 Chunren Meng
Chunren Meng Chufeng Gu
Chufeng Gu Shuai He1,2
Shuai He1,2 Tong Su
Tong Su Thashi Lhamo
Thashi Lhamo Qinghua Qiu
Qinghua Qiu- 1Department of Ophthalmology, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China
- 2National Clinical Research Center for Eye Diseases; Shanghai Key Laboratory of Ocular Fundus Diseases, Shanghai Engineering Center for Visual Science and Photomedicine, Shanghai Engineering Center for Precise Diagnosis and Treatment of Eye Diseases, Shanghai, China
- 3Department of Ophthalmology, Shigatse People’s Hospital, Shigatse, China
Diabetic retinopathy (DR) is prevalent among people with long-term diabetes mellitus (DM) and remains the leading cause of visual impairment in working-aged people. DR is related to chronic low-level inflammatory reactions. Pyroptosis is an emerging type of inflammatory cell death mediated by gasdermin D (GSDMD), NOD-like receptors and inflammatory caspases that promote interleukin-1β (IL-1β) and IL-18 release. In addition, the retinal neurovascular unit (NVU) is the functional basis of the retina. Recent studies have shown that pyroptosis may participate in the destruction of retinal NVU cells in simulated hyperglycemic DR environments. In this review, we will clarify the importance of pyroptosis in the retinal NVU during the development of DR.
1 Introduction
Diabetes mellitus (DM) is a prevalent metabolic disorder syndrome causing multiple systemic complications (1). According to the International Diabetes Federation, the global incidence of DM will increase in the next few decades, from an estimated 9.3% in 2019 to 10.2% by 2030 and 10.9% by 2045 (2). DR is a common and progressive microvascular complication of DM that can cause irreversible retinal damage (3) and it remains the main cause of impaired vision in working-aged people (4). According to a meta-analysis, the global population with moderate or more severe vision impairment due to DR was 2.6 million in 2015, and the number is predicted to increase to 3.2 million by 2020 (5). Thus, DR will impose a heavy economic burden on individuals and society worldwide. DR is mainly caused by hyperglycemia. Long-term hyperglycemia can cause characteristic pathological changes in the retina, such as thickening of the basement membrane of the retinal microvessels, loss of vascular cells, increased vascular permeability, and neovascularization (6). A better understanding of the pathogenesis in the retina is urgently needed to develop interventions. According to the International Council of Ophthalmology, DR falls into two categories: nonproliferative DR (NPDR) and proliferative DR (PDR, Table 1) (7). NPDR is regarded as the early stage of DR and PDR is the advanced stage. When DR affects the macula, it can cause diabetic macular edema (DME). DME can occur in any stage of DR and is the most frequent cause of blindness in diabetic patients (8).
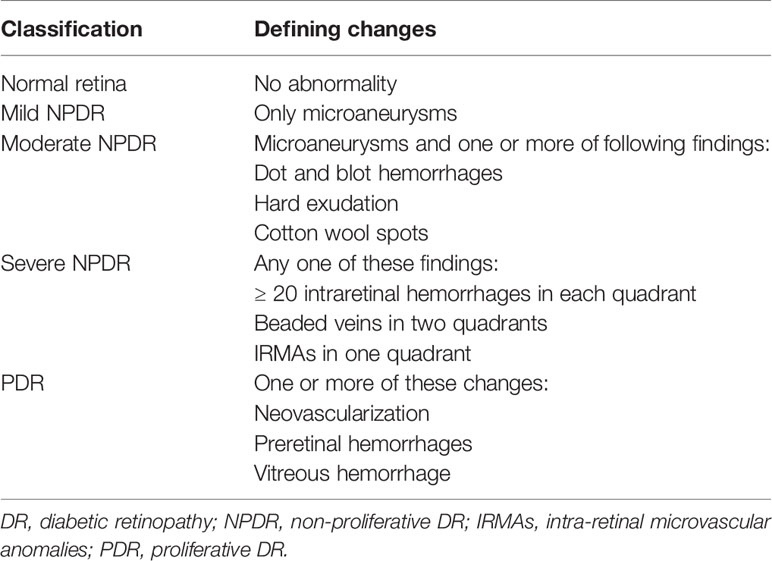
Table 1 Classification of Diabetic Retinopathy.
The retina is a complex system consisting of the retinal pigment epithelium (RPE) and the neurosensory retina. Generally, the retina is comprised of ten layers, from the outside to the inside: RPE, rod and cone layer, outer limited membrane (OLM), outer nuclear layer (ONL), outer plexiform layer (OPL), internal nuclear layer (INL), internal plexiform layer (IPL), ganglion cell layer (GCL), nerve fiber layer (NFL) and internal limited membrane (ILM) (9). Histologically, neurons, glia, and blood cells in the retina are linked together to form an important structure named the retinal neurovascular unit (NVU, Figure 1A) (10). The NVU consists of retinal neurons (photoreceptors: cones and rods, horizontal and bipolar cells, amacrine cells, and ganglion cells), glial cells (Müller cells, astrocytes, and microglia) and blood cells (endothelial cells and pericytes) (11). All components of the retinal NVU have different distributions in the ten-layer structure of the retina (Figure 1B) (9). Accumulating studies have suggested that these factors are related to the pathogenesis of DR, namely, retinal microangiopathy, retinal neurodegeneration and inflammation (6). In recent years, chronic inflammation has been shown to be the key to pathological changes in the NVU (12, 13). Pyroptosis is a novel inflammatory form of regulated cell death that facilitates the release of many proinflammatory factors, including interleukin-1β (IL-1β) and IL-18 (14, 15). Multiple studies have revealed that pyroptosis is relevant to the development of DR. This review describes the currently available studies examining the effect of pyroptosis in the retinal NVU on DR. We will focus on the retinal NVU, pyroptosis, and the effect of the latter on the former.
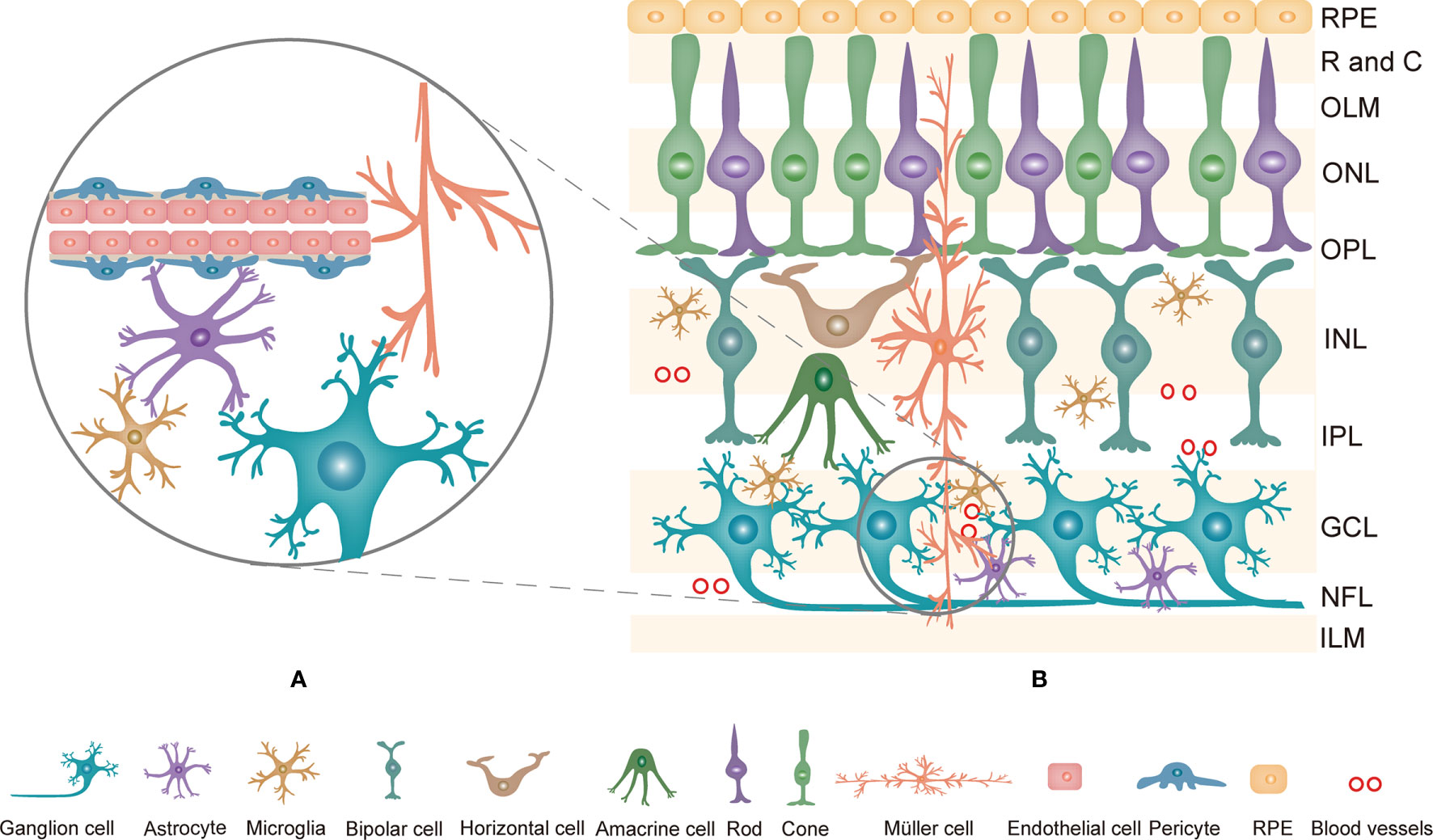
Figure 1 (A) The composition of the retinal NVU. Neurons, Müller cells, astrocytes, microglia, endothelial cells and pericytes are linked together to form the retinal NVU. (B) Structure of the retina and the distribution of retinal NVU components in the retina. The retina is generally divided into ten layers. Retinal neurons, various glial cells and blood cells are distributed in the corresponding layers in the retina. NVU, neurovascular unit; RPE, Retinal pigment epithelial; R and C, rod and cone; OLM, outer limited membrane; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, internal nuclear layer; IPL, internal plexiform layer; GCL, ganglion cell layer; NFL, nerve fiber layer; ILM, internal limited membrane.
2 Retinal NVU in DR
The NVU is an important structure of the retina. As mentioned above, the retinal NVU mainly includes six components: retinal neurons, endothelial cells and pericytes, Müller cells, astrocytes and microglia. Each component of the retinal NVU has different physiological functions, but a close relationship exists among them. As more in-depth research on DR has been conducted, it has come to be regarded as a NVU disease (16). During the development of DR, many mechanisms lead to pathological changes in the retina, including oxidative stress, endoplasmic reticulum stress and inflammation (17–20).
2.1 The Retinal NVU
The retinal microvascular system is an indispensable constituent of the normal retina (21). The distribution of the microvasculature in the retina is specific, not spread through all layers of the retina. Currently, the microvasculature is found in four layers of the retina: (1) the deep part of the INL, (2) the border of the shallow INL and deep IPL, (3) the shallow part of the IPL and the RGCL, and (4) the NFL, and the photoreceptor layer is devoid of retinal blood flow (22, 23). Vascular endothelial cells and pericytes are important components of the retinal microvasculature, sharing a common basement membrane (24). The structure and function of the microvasculature rely on interactions between pericytes and endothelial cells, which are disturbed in some retinal vascular diseases, such as DR, retinal vascular occlusion and retinopathy of prematurity (21). Additionally, pericytes and endothelial cells are important components of the retinal NVU, and interactions between the two types of cells are necessary for the complete structure and normal function of the retinal NVU (21). In addition, the pericyte-endothelial interaction is an essential component of the internal blood retina-barrier (BRB), which is a highly selective barrier protecting the retina from the blood circulation (25, 26).
