kumar P, Graham SF, Buelow KL, et al. Tryptophan Metabolism Contributes to Radiation-Induced Immune Checkpoint Reactivation in Glioblastoma. Clin Cancer Res (2018) 24:3632–43. doi: 10.1158/1078-0432.CCR-18-0041
245. Lowther DE, Goods BA, Lucca LE, Lerner BA, Raddassi K, van Dijk D, et al. Pd-1 Marks Dysfunctional Regulatory T Cells in Malignant Gliomas. JCI Insight (2016) 1:e85935. doi: 10.1172/jci.insight.85935
246. Maes W, Verschuere T, Van Hoylandt A, Boon L, Van Gool S. Depletion of Regulatory T Cells in a Mouse Experimental Glioma Model Through Anti-Cd25 Treatment Results in the Infiltration of non-Immunosuppressive Myeloid Cells in the Brain. Clin Dev Immunol (2013) 2013:952469. doi: 10.1155/2013/952469
247. Poirier MD, Haban H, El Andaloussi A. A Combination of Systemic and Intracranial Anti-Cd25 Immunotherapy Elicits a Long-Time Survival in Murine Model of Glioma. J Oncol (2009) 2009:963037. doi: 10.1155/2009/963037
248. Driessens G, Gordower L, Nuttin L, Stordeur P, Blocklet D, Egrise D, et al. Therapeutic Efficacy of Antitumor Dendritic Cell Vaccinations Correlates With Persistent Th1 Responses, High Intratumor Cd8+ T Cell Recruitment and Low Relative Regulatory T Cell Infiltration. Cancer Immunol Immunother (2008) 57:1745–56. doi: 10.1007/s00262-008-0500-y
249. Maes W, Rosas GG, Verbinnen B, Boon L, De Vleeschouwer S, Ceuppens JL, et al. Dc Vaccination With Anti-Cd25 Treatment Leads to Long-Term Immunity Against Experimental Glioma. Neuro Oncol (2009) 11:529–42. doi: 10.1215/15228517-2009-004
250. Gabrilovich DI. Myeloid-Derived Suppressor Cells. Cancer Immunol Res (2017) 5:3–8. doi: 10.1158/2326-6066.CIR-16-0297
251. Talmadge JE, Gabrilovich DI. History of Myeloid-Derived Suppressor Cells. Nat Rev Cancer (2013) 13:739–52. doi: 10.1038/nrc3581
252. Qu P, Wang LZ, Lin PC. Expansion and Functions of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Cancer Lett (2016) 380:253–6. doi: 10.1016/j.canlet.2015.10.022
253. Mandruzzato S, Brandau S, Britten CM, Bronte V, Damuzzo V, Gouttefangeas C, et al. Toward Harmonized Phenotyping of Human Myeloid-Derived Suppressor Cells by Flow Cytometry: Results From an Interim Study. Cancer Immunol Immunother (2016) 65:161–9. doi: 10.1007/s00262-015-1782-5
254. Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res (2007) 67:425; author reply 6. doi: 10.1158/0008-5472.CAN-06-3037
255. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-Type Oxidized Ldl Receptor-1 Distinguishes Population of Human Polymorphonuclear Myeloid-Derived Suppressor Cells in Cancer Patients. Sci Immunol (2016) 1:aaf8943. doi: 10.1126/sciimmunol.aaf8943
256. Bronte V, Chappell DB, Apolloni E, Cabrelle A, Wang M, Hwu P, et al. Unopposed Production of Granulocyte-Macrophage Colony-Stimulating Factor by Tumors Inhibits Cd8+ T Cell Responses by Dysregulating Antigen-Presenting Cell Maturation. J Immunol (1999) 162:5728–37.
