
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 22 November 2021
Sec. Comparative Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.769204
 Jiaojiao Yang1
Jiaojiao Yang1 Qiaoli Yang1
Qiaoli Yang1 Juanli Zhang1Xiaoli Gao1
Juanli Zhang1Xiaoli Gao1 Ruirui Luo1Kaihui Xie1Wei Wang2Jie Li1
Ruirui Luo1Kaihui Xie1Wei Wang2Jie Li1 Xiaoyu Huang1Zunqiang Yan1Pengfei Wang1
Xiaoyu Huang1Zunqiang Yan1Pengfei Wang1 Shuangbao Gun1,3*
Shuangbao Gun1,3*Background: The n6-methyladenosine (m6A) modification is present widely in mRNAs and long non-coding RNAs (lncRNAs), and is related to the occurrence and development of certain diseases. However, the role of m6A methylation in Clostridium perfringens type C infectious diarrhea remains unclear.
Methods: Here, we treated intestinal porcine jejunum epithelial cells (IPEC-J2 cells) with Clostridium perfringens beta2 (CPB2) toxin to construct an in vitro model of Clostridium perfringens type C (C. perfringens type C) infectious diarrhea, and then used methylated RNA immunoprecipitation sequencing (MeRIP-seq) and RNA sequencing (RNA-seq) to identify the methylation profiles of mRNAs and lncRNAs in IPEC-J2 cells.
Results: We identified 6,413 peaks, representing 5,825 m6A-modified mRNAs and 433 modified lncRNAs, of which 4,356 m6A modified mRNAs and 221 m6A modified lncRNAs were significantly differential expressed between the control group and CPB2 group. The motif GGACU was enriched significantly in both the control group and the CPB2 group. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation analysis showed that the differentially methylated modified mRNAs were mainly enriched in Hippo signaling pathway and Wnt signaling pathway. In addition, the target genes of the differentially m6A modified lncRNAs were related to defense response to virus and immune response. For example, ENSSSCG00000042575, ENSSSCG00000048701 and ENSSSCG00000048785 might regulate the defense response to virus, immune and inflammatory response to resist the harmful effects of viruses on cells.
Conclusion: In summary, this study established the m6A transcription profile of mRNAs and lncRNAs in IPEC-J2 cells treated by CPB2 toxin. Further analysis showed that m6A-modified RNAs were related to defense against viruses and immune response after CPB2 toxin treatment of the cells. Threem6A-modified lncRNAs, ENSSSCG00000042575, ENSSSCG00000048785 and ENSSSCG00000048701, were most likely to play a key role in CPB2 toxin-treated IPEC-J2 cells. The results provide a theoretical basis for further research on the role of m6A modification in piglet diarrhea.
Bacterial diarrhea in piglets, which leads to a decline in piglet survival and seriously affects pig husbandry, is an urgent problem in pig production. Clostridium perfringens type C (C. perfringens type C) is one of the main pathogenic bacteria causing diarrhea in piglets (1). C. perfringens type C attaches to the top of the villi of intestinal epithelial cells and multiplies in the chorionic basement membrane to produce C. perfringens beta2 (CPB2) toxin. CPB2 toxin acts directly on the intestine or is absorbed through the mucosa into the blood circulation, damaging intestinal epithelial cells and triggering an intestinal inflammatory response (2–4). Currently, the prevention and treatment of this diarrhea disease relies mainly on vaccines and antibiotics. Although these measures have reduced and controlled the occurrence of diarrhea to some extent, long-term use of antibiotics has led to increased bacterial resistance and resulted in food safety and human health concerns. In addition, mutation of pathogenic bacteria is a continuous challenge for the research and development of vaccines, making the prevention and treatment of piglet diarrhea more difficult. Therefore, identifying molecular markers related to piglet diarrhea resistance and determining its mechanism of action in the resistance to pathogen infection have become important methods to solve this problem.
RNA methylation modification is one of the important areas of epigenetic research, and n6–methyladenosine (m6A) is a common type of modification in mammalian cells (5). M6A is present widely in eukaryotic mRNAs, long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) (6). The m6A modification regulates protein expression by influencing the processes of mRNA shearing, transport, translation, and degradation, and participates in the process of resisting bacterial and viral infections, inflammatory bowel disease, and immune responses (7–12). With the emergence of methylated RNA immunoprecipitation sequencing (MeRIP-seq) technology, research into m6A methylation modification has entered a stage of rapid development, which has greatly promoted the identification of RNA m6A modifications in different species (13).
LncRNAs are non-coding RNAs with a length of more than 200 nt, which have crucial roles in the occurrence and development of diarrhea caused by pathogen infection in the host. In the process of piglet diarrhea caused by Porcine epidemic diarrhea virus (PEDV) infection, lncRNA-9606 plays a regulatory role by activating the T cell receptor signaling pathway of the ileal immune system and promoting the production of IgA (14). LncRNA-EPS affects the innate immune response by regulating the expression of immune response genes in mouse macrophages (15). Previous studies have shown that lnc_033304 and lnc_001066 might be involved in the defense of piglets against C. perfringens type C infection by regulating the key genes of the nuclear factor kappa B (NF-κB), mitogen-activated protein kinase (MAPK), and Toll-like receptor signaling pathways (16). However, whether m6A methylation–regulated lncRNAs are involved in the process of piglet resistance to diarrhea infection, and the molecular mechanism of their functions, are currently unclear. Therefore, it is necessary to study the function and mechanism of m6A-modified lncRNAs in the process of piglet diarrhea.
In the present study, a cell model of C. perfringens type C infection was constructed, and MeRIP-seq was performed on these cells. We obtained the transcription profiles of m6A-modified mRNAs and lncRNAs in intestinal porcine jejunum epithelial cells (IPEC-J2) treated with CPB2 toxin. The general characteristics, topological patterns, and participation in biological pathways of m6A methylation were compared, thereby providing an improved understanding of the roles of the m6a modification in piglet diarrhea. The results of the present study expand our understanding of the molecular mechanisms underlying the m6A modification in piglet diarrhea.
We used the method described by Gao and Luo (17, 18) to extract and purify the CPB2 toxin protein. Then, the endotoxin was removed using Endotoxin Erasol (Genscript, Nanjing, China). Finally, the integrity of the CPB2 protein was assessed using 12% SDS–PAGE.
IPEC-J2 cells were purchased from the BeNa Culture Collection (BNCC, Beijing, China) and cultured in Dulbecco’s modified Eagle’s medium (DMEM, Grand island, NY, USA)/F12 medium (Gibco), to which 10% FBS (fetal bovine serum; GeminiBio, Sacramento, CA, USA) and antibiotics (penicillin and streptomycin) were added; the cells were maintained at 37°C in a 5% CO2 atmosphere. IPEC-J2 cells were stimulated with CPB2 (20 μg/mL) for 24 h, which was defined as the CPB2 group, and unstimulated IPEC-J2 cells were defined as the control group.
Total RNA was extracted using the TRIzol™ reagent (Invitrogen, Carlsbad, CA, USA). An EpiQuik m6A RNA methylation quantification kit (colorimetric method) (AmyJet Scientific, Wuhan, China) was then used to determine the total m6A content.
Total RNA from IPEC-J2 cells was isolated using the TRIzol™ reagent. The RNA concentration and integrity of IPEC-J2 cells were estimated using a NanoDrop ND-8000 spectrophotometer (NanoDrop, Wilmington, DE, USA) and an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) to select high-quality RNA samples [RNA integrity number (RIN) > 7). A Ribo-Zero rRNA Removal Kit (Illumina, San Diego, CA, USA] was used to remove ribosomal RNA from the total RNA exceeding 25 μg. Then, these RNAs were fragmented into small fragments using a Magnesium RNA Fragmentation Module (NEB, Ipswich, MA, USA; cat no. e6150). Half of the approximately 100 nt fragmented RNAs were used for immunoprecipitation (IP), and the other half was used as the IP control (Input). Then, the cleaved RNA fragments were incubated with an m6A-specific antibody (No. 202003, Synaptic Systems, Göttingen, Germany) to obtain IP RNA, which was then reverse transcribed to generate cDNA, end repaired, added with an A tail and connectors, purified, and PCR enriched, all performed according to the manufacturer’s instructions. MeRIP-Seq and RNA-seq libraries (IP and Input) were then constructed, and 2 × 150 bp paired-end sequencing (PE150) was performed on an Illumina Novaseq™ 6000 instrument by LC-Bio Technology CO., Ltd. (Hangzhou, China).
Fstp software was used to perform quality control on the original data from the IP and Input samples, including removing connectors, repeated sequences, and low-quality sequences (quality scores < Q20) to obtain clean data (19). The clean reads were mapped to the pig reference genome sequence (Sus scrofa 11.1 v101) using HISAT2 (20). The R package exomePeak was used to perform peak calling analysis and genetic difference peak analysis on the IP and Input samples, and then we used ChIPseeker to annotate the peaks (21). Finally, MEME2, HOMER, and ChIPseeker software were used for motif analysis, positioning, and annotation (22–24). StringTie software was used for gene assembly and quantification via FPKM (fragments per kilobase of exon model per million mapped reads) (25).
