 Jiaqi Zhang1,2,3†Mengru Xie1,2,3†Xiaofei Huang1,2,3Guangjin Chen1,2,3Ying Yin1,2,3Xiaofeng Lu1,2,3Guangxia Feng1,2,3Ran Yu1,2,3*
Jiaqi Zhang1,2,3†Mengru Xie1,2,3†Xiaofei Huang1,2,3Guangjin Chen1,2,3Ying Yin1,2,3Xiaofeng Lu1,2,3Guangxia Feng1,2,3Ran Yu1,2,3* Lili Chen1,2,3*
Lili Chen1,2,3*- 1Department of Stomatology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 2School of Stomatology, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Hubei Province Key Laboratory of Oral and Maxillofacial Development and Regeneration, Wuhan, China
Atherosclerosis (AS), one of the most common types of cardiovascular disease, has initially been attributed to the accumulation of fats and fibrous materials. However, more and more researchers regarded it as a chronic inflammatory disease nowadays. Infective disease, such as periodontitis, is related to the risk of atherosclerosis. Porphyromonas gingivalis (P. gingivalis), one of the most common bacteria in stomatology, is usually discovered in atherosclerotic plaque in patients. Furthermore, it was reported that P. gingivalis can promote the progression of atherosclerosis. Elucidating the underlying mechanisms of P. gingivalis in atherosclerosis attracted attention, which is thought to be crucial to the therapy of atherosclerosis. Nevertheless, the pathogenesis of atherosclerosis is much complicated, and many kinds of cells participate in it. By summarizing existing studies, we find that P. gingivalis can influence the function of many cells in atherosclerosis. It can induce the dysfunction of endothelium, promote the formation of foam cells as well as the proliferation and calcification of vascular smooth muscle cells, and lead to the imbalance of regulatory T cells (Tregs) and T helper (Th) cells, ultimately promoting the occurrence and development of atherosclerosis. This article summarizes the specific mechanism of atherosclerosis caused by P. gingivalis. It sorts out the interaction between P. gingivalis and AS-related cells, which provides a new perspective for us to prevent or slow down the occurrence and development of AS by inhibiting periodontal pathogens.
Introduction
Cardiovascular disease (CVD) is the most common cause of death worldwide, which leads to about 16.7 million people losing their lives each year (1). Atherosclerosis (AS), a chronic disease that often occurs in large- and medium-sized arteries, is regarded as the pathogenetic basis of most CVDs (2). Although traditional risk factors for AS, such as hyperlipidemia, hypertension, and smoking, have been effectively reduced, the incidence of atherosclerotic diseases remains high (3). In the past few decades, new evidence that AS is a chronic inflammatory disease emerged (4). A variety of pathogens, such as Chlamydia pneumoniae (C. pneumoniae) (5), P. gingivalis (6), and Helicobacter pylori (7), have been detected in human AS plaque lesions and promote the progression of AS, which suggests that pathogen infection may participate in the formation of AS plaques (8).
Periodontitis, which affects 11.2% of the population worldwide, is the sixth most common disease and a highly and prevalently chronic non-communicable disease (9). Many epidemiological and clinical studies have shown that periodontal disease is related to carotid AS (10, 11). People suffering from periodontitis have a higher risk of AS/CVD, and its risk ratio ranges from 1.074 to 1.213, 95% CI (12–15). However, few studies suggested that the link between these two diseases is not very clear (16). Maybe the clinical association between periodontal disease and AS was unsure, but numerous animal experiments have confirmed the promotion role of periodontal pathogens in the progress of AS (17, 18). Periodontal pathogens, such as P. gingivalis (19), Aggregatibacter actinomycetemcomitans (20), and Tannerella forsythia (21), have been detected in human AS plaque lesions. Among the periodontal bacteria detected, the detection rate of P. gingivalis is particularly high (22–24), and studies about the promotion effect of P. gingivalis on AS are also the most common. It seems that out of oral or periodontal pathogens, P. gingivalis has the advantage in AS pathogenicity.
P. gingivalis is the main component of the subgingival plaque in patients with chronic periodontitis. It is not only involved in inflammation and tissue destruction during periodontal disease (25) but also related to the inflammatory pathology of distal body organs, including AS and Alzheimer’s disease (AD) (26, 27). It can enter the blood system through ulcers in the epithelium and lymphatic vessels after treatment intervention (subgingival scaling, surgical periodontal therapy) or daily activities (brushing, chewing), and then survive and colonize in other organs (28). P. gingivalis (6) and its contents, such as fimbriae (29) and DNA (30), have been detected in human atherosclerotic plaques. In recent years, a lot of studies proved that P. gingivalis could accelerate atherosclerosis (18, 25, 31–35); the underlying mechanisms have also been discussed. Most studies and reviews focus on endothelial cells (ECs), but there are many other kinds of cells involved in the development of AS, like vascular smooth muscle cells (VSMCs), macrophages, and T cells. P. gingivalis can also affect the functions of these cells. In order to give a more systematical and comprehensive understanding of the promoting role of P. gingivalis on AS, here we summarize the effects and internal mechanisms of P. gingivalis on all types of cells related to AS.
Characteristics of P. gingivalis
P. gingivalis is an obligate asaccharolytic gram-negative bacteria. It is the most dominant bacteria in periodontitis and has been proven as the main pathogenic bacteria in patients with chronic periodontitis (36, 37). Researchers have clarified that P. gingivalis can promote the development and aggravation of systemic diseases, such as cardiovascular diseases, largely because of its ability to modulate the entire ecosystem by changing the immune response of the host to survive and persist in host tissues (38), which is related to interacting with various host receptors and changing the inflammation and complement system signal transduction pathways as well as cell cycle and apoptosis (39). Gingipains (40) and outer membrane vesicles (OMVs) (41) secreted by P. gingivalis, with its lipopolysaccharides (LPS) (42), proteins (43), and fimbriae (44), make P. gingivalis highly pathogenic and thus persistent in host tissues and promote the emergence of dysbiosis.
LPS is an important part of the outer layer of P. gingivalis and has a strong pathogenic effect (37). It can induce toll-like receptor (TLR)-specific immune upregulation, in which TLR4 and TLR2 are the main receptors (33, 45), so as to trigger inflammation and immune responses between the host through TLRs. P. gingivalis fimbriae, comprised of FimA and Mfa1 subunits, is a crucial factor in the interaction between bacteria and host tissues, promoting the adhesion and invasion of bacteria to target sites (46). It also can be recognized by TLRs on ECs (22), macrophages (47), and immune cells (48), thereby activating the cells to produce cytokines and adhesion molecules. Experiments showed that infection with the fimbriae-deficient mutant DPG3 of P. gingivalis had a minimal effect on pro-AS (49, 50). The heat shock protein 60 (HSP60) of P. gingivalis is remarkably immunogenic (51), and existing reports indicate that the HSP60 IgG antibody titers in patients with AS and periodontitis are elevated (52). As the main secretory component of P. gingivalis, gingipains consist of arginine-gingipain (Rgp) and lysine-gingipain with hemagglutinin (Hag)-adhesin domain, with 85% of extracellular proteolytic ability coming from it (53), providing P. gingivalis the ability of tissue destruction, and can modulate the expression of cytokines and immunoglobulins and thus affect the immune responses of the host cells (54). With research going on, the OMVs with double-layer, spherical, membrane-like structures secreted by P. gingivalis have been proven, with a size of about 50–250 nm (55), to contain LPS, outer membrane proteins, phospholipids, and DNA inside (56). The OMVs make a large number of pathogenic factors highly concentrated and avoid the degradation and destruction of proteolytic enzymes, thus greatly improving the toxicity from P. gingivalis (41) (Figure 1).
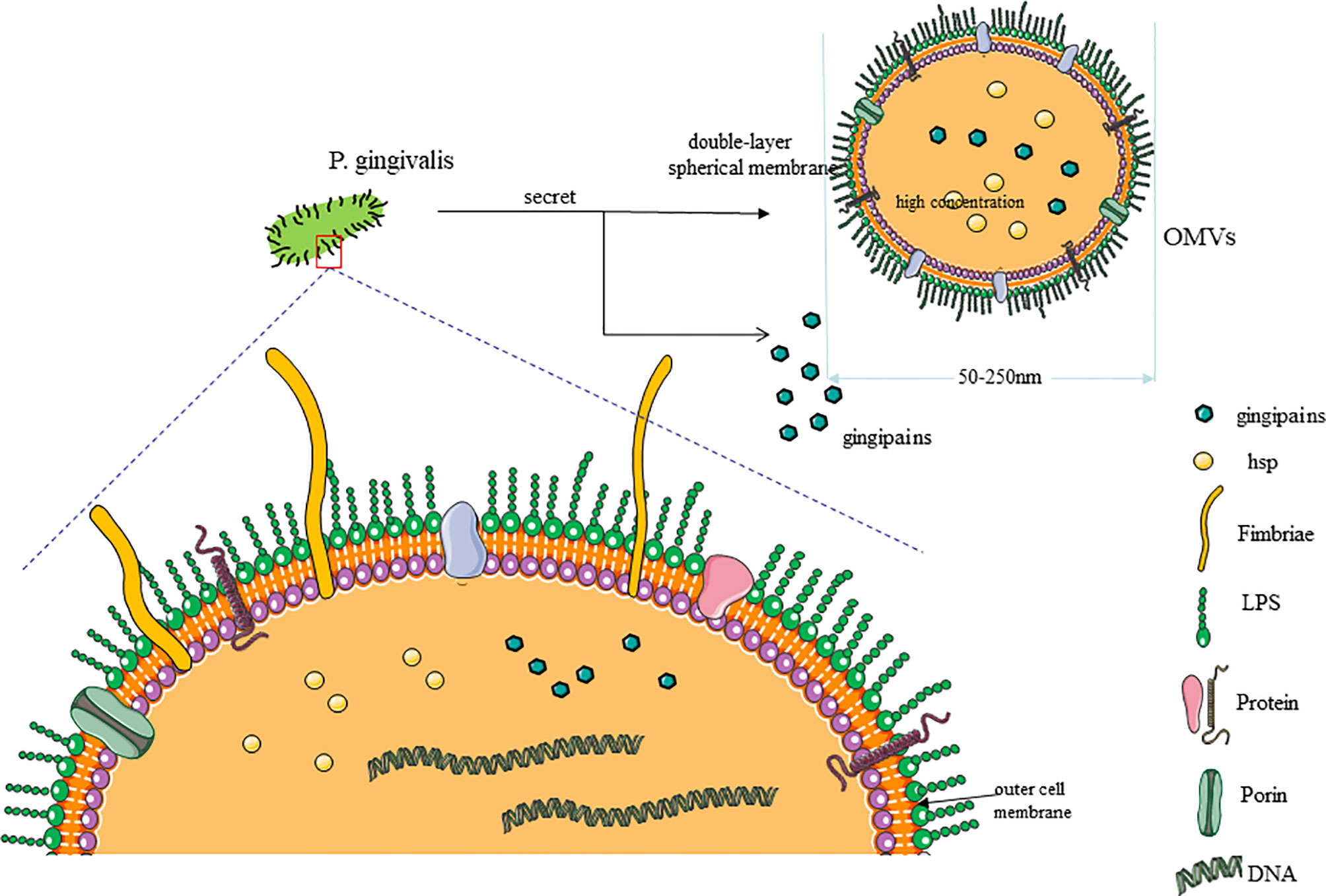
Figure 1 Characteristic of P. gingivalis. (1) As the most common periodontal pathogen, P. gingivalis is composed of cell membrane and genetic material. The outer layer of the cell membrane has a large number of fimbriae, proteins, and channels. (2) The pathogenicity of P. gingivalis mainly comes from its own structural components (lipopolysaccharide, fimbriae, and heat shock proteins) and secretory components (gingipains and outer membrane vesicles, OMVs). (3) OMVs have a double-layer spherical membrane and contain a lot of pathogenic factors with high concentration.
There are some perspectives from new studies that the vasculature can be invaded by P. gingivalis via an ulcerative epithelium (57) and lymphatic vessels (58); then, P. gingivalis could be internalized in gingival epithelial cells and KB cell lines with ECs through the “folding” mechanism which caused severe folds of the host cell membrane at the invasion site and was internalized in the form of spacious vacuoles (59). In addition, studies have shown that P. gingivalis can transmit among different types of cells in vascular tissues (60). All the properties described above support that P. gingivalis invades distant tissues and colonize in parts other than the oral cavity, leading to a more serious outcome of the systemic disease (Figure 1).
Pathogenesis of Atherosclerosis
AS is a continuous course of decades, along with the accumulation of fatty material and plaque formation in the innermost lining of the artery, causing acute coronary syndromes, myocardial infarction, or stroke (16). The pathological process of atherosclerosis is related to the physiological activities and transformation of various cells, including ECs, VSMCs, macrophages, T cells and, dendritic cells (DCs). At the onset, in some atherosclerotic lesions, the vascular endothelium will be abnormally stimulated. As the disease progresses, there will be shed areas in the endothelium, and platelets stick to exposed areas (43, 44). Subsequently, circulating monocytes are recruited from the blood to the subintima; they internalize and modify lipoproteins and finally differentiate into foam cells (49). VSMCs proliferate, migrate, and produce a sizeable extracellular matrix (ECM), which is the main component of the fibrous cap of AS plaques. In addition, VSMCs can also internalize lipids and differentiate into foam cells (50). Last but not least, the immune response caused by T cells and DCs also plays an indispensable role in the pathological development of AS (Figure 2).
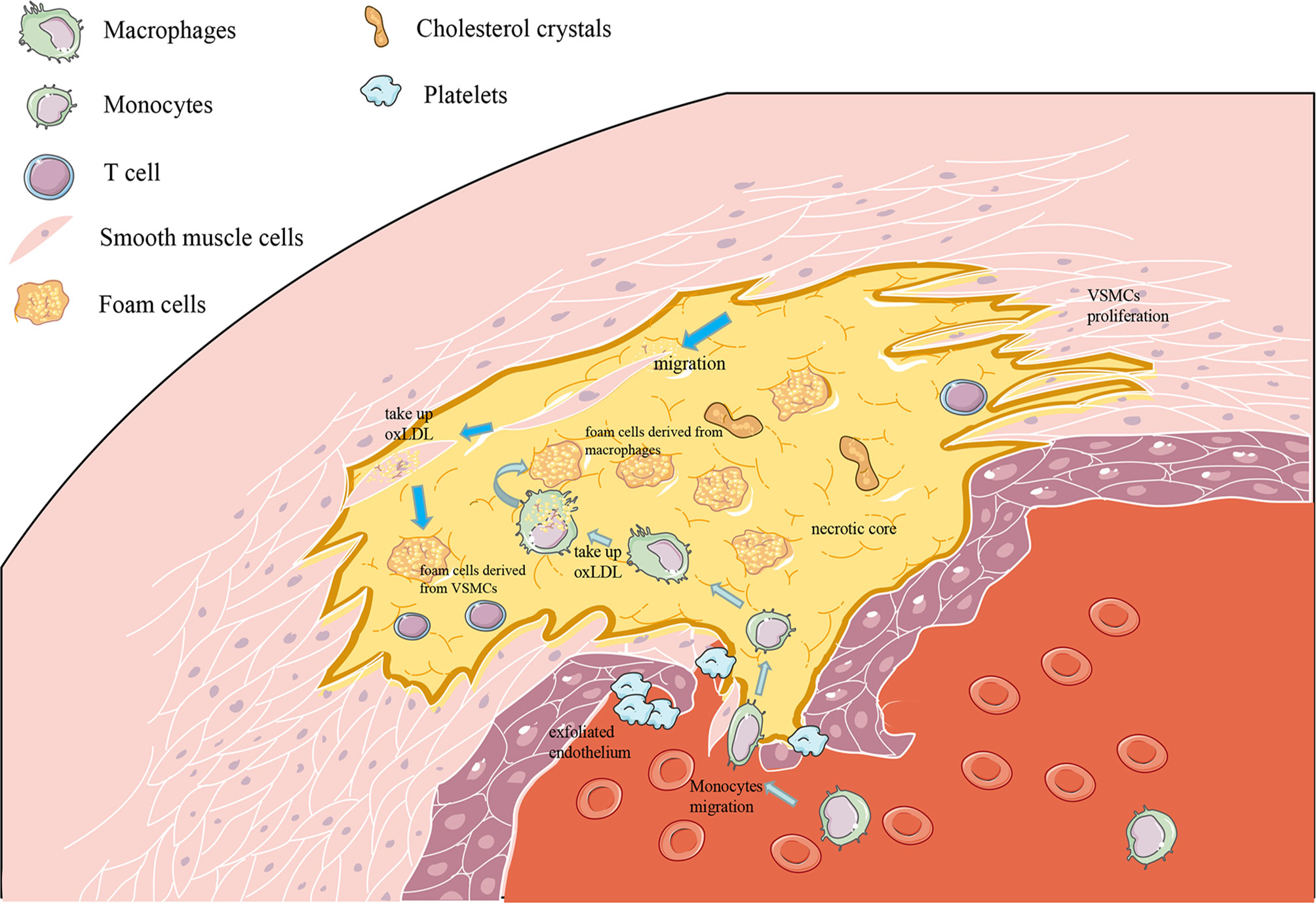
Figure 2 Pathogenesis of atherosclerosis (AS). (1) As the picture shows, AS occurs in the intima, where endothelial cell damage and monocyte migration and adhesion occur. (2) After the monocytes enter the inner membrane, they differentiate into macrophages and increase the uptake of oxLDL to become foam cells. (3) Outside the intima, the media contains vascular smooth muscle cells, which proliferate and migrate to the intima and then differentiate into vascular smooth muscle cell-derived foam cells.