257. Liu Q, Zhang M, Jiang X, Zhang Z, Dai L, Min S, et al. Mir-223 Suppresses Differentiation of Tumor-Induced Cd11b(+) Gr1(+) Myeloid-Derived Suppressor Cells From Bone Marrow Cells. Int J Cancer (2011) 129:2662–73. doi: 10.1002/ijc.25921
258. De Cicco P, Ercolano G, Ianaro A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front Immunol (2020) 11:1680. doi: 10.3389/fimmu.2020.01680
259. Haist M, Stege H, Grabbe S, Bros M. The Functional Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells Within the Immunosuppressive Tumor Microenvironment. Cancers (Basel) (2021) 13:210. doi: 10.3390/cancers13020210
260. Gabrilovich DI, Nagaraj S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat Rev Immunol (2009) 9:162–74. doi: 10.1038/nri2506
261. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of Myeloid-Derived Suppressor Cells Impair T Cell Responses Through Independent Nitric Oxide-Related Pathways. Int J Cancer (2014) 134:2853–64. doi: 10.1002/ijc.28622
262. Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered Recognition of Antigen is a Mechanism of Cd8+ T Cell Tolerance in Cancer. Nat Med (2007) 13:828–35. doi: 10.1038/nm1609
263. Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-Derived Suppressor Cells Down-Regulate L-Selectin Expression on Cd4+ and Cd8+ T Cells. J Immunol (2009) 183:937–44. doi: 10.4049/jimmunol.0804253
264. Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated Regulation of Myeloid Cells by Tumours. Nat Rev Immunol (2012) 12:253–68. doi: 10.1038/nri3175
265. Fujimura T, Kambayashi Y, Aiba S. Crosstalk Between Regulatory T Cells (Tregs) and Myeloid Derived Suppressor Cells (Mdscs) During Melanoma Growth. Oncoimmunology (2012) 1:1433–4. doi: 10.4161/onci.21176
266. Bruno A, Mortara L, Baci D, Noonan DM, Albini A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-Angiogenic Activities: Roles in Tumor Progression. Front Immunol (2019) 10:771. doi: 10.3389/fimmu.2019.00771
267. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Petersen-Baltussen HM, ter Laan M, et al. Increase in Both Cd14-Positive and Cd15-Positive Myeloid-Derived Suppressor Cell Subpopulations in the Blood of Patients With Glioma But Predominance of Cd15-Positive Myeloid-Derived Suppressor Cells in Glioma Tissue. J Neuropathol Exp Neurol (2015) 74:390–400. doi: 10.1097/NEN.0000000000000183
268. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Bossman SA, Ter Laan M, et al. Elevated Levels of Polymorphonuclear Myeloid-Derived Suppressor Cells in Patients With Glioblastoma Highly Express S100a8/9 and Arginase and Suppress T Cell Function. Neuro Oncol (2016) 18:1253–64. doi: 10.1093/neuonc/now034
269. Alban TJ, Bayik D, Otvos B, Rabljenovic A, Leng L, Jia-Shiun L, et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That can be Targeted to Reduce Immune Suppression. Front Immunol (2020) 11:1191. doi: 10.3389/fimmu.2020.01191
270. Otvos B, Silver DJ, Mulkearns-Hubert EE, Alvarado AG, Turaga SM, Sorensen MD, et al. Cancer Stem Cell-Secreted Macrophage Migration Inhibitory Factor Stimulates Myeloid Derived Suppressor Cell Function and Facilitates Glioblastoma Immune Evasion. Stem Cells (2016) 34:2026–39. doi: 10.1002/stem.2393
271. Abad C, Nobuta H, Li J, Kasai A, Yong WH, Waschek JA. Targeted Stat3 Disruption in Myeloid Cells Alters Immunosuppressor Cell Abundance in a Murine Model of Spontaneous Medulloblastoma. J Leukoc Biol (2014) 95:357–67. doi: 10.1189/jlb.1012531
272. Raychaudhuri B, Rayman P, Ireland J, Ko J, Rini B, Borden EC, et al. Myeloid-Derived Suppressor Cell Accumulation and Function in Patients With Newly Diagnosed Glioblastoma. Neuro Oncol (2011) 13:591–9. doi: 10.1093/neuonc/nor042
