
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 19 November 2021
Sec. B Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.765044
This article is part of the Research Topic Insights into Regulatory B Cells View all 11 articles
 Ziwen Zhong1,2†
Ziwen Zhong1,2† Ke Nan1†Meilin Weng1†Ying Yue1Wenchang Zhou1Zhiqiang Wang3Yiwei Chu3,4
Ke Nan1†Meilin Weng1†Ying Yue1Wenchang Zhou1Zhiqiang Wang3Yiwei Chu3,4 Ronghua Liu4*
Ronghua Liu4* Changhong Miao1*
Changhong Miao1*B cells are well known as key mediators of humoral immune responses via the production of antibodies. Immunoglobulin A (IgA) is the most abundantly produced antibody isotype and provides the first line of immune protection at mucosal surfaces. However, IgA has long been a divisive molecule with respect to tumor progression. IgA exerts anti- or pro-tumor effect in different tumor types. In this review, we summarize emerging evidence regarding the production and effects of IgA and IgA+ cells in the tumor microenvironment (TME). Moreover, we discuss that the TME cytokines, host diet, microbiome, and metabolites play a pivotal role in controlling the class-switch recombination (CSR) of IgA. The analysis of intratumoral Ig repertoires and determination of metabolites that influence CSR may help establish novel therapeutic targets for the treatment of cancers.
Immunoglobulin A (IgA) is the most abundant antibody class present on mucosal surfaces. It plays a key role at these surfaces, which are continuously exposed to antigens, food, and commensal microorganisms (1). IgA-producing plasma cells (IgA+ PCs) are derived from B cells and undergo antibody class-switch recombination (CSR). Emerging evidence shows significant tumor infiltration by B cells. However, the role of B cells and their antibodies in tumors has remained elusive, perhaps due to their phenotype, antibody isotype and production, localization, and the tumor type and tumor microenvironment (TME) (2–5). Multiple lines of evidence now leave no doubt that B cells undergo clonal proliferation, selection for high-affinity antibodies, and isotype switching within tumor-associated tertiary lymphoid structures (TLS) and in less organized structures, ultimately converting into effector/memory B cells and antibody-producing PCs (6–9). However, under certain conditions, tumor-infiltrating B cells, PCs, and their antibodies may exert prominent immunosuppressive effects. Studies have provided well-founded evidence of discrete subsets of immunosuppressive or regulatory B cells (Bregs) in both mice and humans. Their function depends on immunosuppressive factors, such as IL-10, PD-L1, and/or TGF-β (10–13). Moreover, IgA has recently been recognized as a key biomarker of immunosuppressive B cells. However, the effect of IgA+ cells and their antibodies on cancer development remains controversial (1, 11, 14–16). In this review, we summarize recent findings related to the production and effect of IgA and IgA+ cells in the TME and discuss their potential roles in novel cancer treatment strategies.
Approximately 80% of all human PCs are located in organized gut-associated lymphoid tissue (GALT). In GALT, IgA is produced at a constant synthesis rate of 50 mg/kg/day, which is more than the total synthesis rate of other Ig isotypes, indicating that continuous secretion of IgA requires an enormous amount of energy in mammalian species (17). In addition, IgA is present in milk and bronchial secretions (18). Serum IgA is predominantly monomeric in humans and is produced by local PCs of the bone marrow, spleen, and lymph nodes (1). IgA at mucosal surfaces is generally produced by local PCs as dimeric molecules, although small amounts of monomers, trimers, tetramers, or polymers can also be present. Dimeric IgA interacts with the polymeric Ig receptor (pIgR), an antibody transporter expressed on the basolateral surface of intestinal epithelial cells (IECs). A secretory IgA (sIgA) complex is formed after a secretory component (SC) of pIgR binds to IgA through a joining (J) chain. SC is a hydrophilic and highly (N- and O-linked) glycosylated negatively charged molecule that protects sIgA from degradation in luminal secretions (19).
It has been confirmed that various structural compartments differ in the mode of IgA induction. Mucosal antigens initiate IgA production through both T cell-dependent (TD) and T cell-independent (TI) reactions (20–22). Most antigens produce IgA by activating follicular B cells in the germinal center of Peyer’s patches (PPs) and mesenteric lymph nodes (MLNs). This TD pathway involves a cognate interaction between follicular B cells and antigen-activated CD4+ T cells, including TH2 cells, regulatory T (Treg) cells, and T follicular helper (TFH) cells (22, 23). Together with follicular dendritic cells (FDCs), this interaction fosters proliferation, differentiation, somatic hypermutation (SHM), and IgA CSR of B cells in the germinal center. Ultimately, this TD pathway leads to the emergence of long-lived memory B cells and PCs that release high-affinity IgA antibodies in the lamina propria (LP) (20). However, TD antibody responses require 5-7 days, which can be too much of a delay to control pathogens. To compensate for this limitation, a TI reaction occurs in PPs, MLNs, isolated lymphoid follicles (ILFs), and LP. Commensals trigger multiple Toll-like receptors (TLRs), which stimulate FDCs, DCs, and stromal cells to release BAFF, APRIL, TGF-β, and other IgA-inducing cytokines, thereby triggering the induction of TI IgA CSR and production in PPs, MLNs, ILFs, and LP (24, 25). Class-switched B cells emerging from these pathways ultimately release both low- and high-affinity IgA antibodies in LP. Moreover, it is likely that the generation of IgA+ PCs depends on the nature of IgA and the initial location of B cells. A precise combination of different leukocyte and mesenchymal cell types may also influence the generation of IgA+ PCs (23) (Figure 1).
