 Lauren W. Kaminsky
Lauren W. Kaminsky Rana Al-Sadi
Rana Al-Sadi Thomas Y. Ma
Thomas Y. Ma
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 25 October 2021
Sec. Cytokines and Soluble Mediators in Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.767456
This article is part of the Research Topic Importance of Cytokines and Receptor Members from the IL-1 Family in the Context of Chronic Autoimmune Inflammatory Diseases View all 7 articles
The intestinal epithelial tight junction (TJ) barrier controls the paracellular permeation of contents from the intestinal lumen into the intestinal tissue and systemic circulation. A defective intestinal TJ barrier has been implicated as an important pathogenic factor in inflammatory diseases of the gut including Crohn’s disease, ulcerative colitis, necrotizing enterocolitis, and celiac disease. Previous studies have shown that pro-inflammatory cytokines, which are produced during intestinal inflammation, including interleukin-1β (IL-1β), tumor necrosis factor-α, and interferon-γ, have important intestinal TJ barrier-modulating actions. Recent studies have shown that the IL-1β-induced increase in intestinal TJ permeability is an important contributing factor of intestinal inflammation. The IL-1β-induced increase in intestinal TJ permeability is mediated by regulatory signaling pathways and activation of nuclear transcription factor nuclear factor-κB, myosin light chain kinase gene activation, and post-transcriptional occludin gene modulation by microRNA and contributes to the intestinal inflammatory process. In this review, the regulatory role of IL-1β on intestinal TJ barrier, the intracellular mechanisms that mediate the IL-1β modulation of intestinal TJ permeability, and the potential therapeutic targeting of the TJ barrier are discussed.
The gastrointestinal (GI) tract is lined by a single cell layer of intestinal epithelial cells (IECs) which serve as a physical barrier against the influx of luminally located noxious substances, large hydrophilic molecules, and bacterial organisms (1, 2). The epithelial cells are interconnected to each other at the apex of the basolateral membranes by the tight junctions (TJs), which serve as both a boundary demarcating apical from basolateral membrane (referred to as the “fence” function) and as a gate or barrier to paracellular permeation of luminal contents (1, 3–5). The TJs are the apical-most intercellular junctions that prevent or regulate invasion by microorganisms, diffusion of toxins, and flux of water soluble molecules between cells, referred to as paracellular permeability (1, 2, 6–9). A complex of transmembrane proteins interact or make contact across the intercellular spaces to form TJs (1, 2, 10–12). These proteins include occludin, members of the claudin family of proteins, and the junctional adhesion molecule (JAM) family of proteins (1, 5, 12–14). Occludin plays a role in the formation and disassembly of the TJ and is involved in the regulation of the “leak pathway” or paracellular flux of large molecules (1, 15–17), whereas claudins have a greater role in controlling the “pore pathway” or paracellular flux of ions and small molecules, and JAMs are important for TJ assembly and facilitating leukocyte adhesion and migration (1, 6, 11, 18–20). The TJ proteins are also directly linked to cytoskeletal actomyosin fibers via cytoplasmic TJ proteins, including the zonula occludens (ZO) family (ZO-1, ZO-2, and ZO-3) (1, 6, 21–23).
A defective intestinal TJ barrier is an important contributing factor to the pathogenesis of various inflammatory conditions of the gut including celiac disease, inflammatory bowel disease (IBD), and necrotizing enterocolitis (NEC) (4–6, 24–26). Patients with Crohn’s disease have increased intestinal permeability and associated alterations in TJ proteins (6, 26). In patients with quiescent Crohn’s disease, increased intestinal permeability has been identified as an important predictor of early disease relapse (25). Therapeutic re-tightening of the intestinal TJ barrier in patients with active Crohn’s disease is associated with more rapid improvement and prolonged clinical remission, while the presence of persistently increased intestinal permeability is associated with early disease relapse (25, 27, 28), suggesting that therapeutic tightening of the intestinal TJ barrier could be an important treatment option in IBD. In addition, alterations in intestinal permeability and TJ proteins have been shown to be associated with the development of colorectal cancer (29–32).
Pro-inflammatory cytokines, including interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), are markedly elevated in inflammatory conditions of the gut, including IBD and NEC (33–38). Previous studies from our laboratory and others have shown that IL-1β and TNF-α at physiological concentrations cause a marked increase in intestinal epithelial TJ permeability (39–48). Recent studies suggest that, in addition to its direct immune activating effects, IL-1β also promotes intestinal inflammation by disrupting the intestinal TJ barrier and allowing increased intestinal penetration of luminal antigens (49). Consistent with such a possibility, it was recently demonstrated that the inhibition of the IL-1β-induced increase in intestinal TJ permeability prevented the dextran sodium sulfate (DSS)-induced intestinal inflammation (49). In this review, we summarize the published studies as they relate to the effect of IL-1β on the intestinal epithelial TJ barrier, the intracellular signaling pathways involved in TJ barrier modulation, the downstream molecular targets of IL-1β, and the clinical implications of IL-1β intestinal barrier modulation in the context of intestinal inflammation.
The IL-1 family consists of 11 distinct members which are comprised of seven immunomodulatory cytokines, IL-1α, IL-1β, IL-18, IL-36α, IL-36β, IL-36γ, IL-33, and four natural antagonists, IL-1 Receptor antagonist (IL-1Ra), IL-36Ra, IL-37, IL-38 (50). The IL-1 subfamily consists of IL-1α, IL-1β, and IL-33 (50). IL-1α and IL-1β are agonists; IL-1Ra functions as a competitive inhibitor of IL-1 (50–52). These three signaling molecules, IL-1α, IL-1β, and IL-1Ra, share 20-25% amino acid sequence homology (51). IL-1α and IL-1β are produced by immune cells such as monocytes and macrophages, but also by non-immune cells, including endothelial cells and epidermal cells, in response to different stimuli including microbes and cytokines (51). Initially produced as precursor proteins, IL-1α and IL-1β precursors undergo cleavage to generate mature proteins (51, 52). IL-1Ra can function intracellularly, such as in keratinocytes and epithelial cells, but in other cell types, such as mononuclear cells, the IL-1Ra is transported outside of the cell as secretory IL-1Ra (sIL-1Ra) where it functions to bind to and inhibit signaling through IL-1 receptors (52, 53).
There are 10 members of the IL-1 family of receptors, including IL-1 receptor type I (IL-1RI) and IL-1 receptor type II (IL-1R2) which mediate signaling by IL-1α and IL-1β with IL-1 receptor type III (IL-1R3) as a co-receptor (50, 51). IL-1R1 also binds IL-1Ra (50). IL-1R2 is released from the cell surface and binds precursor and mature forms of IL-1β, acting as a decoy receptor and inhibitor of IL-1β function (51–54), as IL-1R2 binds IL-1 but does not transduce any signal (51, 52, 54).
IL-1 and IL-1Ra levels have been shown to be elevated in intestinal inflammatory conditions such as Crohn’s disease and ulcerative colitis (33, 34). Although the levels of both IL-1 and IL-1Ra have been shown to be increased in IBD, a shift in the proportions of these proteins compared to a healthy state has been shown, with relatively less IL-1Ra compared to IL-1 (33). This shift in the IL-1Ra:IL-1 ratio correlated with disease severity (33, 53, 55), suggesting that this imbalance may play a role in the pathogenesis of IBD. A decrease in the IL-1Ra : IL-1β ratio has also been observed in canine IBD (56).
Patients with IBD have been further observed to have an abnormal increase in small intestinal permeability as measured by intestinal absorption of permeability markers such as lactulose and mannitol (57–67). Numerous clinical studies examining intestinal permeability in ulcerative colitis and Crohn’s disease have consistently shown patients with ulcerative colitis and Crohn’s disease to have significant increases in small intestinal permeability (57–67). Although patients with ulcerative colitis have intestinal inflammation that is limited to the colon, the studies have shown that these patients, similar to patients with Crohn’s disease, have significant increase in small intestinal permeability (57–67). The increase in small intestinal permeability is likely to represent in part the effect of the circulating inflammatory mediators, such as IL-1β, TNF-α, IFN-γ, and lipopolysaccharide, which cause an increase in intestinal permeability. As intestinal permeability markers are hydrophilic and do not permeate across the bilipid enterocyte plasma membrane, changes in intestinal permeability are reflective of relative “tightness” or “leakiness” of the intestinal TJ barrier or the paracellular pathways (68, 69). The abnormal increase in intestinal permeability in inflammatory diseases of the gut may be due to the intrinsic structural defect of the TJ barrier related to the underlying disease state or secondary to the effects of inflammatory process or inflammatory mediators, including IL-1β and TNF-α (7, 26, 46, 58, 68, 70, 71). IL-1β at physiologically and clinically relevant concentrations, as seen in IBD and NEC, has been shown to cause an increase in intestinal epithelial TJ permeability (33, 34, 38, 72–74). Interestingly, the inhibition of the IL-1β-induced increase in intestinal permeability in an animal model of colitis was found to be protective against the development of colitis (49), suggesting the possibility that IL-1β-induced disruption of the intestinal TJ barrier contributes to the intestinal inflammation process. It should also be noted that in the setting of ischemic injury to the small intestine, polymorphonuclear leukocytes (PMNs) were shown to enhance intestinal epithelial barrier function in an IL-1β-dependent manner (75). In this model system, it was suggested that the PMN enhancement of intestinal epithelial barrier function was mediated by IL-1β-induced upregulation of cyclooxygenase-2 (COX-2) (75). Thus in the setting of ischemic injury of intestine, IL-1β appears to mediate the PMN-protective effect. In a mouse model of colitis, IL-1β was shown to be involved in mediating tissue repair during resolution of colitis (76).
There have been very few published reports related to the role of IL-1α on the intestinal TJ barrier. In the setting of ischemic-reperfusion injury secondary to burn, IL-1α was shown to ameliorate the burn-induced increase in intestinal permeability (77). In a mouse model of colitis, IL-1α was predominantly produced by IECs and functioned as an inflammatory mediator with associated increased colonic inflammation (76). In the following sections, the intracellular and molecular mechanisms of the IL-1β modulation of the intestinal TJ barrier are reviewed.
In mouse models of colitis and in IBD patients, NF-κB has been shown to play an important role in mediating intestinal inflammation (78, 79). NF-κB activation has been found in macrophages and in IECs from biopsy samples from patients with IBD, and the level of activated NF-κB correlated with the severity of inflammation (78). The NF-κB activity was associated with production of the pro-inflammatory cytokines IL-1, IL-6, and TNF-α (79). IL-1 induced nuclear translocation of NF-κB mediated the activation and transcription of inflammatory genes (80, 81), with other studies showing that specifically IL-1β induced the cytoplasmic to nuclear translocation of NF-κB (39, 82–84).
