 Yi Qin
Yi Qin Hui-Zhi Jin
Hui-Zhi Jin Yu-Jing Li
Yu-Jing Li Zhu Chen
Zhu Chen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 18 October 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.764825
This article is part of the Research Topic Th2-Associated Immunity in The Pathogenesis of Systemic Lupus Erythematosus and Rheumatoid Arthritis View all 12 articles
Eosinophils are a minor component of circulating granulocytes, which are classically viewed as end-stage effector cells in host defense against helminth infection and promoting allergic responses. However, a growing body of evidence has emerged showing that eosinophils are versatile leukocytes acting as an orchestrator in the resolution of inflammation. Rheumatoid arthritis (RA) is the most common chronic inflammatory disease characterized by persistent synovitis that hardly resolves spontaneously. Noteworthy, a specific population of eosinophils, that is, regulatory eosinophils (rEos), was identified in the synovium of RA patients, especially in disease remission. Mechanistically, the rEos in the synovium display a unique pro-resolving signature that is distinct from their counterpart in the lung. Herein, we summarize the latest understanding of eosinophils and their emerging role in promoting the resolution of arthritis. This knowledge is crucial to the design of new approaches to rebalancing immune homeostasis in RA, considering that current therapies are centered on inhibiting pro-inflammatory cytokines and mediators rather than fostering the resolution of inflammation.
Eosinophils are leukocytes that normally amount to less than 5% of white blood cells in the peripheral blood. In certain pathological settings, eosinophils significantly expand and count over 1,500 cells/μl blood, which is defined as hypereosinophilia (1). Although previously considered as the end-stage effector cells involved in helminth infection and allergic diseases like asthma, increasing evidence shows that eosinophils are multifunctional granulocytes involved in regulating adaptive immune responses, especially in inflammatory and autoimmune disorders (2).
Rheumatoid arthritis (RA) is the most common chronic immune-mediated inflammatory disease characterized by persistent synovitis that lacks self-remission (3). Although the pathogenesis of RA remains incompletely understood, the general consensus is that self-tolerance breakdown triggers autoantibody production in genetically predisposed individuals before progressing into clinically apparent RA (3). During the transition from asymptomatic autoimmunity to synovial inflammation, a diverse range of pro-inflammatory cytokines produced by immune cells such as CD4+ T cells, macrophages, and fibroblast-like synoviocytes emerge quickly, which eventually contribute to cartilage damage and bone erosion in the joint (4). Notably, once the joint inflammation is established, it tends to be chronic, as evidenced by the insufficiency of regulatory factors that counteract or rebalance aberrant immune responses (4). Hence, ineffective resolution of RA remains a major clinical challenge, although novel anti-inflammatory biological agents have been increasingly introduced (5, 6).
In contrast with the pro-inflammatory properties of eosinophils in asthma that cause structural remodeling of the airways, recent studies by us and collaborators have suggested that, as a crucial component of Th2 immune responses, eosinophils have previously undifferentiated pro-resolving signature in RA (7–10). In this review, the emerging role of eosinophils in promoting the resolution of arthritis is summarized. The potential underlying mechanisms that allow eosinophils to exert anti-inflammatory properties and therapeutic implications of eosinophils in arthritis are also discussed.
Eosinophils are generated in the bone marrow from multipotent hematopoietic stem cells, which give rise to eosinophil-committed progenitors (EoPs). These will eventually differentiate into mature eosinophils in response to several cytokines such as IL-5, IL-3, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-33 (11–13). Eosinophilopoiesis is governed by at least three transcription factors, including GATA-1 (a zinc family finger member), PU.1 (an ETS family member), and C/EBP members (CCAAT/enhancer-binding protein family) (14, 15). Notably, GATA-1 is essential for eosinophil differentiation, since the deletion of a GATA-binding enhancer site in the GATA-1 gene generated a specific eosinophil-deficient ΔdblGATA mouse with no influence on other cell lineages (16). In addition, some microRNAs and long non-coding RNAs have been reported to be involved in eosinophilopoiesis (17–19).
Once eosinophils mature in the bone marrow, they are released into circulation and migrate into peripheral tissues under stimulation of IL-5 and eotaxin-1 (CCL11) (2). In homeostatic conditions, eosinophils are distributed in the spleen, gastrointestinal tract, thymus, adipose tissue, and uterus, indicating that they are likely to be responsible for maintaining homeostasis in different tissues (20). It is well established that eosinophils synthesize a broad range of mediators stored in granules throughout the cytoplasm, including cytotoxic granule proteins such as major basic protein (MBP), eosinophil cationic protein (ECP), eosinophil peroxidase (EPX), and eosinophil-derived neurotoxin (EDN) (21). When encountering the stimulus present in the tissue, eosinophils release granule contents rapidly, which is termed degranulation, to exert host immune defense against pathogens (2). Hence, historically it is considered that the primary effector function of eosinophils was in anti-pathogen responses, especially those involving parasites. Nonetheless, eosinophil granules also contain numerous cytokines, particularly type 2 cytokines, as well as growth factors and resolvins, suggesting their ability to be involved in tissue repair and a wide range of immunological disorders such as allergy and asthma (22, 23).
Although they represent a minor component of innate immune cells, eosinophils are well known to be an important innate immune regulator in pathogen clearance by releasing cytokines and chemokines or by interacting with other innate immune cells (24–28). Meanwhile, increasing evidence has extended understanding that eosinophils are versatile leukocytes capable of modulating adaptive immune responses as well. For example, murine eosinophils can present antigen via MHC class II and promote IL-4, IL-5, and IL-13 production from antigen-specific CD4+ T cells in the context of helminth infection or asthma (23, 29, 30).