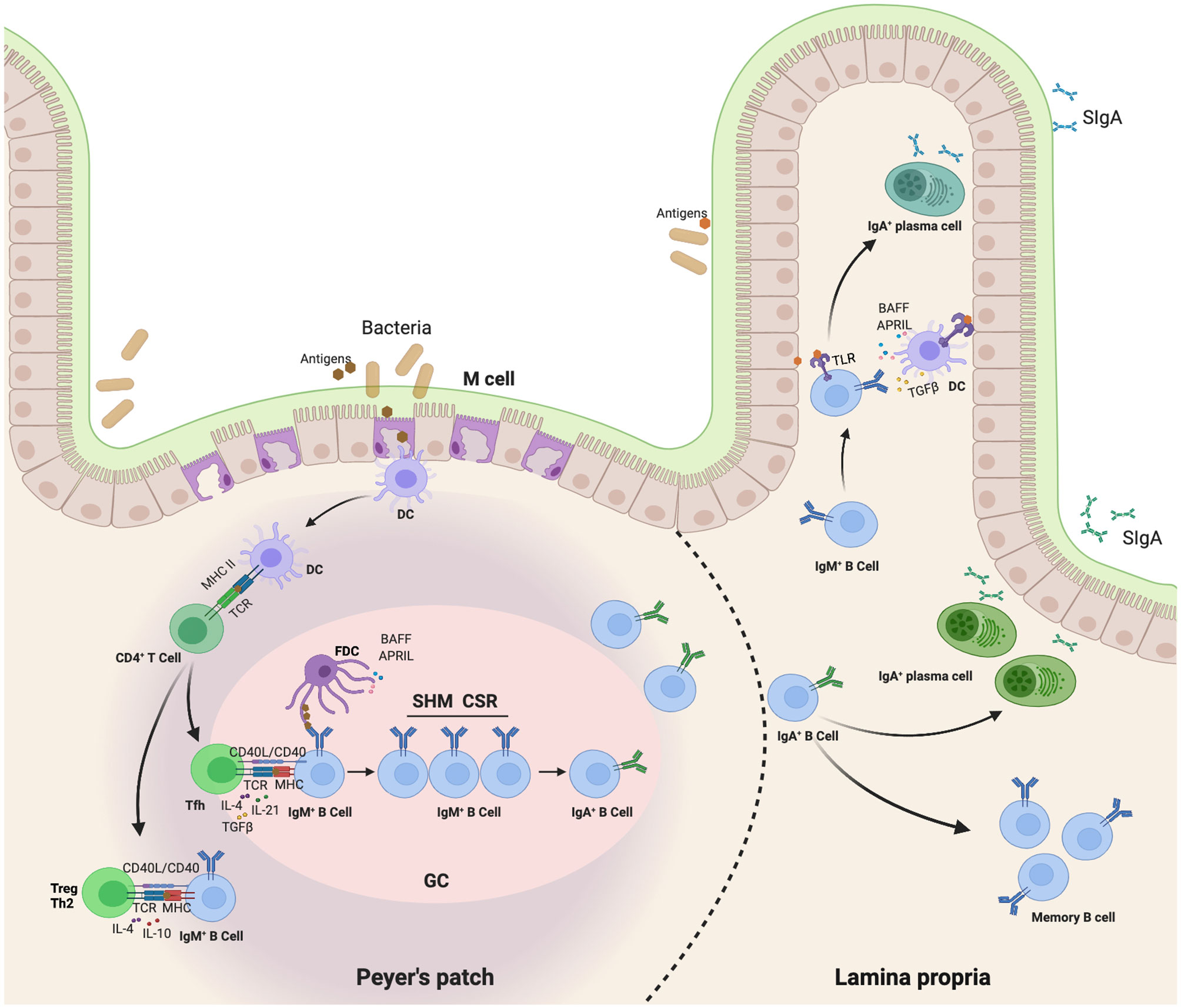
Figure 1 Map of IgA class switching through both TD and TI reaction in the gut. This TD pathway involves a cognate interaction of follicular B cells with antigen-activated CD4+ T cells, including TH2 cells, regulatory T (Treg) cells, and T follicular helper (TFH) cells (22, 23). First, Dendritic cells (DCs) in the subepithelial dome (SED) of the PP capture and present antigen to follicular CD4+ T cells, thereby inducing them to differentiate into effector T cells releasing IgA-inducing cytokines. These T cells also interact with antigen-specific IgM+IgD+ naive B cells. Together with follicular dendritic cells (FDCs), this interaction fosters a proliferation, differentiation, somatic hypermutation (SHM) and IgA class-switch recombination (CSR) of B cells in germinal center. Ultimately, TD pathway leads to the emergence of long-lived memory B cells and plasma cells that release high-affinity IgA antibodies in the lamina propria (20). In addition, commensals trigger multiple Toll-like receptors (TLRs) to stimulate FDC, DC and stromal cells release of BAFF, APRIL, TGFβ and other IgA-inducing cytokines, thereby triggering direct IgA CSR in lamina propria IgM+ B cells in a T-cell-independent manner (24, 25).
The tumor-promoting effects of B cells and PCs are generally associated with the presence of heterogeneous cell populations called Bregs and immunosuppressive plasma cells (ISPCs), which may accelerate tumor progression through various mechanisms. Multiple studies have shown that Bregs suppress tumor immunity through immunosuppressive factors, such as IL-10, TGF-β, PD-L1, FASL, IL-35, and Tim1 (11, 14, 26–28). This type of tumor immunity suppression is activated by signals from CD40, TLRs, and B cell receptors (BCRs) (10, 29). Furthermore, Bregs reportedly cause cytotoxic T lymphocyte (CTL) suppression by inducing M2 macrophage polarization (30, 31) and Tregs in squamous carcinomas (SCCs) and pancreatic ductal adenocarcinoma (PDAC) (32).
However, no specific transcriptional marker has been identified that exclusively defines the Bregs phenotype in mouse or humans. Numerous studies have now demonstrated that Bregs mainly suppress inflammatory responses via IL-10-depengdent and -independent mechanisms (33). Subsets that are enriched for IL-10-producing B cells include transitional-2 marginal zone precursor (T2-MZP) B cells, peritoneal B-1 B cells, and antibody-producing B cell subsets in mice, as well as antibody-producing and immature B cell subsets in humans. In a recent series of studies, recirculating IgA+ regulatory B cells have been shown to suppress inflammation via expression of IL-10 (34, 35). Shalapour et al. discovered in 2015 that IgA+ PCs suppress anti-tumor immunity. These immunosuppressive B cells are IgA-producing PCs that express PD-L1, IL-10, and Fas-L (11). Most tumoral CD19+ B cells in oxaliplatin-treated pancreatic cancer are IgA-positive, and successful eradication of tumors requires removal of IgA+ cells (11). In a murine study of hepatocellular carcinoma, IgA+ cells expressing high levels of PD-L1, IL-10, and TGF-β were shown to directly repress CD8+ T cell proliferation and activation (14). IgA+ PCs within prostate tumors induce CD8+ T cell exhaustion and suppress anti-tumor CTL responses through PD-L1 and IL-10, either of which can induce anergy or exhaustion (36, 37). In general, IgA+ cells promote the expansion of Treg cell populations, whereas Treg cells produce TGF-β, which mediates the isotype class switch to IgA (9, 38). This regulatory loop induces a state of relative immune suppression and may further promote tumor progression in at least some cancer types or subtypes (Figure 2).
