Abstract
Although initial immunophenotypical studies on peripheral blood and bronchoalveolar lavage samples have provided a glimpse into the immunopathology of COVID-19, analyses of pulmonary draining lymph nodes are currently scarce. 22 lethal COVID-19 cases and 28 controls were enrolled in this study. Pulmonary draining lymph nodes (mediastinal, tracheal, peribronchial) were collected at autopsy. Control lymph nodes were selected from a range of histomorphological sequelae [unremarkable histology, infectious mononucleosis, follicular hyperplasia, non-SARS related HLH, extrafollicular plasmablast activation, non-SARS related diffuse alveolar damage (DAD), pneumonia]. Samples were mounted on a tissue microarray and underwent immunohistochemical staining for a selection of immunological markers and in-situ hybridization for Epstein Barr Virus (EBV) and SARS-CoV-2. Gene expression profiling was performed using the HTG EdgeSeq Immune Response Panel. Characteristic patterns of a dysregulated immune response were detected in COVID-19: 1. An accumulation of extrafollicular plasmablasts with a relative paucity or depletion of germinal centers. 2. Evidence of T-cell dysregulation demonstrated by immunohistochemical paucity of FOXP3+, Tbet+ and LEF1+ positive T-cells and a downregulation of key genes responsible for T-cell crosstalk, maturation and migration as well as a reactivation of herpes viruses in 6 COVID-19 lymph nodes (EBV, HSV). 3. Macrophage activation by a M2-polarized, CD163+ phenotype and increased incidence of hemophagocytic activity. 4. Microvascular dysfunction, evidenced by an upregulation of hemostatic (CD36, PROCR, VWF) and proangiogenic (FLT1, TEK) genes and an increase of fibrin microthrombi and CD105+ microvessels. Taken together, these findings imply widespread dysregulation of both innate and adoptive pathways with concordant microvascular dysfunction in severe COVID-19.
Introduction
The COVID-19 (Coronavirus Disease 2019) pandemic, caused by SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2), has rapidly evolved into the greatest public health crisis of the 21st century. Despite extensive scientific progress in diagnosis, treatment and vaccine development, systematic investigations explicating the immunopathology of COVID-19 are urgently needed, since there is cumulative evidence of its instrumental role in acute disease progression but also in “long COVID” or post-acute sequelae of COVID-19 (PASC) (1–3). While extensive immunophenotyping studies performed on peripheral blood have offered an initial glimpse into the dysregulated host response affecting both innate and adaptive immunity (1, 4, 5), in-situ data on the pulmonary draining lymph nodes are currently scarce.
Initial autopsy series have revealed characteristic histomorphology in lymph nodes of severe COVID-19, demarcating it from other viral infections. Predominant features included marked capillary congestion and edema, as well as paracortical increase of plasmablasts, a population of extrafollicular IgG and particularly IgM positive B-blasts, lacking germinal center formation, a predominance of M2-polarized macrophages and hemophagocytic lymphohistiocytosis (HLH) (6, 7). Follicular hyperplasia, the histological correlate of an adequate immune response with immunologic memory, was rarely observed, replaced by an expansion of the abovementioned plasmablasts indicative of a transient humoral response in the absence of germinal center formation (8, 9). Taken together, these features suggest extensive dysregulation in antigen presentation and B-cell activation and response in severe disease, both essential for the development of long-term immunity (10, 11).
Pathological and clinical data have provided ample evidence of an increased incidence of bleeding events and coagulopathies in severe COVID-19 (12–14). In line with these observations, thrombocytopenia was reported to be associated with more severe disease outcomes (15, 16), suggesting that systemic thrombotic microangiopathy plays a central role in COVID-19 pathogenesis. Indeed, a dysregulated interferon gene signature (17–19), resulting in a complex interaction between platelets, endothelium, leucocytes and macrophages, contributes to fibrin deposition, microthrombosis, excess formation of neutrophil extracellular traps (NETosis) (20), cytokine storm (21) and HLH (22, 23). Fibrin microthrombi in multiple capillary beds detected in post-mortem series (7, 12, 24), cytokine-induced complement activation (25), thrombocytopenia with hypersecretion of von Willebrand factor (vWF), angiopoietin 2 and coagulation factor VIII as well as IL-6 and fibrinogen (25–28) all point to a profound dysregulation of the innate immune response leading to thromboinflammation in severe COVID-19. Furthermore, angiotensin converting enzyme 2 (ACE2), the entry receptor for SARS-CoV-2, may play a vital role in platelet activation (29).
An aberrant antigen presentation and resultant disruption of the adaptive immune response is a further immunopathological hallmark of severe COVID-19. Lymphopenia, which has been shown in clinical studies to be negatively associated with disease outcome (30), may be a correlate of systemic T-cell suppression or extensive lymphocyte trafficking into the lungs (31). The abovementioned lack of germinal center formation has been linked to a specific block of BCL6-expressing T-follicular helper cell differentiation, resulting in an accumulation of extrafollicular plasmablasts (9). Due to missing germinal center reaction, these blasts do not undergo somatic hypermutation and class switch recombination, which would typically generate high-affinity antibodies or memory B-cells (8, 32). Instead, incomplete humoral response may result in antibody-dependent enhancement, characterized by the formation of low-affinity, insufficiently neutralizing antibodies. Upon binding to viral antigens, they facilitate direct entry into macrophages (33) or enhance macrophage activation, inducing HLH or Kawasaki-like syndrome as observed in a proportion of patients (23, 34, 35). This process has been previously reported in other viral infections and may be a further pathophysiological principle behind a dysregulation of the adaptive immune response in severe COVID-19, potentially leading to an insubstantial durability of immunity (36). The central role of plasmablasts in COVID-19 has been further substantiated by several immunophenotyping studies of peripheral blood (1, 4, 5, 37, 38).
In order to shed light on the underlying immunopathological mechanisms behind severe disease, we studied the pulmonary draining lymph nodes collected from COVID-19 autopsies by systematic histomorphological, immunohistochemical and transcriptomic analysis in comparison to controls of non-COVID-19 reactive pulmonary lymph nodes.
Materials and Methods
Patient Cohort and Study Design
A total of 22 cases of lethal COVID-19 as well as 28 controls were enrolled in this study (Table 1). Autopsies of lethal COVID-19 were performed as previously reported (7). Draining lymph nodes were extracted from peribronchial, tracheal and/or mediastinal regions and fixed in 4% buffered formalin for 48 hours before further processing. Control lymph nodes (n=28) from autopsies and biopsy material were selected from a relevant range of histomorphological sequelae, encompassing other infectious processes (infectious mononucleosis lymphadenopathy, n=5, draining lymph nodes of pneumonia, n=1), histopathologic changes reminiscent of COVID-19 [non-SARS related HLH, n=2; mucosa-associated lymph nodes with extrafollicular plasmablast activation n=4; non-SARS related diffuse alveolar damage (DAD) n=3] as well as follicular hyperplasia (n=3) and unremarkable mediastinal lymph nodes, (n=10).This study has received approval by the Ethics Committee of Northwestern and Central Switzerland (ID 2020-00629).
Table 1
| COVID-19 (n=22) | Controls (n=28) | p-Value | |
|---|---|---|---|
| Age, median years (IQR) | 76 (16) | 57 (50) | <0.001** |
| Sex, male, N (%) | 11 (50) | 13 (46) | 0.513 |
| BMI, median (IQR) | 29 (10) | 27 (4) | 0.336 |
| Hospitalization time, median (days) | 9 (12) | 9 (9) | 0.958 |
| Number of SARS-CoV-2 positive patients, as determined by CT Value (%) | 12 (55) | n/a | n/a |
| Immunosuppressive therapy, N (%) | 2 (9) | 4 (14) | 0.018* |
| Steroid therapy, N (%) | 7 (32) | 5 (18) | 0.041* |
| High dose dexamethasone (0.4-0.8mg/kg/day) | 1 (5) | 0 (0) | 0.440 |
| Anticoagulation therapy, N (%) | 16 (73) | 11 (39) | 0.009** |
| CRP, mg/L, median (IQR) | 191 (182) | 74 (136) | 0.010** |
| LDH, U/L, median (IQR) | 444 (441) | 280 (251.5) | 0.032* |
| INR, median (IQR) | 1.3 (1.1) | 1.1 (0.1) | 0.150 |
| Thrombocytes, 109/L, median (IQR) | 171.0 (291.0) | 281.0 (174.0) | 0.145 |
| Leucocytes, 109/L, median (IQR) | 11.1 (16.6) | 9.3 (5.3) | 0.773 |
| Neutrophilic granulocytes, 109/L, median (IQR) | 7.3 (12.9) | 5.0 (10.4) | 0.232 |
| Lymphocytes, 109/L, median (IQR) | 0.6 (0.5) | 1.5 (2.0) | 0.018* |
| Monocytes, 109/L, median (IQR) | 0.4 (0.7) | 0.5 (0.6) | 0.307 |
Clinical and Serological Characteristics of COVID-19 Patients versus Controls.
*significant at the 0.05 level, ** significant at the 0.01 level.
Bold means significant values.
Tissue Microarrays, Immunohistochemistry and In Situ Hybridization
Representative regions of histological sequelae were annotated by board-certified pathologists on corresponding hematoxylin and eosin (H&E)-stained sections. These regions were transferred to a recipient block using the TMA Grand Master (3DHistech Ltd., Budapest, Hungary), generating three 1.5 mm diameter cores per patient. Immunohistochemistry and in-situ hybridization (ISH) for Epstein-Barr virus (EBV) small RNA were then performed on the TMAs using the automated staining system Benchmark XT (Roche/Ventana Medical Systems, Tucson, USA). All primary reagents, retrieval and incubation conditions as well as histological scoring criteria are listed in Supplementary Table 1. ISH for SARS-CoV-2 was performed as previously reported (39).
RT-PCR
To detect SARS-CoV-2, RNA was isolated from the embedded tissues using the Maxwell RSC RNA FFPE Kit (Promega, Madison, WI, USA). A TaqMan reverse transcription polymerase chain reaction (RT-PCR) was performed by using the TaqMan 2019-nCoV Control Kit v1 (ThermoFisher Scientific, Catalog Number A47533) to target three different viral genomic regions (ORF1AB, S and N genes) and the human RPPH1 gene (RNAse-P). According to the manufacturer’s protocol, a Cт value below 37 in at least two out of three viral genomic regions was considered positive. A case was considered negative if Cт values were above 40. Values between 37 and 40 were considered undetermined and the assay was repeated. Based on the Cт values and quantitation of RPPH1 transcripts, the viral load was calculated as virus genome copy numbers/106RPPH1 transcripts. Samples were always run in duplicates.
Gene Expression Profiling (GEP)
GEP was performed by HTG according to established protocols (https://www.htgmolecular.com/assets/htg/resources/BR-05-HTG-EdgeSeq-System.pdf). Lysates from samples were run on the HTG EdgeSeq Processor (HTG Molecular Diagnostics, Tucson, AZ, USA) using the HTG EdgeSeq Immune Response Panel with an excess of nuclease protection probes (NPPs) complimentary to their target. S1 nuclease then removed un-hybridized probes, and RNAs leaving behind NPPs hybridized to their targets in a 1-to-1 ratio. Samples were individually barcoded using a 16-cycle PCR reaction to add adapters and molecular barcodes, individually purified using AMPure XP beads (Beckman Coulter, Brea, CA, USA) and quantitated using a KAPA Library Quantification kit (KAPA Biosystems, Wilmington, MA, USA). Libraries were sequenced on the Illumina SEQUENCER platform (Illumina, San Diego, CA, USA) for quantification. Quality control, standardization and normalization were performed by HTG and provided to the investigators. Quality control criteria as determined by the manufacturer (percentage of overall reads allocated to the positive process control probe per sample <28%, read depth ≥750000, relative standard deviation of reads of each probe within a sample >0.094) were met for all samples.