ECs in AS
The endothelium, as the outermost layer between blood and arterial intima, is the initial area of atherosclerotic lesions (61). It is a key factor in regulating vascular homeostasis because of the barrier function of ECs and the ability to regulate the phenotypes of the vascular wall (62). In the prone areas of atherosclerotic lesions, the vascular endothelium firstly leaks, activates, and malfunctions after being stimulated by dyslipidemia, hypertension, or pro-inflammatory mediators, which is also called endothelial dysfunction (63, 64). When the endothelium leaks, the permeability of the endothelium is destroyed, and more circulating low-density lipoprotein (LDL) enters (61). Meanwhile, oxidative stress occurs in the endothelium and produces a lot of superoxide. LDL accumulates in the vascular intima and is oxidized by this superoxide, and then being oxidized low-density lipoprotein (oxLDL), it can induce and bind to cell surface adhesion molecules to activate ECs or be recognized by T cells and drive an autoimmune response (65, 66). As the disease progresses, exfoliated areas appear in the endothelium, and platelets may be adhering to this exposed subendothelial tissue. Before the morphological changes of AS occurred, the endothelial function changed (67). It is a complex pathophysiological event, including endothelial activation, impaired vascular tone, and other endothelial phenotypic changes (61). The pro-inflammatory and procoagulant state of ECs is called endothelial activation (68). In this state, ECs express many chemokines, cytokines, and adhesion molecules, which trigger leukocytes to homing, adhering, and migrating to target tissues. The activated ECs first selectively recruit circulating monocytes from the blood to under the inner membrane, where the monocytes differentiate into macrophages, modify lipoproteins, internalize a large number of lipids, and finally differentiate into foam cells (this is a sign of early fatty streak disease) (69). ECs can also produce perlecan and heparan sulfate proteoglycans under mechanical forces, which are intimately involved in the endothelial inhibition of VSMC proliferation (70). The outer edge of the plaque is rich in inflammatory cells, which further regulates the pro-inflammatory phenotype of ECs and ultimately leads to the instability of the plaque structure (71). ECs can acquire myofibroblast-like properties through endothelial cell–mesenchymal transition (EndMT), which is involved in the occurrence of AS (72).
Vascular Smooth Muscle Cells in AS
VSMCs are the main cell types that exist in various stages of atherosclerotic plaques and can differentiate into various cell phenotypes, including macrophages and foam cells (73). The response of VSMCs to arterial injury and lipid infiltration is the main pathological process of atherosclerotic plaque development (74). VSMCs are the primary source of elastin and interstitial collagen in the inner membrane and a vital part of the ECM, which allows the artery to be compliant and elastically retractable (75, 76). In the pre-AS stage, the proliferation and migration of VSMCs contribute to its migration to the inner membrane from mid-arterial (77). VSMCs secrete a large amount of ECM, which promotes diffuse intimal thickening of the vessel wall (78, 79). In the early stage of AS, VSMCs proliferate, absorb oxLDL, and form foam cells, which promotes pathological intimal thickening (PIT). The arterial intima forms a deep pool of extracellular lipids, and a large number of VSMCs and ECMs accumulate. Microcalcifications (0.5–15-μm spots) are often produced in the lipid pool of PIT, usually near the boundary of the medium, which may be the result of VSMC apoptosis (80–83). At the same time, VSMCs are an essential source of macrophages in AS, and macrophages derived from VSMCs participate in the process from PIT to fibrotic plaques. Finally, the fibrous cap of the AS plaque is formed (84). In the advanced stage of AS, after the death or apoptosis of VSMCs, the accumulated lipids are released outside the cell, forming a necrotic core in the plaque and, at the same time, acting as an antigen to activate T cells to secrete inflammatory mediators, resulting in unstable plaque formation (85).
Macrophages in AS
Macrophages are the main immune cells in atherosclerotic lesions (86). Macrophages are critical and a requisite in every stage of AS (87), from its initiation and expansion to the rupture caused by necrosis and clinical manifestations and to the regression of lesions. The blood monocytes are primarily derived from focal macrophages (88), and circulating monocytes enter the arterial hemodynamic stress site by adhering to the ECs of the susceptible artery lumen (89). The different phenotypes of macrophages enable them to perform different functions (90). Circulating monocytes in the blood bind to adhesion molecules, monocyte chemoattractant protein-1 (MCP-1), and intercellular adhesion molecule-1 (ICAM-1) expressed in activated ECs (91). Then, they enter the plaque through three activities: capture, rolling, and migration. Each step is regulated by multiple molecular factors, sometimes overlapping (89). Once monocytes enter the inner membrane, they can differentiate and mature into macrophages and acquire characteristics associated with repairing or less pro-inflammatory monocyte/macrophage populations (92). The formation of foam cells by macrophages is an important process of pathological changes in AS, and it is also one of the main sources of foam cells in the lipid pool (87). Macrophages increase the uptake of oxLDL while reducing cholesterol efflux, which, in turn, leads to the deposition of intracellular esterified cholesterol and the production of foam cells derived from macrophages. Inflammatory macrophages secrete cytokines and proteases, increasing the expansion of diseased cells, causing changes in plaque morphology, and leading to plaque rupture and acute intraluminal thrombosis. In contrast, dissociated macrophages perform functions related to stabilizing plaques, including removing dead cells (exocytosis) to stabilize plaques and secreting collagen to form protective scars on the lesions (93).
T Cells in AS
The latest research shows that AS is a chronic inflammatory disease (86). Tregs and effector T cells mainly control the adaptive immune process of AS (92). For plaques in AS, various T cell lineages are crucial for their initiation, progression, and stability (94). T helper cells 1 (TH1) can accelerate atherosclerosis, and Tregs can inhibit the progression of atherosclerosis (66). It should be noted that Tregs can become pro-atherogenic cells. The complexity of TH1 and Tregs functions is due to activating or inhibiting the roles of other T cells, promoting the production of high-affinity resistance and cytotoxicity (95). The roles of other T cell subgroups like CD8+ T cells and γδ T cells and TH cell subgroups such as TH2 and TH9 are not much understood (66).
Dendritic Cells in AS
When monocytes are recruited to enter the subendothelial layer in the early stage of AS lesions, another type of immune cell, DCs, also takes the opportunity to invade the subendothelial intima preferentially, forming a structure like the cutaneous Langerhans cell network (96). The role of DCs in the pre-AS stage is a double-edged sword. It can have a protective effect by reducing effector T cell proliferation and inhibiting IFN-γ production (97). On the other side, DCs under the inner membrane has a pro-atherosclerotic effect. It can ingest cholesterol to promote lipid accumulation and foam-like lesions (98). Treg homeostasis can be regulated by DC-derived chemokines, which suggests that DCs control T cell responses by multifarious mechanisms to achieve anti- or pro-inflammatory effects in AS (99). Last but not least, DCs can infiltrate arterial walls, which may destabilize atherosclerotic plaques and contribute to inflammatory development in AS (100) (Figure 2).
The Influence of P. gingivalis on ECs
In the past few years, research has proposed that P. gingivalis has the capability to act and invade ECs, induce endothelial dysfunction, destroy endothelial integrity, and then promote the formation and development of atherosclerotic plaques (101) (Figure 3).
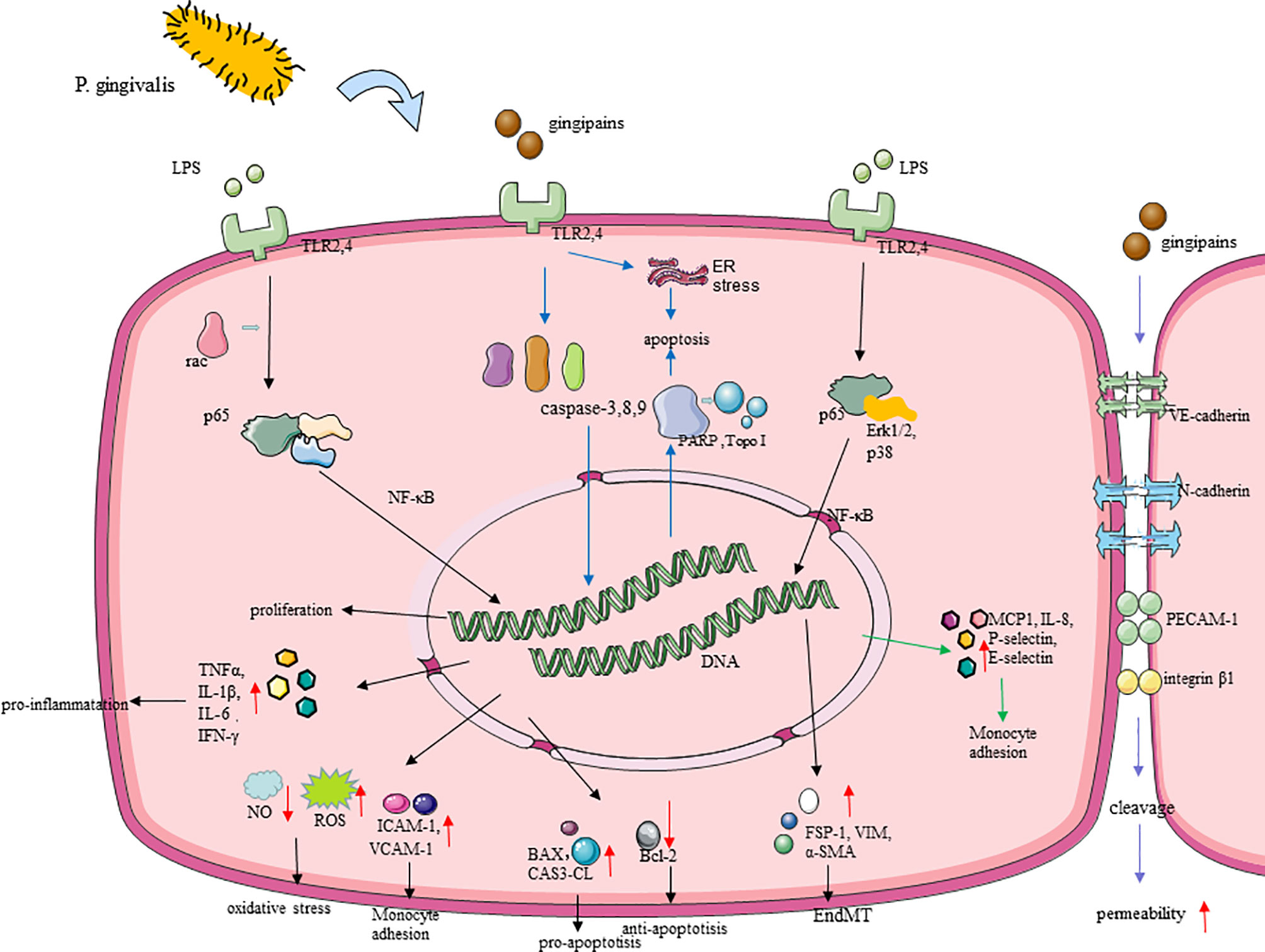
Figure 3 Molecular cascades activated by P. gingivalis in endothelial cells (ECs). (1) TLR is believed to mediate the recognition of P. gingivalis. P. gingivalis promotes EC oxidative stress through the TLRs–NF-κB signal axis and NLRP3 inflammasomes. P. gingivalis leads to nitrifying stress and impaired endothelial function, with upregulating iNOS, downregulating eNOS, and regulating the release of NO in EC (86), which is associated with the change of the GSK-3β/BH4/eNOS/Nrf2 pathway. (2) P. gingivalis gingipains induced endothelial cell (EC) apoptosis by activating caspase-3,8,9, and its gingipains can also induce EC apoptosis mainly through inducing the cleavage of PARP and Topo I. (3) P. gingivalis lipopolysaccharide promoted EndMT through the regulation of p38, Erk1/2, and p65. (4) P. gingivalis enhanced monocyte migration with an increased expression of MCP-1, ICAM-1, IL-8, P-selectin, and E-selectin in ECs. (5) Gingipain destroyed the endothelial cell–cell junction through inducing the cleavage of VE-cadherin, N-cadherin, PECAM-1, and integrin β1.
P. gingivalis Activates Endothelial Oxidative Stress and Promotes Inflammation Response
Oxidative stress is fundamental to AS. Endothelial oxidative stress promotes the adhesion of monocytes and the release of pro-inflammatory cytokines, leading to endothelial dysfunction, which is a precursor to atherosclerotic lesions (61). Recent studies have proved that P. gingivalis induces severe endothelial oxidative stress (102). P. gingivalis can significantly increase the output of total reactive oxygen species (ROS) and superoxide free radicals in vitro, destroying endothelial function. This process is mainly promoted by the TLRs–NF-κB signal axis (22). TLRs mediate the recognition of P. gingivalis LPS and then activate the downstream signaling pathway NF-κB and its active subunit p65, thereby triggering subsequent oxidative stress (22), and peroxisome proliferator-activated receptor may be also involved in modulating oxidative stress during this process (103). Moreover, P. gingivalis activated nucleotide-binding domain leucine-rich repeat (NLR) and promote the production of pyrin domain-containing receptor 3 (NLRP3) inflammasomes in ECs, which depends on ROS and LPS. Then, interleukin-1β (IL-1β) and IL-18 begin to be secreted, thereby promoting further inflammatory processes and oxidative stress in the endothelium (104). It is worth noting that, in the process of P. gingivalis promoting oxidative stress, there is also the influence of circadian clock disruption (22), which can provide new insights on the treatment of P gingivalis-accelerated atherosclerosis. By the way, nitric oxide plays an important role in maintaining homeostasis and anti-oxidative stress through inhibiting the production of ROS (105), and the action of P. gingivalis eventually leads to nitrifying stress and impaired endothelial function, which is achieved by upregulating inducible nitric oxide synthase (iNOS), downregulating endothelial nitric oxide synthase (eNOS), and regulating the release of NO in EC (106), with the change of glycogen synthase kinase-3 (GSK-3β)/tetrahydrobiopterin (BH4)/eNOS/nuclear factor erythroid-derived 2-like 2 (Nrf2) pathways (107). Animal experiments have shown that P gingivalis infection leads to a significant decrease in the bioavailability of BH4, which may be due to the inhibition of the expression of dihydrofolatereductase (DHFR) that predominates the conversion of BH2 to BH4 and the rate-limiting enzyme GCH-1 (GTP cyclohydrolase 1) responsible for the biosynthesis of BH4 (108). NrF2 can protect cells from oxidation by activating antioxidant enzymes, including GSH synthase (GCSc, GCSm) and heme oxygenase-1, which is essential for cell protection. In the vascular tissues of mice infected with P gingivalis, the level of NrF2 was significantly reduced (107). Interestingly, after the intervention of HAECs cells with P. gingivalis, the expression of DHFR was significantly inhibited, but the expression of DHFR did not change significantly in vivo. This may be due to the latter being a variety of periodontal pathogen infections after intervention.
