 Constanca Figueiredo
Constanca Figueiredo Rainer Blasczyk
Rainer Blasczyk- Institute of Transfusion Medicine and Transplant Engineering, Hannover Medical School, Hannover, Germany
Patelet transfusion refractoriness remains a relevant hurdle in the treatment of severe alloimmunized thrombocytopenic patients. Antibodies specific for the human leukocyte antigens (HLA) class I are considered the major immunological cause for PLT transfusion refractoriness. Due to the insufficient availability of HLA-matched PLTs, the development of new technologies is highly desirable to provide an adequate management of thrombocytopenia in immunized patients. Blood pharming is a promising strategy not only to generate an alternative to donor blood products, but it may offer the possibility to optimize the therapeutic effect of the produced blood cells by genetic modification. Recently, enormous technical advances in the field of in vitro production of megakaryocytes (MKs) and PLTs have been achieved by combining progresses made at different levels including identification of suitable cell sources, cell pharming technologies, bioreactors and application of genetic engineering tools. In particular, use of RNA interference, TALEN and CRISPR/Cas9 nucleases or nickases has allowed for the generation of HLA universal PLTs with the potential to survive under refractoriness conditions. Genetically engineered HLA-silenced MKs and PLTs were shown to be functional and to have the capability to survive cell- and antibody-mediated cytotoxicity using in vitro and in vivo models. This review is focused on the methods to generate in vitro genetically engineered MKs and PLTs with the capacity to evade allogeneic immune responses.
Platelet Transfusion Refractoriness
PLT transfusion refractoriness (PTR) remains a major complication for thrombocytopenic patients due to the high risk for unprompted life-threatening bleeding (1). PTR is characterized by unexpectedly insufficient platelet (PLT) count increments after transfusion. Patients with PLT counts lower than 10x109 show increased predisposition for bleeding. Sixty to 80% of the PLT transfusion refractoriness cases are not associated with immunological aspects, but present non-immune etiologies such as massive bleeding, fever, infection, sepsis, drugs, accelerated PLT consumption, splenic sequestration or graft-versus-host disease (2, 3). Ten to 39% of the cases of PLT transfusion refractoriness are triggered by the development of antibodies specific for antigens expressed on the PLT surface or the development of drug-dependent PLT antibodies (3, 4). Immune causes of PTR include the alloimmunization to the human leukocyte antigen (HLA) and or human PLT antigens (HPA) triggered by previous transfusions, pregnancies, transplantation or PLT autoantibodies. Anti-HLA antibodies are the cause for immune PTR in 90% of the cases (5, 6). This review provides an overview on the strategies to downregulate HLA class I expression on in vitro produced Megakaryocytes (MKs) and PLTs as a promising approach to prevent immune PTR due to anti-HLA antibodies.
HLA Class I Antigens
The HLA system comprises the most polymorphic genes of the entire human genome. HLA class I molecules are constituted by a polymorphic heavy chain non-covalently bound to an invariable beta-microglobulin (β2m) light chain. HLA class I molecules are the protein product of HLA-A, -B and –C genes that are expressed on the cell surface of most nucleated cells (7, 8). HLA class I antigens mainly present endogenous peptides to T-cells and are the basis for a highly desirable and effective anti-viral and anti-tumor cell immune responses. In addition, HLA class I antigens allow the distinction of self- from non-self by providing inhibitory signals to natural killer (NK) cells and therefore supporting the elimination of pathogens and cancer cells. However, after the application of off-the-shelf cell products including cells, tissues or organs, mismatched HLA class I molecules reveal the foreigner identity of the allogeneic cell products by being itself recognized by donor-specific antibodies, alloreactive T-cells or by triggering new humoral or cellular responses (9–12). HLA expression turn PLTs to be highly immunogenic. Multiparous women show HLA sensitization rates up to 74% (13) and despite leukoreduction still 20% of leukemia patients become alloimmunized (14). PLTs express mainly HLA-A and HLA-B on their surface. Accordingly, HLA-C appears to play an irrelevant role in PTR, even though some cases have been described (15). As beta2-microglobulin is a common domain to HLA-A, HLA-B and HLA-C, it has been the selected target to knockdown or knockout the expression of those HLA class I proteins (16).
Platelets Gene Therapy
Gene therapy is a promising solution for the treatment of diseases by enabling the proper expression of genes in their correct form and adequate level. In particular, monogenetic diseases are a suitable target for treatment based on gene correction or regulation. PLTs are one of the most frequent cells in blood. In addition, PLTs are multifunctional cells that beside the fundamental roles in hemostasis also serve in the storage and delivery of important regenerative factors and regulation of immune responses. PLTs have been and attractive target for gene therapeutic strategies to treat diseases such as hemophilia A, a recessive X-linked bleeding disorder characterized by the factor VIII (FVIII) deficiency. Several studies showed the phenotypic correction of hemophilia A by the ectopic expression of FVIII on PLTs under the control of MK-specific promoters such as the megakaryocytic/PLT-specific glycoprotein IIb (alphaIIb) promoter (17). Also, ectopically expression of Factor IX (FIX) in PLTs showed to correct hemophilia B phenotype (18). Other gene therapeutic strategies as attempt to treat hemophilia A and B have used lentiviral vectors for the delivery of the activated factor Xa precursor gene sequence under integrin αIIb promoter into hematopoietic stem and progenitor cells (19). Hence, such studies have demonstrated the promising values of gene therapy based on PLT engineering.
PLTs are known to regulate important innate and adaptive immune responses such as by inducing recruitment of macrophages, neutrophil autophagy or differentiation and polarization of CD4 T-cells (20–23). Nevertheless, PLTs are also the end target for immune responses that cause their depletion and leading to life-threatening thrombocytopenia. This review is focused on gene therapeutic approaches to generate PLTs with the capacity to evade allogeneic immune responses.
Genetically Engineered Platelets
Blood pharming is defined by the differentiation of blood cells in vitro using protocols that recapitulate hematopoiesis ex vivo. A branch of blood farming technologies is focused on the in vitro differentiation of PLTs to serve as an alternative to donor PLTs transfusion.
Several groups have demonstrated the feasibility to differentiate MKs and PLTs in vitro. Besides the remarkable value of in vitro pharmed PLTs to meet the increasing demand on this product, in vitro blood pharming technologies enable the optimization of the differentiated cells towards the decrease of their immunogenicity and thereby potentially increasing their therapeutic efficiency. The generation of genetically engineered MKs and PLTs rely on the selection of an appropriate cell source and method for genetic engineering.