273. Peereboom DM, Alban TJ, Grabowski MM, Alvarado AG, Otvos B, Bayik D, et al. Metronomic Capecitabine as an Immune Modulator in Glioblastoma Patients Reduces Myeloid-Derived Suppressor Cells. JCI Insight (2019) 4:e130748. doi: 10.1172/jci.insight.130748
274. Stewart TJ, Smyth MJ. Improving Cancer Immunotherapy by Targeting Tumor-Induced Immune Suppression. Cancer Metastasis Rev (2011) 30:125–40. doi: 10.1007/s10555-011-9280-5
275. Gao X, Sui H, Zhao S, Gao X, Su Y, Qu P. Immunotherapy Targeting Myeloid-Derived Suppressor Cells (Mdscs) in Tumor Microenvironment. Front Immunol (2020) 11:585214. doi: 10.3389/fimmu.2020.585214
276. Kodumudi KN, Woan K, Gilvary DL, Sahakian E, Wei S, Djeu JY. A Novel Chemoimmunomodulating Property of Docetaxel: Suppression of Myeloid-Derived Suppressor Cells in Tumor Bearers. Clin Cancer Res (2010) 16:4583–94. doi: 10.1158/1078-0432.CCR-10-0733
277. Michels T, Shurin GV, Naiditch H, Sevko A, Umansky V, Shurin MR. Paclitaxel Promotes Differentiation of Myeloid-Derived Suppressor Cells Into Dendritic Cells in Vitro in a Tlr4-Independent Manner. J Immunotoxicol (2012) 9:292–300. doi: 10.3109/1547691X.2011.642418
278. Mirza N, Fishman M, Fricke I, Dunn M, Neuger AM, Frost TJ, et al. All-Trans-Retinoic Acid Improves Differentiation of Myeloid Cells and Immune Response in Cancer Patients. Cancer Res (2006) 66:9299–307. doi: 10.1158/0008-5472.CAN-06-1690
279. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-Stimulating Factor 1 Receptor (Csf1r) Inhibitors in Cancer Therapy. J Immunother Cancer (2017) 5:53. doi: 10.1186/s40425-017-0257-y
280. Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic Regulation of Myeloid-Derived Suppressor Cells and Immune Response to Cancer Vaccine in Patients With Extensive Stage Small Cell Lung Cancer. Cancer Immunol Immunother (2013) 62:909–18. doi: 10.1007/s00262-013-1396-8
281. Riva M, Wouters R, Sterpin E, Giovannoni R, Boon L, Himmelreich U, et al. Radiotherapy, Temozolomide, and Antiprogrammed Cell Death Protein 1 Treatments Modulate the Immune Microenvironment in Experimental High-Grade Glioma. Neurosurgery (2021) 88:E205–15. doi: 10.1093/neuros/nyaa421
282. Mosser DM. The Many Faces of Macrophage Activation. J Leukoc Biol (2003) 73:209–12. doi: 10.1189/jlb.0602325
283. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 Potently Enhances Murine Macrophage Mannose Receptor Activity: A Marker of Alternative Immunologic Macrophage Activation. J Exp Med (1992) 176:287–92. doi: 10.1084/jem.176.1.287
284. Mills CD, Ley K. M1 and M2 Macrophages: The Chicken and the Egg of Immunity. J Innate Immun (2014) 6:716–26. doi: 10.1159/000364945
285. Hamilton TA, Zhao C, Pavicic PG Jr, Datta S. Myeloid Colony-Stimulating Factors as Regulators of Macrophage Polarization. Front Immunol (2014) 5:554. doi: 10.3389/fimmu.2014.00554
286. Akagawa KS, Komuro I, Kanazawa H, Yamazaki T, Mochida K, Kishi F. Functional Heterogeneity of Colony-Stimulating Factor-Induced Human Monocyte-Derived Macrophages. Respirology (2006) 11 Suppl:S32–6. doi: 10.1111/j.1440-1843.2006.00805.x
287. Verreck FA, de Boer T, Langenberg DM, Hoeve MA, Kramer M, Vaisberg E, et al. Human Il-23-Producing Type 1 Macrophages Promote But Il-10-Producing Type 2 Macrophages Subvert Immunity to (Myco)Bacteria. Proc Natl Acad Sci U S A (2004) 101:4560–5. doi: 10.1073/pnas.0400983101
288. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J Immunol (2000) 164:6166–73. doi: 10.4049/jimmunol.164.12.6166
289. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The Chemokine System in Diverse Forms of Macrophage Activation and Polarization. Trends Immunol (2004) 25:677–86. doi: 10.1016/j.it.2004.09.015