NF-κB signaling for transcriptional activation can occur through the classical (canonical) or the alternative (non-canonical) pathway, with both pathways leading to the nuclear translocation and activation of NF-κB and binding of DNA by dimers of five proteins: p50, p52, p65 (RelA), RelB, and c-Rel (85–87). In the canonical pathway, degradation of the inhibitory IκB protein occurs following phosphorylation of the IκB kinase (IKK) complex that consists of the catalytic kinases IKKα and IKKβ and the regulatory subunit, IKKγ (NEMO) (86, 87). IKKβ phosphorylates IκB at specific serine residues to mediate degradation of this inhibitory protein (86, 87). Phosphorylated IκB is ubiquitinated and degraded by the proteasome, which allows NF-κB p50/p65 heterodimers to translocate from the cytoplasm to the nucleus to activate the inflammatory genes (86, 87).
Although both the canonical and non-canonical NF-κB pathways were found to be activated in IECs by IL-1β in the context of TJ barrier regulation, only the canonical pathway (NF-κB p50/p65) was involved in the IL-1β-induced dysfunction of the TJ barrier (88). Supporting the requirement for the canonical NF-κB pathway, inhibition of NF-κB p50/p65 activation, but not p52 or p100, prevented the IL-1β-induced increase in intestinal TJ permeability (39). In regards to the canonical NF-κB pathway, IL-1β caused a rapid degradation of IκBα leading to the cytoplasmic-to-nuclear translocation of NF-κB p50/p65 and activation of the target gene, myosin light chain kinase (MLCK) gene (39, 88–90). IL-1β rapidly produced a time-dependent activation of the upstream regulatory protein kinase, mitogen-activated protein kinase kinase kinase-1 (MEKK-1), and NF-κB-inducing kinase (NIK) (88, 91). Although IL-1β caused activation of both MEKK-1 and NIK, the canonical NF-κB pathway (NF-κB p50/p65) activation was mediated by only MEKK-1 and not NIK activation (88). The siRNA induced knockdown of MEKK-1, but not of NIK, inhibited the IL-1β-induced activation of NF-κB p50/p65 (88). Knockdown of p65 also inhibited the IL-1β-induced dysfunction of the TJ barrier (88), confirming the requirement for the canonical NF-κB pathway. IL-1β also caused an activation and increase in nuclear translocation of the p52 subunit (non-canonical NF-κB pathway) with binding of p52 to the κB DNA binding site; however, the p52 subunit was not required for the IL-1β-induced dysfunction of the TJ barrier (88). These findings confirmed that the IL-1β-induced increase in intestinal TJ permeability was mediated by the MEKK-1-dependent activation of NF-κB p50/p65 (Figure 1).
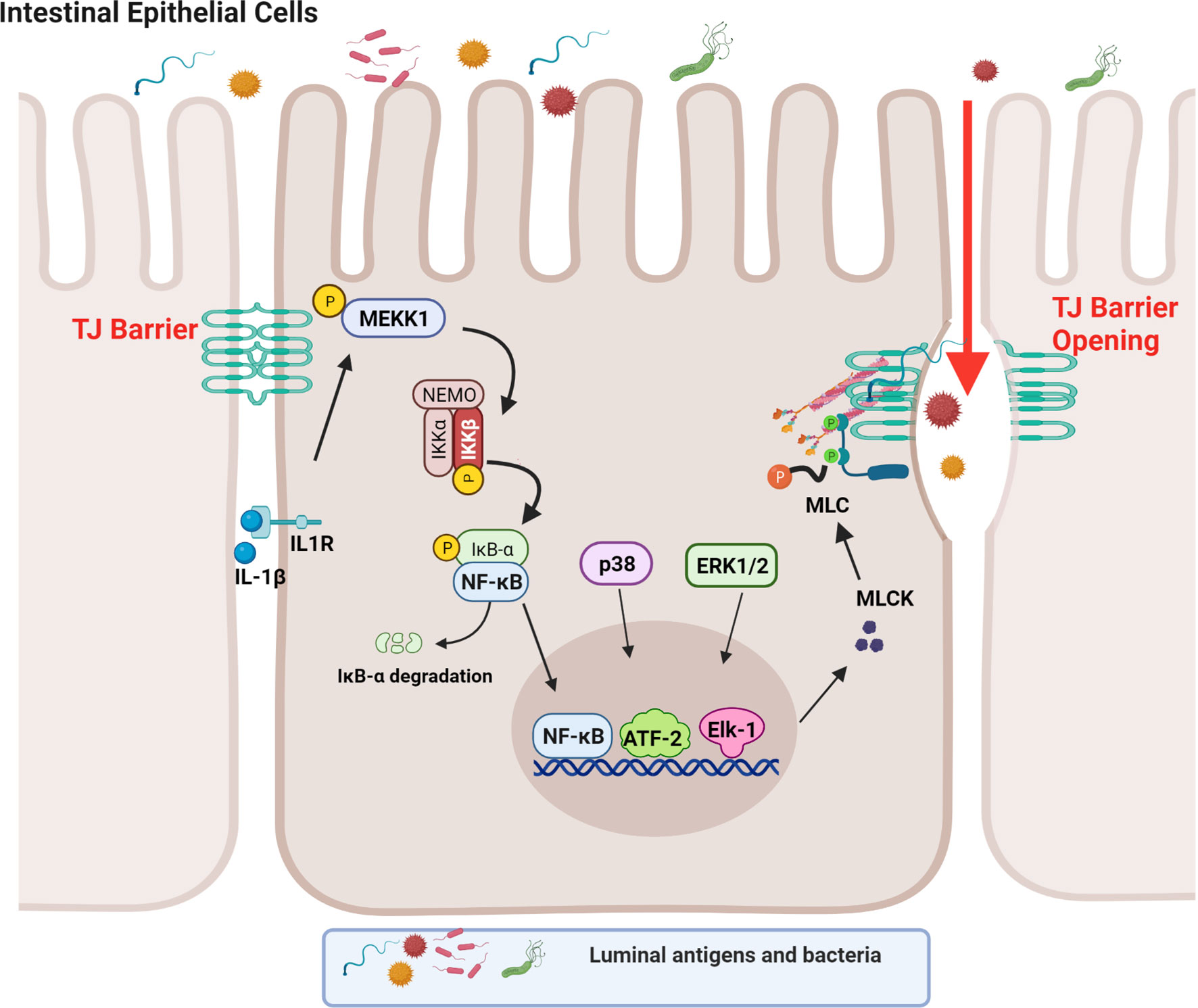
Figure 1 Schematic diagram depicting the involvement of MEKK-1, IKK-β, p38 kinase, ERK1/2, and NF-κB in the interleukin-1β (IL-1β)-induced increase in myosin light chain kinase (MLCK) gene activity in intestinal epithelial tight junction (TJ) permeability. (Created with BioRender.com).
As an integral component of IL-1β regulation of the canonical NF-κB pathway, IL-1β caused a rapid activation of IKK catalytic subunits, IKKα and IKKβ, in Caco-2 intestinal epithelial monolayers and in mouse IECs (88, 92–94). The IL-1β-induced activation of IEC IKKα and IKKβ was dependent on MEKK-1 activation (88). As for the specific IKK subunit involved in the NF-κB p50/p65 activation and the increase in TJ permeability, IKKβ was the dominant subunit that was required for the NF-κB activation and the subsequent increase in TJ permeability in the intestinal monolayers (88). Interestingly, IKKα also played a role in the increase in TJ permeability and the targeted knockdown of IKKα resulted in partial inhibition of NF-κB activation and an increase in TJ permeability (88). Thus, the IL-1β-induced increase in intestinal TJ permeability was mediated by the MEKK-1-induced activation of IKK catalytic subunits IKKβ and IKKα, and IKKβ-induced phosphorylation and degradation of enterocyte inhibitory κB protein and activation of NF-κB p50/p65 (Figure 1) (88).
TNF-α is an additional pro-inflammatory cytokine that has been implicated in IBD and causes increased intestinal epithelial TJ permeability (40, 95, 96). The TNF-α-induced increase in intestinal TJ permeability was also dependent on NF-κB p50/p65 activation (40, 97–103). However, important differences are noted concerning how the enterocyte NF-κB p50/p65 was activated and how the TJ barrier was regulated. The TNF-α-induced activation of NF-κB was mediated by the NIK-induced activation of the IKKα homodimer and not IKKβ catalytic subunit (104); as described above, the IL-1β-induced increase in TJ permeability was regulated by MEKK-1 activation of IKKβ catalytic subunit (88).
It is well-established that MLCK protein level and activity are markedly increased in intestinal tissues obtained from patients with IBD (105). The increase in MLCK protein level was found to correlate with the severity of intestinal inflammation (105). MLCK is a Ca2+-calmodulin activated serine/threonine kinase that regulates actomyosin rearrangement and cell contraction in various cell types including smooth, cardiac, and skeletal muscle and in non-muscle cells (106–111). Two long non-muscle isoforms, MLCK1 (full-length long MLCK) and MLCK2 (which lacks a single exon), are predominantly expressed in intestinal epithelial cells and play a central role in modulating various cell functions (112–114). MLCK1 is also known to regulate the intestinal TJ barrier function via phosphorylation of myosin II regulatory light chain (MLC) at threonine-18 and/or serine-19 leading to peri-junctional actomyosin ring contraction, mechanical retraction of apical membrane and pulling apart of the TJ complex, and opening of the intestinal TJ barrier (109, 115–117). Previous studies have shown that the IL-1β-induced increase in intestinal epithelial TJ permeability in Caco-2 intestinal epithelial monolayers and mouse small intestine in vivo was mediated by an increase in MLCK gene activity and protein expression and an increase in MLCK enzymatic activity (84) (45, 46, 118–120). In these studies, IL-1β caused a rapid activation of MLCK gene activity and increase in MLCK protein expression; inhibition of MLCK activity by pharmacologic inhibitors or by siRNA-induced knockdown prevented the IL-1β-induced increase in intestinal epithelial TJ permeability (84, 121). The IL-1β-induced increase in MLCK gene activity and protein synthesis was mediated by the activation of NF-κB p50/p65, involving the MEKK-1/IKKβ/NF-κB p65 axis as described above (Figure 1) (84). In these studies, targeted knockdown of MEKK-1, IKKβ, or NF-κB p65 prevented the IL-1β-induced increase in MLCK gene activity and increase in intestinal epithelial TJ permeability, confirming the regulatory role of MEKK-1, IKKβ and the NF-κB p50/p65 axis on MLCK gene activation (88). Zhai and colleagues recently showed that low-fat yogurt prevented the IL-1β-induced intestinal barrier disruption by inhibiting the upregulation of MLCK gene expression in Caco-2 cells (122). These studies also showed that the IL-1β-induced increase in NF-κB activation is inhibited by low-fat yogurt application to Caco-2 cells (122). Collectively, these studies show that IL-1β-induced increases in MLCK gene activity and protein synthesis are required for increased intestinal TJ permeability. The NF-κB p50/p65 signaling pathway regulates the TJ barrier by targeting MLCK gene activation (Figure 1).