Retinal neurons are sensitive cells of various types in the retinal NVU. Retinal neurons include five major cell types: photoreceptors, horizontal cells, bipolar cells, amacrine cells and ganglion cells (27). Photoreceptors are light-sensing cells that are categorized into cone and rod cell types (28). The ONL contains the cone and rod photoreceptor cell bodies, the INL contains the cell bodies of amacrine, bipolar and horizontal cells, and the GCL mainly consists of ganglion cell bodies (29). Photoreceptor cells are in contact with secondary neurons (bipolar and horizontal cells), which in turn are in contact with ganglion cells in the IPL. In the retina, ganglion cells are the output neurons integrating information (30). Axons of ganglion cells comprise the NFL that sends the visual signal to the visual cortex through the optic nerve (31, 32).
Müller cells and astrocytes are two types of macroglia in the retina (33). Müller cells are the most abundant, accounting for 90% (34). In addition to their larger number, the distribution of Müller cells is also wide. Müller cells penetrate almost all layers of the retina and contact a variety of retinal cells (35). Due to their unique position, the normal function of Müller cells is necessary to maintain retinal homeostasis. Müller cells participate in structural support and metabolic nutrition in a healthy retina. For instance, Müller cells participate in the regulation of nutrition metabolism and protection of neurons (35). In comparison, retinal astrocytes are found only in the NFL and GCL (36). Astrocytes play a pivotal role in the metabolism and mechanical support of the neurons and serve as an essential component in the internal BRB (34, 36, 37).
In the brain and retina, microglia are resident immune cells that monitor their surroundings (38). Under normal circumstances, microglia spread over the NFL, GCL, IPL, INL, and OPL of the retina (34). According to recent research, microglia are present in the ONL only under pathological conditions (38). As immune cells of the central nervous system (CNS) (39), the functions of microglia are subdivided into six major categories: (1) Phagocytosis: Microglia predominantly clear cellular waste from the retina (40). (2) Immune Functions: Microglia are thought to participate in antigen presentation, inflammatory reactions and complement activation during defense against infectious substances and to facilitate tissue repair and immune regulation in the retina (38). (3) Microglia participate in regulating progenitor cell proliferation, differentiation, and neuronal survival (41). (4) Microglia are necessary to maintain synaptic transmission based on the synaptic structure and normal visual function in the adult retina (42). (5) Microglia have an essential role in angiogenesis. (6) Microglia are necessary to maintain retinal homeostasis (43).
Generally, the retinal NVU participates in retinal nutrition and metabolism and provides an appropriate environment for neural signal transmission (12, 44). Furthermore, RPE cells and the retinal NVU are the core components of the BRB, an important protective barrier that is comprised of two parts: the internal BRB and the outer BRB. Most of the components of the retinal NVU are involved in the composition of the internal BRB (Figure 2) (45). The outer BRB is mainly comprised of tight junctions of RPE cells (46). Functionally, the internal BRB is essential in maintaining the microenvironmental homeostasis of the inner retina layers, and the outer BRB mainly regulates the transfer of solutes and nutrients from the blood to the photoreceptors (47–49).
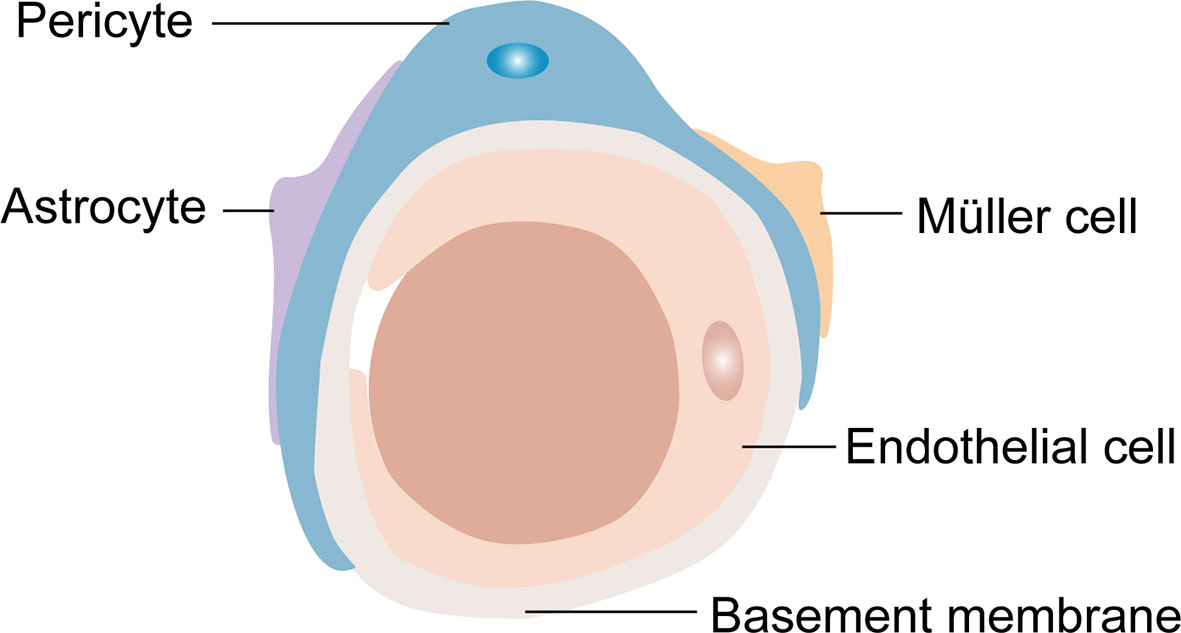
Figure 2 The composition of the internal blood retina barrier (iBRB). Pericytes, endfeet of astrocytes and Müller cells cover the endothelial cells to form the iBRB.
2.2 Retinal NVU Changes in DR
According to previous studies, many components of the retinal NVU are affected in individuals with diabetes (50). The proper function of every element of this retinal NVU is critical for normal retinal function. In individuals with DR, damage to various cells in the retinal NVU leads to dysfunction of every component associated with the development of DR (51). The diabetic environment damages the retinal NVU through various pathways, such as oxidative stress, endoplasmic reticulum stress and inflammation (52).
2.2.1 Microangiopathy in DR
In the early stage of DR, a variety of pathological changes occur in the microvascular system, such as basement membrane thickening and the loss of pericytes and endothelial cells, resulting in the destruction of the BRB and the formation of microaneurysms (53). The collected evidence indicates that retinal microvascular pathology is related to oxidative stress, apoptosis, inflammation and endoplasmic reticulum stress (54, 55). Mitochondria are the main sites for reactive oxygen species (ROS) production. High glucose (HG) increases mitochondrial production of ROS, and excessively produced ROS leads to mitochondrial dysfunction. Mitochondrial dysfunction induces apoptosis of retinal vascular cells (55). Furthermore, hyperglycemia initiates the caspase-3 activation pathway mediated by mitochondrial cytochrome C to induce retinal capillary cell death (56). Moreover, elevated ROS levels promote the expression of proinflammatory cytokines by the ROS/nuclear factor-κB (NF-κB) pathway. These proinflammatory mediators promote BRB disruption leading to microaneurysms and retinal leakage (57). The endoplasmic reticulum (ER) is mainly responsible for protein synthesis and folding in cells. Multiple studies have shown that ER stress is involved in pericyte changes in DR. Intermittent hyperglycemia promotes pericytes to secrete more macrophage chemotactic protein 1 (MCP-1), activated transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP). MCP-1, ATF4 and CHOP are mediators of ER stress related to inflammation and cell death (58). Oxidative stress and ER stress promote the release of proinflammatory mediators. The inflammatory response of the retinal microvascular system is triggered by various factors, such as HG, cytokines and chemokines, and ROS, and plays a crucial role in early DR (59). Endothelial cells are extremely sensitive to proinflammatory factors. Upregulated proinflammatory factors not only induce changes in inflammatory pathways and apoptosis in endothelial cells but also stimulate endothelial cells to produce intracellular adhesion molecules, causing leukocyte stagnation (20).
2.2.2 Neurodegeneration in DR
Neurodegeneration is also an important pathological change in DR that may occur before visible microvascular pathologies (60). Neuronal apoptosis is an important characteristic of neuronal degeneration. A previous study revealed an association between increased levels of protein kinase RNA-like ER kinase (PERK) and CHOP in retinal neurons of diabetic rats with retinal ganglion cell (RGC) apoptosis, similar to the results obtained from nondiabetic rats exposed to HG (61). CHOP promotes protein synthesis in the ER to cause oxidative stress and cell death (62). In addition, the hyperglycemic environment cause oxidative stress. The HG-induced increase in ROS levels promotes the apoptosis of RGCs (63). Inflammation also contributes substantially to neuronal apoptosis. For example, NF-κB activation induced by hyperglycemia is associated with RGC death in DR (64).
As mentioned above, accumulating evidence has suggested that inflammation is associated with the development of DR (65), and sustained inflammation can lead to retinal NVU component injuries (66). Pyroptosis is an emerging type of inflammatory cell death inextricably linked with inflammation. The caspase-1 mediated pathway, which is activated by NLR family pyrin domain containing 3 (NLRP3) and NLRP1 inflammasomes, is the canonical inflammasome pathway that triggers pyroptosis. Recent studies have indicated that these inflammasomes are associated with neurovascular diseases, especially those occurring in the CNS, such as DR, neurodegeneration disease and stroke (65). In other words, pyroptosis may be related to retinal NVU dysfunction under diabetic conditions.
3 Pyroptosis
Pyroptosis is a form of programmed cell death that has been identified in the past decade. Pyroptosis is crucial for innate immune defense, and it occurs in both macrophages and other cells (67). In contrast to apoptosis, pyroptosis is associated with inflammation. Some characteristics of pyroptosis are cell swelling, and IL-1β and IL-18 release from gasdermin pores in membranes (68). In pyroptosis, the caspase-1-dependent pathway is called the canonical inflammasome pathway, and the caspase-4/5/11-dependent pathway is described as the noncanonical pathway (69, 70). Diverse infections and immune challenges activate caspase-1 in cells through different inflammasomes, including NLRP3, NLRP1, apoptotic speck-like protein containing a caspase recruitment domain (ASC), NOD-like receptor family, caspase recruitment domain (CARD) containing 4 (NLRC4) and absent in melanoma 2 (AIM2) (71). Unlike caspase-1, intracellular lipopolysaccharide (LPS) directly interacts with caspase-4/5/11, and then the latter is activated (72–74). The common result of caspase-1/4/5/11 activation is that gasdermin D (GSDMD) becomes a pyroptotic effector of these caspases. Subsequently, GSDMD is cleaved to produce two parts: the N-terminus and the C-terminus. The N-terminus of GSDMD induces pore formation in the membrane, and these pores become the channels through which IL-1β and IL-18 are released, ultimately leading to cell death (Figure 3) (67, 75). More interestingly, caspase-11-mediated maturation of GSDMD triggers caspase-1 activation, accompanied by the secretion of IL-1β (76). In addition, GSDMD is not the only substrate of pyroptosis. In some cases, activation of gasdermin E (GSDME) by caspase-3 has also been shown to induce pyroptotic cell death (77, 78). In the innate immune system, pyroptosis exerts a dual effect. It protects the body from pathogen infection and endogenous threats but causes harmful inflammation in the case of excessive activation (68). As more in-depth research on pyroptosis has been conducted, pyroptosis has been found to be associated with the occurrence and development of many common diseases, including obesity, type 2 diabetes mellitus (T2DM) and complications of diabetes (79–81).