Cell Sources for Platelets Gene Therapy
The major breakthrough in in vitro MK/PLT production was linked with discovery and characterization of thrombopoietin (TPO) and the key role of Mpl receptor in thrombopoiesis in the mid-1990s (24–28). While former attempts to generate primitive MK lines ex vivo have been performed already in the 1980s, the first robust protocol to provide MKs capable of releasing PLTs was developed with introduction of thrombopoietin and published in 1995 (29–31). Several strategies have been explored to improve the quality and quantity of MK/PLT production with the aim to substitute or supplement the demand for donor PLTs in the future therapies.
Several cell sources were evaluated for the capability to differentiate in MKs and PLTs in vitro. Initially, CD34+ hematopoietic progenitor and stem cells (HPSCs) were considered the cell type of choice for development of blood pharming protocols and still serve as a golden standard in this field (29, 32, 33). Peripheral blood, umbilical cord blood, bone marrow and fetal liver provided the sources for of CD34+ cell populations with the further capability to differentiate into MKs and PLTS with varied success (29, 31–41). Some attempts have been made to generate MKs from the stromal cells (42–44). While these cell sources provided a fundamental basis for the progress in methods for ex vivo generation of MKs and PLTs, major drawbacks are associated with the use of HPSCs such as their limited proliferation capacity, which leads to continuous necessity of donor material. Thus, a great potential was identified in application of pluripotent cell lines, which are characterized by high proliferative capacity and high plasticity. Embryonic stem cells (ESCs) provide inherent pluripotency and capability to virtually unlimited proliferation in vitro, which could have been among the main parameters for the clinically-relevant upscaling of donor-independent MK/PLT production on the basis of this source. Numerous groups developed efficient protocols for differentiation of MKs from ESCs (45–51). However, globally accepted ethical issues with the consequent adaptation of legislative regulations basically limit application of ESCs to the narrow research field and prohibit the progress towards clinics. Shinya Yamanaka and Kazutoshi Takahashi provided a great alternative to ESCs by establishing cellular reprogramming and by introducing a rapid advance in generation of induced pluripotent cell lines (iPSCs) (52). Having similar properties to ESCs, but lacking valid ethical concerns, iPSCs became a major focus in the development of efficient and scalable MK/PLT production protocols. With the first report for successful differentiation of iPSCs into MKs only a decade ago, several progresses have been achieved such as improvement of differentiation efficiency, generation of self-proliferative intermediate MK-progenitor lines, possibility to upscale the production, development of GMP-compatible protocols or biobanking technologies (53–60). Both CD34+ HPSCs and iPSCs were also immediately recognized as promising target for genetic engineering to generate “universal” cell sources for the differentiation of blood products with increased survival after transfusion by escaping allogeneic immune responses.
Producing MKs and Platelets In Vitro
Although the majority of the current methods to ex vivo MK and PLTs production are based on induction with thrombopoietin, several strategies were established to achieve in vitro thrombopoiesis (Table 1). These methods differ in efficiency, characteristics and functionality of differentiated lines, as well as in time period required for completing the differentiation and maturation. in vitro MK and PLT production strategies can be classified into three general approaches: differentiation induced by specific cytokine cocktails, transdifferentiation, and forward programming.
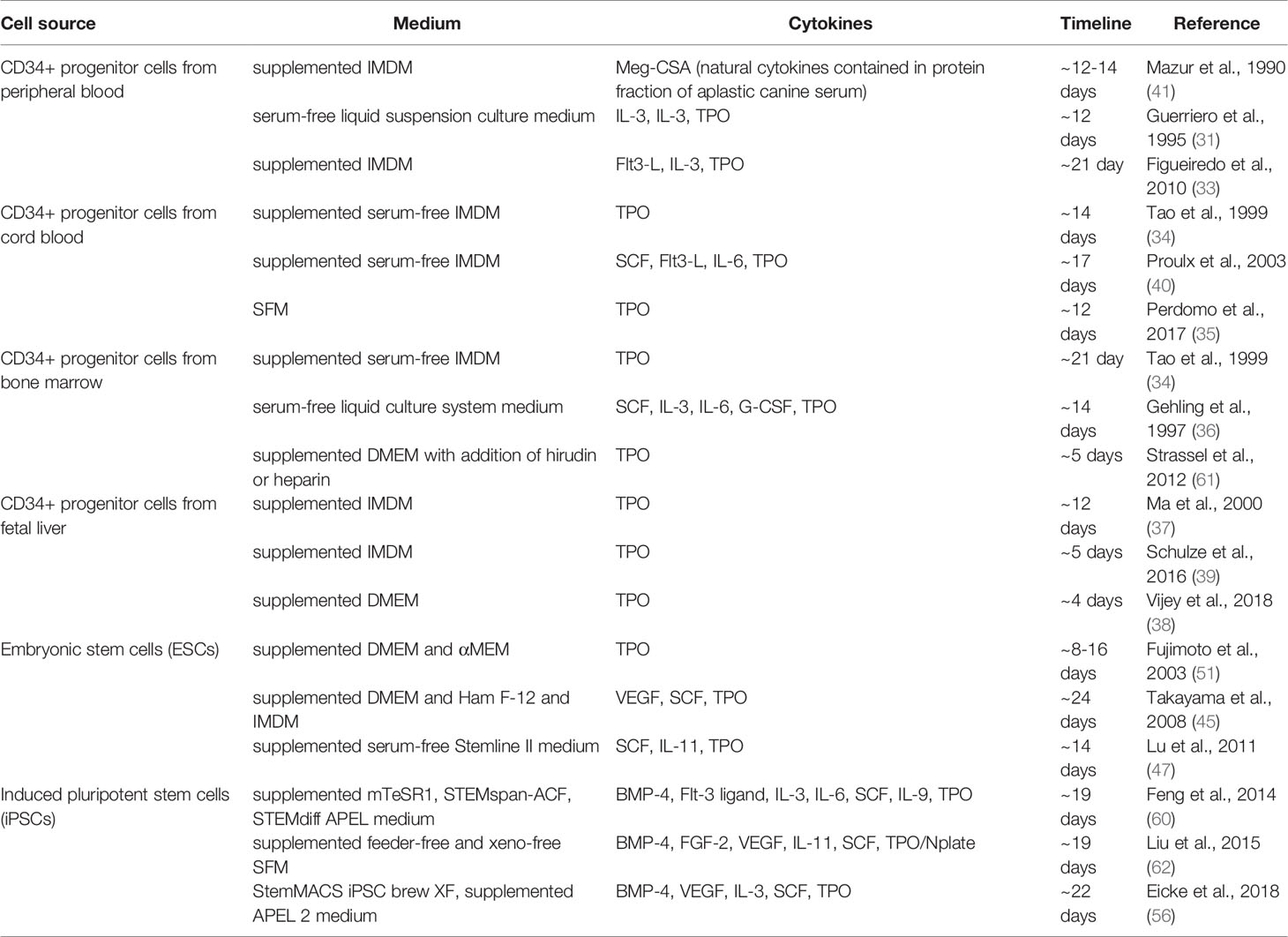
Table 1 Strategies to differentiate MKs and PLTs in vitro.