290. Goerdt S, Orfanos CE. Other Functions, Other Genes: Alternative Activation of Antigen-Presenting Cells. Immunity (1999) 10:137–42. doi: 10.1016/s1074-7613(00)80014-x
291. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage Plasticity, Polarization, and Function in Health and Disease. J Cell Physiol (2018) 233:6425–40. doi: 10.1002/jcp.26429
292. Bashir S, Sharma Y, Elahi A, Khan F. Macrophage Polarization: The Link Between Inflammation and Related Diseases. Inflamm Res (2016) 65:1–11. doi: 10.1007/s00011-015-0874-1
293. Krzyszczyk P, Schloss R, Palmer A, Berthiaume F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-Wound Healing Phenotypes. Front Physiol (2018) 9:419. doi: 10.3389/fphys.2018.00419
294. Spiller KL, Anfang RR, Spiller KJ, Ng J, Nakazawa KR, Daulton JW, et al. The Role of Macrophage Phenotype in Vascularization of Tissue Engineering Scaffolds. Biomaterials (2014) 35:4477–88. doi: 10.1016/j.biomaterials.2014.02.012
295. Ferrante CJ, Leibovich SJ. Regulation of Macrophage Polarization and Wound Healing. Adv Wound Care (New Rochelle) (2012) 1:10–6. doi: 10.1089/wound.2011.0307
296. Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates With Immunological Changes in the Microenvironment. Cancer Cell (2017) 32:42–56.e6. doi: 10.1016/j.ccell.2017.06.003
297. Yue Y, Yang X, Feng K, Wang L, Hou J, Mei B, et al. M2b Macrophages Reduce Early Reperfusion Injury After Myocardial Ischemia in Mice: A Predominant Role of Inhibiting Apoptosis via A20. Int J Cardiol (2017) 245:228–35. doi: 10.1016/j.ijcard.2017.07.085
298. Ambarus CA, Santegoets KC, van Bon L, Wenink MH, Tak PP, Radstake TR, et al. Soluble Immune Complexes Shift the Tlr-Induced Cytokine Production of Distinct Polarized Human Macrophage Subsets Towards Il-10. PloS One (2012) 7:e35994. doi: 10.1371/journal.pone.0035994
299. Anderson CF, Mosser DM. A Novel Phenotype for an Activated Macrophage: The Type 2 Activated Macrophage. J Leukoc Biol (2002) 72:101–6. doi: 10.1189/jlb.72.1.101
300. Lolmede K, Campana L, Vezzoli M, Bosurgi L, Tonlorenzi R, Clementi E, et al. Inflammatory and Alternatively Activated Human Macrophages Attract Vessel-Associated Stem Cells, Relying on Separate Hmgb1- and Mmp-9-Dependent Pathways. J Leukoc Biol (2009) 85:779–87. doi: 10.1189/jlb.0908579
301. Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM. Anti-Inflammatory M2, But Not Pro-Inflammatory M1 Macrophages Promote Angiogenesis in Vivo. Angiogenesis (2014) 17:109–18. doi: 10.1007/s10456-013-9381-6
302. Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-Associated Macrophages: Potential Therapeutic Targets for Anti-Cancer Therapy. Adv Drug Deliv Rev (2016) 99:180–5. doi: 10.1016/j.addr.2015.11.009
303. Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-Resident Macrophages Provide a Pro-Tumorigenic Niche to Early Nsclc Cells. Nature (2021) 595:578–84. doi: 10.1038/s41586-021-03651-8
304. Yang L, Zhang Y. Tumor-Associated Macrophages: From Basic Research to Clinical Application. J Hematol Oncol (2017) 10:58. doi: 10.1186/s13045-017-0430-2
305. Ambade A, Satishchandran A, Saha B, Gyongyosi B, Lowe P, Kodys K, et al. Hepatocellular Carcinoma is Accelerated by Nash Involving M2 Macrophage Polarization Mediated by Hif-1alphainduced Il-10. Oncoimmunology (2016) 5:e1221557. doi: 10.1080/2162402X.2016.1221557
306. Zhao P, Gao D, Wang Q, Song B, Shao Q, Sun J, et al. Response Gene to Complement 32 (Rgc-32) Expression on M2-Polarized and Tumor-Associated Macrophages is M-Csf-Dependent and Enhanced by Tumor-Derived Il-4. Cell Mol Immunol (2015) 12:692–9. doi: 10.1038/cmi.2014.108