After oxidative stress, P. gingivalis triggers the inflammatory response in vasculature. IL-1β, IL-6, TNFα, and interferon-gamma (IFN-γ), as pro-inflammatory factors, were increased by P. gingivalis in ECs (109, 110). It is also reported that P. gingivalis LPS-, fimbriae-, OMV-, and gingipain-stimulated ECs expressed high levels of MCP-1, ICAM-1, IL-8, P-selectin, and E-selectin, as well as their receptors C-C chemokine receptor type 2 and integrin αMβ2, of which all of them enhance monocyte migration and adhesion (50), thereby initiating and promoting inflammation and promoting the development of AS (111, 112). Multiple signaling pathways, including p38, c-Jun N-kinase, NF-κB, and activator protein 1 (AP-1), are involved in this process (113–116). What is more, the inner-out signal transduction of P. gingivalis fimbriae is mediated by Ras-related C3 botulinum toxin substrate 1 (Rac1) and phosphatidylinositol 3-kinase (PI3K) (117). In addition, with P. gingivalis-LPS stimulation in ECs, it secreted anti- chemotaxis and anti-adhesion proteins like Growth arrest-specific 6 (Gas6) and pro-adhesion proteins like ICAM-1 and macrophage migration inhibitory factor (MIF), and these signs of progress can be regulated by LncRNA GAS6-AS2 (118). MIF binds with the MHC class II invariant chain, called Ii/CXC motif chemokine receptor type 4; a matchable receptor complex, it facilitated monocyte adhesion, too (118).
P. gingivalis Destroys the Endothelial Barrier
The permeability of the endothelial barrier is also a part of the inflammatory responses in the development of AS (119), which has been proven to be promoted by P. gingivalis (120). P. gingivalis and its gingipains, LPS, and OMVs may contribute to endothelial barrier destruction at the endothelial cell–cell junction by inducing the decomposition of adhesion molecules like VE-cadherin and N-cadherin. During this process, platelet endothelial cell adhesion molecule-1 (PECAM-1) and integrin beta1 were also cleaved and destroyed by P. gingivalis in ECs (120–123), thus allowing leukocyte transmigration in this system. P. gingivalis LPS can promote the internalization of VE-cadherin in ECs, which play an essential role in regulating EC permeability (123). Evidence have indicated that IL-8 directly increased endothelial permeability (124), and it can be upregulated by LPS of P. gingivalis with the activation of the NF-κB pathway (123). P. gingivalis also promoted vascular coagulation and inflammation which were mainly related to the degradation and inactivation of glycoprotein thrombomodulin on the surface of ECs (120, 125). The permeability of the endothelium increases, allowing bacteria to pass through the endothelium through loose connections. This may explain why P. gingivalis invading ECs is accompanied by a mixed infection of other periodontal pathogens.
P. gingivalis was able to induce ECs apoptosis and EndMT as well as inhibit its proliferation, which decreases the quantity of ECs, leading to the damage of the vascular endothelial barrier. At this time, circulating leukocytes and LDL accumulate under a damaged endothelium, leading to the development of AS (126). After P. gingivalis infection, pro-apoptotic proteins Bcl−2−associated X protein and CAS3-CL (127) expressed by ECs increased, while the anti-apoptotic protein Bcl−2 decreased significantly (128). P. gingivalis gingipains can also induce EC apoptosis, mainly through inducing the cleavage of topoisomerase I (Topo I) and Poly (ADP-ribose) polymerase (PARP), which may regulate the process of cell death to a certain extent (122). The death of apoptotic ECs usually occurs through the stimulation of activated caspase (129). P. gingivalis induces EC apoptosis by activating the caspase-8 death receptor and the caspase-9 mitochondrial-dependent apoptosis pathway as well as activating the DNA fragmentation induced by caspase-3 (130). P. gingivalis can also induce ER stress with the expression of several growth arrest- and DNA damage-inducible gene 153, glucose-regulated protein 78, and caspase-12, thereby promoting EC apoptosis (130). In addition, several pro-atherosclerotic factors, such as modified lipoproteins and tumor necrosis factor-alpha (TNF-α), affect the P. gingivalis-induced death of ECs, as well as other cells, and promote the compound of necrotic cores (107, 131). NLRP3 inflammasome-mediated pyroptosis has been identified as a potential cause of EC death (132), and the production of ROS in ECs with P. gingivalis infection may activate the NLRP3 inflammasome. The induction effect of P. gingivalis and its LPS on EndMT has been noted in recent studies (111). After the intervention of P. gingivalis, the typical paving stone-like ECs become polygonal fibroblast-like cells with enhanced migratory phenotype and an increase happening in long- and spindle-shaped cells, which were suppressed after the use of TLRs–NF-κB pathway inhibitors (127). The expression of EndMT-related proteins have been also changed to cluster of differentiation 31, as the endothelial-specific markers that were downregulated with VE-cadherin were reduced, and α-smooth muscle actin (α-SMA) related with mesenchymal transition was upregulated, which may be mediated by p38, extracellular signal-regulated kinase 1/2 (Erk1/2), and p65 (111). The proliferation of ECs is indispensable in repairing the shedding area of ECs and maintaining the integrity of the endothelium, but it can be significantly inhibited by P. gingivalis (127, 130). P. gingivalis and its OMVs significantly inhibit the proliferation and growth of ECs (133) and suppressed capillary tube formation by ECs, in which NF-κB signaling played a critical role.
P. gingivalis Survives in Endothelium Leading to Constant Stimulation
In the AS initial stage, P. gingivalis can introduce ECs to internalize it and begin autophagy, which was utilized in transporting bacteria or/and toxins. In in vitro experiments, P. gingivalis invaded ECs through ICAM-1-mediated endocytosis (134). After that, EC autophagy induced by P. gingivalis provides a replicative niche where bacteria survive and replicate while suppressing apoptosis (135). The conclusion that the increased endoplasmic reticulum-associated protein Beclin-1 and microtubule-associated protein light chain 3-II can draw is that the endoplasmic reticulum stress induced by P. gingivalis enhances autophagy (130, 136). P. gingivalis interacts with ECs through a variety of adhesin, including FimA (137) and hemagglutinin B (HagB) (138), and is subsequently transformed into phagosomes through the internalization of lipid rafts (139). After invading ECs, P. gingivalis is swallowed by phagosomes to form early autophagosomes. Thereafter, delaying autophagosome–lysosome fusion or redirecting autophagosomes prevent the formation of autolysates to avoid being destroyed (140). In general, P. gingivalis directly forms late autophagosomes from early autophagosomes to survive and persist in ECs, but the mechanism remains unclear. Interestingly, scholars have explained that p38 mitogen-activated protein (MAP) kinase in monocytes can be activated by P. gingivalis, but ECs do not obey this law, which was shown by the fact that P. gingivalis and its LPS have no activation effect on p38 or ERK MAP kinase (141). On the contrary, the effect of MAP kinase in ECs is interfered by P. gingivalis LPS in the progress of modulating host defenses, which may also be helpful for P. gingivalis survival and replication in ECs and lead to the development of AS.
In general, after P. gingivalis reaches the endothelium, it is internalized by ECs and induces autophagy to preserve its virulence (135). Hereafter, P. gingivalis activates oxidative stress of ECs, which release a large amount of ROS and inflammatory factors, amplifying the inflammatory response through TLRs–NF-κB and NLRP3 pathways (22, 102). Apart from this, P. gingivalis can also increase the permeability of the endothelium by destroying the connections between ECs directly (120). Last but not least, P. gingivalis destroys the completeness of the endothelium through the promotion of apoptosis and the inhibition of proliferation in ECs (130). P. gingivalis causes endothelial dysfunction and damage in many ways, which all promote the occurrence and development of AS. This also helps us understand that microbial infection plays a role in the pathological development of AS so as to find more effective treatments (Table 1 and Figure 3).
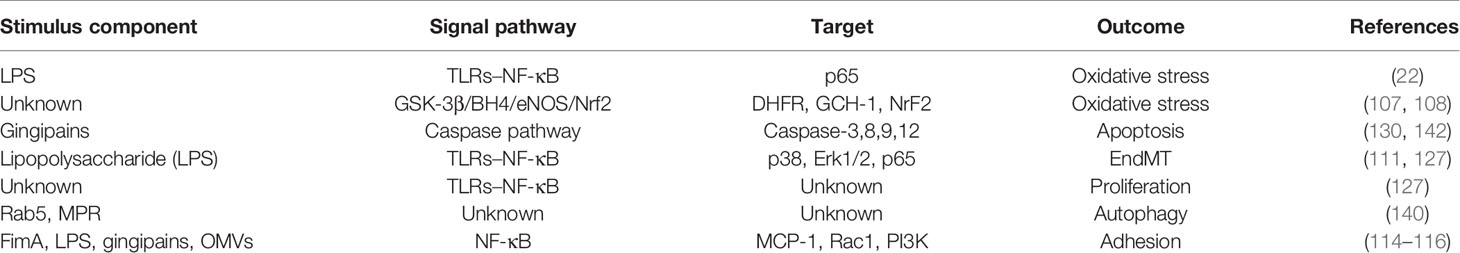
Table 1 The effect of P. gingivalis on endothelial cells.
The Influence of P. gingivalis on Vascular Smooth Muscle Cells
P. gingivalis Promotes the Proliferation and Migration of VSMCs
VSMC proliferation is suggested to contribute to diffuse intimal thickening in AS (143). VSMCs infected with P. gingivalis have shown an increasing trend of cell growth and a significant transition from the contractile phenotype to proliferative phenotype. P. gingivalis and its gingipains upregulate osteopontin (OPN), SMemb, and S100A9 expression, which were contrary to α-SMA and have an important role in cellular proliferation (144, 145). Gingipains may trigger the proliferation of VSMCs by cleaving plasma proteins at the lysine and arginine residues (145). This process may be mainly regulated by the transforming growth factor-beta (TGF-beta)/Notch pathway (146). P. gingivalis mediates the upregulation of connective tissue growth factor [small body size (a Caenorhabditis elegans) mothers against decapentaplegic (a Drosophila protein family)-3 (SMAD3)], which are signaling molecules of the TGF family (147). Hairy/enhancer−of−split related with YRPW motif 1 (HEY1) and Notch1, as two key genes of the Notch pathway (148), were upregulated in VSMCs infected with P. gingivalis. Moreover, a multi-center cohort study in Japan showed that the intima-media thickness was significantly decreased in patients after control of periodontal infection by periodontal treatment (149). This implies that inhibiting P. gingivalis infection can decrease the thickness of plaques to reduce the risk of rupture, which is related to inhibition of the proliferation of VSMCs.
The migration of VSMCs from the middle layer to the inner layer of the blood vessel is a key event of the progression of AS (150). P. gingivalis gingipains enhanced the migration ability of VSMCs by upregulating angiopoietins 2 (Angpt2) and ETS proto-oncogene 1 (ETS1) while inhibiting Angpt1. ETS1 is the transcription factor of Angpt2, which is critical for P. gingivalis to induce Angpt2 (151). Angiopoietins (Angpt1, Angpt2, etc.) regulate vascular maturation, stability, and remodeling by the Tie2 receptor signaling pathway (152), in which Angpt2, particularly, enhanced VSMCs to migrate but had no influence on its proliferation. In addition, after the invasion from P. gingivalis to VSMCs, its LPS could significantly reduce the expression and activity of tissue factor inhibitor, thereby inducing the migration as well as the proliferation of VSMCs through which atherosclerotic plaques have been promoted (153). Unexpectedly, for C. pneumoniae, as another Gram-negative pathogen associated with AS, its infection was found to promote VSMC migration via c-Fos/IL-17C signaling (154). Despite the same outcome from both of them, they promote plaque progression in different pathways.
P. gingivalis Promotes the Calcification of VSMCs
Vascular calcification, described as excessive deposition of calcium-containing phosphate, is one of the signs of AS (83), which might ultimately lead to the hardening of blood vessels and reduction in elasticity (155). P. gingivalis can induce the calcification of VSMCs and promote vascular calcification, which is also induced by LPS and OMVs. The OMVs of P. gingivalis promoted the calcification of VSMCs, along with the involvement of the ERK1/2- Runt-related transcription factor 2 (RUNX2) pathway (156), in a concentration-dependent manner and regulate the process of VSMC osteogenic differentiation and mineralization (157). The key regulator of this progress is Runx2, regulated by the ERK signaling pathway and involved in osteogenic transcription (158). Bone morphogenetic protein 4 was upregulated and mediated by TLR4 and ERK1/2-p38 pathway, and ultimately it promoted vascular calcification in VSMC from one suffering from P. gingivalis infection (159). During vascular calcification, VSMCs change to an osteoblast-like phenotype, which is an important step in mediating vascular media calcification (160), and P. gingivalis LPS significantly promoted the upregulation of osteogenic genes [such as alkaline phosphatase (ALP), core-binding factor, alpha 1, bone sialoprotein, and OPN] (161). Moreover, apoptosis of VSMCs, accompanied by considerable matrix vesicles with the bound calcifying membrane released (162), was increased by P. gingivalis in inorganic phosphate-induced VSMCs, in which the Gas6/Axl/Akt survival pathway was inhibited (163). In addition, TNF-α and IL-1β, as pro-inflammatory cytokines, can upregulate ALP and RUNX2 in VSMCs, contributing to vascular calcification (164). Therefore, P. gingivalis may promote vascular calcification through its structure or secreted substances and, alternatively, through the secretions of VSMCs after infection. Matrix-gla protein (MGP), an effective inhibitor of vascular calcification, and lack of MGP will increase the risk of AS (165, 166). However, the relationship between MGP and P. gingivalis is still unclear, which also suggests our next research direction, that is, whether MGP can be used to alleviate vascular calcification caused by P. gingivalis.
P. gingivalis Promotes VSMCs to Engulf Lipids to Form Foam Cells
As the most indispensable cell in AS lesions, VSMCs contribute to the proportion of more than half of the foam cells in the lesion area (167). In the process of that, the aggregation of LDL and its oxidative modification product oxLDL as well as the immune complexes they induce, such as b2glycoprotein I (b2GPI), anti-b2GPI, plays a key role in promoting the formation of foam cells (168). Recent studies have shown that the lipid uptake pathways of VSMCs include SR-AI/II (class A), CD36 (class B), LOX-1 (class E), and SR-PSOX/CXCL16 (class G) (169). In addition, the presence of macrophages also promotes the transformation of SMCs into foam cells (170). At present, the research on the effect of P. gingivalis on VSMCs is not sufficient and in depth. VSMCs may serve as deposits of lipids from the insudating lipoproteins and become foam cells easily with P. gingivalis, but the in vivo mechanisms remain incompletely understood (85). According to our research, P. gingivalis promotes the accumulation and oxidation of lipids under the endothelium, and P. gingivalis promotes the chemotaxis of macrophages. Therefore, we speculate that P. gingivalis can promote the uptake of lipids by VSMCs and form foam cells through these pathways (Table 2).

Table 2 The effect of P. gingivalis on vascular smooth muscle cells.
The Influence of P. gingivalis on Macrophages
P. gingivalis Achieves Immune Evasion Through Macrophages
P. gingivalis achieves immune evasion through internalization by macrophages, which may result in the preservation of P. gingivalis virulence and chronic infection during AS. The uptake of P. gingivalis by macrophages hinges on complement receptor type 3 [CR3 (CD11b/CD18)] and TLR2 (171). CR3 is a β2 integrin, which can recognize sort of structurally and morphologically unrelated molecules from a pathogen or a host, such as intercellular adhesion molecules, fibrinogen, and so on (172). P. gingivalis selected the TLR2 pre-pathway to bind CR3 and entered the cell (173). The intracellular P. gingivalis stimulates TLR2 through its surface fimbriae and activates the signal pathway from the inside out to induce a distinct conformation of CR3 with high affinity. This pathway can be mediated by Rac1/PI3K, and it requires fimbriae to bind CD14 to promote fimbria–TLR2 interaction (117, 174). What is more, CR3 is utilized as a relatively safe portal of entry by P. gingivalis, and its fimbriae additionally inhibited the production of IL-12 (p70) with biological activity by interacting with CR3 on the surface of macrophages (171, 174, 175), which support that P. gingivalis achieves evasion immune clearance.
P. gingivalis Promotes the Inflammatory Response of Macrophages
Overwhelming experimental and clinical evidence suggest that AS is a chronic inflammatory disease (176). P. gingivalis triggers the inflammatory response of macrophages, thereby promoting different stages of AS. P. gingivalis fimbriae and OMVs stimulated monocytes and macrophages to secret pro-inflammatory cytokines—for instance, IL-1β, IL-18, TNF-α, and NLRP3 inflammasome activation (49, 177). Under the action of TNF-α and IL-6, the activation and antigen presentation ability of macrophages are embellished, and immunity is also modulated by different mechanisms (178). Furthermore, the TNF-α released by macrophages can promote EndMT in ECs (111), which can be promoted by P. gingivalis. Therefore, it is possible for P. gingivalis to induce EndMT of ECs by promoting the secretion of TNF-α from macrophages (111). P. gingivalis can easily interact with activated CR3, activating the outside-in signaling pathway, and lessen IL-12 due to ERK1/2, thereby inhibiting the production of biologically active (p70) IL-12 (174, 179), which mediates immune clearance (180). We can understand from studies that P. gingivalis LPS and gingipains activated the macrophage NLRP3 inflammasomes and then produced powerful inflammatory cell factor IL-1β with the activation of NF-κB signaling (181), which makes M1-Mϕ secrete TNF-α and M2-Mϕ secrete IL-10, along with chemotactic chemokines like knuckle cracking (regulated upon activation, normal T cell expressed and secreted)/CC chemokine ligand 5, eotaxin, and IL-10 from polarized macrophages (182). In particular, P. gingivalis OMVs were also suggested to minimize anti-inflammatory IL-10 secretion (183), and NLRP12 is upregulated, which downregulated TNF-α production and iNOS expression in macrophages infected with P. gingivalis (184). Moreover, P. gingivalis gingipains reduced the expression of CD14 in macrophage to reduce macrophage interactions with apoptotic cells, which could curb TNF-α-induced expression by P. gingivalis LPS (185).
