 Panpan Chang
Panpan Chang Hao Li
Hao Li Hui Hu3
Hui Hu3 Yongqing Li
Yongqing Li- 1Trauma Medicine Center, Peking University People’s Hospital, Key Laboratory of Trauma and Neural Regeneration (Peking University), National Center for Trauma Medicine of China, Beijing, China
- 2Department of Emergency, First Hospital of China Medical University, Shenyang, China
- 3Department of Traumatology, Central Hospital of Chongqing University, Chongqing Emergency Medical Center, Chongqing, China
- 4Department of Surgery, University of Michigan, Ann Arbor, MI, United States
Autophagy fights against harmful stimuli and degrades cytosolic macromolecules, organelles, and intracellular pathogens. Autophagy dysfunction is associated with many diseases, including infectious and inflammatory diseases. Recent studies have identified the critical role of the NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasomes activation in the innate immune system, which mediates the secretion of proinflammatory cytokines IL-1β/IL-18 and cleaves Gasdermin D to induce pyroptosis in response to pathogenic and sterile stimuli. Accumulating evidence has highlighted the crosstalk between autophagy and NLRP3 inflammasome in multifaceted ways to influence host defense and inflammation. However, the underlying mechanisms require further clarification. Histone deacetylase 6 (HDAC6) is a class IIb deacetylase among the 18 mammalian HDACs, which mainly localizes in the cytoplasm. It is involved in two functional deacetylase domains and a ubiquitin-binding zinc finger domain (ZnF-BUZ). Due to its unique structure, HDAC6 regulates various physiological processes, including autophagy and NLRP3 inflammasome, and may play a role in the crosstalk between them. In this review, we provide insight into the mechanisms by which HDAC6 regulates autophagy and NLRP3 inflammasome and we explored the possibility and challenges of HDAC6 in the crosstalk between autophagy and NLRP3 inflammasome. Finally, we discuss HDAC6 inhibitors as a potential therapeutic approach targeting either autophagy or NLRP3 inflammasome as an anti-inflammatory strategy, although further clarification is required regarding their crosstalk.
Introduction
Autophagy is a conservative mechanism for maintaining homeostasis in cells, which degrades misfolded proteins, damaged organelles, and intracellular pathogens (1). It is associated with many diseases, including infectious and inflammatory diseases (2). The NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasomes are oligomeric complexes activated by invading pathogens, endogenous danger signals, and stress signals (3). The activation of NLRP3 inflammasome induces interleukin-1β (IL-1β) and interleukin-18 (IL-18) release and pyroptosis, which is a caspase-1-dependent form of programmed cell death (4). NLRP3 inflammasome is essential for defense against infectious and inflammatory diseases, and its aberrant activation aggravates inflammation and tissue damage (5, 6). Recent studies have suggested that autophagy eliminates the overaction of NLRP3 inflammasome and maintains homeostasis (7–9). Additionally, NLRP3 inflammasome activation can upregulate autophagy to suppress excessive responses and protect the host (10, 11). There is emerging evidence highlighting the importance of crosstalk between autophagy and NLRP3 inflammasome in various inflammatory diseases (12–16).
Histone deacetylase 6 (HDAC6) is a class IIb deacetylase found in 18 mammalian HDACs. It harbors two functional deacetylase catalytic domains and a ubiquitin-binding zinc finger domain (ZnF-BUZ) (17). HDAC6 is a structurally and functionally unique cytoplasmic deacetylase that can deacetylate multiple non-histone proteins such as α-tubulin, cortactin, heat shock protein (HSP90), heat shock transcription factor-1 (HSF-1), peroxiredoxin I (Prx I), and peroxiredoxin II (Prx II) (18–21). In addition, HDAC6 binds to ubiquitinated misfolded proteins through the ZnF-BUZ (22). Therefore, it is essential for multiple physiological and pathological processes. Recent studies have demonstrated that HDAC6 regulates autophagy and NLRP3 inflammasome activation through various mechanisms (14, 23–27). It is suggested that HDAC6 plays a possible role in the crosstalk between autophagy and NLRP3 inflammasome, although there is little direct evidence to date. In this review, we present the distinct roles of HDAC6 in the regulation of autophagy and NLRP3 inflammasome. We then focus on exploring the possibility and challenges of HDAC6 involvement in the crosstalk between autophagy and NLRP3 inflammasome. Finally, we discuss HDAC6 inhibitors as a promising therapeutic target for various diseases and its prospect in the crosstalk between autophagy and NLRP3 inflammasome.
The Role of HDAC6 in Autophagy
Autophagy, specifically macroautophagy, is a conserved self-eating process that is vital for cellular homeostasis and delivery intracellular components, including soluble proteins, aggregated proteins, organelles, macromolecular complexes, and foreign bodies for degradation (28). This process begins with the sequestration of organelles or portions of the cytoplasm into a double-membrane structure, the autophagosome (29). Autophagosomes fuse with lysosomes to form hybrid organelles called autophagolysosomes (30). Autophagolysosomes degrade the contents to achieve cell homeostasis and organelle renewal (31). HDAC6 is involved in the regulation of autophagy at multiple levels, including participation in post-translational modifications (PTM) of autophagy-related transcription factors (32, 33), the formation of aggresomes that are routinely cleaned through the autophagy pathway (22, 34, 35), and the transportation and degradation of autophagosomes (Figure 1) (23, 25).
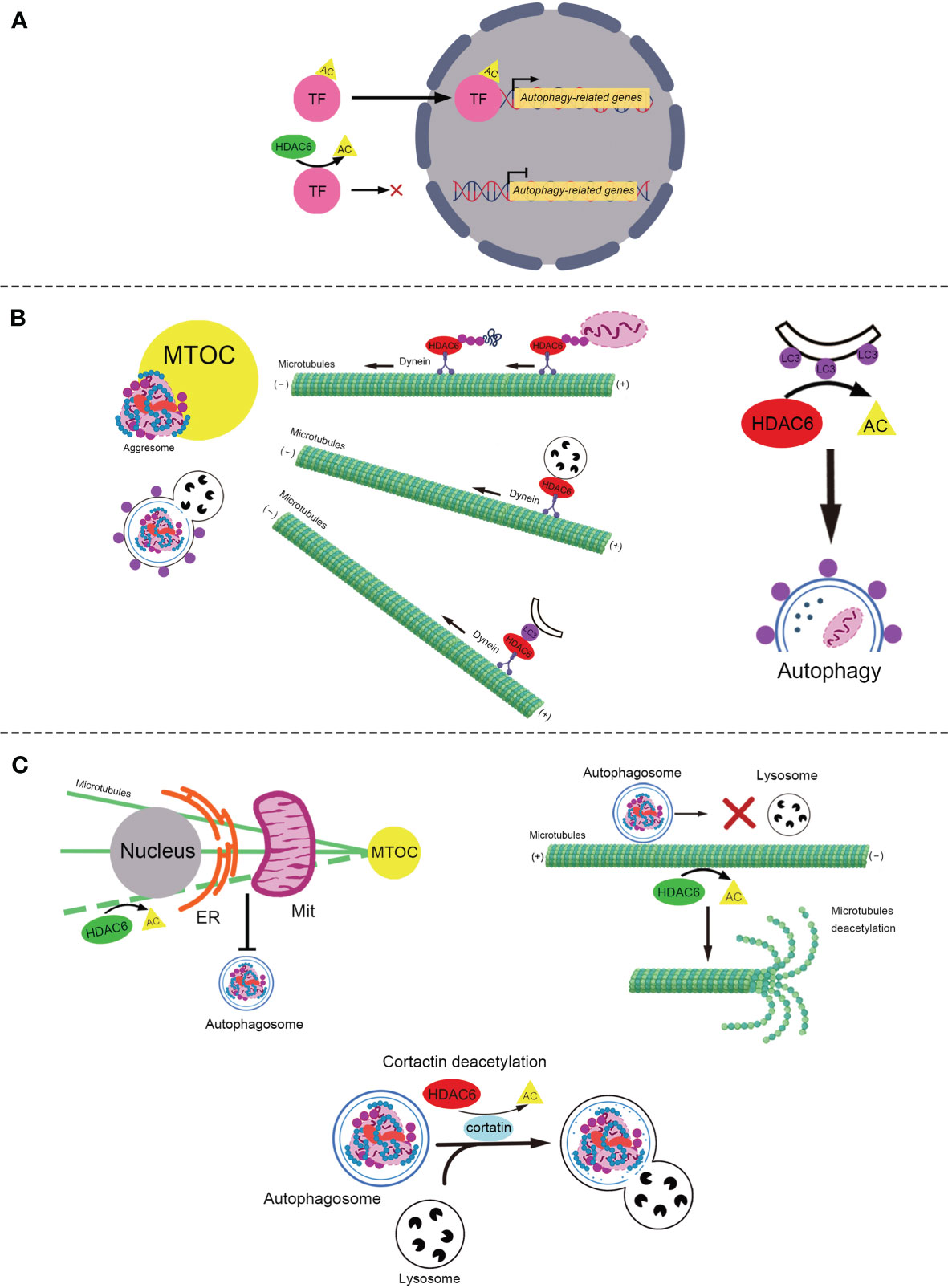
Figure 1 The role of HDAC6 in autophagy. (A) The role of HDAC6 in PTM of autophagy-related transcription factors. HDAC6 deacetylates transcription factors, TFEB and FOXO1, to reduce their transcriptional activity and inhibit autophagy. (B) HDAC6 promotes the autophagic degradation of aggresome in various ways. Left: HDAC6 interacts with the microtubule motor protein dynein to escort the ubiquitinated misfolded protein or ubiquitinated damaged mitochondria to form the aggresome, to transport the lysosome for the degardaion of aggresome, and to deliver LC3-II (the purple point) to promote the formation of the autophagosome containing aggresome. Right: HDAC6 deacetylates LC3-II to promote the formation of the autophagosome. (C) HDAC6 plays various roles in the regulation of autophagy via deacetylating α-tubulin and cortactin (positive and negative roles are marked with red and green respectively). Top-left: HDAC6 deacetylates microtubules to block the ER–Mit contact where autophagosome generates. Top-right: HDAC6 suppresses the transport of autophagosomes through deacetylating and reducing the stability of the microtubules. Bottom: HDAC6 blocks the fusion of the autophagosome that contains the misfolded protein or mitochondria and the lysosome by deacetylating cortactin. HDAC6, Histone deacetylase 6; TF, Transcription factor; PTM, Post-translational modifications; AC, Acetylation; FOXO1, Forkhead Box 1; TFEB, Transcription factor EB; MTOC, Microtubule-organizing center; LC3, Microtubule-associated protein 1 light chain 3; ER, Endoplasmic Reticulum; Mit, Mitochondria.
The Role of HDAC6 in PTM of Autophagy-Related Transcription Factors
PTM of autophagy-related transcription factors (36), such as transcription factor EB (TFEB) and Forkhead Box 1 (FOXO1) affect their activities, which regulate the autophagy-lysosome pathway (37–39). Recently, it was reported that HDAC6 deacetylates TFEB and FOXO1 to decrease their activity and inhibit autophagy (32, 33, 40, 41).
TFEB is a major regulator of the autophagy-lysosomal pathway (42). Acetylation of TFEB causes translocation to the nucleus and enhancement of autophagy and lysosomal gene transcription (32, 40). It was reported that acetylated TFEB accumulates in the nuclei, which is associated with increased transcriptional activity and lysosomal function following treatment with a pan-HDAC inhibitor, SAHA (40). Similarly, in subtotally nephrectomized rats, the HDAC6 inhibitor Tubastatin A (Tub-A) promotes the acetylation of TFEB, which translocates into the nucleus and enhances the expression of autophagy-related protein Beclin 1 (32), a known direct target of TFEB (43). However, Jung et at. showed that HDAC6 overexpression activated c-Jun NH2-terminal kinase (JNK) and increased the phosphorylation of c-Jun, which activated Beclin 1 dependent autophagy in liver cancer (44).