MAPKs are serine-threonine protein kinases that mediate cellular activities including proliferation, differentiation, apoptosis, survival, inflammation, and innate immune responses (123, 124). There are at least three groups of MAPKs identified including extracellular signal-regulated kinases (ERK), the p38 MAPKs, and the cJun NH2-terminal kinases (JNK) (125). It is well-established that IL-1β and other pro-inflammatory cytokines can activate MAPK signal transduction pathways. Previous studies have shown that the p38 kinase signaling cascade also contributes to IL-1β-induced increased MLCK expression and increased intestinal TJ permeability in vitro and in vivo (46). These studies found that IL-1β induced a rapid phosphorylation and activation of p38 kinase and downstream activation of a p38-dependent transcription factor, activating transcription factor-2 (ATF-2); the activated ATF-2 translocated to the nucleus and attached to the binding motif on the MLCK promoter region, leading to MLCK gene activation and protein synthesis, and MLCK-dependent opening of the intestinal TJ barrier (46). The targeted knockdown of p38 kinase and ATF-2 inhibited the IL-1β-induced activation of MLCK gene and increased intestinal TJ permeability in Caco-2 monolayers and in mouse small intestine (46). Thus, in addition to NF-κB, ATF-2 also played an important role in the regulation of the IL-1β-induced activation of MLCK gene and increase in MLCK protein synthesis. Other studies have shown that p38 kinase activation is required for IL-1β-induced regulation of the TJ protein claudin-2 in a rat hepatic injury model (126, 127).
In addition to NF-κB and ATF-2, transcription factor Elk-1 has also been shown to contribute to IL-1β-induced increase in intestinal TJ permeability (46, 128). IL-1β has also been found to cause a rapid activation of Elk-1 in intestinal epithelial cells (128, 129). Upon activation, Elk-1 translocated to the nucleus and attached to the cis-binding motif on the MLCK promoter region, leading to the activation of MLCK gene and an increase in TJ permeability (128). In aggregate, these studies suggest that IL-1β induces the activation and binding of transcription factors NF-κB p50/p65, ATF-2, and Elk-1 to the MLCK minimal promoter region in close proximity to each other, and in concert, activate the MLCK gene and produce an opening of the TJ barrier (Figure 1) (128).
Intestinal epithelial TJ barrier function is maintained by intercellular TJs, including multi-protein complexes that seal the space between adjacent cells at the boundary of the apical and lateral membrane surfaces of adjacent epithelial cells (1, 130–133). TJs, which are the apical-most intercellular junctions, regulate the paracellular flux of ions and solutes, referred to as the “gate function” (8, 130–133). Related to the gate function, disturbances in TJ barrier function result in increased TJ or paracellular permeability and increased exposure of underlying intestinal tissue and other organ systems to noxious luminal antigens.
The TJ complex consists of integral transmembrane proteins such as occludin, claudins, and JAMs, as well as cytosolic scaffold proteins, such as ZO-1, ZO-2, and ZO-3, which in turn anchor the transmembrane proteins to the actin cytoskeleton (5, 11, 134). Previous studies from our laboratory and others have shown that occludin plays an important regulatory role in restricting the paracellular flux of large macromolecules via a non-restrictive or “leak” pathway and that occludin depletion leads to a preferential increase in paracellular flux of macromolecules (15, 131–133, 135, 136). In addition to targeting MLCK gene activation, IL-1β-induced increase in intestinal TJ permeability has also been found to be dependent on post-transcriptional degradation of occludin mRNA (49).
IL-1β treatment has been shown to selectively decrease the occludin level in Caco-2 monolayers and mouse intestinal epithelial cells in live mice without affecting other transmembrane TJ proteins (49). In these studies involving Caco-2 monolayers and live mice, IL-1β caused a rapid increase in miRNAs which bind to occludin mRNA (49). MiRNAs are small, single-stranded non-coding RNAs that bind to miRNA response elements on the 3’ untranslated region (3’-UTR) of mRNA to induce mRNA degradation or suppression of translation (49, 137, 138). It was found that IL-1β caused a rapid and marked increase in miR-200c-3p in IECs (49). Bioinformatics analysis predicted high probability of miR-200c-3p binding to the occludin 3’-UTR and post-transcriptional regulation. These studies also showed that increasing miR-200c-3p expression in IECs was sufficient to cause an increase in intestinal TJ permeability in Caco-2 monolayers and mice and that treatment with antagomirs, which inhibit or silence miR-200c-3p, inhibited the IL-1β-induced decrease in occludin expression and increase in intestinal epithelial TJ permeability (49). This work provides support for the cause-and-effect relationship between post-transcriptional miR-200c-3p degradation of occludin mRNA and increase in TJ permeability (Figure 2) (49).
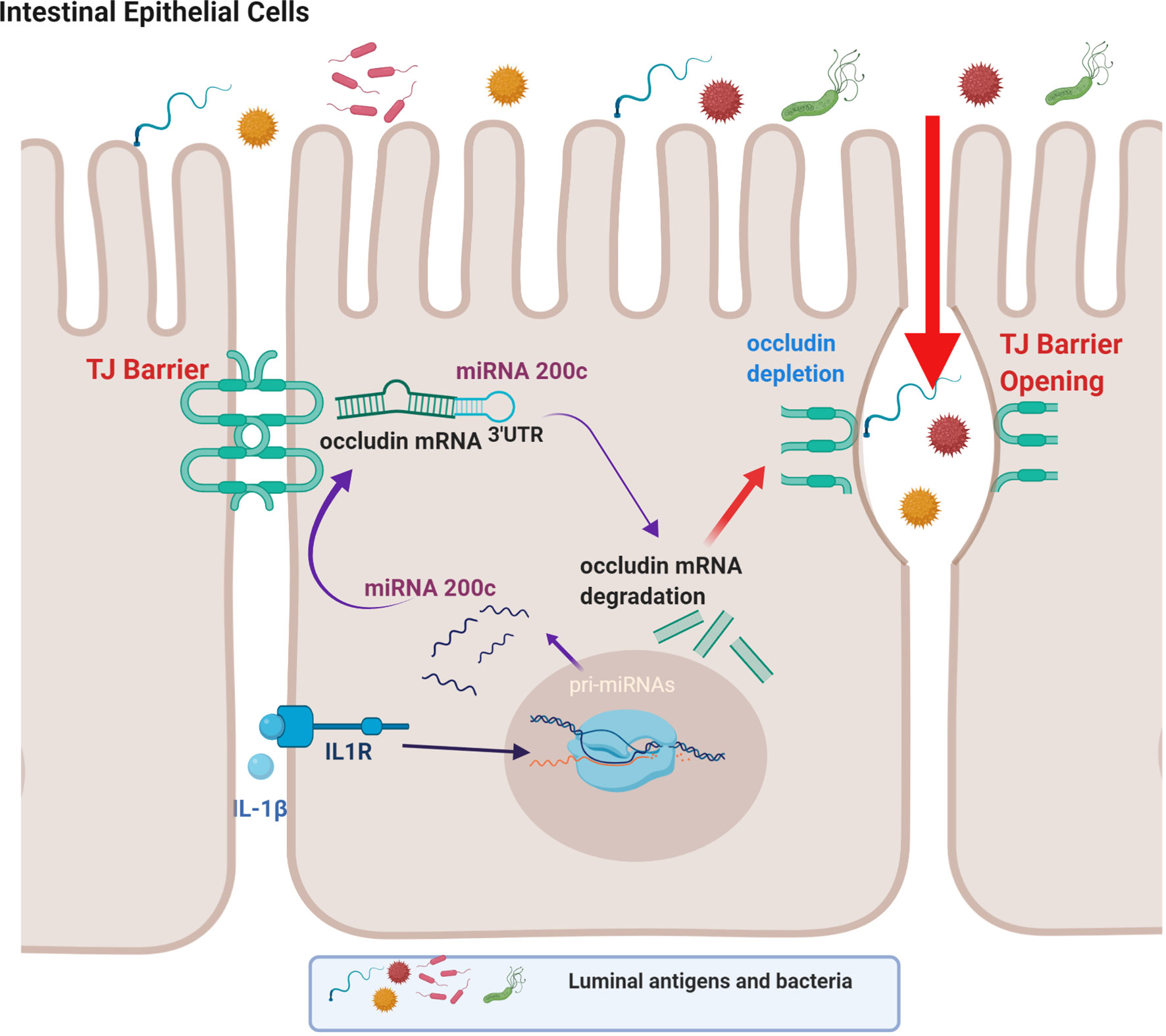
Figure 2 Schematic diagram showing the involvement of the microRNA-200c-3p-induced degradation of occludin mRNA in interleukin-1β (IL-1β)-induced increase in intestinal epithelial tight junction permeability. (Created with BioRender.com).
The defective intestinal TJ barrier is an important pathogenic factor contributing to intestinal inflammatory processes in IBD, NEC, and celiac disease (57, 139–141). Previous work has also investigated the potential role of elevated IL-1β present in intestinal inflammation and its contribution to TJ barrier disturbance or “leaky gut” through targeting of miR-200c-3p expression (49). IL-1β expression was shown to be markedly elevated in colonic tissues of ulcerative colitis patients and directly correlated with an increase in miR-200c-3p expression (49). Interestingly, the organoids generated from colonic tissue from ulcerative colitis patients also showed marked increase in IL-1β and miR-200c-3p expression compared to the organoids from the healthy control patients (49). These studies indicated that IL-1β and miR-200c-3p expression was markedly elevated in colonic tissue and epithelial cells from patients with ulcerative colitis and also suggested the possibility that miR-200c-3p may be targeted to inhibit the IL-1β-associated increase in intestinal TJ permeability and treat intestinal inflammation. This possibility was tested in proof-of-concept studies using DSS-induced colitis as a murine model of colitis (49). DSS oral administration caused a marked increase in intestinal tissue IL-1β and miR-200c-3p expression and was further associated with a decrease in occludin expression and increase in colonic permeability. The oral administration of antogomiR-200c inhibited DSS-induced increase in intestinal tissue miR-200c-3p levels, decrease in occludin levels, increase in colonic permeability, and development of colitis (49). Taken together, these studies demonstrate that therapeutic targeting of the IL-1β-induced increase in miR-200c-3p expression and intestinal permeability is effective in preventing DSS-induced intestinal inflammation (Figure 3). Additional studies have examined the impact of IL-1β on intestinal occludin expression using a dog model of IBD. Ogawa et al. reported an increase in the IL-1β:IL-1Ra ratio in the colonic mucosa of dogs with IBD; this increase was correlated with a decrease in occludin mRNA expression, intestinal barrier dysfunction, and colonic inflammation in a dog model of IBD (73).