Cytokine induced differentiation of MK and PLTs as been applied to a wide variety of cell sources. The range, composition, and time points of application of cytokine cocktails are strongly dependent on initial cell type used for differentiation. In the case of initial cells being of hematopoietic origin, such as CD34+, the process basically implies cell culture followed by stimulation with TPO to induce megakaryopoiesis. In the case of pluripotent stem cells, such as ESCs or iPSCs, the process of cytokine-induced MK differentiation includes the following steps: 1) culturing and expansion of initial cell line, 2) mesoderm induction, 3) forwarding the cell fate into hematopoietic progenitors, and, finally, 4) induction of megakaryopoiesis and thrombopoiesis. Cytokine-induced differentiation showed to be a robust method enabling the possibility to upscale the process in bioreactor systems and delivering MKs and PLTs that show functionality in vivo (32, 47, 56, 60, 63, 64).
Transdifferentiation is a strategy based on the transformation of one cell type to the other, including processes resembling the switch of stem cells from one tissue type switch to the other (65). In vitro, this process can be induced using modern gene engineering tools to promote targeted differentiation of various cell types by direct reprogramming of one cell type to the other circumventing the iPSC stage (66). This approach has been also utilized for transdifferentiation of a range of somatic cell types into MKs. For example, transduction of 3T3 mouse fibroblasts and adult human dermal fibroblasts with nuclear factor erythroid–derived 2 p45 unit and musculoaponeurotic fibrosarcoma oncogene homolog (p45NF-E2/MafG/MafK), followed by culturing in TPO-based MK lineage induction medium, allowed transdifferentiating into MKs with typical morphology and functionality in vivo (43, 67). Another group was able to transdifferentiate CD71+ and GPA+ erythroblast subpopulations into functional MKs capable of PLT release by overexpression of the FLI1 and ERG genes (68). Further advances were made by transdifferentiation of embryonic murine fibroblasts and several lines of human skin fibroblasts into MK-like progenitors by overexpression of a set of six transcription factors: LMO2, GATA1, TAL-1, c-Myc, GATA2, and RUNX1 (69). The cells obtained with this method expressed CD41, were polyploid, formed MK colonies, and produced PLTs in vitro and engrafted in vivo. Moreover, human adipose-derived mesenchymal stromal cells have been transdifferentiated into MKs and PLTs even without the necessity to gene transfer, but with upregulation of endogenous TPO expression by incubation in specific MK lineage induction media (44, 70, 71). Here, the researchers were able to obtain the peak of MK population already at day 8 of cell culture with the peak for PLT release at day 12, also showing functionality in vivo in a mouse model (70).
Another key strategy for generating MKs and PLTs in vitro is forward programming. The strategy is based on the transient ectopic expression of a range of transcription factors and is mainly applied to pluripotent cell lines (57, 72–74). Successful examples include generation of MKs with minimum cytokines and chemically defined culture by combined expression of GATA1, FLI1, and TAL1 in iPSCs (72). Obtained MKs possessed typical morphology and delivered high purity of population with capability to be maintained in culture for several months (57). Forward programming allows developing GMP-compatible protocols and the generated MKs were capable to withstand cryopreservation procedures, which facilitates biobanking and potential clinical application (57, 72). Moreover, PLT release and purity were improved using bioengineered collagen scaffolds, also showing the importance of the 3D microenvironment (74).
Remarkably, bioreactor engineering for PLT production have focused on optimizing shear stress conditions to increase PLT yield from MKs. Different strategies have been proposed to mimic bone marrow stiffness by using different biomaterials such as collagen-1, collagen-4, fibrinogen, fibronectin, type IV collagen, laminin or alginate. The sinusoidal vessels have been reproduced by silk microtubes or chambers in which specific flows are applied to mimic the shear stress that MKs are exposed to produce PLTs in vivo. Also, bottle flasks have been used to produce PTLs using an impeller system to promote the medium flow. This issue has been comprehensively reviewed in Baigger et al. (75–79)
Essential issues such as practicability, quality and safety need to be considered to select the best method of MK/PLT production and ensure translation into clinical application (75). In particular, different methods have been proposed to assess PLT quality including analysis of vitality, morphology, characterization of phenotype (CD34, CD41, CD42a, CD42b, CD49b, CD61) and capacity to aggregate upon stimulation with specific agonists (55, 75, 77, 79–81).
In addition, biodistribution assays have been performed to ensure that in vitro produced and genetically modified MK/PLTs transfused into mouse models do not accumulated in specific tissues and organs. Remarkably, irradiation of in vitro generated MKs did not affect PLT production and may represent an effective strategy to support safety of this iPSC-derived cell products (55, 56, 63).
Strategies for Genetic Engineering
Different strategies have been developed to genetically engineer cell sources for MK and PLT production in vitro (Table 2).

Table 2 Impact of regulation of HLA expression on MKs and PLTs in functionality and survival to allogeneic immune responses.