Other than behaving as antigen-presenting cells, eosinophils are thought to regulate Th2 immune responses in multiple ways. A Notch ligand Jagged1, which constitutes an instructive signal for Th2 differentiation, has been found to express on human eosinophils constitutively, indicating the capability of eosinophils to provide a polarization signal to naïve CD4+ T cells (31, 32). Studies on helminth infection models revealed that eosinophils precede lymphocyte recruitment into inflammatory sites (33, 34). In eosinophil-deficient ΔdblGATA mice infected with Trichinella spiralis, infiltration of Th2 cells into the muscles was highly decreased (35). In another study using IL-5/eotaxin double-knockout mice in which eosinophil counts are severely reduced, significantly decreased IL-13 production by Th2 cells in response to the OVA challenge was observed (36). Notably, this defect can be rescued by the adoptive transfer of eosinophils, suggesting the role of eosinophils in the regulation of Th2 immune responses (36). In addition to the secretion of Th2-related cytokines, eosinophils can also promote Th2 responses through the synthesis of indoleamine 2,3-dioxygenase (IDO), an enzyme that catalyzes the oxidative catabolism of tryptophan to kynurenines (37).
Inflammation is an evolutionary defensive host response to injury, characterized by the recruitment of leukocytes and cytokines from the circulation to the inflamed tissue. Generally, acute inflammation in healthy individuals is self-limited and resolves timely, thus preventing to progress to chronic inflammation (38). During the course of acute inflammation, the migration of polymorphonuclear neutrophils (PMNs) into tissues is the early event, followed by the recruitment of monocytes that will further differentiate into tissue macrophages. It is known that a variety of classic proinflammatory mediators such as prostaglandins (PGs) and leukotrienes (LTs) coordinate these initial events of acute inflammation by regulating vascular permeability and leukocyte infiltration (39). Once the malicious components are removed by phagocytosis, the inflammatory response must be promptly resolved to prevent excessive tissue damage and return to homeostasis. However, uncontrolled or long-lasting inflammation is believed to exist in the pathogenesis of many human autoimmune and inflammatory diseases including RA (40).
In recent years, a growing body of evidence has emerged that shows that resolution of acute inflammation is not a passive but an active process controlled by endogenous resolving mediators, termed specialized pro-resolving mediators (SPMs) such as protectins, resolvins, and maresins, which belong to families of lipid mediators (41). Blocking lipid mediator biosynthesis by either cyclooxygenase (COX)-2 or lipoxygenase (LOX) inhibitors resulted in resolution defect, which is characterized by sustained leukocyte infiltration in inflamed sites and impaired removal of phagocytes to the draining lymph nodes (42), suggesting the critical role of these lipid mediators in regulating the timely resolution of acute inflammation.
Interestingly, eosinophils are an orchestrator in the resolution of inflammation. In a murine zymosan-induced peritonitis model, eosinophils were recruited to the inflamed site during the resolution phase of acute peritonitis (43). Liquid chromatography–tandem mass spectrometry (LC-MS/MS)-based lipidomics analyses revealed that pro-resolving mediators such as protectin D1 (PD1) were increased during the resolution phase in a 12/15-LOX dependent manner (43). PD1 promotes the resolution process by inhibiting PMN influx and stimulating macrophage ingestion of apoptotic PMNs, as well as increasing phagocyte clearance into draining lymph nodes (42). Importantly, the researchers revealed that eosinophils were the main PD1-producing cells in the resolution phase of zymosan-induced peritonitis (43). Depletion of eosinophils or CXCL13 in vivo caused a resolution defect, characterized by impaired lymphatic drainage of inflammatory phagocytes carrying engulfed zymosan in the draining lymph node, and delayed removal of PMNs in the inflamed tissues (44). Notably, administration of PD1, CXCL13, or adoptive transfer of eosinophils from wild-type but not from 12/15-LOX deficient mice reversed the defective phenotype of the resolution process, suggesting that eosinophils promote the resolution of inflammation through pro-resolving mediators and CXCL13 pathway (43, 44). In another experimental colitis model, more severe colitis was observed in eosinophil-deficient mice compared with wild-type controls, accompanied with decreased level of PD1 in the colon (45). Furthermore, administration of exogenous PD1 alleviated the severity of colitis and reduced neutrophil infiltration. All these findings indicated that eosinophils contribute to the resolution of inflammation by producing pro-resolving lipid mediators such as PD1 via the 12/15-LOX-mediated biosynthetic pathway.
Although eosinophils act as a counter regulator in several inflammatory disorders, the association between eosinophils and the development of RA was largely undetermined, probably due to the uncommon clinical manifestation of eosinophilia in RA. Indirect evidence from previous studies reported that RA patients, especially those with high activity and short disease duration, have increased serum levels of ECP, supporting the notion that eosinophils were involved in the inflammatory responses of RA (46, 47). In a prospective multi-center cohort study, the prevalence of eosinophilia in patients with new-onset arthritis (disease duration ranges from 6 weeks to 6 months) is only 3.2% (48). Notably, after 3 years, patients with eosinophilia presented with signs of higher disease activity compared with those without eosinophilia at baseline, suggesting that baseline eosinophilia might be a poor prognosis marker in early arthritis patients (48). A more recent prospective observational study investigated clinical characteristics of RA patients with persistent eosinophilia and compared with the patients without eosinophilia (49). After excluding secondary causes of eosinophilia such as concomitant allergic diseases and intestinal helminth infection, the authors did not find differences in clinical features between RA patients with or without eosinophilia (49). The discrepancies among studies might reflect the complex heterogeneity of eosinophil phenotype or function in inflammatory arthritis.