Transcriptomes from the samples were merged to reconstruct a comprehensive transcriptome using gffcompare (26). First, we removed the known mRNAs and transcripts smaller than 200 bp, and then used CPC (27) and CNCI (28) to predict the coding potential of the remaining transcripts, filtering out the transcripts with CPC scores < 1 and CNCI scores < 0 to obtain lncRNAs. Significantly differentially expressed mRNAs and lncRNAs were selected using |log2 (fold change)| ≥ 1.0 and P-value < 0.05 in the R package edgeR (29).
Cis and trans methods were used to analyze and predict the target genes of the lncRNAs. The cis-regulated target genes were derived from differential expressed mRNAs in the range of 100 kb upstream and downstream of the lncRNA (30). The trans-regulatory target genes were identified using RIsearch (36.3) software (31). The free energy of forming a secondary structure between an lncRNA and an mRNA sequence should be lower than the threshold value of -11, and they should also show expression correlation (the absolute value of Pearson correlation coefficient should be greater than 0.95). Finally, Gene Ontology (GO) (http://www.geneontology.org/) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.kegg.jp/) enrichment analysis were used to annotate the functions of the lncRNA target genes (32, 33).
M6A-modified RNAs from the IP sample and RNAs from the Input sample were reverse–transcribed into cDNA using a reverse transcriptase kit (TransGen Biotech, Beijing, China). Quantitative real-time polymerase chain reaction (qPCR) was performed using the cDNA as the template on a LightCycler 480 instrument (Roche Applied Science, Mannheim, Germany) with the SYBR Green Realtime PCR Master Mix (Accurate Biotechnology, Hunan, China). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. The primer information is presented in Supplementary Table 1. The relative expression levels were calculated using the 2−ΔΔCt method (34).
SPSS v.20 software (IBM Corp, Armonk, NY, USA) and GraphPad Prism 6.0 software (GraphPad Inc., La Jolla, CA, USA) were used for all statistical analyses. Data are displayed as the mean ± SD. The analysis was performed using Student’s t test (two-tailed). P < 0.05 (*) and P < 0.01 (**) were considered statistically significant.
To explore whether CPB2 toxin-treated cells were related to m6A methylation, the overall level of m6A methylation was determined. These results showed that the overall level of m6A methylation increased significantly after CPB2 toxin treatment (Figure 1). Therefore, we speculated that the m6A modification was involved in the physiological changes of IPEC-J2 cells after CPB2 treatment.
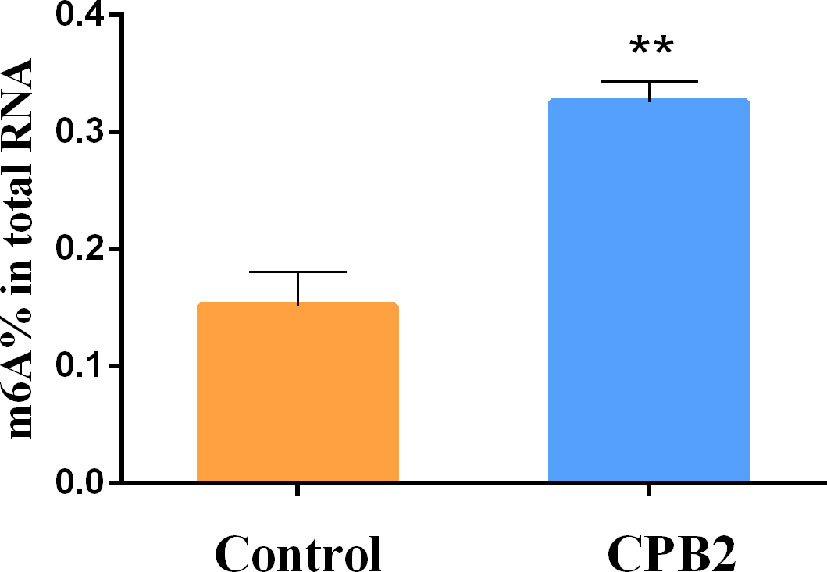
Figure 1 The total m6A content in the CPB2-treated IPEC-J2 cells ** means P < 0.01.
Transcriptome sequencing constructed 12 libraries from six samples, with more than 82,300,026 clean reads per sample in MeRIP-Seq data. The ratio of Q30 (sequencing error rate less than 0.001) for all samples ranged from 90.59 to 91.17%. The average GC content of the twelve libraries was 54.41%. (Supplementary Table 2). About 80.00% of the clean reads could be aligned to the pig reference genome sequence (Sus scrofa 11.1), with above 60.00% of the reads being uniquely aligned (Supplementary Table 3). According to the regional information of the pig reference genome, clean reads that can be matched to the reference genome were defined as exons, introns and intergenic sequences. In the control group, the average proportions of exons, introns, and intergenic sequences of IPEC-J2 cell IP samples were 75.06%, 21.39%, and 3.55%, and were 69.77%, 27.84%, and 2.39%, respectively, in the input samples. In addition, after CPB2 toxin treatment, the average proportions of exon, intron, and intergenic sequences in the IP samples from IPEC-J2 cells were 73.20%, 23.34%, and 3.46%, respectively, and were 64.62%, 32.74%, and 2.64%, respectively, in the Input samples (Supplementary Table 4).
The peak-calling software, R package exomePeak, was used to perform peak scanning of the entire gene range. The peak identification data came from MeRIP-seq sequencing data. Regions with a P value < 0.05 were considered as peaks. All samples were combined to show the enrichment of peaks distribution near the gene transcription start site (TSS) (Figure 2A). A total of 6,413 peaks were simultaneously identified in the two groups. Among them, there were 1,752 significantly different peaks, identified using screening conditions of |log2 (fold change) | ≥ 1, and P < 0.05. Compared with the control group, 521 peaks were upregulated in the CPB2 group involved 78 differentially expressed genes, and 1,231 peaks were downregulated in the CPB2 group involved 143 differentially expressed genes (Supplementary Table 5). The m6A modification can affect mRNA transcription, splicing, localization, translation, stability, and post-transcriptional regulation at the RNA level. To further understand the preferential position of m6A on the transcripts, we divided the transcripts into four areas: 5’ untranslated region (5’ UTR), 1st exon, other exon, and 3′ UTR (Figure 2B). We found that the differential m6A peaks were mainly enriched in the 3′ UTRs. At the same time, the peak density further confirmed that m6A was enriched around the 3’ UTR (Figure 2C). The top 20 significantly different m6A modified peaks are shown in Table 1.
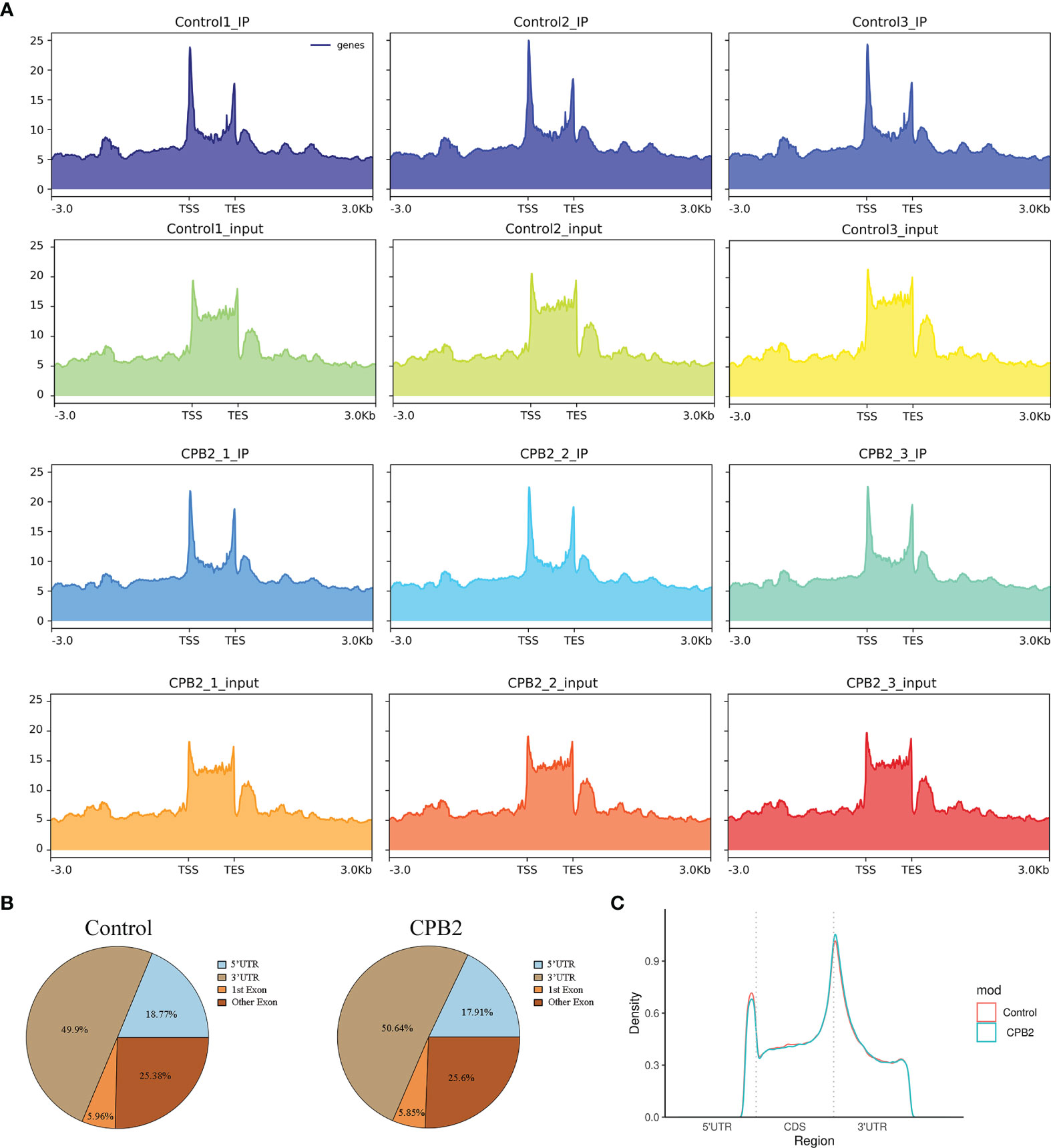
Figure 2 General features of IPEC-J2 cells m6A methylation. (A) The enrichment of peaks near the gene transcription start site. (B) The position distribution of m6A peaks on gene functional elements. (C) The m6A peak density along a metagene.