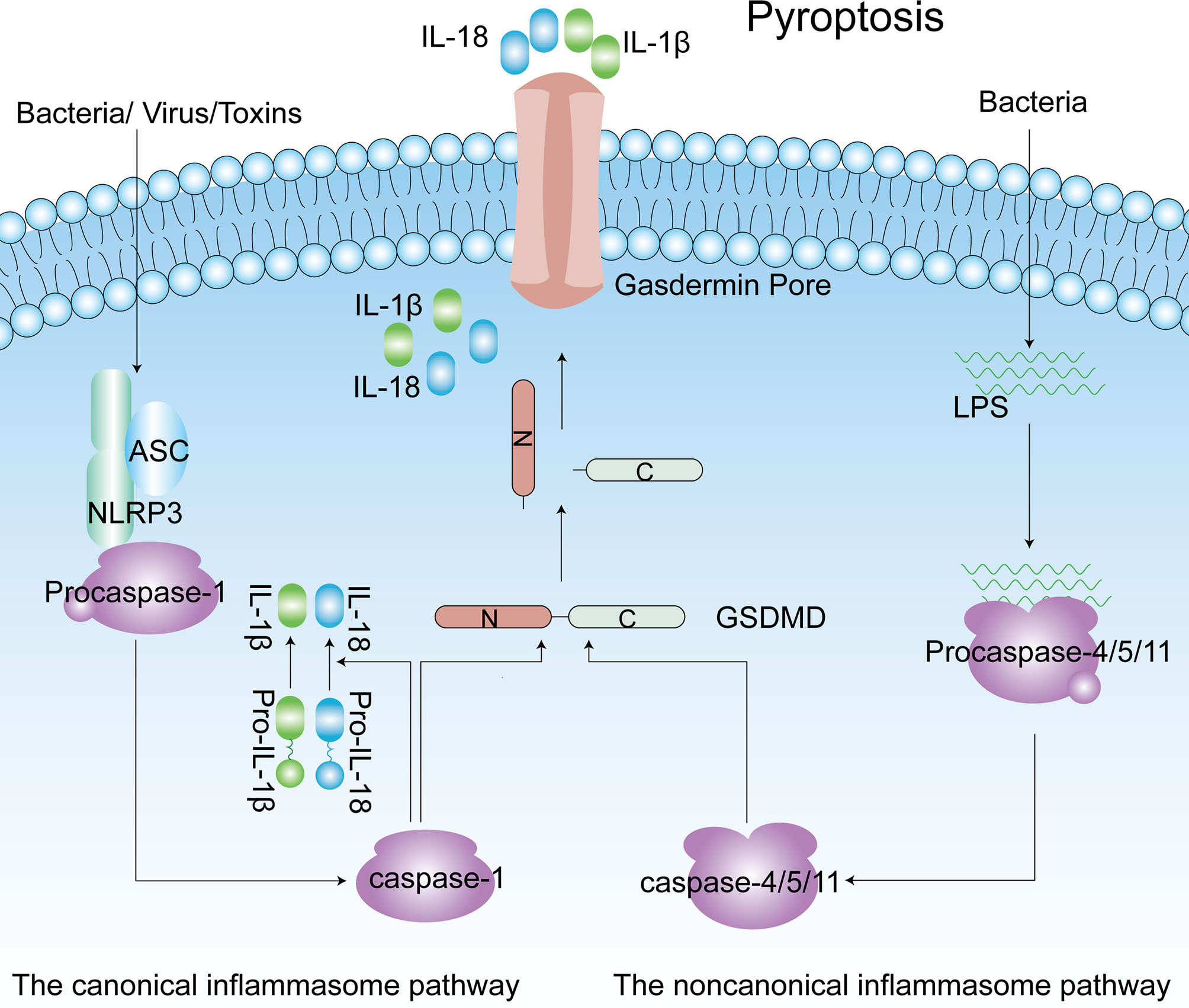
Figure 3 The canonical inflammasome pathway (Caspase-1-dependent) and noncanonical inflammasome pathway (Caspase4/5/11-dependent) of pyroptosis. Caspase-1 is activated by NLRP3 inflammasomes, and caspase4/5/11 are activated by direct interaction with LPS. Active caspase-1 and caspase-4/5/11 cleave the GSDMD to produce the C-terminus and N-terminus. Released gasdermin-N domains form an approximately 12–14 nm inner diameter pore on the plasma membrane. IL-1β and IL-18 are matured by active capase-1 and released from the gasdermin pore. ASC, apoptotic speck-like protein containing a caspase recruitment domain; IL-1β, interleukin-1β; LPS, lipopolysaccharide; GSDMD, gasdermin D; NLRP3, NLR family pyrin domain-containing 3.
3.1 Pyroptosis in Obesity and T2DM
As living standards improve, the number of people with metabolic diseases is increasing annually (82). Obesity and DM are common metabolic diseases worldwide. As mentioned above, NLRP3 inflammasomes activate caspase-1 to trigger pyroptosis. Eventually, IL-1β and IL-18 are secreted by pyroptotic cells. Many studies have found that NLRP3 inflammasomes correlated with pyroptosis participate in the pathogenic mechanism of some metabolic diseases, including obesity and type 2 diabetes mellitus (83).
Obesity is a metabolic disorder with multifactorial pathogenesis. In addition, obesity is also a risk factor for T2DM. Macrophages of monocyte origin infiltrate tissues as one of the pathological changes of obesity. Monocytes in the peripheral blood of obese patients exhibit high inflammatory caspase activity. In monocytes, saturated fatty acids activate caspase-4/5 to induce the production and release of IL-1β and IL-18, eventually leading to obesity-related inflammation (84). Moreover, higher expression of NLRP3 is detected in adipose tissue from obese individuals than in adipose tissue from metabolically healthy individuals (85). Of note, NLRP3 knockout prevented mice fed a high-fat diet from becoming obese (86). Adipocytes not only store energy but also secrete adipokines to regulate metabolism (87). Adipocyte dysfunction (e.g., decreased levels of insulin-sensitive adipokines and increased levels of proinflammatory cytokines) are related to insulin resistance and T2DM (88). Pancreatic β-cell dysfunction and insulin resistance are the main characteristics of T2DM. Previous studies have suggested that the NLRP3 inflammasome is closely associated with the pathogenesis of T2DM (89). HG, free fatty acids and a high-fat diet promote the activation of the NLRP3 inflammasome in patients with T2DM (90). Activation of NLRP3 inflammasomes induces excessive secretion of IL-1β and IL-18. Increased IL-1β levels cause the dysregulation of blood sugar levels by impairing pancreatic β-cells and inducing insulin resistance (83). The high expression of IL-1β receptors on pancreatic β-cells not only contributes to increase production of IL-1β, but also facilitates the spread of inflammatory signals through the NF-κB pathway, eventually leading to pancreatic β-cell dysfunction (91). IL-1β promotes insulin resistance by reducing the tyrosine phosphorylation and mRNA expression of insulin receptor substrate-1, and inducing the expression of tumor necrosis factor α (92). Additionally, IL-1β and IL-18 decrease the insulin sensitivity of target organs by inducing lymphocytes to accumulate in adipose tissue (93). In conclusion, NLRP3 inflammasomes are critical for the development and progression of obesity and T2DM, especially IL-1β, a product of NLRP3 inflammasome activation. Importantly, pyroptosis is associated with the occurrence and development of DM but also with the development of its complications, such as diabetic cardiomyopathy (94), diabetic nephropathy (95) and DR (96).
3.2 Pyroptosis in Diabetic Complications
3.2.1 Diabetic Retinopathy
Diabetic retinopathy is prevalent among people with long-term DM and remains the critical cause of visual impairment in working-aged people. Loukovaara et al. used immunohistochemistry and observed that the levels of caspase-1 and IL-18 were significantly increased in DR patients’ vitreous (97). Simultaneously, they found that NLRP3 inflammasome activation plays an important role in the pathogenesis of proliferative DR (97). Numerous studies have reported that the expression levels of inflammatory components including NLRP3, ASC, procaspase-1, IL-1β, and IL-18, were significantly upregulated in diabetic rat retinal tissues compared to control group (98, 99). In a recent study, the authors documented that HG promoted RPE cell pyroptosis, and methyltransferase-like protein 3 (METTL3) could reverse these changes by targeting the miR-25-3p/PTEN/Akt signaling pathway (100). In addition, researchers have observed that HG promotes NLRP3 inflammasome activation and pyroptosis in HG-induced human retinal microvascular endothelial cells (HRMECs) and human retinal pericytes (HRPs) (101, 102). Several reports have suggested that P2X7 purinergic receptor (P2X7R) promotes DR pathogenesis (103, 104). P2X7R activates the NLRP3 inflammasome and promotes the release of the proinflammatory cytokine IL-1β in retinal pericytes treated by HG (105, 106). JNJ47965567, a P2X7R antagonism, can revert the damage caused by HG in cultured pericytes (104). Similarly, previous researchers have documented that H3 relaxin inhibits AGE-induced HRMEC pyroptosis by attenuating the P2X7R/NLRP3 pathway (107). In addition, researchers found that fenofibrate and sulforaphane provide significant protection against DR by attenuating NLRP3 inflammasome activation and activating the antioxidative Nrf2 pathway (108, 109). A recent study found that vitamin D3 exerts protective effects against DR by inhibiting ROS/TXNIP/NLRP3 inflammasome pathway activation (110). Similarly, a recent study demonstrated that vitamin D3 protects RGCs by reducing inflammatory cytokines and increasing the expression of neuroprotective factors in glaucomatous mice (111). Based on the above studies, pyroptosis may play a crucial role in the changes in retinal cells in the DR environment.
3.2.2 Other Complications
Diabetic cardiomyopathy (DCM) is a crucial complication of DM and can result in heart failure (112). Growing research suggests that pyroptosis may be involved in the pathogenesis of DCM (113). Myocardial ultrastructure studies demonstrated that dying cells exhibited swollen fibril and mitochondria in the myocardium of diabetic rats, similar to the phenotypic features of pyroptosis (114, 115). Protein expression levels of the NLRP3 inflammasome, caspase-1, IL-1β and GSDMD were remarkably elevated in diabetic mouse cardiac tissue (94). In line with this, Ye et al. found that the mRNA levels of NLRP3, caspase-1 and IL-1β were considerably higher in the T2DM mice hearts than in control mouse hearts (116). In addition, a recent study showed that silencing long non-coding RNA (lncRNA) Kcnq1ot1 ameliorated pyroptosis and fibrosis in myocardial tissues of diabetic mice and was related to the Kcnq1ot1/miR-214-3p/caspase-1/TGF-β1 signaling pathway (94). Hyperglycemia induced cardiomyocyte pyroptosis in high-fat diet-induced T2DM mice via the AMPK-TXNIP pathway (117). Furthermore, other studies showed that AIM2 expression was significantly elevated in the heart tissue of diabetic rats compared to the control group (118). AIM2 is involved in HG-induced DCM cell death and fibrosis through the GSDMD pathway (118). These studies illustrate that pyroptosis may be an important contributor to the pathogenesis of DCM.