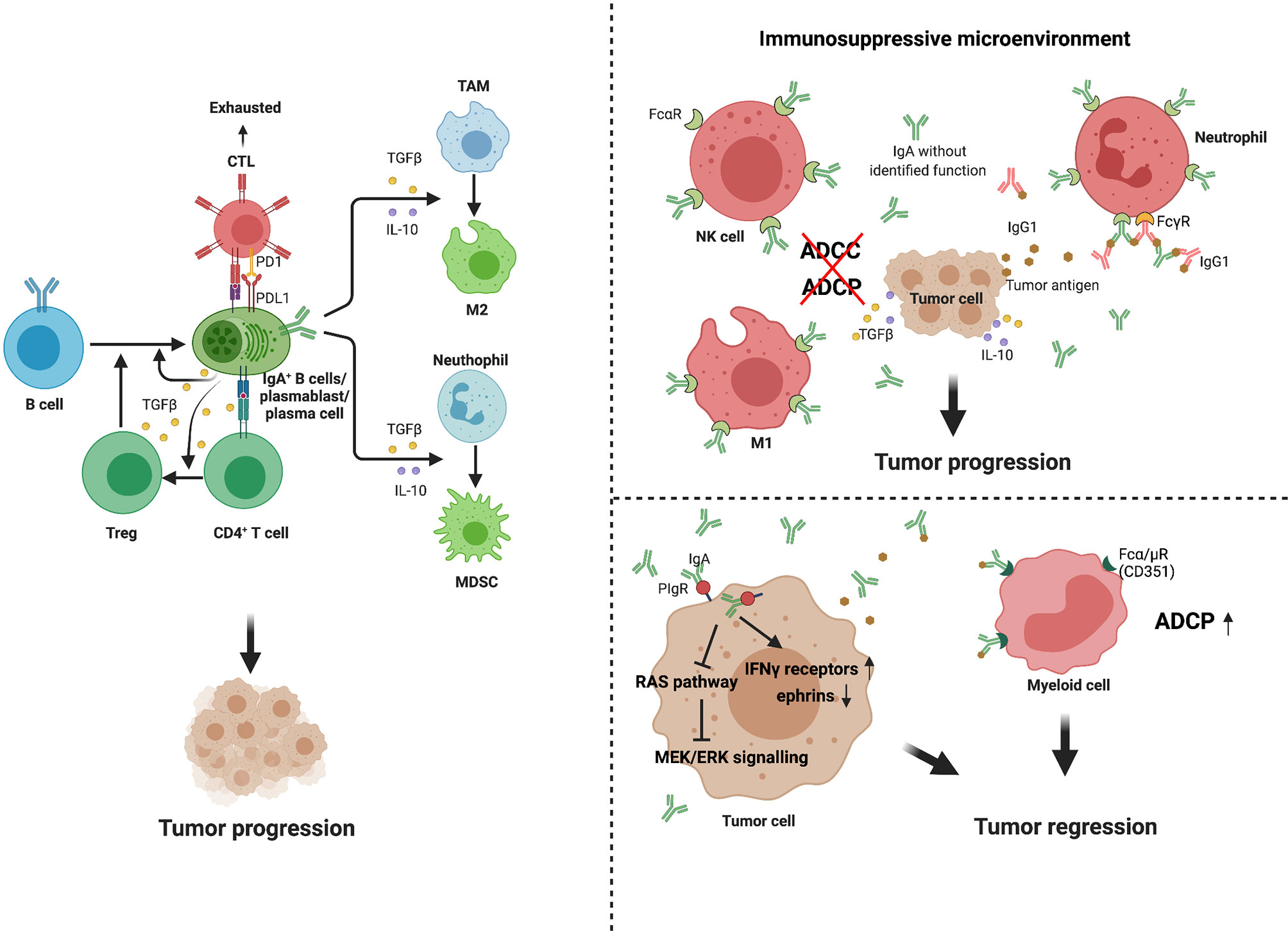
Figure 2 Multiple functions of IgA+ cells and IgA in tumor. IgA+ B cells and plasma cells may promote tumor progression through several mechanisms. First, they can release high level of PD-L1, IL-10 and TGFβ, which directly induce CD8+T cell exhaustion and suppress anti-tumor CTL responses (14, 36, 37). Second, the IgA+ cells promote expansion of Treg cell populations, while Treg cells produce TGFβ, which mediates isotype class switch to IgA (9, 38). In addition, these immunosuppressive cytokines secreted by IgA+ cells, such as IL-10 and TGFβ, can promote immunosuppressive phenotypes in macrophage and myeloid cells. IgA performs dual effects on tumor immunity. In immunosuppressive environment, due to the binding of IgA without identified function and Fcα receptors (FcαR) expressed on macrophages, natural killer (NK) cells and neutrophils, high levels of production of IgA block ADCC and ADCP (39, 40). Meanwhile, such antibodies can form immune complexes with tumor or non-tumor antigens, promoting chronic inflammation, tissue remodeling (41–43). However, in some specific tumor types, IgA transcytosis induced broad transcriptional changes in inflammatory pathways in tumor cells, contributing to hindering malignant progression. In further support of antibody-dependent cellular phagocytosis, tumor-derived IgA redirection of Fcα/µ+(CD351) myeloid cells against extracellular oncogenic drivers, which causes tumor cell death (15).
Although there is more than sufficient evidence indicating that IgA+ B cells and PCs may exert prominent immunosuppressive effects, the antitumor effect of IgA+ cells has also been reported; this may have resulted from the dual effect of IgA. The expression of IgA by tumor-infiltrating PCs has been linked to poor outcomes in colorectal cancer (CRC) (44) and melanoma (45, 46). Elevated levels of intratumoral IgA have been shown to be associated with poor prognosis and shorter survival in patients with bladder cancer (47). A recent meta-analysis indicated a strong association between IgA CSR and solid cancer diagnosis, and confirmed its immunosuppressive and pro-tumorigenic roles (48). The aforementioned study showed that, compared with healthy individuals, patients with solid malignancies had significantly higher serum IgA levels, which further increased in patients with advanced cancer. However, Biswas et al. demonstrated that tumor-antigen-specific and tumor-antigen-independent IgA responses antagonize the growth of ovarian cancer by governing coordinated tumor cell, T cell, and B cell responses (15). These results suggest that IgA may play various roles in different tumor types or in the tumor immune microenvironment (Table 1).

Table 1 Mechanism and prognostic impact of IgA in cancers.
A systematic review and meta-analysis revealed that a 2-fold increase in the standardized mean difference (SMD) of the IgA level occurred in advanced cancer compared with early carcinoma, further supporting the possibility that serum IgA levels may correlate with immune escape and tumor burden (48). A high intratumoral proportion of the IgA isotype has a close relationship with a negative prognosis in the KRAS-mutant subtype of lung adenocarcinoma (4). TCGA RNA-seq data of human melanoma indicate that high proportions of IgA, IgD, and IgE are related to a poor prognosis (2).
The tumor-promoting mechanism of IgA can be summarized as follows. Firstly, it is a tumor immunosuppressive environment and not a tumor antigen that promotes IgA production. IgA with unfocused specificity is ineffective in mediating antitumor responses, for which it cannot facilitate antigen presentation or mediate antibody-dependent cellular cytotoxicity (ADCC) and phagocytosis (ADCP) of tumor cells (39, 40). On the other hand, such antibodies can form immune complexes with tumor or non-tumor antigens, promoting chronic inflammation and tissue remodeling, and thus directly or indirectly favoring the myeloid-derived suppressor cell phenotype (41–43) (Figure 2). Another possible role of mucosal IgA in tumor immunity depends on its dynamic relationship with environmental factors, such as microbiota or diet (56). Tissue-specific immunoregulation may determine the ability of different tumors to exploit the IgA–microbiota axis to facilitate immune escape (56, 57). Furthermore, IgA fails to activate the complement system but mediates the regulatory effects of its main inducer, TGF-β (58). In particular, monomeric IgA exerts inhibitory effects on many immune cell subsets by activating the myeloid-cell-specific type I Fc receptor for IgA (FcαRI; also known as CD89) (59, 60) and inducing IL-10 production (61), as well as by regulating proinflammatory cytokines (62).
IgA also plays an anti-tumor role in some specific cancers. Recent studies have demonstrated that tumor-derived IgA abrogates tumor growth through antigen-dependent redirection of Fcα/µ+ myeloid cells and antigen-independent, pIgR-mediated transcytosis (15). IgA can bind to polymeric IgA receptors that are universally expressed on ovarian cancer cells. IgA transcytosis induces broad transcriptional changes in inflammatory pathways in tumor cells, including the upregulation of IFN-γ receptors and downregulation of tumor-promoting ephrins. In addition, IgA transcytosis through malignant epithelial cells can antagonize the RAS pathway and sensitize tumor cells to cytolytic killing by T cells, which also helps prevent malignant progression (Figure 2). Thus, tumor-antigen-specific and tumor-antigen-independent IgA responses inhibit the growth of ovarian cancer cells by regulating coordinated B cell, T cell, and tumor cell responses.
These results indicate that IgA plays a completely different role in governing ovarian cancer than that in some other types of tumors. The causes may be related to the tumor type and TME. It can be hypothesized that most solid tumor cells do not express polymeric IgA receptors that are universally expressed on ovarian cancer cells, and hence IgA fails to exert anti-tumor effects through transcytosis. Furthermore, some tumor antigens can be specifically recognized by IgA in ovarian cancer. However, in some other tumor types, tumor antigens may not be exposed and thus cannot be recognized on the surface of tumor cells, or do not contain immunogenic peptides suitable for presentation to T cells on major histocompatibility complex class (MHC) molecules, which may lead to failure of recognition by IgA and thus mislead the immune response and favor pro-tumor processes (40, 63). Therefore, the analysis of intratumoral IgA expression and clonality is crucial for evaluating the role of IgA in tumors.