Gene Regulation
After the discovery of RNAi interference (RNAi) in 1998 by Craig and Mello (84) showing its potential as promising tool for the development of cell and gene-therapeutic approaches, RNA interference is a gene regulatory mechanism that prevents protein translation by activating sequence-specific RNA degradation. Hence, RNAi is a highly specific and selective process enabling the control of protein expression. RNAi is initiated by double strand RNA (dsRNA) that after being processed and integrated into a multimeric protein complex lead to the sequence-specific degradation of a target mRNA. First, the RNase III Dicer cleaves the long dsRNA into 21-23-nucleotide small interfering RNA (siRNA). Then, the siRNA is incorporated in the RNA-inducing silencing complex (RISC) which is associated with the Argonaute 2 (Ago2) cleaving enzyme. The helicase activity of RISC unwind the siRNA allowing one of the siRNA strands to serve as guiding RNA sequence to RISC/Ago2 for the recognition of the target mRNA. After hybridization with the target mRNA, the RISC/Ago2 complex promotes the target mRNA cleavage which causes post-transcriptional gene silencing (85). RNAi has been widespread used in proof-of-concept studies and clinical trials (86). In previous studies, we have shown the feasibility to silence HLA class I expression on the surface of MKs and PLTs by lentiviral vector-mediated transduction of CD34+ HPSCs or iPSCs for expression of shRNAs targeting β2-microglobulin transcripts (33, 55, 63). Silencing HLA class I on the progenitor/stem cells did not impair their capacity to be differentiated into MKs and PLTs. In iPSCs, the expression of HLA class I antigens was silenced by up to 82%. The HLA class I silencing effect was maintained during several passages of iPSC culture. iPSC culture and differentiation was performed under feeder-and xeno-free conditions. HLA class I-silenced MKs showed typical morphology and increase of ploidy. HLA class I silenced PLTs were capable to upregulate the expression of activation markers such as CD62P and to aggregate upon stimulation with ADP and thrombin, indicating that HLA silencing does not impair the functionality of PLTs. Remarkably, in contrast to MKs and PLTs derived from fully HLA class I expression cell sources, HLA class I-silenced MK and PLTs showed significantly lower lysis rates in in vitro complement-dependent cytotoxic assays as well as antibody mediated cellular-dependent cytotoxic assays suggesting that HLA class I silenced MK and PLTs are capable to survive under refractoriness conditions. This was confirmed using in vivo PLT transfusion refractoriness mouse model showing that HLA class I-silenced MKs and PLTs prevail in the mouse circulation for longer periods than HLA-expressing MK/PLTs (55, 63). Hence, the use of RNA interference to downregulate HLA class I expression on MKs and PLTs demonstrated to be an efficient strategy to decrease their immunogenicity and improve cell survival after application in PLT transfusion refractory recipients. The use of RNAi to silence HLA class I expression is associated with intrinsic advantages and disadvantages. The efficiency of the delivery of the RNAi cassettes encoding for shRNA using lentiviral vectors into the MK/PLT cell source is depending on the type of the cell. Although the efficiency of the genetic modification of CD34+ cells is moderate to low, it is very high in high proliferative cell sources such as iPSCs. Furthermore, it is necessary to monitor the silencing effect during the culture of the cell source and during differentiation to ensure a sufficient HLA-silencing effect (33, 55). The residual HLA class I expression may be beneficial to prevent undesired NK cell cytotoxicity towards HLA class I-silenced MKs after transfusion.
Gene Editing
Site-specific gene editing technologies have also been used in PLT gene therapy and are mainly based on the use of Transcription-Activator Like Effector Nucleases (TALEN) and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-Associated 9 (CRISPR-Cas9) including its variants such as nickase Cas9 (60, 82, 83). Targeted genome editing relies on the generation of nuclease-induced double-stranded breaks (DSBs) which support highly efficient recombination mechanisms of DNA. Nuclease-induced DNA DSBs can be repaired by homology-directed repair (HDR) and nonhomologous end-joining (NHEJ) resulting in targeted integration or gene disruptions, respectively.
TALENs result from the fusion of transcription activator-like (TAL) proteins with a FokI nuclease. TAL proteins are composed of 33-35 amino acid repeating motifs with two variable positions that recognize single nucleotides. Hence, TALENs induce DSBs into specific DNA sites, which are then repaired (87). In 2014, Feng and colleagues have disrupted the β2-microglobulin gene to generate HLA class I knockout iPSCs under feeder- and xeno-free conditions. Furthermore, they show the feasibility to generate HLA class I knockout MK and PLTs from those source. The differentiated PLTs showed to respond in vitro to thrombin in PAC-1 binding assays (60). Suzuki and colleagues have knockout β2m using CRISPR/Cas9 nuclease technology. The group has re-reprogramed the previously immortalized MK cell lines (imMKCLs) into iPSCs (MK-iPSCs) prior the generation of β2m knockout. The group confirmed that the generated MK-iPSCs without HLA class I expression still carry the DOX-inducible c-MYC, BCL-XL and BMI1 transgenes of the original iMKCL and also could be re-induced, expanded and used for PLT production (iPLATs). Furthermore, the authors showed in vitro that the HLA KO iPLATs were not a target for NK cell cytotoxicity. They have confirmed those results in vivo using a NK cell reconstituted humanized mice treated with anti-HLA-A2 antibodies. The group propose that NK cell tolerance against HLA KO iPLATs might be due to the absence of NK cell activating ligands expression (82). In 2013, Ran et al. proposed the use of a D10A mutant nickase version of Cas9 with a pair of guide RNAs complementary to opposite strands of the target to generate DSBs. The group shows that individual nicks may increase genome editing specificity (88). Therefore, Norbnop and colleagues proposed the use of CRISPR/Cas9 nickase strategies to knockout β2m in iPSCs to be used as cell source for PLTs. After genetic modification, the iPSCs presented a normal karyotype. They differentiate β2m KO CD34+ HSCs from iPSCs using culture with 10T1/2 feeder cells. Isolated HLA KO iPSC-derived HSCs were used to differentiate MKs which showed typical proPLT and PLT formation. β2m KO PLTs showed to respond in vitro to thrombin with the upregulation of CD62P expression (83). These studies showed the feasibility to generate gene edited HLA-universal PLTs with the capacity to survive alloimmune responses. The efficiency of editing HLA may vary depending on the cell type and on the strategy to bring the gRNA and enzymes into the target cell. In case of bi-allelic gene editing, the cells will completely lack HLA class I expression, which may completely prevent the binding of anti-HLA class I antibodies.
Genetic engineering of MKs and PLTs using the above mentioned technologies is associated with the risk for off-target effects caused by unspecific cutting which may create undesired mutations. Several approaches have been designed to improve gRNA design and increase the specificity of the Cas. Current studies have demonstrated the absence or only rare off-target sites, long-size deletions or chromossomal rearrangements were observed. In addition, the number of such off-target effects has decreased with time. Nevertheless, precise methods including high-throughput sequencing and multi-omics analyses are highly desirable to detect sporadic off-target effects. Also, anti-Cas antibodies and anti-Cas T-cells have been detected (89, 90). Blood pharming supports the safe application of genetic engineered products, as the target for genetic engineering is cell source (e.g. iPSC line) and not the applied product (e.g. PLTs). Hence, there is the time and possibility for the completely characterization and evaluation of potential off-target effects including the formation of neo-antigens that could trigger unwanted immune responses towards the transfused PLTs. Only the genetically engineered iPSC that show no off-target effect will be selected for in vitro production of MKs and PLTs. This has the potential to abrogate major safety concerns.
Conclusion
The feasibility to improve the therapeutic effect of in vitro generated MKs and PLTs has laid the foundation for the “next generation” of blood pharming. Genetic engineering may not only be applied to facilitate the in vitro differentiation of PLTs from different cells sources, but also to regulate the expression of antigens on their surface such as HLA class I and thereby supporting their survival after transfusion. The translation of this technology into clinical application is tightly associated with the development of effective bioreactors and production strategies that allow the fabrication of enough numbers of MKs and PLTs as well as reducing the production costs. HLA universal PLTs may become an important biotherapeutic product for the treatment of severe immunized thrombocytopenic patients.