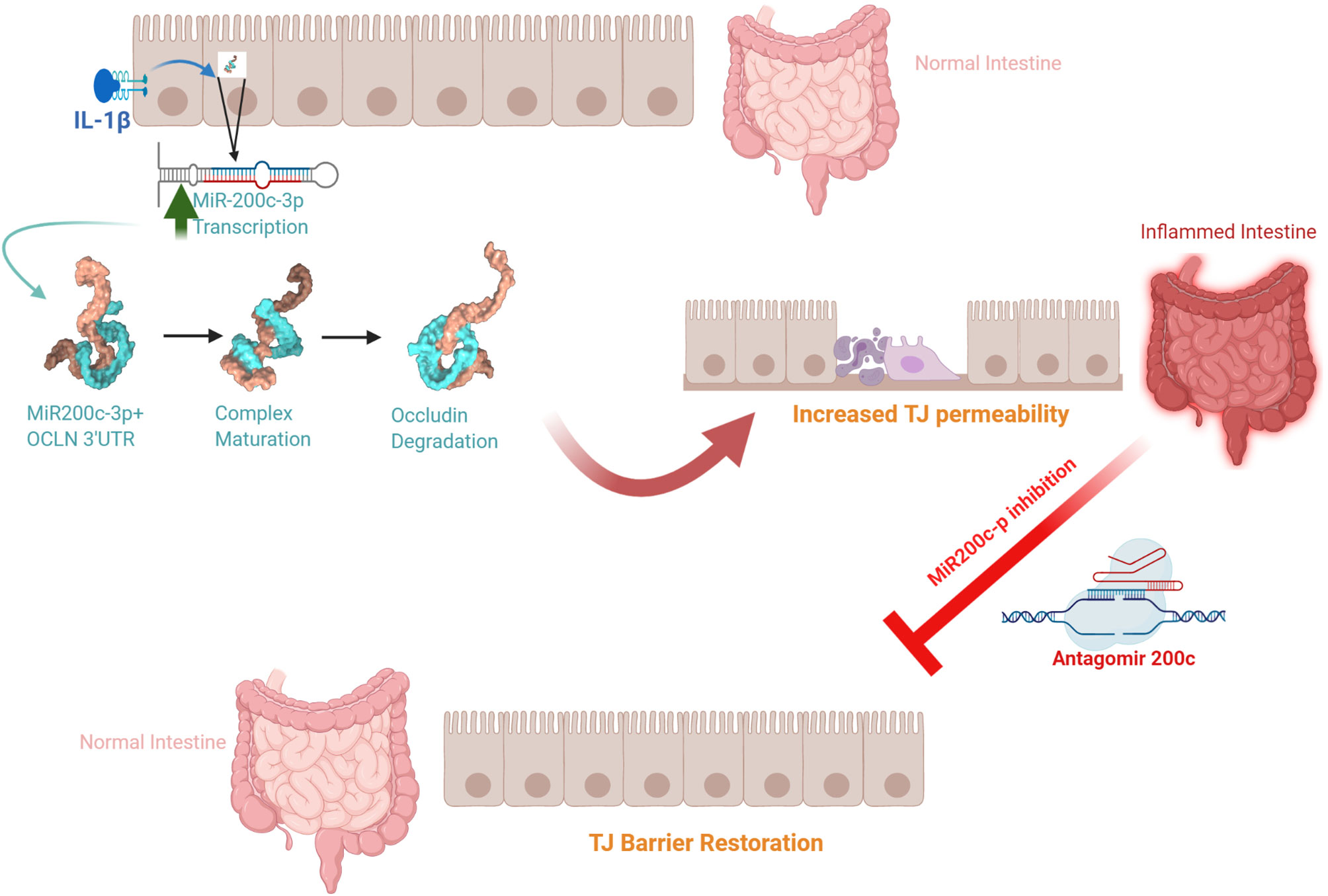
Figure 3 Schematic diagram of the potential role of the interleukin-1β (IL-1β)-induced increase in microRNA-200c-3p expression and intestinal tight junction (TJ) permeability in modulating intestinal inflammation. (Created with BioRender.com).
Although dysregulation of different types of claudin proteins has been linked to the pathogenesis of IBD, the observed effect of IL-1β on claudin proteins has been shown to be limited and inconsistent (30, 142–148). Claudins are a transmembrane protein family comprised of at least 27 members (149). In the GI tract, only a few of the claudins are expressed and known to regulate intestinal TJ barrier function, including claudin-1, claudin-2, claudin-3, claudin-4, claudin-5, claudin-8, claudin-12, and claudin-18 (150–153). A previous study showed that IL-1β did not affect the expression or the distribution of claudin-1, claudin-2, claudin-3, claudin-4, or claudin-5 (39). In contrast, Maria-Ferreira et al. showed that IL-1β induced an increase in Caco-2 TJ permeability that was associated with a decrease in claudin-1 expression and disruption in occludin junctional localization (154). Haines et al. reported that the IL-1β-induced increase in intestinal TJ permeability was mediated in part by claudin-3 downregulation and translocation from the junctional localization into the nucleus; however, expression levels of ZO-1, occludin, claudin-1, claudin-4, and claudin-15 remained unchanged (121). This study also found that IL-1β induced an association of β-catenin with the claudin-3 promoter in an MLCK-dependent manner (121). Consistent with these findings, other reports have shown that Wnt signaling (and subsequent β-catenin nuclear accumulation) induced claudin-3 downregulation, and subsequently led to an increase in intestinal permeability (155). Although Guo and colleagues found that there was no change in ZO-1 protein level in response to IL-1β, they found an increase in claudin-1 expression and a decrease in occludin expression (82). Wang et al. showed that ZO-1, claudin-1, and claudin-7 junctional localization was also disrupted by IL-1β treatment in Caco-2 cells, and was associated with an increase in intestinal TJ permeability (156). The effect of IL-1β-induced increase in Caco-2 TJ permeability was also proposed to be mediated by an increase in p38 kinase phosphorylation, leading to an increase in MLCK gene and protein expression and indirectly influencing the junctional localization of ZO-1, claudin-1, and claudin-7 (156). The results of studies evaluating the effect of IL-1β on claudin proteins have been inconsistent; some studies suggested that IL-1β has no effect, while others have suggested that IL-1β downregulates claudin-3 or causes an alteration in ZO-1, claudin-1, and claudin-7 junctional localization in a MLCK-dependent manner.
IL-1β is a pluripotent pro-inflammatory cytokine that is elevated in various inflammatory diseases, including NEC, irritable bowel syndrome, infectious diarrhea, and IBD. Accumulating evidence has shown that IL-1β plays an important pathogenic role in the intestinal inflammation process in these diseases (51, 157, 158). A key hallmark of IBD is intestinal epithelial TJ barrier disruption manifested by an increase in intestinal permeability (71, 140, 159); however, the precise factors which contribute to the defective intestinal TJ barrier and the role IL-1β plays in the barrier defect remains unclear. In this review, we highlight recent scientific advances that demonstrate the disruptive influence of IL-1β on the intestinal TJ barrier. We further provide insight into the intracellular signaling process, the effector and the TJ proteins involved, and the potential for therapeutic targeting of IL-1β-induced disruption of the intestinal TJ barrier to prevent or treat intestinal inflammation. In summary, IL-1β is markedly elevated in intestinal inflammation associated with ulcerative colitis and other inflammatory diseases, IL-1β causes a rapid and marked increase in intestinal TJ permeability, IL-1β-induced increase in TJ permeability is mediated in part by the activation of the canonical NF-κB pathway, MLCK gene activation and post-transcriptional degradation of occludin mRNA, and therapeutic targeting of the IL-1β-induced increase in intestinal TJ permeability is effective in preventing colitis in a murine model of colitis. Thus, IL-1β modulation of the intestinal TJ barrier represents an important potential pathogenic mechanism contributing to the observed increase in intestinal permeability in various inflammatory conditions of the gut and is a potential therapeutic target to prevent or treat intestinal inflammation.
LWK wrote and edited the manuscript. RA-S edited and revised the manuscript and created the figures. TYM conceived, edited and revised the manuscript and approved final version of this manuscript. All authors contributed to the article and approved the submitted version.
This research project was supported by the National Institute of Diabetes and Digestive and Kidney Diseases grants R0-1-DK-64165-01 (TYM), R0-1-DK-106072-01 (TYM), R01-DK-121073-01 (TYM), R01-DK-081429 (TYM), the J. Lloyd Huck Chair in Medicine endowment from the Penn State College of Medicine (TYM), and the Department of Medicine Inspiration Award from the Penn State College of Medicine (RA-S).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ma TY, Nighot P, Al-Sadi R. Tight Junctions and the Intestinal Barrier. In: Physiology of the Gastrointestinal Tract: Sixth Edition. London, United Kingdom, San Diego, CA, United States, Cambridge, MA, United States, Kidlington, United Kingdom: Elsevier Inc (2018). p. 587–639. 1-2.