Emerging evidence points that a combination of host, environmental, and tumor factors mediates IgA class switching and determines the efficiency of cancer surveillance or promotion (48, 64). At the host level, this implies that increased serum IgA levels are related to older age, male gender, metabolic syndrome, etc. (48, 64, 65). Recent study has shown that environmental factors, including commensal bacteria and diet, and tumor immunosuppressive cytokines, including IL-10 and TGF-β, affect the production of IgA (66–68).
In the TME, most tumor-infiltrating TH/TFH cells present a regulatory phenotype, which secretes immunosuppressive cytokines and supports the development of immunosuppressive and anti-inflammatory isotypes (69). Furthermore, activated cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs) in the TME can produce TGF-β, which represses NK cell and CTL activation, promotes Tregs, and supports IgA CSR (38, 70, 71). More precisely, naïve B cells are recruited into the TME by CAFs via CXCL13 and CXCL12 and further exposed to TGF-β, IL-21, IL-33, lymphotoxin β (LTβ), and IL-10, which favors IgM-naïve BCR class switching to IgA (11, 70). Emerging evidence shows that tumors can also induce IgA CSR through a regulatory loop formed by myeloid-derived suppressor cells (MDSCs) and IgA. A recent study showed that CD11b+ MDSCs induced the differentiation of B cells into IgA+ PCs by secreting IL-10 and TGF-β (72). IgA with unfocused specificity can form immune complexes simultaneously with tumor or non-tumor antigens, promoting immunosuppressive phenotypes in myeloid cells (40).
Diet is universally considered a mechanism that can affect energy metabolism and immune responses, including intestinal IgA production (73). Among the various dietary components, vitamins are the most important factors for immune responses. For example, vitamin B1 has been confirmed to play a pivotal role in intestinal IgA responses as a cofactor of pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, which are essential enzymes in the TCA cycle (74). Similar to vitamins, dietary fatty acids influence B cell metabolism and the production of IgA (73, 75). A study showed that intestinal IgA production increased in mice maintained on a palmitic acid-enriched oil (76). Palmitoyl-CoA and serine act as substrates of serine palmitoyltransferase (SPT) and are subsequently converted into sphingolipids, such as ceramide, sphingosine, sphingomyelin, and S1P (77). Sphingolipids are a class of membrane lipids that also have biologic functions (78). For instance, an increase in ceramide concentrations may induce the proliferation of IgA PCs. Moreover, S1P can recruit IgA PCs into the intestine. These activation pathways may coincide with the changes in energy metabolism that occur in intestinal IgA PCs (76). Furthermore, metabolic intermediates of glycolysis, such as glucose monophosphate and fructose bis-phosphate, were detected preferentially in IgA PCs compared with naïve B cells, which implied that the shift to glycolysis-mediated energy metabolism likely is useful for the generation and production of IgA (73). For example, the glucose monophosphate and 3-phosphoglycerate generated through glycolysis are used in the pentose phosphate and serine biosynthetic pathways for nucleotide and amino acid synthesis, respectively (79). In addition, pyruvate, the metabolic product of glycolysis, can subsequently be converted to acetyl coenzyme A (acetyl-CoA) and used in fatty acid synthesis, which is required for B cell differentiation (80) (Figure 3).
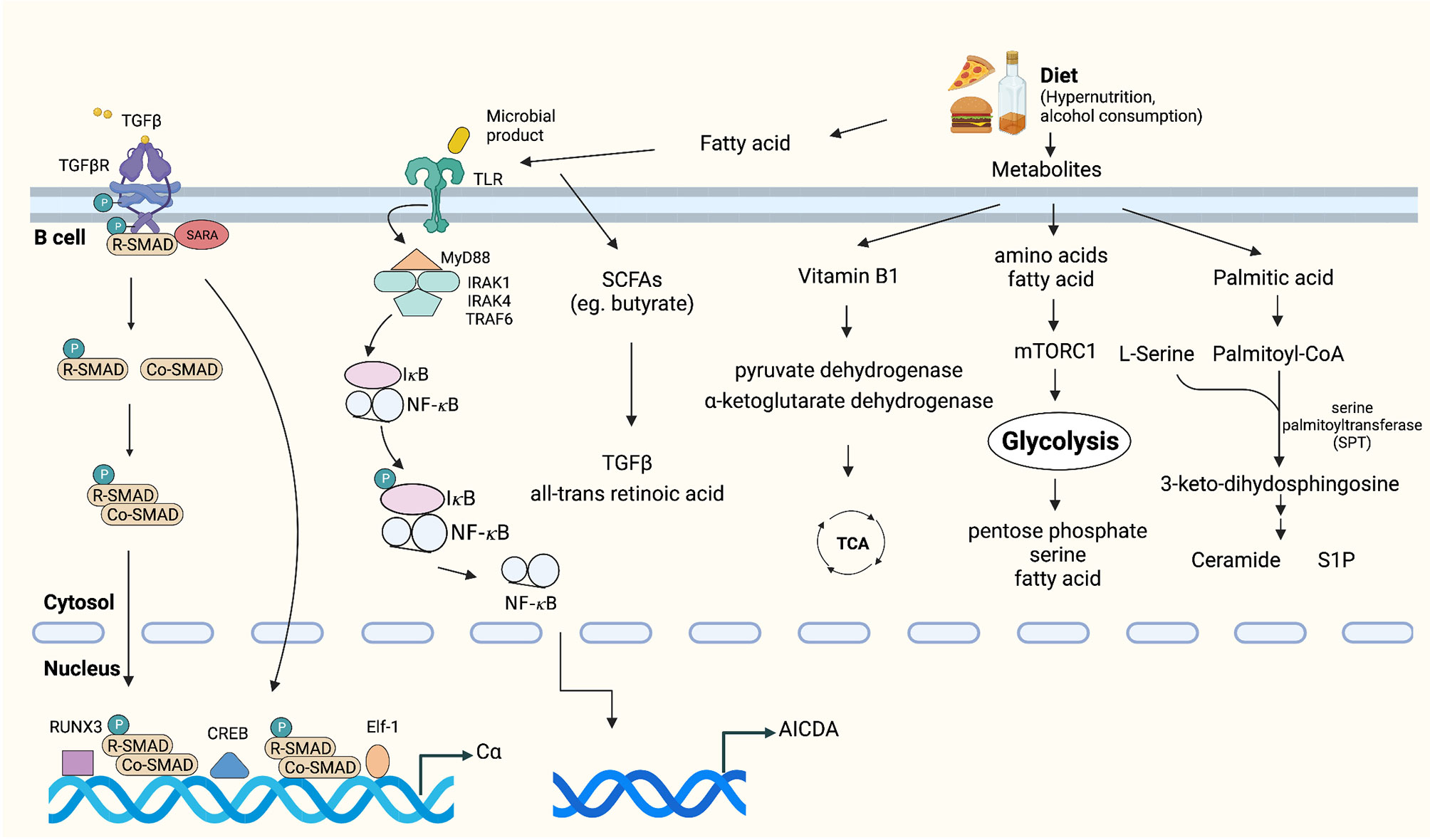
Figure 3 Tumor microenvironmental stimuli that induce IgA class switching. TGFβ is highly expressed in the tumor microenvironment, which activates the constant heavy chain α (Cα) gene promoters (38, 70, 71). Exposure to dietary extracellular metabolites within the immediate microenvironment may play an important role in controlling the production of IgA (73–75, 77). Fatty acid or microbial products can activate TLRs, which promote activation of IκB kinase (IKK) complex, phosphorylation and degradation of IκB [inhibitor of nuclear factor-κB (NF-κB)]. Subsequently, IκB-free NF-κB translocates to the nucleus and initiates CSR by binding to κB motifs on the AID gene promoter (81–83).