P. gingivalis Promotes Macrophages to Form Foam Cells
Foam cells are critically important to the development and progression of AS, and P. gingivalis promotes macrophages to form foam cells. With LDL, low-concentration P. gingivalis OMVs also induce foam cell formation, which is in a dose-dependent manner (186). Firstly, P. gingivalis and OMVs can provoke LDL modification (187), which is vital in AS by acting on multiple cells (188), and can be easily transported to macrophages to form foam cells. P. gingivalis OMVs stimulated the macrophages to produce matrix metalloproteinases (MMP), and a few types of them were able to cut apolipoprotein B-100 (apoB-100) from LDL particles and then lead to the aggregation and modification of LDL (189, 190). P. gingivalis induced the conversion of macrophage metabolism from oxidative phosphorylation to glycolysis, which enhances the release of lactic acid, reduces mitochondrial oxygen consumption, and increases ROS (191), so that modification of LDL may be increased by ROS in macrophages with P. gingivalis. Secondly, P. gingivalis fimbriae significantly promotes the uptake of LDL by macrophages to form foam cells (187), with P. gingivalis LPS enhancing lipid accumulation in macrophages and reducing cholesterol efflux (192). The clearance receptors of macrophages, such as the class A clearance receptor (SR-A) and CD36, mediate the internalization of oxLDL, thereby promoting the accumulation of intracellular cholesterol (193). In contrast, reverse cholesterol transporters including scavenger receptor class B type I (SR-BI) and adenosine-binding cassette transporters A1 and G1 (ABCA1/ABCG1) are responsible for cholesterol efflux (194). P. gingivalis LPS reduced ABCA1 in macrophages but increased CD36 through the c-Jun-AP-1 pathway, while it did not affect SR-A, SR-BI, and ABCG1 (195). These outcomes are partially associated with the activation of protein kinase C and c-Jun N-terminal kinase 1/2 phosphorylation, which promotes NF-κB to activate (110). At the same time, P. gingivalis LPS reduces the stability of ABCA1 protein by increasing calpain activity (195). Moreover, the TLR signal transduction in macrophages is mainly composed of MyD88 and Toll/IL-1R domain-containing adaptor-inducing IFN-β, and it exercises function in foam cells caused by P. gingivalis (196) (Figure 4).
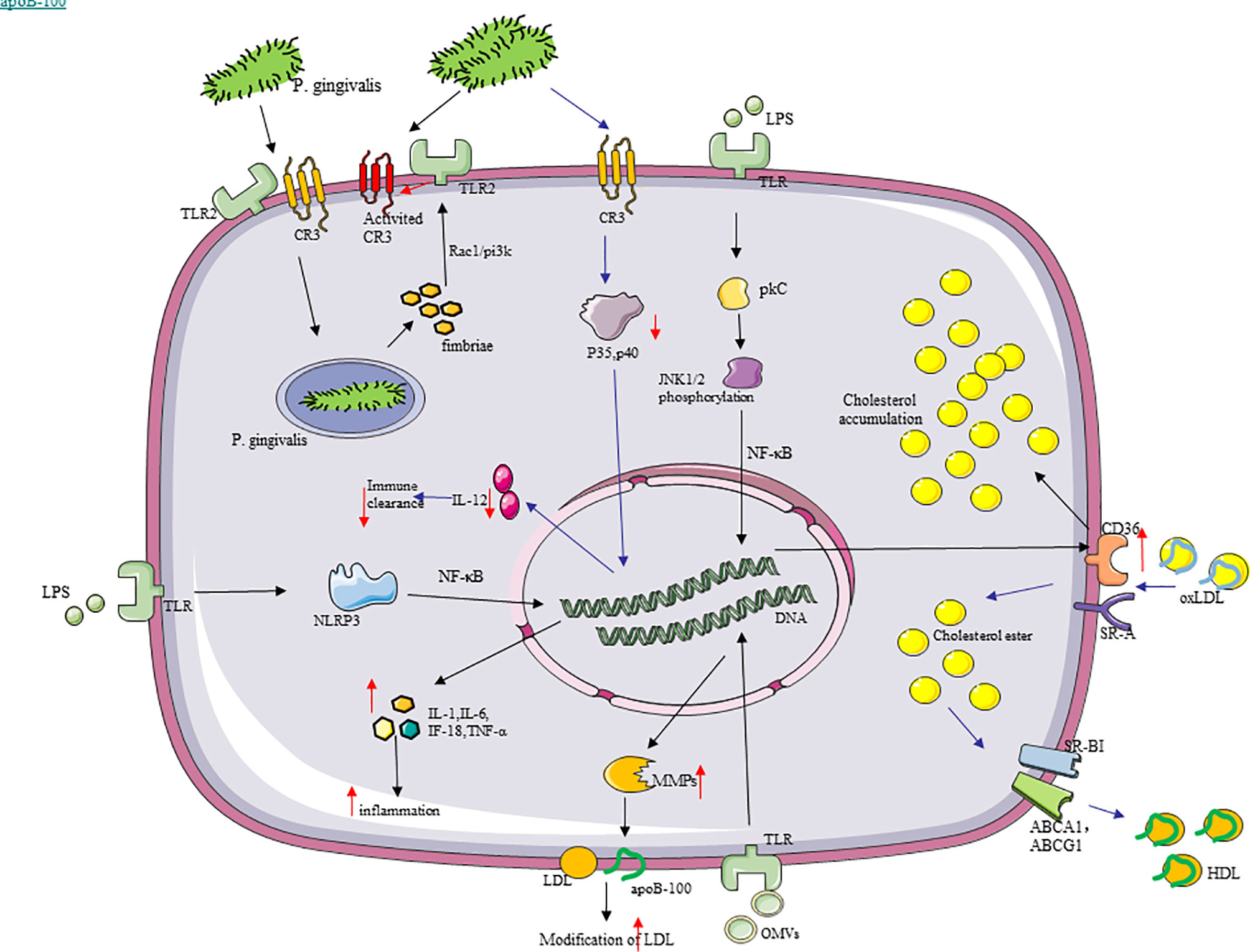
Figure 4 Molecular cascades activated by P. gingivalis in macrophages. (1) P. gingivalis recognizes TLR2, binds to CR3, and enters macrophages. The intracellular P. gingivalis stimulates TLR2 through its surface fimbriae, thereby inducing the high-affinity conformation of CR3, which is conducive to the uptake of more P. gingivalis by macrophages. (2) SR-A and CD36, as the clearance receptors of macrophages, mediate the internalization of oxLDL, thereby promoting the accumulation of intracellular cholesterol. In contrast, SR-BI and ABCA1/ABCG1 are responsible for cholesterol efflux. P. gingivalis lipopolysaccharide increased the expression of CD36 through the c-Jun-AP-1 pathway and promoted cholesterol accumulation in macrophages. (3) P. gingivalis outer membrane vesicles stimulated the macrophages to produce matrix metalloproteinases capable of cleaving the apoB-100 of low-density lipoprotein (LDL) particles to increase the modification of LDL. (4) P. gingivalis promoted the production of pro-inflammatory cytokines, like IL-1, IL-18, IL-6, and TNF-α in macrophages, with the activation of NLRP3 inflammasomes.
Hence, chronic inflammation caused by P. gingivalis might gradually worsen in this way. In addition, macrophages can generate inducible proteases under the action of P. gingivalis, such as MMP, which can crack cytokine precursors, growth factors, cytokine receptors, and cell adhesion molecules (122). In summary, P. gingivalis has a considerable immunomodulatory impact and can act on monocytes and macrophages, producing various inflammatory mediators and enzymes through these pathways to promote inflammation and tissue damage in the process of AS (Figure 4).
The Influence of P. gingivalis on T Cells
P. gingivalis Causes Th17/Treg Imbalance
The immune balance between Th cells and Tregs has an important regulatory role in AS (71). With the development of AS, the number and response of Th17 will increase accordingly with P. gingivalis infection, reducing the number and inhibiting the regulatory function of Tregs and causing Th17/Treg imbalance, which may lead to plaque instability (197, 198). P. gingivalis and its LPS and gingipains can activate monocytes, promote a Th17/IL-17 response, and then make TNF-α, IL-1β, IL-6, and IL-17 increase, mediating by TLR2/TLR4 signaling and inducing atherosclerotic plaque formation through an inflammatory response (199, 200). The Th17-related genes like IL-6, retinoid-related orphan receptor-gammat (RORγt), and STAT3 were elevated, with TGF- β and IL-10 decreasing in P. gingivalis-challenged mice (201). P. gingivalis used gingipain to highly specifically induce Th17 cells, by which IL-6 signaling was blocked (202). Furthermore, P. gingivalis infection promotes IL-6 to secret in DCs, and increased IL-6 may be good for Th17 cell proliferation and may inhibit the production and effect of Tregs (203). In general, pro-inflammatory Th17 cell responses were strengthened by P. gingivalis, thereby accelerating AS. Tregs can inhibit effector T cell proliferation and the production of cytokine (mainly Th1 and Th17 lymphocytes) and are vital in maintaining the homeostasis of the immune-inflammatory response of the host (204). P. gingivalis infection reduces the number and inhibits the regulatory function of Tregs. Compared with people in healthy conditions, AS patients with P. gingivalis have fewer Tregs (198). Tregs themselves can come into contact with other effector cells and can also secrete anti-inflammatory IL-10 and TGF-β1, thereby directly or indirectly inhibiting inflammation (197, 205). TGF-β1 has contributed much to the development of Tregs (206). IL-10 is a cytokine that contributed a lot to anti-inflammatory effects (207). Experiments have shown that P. gingivalis infection reduces IL-10. TLR2/1 signaling is the main mechanism of IL-10 production. Thirdly, the reaction caused by the main surface protein of P. gingivalis FimA is also involved among them (48), and the concentration of Treg-related factors like TGF-β1 and FoxP3 was reduced in P. gingivalis-positive patients (198). According to reports, there is Th17/Treg imbalance in AS patients, and immune answer induced by T cell is principal in plaque instability (208, 209). As a result, P. gingivalis ultimately induces an increasing inflammation reaction in AS plaque and plaque instability by promoting Th17/Treg unevenness (Figure 5).
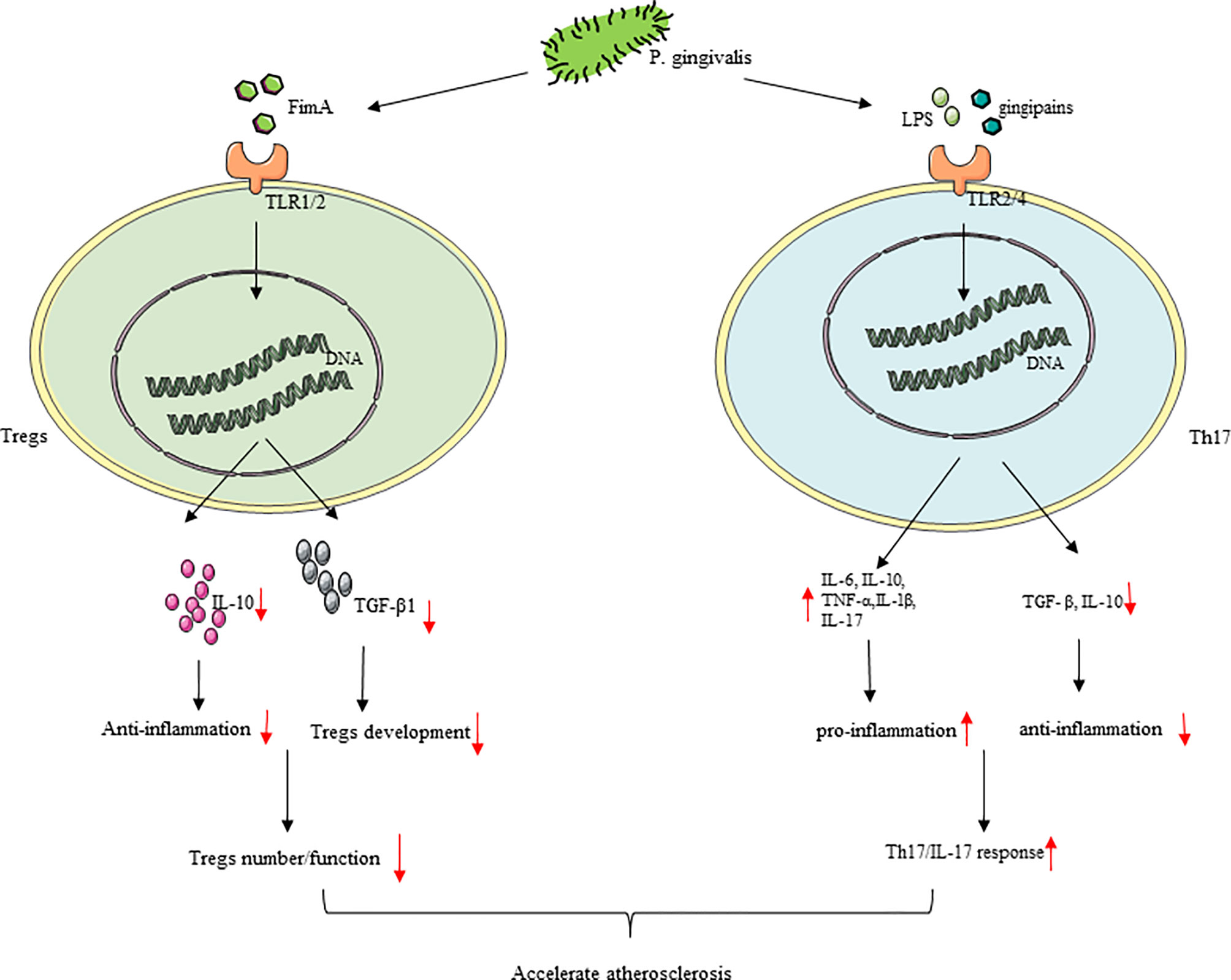
Figure 5 P. gingivalis infection cause Th17/Treg imbalance. (1) In the process of atherosclerosis, P. gingivalis infection increased the number and response of Th17, inhibited Tregs with regulatory effects, and cause Th17/Treg imbalance. (2) P. gingivalis reduced the number and inhibited the regulatory function of Tregs with the downregulation of IL-10 and TGF-β1. P. gingivalis promoted a Th17/IL-17 response resulting in increased TNF-α, IL-1β, IL-6, and IL-17 production by T cells, which may be mediated by TLR2/TLR4 signaling.
P. gingivalis Inhibits T Cell Answer
P. gingivalis can inhibit T cell answer through the suppression of differentiation and activation caused by chemokines, proliferation, and communication in T cells. Firstly, the T cell chemokine interferon-inducible protein 10 or CXC motif chemokine 10, which comes from neutrophils and monocytes, were not influenced by P. gingivalis, which makes a T cell respond weakly and achieve local immune evasion (210). Secondly, the CD4 and CD8 proteins on human T cells can be destroyed by P. gingivalis, thereby inhibiting the activation of T cells. It is also through this mechanism that P. gingivalis can protect itself from the immune system. The establishment and proliferation of the bacteria in the host is achieved (211). P. gingivalis HSP60 can have different effects on T cell polarization by different mechanisms and then make atherosclerotic occur or not (212). Last but not least, P. gingivalis inhibits the expression of IL-2 that promotes the proliferation and communication of T cells (101). The activity of NF-kB and AP-1 is downregulated by P. gingivalis and its Rgp protease, so IL-2 cannot be transcribed and expressed (213), and then IL-2 cannot stably accumulate in T cells, which resulted in T cells without energy (214). This weakens the inflammatory response, which is connected with T- and B-cell activation, and subsequently IFN-γ from T cells (101). We have a clearer understanding of how P. gingivalis can prevent itself from being cleared by the immune system by inhibiting T cell response and thus surviving and persisting in AS lesions to promote AS development (Figure 5).