Diabetic nephropathy is a microvascular complication of DM and remains the major cause of chronic kidney disease throughout the world (119). Accumulating evidence demonstrates that pyroptosis plays a pivotal role in the progression of diabetic nephropathy (120). Recent studies have revealed that the protein levels of the NLRP3 inflammasome, GSDMD, caspase-1, and IL-1β in the kidney were significantly increased in diabetic rats and mouse models compared to the control group (95, 121). In addition, the expression levels of pyroptosis-associated proteins, such as caspase-11 or caspase-4, GSDMD, IL-1β and IL-18, in human and mouse podocytes cultured in HG are augmented (122). Caspase-4 or GSDMD knockdown considerably reversed these changes (122). Studies have confirmed that hyperglycemia promotes HK-2 cell pyroptosis (123). The lncRNA MALAT1 promotes hyperglycemia-induced HK-2 cell pyroptosis by inhibiting the expression of miR-23c, leading to the activation of the ELAVL1/NLRP3 pathway (123). Current studies have documented that the expression of lncRNA GAS5 in HG-stimulated HK-2 cells is repressed (124). In addition, GAS5 suppression significantly increased the expression of NLRP3, caspase1, IL-1β and GSDMD, and GAS5 overexpression reversed these changes (124). These notable findings indicate that pyroptosis may promote diabetic nephropathy pathogenesis.
4 Effect of Pyroptosis on the Retinal NVU in DR
4.1 Pyroptosis in Retinal Pericytes and Endothelial Cells
One of the earliest hallmarks of DR is microvascular changes, accompanied by the loss of pericytes, basement membrane thickening and the destruction of tight junctions between endothelial cells, together with hyperpermeability, capillary nonperfusion, microaneurysms, and the subsequent loss of endothelial cells (125, 126). Pericyte and endothelial cell death is a fatal blow to the retinal microvasculature. Multiple forms of cell death have been observed in diabetes-induced pericytes and endothelial cell death. Based on previous evidence, pericytes may die due to apoptosis and necrosis, and endothelial cells predominantly undergo apoptosis during the development of DR (127). However, some studies have found that pyroptosis might participate in the death of vascular cells in the retina and the pathological changes in DR.
Recently, HG was revealed to significantly induce the release of inflammatory cytokines and pore formation HRPs, resulting in pericyte lysis (102). Based on these findings, HG induces inflammation and pyroptosis in HRPs. Furthermore, HG induces retinal pericyte pyroptosis through the NLRP3-caspase-1 pathway (102). Coincidentally, HRPs undergo caspase-1-dependent pyroptosis after treatment with advanced glycation end product modified bovine serum albumin (AGE-BSA), which often appears in the diabetic environment (128). LncRNA myocardial infarction-associated transcript (MIAT) regulates caspase-1 expression by sponging miR-342–3p, ultimately resulting in the pyroptosis of HRPs treated with AGE-BSA (128). Notably, the authors used immunofluorescence staining and observed that AGE-BSA-induced HRPs exhibited phenotypic features of pyroptosis, including pyknosis, cell swelling, and hyperpermeability in the plasma membrane (128). Chen et al. documented that the protein expression of caspase-1, NLRP3, ASC, IL-1β, and IL-18 was significantly upregulated in retinal tissues of streptozotocin-induced diabetic rats (129). Furthermore, HG activated the NLRP3 inflammasome in HG-exposed HRMECs by the ROS-TXNIP pathway (129). Other studies have also shown that HRMECs undergo pyroptotic cell death under diabetic-like conditions (107). Platania et al. suggested that some miRNAs, such as miR-20a-5p, miR-20b and miR-106a-5p, are dysregulated in the retina and blood circulation of diabetic mice. These miRNAs can modulate the expression of DR-related factors, such as vascular endothelial growth factor (VEGF), participating in the progression of DR (130). Our group has reported that HRMECs cultured under HG conditions suffer from pyroptosis. Notably, miR-590-3p targets NLRP1 and inactivates the NOX4 signaling pathway to inhibit pyroptosis in HRMECs (101). In addition, prostaglandin E2 (an inflammatory mediator) participates in the activation of NLRP3 inflammasomes in HRMECs (131). Mcc950 selectively inhibits NLRP3 inflammasomes, thereby inhibiting human retinal endothelial cell (HREC) dysfunction under HG conditions (132).
The presence of gasdermin pores on the cell membrane is one of the characteristics of pyroptotic death. These pores destroy the osmotic potential, causing the cells to swell and eventually lyse (133). Diabetic environments such as HG and AGE-BSA promote retinal microvascular cell loss through pyroptosis (107). The loss of these two types of cells results in decreased pericyte-endothelial interactions and their miscommunication and contributes to microvascular instability. In addition, the loss of pericytes contributes to the formation of acellular capillaries, capillary occlusions, microaneurysms and hemorrhage (134). These significant pathological changes occur in DR. Vascular occlusion may lead to perfusion failure and retinal ischemic-hypoxic injury. The latter increases the expression of VEGF in glial cells and endothelial cells. Moreover, vascular occlusion may lead to retinal neuron dysfunction and even neuronal death. Furthermore, vascular cell death causes the destruction of the BRB (135), which increases vascular permeability and the possibility of inflammatory cells entering the retinal microenvironment. These processes are also the main features of DR (25). Alterations in BRB integrity lead to diabetic macular edema, eventually resulting in a severe visual impairment without timely intervention (136). Hyperglycemia-induced pyroptosis in the retinal microvasculature not only causes the death of pericytes and endothelial cells but also increases the number of inflammatory mediators, including IL-18 and IL-1β (102, 107). Researchers found increased levels of proinflammatory mediators in the serum or aqueous humor in patients with DR compared with normal controls (20). These inflammatory cytokines participate in triggering an even more excessive inflammatory reaction, promoting the development of DR (66, 137).
4.2 Pyroptosis in Retinal Neurons
Neurons in the retinal NVU are the major cells that transmit light signals and form vision. Extensive studies have shown the presence of retinal neuronal degeneration in the early stage of DR, even earlier than visible vasculopathy (138, 139). Two features of neurodegeneration in the retina are neuronal death and reactive gliosis (9). Many researchers have provided a description of neuronal apoptosis in DR. Pyroptosis, a form of programmed cell death involved in inflammation, can occur in retinal ganglion cells (140). DM causes hypoxia in retinal tissue and leads to an imbalance in retinal immune responses (141). Hypoxia-induced factor-1 (HIF-1) is continuously produced and degraded under hypoxic conditions. Then, HIF-1α functions as a transcription factor to activate the genes encoding the proangiogenic growth factors, IL-6 and IL-8 (141). Moreover, loss of pericytes potentially leads to acellular capillary formation, which is associated with vascular occlusion and leads to nonperfusion and retinal ischemic-hypoxic injury. Ischemia-hypoxia upregulates the expression of HIF-1 (25). Pyroptosis participates in retinal ischemic damage and promotes retinal ganglion cell death in acute glaucoma (142). The caspase-8-HIF-1α-NLRP12/NLRP3/NLRC4 pathway initiates neuroinflammation and pyroptosis (142). Moreover, pyroptosis is an alternative pathway through which photoreceptors degenerate after retinal detachment (143). Additionally, caspase-1/3/4/5 activities were found to be increased in a streptozotocin-induced diabetes mouse model (140). A previous study reported that NLRP3, ASC, and caspase-1 were specifically located in the GCL and the INL and ONL in the retinas of diabetic rats according to immunohistochemical results (99). Simultaneously, other authors have also documented that the expression levels of NLRP3, ASC, and caspase-1 were increased in retinal cells of diabetic rats (99).
4.3 Pyroptosis in Müller Cells
Müller cells and astrocytes of the retinal NVU participate in retinal structural support and maintain retinal homeostasis. Studies have found that approximately 15% of Müller cells die after 7 months in retinas of the diabetic mice (127). After exposure to HG, caspase-1 activity and IL-1β production in Müller cells increase and subsequently induce cell death (144). Furthermore, inhibition of the caspase-1/IL-1β pathway prevents the loss of Müller cells under diabetic conditions (127). The aforementioned evidence revealed that pyroptotic death may be responsible for Müller cell death under diabetic conditions. Consistently, HG-induced nuclear accumulation of GAPDH in Müller cells relies on activating the caspase-1/IL-1β pathway. More interestingly, the accumulation of GAPDH in the nucleus is associated with the induction of cell death (145). Due to their important locations and functions, Müller cell loss will lead to an incomplete retinal structure. For example, Müller cell loss promotes the destruction of the internal BRB integrity and increases the vascular permeability and the loss of neuroprotective effects, affecting both neurons and blood cells (35). Specific removal of Müller cells from the retina leads to retinoschisis, showing that Müller cells hold the neural layers together to protect the neural tissue from ripping apart (146). Previous studies have reported that selective removal of Müller cells results in photoreceptor apoptosis, BRB breakdown, and vascular telangiectasis (147). In addition, destruction of the cell-to-cell communication between Müller cells and retinal pericytes promotes pericyte death (148). The loss of Müller cells in DM is also related to the formation of aneurysms, a clinical feature of DR (35). Furthermore, Müller cells are the major source of IL-1β (149). Long-term production of IL-1β from Müller cells affects the viability of endothelial cells in a paracrine manner (150). Due to the high sensitivity of endothelial cells to IL-1β, endothelial cells die after responding to this inflammatory cytokine (150). In addition, the death of endothelial cells promotes the formation of acellular capillaries, which are the main hallmark of DR pathology (151).
4.4 Pyroptosis in Retinal Microglia
Microglia are specific innate immune cells of the retina that monitor the environment and remove metabolic waste. As more in-depth research on pyroptosis has been conducted, pyroptosis has also been identified in microglia. Microglial pyroptosis occurs in various disease states, such as after spinal cord injury (152), ischemic brain injury (153), retinal ischemia and reperfusion injury (154), and DR. Retinal ischemia and reperfusion injury (I/R) is the basis of multiple retinal diseases, including DR, glaucoma, and retinal artery occlusion (155). Accumulating studies have shown that I/R promotes retinal microglial pyroptotic death, which is associated with lncRNA H19 (154). In addition, S100A12 is closely related to the incidence and severity of DR. S100A12 represents a proinflammatory trigger for retinal microglial activation by activating the NLRP3 inflammasome in a diabetic environment (156). Moreover, HG was recently shown to induce retinal microglial pyroptosis through NLPR3 inflammasome signaling (157). HG upregulated the protein expression of NLPR3, caspase-1, GSDMD, and IL-1β in retinal microglia (157). In another study, HG-induced retinal cells produced more IL-1β, and the IL-1β induced microglial proliferation (158). Moreover, IL-1β is mainly produced by microglia under diabetic conditions (159). Overactive microglia produce various proinflammatory and cytotoxic factors, including IL-1β, TNF-α, and ROS, which lead to chronic inflammation and contribute to the destruction of hemostasis in the NVU, BRB breakdown and worsening of the pathology of DR (160).
4.5 Proinflammatory Mediators Produced From Pyroptosis in the NVU
Accumulating research suggests that IL-1β and IL-18 may partly come from the pyroptosis-mediated cell death of retinal cells in diabetic rats and mice (99). IL-1β and IL-18 are the two key cytokines that undergo maturation through cleavage by active caspase-1 and are released through pyroptosis. In addition, IL-1β is the most studied IL-1 family member in retinopathy, such as DR (161). These proinflammatory mediators lead to persistent low-grade inflammation, affecting the hemostasis of the retinal NVU. Under HG conditions, IL-1β induces pericyte apoptosis by activating NF-κB, thereby increasing vascular permeability (162). Additionally, IL-1β affects glial cells (microglia and macroglia) and ultimately results in neural changes (158). Moreover, the increased levels of IL-1β coincide with increasing retinal neovascularization (163). Coincidentally, IL-18 may promote retinal angiogenesis in active PDR together with VEGF or through VEGF (164). Accumulated studies indicate that glucocorticoids show anti-inflammatory efficacy by inhibiting key proinflammatory mediators such as IL-1β (165). Thus, glucocorticoids may have potential use in modulating pyroptosis. Collectively, IL-1β and IL-18 produced during pyroptosis participate in inflammation, promoting the progression of DR.