307. Schmidt A, Zhang XM, Joshi RN, Iqbal S, Wahlund C, Gabrielsson S, et al. Human Macrophages Induce Cd4(+)Foxp3(+) Regulatory T Cells via Binding and Re-Release of Tgf-Beta. Immunol Cell Biol (2016) 94:747–62. doi: 10.1038/icb.2016.34
308. Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. Pge(2)-Induced Cxcl12 Production and Cxcr4 Expression Controls the Accumulation of Human Mdscs in Ovarian Cancer Environment. Cancer Res (2011) 71:7463–70. doi: 10.1158/0008-5472.CAN-11-2449
309. Inaba T, Sano H, Kawahito Y, Hla T, Akita K, Toda M, et al. Induction of Cyclooxygenase-2 in Monocyte/Macrophage by Mucins Secreted From Colon Cancer Cells. Proc Natl Acad Sci U S A (2003) 100:2736–41. doi: 10.1073/pnas.0435410100
310. Liu J, Zhang N, Li Q, Zhang W, Ke F, Leng Q, et al. Tumor-Associated Macrophages Recruit Ccr6+ Regulatory T Cells and Promote the Development of Colorectal Cancer via Enhancing Ccl20 Production in Mice. PloS One (2011) 6:e19495. doi: 10.1371/journal.pone.0019495
311. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific Recruitment of Regulatory T Cells in Ovarian Carcinoma Fosters Immune Privilege and Predicts Reduced Survival. Nat Med (2004) 10:942–9. doi: 10.1038/nm1093
312. Cendrowicz E, Sas Z, Bremer E, Rygiel TP. The Role of Macrophages in Cancer Development and Therapy. Cancers (Basel) (2021) 13:1946. doi: 10.3390/cancers13081946
313. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-Associated Macrophages: An Accomplice in Solid Tumor Progression. J BioMed Sci (2019) 26:78. doi: 10.1186/s12929-019-0568-z
314. Roesch S, Rapp C, Dettling S, Herold-Mende C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int J Mol Sci (2018) 19:436. doi: 10.3390/ijms19020436
315. Liguori M, Buracchi C, Pasqualini F, Bergomas F, Pesce S, Sironi M, et al. Functional Trail Receptors in Monocytes and Tumor-Associated Macrophages: A Possible Targeting Pathway in the Tumor Microenvironment. Oncotarget (2016) 7:41662–76. doi: 10.18632/oncotarget.9340
316. Popovic PJ, Zeh HJ 3rd, Ochoa JB. Arginine and Immunity. J Nutr (2007) 137:1681S–6S. doi: 10.1093/jn/137.6.1681S
317. Wang XF, Wang HS, Wang H, Zhang F, Wang KF, Guo Q, et al. The Role of Indoleamine 2,3-Dioxygenase (Ido) in Immune Tolerance: Focus on Macrophage Polarization of Thp-1 Cells. Cell Immunol (2014) 289:42–8. doi: 10.1016/j.cellimm.2014.02.005
318. Han Y, Liu D, Li L. Pd-1/Pd-L1 Pathway: Current Researches in Cancer. Am J Cancer Res (2020) 10:727–42.
319. Yu GT, Bu LL, Zhao YY, Mao L, Deng WW, Wu TF, et al. Ctla4 Blockade Reduces Immature Myeloid Cells in Head and Neck Squamous Cell Carcinoma. Oncoimmunology (2016) 5:e1151594. doi: 10.1080/2162402X.2016.1151594
320. Dockrell DH, Badley AD, Villacian JS, Heppelmann CJ, Algeciras A, Ziesmer S, et al. The Expression of Fas Ligand by Macrophages and its Upregulation by Human Immunodeficiency Virus Infection. J Clin Invest (1998) 101:2394–405. doi: 10.1172/JCI1171
321. Landry AP, Balas M, Alli S, Spears J, Zador Z. Distinct Regional Ontogeny and Activation of Tumor Associated Macrophages in Human Glioblastoma. Sci Rep (2020) 10:19542. doi: 10.1038/s41598-020-76657-3
322. Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, et al. Increased Microglia/Macrophage Gene Expression in a Subset of Adult and Pediatric Astrocytomas. PloS One (2012) 7:e43339. doi: 10.1371/journal.pone.0043339
323. Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The Brain Tumor Microenvironment. Glia (2012) 60:502–14. doi: 10.1002/glia.21264
324. Hambardzumyan D, Gutmann DH, Kettenmann H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat Neurosci (2016) 19:20–7. doi: 10.1038/nn.4185