Besides TFEB, HDAC6 also deacetylates the transcription factor FOXO1 (33), which is a conserved transcription factor that modulates autophagy (45). It has been reported that HDAC6 binds to and deacetylates cytosolic FOXO1, which is required for nuclear translocation and stabilization of interleukin-17 (IL-17)-producing helper T cells (46). Zhang et al. found that trichostatin A (TSA), an HDAC inhibitor, enhances the transcriptional activity of FOXO1 by increasing its acetylation, which enhances the process of autophagy (41). Recently, another study reported that HDAC6 was suppressed by the calcium binding protein S100 calcium binding protein A11 (S100A11) in hepatocytes, which leads to the upregulation of FOXO1 acetylation to enhance its transactional activity and activate autophagy (33).
The Role of HDAC6 in Aggresome Degradation Mediated by Autophagy
Under physiological conditions, misfolded and aggregated proteins are cleaned through ubiquitylation and proteasome-mediated degradation (47, 48). When the degrading capacity is overwhelmed (47), misfolded or aggregated proteins are generally transported along microtubules towards the microtubule-organizing center (MTOC) through motor protein dynein (49). Once at the MTOC, they are packaged into a single aggresome (49), which is eventually degraded by autophagy (50). Aggresomes are crucial for the clearance of accumulated misfolded proteins and cellular death (51). HDAC6 is a component of aggresomes induced by misfolded proteins. In the process of forming aggresomes containing ubiquitinated proteins, HDAC6 works as a bridge between ubiquitinated-misfolded proteins and the dynein motor (22). It binds to polyubiquitinated misfolded CFTR-ΔF508 via its C-terminus ubiquitin binding ZnF-BUZ domain, and it binds to the dynein motor through a separate domain, dynein motor domain (DMB) (22). However, HDAC6 may not recognize protein aggregates and may not bind directly to polyubiquitinated proteins. A recent study indicated that the ZnF-UBP domain of HDAC6 binds to unconjugated C-terminal diglycine motifs of ubiquitin, and this interaction is important for the binding and transport of polyubiquitinated protein aggregates (35). In addition, small-molecule inhibition of HDAC6 has been shown to inhibit the formation of aggresomes in multiple myeloma and lymphoma models (52–54). Recently, HDAC6 was found to be involved in the formation of aggresomes of α-synuclein, TAR DNA-binding protein 43, and Tau (34, 55, 56). It has been suggested that HDAC6 acts as a scaffold for a variety of ubiquitinated proteins. Strikingly, although HDAC6 was initially concentrated at the aggresome as previously reported (22), it was no longer detectable in the ubiquitin-positive structures once aggresomes were cleared by autophagy (57). As the HDAC6 protein levels remained stable during the biological process of aggresome formation and clearance, HDAC6 is not degraded together with aggresomes (57). HDAC6 seems recycled during aggresome-autophagy.
Other studies have shown that HDAC6 is required for lysosomes to form aggregates. Lysosomes are generated in the cell periphery and transported to MTOC to degrade aggresomes (58). HDAC6 and dynein transport lysosomes along microtubules to promote autophagic degradation of aggresomes (59, 60). Lee et al. found that lysosomes in HDAC6 knockout mouse embryonic fibroblasts were dispersed to the cell periphery and not concentrated to protein aggregates (59). Similarly, Iwata et al. also showed that HDAC6 knockdown leads to the periplasmic dispersion of lysosomes (60). This indicates that the targeting of lysosomes to autophagic substrates is regulated by HDAC6.
Microtubule-associated protein 1 light chain 3 (LC3) is a well-known regulator of autophagy (61). LC3-I is conjugated to phosphatidylethanolamine to form LC3-PE conjugate (LC3-II), which is recruited to autophagosomal membranes to promote its formation (62, 63). HDAC6 transports LC3 to the MTOC to promote autophagosome formation (60). The knockdown of HDAC6 attenuates the recruitment of LC3 to aggregated Huntingtin protein for degradation in Neuro2a cells and HeLa cells (60). However, the mechanism by which HDAC6 regulates LC3 needs to be further elucidated. In addition, the deacetylation of LC3 influences autophagy in starvation-induced cells (64). Liu et al. reported that the deacetylation of LC3-II modulated by HDAC6 promotes autophagic flux in starvation-induced HeLa cells (65). The acetylation of LC3-II increases in HDAC6 siRNA Hela cells, which blocks autophagy flux (65). These studies suggested HDAC6 works as a scaffold protein or deacetylase to regulate LC3, which promotes autophagy.
HDAC6 Deacetylates α-Tubulin and Cortactin to Mediate Autophagy
HDAC6 associates with microtubules and filamentous actin (F-actin) by deacetylating α-tubulin (66–68), and cortactin (19), both of which play important roles in autophagy (69–71). As the first reported and most studied physiological substrate of HADC6, α-tubulin is deacetylated by HDAC6 at lysine 40 (72). Additionally, acetylation of cortactin following inhibition of HDAC6 reduces its interaction with F-actin (19).
Microtubules, composed of α- and β-tubulin heterodimers (73), are essential for cell division, shaping, motility, and intracellular transport (74). Accumulating evidence indicates that microtubules participate in the mediation of autophagosome formation (75, 76), autophagosome transport across the cytoplasm (77, 78), and the formation of autolysosomes (79, 80). Lei et al. demonstrated that HDAC6 decreases the acetylation of microtubules to inhibit the formation of autophagosomes in acidic pH-mediated rat cardiomyocytes (81). The possible underlying mechanism is that acetylation of α-tubulin enhances the endoplasmic reticulum-mitochondria contact, which promotes the formation of autophagosomes (82, 83). Additionally, other studies have reported that HDAC6 mediates α-tubulin deacetylation to suppress autophagy in podocytes and human embryonic kidney 293 cells (84, 85). However, the underlying mechanisms remain unclear. It has been suggested that HDAC6 impairs stable acetylated microtubules via deacetylating α-tubulin, which leads to the blockade of autophagosome-lysosome fusion and accumulation of autophagosomes (86). In mouse embryonic fibroblasts, bpV(phen), an insulin mimic and a PTEN inhibitor, blocked autophagosomal degradation by reducing the stability of p62 to activate HDAC6 to impair the fusion of autophagosomes and lysosomes, followed by acetylation of microtubules (86). Furthermore, Li et at. found that HDAC6 inhibited the transportation of autophagosomes to fuse with lysosomes through the deacetylation of α-tubulin, resulting in the depolymerization of microtubules (25). In conclusion, HDAC6 suppresses the formation and degradation of autophagosome via deacetylation the microtubules.
As an important part of the cytoskeleton, the F-actin network plays an important role in cell movement, adhesion, morphology, and intracellular material transport (87). Additionally, the F-actin network is essential for the fusion of autophagosomes and lysosomes (70). Lee et al. found that HDAC6 promotes autophagy by recruiting a cortactin-dependent, actin-remodeling machinery, which in turn assembles an F-actin network that stimulates autophagosome-lysosome fusion and substrate degradation (23). However, this mechanism has been demonstrated in quality control autophagy but not in starvation-induced autophagy (23). It is possible that substrates of starvation-induced autophagy are widely distributed in the cell and encounter lysosomes more easily (23). Recently, another study reported that HDAC6 was recruited by ATP13A2, whose mutations are associated with Kufor-Rakeb syndrome (KRS), an autosomal recessive form of juvenile-onset atypical Parkinson’s disease (PD), which is known as Parkinson’s disease-9, to deacetylate cortactin and promote autophagosome-lysosome fusion and autophagy (88). Impaired ATP13A2/HDAC6/cortactin signaling likely contributes to KRS and PD pathogenesis by disrupting the clearance of protein aggregates and damaged mitochondria (88). These results indicate HDAC6 deacetylates cortactin which enhances the activity of the F-actin network to promote the fusion of autophagosomes and lysosomes.
The Role of HDAC6 in Mitophagy
Mitophagy is an autophagic response that specifically targets damaged and potentially cytotoxic mitochondria (89, 90). HDAC6 has also been reported to mediate mitophagy (88, 91, 92). The underlying mechanisms may include the formation of mitochondrial aggregates (mito-aggresomes) (91–94), and degradation of mitophagosomes through cortactin or α-tubulin action (88, 92, 93, 95). Parkin, a ubiquitin ligase, promotes mitophagy by catalyzing mitochondrial ubiquitination, which in turn recruits ubiquitin-binding autophagic components, HDAC6 and p62, leading to mitochondrial clearance (91, 92). Similar to the aggresome, the formation of mito-aggresomes depends on the transportation of microtubule dynein motors mediated by HDAC6 to MTOC (91, 92). HDAC6 deacetylates cortactin to promote the fusion of mitophagosomes and lysosomes (91, 93). Mito-aggresomes are then degraded by the conventional autophagy pathway (88, 91, 93). Conversely, Pedro et al. found that pharmacological inhibition of the HDAC6 deacetylase activity with Tub-A, did not block striatal neuronal autophagosome-lysosome fusion, suggesting no impairment in mitophagy (95). Interestingly, that HDAC6 inhibition increased acetylated α-tubulin levels, and induced mitophagy in striatal neurons (95). Overall, the effects and mechanisms of HDAC6 in mitophagy remain to be elucidated.
The Relationship of HDAC6 and p62 in Autophagy
P62 is the first selective autophagy adaptor protein discovered in mammals (96, 97), and plays multiple roles in autophagy, including participating in the formation of aggresomes (98, 99), anchoring the aggresomes to the autophagosome (100), and the degradation of aggresomes in selective autophagy (101, 102). Accumulating evidence indicates that the interaction between HDAC6 and p62 is curial for autophagy (23, 39, 86, 91, 103–108). As mentioned above, HDAC6 and p62 work as two ubiquitin-binding proteins required for efficient autophagy that target protein aggregates and damaged mitochondria (23, 91). Cyclin-dependent kinase 1 (CDK1) in human breast cancer is degraded by p62- and HDAC6- mediated selective autophagy (104). Additionally, interferon-stimulated gene 15 (ISG15) interacts with HDAC6 and p62 independently to be degraded through autophagy (105). These studies suggest that HDAC6 and p62 may mediate autophagy in parallel. However, other studies have indicated that HDAC6 and p62 may regulate autophagy synergistically. Yan et al. reported that HDAC6 regulates lipid droplet turnover in response to nutrient deprivation via p62-mediated aggresome formation (107). Interestingly, some studies have indicated that p62 inhibits the deacetylase activity of HDAC6 to enhance the acetylation of microtubules or cortactin, promoting autophagic flux (86, 103, 108). In contrast, Jiang et al. showed that p62 promotes the expression of HDAC6, reducing the acetylation level of microtubules and inhibiting autophagy in hormone-independent prostate adenocarcinoma cell lines (109). However, the mechanisms by which p62 regulates HDAC6 remain to be clarified. The relationship between HDAC6 and p62 is complicated. Thus, further research is required to elucidate the underlying mechanisms.
It is interesting that HDAC6 differentially regulates autophagy via multiple mechanisms. It may depend on the specific cell type, disease, and autophagy inducer/inhibitor. The mechanisms of HDAC6 regulation in autophagy require further investigation.
The Role of HDAC6 in NLRP3 Inflammasome
The canonical NLRP3 inflammasome consists of NLRP3 (the sensors), apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) (the adaptor), and protein-caspase-1 (the effector) (4). It is critical for the innate immune system to mediate caspase-1 activation to release proinflammatory cytokines IL-1β/IL-18 and cleave Gasdermin D to induce pyroptosis in response to microbial infection and cellular damage (110–112). The mechanism of the canonical NLRP3 inflammasome is currently considered to include the following: priming, activation, and PTM- interacting components. The primary signal induces the activation of Toll-like-receptors (TLRs) and nuclear factor-kappa B (NF-κB), leading to transcriptional upregulation of NLRP3, pro-IL-1β, and pro-IL-18 (112). The secondary signal is provided by multiple molecular or cellular events, including ionic flux, mitochondrial dysfunction, and reactive oxygen species (ROS) generation (113). The aberrant activation of NLRP3 inflammasome is responsible for a wide range of inflammatory diseases such as sepsis, trauma and gout (3, 114–116). HDAC6 plays various roles in the priming, activation and PTM of NLRP3 inflammasome (Figure 2) (14, 26, 27, 117).