Recently, we have shown that the expression of synovium EPX was elevated in RA patients compared with osteoarthritis (OA) patients (7). Consistently, serum EPX level was higher in RA patients than pre-RA and healthy controls (7). In the K/BxN serum-induced arthritis model, IL-5 transgenic (IL-5tg) mice that have extraordinary hypereosinophilia showed a significant reduction of arthritis score, whereas eosinophil-deficient ΔdblGATA mice presented with higher disease activity (7). In addition, the adoptive transfer of eosinophils into collagen-induced arthritic mice led to the alleviation of arthritis, accompanied by reduced joint inflammation and bone erosion in histology evaluation (9). Altogether, these data suggested that eosinophils have previous unknown pro-resolving properties in promoting the resolution of inflammatory arthritis.
Considering the dual nature of eosinophils as pro-inflammatory and pro-resolving cells, it is reasonable to speculate that eosinophils have different subsets responsible for different biological functions. Indeed, this is supported by the research that revealed two distinct eosinophil subsets, lung resident eosinophils and recruited inflammatory eosinophils, in asthmatic lungs (50). Remarkably, a very recent study described a specific population of eosinophils, named regulatory eosinophils (rEos), was present in the joints of arthritic mice as well as synovium of RA patients (8). OVA allergen challenge triggered an earlier resolution of K/BxN serum-induced arthritis, accompanied with increased eosinophils in the arthritic joints. Strikingly, this protective manifestation was only observed in wild-type but not eosinophil-deficient ΔdblGATA mice, indicating the essential role of eosinophils in the asthma-induced resolution of joint inflammation (8). In further analyses, both single-cell RNA sequencing and proteome profile analyses confirmed that the rEos in the joint display a unique pro-resolving characteristic, which is distinct from their counterpart in the lung. For example, joint rEos have strongly upregulated expression of 5-LOX and 12/15-LOX (8), which could explain the anti-inflammatory and pro-resolving effector function of rEos, since the deletion of 12/15-LOX was linked to uncontrolled inflammation and tissue damage in chronic arthritis (51). Moreover, rEos has a specific secretion pattern compared with its inflammatory counterpart in the lung, characterized by the production of MMP-3, osteopontin, and serpin E1, suggesting that rEos might also foster synovial tissue recovery besides ceasing inflammation (8). Interestingly, rEos was infiltrated more frequently in RA patients in remission than patients in the active stage. Inactive RA patients with concomitant asthma developed a flare of disease after anti-IL-5 monoclonal antibody treatment, which might be explained reasonably by the depletion of rEos (8).
As a messenger between the innate and adaptive immune systems, innate lymphoid cells (ILCs) have been realized to be intensely involved in the pathogenesis of inflammatory arthritis (52, 53). In particular, ILC2 acts as a crucial regulator in damping joint inflammation in contrast to proinflammatory ILC1/ILC3. Circulating ILC2 count was inversely correlated with disease activity index in RA patients and increased after receiving anti-rheumatic treatment (54). In line with these human data, genetic deletion of ILC2 in mice aggravated K/BxN serum-induced arthritis whereas expansion of ILC2 by IL-25/IL-33 mini-circles or adoptive transfer of ILC2 from wild-type but not IL-4-/-IL-13-/- mice attenuated arthritis (55). Furthermore, ILC2 was found to inhibit osteoclast differentiation and bone loss independently of inflammation (56).
Interestingly, it has been established that tissue-resident ILC2 regulates eosinophil homeostasis and accumulation into tissues through constitutive secretion of IL-5 (57). In asthmatic lung, ILC2 was the main producer of IL-5, which consequently drives the expansion and infiltration of rEos into the arthritic joints (8). Another recent work by us showed that activation of ILC2 by a small neuropeptide significantly suppressed the development of collagen-induced arthritis, accompanied by the expansion of eosinophils in the arthritic joints (10). In addition, induction of ILC2 by administration of IL-25/IL-33 accelerated the resolution of K/BxN serum-induced arthritis in wild-type but not eosinophil-deficient ΔdblGATA mice (8). On the contrary, neutralization of IL-5 by a monoclonal antibody blocked asthma-induced resolution of arthritis, with a reduced expansion of rEos in the joints (8). All these results supported the perspective that eosinophils are indispensable for ILC2-mediated resolution of arthritis (8).
Besides secreting a variety of pro-resolving lipid mediators that are crucial for the resolution of inflammation, the role of eosinophils in the suppression of arthritis could also include switching macrophages from pro-inflammatory M1 to anti-inflammatory M2 phenotype. As is well known, synovial macrophages act as central effector cells in the development of synovitis (3). The abundant presence of pro-inflammatory cytokines such as TNFα, IL-6, and IL-1β in the inflamed synovium suggests a predominant M1 macrophage phenotype in RA. However, the phenotype of synovial macrophages in vivo is highly complex and often exhibit a mixed polarization state (58). Previously it has been reported that even the same M1 macrophages recruited during the initial phase of arthritis can switch their phenotype toward M2 macrophages (59). In contrast to M1 macrophages that facilitate the inflammatory cascade in the synovium, M2 macrophages halt joint inflammation by removing dead cells (efferocytosis) and producing pro-resolving lipid mediators (58). It has been demonstrated that eosinophils induce polarization of macrophages toward M2 phenotype through secretion of IL-4, IL-13, and 12/15-LOX-derived lipid mediators (43). Eosinophil deficiency was associated with the impaired distribution of anti-inflammatory MHC-II- macrophages in the steady state as well as in arthritis (7). Both in vitro and in vivo studies showed that eosinophils foster the polarization of M1 to M2 macrophages in the synovial tissue, partly via the IκB/P38 MAPK signaling pathway (8, 9). This is consistent with previous studies that reported that eosinophils in adipose tissue mediate macrophage differentiation into M2 phenotype, which are required for glucose homeostasis (60, 61). Taken together, the ILC2–eosinophil–M2 macrophage axis represents a novel and important immunological pathway counteracting joint inflammation and eliciting resolution of arthritis (Figure 1).