Data was first analyzed by the HTG online tool (https://reveal.htgmolecular.com/), including analysis of the top 20 up- and downregulated genes, differential expression of gene sets according to functional groups and pathways and a manual analysis of the corresponding genes of immunohistochemical markers. A principal component analysis (PCA) was then performed using the pcomp function in R©, version 4.0.3 (R-Project for Statistical Computing, Vienna, Austria) and differential expression analysis of COVID-19 cases against controls was conducted with the DESeq2 package using default settings. Sex and age were included in the design formula as additional variables. Count estimates were normalized with the median ratio method, and low-quality samples were excluded from analysis. Prior to the heatmap visualization, the normalized counts were further log2-transformed using a robust variance stabilization. The heatmap including patients’ sex, age and logarithmic viral load was produced with the pheatmap package. The column clusters of the samples as well as the row clusters of the significant genes were obtained by hierarchical clustering with complete linkage and a Euclidean distance metric. The Wald test-statistic was utilized and p-values were adjusted for false discoveries. Adjusted p-values <0.05 and | log2 (fold change) | > 1 were considered significant and included in the analysis.
All expression values were pre-ranked by means of gene set enrichment analysis (GSEA) and their positions compared with the KEGG (Kyoto Encyclopedia of Genes and Genomes), Gene Ontology, GSEA c7 immunology and Hallmark databases. Normalized enrichment scores (NES) > 2 or -2 and adjusted p-values <0.05 were used as preselection criteria for enriched pathways.
Statistical Analysis
Descriptive statistical methods used to analyze immunohistochemical markers and clinical characteristics are described in the Supplementary Methods.
Results
Clinical Characteristics
An overview of demographics and clinical history of both COVID-19 patients and controls can be found in Table 1.
Histomorphology, Immunohistochemical Profiling, ISH, RT-PCR
As previously described in systematic histomorphological analyses on the same autopsy cohort (6), pulmonary draining lymph nodes of lethal COVID-19 displayed the following characteristics compared to controls: 1. vascular changes such as capillary stasis and edema (moderate-extensive 20/21 vs. 5/14; p=0.015) and presence of fibrin microthrombi (6/19 vs. 0/14; p=0.027) (Figure 1A); 2. absence or paucity of germinal centers (16/21 vs. 4/14; p=0.013) (Figure 1B); 3. increased extrafollicular accumulation of plasmablasts (accumulation in 8/21 vs. 1/14; p=0.05) (Figure 1C); 4. an overall increased presence of histiocytes with hemophagocytic activity (moderate-extensive 6/21 vs. 3/14; p=0.037) (Figure 1D).
Figure 1
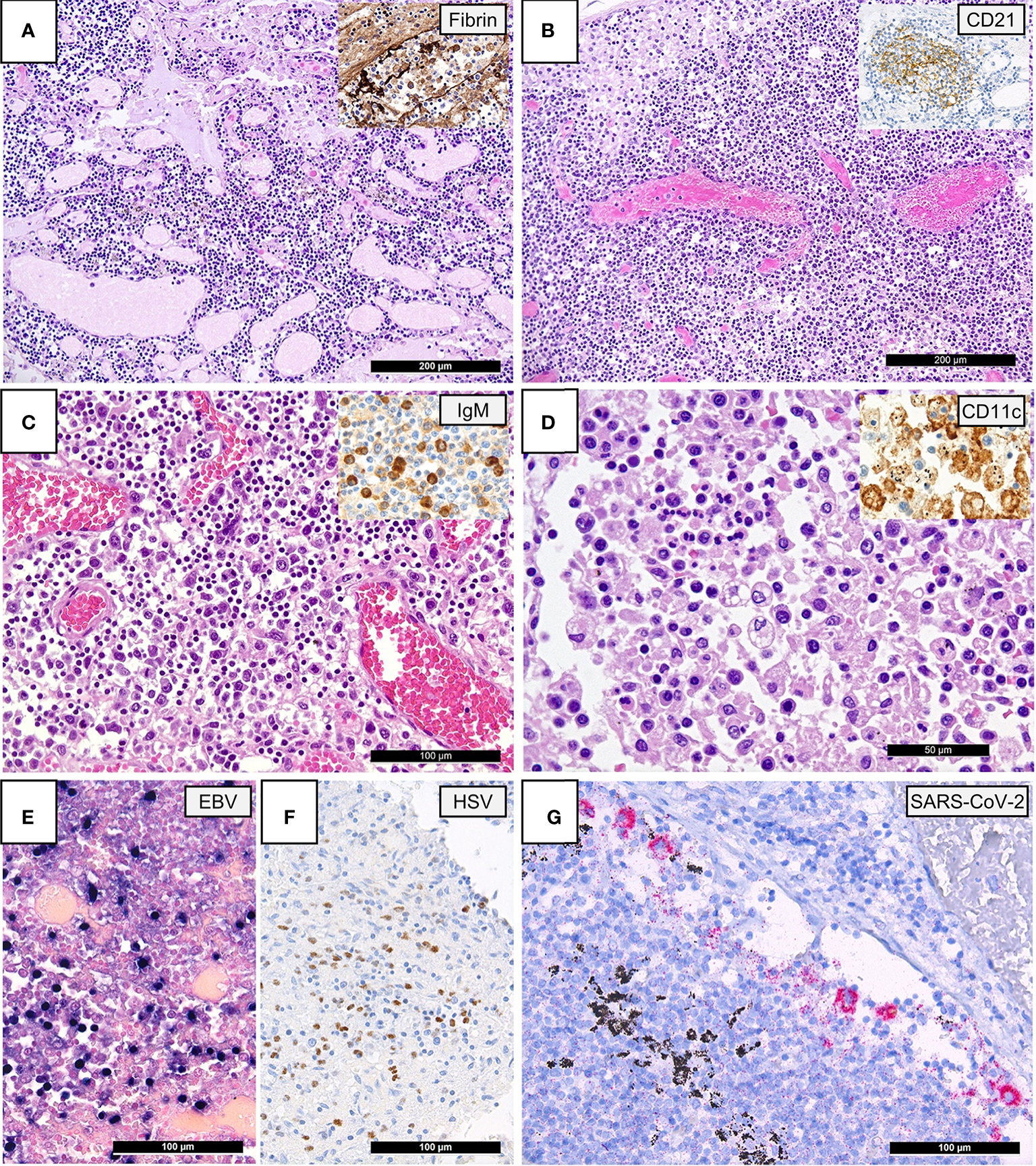
Lymph Node Histomorphology in COVID-19. (A) Overview of a lymph node draining a COVID-19 lung with edema and capillary stasis (H&E; 100x); inset: fibrin microthrombus in a dilated subcapsular sinus (immunoperoxidase; 100x). (B) Severe capillary stasis and expansion of the paracortex without discernable germinal centers (H&E; 100x); inset: disrupted, CD21+ germinal center network (immunoperoxidase; 100x). (C) Proliferation of extrafollicular plasmablasts (H&E; 200x); inset: expression of IgM by plasmablasts (immunoperoxidase; 360x). (D) Hemophagocytosis in the sinus of a lymph node (H&E; 360x); inset: positivity for CD11c in histiocytes with hemophagocytosis (immunoperoxidase; 400x). (E) Increased amount of EBV-infected B-cells in a lymph node of a COVID-19 patient (EBER ISH; 280x). (F) Increased amount of HSV infected B-cells (immunoperoxidase; 280x). (G) SARS-CoV-2 positivity in sinus histiocytes as detected by immunohistochemistry for the SARS-CoV-2 N-antigen (immunoperoxidase with 3-amino-9-ethylcarbazole used as a chromogen; 280x).
A reactivation of various herpes viruses was observed in a subset of COVID-19 patients by means of immunohistochemistry and ISH [EBV: 4/21 (19%), HSV: 2/21 (9.5%); Figure 1E]. The incidence of herpes reactivation was higher in COVID-19 patients than controls [EBV: 2/28 (7%) excluding cases with infectious mononucleosis; HSV: 0/28 (0%), p=0.045]. Importantly, among the 4 COVID-19 cases with EBV reactivation, only one patient was documented as immunocompromised, suffering from therapy-associated myelodysplastic syndrome. VZV was detectable in a lymph node of one non-SARS related DAD, but not in COVID-19. There was no in-situ evidence of re-/infection by adenovirus, CMV, HHV8, parvovirus B-19, SV40, nor could SARS-CoV-2 RNA be detected by ISH in the analyzed cohorts. SARS-CoV-2 N-antigen positive macrophages were observed in the sinus of one COVID-19 patient (Figure 1F).
Immunohistochemical findings are shown in Figure 2, while histological and immunohistochemical descriptive data are shown in Figure 3. Compared to controls, COVID-19 cases displayed a considerably shrunken follicular B-cell compartment (paucity of germinal centers, see above) with a disruption of CD21-positive follicular dendritic cell (FDC) networks (19/21 disrupted in COVID-19 vs. 11/14 in controls; p<0.001) (Figures 1B and 3), while showing a median of 15% of MUM1p-positive plasmablasts compared to 5% in controls (p=0.001) (Figure 3); these plasmablasts expressed mainly IgM and IgG, and the amount of IgA-positive plasmablasts and plasma cells was decreased in COVID-19 (data not shown). Major differences applied to the paracortical T-cell zones, to the histiocytic and vascular compartment of the lymph nodes. While the relative amount of T-cells was not decreased in COVID-19, a particular paucity of FOXP3-positive regulatory T-cell-equivalents (Treg) (median 0.75% compared to 12%, p<0.001) (Figures 2A and 3), Tbet-positive TH1-equivalents (median 2% compared to 12%, p=0.001) (Figures 2B and 3) and decreased amounts of LEF1-positive T-cells (median 15% compared to 25%, p=0.009) were observed (Figure 3). There were no perceptible immunohistochemical differences of ACE2, CD25, T-cell receptor (TCR)-βF1, TCRδ and RORγt between controls and COVID-19 lymph nodes (data not shown). Additionally, an increased amount of especially sinus-associated CD163 (and CD206)-positive M2-polarized macrophages (median 30% compared to 0%, p=0.011) (Figures 2D and 3) and a decreased amount of both sinus-associated and paracortical lysozyme (and HLA-DR)-positive M1-polarized macrophages was noted in COVID-19 (median 4% compared to 15%, p=0.027) (Figure 2C). While CD4:CD8 and TH1:TH2 ratios were not significantly skewed in COVID-19 compared to controls, a significantly lower M1:M2 ratio was observed in COVID-19 [median 0.17 (mean 0.32) in COVID-19 compared to 0.76 (1.34) in controls, p=0.001] (Figure 3). COVID-19 lymph nodes contained more CD105-positive (partially newly formed) vessels (median 68/1.33mm² vs. 44/1.33mm², p=0.002), showed significantly higher VEGF expression (median 3 vs. 2, p=0.045) as well as a higher presence of vWF-positive mononuclear cells in the paracortical zones (8/20 cases vs. 3/14 cases with >5% positive cells, p=0.038) (Figure 3). Of all above, only CD105-positive vessel density was linked to outcome, with patients dying earlier showing densities over the ROC-determined cut-off score of >67/1.33mm² (Supplementary Figure 1). Detailed correlation plots between each immunohistochemical marker and clinical characteristics are provided in Supplementary Figure 2.