Author Contributions
CF and RB: conception, design, and writing of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
We acknowledge the Foundation of the German Red Cross - Service of blood donation NSTOB (Project: StiBSD/V2871) for funding part of the authors’ work mentioned in this review.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rebulla P. Refractoriness to Platelet Transfusion. Curr Opin Hematol (2002) 9(6):516–20. doi: 10.1097/00062752-200211000-00009
2. Schmidt AE, Refaai MA, Coppage M. HLA-Mediated Platelet Refractoriness. Am J Clin Pathol (2019) 151(4):353–63. doi: 10.1093/ajcp/aqy121
3. Stanworth SJ, Navarrete C, Estcourt L, Marsh J. Platelet Refractoriness–Practical Approaches and Ongoing Dilemmas in Patient Management. Br J Haematol (2015) 171(3):297–305. doi: 10.1111/bjh.13597
4. Cohn CS. Platelet Transfusion Refractoriness: How do I Diagnose and Manage? Hematol Am Soc Hematol Educ Program (2020) 2020(1):527–32. doi: 10.1182/hematology.2020000137
5. Schiffer CA. Diagnosis and Management of Refractoriness to Platelet Transfusion. Blood Rev (2001) 15(4):175–80. doi: 10.1054/blre.2001.0164
6. Barbagallo NBA, Costa TH, Bastos E, Aravechia MG, Kutner JM, Bonet-Bub C. The Relevance of a Bank With Genotyped Platelets Donors. Hematol Transfus Cell Ther (2021). doi: 10.1016/j.htct.2021.03.006
7. Choo SY. The HLA System: Genetics, Immunology, Clinical Testing, and Clinical Implications. Yonsei Med J (2007) 48(1):11–23. doi: 10.3349/ymj.2007.48.1.11
8. Leen G, Stein JE, Robinson J, Maldonado Torres H, Marsh SGE. The HLA Diversity of the Anthony Nolan Register. HLA (2021) 97(1):15–29. doi: 10.1111/tan.14127
9. Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC, et al. Receptors for HLA Class-I Molecules in Human Natural Killer Cells. Annu Rev Immunol (1996) 14:619–48. doi: 10.1146/annurev.immunol.14.1.619
10. Del Vecchio F, Martinez-Rodriguez V, Schukking M, Cocks A, Broseghini E, Fabbri M. Professional Killers: The Role of Extracellular Vesicles in the Reciprocal Interactions Between Natural Killer, CD8+ Cytotoxic T-Cells and Tumour Cells. J Extracell Vesicles (2021) 10(6):e12075. doi: 10.1002/jev2.12075
11. Quach DH, Becerra-Dominguez L, Rouce RH, Rooney CM. A Strategy to Protect Off-the-Shelf Cell Therapy Products Using Virus-Specific T-Cells Engineered to Eliminate Alloreactive T-Cells. J Transl Med (2019) 17(1):240. doi: 10.1186/s12967-019-1988-y
12. Zachary AA, Leffell MS. HLA Mismatching Strategies for Solid Organ Transplantation - A Balancing Act. Front Immunol (2016) 7:575. doi: 10.3389/fimmu.2016.00575
13. Masson E, Vidal C, Deschamps M, Bongain S, Thevenin C, Dupont I, et al. Incidence and Risk Factors of Anti-HLA Immunization After Pregnancy. Hum Immunol (2013) 74(8):946–51. doi: 10.1016/j.humimm.2013.04.025
14. Hod E, Schwartz J. Platelet Transfusion Refractoriness. Br J Haematol (2008) 142(3):348–60. doi: 10.1111/j.1365-2141.2008.07189.x
15. Saito S, Ota S, Seshimo H, Yamazaki Y, Nomura S, Ito T, et al. Platelet Transfusion Refractoriness Caused by a Mismatch in HLA-C Antigens. Transfusion (2002) 42(3):302–8. doi: 10.1046/j.1537-2995.2002.00051.x
16. Figueiredo C, Seltsam A, Blasczyk R. Class-, Gene-, and Group-Specific HLA Silencing by Lentiviral shRNA Delivery. J Mol Med (Berl) (2006) 84(5):425–37. doi: 10.1007/s00109-005-0024-2
17. Shi Q, Wilcox DA, Fahs SA, Kroner PA, Montgomery RR. Expression of Human Factor VIII Under Control of the Platelet-Specific alphaIIb Promoter in Megakaryocytic Cell Line as Well as Storage Together With VWF. Mol Genet Metab (2003) 79(1):25–33. doi: 10.1016/s1096-7192(03)00049-0
18. Zhang G, Shi Q, Fahs SA, Kuether EL, Walsh CE, Montgomery RR. Factor IX Ectopically Expressed in Platelets can be Stored in Alpha-Granules and Corrects the Phenotype of Hemophilia B Mice. Blood (2010) 116(8):1235–43. doi: 10.1182/blood-2009-11-255612
19. Wang D, Shao X, Wang Q, Pan X, Dai Y, Yao S, et al. Activated Factor X Targeted Stored in Platelets as an Effective Gene Therapy Strategy for Both Hemophilia A and B. Clin Transl Med (2021) 11(3):e375. doi: 10.1002/ctm2.375
20. Gerdes N, Zhu L, Ersoy M, Hermansson A, Hjemdahl P, Hu H, et al. Platelets Regulate CD4(+) T-Cell Differentiation via Multiple Chemokines in Humans. Thromb Haemost (2011) 106(2):353–62. doi: 10.1160/TH11-01-0020
21. Tan S, Zhang J, Sun Y, Gistera A, Sheng Z, Malmstrom RE, et al. Platelets Enhance CD4+ Central Memory T Cell Responses via Platelet Factor 4-Dependent Mitochondrial Biogenesis and Cell Proliferation. Platelets (2021), 1–11. doi: 10.1080/09537104.2021.1936479
22. Zhu L, Huang Z, Stalesen R, Hansson GK, Li N. Platelets Provoke Distinct Dynamics of Immune Responses by Differentially Regulating CD4+ T-Cell Proliferation. J Thromb Haemost (2014) 12(7):1156–65. doi: 10.1111/jth.12612
23. Ribeiro LS, Migliari Branco L, Franklin BS. Regulation of Innate Immune Responses by Platelets. Front Immunol (2019) 10:1320. doi: 10.3389/fimmu.2019.01320
24. Kuter DJ, Beeler DL, Rosenberg RD. The Purification of Megapoietin: A Physiological Regulator of Megakaryocyte Growth and Platelet Production. Proc Natl Acad Sci USA (1994) 91(23):11104–8. doi: 10.1073/pnas.91.23.11104