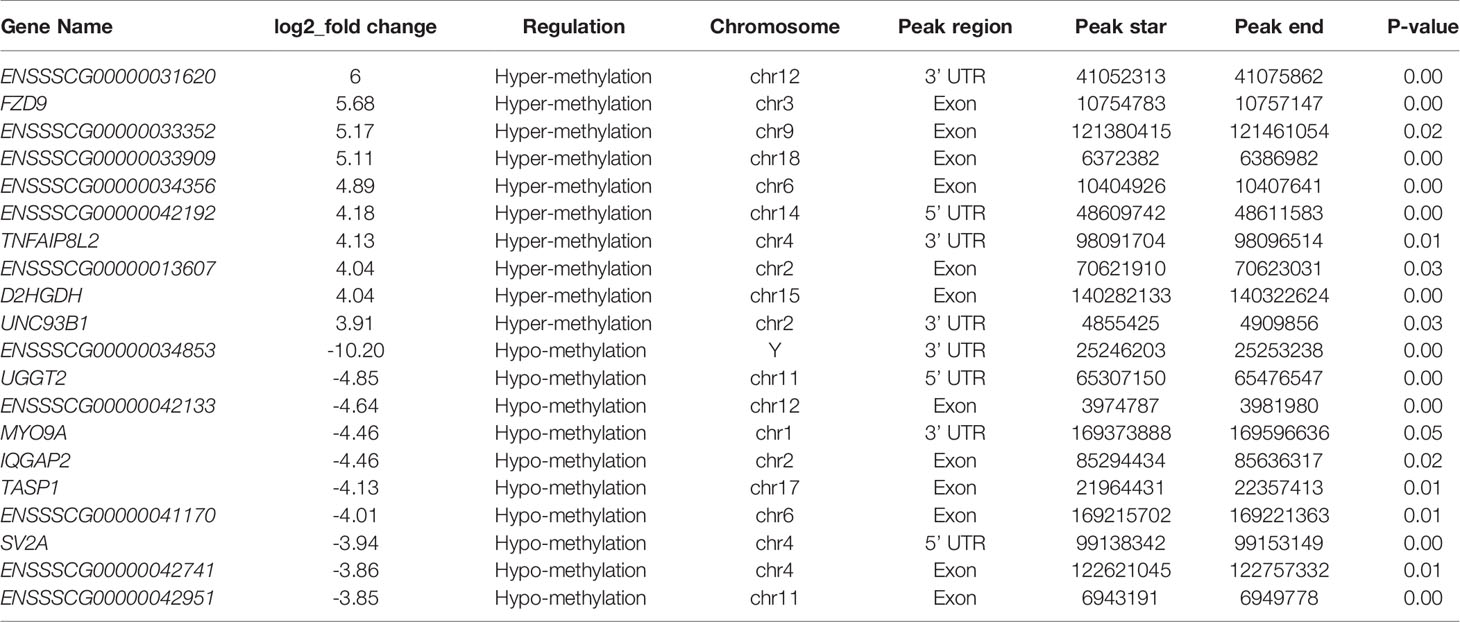
Table 1 The top 20 significantly differentially expressed m6A peaks.
Motifs are nucleic acid sequence patterns with biological significance, and RNA methylation–related enzymes can recognize and bind these motifs to regulate gene expression. To determine whether the identified m6A peaks contained the RRACH (R = A or G, H = A, C or U) conserved sequence motif, HOMER (motif analysis software) was used to find credible motifs in the peak areas. These results showed that both the control group and the CPB2 group contained the motif GGACU (Figure 3).
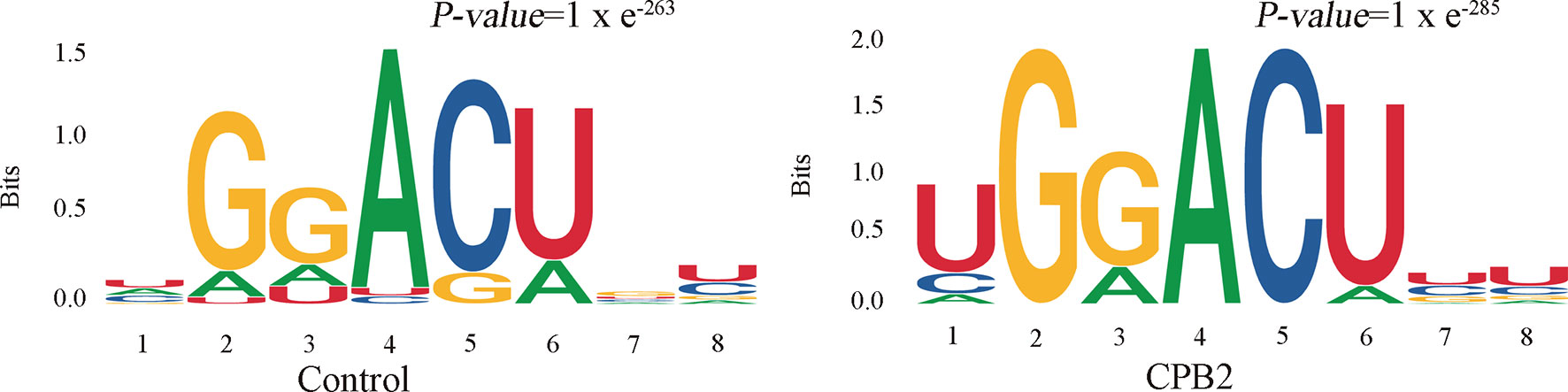
Figure 3 The RRACH conserved sequence motif for m6A peak regions.
To investigate the effect of CPB2 toxin treatment on IPEC-J2 cells, we analyzed the levels of differentially expressed mRNAs and lncRNAs in the cells between the control group and CPB2 group using the edge R software. The expression level and quantity of mRNAs and lncRNAs are shown in Figures 4A, B. The volcano map shows the differentially expressed lncRNAs and mRNAs (Figures 4C, D). There were 760 mRNAs and 49 lncRNAs showing significant differential expression between the control group and the CPB2 group. Compared with the control group, 39 lncRNAs and 596 mRNAs were significantly upregulated in the CPB2 group, while 10 lncRNAs and 164 mRNAs were significantly downregulated in the CPB2 group.
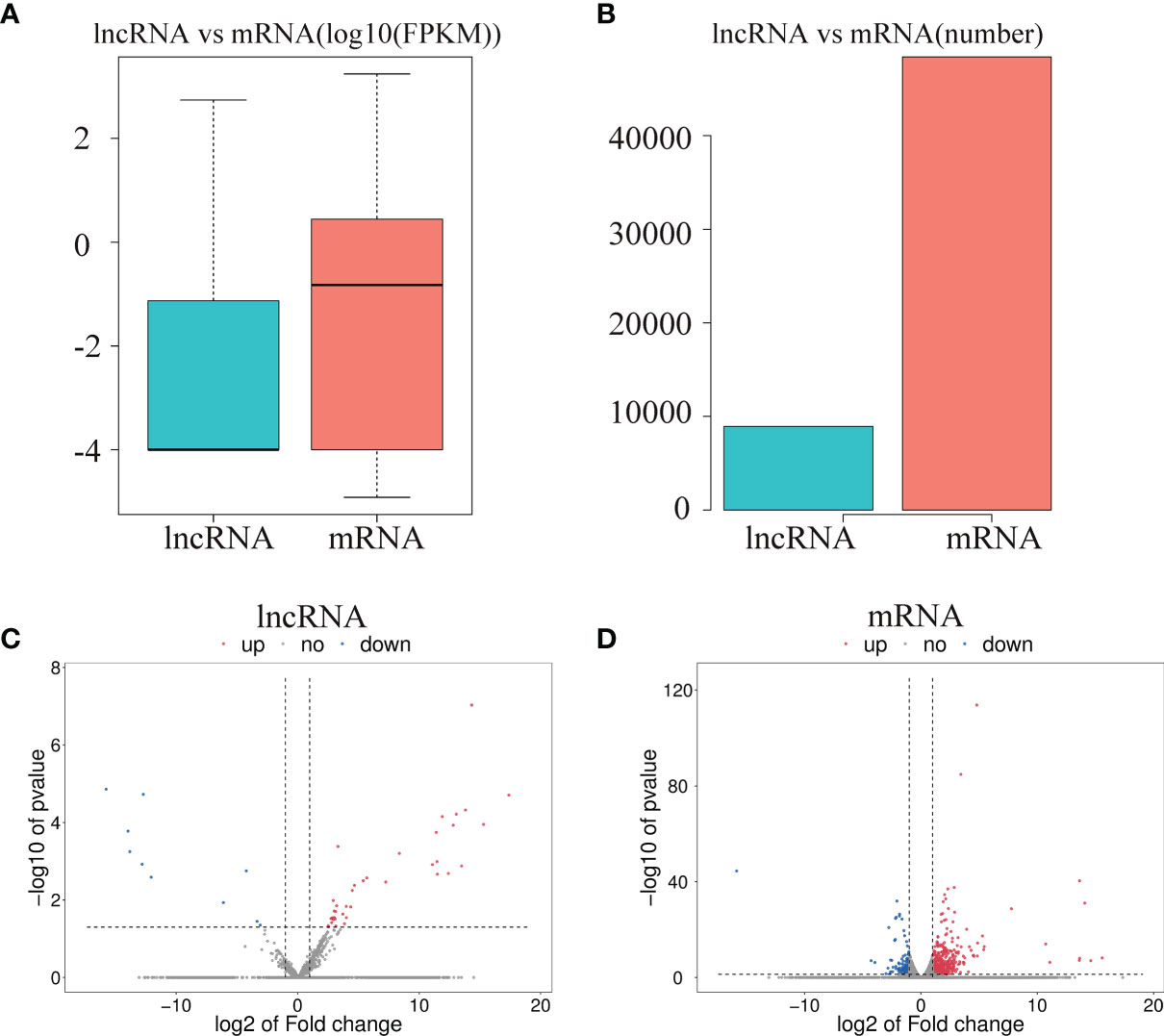
Figure 4 The differential expression of lncRNAs and mRNAs. (A) The significantly differentially expressed lncRNAs and mRNAs between the CPB2 group and the control group (using the log10 FPKM method) box plots. (B) The number of significantly differentially expressed lncRNAs and mRNAs. (C) The differentially expressed lncRNAs in the control group and CPB2 group. (D) The differentially expression of mRNAs in the control group and the CPB2 group. Blue indicates a significant increase, and red indicates a significant decrease.