Figure 2
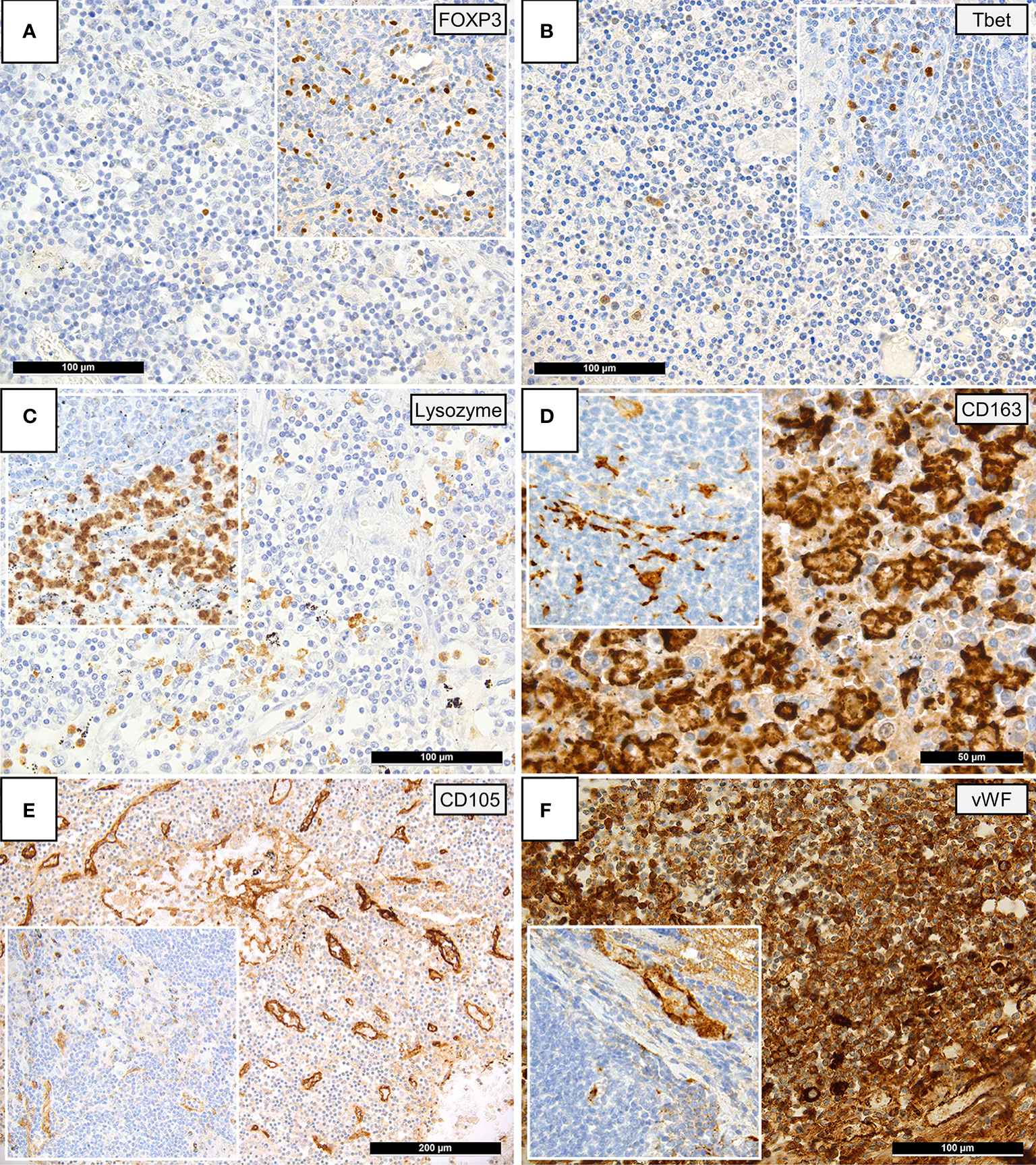
Immunohistochemical Patterns of a Dysregulated Immune Response in COVID-19. Insets display immunohistochemical stainings in controls. (A) Paucity of FOXP3-positive Tregs in COVID-19 patients compared to controls (immunoperoxidase; 280x). (B) Reduced amounts of Tbet positive T-helper 1 (TH1) cells in COVID-19 versus controls (immunoperoxidase; 280x). (C) Reduced amounts of lysozyme-positive histiocytes/macrophages in the paracortex of COVID-19 lymph nodes versus controls (immunoperoxidase; 280x). (D) Increased amounts of CD163-positive histiocytes/macrophages in the paracortex of COVID-19 lymph nodes versus controls (immunoperoxidase; 400x). (E) Increased CD105-positive microvessel density in COVID-19 lymph nodes versus controls (immunoperoxidase; 50x). (F) Increased amount of vWF expression in COVID-19 lymph nodes, vWF positivity in controls is limited to the vascular wall (inset) (immunoperoxidase; 280x).
Figure 3
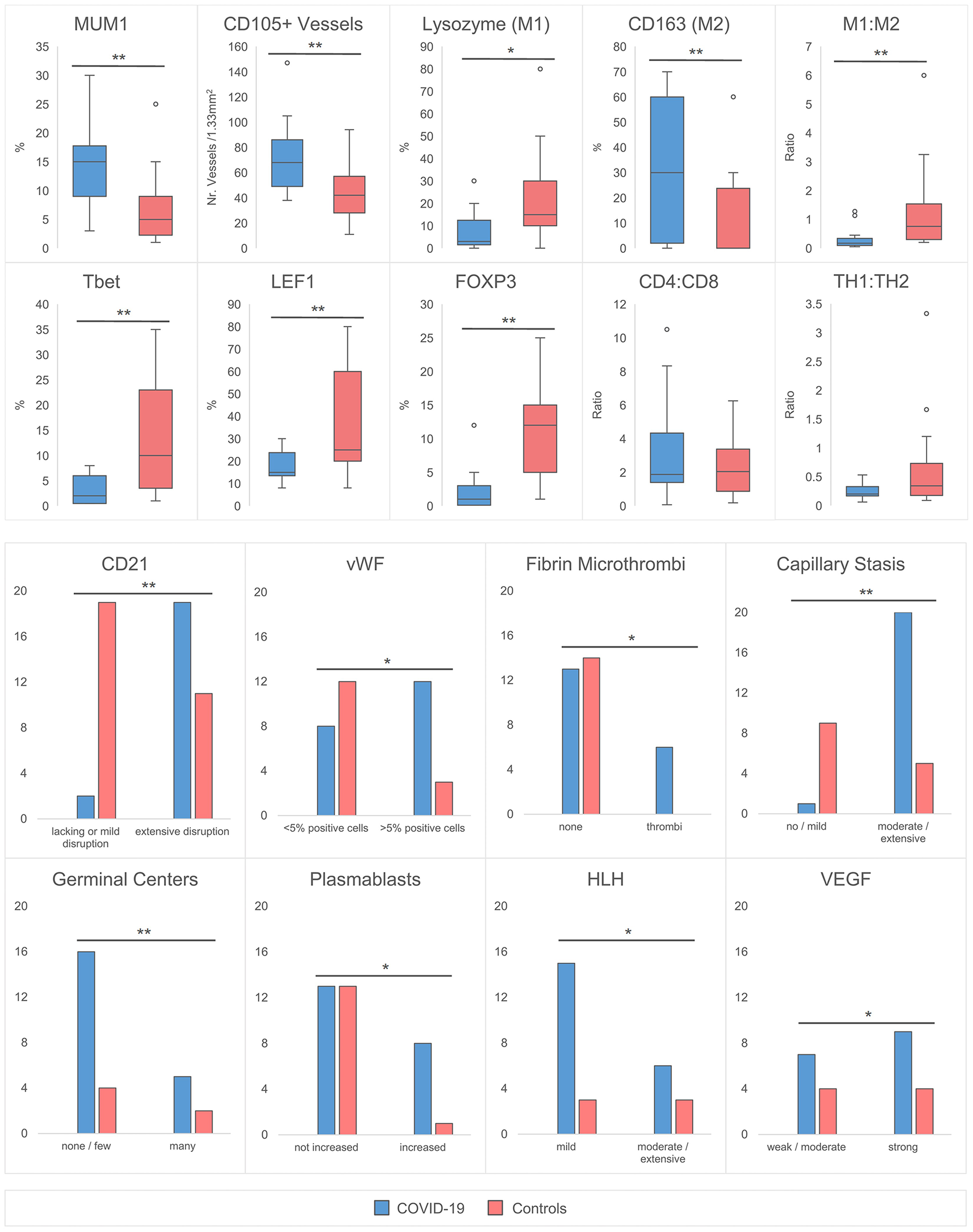
Quantitative Histomorphological and Immunohistochemical Characteristics between COVID-19 Cases and Controls. A significantly higher % of MUM1+ plasmablasts, CD105+ capillaries and CD163+ macrophages is observed in COVID-19. In contrast, the % of lysozyme positive macrophages, Tbet, LEF1 and FOXP3+ T-cells was decreased. The M1:M2 (lysozyme:CD163) ratio is decreased, while CD4:CD8 and TH1:TH2 (Tbet : GATA3) ratios remain unchanged. An increased incidence of CD21+ FDC network disruption with an accompanied lack of germinal centers, increase of plasmablasts, microthrombosis, stasis/edema and moderate/extensive hemophagocytosis is observed in COVID-19 versus controls. The incidence of >5% vWF positive cells and the expression of VEGF is increased. *significant at the 0.05 level, ** significant at the 0.01 level. HLH designates increased hemophagocytic activity of histiocytes, but not hemophagocytic lymphohistiocytosis.
Gene Expression Profiling, Pathway Enrichment and Cluster Analysis
A characteristic gene expression pattern in COVID-19 patients compared to controls is demonstrated in Figure 4. A PCA biplot of gene expression profiling between COVID-19 patients and control lymph node tissue is shown in Figure 4A. COVID-19 patients distinctly cluster apart from controls with no significant overlap, indicative of a characteristic gene expression profile. This is driven by a group of top up- and downregulated genes (as shown by the collective bundling of arrows in the distinct directions of the clusters). Gene expression profiles in controls were more heterogeneous; cases mapped in proximity to the COVID-19 cluster included one case each of infectious mononucleosis, DAD and draining lymph node of pneumococcus pneumonia. COVID-19 cases in proximity to the control cluster featured cases with longer hospitalization time, notably the one case with HLH (Figure 1D) (35).