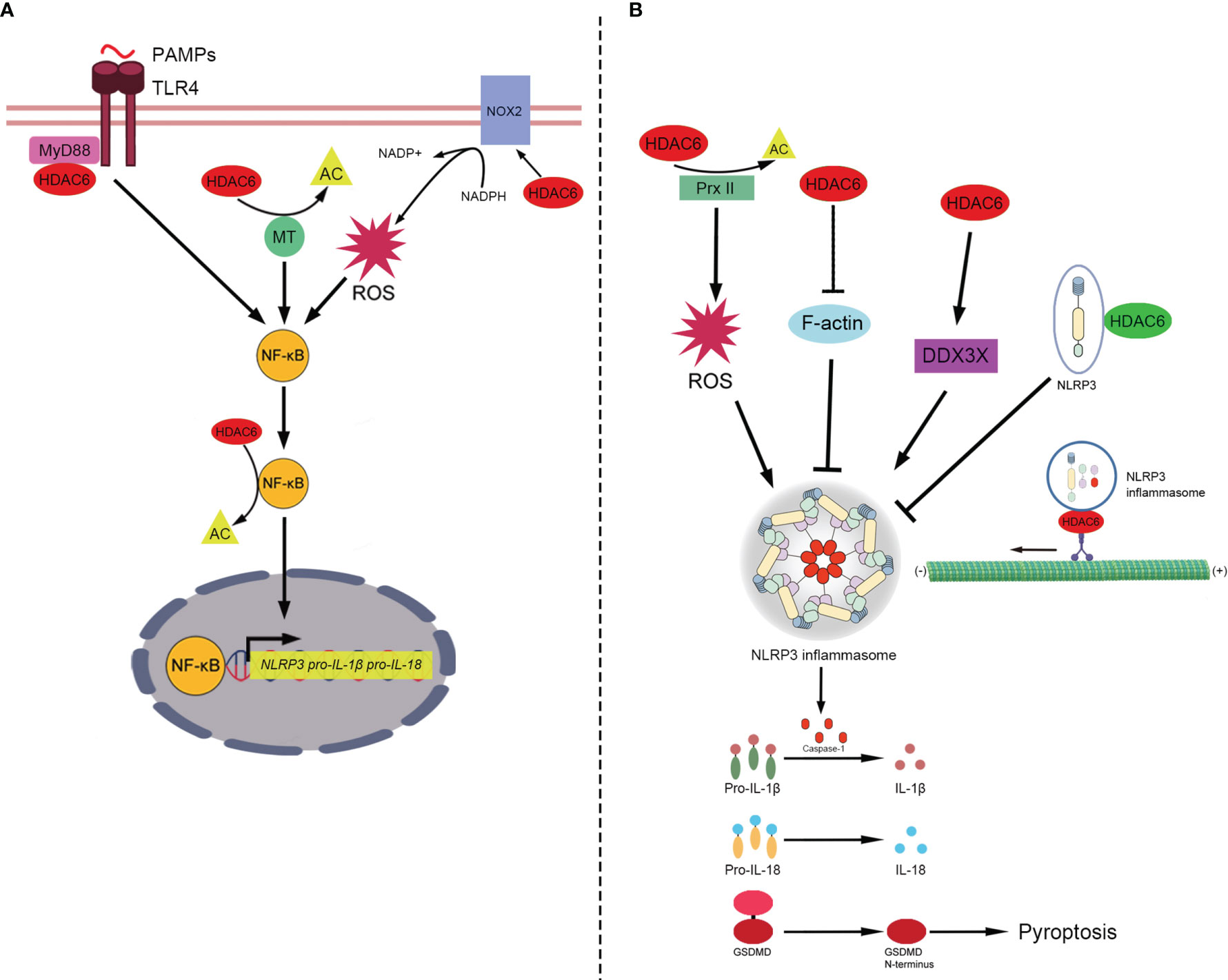
Figure 2 The role of HDAC6 in NLRP3 inflammasome. (A) In the priming of NLRP3 inflammasome, HDAC6 promotes NF-κB to enhance the transcription of NLRP3, pro-IL-1β and pro-IL-18. HDAC6 promotes NF-κB in a number of mechanisms. (1) TLR4 senses PAMPs and recruits the downstream adapter proteins MyD88. HDAC6 interacts with MyD88 to enhance the activation of NF-κB. (2) HDAC6 deacetylates microtubules to promote the activity of NF-κB. (3) HDAC6 elevates the expression of NOX2, the component of NADPH oxidase, to promote the level of ROS which upregulates NF-κB activity. (4) HDAC6 directly deacetylates NF-κB. Then, NF-κB upregulates the transcription of NLRP3, pro-IL-1β, and pro-IL-18. (B) The role of HDAC6 in the activation and PTM of NLRP3 inflammasome includes a variety of signaling mechanisms (positive and negative roles are marked with red and green respectively). HDAC6 regulates the activation of NLRP3 inflammasome in different ways. (1) HDAC6 suppresses the activity of Prx II via deacetylation and increase the level of ROS which is vital for the activation of NLRP3 inflammasome. (2) HDAC6 promotes the activation of NLRP3 inflammasome via suppressing F-actin, a negative factor of NLRP3 assembly. (3) HDAC6 enhances the expression of DDX3X. And DDX3X facilitates NLRP3 assembly. In addition, HDAC6 plays both the negative and positive roles in the PTM of NLRP3 inflammasome. The negative one: HDAC6 interacts with ubquitinated NLRP3 protein directly to prevents the activation of NLRP3 inflammasome. The positive one: In an aggresome-like way, HDAC6 works as a dynein adapter to facilitate retrograde transport of NLRP3 inflammasome for activation. Finally, NLRP3 inflammasome releases active caspase-1, which can promote pro-IL-1β/IL-18 to IL-1β/IL-18 and cleave GSDMD to induce pyroptosis. HDAC6, Histone deacetylase 6; NF-κB, Nuclear factor-kappaB; NLRP3, NACHT, LRR, and PYD domains-containing protein 3; Pro-IL-1β, Pro-interleukin-1β; Pro-IL-18, Pro-interleukin-18; PAMPs, Pathogen-associated molecular patterns; TLR4, Toll-like-receptor 4; MyD88, Myeloid differentiation primary response protein 88; AC, Acetylation; MT, microtubule; NADPH, nicotinamide adenine dinucleotide phosphate; NOX2, NADPH oxidase 2; ROS, Reactive oxygen species; Prx II, Peroxiredoxin II; DDX3X, DEAD-Box Helicase 3 X-Linked; F-actin, Filamentous actin; GSDMD, Gasdermin D.
The Role of HDAC6 in the Priming of NLRP3 Inflammasome
NF-κB, activated by the primary signal, promotes the transcription of NLRP3, pro-IL-1β, and pro-IL-18 (112). The NF-κB transcription factor complex plays a central role in regulating the inducible expression of inflammatory genes in response to immune and inflammatory stimuli. Acetylation of p65, a subunit of NF-κB, has been found to regulate its translocation (118, 119). Jia et al. found that HDAC6 inhibition induces the acetylation of p65 to inhibit its nuclear translocation in diffuse large B-cell lymphoma (120). Xu et al. showed that HDAC6 inhibition upregulated p65 expression in the cytoplasm and reduced p65 expression in the macrophage nucleus to attenuate the transcription of NLRP3 and reduce pyroptosis (27). The inhibition of HDAC6 also reduces p65 expression levels in the nucleus after high glucose stimulation of human retinal pigment epithelium cells, thereby inhibiting the expression of NLRP3 protein and attenuating inflammation (121). These studies suggest HDAC6 deacetylates p65 to upregulate the priming of NLRP3 inflammasome.
Additionally, HDAC6 has been reported to promote the expression of NF-κB to enhance the transcription of pro-IL-1β, increase the release of IL-1β, and aggravate inflammation via the interaction of upstream activators of NF-κB, including myeloid differentiation primary response protein 88 (Myd88), α-tubulin, and ROS (93, 122, 123). Gonzalo et al. found that HDAC6 interacts with the TLR adaptor molecule Myd88 (93). The absence of HDAC6 appears to diminish NF-κB induction by TLR4 stimulation and decrease the release of inflammatory factors, including IL-1β (93). Inhibition of HDAC6 upregulates the acetylation of α-tubulin, which decreases the depolymerization of microtubules, to attenuate the activation of NF-κB by blocking IκBα phosphorylation and IL-1β release in mouse lung tissues challenged with lipopolysaccharide (LPS) (122). ROS are mainly produced by NADPH oxidases (124, 125), which are composed of two membrane-bound subunits (p22phox and gp91phox/Nox2), three cytosolic subunits (p67phox, p47phox, and p40phox), and a small G-protein Rac (Rac1 and Rac2) (126). HDAC6 upregulates the expression of Nox2-based NADPH oxidase subunits to increase the production of ROS (123, 127–129), which promotes NF-κB activation and IL-1β release (123, 127). Given that the maturation and release of pro-IL-1β are mainly mediated through inflammasome-activating caspase-1 (130, 131), it is possible that HDAC6 stimulates NF-κB activation via Myd88, microtubules or ROS to activate NLRP3 inflammasomes. However, the underlying mechanisms remain to be elucidated.
The Role of HDAC6 in the Activation of NLRP3 Inflammasome
Following the primary signal that licenses the cell, the secondary signal occurs following the recognition of an NLRP3 activator and induces full activation and inflammasome formation (113). NLRP3 is activated by a wide variety of stimuli including ROS (132–134). The crystal structure of NLRP3 contains a highly conserved disulfide bond connecting the PYD domain and the nucleotide-binding site domain, which is highly sensitive to altered redox states (135). Redox regulatory proteins, Prx I and Prx II, are highly homologous 2-cysteine members of the Prx protein family that function as antioxidants at low resting levels of H2O2, an ROS (136). Prx I and Prx II are specific targets of HDAC6 deacetylases. Inhibition of HDAC6 increases the levels of acetylated Prx I and Prx II (20, 137). Recently, Yan et al. reported that pharmacological inhibition of HDAC6 attenuates the expression of NLRP3 and mature caspase-1 and IL-1β, and protects dopaminergic neurons via Prx II acetylation, which reduces ROS production (26). These studies suggest that HDAC6 also mediates the activation of NLRP3 inflammasome, probably through Prx I and Prx II deacetylation which upregulates ROS production. However, with the treatment of LPS, ZnF-BUZ but not deacetylase domains facilitates the activation of NLRP3 inflammasome in mouse bone marrow-derived macrophages (iBMDM) (14). Hence, the role of deacetylase domains in the activation of NLRP3 inflammasome remain to be elucidated.
Additionally, HDAC6 inhibitor ACY1215 downregulates the activation of NLRP3 inflammasome via modulating F-actin and DEAD-Box Helicase 3 X-Linked (DDX3X) (138, 139). F-actin acts as a negative regulator by interacting directly with NLRP3 and ASC, following the activation of NLRP3 inflammasome (140). Flightless-I (FliI) and leucine-rich repeat FliI-interaction protein 2 (LRRFIP2) are required for the co-localization of NLRP3, ASC, and F-actin (140). Recently, Chen et al. reported that the HDAC6 inhibitor ACY1215 decreases the activation of NLRP3 inflammasome in acute liver failure (ALF) by increasing the expression of F-actin (138). However, the mechanism underlying HDAC6 inhibition that upregulates the expression of F-actin still needs to be elucidated. Interestingly, another study also found similar results that ACY1215 inhibits the activation of M1 macrophages by regulating NLRP3 inflammasome in ALF, but by a different mechanism (141). In LPS-stimulated ALF mice, ACY1215 decreased the expression of NLRP3 and increased the expression of DEAD-Box Helicase 3 X-Linked (DDX3X) (141), a critical factor for NLRP3 inflammasome assembly (139). It is suggested that the DDX3X/NLRP3 pathway is involved in the protective effects of the HDAC6 inhibitor ALF, but the interaction of HDAC6 and DDX3X needs to be further studied.