The Influence of P. gingivalis on DCs
DCs can promote lipid accumulation, promote inflammation, and destroy plaque stability during various stages of AS (99, 215). It is worth noting that there are many connections between DCs and P. gingivalis. DCs can be used as carriers to transport pathogenic bacteria such as P. gingivalis from the oral cavity with serologic exposure through the bloodstream to reach the arteries in the pre-AS lesions (216–218). The minor fimbria of P. gingivalis binds with a cell adhesion molecule on DCs called CD209 to enable it to escape immune surveillance (219). P. gingivalis promotes both its own survival and the survival of its host DCs through manipulating dendritic cell signaling to perturb both autophagy and apoptosis, in which activation of the Akt/mTOR axis was linked. There was also the induction of the anti-apoptotic protein Bcl2 and decrease of caspase-3 cleavage and pro-apoptotic proteins Bax and Bim in this progress (220).
Conclusion
In recent years, the promotion of P. gingivalis in the pathological process of AS has received more attention. P. gingivalis has the capability of leading to arterial endothelial dysfunction, inducing foam cell formation, and making vascular smooth muscle cells proliferate and calcify, causing T helper cells and Tregs imbalance. Accompanied with the progression of endothelial activation, lipid accumulation, plaque formation, and rupture, P. gingivalis eventually aggravates the process of AS. Here we summarized and provided several of the latest research findings on the effects of P. gingivalis on AS-related cells as well as the underlying mechanisms, which may help to provide new insights on the targets for the effective treatment and prevention of AS.
Author Contributions
JZ and MX performed the original draft preparation and revision, created the tables and figures, and were the major contributors in writing the manuscript. XH, GC, and YY made suggestions to the writing of the manuscript and revisions to tables and figures. XL and GF participated in conceptualization and methodology. GC supervised the work and acquired funding. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China for Key Program Projects (no. 82030070), the National Natural Science Foundation of China for Distinguished Young Scholars (no. 31725011), Hubei Provincial Natural Science Fund for Creative Research Groups (2020CFA014), National Natural Science Foundation of China for Young Scientists (no. 82101025), and Key Supporting Program by the Health Commission of Hubei Province (WJ2019C001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary

References
2. Mendis S, Davis S, Norrving B. Organizational Update: The World Health Organization Global Status Report on Noncommunicable Diseases 2014; One More Landmark Step in the Combat Against Stroke and Vascular Disease. Stroke (2015) 46(5):e121–2. doi: 10.1161/STROKEAHA.115.008097
3. Lechner K, von Schacky C, McKenzie AL, Worm N, Nixdorff U, Lechner B, et al. Lifestyle Factors and High-Risk Atherosclerosis: Pathways and Mechanisms Beyond Traditional Risk Factors. Eur J Prev Cardiol (2020) 27(4):394–406. doi: 10.1177/2047487319869400
4. Libby P, Loscalzo J, Ridker PM, Farkouh ME, Hsue PY, Fuster V, Hasan AA, et al. Inflammation, Immunity, and Infection in Atherothrombosis: JACC Review Topic of the Week. J Am Coll Cardiol (2018) 72(17):2071–81. doi: 10.1016/j.jacc.2018.08.1043
5. Tumurkhuu G, Dagvadorj J, Porritt RA, Crother TR, Shimada K, Tarling EJ, et al. Chlamydia Pneumoniae Hijacks a Host Autoregulatory IL-1β Loop to Drive Foam Cell Formation and Accelerate Atherosclerosis. Cell Metab (2018) 28(3):432–448.e4. doi: 10.1016/j.cmet.2018.05.027
6. Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of Periodontal Pathogens in Atheromatous Plaques. J Periodontol (2000) 71(10):1554–60. doi: 10.1902/jop.2000.71.10.1554
7. Xu Z, Li J, Wang H, Xu G. Helicobacter Pylori Infection and Atherosclerosis: Is There a Causal Relationship? Eur J Clin Microbiol Infect Dis (2017) 36(12):2293–301. doi: 10.1007/s10096-017-3054-0
8. Li B, Xia Y, Hu B. Infection and Atherosclerosis: TLR-Dependent Pathways. Cell Mol Life Sci (2020) 77(14):2751–69. doi: 10.1007/s00018-020-03453-7
9. Slots J. Periodontitis: Facts, Fallacies and the Future. Periodontol 2000 (2017) 75(1):7–23. doi: 10.1111/prd.12221
10. Zeng XT, et al. Periodontal Disease and Carotid Atherosclerosis: A Meta-Analysis of 17,330 Participants. Int J Cardiol (2016) 203:1044–51. doi: 10.1016/j.ijcard.2015.11.092
11. Tonetti MS. Periodontitis and Risk for Atherosclerosis: An Update on Intervention Trials. J Clin Periodontol (2009) 36 Suppl 10:15–9. doi: 10.1111/j.1600-051X.2009.01417.x
12. Kolltveit KM, Eriksen HM. Is the Observed Association Between Periodontitis and Atherosclerosis Causal? Eur J Oral Sci (2001) 109(1):2–7. doi: 10.1034/j.1600-0722.2001.109001002.x
13. Rydén L, Buhlin K, Ekstrand E, de Faire U, Gustafsson A, Holmer J, et al. Periodontitis Increases the Risk of a First Myocardial Infarction: A Report From the PAROKRANK Study. Circulation (2016) 133(6):576–83. doi: 10.1161/CIRCULATIONAHA.115.020324
14. Söder B, Meurman JH, Söder P. Gingival Inflammation Associates With Stroke–a Role for Oral Health Personnel in Prevention: A Database Study. PloS One (2015) 10(9):e0137142. doi: 10.1371/journal.pone.0137142
15. Carrizales-Sepúlveda EF, Ordaz-Farías A, Vera-Pineda R, Flores-Ramírez R. Periodontal Disease, Systemic Inflammation and the Risk of Cardiovascular Disease. Heart Lung Circ (2018) 27(11):1327–34. doi: 10.1016/j.hlc.2018.05.102
16. Lockhart PB, Bolger AF, Papapanou PN, Osinbowale O, Trevisan M, Levison ME, et al. Periodontal Disease and Atherosclerotic Vascular Disease: Does the Evidence Support an Independent Association?: A Scientific Statement From the American Heart Association. Circulation (2012) 125(20):2520–44. doi: 10.1161/CIR.0b013e31825719f3
17. Miyauchi S, Maekawa T, Aoki Y, Miyazawa H, Tabeta K, Nakajima T, et al. Oral Infection With Porphyromonas Gingivalis and Systemic Cytokine Profile in C57BL/6.KOR-Apoe Shl Mice. J Periodontal Res (2012) 47(3):402–8. doi: 10.1111/j.1600-0765.2011.01441.x
18. Brodala N, Merricks EP, Bellinger DA, Damrongsri D, Offenbacher S, Beck J, et al. Porphyromonas Gingivalis Bacteremia Induces Coronary and Aortic Atherosclerosis in Normocholesterolemic and Hypercholesterolemic Pigs. Arterioscler Thromb Vasc Biol (2005) 25(7):1446–51. doi: 10.1161/01.ATV.0000167525.69400.9c
19. Kozarov EV, Dorn BR, Shelburne CE, Dunn WA Jr, Progulske-Fox A. Human Atherosclerotic Plaque Contains Viable Invasive Actinobacillus Actinomycetemcomitans and Porphyromonas Gingivalis. Arterioscler Thromb Vasc Biol (2005) 25(3):e17–8. doi: 10.1161/01.ATV.0000155018.67835.1a
20. Jia R, Kurita-Ochiai T, Oguchi S, Yamamoto M. Periodontal Pathogen Accelerates Lipid Peroxidation and Atherosclerosis. J Dent Res (2013) 92(3):247–52. doi: 10.1177/0022034513475625
21. Pucar A, Milasin J, Lekovic V, Vukadinovic M, Ristic M, Putnik S, et al. Correlation Between Atherosclerosis and Periodontal Putative Pathogenic Bacterial Infections in Coronary and Internal Mammary Arteries. J Periodontol (2007) 78(4):677–82. doi: 10.1902/jop.2007.060062
22. Xie M, Tang Q, Nie J, Zhang C, Zhou X, Yu S, et al. BMAL1-Downregulation Aggravates Porphyromonas Gingivalis-Induced Atherosclerosis by Encouraging Oxidative Stress. Circ Res (2020) 126(6):e15–29. doi: 10.1161/CIRCRESAHA.119.315502
23. Figuero E, Sánchez-Beltrán M, Cuesta-Frechoso S, Tejerina JM, del Castro JA, Gutiérrez JM, et al. Detection of Periodontal Bacteria in Atheromatous Plaque by Nested Polymerase Chain Reaction. J Periodontol (2011) 82(10):1469–77. doi: 10.1902/jop.2011.100719
24. Gaetti-Jardim E, Marcelino SL, Feitosa ACR, Romito GA, Avila-Campos MJ. Quantitative Detection of Periodontopathic Bacteria in Atherosclerotic Plaques From Coronary Arteries. J Med Microbiol (2009) 58(Pt 12):1568–75. doi: 10.1099/jmm.0.013383-0
25. Hussain M, Stover CM, Dupont A. P. Gingivalis in Periodontal Disease and Atherosclerosis - Scenes of Action for Antimicrobial Peptides and Complement. Front Immunol (2015) 6:45. doi: 10.3389/fimmu.2015.00045
26. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas Gingivalis in Alzheimer's Disease Brains: Evidence for Disease Causation and Treatment With Small-Molecule Inhibitors. Sci Adv (2019) 5(1):eaau3333. doi: 10.1126/sciadv.aau3333
27. Kato T, Yamazaki K, Nakajima M, Date Y, Kikuchi J, Hase K, et al. Oral Administration of Porphyromonas Gingivalis Alters the Gut Microbiome and Serum Metabolome. mSphere (2018) 3(5):e00460–18. doi: 10.1128/mSphere.00460-18
28. Gibson FC 3rd, Hong C, Chou HH, Yumoto H, Chen J, Lien E, Wong J, et al. Innate Immune Recognition of Invasive Bacteria Accelerates Atherosclerosis in Apolipoprotein E-Deficient Mice. Circulation (2004) 109(22):2801–6. doi: 10.1161/01.CIR.0000129769.17895.F0
29. Nakano K, Inaba H, Nomura R, Nemoto H, Takeuchi H, Yoshioka H, et al. Distribution of Porphyromonas Gingivalis Fima Genotypes in Cardiovascular Specimens From Japanese Patients. Oral Microbiol Immunol (2008) 23(2):170–2. doi: 10.1111/j.1399-302X.2007.00406.x
30. Atarbashi-Moghadam F, Havaei SR, Havaei SA, Hosseini NS, Behdadmehr G, Atarbashi-Moghadam S. Periopathogens in Atherosclerotic Plaques of Patients With Both Cardiovascular Disease and Chronic Periodontitis. ARYA Atheroscler (2018) 14(2):53–7. doi: 10.22122/arya.v14i2.1504
31. Li L, Messas E, Batista EL Jr, Levine RA, Amar S. Porphyromonas Gingivalis Infection Accelerates the Progression of Atherosclerosis in a Heterozygous Apolipoprotein E-Deficient Murine Model. Circulation (2002) 105(7):861–7. doi: 10.1161/hc0702.104178
32. Gibson FC 3rd, Ukai T, Genco CA. Engagement of Specific Innate Immune Signaling Pathways During Porphyromonas Gingivalis Induced Chronic Inflammation and Atherosclerosis. Front Biosci (2008) 13:2041–59. doi: 10.2741/2822
33. Gibson FC 3rd, Yumoto H, Takahashi Y, Chou HH, Genco CA. Innate Immune Signaling and Porphyromonas Gingivalis-Accelerated Atherosclerosis. J Dent Res (2006) 85(2):106–21. doi: 10.1177/154405910608500202
34. Hayashi C, Viereck J, Hua N, Phinikaridou A, Madrigal AG, Gibson FC 3rd, et al. Porphyromonas Gingivalis Accelerates Inflammatory Atherosclerosis in the Innominate Artery of Apoe Deficient Mice. Atherosclerosis (2011) 215(1):52–9. doi: 10.1016/j.atherosclerosis.2010.12.009
35. Pussinen PJ, Mattila K. Periodontal Infections and Atherosclerosis: Mere Associations? Curr Opin Lipidol (2004) 15(5):583–8. doi: 10.1097/00041433-200410000-00013
36. Mulhall H, Huck O, Amar S. Porphyromonas Gingivalis, a Long-Range Pathogen: Systemic Impact and Therapeutic Implications. Microorganisms (2020) 8(6):869. doi: 10.3390/microorganisms8060869
37. Xu W, Zhou W, Wang H, Liang S. Roles of Porphyromonas Gingivalis and Its Virulence Factors in Periodontitis. Adv Protein Chem Struct Biol (2020) 120:45–84. doi: 10.1016/bs.apcsb.2019.12.001
38. Mei F, Xie M, Huang X, Long Y, Lu X, Wang X. Porphyromonas Gingivalis and Its Systemic Impact: Current Status. Pathogens (2020) 9(11):944. doi: 10.3390/pathogens9110944
39. Chopra A, Bhat SG, Sivaraman K. Porphyromonas Gingivalis Adopts Intricate and Unique Molecular Mechanisms to Survive and Persist Within the Host: A Critical Update. J Oral Microbiol (2020) 12(1):1801090. doi: 10.1080/20002297.2020.1801090
40. Guo Y, Nguyen KA, Potempa J. Dichotomy of Gingipains Action as Virulence Factors: From Cleaving Substrates With the Precision of a Surgeon's Knife to a Meat Chopper-Like Brutal Degradation of Proteins. Periodontol 2000 (2010) 54(1):15–44. doi: 10.1111/j.1600-0757.2010.00377.x
41. Gui MJ, Dashper SG, Slakeski N, Chen YY, Reynolds EC. Spheres of Influence: Porphyromonas Gingivalis Outer Membrane Vesicles. Mol Oral Microbiol (2016) 31(5):365–78. doi: 10.1111/omi.12134
42. Viafara-García SM, Morantes SJ, Chacon-Quintero Y, Castillo DM, Lafaurie GI, Buitrago DM. Repeated Porphyromonas Gingivalis W83 Exposure Leads to Release Pro-Inflammatory Cytokynes and Angiotensin II in Coronary Artery Endothelial Cells. Sci Rep (2019) 9(1):19379. doi: 10.1038/s41598-019-54259-y
43. Jeong E, Lee JY, Kim SJ, Choi J. Predominant Immunoreactivity of Porphyromonas Gingivalis Heat Shock Protein in Autoimmune Diseases. J Periodontal Res (2012) 47(6):811–6. doi: 10.1111/j.1600-0765.2012.01501.x
44. Inaba H, Kawai S, Kato T, Nakagawa I, Amano A. Association Between Epithelial Cell Death and Invasion by Microspheres Conjugated to Porphyromonas Gingivalis Vesicles With Different Types of Fimbriae. Infect Immun (2006) 74(1):734–9. doi: 10.1128/IAI.74.1.734-739.2006
45. Palaska I, Gagari E, Theoharides TC. The Effects of P. Gingivalis and E. Coli LPS on the Expression of Proinflammatory Mediators in Human Mast Cells and Their Relevance to Periodontal Disease. J Biol Regul Homeost Agents (2016) 30(3):655–64. doi: 10.7705/biomedica.v29i2.31