325. Hu Y, He MY, Zhu LF, Yang CC, Zhou ML, Wang Q, et al. Tumor-Associated Macrophages Correlate With the Clinicopathological Features and Poor Outcomes via Inducing Epithelial to Mesenchymal Transition in Oral Squamous Cell Carcinoma. J Exp Clin Cancer Res (2016) 35:12. doi: 10.1186/s13046-015-0281-z
326. Liu S, Zhang C, Maimela NR, Yang L, Zhang Z, Ping Y, et al. Molecular and Clinical Characterization of Cd163 Expression via Large-Scale Analysis in Glioma. Oncoimmunology (2019) 8:1601478. doi: 10.1080/2162402X.2019.1601478
327. Nishie A, Ono M, Shono T, Fukushi J, Otsubo M, Onoue H, et al. Macrophage Infiltration and Heme Oxygenase-1 Expression Correlate With Angiogenesis in Human Gliomas. Clin Cancer Res (1999) 5:1107–13. doi: 10.1101/cshperspect.a020537
328. Ginhoux F, Prinz M. Origin of Microglia: Current Concepts and Past Controversies. Cold Spring Harb Perspect Biol (2015) 7:a020537. doi: 10.1101/cshperspect.a020537
329. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res (2017) 77:2266–78. doi: 10.1158/0008-5472.CAN-16-2310
330. Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep (2016) 17:2445–59. doi: 10.1016/j.celrep.2016.10.052
331. Sevenich L. Brain-Resident Microglia and Blood-Borne Macrophages Orchestrate Central Nervous System Inflammation in Neurodegenerative Disorders and Brain Cancer. Front Immunol (2018) 9:697. doi: 10.3389/fimmu.2018.00697
332. Desbaillets I, Tada M, de Tribolet N, Diserens AC, Hamou MF, Van Meir EG. Human Astrocytomas and Glioblastomas Express Monocyte Chemoattractant Protein-1 (Mcp-1) in Vivo and in Vitro. Int J Cancer (1994) 58:240–7. doi: 10.1002/ijc.2910580216
333. de Vrij J, Maas SL, Kwappenberg KM, Schnoor R, Kleijn A, Dekker L, et al. Glioblastoma-Derived Extracellular Vesicles Modify the Phenotype of Monocytic Cells. Int J Cancer (2015) 137:1630–42. doi: 10.1002/ijc.29521
334. Leblond MM, Gerault AN, Corroyer-Dulmont A, MacKenzie ET, Petit E, Bernaudin M, et al. Hypoxia Induces Macrophage Polarization and Re-Education Toward an M2 Phenotype in U87 and U251 Glioblastoma Models. Oncoimmunology (2016) 5:e1056442. doi: 10.1080/2162402X.2015.1056442
335. Gabrusiewicz K, Ellert-Miklaszewska A, Lipko M, Sielska M, Frankowska M, Kaminska B. Characteristics of the Alternative Phenotype of Microglia/Macrophages and its Modulation in Experimental Gliomas. PloS One (2011) 6:e23902. doi: 10.1371/journal.pone.0023902
336. da Fonseca AC, Badie B. Microglia and Macrophages in Malignant Gliomas: Recent Discoveries and Implications for Promising Therapies. Clin Dev Immunol (2013) 2013:264124. doi: 10.1155/2013/264124
337. Chen Z, Hambardzumyan D. Immune Microenvironment in Glioblastoma Subtypes. Front Immunol (2018) 9:1004. doi: 10.3389/fimmu.2018.01004
338. Choi J, Mai N, Jackson C, Belcaid Z, Lim M. It Takes Two: Potential Therapies and Insights Involving Microglia and Macrophages in Glioblastoma. Neuroimmunol Neuroinflamm (2018) 5:42. doi: 10.20517/2347-8659.2018.47
339. van den Bossche WBL, Kleijn A, Teunissen CE, Voerman JSA, Teodosio C, Noske DP, et al. Oncolytic Virotherapy in Glioblastoma Patients Induces a Tumor Macrophage Phenotypic Shift Leading to an Altered Glioblastoma Microenvironment. Neuro Oncol (2018) 20:1494–504. doi: 10.1093/neuonc/noy082
340. Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell (2017) 32:253–67 e5. doi: 10.1016/j.ccell.2017.07.006
341. Vandenberk L, Garg AD, Verschuere T, Koks C, Belmans J, Beullens M, et al. Irradiation of Necrotic Cancer Cells, Employed for Pulsing Dendritic Cells (Dcs), Potentiates Dc Vaccine-Induced Antitumor Immunity Against High-Grade Glioma. Oncoimmunology (2016) 5:e1083669. doi: 10.1080/2162402X.2015.1083669