Figure 4
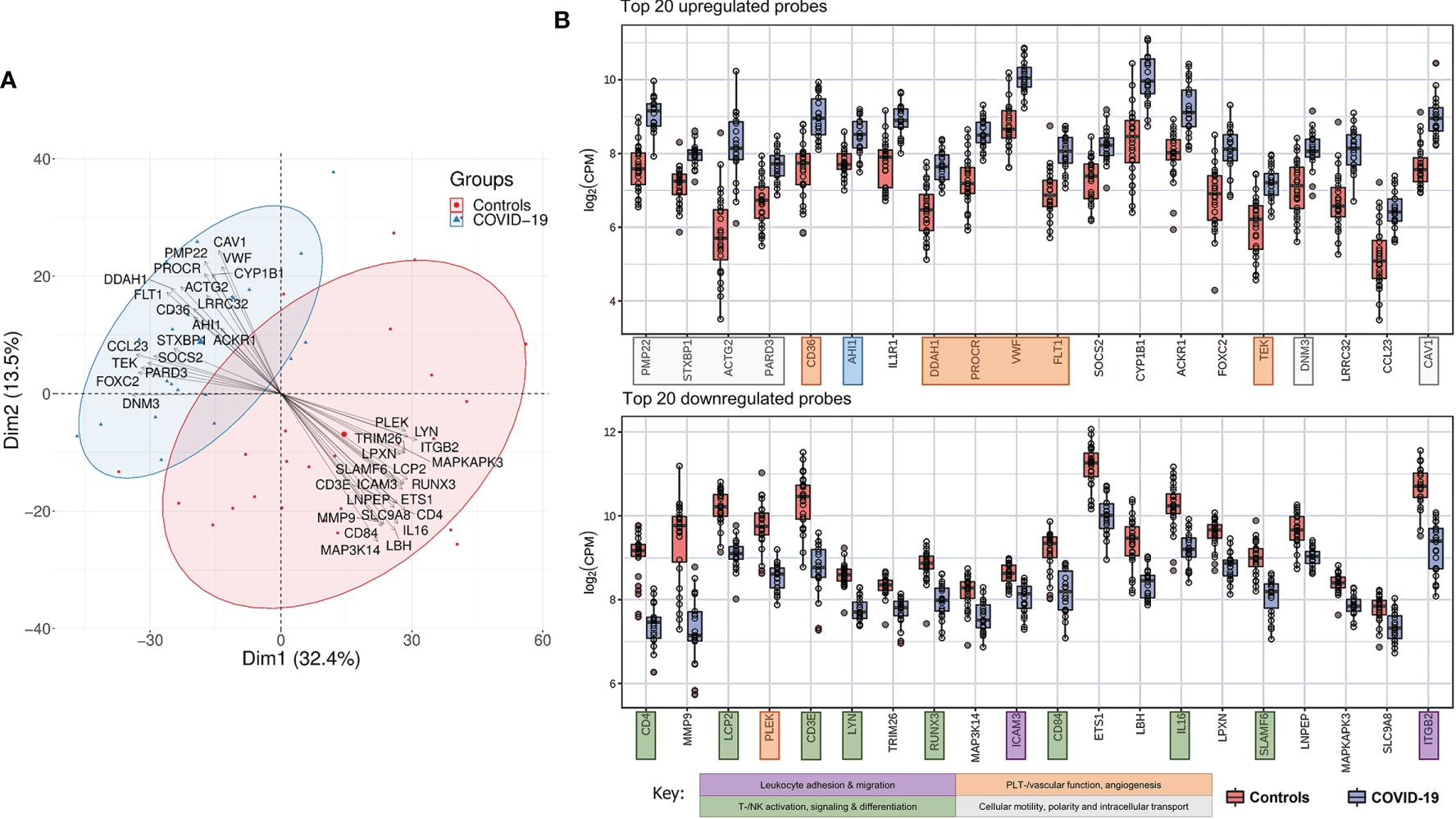
Characteristic Gene Expression Profiling of COVID-19 Lymph Nodes. (A) Principal component analysis biplot showing distinct clustering of COVID-19 and controls in whole tissue analysis. Top up- and downregulated genes are superimposed as vectors determining the individual clusters. (B) Top 20 up-regulated genes in COVID-19 versus controls: genes related to cellular structure, motility and intracellular transport, proangiogenic and prothrombotic genes were predominantly upregulated in COVID-19. Genes involved in T-/NK- and B-cell differentiation and signaling and leukocyte trafficking were predominantly down-regulated in COVID-19.
Top up- and downregulated genes in COVID-19 patients versus controls are shown in Figure 4B. Genes encoding for cellular structure, motility and intracellular transport were predominantly found amongst the 20 most upregulated (genes marked in pale grey). PMP22 (peripheral myelin protein 22), ACTG2 (actin gamma 2, smooth muscle), PARD3 (par-3 family cell polarity regulator) and CAV1 (caveolin 1) encode for cytoskeletal and/or membrane proteins responsible for cell motility, division and structure. Essential genes involved in intracellular transport and vesicle formation were similarly upregulated; these include STXBP1 (syntaxin binding protein 1), AHI1 (Abelson helper integration site 1) and DNM3 (dynamin 3). An increased expression of genes associated with vascular function, development and hemostasis likely reflects the previously described microangiopathy in severe COVID-19 (genes marked in orange). This is exemplified by a significantly upregulated expression of CD36, encoding for platelet glycoprotein 4, and vWF, both essential for efficient platelet adhesion and function, a significant downregulation of PLEK(-strin), and by the increased expression of genes involved in angiogenesis and vascular development (PROCR, protein C receptor; FLT1, Fms related receptor tyrosine kinase 1/vascular endothelial growth factor receptor 2; TEK, angiopoeitin 1 receptor). An upregulation of CYP1B1 (cytochrome P450 family 1 subfamily B member 1) indicates a dysregulation in arachidonic acid metabolism (40). An additionally upregulated gene, DDAH1 (dimethylarginine dimethylaminohydrolase 1), plays an important role in nitric oxide (NO) synthesis, and implies a state of oxidative stress in chronically inflamed tissue, in line with previous reports of NO/ROS imbalance and decreased NO bioavailability in critically ill COVID-19 patients (41, 42). Furthermore, an upregulation of IL1R1 (interleukin 1 receptor type 1), SOCS2 (suppressor of cytokine signaling 2) and ACKR1 [atypical chemokine receptor 1 (Duffy blood group)] implies a dysregulated cytokine/chemokine response, which may be the correlate of the cytokine storm observed in severe COVID-19 (28).
A severely dysregulated adaptive immune response (genes marked in green) is reflected in the downregulation of genes predominantly responsible for T-/NK- and B-cell differentiation and signaling. These include CD4, LCP2 (lymphocyte cytosolic protein 2), CD3E, LYN, CD84, IL16 and SLAMF6. Many of these genes are essential regulative players in leucocyte differentiation and activation, cytokine and integrin signaling and TCR signal transduction. Additionally, a significant downregulation of ITGB2 (integrin subunit beta 2) and its ligand ICAM3 (intercellular adhesion molecule 3) implies an inhibition of leukocyte adhesion and migration (genes marked in purple), further supported by a significantly lower expression of MMP9 (matrix metalloprotease 9), an enzyme previously shown to be involved in local proteolysis of the extracellular matrix essential for leucocyte and macrophage movement and chemotaxis (43–45).
Pathway enrichment analysis by means of GSEA, Gene Ontology and KEGG pathway sets revealed cell cycle related pathways, influenza vaccine mediated memory T-cell-response, MYC targets, mTORC1-signaling, epithelial-mesenchymal transition and fatty acid metabolism related pathways as the most significantly upregulated in COVID-19 versus controls. Pathways involving hematopoietic cell lineage development, B-cell receptor signaling, lysosome function, leucocyte adhesion, TNFα signaling via NF-κB and allograft rejection were the most downregulated (Figure 5). These results correspond to immunohistochemical results (Figures 2, 3). A full list of pathway names and links can be found in Supplementary Table 3.
Figure 5
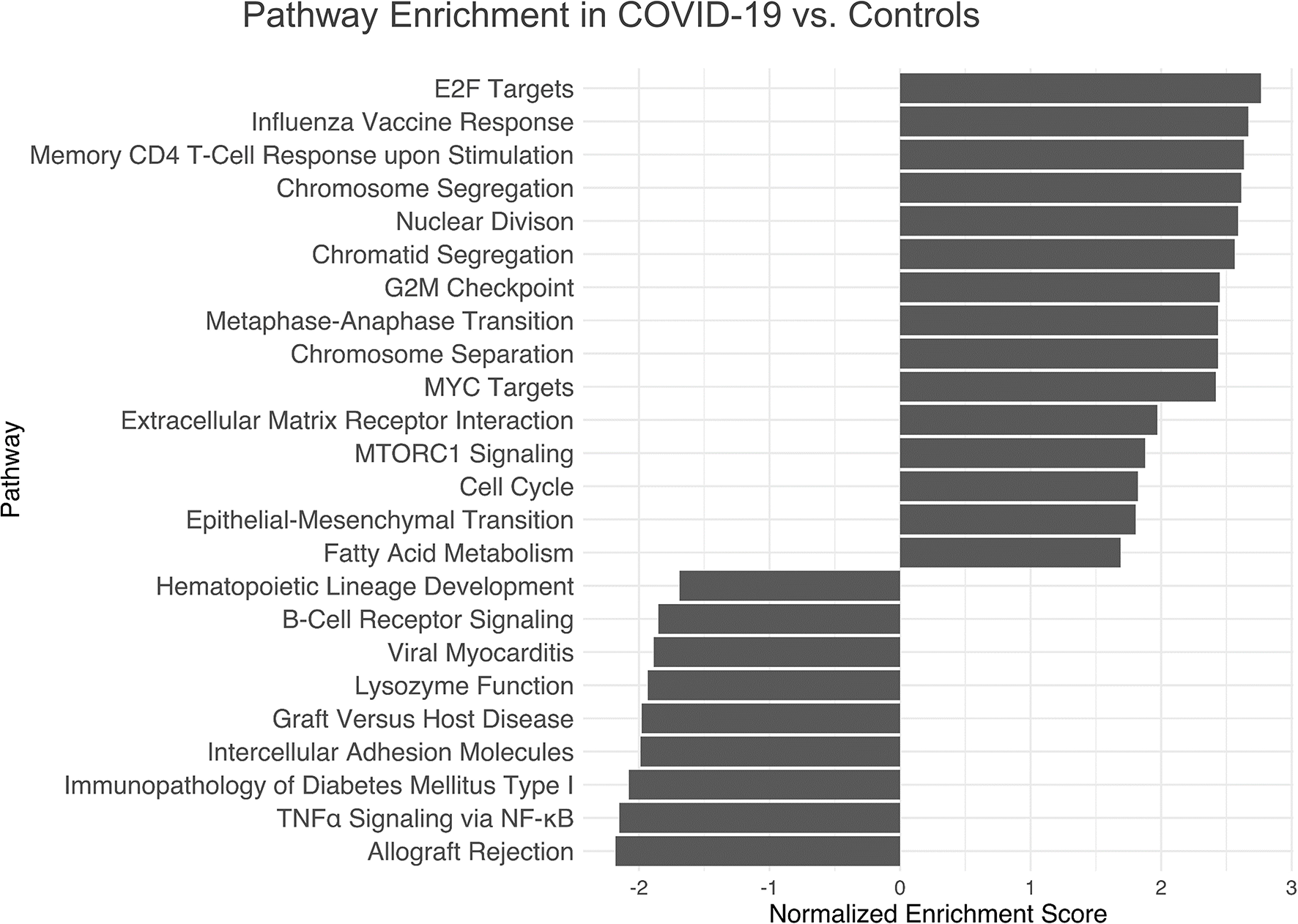
Pathway Enrichment Analysis of Gene Expression Profiling in COVID-19 Immunology. Top up- and downregulated pathways of the GSEA, Hallmark and KEGG database with an abs(NES) > 1. Cell cycle and memory T-cell-response upon influenza vaccine-stimulation, MYC targets and mTORC1-signaling as well as epithelial-mesenchymal transition and fatty acid metabolism related pathways are significantly upregulated in COVID-19, while pathways involving hematopoietic cell lineage development, B-cell receptor signaling, lysosome functions, leucocyte adhesion, TNFα signaling via NF-κB and allograft rejection are downregulated. Further details are found in Supplementary Materials, Table 2.