To explore the function of m6A-modified mRNAs, GO and KEGG pathway analyses were performed for the 4,356 significantly differentially expressed m6A-modified mRNAs. The screening threshold was |(log2 (fold change)| ≥ 1, and P < 0.05. These mRNAs were enriched for 2,801 GO terms and 244 KEGG pathways (Supplementary Table 6). The significantly enriched GO terms included defense response to positive regulation of histone methylation and proximal promoter sequence-specific DNA binding (Figures 5A, B). KEGG Pathway analysis recognized that the differentially m6A-modified mRNAs were significantly associated with Herpes simplex virus 1 infection, Regulating pluripotency of stem cells, the Wnt signaling pathway, and the Hippo signaling pathway (Figure 5C).
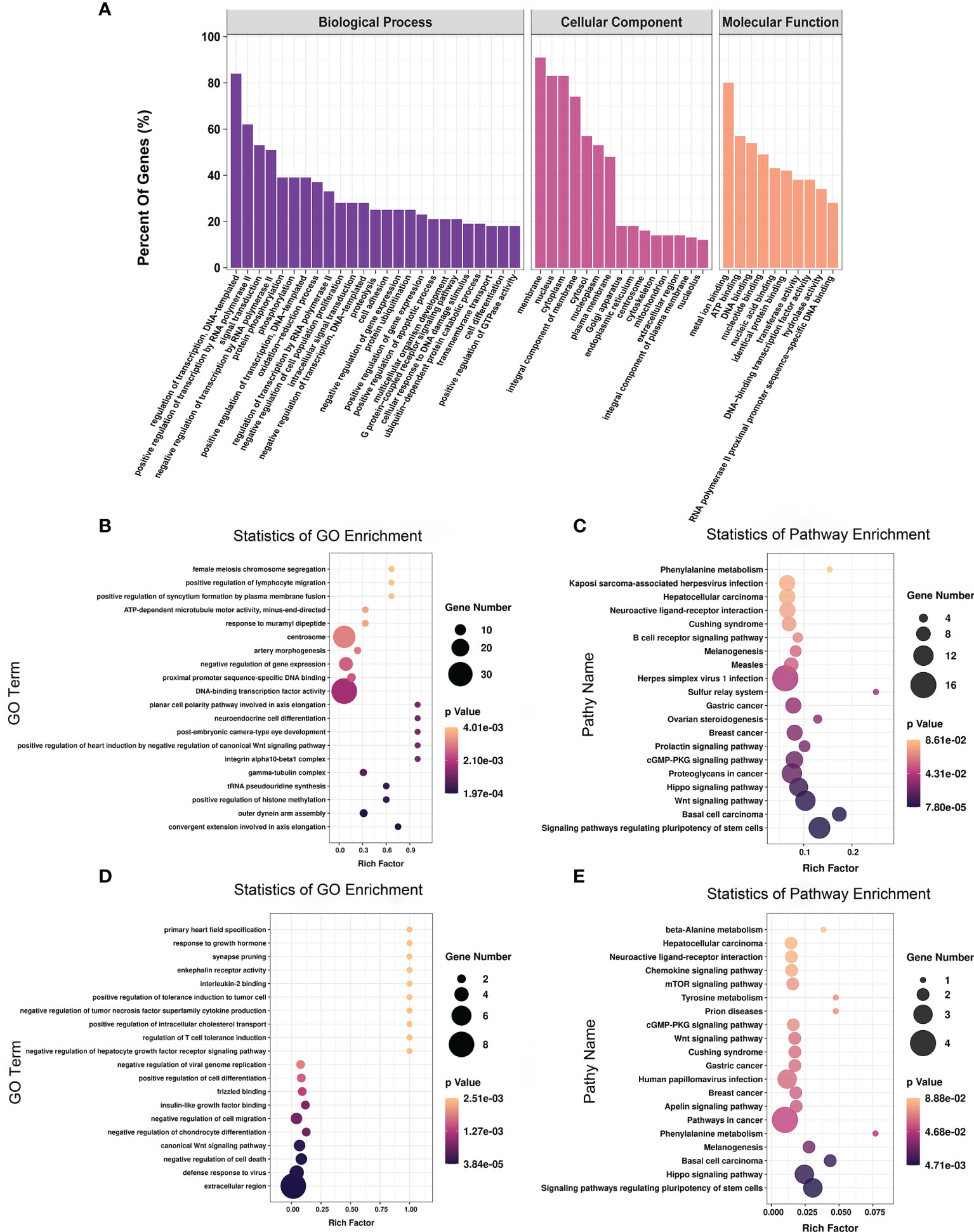
Figure 5 The distribution and functional analysis for differential m6A-modified mRNAs. GO enrichment (A, B) and KEGG signaling pathway analysis (C) of differential m6A-modified mRNAs. GO enrichment (D) and KEGG signaling pathway analysis (E) of mRNAs with simultaneous differences in m6A and transcription levels.
To further investigate the effect of m6A-modified mRNAs on CPB2-induced IPEC-J2 cells, we conducted a combined analysis of MeRIP-seq and RNA-seq data. We identified 36 mRNAs with significant differences in their methylation level and transcription level (Supplementary Table 7). The screening criteria were |(log2 (fold change)| ≥ 1, and P < 0.05. GO enrichment analysis of these 36 genes found that extracellular region, defense response to virus, and negative regulation of cell death were all significantly enriched terms (Figure 5D). In the KEGG analysis, signaling pathways regulating the pluripotency of stem cells, the Hippo signaling pathway, and pathways in cancer were the most significant enriched pathways (Figure 5E).
To further explore the functions of the 153 significantly differentially expressed m6A-modified lncRNAs, we used the screening conditions |(log2 (fold change)| ≥ 0.585, and P < 0.05 to predict the cis–regulated and trans-regulated target genes of these lncRNAs. The results showed that a total of 140 target genes could be predicted (Supplementary Table 8), and then GO and KEGG enrichment analyses were used to analyze the target genes. These target genes were enriched for 1,148 GO terms and 166 KEGG pathways. The significantly enriched GO terms included defense response to virus, negative regulation of viral genome replication, response to virus, response to interferon-alpha, and regulation of ribonuclease activity (Figures 6A, B). KEGG Pathway analysis showed that the target genes of the differentially expressed m6A-modified lncRNAs were significantly associated with Human papillomavirus infection, Influenza A, Hepatitis C, and the NF-kappa B signaling pathway (Figure 6C).