342. Dammeijer F, Lievense LA, Kaijen-Lambers ME, van Nimwegen M, Bezemer K, Hegmans JP, et al. Depletion of Tumor-Associated Macrophages With a Csf-1r Kinase Inhibitor Enhances Antitumor Immunity and Survival Induced by Dc Immunotherapy. Cancer Immunol Res (2017) 5:535–46. doi: 10.1158/2326-6066.CIR-16-0309
343. Mueller SN, Gebhardt T, Carbone FR, Heath WR. Memory T Cell Subsets, Migration Patterns, and Tissue Residence. Annu Rev Immunol (2013) 31:137–61. doi: 10.1146/annurev-immunol-032712-095954
344. Farber DL, Yudanin NA, Restifo NP. Human Memory T Cells: Generation, Compartmentalization and Homeostasis. Nat Rev Immunol (2014) 14:24–35. doi: 10.1038/nri3567
345. Sallusto F, Geginat J, Lanzavecchia A. Central Memory and Effector Memory T Cell Subsets: Function, Generation, and Maintenance. Annu Rev Immunol (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
346. Park J, Kwon M, Kim KH, Kim TS, Hong SH, Kim CG, et al. Immune Checkpoint Inhibitor-Induced Reinvigoration of Tumor-Infiltrating Cd8(+) T Cells is Determined by Their Differentiation Status in Glioblastoma. Clin Cancer Res (2019) 25:2549–59. doi: 10.1158/1078-0432.CCR-18-2564
347. Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A Novel Transcription Factor, T-Bet, Directs Th1 Lineage Commitment. Cell (2000) 100:655–69. doi: 10.1016/s0092-8674(00)80702-3
349. Schnell A, Bod L, Madi A, Kuchroo VK. The Yin and Yang of Co-Inhibitory Receptors: Toward Anti-Tumor Immunity Without Autoimmunity. Cell Res (2020) 30:285–99. doi: 10.1038/s41422-020-0277-x
350. Garber ST, Hashimoto Y, Weathers SP, Xiu J, Gatalica Z, Verhaak RG, et al. Immune Checkpoint Blockade as a Potential Therapeutic Target: Surveying Cns Malignancies. Neuro Oncol (2016) 18:1357–66. doi: 10.1093/neuonc/now132
351. Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, et al. Cd8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu Rev Med (2018) 69:301–18. doi: 10.1146/annurev-med-012017-043208
352. Philip M, Schietinger A. Cd8(+) T Cell Differentiation and Dysfunction in Cancer. Nat Rev Immunol (2021). doi: 10.1038/s41577-021-00574-3
353. Collier JL, Weiss SA, Pauken KE, Sen DR, Sharpe AH. Not-So-Opposite Ends of the Spectrum: Cd8(+) T Cell Dysfunction Across Chronic Infection, Cancer and Autoimmunity. Nat Immunol (2021) 22:809–19. doi: 10.1038/s41590-021-00949-7
354. Dubinski D, Wolfer J, Hasselblatt M, Schneider-Hohendorf T, Bogdahn U, Stummer W, et al. Cd4+ T Effector Memory Cell Dysfunction is Associated With the Accumulation of Granulocytic Myeloid-Derived Suppressor Cells in Glioblastoma Patients. Neuro Oncol (2016) 18:807–18. doi: 10.1093/neuonc/nov280
355. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A, et al. Programmed Death Ligand 1 Expression and Tumor-Infiltrating Lymphocytes in Glioblastoma. Neuro Oncol (2015) 17:1064–75. doi: 10.1093/neuonc/nou307
356. Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, et al. Pd-L1 Expression and Prognostic Impact in Glioblastoma. Neuro Oncol (2016) 18:195–205. doi: 10.1093/neuonc/nov172
357. Dejaegher J, Verschuere T, Vercalsteren E, Boon L, Cremer J, Sciot R, et al. Characterization of Pd-1 Upregulation on Tumor-Infiltrating Lymphocytes in Human and Murine Gliomas and Preclinical Therapeutic Blockade. Int J Cancer (2017) 141:1891–900. doi: 10.1002/ijc.30877
358. Goods BA, Hernandez AL, Lowther DE, Lucca LE, Lerner BA, Gunel M, et al. Functional Differences Between Pd-1+ and Pd-1- Cd4+ Effector T Cells in Healthy Donors and Patients With Glioblastoma Multiforme. PloS One (2017) 12:e0181538. doi: 10.1371/journal.pone.0181538