The Role of HDAC6 in the PTM of NLRP3 Inflammasome
PTM, including ubiquitination, deubiquitination, phosphorylation, and degradation, occurs in almost every aspect of inflammasome activity, and can either lead to the activation of the inflammasome or suppression of inflammasome activation (142). Recently, Magupalli et al. proved that NLRP3 inflammasome activation depends on regulated ubiquitination (143, 144) and engagement of the dynein adaptor HDAC6 to transport NLRP3 inflammasome to the MTOC for activation in a ubiquitin-misfolded protein-like manner (14). However, it is unknown which inflammasome components need to be ubiquitinated. Hwang et al. previously reported that HDAC6 negatively regulates NLRP3 inflammasome activation through its interaction with ubiquitinated NLRP3 (117). Co-immunoprecipitation data revealed a specific association between HDAC6 and NLRP3 (117). PR619 treatment (deubiquitinase inhibitor) resulted in an increase in the interaction of NLRP3 with HDAC6 and a decrease in NLRP3-dependent caspase-1 activation (117). This indicates that the Zn-BUZ domain of HDAC6 might interact with ubiquitinated NLRP3 (117). The effect of HDAC6 on the PTM of NLRP3 inflammasome is controversial, although previous studies indicated that the HDAC6 ubiquitin-binding domain but not deacetylase activity, is required for NLRP3 activation.
Discussion
The association between autophagy and inflammasomes was discovered more than ten years ago. Satioh et al. first reported the interplay between autophagy and the endotoxin-induced inflammatory immune response through activation of the inflammasome and release of cytokines (145). In LPS-stimulated macrophages, autophagy-related protein Atg16L1 (autophagy-related 16-like 1) deficiency resulted in increased caspase-1 activation, leading to increased IL-1β production (145). Since then, Nakahira et al. indicated that autophagic proteins regulate NLRP3-dependent inflammation by preserving mitochondrial integrity (146). LC3B-deficient mice produced more caspase-1-dependent cytokines in sepsis models and were susceptible to LPS-induced mortality than controls (146). In the last decade, numerous studies have further indicated that autophagy can affect NLRP3 inflammasome activation through various mechanisms (147). Autophagy can suppress NLRP3 inflammasome activation by removing endogenous inflammasome activators, such as ROS-producing damaged mitochondria (148) and removing inflammasome components (149) and cytokines (150). Additionally, NLRP3 inflammasome activation regulates autophagosome formation through various mechanisms. Silencing NLRP3 downregulated autophagy (151, 152). Interestingly, caspase-1 also regulates the autophagic process through cleavage of other substrates (153, 154). Interplay between autophagy and NLRP3 inflammasomes is essential for the balance between the required host defense inflammatory response and prevention of excessive inflammation. As mentioned above, previous studies have shown that HDAC6 mediates the process of autophagy and the functioning of NLRP3 inflammasomes via multiple mechanisms. However, the role of HDAC6 in the crosstalk between autophagy and NLRP3 inflammasome is poorly understood. In the following sections, we will discuss the possible link between HDAC6 and the interplay between autophagy and inflammasomes, considering the current evidence (Figure 3).
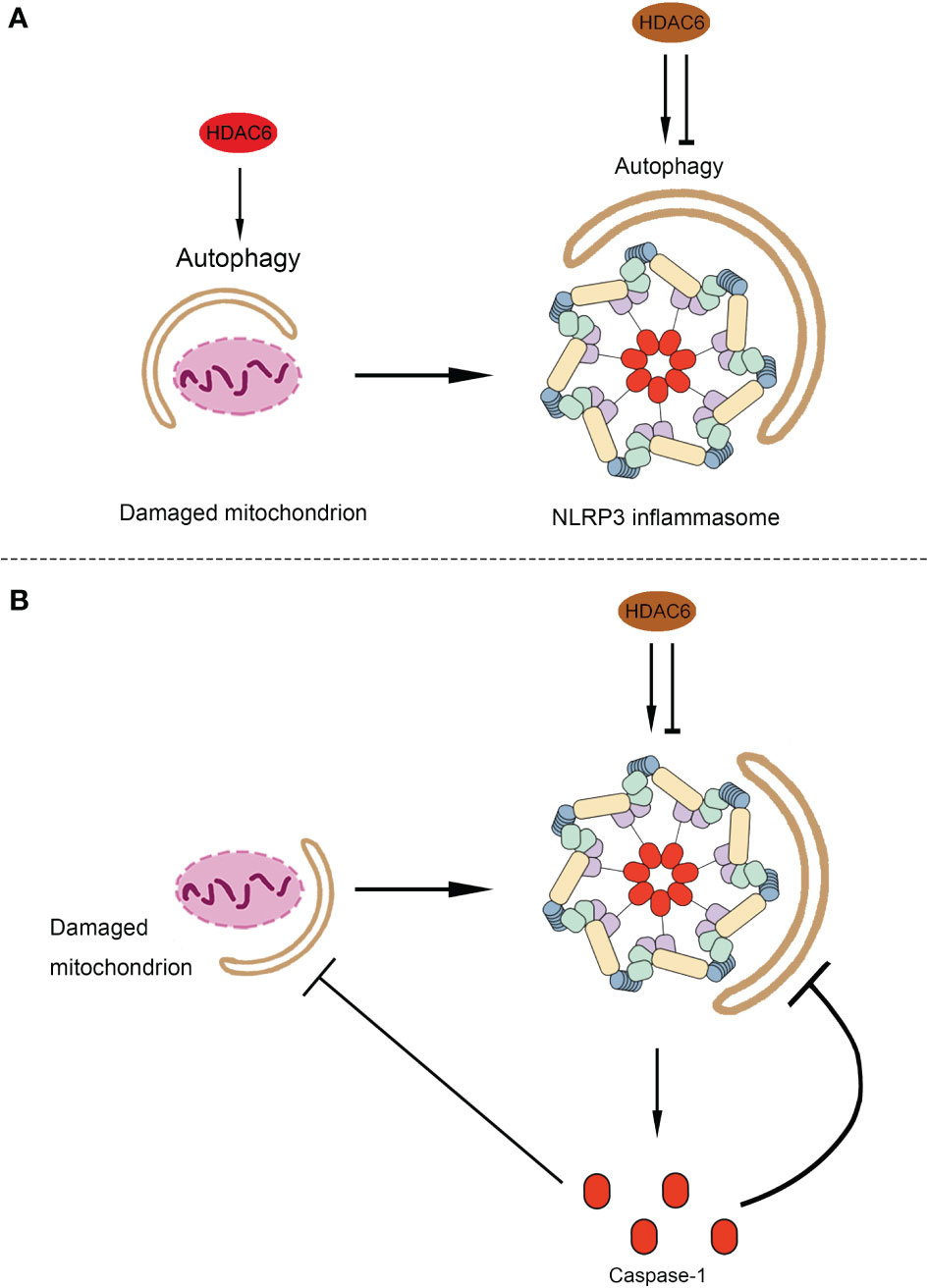
Figure 3 The possible role of HDAC6 in the crosstalk between autophagy and NLRP3 inflammasome. (A) The possible role of HDAC6 in the autophagic degradation of the component of NLRP3 inflammasomes and endogenous inflammasome activators. As a source of endogenous inflammasome activators, damaged mitochondrion promotes the activation of NLRP3 inflammasome. Both the damaged mitochondrion and the component of NLRP3 inflammasome can be limited by autophagy. HDAC6 may promote the mitophagy and then inhibit the activation of NLRP3 inflammasome indirectly. Moreover, it is possible that the HDAC6 upregulate or downregulate autophagy to affect NLRP3 inflammasome. (B) The possible role of HDAC6 in the regulation of autophagy by NLRP3 inflammasome. HDAC6 plays a dual role in the activation of NLRP3 inflammasome, which release the caspase-1. Caspase-1 inhibits the autophagic degradation of the damaged mitochondrion or the component of NLRP3 inflammasome. Hence, HDAC6 may regulate autophagy via the activation of NLRP3 inflammasome. HDAC6, Histone deacetylase 6; NLRP3, NACHT, LRR, and PYD domain-containing protein 3.
The Possible Role of HDAC6 in the Autophagic Degradation of the Component of NLRP3 Inflammasomes and Endogenous Inflammasome Activators
Components of NLRP3 inflammasome, including NLRP3 and ASC, are recognized by p62, a ubiquitin-binding protein, that forms aggresomes and is degraded by autophagy (149). Similarly, a recent study by Han et al. showed that small molecules (kaempferol-Ka) induced autophagy to promote the degradation of inflammasome components and reduce inflammasome activation in an LPS-induced Parkinson disease mouse model (155). As described previously, HDAC6 can also function as a ubiquitin-binding protein to participate in aggresome formation (22). Additionally, HDAC6 can also mediate the acetylation of cortactin and microtubules to regulate autophagy via autophagosome-lysosome fusion and autophagosome transportation (23, 86, 156). Furthermore, HDAC6 interacts with p62 to regulate autophagy via various mechanisms (23, 91, 104, 107). Although, there are no studies indicating that HDAC6 promotes autophagy to reduce the activation of NLRP3 inflammasomes directly, according to the current evidence, it is possible that HDAC6 participates in the autophagic degradation of the components of NLRP3 inflammasomes to regulate its activation.
On the other hand, autophagy removes damaged organelles, such as mitochondria, leading to a reduction in the release of mitochondrial-derived damage-associated molecular patterns (DAMPs), mitochondrial ROS (mtROS), and mitochondrial DNA (mtDNA) (148, 157). Numerous studies have shown that Parkin-mediated mitochondrial autophagy suppresses the production of mtROS and mtDNA, which inhibits the activation of NLRP3 inflammasomes (158–162). As mentioned above, following the decoration of mitochondria with ubiquitin by Parkin, HDAC6 is recruited as a ubiquitin-binding autophagic component that causes mitochondrial clearance (91–93). This evidence suggests that HDAC6 may mediate the functioning of NLRP3 inflammasome via mitophagy eliminating mtROS and mtDNA.
The Possible Role of HDAC6 in the Regulation of Autophagy by NLRP3 Inflammasome
Following the activation of the NLPR3 inflammasome, caspase-1 cleaves some components of autophagy to block this process (153, 154). Yu et al. showed that caspase-1 triggers mitochondrial damage via cleavage of Parkin inhibiting mitophagy, following its activation by NLRP3 and melanoma 2 (AIM2) inflammasomes (153). Furthermore, caspase-1 mediated cleavage of the signaling intermediate Toll-interleukin-1 receptor (TIR)-domain-containing adaptor-inducing interferon-β (TRIF), an essential part of the TLR4-mediated signaling pathway, leading to the promotion of autophagy (154). As HDAC6 regulates the priming, activation, and PTM of NLRP3 inflammasome (14, 26, 27, 117), it is possible that HDAC6 may regulate autophagy through the activation of NLRP3 inflammasomes. The regulation of the crosstalk between autophagy and NLRP3 inflammasome machinery by HDAC6 is obviously complex and requires further investigation, and may be dependent on specific conditions such as cell type, model of disease, inflammasome activator, and autophagy inducer/inhibitor.