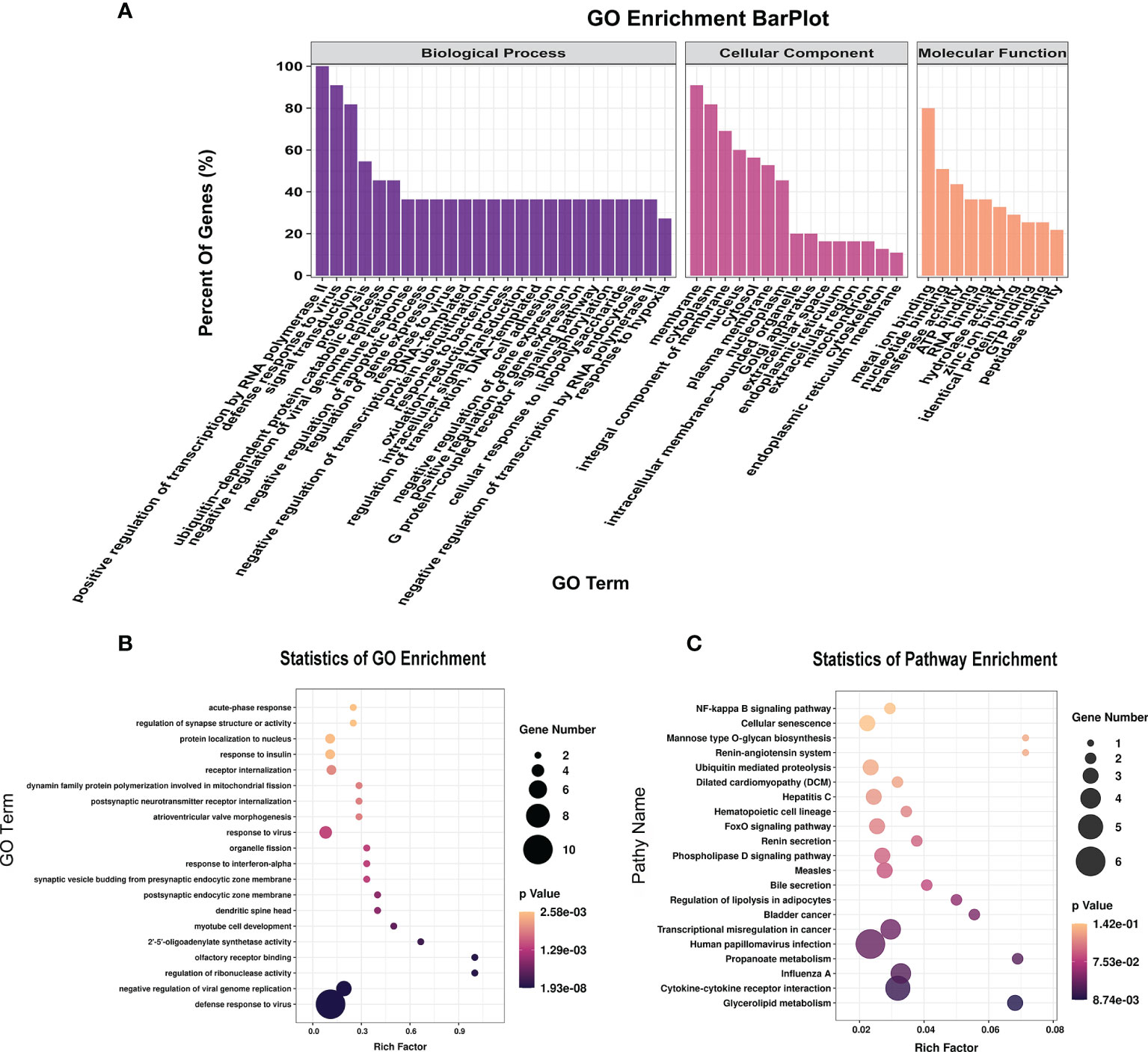
Figure 6 The distribution and functional analysis for differential m6A-modified lncRNAs. GO enrichment (A, B) and KEGG signaling pathway analysis (C) of the target genes of differentially m6A modified lncRNAs.
To study the effect of m6A-modified lncRNAs on CPB2-induced IPEC-J2 cells, we performed correlation analysis of the MeRIP-seq and RNA-seq data, and screened out six lncRNAs with the most significant differences in m6A modification level and transcription level (Supplementary Table 9). The screening threshold was |(log2 (fold change)| ≥ 0.585, and P < 0.05. Then these six lncRNAs and their corresponding target mRNAs were constructed into an lncRNA-mRNA network (Figure 7A). GO enrichment analysis of the target genes of these six lncRNAs found that defense response to virus was the most significant enrichment item, and immune response-activating signal transduction and Interleukin-8 receptor binding were significantly enriched terms (Figure 7B). In the KEGG analysis, cytokine-cytokine receptor interaction and NF-kappa B signaling pathway were the most significantly enriched pathways (Figure 7C). We found that there was a common differential expression target gene, CXCL8 (encoding C-X-C motif chemokine ligand 8), in these two pathways (Supplementary Table 10).
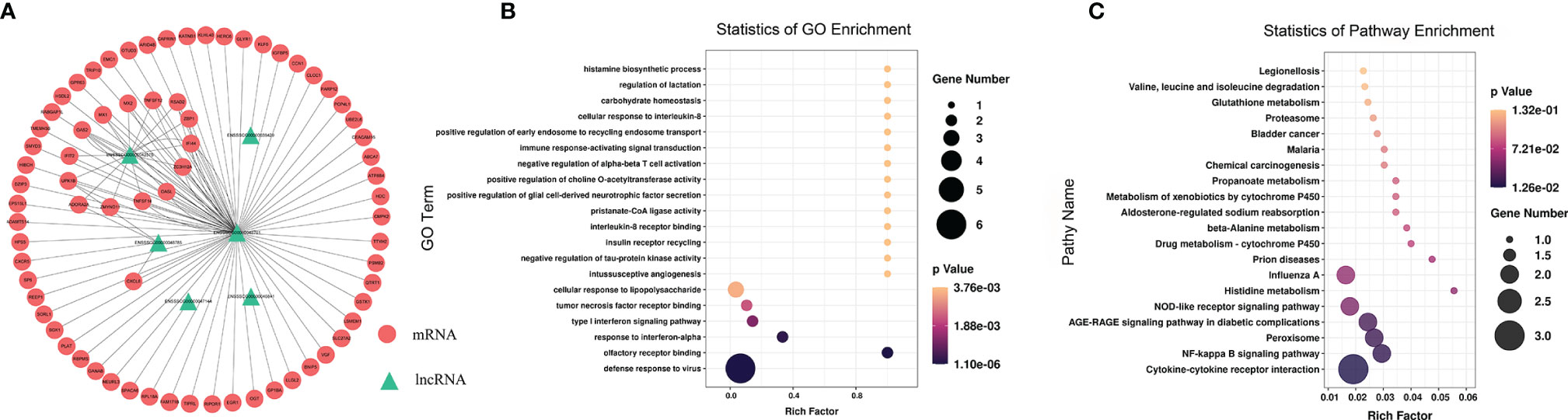
Figure 7 Combined analysis of lncRNAs in the RNA-seq and MeRIP-seq data. The network between differentially expressed m6A-modified lncRNAs and differentially expressed mRNAs (A). The red circles represent significantly differentially expressed mRNAs, and the green triangles represent significantly differentially expressed m6A-modified lncRNAs. GO enrichment (B) and KEGG signaling pathway analysis (C) of differentially expressed target genes of differentially m6A-modified lncRNAs.
To validate the reliability of our RNA-seq and MeRIP-seq results, six differentially expressed lncRNAs and five differentially expressed m6A-modified lncRNAs were selected randomly and subjected to qRT-PCR and MeRIP-qPCR analysis. In the qRT-PCR results, the expression levels of ENSSSCG00000042386, ENSSSCG00000045416, and ENSSSCG00000042575 were lower in the CPB2 group than in the control cells, while the expression levels of ENSSSCG00000048785, ENSSSCG00000041817, and ENSSSCG00000048701 were higher in the CPB2 group cells than in control cells (Figure 8A). In the MeRIP-qPCR results, the expression m6A level of m6A-modified ENSSSCG00000042386 was increased after CPB2 treated cells, while the expression of m6A-modified ENSSSCG00000048785, ENSSSCG00000042575, ENSSSCG00000048701, and ENSSSCG00000040169 were reduced after CPB2 treated cells (Figure 8B). These results indicated that the regulatory direction and expression pattern of these lncRNAs, as verified by RT-qPCR and MeRIP–qPCR, were consistent with the sequencing results, thus confirming the reliability and reproducibility of RNA-seq and MeRIP-seq methods.