25. de Sauvage FJ, Hass PE, Spencer SD, Malloy BE, Gurney AL, Spencer SA, et al. Stimulation of Megakaryocytopoiesis and Thrombopoiesis by the C-Mpl Ligand. Nature (1994) 369(6481):533–8. doi: 10.1038/369533a0
26. Lok S, Kaushansky K, Holly RD, Kuijper JL, Lofton-Day CE, Oort PJ, et al. Cloning and Expression of Murine Thrombopoietin cDNA and Stimulation of Platelet Production In Vivo. Nature (1994) 369(6481):565–8. doi: 10.1038/369565a0
27. Sohma Y, Akahori H, Seki N, Hori T, Ogami K, Kato T, et al. Molecular Cloning and Chromosomal Localization of the Human Thrombopoietin Gene. FEBS Lett (1994) 353(1):57–61. doi: 10.1016/0014-5793(94)01008-0
28. Wendling F, Maraskovsky E, Debili N, Florindo C, Teepe M, Titeux M, et al. Cmpl Ligand is a Humoral Regulator of Megakaryocytopoiesis. Nature (1994) 369(6481):571–4. doi: 10.1038/369571a0
29. Choi ES, Nichol JL, Hokom MM, Hornkohl AC, Hunt P. Platelets Generated In Vitro From Proplatelet-Displaying Human Megakaryocytes are Functional. Blood (1995) 85(2):402–13. doi: 10.1182/blood.V85.2.402.402
30. Kaushansky K. Thrombopoiesis. Semin Hematol (2015) 52(1):4–11. doi: 10.1053/j.seminhematol.2014.10.003
31. Guerriero R, Testa U, Gabbianelli M, Mattia G, Montesoro E, Macioce G, et al. Unilineage Megakaryocytic Proliferation and Differentiation of Purified Hematopoietic Progenitors in Serum-Free Liquid Culture. Blood (1995) 86(10):3725–36. doi: 10.1182/blood.V86.10.3725.bloodjournal86103725
32. Figueiredo C, Blaszczyk R. Genetically Engineered Blood Pharming: Generation of HLA-Universal Platelets Derived From CD34+ Progenitor Cells. J Stem Cells (2014) 9(3):149–61. doi: jsc.2014.9.3.149
33. Figueiredo C, Goudeva L, Horn PA, Eiz-Vesper B, Blasczyk R, Seltsam A. Generation of HLA-Deficient Platelets From Hematopoietic Progenitor Cells. Transfusion (2010) 50(8):1690–701. doi: 10.1111/j.1537-2995.2010.02644.x
34. Tao H, Gaudry L, Rice A, Chong B. Cord Blood is Better Than Bone Marrow for Generating Megakaryocytic Progenitor Cells. Exp Hematol (1999) 27(2):293–301. doi: 10.1016/s0301-472x(98)00050-2
35. Perdomo J, Yan F, Leung HHL, Chong BH. Megakaryocyte Differentiation and Platelet Formation From Human Cord Blood-Derived CD34+ Cells. J Vis Exp (2017) 130:1–8. doi: 10.3791/56420
36. Gehling UM, Ryder JW, Hogan CJ, Hami L, McNiece I, Franklin W, et al. Ex Vivo Expansion of Megakaryocyte Progenitors: Effect of Various Growth Factor Combinations on CD34+ Progenitor Cells From Bone Marrow and G-CSF-Mobilized Peripheral Blood. Exp Hematol (1997) 25(11):1125–39. doi: 10.3390/cells9061350
37. Ma DC, Sun YH, Zuo W, Chang KZ, Chu JJ, Liu YG. CD34+ Cells Derived From Fetal Liver Contained a High Proportion of Immature Megakaryocytic Progenitor Cells. Eur J Haematol (2000) 64(5):304–14. doi: 10.1034/j.1600-0609.2000.90038.x
38. Vijey P, Posorske B, Machlus KR. In Vitro Culture of Murine Megakaryocytes From Fetal Liver-Derived Hematopoietic Stem Cells. Platelets (2018) 29(6):583–8. doi: 10.1080/09537104.2018.1492107
39. Schulze H. Culture, Expansion, and Differentiation of Murine Megakaryocytes From Fetal Liver, Bone Marrow, and Spleen. Curr Protoc Immunol (2016) 112:22F 6 1–22F 6 15. doi: 10.1002/0471142735.im22f06s112
40. Proulx C, Boyer L, Hurnanen DR, Lemieux R. Preferential Ex Vivo Expansion of Megakaryocytes From Human Cord Blood CD34+-Enriched Cells in the Presence of Thrombopoietin and Limiting Amounts of Stem Cell Factor and Flt-3 Ligand. J Hematother Stem Cell Res (2003) 12(2):179–88. doi: 10.1089/152581603321628322
41. Mazur EM, Basilico D, Newton JL, Cohen JL, Charland C, Sohl PA, et al. Isolation of Large Numbers of Enriched Human Megakaryocytes From Liquid Cultures of Normal Peripheral Blood Progenitor Cells. Blood (1990) 76(9):1771–82. doi: 10.1182/blood.V76.9.1771.1771
42. Matsubara Y, Ono Y, Suzuki H, Arai F, Suda T, Murata M, et al. OP9 Bone Marrow Stroma Cells Differentiate Into Megakaryocytes and Platelets. PloS One (2013) 8(3):e58123. doi: 10.1371/journal.pone.0058123
43. Ono Y, Wang Y, Suzuki H, Okamoto S, Ikeda Y, Murata M, et al. Induction of Functional Platelets From Mouse and Human Fibroblasts by P45nf-E2/Maf. Blood (2012) 120(18):3812–21. doi: 10.1182/blood-2012-02-413617
44. Ono-Uruga Y, Tozawa K, Horiuchi T, Murata M, Okamoto S, Ikeda Y, et al. Human Adipose Tissue-Derived Stromal Cells can Differentiate Into Megakaryocytes and Platelets by Secreting Endogenous Thrombopoietin. J Thromb Haemost (2016) 14(6):1285–97. doi: 10.1111/jth.13313
45. Takayama N, Nishikii H, Usui J, Tsukui H, Sawaguchi A, Hiroyama T, et al. Generation of Functional Platelets From Human Embryonic Stem Cells In Vitro via ES-Sacs, VEGF-Promoted Structures That Concentrate Hematopoietic Progenitors. Blood (2008) 111(11):5298–306. doi: 10.1182/blood-2007-10-117622
46. Gaur M, Kamata T, Wang S, Moran B, Shattil SJ, Leavitt AD. Megakaryocytes Derived From Human Embryonic Stem Cells: A Genetically Tractable System to Study Megakaryocytopoiesis and Integrin Function. J Thromb Haemost (2006) 4(2):436–42. doi: 10.1111/j.1538-7836.2006.01744.x