Gene set enrichment and clustering analysis according to the HTG Immune Response Panel revealed a general upregulation of angiogenesis related genes in COVID-19 (Figure 6A): these include ANG (angiogenin) (fold change 1.52; p<0.01), ANGPT2 (angiopoietin 2) (fold change 1.61; p<0.01), AGT (angiotensinogen) (fold change 2.03; p<0.01), ANGPTL1 (angiopoietin like 1) (fold change 2.05; p<0.01), ANGPT1 (angiopoietin 1) (fold change 1.99; p<0.01), BMPER (bone morphogenic protein binding endothelial regulator protein) (fold change 1.68; p<0.01), SERPINF1 (serpin family F member 1, encoding for α2-antiplasmin) (fold change 1.89; p<0.01) and PECAM1 (platelet and endothelial cell adhesion molecule 1) (fold change 1.25; p=0.05), in line with the vascular dysfunction and hypercoaguable state observed in COVID-19 (12–14). Individual p-values can be found in Supplementary Table 2. An upregulation of complement-related genes and a downregulation of T-cell checkpoint related genes was observed (see Supplementary Figures 3, 4), in line with previous observations (17).
Figure 6
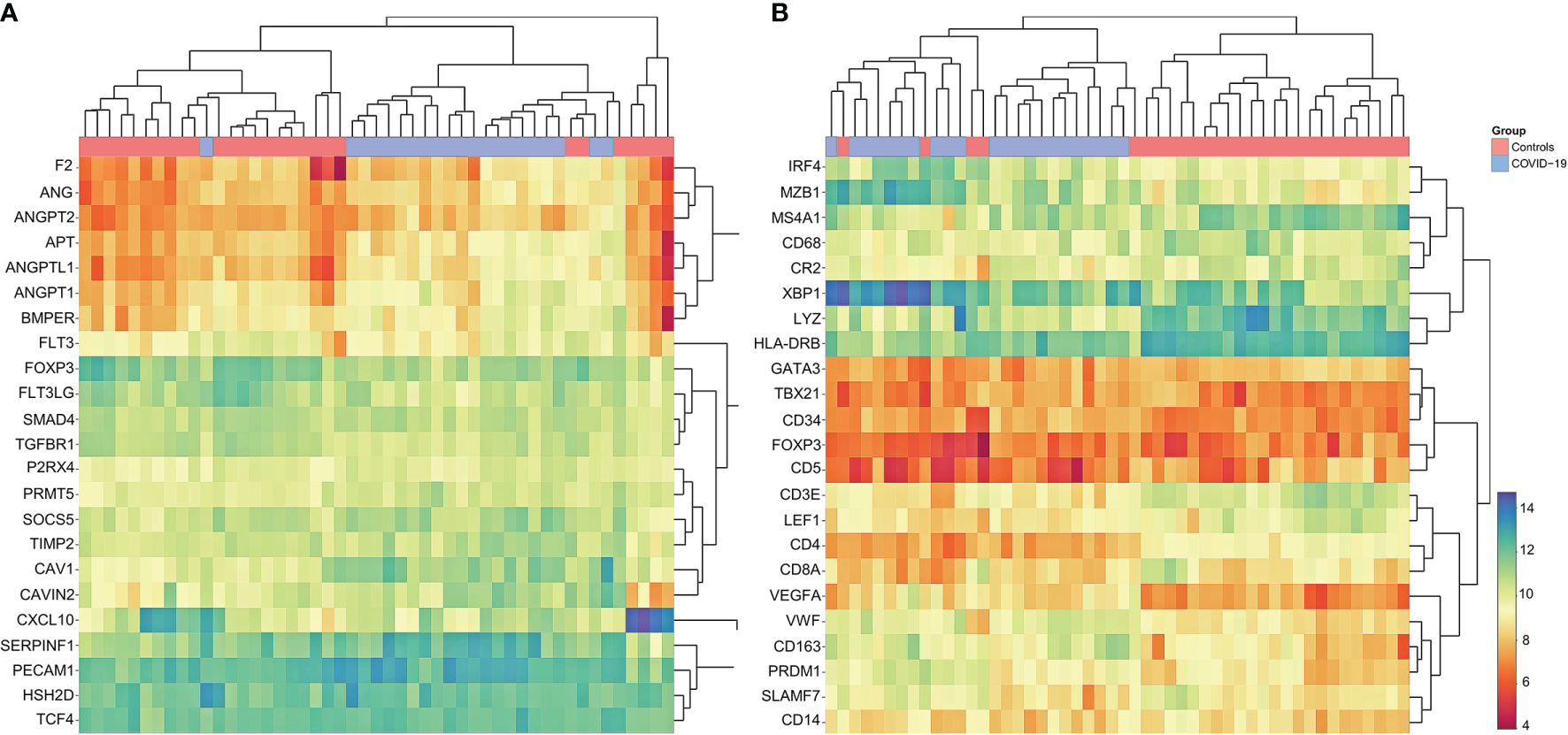
Cluster Analysis of Gene Expression Profiling in COVID-19. (A) Marked dysregulation of angiogenesis-related genes in COVID-19 compared to controls; upregulation of ANG, ANGPT2, AGT, ANGPTL1 ANGPT1, BMPER, SERPINF1 and PECAM1; note that COVID-19 cases cluster together, indicating concordance between individual cases. Individual p-values to be found in Supplementary Materials 2.5. (B) Customized gene set enrichment and clustering analysis of related markers analyzed also by immunohistochemistry shows distinct clustering between COVID-19 and controls: downregulation of CR2 (encodes CD21), TBX21 (encodes Tbet) and LEF1; upregulation of plasmablast genes IRF4, MZB1, XBP1, PRDM1 and SLAMF7; upregulation of angiogenesis related genes (CD34 and VEGF) and of VWF; downregulation of LYZ (encoding for lysozyme) and upregulation of CD163 indicating M2 polarization of macrophages. Note that COVID-19 cases cluster together, indicating concordance between individual cases.
In an effort to correlate phenotypic data (Figures 2, 3) with transcriptomics, supervised manual clustering analysis of immunohistochemically analyzed markers was performed (Figure 6B). TBX21 (T-Box transcription factor 1), which encodes Tbet, was downregulated in COVID-19, mirroring the significant decrease of Tbet positive cells as shown in Figures 2B and 3. Analogous observations were made for FOXP3 and LEF1 (Figure 3). CR2 (Complement receptor 2) encoding for CD21 was decreased, reflecting the disruption of CD21+ FDC networks (Figures 1B and 3). Genes involved in plasmablast differentiation [IRF4 (interferon regulatory factor 4), MZB1 (marginal zone B and B1 cell specific protein), XBP1 (X-box binding protein 1), PRDM1 (PR/SET domain 1) and SLAMF7 (SLAM family member)] were upregulated, reflecting the accumulation of plasmablasts seen histomorphologically (Figure 1C). The upregulation of angiogenesis-related genes (CD34 and VEGF) and of VWF mirrored immunohistochemical results (Figures 2E, F and 3). Macrophages were M2-polarized, as reflected by a downregulation of LYZ (encoding for lysozyme), and an upregulation of CD163 (mirroring results of Figures 2C, D and 3).
Unsupervised cluster analysis revealed a unique differential gene expression of 46 genes in the draining lymph nodes of COVID-19, except for two control cases clustering together with the COVID-19 cases (a case of non-SARS related DAD and a case of pneumococcus pneumonia) (Figure 7). Compared to control cases, and in line with above mentioned phenotypic differences (Figures 1–3) and the results of the pathway enrichment analysis (Figure 5), there was a downregulation of multiple genes related to T-cell function and differentiation (LCP2, CD3E, LEF1, CD6, MAL, TCL1A, TYROBP, CD84, CD5), particularly related to CD4 cells (IL16, CD4), macrophage function (LYZ, SIGLEC1), genes encoding for immunoglobulin receptors (FCER2, PIGR) and transcription factors (ETS1, EGR1, FOS) in COVID-19. An upregulation of genes related to complement and fluid homeostasis and vascular functions (SERPING1, AQP1, ADM, ANGPTL4) was observed. Furthermore, there was evidence of aberrant leukocyte adhesion and motility (downregulation of IRGB2, upregulation of ACTG2 and DNASH12). Genes encoding for enzymes involved in remodeling, such as MMP9, were downregulated (similarly shown in Figure 4B), while its inhibitor TIMP1 as well as ELN, PDGFRA and FN1 were upregulated. Expression patterns suggested a dysregulation in arachidonic acid metabolism: PTGDS (prostaglandin D2 synthase), was downregulated, while CYP1B1 was upregulated.
Figure 7
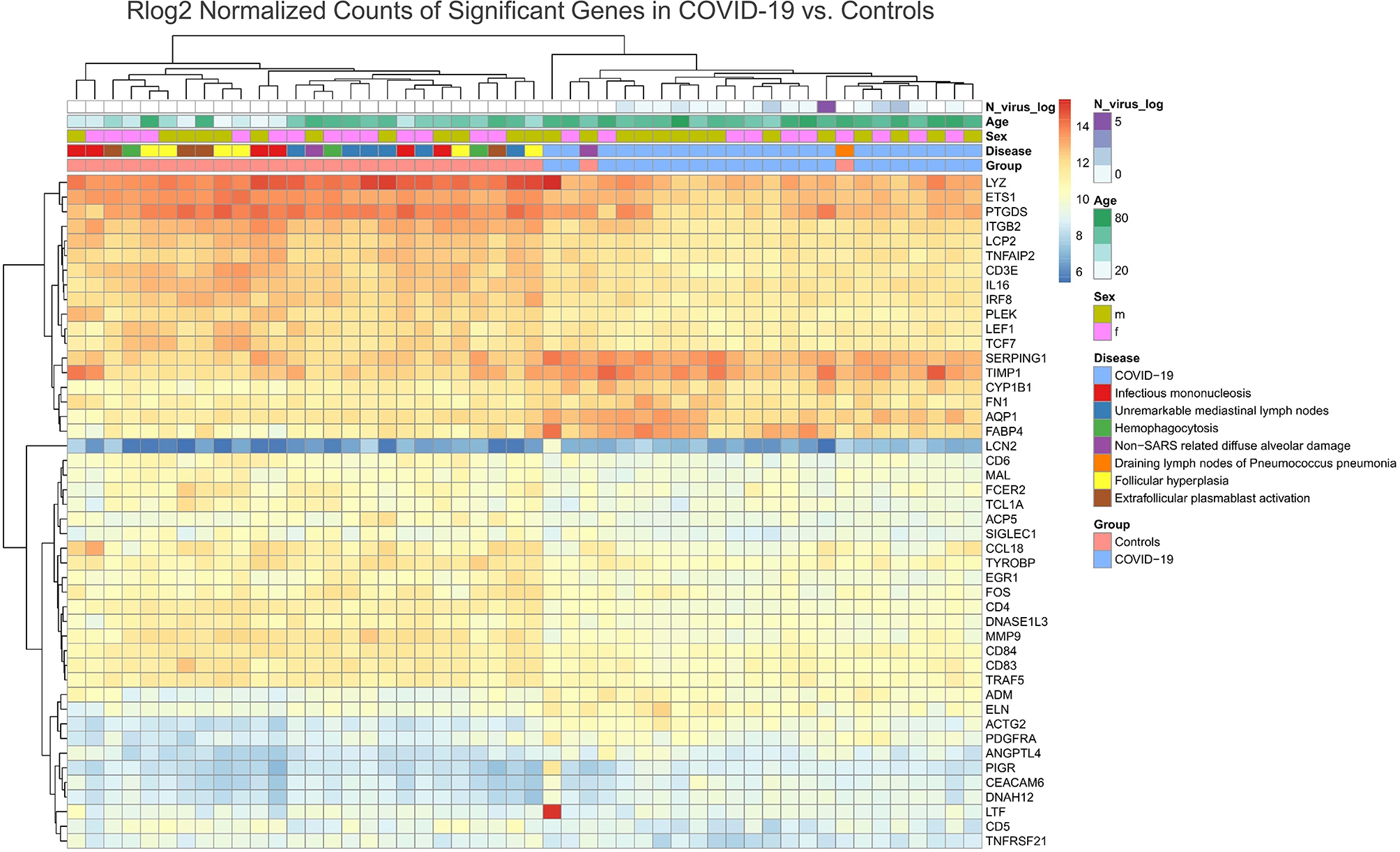
Unsupervised Cluster Analysis of Differentially Expressed Genes in the Draining Lymph Nodes of COVID-19 Compared to Control Cases. A downregulation of genes related to T-cell function and differentiation (LCP2, CD3E, LEF1, CD6, MAL, TCL1A, TYROBP, CD84, CD5), in particular CD4 associated (IL16, CD4) and genes encoding for immunoglobulin receptors (FCER2, PIGR) and transcription factors (ETS1, EGR1, FOS) was observed in COVID-19 patients versus controls. A downregulation of LYZ and SIGLEC1 indicates an altered macrophage phenotype (less prevalent M1 polarization). Genes related to complement and fluid homeostasis and vascular functions (SERPING1, AQP1, ADM, ANGPTL4) are upregulated. A dysregulation in expression amongst genes encoding for leukocyte adhesion and motility was observed (downregulation of IRGB2, upregulation of ACTG2 and DNASH12). MMP9 is downregulated while its inhibitor TIMP1 is upregulated. Other genes involved in remodeling (ELN, PDGFRA and FN1) are upregulated. A downregulation of PTGDS and upregulation of CYP1B1 indicates an altered arachidonic acid metabolism.