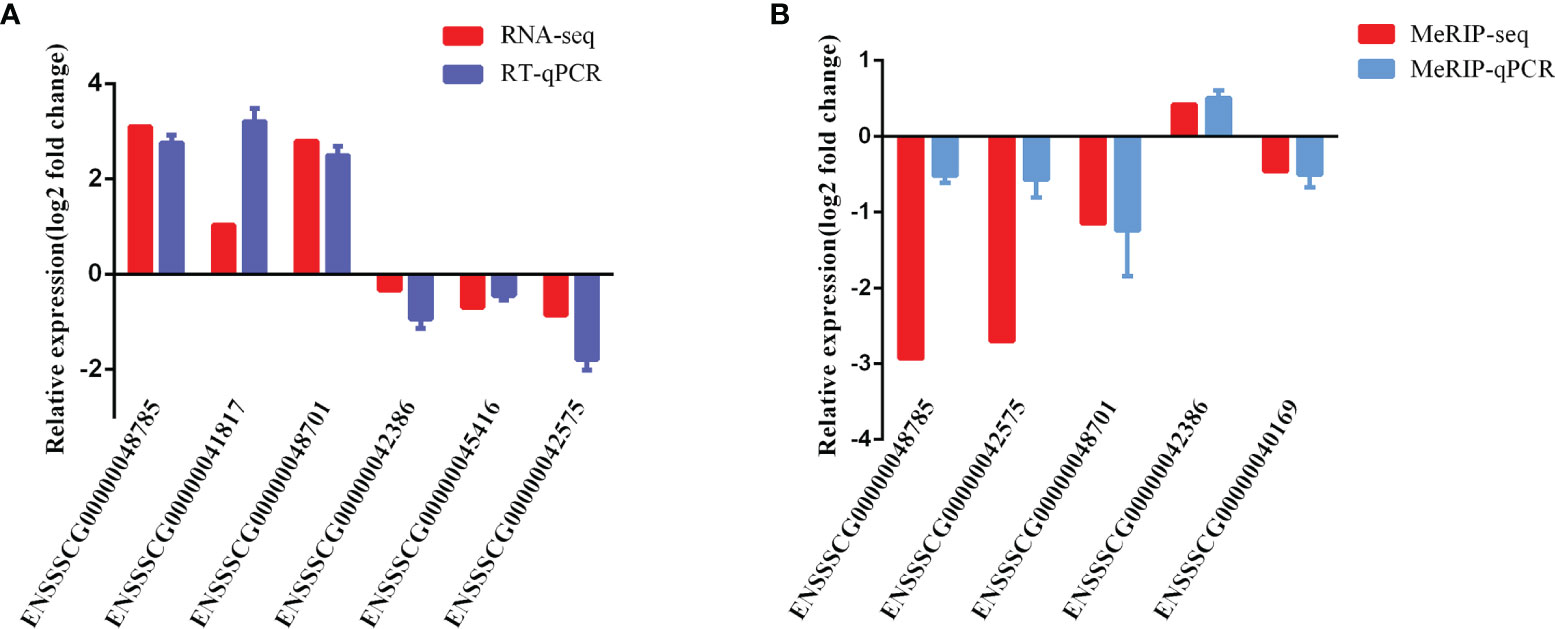
Figure 8 Validation of MeRIP-seq and RNA-seq by qRT-PCR and MeRIP-qPCR. (A) RT-qPCR validation of six lncRNAs. (B) MeRIP-qPCR validation of five m6A-modified lncRNAs.
C. perfringens type C is a common pathogen of newborn livestock. Animals infected with this pathogen might suffer from necrotizing enteritis, accompanied by segmental hemorrhagic and necrotizing jejunitis (35). CPB2 toxin is a strongly necrotic and deadly toxin produced by C. perfringens type C, which plays an important role in the occurrence and development of piglet diarrhea (36). However, the relationship between piglet diarrhea caused by C. perfringens type C infection and m6A methylation modification is rarely studied. In this study, IPEC-J2 cells were treated with CPB2 toxin to construct an in vitro model of C. perfringens type C infectious piglet diarrhea. MeRIP-seq and RNA-seq technologies were used to determine the presence of m6A-modified mRNAs and lncRNAs, and explore the functions of these m6A-modified mRNAs and lncRNAs, which would provide a reference to analyze piglet diarrhea from the perspective of m6A methylation.
The overall level of m6A methylation is associated with disease. Reducing m6A levels can promote the proliferation of cancer cells, and increasing m6A levels can significantly inhibit tumor development, both in vivo and in vitro (37). The total modification level of m6A in human lens epithelial cells (HLECs) was increased by high glucose, which promoted HLEC proliferation and inhibited their apoptosis (38). The protective effect of curcumin on lipopolysaccharide (LPS)-induced liver injury and the destruction of liver lipid metabolism might be the result of an increase in m6A RNA methylation (39). Treatment of IPEC-J2 cells with CPB2 toxin can induce apoptosis, inflammation, and barrier function damage (17, 18). In this study, the overall m6A RNA methylation level was significantly upregulated after CPB2 toxin treatment of IPEC-J2 cells, suggesting that m6A methylation is involved in the CPB2 toxin-induced damage to cells.
Many studies related to m6A have been carried out in mammals. For example, 8,306 m6A peaks related to 2,697 genes were detected in the sheep heat stress group, and 12,958 m6A peaks related to 5,494 genes were detected in the control group (40). In the present study, 6,413 different peaks were detected, of which 521 were significantly upregulated and 1,231 were significantly downregulated. From the different peaks, 4,356 mRNAs and 153 lncRNAs with significant differences were identified. M6A is most often found in proximity to stop codons or the 3’ UTR, and has been broadly linked to diverse aspects of mRNA metabolism and function, including altered splicing, decreased mRNA stability, and altered translational efficiency (41). A previous study noted that m6A peaks were enriched in the 3’ UTR and near the stop codons of mature polyadenylated mRNAs (42), which was similar to the results of our study. The m6A sites in the 3’UTR region of human immunodeficiency virus 1 (HIV-1) strongly enhanced the expression of mRNAs by recruiting the cell reader protein YTHDF (43). In this study, m6A peaks, which were mainly enriched in 3’ UTRs, might be involved in regulating the metabolism and function of mRNA.
RRACH is a highly conserved m6A motif in plants and animals (44, 45). RNAs containing the GGACU motif in Hela cell were good substrates for methylases (46). In oligodendrocyte progenitor cells (OPCs), the m6A reader PRRC2A (Proline rich coiled-coil 2 A) binds to the consensus GGACU motif in the Olig2 CDS (coding sequence) in an m6A-dependent manner to stabilize Olig2 mRNA, thus regulating cell proliferation (47). In our sequencing results, the motif sequence GGACU was also significantly enriched. However, whether stability of the RNAs with the GGACU motif site can change the stability of the mRNAs by binding to methylation-related enzymes, and whether they regulate the fate of IPEC-J2 cells induced by CPB2, remains to be further studied.
To explore the role of m6A-modified mRNAs in CPB2 toxin-induced IPEC-J2 cells, we performed statistical analysis of m6A-modified differentially expressed mRNA. We found that whether it was functional enrichment analysis of m6A-modified mRNAs or mRNAs obtained after combined analysis, the Hippo signaling pathway, Wnt signaling pathway and cancer-related signaling pathways were always significantly enriched. These signaling pathways are related to the occurrence of diseases, especially cancer (48, 49). These results indicated that methylated mRNAs play a regulatory role in the pathogenesis of the cells after CPB2 treatment.
To date, there has been no report on the pathway analysis of m6A-modified lncRNAs target genes in IPEC-J2 cells treated with CPB2 toxin. Therefore, in this study, we used GO and KEGG pathway annotation to analyze 153 m6A-modified lncRNA target genes that were significantly differentially expressed in IPEC-J2 cells exposed to 20 μg/mL CPB2 toxin. We attempted to identify the key functions and pathways by which CPB2 treatment may cause IPEC-J2 damage. Comparing the functional enrichment analysis of the significantly different m6A-modified lncRNAs target genes with that of the six lncRNAs from combined analysis showed that the defense response to virus, three pathways of NF-kappa B signaling pathway, cytokine-cytokine receptor interaction, and Influenza A were always significantly enriched. Further analysis revealed that the target gene CXCL8 coexisted in these three pathways. Target genes such as MX2 (encoding MX dynamin like GTPase 2), OAS2 (encoding 2’-5’-oligoadenylate synthetase 2), and IFIT2 (encoding interferon induced protein with tetratricopeptide repeats 2) were related to the defense response to viruses. MX2 is a cytokine induced by interferon IFN-α, which resists viral infection by inhibiting the accumulation of viral nuclear DNA (50, 51). As a new potentially sensitive biomarker, OAS2 is significantly up-regulated in psoriasis, a chronic inflammatory systemic disease, and can predict the severity and activity of the disease (52). The antiviral protein IFIT2 was found to be an important limiting factor for rabies virus, which can directly or indirectly resist virus replication (53). In addition, we also found that the three genes MX2, OAS2 and IFIT2, which were highly related to the defense response of the virus, were all target genes of lncRNA ENSSSCG00000042575. In summary, the differentially methylated lncRNA ENSSSCG00000042575 might resist virus invasion of cells by regulating target genes related to virus defense. Furthermore, we found that two target genes, ZC3H12A (encoding zinc finger CCCH-type containing 12A) and CXCL8, are related to immune response activation signal transduction and interleukin-8 receptor binding. ZC3H12A is an essential RNase that prevents immune disorders by directly controlling the stability of a group of inflammatory genes (54). There is compelling evidence suggesting that the m6A modification is particularly important in various pathological and physiological immune responses, including T cell homeostasis and differentiation, inflammation, and type I interferon production (55–58). We speculated that after CPB2 toxin treatment of cells, m6A modification might play an important role in the process of resistance to pathogen invasion by regulating virus defense-related genes, inflammation-related genes, and immune response-related genes.
CXCL8 (also known as interleukin IL-8), an activator of neutrophils, is associated with the initiation and maintenance of inflammation (59). Knockdown of lncRNA MALAT1 inhibited the expression of IL8 and reduced the activation of NF-κB, thereby reducing airway and lung inflammation caused by Mycoplasma pneumonia infection (60). In this work, the CXCL8 gene was enriched for multiple GO terms, such as inflammation, immune response, and cell cycle arrest. Moreover, CXCL8 was also enriched in many pathways, such as the NF-κB signaling pathway, the Toll-like receptor signaling pathway, salmonella infection, and influenza A infection. IL-8 expression was significantly upregulated in CPB2-treated IPEC-J2 cells (18), and our sequencing results also showed that CXCL8 was significantly upregulated after CPB2 toxin treatment. Interestingly, we found that CXCL8 has a targeting relationship with the two lncRNAs ENSSSCG00000048785 and ENSSSCG00000048701, which belong to the group of six significantly differentially expressed m6A-modified lncRNAs identified in this study. The results indicated that m6A-modified lncRNAs might affect the inflammation and immune response of CPB2 toxin-treated cells by regulating the expression of CXCL8/IL-8.