Conclusion and Perspective
An increasing number of studies have reported the crosstalk between NLRP3 inflammasome and autophagy in various models and diseases in the last ten years. Numerous studies have indicated that autophagy suppresses NLRP3 inflammasome activation, through various mechanisms. In addition, NLRP3 inflammasome activation regulates autophagosome formation via multiple mechanisms. The crosstalk between autophagy and NLRP3 inflammasome is essential for host defense and the inflammatory response. On the other hand, accumulating evidence indicates that HDAC6 plays important roles in the mediation of autophagy and functioning of NLRP3 inflammasome via differential mechanisms. However, the role of HDAC6 in the crosstalk between autophagy and NLRP3 inflammasome remains poorly understood. In this review, we explored the possible link between HDAC6 and the interplay between autophagy and inflammasomes, considering the current evidence. HDAC6 is a promising therapeutic target in multiple diseases including inflammatory diseases, cancer, and autoimmune diseases. With the development of small molecules inhibiting HDAC6, some clinical trials have shown that selective HDAC6 inhibitors are effective in tumor treatment (163–166). It is worth noting that the effects of HDAC6 differ in specific cell types and conditions. Considering the role of HDAC6 in autophagy and NLRP3 inflammasome, HDAC6 inhibitors have broad prospects and should be studied further deserves to pursue in future research.
Author Contributions
Conception and design – PC and TW. Manuscript preparation – PC and HL. Critical revisions – PC, HL, HH, YL, and TW. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by National Natural Science Foundation of China, 82102315 (PC), Beijing Natural Science Foundation, 7214265 (PC), National Natural Science Foundation of China, 31771326 (TW), and UMHS-PUHSC Joint Institute for Translational and Clinical Research, BMU2020JI007 (TW).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Dr. Yao Yongming for the suggestions and help of this manuscript.
References
1. Glick D, Barth S, Macleod KF. Autophagy: Cellular and Molecular Mechanisms. J Pathol (2010) 221(1):3–12. doi: 10.1002/path.2697
2. Matsuzawa-Ishimoto Y, Hwang S, Cadwell K. Autophagy and Inflammation. Annu Rev Immunol (2018) 36:73–101. doi: 10.1146/annurev-immunol-042617-053253
3. Wang L, Hauenstein AV. NLRP3 Inflammasome: Mechanism of Action, Role in Disease and Therapies. Mol Aspects Med (2020) 76:100889. doi: 10.1016/j.mam.2020.100889
4. Malik A, Kanneganti TD. Inflammasome Activation and Assembly at a Glance. J Cell Sci (2017) 130(23):3955–63. doi: 10.1242/jcs.207365
5. Fusco R, Siracusa R, Genovese T, Cuzzocrea S, Di Paola R. Focus on the Role of NLRP3 Inflammasome in Diseases. Int J Mol Sci (2020) 21(12):4223. doi: 10.3390/ijms21124223
6. Zhao C, Zhao W. NLRP3 Inflammasome-A Key Player in Antiviral Responses. Front Immunol (2020) 11:211. doi: 10.3389/fimmu.2020.00211
7. Su S-H, Wu Y-F, Lin Q, Wang D-P, Hai J. URB597 Protects Against NLRP3 Inflammasome Activation by Inhibiting Autophagy Dysfunction in a Rat Model of Chronic Cerebral Hypoperfusion. J Neuroinflamm (2019) 16(1):260. doi: 10.1186/s12974-019-1668-0
8. Ou Z, Zhou Y, Wang L, Xue L, Zheng J, Chen L, et al. NLRP3 Inflammasome Inhibition Prevents α-Synuclein Pathology by Relieving Autophagy Dysfunction in Chronic MPTP-Treated NLRP3 Knockout Mice. Mol Neurobiol (2021) 58(4):1303–11. doi: 10.1007/s12035-020-02198-5
9. Peng W, Peng F, Lou Y, Li Y, Zhao N, Shao Q, et al. Autophagy Alleviates Mitochondrial DAMP-Induced Acute Lung Injury by Inhibiting NLRP3 Inflammasome. Life Sci (2021) 265:118833. doi: 10.1016/j.lfs.2020.118833
10. Sun Q, Fan J, Billiar TR, Scott MJ. Inflammasome and Autophagy Regulation - A Two-Way Street. Mol Med (2017) 23:188–95. doi: 10.2119/molmed.2017.00077
11. Cadwell K. Crosstalk Between Autophagy and Inflammatory Signalling Pathways: Balancing Defence and Homeostasis. Nat Rev Immunol (2016) 16(11):661–75. doi: 10.1038/nri.2016.100
12. Xue Z, Zhang Z, Liu H, Li W, Guo X, Zhang Z, et al. lincRNA-Cox2 Regulates NLRP3 Inflammasome and Autophagy Mediated Neuroinflammation. Cell Death Differ (2019) 26(1):130–45. doi: 10.1038/s41418-018-0105-8
13. Wang H, Guo Y, Qiao Y, Zhang J, Jiang P. Nobiletin Ameliorates NLRP3 Inflammasome-Mediated Inflammation Through Promoting Autophagy via the AMPK Pathway. Mol Neurobiol (2020) 57(12):5056–68. doi: 10.1007/s12035-020-02071-5
14. Magupalli VG, Negro R, Tian Y, Hauenstein AV, Di Caprio G, Skillern W, et al. HDAC6 Mediates an Aggresome-Like Mechanism for NLRP3 and Pyrin Inflammasome Activation. Science (2020) 369(6510):eaas8995. doi: 10.1126/science.aas8995
15. Seveau S, Turner J, Gavrilin MA, Torrelles JB, Hall-Stoodley L, Yount JS, et al. Checks and Balances Between Autophagy and Inflammasomes During Infection. J Mol Biol (2018) 430(2):174–92. doi: 10.1016/j.jmb.2017.11.006
16. Harris J, Lang T, Thomas JPW, Sukkar MB, Nabar NR, Kehrl JH. Autophagy and Inflammasomes. Mol Immunol (2017) 86:10–5. doi: 10.1016/j.molimm.2017.02.013
17. Micelli C, Rastelli G. Histone Deacetylases: Structural Determinants of Inhibitor Selectivity. Drug Discov Today (2015) 20(6):718–35. doi: 10.1016/j.drudis.2015.01.007
18. Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-Selective Small-Molecule Inhibitor of Histone Deacetylase 6 (HDAC6)-Mediated Tubulin Deacetylation. Proc Natl Acad Sci USA (2003) 100(8):4389–94. doi: 10.1073/pnas.0430973100
19. Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J, et al. HDAC6 Modulates Cell Motility by Altering the Acetylation Level of Cortactin. Mol Cell (2007) 27(2):197–213. doi: 10.1016/j.molcel.2007.05.033
20. Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, et al. HDAC6 is a Specific Deacetylase of Peroxiredoxins and Is Involved in Redox Regulation. P Natl Acad Sci USA (2008) 105(28):9633–8. doi: 10.1073/pnas.0803749105
21. Li Y, Shin D, Kwon SH. Histone Deacetylase 6 Plays a Role as a Distinct Regulator of Diverse Cellular Processes. FEBS J (2013) 280(3):775–93. doi: 10.1111/febs.12079
22. Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The Deacetylase HDAC6 Regulates Aggresome Formation and Cell Viability in Response to Misfolded Protein Stress. Cell (2003) 115(6):727–38. doi: 10.1016/s0092-8674(03)00939-5
23. Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, et al. HDAC6 Controls Autophagosome Maturation Essential for Ubiquitin-Selective Quality-Control Autophagy. EMBO J (2010) 29(5):969–80. doi: 10.1038/emboj.2009.405
24. Nanduri P, Hao R, Fitzpatrick T, Yao TP. Chaperone-Mediated 26S Proteasome Remodeling Facilitates Free K63 Ubiquitin Chain Production and Aggresome Clearance. J Biol Chem (2015) 290(15):9455–64. doi: 10.1074/jbc.M114.627950
25. Li Z, Liu S, Fu T, Peng Y, Zhang J. Microtubule Destabilization Caused by Silicate via HDAC6 Activation Contributes to Autophagic Dysfunction in Bone Mesenchymal Stem Cells. Stem Cell Res Ther (2019) 10(1):351. doi: 10.1186/s13287-019-1441-4
26. Yan S, Wei X, Jian W, Qin Y, Liu J, Zhu S, et al. Pharmacological Inhibition of HDAC6 Attenuates NLRP3 Inflammatory Response and Protects Dopaminergic Neurons in Experimental Models of Parkinson's Disease. Front Aging Neurosci (2020) 12:78. doi: 10.3389/fnagi.2020.00078
27. Xu S, Chen H, Ni H, Dai Q. Targeting HDAC6 Attenuates Nicotine-Induced Macrophage Pyroptosis via NF-Kappab/NLRP3 Pathway. Atherosclerosis (2021) 317:1–9. doi: 10.1016/j.atherosclerosis.2020.11.021
28. Yu L, Chen Y, Tooze SA. Autophagy Pathway: Cellular and Molecular Mechanisms. Autophagy (2018) 14(2):207–15. doi: 10.1080/15548627.2017.1378838
29. Cao W, Li J, Yang K, Cao D. An Overview of Autophagy: Mechanism, Regulation and Research Progress. Bull Cancer (2021) 108(3):304–22. doi: 10.1016/j.bulcan.2020.11.004
30. Parzych KR, Klionsky DJ. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid Redox Signal (2014) 20(3):460–73. doi: 10.1089/ars.2013.5371
31. Dikic I, Elazar Z. Mechanism and Medical Implications of Mammalian Autophagy. Nat Rev Mol Cell Biol (2018) 19(6):349–64. doi: 10.1038/s41580-018-0003-4
32. Brijmohan AS, Batchu SN, Majumder S, Alghamdi TA, Thieme K, McGaugh S, et al. HDAC6 Inhibition Promotes Transcription Factor EB Activation and Is Protective in Experimental Kidney Disease. Front Pharmacol (2018) 9:34. doi: 10.3389/fphar.2018.00034
33. Zhang L, Zhang Z, Li C, Zhu T, Gao J, Zhou H, et al. S100A11 Promotes Liver Steatosis via FOXO1-Mediated Autophagy and Lipogenesis. Cell Mol Gastroenterol Hepatol (2021) 11(3):697–724. doi: 10.1016/j.jcmgh.2020.10.006
34. Su M, Shi JJ, Yang YP, Li J, Zhang YL, Chen J, et al. HDAC6 Regulates Aggresome-Autophagy Degradation Pathway of Alpha-Synuclein in Response to MPP+-Induced Stress. J Neurochem (2011) 117(1):112–20. doi: 10.1111/j.1471-4159.2011.07180.x
35. Ouyang H, Ali YO, Ravichandran M, Dong A, Qiu W, MacKenzie F, et al. Protein Aggregates are Recruited to Aggresome by Histone Deacetylase 6 via Unanchored Ubiquitin C Termini. J Biol Chem (2012) 287(4):2317–27. doi: 10.1074/jbc.M111.273730
36. Filtz TM, Vogel WK, Leid M. Regulation of Transcription Factor Activity by Interconnected Post-Translational Modifications. Trends Pharmacol Sci (2014) 35(2):76–85. doi: 10.1016/j.tips.2013.11.005
37. Zhu L, Duan W, Wu G, Zhang D, Wang L, Chen D, et al. Protective Effect of Hydrogen Sulfide on Endothelial Cells Through Sirt1-FoxO1-Mediated Autophagy. Ann Transl Med (2020) 8(23):1586. doi: 10.21037/atm-20-3647