2. Buckley A, Turner JR. Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease. Cold Spring Harb Perspect Biol (2018) 10(1):1–16. doi: 10.1101/cshperspect.a029314
3. Madara JL. Loosening Tight Junctions. Lessons From the Intestine. J Clin Invest. (1989) 83(4):1089–94. doi: 10.1172/JCI113987
4. Turner JR. Intestinal Mucosal Barrier Function in Health and Disease. Nat Rev Immunol (2009) 9(11):799–809. doi: 10.1038/nri2653
5. König J, Wells J, Cani PD, García-Ródenas CL, MacDonald T, Mercenier A, et al. Human Intestinal Barrier Function in Health and Disease. Clin Transl Gastroenterol (2016) 7(10):e196. doi: 10.1038/ctg.2016.54
6. Salvo Romero E, Alonso Cotoner C, Pardo Camacho C, Casado Bedmar M, Vicario M. The Intestinal Barrier Function and its Involvement in Digestive Disease. Rev Esp Enferm Dig. (2015) 107(11):686–96. doi: 10.17235/reed.2015.3846/2015
7. Zuo L, Kuo WT, Turner JR. Tight Junctions as Targets and Effectors of Mucosal Immune Homeostasis. Cell Mol Gastroenterol Hepatol (2020) 10(2):327–40. doi: 10.1016/j.jcmgh.2020.04.001
8. Turner JR. ‘Putting the Squeeze’ on the Tight Junction: Understanding Cytoskeletal Regulation. Semin Cell Dev Biol (2000) 11(4):301–8. doi: 10.1006/scdb.2000.0180
9. Turner JR. Show Me the Pathway! Regulation of Paracellular Permeability by Na(+)-Glucose Cotransport. Adv Drug Deliv Rev (2000) 41(3):265–81. doi: 10.1016/S0169-409X(00)00046-6
10. Elkouby-Naor L, Ben-Yosef T. Functions of Claudin Tight Junction Proteins and Their Complex Interactions in Various Physiological Systems. Int Rev Cell Mol Biol (2010) 279:1–32. doi: 10.1016/S1937-6448(10)79001-8
11. Van Itallie CM, Anderson JM. Claudins and Epithelial Paracellular Transport. Annu Rev Physiol (2006) 68:403–29. doi: 10.1146/annurev.physiol.68.040104.131404
12. Mitic LL, Van Itallie CM, Anderson JM. Molecular Physiology and Pathophysiology of Tight Junctions I. Tight Junction Structure and Function: Lessons From Mutant Animals and Proteins. Am J Physiol Gastrointest Liver Physiol (2000) 279(2):G250–4. doi: 10.1152/ajpgi.2000.279.2.G250
13. Van Itallie CM, Anderson JM. The Molecular Physiology of Tight Junction Pores. Physiol (Bethesda) (2004) 19:331–8. doi: 10.1152/physiol.00027.2004
14. Van Itallie CM, Anderson JM. Architecture of Tight Junctions and Principles of Molecular Composition. Semin Cell Dev Biol (2014) 36:157–65. doi: 10.1016/j.semcdb.2014.08.011
15. Al-Sadi R, Khatib K, Guo S, Ye D, Youssef M, Ma T. Occludin Regulates Macromolecule Flux Across the Intestinal Epithelial Tight Junction Barrier. Am J Physiol Gastrointest Liver Physiol (2011) 300(6):G1054–64. doi: 10.1152/ajpgi.00055.2011
16. Almansour K, Taverner A, Turner JR, Eggleston IM, Mrsny RJ. An Intestinal Paracellular Pathway Biased Toward Positively-Charged Macromolecules. J Control Release (2018) 288:111–25. doi: 10.1016/j.jconrel.2018.09.003
17. Buschmann MM, Shen L, Rajapakse H, Raleigh DR, Wang Y, Wang Y, et al. Occludin OCEL-Domain Interactions are Required for Maintenance and Regulation of the Tight Junction Barrier to Macromolecular Flux. Mol Biol Cell (2013) 24(19):3056–68. doi: 10.1091/mbc.e12-09-0688
18. Monaco A, Ovryn B, Axis J, Amsler K. The Epithelial Cell Leak Pathway. Int J Mol Sci (2021) 22(14):1–26. doi: 10.3390/ijms22147677
19. Pearce SC, Al-Jawadi A, Kishida K, Yu S, Hu M, Fritzky LF, et al. Marked Differences in Tight Junction Composition and Macromolecular Permeability Among Different Intestinal Cell Types. BMC Biol (2018) 16(1):19. doi: 10.1186/s12915-018-0481-z
20. Krug SM, Gunzel D, Conrad MP, Lee IF, Amasheh S, Fromm M, et al. Charge-Selective Claudin Channels. Ann N Y Acad Sci (2012) 1257:20–8. doi: 10.1111/j.1749-6632.2012.06555.x
21. Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, et al. ZO-1 Controls Endothelial Adherens Junctions, Cell-Cell Tension, Angiogenesis, and Barrier Formation. J Cell Biol (2015) 208(6):821–38. doi: 10.1083/jcb.201404140
22. Odenwald MA, Choi W, Kuo WT, Singh G, Sailer A, Wang Y, et al. The Scaffolding Protein ZO-1 Coordinates Actomyosin and Epithelial Apical SpecializationsIn Vitro and In Vivo. J Biol Chem (2018) 293(45):17317–35. doi: 10.1074/jbc.RA118.003908
23. Cordenonsi M, D’Atri F, Hammar E, Parry DA, Kendrick-Jones J, Shore D, et al. Cingulin Contains Globular and Coiled-Coil Domains and Interacts With ZO-1, ZO-2, ZO-3, and Myosin. J Cell Biol (1999) 147(7):1569–82. doi: 10.1083/jcb.147.7.1569
24. Arrieta MC, Madsen K, Doyle J, Meddings J. Reducing Small Intestinal Permeability Attenuates Colitis in the IL10 Gene-Deficient Mouse. Gut (2009) 58(1):41–8. doi: 10.1136/gut.2008.150888
25. Wyatt J, Vogelsang H, Hübl W, Waldhöer T, Lochs H. Intestinal Permeability and the Prediction of Relapse in Crohn’s Disease. Lancet (1993) 341(8858):1437–9. doi: 10.1016/0140-6736(93)90882-H
26. Ma TY. Intestinal Epithelial Barrier Dysfunction in Crohn’s Disease. Proc Soc Exp Biol Med (1997) 214(4):318–27. doi: 10.3181/00379727-214-44099
27. Miehsler W, Püspök A, Oberhuber T, Vogelsang H. Impact of Different Therapeutic Regimens on the Outcome of Patients With Crohn’s Disease of the Upper Gastrointestinal Tract. Inflamm Bowel Dis (2001) 7(2):99–105. doi: 10.1097/00054725-200105000-00004
28. Wild GE, Waschke KA, Bitton A, Thomson AB. The Mechanisms of Prednisone Inhibition of Inflammation in Crohn’s Disease Involve Changes in Intestinal Permeability, Mucosal Tnfalpha Production and Nuclear Factor Kappa B Expression. Aliment Pharmacol Ther (2003) 18(3):309–17. doi: 10.1046/j.1365-2036.2003.01611.x
29. Landy J, Ronde E, English N, Clark SK, Hart AL, Knight SC, et al. Tight Junctions in Inflammatory Bowel Diseases and Inflammatory Bowel Disease Associated Colorectal Cancer. World J Gastroenterol (2016) 22(11):3117–26. doi: 10.3748/wjg.v22.i11.3117
30. Zhu L, Han J, Li L, Wang Y, Li Y, Zhang S. Claudin Family Participates in the Pathogenesis of Inflammatory Bowel Diseases and Colitis-Associated Colorectal Cancer. Front Immunol (2019) 10:1441. doi: 10.3389/fimmu.2019.01441
31. Jobin C. Colorectal Cancer: CRC–All About Microbial Products and Barrier Function? Nat Rev Gastroenterol Hepatol (2012) 9(12):694–6. doi: 10.1038/nrgastro.2012.220
32. Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, et al. Adenoma-Linked Barrier Defects and Microbial Products Drive IL-23/IL-17-Mediated Tumour Growth. Nature (2012) 491(7423):254–8. doi: 10.1038/nature11465
33. Casini-Raggi V, Kam L, Chong YJ, Fiocchi C, Pizarro TT, Cominelli F. Mucosal Imbalance of IL-1 and IL-1 Receptor Antagonist in Inflammatory Bowel Disease. A Novel Mechanism of Chronic Intestinal Inflammation. J Immunol (1995) 154(5):2434–40.
34. Ludwiczek O, Vannier E, Borggraefe I, Kaser A, Siegmund B, Dinarello CA, et al. Imbalance Between Interleukin-1 Agonists and Antagonists: Relationship to Severity of Inflammatory Bowel Disease. Clin Exp Immunol (2004) 138(2):323–9. doi: 10.1111/j.1365-2249.2004.02599.x
35. Al-Sadi R, Boivin M, Ma T. Mechanism of Cytokine Modulation of Epithelial Tight Junction Barrier. Front Biosci (Landmark Ed). (2009) 14:2765–78. doi: 10.2741/3413
36. Yang B, Fu L, Privratsky JR, Lu X, Ren J, Mei C, et al. Interleukin-1 Receptor Activation Aggravates Autosomal Dominant Polycystic Kidney Disease by Modulating Regulated Necrosis. Am J Physiol Renal Physiol (2019) 317(2):F221–F8. doi: 10.1152/ajprenal.00104.2019
37. Bhatia AM, Stoll BJ, Cismowski MJ, Hamrick SE. Cytokine Levels in the Preterm Infant With Neonatal Intestinal Injury. Am J Perinatol (2014) 31(6):489–96. doi: 10.1055/s-0033-1353437
38. Nanthakumar NN, Fusunyan RD, Sanderson I, Walker WA. Inflammation in the Developing Human Intestine: A Possible Pathophysiologic Contribution to Necrotizing Enterocolitis. Proc Natl Acad Sci USA (2000) 97(11):6043–8. doi: 10.1073/pnas.97.11.6043
39. Al-Sadi RM, Ma TY. IL-1beta Causes an Increase in Intestinal Epithelial Tight Junction Permeability. J Immunol (2007) 178(7):4641–9. doi: 10.4049/jimmunol.178.7.4641
40. Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, et al. TNF-Alpha-Induced Increase in Intestinal Epithelial Tight Junction Permeability Requires NF-Kappa B Activation. Am J Physiol Gastrointest Liver Physiol (2004) 286(3):G367–76. doi: 10.1152/ajpgi.00173.2003
41. Fukuda T, Majumder K, Zhang H, Turner PV, Matsui T, Mine Y. Adenine Inhibits TNF-Alpha Signaling in Intestinal Epithelial Cells and Reduces Mucosal Inflammation in a Dextran Sodium Sulfate-Induced Colitis Mouse Model. J Agric Food Chem (2016) 64(21):4227–34. doi: 10.1021/acs.jafc.6b00665
42. Su L, Nalle SC, Shen L, Turner ES, Singh G, Breskin LA, et al. TNFR2 Activates MLCK-Dependent Tight Junction Dysregulation to Cause Apoptosis-Mediated Barrier Loss and Experimental Colitis. Gastroenterology (2013) 145(2):407–15. doi: 10.1053/j.gastro.2013.04.011
43. Turner JR. Molecular Basis of Epithelial Barrier Regulation: From Basic Mechanisms to Clinical Application. Am J Pathol (2006) 169(6):1901–9. doi: 10.2353/ajpath.2006.060681