Discussion
To our knowledge, this is the first paper in the literature systematically characterizing COVID-19 lymph nodes in an effort to better understand the complex immunopathological changes of severe disease, both from a morphological and transcriptional level. Additionally, our dataset comprises COVID-19 patients from the first wave (March-July 2020) in Switzerland during which the use of high dose dexamethasone, which could have potentially served as a confounder, was limited (given to only 1/21 patients, presenting with clinical symptoms of HLH), thus potentially generating more robust evidence, and utilizes representative controls with a carefully selected spectrum of histological sequelae.
Our data demonstrates characteristic reaction patterns typical for severe COVID-19. Firstly, an unspecific albeit distinctive morphological response was observed, encompassing a three-fold accumulation of extrafollicular plasmablasts and a paucity and/or absence of germinal centers (Figures 1B, C). An accumulation of extrafollicular plasmablasts is a generally rarely observed, pathogenetically heterogeneous histological pattern most characteristically triggered by primary immune responses in mucosa-associated lymph nodes, occurring independently from germinal center formation and without establishment of an immunological memory (8). An accumulation of plasmablasts has previously been described in COVID-19 (5, 46–49). Its significance in this context is still unclear, although it may generate production of non- or sub-neutralizing antibodies which could result in antibody-mediated enhancement (AME), either by viral uptake into Fc-γ receptor IIa (FcγRIIa)-expressing phagocytic cells or by excessive antibody Fc-mediated immune complex formation. This would precipitate increased viral infection and replication in macrophages as well as an augmented inflammatory response, which could be one of the pathophysiological mechanisms contributing to severe COVID-19 (36). AME is a well-documented phenomenon in other viral diseases such as dengue fever and influenza (50, 51), although its role in SARS-CoV-2 immunology is still unknown. Potential evidence of AME in this study is demonstrated in Figure 1F, which features aggregates of SARS-CoV-2 N-protein in macrophages.
Secondly, several levels of evidence in this study suggest a dysregulation and/or exhaustion of T-cells. Six patients demonstrated a histologically evident reactivation of herpes viruses (EBV, HSV), which has also previously been described to have occurred in severe COVID-19 (52). T-cell dysfunction is known to play a critical role in herpes virus-family reactivation (53). Additionally, a significant paucity of FOXP3+, Tbet+ and LEF1+ lymphocytes in COVID-19 suggest a depletion of Treg and TH1 equivalents (Figures 2A, B and 3); this is mirrored by the downregulation of key genes responsible for T-cell maturation and TCR signal transduction (Figure 4B, genes in green; Figure 6B) as well as ICAM3, ITGB2 and MMP9, which are involved in leucocyte adhesion and migration. Pathway enrichment and clustering analysis similarly confirms these observations (Figures 5, 6B and 7). Taken together, these data imply a markedly deregulated and exhausted cellular immune response in severe COVID-19, particularly discernible in several T-cell compartments and checkpoints (54). It may also explain the high incidence of lymphopenia in severe COVID-19 (30, 55). Interestingly, neuropilin-1 which also serves as an entry factor for SARS-CoV-2, is highly expressed in mature FOXP3+ Tregs; this may imply viral tropism for Tregs, which may contribute to the T-cell dysregulation and decreased Treg equivalents observed here (56), warranting further study.
Thirdly, a pathological activation and phenotypic switch of macrophages, most notably reflected in situ by an increased hemophagocytic activity as well as clinically manifest HLH in one patient, was observed in a subset of our cohort (Figure 1D). As mentioned above and supported by previous evidence, HLH may be linked to the cytokine storm and aberrant antigen presentation in severe COVID-19 (12, 26). An increased fraction of a putative M2-polarized phenotype, as exemplified by an increase of CD163+ (and CD206+) cells in the lymph nodes, was similarly observed in lung samples of COVID-19 patients (57, 58). CD163+ macrophages, involved in the endocytotic uptake of free hemoglobin and haptoglobin, were previously shown to be increased in a broad spectrum of inflammatory disorders (59, 60). Soluble CD163+ (sCD163) is considered an effective biomarker of macrophage activation, which is frequently associated with hypercytokinemia (61). Accordingly, increased sCD163 was observed in COVID-19 patients (62), and a potential pathophysiological association between CD163+ macrophages and COVID-19-associated cytokine storm has recently been proposed by RNA-seq transcriptome analyses (57). Furthermore, an overall scarcity and downregulation of lysozyme, observed in the histological, gene expression and pathway enrichment data (Figure 5) of this study, could indicate an increased outflux and secretory activity of macrophages in the periphery (63). This would have to be confirmed by measuring the corresponding serum lysozyme (which would then be increased) (64), an issue requiring further investigation. Importantly, prior studies have shown that lysozyme can play a role in antiviral activity and suggest that low lysozyme levels (as found in COVID-19) may increase virulence (65, 66). SIGLEC-1, which negatively regulates viral infection-triggered type I IFN production (67) in macrophages, was downregulated in our dataset - this further underscores a macrophage-mediated dysregulated IFN-response as a hallmark of severe disease (68), as previously observed in lung tissue of the same autopsy cohort (17). In line with these findings, early clinical trials of otilimab, an anti-GM-CSF (granulocyte-macrophage colony stimulating factor) monoclonal antibody, showed a reduction of all-cause mortality in older patients with severe COVID-19 (69).
Finally, we present comprehensive evidence of widespread microvascular pathology affecting lymph nodes. An increased incidence of capillary stasis, accompanied by fibrin microthrombi (Figure 1A) was detected, findings consistent with systemic microthrombosis described in previous studies (12, 24, 70) and also our own on cardiac tissues (71) of the same cohort. Our findings imply that microangiopathy and thromboinflammation extends into lymphatic tissues, and its extent may be also linked to outcome (in the case of CD105, see Supplementary Figure 1). These histomorphological observations are further supported by GEP (Figure 4B, genes marked in red), which features a significant upregulation of genes essential for hemostasis (CD36, PROCR, VWF) and angiogenesis (FLT1, TEK), and in gene set enrichment and clustering analyses (Figure 6A). These findings engender likely therapeutic targets in severe COVID-19-associated thrombotic disease such as caplacizumab, an anti-vWF humanized immunoglobulin, which is currently approved for the use in thrombotic thrombocytopenic purpura (TTP) (72). Additionally, evidence of an upregulation of complement-associated genes (Supplementary Figure 3) imply a potential therapeutic use of C5a receptor inhibitors such as eculizumab or avacopan in severe disease (73, 74).
A dysregulation of arachidonic acid metabolism was observed in this cohort, evident in the upregulation of CYP1B1 and the downregulation of PTDGS (Figure 7). PTDGS catalyzes the conversion of prostaglandin H2 (PGH2) to prostaglandin D2, while CYP1B1 converts arachidonic acid to epoxyeicosatrienoic acid regioisomers [EpETrE; functioning as lipid mediators in the vascular system (75)]. This raises questions regarding pharmacological agents, such as COX2 inhibitors, in the treatment of COVID-19, especially due to initial reports demonstrating activated COX2 expression by the SARS-CoV N-protein (76). This subsequently led to investigations on COX2 inhibitors such as celecoxib in SARS and COVID-19 with some promising results (77). Notably, the downregulation/knock-out of PTGDS is known to increase vascular permeability, neutrophilic influx and lung damage, in line with the clinical presentation of severe COVID-19 (78), which might be counteracted by COX2 inhibition (79). Thus, the therapeutic significance of pharmaceutic agents interfering with arachidonic acid metabolism (68) requires further investigation in adequately powered, randomized controlled trials.
This study has several limitations. Its retrospective design may not allow sufficiently accurate measurement of potential confounders, which may have influenced GEP or histological data. Furthermore, a sample size of 20 patients may not adequately capture the heterogeneity of COVID-19 immunology; thus, results such as the incidence of herpes-virus-family reactivation in the COVID-19 subgroup should be interpreted with caution. Future studies may provide further insight by comparing lymph nodes with peripheral blood, and other lymphatic organs such as bone marrow, spleen and tonsils, and also making use of single cell sequencing to unravel the subtle expression differences between different leucocyte populations at various sites.
In summary, the immunopathological effects of SARS-CoV-2 are heterogeneous, affecting both innate and adoptive pathways. Thromboinflammatory effects of COVID-19 extend to the lymphatic tissue, highlighting the central role of vascular dysfunction in severe disease. The data in this study thus underscore that severe/lethal COVID-19 is a predominantly angiocentric disease with profound immune dysregulation, possibly without the formation of an immunological memory.