47. Lu SJ, Li F, Yin H, Feng Q, Kimbrel EA, Hahm E, et al. Platelets Generated From Human Embryonic Stem Cells are Functional In Vitro and in the Microcirculation of Living Mice. Cell Res (2011) 21(3):530–45. doi: 10.1038/cr.2011.8
48. Kawaguchi M, Kitajima K, Kanokoda M, Suzuki H, Miyashita K, Nakajima M, et al. Efficient Production of Platelets From Mouse Embryonic Stem Cells by Enforced Expression of Gata2 in Late Hemogenic Endothelial Cells. Biochem Biophys Res Commun (2016) 474(3):462–8. doi: 10.1016/j.bbrc.2016.04.140
49. Toscano MG, Munoz P, Sanchez-Gilabert A, Cobo M, Benabdellah K, Anderson P, et al. Absence of WASp Enhances Hematopoietic and Megakaryocytic Differentiation in a Human Embryonic Stem Cell Model. Mol Ther (2016) 24(2):342–53. doi: 10.1038/mt.2015.196
50. Canver MC, Bauer DE, Orkin SH. Embryonic Stem Cells as Sources of Donor-Independent Platelets. J Clin Invest (2015) 125(6):2261–3. doi: 10.1172/JCI82348
51. Fujimoto TT, Kohata S, Suzuki H, Miyazaki H, Fujimura K. Production of Functional Platelets by Differentiated Embryonic Stem (ES) Cells In Vitro. Blood (2003) 102(12):4044–51. doi: 10.1182/blood-2003-06-1773
52. Takahashi K, Yamanaka S. Induction of Pluripotent Stem Cells From Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell (2006) 126(4):663–76. doi: 10.1016/j.cell.2006.07.024
53. Takayama N, Nishimura S, Nakamura S, Shimizu T, Ohnishi R, Endo H, et al. Transient Activation of C-MYC Expression is Critical for Efficient Platelet Generation From Human Induced Pluripotent Stem Cells. J Exp Med (2010) 207(13):2817–30. doi: 10.1084/jem.20100844
54. Nakamura S, Takayama N, Hirata S, Seo H, Endo H, Ochi K, et al. Expandable Megakaryocyte Cell Lines Enable Clinically Applicable Generation of Platelets From Human Induced Pluripotent Stem Cells. Cell Stem Cell (2014) 14(4):535–48. doi: 10.1016/j.stem.2014.01.011
55. Borger AK, Eicke D, Wolf C, Gras C, Aufderbeck S, Schulze K, et al. Generation of HLA-Universal iPSC-Derived Megakaryocytes and Platelets for Survival Under Refractoriness Conditions. Mol Med (2016) 22:274–85. doi: 10.2119/molmed.2015.00235
56. Eicke D, Baigger A, Schulze K, Latham SL, Halloin C, Zweigerdt R, et al. Large-Scale Production of Megakaryocytes in Microcarrier-Supported Stirred Suspension Bioreactors. Sci Rep (2018) 8(1):10146. doi: 10.1038/s41598-018-28459-x
57. Moreau T, Evans AL, Vasquez L, Tijssen MR, Yan Y, Trotter MW, et al. Large-Scale Production of Megakaryocytes From Human Pluripotent Stem Cells by Chemically Defined Forward Programming. Nat Commun (2016) 7:11208. doi: 10.1038/ncomms11208
58. Sugimoto N, Eto K. Platelet Production From Induced Pluripotent Stem Cells. J Thromb Haemost (2017) 15(9):1717–27. doi: 10.1111/jth.13736
59. Sugimoto N, Eto K. Development of iPS Cell-Derived Blood Products and Production Guidelines. Rinsho Ketsueki (2017) 58(10):2150–9. doi: 10.11406/rinketsu.58.2150
60. Feng Q, Shabrani N, Thon JN, Huo H, Thiel A, Machlus KR, et al. Scalable Generation of Universal Platelets From Human Induced Pluripotent Stem Cells. Stem Cell Rep (2014) 3(5):817–31. doi: 10.1016/j.stemcr.2014.09.010
61. Strassel C, Eckly A, Leon C, Moog S, Cazenave JP, Gachet C, et al. Hirudin and Heparin Enable Efficient Megakaryocyte Differentiation of Mouse Bone Marrow Progenitors. Exp Cell Res (2012) 318(1):25–32. doi: 10.1016/j.yexcr.2011.10.003
62. Liu Y, Wang Y, Gao Y, Forbes JA, Qayyum R, Becker L, et al. Efficient Generation of Megakaryocytes From Human Induced Pluripotent Stem Cells Using Food and Drug Administration-Approved Pharmacological Reagents. Stem Cells Transl Med (2015) 4(4):309–19. doi: 10.5966/sctm.2014-0183
63. Gras C, Schulze K, Goudeva L, Guzman CA, Blasczyk R, Figueiredo C. HLA-Universal Platelet Transfusions Prevent Platelet Refractoriness in a Mouse Model. Hum Gene Ther (2013) 24(12):1018–28. doi: 10.1089/hum.2013.074
64. Patel A, Clementelli CM, Jarocha D, Mosoyan G, Else C, Kintali M, et al. Pre-Clinical Development of a Cryopreservable Megakaryocytic Cell Product Capable of Sustained Platelet Production in Mice. Transfusion (2019) 59(12):3698–713. doi: 10.1111/trf.15546
65. Shen CN, Burke ZD, Tosh D. Transdifferentiation, Metaplasia and Tissue Regeneration. Organogenesis (2004) 1(2):36–44. doi: 10.4161/org.1.2.1409
66. Grath A, Dai G. Direct Cell Reprogramming for Tissue Engineering and Regenerative Medicine. J Biol Eng (2019) 13:14. doi: 10.1186/s13036-019-0144-9
67. Masuda S, Li M, Izpisua Belmonte JC. In Vitro Generation of Platelets Through Direct Conversion: First Report in My Knowledge (iMK). Cell Res (2013) 23(2):176–8. doi: 10.1038/cr.2012.142
68. Siripin D, Kheolamai P, Supokawej U. P. Y, A., Wattanapanitch M, Klincumhom N, Laowtammathron C, et al. Transdifferentiation of Erythroblasts to Megakaryocytes Using FLI1 and ERG Transcription Factors. Thromb Haemost (2015) 114(3):593–602. doi: 10.1160/TH14-12-1090
69. Pulecio J, Alejo-Valle O, Capellera-Garcia S, Vitaloni M, Rio P, Mejia-Ramirez E, et al. Direct Conversion of Fibroblasts to Megakaryocyte Progenitors. Cell Rep (2016) 17(3):671–83. doi: 10.1016/j.celrep.2016.09.036