In addition, dysbiosis and changes in microbiome diversity were recently shown to regulate the IgA–microbiota axis and influence multiple aspects of antitumor immunity by altering the risk of developing cancer and regulating responses to immunotherapy (81–83). It was shown that gut and lung microbiota themselves are capable of promoting IgA CSR through antigen presentation by CD103+ DCs and induction of IL-10 and TGF-β (84). Conversely, a decrease in intestinal IgA responses and structural immaturity of PPs occurred in germ-free mice (85). Gut commensal microbes can produce biologically active metabolites, represented by the short-chain fatty acids (SCFAs) acetate, propionate and butyrate, which are all reported to promote IgA CSR (86, 87). Recently, Isobe et al. discovered that commensal-bacteria-derived butyrate up-regulated the production of TGF-β and all-trans retinoic acid by CD103+CD11b+DCs, both of which promote the T-cell-independent IgA response in the colon (88) (Figure 3).
A possible role of IgA in tumor immunity arises from its dynamic relationship with diet and the microbiota. Hyper-nutrition and alcohol consumption not only regulate the magnitude of B cell activity and effector function but also change the levels and diversity of IgA, which may further enhance tumor development (14, 89, 90). In addition, Piper et al. identified that the aryl-hydrocarbon receptor (AhR), a ligand-activated transcription factor that senses dietary, microbial, and metabolic cues, binds to the IL-10 locus in murine cells (91). The supplementation with microbially derived SCFA butyrate supports Breg to produce IL-10 by amplifying AhR activation (91). IL-10 upregulates the expression of AID and triggers the induction of SHM and CSR from IgM to IgA, which may mediate tumor progression (20, 33).
RNA- and DNA-based analyses of Ig repertoires and intratumoral T-cell receptors have received considerable attention as powerful tools for identifying prognostic and predictive cancer biomarkers, such as clonality metrics (2, 92). In particular, assessment of the degree of clonality and the amount of somatic hypermutations in Ig gene segments can offer prognostic value for some types of cancer (93). Clonality reflects mainly the presence of plasma cell clonal expansion. TCGA RNA-seq data on melanoma samples have demonstrated that a high IgG1 proportion is strongly associated with the presence of large clonal expansions, contributing to a better prognosis. In contrast, a high IgA proportion correlates with low clonality, resulting in a negative outcome (2). These results can be explained by a focused immune response leading to the production of high-affinity, tumor-specific IgG1 antibodies; conversely, switching of B cells to the IgA isotype is not driven by particular antigens but is a passive consequence of the intratumoral suppressive cytokine environment (40). Therefore, it can be hypothesized that dual effect of IgA on tumor immunity is highly associated with clonal expansion of PCs. High IgA expression and high clonality, implying that IgA may be driven by a focused immune response and may result in a better prognosis, whereas high IgA expression and low clonality mean that an immunosuppressive microenvironment drives IgA production and is likely to contribute to poor outcomes in some specific malignancies.
IgA and IgA+ cells are active participants that can fundamentally orchestrate the immune response in the TME. IgA can exert pro-tumorigenic roles in cancer by inducing the release of immunosuppressive cytokines, forming immune complexes with tumor or non-tumor antigens, and interacting with immunosuppressive cells, such as MDSCs or Tregs. However, IgA can also promote anticancer immunity and suppress tumor growth. These opposing effects can be explained by the influence of different tumor types, environmental factors, and host factors, which elicit a different antigen specificity of IgA, as well as the inconsistent interaction between IgA and tumor cells. Decisions on the application of IgA-targeted therapies and their combination with other therapies should be based on an advanced understanding of the prevailing nature of IgA involvement in tumor–immune interactions in each cancer subtype or even in each patient. Therefore, analysis of intratumoral IgA repertoires and determination of their metabolites are becoming powerful tools for identifying prognostic and predictive cancer biomarkers.
Conceptualization: ZZ, RL, and CM. Writing original draft: ZZ, KN, and MW. Supervision: YC, RL, and CM. All authors contributed to the article and approved the submitted version.
This work was supported by the Shanghai Rising-Star Program (19QA1406900), the National Science Foundation of China (81873948, 82072213, 81972666, 81730045), and the National Science and Technology Major Project of China (2020YFC2008402).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Breedveld A, van Egmond M. IgA and Fcαri: Pathological Roles and Therapeutic Opportunities. Front Immunol (2019) 10:553. doi: 10.3389/fimmu.2019.00553
2. Bolotin DA, Poslavsky S, Davydov AN, Frenkel FE, Fanchi L, Zolotareva OI, et al. Antigen Receptor Repertoire Profiling From RNA-Seq Data. Nat Biotechnol (2017) 35(10):908–11. doi: 10.1038/nbt.3979
3. Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The Prognostic Landscape of Genes and Infiltrating Immune Cells Across Human Cancers. Nat Med (2015) 21(8):938–45. doi: 10.1038/nm.3909
4. Isaeva OI, Sharonov GV, Serebrovskaya EO, Turchaninova MA, Zaretsky AR, Shugay M, et al. Intratumoral Immunoglobulin Isotypes Predict Survival in Lung Adenocarcinoma Subtypes. J Immunother Cancer (2019) 7(1):279. doi: 10.1186/s40425-019-0747-1
5. Lohr M, Edlund K, Botling J, Hammad S, Hellwig B, Othman A, et al. The Prognostic Relevance of Tumour-Infiltrating Plasma Cells and Immunoglobulin Kappa C Indicates an Important Role of the Humoral Immune Response in Non-Small Cell Lung Cancer. Cancer Lett (2013) 333(2):222–8. doi: 10.1016/j.canlet.2013.01.036
6. Teillaud JL, Dieu-Nosjean MC. Tertiary Lymphoid Structures: An Anti-Tumor School for Adaptive Immune Cells and an Antibody Factory to Fight Cancer? Front Immunol (2017) 8:830. doi: 10.3389/fimmu.2017.00830
7. Germain C, Gnjatic S, Tamzalit F, Knockaert S, Remark R, Goc J, et al. Presence of B Cells in Tertiary Lymphoid Structures Is Associated With a Protective Immunity in Patients With Lung Cancer. Am J Respir Crit Care Med (2014) 189(7):832–44. doi: 10.1164/rccm.201309-1611OC
8. Kroeger DR, Milne K, Nelson BH. Tumor-Infiltrating Plasma Cells Are Associated With Tertiary Lymphoid Structures, Cytolytic T-Cell Responses, and Superior Prognosis in Ovarian Cancer. Clin Cancer Res (2016) 22(12):3005–15. doi: 10.1158/1078-0432.CCR-15-2762
9. Sautès-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary Lymphoid Structures in the Era of Cancer Immunotherapy. Nat Rev Cancer (2019) 19(6):307–25. doi: 10.1038/s41568-019-0144-6
10. Tokunaga R, Naseem M, Lo JH, Battaglin F, Soni S, Puccini A, et al. B Cell and B Cell-Related Pathways for Novel Cancer Treatments. Cancer Treat Rev (2019) 73:10–9. doi: 10.1016/j.ctrv.2018.12.001
11. Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, et al. Immunosuppressive Plasma Cells Impede T-Cell-Dependent Immunogenic Chemotherapy. Nature (2015) 521(7550):94–8. doi: 10.1038/nature14395
12. Mauri C, Bosma A. Immune Regulatory Function of B Cells. Annu Rev Immunol (2012) 30:221–41. doi: 10.1146/annurev-immunol-020711-074934
13. Rosser EC, Mauri C. Regulatory B Cells: Origin, Phenotype, and Function. Immunity (2015) 42(4):607–12. doi: 10.1016/j.immuni.2015.04.005
14. Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, et al. Inflammation-Induced IgA+ Cells Dismantle Anti-Liver Cancer Immunity. Nature (2017) 551(7680):340–5. doi: 10.1038/nature24302
15. Biswas S, Mandal G, Payne KK, Anadon CM, Gatenbee CD, Chaurio RA, et al. IgA Transcytosis and Antigen Recognition Govern Ovarian Cancer Immunity. Nature (2021) 591(7850):464–70. doi: 10.1038/s41586-020-03144-0
16. Sterlin D, Gorochov G. When Therapeutic IgA Antibodies Might Come of Age. Pharmacology (2021) 106(1–2):9–19. doi: 10.1159/000510251
17. Brandtzaeg P, Johansen FE. Mucosal B Cells: Phenotypic Characteristics, Transcriptional Regulation, and Homing Properties. Immunol Rev (2005) 206:32–63. doi: 10.1111/j.0105-2896.2005.00283.x
18. Macpherson AJ, Geuking MB, McCoy KD. Homeland Security: IgA Immunity at the Frontiers of the Body. Trends Immunol (2012) 33(4):160–7. doi: 10.1016/j.it.2012.02.002
19. Brandtzaeg P, Prydz H. Direct Evidence for an Integrated Function of J Chain and Secretory Component in Epithelial Transport of Immunoglobulins. Nature (1984) 311(5981):71–3. doi: 10.1038/311071a0
20. Puga I, Cols M, Cerutti A. Innate Signals in Mucosal Immunoglobulin Class Switching. J Allergy Clin Immunol (2010) 126(5):889–95; quiz 96-7. doi: 10.1016/j.jaci.2010.09.026
21. Cerutti A. The Regulation of IgA Class Switching. Nat Rev Immunol (2008) 8(6):421–34. doi: 10.1038/nri2322
22. Chorny A, Puga I, Cerutti A. Regulation of Frontline Antibody Responses by Innate Immune Signals. Immunol Res (2012) 54(1-3):4–13. doi: 10.1007/s12026-012-8307-5
23. Gommerman JL, Rojas OL, Fritz JH. Re-Thinking the Functions of IgA(+) Plasma Cells. Gut Microbes (2014) 5(5):652–62. doi: 10.4161/19490976.2014.969977
24. Litinskiy MB, Nardelli B, Hilbert DM, He B, Schaffer A, Casali P, et al. DCs Induce CD40-Independent Immunoglobulin Class Switching Through BLyS and APRIL. Nat Immunol (2002) 3(9):822–9. doi: 10.1038/ni829
25. He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, et al. Intestinal Bacteria Trigger T Cell-Independent Immunoglobulin A(2) Class Switching by Inducing Epithelial-Cell Secretion of the Cytokine APRIL. Immunity (2007) 26(6):812–26. doi: 10.1016/j.immuni.2007.04.014
26. Pylayeva-Gupta Y, Das S, Handler JS, Hajdu CH, Coffre M, Koralov SB, et al. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov (2016) 6(3):247–55. doi: 10.1158/2159-8290.CD-15-0843
27. Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TF, et al. Interleukin 21-Induced Granzyme B-Expressing B Cells Infiltrate Tumors and Regulate T Cells. Cancer Res (2013) 73(8):2468–79. doi: 10.1158/0008-5472.CAN-12-3450
28. Ye L, Zhang Q, Cheng Y, Chen X, Wang G, Shi M, et al. Tumor-Derived Exosomal HMGB1 Fosters Hepatocellular Carcinoma Immune Evasion by Promoting TIM-1(+) Regulatory B Cell Expansion. J Immunother Cancer (2018) 6(1):145. doi: 10.1186/s40425-018-0451-6
29. Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T, Baba Y. The Calcium Sensors STIM1 and STIM2 Control B Cell Regulatory Function Through Interleukin-10 Production. Immunity (2011) 34(5):703–14. doi: 10.1016/j.immuni.2011.03.016
30. Affara NI, Ruffell B, Medler TR, Gunderson AJ, Johansson M, Bornstein S, et al. B Cells Regulate Macrophage Phenotype and Response to Chemotherapy in Squamous Carcinomas. Cancer Cell (2014) 25(6):809–21. doi: 10.1016/j.ccr.2014.04.026
31. Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-Talk Drives Pancreas Cancer. Cancer Discov (2016) 6(3):270–85. doi: 10.1158/2159-8290.CD-15-0827
32. Olkhanud PB, Damdinsuren B, Bodogai M, Gress RE, Sen R, Wejksza K, et al. Tumor-Evoked Regulatory B Cells Promote Breast Cancer Metastasis by Converting Resting CD4⁺ T Cells to T-Regulatory Cells. Cancer Res (2011) 71(10):3505–15. doi: 10.1158/0008-5472.CAN-10-4316
33. Rosser EC, Mauri C. The Emerging Field of Regulatory B Cell Immunometabolism. Cell Metab (2021) 33(6):1088–97. doi: 10.1016/j.cmet.2021.05.008
34. Rojas OL, Pröbstel AK, Porfilio EA, Wang AA, Charabati M, Sun T, et al. Recirculating Intestinal IgA-Producing Cells Regulate Neuroinflammation via IL-10. Cell (2019) 176(3):610–24.e18. doi: 10.1016/j.cell.2018.11.035
35. Fehres CM, van Uden NO, Yeremenko NG, Fernandez L, Franco Salinas G, van Duivenvoorde LM, et al. APRIL Induces a Novel Subset of IgA(+) Regulatory B Cells That Suppress Inflammation via Expression of IL-10 and PD-L1. Front Immunol (2019) 10:1368. doi: 10.3389/fimmu.2019.01368
36. Schietinger A, Greenberg PD. Tolerance and Exhaustion: Defining Mechanisms of T Cell Dysfunction. Trends Immunol (2014) 35(2):51–60. doi: 10.1016/j.it.2013.10.001
37. Pardoll DM. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat Rev Cancer (2012) 12(4):252–64. doi: 10.1038/nrc3239
38. Stavnezer J, Kang J. The Surprising Discovery That TGF Beta Specifically Induces the IgA Class Switch. J Immunol (2009) 182(1):5–7. doi: 10.4049/jimmunol.182.1.5
39. Pinto D, Montani E, Bolli M, Garavaglia G, Sallusto F, Lanzavecchia A, et al. A Functional BCR in Human IgA and IgM Plasma Cells. Blood (2013) 121(20):4110–4. doi: 10.1182/blood-2012-09-459289
40. Sharonov GV, Serebrovskaya EO, Yuzhakova DV, Britanova OV, Chudakov DM. B Cells, Plasma Cells and Antibody Repertoires in the Tumour Microenvironment. Nat Rev Immunol (2020) 20(5):294–307. doi: 10.1038/s41577-019-0257-x
41. Gunderson AJ, Coussens LM. B Cells and Their Mediators as Targets for Therapy in Solid Tumors. Exp Cell Res (2013) 319(11):1644–9. doi: 10.1016/j.yexcr.2013.03.005
42. Kumar S, Mohan A, Guleria R. Prognostic Implications of Circulating Anti-P53 Antibodies in Lung Cancer–A Review. Eur J Cancer Care (Engl) (2009) 18(3):248–54. doi: 10.1111/j.1365-2354.2008.01019.x
43. Wieland A, Shashidharamurthy R, Kamphorst AO, Han JH, Aubert RD, Choudhury BP, et al. Antibody Effector Functions Mediated by Fcγ-Receptors Are Compromised During Persistent Viral Infection. Immunity (2015) 42(2):367–78. doi: 10.1016/j.immuni.2015.01.009
44. Liu R, Lu Z, Gu J, Liu J, Huang E, Liu X, et al. MicroRNAs 15A and 16-1 Activate Signaling Pathways That Mediate Chemotaxis of Immune Regulatory B Cells to Colorectal Tumors. Gastroenterology (2018) 154(3):637–51.e7. doi: 10.1053/j.gastro.2017.09.045
45. Bosisio FM, Wilmott JS, Volders N, Mercier M, Wouters J, Stas M, et al. Plasma Cells in Primary Melanoma. Prognostic Significance and Possible Role of IgA. Mod Pathol (2016) 29(4):347–58. doi: 10.1038/modpathol.2016.28
46. Chiaruttini G, Mele S, Opzoomer J, Crescioli S, Ilieva KM, Lacy KE, et al. B Cells and the Humoral Response in Melanoma: The Overlooked Players of the Tumor Microenvironment. Oncoimmunology (2017) 6(4):e1294296. doi: 10.1080/2162402X.2017.1294296
47. Welinder C, Jirström K, Lehn S, Nodin B, Marko-Varga G, Blixt O, et al. Intra-Tumour IgA1 Is Common in Cancer and Is Correlated With Poor Prognosis in Bladder Cancer. Heliyon (2016) 2(8):e00143. doi: 10.1016/j.heliyon.2016.e00143
48. Peppas I, George G, Sollie S, Josephs DH, Hammar N, Walldius G, et al. Association of Serum Immunoglobulin Levels With Solid Cancer: A Systematic Review and Meta-Analysis. Cancer Epidemiol Biomarkers Prev (2020) 29(3):527–38. doi: 10.1158/1055-9965.EPI-19-0953
49. Cipponi A, Mercier M, Seremet T, Baurain JF, Théate I, van den Oord J, et al. Neogenesis of Lymphoid Structures and Antibody Responses Occur in Human Melanoma Metastases. Cancer Res (2012) 72(16):3997–4007. doi: 10.1158/0008-5472.CAN-12-1377
50. Zhang J, Hu X, Wang J, Sahu AD, Cohen D, Song L, et al. Immune Receptor Repertoires in Pediatric and Adult Acute Myeloid Leukemia. Genome Med (2019) 11(1):73. doi: 10.1186/s13073-019-0681-3
51. Garaud S, Zayakin P, Buisseret L, Rulle U, Silina K, de Wind A, et al. Antigen Specificity and Clinical Significance of IgG and IgA Autoantibodies Produced In Situ by Tumor-Infiltrating B Cells in Breast Cancer. Front Immunol (2018) 9:2660. doi: 10.3389/fimmu.2018.02660
52. Frontera ED, Khansa RM, Schalk DL, Leakan LE, Guerin-Edbauer TJ, Ratnam M, et al. IgA Fc-Folate Conjugate Activates and Recruits Neutrophils to Directly Target Triple-Negative Breast Cancer Cells. Breast Cancer Res Treat (2018) 172(3):551–60. doi: 10.1007/s10549-018-4941-5
53. Schantz SP, Liu FJ, Taylor D, Beddingfield N, Weber RS. The Relationship of Circulating IgA to Cellular Immunity in Head and Neck Cancer Patients. Laryngoscope (1988) 98(6 Pt 1):671–8. doi: 10.1288/00005537-198806000-00019
54. Vinzenz K, Pavelka R, Schönthal E, Zekert F. Serum Immunoglobulin Levels in Patients With Head and Neck Cancer (IgE, IgA, IgM, IgG). Oncology (1986) 43(5):316–22. doi: 10.1159/000226390
55. Sollie S, Santaolalla A, Michaud DS, Sarker D, Karagiannis SN, Josephs DH, et al. Serum Immunoglobulin G Is Associated With Decreased Risk of Pancreatic Cancer in the Swedish AMORIS Study. Front Oncol (2020) 10:263. doi: 10.3389/fonc.2020.00263
56. Pao W, Ooi CH, Birzele F, Ruefli-Brasse A, Cannarile MA, Reis B, et al. Tissue-Specific Immunoregulation: A Call for Better Understanding of the "Immunostat" in the Context of Cancer. Cancer Discovery (2018) 8(4):395–402. doi: 10.1158/2159-8290.CD-17-1320
57. Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, et al. Foxp3(+) T Cells Regulate Immunoglobulin a Selection and Facilitate Diversification of Bacterial Species Responsible for Immune Homeostasis. Immunity (2014) 41(1):152–65. doi: 10.1016/j.immuni.2014.05.016
58. Karagiannis P, Gilbert AE, Josephs DH, Ali N, Dodev T, Saul L, et al. IgG4 Subclass Antibodies Impair Antitumor Immunity in Melanoma. J Clin Invest (2013) 123(4):1457–74. doi: 10.1172/JCI65579
59. Mkaddem SB, Christou I, Rossato E, Berthelot L, Lehuen A, Monteiro RC. IgA, IgA Receptors, and Their Anti-Inflammatory Properties. Curr Top Microbiol Immunol (2014) 382:221–35. doi: 10.1007/978-3-319-07911-0_10
60. Ben Mkaddem S, Rossato E, Heming N, Monteiro RC. Anti-Inflammatory Role of the IgA Fc Receptor (CD89): From Autoimmunity to Therapeutic Perspectives. Autoimmun Rev (2013) 12(6):666–9. doi: 10.1016/j.autrev.2012.10.011
61. Pilette C, Detry B, Guisset A, Gabriels J, Sibille Y. Induction of Interleukin-10 Expression Through Fcalpha Receptor in Human Monocytes and Monocyte-Derived Dendritic Cells: Role of P38 MAPKinase. Immunol Cell Biol (2010) 88(4):486–93. doi: 10.1038/icb.2009.120
62. Saha C, Das M, Patil V, Stephen-Victor E, Sharma M, Wymann S, et al. Monomeric Immunoglobulin A From Plasma Inhibits Human Th17 Responses In Vitro Independent of Fcαri and DC-SIGN. Front Immunol (2017) 8:275. doi: 10.3389/fimmu.2017.00275
63. Yuen GJ, Demissie E, Pillai S. B Lymphocytes and Cancer: A Love-Hate Relationship. Trends Cancer (2016) 2(12):747–57. doi: 10.1016/j.trecan.2016.10.010
64. Shalapour S, Karin M. The Neglected Brothers Come of Age: B Cells and Cancer. Semin Immunol (2021) 52:101479. doi: 10.1016/j.smim.2021.101479
65. Gonzalez-Quintela A, Alende R, Gude F, Campos J, Rey J, Meijide LM, et al. Serum Levels of Immunoglobulins (IgG, IgA, IgM) in a General Adult Population and Their Relationship With Alcohol Consumption, Smoking and Common Metabolic Abnormalities. Clin Exp Immunol (2008) 151(1):42–50. doi: 10.1111/j.1365-2249.2007.03545.x
66. Goedert JJ, Hua X, Bielecka A, Okayasu I, Milne GL, Jones GS, et al. Postmenopausal Breast Cancer and Oestrogen Associations With the IgA-Coated and IgA-Noncoated Faecal Microbiota. Br J Cancer (2018) 118(4):471–9. doi: 10.1038/bjc.2017.435
67. Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF. The Microbiome in Cancer Immunotherapy: Diagnostic Tools and Therapeutic Strategies. Science (2018) 359(6382):1366–70. doi: 10.1126/science.aar6918
68. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut Microbiome Influences Efficacy of PD-1-Based Immunotherapy Against Epithelial Tumors. Science (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
69. Song H, Liu A, Liu G, Wu F, Li Z. T Follicular Regulatory Cells Suppress Tfh-Mediated B Cell Help and Synergistically Increase IL-10-Producing B Cells in Breast Carcinoma. Immunol Res (2019) 67(4-5):416–23. doi: 10.1007/s12026-019-09090-y
70. Ammirante M, Shalapour S, Kang Y, Jamieson CA, Karin M. Tissue Injury and Hypoxia Promote Malignant Progression of Prostate Cancer by Inducing CXCL13 Expression in Tumor Myofibroblasts. Proc Natl Acad Sci USA (2014) 111(41):14776–81. doi: 10.1073/pnas.1416498111
71. Batlle E, Massagué J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity (2019) 50(4):924–40. doi: 10.1016/j.immuni.2019.03.024
72. Xu X, Meng Q, Erben U, Wang P, Glauben R, Kühl AA, et al. Myeloid-Derived Suppressor Cells Promote B-Cell Production of IgA in a TNFR2-Dependent Manner. Cell Mol Immunol (2017) 14(7):597–606. doi: 10.1038/cmi.2015.103
73. Kunisawa J. Metabolic Changes During B Cell Differentiation for the Production of Intestinal IgA Antibody. Cell Mol Life Sci (2017) 74(8):1503–9. doi: 10.1007/s00018-016-2414-8
74. Frank RA, Leeper FJ, Luisi BF. Structure, Mechanism and Catalytic Duality of Thiamine-Dependent Enzymes. Cell Mol Life Sci (2007) 64(7-8):892–905. doi: 10.1007/s00018-007-6423-5
75. Kunisawa J, Kiyono H. Sphingolipids and Epoxidized Lipid Metabolites in the Control of Gut Immunosurveillance and Allergy. Front Nutr (2016) 3:3. doi: 10.3389/fnut.2016.00003
76. Kunisawa J, Hashimoto E, Inoue A, Nagasawa R, Suzuki Y, Ishikawa I, et al. Regulation of Intestinal IgA Responses by Dietary Palmitic Acid and its Metabolism. J Immunol (2014) 193(4):1666–71. doi: 10.4049/jimmunol.1302944
77. Gault CR, Obeid LM, Hannun YA. An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown. Adv Exp Med Biol (2010) 688:1–23. doi: 10.1007/978-1-4419-6741-1_1
78. Fyrst H, Saba JD. An Update on Sphingosine-1-Phosphate and Other Sphingolipid Mediators. Nat Chem Biol (2010) 6(7):489–97. doi: 10.1038/nchembio.392
79. Vander Heiden MG, Lunt SY, Dayton TL, Fiske BP, Israelsen WJ, Mattaini KR, et al. Metabolic Pathway Alterations That Support Cell Proliferation. Cold Spring Harb Symp Quant Biol (2011) 76:325–34. doi: 10.1101/sqb.2012.76.010900
80. Dufort FJ, Gumina MR, Ta NL, Tao Y, Heyse SA, Scott DA, et al. Glucose-Dependent De Novo Lipogenesis in B Lymphocytes: A Requirement for Atp-Citrate Lyase in Lipopolysaccharide-Induced Differentiation. J Biol Chem (2014) 289(10):7011–24. doi: 10.1074/jbc.M114.551051
81. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science (2018) 359(6371):97–103. doi: 10.1126/science.aan4236
82. Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, Wargo JA. The Microbiome, Cancer, and Cancer Therapy. Nat Med (2019) 25(3):377–88. doi: 10.1038/s41591-019-0377-7
83. Sims TT, El Alam MB, Karpinets TV, Dorta-Estremera S, Hegde VL, Nookala S, et al. Gut Microbiome Diversity Is an Independent Predictor of Survival in Cervical Cancer Patients Receiving Chemoradiation. Commun Biol (2021) 4(1):237. doi: 10.1038/s42003-021-01741-x
84. Ruane D, Chorny A, Lee H, Faith J, Pandey G, Shan M, et al. Microbiota Regulate the Ability of Lung Dendritic Cells to Induce IgA Class-Switch Recombination and Generate Protective Gastrointestinal Immune Responses. J Exp Med (2016) 213(1):53–73. doi: 10.1084/jem.20150567
85. Weinstein PD, Cebra JJ. The Preference for Switching to IgA Expression by Peyer's Patch Germinal Center B Cells Is Likely Due to the Intrinsic Influence of Their Microenvironment. J Immunol (1991) 147(12):4126–35.
86. Wu W, Sun M, Chen F, Cao AT, Liu H, Zhao Y, et al. Microbiota Metabolite Short-Chain Fatty Acid Acetate Promotes Intestinal IgA Response to Microbiota Which Is Mediated by GPR43. Mucosal Immunol (2017) 10(4):946–56. doi: 10.1038/mi.2016.114
87. Kim M, Qie Y, Park J, Kim CH. Gut Microbial Metabolites Fuel Host Antibody Responses. Cell Host Microbe (2016) 20(2):202–14. doi: 10.1016/j.chom.2016.07.001
88. Isobe J, Maeda S, Obata Y, Iizuka K, Nakamura Y, Fujimura Y, et al. Commensal-Bacteria-Derived Butyrate Promotes the T-Cell-Independent IgA Response in the Colon. Int Immunol (2020) 32(4):243–58. doi: 10.1093/intimm/dxz078
89. Lindner C, Wahl B, Föhse L, Suerbaum S, Macpherson AJ, Prinz I, et al. Age, Microbiota, and T Cells Shape Diverse Individual IgA Repertoires in the Intestine. J Exp Med (2012) 209(2):365–77. doi: 10.1084/jem.20111980
90. McPherson S, Henderson E, Burt AD, Day CP, Anstee QM. Serum Immunoglobulin Levels Predict Fibrosis in Patients With Non-Alcoholic Fatty Liver Disease. J Hepatol (2014) 60(5):1055–62. doi: 10.1016/j.jhep.2014.01.010
91. Piper CJM, Rosser EC, Oleinika K, Nistala K, Krausgruber T, Rendeiro AF, et al. Aryl Hydrocarbon Receptor Contributes to the Transcriptional Program of IL-10-Producing Regulatory B Cells. Cell Rep (2019) 29(7):1878–92.e7. doi: 10.1016/j.celrep.2019.10.018
92. DeFalco J, Harbell M, Manning-Bog A, Baia G, Scholz A, Millare B, et al. Non-Progressing Cancer Patients Have Persistent B Cell Responses Expressing Shared Antibody Paratopes That Target Public Tumor Antigens. Clin Immunol (2018) 187:37–45. doi: 10.1016/j.clim.2017.10.002
Keywords: B cells, plasma cells, immunoglobulin A, tumor immunity, class switching
Citation: Zhong Z, Nan K, Weng M, Yue Y, Zhou W, Wang Z, Chu Y, Liu R and Miao C (2021) Pro- and Anti- Effects of Immunoglobulin A- Producing B Cell in Tumors and Its Triggers. Front. Immunol. 12:765044. doi: 10.3389/fimmu.2021.765044
Received: 26 August 2021; Accepted: 01 November 2021;
Published: 19 November 2021.
Edited by:
Damo Xu, Shenzhen University, ChinaReviewed by:
Wei Luo, Purdue University Indianapolis, United StatesCopyright © 2021 Zhong, Nan, Weng, Yue, Zhou, Wang, Chu, Liu and Miao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ronghua Liu, cm9uZ2h1YWxpdUBmdWRhbi5lZHUuY24=; Changhong Miao, bWlhb19jaGhAMTI2LmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.