5 Conclusions
Pyroptosis is an emerging type of inflammatory cell death. Recent studies suggest that pyroptosis is involved in the pathogeneses of many diseases, such as obesity, T2DM and complications of diabetes. The retinal NVU is the functional basis of the retina. The proper function of every element of this retinal NVU is critical for normal retinal function. Impairment of the retinal NVU may result in abnormal physiological functions and even retinal disorders. Under diabetic conditions, most retinal NVU cells undergo pyroptotic cell death. Pyroptosis leads to cell death and promotes damaged cells to release various proinflammatory mediators, including IL-1β and IL-18. The accumulation of these proinflammatory factors promotes the formation of an inflammatory environment, further damaging the retinal cells and aggravating retinopathy. Inhibition of pyroptosis in retinal cells may be a treatment strategy for DR. The development of drugs targeting pyroptosis may provide benefits to the vast number of patients with DR. However, published research about the potential molecular mechanism and underlying role of pyroptosis-mediated cell death in retinal NVU cells is currently limited. Additional studies are necessary to investigate the fundamental role of pyroptosis in DR.
Author Contributions
All authors contributed to the literature search writing and design of the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported by the National Natural Science Foundation of China (81970811), Domestic Science and Technology Cooperation Project of Shanghai Municipal Science and Technology Commission (21015800700) and National Key R&D Program of China (2019YFC0840607).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lunder M, Janić M, Japelj M, Juretič A, Janež A, Šabovič M. Empagliflozin on Top of Metformin Treatment Improves Arterial Function in Patients With Type 1 Diabetes Mellitus. Cardiovasc Diabetol (2018) 171:153. doi: 10.1186/s12933-018-0797-6
2. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and Regional Diabetes Prevalence Estimates for 2019 and Projections for 2030 and 2045: Results From the International Diabetes Federation Diabetes Atlas, 9(Th) Edition. Diabetes Res Clin Pract (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
3. Rodríguez ML, Pérez S, Mena-Mollá S, Desco MC, Ortega ÁL. Oxidative Stress and Microvascular Alterations in Diabetic Retinopathy: Future Therapies. Oxid Med Cell Longev (2019) 2019:4940825. doi: 10.1155/2019/4940825
4. Cheung N, Mitchell P, Wong TY. Diabetic Retinopathy. Lancet (Lond Engl) (2010) 3769735:124–36. doi: 10.1016/s0140-67360962124-3
5. Flaxman SR, Bourne RRA, Resnikoff S, Ackland P, Braithwaite T, Cicinelli MV, et al. Global Causes of Blindness and Distance Vision Impairment 1990-2020: A Systematic Review and Meta-Analysis. Lancet Global Health (2017) 512:e1221–34. doi: 10.1016/s2214-109x1730393-5
6. Wang W, Lo ACY. Diabetic Retinopathy: Pathophysiology and Treatments. Int J Mol Sci (2018) 196:1816. doi: 10.3390/ijms19061816
7. Wong TY, Sun J, Kawasaki R, Ruamviboonsuk P, Gupta N, Lansingh VC, et al. Guidelines on Diabetic Eye Care: The International Council of Ophthalmology Recommendations for Screening, Follow-Up, Referral, and Treatment Based on Resource Settings. Ophthalmology (2018) 12510:1608–22. doi: 10.1016/j.ophtha.2018.04.007
8. Stitt AW, Curtis TM, Chen M, Medina RJ, McKay GJ, Jenkins A, et al. The Progress in Understanding and Treatment of Diabetic Retinopathy. Prog Retin Eye Res (2016) 51:156–86. doi: 10.1016/j.preteyeres.2015.08.001
9. Wong TY, Cheung CM, Larsen M, Sharma S, Simó R. Diabetic Retinopathy. Nat Rev Dis Primers (2016) 2:16012. doi: 10.1038/nrdp.2016.12
10. Spaide RF. Measurable Aspects of the Retinal Neurovascular Unit in Diabetes, Glaucoma, and Controls. Am J Ophthalmol (2019) 207:395–409. doi: 10.1016/j.ajo.2019.04.035
11. Hammes HP. Diabetic Retinopathy: Hyperglycaemia, Oxidative Stress and Beyond. Diabetologia (2018) 611:29–38. doi: 10.1007/s00125-017-4435-8
12. Spencer BG, Estevez JJ, Liu E, Craig JE, Finnie JW. Pericytes, Inflammation, and Diabetic Retinopathy. Inflammopharmacology (2020) 283:697–709. doi: 10.1007/s10787-019-00647-9
13. Arroba AI, Campos-Caro A, Aguilar-Diosdado M, Valverde ÁM. IGF-1, Inflammation and Retinal Degeneration: A Close Network. Front Aging Neurosci (2018) 10:203. doi: 10.3389/fnagi.2018.00203
14. Tsuchiya K. Inflammasome-Associated Cell Death: Pyroptosis, Apoptosis, and Physiological Implications. Microbiol Immunol (2020) 644:252–69. doi: 10.1111/1348-0421.12771
15. Liang F, Zhang F, Zhang L, Wei W. The Advances in Pyroptosis Initiated by Inflammasome in Inflammatory and Immune Diseases. Inflammation Res (2020) 692:159–66. doi: 10.1007/s00011-020-01315-3
16. Duh EJ, Sun JK, Stitt AW. Diabetic Retinopathy: Current Understanding, Mechanisms, and Treatment Strategies. JCI Insight (2017) 214:e93751. doi: 10.1172/jci.insight.93751
17. Kang Q, Yang C. Oxidative Stress and Diabetic Retinopathy: Molecular Mechanisms, Pathogenetic Role and Therapeutic Implications. Redox Biol (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
18. Kong DQ, Li L, Liu Y, Zheng GY. Association Between Endoplasmic Reticulum Stress and Risk Factors of Diabetic Retinopathy. Int J Ophthalmol (2018) 1110:1704–10. doi: 10.18240/ijo.2018.10.20
19. Barber AJ, Gardner TW, Abcouwer SF. The Significance of Vascular and Neural Apoptosis to the Pathology of Diabetic Retinopathy. Invest Ophthalmol Vis Sci (2011) 522:1156–63. doi: 10.1167/iovs.10-6293
20. Rübsam A, Parikh S, Fort PE. Role of Inflammation in Diabetic Retinopathy. Int J Mol Sci (2018) 194:942. doi: 10.3390/ijms19040942
21. Huang H. Pericyte-Endothelial Interactions in the Retinal Microvasculature. Int J Mol Sci (2020) 2119:7413. doi: 10.3390/ijms21197413
22. Chan G, Balaratnasingam C, Yu PK, Morgan WH, McAllister IL, Cringle SJ, et al. Quantitative Morphometry of Perifoveal Capillary Networks in the Human Retina. Invest Ophthalmol Visual Sci (2012) 539:5502–14. doi: 10.1167/iovs.12-10265
23. Tawfik A, Samra YA, Elsherbiny NM, Al-Shabrawey M. Implication of Hyperhomocysteinemia in Blood Retinal Barrier (BRB) Dysfunction. Biomolecules (2020) 108:1119. doi: 10.3390/biom10081119
24. Trost A, Lange S, Schroedl F, Bruckner D, Motloch KA, Bogner B, et al. Brain and Retinal Pericytes: Origin, Function and Role. Front Cell Neurosci (2016) 10:20. doi: 10.3389/fncel.2016.00020
25. Ferland-McCollough D, Slater S, Richard J, Reni C, Mangialardi G. Pericytes, an Overlooked Player in Vascular Pathobiology. Pharmacol Ther (2017) 171:30–42. doi: 10.1016/j.pharmthera.2016.11.008
26. Hudson N, Campbell M. Inner Blood-Retinal Barrier Regulation in Retinopathies. Adv Exp Med Biol (2019) 1185:329–33. doi: 10.1007/978-3-030-27378-1_54
27. Yan RJ, Gong HQ, Zhang PM, Liang PJ. Information Coding in Retinal Ganglion Cells. Sheng li xue bao [Acta Physiol Sinica] (2016) 684:414–22.
28. Hoon M, Okawa H, Della Santina L, Wong RO. Functional Architecture of the Retina: Development and Disease. Prog Retinal Eye Res (2014) 42:44–84. doi: 10.1016/j.preteyeres.2014.06.003
29. Moran EP, Wang Z, Chen J, Sapieha P, Smith LE, Ma JX. Neurovascular Cross Talk in Diabetic Retinopathy: Pathophysiological Roles and Therapeutic Implications. Am J Physiol Heart Circulatory Physiol (2016) 3113:H738–49. doi: 10.1152/ajpheart.00005.2016
30. Völgyi B. Molecular Biology of Retinal Ganglion Cells. Cells (2020) 911:2483. doi: 10.3390/cells9112483
31. Masland RH. The Neuronal Organization of the Retina. Neuron (2012) 762:266–80. doi: 10.1016/j.neuron.2012.10.002
32. Lechner J, O'Leary OE, Stitt AW. The Pathology Associated With Diabetic Retinopathy. Vision Res (2017) 139:7–14. doi: 10.1016/j.visres.2017.04.003
33. Altmann C, Schmidt MHH. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int J Mol Sci (2018) 191:110. doi: 10.3390/ijms19010110
34. Vecino E, Rodriguez FD, Ruzafa N, Pereiro X, Sharma SC. Glia-Neuron Interactions in the Mammalian Retina. Prog Retinal Eye Res (2016) 51:1–40. doi: 10.1016/j.preteyeres.2015.06.003
35. Coughlin BA, Feenstra DJ, Mohr S. Müller Cells and Diabetic Retinopathy. Vision Res (2017) 139:93–100. doi: 10.1016/j.visres.2017.03.013
36. Fernández-Sánchez L, Lax P, Campello L, Pinilla I, Cuenca N. Astrocytes and Müller Cell Alterations During Retinal Degeneration in a Transgenic Rat Model of Retinitis Pigmentosa. Front Cell Neurosci (2015) 9:484. doi: 10.3389/fncel.2015.00484
37. Sorrentino FS, Allkabes M, Salsini G, Bonifazzi C, Perri P. The Importance of Glial Cells in the Homeostasis of the Retinal Microenvironment and Their Pivotal Role in the Course of Diabetic Retinopathy. Life Sci (2016) 162:54–9. doi: 10.1016/j.lfs.2016.08.001
38. Rathnasamy G, Foulds WS, Ling EA, Kaur C. Retinal Microglia - A Key Player in Healthy and Diseased Retina. Prog Neurobiol (2019) 173:18–40. doi: 10.1016/j.pneurobio.2018.05.006
39. Ramirez AI, de Hoz R, Salobrar-Garcia E, Salazar JJ, Rojas B, Ajoy D, et al. The Role of Microglia in Retinal Neurodegeneration: Alzheimer's Disease, Parkinson, and Glaucoma. Front Aging Neurosci (2017) 9:214. doi: 10.3389/fnagi.2017.00214
40. Zabel MK, Zhao L, Zhang Y, Gonzalez SR, Ma W, Wang X, et al. Microglial Phagocytosis and Activation Underlying Photoreceptor Degeneration is Regulated by CX3CL1-CX3CR1 Signaling in a Mouse Model of Retinitis Pigmentosa. Glia (2016) 649:1479–91. doi: 10.1002/glia.23016
41. Anderson SR, Zhang J, Steele MR, Romero CO, Kautzman AG, Schafer DP, et al. Complement Targets Newborn Retinal Ganglion Cells for Phagocytic Elimination by Microglia. J Neurosci (2019) 3911:2025–40. doi: 10.1523/jneurosci.1854-18.2018
42. Wang X, Zhao L, Zhang J, Fariss RN, Ma W, Kretschmer F, et al. Requirement for Microglia for the Maintenance of Synaptic Function and Integrity in the Mature Retina. J Neurosci (2016) 369:2827–42. doi: 10.1523/jneurosci.3575-15.2016
43. O'Koren EG, Yu C, Klingeborn M, Wong AYW, Prigge CL, Mathew R, et al. Microglial Function Is Distinct in Different Anatomical Locations During Retinal Homeostasis and Degeneration. Immunity (2019) 503:723–37.e7. doi: 10.1016/j.immuni.2019.02.007
44. Yang S, Zhang J, Chen L. The Cells Involved in the Pathological Process of Diabetic Retinopathy. Biomed Pharmacother = Biomed Pharmacother (2020) 132:110818. doi: 10.1016/j.biopha.2020.110818