38. Li C, Wang X, Li X, Qiu K, Jiao F, Liu Y, et al. Proteasome Inhibition Activates Autophagy-Lysosome Pathway Associated With TFEB Dephosphorylation and Nuclear Translocation. Front Cell Dev Biol (2019) 7:170. doi: 10.3389/fcell.2019.00170
39. Shen M, Cao Y, Jiang Y, Wei Y, Liu H. Melatonin Protects Mouse Granulosa Cells Against Oxidative Damage by Inhibiting FOXO1-Mediated Autophagy: Implication of an Antioxidation-Independent Mechanism. Redox Biol (2018) 18:138–57. doi: 10.1016/j.redox.2018.07.004
40. Zhang J, Wang J, Zhou Z, Park JE, Wang L, Wu S, et al. Importance of TFEB Acetylation in Control of Its Transcriptional Activity and Lysosomal Function in Response to Histone Deacetylase Inhibitors. Autophagy (2018) 14(6):1043–59. doi: 10.1080/15548627.2018.1447290
41. Zhang J, Ng S, Wang J, Zhou J, Tan SH, Yang N, et al. Histone Deacetylase Inhibitors Induce Autophagy Through FOXO1-Dependent Pathways. Autophagy (2015) 11(4):629–42. doi: 10.1080/15548627.2015.1023981
42. Song JX, Liu J, Jiang Y, Wang ZY, Li M. Transcription Factor EB: An Emerging Drug Target for Neurodegenerative Disorders. Drug Discov Today (2021) 26(1):164–72. doi: 10.1016/j.drudis.2020.10.013
43. Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR Network Reveals an Integrated Control of Cellular Clearance Pathways. Hum Mol Genet (2011) 20(19):3852–66. doi: 10.1093/hmg/ddr306
44. Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ, Chang YG, et al. Histone Deacetylase 6 Functions as a Tumor Suppressor by Activating C-Jun NH2-Terminal Kinase-Mediated Beclin 1-Dependent Autophagic Cell Death in Liver Cancer. Hepatology (2012) 56(2):644–57. doi: 10.1002/hep.25699
45. Xing YQ, Li A, Yang Y, Li XX, Zhang LN, Guo HC. The Regulation of FOXO1 and its Role in Disease Progression. Life Sci (2018) 193:124–31. doi: 10.1016/j.lfs.2017.11.030
46. Qiu W, Wang B, Gao Y, Tian Y, Tian M, Chen Y, et al. Targeting Histone Deacetylase 6 Reprograms Interleukin-17-Producing Helper T Cell Pathogenicity and Facilitates Immunotherapies for Hepatocellular Carcinoma. Hepatology (2020) 71(6):1967–87. doi: 10.1002/hep.30960
47. Johnston HE, Samant RS. Alternative Systems for Misfolded Protein Clearance: Life Beyond the Proteasome. FEBS J (2021) 288(15):4464–87. doi: 10.1111/febs.15617
48. Hyttinen JM, Amadio M, Viiri J, Pascale A, Salminen A, Kaarniranta K. Clearance of Misfolded and Aggregated Proteins by Aggrephagy and Implications for Aggregation Diseases. Ageing Res Rev (2014) 18:16–28. doi: 10.1016/j.arr.2014.07.002
49. Garcia-Mata R, Gao YS, Sztul E. Hassles With Taking Out the Garbage: Aggravating Aggresomes. Traffic (2002) 3(6):388–96. doi: 10.1034/j.1600-0854.2002.30602.x
50. Park Y, Park J, Kim YK. Crosstalk Between Translation and the Aggresome-Autophagy Pathway. Autophagy (2018) 14(6):1079–81. doi: 10.1080/15548627.2017.1358849
51. Markossian KA, Kurganov BI. Protein Folding, Misfolding, and Aggregation. Formation of Inclusion Bodies and Aggresomes. Biochem (Mosc) (2004) 69(9):971–84. doi: 10.1023/b:biry.0000043539.07961.4c
52. Amengual JE, Johannet P, Lombardo M, Zullo K, Hoehn D, Bhagat G, et al. Dual Targeting of Protein Degradation Pathways With the Selective HDAC6 Inhibitor ACY-1215 and Bortezomib Is Synergistic in Lymphoma. Clin Cancer Res (2015) 21(20):4663–75. doi: 10.1158/1078-0432.CCR-14-3068
53. Mishima Y, Santo L, Eda H, Cirstea D, Nemani N, Yee AJ, et al. Ricolinostat (ACY-1215) Induced Inhibition of Aggresome Formation Accelerates Carfilzomib-Induced Multiple Myeloma Cell Death. Br J Haematol (2015) 169(3):423–34. doi: 10.1111/bjh.13315
54. Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, et al. Small-Molecule Inhibition of Proteasome and Aggresome Function Induces Synergistic Antitumor Activity in Multiple Myeloma. Proc Natl Acad Sci USA (2005) 102(24):8567–72. doi: 10.1073/pnas.0503221102
55. Watanabe S, Inami H, Oiwa K, Murata Y, Sakai S, Komine O, et al. Aggresome Formation and Liquid-Liquid Phase Separation Independently Induce Cytoplasmic Aggregation of TAR DNA-Binding Protein 43. Cell Death Dis (2020) 11(10):909. doi: 10.1038/s41419-020-03116-2
56. Mazzetti S, De Leonardis M, Gagliardi G, Calogero AM, Basellini MJ, Madaschi L, et al. Phospho-HDAC6 Gathers Into Protein Aggregates in Parkinson's Disease and Atypical Parkinsonisms. Front Neurosci (2020) 14:624. doi: 10.3389/fnins.2020.00624
57. Hao R, Nanduri P, Rao Y, Panichelli RS, Ito A, Yoshida M, et al. Proteasomes Activate Aggresome Disassembly and Clearance by Producing Unanchored Ubiquitin Chains. Mol Cell (2013) 51(6):819–28. doi: 10.1016/j.molcel.2013.08.016
58. Hilverling A, Szego EM, Dinter E, Cozma D, Saridaki T, Falkenburger BH. Maturing Autophagosomes are Transported Towards the Cell Periphery. Cell Mol Neurobiol (2021). doi: 10.1007/s10571-021-01116-0
59. Lee JY, Kawaguchi Y, Li M, Kapur M, Choi SJ, Kim HJ, et al. Uncoupling of Protein Aggregation and Neurodegeneration in a Mouse Amyotrophic Lateral Sclerosis Model. Neurodegener Dis (2015) 15(6):339–49. doi: 10.1159/000437208
60. Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and Microtubules Are Required for Autophagic Degradation of Aggregated Huntingtin. J Biol Chem (2005) 280(48):40282–92. doi: 10.1074/jbc.M508786200
61. Mizushima N, Yoshimori T. How to Interpret LC3 Immunoblotting. Autophagy (2007) 3(6):542–5. doi: 10.4161/auto.4600
62. Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol (2008) 445:77–88. doi: 10.1007/978-1-59745-157-4_4
63. Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 Localize to Autophagosomal Membrane Depending on Form-II Formation. J Cell Sci (2004) 117(Pt 13):2805–12. doi: 10.1242/jcs.01131
64. Huang R, Liu W. Identifying an Essential Role of Nuclear LC3 for Autophagy. Autophagy (2015) 11(5):852–3. doi: 10.1080/15548627.2015.1038016
65. Liu KP, Zhou D, Ouyang DY, Xu LH, Wang Y, Wang LX, et al. LC3B-II Deacetylation by Histone Deacetylase 6 is Involved in Serum-Starvation-Induced Autophagic Degradation. Biochem Biophys Res Commun (2013) 441(4):970–5. doi: 10.1016/j.bbrc.2013.11.007
66. Brush MH, Guardiola A, Connor JH, Yao TP, Shenolikar S. Deactylase Inhibitors Disrupt Cellular Complexes Containing Protein Phosphatases and Deacetylases. J Biol Chem (2004) 279(9):7685–91. doi: 10.1074/jbc.M310997200
67. Zhang Y, Li N, Caron C, Matthias G, Hess D, Khochbin S, et al. HDAC-6 Interacts With and Deacetylates Tubulin and Microtubules In Vivo. EMBO J (2003) 22(5):1168–79. doi: 10.1093/emboj/cdg115
68. Matsuyama A, Shimazu T, Sumida Y, Saito A, Yoshimatsu Y, Seigneurin-Berny D, et al. In Vivo Destabilization of Dynamic Microtubules by HDAC6-Mediated Deacetylation. EMBO J (2002) 21(24):6820–31. doi: 10.1093/emboj/cdf682
69. Mackeh R, Perdiz D, Lorin S, Codogno P, Pous C. Autophagy and Microtubules - New Story, Old Players. J Cell Sci (2013) 126(Pt 5):1071–80. doi: 10.1242/jcs.115626
70. Kruppa AJ, Kendrick-Jones J, Buss F. Myosins, Actin and Autophagy. Traffic (2016) 17(8):878–90. doi: 10.1111/tra.12410
71. Kast DJ, Dominguez R. The Cytoskeleton-Autophagy Connection. Curr Biol (2017) 27(8):R318–R26. doi: 10.1016/j.cub.2017.02.061
72. Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. HDAC6 is a Microtubule-Associated Deacetylase. Nature (2002) 417(6887):455–8. doi: 10.1038/417455a
73. Wloga D, Joachimiak E, Fabczak H. Tubulin Post-Translational Modifications and Microtubule Dynamics. Int J Mol Sci (2017) 18(10):2207. doi: 10.3390/ijms18102207
74. Janke C, Magiera MM. The Tubulin Code and its Role in Controlling Microtubule Properties and Functions. Nat Rev Mol Cell Biol (2020) 21(6):307–26. doi: 10.1038/s41580-020-0214-3
75. Kochl R, Hu XW, Chan EY, Tooze SA. Microtubules Facilitate Autophagosome Formation and Fusion of Autophagosomes With Endosomes. Traffic (2006) 7(2):129–45. doi: 10.1111/j.1600-0854.2005.00368.x
76. Geeraert C, Ratier A, Pfisterer SG, Perdiz D, Cantaloube I, Rouault A, et al. Starvation-Induced Hyperacetylation of Tubulin is Required for the Stimulation of Autophagy by Nutrient Deprivation. J Biol Chem (2010) 285(31):24184–94. doi: 10.1074/jbc.M109.091553
77. Jahreiss L, Menzies FM, Rubinsztein DC. The Itinerary of Autophagosomes: From Peripheral Formation to Kiss-and-Run Fusion With Lysosomes. Traffic (2008) 9(4):574–87. doi: 10.1111/j.1600-0854.2008.00701.x
78. Nowosad A, Creff J, Jeannot P, Culerrier R, Codogno P, Manenti S, et al. P27 Controls Autophagic Vesicle Trafficking in Glucose-Deprived Cells via the Regulation of ATAT1-Mediated Microtubule Acetylation. Cell Death Dis (2021) 12(5):481. doi: 10.1038/s41419-021-03759-9
79. Xie R, Nguyen S, McKeehan WL, Liu L. Acetylated Microtubules are Required for Fusion of Autophagosomes With Lysosomes. BMC Cell Biol (2010) 11:89. doi: 10.1186/1471-2121-11-89
80. Hać A, Pierzynowska K, Herman-Antosiewicz A. S6K1 Is Indispensible for Stress-Induced Microtubule Acetylation and Autophagic Flux. Cells (2021) 10(4):929. doi: 10.3390/cells10040929
81. Yang L, Zhao L, Cui L, Huang Y, Ye J, Zhang Q, et al. Decreased Alpha-Tubulin Acetylation Induced by an Acidic Environment Impairs Autophagosome Formation and Leads to Rat Cardiomyocyte Injury. J Mol Cell Cardiol (2019) 127:143–53. doi: 10.1016/j.yjmcc.2018.12.011
82. Gomez-Suaga P, Paillusson S, Miller CCJ. ER-Mitochondria Signaling Regulates Autophagy. Autophagy (2017) 13(7):1250–1. doi: 10.1080/15548627.2017.1317913
83. Friedman JR, Webster BM, Mastronarde DN, Verhey KJ, Voeltz GK. ER Sliding Dynamics and ER-Mitochondrial Contacts Occur on Acetylated Microtubules. J Cell Biol (2010) 190(3):363–75. doi: 10.1083/jcb.200911024