44. Graham WV, Wang F, Clayburgh DR, Cheng JX, Yoon B, Wang Y, et al. Tumor Necrosis Factor-Induced Long Myosin Light Chain Kinase Transcription is Regulated by Differentiation-Dependent Signaling Events. Characterization of the Human Long Myosin Light Chain Kinase Promoter. J Biol Chem (2006) 281(36):26205–15. doi: 10.1074/jbc.M602164200
45. Al-Sadi R, Guo S, Dokladny K, Smith MA, Ye D, Kaza A, et al. Mechanism of Interleukin-1β Induced-Increase in Mouse Intestinal Permeability In Vivo. J Interferon Cytokine Res (2012) 32(10):474–84. doi: 10.1089/jir.2012.0031
46. Al-Sadi R, Guo S, Ye D, Dokladny K, Alhmoud T, Ereifej L, et al. Mechanism of IL-1β Modulation of Intestinal Epithelial Barrier Involves P38 Kinase and Activating Transcription Factor-2 Activation. J Immunol (2013) 190(12):6596–606. doi: 10.4049/jimmunol.1201876
47. Al-Sadi R, Guo S, Ye D, Rawat M, Ma TY. Tnf-α Modulation of Intestinal Tight Junction Permeability is Mediated by NIK/IKK-α Axis Activation of the Canonical NF-κb Pathway. Am J Pathol (2016) 186(5):1151–65. doi: 10.1016/j.ajpath.2015.12.016
48. Haines RJ, Beard RS Jr., Chen L, Eitnier RA, Wu MH. Interleukin-1beta Mediates Beta-Catenin-Driven Downregulation of Claudin-3 and Barrier Dysfunction in Caco2 Cells. Dig Dis Sci (2016) 61(8):2252–61. doi: 10.1007/s10620-016-4145-y
49. Rawat M, Nighot M, Al-Sadi R, Gupta Y, Viszwapriya D, Yochum G, et al. IL1B Increases Intestinal Tight Junction Permeability by Up-Regulation of MIR200C-3p, Which Degrades Occludin Mrna. Gastroenterology (2020) 159(4):1375–89. doi: 10.1053/j.gastro.2020.06.038
50. Dinarello CA. Overview of the IL-1 Family in Innate Inflammation and Acquired Immunity. Immunol Rev (2018) 281(1):8–27. doi: 10.1111/imr.12621
51. Stylianou E, Saklatvala J. Interleukin-1. Int J Biochem Cell Biol (1998) 30(10):1075–9. doi: 10.1016/S1357-2725(98)00081-8
52. Dinarello CA. Biologic Basis for Interleukin-1 in Disease. Blood (1996) 87(6):2095–147. doi: 10.1182/blood.V87.6.2095.bloodjournal8762095
53. Cominelli F, Pizarro TT. Interleukin-1 and Interleukin-1 Receptor Antagonist in Inflammatory Bowel Disease. Aliment Pharmacol Ther (1996) 10(Suppl 2):49–53; discussion 4. doi: 10.1046/j.1365-2036.1996.22164020.x
54. O’Neill LA, Dinarello CA. The IL-1 Receptor/Toll-Like Receptor Superfamily: Crucial Receptors for Inflammation and Host Defense. Immunol Today (2000) 21(5):206–9. doi: 10.1016/S0167-5699(00)01611-X
55. Hyams JS, Fitzgerald JE, Wyzga N, Muller R, Treem WR, Justinich CJ, et al. Relationship of Interleukin-1 Receptor Antagonist to Mucosal Inflammation in Inflammatory Bowel Disease. J Pediatr Gastroenterol Nutr (1995) 21(4):419–25. doi: 10.1097/00005176-199511000-00008
56. Maeda S, Ohno K, Nakamura K, Uchida K, Nakashima K, Fukushima K, et al. Mucosal Imbalance of Interleukin-1β and Interleukin-1 Receptor Antagonist in Canine Inflammatory Bowel Disease. Vet J (2012) 194(1):66–70. doi: 10.1016/j.tvjl.2012.02.026
57. Michielan A, D’Inca R. Intestinal Permeability in Inflammatory Bowel Disease: Pathogenesis, Clinical Evaluation, and Therapy of Leaky Gut. Mediators Inflammation (2015) 2015:628157. doi: 10.1155/2015/628157
58. Turpin W, Lee SH, Raygoza Garay JA, Madsen KL, Meddings JB, Bedrani L, et al. Increased Intestinal Permeability is Associated With Later Development of Crohn’s Disease. Gastroenterology (2020) 159(6):2092–100.e5. doi: 10.1053/j.gastro.2020.08.005
59. Halme L, Turunen U, Tuominen J, Forsstrom T, Turpeinen U. Comparison of Iohexol and Lactulose-Mannitol Tests as Markers of Disease Activity in Patients With Inflammatory Bowel Disease. Scand J Clin Lab Invest (2000) 60(8):695–701. doi: 10.1080/00365510050216420
60. Teshima CW, Dieleman LA, Meddings JB. Abnormal Intestinal Permeability in Crohn’s Disease Pathogenesis. Ann N Y Acad Sci (2012) 1258:159–65. doi: 10.1111/j.1749-6632.2012.06612.x
61. Chang J, Leong RW, Wasinger VC, Ip M, Yang M, Phan TG. Impaired Intestinal Permeability Contributes to Ongoing Bowel Symptoms in Patients With Inflammatory Bowel Disease and Mucosal Healing. Gastroenterology (2017) 153(3):723–31. doi: 10.1053/j.gastro.2017.05.056
62. Keita Å, Lindqvist CM, Öst Å, Magana CDL, Schoultz I, Halfvarson J. Gut Barrier Dysfunction-a Primary Defect in Twins With Crohn’s Disease Predominantly Caused by Genetic Predisposition. J Crohns Colitis (2018) 12(10):1200–9. doi: 10.1093/ecco-jcc/jjy045
63. Benjamin J, Makharia GK, Ahuja V, Kalaivani M, Joshi YK. Intestinal Permeability and its Association With the Patient and Disease Characteristics in Crohn’s Disease. World J Gastroenterol (2008) 14(9):1399–405. doi: 10.3748/wjg.14.1399
64. Vilela EG, Torres HO, Ferrari ML, Lima AS, Cunha AS. Gut Permeability to Lactulose and Mannitol Differs in Treated Crohn’s Disease and Celiac Disease Patients and Healthy Subjects. Braz J Med Biol Res (2008) 41(12):1105–9. doi: 10.1590/S0100-879X2008001200010
65. Dastych M, Novotná H, Cíhalová J. Lactulose/Mannitol Test and Specificity, Sensitivity, and Area Under Curve of Intestinal Permeability Parameters in Patients With Liver Cirrhosis and Crohn’s Disease. Dig Dis Sci (2008) 53(10):2789–92. doi: 10.1007/s10620-007-0184-8
66. Büning C, Geissler N, Prager M, Sturm A, Baumgart DC, Büttner J, et al. Increased Small Intestinal Permeability in Ulcerative Colitis: Rather Genetic Than Environmental and a Risk Factor for Extensive Disease? Inflamm Bowel Dis (2012) 18(10):1932–9. doi: 10.1002/ibd.22909
67. Miele E, Pascarella F, Quaglietta L, Giannetti E, Greco L, Troncone R, et al. Altered Intestinal Permeability is Predictive of Early Relapse in Children With Steroid-Responsive Ulcerative Colitis. Aliment Pharmacol Ther (2007) 25(8):933–9. doi: 10.1111/j.1365-2036.2007.03291.x
68. Kong W, Wang J, Ping X, Shen J, Ni X, Liu F, et al. Biomarkers for Assessing Mucosal Barrier Dysfunction Induced by Chemotherapy: Identifying a Rapid and Simple Biomarker. Clin Lab (2015) 61(3-4):371–8. doi: 10.7754/Clin.Lab.2014.140712
69. Kevans D, Turpin W, Madsen K, Meddings J, Shestopaloff K, Xu W, et al. Determinants of Intestinal Permeability in Healthy First-Degree Relatives of Individuals With Crohn’s Disease. Inflamm Bowel Dis (2015) 21(4):879–87. doi: 10.1097/MIB.0000000000000323
70. Araujo G, Yunta C, Terre M, Mereu A, Ipharraguerre I, Bach A. Intestinal Permeability and Incidence of Diarrhea in Newborn Calves. J Dairy Sci (2015) 98(10):7309–17. doi: 10.3168/jds.2015-9666
71. Odenwald MA, Turner JR. Intestinal Permeability Defects: Is it Time to Treat? Clin Gastroenterol Hepatol (2013) 11(9):1075–83. doi: 10.1016/j.cgh.2013.07.001
72. Hügle B, Speth F, Haas JP. Inflammatory Bowel Disease Following Anti-Interleukin-1-Treatment in Systemic Juvenile Idiopathic Arthritis. Pediatr Rheumatol Online J (2017) 15(1):16. doi: 10.1186/s12969-017-0147-3
73. Ogawa M, Osada H, Hasegawa A, Ohno H, Yanuma N, Sasaki K, et al. Effect of Interleukin-1β on Occludin Mrna Expression in the Duodenal and Colonic Mucosa of Dogs With Inflammatory Bowel Disease. J Vet Intern Med (2018) 32(3):1019–25. doi: 10.1111/jvim.15117
74. Hou Y, Lu X, Zhang Y. IRAK Inhibitor Protects the Intestinal Tract of Necrotizing Enterocolitis by Inhibiting the Toll-Like Receptor (TLR) Inflammatory Signaling Pathway in Rats. Med Sci Monit (2018) 24:3366–73. doi: 10.12659/MSM.910327
75. Shifflett DE, Bottone FG, Young KM, Moeser AJ, Jones SL, Blikslager AT. Neutrophils Augment Recovery of Porcine Ischemia-Injured Ileal Mucosa by an IL-1beta- and COX-2-Dependent Mechanism. Am J Physiol Gastrointest Liver Physiol (2004) 287(1):G50–7. doi: 10.1152/ajpgi.00076.2003
76. Bersudsky M, Luski L, Fishman D, White RM, Ziv-Sokolovskaya N, Dotan S, et al. Non-Redundant Properties of IL-1α and IL-1β During Acute Colon Inflammation in Mice. Gut (2014) 63(4):598–609. doi: 10.1136/gutjnl-2012-303329
77. Tadros T, Traber DL, Heggers JP, Herndon DN. Effects of Interleukin-1alpha Administration on Intestinal Ischemia and Reperfusion Injury, Mucosal Permeability, and Bacterial Translocation in Burn and Sepsis. Ann Surg (2003) 237(1):101–9. doi: 10.1097/00000658-200301000-00014
78. Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, et al. Nuclear Factor Kappab is Activated in Macrophages and Epithelial Cells of Inflamed Intestinal Mucosa. Gastroenterology (1998) 115(2):357–69. doi: 10.1016/S0016-5085(98)70202-1
79. Neurath MF, Pettersson S, Meyer zum Büschenfelde KH, Strober W. Local Administration of Antisense Phosphorothioate Oligonucleotides to the P65 Subunit of NF-Kappa B Abrogates Established Experimental Colitis in Mice. Nat Med (1996) 2(9):998–1004. doi: 10.1038/nm0996-998
80. Delhalle S, Blasius R, Dicato M, Diederich M. A Beginner’s Guide to NF-Kappab Signaling Pathways. Ann N Y Acad Sci (2004) 1030:1–13. doi: 10.1196/annals.1329.002
81. O’Neill LA. Signal Transduction Pathways Activated by the IL-1 Receptor/Toll-Like Receptor Superfamily. Curr Top Microbiol Immunol (2002) 270:47–61.