45. Wareham LK, Calkins DJ. The Neurovascular Unit in Glaucomatous Neurodegeneration. Front Cell Dev Biol (2020) 8:452. doi: 10.3389/fcell.2020.00452
46. Jo DH, Yun JH, Cho CS, Kim JH, Kim JH, Cho CH. Interaction Between Microglia and Retinal Pigment Epithelial Cells Determines the Integrity of Outer Blood-Retinal Barrier in Diabetic Retinopathy. Glia (2019) 672:321–31. doi: 10.1002/glia.23542
47. Naylor A, Hopkins A, Hudson N, Campbell M. Tight Junctions of the Outer Blood Retina Barrier. Int J Mol Sci (2019) 211:211. doi: 10.3390/ijms21010211
48. Díaz-Coránguez M, Ramos C, Antonetti DA. The Inner Blood-Retinal Barrier: Cellular Basis and Development. Vision Res (2017) 139:123–37. doi: 10.1016/j.visres.2017.05.009
49. Cunha-Vaz J, Bernardes R, Lobo C. Blood-Retinal Barrier. Eur J Ophthalmol (2011) 21 Suppl 6:S3–9. doi: 10.5301/ejo.2010.6049
50. Gardner TW, Davila JR. The Neurovascular Unit and the Pathophysiologic Basis of Diabetic Retinopathy. Graefe's Arch Clin Exp Ophthalmol = Albrecht von Graefes Archiv fur Klin und Exp Ophthalmol (2017) 2551:1–6. doi: 10.1007/s00417-016-3548-y
51. Sinclair SH, Schwartz SS. Diabetic Retinopathy-An Underdiagnosed and Undertreated Inflammatory, Neuro-Vascular Complication of Diabetes. Front Endocrinol (2019) 10:843. doi: 10.3389/fendo.2019.00843
52. Ma JH, Wang JJ, Zhang SX. The Unfolded Protein Response and Diabetic Retinopathy. J Diabetes Res (2014) 2014:160140. doi: 10.1155/2014/160140
53. Nian S, Lo ACY, Mi Y, Ren K, Yang D. Neurovascular Unit in Diabetic Retinopathy: Pathophysiological Roles and Potential Therapeutical Targets. Eye Vision (Lond Engl) (2021) 81:15. doi: 10.1186/s40662-021-00239-1
54. Taurone S, Ralli M, Nebbioso M, Greco A, Artico M, Attanasio G, et al. The Role of Inflammation in Diabetic Retinopathy: A Review. Eur Rev Med Pharmacol Sci (2020) 2420:10319–29. doi: 10.26355/eurrev_202010_23379
55. Dehdashtian E, Mehrzadi S, Yousefi B, Hosseinzadeh A, Reiter RJ, Safa M, et al. Diabetic Retinopathy Pathogenesis and the Ameliorating Effects of Melatonin; Involvement of Autophagy, Inflammation and Oxidative Stress. Life Sci (2018) 193:20–33. doi: 10.1016/j.lfs.2017.12.001
56. Kowluru RA, Abbas SN. Diabetes-Induced Mitochondrial Dysfunction in the Retina. Invest Ophthalmol Visual Sci (2003) 4412:5327–34. doi: 10.1167/iovs.03-0353
57. Cecilia OM, José Alberto CG, José NP, Ernesto Germán CM, Ana Karen LC, Luis Miguel RP, et al. Oxidative Stress as the Main Target in Diabetic Retinopathy Pathophysiology. J Diabetes Res (2019) 2019:8562408. doi: 10.1155/2019/8562408
58. Zhong Y, Wang JJ, Zhang SX. Intermittent But Not Constant High Glucose Induces ER Stress and Inflammation in Human Retinal Pericytes. Adv Exp Med Biol (2012) 723:285–92. doi: 10.1007/978-1-4614-0631-0_37
59. Al-Kharashi AS. Role of Oxidative Stress, Inflammation, Hypoxia and Angiogenesis in the Development of Diabetic Retinopathy. Saudi J Ophthalmol (2018) 324:318–23. doi: 10.1016/j.sjopt.2018.05.002
60. Simó R, Stitt AW, Gardner TW. Neurodegeneration in Diabetic Retinopathy: Does it Really Matter? Diabetologia (2018) 619:1902–12. doi: 10.1007/s00125-018-4692-1
61. Oshitari T, Yoshida-Hata N, Yamamoto S. Effect of Neurotrophin-4 on Endoplasmic Reticulum Stress-Related Neuronal Apoptosis in Diabetic and High Glucose Exposed Rat Retinas. Neurosci Lett (2011) 5012:102–6. doi: 10.1016/j.neulet.2011.06.057
62. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, et al. ER-Stress-Induced Transcriptional Regulation Increases Protein Synthesis Leading to Cell Death. Nat Cell Biol (2013) 155:481–90. doi: 10.1038/ncb2738
63. Potilinski MC, Lorenc V, Perisset S, Gallo JE. Mechanisms Behind Retinal Ganglion Cell Loss in Diabetes and Therapeutic Approach. Int J Mol Sci (2020) 217:2351. doi: 10.3390/ijms21072351
64. Kim SJ, Yoo WS, Choi M, Chung I, Yoo JM, Choi WS. Increased O-GlcNAcylation of NF-κb Enhances Retinal Ganglion Cell Death in Streptozotocin-Induced Diabetic Retinopathy. Curr Eye Res (2016) 412:249–57. doi: 10.3109/02713683.2015.1006372
65. Mohamed IN, Ishrat T, Fagan SC, El-Remessy AB. Role of Inflammasome Activation in the Pathophysiology of Vascular Diseases of the Neurovascular Unit. Antioxid Redox Signal (2015) 2213:1188–206. doi: 10.1089/ars.2014.6126
66. Mesquida M, Drawnel F, Fauser S. The Role of Inflammation in Diabetic Eye Disease. Semin Immunopathol (2019) 414:427–45. doi: 10.1007/s00281-019-00750-7
67. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature (2015) 5267575:660–5. doi: 10.1038/nature15514
68. Lu F, Lan Z, Xin Z, He C, Guo Z, Xia X, et al. Emerging Insights Into Molecular Mechanisms Underlying Pyroptosis and Functions of Inflammasomes in Diseases. J Cell Physiol (2020) 2354:3207–21. doi: 10.1002/jcp.29268
69. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci (2017) 424:245–54. doi: 10.1016/j.tibs.2016.10.004
70. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature (2016) 5357610:153–8. doi: 10.1038/nature18629
71. Lamkanfi M, Dixit VM. Mechanisms and Functions of Inflammasomes. Cell (2014) 1575:1013–22. doi: 10.1016/j.cell.2014.04.007
72. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science (New York NY) (2013) 3416151:1246–9. doi: 10.1126/science.1240248
73. Yang J, Zhao Y, Shao F. Non-Canonical Activation of Inflammatory Caspases by Cytosolic LPS in Innate Immunity. Curr Opin Immunol (2015) 32:78–83. doi: 10.1016/j.coi.2015.01.007
74. Kovacs SB, Miao EA. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol (2017) 279:673–84. doi: 10.1016/j.tcb.2017.05.005
75. Frank D, Vince JE. Pyroptosis Versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death Differ (2019) 261:99–114. doi: 10.1038/s41418-018-0212-6
76. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature (2015) 5267575:666–71. doi: 10.1038/nature15541
77. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy Drugs Induce Pyroptosis Through Caspase-3 Cleavage of a Gasdermin. Nature (2017) 5477661:99–103. doi: 10.1038/nature22393
78. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by Caspase-3 During Apoptosis Mediates Progression to Secondary Necrotic/Pyroptotic Cell Death. Nat Commun (2017) 8:14128. doi: 10.1038/ncomms14128
79. Engin A. Fat Cell and Fatty Acid Turnover in Obesity. Adv Exp Med Biol (2017) 960:135–60. doi: 10.1007/978-3-319-48382-5_6
80. Ding S, Xu S, Ma Y, Liu G, Jang H, Fang J. Modulatory Mechanisms of the NLRP3 Inflammasomes in Diabetes. Biomolecules (2019) 912:850. doi: 10.3390/biom9120850
81. Yu ZW, Zhang J, Li X, Wang Y, Fu YH, Gao XY. A New Research Hot Spot: The Role of NLRP3 Inflammasome Activation, a Key Step in Pyroptosis, in Diabetes and Diabetic Complications. Life Sci (2020) 240:117138. doi: 10.1016/j.lfs.2019.117138
82. Saklayen MG. The Global Epidemic of the Metabolic Syndrome. Curr Hypertens Rep (2018) 202:12. doi: 10.1007/s11906-018-0812-z
83. Jiang D, Chen S, Sun R, Zhang X, Wang D. The NLRP3 Inflammasome: Role in Metabolic Disorders and Regulation by Metabolic Pathways. Cancer Lett (2018) 419:8–19. doi: 10.1016/j.canlet.2018.01.034
84. Pillon NJ, Chan KL, Zhang S, Mejdani M, Jacobson MR, Ducos A, et al. Saturated Fatty Acids Activate Caspase-4/5 in Human Monocytes, Triggering IL-1β and IL-18 Release. Am J Physiol Endocrinol Metab (2016) 3115:E825–35. doi: 10.1152/ajpendo.00296.2016
85. Esser N, L'Homme L, De Roover A, Kohnen L, Scheen AJ, Moutschen M, et al. Obesity Phenotype is Related to NLRP3 Inflammasome Activity and Immunological Profile of Visceral Adipose Tissue. Diabetologia (2013) 5611:2487–97. doi: 10.1007/s00125-013-3023-9
86. Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat Med (2011) 172:179–88. doi: 10.1038/nm.2279
87. Francisco V, Pino J, Gonzalez-Gay MA, Mera A, Lago F, Gómez R, et al. Adipokines and Inflammation: Is it a Question of Weight? Br J Pharmacol (2018) 17510:1569–79. doi: 10.1111/bph.14181
88. Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte Dysfunctions Linking Obesity to Insulin Resistance and Type 2 Diabetes. Nat Rev Mol Cell Biol (2008) 95:367–77. doi: 10.1038/nrm2391
89. Rheinheimer J, de Souza BM, Cardoso NS, Bauer AC, Crispim D. Current Role of the NLRP3 Inflammasome on Obesity and Insulin Resistance: A Systematic Review. Metab: Clin Exp (2017) 74:1–9. doi: 10.1016/j.metabol.2017.06.002
90. Zhang X, Xu A, Lv J, Zhang Q, Ran Y, Wei C, et al. Development of Small Molecule Inhibitors Targeting NLRP3 Inflammasome Pathway for Inflammatory Diseases. Eur J Med Chem (2020) 185:111822. doi: 10.1016/j.ejmech.2019.111822
91. Tsalamandris S, Antonopoulos AS, Oikonomou E, Papamikroulis GA, Vogiatzi G, Papaioannou S, et al. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. Eur Cardiol (2019) 141:50–9. doi: 10.15420/ecr.2018.33.1
92. Pirzada RH, Javaid N, Choi S. The Roles of the NLRP3 Inflammasome in Neurodegenerative and Metabolic Diseases and in Relevant Advanced Therapeutic Interventions. Genes (2020) 112:131. doi: 10.3390/genes11020131
93. Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ Effector T Cells Contribute to Macrophage Recruitment and Adipose Tissue Inflammation in Obesity. Nat Med (2009) 158:914–20. doi: 10.1038/nm.1964
94. Yang F, Qin Y, Lv J, Wang Y, Che H, Chen X, et al. Silencing Long non-Coding RNA Kcnq1ot1 Alleviates Pyroptosis and Fibrosis in Diabetic Cardiomyopathy. Cell Death Dis (2018) 910:1000. doi: 10.1038/s41419-018-1029-4
95. An X, Zhang Y, Cao Y, Chen J, Qin H, Yang L. Punicalagin Protects Diabetic Nephropathy by Inhibiting Pyroptosis Based on TXNIP/NLRP3 Pathway. Nutrients (2020) 125:1516. doi: 10.3390/nu12051516
96. Al Mamun A, Mimi AA, Zaeem M, Wu Y, Monalisa I, Akter A, et al. Role of Pyroptosis in Diabetic Retinopathy and Its Therapeutic Implications. Eur J Pharmacol (2021) 904:174166. doi: 10.1016/j.ejphar.2021.174166
97. Loukovaara S, Piippo N, Kinnunen K, Hytti M, Kaarniranta K, Kauppinen A. NLRP3 Inflammasome Activation Is Associated With Proliferative Diabetic Retinopathy. Acta Ophthalmol (2017) 958:803–8. doi: 10.1111/aos.13427
98. Hao J, Zhang H, Yu J, Chen X, Yang L. Methylene Blue Attenuates Diabetic Retinopathy by Inhibiting NLRP3 Inflammasome Activation in STZ-Induced Diabetic Rats. Ocul Immunol Inflamm (2019) 275:836–43. doi: 10.1080/09273948.2018.1450516
99. Yin Y, Chen F, Wang W, Wang H, Zhang X. Resolvin D1 Inhibits Inflammatory Response in STZ-Induced Diabetic Retinopathy Rats: Possible Involvement of NLRP3 Inflammasome and NF-κb Signaling Pathway. Mol Vis (2017) 23:242–50.