359. Mohme M, Schliffke S, Maire CL, Runger A, Glau L, Mende KC, et al. Immunophenotyping of Newly Diagnosed and Recurrent Glioblastoma Defines Distinct Immune Exhaustion Profiles in Peripheral and Tumor-Infiltrating Lymphocytes. Clin Cancer Res (2018) 24:4187–200. doi: 10.1158/1078-0432.CCR-17-2617
360. Luo XH, Meng Q, Rao M, Liu Z, Paraschoudi G, Dodoo E, et al. The Impact of Inflationary Cytomegalovirus-Specific Memory T Cells on Anti-Tumour Immune Responses in Patients With Cancer. Immunology (2018) 155:294–308. doi: 10.1111/imm.12991
361. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov (2018) 8:1069–86. doi: 10.1158/2159-8290.CD-18-0367
362. Marin-Acevedo JA, Chirila RM, Dronca RS. Immune Checkpoint Inhibitor Toxicities. Mayo Clin Proc (2019) 94:1321–9. doi: 10.1016/j.mayocp.2019.03.012
363. Lukas RV, Rodon J, Becker K, Wong ET, Shih K, Touat M, et al. Clinical Activity and Safety of Atezolizumab in Patients With Recurrent Glioblastoma. J Neurooncol (2018) 140:317–28. doi: 10.1007/s11060-018-2955-9
364. Reiss SN, Yerram P, Modelevsky L, Grommes C. Retrospective Review of Safety and Efficacy of Programmed Cell Death-1 Inhibitors in Refractory High Grade Gliomas. J Immunother Cancer (2017) 5:99. doi: 10.1186/s40425-017-0302-x
365. Filley AC, Henriquez M, Dey M. Recurrent Glioma Clinical Trial, Checkmate-143: The Game Is Not Over Yet. Oncotarget (2017) 8:91779–94. doi: 10.18632/oncotarget.21586
366. Chamberlain MC, Kim BT. Nivolumab for Patients With Recurrent Glioblastoma Progressing on Bevacizumab: A Retrospective Case Series. J Neurooncol (2017) 133:561–9. doi: 10.1007/s11060-017-2466-0
367. Blumenthal DT, Yalon M, Vainer GW, Lossos A, Yust S, Tzach L, et al. Pembrolizumab: First Experience With Recurrent Primary Central Nervous System (Cns) Tumors. J Neurooncol (2016) 129:453–60. doi: 10.1007/s11060-016-2190-1
368. de Groot J, Penas-Prado M, Alfaro-Munoz KD, Hunter K, Pei B, O’Brien B, et al. Window-of-Opportunity Clinical Trial of Pembrolizumab in Patients With Recurrent Glioblastoma Reveals Predominance of Immune-Suppressive Macrophages. Neuro Oncol (2019) 22:539–49. doi: 10.1093/neuonc/noz185
369. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant Nivolumab Modifies the Tumor Immune Microenvironment in Resectable Glioblastoma. Nat Med (2019) 25:470–6. doi: 10.1038/s41591-018-0339-5
370. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant Anti-Pd-1 Immunotherapy Promotes a Survival Benefit With Intratumoral and Systemic Immune Responses in Recurrent Glioblastoma. Nat Med (2019) 25:477–86. doi: 10.1038/s41591-018-0337-7
371. Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, et al. Intratumoral Tcf1(+)Pd-1(+)Cd8(+) T Cells With Stem-Like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity (2019) 50:195–211.e10. doi: 10.1016/j.immuni.2018.12.021
372. Antonios JP, Soto H, Everson RG, Orpilla J, Moughon D, Shin N, et al. Pd-1 Blockade Enhances the Vaccination-Induced Immune Response in Glioma. JCI Insight (2016) 1:e87059. doi: 10.1172/jci.insight.87059
Keywords: dendritic cells, vaccination, immunotherapy, glioblastoma, review (article), glioma, brain tumor
Citation: Datsi A and Sorg RV (2021) Dendritic Cell Vaccination of Glioblastoma: Road to Success or Dead End. Front. Immunol. 12:770390. doi: 10.3389/fimmu.2021.770390
Received: 03 September 2021; Accepted: 11 October 2021;
Published: 02 November 2021.
Edited by:
I. Jolanda M. De Vries, Radboud University Nijmegen Medical Centre, NetherlandsReviewed by:
Evelien Smits, University of Antwerp, BelgiumCopyright © 2021 Datsi and Sorg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rüdiger V. Sorg, cnVlZGlnZXIuc29yZ0BtZWQudW5pLWR1ZXNzZWxkb3JmLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.