In summary, we comprehensively analyzed the m6A methylation profile of RNAs in the control group and the CPB2 group. These results revealed the general characteristics and topological patterns of m6A modification in IPEC-J2 cells, as well as the difference in methylation profiles between the CPB2 group and the control group. Further functional enrichment analysis found that m6A-modified RNAs were related to defense against viruses and inflammation after CPB2 toxin treatment of the cells. The results suggested that three m6A-modified lncRNAs, ENSSSCG00000048785, ENSSSCG00000042575 and ENSSSCG00000048701, are most likely to play a key role in CPB2 toxin-treated IPEC-J2 cells. In conclusion, this study provides a reference for exploring C. perfringens type C diarrhea from the perspective of methylation modification.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, SRR15275162-SRR15275173.
Conceptualization: JY and SG. Data curation: JZ and XG. Formal analysis: QY. Funding acquisition: QY. Investigation: XH. Methodology: ZY and RL. Project administration: SG. Validation: KX, WW, and JL. Visualization: PW. Writing - original draft: JY. Writing - review and editing: SG. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (31960646), Education Department of Gansu Province: Excellent Graduate student “Innovation Star” project (2021CXZX-351), the Education Science and Technology Innovation Project of Gansu Province, China (GSSYLXM-02), and the Chief Special Project of Pig and Chicken Industry of Gansu Province Modern Agricultural Industrial Technology System (GARS-ZJ-1).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank LC-BIO Bio-tech ltd (Hangzhou, China) for their assistance with sequencing and data processing.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.769204/full#supplementary-material
1. Silva R, Junior CO, Guedes R, Lobato F. Clostridium Perfringens: A Review of the Disease in Pigs, Horses and Broiler Chickens. Ciec Rural (2015) 45:1027–34. doi: 10.1590/0103-8478cr20140927
2. Waters M, Savoie A, Garmory HS, Bueschel D, Popoff MR, Songer JG, et al. Genotyping and Phenotyping of Beta2-Toxigenic Clostridium Perfringens Fecal Isolates Associated With Gastrointestinal Diseases in Piglets. J Clin Microbiol (2003) 41:3584–91. doi: 10.1128/JCM.41.8.3584-3591.2003
3. Lin HY, Tang CH, Chen JH, Chuang JY, Huang SM, Tan TW, et al. Peptidoglycan Induces Interleukin-6 Expression Through the TLR2 Receptor, JNK, C-Jun, and AP-1 Pathways in Microglia. J Cell Physiol (2011) 226:1573–82. doi: 10.1002/jcp.22489
4. Nagahama M, Shibutani M, Seike S, Yonezaki M, Sakurai J. The P38 MAPK and JNK Pathways Protect Host Cells Against Clostridium Perfringens Beta-Toxin. Infect Immunity (2013) 81:3703–8. doi: 10.1128/IAI.00579-13
5. Xia Z, Tang M, Ma J, Zhang H, Gimple RC, Prager BC, et al. Epitranscriptomic Editing of the RNA N6-Methyladenosine Modification by Dcasrx Conjugated Methyltransferase and Demethylase. Nucleic Acids Res (2021) 49:7361–74. doi: 10.1093/nar/gkab517
6. Fu Y, Dominissini D, Rechavi G, He C. Gene Expression Regulation Mediated Through Reversible m6A RNA Methylation. Nat Rev Genet (2014) 15:293–306. doi: 10.1038/nrg3724
7. Sebastian-Delacruz M, Olazagoitia-Garmendia A, Gonzalez-Moro I, Santin I, Castellanos-Rubio A. Implication of m6A mRNA Methylation in Susceptibility to Inflammatory Bowel Disease. Epigenomes (2020) 4:16. doi: 10.3390/epigenomes4030016
8. Wu C, Chen W, He J, Jin S, Liu Y, Yi Y, et al. Interplay of m6A and H3K27 Trimethylation Restrains Inflammation During Bacterial Infection. Sci Advances (2020) 6:eaba0647. doi: 10.1126/sciadv.aba0647
9. Anderson SJ, Kramer MC, Gosai SJ, Yu X, Vandivier LE, Nelson A, et al. N6-Methyladenosine Inhibits Local Ribonucleolytic Cleavage to Stabilize mRNAs in Arabidopsis. Cell Rep (2018) 25:1146–57.e3. doi: 10.1016/j.celrep.2018.10.020
10. Adhikari S, Xiao W, Zhao Y-L, Yang Y. m6A: Signaling for mRNA Splicing. RNA Biol (2016) 13:756–9. doi: 10.1080/15476286.2016.1201628
11. Shi H, Xiao W, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 Facilitates Translation and Decay of N6-Methyladenosine-Modified RNA. Cell Res (2017) 27:315–28. doi: 10.1038/cr.2017.15
12. Slobodin B, Han R, Calderone V, Vrielink J, Loayza-Puch F, Elkon R, et al. Transcription Impacts the Efficiency of mRNA Translation via Co-Transcriptional N6-Adenosine Methylation. Cell (2017) 169:326–37. doi: 10.1016/j.cell.2017.03.031
13. Motorin Y, Helm M. Methods for RNA Modification Mapping Using Deep Sequencing: Established and New Emerging Technologies. Genes (2019) 10:35. doi: 10.3390/genes10010035
14. Augustino S, Xu Q, Liu X, Mi S, Yu Y. Integrated Analysis of lncRNAs and mRNAs Reveals Key Trans-Target Genes Associated With ETEC-F4ac Adhesion Phenotype in Porcine Small Intestine Epithelial Cells. BMC Genomics (2020) 21:780–94. doi: 10.1186/s12864-020-07192-8
15. Atianand M, Hu W, Satpathy A, Shen Y, Ricci E, Alvarez-Dominguez J, et al. A Long Noncoding RNA lincRNA-EPS Acts as a Transcriptional Brake to Restrain Inflammation. Cell (2016) 165:1672–85. doi: 10.1016/j.cell.2016.05.075
16. Huang X, Sun W, Yan Z, Shi H, Yang Q, Wang P, et al. Integrative Analyses of Long Non-Coding RNA and mRNA Involved in Piglet Ileum Immune Response to Clostridium Perfringens Type C Infection. Front Cell Infect Microbiol (2019) 9:130. doi: 10.3389/fcimb.2019.00130
17. Gao X, Yang Q, Huang X, Yan Z, Gun S. Effects of Clostridium Perfringens Beta2 Toxin on Apoptosis, Inflammation, and Barrier Function of Intestinal Porcine Jejunum Epithelial Cells. Microb Pathogen (2020) 147:104379. doi: 10.1016/j.micpath.2020.104379
18. Rl A, Qy A, Xh A, Zy A, Xg A, Wei WA, et al. Clostridium Perfringens Beta2 Toxin Induced In Vitro Oxidative Damage and its Toxic Assessment in Porcine Small Intestinal Epithelial Cell Lines. Gene (2020) 759:144999. doi: 10.1016/j.gene.2020.144999
19. Chen S, Zhou Y, Chen Y, Jia G. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics (2018) 34:i884–i90. doi: 10.1093/bioinformatics/bty560
20. Kim D, Langmead B, Salzberg SL. HISAT: A Fast Spliced Aligner With Low Memory Requirements. Nat Methods (2015) 12:357–60. doi: 10.1038/nmeth.3317
21. Meng J, Lu Z, Liu H, Zhang L, Zhang S, Chen Y, et al. A Protocol for RNA Methylation Differential Analysis With MeRIP-Seq Data and Exomepeak R/Bioconductor Package. Methods (2014) 69:274–81. doi: 10.1016/j.ymeth.2014.06.008
22. Bailey TL, Mikael B, Buske FA, Martin F, Grant CE, Luca C, et al. MEME Suite: Tools for Motif Discovery and Searching. Nucleic Acids Res (2009) 37:W202–8. doi: 10.1093/nar/gkp335
23. Yu G, Wang L-G, He Q-Y. ChIPseeker: An R/Bioconductor Package for ChIP Peak Annotation, Comparison and Visualization. Bioinformatics (2015) 31:2382–3. doi: 10.1093/bioinformatics/btv145
24. Heinz S, Benner C, Spann N, Bertolino E, Lin Y, Laslo P, et al. Simple Combinations of Lineage-Determining Transcription Factors Prime Cis-Regulatory Elements Required for Macrophage and B Cell Identities - ScienceDirect. Mol Cell (2010) 38:576–89. doi: 10.1016/j.molcel.2010.05.004
25. Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie Enables Improved Reconstruction of a Transcriptome From RNA-Seq Reads. Nat Biotechnol (2015) 33:290–5. doi: 10.1038/nbt.3122
26. Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, Baren M, et al. Transcript Assembly and Quantification by RNA-Seq Reveals Unannotated Transcripts and Isoform Switching During Cell Differentiation. Nat Biotechnol (2010) 28:511–5. doi: 10.1038/nbt.1621