100. Zha X, Xi X, Fan X, Ma M, Zhang Y, Yang Y. Overexpression of METTL3 Attenuates High-Glucose Induced RPE Cell Pyroptosis by Regulating miR-25-3p/PTEN/Akt Signaling Cascade Through DGCR8. Aging (Albany NY) (2020) 129:8137–50. doi: 10.18632/aging.103130
101. Gu C, Draga D, Zhou C, Su T, Zou C, Gu Q, et al. miR-590-3p Inhibits Pyroptosis in Diabetic Retinopathy by Targeting NLRP1 and Inactivating the NOX4 Signaling Pathway. Invest Ophthalmol Vis Sci (2019) 6013:4215–23. doi: 10.1167/iovs.19-27825
102. Gan J, Huang M, Lan G, Liu L, Xu F. High Glucose Induces the Loss of Retinal Pericytes Partly via NLRP3-Caspase-1-GSDMD-Mediated Pyroptosis. BioMed Res Int (2020) 2020:4510628. doi: 10.1155/2020/4510628
103. Portillo JC, Lopez Corcino Y, Miao Y, Tang J, Sheibani N, Kern TS, et al. CD40 in Retinal Müller Cells Induces P2X7-Dependent Cytokine Expression in Macrophages/Microglia in Diabetic Mice and Development of Early Experimental Diabetic Retinopathy. Diabetes (2017) 662:483–93. doi: 10.2337/db16-0051
104. Platania CBM, Giurdanella G, Di Paola L, Leggio GM, Drago F, Salomone S, et al. P2X7 Receptor Antagonism: Implications in Diabetic Retinopathy. Biochem Pharmacol (2017) 138:130–9. doi: 10.1016/j.bcp.2017.05.001
105. Kowluru RA, Zhong Q, Kanwar M. Metabolic Memory and Diabetic Retinopathy: Role of Inflammatory Mediators in Retinal Pericytes. Exp Eye Res (2010) 905:617–23. doi: 10.1016/j.exer.2010.02.006
106. Karmakar M, Katsnelson MA, Dubyak GR, Pearlman E. Neutrophil P2X7 Receptors Mediate NLRP3 Inflammasome-Dependent IL-1β Secretion in Response to ATP. Nat Commun (2016) 7:10555. doi: 10.1038/ncomms10555
107. Yang K, Liu J, Zhang X, Ren Z, Gao L, Wang Y, et al. H3 Relaxin Alleviates Migration, Apoptosis and Pyroptosis Through P2X7R-Mediated Nucleotide Binding Oligomerization Domain-Like Receptor Protein 3 Inflammasome Activation in Retinopathy Induced by Hyperglycemia. Front Pharmacol (2020) 11:603689. doi: 10.3389/fphar.2020.603689
108. Qiu F, Meng T, Chen Q, Zhou K, Shao Y, Matlock G, et al. Fenofibrate-Loaded Biodegradable Nanoparticles for the Treatment of Experimental Diabetic Retinopathy and Neovascular Age-Related Macular Degeneration. Mol Pharm (2019) 165:1958–70. doi: 10.1021/acs.molpharmaceut.8b01319
109. Li S, Yang H, Chen X. Protective Effects of Sulforaphane on Diabetic Retinopathy: Activation of the Nrf2 Pathway and Inhibition of NLRP3 Inflammasome Formation. Exp Anim (2019) 682:221–31. doi: 10.1538/expanim.18-0146
110. Lu L, Lu Q, Chen W, Li J, Li C, Zheng Z. Vitamin D3 Protects Against Diabetic Retinopathy by Inhibiting High-Glucose-Induced Activation of the ROS/TXNIP/NLRP3 Inflammasome Pathway. J Diabetes Res (2018) 2018:8193523. doi: 10.1155/2018/8193523
111. Lazzara F, Amato R, Platania CBM, Conti F, Chou TH, Porciatti V, et al. 1α,25-Dihydroxyvitamin D3 Protects Retinal Ganglion Cells in Glaucomatous Mice. J Neuroinflamm (2021) 181:206. doi: 10.1186/s12974-021-02263-3
112. Dillmann WH. Diabetic Cardiomyopathy. Circ Res (2019) 1248:1160–2. doi: 10.1161/circresaha.118.314665
113. Zeng C, Wang R, Tan H. Role of Pyroptosis in Cardiovascular Diseases and Its Therapeutic Implications. Int J Biol Sci (2019) 157:1345–57. doi: 10.7150/ijbs.33568
114. Van Linthout S, Spillmann F, Riad A, Trimpert C, Lievens J, Meloni M, et al. Human Apolipoprotein A-I Gene Transfer Reduces the Development of Experimental Diabetic Cardiomyopathy. Circulation (2008) 11712:1563–73. doi: 10.1161/circulationaha.107.710830
115. Yu W, Wu J, Cai F, Xiang J, Zha W, Fan D, et al. Curcumin Alleviates Diabetic Cardiomyopathy in Experimental Diabetic Rats. PLoS One (2012) 712:e52013. doi: 10.1371/journal.pone.0052013
116. Ye Y, Bajaj M, Yang HC, Perez-Polo JR, Birnbaum Y. SGLT-2 Inhibition With Dapagliflozin Reduces the Activation of the Nlrp3/ASC Inflammasome and Attenuates the Development of Diabetic Cardiomyopathy in Mice With Type 2 Diabetes. Further Augmentation of the Effects With Saxagliptin, a DPP4 Inhibitor. Cardiovasc Drugs Ther (2017) 312:119–32. doi: 10.1007/s10557-017-6725-2
117. Wei H, Bu R, Yang Q, Jia J, Li T, Wang Q, et al. Exendin-4 Protects Against Hyperglycemia-Induced Cardiomyocyte Pyroptosis via the AMPK-TXNIP Pathway. J Diabetes Res (2019) 2019:8905917. doi: 10.1155/2019/8905917
118. Wang X, Pan J, Liu H, Zhang M, Liu D, Lu L, et al. AIM2 Gene Silencing Attenuates Diabetic Cardiomyopathy in Type 2 Diabetic Rat Model. Life Sci (2019) 221:249–58. doi: 10.1016/j.lfs.2019.02.035
119. Xiong Y, Zhou L. The Signaling of Cellular Senescence in Diabetic Nephropathy. Oxid Med Cell Longev (2019) 2019:7495629. doi: 10.1155/2019/7495629
120. Zhang KJ, Wu Q, Jiang SM, Ding L, Liu CX, Xu M, et al. Pyroptosis: A New Frontier in Kidney Diseases. Oxid Med Cell Longev (2021) 2021:6686617. doi: 10.1155/2021/6686617
121. Zhan JF, Huang HW, Huang C, Hu LL, Xu WW. Long Non-Coding RNA NEAT1 Regulates Pyroptosis in Diabetic Nephropathy via Mediating the miR-34c/NLRP3 Axis. Kidney Blood Press Res (2020) 454:589–602. doi: 10.1159/000508372
122. Cheng Q, Pan J, Zhou ZL, Yin F, Xie HY, Chen PP, et al. Caspase-11/4 and Gasdermin D-Mediated Pyroptosis Contributes to Podocyte Injury in Mouse Diabetic Nephropathy. Acta Pharmacol Sin (2021) 426:954–63. doi: 10.1038/s41401-020-00525-z
123. Li X, Zeng L, Cao C, Lu C, Lian W, Han J, et al. Long Noncoding RNA MALAT1 Regulates Renal Tubular Epithelial Pyroptosis by Modulated miR-23c Targeting of ELAVL1 in Diabetic Nephropathy. Exp Cell Res (2017) 3502:327–35. doi: 10.1016/j.yexcr.2016.12.006
124. Xie C, Wu W, Tang A, Luo N, Tan Y. lncRNA GAS5/miR-452-5p Reduces Oxidative Stress and Pyroptosis of High-Glucose-Stimulated Renal Tubular Cells. Diabetes Metab Syndr Obes (2019) 12:2609–17. doi: 10.2147/dmso.s228654
125. Beltramo E, Porta M. Pericyte Loss in Diabetic Retinopathy: Mechanisms and Consequences. Curr Med Chem (2013) 2026:3218–25. doi: 10.2174/09298673113209990022
126. Betts-Obregon BS, Mondragon AA, Mendiola AS, LeBaron RG, Asmis R, Zou T, et al. Tgfβ Induces BIGH3 Expression and Human Retinal Pericyte Apoptosis: A Novel Pathway of Diabetic Retinopathy. Eye (2016) 3012:1639–47. doi: 10.1038/eye.2016.179
127. Feenstra DJ, Yego EC, Mohr S. Modes of Retinal Cell Death in Diabetic Retinopathy. J Clin Exp Ophthalmol (2013) 45:298. doi: 10.4172/2155-9570.1000298
128. Yu X, Ma X, Lin W, Xu Q, Zhou H, Kuang H. Long Noncoding RNA MIAT Regulates Primary Human Retinal Pericyte Pyroptosis by Modulating miR-342-3p Targeting of CASP1 in Diabetic Retinopathy. Exp Eye Res (2021) 202:108300. doi: 10.1016/j.exer.2020.108300
129. Chen W, Zhao M, Zhao S, Lu Q, Ni L, Zou C, et al. Activation of the TXNIP/NLRP3 Inflammasome Pathway Contributes to Inflammation in Diabetic Retinopathy: A Novel Inhibitory Effect of Minocycline. Inflamm Res (2017) 662:157–66. doi: 10.1007/s00011-016-1002-6
130. Platania CBM, Maisto R, Trotta MC, D'Amico M, Rossi S, Gesualdo C, et al. Retinal and Circulating miRNA Expression Patterns in Diabetic Retinopathy: An in Silico and In Vivo Approach. Br J Pharmacol (2019) 17613:2179–94. doi: 10.1111/bph.14665
131. Wang Y, Tao J, Yao Y. Prostaglandin E2 Activates NLRP3 Inflammasome in Endothelial Cells to Promote Diabetic Retinopathy. Hormone Metab Res = Hormon- und Stoffwechselforschung = Hormones Metab (2018) 509:704–10. doi: 10.1055/a-0664-0699
132. Zhang Y, Lv X, Hu Z, Ye X, Zheng X, Ding Y, et al. Protection of Mcc950 Against High-Glucose-Induced Human Retinal Endothelial Cell Dysfunction. Cell Death Dis (2017) 87:e2941. doi: 10.1038/cddis.2017.308
133. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD Membrane Pore Formation Constitutes the Mechanism of Pyroptotic Cell Death. EMBO J (2016) 3516:1766–78. doi: 10.15252/embj.201694696
134. Arboleda-Velasquez JF, Valdez CN, Marko CK, D'Amore PA. From Pathobiology to the Targeting of Pericytes for the Treatment of Diabetic Retinopathy. Curr Diabetes Rep (2015) 152:573. doi: 10.1007/s11892-014-0573-2
135. Omori K, Nagata N, Kurata K, Fukushima Y, Sekihachi E, Fujii N, et al. Inhibition of Stromal Cell-Derived Factor-1α/CXCR4 Signaling Restores the Blood-Retina Barrier in Pericyte-Deficient Mouse Retinas. JCI Insight (2018) 323:e120706. doi: 10.1172/jci.insight.120706
136. Rudraraju M, Narayanan SP, Somanath PR. Regulation of Blood-Retinal Barrier Cell-Junctions in Diabetic Retinopathy. Pharmacol Res (2020) 161:105115. doi: 10.1016/j.phrs.2020.105115
137. Capitão M, Soares R. Angiogenesis and Inflammation Crosstalk in Diabetic Retinopathy. J Cell Biochem (2016) 11711:2443–53. doi: 10.1002/jcb.25575
138. Jonsson KB, Frydkjaer-Olsen U, Grauslund J. Vascular Changes and Neurodegeneration in the Early Stages of Diabetic Retinopathy: Which Comes First? Ophthalmic Res (2016) 561:1–9. doi: 10.1159/000444498
139. Srinivasan S, Dehghani C, Pritchard N, Edwards K, Russell AW, Malik RA, et al. Corneal and Retinal Neuronal Degeneration in Early Stages of Diabetic Retinopathy. Invest Ophthalmol Visual Sci (2017) 5814:6365–73. doi: 10.1167/iovs.17-22736
140. Thomas CN, Berry M, Logan A, Blanch RJ, Ahmed Z. Caspases in Retinal Ganglion Cell Death and Axon Regeneration. Cell Death Discov (2017) 3:17032. doi: 10.1038/cddiscovery.2017.32
141. Semeraro F, Cancarini A, dell'Omo R, Rezzola S, Romano MR, Costagliola C. Diabetic Retinopathy: Vascular and Inflammatory Disease. J Diabetes Res (2015) 2015:582060. doi: 10.1155/2015/582060
142. Chen H, Deng Y, Gan X, Li Y, Huang W, Lu L, et al. NLRP12 Collaborates With NLRP3 and NLRC4 to Promote Pyroptosis Inducing Ganglion Cell Death of Acute Glaucoma. Mol Neurodegeneration (2020) 151:26. doi: 10.1186/s13024-020-00372-w
143. Li X, Liu Y, Sun M, Gao M, Li T, Liang J, et al. Photoreceptors Degenerate Through Pyroptosis After Experimental Retinal Detachment. Invest Ophthalmol Visual Sci (2020) 618:31. doi: 10.1167/iovs.61.8.31
144. Trueblood KE, Mohr S, Dubyak GR. Purinergic Regulation of High-Glucose-Induced Caspase-1 Activation in the Rat Retinal Müller Cell Line rMC-1. Am J Physiol Cell Physiol (2011) 3015:C1213–23. doi: 10.1152/ajpcell.00265.2011
145. Yego EC, Vincent JA, Sarthy V, Busik JV, Mohr S. Differential Regulation of High Glucose-Induced Glyceraldehyde-3-Phosphate Dehydrogenase Nuclear Accumulation in Müller Cells by IL-1beta and IL-6. Invest Ophthalmol Visual Sci (2009) 504:1920–8. doi: 10.1167/iovs.08-2082
146. MacDonald RB, Randlett O, Oswald J, Yoshimatsu T, Franze K, Harris WA. Müller Glia Provide Essential Tensile Strength to the Developing Retina. J Cell Biol (2015) 2107:1075–83. doi: 10.1083/jcb.201503115
147. Shen W, Fruttiger M, Zhu L, Chung SH, Barnett NL, Kirk JK, et al. Conditional Müllercell Ablation Causes Independent Neuronal and Vascular Pathologies in a Novel Transgenic Model. J Neurosci (2012) 3245:15715–27. doi: 10.1523/jneurosci.2841-12.2012
148. Muto T, Tien T, Kim D, Sarthy VP, Roy S. High Glucose Alters Cx43 Expression and Gap Junction Intercellular Communication in Retinal Müller Cells: Promotes Müller Cell and Pericyte Apoptosis. Invest Ophthalmol Visual Sci (2014) 557:4327–37. doi: 10.1167/iovs.14-14606
149. Lei X, Zhang J, Shen J, Hu LM, Wu Y, Mou L, et al. EPO Attenuates Inflammatory Cytokines by Muller Cells in Diabetic Retinopathy. Front Biosci (Elite Ed) (2011) 3:201–11. doi: 10.2741/e234
150. Busik JV, Mohr S, Grant MB. Hyperglycemia-Induced Reactive Oxygen Species Toxicity to Endothelial Cells Is Dependent on Paracrine Mediators. Diabetes (2008) 577:1952–65. doi: 10.2337/db07-1520
151. Wu JH, Li YN, Chen AQ, Hong CD, Zhang CL, Wang HL, et al. Inhibition of Sema4D/PlexinB1 Signaling Alleviates Vascular Dysfunction in Diabetic Retinopathy. EMBO Mol Med (2020) 122:e10154. doi: 10.15252/emmm.201810154
152. Li X, Yu Z, Zong W, Chen P, Li J, Wang M, et al. Deficiency of the Microglial Hv1 Proton Channel Attenuates Neuronal Pyroptosis and Inhibits Inflammatory Reaction After Spinal Cord Injury. J Neuroinflamm (2020) 171:263. doi: 10.1186/s12974-020-01942-x
153. Hu J, Zeng C, Wei J, Duan F, Liu S, Zhao Y, et al. The Combination of Panax Ginseng and Angelica Sinensis Alleviates Ischemia Brain Injury by Suppressing NLRP3 Inflammasome Activation and Microglial Pyroptosis. Phytomedicine (2020) 76:153251. doi: 10.1016/j.phymed.2020.153251
154. Wan P, Su W, Zhang Y, Li Z, Deng C, Li J, et al. LncRNA H19 Initiates Microglial Pyroptosis and Neuronal Death in Retinal Ischemia/Reperfusion Injury. Cell Death Differ (2020) 271:176–91. doi: 10.1038/s41418-019-0351-4
155. Hartsock MJ, Cho H, Wu L, Chen WJ, Gong J, Duh EJ. A Mouse Model of Retinal Ischemia-Reperfusion Injury Through Elevation of Intraocular Pressure. J Vis Exp JoVE (2016) 113:54065. doi: 10.3791/54065
156. Dong N, Wang Y. MiR-30a Regulates S100A12-Induced Retinal Microglial Activation and Inflammation by Targeting Nlrp3. Curr Eye Res (2019) 4411:1236–43. doi: 10.1080/02713683.2019.1632350
157. Huang L, You J, Yao Y, Xie M. High Glucose Induces Pyroptosis of Retinal Microglia Through NLPR3 Inflammasome Signaling. Arq Bras Oftalmol (2021) 841:67–73. doi: 10.5935/0004-2749.20210010
158. Baptista FI, Aveleira CA, Castilho ÁF, Ambrósio AF. Elevated Glucose and Interleukin-1β Differentially Affect Retinal Microglial Cell Proliferation. Mediators Inflamm (2017) 2017:4316316. doi: 10.1155/2017/4316316
159. Mendiola AS, Cardona AE. The IL-1β Phenomena in Neuroinflammatory Diseases. J Neural Transm (Vienna Austria 1996) (2018) 1255:781–95. doi: 10.1007/s00702-017-1732-9
160. Scholz R, Caramoy A, Bhuckory MB, Rashid K, Chen M, Xu H, et al. Targeting Translocator Protein (18 kDa) (TSPO) Dampens Pro-Inflammatory Microglia Reactivity in the Retina and Protects From Degeneration. J Neuroinflamm (2015) 12:201. doi: 10.1186/s12974-015-0422-5
161. Chatziralli I, Sergentanis TN, Crosby-Nwaobi R, Winkley K, Eleftheriadis H, Ismail K, et al. Model for Risk-Based Screening of Diabetic Retinopathy in People With Newly-Diagnosed Type 2 Diabetes Mellitus. Invest Ophthalmol Visual Sci (2017) 586:BIO99–BIO105. doi: 10.1167/iovs.17-21713
162. Yun JH. Interleukin-1β Induces Pericyte Apoptosis via the NF-κb Pathway in Diabetic Retinopathy. Biochem Biophys Res Commun (2021) 546:46–53. doi: 10.1016/j.bbrc.2021.01.108
163. Rivera JC, Sitaras N, Noueihed B, Hamel D, Madaan A, Zhou T, et al. Microglia and Interleukin-1β in Ischemic Retinopathy Elicit Microvascular Degeneration Through Neuronal Semaphorin-3A. Arterioscler Thromb Vasc Biol (2013) 338:1881–91. doi: 10.1161/atvbaha.113.301331
164. Song Z, Sun M, Zhou F, Huang F, Qu J, Chen D. Increased Intravitreous Interleukin-18 Correlated to Vascular Endothelial Growth Factor in Patients With Active Proliferative Diabetic Retinopathy. Graefe's Arch Clin Exp Ophthalmol = Albrecht von Graefes Archiv fur Klin und Exp Ophthalmol (2014) 2528:1229–34. doi: 10.1007/s00417-014-2586-6
Keywords: diabetic retinopathy, retinal neurovascular unit, pyroptosis, inflammation, IL-1β and IL-18
Citation: Meng C, Gu C, He S, Su T, Lhamo T, Draga D and Qiu Q (2021) Pyroptosis in the Retinal Neurovascular Unit: New Insights Into Diabetic Retinopathy. Front. Immunol. 12:763092. doi: 10.3389/fimmu.2021.763092
Received: 23 August 2021; Accepted: 28 September 2021;
Published: 19 October 2021.
Edited by:
Kai Fang, University of California, Los Angeles, United StatesReviewed by:
Claudio Bucolo, University of Catania, ItalyDaiane Maria Cerqueira, Federal University of Minas Gerais, Brazil
Copyright © 2021 Meng, Gu, He, Su, Lhamo, Draga and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qinghua Qiu, cWluZ2h1YXFpdUAxNjMuY29t
†These authors have contributed equally to this work