82. Guo S, Gillingham T, Guo Y, Meng D, Zhu W, Walker WA, et al. Secretions of Bifidobacterium Infantis and Lactobacillus Acidophilus Protect Intestinal Epithelial Barrier Function. J Pediatr Gastroenterol Nutr (2017) 64(3):404–12. doi: 10.1097/MPG.0000000000001310
83. Gentile C, Perrone A, Attanzio A, Tesoriere L, Livrea MA. Sicilian Pistachio (Pistacia Vera L.) Nut Inhibits Expression and Release of Inflammatory Mediators and Reverts the Increase of Paracellular Permeability in IL-1β-Exposed Human Intestinal Epithelial Cells. Eur J Nutr (2015) 54(5):811–21. doi: 10.1007/s00394-014-0760-6
84. Al-Sadi R, Ye D, Dokladny K, Ma TY. Mechanism of IL-1beta-Induced Increase in Intestinal Epithelial Tight Junction Permeability. J Immunol (2008) 180(8):5653–61. doi: 10.4049/jimmunol.180.8.5653
85. Lawrence T. The Nuclear Factor NF-Kappab Pathway in Inflammation. Cold Spring Harb Perspect Biol (2009) 1(6):a001651. doi: 10.1101/cshperspect.a001651
86. Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κb Signaling Pathways. Nat Immunol (2011) 12(8):695–708. doi: 10.1038/ni.2065
87. Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-κb: A Blossoming of Relevance to Human Pathobiology. Cell (2017) 168(1-2):37–57. doi: 10.1016/j.cell.2016.12.012
88. Al-Sadi R, Ye D, Said HM, Ma TY. IL-1beta-Induced Increase in Intestinal Epithelial Tight Junction Permeability is Mediated by MEKK-1 Activation of Canonical NF-Kappab Pathway. Am J Pathol (2010) 177(5):2310–22. doi: 10.2353/ajpath.2010.100371
89. Nalli AD, Kumar DP, Mahavadi S, Al-Shboul O, Alkahtani R, Kuemmerle JF, et al. Hypercontractility of Intestinal Longitudinal Smooth Muscle Induced by Cytokines is Mediated by the Nuclear Factor-Kappab/AMP-Activated Kinase/Myosin Light Chain Kinase Pathway. J Pharmacol Exp Ther (2014) 350(1):89–98. doi: 10.1124/jpet.113.212522
90. Al-Sadi R, Guo S, Dokladny K, Smith MA, Ye D, Kaza A, et al. Mechanism of Interleukin-1beta Induced-Increase in Mouse Intestinal Permeability In Vivo. J Interferon Cytokine Res (2012) 32(10):474–84. doi: 10.1089/jir.2012.0031
91. Rodriguez-Yoldi MJ, Gascon S, Barranquero C, Garcia-Barrios A, Osada J. Involvement of Intracellular Signaling in the IL-1beta Inhibitory Effect on Fructose Intestinal Absorption. J Cell Physiol (2015) 230(4):896–902. doi: 10.1002/jcp.24820
92. Tarantino N, Tinevez JY, Crowell EF, Boisson B, Henriques R, Mhlanga M, et al. TNF and IL-1 Exhibit Distinct Ubiquitin Requirements for Inducing NEMO-IKK Supramolecular Structures. J Cell Biol (2014) 204(2):231–45. doi: 10.1083/jcb.201307172
93. Wu Y, Liu J, Zhang Z, Huang H, Shen J, Zhang S, et al. HSP27 Regulates IL-1 Stimulated IKK Activation Through Interacting With TRAF6 and Affecting its Ubiquitination. Cell Signal (2009) 21(1):143–50. doi: 10.1016/j.cellsig.2008.10.001
94. Funakoshi-Tago M, Tago K, Andoh K, Sonoda Y, Tominaga S, Kasahara T. Functional Role of C-Src in IL-1-Induced NF-Kappa B Activation: C-Src is a Component of the IKK Complex. J Biochem (2005) 137(2):189–97. doi: 10.1093/jb/mvi018
95. Marano CW, Lewis SA, Garulacan LA, Soler AP, Mullin JM. Tumor Necrosis Factor-Alpha Increases Sodium and Chloride Conductance Across the Tight Junction of CACO-2 BBE, a Human Intestinal Epithelial Cell Line. J Membr Biol (1998) 161(3):263–74. doi: 10.1007/s002329900333
96. Wong M, Ziring D, Korin Y, Desai S, Kim S, Lin J, et al. Tnfalpha Blockade in Human Diseases: Mechanisms and Future Directions. Clin Immunol (2008) 126(2):121–36. doi: 10.1016/j.clim.2007.08.013
97. Zhong J, Yu R, Zhou Q, Liu P, Liu Z, Bian Y. Naringenin Prevents TNF-Alpha-Induced Gut-Vascular Barrier Disruption Associated With Inhibiting the NF-Kappab-Mediated MLCK/P-MLC and NLRP3 Pathways. Food Funct (2021) 12(6):2715–25. doi: 10.1039/D1FO00155H
98. Chen SW, Zhu J, Zuo S, Zhang JL, Chen ZY, Chen GW, et al. Protective Effect of Hydrogen Sulfide on TNF-Alpha and IFN-Gamma-Induced Injury of Intestinal Epithelial Barrier Function in Caco-2 Monolayers. Inflammation Res (2015) 64(10):789–97. doi: 10.1007/s00011-015-0862-5
99. Feng Y, Teitelbaum DH. Tumour Necrosis Factor–Induced Loss of Intestinal Barrier Function Requires TNFR1 and TNFR2 Signalling in a Mouse Model of Total Parenteral Nutrition. J Physiol (2013) 591(15):3709–23. doi: 10.1113/jphysiol.2013.253518
100. Cao M, Wang P, Sun C, He W, Wang F. Amelioration of IFN-Gamma and TNF-Alpha-Induced Intestinal Epithelial Barrier Dysfunction by Berberine via Suppression of MLCK-MLC Phosphorylation Signaling Pathway. PloS One (2013) 8(5):e61944. doi: 10.1371/journal.pone.0061944
101. Gadjeva M, Wang Y, Horwitz BH. NF-Kappab P50 and P65 Subunits Control Intestinal Homeostasis. Eur J Immunol (2007) 37(9):2509–17. doi: 10.1002/eji.200737186
102. Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-Gamma and Tumor Necrosis Factor-Alpha Synergize to Induce Intestinal Epithelial Barrier Dysfunction by Up-Regulating Myosin Light Chain Kinase Expression. Am J Pathol (2005) 166(2):409–19. doi: 10.1016/S0002-9440(10)62264-X
103. Ye D, Ma I, Ma TY. Molecular Mechanism of Tumor Necrosis Factor-Alpha Modulation of Intestinal Epithelial Tight Junction Barrier. Am J Physiol Gastrointest Liver Physiol (2006) 290(3):G496–504. doi: 10.1152/ajpgi.00318.2005
104. Al-Sadi R, Guo S, Ye D, Rawat M, Ma TY. TNF-Alpha Modulation of Intestinal Tight Junction Permeability is Mediated by NIK/IKK-Alpha Axis Activation of the Canonical NF-Kappab Pathway. Am J Pathol (2016) 186(5):1151–65. doi: 10.1016/j.ajpath.2015.12.016
105. Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial Myosin Light Chain Kinase Expression and Activity are Upregulated in Inflammatory Bowel Disease. Lab Invest. (2006) 86(2):191–201. doi: 10.1038/labinvest.3700373
106. Rigor RR, Shen Q, Pivetti CD, Wu MH, Yuan SY. Myosin Light Chain Kinase Signaling in Endothelial Barrier Dysfunction. Med Res Rev (2013) 33(5):911–33. doi: 10.1002/med.21270
107. Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of Endothelial Junctional Permeability. Ann N Y Acad Sci (2008) 1123:134–45. doi: 10.1196/annals.1420.016
108. Herring BP, El-Mounayri O, Gallagher PJ, Yin F, Zhou J. Regulation of Myosin Light Chain Kinase and Telokin Expression in Smooth Muscle Tissues. Am J Physiol Cell Physiol (2006) 291(5):C817–27. doi: 10.1152/ajpcell.00198.2006
109. Ma TY, Hoa NT, Tran DD, Bui V, Pedram A, Mills S, et al. Cytochalasin B Modulation of Caco-2 Tight Junction Barrier: Role of Myosin Light Chain Kinase. Am J Physiol Gastrointest Liver Physiol (2000) 279(5):G875–85. doi: 10.1152/ajpgi.2000.279.5.G875
110. Tinsley JH, De Lanerolle P, Wilson E, Ma W, Yuan SY. Myosin Light Chain Kinase Transference Induces Myosin Light Chain Activation and Endothelial Hyperpermeability. Am J Physiol Cell Physiol (2000) 279(4):C1285–9. doi: 10.1152/ajpcell.2000.279.4.C1285
111. Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N. Ethanol Modulation of Intestinal Epithelial Tight Junction Barrier. Am J Physiol (1999) 276(4):G965–74. doi: 10.1152/ajpgi.1999.276.4.G965
112. Graham WV, Magis AT, Bailey KM, Turner JR, Ostrov DA. Crystallization and Preliminary X-Ray Analysis of the Human Long Myosin Light-Chain Kinase 1-Specific Domain Igcam3. Acta Crystallogr Sect F Struct Biol Cryst Commun (2011) 67(Pt 2):221–3. doi: 10.1107/S1744309110050323
113. Chen M, Zhang W, Lu X, Hoggatt AM, Gunst SJ, Kassab GS, et al. Regulation of 130-Kda Smooth Muscle Myosin Light Chain Kinase Expression by an Intronic Carg Element. J Biol Chem (2013) 288(48):34647–57. doi: 10.1074/jbc.M113.510362
114. Clayburgh DR, Rosen S, Witkowski ED, Wang F, Blair S, Dudek S, et al. A Differentiation-Dependent Splice Variant of Myosin Light Chain Kinase, MLCK1, Regulates Epithelial Tight Junction Permeability. J Biol Chem (2004) 279(53):55506–13. doi: 10.1074/jbc.M408822200
115. He WQ, Wang J, Sheng JY, Zha JM, Graham WV, Turner JR. Contributions of Myosin Light Chain Kinase to Regulation of Epithelial Paracellular Permeability and Mucosal Homeostasis. Int J Mol Sci (2020) 21(3):1–17. doi: 10.3390/ijms21030993
116. Turner JR, Angle JM, Black ED, Joyal JL, Sacks DB, Madara JL. PKC-Dependent Regulation of Transepithelial Resistance: Roles of MLC and MLC Kinase. Am J Physiol (1999) 277(3):C554–62. doi: 10.1152/ajpcell.1999.277.3.C554
117. Zolotarevsky Y, Hecht G, Koutsouris A, Gonzalez DE, Quan C, Tom J, et al. A Membrane-Permeant Peptide That Inhibits MLC Kinase Restores Barrier Function in In Vitro Models of Intestinal Disease. Gastroenterology (2002) 123(1):163–72. doi: 10.1053/gast.2002.34235
118. Xu T, Dong Z, Wang X, Qi S, Li X, Cheng R, et al. IL-1beta Induces Increased Tight Junction Permeability in Bovine Mammary Epithelial Cells via the IL-1beta-ERK1/2-MLCK Axis Upon Blood-Milk Barrier Damage. J Cell Biochem (2018) 119(11):9028–41. doi: 10.1002/jcb.27160
119. Lin CY, Zu CH, Yang CC, Tsai PJ, Shyu JF, Chen CP, et al. IL-1beta-Induced Mesenchymal Stem Cell Migration Involves MLCK Activation via PKC Signaling. Cell Transplant (2015) 24(10):2011–28. doi: 10.3727/096368914X685258
120. Beard RS Jr., Haines RJ, Wu KY, Reynolds JJ, Davis SM, Elliott JE, et al. Non-Muscle Mlck is Required for Beta-Catenin- and Foxo1-Dependent Downregulation of Cldn5 in IL-1beta-Mediated Barrier Dysfunction in Brain Endothelial Cells. J Cell Sci (2014) 127(Pt 8):1840–53. doi: 10.1242/jcs.144550
121. Haines RJ, Beard RS, Chen L, Eitnier RA, Wu MH. Interleukin-1β Mediates β-Catenin-Driven Downregulation of Claudin-3 and Barrier Dysfunction in Caco2 Cells. Dig Dis Sci (2016) 61(8):2252–61. doi: 10.1007/s10620-016-4145-y
122. Zhai Z, Wang J, Huang B, Yin S. Low-Fat Yogurt Alleviates the Pro-Inflammatory Cytokine IL-1β-Induced Intestinal Epithelial Barrier Dysfunction. J Dairy Sci (2019) 102(2):976–84. doi: 10.3168/jds.2018-15226
123. Arthur JS, Ley SC. Mitogen-Activated Protein Kinases in Innate Immunity. Nat Rev Immunol (2013) 13(9):679–92. doi: 10.1038/nri3495
124. Peti W, Page R. Molecular Basis of MAP Kinase Regulation. Protein Sci (2013) 22(12):1698–710. doi: 10.1002/pro.2374
125. Sabio G, Davis RJ. TNF and MAP Kinase Signalling Pathways. Semin Immunol (2014) 26(3):237–45. doi: 10.1016/j.smim.2014.02.009
126. Tang N, Zhang YP, Ying W, Yao XX. Interleukin-1beta Upregulates Matrix Metalloproteinase-13 Gene Expression via C-Jun N-Terminal Kinase and P38 MAPK Pathways in Rat Hepatic Stellate Cells. Mol Med Rep (2013) 8(6):1861–5. doi: 10.3892/mmr.2013.1719
127. Yamamoto T, Kojima T, Murata M, Takano K, Go M, Chiba H, et al. IL-1beta Regulates Expression of Cx32, Occludin, and Claudin-2 of Rat Hepatocytes via Distinct Signal Transduction Pathways. Exp Cell Res (2004) 299(2):427–41. doi: 10.1016/j.yexcr.2004.06.011
128. Al-Sadi R, Ye D, Said HM, Ma TY. Cellular and Molecular Mechanism of Interleukin-1β Modulation of Caco-2 Intestinal Epithelial Tight Junction Barrier. J Cell Mol Med (2011) 15(4):970–82. doi: 10.1111/j.1582-4934.2010.01065.x
129. Awane M, Andres PG, Li DJ, Reinecker HC. NF-Kappa B-Inducing Kinase is a Common Mediator of IL-17-, TNF-Alpha-, and IL-1 Beta-Induced Chemokine Promoter Activation in Intestinal Epithelial Cells. J Immunol (1999) 162(9):5337–44.