27. Kong L, Yong Z, Ye ZQ, Liu XQ, Ge G. CPC: Assess the Protein-Coding Potential of Transcripts Using Sequence Features and Support Vector Machine. Nucleic Acids Res (2007) 35:W345–9. doi: 10.1093/nar/gkm391
28. Sun L, Luo H, Bu D, Zhao G, Yu K, Zhang C, et al. Utilizing Sequence Intrinsic Composition to Classify Protein-Coding and Long non-Coding Transcripts. Nucleic Acids Res (2013) 41:e166–e. doi: 10.1093/nar/gkt646
29. Smyth GK. Edger: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics (2010) 26:139. doi: 10.1093/bioinformatics/btp616
30. Guil S, Esteller M. Cis-Acting Noncoding RNAs: Friends and Foes. Nat Struct Mol Biol (2012) 19:1068–75. doi: 10.1038/nsmb.2428
31. Wenzel A, Akbasli Erdinç, Gorodkin J. RIsearch: Fast RNA-RNA Interaction Search Using a Simplified Nearest-Neighbor Energy Model. Bioinf (Oxford England) (2012) 28:2738–46. doi: 10.1093/bioinformatics/bts519
32. Minoru K, Michihiro A, Susumu G, Masahiro H, Mika H, Masumi I, et al. KEGG for Linking Genomes to Life and the Environment. Nucleic Acids Res (2008) 36:480–4. doi: 10.1093/nar/gkm882
33. Young M, Wakefield MJ, Smyth GK, Oshlack A. Gene Ontology Analysis for RNA-Seq: Accounting for Selection Bias. Genome Biol (2010) 11:R14–R. doi: 10.1186/gb-2010-11-2-r14
34. KJL A, TDS B. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2 ΔΔ C T Method. Methods (2001) 25:402–8. doi: 10.1006/meth.2001
35. Songer JG. Clostridia as Agents of Zoonotic Disease. Vet Microbiol (2010) 140:399–404. doi: 10.1016/j.vetmic.2009.07.003
36. Fisher DJ, Miyamoto K, Harrison B, Akimoto S, Sarker MR, Mcclane BA. Association of Beta2 Toxin Production With Clostridium Perfringens Type A Human Gastrointestinal Disease Isolates Carrying a Plasmid Enterotoxin Gene. Mol Microbiol (2010) 56:747–62. doi: 10.1111/j.1365-2958.2005.04573.x
37. Wang X, Li Z, Kong B, Song C, Cong J, Hou J, et al. Reduced M 6 A mRNA Methylation is Correlated With the Progression of Human Cervical Cancer. Oncotarget (2017) 8:98918–30. doi: 10.18632/oncotarget.22041
38. Yang J, Liu J, Zhao S, Tian F. N6-Methyladenosine METTL3 Modulates the Proliferation and Apoptosis of Lens Epithelial Cells in Diabetic Cataract. Mol Therapy Nucleic Acids (2020) 20:111–6. doi: 10.1016/j.omtn.2020.02.002
39. Lu Na, Li X, Yu J, Li Y, Wang C, Zhang L, et al. Curcumin Attenuates Lipopolysaccharide-Induced Hepatic Lipid Metabolism Disorder by Modification of m6A RNA Methylation in Piglets. Lipids (2018) 53:53–63. doi: 10.1002/lipd.12023
40. Lu Z, Liu J, Yuan C, Jin M, Wei C. m6A mRNA Methylation Analysis Provides Novel Insights Into Heat Stress Responses in the Liver Tissue of Sheep. Genomics (2020) 9:9750. doi: 10.1016/j.ygeno.2020.09.038
41. Bushkin GG, Pincus D, Morgan J, Richardson K, Lewis C, Chan SH, et al. m6A Modification of a 3’ UTR Site Reduces RME1 mRNA Levels to Promote Meiosis. Nat Commun (2019) 10(1):3414–27. doi: 10.1038/s41467-019-11232-7
42. Yue Y, Liu J, Cui X. VIRMA Mediates Preferential mA mRNA Methylation in 3 UTR and Near Stop Codon and Associates With Alternative Polyadenylation. Cell Discov (2018) 4:10–27. doi: 10.1038/s41421-018-0019-0
43. Kennedy EM, Bogerd HP, Kornepati AVR, Kang D, Ghoshal D, Marshall JB, et al. Posttranscriptional M(6)A Editing of HIV-1 mRNAs Enhances Viral Gene Expression. Cell Host Microbe (2016) 4(2):675–85. doi: 10.1016/j.chom.2016.04.002
44. Merkurjev D, Hong W-T, Iida K, Oomoto I, Goldie BJ, Yamaguti H, et al. Synaptic N 6 -Methyladenosine (M 6 A) Epitranscriptome Reveals Functional Partitioning of Localized Transcripts. Nat Neurosci (2018) 21:1004. doi: 10.1038/s41593-018-0173-6
45. Duan HC, Wei LH, Zhang C, Wang Y, Chen L, Lu Z, et al. ALKBH10B is An RNA N6-Methyladenosine Demethylase Affecting Arabidopsis Floral Transition. Plant Cell (2017) 29:2995–3011. doi: 10.1105/tpc.16.00912
46. Harper JE, Miceli SM, Roberts RJ, Manley JL. Sequence Specificity of the Human mRNA N6-Adenosine Methylase In Vitro. Nucleic Acids Res (1990) 18:5735–41. doi: 10.1093/nar/18.19.5735
47. Rong W, Li A, Sun B, Sun JG, Yuan Z. A Novel m6A Reader Prrc2a Controls Oligodendroglial Specification and Myelination. Cell Res (2019) 29:1–19. doi: 10.1038/s41422-018-0113-8
48. Duchartre Y, Kim YM, Kahn M. The Wnt Signaling Pathway in Cancer. Crit Rev Oncol (2016) 12(5):141–9. doi: 10.1016/j.critrevonc.2015.12.005
49. Mia MM, Singh MK. The Hippo Signaling Pathway in Cardiac Development and Diseases. Front Cell Dev Biol (2019) 7:211. doi: 10.3389/fcell.2019.00211
50. Betancor G, Dicks M, Jimenez-Guardeo JM, Ali NH, Apolonia L, Malim MH. The GTPase Domain of MX2 Interacts With the HIV-1 Capsid, Enabling Its Short Isoform to Moderate Antiviral Restriction. Cell Rep (2019) 29(7):1923–1933.e1923. doi: 10.1016/j.celrep.2019.10.009
51. Wang YX, Niklasch M, Liu T, Wang Y, Wen YM. Interferon-Inducible MX2 is a Host Restriction Factor of Hepatitis B Virus Replication. J Hepatol (2019) 72(5):865–76. doi: 10.1016/j.jhep.2019.12.009
52. Zhou Y, Wang P, Yan BX, Chen XY, Man XY. Quantitative Proteomic Profile of Psoriatic Epidermis Identifies OAS2 as a Novel Biomarker for Disease Activity. Front Immunol (2020) 11:1432. doi: 10.3389/fimmu.2020.01432
53. Davis BM, Fensterl V, Lawrence TM, Hudacek AW, Sen GC, Schnell MJ. Ifit2 Is a Restriction Factor in Rabies Virus Pathogenicity. J Virol (2017) JVI.00889–00817. doi: 10.1128/jvi.00889-17
54. Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase Essential for Controlling Immune Responses by Regulating mRNA Decay. Nature (2009) 458(7242):1185–90. doi: 10.1038/nature07924
55. Feng Z, Li Q, Meng R, Yi B, Xu Q. METTL3 Regulates Alternative Splicing of MyD88 Upon the Lipopolysaccharide-Induced Inflammatory Response in Human Dental Pulp Cells. J Cell Mol Med (2018) 22:2558–68. doi: 10.1111/jcmm.13491
56. Li HB, Tong J, Shu Z, Batista PJ, Flavell RA. m6A mRNA Methylation Controls T Cell Homeostasis by Targeting the IL-7/STAT5/SOCS Pathways. Nature (2017) 548:338–42. doi: 10.1038/nature23450
57. Zheng Q, Hou J, Zhou Y, Li Z, Cao X. The RNA Helicase DDX46 Inhibits Innate Immunity by Entrapping m6A-Demethylated Antiviral Transcripts in the Nucleus. Nat Immunol (2017) 18:1094–103. doi: 10.1038/ni.3830
58. Zhang C, Fu J, Zhou Y. A Review in Research Progress Concerning m6A Methylation and Immunoregulation. Front Immunol (2019) 10:922. doi: 10.3389/fimmu.2019.00922
59. Pease J, Sabroe I. The Role of Interleukin-8 and its Receptors in Inflammatory Lung Disease: Implications for Therapy. Am J Respir Med Drugs Devices Other Interventions (2002) 1:19–25. doi: 10.1007/BF03257159
Keywords: M6A methylation, MeRIP-seq, RNA-seq, IPEC-J2, CPB2
Citation: Yang J, Yang Q, Zhang J, Gao X, Luo R, Xie K, Wang W, Li J, Huang X, Yan Z, Wang P and Gun S (2021) N6-Methyladenosine Methylation Analysis of Long Noncoding RNAs and mRNAs in IPEC-J2 Cells Treated With Clostridium perfringens beta2 Toxin. Front. Immunol. 12:769204. doi: 10.3389/fimmu.2021.769204
Received: 14 September 2021; Accepted: 04 November 2021;
Published: 22 November 2021.
Edited by:
Anastasia N. Vlasova, The Ohio State University, United StatesReviewed by:
Zhangbin Yu, Nanjing Medical University, ChinaCopyright © 2021 Yang, Yang, Zhang, Gao, Luo, Xie, Wang, Li, Huang, Yan, Wang and Gun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuangbao Gun, Z3Vuc2JhbzA1NkAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.