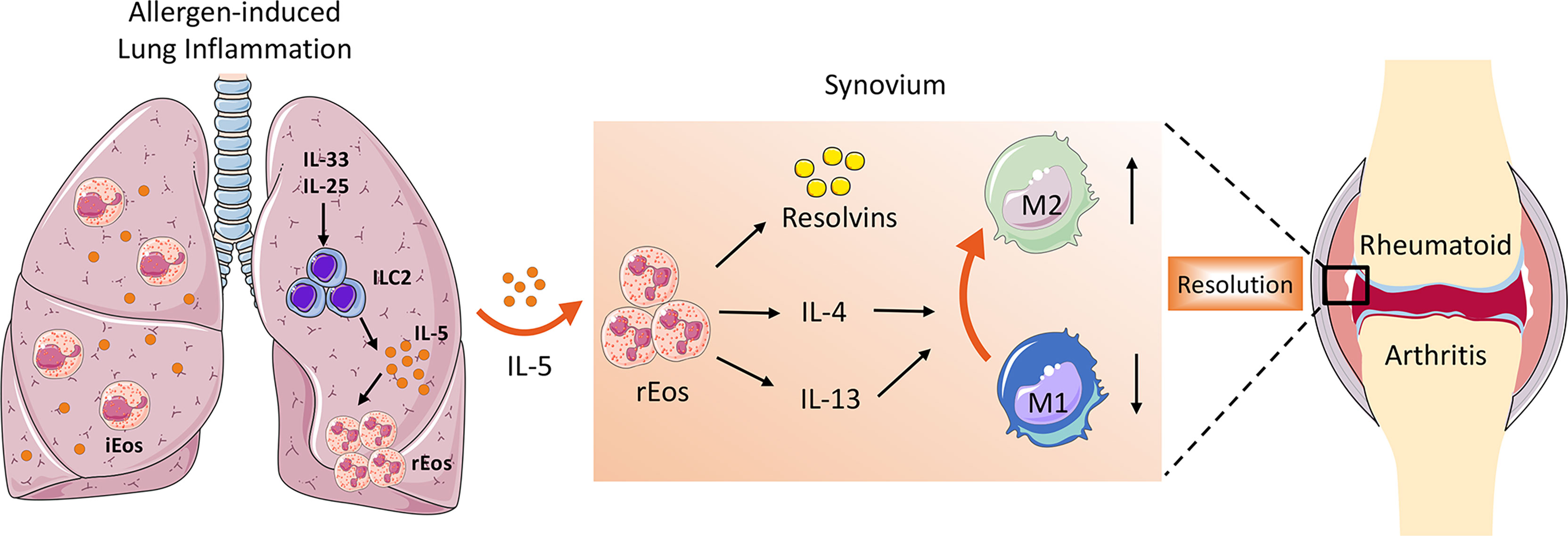
Figure 1 The role of ILC2–eosinophil–M2 macrophage axis in promoting resolution of arthritis. In allergen-triggered lung inflammation, ILC2 was expanded and activated in response to stimulators such as IL-33 and IL-25. Systemically elevated IL-5 secreted by ILC2 drives proliferation and recruitment of rEos into the inflamed joints, where they produce a variety of lipid mediators and anti-inflammatory type 2 cytokines, which facilitate M2 macrophage priming and eventually contribute to the resolution of arthritis.
With accumulating evidence and advancing technologies, the classical view of eosinophils has changed from a pro-inflammatory cell in helminth infection and allergy to a cell type aggressively involved in anti-inflammatory responses in the resolution of chronic inflammation. The existence of rEos in the synovium of RA patients extended previous understanding that eosinophils are critical in counteracting joint inflammation and facilitating the resolution of disease. These findings are crucial for designing new approaches to rebalancing immune homeostasis in inflammatory arthritis, considering that current therapies are centered on inhibiting pro-inflammatory cytokines and mediators rather than fostering the resolution of inflammation. Hence, understanding how rEos are activated and expanded will offer a novel strategy for the development of safe and effective treatment for arthritis.
Indeed, it is well known that the major extrinsic driver of eosinophil expansion was helminth infection. Numerous previous studies in experimental mouse models have demonstrated clinical improvement of inflammatory activity in a variety of autoimmune and inflammatory diseases including RA (7, 62–67). These observations led to the proposal that harness helminth and their secreted products would represent a promising interventional approach for the treatment of RA, as supported by the findings that a filarial nematode-derived glycoprotein, ES-62, has been proved to exert an anti-inflammatory and anti-osteoclastogenic effect in mouse arthritis models (68–71). In addition, several clinical trials have been performed to evaluate the immune-regulatory effect of helminth on autoimmune diseases, especially in inflammatory bowel disease (72). The mechanisms by which helminth and their derivatives modulate the host’s immune system were attributed to shifting immune responses from Th1 to Th2, induction of regulatory T and B cell subsets, as well as downregulation of IFN-γ and IL-17 (73). This helminth-based immunotherapy is becoming of major interest since current conventional management of RA relies generally on nonspecific inhibition of the immune system, which often results in severe infections and malignancies. Consistent with this concept, one of our recent studies showed that a small neuropeptide named Neuromedin U successfully alleviated collagen-induced arthritis, with evidence of ILC2-eosinophil activation (10). On the other hand, the plasticity of eosinophils offers another strategy that induces differentiation of pro-inflammatory eosinophils into regulatory phenotypes.