Funding
This study was funded by Botnar Research Centre for Child Health (BRCCH), Grant Nr: FTC-2020-10.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE189199.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics Committee of Northwestern and Central Switzerland. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
JH, AT, and MM designed the study. JH and AT compiled the manuscript. Statistical analysis by CZ. Clinical data acquisition by SB, AS, JH, PW, and KM. RT-PCR and GEP assays by JS. Histomorphology and immunohistochemistry scoring by MM, JH, AT, and TM. Critical revision of the manuscript by KM, PW, SB, MM, CZ, AS, and JS. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors would like to thank all patients and their relatives, as well as Petra Hirschmann, Michèle Baumann, Chantal Tresch and Susanne Grieshaber for performing immunohistochemical analyses.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.763098/full#supplementary-material
References
1
Mathew D Giles JR Baxter AE Oldridge DA Greenplate AR Wu JE et al . Deep Immune Profiling of COVID-19 Patients Reveals Distinct Immunotypes With Therapeutic Implications. Science (2020) 369:eabc8511. doi: 10.1126/science.abc8511
2
Brodin P . Immune Determinants of COVID-19 Disease Presentation and Severity. Nat Med (2021) 27:28–33. doi: 10.1038/s41591-020-01202-8
3
Townsend L Dyer AH Naughton A Kiersey R Holden D Gardiner M et al . Longitudinal Analysis of COVID-19 Patients Shows Age-Associated T Cell Changes Independent of Ongoing Ill-Health. Front Immunol (2021) 12:676932. doi: 10.3389/fimmu.2021.676932
4
Kuri-Cervantes L Pampena MB Meng W Rosenfeld AM Ittner CAG Weisman AR et al . Comprehensive Mapping of Immune Perturbations Associated With Severe COVID-19. Sci Immunol (2020) 5:1–15. doi: 10.1126/sciimmunol.abd7114
5
De Biasi S Lo Tartaro D Meschiari M Gibellini L Bellinazzi C Borella R et al . Expansion of Plasmablasts and Loss of Memory B Cells in Peripheral Blood From COVID-19 Patients With Pneumonia. Eur J Immunol (2020) 50:1283–94. doi: 10.1002/eji.202048838
6
Haslbauer JD Matter MS Stalder AK Tzankov A . Reaktionsmuster Der Lokoregionären Lymphknoten Im Abflussgebiet Von COVID-19-Lungen. Pathol (2021) 42:89–97. doi: 10.1007/s00292-021-00914-z
7
Menter T Haslbauer JD Nienhold R Savic S Hopfer H Deigendesch N et al . Post-Mortem Examination of COVID19 Patients Reveals Diffuse Alveolar Damage With Severe Capillary Congestion and Variegated Findings of Lungs and Other Organs Suggesting Vascular Dysfunction. Histopathology (2020) 77:198–209. doi: 10.1111/his.14134
8
Brighenti A Andrulis M Geissinger E Roth S Müller-Hermelink HK Rüdiger T . Extrafollicular Proliferation of B Cells in the Absence of Follicular Hyperplasia: A Distinct Reaction Pattern in Lymph Nodes Correlated With Primary or Recall Type Responses. Histopathology (2005) 47:90–100. doi: 10.1111/j.1365-2559.2005.02173.x
9
Kaneko N Kuo H-H Boucau J Farmer JR Allard-Chamard H Mahajan VS et al . Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell (2020) 183:1–15. doi: 10.1016/j.cell.2020.08.025
10
Dan JM Mateus J Kato Y Hastie KM Yu ED Faliti CE et al . Immunological Memory to SARS-CoV-2 Assessed for Up to 8 Months After Infection. Science (2021) 371:eabf4063. doi: 10.1126/science.abf4063
11
Sosa-Hernández VA Torres-Ruíz J Cervantes-Díaz R Romero-Ramírez S Páez-Franco JC Meza-Sánchez DE et al . B Cell Subsets as Severity-Associated Signatures in COVID-19 Patients. Front Immunol (2020) 11:611004. doi: 10.3389/fimmu.2020.611004
12
Ackermann M Verleden SE Kuehnel M Haverich A Welte T Laenger F et al . Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
13
Bikdeli B Madhavan MV Jimenez D Chuich T Dreyfus I Driggin E et al . COVID-19 and Thrombotic or Thromboembolic Disease: Implications for Prevention, Antithrombotic Therapy, and Follow-Up: JACC State-Of-the-Art Review. J Am Coll Cardiol (2020) 75:2950–73. doi: 10.1016/j.jacc.2020.04.031
14
Helms J Tacquard C Severac F Leonard-Lorant I Ohana M Delabranche X et al . High Risk of Thrombosis in Patients With Severe SARS-CoV-2 Infection: A Multicenter Prospective Cohort Study. Intensive Care Med (2020) 46:1089–98. doi: 10.1007/s00134-020-06062-x
15
Lippi G Plebani M Henry BM . Thrombocytopenia Is Associated With Severe Coronavirus Disease 2019 (COVID-19) Infections: A Meta-Analysis. Clin Chim Acta Int J Clin Chem (2020) 506:145–8. doi: 10.1016/j.cca.2020.03.022
16
Liu Y Sun W Guo Y Chen L Zhang L Zhao S et al . Association Between Platelet Parameters and Mortality in Coronavirus Disease 2019: Retrospective Cohort Study. Platelets (2020) 31:490–6. doi: 10.1080/09537104.2020.1754383
17
Nienhold R Ciani Y Koelzer VH Tzankov A Haslbauer JD Menter T et al . Two Distinct Immunopathological Profiles in Autopsy Lungs of COVID-19. Nat Commun (2020) 11:5086. doi: 10.1038/s41467-020-18854-2
18
Bastard P Rosen LB Zhang Q Michailidis E Hoffmann H-H Zhang Y et al . Autoantibodies Against Type I IFNs in Patients With Life-Threatening COVID-19. Science (2020) 370:423. doi: 10.1126/science.abd4585
19
Zhang Q Bastard P Liu Z Pen JL Moncada-Velez M Chen J et al . Inborn Errors of Type I IFN Immunity in Patients With Life-Threatening COVID-19. Science (2020) 370:422. doi: 10.1126/science.abd4570
20
Obermayer A Jakob L-M Haslbauer JD Matter MS Tzankov A Stoiber W . Neutrophil Extracellular Traps in Fatal COVID-19-Associated Lung Injury. Dis Markers (2021) 2021:e5566826. doi: 10.1155/2021/5566826
21
Channappanavar R Perlman S . Pathogenic Human Coronavirus Infections: Causes and Consequences of Cytokine Storm and Immunopathology. Semin Immunopathol (2017) 39:529–39. doi: 10.1007/s00281-017-0629-x
22
Prilutskiy A Kritselis M Shevtsov A Yambayev I Vadlamudi C Zhao Q et al . SARS-CoV-2 Infection-Associated Hemophagocytic Lymphohistiocytosis. Am J Clin Pathol (2020) 154:466–74. doi: 10.1093/ajcp/aqaa124
23
McGonagle D Sharif K O’Regan A Bridgewood C . The Role of Cytokines Including Interleukin-6 in COVID-19 Induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun Rev (2020) 19:1025–37. doi: 10.1016/j.autrev.2020.102537
24
Wichmann D Sperhake J-P Lütgehetmann M Steurer S Edler C Heinemann A et al . Autopsy Findings and Venous Thromboembolism in Patients With COVID-19. Ann Intern Med (2020) 173(12):1028–30. doi: 10.7326/M20-2003
25
Magro C Mulvey JJ Berlin D Nuovo G Salvatore S Harp J et al . Complement Associated Microvascular Injury and Thrombosis in the Pathogenesis of Severe COVID-19 Infection: A Report of Five Cases. Transl Res J Lab Clin Med (2020) 220:1–13. doi: 10.1016/j.trsl.2020.04.007
26
Zhang F Gan R Zhen Z Hu X Li X Zhou F et al . Adaptive Immune Responses to SARS-CoV-2 Infection in Severe Versus Mild Individuals. Signal Transduct Target Ther (2020) 5:1–11. doi: 10.1038/s41392-020-00263-y
27
Ranucci M Ballotta A Di Dedda U Bayshnikova E Dei Poli M Resta M et al . The Procoagulant Pattern of Patients With COVID-19 Acute Respiratory Distress Syndrome. J Thromb Haemost JTH (2020) 18:1747–51. doi: 10.1111/jth.14854
28
Mehta P McAuley DF Brown M Sanchez E Tattersall RS Manson JJ . COVID-19: Consider Cytokine Storm Syndromes and Immunosuppression. Lancet Lond Engl (2020) 395:1033–4. doi: 10.1016/S0140-6736(20)30628-0
29
Zhang S Liu Y Wang X Yang L Li H Wang Y et al . SARS-CoV-2 Binds Platelet ACE2 to Enhance Thrombosis in COVID-19. J Hematol OncolJ Hematol Oncol (2020) 13:120. doi: 10.1186/s13045-020-00954-7
30
Henry B Cheruiyot I Vikse J Mutua V Kipkorir V Benoit J et al . Lymphopenia and Neutrophilia at Admission Predicts Severity and Mortality in Patients With COVID-19: A Meta-Analysis. Acta Bio Med Atenei Parm (2020) 91:e2020008–e2020008. doi: 10.23750/abm.v91i3.10217
31
Alon R Sportiello M Kozlovski S Kumar A Reilly EC Zarbock A et al . Leukocyte Trafficking to the Lungs and Beyond: Lessons From Influenza for COVID-19. Nat Rev Immunol (2021) 21:49–64. doi: 10.1038/s41577-020-00470-2
32
Chappell CP Draves KE Giltiay NV Clark EA . Extrafollicular B Cell Activation by Marginal Zone Dendritic Cells Drives T Cell-Dependent Antibody Responses. J Exp Med (2012) 209:1825–40. doi: 10.1084/jem.20120774
33
Iwasaki A Yang Y . The Potential Danger of Suboptimal Antibody Responses in COVID-19. Nat Rev Immunol (2020) 20:339–41. doi: 10.1038/s41577-020-0321-6
34
Pouletty M Borocco C Ouldali N Caseris M Basmaci R Lachaume N et al . Paediatric Multisystem Inflammatory Syndrome Temporally Associated With SARS-CoV-2 Mimicking Kawasaki Disease (Kawa-COVID-19): A Multicentre Cohort. Ann Rheum Dis (2020) 79:999–1006. doi: 10.1136/annrheumdis-2020-217960
35
Zellweger NM Huber J Tsakiris DA Tzankov A Gebhard CE Siegemund M . Haemophagocytic Lymphohistiocytosis and Liver Failure-Induced Massive Hyperferritinaemia in a Male COVID-19 Patient. Swiss Med Wkly (2021) 151:w20420. doi: 10.4414/smw.2021.20420
36
Lee WS Wheatley AK Kent SJ DeKosky BJ . Antibody-Dependent Enhancement and SARS-CoV-2 Vaccines and Therapies. Nat Microbiol (2020) 5:1185–91. doi: 10.1038/s41564-020-00789-5
37
Wilk AJ Rustagi A Zhao NQ Roque J Martínez-Colón GJ McKechnie JL et al . A Single-Cell Atlas of the Peripheral Immune Response in Patients With Severe COVID-19. Nat Med (2020) 26:1070–6. doi: 10.1038/s41591-020-0944-y
38
Liao M Liu Y Yuan J Wen Y Xu G Zhao J et al . Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients With COVID-19. Nat Med (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
39
Reinhold A Tzankov A Matter MS Mihic-Probst D Scholl HPN Meyer P . Ocular Pathology and Occasionally Detectable Intraocular SARS-CoV-2 RNA in Five Fatal COVID-19 Cases. Ophthalmic Res (2021) 64:785–92. doi: 10.1159/000514573
40
Spector AA Norris AW . Action of Epoxyeicosatrienoic Acids on Cellular Function. Am J Physiol-Cell Physiol (2007) 292:C996–1012. doi: 10.1152/ajpcell.00402.2006
41