130. Choi W, Yeruva S, Turner JR. Contributions of Intestinal Epithelial Barriers to Health and Disease. Exp Cell Res (2017) 358(1):71–7. doi: 10.1016/j.yexcr.2017.03.036
131. Clayburgh DR, Shen L, Turner JR. A Porous Defense: The Leaky Epithelial Barrier in Intestinal Disease. Lab Invest (2004) 84(3):282–91. doi: 10.1038/labinvest.3700050
132. Nusrat A, Turner JR, Madara JL. Molecular Physiology and Pathophysiology of Tight Junctions. IV. Regulation of Tight Junctions by Extracellular Stimuli: Nutrients, Cytokines, and Immune Cells. Am J Physiol Gastrointest Liver Physiol (2000) 279(5):G851–7. doi: 10.1152/ajpgi.2000.279.5.G851
133. Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight Junction Pore and Leak Pathways: A Dynamic Duo. Annu Rev Physiol (2011) 73:283–309. doi: 10.1146/annurev-physiol-012110-142150
134. Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1: A High Molecular Weight Polypeptide Associated With the Tight Junction (Zonula Occludens) in a Variety of Epithelia. J Cell Biol (1986) 103(3):755–66. doi: 10.1083/jcb.103.3.755
135. France MM, Turner JR. The Mucosal Barrier at a Glance. J Cell Sci (2017) 130(2):307–14. doi: 10.1242/jcs.193482
136. Shen L, Turner JR. Role of Epithelial Cells in Initiation and Propagation of Intestinal Inflammation. Eliminating the Static: Tight Junction Dynamics Exposed. Am J Physiol Gastrointest Liver Physio (2006) 290(4):G577–82. doi: 10.1152/ajpgi.00439.2005
137. Mohr AM, Mott JL. Overview of MicroRNA Biology. Semin Liver Dis (2015) 35(1):3–11. doi: 10.1055/s-0034-1397344
138. Ye D, Guo S, Al-Sadi R, Ma TY. MicroRNA Regulation of Intestinal Epithelial Tight Junction Permeability. Gastroenterology (2011) 141(4):1323–33. doi: 10.1053/j.gastro.2011.07.005
139. Cho SX, Rudloff I, Lao JC, Pang MA, Goldberg R, Bui CB, et al. Characterization of the Pathoimmunology of Necrotizing Enterocolitis Reveals Novel Therapeutic Opportunities. Nat Commun (2020) 11(1):5794. doi: 10.1038/s41467-020-19400-w
140. Camilleri M, Madsen K, Spiller R, Greenwood-Van Meerveld B, Van Meerveld BG, Verne GN. Intestinal Barrier Function in Health and Gastrointestinal Disease. Neurogastroenterol Motil (2012) 24(6):503–12. doi: 10.1111/j.1365-2982.2012.01921.x
141. Munkholm P, Langholz E, Hollander D, Thornberg K, Orholm M, Katz KD, et al. Intestinal Permeability in Patients With Crohn’s Disease and Ulcerative Colitis and Their First Degree Relatives. Gut (1994) 35(1):68–72. doi: 10.1136/gut.35.1.68
142. Fiorentino M, Sapone A, Senger S, Camhi SS, Kadzielski SM, Buie TM, et al. Blood-Brain Barrier and Intestinal Epithelial Barrier Alterations in Autism Spectrum Disorders. Mol Autism (2016) 7:49. doi: 10.1186/s13229-016-0110-z
143. Li N, Russell WM, Douglas-escobar M, Hauser N, Lopez M, Neu J. Live and Heat-Killed Lactobacillus Rhamnosus GG: Effects on Proinflammatory and Anti-Inflammatory Cytokines/Chemokines in Gastrostomy-Fed Infant Rats. Pediatr Res (2009) 66(2):203–7. doi: 10.1203/PDR.0b013e3181aabd4f
144. Ma X, Yu X, Zhou Q. The IL1beta-HER2-CLDN18/CLDN4 Axis Mediates Lung Barrier Damage in ARDS. Aging (Albany NY) (2020) 12(4):3249–65. doi: 10.18632/aging.102804
145. Weber CR, Nalle SC, Tretiakova M, Rubin DT, Turner JR. Claudin-1 and Claudin-2 Expression is Elevated in Inflammatory Bowel Disease and may Contribute to Early Neoplastic Transformation. Lab Invest (2008) 88(10):1110–20. doi: 10.1038/labinvest.2008.78
146. Xing T, Camacho Salazar R, Chen YH. Animal Models for Studying Epithelial Barriers in Neonatal Necrotizing Enterocolitis, Inflammatory Bowel Disease and Colorectal Cancer. Tissue Barriers (2017) 5(4):e1356901. doi: 10.1080/21688370.2017.1356901
147. Hering NA, Schulzke JD. Therapeutic Options to Modulate Barrier Defects in Inflammatory Bowel Disease. Dig Dis (2009) 27(4):450–4. doi: 10.1159/000233283
148. Ivanov AI, Nusrat A, Parkos CA. The Epithelium in Inflammatory Bowel Disease: Potential Role of Endocytosis of Junctional Proteins in Barrier Disruption. Novartis Found Symp (2004) 263:115–24; discussion 24-32, 211-8. doi: 10.1002/0470090480.ch9
149. Piontek J, Krug SM, Protze J, Krause G, Fromm M. Molecular Architecture and Assembly of the Tight Junction Backbone. Biochim Biophys Acta Biomembr (2020) 1862(7):183279. doi: 10.1016/j.bbamem.2020.183279
150. Garcia-Hernandez V, Quiros M, Nusrat A. Intestinal Epithelial Claudins: Expression and Regulation in Homeostasis and Inflammation. Ann N Y Acad Sci (2017) 1397(1):66–79. doi: 10.1111/nyas.13360
151. Barmeyer C, Fromm M, Schulzke JD. Active and Passive Involvement of Claudins in the Pathophysiology of Intestinal Inflammatory Diseases. Pflugers Arch (2017) 469(1):15–26. doi: 10.1007/s00424-016-1914-6
152. Lu Z, Ding L, Lu Q, Chen YH. Claudins in Intestines: Distribution and Functional Significance in Health and Diseases. Tissue Barriers (2013) 1(3):e24978. doi: 10.4161/tisb.24978
153. Amasheh S, Fromm M, Gunzel D. Claudins of Intestine and Nephron - a Correlation of Molecular Tight Junction Structure and Barrier Function. Acta Physiol (Oxf) (2011) 201(1):133–40. doi: 10.1111/j.1748-1716.2010.02148.x
154. Maria-Ferreira D, Nascimento AM, Cipriani TR, Santana-Filho AP, Watanabe PDS, Sant Ana DMG, et al. Rhamnogalacturonan, a Chemically-Defined Polysaccharide, Improves Intestinal Barrier Function in DSS-Induced Colitis in Mice and Human Caco-2 Cells. Sci Rep (2018) 8(1):12261. doi: 10.1038/s41598-018-30526-2
155. Ahmad R, Kumar B, Chen Z, Chen X, Muller D, Lele SM, et al. Loss of Claudin-3 Expression Induces IL6/Gp130/Stat3 Signaling to Promote Colon Cancer Malignancy by Hyperactivating Wnt/Beta-Catenin Signaling. Oncogene (2017) 36(47):6592–604. doi: 10.1038/onc.2017.259
156. Wang J, Ghosh SS, Ghosh S. Curcumin Improves Intestinal Barrier Function: Modulation of Intracellular Signaling, and Organization of Tight Junctions. Am J Physiol Cell Physiol (2017) 312(4):C438–C45. doi: 10.1152/ajpcell.00235.2016
157. Kim JM. Inflammatory Bowel Diseases and Inflammasome. Korean J Gastroenterol (2011) 58(6):300–10. doi: 10.4166/kjg.2011.58.6.300
158. Zambon CF, Basso D, Navaglia F, Belluco C, Falda A, Fogar P, et al. Pro- and Anti-Inflammatory Cytokines Gene Polymorphisms and Helicobacter Pylori Infection: Interactions Influence Outcome. Cytokine (2005) 29(4):141–52. doi: 10.1016/j.cyto.2004.10.013
Keywords: interleukin-1β (IL-1β), intestinal tight junction (TJ) barrier, myosin light chain kinase (MLCK), microRNA, intestinal inflammation, NF-kappaB (NF-κB)
Citation: Kaminsky LW, Al-Sadi R and Ma TY (2021) IL-1β and the Intestinal Epithelial Tight Junction Barrier. Front. Immunol. 12:767456. doi: 10.3389/fimmu.2021.767456
Received: 30 August 2021; Accepted: 11 October 2021;
Published: 25 October 2021.
Edited by:
Remo Castro Russo, Federal University of Minas Gerais, BrazilReviewed by:
Anthony Blikslager, North Carolina State University, United StatesCopyright © 2021 Kaminsky, Al-Sadi and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Y. Ma, dGhvbWFzbWFAcGVubnN0YXRlaGVhbHRoLnBzdS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.