In summary, emerging evidence has shown that eosinophils not only act as a pro-inflammatory effector cell but also display a pro-resolving feature in RA. They consistently reside in the synovium of RA patients in remission and proliferate under stimulation of ILC2-derived IL-5. Mechanistically, the rEos promote the resolution of arthritis through secreting resolvins in a 12/15-LOX-dependent manner and switching synovial macrophages into the M2 phenotype.
YQ, H-ZJ, and Y-JL drafted the manuscript. ZC revised the manuscript. All authors contributed to the discussion and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant Nos. 81871227 and 81501344).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bain BJ. Hypereosinophilia. Curr Opin Hematol (2000) 7(1):21–5. doi: 10.1097/00062752-200001000-00005
2. Klion AD, Ackerman SJ, Bochner BS. Contributions of Eosinophils to Human Health and Disease. Annu Rev Pathol (2020) 15:179–209. doi: 10.1146/annurev-pathmechdis-012419-032756
3. Smolen JS, Aletaha D, Barton A, Burmester GR, Emery P, Firestein GS, et al. Rheumatoid Arthritis. Nat Rev Dis Primers (2018) 4:18001. doi: 10.1038/nrdp.2018.1
4. Weyand CM, Goronzy JJ. The Immunology of Rheumatoid Arthritis. Nat Immunol (2021) 22(1):10–8. doi: 10.1038/s41590-020-00816-x
5. Kerschbaumer A, Sepriano A, Smolen JS, van der Heijde D, Dougados M, van Vollenhoven R, et al. Efficacy of Pharmacological Treatment in Rheumatoid Arthritis: A Systematic Literature Research Informing the 2019 Update of the EULAR Recommendations for Management of Rheumatoid Arthritis. Ann Rheum Dis (2020) 79(6):744–59. doi: 10.1136/annrheumdis-2019-216656
6. Conigliaro P, Triggianese P, De Martino E, Fonti GL, Chimenti MS, Sunzini F, et al. Challenges in the Treatment of Rheumatoid Arthritis. Autoimmun Rev (2019) 18(7):706–13. doi: 10.1016/j.autrev.2019.05.007
7. Chen Z, Andreev D, Oeser K, Krljanac B, Hueber A, Kleyer A, et al. Th2 and Eosinophil Responses Suppress Inflammatory Arthritis. Nat Commun (2016) 7:11596. doi: 10.1038/ncomms11596
8. Andreev D, Liu M, Kachler K, Llerins Perez M, Kirchner P, Kolle J, et al. Regulatory Eosinophils Induce the Resolution of Experimental Arthritis and Appear in Remission State of Human Rheumatoid Arthritis. Ann Rheum Dis (2020) 80:451–68. doi: 10.1136/annrheumdis-2020-218902
9. Liu L, Zhang Y, Zheng X, Jin L, Xiang N, Zhang M, et al. Eosinophils Attenuate Arthritis by Inducing M2 Macrophage Polarization via Inhibiting the IkappaB/P38 MAPK Signaling Pathway. Biochem Biophys Res Commun (2019) 508(3):894–901. doi: 10.1016/j.bbrc.2018.12.010
10. Zhang Y, Qin Y, Chen Z. Neuromedin U Suppresses Collagen-Induced Arthritis Through ILC2-Th2 Activation. J Immunol Res (2021) 2021:5599439. doi: 10.1155/2021/5599439
11. Rothenberg ME, Pomerantz JL, Owen WF Jr., Avraham S, Soberman RJ, Austen KF, et al. Characterization of a Human Eosinophil Proteoglycan, and Augmentation of Its Biosynthesis and Size by Interleukin 3, Interleukin 5, and Granulocyte/Macrophage Colony Stimulating Factor. J Biol Chem (1988) 263(27):13901–8. doi: 10.1016/S0021-9258(18)68329-2
12. Shalit M, Sekhsaria S, Malech HL. Modulation of Growth and Differentiation of Eosinophils From Human Peripheral Blood CD34+ Cells by IL5 and Other Growth Factors. Cell Immunol (1995) 160(1):50–7. doi: 10.1016/0008-8749(95)80008-7
13. Johnston LK, Hsu CL, Krier-Burris RA, Chhiba KD, Chien KB, McKenzie A, et al. IL-33 Precedes IL-5 in Regulating Eosinophil Commitment and Is Required for Eosinophil Homeostasis. J Immunol (2016) 197(9):3445–53. doi: 10.4049/jimmunol.1600611
14. Hirasawa R, Shimizu R, Takahashi S, Osawa M, Takayanagi S, Kato Y, et al. Essential and Instructive Roles of GATA Factors in Eosinophil Development. J Exp Med (2002) 195(11):1379–86. doi: 10.1084/jem.20020170
15. Bedi R, Du J, Sharma AK, Gomes I, Ackerman SJ. Human C/EBP-Epsilon Activator and Repressor Isoforms Differentially Reprogram Myeloid Lineage Commitment and Differentiation. Blood (2009) 113(2):317–27. doi: 10.1182/blood-2008-02-139741
16. Yu C, Cantor AB, Yang H, Browne C, Wells RA, Fujiwara Y, et al. Targeted Deletion of a High-Affinity GATA-Binding Site in the GATA-1 Promoter Leads to Selective Loss of the Eosinophil Lineage In Vivo. J Exp Med (2002) 195(11):1387–95. doi: 10.1084/jem.20020656
17. Lu TX, Lim EJ, Itskovich S, Besse JA, Plassard AJ, Mingler MK, et al. Targeted Ablation of miR-21 Decreases Murine Eosinophil Progenitor Cell Growth. PloS One (2013) 8(3):e59397. doi: 10.1371/journal.pone.0059397
18. Lu TX, Lim EJ, Besse JA, Itskovich S, Plassard AJ, Fulkerson PC, et al. MiR-223 Deficiency Increases Eosinophil Progenitor Proliferation. J Immunol (2013) 190(4):1576–82. doi: 10.4049/jimmunol.1202897
19. Wagner LA, Christensen CJ, Dunn DM, Spangrude GJ, Georgelas A, Kelley L, et al. EGO, A Novel, Noncoding RNA Gene, Regulates Eosinophil Granule Protein Transcript Expression. Blood (2007) 109(12):5191–8. doi: 10.1182/blood-2006-06-027987
20. Weller PF, Spencer LA. Functions of Tissue-Resident Eosinophils. Nat Rev Immunol (2017) 17(12):746–60. doi: 10.1038/nri.2017.95
21. Acharya KR, Ackerman SJ. Eosinophil Granule Proteins: Form and Function. J Biol Chem (2014) 289(25):17406–15. doi: 10.1074/jbc.R113.546218
22. MacKenzie JR, Mattes J, Dent LA, Foster PS. Eosinophils Promote Allergic Disease of the Lung by Regulating CD4(+) Th2 Lymphocyte Function. J Immunol (2001) 167(6):3146–55. doi: 10.4049/jimmunol.167.6.3146
23. Wang HB, Ghiran I, Matthaei K, Weller PF. Airway Eosinophils: Allergic Inflammation Recruited Professional Antigen-Presenting Cells. J Immunol (2007) 179(11):7585–92. doi: 10.4049/jimmunol.179.11.7585
24. Phipps S, Lam CE, Mahalingam S, Newhouse M, Ramirez R, Rosenberg HF, et al. Eosinophils Contribute to Innate Antiviral Immunity and Promote Clearance of Respiratory Syncytial Virus. Blood (2007) 110(5):1578–86. doi: 10.1182/blood-2007-01-071340
25. Soukup JM, Becker S. Role of Monocytes and Eosinophils in Human Respiratory Syncytial Virus Infection In Vitro. Clin Immunol (2003) 107(3):178–85. doi: 10.1016/S1521-6616(03)00038-X
26. Ueki S, Melo RC, Ghiran I, Spencer LA, Dvorak AM, Weller PF. Eosinophil Extracellular DNA Trap Cell Death Mediates Lytic Release of Free Secretion-Competent Eosinophil Granules in Humans. Blood (2013) 121(11):2074–83. doi: 10.1182/blood-2012-05-432088
27. Specht S, Saeftel M, Arndt M, Endl E, Dubben B, Lee NA, et al. Lack of Eosinophil Peroxidase or Major Basic Protein Impairs Defense Against Murine Filarial Infection. Infect Immun (2006) 74(9):5236–43. doi: 10.1128/IAI.00329-06
28. Wong CK, Cheung PF, Ip WK, Lam CW. Intracellular Signaling Mechanisms Regulating Toll-Like Receptor-Mediated Activation of Eosinophils. Am J Respir Cell Mol Biol (2007) 37(1):85–96. doi: 10.1165/rcmb.2006-0457OC
29. Padigel UM, Lee JJ, Nolan TJ, Schad GA, Abraham D. Eosinophils can Function as Antigen-Presenting Cells to Induce Primary and Secondary Immune Responses to Strongyloides Stercoralis. Infect Immun (2006) 74(6):3232–8. doi: 10.1128/IAI.02067-05
30. Shi HZ, Humbles A, Gerard C, Jin Z, Weller PF. Lymph Node Trafficking and Antigen Presentation by Endobronchial Eosinophils. J Clin Invest (2000) 105(7):945–53. doi: 10.1172/JCI8945
31. Radke AL, Reynolds LE, Melo RC, Dvorak AM, Weller PF, Spencer LA. Mature Human Eosinophils Express Functional Notch Ligands Mediating Eosinophil Autocrine Regulation. Blood (2009) 113(13):3092–101. doi: 10.1182/blood-2008-05-155937
32. Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of Distinct CD4 T Helper Cell Fates by Different Notch Ligands on Antigen-Presenting Cells. Cell (2004) 117(4):515–26. doi: 10.1016/S0092-8674(04)00451-9
33. Sabin EA, Kopf MA, Pearce EJ. Schistosoma Mansoni Egg-Induced Early IL-4 Production Is Dependent Upon IL-5 and Eosinophils. J Exp Med (1996) 184(5):1871–8. doi: 10.1084/jem.184.5.1871
34. Voehringer D, Shinkai K, Locksley RM. Type 2 Immunity Reflects Orchestrated Recruitment of Cells Committed to IL-4 Production. Immunity (2004) 20(3):267–77. doi: 10.1016/S1074-7613(04)00026-3
35. Gebreselassie NG, Moorhead AR, Fabre V, Gagliardo LF, Lee NA, Lee JJ, et al. Eosinophils Preserve Parasitic Nematode Larvae by Regulating Local Immunity. J Immunol (2012) 188(1):417–25. doi: 10.4049/jimmunol.1101980
36. Mattes J, Yang M, Mahalingam S, Kuehr J, Webb DC, Simson L, et al. Intrinsic Defect in T Cell Production of Interleukin (IL)-13 in the Absence of Both IL-5 and Eotaxin Precludes the Development of Eosinophilia and Airways Hyperreactivity in Experimental Asthma. J Exp Med (2002) 195(11):1433–44. doi: 10.1084/jem.20020009
37. Odemuyiwa SO, Ghahary A, Li Y, Puttagunta L, Lee JE, Musat-Marcu S, et al. Cutting Edge: Human Eosinophils Regulate T Cell Subset Selection Through Indoleamine 2,3-Dioxygenase. J Immunol (2004) 173(10):5909–13. doi: 10.4049/jimmunol.173.10.5909
38. Sugimoto MA, Sousa LP, Pinho V, Perretti M, Teixeira MM. Resolution of Inflammation: What Controls Its Onset? Front Immunol (2016) 7:160. doi: 10.3389/fimmu.2016.00160
39. Serhan CN. Pro-Resolving Lipid Mediators Are Leads for Resolution Physiology. Nature (2014) 510(7503):92–101. doi: 10.1038/nature13479
40. Schett G. Resolution of Inflammation in Arthritis. Semin Immunopathol (2019) 41(6):675–9. doi: 10.1007/s00281-019-00768-x
41. Serhan CN, Levy BD. Resolvins in Inflammation: Emergence of the Pro-Resolving Superfamily of Mediators. J Clin Invest (2018) 128(7):2657–69. doi: 10.1172/JCI97943
42. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and Protectin D1 Activate Inflammation-Resolution Programmes. Nature (2007) 447(7146):869–74. doi: 10.1038/nature05877
43. Yamada T, Tani Y, Nakanishi H, Taguchi R, Arita M, Arai H. Eosinophils Promote Resolution of Acute Peritonitis by Producing Proresolving Mediators in Mice. FASEB J (2011) 25(2):561–8. doi: 10.1096/fj.10-170027
44. Tani Y, Isobe Y, Imoto Y, Segi-Nishida E, Sugimoto Y, Arai H, et al. Eosinophils Control the Resolution of Inflammation and Draining Lymph Node Hypertrophy Through the Proresolving Mediators and CXCL13 Pathway in Mice. FASEB J (2014) 28(9):4036–43. doi: 10.1096/fj.14-251132
45. Masterson JC, McNamee EN, Fillon SA, Hosford L, Harris R, Fernando SD, et al. Eosinophil-Mediated Signalling Attenuates Inflammatory Responses in Experimental Colitis. Gut (2015) 64(8):1236–47. doi: 10.1136/gutjnl-2014-306998
46. Hallgren R, Feltelius N, Svenson K, Venge P. Eosinophil Involvement in Rheumatoid Arthritis as Reflected by Elevated Serum Levels of Eosinophil Cationic Protein. Clin Exp Immunol (1985) 59(3):539–46.
47. Mertens AV, De Clerck LS, Moens MM, Bridts CH, Stevens WJ. Study of Eosinophil-Endothelial Adhesion, Production of Oxygen Radicals and Release of Eosinophil Cationic Protein by Peripheral Blood Eosinophils of Patients With Rheumatoid Arthritis. Clin Exp Allergy (1993) 23(10):868–73. doi: 10.1111/j.1365-2222.1993.tb00266.x
48. Guellec D, Milin M, Cornec D, Tobon GJ, Marhadour T, Jousse-Joulin S, et al. Eosinophilia Predicts Poor Clinical Outcomes in Recent-Onset Arthritis: Results From the ESPOIR Cohort. RMD Open (2015) 1(1):e000070. doi: 10.1136/rmdopen-2015-000070
49. Emmanuel D, Parija SC, Jain A, Misra DP, Kar R, Negi VS. Persistent Eosinophilia in Rheumatoid Arthritis: A Prospective Observational Study. Rheumatol Int (2019) 39(2):245–53. doi: 10.1007/s00296-018-4191-1
50. Mesnil C, Raulier S, Paulissen G, Xiao X, Birrell MA, Pirottin D, et al. Lung-Resident Eosinophils Represent a Distinct Regulatory Eosinophil Subset. J Clin Invest (2016) 126(9):3279–95. doi: 10.1172/JCI85664
51. Kronke G, Katzenbeisser J, Uderhardt S, Zaiss MM, Scholtysek C, Schabbauer G, et al. 12/15-Lipoxygenase Counteracts Inflammation and Tissue Damage in Arthritis. J Immunol (2009) 183(5):3383–9. doi: 10.4049/jimmunol.0900327
52. Fang W, Zhang Y, Chen Z. Innate Lymphoid Cells in Inflammatory Arthritis. Arthritis Res Ther (2020) 22(1):25. doi: 10.1186/s13075-020-2115-4
53. Wu X. Innate Lymphocytes in Inflammatory Arthritis. Front Immunol (2020) 11:565275. doi: 10.3389/fimmu.2020.565275
54. Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, et al. Resolution of Inflammation by Interleukin-9-Producing Type 2 Innate Lymphoid Cells. Nat Med (2017) 23(8):938–44. doi: 10.1038/nm.4373
55. Omata Y, Frech M, Primbs T, Lucas S, Andreev D, Scholtysek C, et al. Group 2 Innate Lymphoid Cells Attenuate Inflammatory Arthritis and Protect From Bone Destruction in Mice. Cell Rep (2018) 24(1):169–80. doi: 10.1016/j.celrep.2018.06.005
56. Omata Y, Frech M, Lucas S, Primbs T, Knipfer L, Wirtz S, et al. Type 2 Innate Lymphoid Cells Inhibit the Differentiation of Osteoclasts and Protect From Ovariectomy-Induced Bone Loss. Bone (2020) 136:115335. doi: 10.1016/j.bone.2020.115335
57. Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, et al. Type 2 Innate Lymphoid Cells Control Eosinophil Homeostasis. Nature (2013) 502(7470):245–8. doi: 10.1038/nature12526
58. Kennedy A, Fearon U, Veale DJ, Godson C. Macrophages in Synovial Inflammation. Front Immunol (2011) 2:52. doi: 10.3389/fimmu.2011.00052
59. Misharin AV, Cuda CM, Saber R, Turner JD, Gierut AK, Haines GK 3rd, et al. Nonclassical Ly6C(-) Monocytes Drive the Development of Inflammatory Arthritis in Mice. Cell Rep (2014) 9(2):591–604. doi: 10.1016/j.celrep.2014.09.032
60. Wu D, Molofsky AB, Liang HE, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils Sustain Adipose Alternatively Activated Macrophages Associated With Glucose Homeostasis. Science (2011) 332(6026):243–7. doi: 10.1126/science.1201475
61. Qiu Y, Nguyen KD, Odegaard JI, Cui X, Tian X, Locksley RM, et al. Eosinophils and Type 2 Cytokine Signaling in Macrophages Orchestrate Development of Functional Beige Fat. Cell (2014) 157(6):1292–308. doi: 10.1016/j.cell.2014.03.066
62. Cheng Y, Zhu X, Wang X, Zhuang Q, Huyan X, Sun X, et al. Trichinella Spiralis Infection Mitigates Collagen-Induced Arthritis via Programmed Death 1-Mediated Immunomodulation. Front Immunol (2018) 9:1566. doi: 10.3389/fimmu.2018.01566
63. Sarter K, Kulagin M, Schett G, Harris NL, Zaiss MM. Inflammatory Arthritis and Systemic Bone Loss Are Attenuated by Gastrointestinal Helminth Parasites. Autoimmunity (2017) 50(3):151–7. doi: 10.1080/08916934.2016.1261837
64. Osada Y, Morita K, Tahara S, Ishihara T, Wu Z, Nagano I, et al. Th2 Signals Are Not Essential for the Anti-Arthritic Effects of Trichinella Spiralis in Mice. Parasite Immunol (2020) 42(1):e12677. doi: 10.1111/pim.12677
65. Osada Y, Shimizu S, Kumagai T, Yamada S, Kanazawa T. Schistosoma Mansoni Infection Reduces Severity of Collagen-Induced Arthritis via Down-Regulation of Pro-Inflammatory Mediators. Int J Parasitol (2009) 39(4):457–64. doi: 10.1016/j.ijpara.2008.08.007
66. Shi M, Wang A, Prescott D, Waterhouse CC, Zhang S, McDougall JJ, et al. Infection With an Intestinal Helminth Parasite Reduces Freund's Complete Adjuvant-Induced Monoarthritis in Mice. Arthritis Rheum (2011) 63(2):434–44. doi: 10.1002/art.30098
67. Song X, Shen J, Wen H, Zhong Z, Luo Q, Chu D, et al. Impact of Schistosoma Japonicum Infection on Collagen-Induced Arthritis in DBA/1 Mice: A Murine Model of Human Rheumatoid Arthritis. PloS One (2011) 6(8):e23453. doi: 10.1371/journal.pone.0023453
68. Doonan J, Lumb FE, Pineda MA, Tarafdar A, Crowe J, Khan AM, et al. Protection Against Arthritis by the Parasitic Worm Product ES-62, and Its Drug-Like Small Molecule Analogues, Is Associated With Inhibition of Osteoclastogenesis. Front Immunol (2018) 9:1016. doi: 10.3389/fimmu.2018.01016
69. Rzepecka J, Pineda MA, Al-Riyami L, Rodgers DT, Huggan JK, Lumb FE, et al. Prophylactic and Therapeutic Treatment With a Synthetic Analogue of a Parasitic Worm Product Prevents Experimental Arthritis and Inhibits IL-1beta Production via NRF2-Mediated Counter-Regulation of the Inflammasome. J Autoimmun (2015) 60:59–73. doi: 10.1016/j.jaut.2015.04.005
70. Rodgers DT, Pineda MA, McGrath MA, Al-Riyami L, Harnett W, Harnett MM. Protection Against Collagen-Induced Arthritis in Mice Afforded by the Parasitic Worm Product, ES-62, Is Associated With Restoration of the Levels of Interleukin-10-Producing B Cells and Reduced Plasma Cell Infiltration of the Joints. Immunology (2014) 141(3):457–66. doi: 10.1111/imm.12208
71. Pineda MA, McGrath MA, Smith PC, Al-Riyami L, Rzepecka J, Gracie JA, et al. The Parasitic Helminth Product ES-62 Suppresses Pathogenesis in Collagen-Induced Arthritis by Targeting the Interleukin-17-Producing Cellular Network at Multiple Sites. Arthritis Rheum (2012) 64(10):3168–78. doi: 10.1002/art.34581
72. Fleming JO, Weinstock JV. Clinical Trials of Helminth Therapy in Autoimmune Diseases: Rationale and Findings. Parasite Immunol (2015) 37(6):277–92. doi: 10.1111/pim.12175
Keywords: eosinophil, rheumatoid arthritis, resolution, innate lymphoid cells, alternatively activated macrophages
Citation: Qin Y, Jin HZ, Li YJ and Chen Z (2021) Emerging Role of Eosinophils in Resolution of Arthritis. Front. Immunol. 12:764825. doi: 10.3389/fimmu.2021.764825
Received: 26 August 2021; Accepted: 24 September 2021;
Published: 18 October 2021.
Edited by:
Qingjun Pan, Affiliated Hospital of Guangdong Medical University, ChinaReviewed by:
Amir Abdoli, Jahrom University of Medical Sciences, IranCopyright © 2021 Qin, Jin, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhu Chen, ZG9jemNoZW5AdXN0Yy5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.