84. Liang T, Qi C, Lai Y, Xie J, Wang H, Zhang L, et al. HDAC6-Mediated Alpha-Tubulin Deacetylation Suppresses Autophagy and Enhances Motility of Podocytes in Diabetic Nephropathy. J Cell Mol Med (2020) 24(19):11558–72. doi: 10.1111/jcmm.15772
85. Li Z, Liu X, Ma J, Zhang T, Gao X, Liu L. hnRNPK Modulates Selective Quality-Control Autophagy by Downregulating the Expression of HDAC6 in 293 Cells. Int J Oncol (2018) 53(5):2200–12. doi: 10.3892/ijo.2018.4517
86. Chen Q, Yue F, Li W, Zou J, Xu T, Huang C, et al. Potassium Bisperoxo(1,10-Phenanthroline)Oxovanadate (Bpv(Phen)) Induces Apoptosis and Pyroptosis and Disrupts the P62-HDAC6 Protein Interaction to Suppress the Acetylated Microtubule-Dependent Degradation of Autophagosomes. J Biol Chem (2015) 290(43):26051–8. doi: 10.1074/jbc.M115.653568
87. Haraszti T, Clemen AE, Spatz JP. Biomimetic F-Actin Cortex Models. Chemphyschem (2009) 10(16):2777–86. doi: 10.1002/cphc.200900581
88. Wang R, Tan J, Chen T, Han H, Tian R, Tan Y, et al. ATP13A2 Facilitates HDAC6 Recruitment to Lysosome to Promote Autophagosome-Lysosome Fusion. J Cell Biol (2019) 218(1):267–84. doi: 10.1083/jcb.201804165
89. Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and Mitophagy in Cardiovascular Disease. Circ Res (2017) 120(11):1812–24. doi: 10.1161/CIRCRESAHA.117.311082
90. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular Definitions of Autophagy and Related Processes. EMBO J (2017) 36(13):1811–36. doi: 10.15252/embj.201796697
91. Lee JY, Nagano Y, Taylor JP, Lim KL, Yao TP. Disease-Causing Mutations in Parkin Impair Mitochondrial Ubiquitination, Aggregation, and HDAC6-Dependent Mitophagy. J Cell Biol (2010) 189(4):671–9. doi: 10.1083/jcb.201001039
92. Radomski N, Kagebein D, Liebler-Tenorio E, Karger A, Rufer E, Tews BA, et al. Mito-Xenophagic Killing of Bacteria is Coordinated by a Metabolic Switch in Dendritic Cells. Sci Rep (2017) 7(1):3923. doi: 10.1038/s41598-017-04142-5
93. Moreno-Gonzalo O, Ramirez-Huesca M, Blas-Rus N, Cibrian D, Saiz ML, Jorge I, et al. HDAC6 Controls Innate Immune and Autophagy Responses to TLR-Mediated Signalling by the Intracellular Bacteria Listeria Monocytogenes. PloS Pathog (2017) 13(12):e1006799. doi: 10.1371/journal.ppat.1006799
94. Stoner MW, Thapa D, Zhang M, Gibson GA, Calderon MJ, St Croix CM, et al. Alpha-Lipoic Acid Promotes Alpha-Tubulin Hyperacetylation and Blocks the Turnover of Mitochondria Through Mitophagy. Biochem J (2016) 473(12):1821–30. doi: 10.1042/BCJ20160281
95. Guedes-Dias P, de Proenca J, Soares TR, Leitao-Rocha A, Pinho BR, Duchen MR, et al. HDAC6 Inhibition Induces Mitochondrial Fusion, Autophagic Flux and Reduces Diffuse Mutant Huntingtin in Striatal Neurons. Biochim Biophys Acta (2015) 1852(11):2484–93. doi: 10.1016/j.bbadis.2015.08.012
96. Islam MA, Sooro MA, Zhang P. Autophagic Regulation of P62 is Critical for Cancer Therapy. Int J Mol Sci (2018) 19(5):1405. doi: 10.3390/ijms19051405
97. Cao Y, Luo Y, Zou J, Ouyang J, Cai Z, Zeng X, et al. Autophagy and its Role in Gastric Cancer. Clin Chim Acta (2019) 489:10–20. doi: 10.1016/j.cca.2018.11.028
98. Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and P62 in Cardiac Proteinopathy. Circ Res (2011) 109(3):296–308. doi: 10.1161/CIRCRESAHA.111.244707
99. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J Biol Chem (2007) 282(33):24131–45. doi: 10.1074/jbc.M702824200
100. Lee Y, Weihl CC. Regulation of SQSTM1/p62 via UBA Domain Ubiquitination and its Role in Disease. Autophagy (2017) 13(9):1615–6. doi: 10.1080/15548627.2017.1339845
101. Lin X, Li S, Zhao Y, Ma X, Zhang K, He X, et al. Interaction Domains of P62: A Bridge Between P62 and Selective Autophagy. DNA Cell Biol (2013) 32(5):220–7. doi: 10.1089/dna.2012.1915
102. Jeong SJ, Zhang X, Rodriguez-Velez A, Evans TD, Razani B. P62/SQSTM1 and Selective Autophagy in Cardiometabolic Diseases. Antioxid Redox Signal (2019) 31(6):458–71. doi: 10.1089/ars.2018.7649
103. Yan J, Seibenhener ML, Calderilla-Barbosa L, Diaz-Meco MT, Moscat J, Jiang J, et al. SQSTM1/p62 Interacts With HDAC6 and Regulates Deacetylase Activity. PloS One (2013) 8(9):e76016. doi: 10.1371/journal.pone.0076016
104. Galindo-Moreno M, Giraldez S, Saez C, Japon MA, Tortolero M, Romero F. Both P62/SQSTM1-HDAC6-Dependent Autophagy and the Aggresome Pathway Mediate CDK1 Degradation in Human Breast Cancer. Sci Rep (2017) 7(1):10078. doi: 10.1038/s41598-017-10506-8
105. Nakashima H, Nguyen T, Goins WF, Chiocca EA. Interferon-Stimulated Gene 15 (ISG15) and ISG15-Linked Proteins Can Associate With Members of the Selective Autophagic Process, Histone Deacetylase 6 (HDAC6) and SQSTM1/P62. J Biol Chem (2015) 290(3):1485–95. doi: 10.1074/jbc.M114.593871
106. Zheng Y, Zhu G, Tang Y, Yan J, Han S, Yin J, et al. HDAC6, A Novel Cargo for Autophagic Clearance of Stress Granules, Mediates the Repression of the Type I Interferon Response During Coxsackievirus A16 Infection. Front Microbiol (2020) 11:78. doi: 10.3389/fmicb.2020.00078
107. Yan Y, Wang H, Wei C, Xiang Y, Liang X, Phang CW, et al. HDAC6 Regulates Lipid Droplet Turnover in Response to Nutrient Deprivation via P62-Mediated Selective Autophagy. J Genet Genomics (2019) 46(4):221–9. doi: 10.1016/j.jgg.2019.03.008
108. Li W, Dai Y, Shi B, Yue F, Zou J, Xu G, et al. LRPPRC Sustains Yap-P27-Mediated Cell Ploidy and P62-HDAC6-Mediated Autophagy Maturation and Suppresses Genome Instability and Hepatocellular Carcinomas. Oncogene (2020) 39(19):3879–92. doi: 10.1038/s41388-020-1257-9
109. Jiang X, Huang Y, Liang X, Jiang F, He Y, Li T, et al. Metastatic Prostate Cancer-Associated P62 Inhibits Autophagy Flux and Promotes Epithelial to Mesenchymal Transition by Sustaining the Level of HDAC6. Prostate (2018) 78(6):426–34. doi: 10.1002/pros.23487
110. Kelley N, Jeltema D, Duan Y, He Y. NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci (2019) 20(13):3328. doi: 10.3390/ijms20133328
111. Hughes MM, O'Neill LAJ. Metabolic Regulation of NLRP3. Immunol Rev (2018) 281(1):88–98. doi: 10.1111/imr.12608
112. Pellegrini C, Antonioli L, Lopez-Castejon G, Blandizzi C, Fornai M. Canonical and Non-Canonical Activation of NLRP3 Inflammasome at the Crossroad Between Immune Tolerance and Intestinal Inflammation. Front Immunol (2017) 8:36. doi: 10.3389/fimmu.2017.00036
113. Swanson KV, Deng M, Ting JP. NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat Rev Immunol (2019) 19(8):477–89. doi: 10.1038/s41577-019-0165-0
114. Dalbeth N, Choi HK, Joosten LAB, Khanna PP, Matsuo H, Perez-Ruiz F, et al. Gout. Nat Rev Dis Primers (2019) 5(1):69. doi: 10.1038/s41572-019-0115-y
115. Danielski LG, Giustina AD, Bonfante S, Barichello T, Petronilho F. NLRP3 Inflammasome and Its Role in Sepsis Development. Inflammation (2020) 43(1):24–31. doi: 10.1007/s10753-019-01124-9
116. Irrera N, Russo M, Pallio G, Bitto A, Mannino F, Minutoli L, et al. The Role of NLRP3 Inflammasome in the Pathogenesis of Traumatic Brain Injury. Int J Mol Sci (2020) 21(17):6204. doi: 10.3390/ijms21176204
117. Hwang I, Lee E, Jeon SA, Yu JW. Histone Deacetylase 6 Negatively Regulates NLRP3 Inflammasome Activation. Biochem Biophys Res Commun (2015) 467(4):973–8. doi: 10.1016/j.bbrc.2015.10.033
118. Chen LF, Mu Y, Greene WC. Acetylation of RelA at Discrete Sites Regulates Distinct Nuclear Functions of NF-kappaB. EMBO J (2002) 21(23):6539–48. doi: 10.1093/emboj/cdf660
119. Kiernan R, Brès V, Ng RW, Coudart MP, El Messaoudi S, Sardet C, et al. Post-Activation Turn-Off of NF-Kappa B-Dependent Transcription is Regulated by Acetylation of P65. J Biol Chem (2003) 278(4):2758–66. doi: 10.1074/jbc.M209572200
120. Jia YJ, Liu ZB, Wang WG, Sun CB, Wei P, Yang YL, et al. HDAC6 Regulates microRNA-27b That Suppresses Proliferation, Promotes Apoptosis and Target MET in Diffuse Large B-Cell Lymphoma. Leukemia (2018) 32(3):703–11. doi: 10.1038/leu.2017.299
121. Yang Q, Li S, Zhou Z, Fu M, Yang X, Hao K, et al. HDAC6 Inhibitor Cay10603 Inhibits High Glucose-Induced Oxidative Stress, Inflammation and Apoptosis in Retinal Pigment Epithelial Cells via Regulating NF-kappaB and NLRP3 Inflammasome Pathway. Gen Physiol Biophys (2020) 39(2):169–77. doi: 10.4149/gpb_2019058
122. Liu L, Zhou X, Shetty S, Hou G, Wang Q, Fu J. HDAC6 Inhibition Blocks Inflammatory Signaling and Caspase-1 Activation in LPS-Induced Acute Lung Injury. Toxicol Appl Pharmacol (2019) 370:178–83. doi: 10.1016/j.taap.2019.03.017
123. Youn GS, Lee KW, Choi SY, Park J. Overexpression of HDAC6 Induces Pro-Inflammatory Responses by Regulating ROS-MAPK-NF-Kappab/AP-1 Signaling Pathways in Macrophages. Free Radic Biol Med (2016) 97:14–23. doi: 10.1016/j.freeradbiomed.2016.05.014
124. Nauseef WM. The Phagocyte NOX2 NADPH Oxidase in Microbial Killing and Cell Signaling. Curr Opin Immunol (2019) 60:130–40. doi: 10.1016/j.coi.2019.05.006
125. Buvelot H, Jaquet V, Krause KH. Mammalian NADPH Oxidases. Methods Mol Biol (2019) 1982:17–36. doi: 10.1007/978-1-4939-9424-3_2
126. Kleniewska P, Piechota A, Skibska B, Gorąca A. The NADPH Oxidase Family and its Inhibitors. Archivum Immunol Ther Exp (2012) 60(4):277–94. doi: 10.1007/s00005-012-0176-z
127. Wang J, Zhao L, Wei Z, Zhang X, Wang Y, Li F, et al. Inhibition of Histone Deacetylase Reduces Lipopolysaccharide-Induced-Inflammation in Primary Mammary Epithelial Cells by Regulating ROS-NF-Small Ka, CyrillicB Signaling Pathways. Int Immunopharmacol (2018) 56:230–4. doi: 10.1016/j.intimp.2018.01.039