46. Mantri CK, Chen CH, Dong X, Goodwin JS, Pratap S, Paromov V, et al. Fimbriae-Mediated Outer Membrane Vesicle Production and Invasion of Porphyromonas Gingivalis. Microbiologyopen (2015) 4(1):53–65. doi: 10.1002/mbo3.221
47. Wilensky A, Tzach-Nahman R, Potempa J, Shapira L, Nussbaum G. Porphyromonas Gingivalis Gingipains Selectively Reduce CD14 Expression, Leading to Macrophage Hyporesponsiveness to Bacterial Infection. J Innate Immun (2015) 7(2):127–35. doi: 10.1159/000365970
48. Gaddis DE, Maynard CL, Weaver CT, Michalek SM, Katz J. Role of TLR2-Dependent IL-10 Production in the Inhibition of the Initial IFN-γ T Cell Response to Porphyromonas Gingivalis. J Leukoc Biol (2013) 93(1):21–31. doi: 10.1189/jlb.0512220
49. Pollreisz A, Huang Y, Roth GA, Cheng B, Kebschull M, Papapanou PN, et al. Enhanced Monocyte Migration and Pro-Inflammatory Cytokine Production by Porphyromonas Gingivalis Infection. J Periodontal Res (2010) 45(2):239–45. doi: 10.1111/j.1600-0765.2009.01225.x
50. Nassar H, Chou HH, Khlgatian M, Gibson FC 3rd, Van Dyke TE, Genco CA. Role for Fimbriae and Lysine-Specific Cysteine Proteinase Gingipain K in Expression of Interleukin-8 and Monocyte Chemoattractant Protein in Porphyromonas Gingivalis-Infected Endothelial Cells. Infect Immun (2002) 70(1):268–76. doi: 10.1128/IAI.70.1.268-276.2002
51. Ford PJ, Gemmell E, Timms P, Chan A, Preston FM, Seymour GJ. Anti-P. Gingivalis Response Correlates With Atherosclerosis. J Dent Res (2007) 86(1):35–40. doi: 10.1177/154405910708600105
52. Choi JI, Chung SW, Kang HS, Rhim BY, Park YM, Kim US. Epitope Mapping of Porphyromonas Gingivalis Heat-Shock Protein and Human Heat-Shock Protein in Human Atherosclerosis. J Dent Res (2004) 83(12):936–40. doi: 10.1177/154405910408301209
53. de Diego I, Veillard F, Sztukowska MN, Guevara T, Potempa B, Pomowski A, et al. Structure and Mechanism of Cysteine Peptidase Gingipain K (Kgp), a Major Virulence Factor of Porphyromonas Gingivalis in Periodontitis. J Biol Chem (2014) 289(46):32291–302. doi: 10.1074/jbc.M114.602052
54. Khalaf H, Lönn J, Bengtsson T. Cytokines and Chemokines Are Differentially Expressed in Patients With Periodontitis: Possible Role for TGF-β1 as a Marker for Disease Progression. Cytokine (2014) 67(1):29–35. doi: 10.1016/j.cyto.2014.02.007
55. Beveridge TJ. Structures of Gram-Negative Cell Walls and Their Derived Membrane Vesicles. J Bacteriol (1999) 181(16):4725–33. doi: 10.1128/JB.181.16.4725-4733.1999
56. Cecil JD, Sirisaengtaksin N, O'Brien-Simpson NM, Krachler AM. Outer Membrane Vesicle-Host Cell Interactions. Microbiol Spectr (2019) 7(1):10. doi: 10.1128/microbiolspec.PSIB-0001-2018
57. Forner L, Larsen T, Kilian M, Holmstrup P. Incidence of Bacteremia After Chewing, Tooth Brushing and Scaling in Individuals With Periodontal Inflammation. J Clin Periodontol (2006) 33(6):401–7. doi: 10.1111/j.1600-051X.2006.00924.x
58. Berggreen E, Wiig H. Lymphangiogenesis and Lymphatic Function in Periodontal Disease. J Dent Res (2013) 92(12):1074–80. doi: 10.1177/0022034513504589
59. Olsen I, Progulske-Fox A. Invasion of Porphyromonas Gingivalis Strains Into Vascular Cells and Tissue. J Oral Microbiol (2015) 7:28788. doi: 10.3402/jom.v7.28788
60. Li L, Michel R, Cohen J, Decarlo A, Kozarov E. Intracellular Survival and Vascular Cell-to-Cell Transmission of Porphyromonas Gingivalis. BMC Microbiol (2008) 8:26. doi: 10.1186/1471-2180-8-26
61. Gimbrone MA Jr., Garcia-Cardena G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res (2016) 118(4):620–36. doi: 10.1161/CIRCRESAHA.115.306301
62. Dejana E, Lampugnani MG. Endothelial Cell Transitions. Science (2018) 362(6416):746–7. doi: 10.1126/science.aas9432
63. Gimbrone MA Jr., García-Cardeña G. Vascular Endothelium, Hemodynamics, and the Pathobiology of Atherosclerosis. Cardiovasc Pathol (2013) 22(1):9–15. doi: 10.1016/j.carpath.2012.06.006
64. Charo IF, Taub R. Anti-Inflammatory Therapeutics for the Treatment of Atherosclerosis. Nat Rev Drug Discov (2011) 10(5):365–76. doi: 10.1038/nrd3444
65. Di Pietro N, Formoso G, Pandolfi A. Physiology and Pathophysiology of Oxldl Uptake by Vascular Wall Cells in Atherosclerosis. Vascul Pharmacol (2016) 84:1–7. doi: 10.1016/j.vph.2016.05.013
66. Saigusa R, Winkels H, Ley K. T Cell Subsets and Functions in Atherosclerosis. Nat Rev Cardiol (2020) 17(7):387–401. doi: 10.1038/s41569-020-0352-5
67. Mudau M, Genis A, Lochner A, Strijdom H. Endothelial Dysfunction: The Early Predictor of Atherosclerosis. Cardiovasc J Afr (2012) 23(4):222–31. doi: 10.5830/CVJA-2011-068
68. Incalza MA, D'Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative Stress and Reactive Oxygen Species in Endothelial Dysfunction Associated With Cardiovascular and Metabolic Diseases. Vascul Pharmacol (2018) 100:1–19. doi: 10.1016/j.vph.2017.05.005
69. Sprague AH, Khalil RA. Inflammatory Cytokines in Vascular Dysfunction and Vascular Disease. Biochem Pharmacol (2009) 78(6):539–52. doi: 10.1016/j.bcp.2009.04.029
70. Baker AB, Ettenson DS, Jonas M, Nugent MA, Iozzo RV, Edelman ER. Endothelial Cells Provide Feedback Control for Vascular Remodeling Through a Mechanosensitive Autocrine TGF-Beta Signaling Pathway. Circ Res (2008) 103(3):289–97. doi: 10.1161/CIRCRESAHA.108.179465
71. Zhu Y, Xian X, Wang Z, Bi Y, Chen Q, Han X, et al. Research Progress on the Relationship Between Atherosclerosis and Inflammation. Biomolecules (2018) 8(3):80. doi: 10.3390/biom8030080
72. Souilhol C, Harmsen MC, Evans PC, Krenning G. Endothelial-Mesenchymal Transition in Atherosclerosis. Cardiovasc Res (2018) 114(4):565–77. doi: 10.1093/cvr/cvx253
73. Owens GK, Kumar MS, Wamhoff BR. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol Rev (2004) 84(3):767–801. doi: 10.1152/physrev.00041.2003
74. Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML. Smooth Muscle Cell Fate and Plasticity in Atherosclerosis. Cardiovasc Res (2018) 114(4):540–50. doi: 10.1093/cvr/cvy022
75. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular Smooth Muscle Cells in Atherosclerosis. Nat Rev Cardiol (2019) 16(12):727–44. doi: 10.1038/s41569-019-0227-9
76. Wang G, Jacquet L, Karamariti E, Xu Q. Origin and Differentiation of Vascular Smooth Muscle Cells. J Physiol (2015) 593(14):3013–30. doi: 10.1113/JP270033
77. Bennett MR, Sinha S, Owens GK. Vascular Smooth Muscle Cells in Atherosclerosis. Circ Res (2016) 118(4):692–702. doi: 10.1161/CIRCRESAHA.115.306361
78. Insull W Jr. The Pathology of Atherosclerosis: Plaque Development and Plaque Responses to Medical Treatment. Am J Med (2009) 122(1 Suppl):S3–s14. doi: 10.1016/j.amjmed.2008.10.013
79. Nakagawa K, Nakashima Y. Pathologic Intimal Thickening in Human Atherosclerosis is Formed by Extracellular Accumulation of Plasma-Derived Lipids and Dispersion of Intimal Smooth Muscle Cells. Atherosclerosis (2018) 274:235–42. doi: 10.1016/j.atherosclerosis.2018.03.039
80. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr, et al. A Definition of Advanced Types of Atherosclerotic Lesions and a Histological Classification of Atherosclerosis. A Report From the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation (1995) 92(5):1355–74. doi: 10.1161/01.cir.92.5.1355
81. Orlandi A, Bochaton-Piallat ML, Gabbiani G, Spagnoli LG. Aging, Smooth Muscle Cells and Vascular Pathobiology: Implications for Atherosclerosis. Atherosclerosis (2006) 188(2):221–30. doi: 10.1016/j.atherosclerosis.2006.01.018
82. Pirillo A, Norata GD, Catapano AL. LOX-1, Oxldl, and Atherosclerosis. Mediators Inflamm (2013) 2013:152786. doi: 10.1155/2013/152786
83. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of Smooth Muscle Cells in Vascular Calcification: Implications in Atherosclerosis and Arterial Stiffness. Cardiovasc Res (2018) 114(4):590–600. doi: 10.1093/cvr/cvy010
84. Grootaert MOJ, Moulis M, Roth L, Martinet W, Vindis C, Bennett MR, et al. Vascular Smooth Muscle Cell Death, Autophagy and Senescence in Atherosclerosis. Cardiovasc Res (2018) 114(4):622–34. doi: 10.1093/cvr/cvy007
85. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of Plaque Formation and Rupture. Circ Res (2014) 114(12):1852–66. doi: 10.1161/CIRCRESAHA.114.302721
86. Hansson GK, Hermansson A. The Immune System in Atherosclerosis. Nat Immunol (2011) 12(3):204–12. doi: 10.1038/ni.2001
87. Tabas I, Bornfeldt KE. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ Res (2016) 118(4):653–67. doi: 10.1161/CIRCRESAHA.115.306256
88. Guilliams M, Mildner A, Yona S. Developmental and Functional Heterogeneity of Monocytes. Immunity (2018) 49(4):595–613. doi: 10.1016/j.immuni.2018.10.005
89. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in Atherosclerosis: A Dynamic Balance. Nat Rev Immunol (2013) 13(10):709–21. doi: 10.1038/nri3520
90. Jinnouchi H, Guo L, Sakamoto A, Torii S, Sato Y, Cornelissen A, et al. Diversity of Macrophage Phenotypes and Responses in Atherosclerosis. Cell Mol Life Sci (2020) 77(10):1919–32. doi: 10.1007/s00018-019-03371-3
91. Marchio P, Guerra-Ojeda S, Vila JM, Aldasoro M, Victor VM, Mauricio MD. Targeting Early Atherosclerosis: A Focus on Oxidative Stress and Inflammation. Oxid Med Cell Longev (2019) 2019:8563845. doi: 10.1155/2019/8563845
92. Tabas I, Lichtman AH. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity (2017) 47(4):621–34. doi: 10.1016/j.immuni.2017.09.008
93. Barrett TJ. Macrophages in Atherosclerosis Regression. Arterioscler Thromb Vasc Biol (2020) 40(1):20–33. doi: 10.1161/ATVBAHA.119.312802
94. Bartlett B, Ludewick HP, Misra A, Lee S, Dwivedi G. Macrophages and T Cells in Atherosclerosis: A Translational Perspective. Am J Physiol Heart Circ Physiol (2019) 317(2):H375–h386. doi: 10.1152/ajpheart.00206.2019
95. Albany CJ, Trevelin SC, Giganti G, Lombardi G, Scottà C. Getting to the Heart of the Matter: The Role of Regulatory T-Cells (Tregs) in Cardiovascular Disease (CVD) and Atherosclerosis. Front Immunol (2019) 10:2795. doi: 10.3389/fimmu.2019.02795
96. Worbs T, Hammerschmidt SI, Förster R. Dendritic Cell Migration in Health and Disease. Nat Rev Immunol (2017) 17(1):30–48. doi: 10.1038/nri.2016.116
97. Zernecke A. Dendritic Cells in Atherosclerosis: Evidence in Mice and Humans. Arterioscler Thromb Vasc Biol (2015) 35(4):763–70. doi: 10.1161/ATVBAHA.114.303566
98. Paulson KE, Zhu SN, Chen M, Nurmohamed S, Jongstra-Bilen J, Cybulsky MI. Resident Intimal Dendritic Cells Accumulate Lipid and Contribute to the Initiation of Atherosclerosis. Circ Res (2010) 106(2):383–90. doi: 10.1161/CIRCRESAHA.109.210781
99. Weber C, Meiler S, Döring Y, Koch M, Drechsler M, Megens RT, et al. CCL17-Expressing Dendritic Cells Drive Atherosclerosis by Restraining Regulatory T Cell Homeostasis in Mice. J Clin Invest (2011) 121(7):2898–910. doi: 10.1172/JCI44925
100. Zeituni AE, Carrion J, Cutler CW. Porphyromonas Gingivalis-Dendritic Cell Interactions: Consequences for Coronary Artery Disease. J Oral Microbiol (2010) 2:10. doi: 10.3402/jom.v2i0.5782
101. Olsen I, Taubman MA, Singhrao SK. Porphyromonas Gingivalis Suppresses Adaptive Immunity in Periodontitis, Atherosclerosis, and Alzheimer's Disease. J Oral Microbiol (2016) 8:33029. doi: 10.3402/jom.v8.33029
102. Di Pietro M, Filardo S, Falasca F, Turriziani O, Sessa R. Infectious Agents in Atherosclerotic Cardiovascular Diseases Through Oxidative Stress. Int J Mol Sci (2017) 18(11):2459. doi: 10.3390/ijms18112459
103. Mukohda M, Stump M, Ketsawatsomkron P, Hu C, Quelle FW, Sigmund CD. Endothelial PPAR-γ Provides Vascular Protection From IL-1β-Induced Oxidative Stress. Am J Physiol Heart Circ Physiol (2016) 310(1):H39–48. doi: 10.1152/ajpheart.00490.2015
104. Tang YS, Zhao YH, Zhong Y, Li XZ, Pu JX, Luo YC, et al. Neferine Inhibits LPS-ATP-Induced Endothelial Cell Pyroptosis via Regulation of ROS/NLRP3/Caspase-1 Signaling Pathway. Inflamm Res (2019) 68(9):727–38. doi: 10.1007/s00011-019-01256-6
105. Förstermann U, Xia N, Li H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ Res (2017) 120(4):713–35. doi: 10.1161/CIRCRESAHA.116.309326
106. Sun W, Wu J, Lin L, Huang Y, Chen Q, Ji Y. Porphyromonas Gingivalis Stimulates the Release of Nitric Oxide by Inducing Expression of Inducible Nitric Oxide Synthases and Inhibiting Endothelial Nitric Oxide Synthases. J Periodontal Res (2010) 45(3):381–8. doi: 10.1111/j.1600-0765.2009.01249.x
107. Sampath C, Okoro EU, Gipson MJ, Chukkapalli SS, Farmer-Dixon CM, Gangula PR. Porphyromonas Gingivalis Infection Alters Nrf2-Phase II Enzymes and Nitric Oxide in Primary Human Aortic Endothelial Cells. J Periodontol (2021) 92(7):54–65. doi: 10.1002/JPER.20-0444
108. Gangula P, Ravella K, Chukkapalli S, Rivera M, Srinivasan S, Hale A, et al. Polybacterial Periodontal Pathogens Alter Vascular and Gut BH4/nNOS/NRF2-Phase II Enzyme Expression. PloS One (2015) 10(6):e0129885. doi: 10.1371/journal.pone.0129885
109. Wan M, Liu J, Ouyang X. Nucleotide-Binding Oligomerization Domain 1 Regulates Porphyromonas Gingivalis-Induced Vascular Cell Adhesion Molecule 1 and Intercellular Adhesion Molecule 1 Expression in Endothelial Cells Through NF-κb Pathway. J Periodontal Res (2015) 50(2):189–96. doi: 10.1111/jre.12192
110. Yu XH, Zheng XL, Tang CK. Nuclear Factor-κB Activation as a Pathological Mechanism of Lipid Metabolism and Atherosclerosis. Adv Clin Chem (2015) 70:1–30. doi: 10.1016/bs.acc.2015.03.004
111. Suh JS, Kim S, Boström KI, Wang CY, Kim RH, Park NH. Periodontitis-Induced Systemic Inflammation Exacerbates Atherosclerosis Partly via Endothelial-Mesenchymal Transition in Mice. Int J Oral Sci (2019) 11(3):21. doi: 10.1038/s41368-019-0054-1
112. Soult MC, Dobrydneva Y, Wahab KH, Britt LD, Sullivan CJ. Outer Membrane Vesicles Alter Inflammation and Coagulation Mediators. J Surg Res (2014) 192(1):134–42. doi: 10.1016/j.jss.2014.05.007
113. Li Q, Liu J, Liu W, Chu Y, Zhong J, Xie Y, et al. LOX-1 Regulates P. Gingivalis-Induced Monocyte Migration and Adhesion to Human Umbilical Vein Endothelial Cells. Front Cell Dev Biol (2020) 8:596. doi: 10.3389/fcell.2020.00596
114. Sato N, Matsumoto T, Kawaguchi S, Seya K, Matsumiya T, Ding J, et al. Porphyromonas Gingivalis Lipopolysaccharide Induces Interleukin-6 and C-C Motif Chemokine Ligand 2 Expression in Cultured Hcmec/D3 Human Brain Microvascular Endothelial Cells. Gerodontology (2021). doi: 10.1111/ger.12545
115. Wan M, Liu JR, Wu D, Chi XP, Ouyang XY. E-Selectin Expression Induced by Porphyromonas Gingivalis in Human Endothelial Cells via Nucleotide-Binding Oligomerization Domain-Like Receptors and Toll-Like Receptors. Mol Oral Microbiol (2015) 30(5):399–410. doi: 10.1111/omi.12102
116. Choi EK, Park SA, Oh WM, Kang HC, Kuramitsu HK, Kim BG, et al. Mechanisms of Porphyromonas Gingivalis-Induced Monocyte Chemoattractant Protein-1 Expression in Endothelial Cells. FEMS Immunol Med Microbiol (2005) 44(1):51–8. doi: 10.1016/j.femsim.2004.12.003
117. Harokopakis E, Albzreh MH, Martin MH, Hajishengallis G. TLR2 Transmodulates Monocyte Adhesion and Transmigration via Rac1- and PI3K-Mediated Inside-Out Signaling in Response to Porphyromonas Gingivalis Fimbriae. J Immunol (2006) 176(12):7645–56. doi: 10.4049/jimmunol.176.12.7645
118. Wu Y, Xu W, Hou J, Liu Y, Li R, Liu J, et al. Porphyromonas Gingivalis-Induced MIF Regulates Intercellular Adhesion Molecule-1 Expression in EA.Hy926 Cells and Monocyte-Endothelial Cell Adhesion Through the Receptors CD74 and CXCR4. Inflammation (2019) 42(3):874–83. doi: 10.1007/s10753-018-0942-0
119. Fang Y, Wu D, Birukov KG. Mechanosensing and Mechanoregulation of Endothelial Cell Functions. Compr Physiol (2019) 9(2):873–904. doi: 10.1002/cphy.c180020
120. Farrugia C, Stafford GP, Murdoch C. Porphyromonas Gingivalis Outer Membrane Vesicles Increase Vascular Permeability. J Dent Res (2020) 99(13):1494–501. doi: 10.1177/0022034520943187
121. Yun PL, Decarlo AA, Chapple CC, Hunter N. Functional Implication of the Hydrolysis of Platelet Endothelial Cell Adhesion Molecule 1 (CD31) by Gingipains of Porphyromonas Gingivalis for the Pathology of Periodontal Disease. Infect Immun (2005) 73(3):1386–98. doi: 10.1128/IAI.73.3.1386-1398.2005
122. Sheets SM, Potempa J, Travis J, Casiano CA, Fletcher HM. Gingipains From Porphyromonas Gingivalis W83 Induce Cell Adhesion Molecule Cleavage and Apoptosis in Endothelial Cells. Infect Immun (2005) 73(3):1543–52. doi: 10.1128/IAI.73.3.1543-1552.2005
123. Kim SR, Jeon HJ, Park HJ, Kim MK, Choi WS, Jang HO, et al. Glycyrrhetinic Acid Inhibits Porphyromonas Gingivalis Lipopolysaccharide-Induced Vascular Permeability via the Suppression of Interleukin-8. Inflamm Res (2013) 62(2):145–54. doi: 10.1007/s00011-012-0560-5
124. Fukumoto T, Matsukawa A, Yoshimura T, Edamitsu S, Ohkawara S, Takagi K, et al. IL-8 is an Essential Mediator of the Increased Delayed-Phase Vascular Permeability in LPS-Induced Rabbit Pleurisy. J Leukoc Biol (1998) 63(5):584–90. doi: 10.1002/jlb.63.5.584
125. Inomata M, Ishihara Y, Matsuyama T, Imamura T, Maruyama I, Noguchi T, et al. Degradation of Vascular Endothelial Thrombomodulin by Arginine- and Lysine-Specific Cysteine Proteases From Porphyromonas Gingivalis. J Periodontol (2009) 80(9):1511–7. doi: 10.1902/jop.2009.090114
126. Paone S, Baxter AA, Hulett MD, Poon IKH. Endothelial Cell Apoptosis and the Role of Endothelial Cell-Derived Extracellular Vesicles in the Progression of Atherosclerosis. Cell Mol Life Sci (2019) 76(6):1093–106. doi: 10.1007/s00018-018-2983-9
127. Xie M, Tang Q, Yu S, Sun J, Mei F, Zhao J, et al. Porphyromonas Gingivalis Disrupts Vascular Endothelial Homeostasis in a TLR-NF-κB Axis Dependent Manner. Int J Oral Sci (2020) 12(1):28. doi: 10.1038/s41368-020-00096-z
128. Chen Y, Zhou R, Yi Z, Li Y, Fu Y, Zhang Y, et al. Porphyromonas Gingivalis Induced Inflammatory Responses and Promoted Apoptosis in Lung Epithelial Cells Infected With H1N1 via the Bcl−2/Bax/Caspase−3 Signaling Pathway. Mol Med Rep (2018) 18(1):97–104. doi: 10.3892/mmr.2018.8983
129. Creagh EM, Conroy H, Martin SJ. Caspase-Activation Pathways in Apoptosis and Immunity. Immunol Rev (2003) 193:10–21. doi: 10.1034/j.1600-065X.2003.00048.x
130. Hirasawa M, Kurita-Ochiai T. Porphyromonas Gingivalis Induces Apoptosis and Autophagy via ER Stress in Human Umbilical Vein Endothelial Cells. Mediators Inflamm (2018) 2018:1967506. doi: 10.1155/2018/1967506
131. Komatsu T, Nagano K, Sugiura S, Hagiwara M, Tanigawa N, Abiko Y, et al. E-Selectin Mediates Porphyromonas Gingivalis Adherence to Human Endothelial Cells. Infect Immun (2012) 80(7):2570–6. doi: 10.1128/IAI.06098-11
132. Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y, et al. NLRP3 Inflammasome in Endothelial Dysfunction. Cell Death Dis (2020) 11(9):776. doi: 10.1038/s41419-020-02985-x
133. Bartruff JB, Yukna RA, Layman DL. Outer Membrane Vesicles From Porphyromonas Gingivalis Affect the Growth and Function of Cultured Human Gingival Fibroblasts and Umbilical Vein Endothelial Cells. J Periodontol (2005) 76(6):972–9. doi: 10.1902/jop.2005.76.6.972
134. Reyes L, Getachew H, Dunn WA, Progulske-Fox A. Porphyromonas Gingivalis W83 Traffics via ICAM1 in Microvascular Endothelial Cells and Alters Capillary Organization In Vivo. J Oral Microbiol (2020) 12(1):1742528. doi: 10.1080/20002297.2020.1742528
135. Bélanger M, Rodrigues PH, Dunn WA Jr, Progulske-Fox A. Autophagy: A Highway for Porphyromonas Gingivalis in Endothelial Cells. Autophagy (2006) 2(3):165–70. doi: 10.4161/auto.2828
136. Parzych KR, Klionsky DJ. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid Redox Signal (2014) 20(3):460–73. doi: 10.1089/ars.2013.5371
137. Amano A. Molecular Interaction of Porphyromonas Gingivalis With Host Cells: Implication for the Microbial Pathogenesis of Periodontal Disease. J Periodontol (2003) 74(1):90–6. doi: 10.1902/jop.2003.74.1.90
138. Song H, Bélanger M, Whitlock J, Kozarov E, Progulske-Fox A. Hemagglutinin B is Involved in the Adherence of Porphyromonas Gingivalis to Human Coronary Artery Endothelial Cells. Infect Immun (2005) 73(11):7267–73. doi: 10.1128/IAI.73.11.7267-7273.2005
139. Rodrigues PH, Bélanger M, Dunn W Jr, Progulske-Fox A. Porphyromonas Gingivalis and the Autophagic Pathway: An Innate Immune Interaction? Front Biosci (2008) 13:178–87. doi: 10.2741/2668
140. Dorn BR, Dunn WA Jr., Progulske-Fox A. Porphyromonas Gingivalis Traffics to Autophagosomes in Human Coronary Artery Endothelial Cells. Infect Immun (2001) 69(9):5698–708. doi: 10.1128/IAI.69.9.5698-5708.2001
141. Bainbridge BW, Darveau RP. Porphyromonas Gingivalis Lipopolysaccharide: An Unusual Pattern Recognition Receptor Ligand for the Innate Host Defense System. Acta Odontol Scand (2001) 59(3):131–8. doi: 10.1080/000163501750266710
142. Sheets SM, Potempa J, Travis J, Fletcher HM, Casiano CA. Gingipains From Porphyromonas Gingivalis W83 Synergistically Disrupt Endothelial Cell Adhesion and can Induce Caspase-Independent Apoptosis. Infect Immun (2006) 74(10):5667–78. doi: 10.1128/IAI.01140-05
143. Wada K, Kamisaki Y. Roles of Oral Bacteria in Cardiovascular Diseases–From Molecular Mechanisms to Clinical Cases: Involvement of Porphyromonas Gingivalis in the Development of Human Aortic Aneurysm. J Pharmacol Sci (2010) 113(2):115–9. doi: 10.1254/jphs.09R22FM
144. Cao C, Luo X, Ji X, Wang Y, Zhang Y, Zhang P, et al. Osteopontin Regulates the Proliferation of Rat Aortic Smooth Muscle Cells in Response to Gingipains Treatment. Mol Cell Probes (2017) 33:51–6. doi: 10.1016/j.mcp.2017.03.003
145. Inaba H, Tagashira M, Kanda T, Amano A. Proliferation of Smooth Muscle Cells Stimulated by Porphyromonas Gingivalis is Inhibited by Apple Polyphenol. J Periodontol (2011) 82(11):1616–22. doi: 10.1902/jop.2011.100785
146. Zhang B, Elmabsout AA, Khalaf H, Basic VT, Jayaprakash K, Kruse R, et al. The Periodontal Pathogen Porphyromonas Gingivalis Changes the Gene Expression in Vascular Smooth Muscle Cells Involving the Tgfbeta/Notch Signalling Pathway and Increased Cell Proliferation. BMC Genomics (2013) 14:770. doi: 10.1186/1471-2164-14-770
147. Mao L, Liu L, Zhang T, Wu X, Zhang T, Xu Y. MKL1 Mediates TGF-β-Induced CTGF Transcription to Promote Renal Fibrosis. J Cell Physiol (2020) 235(5):4790–803. doi: 10.1002/jcp.29356
148. Bray SJ. Notch Signalling: A Simple Pathway Becomes Complex. Nat Rev Mol Cell Biol (2006) 7(9):678–89. doi: 10.1038/nrm2009
149. Kudo C, Shin WS, Sasaki N, Harai K, Kato K, Seino H, et al. Effects of Periodontal Treatment on Carotid Intima-Media Thickness in Patients With Lifestyle-Related Diseases: Japanese Prospective Multicentre Observational Study. Odontology (2018) 106(3):316–27. doi: 10.1007/s10266-017-0331-4
150. Chistiakov DA, Orekhov AN, Bobryshev YV. Vascular Smooth Muscle Cell in Atherosclerosis. Acta Physiol (Oxf) (2015) 214(1):33–50. doi: 10.1111/apha.12466
151. Zhang B, Khalaf H, Sirsjö A, Bengtsson T. Gingipains From the Periodontal Pathogen Porphyromonas Gingivalis Play a Significant Role in Regulation of Angiopoietin 1 and Angiopoietin 2 in Human Aortic Smooth Muscle Cells. Infect Immun (2015) 83(11):4256–65. doi: 10.1128/IAI.00498-15
152. Fagiani E, Christofori G. Angiopoietins in Angiogenesis. Cancer Lett (2013) 328(1):18–26. doi: 10.1016/j.canlet.2012.08.018
153. Roth GA, Aumayr K, Giacona MB, Papapanou PN, Schmidt AM, Lalla E. Porphyromonas Gingivalis Infection and Prothrombotic Effects in Human Aortic Smooth Muscle Cells. Thromb Res (2009) 123(5):780–4. doi: 10.1016/j.thromres.2008.07.008
154. Zheng N, Zhang L, Wang B, Wang G, Liu J, Miao G, et al. Chlamydia Pneumoniae Infection Promotes Vascular Smooth Muscle Cell Migration via C-Fos/Interleukin-17C Signaling. Int J Med Microbiol (2019) 309(8):151340. doi: 10.1016/j.ijmm.2019.151340
155. Lee SJ, Lee IK, Jeon JH. Vascular Calcification-New Insights Into Its Mechanism. Int J Mol Sci (2020) 21(8):2685. doi: 10.3390/ijms21082685
156. Yang WW, Guo B, Jia WY, Jia Y. Porphyromonas Gingivalis-Derived Outer Membrane Vesicles Promote Calcification of Vascular Smooth Muscle Cells Through ERK1/2-RUNX2. FEBS Open Bio (2016) 6(12):1310–9. doi: 10.1002/2211-5463.12151
157. Clemente A, Traghella I, Mazzone A, Sbrana S, Vassalle C. Vascular and Valvular Calcification Biomarkers. Adv Clin Chem (2020) 95:73–103. doi: 10.1016/bs.acc.2019.08.002
158. Kim JM, Yang YS, Park KH, Oh H, Greenblatt MB, Shim JH. The ERK MAPK Pathway is Essential for Skeletal Development and Homeostasis. Int J Mol Sci (2019) 20(8):1803. doi: 10.3390/ijms20081803
159. Chen TC, Lin CT, Chien SJ, Chang SF, Chen CN. Regulation of Calcification in Human Aortic Smooth Muscle Cells Infected With High-Glucose-Treated Porphyromonas Gingivalis. J Cell Physiol (2018) 233(6):4759–69. doi: 10.1002/jcp.26268
160. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, et al. Smooth Muscle Cell Phenotypic Transition Associated With Calcification: Upregulation of Cbfa1 and Downregulation of Smooth Muscle Lineage Markers. Circ Res (2001) 89(12):1147–54. doi: 10.1161/hh2401.101070
161. Liu G, Deng J, Zhang Q, Song W, Chen S, Lou X, et al. Porphyromonas Gingivalis Lipopolysaccharide Stimulation of Vascular Smooth Muscle Cells Activates Proliferation and Calcification. J Periodontol (2016) 87(7):828–36. doi: 10.1902/jop.2016.150602
162. Proudfoot D, Skepper JN, Hegyi L, Bennett MR, Shanahan CM, Weissberg PL. Apoptosis Regulates Human Vascular Calcification In Vitro: Evidence for Initiation of Vascular Calcification by Apoptotic Bodies. Circ Res (2000) 87(11):1055–62. doi: 10.1161/01.RES.87.11.1055
163. Park HJ, Kim Y, Kim MK, Park HR, Kim HJ, Bae SK, et al. Infection of Porphyromonas Gingivalis Increases Phosphate-Induced Calcification of Vascular Smooth Muscle Cells. Cells (2020) 9(12):2694. doi: 10.3390/cells9122694
164. Lencel P, Delplace S, Pilet P, Leterme D, Miellot F, Sourice S, et al. Cell-Specific Effects of TNF-α and IL-1β on Alkaline Phosphatase: Implication for Syndesmophyte Formation and Vascular Calcification. Lab Invest (2011) 91(10):1434–42. doi: 10.1038/labinvest.2011.83
165. Barrett H, O'Keeffe M, Kavanagh E, Walsh M, O'Connor EM. Is Matrix Gla Protein Associated With Vascular Calcification? A Systematic Review. Nutrients (2018) 10(4):415. doi: 10.3390/nu10040415
166. Roumeliotis S, Dounousi E, Eleftheriadis T, Liakopoulos V. Association of the Inactive Circulating Matrix Gla Protein With Vitamin K Intake, Calcification, Mortality, and Cardiovascular Disease: A Review. Int J Mol Sci (2019) 20(3):628. doi: 10.3390/ijms20030628
167. Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE, et al. Smooth Muscle Cells Contribute the Majority of Foam Cells in Apoe (Apolipoprotein E)-Deficient Mouse Atherosclerosis. Arterioscler Thromb Vasc Biol (2019) 39(5):876–87. doi: 10.1161/ATVBAHA.119.312434
168. Wang T, Ouyang H, Zhou H, Xia L, Wang X, Wang T. Pro−Atherogenic Activation of A7r5 Cells Induced by the OxLDL/β2GPI/Anti−β2GPI Complex. Int J Mol Med (2018) 42(4):1955–66. doi: 10.3892/ijmm.2018.3805
169. Li H, Freeman MW, Libby P. Regulation of Smooth Muscle Cell Scavenger Receptor Expression In Vivo by Atherogenic Diets and In Vitro by Cytokines. J Clin Invest (1995) 95(1):122–33. doi: 10.1172/JCI117628
170. Pryma CS, Ortega C, Dubland JA, Francis GA. Pathways of Smooth Muscle Foam Cell Formation in Atherosclerosis. Curr Opin Lipidol (2019) 30(2):117–24. doi: 10.1097/MOL.0000000000000574
171. Hajishengallis G, Wang M, Liang S, Shakhatreh MA, James D, Nishiyama S, et al. Subversion of Innate Immunity by Periodontopathic Bacteria via Exploitation of Complement Receptor-3. Adv Exp Med Biol (2008) 632:203–19. doi: 10.1007/978-0-387-78952-1_15
172. Vorup-Jensen T, Jensen RK. Structural Immunology of Complement Receptors 3 and 4. Front Immunol (2018) 9:2716. doi: 10.3389/fimmu.2018.02716
173. Hajishengallis G, Shakhatreh MA, Wang M, Liang S. Complement Receptor 3 Blockade Promotes IL-12-Mediated Clearance of Porphyromonas Gingivalis and Negates Its Virulence In Vivo. J Immunol (2007) 179(4):2359–67. doi: 10.4049/jimmunol.179.4.2359
174. Hajishengallis G, Harokopakis E. Porphyromonas Gingivalis Interactions With Complement Receptor 3 (CR3): Innate Immunity or Immune Evasion? Front Biosci (2007) 12:4547–57. doi: 10.2741/2409
175. Hajishengallis G, Wang M, Harokopakis E, Triantafilou M, Triantafilou K. Porphyromonas Gingivalis Fimbriae Proactively Modulate Beta2 Integrin Adhesive Activity and Promote Binding to and Internalization by Macrophages. Infect Immun (2006) 74(10):5658–66. doi: 10.1128/IAI.00784-06
176. Wolf D, Ley K. Immunity and Inflammation in Atherosclerosis. Circ Res (2019) 124(2):315–27. doi: 10.1161/CIRCRESAHA.118.313591
177. Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T, Ando T. Activation of the NLRP3 Inflammasome in Porphyromonas Gingivalis-Accelerated Atherosclerosis. Pathog Dis (2015) 73(4):ftv011. doi: 10.1093/femspd/ftv011
178. Wang T, He C. TNF-α and IL-6: The Link Between Immune and Bone System. Curr Drug Targets (2020) 21(3):213–27. doi: 10.2174/1389450120666190821161259
179. Yu R, Li Q, Feng Z, Cai L, Xu Q. M6a Reader YTHDF2 Regulates LPS-Induced Inflammatory Response. Int J Mol Sci (2019) 20(6):1323. doi: 10.3390/ijms20061323
180. Yang X, Pan Y, Xu X, Tong T, Yu S, Zhao Y, et al. Sialidase Deficiency in Porphyromonas Gingivalis Increases IL-12 Secretion in Stimulated Macrophages Through Regulation of CR3, IncRNA GAS5 and MiR-21. Front Cell Infect Microbiol (2018) 8:100. doi: 10.3389/fcimb.2018.00100
181. Nie R, Wu Z, Ni J, Zeng F, Yu W, Zhang Y, et al. Porphyromonas Gingivalis Infection Induces Amyloid-β Accumulation in Monocytes/Macrophages. J Alzheimers Dis (2019) 72(2):479–94. doi: 10.3233/JAD-190298
182. Holden JA, Attard TJ, Laughton KM, Mansell A, O'Brien-Simpson NM, Reynolds EC. Porphyromonas Gingivalis Lipopolysaccharide Weakly Activates M1 and M2 Polarized Mouse Macrophages But Induces Inflammatory Cytokines. Infect Immun (2014) 82(10):4190–203. doi: 10.1128/IAI.02325-14
183. Cecil JD, O'Brien-Simpson NM, Lenzo JC, Holden JA, Singleton W, Perez-Gonzalez A, et al. Outer Membrane Vesicles Prime and Activate Macrophage Inflammasomes and Cytokine Secretion In Vitro and In Vivo. Front Immunol (2017) 8:1017. doi: 10.3389/fimmu.2017.01017
184. Pudla M, Srisaowakarn C, Utaisincharoen P. NLRP12 Negatively Modulates Inducible Nitric Oxide Synthase (iNOS) Expression and Tumor Necrosis Factor-α Production in Porphyromonas Gingivalis LPS-Treated Mouse Macrophage Cell Line (RAW264.7). Inflamm Res (2019) 68(10):841–4. doi: 10.1007/s00011-019-01267-3
185. Castro SA, Collighan R, Lambert PA, Dias IH, Chauhan P, Bland CE, et al. Porphyromonas Gingivalis Gingipains Cause Defective Macrophage Migration Towards Apoptotic Cells and Inhibit Phagocytosis of Primary Apoptotic Neutrophils. Cell Death Dis (2017) 8(3):e2644. doi: 10.1038/cddis.2016.481
186. Miyakawa H, Honma K, Qi M, Kuramitsu HK. Interaction of Porphyromonas Gingivalis With Low-Density Lipoproteins: Implications for a Role for Periodontitis in Atherosclerosis. J Periodontal Res (2004) 39(1):1–9. doi: 10.1111/j.1600-0765.2004.00697.x
187. Qi M, Miyakawa H, Kuramitsu HK. Porphyromonas Gingivalis Induces Murine Macrophage Foam Cell Formation. Microb Pathog (2003) 35(6):259–67. doi: 10.1016/j.micpath.2003.07.002
188. Kattoor AJ, Kanuri SH, Mehta JL. Role of Ox-LDL and LOX-1 in Atherogenesis. Curr Med Chem (2019) 26(9):1693–700. doi: 10.2174/0929867325666180508100950
189. Oörni K, Pentikäinen MO, Ala-Korpela M, Kovanen PT. Aggregation, Fusion, and Vesicle Formation of Modified Low Density Lipoprotein Particles: Molecular Mechanisms and Effects on Matrix Interactions. J Lipid Res (2000) 41(11):1703–14. doi: 10.1016/S0022-2275(20)31964-7
190. Edelstein C, Nakajima K, Pfaffinger D, Scanu AM. Oxidative Events Cause Degradation of Apob-100 But Not of Apo[a] and Facilitate Enzymatic Cleavage of Both Proteins. J Lipid Res (2001) 42(10):1664–70. doi: 10.1016/S0022-2275(20)32221-5
191. Fleetwood AJ, Lee MKS, Singleton W, Achuthan A, Lee MC, O'Brien-Simpson NM, et al. Metabolic Remodeling, Inflammasome Activation, and Pyroptosis in Macrophages Stimulated by Porphyromonas Gingivalis and Its Outer Membrane Vesicles. Front Cell Infect Microbiol (2017) 7:351. doi: 10.3389/fcimb.2017.00351
192. Liu F, Wang Y, Xu J, Liu F, Hu R, Deng H. Effects of Porphyromonas Gingivalis Lipopolysaccharide on the Expression of Key Genes Involved in Cholesterol Metabolism in Macrophages. Arch Med Sci (2016) 12(5):959–67. doi: 10.5114/aoms.2016.61909
193. Yu XH, Fu YC, Zhang DW, Yin K, Tang CK. Foam Cells in Atherosclerosis. Clin Chim Acta (2013) 424:245–52. doi: 10.1016/j.cca.2013.06.006
194. Frambach S, de Haas R, Smeitink JAM, Rongen GA, Russel FGM, Schirris TJJ. Brothers in Arms: ABCA1- and ABCG1-Mediated Cholesterol Efflux as Promising Targets in Cardiovascular Disease Treatment. Pharmacol Rev (2020) 72(1):152–90. doi: 10.1124/pr.119.017897
195. Li XY, Wang C, Xiang XR, Chen FC, Yang CM, Wu J. Porphyromonas Gingivalis Lipopolysaccharide Increases Lipid Accumulation by Affecting CD36 and ATP-Binding Cassette Transporter A1 in Macrophages. Oncol Rep (2013) 30(3):1329–36. doi: 10.3892/or.2013.2600
196. Takeda K, Akira S. TLR Signaling Pathways. Semin Immunol (2004) 16(1):3–9. doi: 10.1016/j.smim.2003.10.003
197. Yang J, Wu J, Zhang R, Yao M, Liu Y, Miao L, et al. Porphyromonas Gingivalis Oral Infection Promote T Helper 17/Treg Imbalance in the Development of Atherosclerosis. J Dent Sci (2017) 12(1):60–9. doi: 10.1016/j.jds.2016.10.003
198. Yang J, Wu J, Liu Y, Huang J, Lu Z, Xie L, et al. Porphyromonas Gingivalis Infection Reduces Regulatory T Cells in Infected Atherosclerosis Patients. PloS One (2014) 9(1):e86599. doi: 10.1371/journal.pone.0086599
199. Cheng WC, van Asten SD, Burns LA, Evans HG, Walter GJ, Hashim A, et al. Periodontitis-Associated Pathogens P. Gingivalis and a. Actinomycetemcomitans Activate Human CD14(+) Monocytes Leading to Enhanced Th17/IL-17 Responses. Eur J Immunol (2016) 46(9):2211–21. doi: 10.1002/eji.201545871
200. Zhang L, Gao L, Xu C, Li X, Wang P, Zhang C, et al. Porphyromonas Gingivalis Lipopolysaccharide Promotes T- Helper 17 Cell Differentiation From Human CD4(+) Naïve T Cells via Toll-Like Receptor-2 In Vitro. Arch Oral Biol (2019) 107:104483. doi: 10.1016/j.archoralbio.2019.104483
201. Cai Y, Kobayashi R, Hashizume-Takizawa T, Kurita-Ochiai T. Porphyromonas Gingivalis Infection Enhances Th17 Responses for Development of Atherosclerosis. Arch Oral Biol (2014) 59(11):1183–91. doi: 10.1016/j.archoralbio.2014.07.012
202. Glowczyk I, Wong A, Potempa B, Babyak O, Lech M, Lamont RJ, et al. Inactive Gingipains From P. Gingivalis Selectively Skews T Cells Toward a Th17 Phenotype in an IL-6 Dependent Manner. Front Cell Infect Microbiol (2017) 7:140. doi: 10.3389/fcimb.2017.00140
203. Rajendran M, Looney S, Singh N, Elashiry M, Meghil MM, El-Awady AR, et al. Systemic Antibiotic Therapy Reduces Circulating Inflammatory Dendritic Cells and Treg-Th17 Plasticity in Periodontitis. J Immunol (2019) 202(9):2690–9. doi: 10.4049/jimmunol.1900046
204. Cafferata EA, Jerez A, Vernal R, Monasterio G, Pandis N, Faggion CM Jr.. The Therapeutic Potential of Regulatory T Lymphocytes in Periodontitis: A Systematic Review. J Periodontal Res (2019) 54(3):207–17. doi: 10.1111/jre.12629
205. Cen S, Wang P, Xie Z, Yang R, Li J, Liu Z, et al. Autophagy Enhances Mesenchymal Stem Cell-Mediated CD4(+) T Cell Migration and Differentiation Through CXCL8 and TGF-β1. Stem Cell Res Ther (2019) 10(1):265. doi: 10.1186/s13287-019-1380-0
206. Turner JA, Stephen-Victor E, Wang S, Rivas MN, Abdel-Gadir A, Harb H, et al. Regulatory T Cell-Derived TGF-β1 Controls Multiple Checkpoints Governing Allergy and Autoimmunity. Immunity (2020) 53(6):1202–1214.e6. doi: 10.1016/j.immuni.2020.10.002
207. Ip WKE, Hoshi N, Shouval DS, Snapper S, Medzhitov R. Anti-Inflammatory Effect of IL-10 Mediated by Metabolic Reprogramming of Macrophages. Science (2017) 356(6337):513–9. doi: 10.1126/science.aal3535
208. Gao Q, Jiang Y, Ma T, Zhu F, Gao F, Zhang P, et al. A Critical Function of Th17 Proinflammatory Cells in the Development of Atherosclerotic Plaque in Mice. J Immunol (2010) 185(10):5820–7. doi: 10.4049/jimmunol.1000116
209. He X, Liang B, Gu N. Th17/Treg Imbalance and Atherosclerosis. Dis Markers (2020) 2020:8821029. doi: 10.1155/2020/8821029
210. Jauregui CE, Wang Q, Wright CJ, Takeuchi H, Uriarte SM, Lamont RJ. Suppression of T-Cell Chemokines by Porphyromonas Gingivalis. Infect Immun (2013) 81(7):2288–95. doi: 10.1128/IAI.00264-13
211. Kitamura Y, Matono S, Aida Y, Hirofuji T, Maeda K. Gingipains in the Culture Supernatant of Porphyromonas Gingivalis Cleave CD4 and CD8 on Human T Cells. J Periodontal Res (2002) 37(6):464–8. doi: 10.1034/j.1600-0765.2002.01364.x
212. Jeong E, Kim K, Kim JH, Cha GS, Kim SJ, Kang HS, et al. Porphyromonas Gingivalis HSP60 Peptides Have Distinct Roles in the Development of Atherosclerosis. Mol Immunol (2015) 63(2):489–96. doi: 10.1016/j.molimm.2014.10.004
213. Khalaf H, Bengtsson T. Altered T-Cell Responses by the Periodontal Pathogen Porphyromonas Gingivalis. PloS One (2012) 7(9):e45192. doi: 10.1371/journal.pone.0045192
214. Yun PL, Decarlo AA, Chapple CC, Collyer CA, Hunter N. Binding of Porphyromonas Gingivalis Gingipains to Human CD4(+) T Cells Preferentially Down-Regulates Surface CD2 and CD4 With Little Affect on Co-Stimulatory Molecule Expression. Microb Pathog (2005) 38(2-3):85–96. doi: 10.1016/j.micpath.2005.01.001
215. Koltsova EK, Garcia Z, Chodaczek G, Landau M, McArdle S, Scott SR, et al. Dynamic T Cell-APC Interactions Sustain Chronic Inflammation in Atherosclerosis. J Clin Invest (2012) 122(9):3114–26. doi: 10.1172/JCI61758
216. Pérez-Chaparro PJ, Lafaurie GI, Gracieux P, Meuric V, Tamanai-Shacoori Z, Castellanos JE, et al. Distribution of Porphyromonas Gingivalis Fima Genotypes in Isolates From Subgingival Plaque and Blood Sample During Bacteremia. Biomedica (2009) 29(2):298–306
217. Tonetti MS, D'Aiuto F, Nibali L, Donald A, Storry C, Parkar M, et al. Treatment of Periodontitis and Endothelial Function. N Engl J Med (2007) 356(9):911–20. doi: 10.1056/NEJMoa063186
218. Lafaurie GI, Mayorga-Fayad I, Torres MF, Castillo DM, Aya MR, Barón A, et al. Periodontopathic Microorganisms in Peripheric Blood After Scaling and Root Planing. J Clin Periodontol (2007) 34(10):873–9. doi: 10.1111/j.1600-051X.2007.01125.x
219. Meghil MM, Cutler CW. Oral Microbes and Mucosal Dendritic Cells, "Spark and Flame" of Local and Distant Inflammatory Diseases. Int J Mol Sci (2020) 21(5):1643. doi: 10.3390/ijms21051643
Keywords: Porphyromonas gingivalis, atherosclerosis, endothelial dysfunction, foam cell, T cell
Citation: Zhang J, Xie M, Huang X, Chen G, Yin Y, Lu X, Feng G, Yu R and Chen L (2021) The Effects of Porphyromonas gingivalis on Atherosclerosis-Related Cells. Front. Immunol. 12:766560. doi: 10.3389/fimmu.2021.766560
Received: 29 August 2021; Accepted: 30 November 2021;
Published: 23 December 2021.
Edited by:
Craig Murdoch, The University of Sheffield, United KingdomReviewed by:
Hiroaki Inaba, Okayama University, JapanTomomi Hashizume-Takizawa, Nihon University, Japan
Copyright © 2021 Zhang, Xie, Huang, Chen, Yin, Lu, Feng, Yu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lili Chen, Y2hlbmxpbGkxMDMwQGh1c3QuZWR1LmNu; Ran Yu, eXJoYnljQGh1c3QuZWR1LmNu
†These authors have contributed equally to this work