70. Tozawa K, Ono-Uruga Y, Yazawa M, Mori T, Murata M, Okamoto S, et al. Megakaryocytes and Platelets From a Novel Human Adipose Tissue-Derived Mesenchymal Stem Cell Line. Blood (2019) 133(7):633–43. doi: 10.1182/blood-2018-04-842641
71. Matsubara Y, Murata M, Ikeda Y. Culture of Megakaryocytes and Platelets From Subcutaneous Adipose Tissue and a Preadipocyte Cell Line. Methods Mol Biol (2012) 788:249–58. doi: 10.1007/978-1-61779-307-3_17
72. Moreau T, Evans AL, Ghevaert CJG. Differentiation of Human Pluripotent Stem Cells to Megakaryocytes by Transcription Factor-Driven Forward Programming. Methods Mol Biol (2018) 1812:155–76. doi: 10.1007/978-1-4939-8585-2_10
73. Dalby A, Ballester-Beltran J, Lincetto C, Mueller A, Foad N, Evans A, et al. Transcription Factor Levels After Forward Programming of Human Pluripotent Stem Cells With GATA1, FLI1, and TAL1 Determine Megakaryocyte Versus Erythroid Cell Fate Decision. Stem Cell Rep (2018) 11(6):1462–78. doi: 10.1016/j.stemcr.2018.11.001
74. Shepherd JH, Howard D, Waller AK, Foster HR, Mueller A, Moreau T, et al. Structurally Graduated Collagen Scaffolds Applied to the Ex Vivo Generation of Platelets From Human Pluripotent Stem Cell-Derived Megakaryocytes: Enhancing Production and Purity. Biomaterials (2018) 182:135–44. doi: 10.1016/j.biomaterials.2018.08.019
75. Baigger A, Blasczyk R, Figueiredo C. Towards the Manufacture of Megakaryocytes and Platelets for Clinical Application. Transfus Med Hemother (2017) 44(3):165–73. doi: 10.1159/000477261
76. Di Buduo CA, Wray LS, Tozzi L, Malara A, Chen Y, Ghezzi CE, et al. Programmable 3D Silk Bone Marrow Niche for Platelet Generation Ex Vivo and Modeling of Megakaryopoiesis Pathologies. Blood (2015) 125(14):2254–64. doi: 10.1182/blood-2014-08-595561
77. Nakagawa Y, Nakamura S, Nakajima M, Endo H, Dohda T, Takayama N, et al. Two Differential Flows in a Bioreactor Promoted Platelet Generation From Human Pluripotent Stem Cell-Derived Megakaryocytes. Exp Hematol (2013) 41(8):742–8. doi: 10.1016/j.exphem.2013.04.007
78. Thon JN, Dykstra BJ, Beaulieu LM. Platelet Bioreactor: Accelerated Evolution of Design and Manufacture. Platelets (2017) 28(5):472–7. doi: 10.1080/09537104.2016.1265922
79. Thon JN, Mazutis L, Wu S, Sylman JL, Ehrlicher A, Machlus KR, et al. Platelet Bioreactor-on-a-Chip. Blood (2014) 124(12):1857–67. doi: 10.1182/blood-2014-05-574913
80. Avanzi MP, Oluwadara OE, Cushing MM, Mitchell ML, Fischer S, Mitchell WB. A Novel Bioreactor and Culture Method Drives High Yields of Platelets From Stem Cells. Transfusion (2016) 56(1):170–8. doi: 10.1111/trf.13375
81. Blin A, Le Goff A, Magniez A, Poirault-Chassac S, Teste B, Sicot G, et al. Microfluidic Model of the Platelet-Generating Organ: Beyond Bone Marrow Biomimetics. Sci Rep (2016) 6:21700. doi: 10.1038/srep21700
82. Suzuki D, Flahou C, Yoshikawa N, Stirblyte I, Hayashi Y, Sawaguchi A, et al. iPSC-Derived Platelets Depleted of HLA Class I Are Inert to Anti-HLA Class I and Natural Killer Cell Immunity. Stem Cell Rep (2020) 14(1):49–59. doi: 10.1016/j.stemcr.2019.11.011
83. Norbnop P, Ingrungruanglert P, Israsena N, Suphapeetiporn K, Shotelersuk V. Generation and Characterization of HLA-Universal Platelets Derived From Induced Pluripotent Stem Cells. Sci Rep (2020) 10(1):8472. doi: 10.1038/s41598-020-65577-x
84. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and Specific Genetic Interference by Double-Stranded RNA in Caenorhabditis Elegans. Nature (1998) 391(6669):806–11. doi: 10.1038/35888
85. Agrawal N, Dasaradhi PV, Mohmmed A, Malhotra P, Bhatnagar RK, Mukherjee SK. RNA Interference: Biology, Mechanism, and Applications. Microbiol Mol Biol Rev (2003) 67(4):657–85. doi: 10.1128/MMBR.67.4.657-685.2003
86. Setten RL, Rossi JJ, Han SP. The Current State and Future Directions of RNAi-Based Therapeutics. Nat Rev Drug Discovery (2019) 18(6):421–46. doi: 10.1038/s41573-019-0017-4
87. Maeder ML, Gersbach CA. Genome-Editing Technologies for Gene and Cell Therapy. Mol Ther (2016) 24(3):430–46. doi: 10.1038/mt.2016.10
88. Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell (2013) 154(6):1380–9. doi: 10.1016/j.cell.2013.08.021
89. Ou X, Ma Q, Yin W, Ma X, He Z. CRISPR/Cas9 Gene-Editing in Cancer Immunotherapy: Promoting the Present Revolution in Cancer Therapy and Exploring More. Front Cell Dev Biol (2021) 9:674467. doi: 10.3389/fcell.2021.674467
Keywords: HLA, megakaryocytes, platelets, gene therapy, gene editing, RNAi
Citation: Figueiredo C and Blasczyk R (2021) Generation of HLA Universal Megakaryocytes and Platelets by Genetic Engineering. Front. Immunol. 12:768458. doi: 10.3389/fimmu.2021.768458
Received: 31 August 2021; Accepted: 11 October 2021;
Published: 27 October 2021.
Edited by:
Reem Al-Daccak, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Stephan Meinke, Karolinska Institutet (KI), SwedenRaymond Goodrich, Colorado State University, United States
Seema Patel, Emory University, United States
Copyright © 2021 Figueiredo and Blasczyk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Constanca Figueiredo, RmlndWVpcmVkby5Db25zdGFuY2FAbWgtaGFubm92ZXIuZGU=