Alamdari DH Moghaddam AB Amini S Keramati MR Zarmehri AM Alamdari AH et al . Application of Methylene Blue -Vitamin C –N-Acetyl Cysteine for Treatment of Critically Ill COVID-19 Patients, Report of a Phase-I Clinical Trial. Eur J Pharmacol (2020) 885:173494. doi: 10.1016/j.ejphar.2020.173494
42
Fang W Jiang J Su L Shu T Liu H Lai S et al . The Role of NO in COVID-19 and Potential Therapeutic Strategies. Free Radic Biol Med (2021) 163:153–62. doi: 10.1016/j.freeradbiomed.2020.12.008
43
Kluger MA Zahner G Paust H-J Schaper M Magnus T Panzer U et al . Leukocyte-Derived MMP9 Is Crucial for the Recruitment of Proinflammatory Macrophages in Experimental Glomerulonephritis. Kidney Int (2013) 83:865–77. doi: 10.1038/ki.2012.483
44
Khandoga A Kessler JS Hanschen M Khandoga AG Burggraf D Reichel C et al . Matrix Metalloproteinase-9 Promotes Neutrophil and T Cell Recruitment and Migration in the Postischemic Liver. J Leukoc Biol (2006) 79:1295–305. doi: 10.1189/jlb.0805468
45
Bar-Or A Nuttall RK Duddy M Alter A Kim HJ Ifergan I et al . Analyses of All Matrix Metalloproteinase Members in Leukocytes Emphasize Monocytes as Major Inflammatory Mediators in Multiple Sclerosis. Brain J Neurol (2003) 126:2738–49. doi: 10.1093/brain/awg285
46
Bernardes JP Mishra N Tran F Bahmer T Best L Blase JI et al . Longitudinal Multi-Omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity (2020) 53:1296–1314.e9. doi: 10.1016/j.immuni.2020.11.017
47
Ferreira-Gomes M Kruglov A Durek P Heinrich F Tizian C Heinz GA et al . SARS-CoV-2 in Severe COVID-19 Induces a TGF-β-Dominated Chronic Immune Response That Does Not Target Itself. Nat Commun (2021) 12:1961. doi: 10.1038/s41467-021-22210-3
48
Hoehn KB Ramanathan P Unterman A Sumida TS Asashima H Hafler DA et al . Cutting Edge: Distinct B Cell Repertoires Characterize Patients With Mild and Severe COVID-19. J Immunol (2021) 206(12):2785–90. doi: 10.4049/jimmunol.2100135
49
Stephenson E Reynolds G Botting RA Calero-Nieto FJ Morgan MD Tuong ZK et al . Single-Cell Multi-Omics Analysis of the Immune Response in COVID-19. Nat Med (2021) 27:904–16. doi: 10.1038/s41591-021-01329-2
50
Halstead SB O’Rourke EJ . Dengue Viruses and Mononuclear Phagocytes. I. Infection Enhancement by non-Neutralizing Antibody. J Exp Med (1977) 146:201–17. doi: 10.1084/jem.146.1.201
51
Winarski KL Tang J Klenow L Lee J Coyle EM Manischewitz J et al . Antibody-Dependent Enhancement of Influenza Disease Promoted by Increase in Hemagglutinin Stem Flexibility and Virus Fusion Kinetics. Proc Natl Acad Sci USA (2019) 116:15194–9. doi: 10.1073/pnas.1821317116
52
Le Balc’h P Pinceaux K Pronier C Seguin P Tadié J-M Reizine F . Herpes Simplex Virus and Cytomegalovirus Reactivations Among Severe COVID-19 Patients. Crit Care (2020) 24:530. doi: 10.1186/s13054-020-03252-3
53
Yu W Geng S Suo Y Wei X Cai Q Wu B et al . Critical Role of Regulatory T Cells in the Latency and Stress-Induced Reactivation of HSV-1. Cell Rep (2018) 25:2379–89.e3. doi: 10.1016/j.celrep.2018.10.105
54
Kalfaoglu B Almeida-Santos J Tye CA Satou Y Ono M . T-Cell Hyperactivation and Paralysis in Severe COVID-19 Infection Revealed by Single-Cell Analysis. Front Immunol (2020) 11:589380. doi: 10.3389/fimmu.2020.589380
55
Diao B Wang C Tan Y Chen X Liu Y Ning L et al . Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol (2020) 11:827. doi: 10.3389/fimmu.2020.00827
56
Gudowska-Sawczuk M Mroczko B . The Role of Neuropilin-1 (NRP-1) in SARS-CoV-2 Infection: Review. J Clin Med (2021) 10:2772. doi: 10.3390/jcm10132772
57
Zhang J Wu H Yao X Zhang D Zhou Y Fu B et al . Pyroptotic Macrophages Stimulate the SARS-CoV-2-Associated Cytokine Storm. Cell Mol Immunol (2021) 18:1305–7. doi: 10.1038/s41423-021-00665-0
58
Roussel M Ferrant J Reizine F Le Gallou S Dulong J Carl S et al . Comparative Immune Profiling of Acute Respiratory Distress Syndrome Patients With or Without SARS-CoV-2 Infection. Cell Rep Med (2021) 2:100291. doi: 10.1016/j.xcrm.2021.100291
59
Etzerodt A Moestrup SK . CD163 and Inflammation: Biological, Diagnostic, and Therapeutic Aspects. Antioxid Redox Signal (2013) 18:2352–63. doi: 10.1089/ars.2012.4834
60
Skytthe MK Graversen JH Moestrup SK . Targeting of CD163+ Macrophages in Inflammatory and Malignant Diseases. Int J Mol Sci (2020) 21:5497. doi: 10.3390/ijms21155497
61
Crayne CB Albeituni S Nichols KE Cron RQ . The Immunology of Macrophage Activation Syndrome. Front Immunol (2019) 10:119. doi: 10.3389/fimmu.2019.00119
62
Gómez-Rial J Currás-Tuala MJ Rivero-Calle I Gómez-Carballa A Cebey-López M Rodríguez-Tenreiro C et al . Increased Serum Levels of Scd14 and Scd163 Indicate a Preponderant Role for Monocytes in COVID-19 Immunopathology. Front Immunol (2020) 11:560381. doi: 10.3389/fimmu.2020.560381
63
Ragland SA Criss AK . From Bacterial Killing to Immune Modulation: Recent Insights Into the Functions of Lysozyme. PloS Pathog (2017) 13:e1006512. doi: 10.1371/journal.ppat.1006512
64
Torsteinsdóttir I Håkansson L Hällgren R Gudbjörnsson B Arvidson N-G Venge P . Serum Lysozyme: A Potential Marker of Monocyte/Macrophage Activity in Rheumatoid Arthritis. Rheumatology (1999) 38:1249–54. doi: 10.1093/rheumatology/38.12.1249
65
Mann JK Ndung’u T . The Potential of Lactoferrin, Ovotransferrin and Lysozyme as Antiviral and Immune-Modulating Agents in COVID-19. Future Virol (2020) 15:609–24. doi: 10.2217/fvl-2020-0170
66
Pang G Clancy R Cong M Ortega M Zhigang R Reeves G . Influenza Virus Inhibits Lysozyme Secretion by Sputum Neutrophils in Subjects With Chronic Bronchial Sepsis. Am J Respir Crit Care Med (2000) 161:718–22. doi: 10.1164/ajrccm.161.3.9812047
67
Zheng Q Hou J Zhou Y Yang Y Xie B Cao X . Siglec1 Suppresses Antiviral Innate Immune Response by Inducing TBK1 Degradation. Via Ubiquitin Ligase TRIM27 Cell Res (2015) 25:1121–36. doi: 10.1038/cr.2015.108
68
Sahanic S Löffler-Ragg J Tymoszuk P Hilbe R Demetz E Massanetz RK et al . The Role of Innate Immunity and Bioactive Lipid Mediators in COVID-19 and Influenza. Front Physiol (2021) 0:688946. doi: 10.3389/fphys.2021.688946
69
Patel J Beishuizen A Ruiz XB Boughanmi H Cahn A Criner GJ et al . A Randomized Trial of Otilimab in Severe COVID-19 Pneumonia (OSCAR). medRxiv (2021) 2021.04.14.21255475. doi: 10.1101/2021.04.14.21255475
70
Rapkiewicz AV Mai X Carsons SE Pittaluga S Kleiner DE Berger JS et al . Megakaryocytes and Platelet-Fibrin Thrombi Characterize Multi-Organ Thrombosis at Autopsy in COVID-19: A Case Series. EClinicalMedicine (2020) 24. doi: 10.1016/j.eclinm.2020.100434
71
Haslbauer JD Tzankov A Mertz KD Schwab N Nienhold R Twerenbold R et al . Characterisation of Cardiac Pathology in 23 Autopsies of Lethal COVID-19. J Pathol Clin Res (2021) 7:326–37. doi: 10.1002/cjp2.212
72
Scully M Cataland SR Peyvandi F Coppo P Knöbl P Kremer Hovinga JA et al . Caplacizumab Treatment for Acquired Thrombotic Thrombocytopenic Purpura. N Engl J Med (2019) 380:335–46. doi: 10.1056/NEJMoa1806311
73
Annane D Heming N Grimaldi-Bensouda L Frémeaux-Bacchi V Vigan M Roux A-L et al . Eculizumab as an Emergency Treatment for Adult Patients With Severe COVID-19 in the Intensive Care Unit: A Proof-of-Concept Study. EClinicalMedicine (2020) 28. doi: 10.1016/j.eclinm.2020.100590
74
Maglakelidze N Manto KM Craig TJ . A Review: Does Complement or the Contact System Have a Role in Protection or Pathogenesis of COVID-19? Pulm Ther (2020) 6:169–76. doi: 10.1007/s41030-020-00118-5
75
Mesaros C Lee SH Blair IA . Analysis of Epoxyeicosatrienoic Acids by Chiral Liquid Chromatography/Electron Capture Atmospheric Pressure Chemical Ionization Mass Spectrometry Using [13C]-Analog Internal Standards. Rapid Commun Mass Spectrom RCM (2010) 24:3237–47. doi: 10.1002/rcm.4760
76
Yan X Hao Q Mu Y Timani KA Ye L Zhu Y et al . Nucleocapsid Protein of SARS-CoV Activates the Expression of Cyclooxygenase-2 by Binding Directly to Regulatory Elements for Nuclear Factor-Kappa B and CCAAT/enhancer Binding Protein. Int J Biochem Cell Biol (2006) 38:1417–28. doi: 10.1016/j.biocel.2006.02.003
77
Ong SWX Tan WYT Chan Y Fong S Renia L Ng LF et al . Safety and Potential Efficacy of Cyclooxygenase-2 Inhibitors in Coronavirus Disease 2019. Clin Transl Immunol (2020) 9:e1159. doi: 10.1002/cti2.1159
78
Rittchen S Heinemann A . Therapeutic Potential of Hematopoietic Prostaglandin D2 Synthase in Allergic Inflammation. Cells (2019) 8:619. doi: 10.3390/cells8060619
79
Matsumoto H Naraba H Murakami M Kudo I Yamaki K Ueno A et al . Concordant Induction of Prostaglandin E2 Synthase With Cyclooxygenase-2 Leads to Preferred Production of Prostaglandin E2 Over Thromboxane and Prostaglandin D2 in Lipopolysaccharide-Stimulated Rat Peritoneal Macrophages. Biochem Biophys Res Commun (1997) 230:110–4. doi: 10.1006/bbrc.1996.5894
Summary
Keywords
COVID-19, immunopathology, lymph nodes, macrophage activation, plasmablasts, thrombosis, thromboinflammation, T-cell dysregulation
Citation
Haslbauer JD, Zinner C, Stalder AK, Schneeberger J, Menter T, Bassetti S, Mertz KD, Went P, Matter MS and Tzankov A (2021) Vascular Damage, Thromboinflammation, Plasmablast Activation, T-Cell Dysregulation and Pathological Histiocytic Response in Pulmonary Draining Lymph Nodes of COVID-19. Front. Immunol. 12:763098. doi: 10.3389/fimmu.2021.763098
Received
23 August 2021
Accepted
18 November 2021
Published
13 December 2021
Volume
12 - 2021
Edited by
Gennady Bocharov, Institute of Numerical Mathematics (RAS), Russia
Reviewed by
Paul Klenerman, University of Oxford, United Kingdom; Carl-Philipp Hackstein, University of Oxford, United Kingdom, in collaboration with reviewer PK; Elke Scandella, Kantonsspital St. Gallen, Switzerland
Updates
Copyright
© 2021 Haslbauer, Zinner, Stalder, Schneeberger, Menter, Bassetti, Mertz, Went, Matter and Tzankov.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandar Tzankov, alexandar.tzankov@usb.ch
This article was submitted to Viral Immunology, a section of the journal Frontiers in Immunology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.