128. Jo H, Jang HY, Youn GS, Kim D, Lee CY, Jang JH, et al. Hindsiipropane B Alleviates HIV-1 Tat-Induced Inflammatory Responses by Suppressing HDAC6-NADPH Oxidase-ROS Axis in Astrocytes. BMB Rep (2018) 51(8):394–9. doi: 10.5483/bmbrep.2018.51.8.061
129. Youn GS, Cho H, Kim D, Choi SY, Park J. Crosstalk Between HDAC6 and Nox2-Based NADPH Oxidase Mediates HIV-1 Tat-Induced Pro-Inflammatory Responses in Astrocytes. Redox Biol (2017) 12:978–86. doi: 10.1016/j.redox.2017.05.001
130. Yazdi AS, Ghoreschi K. The Interleukin-1 Family. Adv Exp Med Biol (2016) 941:21–9. doi: 10.1007/978-94-024-0921-5_2
131. Lopez-Castejon G, Brough D. Understanding the Mechanism of IL-1beta Secretion. Cytokine Growth Factor Rev (2011) 22(4):189–95. doi: 10.1016/j.cytogfr.2011.10.001
132. Minutoli L, Puzzolo D, Rinaldi M, Irrera N, Marini H, Arcoraci V, et al. ROS-Mediated NLRP3 Inflammasome Activation in Brain, Heart, Kidney, and Testis Ischemia/Reperfusion Injury. Oxid Med Cell Longev (2016) 2016:2183026. doi: 10.1155/2016/2183026
133. Abais JM, Xia M, Zhang Y, Boini KM, Li PL. Redox Regulation of NLRP3 Inflammasomes: ROS as Trigger or Effector? Antioxid Redox Signal (2015) 22(13):1111–29. doi: 10.1089/ars.2014.5994
134. Sho T, Xu J. Role and Mechanism of ROS Scavengers in Alleviating NLRP3-Mediated Inflammation. Biotechnol Appl Biochem (2019) 66(1):4–13. doi: 10.1002/bab.1700
135. Bae JY, Park HH. Crystal Structure of NALP3 Protein Pyrin Domain (PYD) and its Implications in Inflammasome Assembly. J Biol Chem (2011) 286(45):39528–36. doi: 10.1074/jbc.M111.278812
136. Cao Z, Lindsay JG. The Peroxiredoxin Family: An Unfolding Story. Subcell Biochem (2017) 83:127–47. doi: 10.1007/978-3-319-46503-6_5
137. Jian W, Wei X, Chen L, Wang Z, Sun Y, Zhu S, et al. Inhibition of HDAC6 Increases Acetylation of Peroxiredoxin1/2 and Ameliorates 6-OHDA Induced Dopaminergic Injury. Neurosci Lett (2017) 658:114–20. doi: 10.1016/j.neulet.2017.08.029
138. Chen Q, Wang Y, Jiao F, Cao P, Shi C, Pei M, et al. HDAC6 Inhibitor ACY1215 Inhibits the Activation of NLRP3 Inflammasome in Acute Liver Failure by Regulating the ATM/F-Actin Signalling Pathway. J Cell Mol Med (2021) 25(15):7218–28. doi: 10.1111/jcmm.16751
139. Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, et al. DDX3X Acts as a Live-or-Die Checkpoint in Stressed Cells by Regulating NLRP3 Inflammasome. Nature (2019) 573(7775):590–4. doi: 10.1038/s41586-019-1551-2
140. Burger D, Fickentscher C, de Moerloose P, Brandt KJ. F-Actin Dampens NLRP3 Inflammasome Activity via Flightless-I and LRRFIP2. Sci Rep (2016) 6:29834. doi: 10.1038/srep29834
141. Wang Y, Li X, Chen Q, Jiao F, Shi C, Pei M, et al. Histone Deacetylase 6 Regulates the Activation of M1 Macrophages by the Glycolytic Pathway During Acute Liver Failure. J Inflammation Res (2021) 14:1473–85. doi: 10.2147/jir.s302391
142. Man SM, Kanneganti TD. Regulation of Inflammasome Activation. Immunol Rev (2015) 265(1):6–21. doi: 10.1111/imr.12296
143. Guan K, Wei C, Zheng Z, Song T, Wu F, Zhang Y, et al. MAVS Promotes Inflammasome Activation by Targeting ASC for K63-Linked Ubiquitination via the E3 Ligase Traf3. J Immunol (Baltimore Md 1950) (2015) 194(10):4880–90. doi: 10.4049/jimmunol.1402851
144. Duong BH, Onizawa M, Oses-Prieto JA, Advincula R, Burlingame A, Malynn BA, et al. A20 Restricts Ubiquitination of Pro-Interleukin-1β Protein Complexes and Suppresses NLRP3 Inflammasome Activity. Immunity (2015) 42(1):55–67. doi: 10.1016/j.immuni.2014.12.031
145. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, et al. Loss of the Autophagy Protein Atg16L1 Enhances Endotoxin-Induced IL-1beta Production. Nature (2008) 456(7219):264–8. doi: 10.1038/nature07383
146. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat Immunol (2011) 12(3):222–30. doi: 10.1038/ni.1980
147. Biasizzo M, Kopitar-Jerala N. Interplay Between NLRP3 Inflammasome and Autophagy. Front Immunol (2020) 11:591803. doi: 10.3389/fimmu.2020.591803
148. Zhou R, Yazdi AS, Menu P, Tschopp J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature (2011) 469(7329):221–5. doi: 10.1038/nature09663
149. Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of Autophagy by Inflammatory Signals Limits IL-1β Production by Targeting Ubiquitinated Inflammasomes for Destruction. Nat Immunol (2012) 13(3):255–63. doi: 10.1038/ni.2215
150. Harris J, Hartman M, Roche C, Zeng SG, O'Shea A, Sharp FA, et al. Autophagy Controls IL-1beta Secretion by Targeting Pro-IL-1beta for Degradation. J Biol Chem (2011) 286(11):9587–97. doi: 10.1074/jbc.M110.202911
151. Deng Q, Wang Y, Zhang Y, Li M, Li D, Huang X, et al. Pseudomonas Aeruginosa Triggers Macrophage Autophagy To Escape Intracellular Killing by Activation of NLRP3 Inflammasome. Infect Immun (2016) 84(1):56–66. doi: 10.1128/iai.00945-15
152. Allaeys I, Marceau F, Poubelle PE. NLRP3 Promotes Autophagy of Urate Crystals Phagocytized by Human Osteoblasts. Arthritis Res Ther (2013) 15(6):R176. doi: 10.1186/ar4365
153. Yu J, Nagasu H, Murakami T, Hoang H, Broderick L, Hoffman HM, et al. Inflammasome Activation Leads to Caspase-1-Dependent Mitochondrial Damage and Block of Mitophagy. Proc Natl Acad Sci USA (2014) 111(43):15514–9. doi: 10.1073/pnas.1414859111
154. Jabir MS, Ritchie ND, Li D, Bayes HK, Tourlomousis P, Puleston D, et al. Caspase-1 Cleavage of the TLR Adaptor TRIF Inhibits Autophagy and β-Interferon Production During Pseudomonas Aeruginosa Infection. Cell Host Microbe (2014) 15(2):214–27. doi: 10.1016/j.chom.2014.01.010
155. Han X, Sun S, Sun Y, Song Q, Zhu J, Song N, et al. Small Molecule-Driven NLRP3 Inflammation Inhibition via Interplay Between Ubiquitination and Autophagy: Implications for Parkinson Disease. Autophagy (2019) 15(11):1860–81. doi: 10.1080/15548627.2019.1596481
156. Zhan Y, Wang H, Zhang L, Pei F, Chen Z. HDAC6 Regulates the Fusion of Autophagosome and Lysosome to Involve in Odontoblast Differentiation. Front Cell Dev Biol (2020) 8:605609. doi: 10.3389/fcell.2020.605609
157. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized Mitochondrial DNA Activates NLRP3 Inflammasome During Apoptosis. Immunity (2012) 36(3):401–14. doi: 10.1016/j.immuni.2012.01.009
158. Wu X, Gong L, Xie L, Gu W, Wang X, Liu Z, et al. NLRP3 Deficiency Protects Against Intermittent Hypoxia-Induced Neuroinflammation and Mitochondrial ROS by Promoting the PINK1-Parkin Pathway of Mitophagy in a Murine Model of Sleep Apnea. Front Immunol (2021) 12:628168. doi: 10.3389/fimmu.2021.628168
159. Xu Y, Tang Y, Lu J, Zhang W, Zhu Y, Zhang S, et al. PINK1-Mediated Mitophagy Protects Against Hepatic Ischemia/Reperfusion Injury by Restraining NLRP3 Inflammasome Activation. Free Radic Biol Med (2020) 160:871–86. doi: 10.1016/j.freeradbiomed.2020.09.015
160. Ye JS, Chen L, Lu YY, Lei SQ, Peng M, Xia ZY. Honokiol-Mediated Mitophagy Ameliorates Postoperative Cognitive Impairment Induced by Surgery/Sevoflurane via Inhibiting the Activation of NLRP3 Inflammasome in the Hippocampus. Oxid Med Cell Longev (2019) 2019:8639618. doi: 10.1155/2019/8639618
161. Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin H, et al. PINK1-Parkin Pathway of Mitophagy Protects Against Contrast-Induced Acute Kidney Injury via Decreasing Mitochondrial ROS and NLRP3 Inflammasome Activation. Redox Biol (2019) 26:101254. doi: 10.1016/j.redox.2019.101254
162. Cao S, Shrestha S, Li J, Yu X, Chen J, Yan F, et al. Melatonin-Mediated Mitophagy Protects Against Early Brain Injury After Subarachnoid Hemorrhage Through Inhibition of NLRP3 Inflammasome Activation. Sci Rep (2017) 7(1):2417. doi: 10.1038/s41598-017-02679-z
163. Tu Y, Hershman DL, Bhalla K, Fiskus W, Pellegrino CM, Andreopoulou E, et al. A Phase I-II Study of the Histone Deacetylase Inhibitor Vorinostat Plus Sequential Weekly Paclitaxel and Doxorubicin-Cyclophosphamide in Locally Advanced Breast Cancer. Breast Cancer Res Treat (2014) 146(1):145–52. doi: 10.1007/s10549-014-3008-5
164. Dai Y, Chen S, Wang L, Pei XY, Kramer LB, Dent P, et al. Bortezomib Interacts Synergistically With Belinostat in Human Acute Myeloid Leukaemia and Acute Lymphoblastic Leukaemia Cells in Association With Perturbations in NF-kappaB and Bim. Br J Haematol (2011) 153(2):222–35. doi: 10.1111/j.1365-2141.2011.08591.x
165. Yee AJ, Bensinger WI, Supko JG, Voorhees PM, Berdeja JG, Richardson PG, et al. Ricolinostat Plus Lenalidomide, and Dexamethasone in Relapsed or Refractory Multiple Myeloma: A Multicentre Phase 1b Trial. Lancet Oncol (2016) 17(11):1569–78. doi: 10.1016/s1470-2045(16)30375-8
166. Vogl DT, Raje N, Jagannath S, Richardson P, Hari P, Orlowski R, et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination With Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin Cancer Res (2017) 23(13):3307–15. doi: 10.1158/1078-0432.CCR-16-2526
Keywords: HDAC6, autophagy, NLRP3 inflammasome, inflammation, post-translational modification
Citation: Chang P, Li H, Hu H, Li Y and Wang T (2021) The Role of HDAC6 in Autophagy and NLRP3 Inflammasome. Front. Immunol. 12:763831. doi: 10.3389/fimmu.2021.763831
Received: 24 August 2021; Accepted: 28 September 2021;
Published: 27 October 2021.
Edited by:
Rui Li, University of Pennsylvania, United StatesReviewed by:
Chunying Pei, Harbin Medical University, ChinaJie Xu, University of Michigan, United States
Copyright © 2021 Chang, Li, Hu, Li and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tianbing Wang, d2FuZ3RpYW5iaW5nQHBrdXBoLmVkdS5jbg==; Panpan Chang, Y2hhbmdwYW5wYW5AYmptdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship