 Paolo Casali
Paolo Casali Shili Li
Shili Li Grecia Morales
Grecia Morales Daniel P. Chupp
Daniel P. Chupp Hong Zan
Hong Zan- Department of Microbiology, Immunology & Molecular Genetics, University of Texas Long School of Medicine, UT Health Science Center, San Antonio, TX, United States
IgA is the predominant antibody isotype at intestinal mucosae, where it plays a critical role in homeostasis and provides a first line of immune protection. Dysregulation of IgA production, however, can contribute to immunopathology, particularly in kidneys in which IgA deposition can cause nephropathy. Class-switch DNA recombination (CSR) to IgA is directed by TGF-β signaling, which activates Smad2 and Smad3. Activated Smad2/Smad3 dimers are recruited together with Smad4 to the IgH α locus Iα promoter to activate germline Iα-Cα transcription, the first step in the unfolding of CSR to IgA. Epigenetic factors, such as non-coding RNAs, particularly microRNAs, have been shown to regulate T cells, dendritic cells and other immune elements, as well as modulate the antibody response, including CSR, in a B cell-intrinsic fashion. Here we showed that the most abundant miRNA in resting B cells, miR-146a targets Smad2, Smad3 and Smad4 mRNA 3’UTRs and keeps CSR to IgA in check in resting B cells. Indeed, enforced miR-146a expression in B cells aborted induction of IgA CSR by decreasing Smad levels. By contrast, upon induction of CSR to IgA, as directed by TGF-β, B cells downregulated miR-146a, thereby reversing the silencing of Smad2, Smad3 and Smad4, which, once expressed, led to recruitment of Smad2, Smad3 and Smad4 to the Iα promoter for activation of germline Iα-Cα transcription. Deletion of miR-146a in miR-146a–/– mice significantly increased circulating levels of steady state total IgA, but not IgM, IgG or IgE, and heightened the specific IgA antibody response to OVA. In miR-146a–/– mice, the elevated systemic IgA levels were associated with increased IgA+ B cells in intestinal mucosae, increased amounts of fecal free and bacteria-bound IgA as well as kidney IgA deposition, a hallmark of IgA nephropathy. Increased germline Iα-Cα transcription and CSR to IgA in miR-146a –/– B cells in vitro proved that miR-146a-induced Smad2, Smad3 and Smad4 repression is B cell intrinsic. The B cell-intrinsic role of miR-146a in the modulation of CSR to IgA was formally confirmed in vivo by construction and OVA immunization of mixed bone marrow μMT/miR-146a–/– chimeric mice. Thus, by inhibiting Smad2, Smad3 and Smad4 expression, miR-146a plays an important and B cell intrinsic role in modulation of CSR to IgA and the IgA antibody response.
Introduction
IgA is the predominant antibody isotype at mucosal surfaces and the most abundant immunoglobulin (Ig) isotype in the body (1–5). It plays a critical role in mucosal homeostasis in the gastrointestinal, respiratory and urogenital tracts. IgA also provides a first line of immune protection against harmful commensals as well as airborne, ingested and sexually transmitted pathogens that come into contact with such mucosal surfaces (6). In the gut, IgA dimers are selectively transported across epithelia into the intestinal lumen, where they neutralize toxins and block the entry of pathogenic bacteria and viruses across the intestinal epithelium (4, 7). Gut IgA effect both immune protection and immune exclusion in a non-inflammatory manner, thereby playing a central role in host-microbial interaction. Intestinal IgA comprise low-affinity antibodies, mostly polyreactive, that prevent commensal bacteria from penetrating the mucosal barrier, as well as high-affinity IgA that neutralize toxins and microbial pathogens (8). Low affinity IgA would be generated in a T cell-independent fashion, generally in the lamina propria, while high affinity IgA would be generated in a T cell-dependent way, mainly in germinal centers of Peyer’s patches.
Whether in T cell-dependent or independent fashion, IgA are produced through class-switch DNA recombination (CSR), a process that requires germline transcription of Ig heavy chain constant α gene (Igα), as activated by induction of the Iα promoter, and expression of activation-induced cytidine deaminase (AID, encoded by Aicda gene) (8–11). Like CSR to other isotypes, CSR to IgA is a tightly controlled process. It is initiated by TLR ligand:TLR or CD154:CD40 engagement and TGF-β engagement of the TGF-β receptor. The latter activates (by phosphorylation) Smad2 and Smad3 transcription factors (12, 13). Activated Smad2/Smad3 heterodimers form complexes with Smad4 for nuclear translocation and docking onto Smad-binding elements (SBEs) in the Iα promoter (8, 14–16). Here, in conjunction with co-factors, such as Runx3 and Pu.1, Smad2, Smad3 and Smad4 activate germline Iα-Cα transcription, which critically initiates CSR to IgA (17–20). Accordingly, overexpression of Smad3 and Smad4 in B cells increased IgA expression (15) and B cell-specific Smad2 deletion reduced CSR to IgA (20). Other cytokines, such as IL-4 and IL-5, can enhance CSR to IgA, generally by boosting secretion of endogenous B cell TGF-β (21). Retinoic acid (RA), a vitamin A metabolite that is abundant in the gut, has been shown to induce gut homing receptors on activated T cells and CSR to IgA throughout gut-associated lymphoid tissue (GALT). Indeed, impaired GALT dendritic cells conversion of retinol to RA leads to decreased intestinal IgA, resulting in a noticeably decreased IgA in the circulating blood (22).
Epigenetic factors and modifications, such as non-coding RNAs, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), DNA methylation and histone post-translational modifications effect heritable changes in gene expression that are independent from genomic DNA sequence. As we have shown, epigenetic factors and modifications act in concert with transcription factors to regulate B cell CSR, somatic hypermutation (SHM), plasma cell differentiation and generation of memory B cells, thereby shaping the antibody response to foreign- and self-antigens (11, 23–27). In particular, select miRNAs silence B cell Aicda and Prdm1 genes, thereby downregulating AID enzyme and Blimp1 transcription factor, which critically mediate CSR/SHM and plasma cell differentiation (11, 23, 25, 28–30). Prior to modulating these peripheral B cell differentiation processes, miRNAs regulate B development by modulating the expression of genes at the pro-B and pre-B cell stages (31, 32).
miR-146a is one of a small number of miRNAs, whose cell expression is strongly induced by challenge with bacterial endotoxins, and whose prolonged expression has been linked to immune tolerance, implying that it acts as a fine-tuning mechanism to prevent an overstimulation of the immune system and, possibly, inflammatory response (33–35). Indeed, miR-146a has been suggested to regulate immune cells, such as T cell, dendritic cells and macrophages, as well as myeloid cells by exerting immunosuppressive and anti-inflammatory functions (36–42). These would include suppression of gut immunity, autoimmunity and myeloproliferation (43–45). miR-146a is one of the most abundant miRNAs in B cells, particularly resting B cells (38, 44), and its role in B cell function just starts getting attention. miR-146a has been shown to control germinal center responses by targeting multiple components of the CD40 signaling pathway in B cells (39). miR-146a expression has been suggested to reduce IgA production in the gut, and miR-146a deficiency has led to development of immune complex glomerulonephritis in mice (45, 46). miR-146a G/C polymorphism rs2910164 has been shown to associate with pediatric hyper IgA and IgA nephropathy, a syndrome that is characterized by high serum IgA levels, and IgA deposition in glomeruli, as well as with diabetic nephropathy (47, 48). Such a miR-146a G/C polymorphism seemingly reduced the expression level of pre-miR-146a, likely by affecting the binding efficiency of the Drosha/DGCR8 complex to pri-miR-146a, thereby eventually reducing the amount of mature miR-146a (49). Further, miR-146a was found to act as a negative regulator of TGF-β signaling in skeletal muscle cells (50). Finally, it was shown to target Smad4, as induced by the TGF-β signaling pathway in promyelocytic leukemia cells (51).
The above findings and considerations led us to hypothesize that miR-146a plays an important role in regulating B cell CSR to IgA by modulating TGF-β-induced Smad2, Smad3 and Smad4. We tested our hypothesis by analyzing miR-146a–/– mice for basal level of circulating IgA as well as frequency of IgA+ B cells in spleen, bone marrow, mesenteric lymph nodes (MLNs), lamina propria and Peyer’s patches, fecal bacteria-bound and free IgA as well as IgA deposition in kidney glomeruli. We further analyzed the magnitude of a specific IgA antibody response in miR-146a-/- mice as well as constructed μMT/miR-146a–/– mixed bone marrow chimera mice to prove that any miR-146a related dysregulation in IgA production was due to intrinsic B cell miR-146a. We also tested the miR-146a–/– B cell intrinsic ability to undergo CSR to IgA in vitro in response to T-dependent and T-independent CSR-inducing stimuli. In these in vitro CSR-induced B cells, we measured the expression of Smad2, Smad3, Smad4 transcripts, the recruitment of Smad2, Smad3 and Smad4 transcription factors to the Iα promoter and consequent germline Iα-Cα transcription. Finally, we enforced expressed miR-146 in B cells to analyze the impact of this miRNA on Smad2, Samd3 and Smad4 expression levels. We concluded that miR-146a plays an important role in keeping in check Smad2, Smad3 and Smad4 transcription in resting B cells, and inhibiting CSR to IgA. Upon exposure to IgA CSR-inducing stimuli, such as CD154 or LPS plus TGF-β, B cells physiologically downregulate miR-146a, thereby allowing for increased Smad2, Smad3 and Smad4 expression, germline Iα-Cα transcription and unfolding of IgA CSR.
Materials and Methods
Mice
C57BL/6 (CD45.2+, stock No. 000664), C57/CD45.1+ (B6.SJL-PtprcaPepcb/BoyJ, stock No. 002014), miR-146a–/– (B6.Cg-Mir146tm1.1Bal/J, CD45.2+, stock No. 016239) (52) and μMT (B6.129S2-Ighmtm1Cgn/J, CD45.2+, stock No. 002288) (53) mice were purchased from the Jackson Laboratory. miR-146a–/– and miR-146a+/+ mice (8-12 wk of age) were analyzed for fecal free and bacteria-bound IgA using our published procedure (23). They were also analyzed for spleen, bone marrow, MLNs, lamina propria and Peyer’s patches IgA+ B cells using an anti-mouse IgA mAb (clone C10-3, BD Bioscience) and mAb to mouse CD19 (clone 1D3, BD Bioscience). All mice were housed and maintained in the University of Texas Health Science Center San Antonio pathogen–free vivarium and provided with autoclaved food and deionized water. The Institutional Animal Care and Use Committees (IACUC) of the University of Texas Health San Antonio approved all animal research protocols.
To construct mixed bone marrow chimeric B cell-specific miR-146a–/– (μMT/miR-146a–/–) mice and controls μMT/miR-146a+/+ mice, sex-matched recipient C57/CD45.1+ mice (8-12 wks of age) from the same breeding batch were treated with neomycin sulfate (2 mg/ml in drinking water) for one week before being γ-irradiated for complete myeloablation (1000 Rad or 10 Gy, 12.5 min from a 137cesium source). After 24 h, the mice were randomized into two groups and injected through the tail vein with 2.5 x 106 mixed bone marrow cells as comprised of 80% (2 x 106) cells from a μMT mouse and 20% (0.5 x 106) cells from either a miR-146a–/– mouse or its miR-146a+/+ littermate. Prior to mixing, bone marrow cells isolated form tibia and fibula of donor miR-146a–/– or miR-146a+/+ mice were depleted of T cells by incubation with biotinylated anti-CD3 mAb (Magnisort® Streptavidin Negative Selection Beads (eBioscience). Chimeric mice were monitored by flow cytometry for CD45.1 (clone A20, 110471, BioLegend), CD45.2 (clone 104, 109830, BioLegend), CD19 (clone 1D3, 20-0193, Tonbo), CD4 (clone RM4-5, 45-0042-82, eBioscience), CD8 (clone H35-17.2, 12-0083-81, eBioscience), CD11b (clone M1/70, 101212, BioLegend), and/or CD11c (clone N418, 117323, BioLegend) expression on circulating mononuclear cells for at least 6 wks to confirm the immune system reconstitution.
For antigen-specific IgA response experiments, miR-146a–/– and miR-146a+/+ mice as well as μMT/miR-146a–/– and μMT/miR-146a+/+ mice were given ovalbumin (OVA, 20 mg) with cholera toxin (OVA CT) via intragastric gavage (23) at d 0, 7 and 14. Sera and feces were collected at d 21 (for total and OVA-specific IgM, IgG1, IgA and IgE analysis), at which time mice were sacrificed for all other studies.
ELISAs and ELISPOTs
Titers of IgG1 and IgA in cell culture supernatants of in vitro-stimulated B cells or IgM, IgG1, IgA or IgE in vivo titers of circulating/fecal total and/or OVA-specific-IgM, IgG1, IgA and IgE were measured using specific ELISAs, as we described (28, 54–56). For antibody secreting cell (ASC) analysis, MultiScreen® ELISPOT plates (MAIPS4510; Millipore) were activated with ethanol (35%), washed four times with PBS and coated with unlabeled rabbit polyclonal antibodies against mouse IgM, IgG1 or IgA in PBS overnight at 4°C. The plates were then washed six times with PBS, blocked with BSA (0.5%) in RPMI/HEPES plus L-glutamine for 1 h at room temperature. The plates were then used to culture single cell suspensions from MLNs, Peyer’s patches, bone marrow and spleen cells at 37°C for 16 h in 10% FBS RPMI medium containing 50 mM 2-ME and 1× antibiotic-antimycotic mixture (15240-062; Invitrogen) (FBS-RPMI) at 150,000 cells/ml. The culture fluids were then removed, the plates were washed six times, incubated with biotin-anti-IgM, IgG1, or IgA Ab for 2 h on a shaker at room temperature, washed, incubated with horseradish peroxidase (HRP)-streptavidin (Santa Cruz Biotech) for 1 h on a shaker at room temperature, washed again and developed using the Vectastain AEC peroxidase substrate kit (SK-4200, Vector Laboratories) following manufacturer’s protocol. ASCs were imaged and quantified using a CTL-ImmunoSpot Analyzer and software.
B Cells, CSR and Plasma Cell Differentiation
Naive IgM+IgD+ B cells were isolated from 8- to 12-wk-old C57BL/6 mice, as described (9). B cells were resuspended in FBS-RPMI at 37°C in 48-well plates and stimulated with LPS (5 μg/ml) from Escherichia coli (055:B5; Sigma-Aldrich) for CSR to IgG3; LPS (3 μg/ml) or CD154 [1 U/ml; obtained from membrane fragments of baculovirus-infected Sf21 insect cells (45)] plus IL-4 (5 ng/ml; R&D Systems) for CSR to IgG1/IgE and plasma cell differentiation; or LPS (3 μg/ml) or CD154 (1 U/ml) plus TGF-β (2 ng/ml; R&D Systems), IL-4 (5 ng/ml), IL-5 (3 ng/ml; R&D Systems), RA (10 nM, Sigma) and anti-Igδ mAb for CSR to IgA. After 96 h, the cells were stained with FITC-labeled rat mAb to mouse IgG1 (clone A85-1), mouse IgG2a/c (clone R19-15), mouse IgG3 (clone R40-82), mouse IgA (clone C10-3), or PE-labeled rat mAb to mouse CD19 (clone 1D3), all from BD Biosciences, 7-Aminoactinomycin D (7-AAD; A1310, Invitrogen) and PE-Cy7-anti-CD138 mAb (142513, Biolegend). The same mAbs were used to stain mouse mononuclear cells ex vivo. Cell analyses were performed using a LSR-II flow cytometer (BD Biosciences), and data were analyzed using FlowJo software (TreeStar). All experiments were performed in triplicates.
Quantitative RT-PCR (qRT-PCR) of mRNAs and miRNAs
For quantification of germline IH-CH, post-recombination Iμ-Cα and circular Iα-Cμ transcripts, as well as Smad2, Smad3, Smad4, Aicda, Prdm1, Traf6 and Irak1 transcripts, RNA was extracted from 0.2-5.0 × 106 cells using either Trizol® Reagent (Invitrogen) or RNeasy Plus Mini Kit (Qiagen). Residual DNA was removed from using gDNA eliminator columns (Qiagen). cDNA was synthesized from total RNA with the SuperScript™ III First-Strand Synthesis System (Invitrogen) using oligo-dT primer. Transcript expression was measured by qRT-PCR with the appropriate primers (Supplementary Table 1) using a Bio-Rad MyiQ™ Real-Time PCR Detection System (Bio-Rad Laboratories) to measure SYBR Green (IQ™ SYBR® Green Supermix, Bio-Rad Laboratories) incorporation with the following protocol: 95°C for 15. sec, 40 cycles of 94°C for 10 sec, 60°C for 30 sec, 72°C for 30 sec. Data acquisition was performed during 72°C extension step. Melting curve analysis was performed from 72-95°C.
For quantification of mature miRNA transcripts, RNA was extracted from 0.2-5.0 × 106 cells using miRNeasy® Mini Kit (Qiagen) and reverse-transcribed with miScript II RT Kit (Qiagen) using miScript HiSpec buffer. A Bio-Rad MyiQ™ Real-Time PCR Detection System was used to measure SYBR Green (miScript SYBR Green PCR Kit; Qiagen) incorporation according to manufacturer’s instructions. Mature miRNA forward primers (Supplementary Table 1) were used at 250 nM in conjunction with the Qiagen miScript Universal Primer and normalized to expression of small nuclear/nucleolar RNAs Rnu6/RNU61/2, Snord61/SNORD61, Snord68/SNORD68, and Snord70/SNORD70. The ΔΔCt method was used for miRNA qRT-PCR data analysis.
Fluorescence Microscopy
To analyze IgM and IgA-producing cells in the lamina propria, the intestine was folded into a “Swiss-roll”, fixed with PFA (4%), and embedded in paraffin. Ten μm sections were cut and heated at 80°C to adhere to the slide, washed four times in xylene for 2 min, dehydrated two times with 100% ethanol for 1 min, two times with 95% ethanol for 1 min and washed two times in water for 1 min. Antigens were unmasked using 2 mM EDTA in 100°C for 40 mins followed by a cooling step at 25°C on the bench top, 3 times washing with 1x TBS and blocking using 10% BSA for 15 min. Slides were again washed three times with 1x TBS and stained with PE-anti-IgM, primary-rabbit anti mouse-IgA mAb (PA-1-30826, Thermo fisher) followed by Alexa Fluor 488®-anti rabbit-IgG (H+L) F(ab’)2 mAb (4412; Cell Signaling) for 2 h in a moist dark chamber. To analyze IgA-producing cells in MLNs, 10 μm MLN sections were prepared by cryostat and loaded onto positively charged slides, fixed in cold acetone and stained with rabbit anti mouse-IgA mAb (PA-1-30826, Thermo fisher) followed by Alexa Fluor 488®-anti rabbit-IgG (H+L) F(ab’)2 mAb (4412, Cell Signaling) for 1 h in a moist dark chamber. After washing three times with Triton X-100 (0.1%) in TBS, slides were air-dried and coverslips were mounted with ProLong® Gold Antifade Reagent using DAPI (Invitrogen). Fluorescence images were captured using a 10 x objective lens with a Zeiss Axio Imager Z1 fluorescence microscope.
Kidney Pathology
To analyze kidney IgA deposition, kidneys from miR-146a–/– and miR-146a+/+ mice were fixed in 4% formaldehyde and paraffin-embedded for hematoxylin and eosin staining. For immunofluorescence analysis, 5-μm cryostat sections were loaded onto positively charged slides, fixed in cold acetone, and stained with FITC-labeled rat anti-mouse IgA mAb (clone C10-3, BD Biosciences). Cover slips were mounted using ProLong Gold Antifade Reagent for microscopy analysis.
Microbiota Analysis
Microbial DNA were extracted from mouse feces using Quick-DNA™ Fecal/Soil Microbe Microprep Kit (Zymo Research) according to the manufacturer’s instructions. To determine the composition of the bacterial phyla present in mouse feces, isolated bacterial DNA was tagged and sequenced using miSeq platform. The V3-V4 hypervariable region of the bacteria 16S rRNA gene was amplified by PCR using tagged bact-341F primer 5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3’, bact-805R primer 5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3’ and Phusion DNA polymerase. Multiplexing indices and Illumina sequencing adapters were then added to the amplicons by limited-cycle amplification using the Nextera XT Index Kit (Illumina). The libraries were normalized, pooled and sequenced using the MiSeq system (Illumina). Sequencing and quality assessment were performed by the UT Health Genome Sequencing Facility. Bacterial taxonomy was assigned using the Ribosomal Database Project (RDP) classifier (57).
Immunoblotting
After stimulation by CSR-inducing stimuli and culture, B cells were lysed in Laemmli buffer. Cell extracts containing equal amounts of protein (20 μg) were fractionated through SDS-PAGE (10%). The fractionated proteins were transferred onto polyvinylidene difluoride membranes (Bio-Rad Laboratories) overnight (30 V) at 4°C. After blocking and overnight incubation at 4°C with anti-Smad2 (A7699, Abclonal), anti-Smad3 (A19115, Abclonal) and anti-Smad4 (A5657, Abclonal) rabbit Abs, or anti-β-Actin mAb (AC-15, Sigma-Aldrich), the membranes were incubated with HRP-conjugated secondary Abs. After washing with PBS-Tween 20 (0.05%), bound HRP-conjugated Abs were revealed using Western Lightning® Plus-Enhanced Chemiluminescence reagents (PerkinElmer Life and Analytical Sciences).
Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed as described (23, 24, 30). Briefly, B cells (5 × 106) were treated with 1% formaldehyde for 10 min at 25°C to cross-link chromatin. After washing with cold PBS containing protease inhibitors (Roche Applied Science), chromatin was separated using nuclear lysis buffer (10 mM Tris-HCl, 1.0 mM EDTA, 0.5 M NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.5% Sarkosyl, pH 8.0) and resuspended in IP-1 buffer (20 mM Tris-HCl, 200 mM NaCl, 2.0 mM EDTA, 0.1% sodium deoxycholate, 0.1% SDS, protease inhibitors). Chromatin was sonicated to yield ~0.2 – 1.0 kb DNA fragments, precleared with agarose beads bearing protein G (Santa Cruz Biotechnology), and then incubated with anti-Smad2 (A7699, Abclonal), anti-Smad3 (A19115, Abclonal) or anti-Smad4 (A5657, Abclonal) rabbit Abs at 4°C. After overnight incubation, immune complexes were isolated using agarose beads bearing protein G, eluted with elution buffer (50 mM Tris-HCl, 0.5% SDS, 200 mM NaCl, 100 μg/ml Proteinase K, pH 8.0), and then incubated at 65°C overnight to reverse formaldehyde cross-links. DNA was purified using QIAquick PCR Purification Kit (Qiagen). The purified DNA was used as a template for qPCR analysis of the Iα promoter using specific primers (Supplemental Table 1).
Enforced Expression of miR-146a
The miR-146a expression retroviral construct pMSCV-PIG-miR-146a (58) was a gift from Joshua Mendell (Addgene plasmid # 64234) and the pMSCV-PIG (Puro IRES GFP) empty vector was a gift from David Bartel (Addgene plasmid # 21654). To generate the retrovirus, pMSCV-PIG-miR-146a or empty pMSCV-PIG vector were used together with the pCL-Eco retrovirus-packaging vector (Imgenex) to transfect HEK293T cells by the calcium phosphate-mediated method (ProFection Mammalian Transfection System; Promega). Viral supernatants were harvested and used to transduce spleen B cells from C57BL/6 mice, as we reported (24, 30, 54), after a 12 h activation by LPS. Transduced B cells were then stimulated by LPS plus TGF-β, IL-4, IL-5, RA and anti-Igδ mAb for 96 h before analyzing GFP+ B cells for surface IgA expression as well as intracellular AID, Smad2, Smad3 and Smad4 expression.
Statistical Analyses
All statistical analyses were performed using Excel (Microsoft) or GraphPad Prism® software. Differences in Ig titers, CSR and RNA transcript expression were analyzed by Student’s paired (in vitro) and unpaired (in vivo) t-test assuming two-tailed distributions.
Results
miR-146a Deficient Mice Show High IgA Levels, Increased IgA+ B Cells in Multiple Body Districts and Kidney IgA Deposition
We investigated the impact of miR-146a deficiency on IgA expression and IgA-producing cells in non-intentionally immunized 22-wk-old miR-146a–/– mice. These mice showed higher levels of serum IgA (n = 6, 146.0 ± 10.2 vs. 78.4 ± 16.6 μg eq/ml, p = 0.014) and fecal IgA (n = 6, 271.3 ± 134.2 vs. 115.1 ± 28.2 μg eq/ml, p = 0.032) as well as fecal bacteria-bound IgA (68.25 vs. 23.2%) as compared to their miR-146a+/+ mouse counterparts. miR-146a–/– mice also showed higher frequency of IgA+ B cells in spleen, bone marrow, mesenteric lymph nodes (MLNs), intestinal lamina propria and Peyer’s patches (Figures 1A–C). In addition, they showed a higher frequency of IgA- but not IgM- or IgG-secreting cells in spleen and MLNs (Figure 1D) as well as IgA deposition in kidney glomeruli and glomerular sclerosis (Figure 1E). Thus, deletion of miR-146a increases systemic and local IgA.
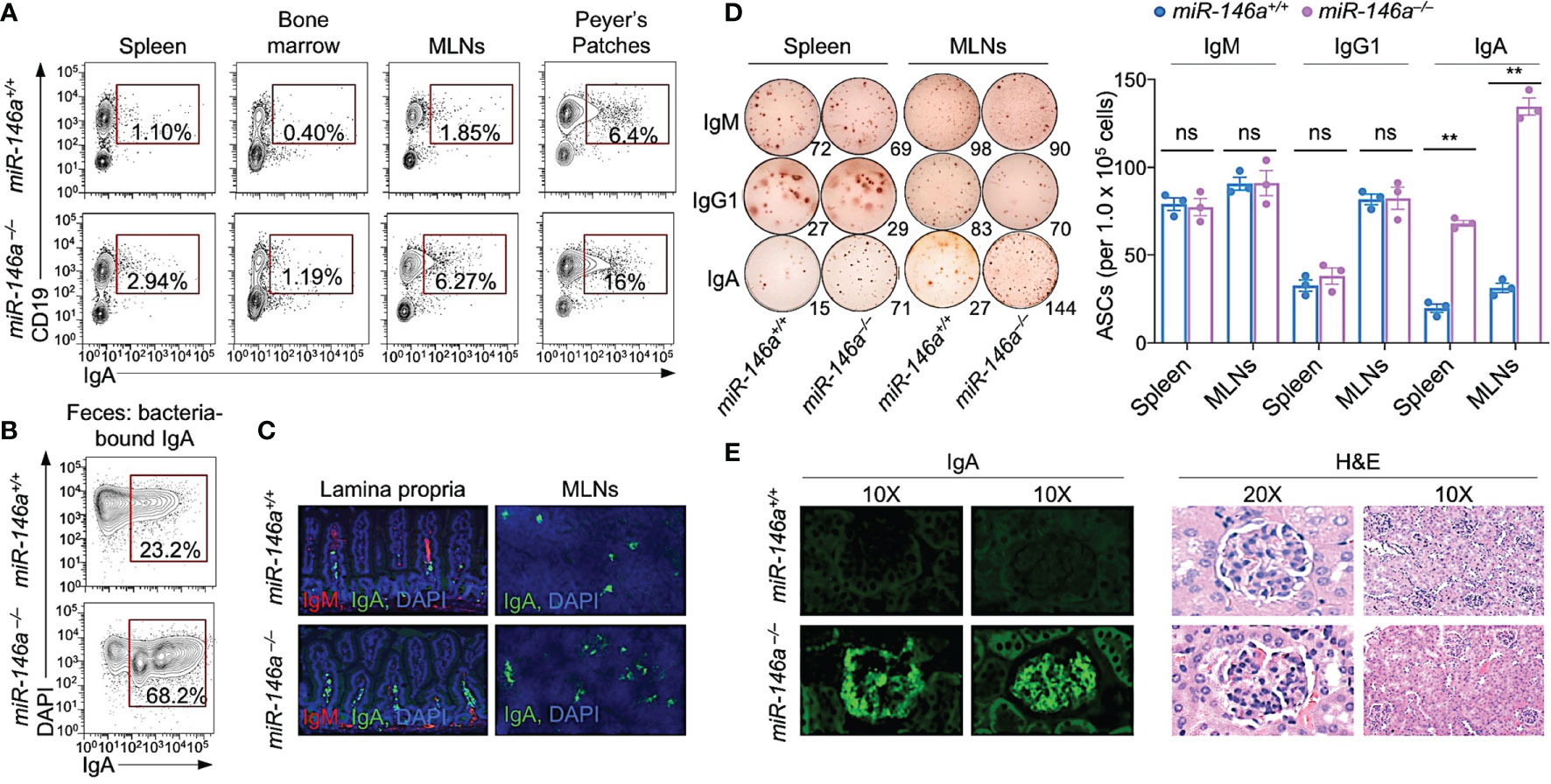
Figure 1 miR-146a–/– mice (12 wk of age) displayed elevated systemic and gut IgA+ B cells, IgA-secreting cells, and kidney IgA deposition, as hallmark of IgA nephropathy. (A) IgA+ B cells in spleen, bone marrow, mesenteric lymph nodes (MLNs) and Peyer’s patches as analyzed by flow cytometry. (B) Fecal bacteria-bound IgA as analyzed by flow cytometry. (C) IgM+ and IgA+ B cells in the lamina propria and MLNs of miR-146a and miR-146a+/+ mice as visualized by fluorescence microscopy. (D) ELISPOT analysis of IgM-, IgG1- and IgA-secreting cells (ASCs) in spleen and MLNs. (E) Photomicrographs of kidney sections from miR-146a–/– and miR-146a+/+ mice, after fluorescence staining for mouse IgA (left) or hematoxylin and eosin (H&E) staining (right). Data are from one representative of three independent experiments yielding similar results; or mean ± SEM of 3-4 miR-146a–/– or miR-146a+/+ mice from three independent experiments (D, right panel). **p < 0.01, ns, not significant, unpaired t-test.
miR-146a Knockout Increases Total and OVA-Specific IgA in Blood and Feces
To define the role of miR-146a in the modulation of an antigen-specific response, miR-146a+/+ and miR-146a–/– mice were administered OVA via intragastric route once a week for three consecutive weeks. Serum and feces were collected from each mouse one week after the third OVA administration for analysis of systemic and fecal total and OVA-specific antibody titers, at which time mice were sacrificed for all other studies. miR-146a–/– mice displayed significantly higher total and OVA-specific IgA (p < 0.05) in serum and feces than their miR-146a+/+ counterparts (Figures 2A–C). By contrast, miR-146a–/– mice showed no significant changes in total or OVA-specific IgM, IgG1 or IgE titers in serum or feces. Thus, miR-146a deletion increases total and antigen-specific IgA in the circulation and the gut.
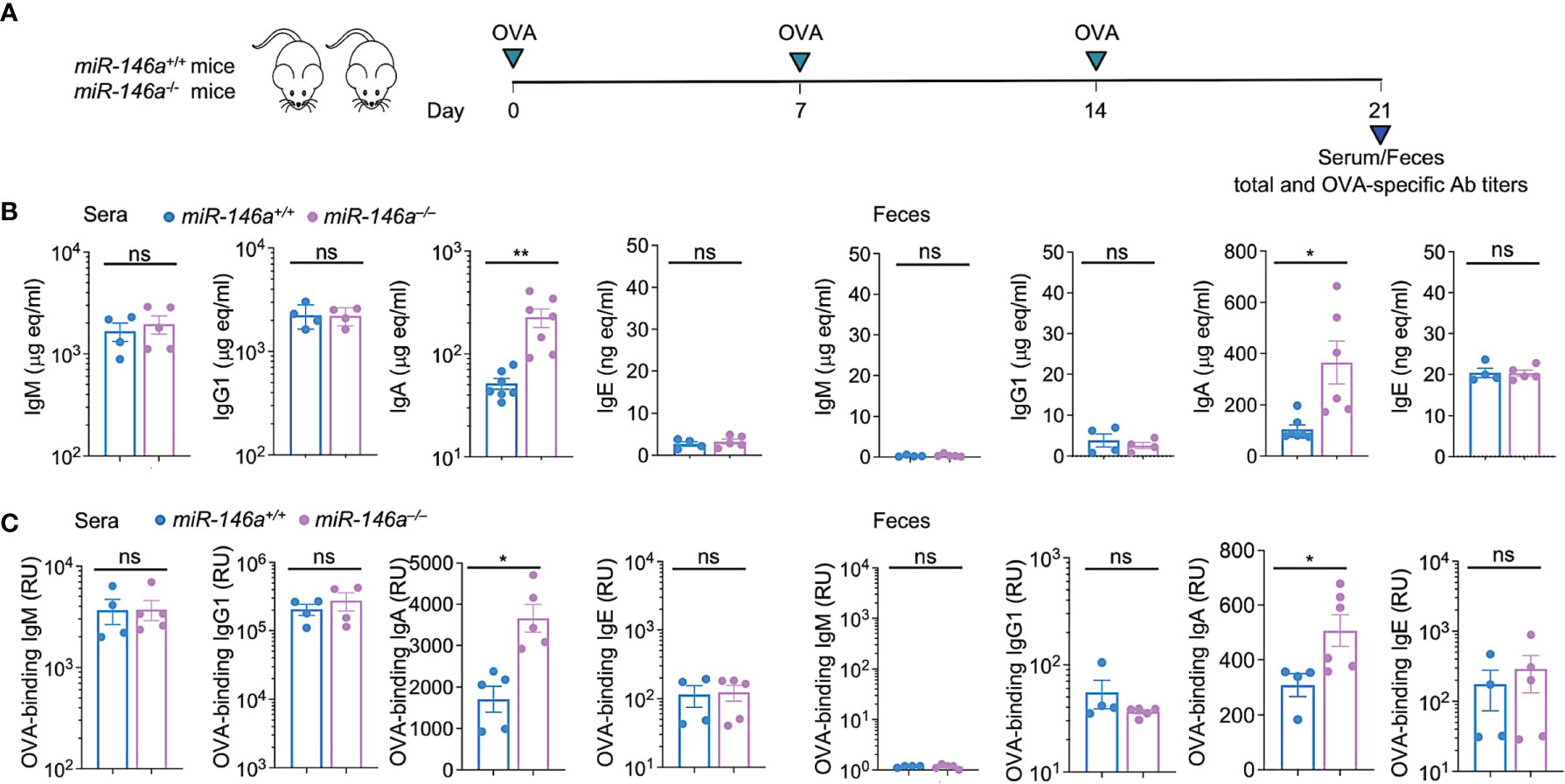
Figure 2 Increased IgA production in miR-146a–/– mice. (A) miR-146a–/– and miR-146a+/+ mice were administered OVA together with CT via intragastric gavage once a week for 3 wks and sacrificed one wk after the last OVA administration for analysis of antibodies, B cells and ASCs. Total (B) and OVA-binding (C) IgM, IgG1, IgA and IgE titers, as measured by ELISA in serum and feces. **p < 0.01, *p < 0.05, ns, not significant, unpaired t-test. Data are mean ± SEM of 4-7 miR-146a–/– or miR-146a+/+ mice from three independent experiments.
Increased CSR to IgA in Intrinsic B Cell miR-146a Deficient (µMT/miR-146a–/–) Chimeric Mice
To determine whether the increased IgA production in miR-146a–/– mice stemmed solely from B cell-intrinsic miR-146a deficiency, we constructed µMT/miR-146a+/+ and µMT/miR-146a–/– chimera mice using myeloablated C57BL/6 mice grafted with (80%) bone marrow cells from µMT (B6.129S2-Ighmtm1Cgn/J) mice specifically deficient in B cells and (20%) purified bone marrow B cells from µMT/miR-146a–/– mice. Chimeric µMT/miR-146a+/+ and µMT/miR-146a–/– mice were administered OVA via intragastric route once a week for three consecutive weeks. Serum and feces were collected one week after the third administration for systemic and gut titles of total and OVA-specific antibodies analysis, at which time mice were sacrificed (6 wks after bone marrow transplantation) for all other analyses. µMT/miR-146a–/– mice show significantly elevated systemic and gut IgA (fecal) total and OVA-specific IgA but not IgM, IgG1 or IgE as compared to their µMT/miR-146a+/+ counterparts (Figures 3A–C). Accordingly, µMT/miR-146a–/– mice showed significantly greater amounts of bacteria-bound IgA in feces, greater proportions of IgA+ B cells in peripheral blood and Peyer’s patches as well as higher numbers of OVA-specific ASCs in bone marrow, spleen and MLNs with no changes in the proportion of CD19+IgG1+ B cells or CD138+ plasma cells (Figures 3D–F). Thus, intrinsic B cell miR-146a deficiency per se underpins increased IgA CSR and increased IgA levels in vivo.
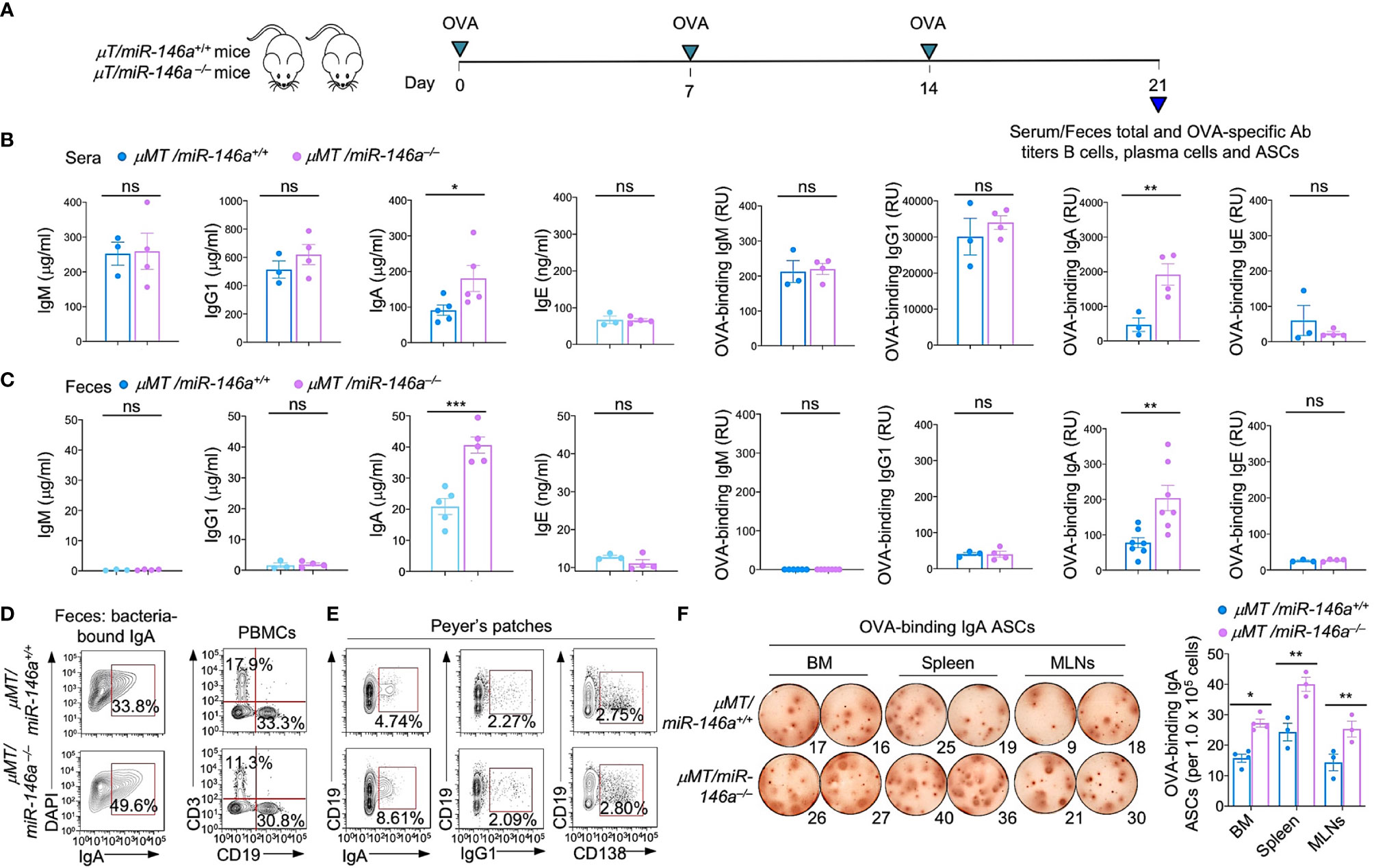
Figure 3 Increased IgA production, IgA+ B cells and IgA-ASCs in μMT/miR-146a–/– chimeric mice. μMT/miR-146a–/– and μMT/miR-146a+/+ chimeric mice were constructed by mixed bone marrow adaptive transfer. (A) All mice were administered OVA together with CT via intragastric gavage once a week for 3 wks and sacrificed one wk after the last OVA administration for analysis of antibodies, B cells and ASCs. Total and OVA-binding IgM, IgG1, IgA and IgE titers in sera (B) and feces (C) as analyzed by ELISAs. (D) Fecal bacteria-bound IgA as analyzed by flow cytometry. (E) Circulating T (CD3+) and B (CD19+) cells, Peyer’s patched IgA+ B cells, IgG1+ B cells and CD138+ plasma cells, as analyzed by flow cytometry. (F) ELISPOT analysis of OVA-binding IgA ASCs in the bone marrow (BM), spleen and MLNs. ***p < 0.001, **p < 0.01, *p < 0.05, ns, not significant, unpaired t-test. Data in (B, C) and (F, right panel) are mean ± SEM of 3-7 μMT/miR-146a–/– or μMT/miR-146a+/+ from three independent experiments. Data in (D, E) and (F, left panel) are from one representative of three independent experiments yielding similar results.
Altered Gut Microbiota in Intrinsic B Cell miR-146a Deficient (µMT/miR-146a–/–) Chimeric Mice
As IgA is critical for microbiome homeostasis at the intestinal mucosa and as miR-146a deletion resulted in major increases of IgA-producing B cells as well as total and specific IgA, we hypothesized that miR-146a deficiency would impact the composition of gut bacteria, thereby further emphasizing the role of miR-146a in IgA response. We analyzed the fecal microbiota in µMT/miR-146a–/– mice by 16S rRNA gene amplicon sequencing. In all three µMT/miR-146a–/– mice analyzed, the increased IgA production associated with an altered gut bacterial composition as compared to the three µMT/miR-146a+/+ mouse counterparts. We found that Verrucomicrobiaceae, Bacteroidaceae, Porphyromonadaceae and Lachnospiraceae, which are among the most abundant bacterial taxa in feces, were the major families represented in the gut (fecal) microbiome of µMT/miR-146a+/+ mice (Figures 4A–C). Verrucomicrobiaceae and particularly its Akkermansia genus bacteria have been shown to be physiologically bound by IgA and to positively correlate with IgA level in the gut (59–61). Accordingly, Verrucomicrobiaceae and Akkermansia bacteria were significantly increased in µMT/miR-146a–/– mice, in which IgA were elevated. Bacteria of the Bacteroidaceae family and particularly its Bacteroides genus bacteria, which are typically abundant in the colon but not generally bound by IgA (59, 60), were decreased in µMT/miR-146a–/– mice (Figures 4A–C). Bacteria of the Clostridia class, including those of the Lachnospiraceae family and the Blautia genus, members of the phyla Firmicutes, which are also not bound by IgA in vivo (59, 60) were not altered in µMT/miR-146a–/– mice (Figures 4A–C). Rikenellaceae and Prevotellaceae family bacteria, which may or may not be bound by IgA, were decreased, while Sutterellaceae and Enterobacteriaceae, which also may or may not be bound by IgA, were increased in µMT/miR-146a–/– mice. Overall, fecal bacteria families and genera were consistently different in µMT/miR-146a–/– and µMT/miR-146a+/+ mice, as revealed by principal component analysis (PCA) (Figure 4D). Thus, intrinsic B cell miR-146a deficiency, as in µMT/miR-146a–/– chimeric mice, leads to increased IgA levels and altered composition of the gut microbiota.
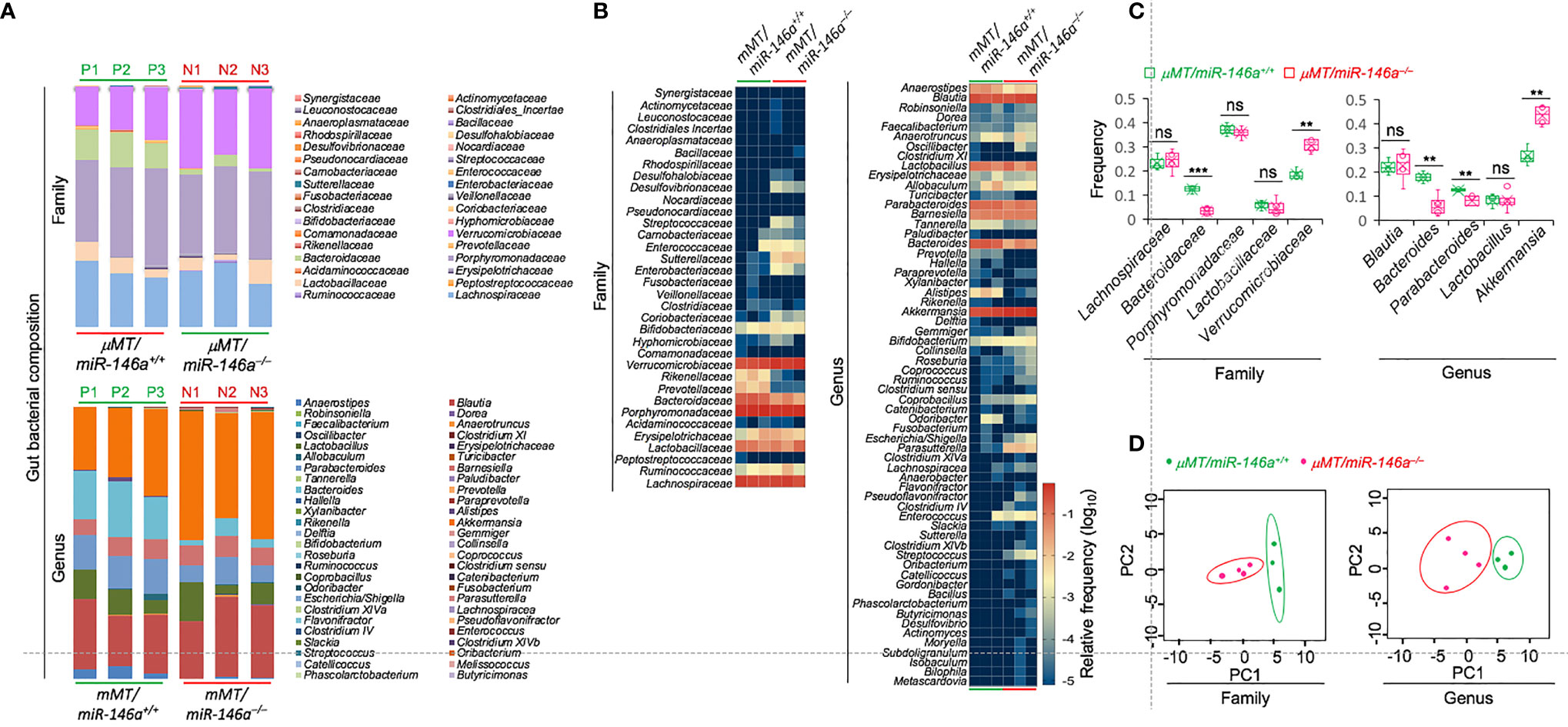
Figure 4 B cell miR-146a deficiency alters gut bacterial microbiome in μMT/miR-146a–/– mice. Relative abundance of fecal bacteria families and genera (A, B) in μMT/miR-146a–/– and μMT/miR-146a+/+ chimeric mice (n = 3 per group) as determined by high-throughput 16s rRNA gene miSeq amplicon sequencing. (C) Frequency of five relatively abundant fecal bacteria families and genera in these mice. ***p < 0.001, **p < 0.01, ns, not significant, unpaired t-test. (D) Principal component analysis of gut bacterial composition of the fecal bacteria families and genera in μMT/miR-146a–/– and μMT/miR-146a+/+ chimeric mice. Data are from three independent experiments.
B Cell Intrinsic miR-146a Deficiency Increases CSR to IgA
To further define the role of miR-146a in CSR, we stimulated miR-146a–/– and miR-146a+/+ B cells with LPS or CD154 plus IL-4 (to induce CSR to IgG1), LPS alone (CSR to IgG3), LPS or CD154 plus IL-4, IL-5, TGF-β, anti-δ mAb and RA (CSR to IgA). In addition to CSR, all these stimuli also induce plasma cell differentiation, albeit at different degrees. After a 96 h culture, CSR to IgA in miR-146a–/– B cells was significantly increased as compared to miR-146+/+ B cells (27.0% vs 19.3% upon stimulation by LPS plus IL-4, IL-5, TGF-β, anti-δ mAb and RA; 26.6% vs 18.4% upon stimulation with CD154 plus IL-4, IL-5, TGF-β, anti-δ mAb and RA) (Figure 5A). Increased CSR to IgA in miR-146a–/– B cells was specific, as these B cells showed no alteration in CSR to IgG1 or differentiation to plasmablasts/plasma cells (as determined by proportion of CD19+ IgG+ B cells and CD138+ cells). The specificity of increased IgA CSR in miR-146a–/– B cells stimulated by LPS or CD154 plus IL-4, IL-5, TGF-β, anti-δ mAb and RA was emphasized by the increased germline Iα-Cα transcripts, post-recombination Iμ-Cα transcripts, post-recombination circle Iα-Cμ transcripts and increased IgA secretion as compared to miR-146a+/+ B cells, as well as by the unchanged IgG1 secretion, and germline Iγ1-Cγ1 and Iϵ-Cϵ transcripts in the very miR-146a–/– B cells (Figures 5B, C). Finally, the specific impact of the miR-146a on Igα locus transcription and recombination was further underlined by the normal level of Aicda transcripts in miR-146a–/– B cells undergoing CSR to IgA. Thus, intrinsic B cell miR-146a deficiency specifically enhances CSR to IgA while not affecting Aicda expression or plasma cell differentiation.
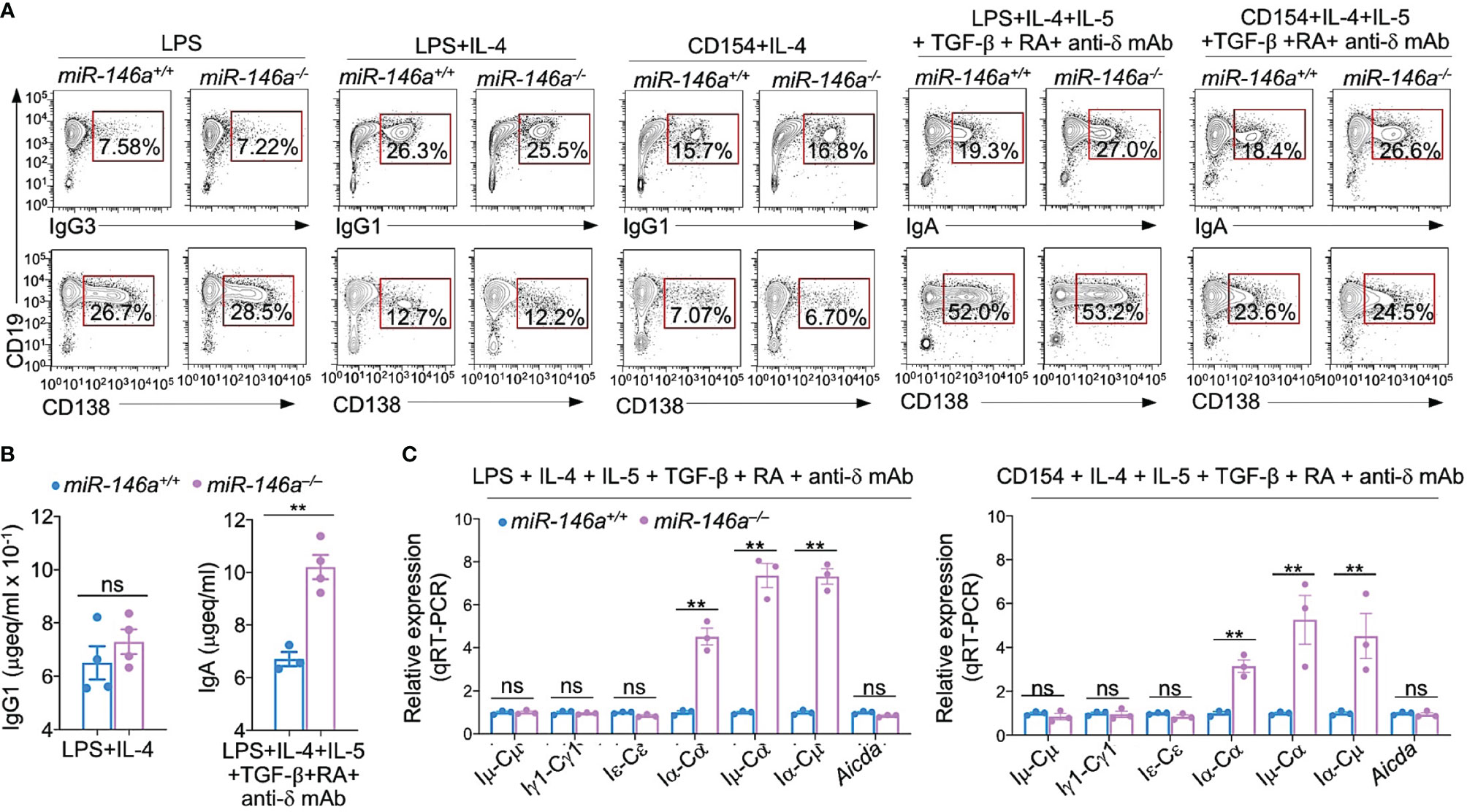
Figure 5 miR-146a ablation results in increased germline Iα-Cα transcripts and enhanced CSR to IgA. (A) miR-146a–/– and miR-146a+/+ B cells were stimulated with LPS, LPS or CD154 plus IL-4, LPS or CD154 plus IL-4, IL-5, TGF-β, RA and anti-δ mAb, and cultured for 96 h. IgG1+, IgG3+ and IgA+ B cells and CD138+ plasma cells as analyzed by flow cytometry. Data are one representative of three independent experiments yielding similar results. (B) IgG1 and IgA concentrations in culture fluids from 146a–/– and miR-146a+/+ B cells stimulated by LPS plus IL-4 or LPS plus IL-4, IL-5, TGF-β, RA and anti-δ mAb, as measured by ELISA. (C) miR-146a–/– and miR-146a+/+ B cells were stimulated by LPS or CD154 plus IL-4, IL-5, TGF-β, anti-δ mAb and RA, and cultured for 72 h. Expression of germline Iμ-Cμ, Iγ1-Cγ1, Iα-Cα and Iϵ-Cϵ transcripts, circle Iα-Cμ and post-recombination Iμ-Cα transcripts, as well as Aicda transcripts as analyzed by qRT-PCR and normalized to β-Actin. Data are ratios to miR-146a+/+ B cells (set as 1; means ± SEM of three independent experiments). **p < 0.01, ns, not significant, paired t-test.
The Stimuli That Induce CSR to IgA Repress B Cell miR-146a, Thereby Upregulating Smad2, Smad3 and Smad4 Expression
Initiation of CSR to IgA requires TGF-β signaling which induces expression of Smad2, Smad3 and Smad4. Activated Smad2 and Smad3 dimerize and together with Smad4 bind to SBEs in the Iα promoter, thereby initiating germline Iα-Cα transcription, the first step in CSR to IgA (62). Prompted by luciferase reporter assays showing miR-146a targeting of SMAD4 mRNA 3’UTR (63–65), we used miRNA-mRNA complementarity prediction tools (TargetScan.org, miRNA.org and miRbase.org) to identify miR-146a-specific target sites in the 3’UTRs of human and mouse Smad2/SMAD2, Smad3/SMAD3 and Smad4/SMAD4 (Figure 6A), suggesting a potentially silencing of Smad2, Smad3 and Smad4 by miR-146a in both human and mouse B cells. This implies that the miR-146a expression level, which is high in resting B cells, must be dampened by those stimuli that induce CSR to IgA to free Smad2/SMAD2, Smad3/SMAD3 and Smad4/SMAD4 genes from miR-146a silencing for CSR to IgA to unfold. Toward addressing this postulated miR-146a modulatory function on CSR to IgA, C57BL/6 mouse B cells were stimulated with LPS or CD154 plus IL-4, TGF-β with or without RA, as well as human B cells with CD154 or CpG plus TGF-β to induce CSR to IgA. miR-146a expression was significantly downregulated (p < 0.01) in such mouse and human B cells stimulated to undergo CSR to IgA as compared to (mouse) B cells stimulated by nil or (human) B cells stimulated by CD154 or CpG alone (in absence of TGF-β), which does not induce CSR to IgA (Figure 6B). The downregulation of miR-146a in B cells undergoing CSR to IgA in vitro mimicked the downregulation of miR-146a in B cells undergoing IgA CSR in vivo, as in MLNs and Peyer’s patches, as opposed to the high level of miR-146a in spleen B cells among which the B cells switching to IgA are a minor proportion of the overall B cell population (in non-intentionally immunized mice) (Figure 6B). In mouse B cells induced to undergo CSR to IgA by LPS plus IL-4, TGF-β and RA, miR-146a downregulation was concomitant with increased Smad2, Smad3 and Smad4 expression (Figure 6C). Thus, the stimuli that induce B cells to undergo CSR to IgA downregulate miR-146a, thereby freeing Smad2/SMAD2, Smad3/SMAD3 and Smad4/SMAD4 from miR-146a inhibition and promoting expression of these transcription factors.
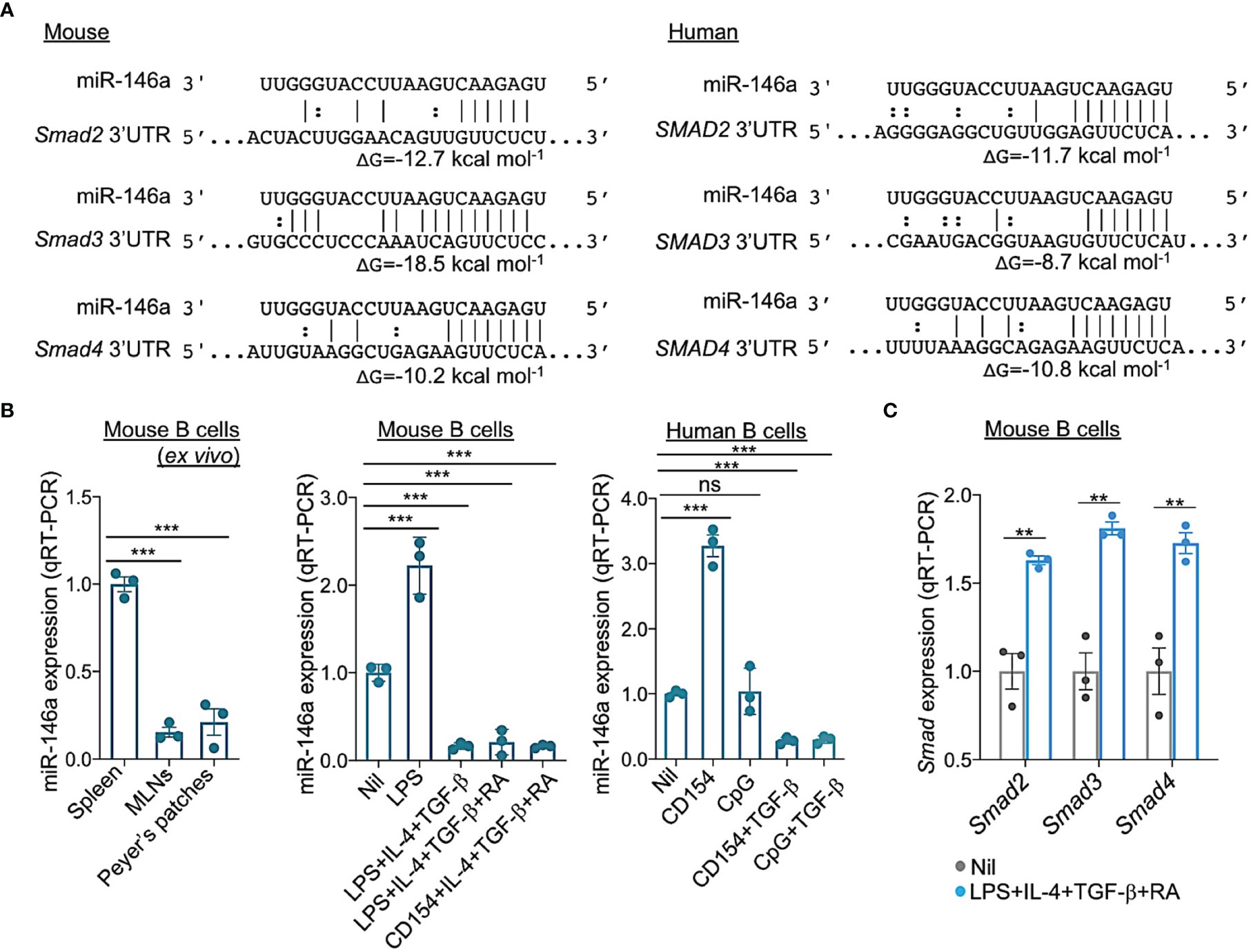
Figure 6 miR-146a targets 3’UTR of Smad2, Smad3 and Smad4 mRNAs and is downregulated by the stimuli that induce CSR to IgA in human and mouse B cells. (A) Alignment of miR-146a with its target sites in the 3′ UTR of human and mouse SMAD2/Smad2, SMAD3/Smad3 and SMAD4/Smad4 mRNAs. The complementary base pairing between miRNA-146a and seed sequences within the 3’ UTRs of SMAD2/Smad2, SMAD3/Smad3 and SMAD4/Smad4, indicating miR-146a ability to silence SMAD2/Smad2, SMAD3/Smad3 and SMAD4/Smad4 transcripts. Gibbs free energy (ΔG) of nucleic acid folding and hybridization calculated by Mfold. Watson–Crick base-pairing (“|”); wobble base-pairing (“:”). (B) Expression of miRNA-146a in mouse spleen, MLN and Peyer’s patch B cells ex vivo; and in mouse spleen B cells stimulated for 72 h with Nil, LPS, LPS plus IL-4 and TGF-β, LPS or CD154 plus IL-4, TGF-β and RA, or human B cells stimulated for 72 h with Nil, CD154, CpG, and CD154 or CpG plus TGF-β, as analyzed by qRT-PCR. Values were normalized to expression of small nuclear/nucleolar RNAs Rnu6, Snord61, Snord68, and Snord70, and depicted as relative to the expression of miRNA-146a in spleen B cells stimulated with Nil, set as 1. (C) Expression of Smad2, Smad3 and Smad4 transcripts in mouse B cells stimulated for 72 h with Nil or LPS plus IL-4, TGF-β and RA, as analyzed by qRT-PCR and normalized to β-Actin, and depicted as relative to the expression of these genes in spleen B cells stimulated with nil, set as 1. Data are mean ± SEM from three independent experiments. ***p < 0.001, **p < 0.01, ns, not significant, unpaired t-test.
miR-146a Ablation Leads to Further Smad2, Smad3 and Smad4 Upregulation in B Cells Induced to Undergo CSR to IgA
Having shown that IgA CSR-inducing stimuli downregulated miR-146a resulting in increased Smad2, Smad3 and Smad4 expression, germline Iα-Cα transcription, followed by post-recombination Iμ-Cα, circle Iα-Cμ transcripts, and CSR to IgA, we wanted to prove that ablation of miR-146a leads to further Smad2, Smad3 and Smad4 expression (critical for Iα-Cα transcription and initiation of CSR to IgA). To this end, we stimulated miR-146a–/– B cells and for comparison miR-146a+/+ B cells with IgA CSR-inducing stimuli LPS or CD154 plus IL-4, IL-5, TGF-β, anti-δ mAb and RA. Whether stimulated by LPS or CD154, miR-146a–/– B cells upregulated Smad2, Smad3 and Smad4 transcripts as well as Smad2, Smad3 and Smad4 proteins at higher levels than their equally stimulated miR-146a+/+ B cell counterparts (Figures 7A, B). They also underwent greater CSR to IgA, as indicated by the significantly higher expression of germline Iα-Cα and post-recombination Iμ-Cα transcripts, as well as Traf6 and Irak1, both genes whose proteins are involved in TGF-β activation signaling pathways (66). miR-146a specificity for Igα locus CSR regulation was further emphasized by the comparable levels of induced Aicda and Prdm1 expression in miR-146a–/– and miR-146a+/+ B cells (Figures 7A, B). Thus, ablation of miR-146a significantly increases Smad2, Smad3 and Smad4 expression.
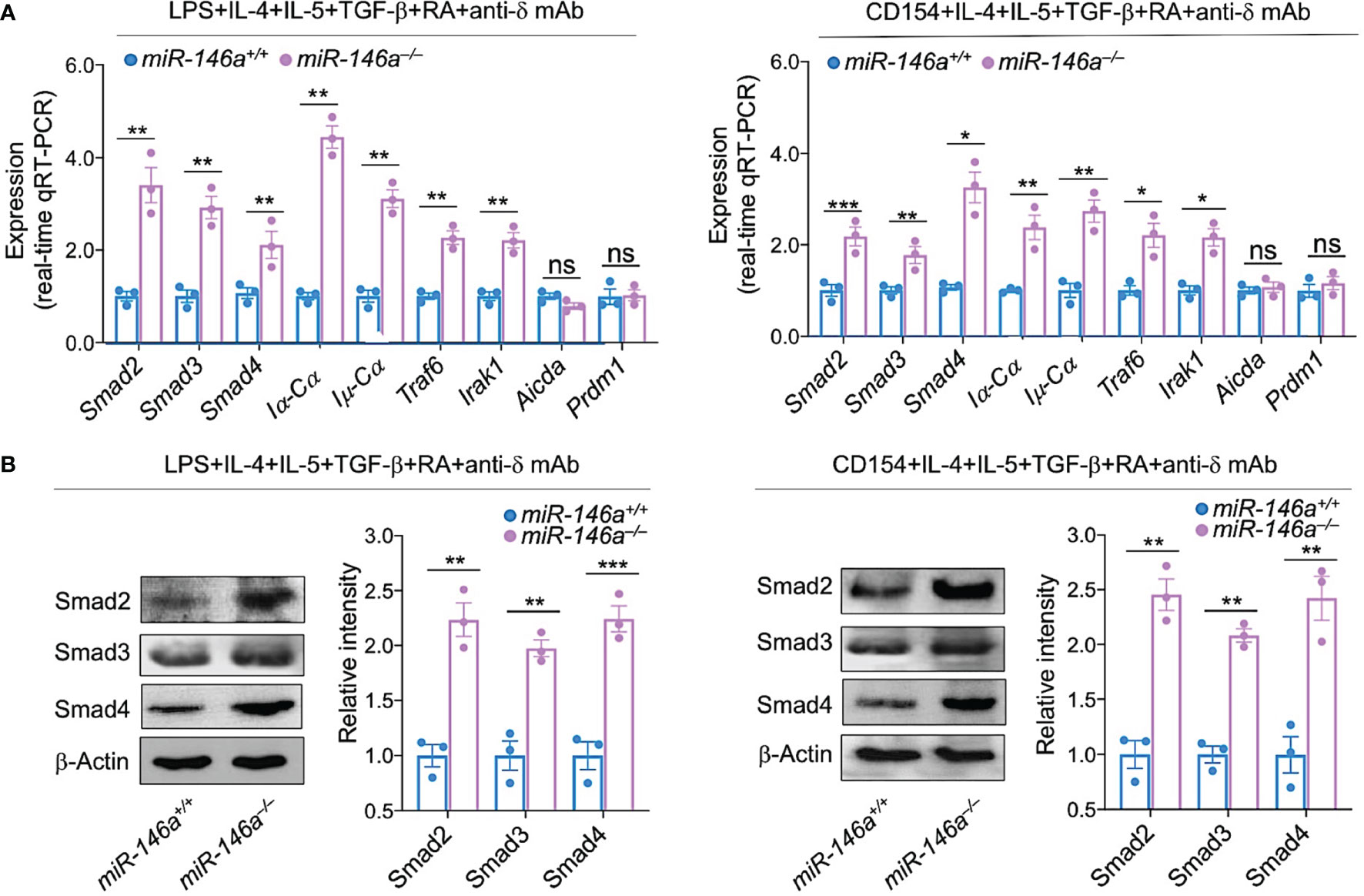
Figure 7 miR-146a deletion increases expression of Smad2, Smad3 and Smad4, germline Iα-Cα and post-recombination Iμ-Cα transcripts. miR-146a–/– and miR-146a+/+ B cells were stimulated with LPS or CD154 plus IL-4, IL-5, TGF-β, RA and anti-δ mAb, and cultured for 72 h. (A) The expression of Smad2, Smad3 and Smad4 as well as Iα-Cα, Iμ-Cα, Traf6, Irak1, Aicda and Prdm1 transcripts was analyzed by qRT-PCR performed in triplicate and normalized to β-Actin. (B) Expression of Smad2, Smad3 and Smad4 proteins as analyzed by immunoblotting. Densitometry quantification of immunoblotting signals was normalized to β-Actin levels and depicted as ratios of readings in miR-146a–/– and miR-146a+/+ B cells to the average value in miR-146a+/+ B cells. Data are ratios to stimulated miR-146a+/+ B cells (set as 1; means ± SEM of three independent experiments). ***p < 0.001, **p < 0.01, *p < 0.05, ns, not significant, unpaired t-test.
Increased Smad2, Smad3 and Smad4 Expression by miR-146a Deletion Leads to Increased Smad2, Smad3 and Smad4 Recruitment to the Igα Locus Iα Promoter
Having shown that induction of CSR to IgA in miR-146a ablated B cells leads to even higher levels of Smad2, Smad3 and Smad4, we hypothesized that this would result in increased Smad2, Smad3 and Smad4 recruitment to the Iα promoter, a requirement for activation of germline Iα-Cα transcription and CSR to IgA. Using TargetScan.org, miRNA.org and miRbase.org, we identified five SBEs [CAGAC and GGC(GC)|(CG) (67)] in the Iα promoter, the DNA region immediately upstream of the Iα transcription initiation site. Of such five SBEs, four were CAGAC and one was a GGCCG, overall, highly conserved in mouse and human Iα promoter region (Figure 8A). As we hypothesized, ablation of miR-146a, which resulted in significantly increased Smad2, Smad3 and Smad4 levels, led to significantly increased recruitment of Smad2, Smad3 and Smad4 transcription factors to the Iα promoter in B cells activated by LPS or CD154 plus IL-4, IL-5, TGF-β, anti-δ mAb and RA, and as analyzed by specific ChIP (Figure 8B). Thus, as freed from miR-146a silencing, increased Smad2, Smad3 and Smad4 expression led to significantly increased recruitment of Smad2, Smad3 and Smad4 to the Iα promoter SBEs (Figure 8C).
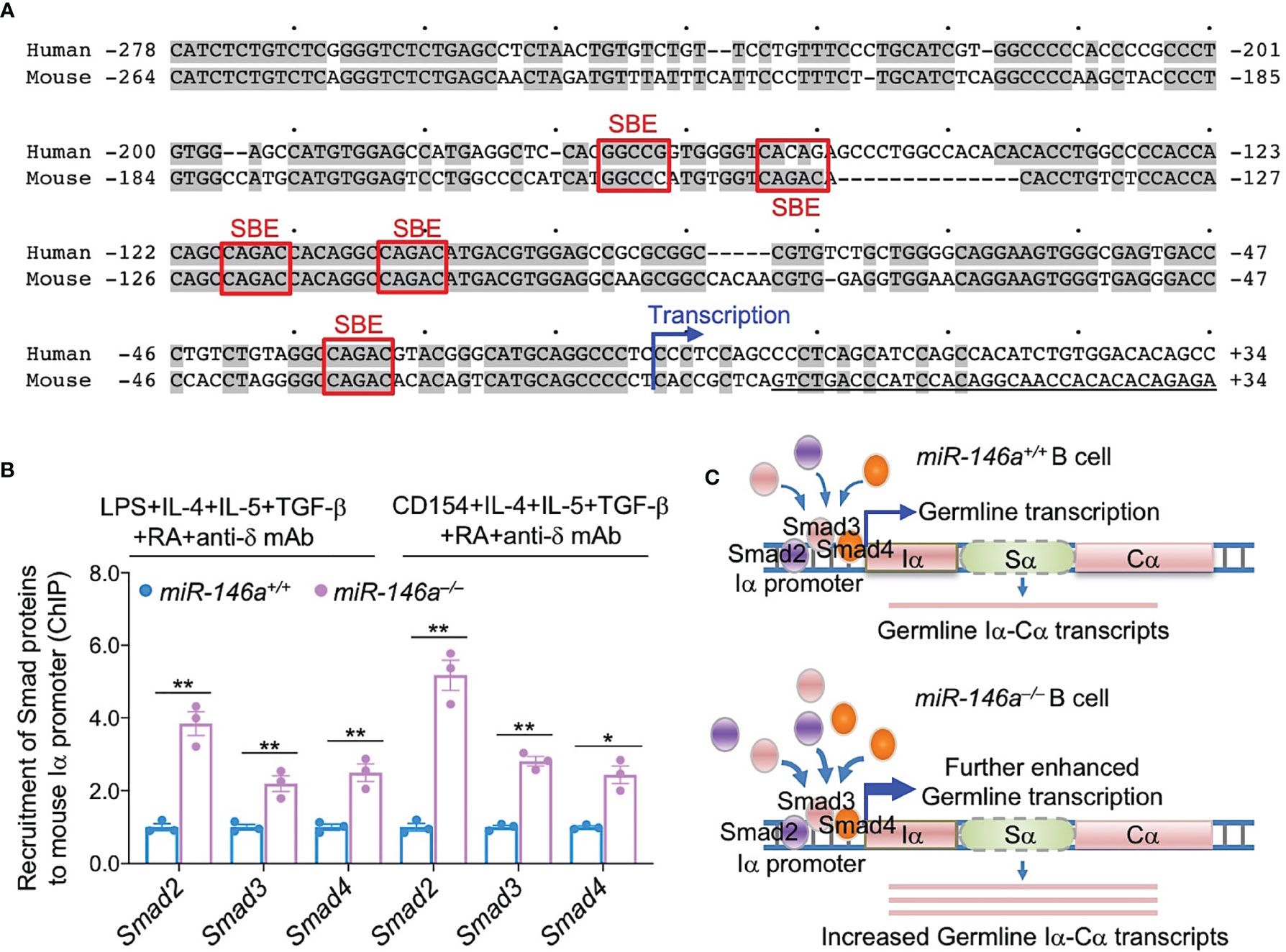
Figure 8 miR-146a deletion results in increased Smad2, Smad3 and Smad4 recruitment to Iα promoter. (A) Human and mouse Iα promoters share five SBEs, three of which are identical and conserved in mouse and human. (B) miR-146a–/– and miR-146a+/+ B cells were stimulated with LPS or CD154 plus IL-4, IL-5, TGF-β, anti-δ mAb and RA for 72 h. The recruitment of Smad2, Smad3 and Smad4 to the Iα promoter was analyzed by specific ChIP assays. Data are mean ± SEM from three independent experiments. **p < 0.01, *p < 0.05, unpaired t-test. (C) Cartoon depicting the increased availability of Smad2, Smad3 and Smad4 in miR-146a–/– B cells and increased recruitment of these transcription factors to the IgH locus Iα promoter.
Enforced Expression of B Cell miR-146a Reduces CSR to IgA
Having shown that mouse and human B cells physiologically downregulate miR-146a in response to IgA CSR-inducing stimuli to free Smad2, Smad3 and Smad4 genes from silencing by this miRNA and, therefore, allowing for greater expression of Smad2, Smad3 and Smad4 and recruitment of these transcription factors to the Iα promoter to effect germline Iα-Cα transcription, we reasoned that CSR to IgA would be inhibited by high levels of miR-146a. To this end, we enforced expression of miR-146a in LPS-primed B cells by transducing them with retroviral vector pMSCV-PIG-miR-146a (58) expressing GFP and miR-146a or a control pMSCV-PIG vector expressing GFP only. The transduced B cells were subsequently activated by LPS plus IL-4, IL-5, TGF-β, anti-δ mAb and RA, and cultured before being analyzed for expression of IgA, AID, Smad2, Smad3 and Smad4 proteins. The pMSCV-PIG-miR-146a retroviral vector-transduced B cells showed a significant reduction in Smad2, Smad3 and Smad4 expression and CSR to IgA as compared to the pMSCV-PIG retroviral vector-transduced B cells in the face of unchanged AID levels (Figure 9). Thus, miR-146a inhibits Smad2, Smad3 and Smad4 expression, thereby dampening CSR to IgA.
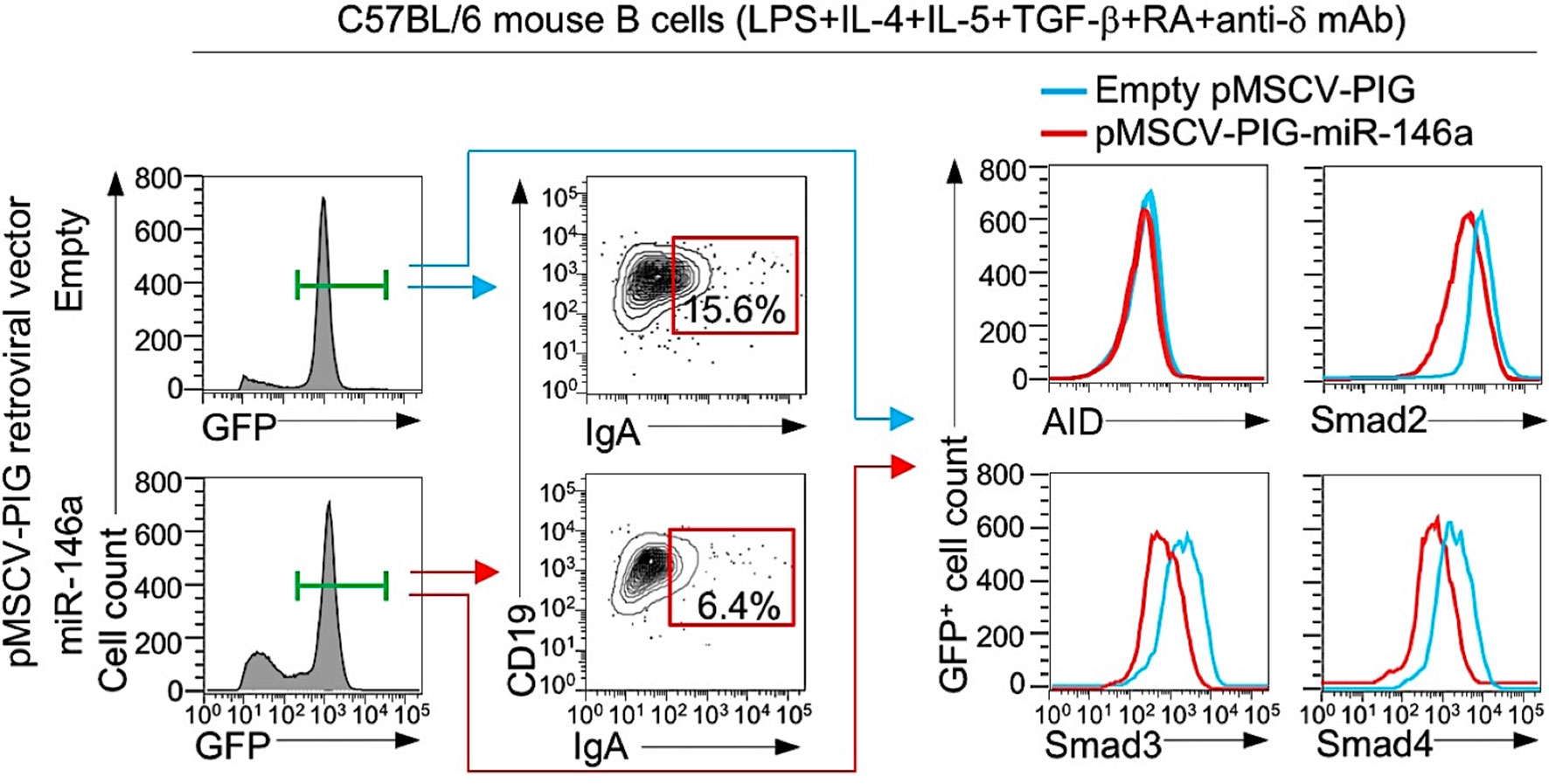
Figure 9 Enforced expression of miR-146a in B cells reduces Smad2, Smad3 and Smad4 expression and CSR to IgA. B cells isolated from C57BL/6 mice were transduced with pMSCV-PIG-miR-146a retroviral vector expressing GFP and miR-146a or empty pMSCV-PIG retroviral vector that expression GFP, then stimulated with LPS plus IL-4, IL-5, TGF-β, anti-δ mAb and RA and cultured for 96 h. Proportions of surface IgA+ B cells, and intracellular AID, Smad2, Smad3 and Smad4 levels among the retroviral vector-transduced (B220+GFP+) B cells were analyzed by flow cytometry. Data are from one representative of three independent experiments.
Discussion
IgA is the predominant antibody isotype found in mucous secretions. By maintaining a homeostatic environment at the mucosal surfaces, IgA provides a protective barrier against microbial pathogens and immune evasion. Dysregulation of CSR to IgA, however, can lead to IgA overproduction, such as in hyper IgA syndrome in which IgA kidney deposition leads to immunopathology. While many factors and trans-regulatory elements have been shown to control CSR, there is more to be learned about direct regulation of CSR to IgA, particularly with respect to the role of epigenetics factors, such as non-coding RNAs. Regulation of CSR to IgA must involve Smad2, Smad3 and Smad4. Indeed, these transcription factors provide the critical link between TGF-β signaling and the downstream activation of germline Iα-Cα transcription, the initiating event of CSR to IgA. Here we showed that miR-146a plays an important role in the epigenetic modulation of CSR to IgA by inhibiting Smad2, Smad3 and Smad4 expression. By using miR-146a–/– mice, we showed the lack of miR-146a leads to an enhancement of IgA responses at intestinal and systemic levels. By using mixed bone marrow μMT/miR-146a–/– chimeric mice, we further showed that the modulation of CSR to IgA by miR-146a is B cell-intrinsic. As miR-146a can be downregulated by Smad proteins (50), TGF-β-mediated activation of Smad proteins in B cells would lead to miR-146 reduction, which in turn would result in further expression of Smad proteins themselves. This positive Smads-miR-146a feedback would amplify TGF-β-directed CSR to IgA.
miR-146a would regulate the proliferation of certain immune cells, as suggested by the development of tumors in secondary lymphoid organs and myeloproliferation in aged miR-146a knock-out mice (44). Aberrant miR-146a expression has been identified in autoimmune diseases and disorders, including systemic lupus erythematosus, rheumatoid arthritis, Sjögren’s syndrome and inflammatory bowel disease (68). miR-146a would play a physiological role in cells of the innate and adaptive response (34, 38). It is highly expressed in Tfh and germinal center B cells and thought to restrain the responsiveness of these two immune cell populations (69). miR-146a has been shown to regulate germinal center response by targeting multiple genes associated with CD40 signaling pathway in B cells (39). In aged CD21-cre miR-146afl/fl mice, or CD21-cre miR-146afl/fl mice immunized (i.p.) with SRBCs or NP-KLH, miR-146a ablation in B cells led to increased germinal center formation and enhanced IgG production. miR-146a has been suggested to restrict the expansion of intestinal T cells and to reduce gut luminal IgA production (45). Indeed, as we showed here, miR-146a expression is lower in gut-associated lymphoid tissues, such as Peyer’s patches and mesenteric lymph nodes, which contain a large portion of IgA-producing cells, than in the spleen which is poorer in IgA-producing cells. miR-146a is generally an LPS-responsive gene, as induced by the NF-κB signaling pathway, particularly in innate immune cells, such as granulocytes, monocytes, macrophages and NK cells. Excessive induction miR-146a dampens Irak1 and Traf6 (34, 68, 70). Irak1 and Traf6 play a significant role in TLR signaling and, possibly, NF-κB activation (71, 72), leading to expression of AID or other genes central to peripheral B cell differentiation. Consistent with a role of miR-146a in targeting Irak1 and Traf6 mRNAs, our data showed that expression of Irak1 and Traf6 was increased in miR-146a–/– B cells. This, however, did not lead to alteration in Aicda or Prdm1 expression or germline Iγ1-Cγ1 and Iϵ-Cϵ transcripts, further emphasizing that miR-146a directly regulates CSR to IgA but not IgG1 or IgE. The direct modulation of CSR to IgA by miR-146a would provide a novel mechanism in miR-146a-mediated regulation of the antibody response in addition to miRNA146a role in regulation of germinal center formation (39). Upon immunization with OVA via intragastric gavage, miR-146a–/– or μMT/miR-146a–/– mice displayed significant increased IgA but not IgG1. In these miR-146a deficient mice, the lack of alteration in IgG1 production was associated with unchanged germline Iγ1-Cγ1 transcripts and might be due to the type of immunization, which is known to skew the antibody response toward IgA and not to elicit full spleen germinal centers.
TGF-β directs CSR to IgA by activating Smad2 and Smad3 transcription factors, which dimerize to Smad3/Smad3. Activated Smad3/Smad3 dimers recruit Smad4 and in conjunction with co-factors, such as Runx3 and Pu.1, would engage the Iα promoter of the IgH Cα gene through the five conserved SBEs we identified in this study, thereby inducing germline Iα-Cα transcription, the first and critical step leading to CSR to IgA (15, 17–21). Here we showed that miR-146a silences Smad2, Smad3 and Smad4 expression in B cells, thereby inhibiting CSR to IgA. We also showed that B cell miR-146a is profoundly downregulated by the stimulus that specifically directs CSR to IgA, i.e., TGF-β, thereby increasing Smad2/Smad3 and Smad4. The specificity of miR-146a downregulation by stimuli that induce Smad proteins and CSR to IgA was emphasized by high levels of miR-146a in human and mouse resting B cells as well as B cells activated by stimuli that are known not to direct CSR to IgA. It was further confirmed by the low levels of miR-146a in B cells undergoing CSR to IgA in vivo, as in MLNs and Peyer’s patches, as opposed to the high level of miR-146a in spleen B cells, among which B cells switching to IgA are a minor proportion of the overall B cell population. Besides miR-146a, Smad2, Smad3 and Smad4 may also be targeted by other miRNAs, such as miR-23a, miR-23b, miR-18, miR-27, miR-214 and miR-21 (73). However, virtually all these miRNAs are expressed at low level and are unchanged in B cells undergoing CSR to IgA (our unpublished high throughput miRNA-Seq data), and, therefore, may not play a significant role in the regulation of such CSR.
The mechanism underlying TGF-β-mediated downregulation of miR-146a is not clear. We suggest here that such downregulation is effected by Smad proteins. These have been shown to modulate miRNA expression through both transcriptional and post-transcriptional mechanisms (74–76), and overexpression of Smad3 and Samd4 has been reported to suppress miR-146 (50). We would argue that in B cells undergoing CSR to IgA, Smad2/Smad3 and Smad4 suppression of miR-146 levels frees Smad2, Smad3 and Smad4 from this miRNA-mediated gene silencing, thereby further increasing Smad2, Smad3 and Smad4 expression. This would create an actively self-potentiating Smads-miR-146a loop starting with TGF-β-induction of activated Smads, Smads-mediated miR-146 repression, which results in increased Smad expression, leading to further miR-146a repression, increased Smad expression, and further potentiation of germline Iα-Cα transcription and CSR to IgA (Figure 10). This actively self-potentiating Smads-miR-146a loop would not rule out the possibility that repression of miR-146a in B cells induced to undergo CSR to IgA may also occur independently of Smad proteins, perhaps through “non-canonical” TGF-β signaling factors such as ERK, p38, RhoA and phosphoinositide 3 kinase (PI3K), which can also be activated by TGF-β (66, 77, 78). Smad-mediated and Smad-independent pathways of repression of miR-146 would not be mutually exclusive, while both dependent on and initiated by TGF-β signaling.
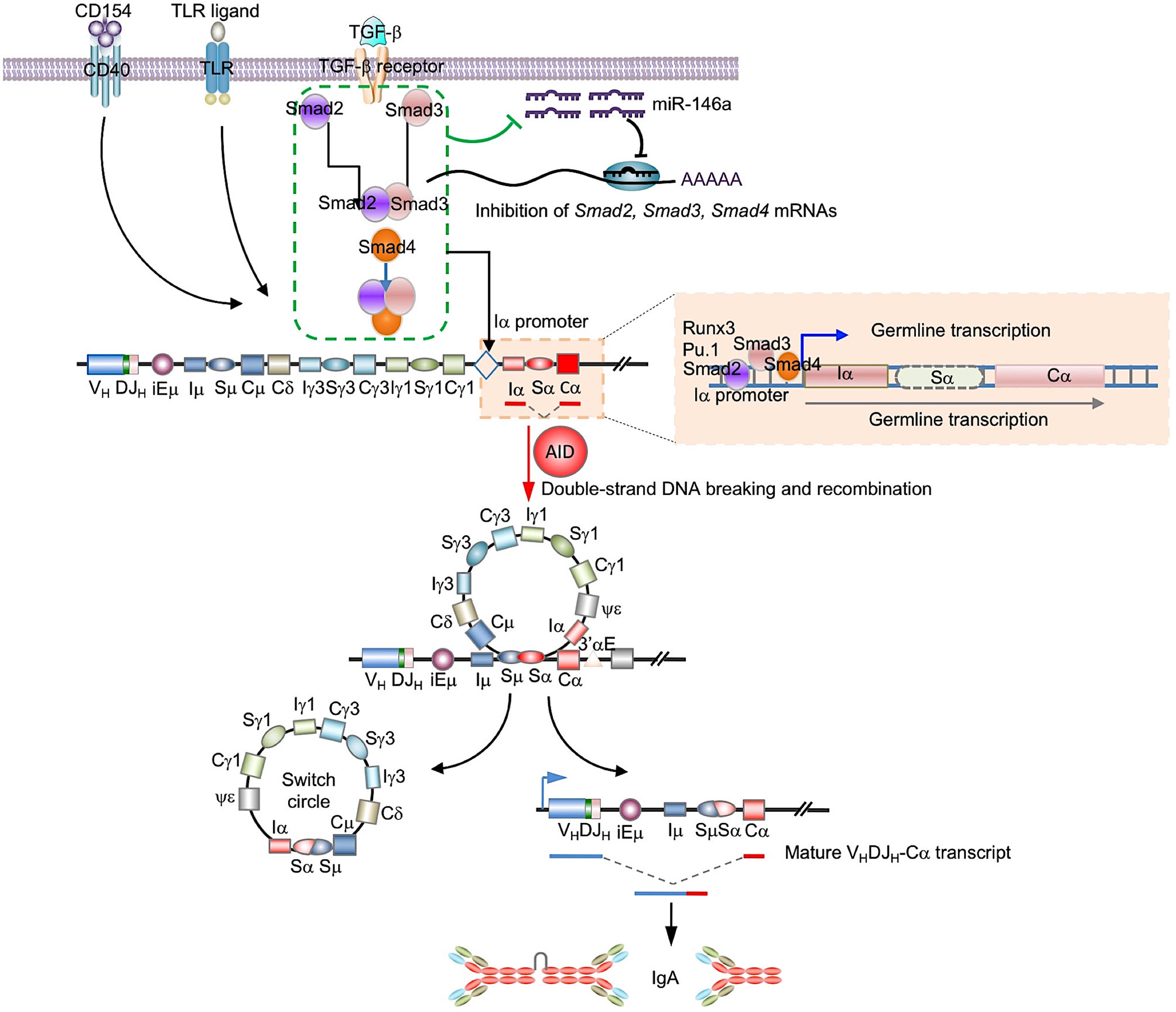
Figure 10 Epigenetic regulation of CSR to IgA by miR-146a. miR-146a targets the 3’UTRs of Smad2, Smad3 and Smad4 mRNAs to downregulate the expression of Smad2, Smad3 and Smad4, and reduce germline Iα-Cα transcription leading to a decreased CSR to IgA and IgA production. In B cells stimulation by TGF-β, activated Smad proteins lead to miR-146 reduction, which results in enhancement of expression of Smad proteins themselves. This miR-146a/Smad loop can amplify TGF-β-induced Smads promotion of CSR to IgA.
In the intestinal lumen, secretory IgA exist in dimeric form. Dimeric IgA bind to and ‘coat’ commensal bacteria, thereby playing a critical role in shaping the composition and functions of the gut microbiome (79). IgA can limit the access of bacteria to intestinal epithelial cells, facilitate bacterial clearance, regulate bacterial colonization and neutralize disease-associated bacteria (80). Humans and mice lacking IgA display gut microbiota dysbiosis, including reduced overall microbial diversity, altered bacterial composition and increased bacterial translocation (60, 81–84). The role of IgA in shaping the gut microbiota was originally thought to be limited to restriction of growth of specific organisms. Recent data, however, have suggested that IgA can also promote colonization and expansion of select bacterial species (85, 86). We show here that B cell-intrinsic miR-146a ablation, as in µMT/miR-146a–/– mice, led to increased production of systemic and intestinal IgA including bacteria-bound IgA (in feces), as compared to µMT/miR-146a+/+ mice, thereby resulting in a skwesed selection of gut microbial populations in the absence of miR-146a. Verrucomicrobiae class bacteria, in particularly, those of the Akkermansia genus, are common members of the human intestinal microbiota and were significantly increased in µMT/miR-146a–/– mice, in which IgA were elevated. Indeed, such bacteria and have been shown to be positively correlate with IgA level and to be highly coated with IgA (59–61, 83). However, Bacteroidaceae family bacteria, particularly those of the Bacteroides genus, which, in contrast with Akkermansia bacteria, are not generally bound by IgA and are typically abundant in the colon (59, 60), were also decreased in µMT/miR-146a–/– mice – some Bacteroides species have been shown to effectively induce production of gut IgA (87), which in turn, could possibly create a negative feedback to the very IgA-inducing bacteria. Similarly, the class Clostridia bacteria, including those of the Lachnospiraceae family and the Blautia genus, members of the phyla Firmicutes, which are also not bound by IgA antibodies in vivo (59, 60), were also not altered in µMT/miR-146a–/– mice. Thus, through regulation of IgA levels, B cell-intrinsic miR-146a can play an important role in modulating the steady state of the gut microbiome.
Dysregulated IgA levels and antibody responses are often associated with disease (88). IgA deficiency can lead to severe respiratory tract infections or increase the risk of adverse reactions to blood products (89). Abnormally high IgA levels may cause immune disorders, such as chronic rheumatic disease or chronic gastrointestinal inflammation (88). Here, we demonstrated that miR-146a plays an important role in keeping in check Smad2, Smad3 and Smad4 expression and, thereby, inhibiting the TGF-β signaling pathway-dependent induction of CSR to IgA, which results in a significant modulation of the IgA antibody response. Conversely, deletion of miR-146a, as in miR-146a−/− mice, increased systemic and gut IgA, thereby leading to IgA deposition in kidney glomeruli and immunopathology. This is highly evocative of the occurrence of miR-146a single nucleotide polymorphism rs2910164 in childhood hyper IgA syndrome and IgA nephropathy (47). Similarly, high serum IgA levels, as in “hyper IgA” mice, have been shown to result in IgA nephropathy, due to IgA kidney deposition leading to chronic inflammation (90). Interestingly, high IgA levels also occur in most cases of hyperimmunoglobulinemia D syndrome (HIDS), an autosomal recessive disorder characterized by abdominal, articular and skin manifestations in conjunction with recurrent febrile attacks (91). In HIDS patients, high IgA concentrations stem from abnormally high IgA1 levels, which correlate with systemically high IgD levels. While mutations in the mevalonate kinase gene underpin the vast majority of HIDS cases, the root of systemic hyper IgA in HIDS patients remains unknown. By providing a first mechanistic and important insight into epigentic regulation of CSR to IgA expression, our findings open new avenues of investigation of the role of miR-146a-mediated IgA dysregulation in a variety of hyper IgA conditions, such as those in HIDS, “idiopathic” hyper IgA, autoimmune and non-autoimmune inflammatory diseases, cancer and select metabolic disorders.
Data Availability Statement
16S rRNA gene-sequencing data have been deposited in NCBI Sequence Read Archive (SRA) under BioProject PRJNA777603. All the other data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by The Institutional Animal Care and Use Committees (IACUC) of the University of Texas Health San Antonio.
Author Contributions
PC conceived and designed the study, planned the experiments, analyzed data, supervised the work, implemented the execution of the overall experimental plan, created figures and wrote the manuscript. SL, GM, CD, DC, and AF, each performed one-two experiments. HZ designed the study, planned the experiments, analyzed data, supervised the work, created figures and wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH grants AI 105813, AI 079705, AI 138944, AI 167416 and the Lupus Research Alliance Target Identification in Lupus Grant ALR 641363 to PC.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to thank Dr. Justin B. Moroney and Dr. Helia N. Sanchez for their assistance in some experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.761450/full#supplementary-material
Supplementary Table 1 | PCR primers used in this study.
Abbreviations
AID, activation-induced cytidine deaminase; ASC, antibody secreting cells; CSR, class switch DNA recombination; IgH, immunoglobulin heavy chain; miRNA, microRNA; OVA, ovalbumin; SBE, Smad-binding element.
References
1. Cerutti A, Chen K, Chorny A. Immunoglobulin Responses at the Mucosal Interface. Annu Rev Immunol (2011) 29:273–93. doi: 10.1146/annurev-immunol-031210-101317
2. Macpherson AJ, Geuking MB, McCoy KD. Homeland Security: IgA Immunity at the Frontiers of the Body. Trends Immunol (2012) 33:160–7. doi: 10.1016/j.it.2012.02.002
3. Cerutti A, Cols M, Gentile M, Cassis L, Barra CM, He B, et al. Regulation of Mucosal IgA Responses: Lessons From Primary Immunodeficiencies. Ann NY Acad Sci (2011) 1238:132–44. doi: 10.1111/j.1749-6632.2011.06266.x
4. Gutzeit C, Magri G, Cerutti A. Intestinal IgA Production and Its Role in Host-Microbe Interaction. Immunol Rev (2014) 260:76–85. doi: 10.1111/imr.12189
5. Chen K, Magri G, Grasset EK, Cerutti A. Rethinking Mucosal Antibody Responses: IgM, IgG and IgD Join IgA. Nat Rev Immunol (2020) 20:427–41. doi: 10.1038/s41577-019-0261-1
6. Danger R, Braza F, Giral M, Soulillou JP, Brouard S. MicroRNAs, Major Players in B Cells Homeostasis and Function. Front Immunol (2014) 5:98. doi: 10.3389/fimmu.2014.00098
7. Macpherson AJ, McCoy KD, Johansen FE, Brandtzaeg P. The Immune Geography of IgA Induction and Function. Mucosal Immunol (2008) 1:11–22. doi: 10.1038/mi.2007.6
8. Cerutti A. The Regulation of IgA Class Switching. Nat Rev Immunol (2008) 8:421–34. doi: 10.1038/nri2322
9. Xu Z, Zan H, Pone EJ, Mai T, Casali P. Immunoglobulin Class-Switch DNA Recombination: Induction, Targeting and Beyond. Nat Rev Immunol (2012) 12:517–31. doi: 10.1038/nri3216
10. Zan H, Casali P. Regulation of Aicda Expression and AID Activity. Autoimmunity (2013) 46:83–101. doi: 10.3109/08916934.2012.749244
11. Zan H, Casali P. Epigenetics of Peripheral B-Cell Differentiation and the Antibody Response. Front Immunol (2015) 6:631. doi: 10.3389/fimmu.2015.00631
12. Shi Y, Massague J. Mechanisms of TGF-β Signaling From Cell Membrane to the Nucleus. Cell (2003) 113:685–700. doi: 10.1016/s0092-8674(03)00432-x
13. Batlle E, Massague J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
14. Shi MJ, Stavnezer J. CBF α3 (AML2) Is Induced by TGF-β1 to Bind and Activate the Mouse Germline Ig α Promoter. J Immunol (1998) 161:6751–60.
15. Park SR, Lee JH, Kim PH. Smad3 and Smad4 Mediate Transforming Growth Factor-β1-Induced IgA Expression in Murine B Lymphocytes. Eur J Immunol (2001) 31:1706–15. doi: 10.1002/1521-4141(200106)31:6<1706::AID-IMMU1706>3.0.CO;2-Z
16. Budi EH, Duan D, Derynck R. Transforming Growth Factor-β Receptors and Smads: Regulatory Complexity and Functional Versatilit. Trends Cell Biol (2017) 27:658–72. doi: 10.1016/j.tcb.2017.04.005
17. Shi MJ, Park SR, Kim PH, Stavnezer J. Roles of Ets Proteins, NF-κb and Nocodazole in Regulating Induction of Transcription of Mouse Germline Igα RNA by Transforming Growth Factor-β1. Int Immunol (2001) 13:733–46. doi: 10.1093/intimm/13.6.733
18. Zhang Y, Derynck R. Transcriptional Regulation of the Transforming Growth Factor-β-Inducible Mouse Germ Line Ig α Constant Region Gene by Functional Cooperation of Smad, CREB, and AML Family Members. J Biol Chem (2000) 275:16979–85. doi: 10.1074/jbc.M001526200
19. Lin YC, Stavnezer J. Regulation of Transcription of the Germ-Line Igα Constant Region Gene by an ATF Element and by Novel Transforming Growth Factor-β1-Responsive Elements. J Immunol (1992) 149:2914–25.
20. Klein J, Ju W, Heyer J, Wittek B, Haneke T, Knaus P, et al. B Cell-Specific Deficiency for Smad2 In Vivo Leads to Defects in TGF-β-Directed IgA Switching and Changes in B Cell Fate. J Immunol (2006) 176:2389–96. doi: 10.4049/jimmunol.176.4.2389
21. Zan H, Cerutti A, Dramitinos P, Schaffer A, Casali P. CD40 Engagement Triggers Switching to IgA1 and IgA2 in Human B Cells Through Induction of Endogenous TGF-β: Evidence for TGF-β But Not IL-10-Dependent Direct Sμ➝Sα and Sequential Sμ➝Sγ, Sγ➝Sα DNA Recombination. J Immunol (1998) 161:5217–25.
22. Hall JA, Grainger JR, Spencer SP, Belkaid Y. The Role of Retinoic Acid in Tolerance and Immunity. Immunity (2011) 35:13–22. doi: 10.1016/j.immuni.2011.07.002
23. Sanchez HN, Moroney JB, Gan H, Shen T, Im JL, Li T, et al. B Cell-Intrinsic Epigenetic Modulation of Antibody Responses by Dietary Fiber-Derived Short-Chain Fatty Acids. Nat Commun (2020) 11:60. doi: 10.1038/s41467-019-13603-6
24. Gan H, Shen T, Chupp DP, Taylor JR, Sanchez HN, Li X, et al. B Cell Sirt1 Deacetylates Histone and Non-Histone Proteins for Epigenetic Modulation of AID Expression and the Antibody Response. Sci Adv (2020) 6:eaay2793. doi: 10.1126/sciadv.aay2793
25. Moroney JB, Vasudev A, Pertsemlidis A, Zan H, Casali P. Integrative Transcriptome and Chromatin Landscape Analysis Reveals Distinct Epigenetic Regulations in Human Memory B Cells. Nat Commun (2020) 11:5435. doi: 10.1038/s41467-020-19242-6
26. Moroney JB, Chupp DP, Xu Z, Zan H, Casali P. Epigenetics of the Antibody and Autoantibody Response. Curr Opin Immunol (2020) 67:75–86. doi: 10.1016/j.coi.2020.09.004
27. Zan H, Tat C, Casali P. MicroRNAs in Lupus. Autoimmunity (2014) 47:272–85. doi: 10.3109/08916934.2014.915955
28. White CA, Pone EJ, Lam T, Tat C, Hayama KL, Li G, et al. Histone Deacetylase Inhibitors Upregulate B Cell microRNAs That Silence AID and Blimp-1 Expression for Epigenetic Modulation of Antibody and Autoantibody Responses. J Immunol (2014) 193:5933–50. doi: 10.4049/jimmunol.1401702
29. Shen T, Sanchez HN, Zan H, Casali P. Genome-Wide Analysis Reveals Selective Modulation of microRNAs and mRNAs by Histone Deacetylase Inhibitor in B Cells Induced to Undergo Class-Switch DNA Recombination and Plasma Cell Differentiation. Front Immunol (2015) 6:627. doi: 10.3389/fimmu.2015.00627
30. Casali P, Shen T, Xu Y, Qiu Z, Chupp DP, Im J, et al. Estrogen Reverses HDAC Inhibitor-Mediated Repression of Aicda and Class-Switching in Antibody and Autoantibody Responses by Downregulation of miR-26a. Front Immunol (2020) 11:491. doi: 10.3389/fimmu.2020.00491
31. de Yebenes VG, Bartolome-Izquierdo N, Ramiro AR. Regulation of B-Cell Development and Function by microRNAs. Immunol Rev (2013) 253:25–39. doi: 10.1111/imr.12046
32. Zheng B, Xi Z, Liu R, Yin W, Sui Z, Ren B, et al. The Function of microRNAs in B-Cell Development, Lymphoma, and Their Potential in Clinical Practice. Front Immunol (2018) 9:936. doi: 10.3389/fimmu.2018.00936
33. O'Connell RM, Rao DS, Baltimore D. microRNA Regulation of Inflammatory Responses. Annu Rev Immunol (2012) 30:295–312. doi: 10.1146/annurev-immunol-020711-075013
34. Saba R, Sorensen DL, Booth SA. MicroRNA-146a: A Dominant, Negative Regulator of the Innate Immune Response. Front Immunol (2014) 5:578. doi: 10.3389/fimmu.2014.00578
35. Mehta A, Baltimore D. MicroRNAs as Regulatory Elements in Immune System Logic. Nat Rev Immunol (2016) 16:279–94. doi: 10.1038/nri.2016.40
36. Yang L, Boldin MP, Yu Y, Liu CS, Ea CK, Ramakrishnan P, et al. miR-146a Controls the Resolution of T Cell Responses in Mice. J Exp Med (2012) 209:1655–70. doi: 10.1084/jem.20112218
37. Rusca N, Deho L, Montagner S, Zielinski CE, Sica A, Sallusto F, et al. MiR-146a and NF-κb1 Regulate Mast Cell Survival and T Lymphocyte Differentiat. Mol Cell Biol (2012) 32:4432–44. doi: 10.1128/MCB.00824-12
38. Testa U, Pelosi E, Castelli G, Labbaye C. miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development. Noncoding RNA (2017) 3:22. doi: 10.3390/ncrna3030022
39. Cho S, Lee HM, Yu IS, Choi YS, Huang HY, Hashemifar SS, et al. Differential Cell-Intrinsic Regulations of Germinal Center B and T Cells by miR-146a and miR-146b. Nat Commun (2018) 9:2757. doi: 10.1038/s41467-018-05196-3
40. Li B, Wang X, Choi IY, Wang YC, Liu S, Pham AT, et al. miR-146a Modulates Autoreactive Th17 Cell Differentiation and Regulates Organ-Specific Autoimmunity. J Clin Invest (2017) 127:3702–16. doi: 10.1172/JCI94012
41. Li Z, Zhang S, Wan Y, Cai M, Wang W, Zhu Y, et al. MicroRNA-146a Overexpression Impairs the Positive Selection During T Cell Development. Front Immunol (2017) 8:2006. doi: 10.3389/fimmu.2017.02006
42. Schmolka N, Papotto PH, Romero PV, Amado T, Enguita FJ, Amorim A, et al. MicroRNA-146a Controls Functional Plasticity in Gammadelta T Cells by Targeting NOD1. Sci Immunol (2018) 3:eaao1392. doi: 10.1126/sciimmunol.aao1392
43. Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-κb-Dependent Induction of microRNA miR-146, an Inhibitor Targeted to Signaling Proteins of Innate Immune Responses. Proc Natl Acad Sci USA (2006) 103:12481–6. doi: 10.1073/pnas.0605298103
44. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a Is a Significant Brake on Autoimmunity, Myeloproliferation, and Cancer in Mice. J Exp Med (2011) 208:1189–201. doi: 10.1084/jem.20101823
45. Runtsch MC, Hu R, Alexander M, Wallace J, Kagele D, Petersen C, et al. MicroRNA-146a Constrains Multiple Parameters of Intestinal Immunity and Increases Susceptibility to DSS Colitis. Oncotarget (2015) 6:28556–72. doi: 10.18632/oncotarget.5597
46. Amrouche L, You S, Sauvaget V, Manda V, Lamarthee B, Desbuissons G, et al. MicroRNA-146a-Deficient Mice Develop Immune Complex Glomerulonephritis. Sci Rep (2019) 9:15597. doi: 10.1038/s41598-019-51985-1
47. Lin J, Huang Y, Zhang X, Chen J, Sheng H. Association of miR-146a Rs2910164 With Childhood IgA Nephropathy. Pediatr Nephrol (2014) 29:1979–86. doi: 10.1007/s00467-014-2818-3
48. Kaidonis G, Gillies MC, Abhary S, Liu E, Essex RW, Chang JH, et al. A Single-Nucleotide Polymorphism in the microRNA-146a Gene Is Associated With Diabetic Nephropathy and Sight-Threatening Diabetic Retinopathy in Caucasian Patients. Acta Diabetol (2016) 53:643–50. doi: 10.1007/s00592-016-0850-4
49. Jazdzewski K, Murray EL, Franssila K, Jarzab B, Schoenberg DR, de la Chapelle A. Common SNP in pre-miR-146a Decreases Mature miR Expression and Predisposes to Papillary Thyroid Carcinoma. Proc Natl Acad Sci USA (2008) 105:7269–74. doi: 10.1073/pnas.0802682105
50. Sun Y, Li Y, Wang H, Li H, Liu S, Chen J, et al. miR-146a-5p Acts as a Negative Regulator of TGF-β Signaling in Skeletal Muscle After Acute Contusion. Acta Biochim Biophys Sin (2017) 49:628–34. doi: 10.1093/abbs/gmx052
51. Zhong H, Wang HR, Yang S, Zhong JH, Wang T, Wang C, et al. Targeting Smad4 Links microRNA-146a to the TGF-β Pathway During Retinoid Acid Induction in Acute Promyelocytic Leukemia Cell Line. Int J Hematol (2010) 92:129–35. doi: 10.1007/s12185-010-0626-5
52. Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, et al. Function of miR-146a in Controlling Treg Cell-Mediated Regulation of Th1 Responses. Cell (2010) 142:914–29. doi: 10.1016/j.cell.2010.08.012
53. Kitamura D, Roes J, Kuhn R, Rajewsky K. A B Cell-Deficient Mouse by Targeted Disruption of the Membrane Exon of the Immunoglobulin Mu Chain Gene. Nature (1991) 350:423–6. doi: 10.1038/350423a0
54. Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, et al. HoxC4 Binds to the Promoter of the Cytidine Deaminase AID Gene to Induce AID Expression, Class-Switch DNA Recombination and Somatic Hypermutation. Nat Immunol (2009) 10:540–50. doi: 10.1038/ni.1725
55. Zan H, White CA, Thomas LM, Mai T, Li G, Xu Z, et al. Rev1 Recruits Ung to Switch Regions and Enhances dU Glycosylation for Immunoglobulin Class Switch DNA Recombination. Cell Rep (2012) 2:1220–32. doi: 10.1016/j.celrep.2012.09.029
56. Zan H, Tat C, Qiu Z, Taylor JR, Guerrero JA, Shen T, et al. Rad52 Competes With Ku70/Ku86 for Binding to S-Region DSB Ends to Modulate Antibody Class-Switch DNA Recombination. Nat Commun (2017) 8:14244. doi: 10.1038/ncomms14244
57. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences Into the New Bacterial Taxonomy. Appl Environ Microbiol (2007) 73:5261–7. doi: 10.1128/AEM.00062-07
58. Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, et al. Widespread microRNA Repression by Myc Contributes to Tumorigenesis. Nat Genet (2008) 40:43–50. doi: 10.1038/ng.2007.30
59. Planer JD, Peng Y, Kau AL, Blanton LV, Ndao IM, Tarr PI, et al. Development of the Gut Microbiota and Mucosal IgA Responses in Twins and Gnotobiotic Mice. Nature (2016) 534:263–6. doi: 10.1038/nature17940
60. Bunker JJ, Bendelac A. IgA Responses to Microbiota. Immunity (2018) 49:211–24. doi: 10.1016/j.immuni.2018.08.011
61. Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A, et al. What Is the Healthy Gut Microbiota Composition? A Changing Ecosystem Across Age, Environment, Diet, and Diseases. Microorganisms (2019) 7:14. doi: 10.3390/microorganisms7010014
62. Stavnezer J, Kang J. The Surprising Discovery That TGF-β Specifically Induces the IgA Class Switch. J Immunol (2009) 182:5–7. doi: 10.4049/jimmunol.182.1.5
63. Li J, Huang J, Dai L, Yu D, Chen Q, Zhang X, et al. miR-146a, an IL-1β Responsive miRNA, Induces Vascular Endothelial Growth Factor and Chondrocyte Apoptosis by Targeting Smad4. Arthritis Res Ther (2012) 14:R75. doi: 10.1186/ar3798
64. Liu Z, Lu CL, Cui LP, Hu YL, Yu Q, Jiang Y, et al. MicroRNA-146a Modulates TGF-β1-Induced Phenotypic Differentiation in Human Dermal Fibroblasts by Targeting SMAD4. Arch Dermatol Res (2012) 304:195–202. doi: 10.1007/s00403-011-1178-0
65. Zhang Q, Cai R, Tang G, Zhang W, Pang W. MiR-146a-5p Targeting SMAD4 and TRAF6 Inhibits Adipogenensis Through TGF-β and AKT/mTORC1 Signal Pathways in Porcine Intramuscular Preadipocytes. J Anim Sci Biotechnol (2021) 12:12. doi: 10.1186/s40104-020-00525-3
66. Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE. TRAF6 Mediates Smad-Independent Activation of JNK and P38 by TGF-β. Mol Cell (2008) 31:918–24. doi: 10.1016/j.molcel.2008.09.002
67. Martin-Malpartida P, Batet M, Kaczmarska Z, Freier R, Gomes T, Aragon E, et al. Structural Basis for Genome Wide Recognition of 5-Bp GC Motifs by SMAD Transcription Factors. Nat Commun (2017) 8:2070. doi: 10.1038/s41467-017-02054-6
68. Mortazavi-Jahromi SS, Aslani M, Mirshafiey A. A Comprehensive Review on miR-146a Molecular Mechanisms in a Wide Spectrum of Immune and Non-Immune Inflammatory Diseases. Immunol Lett (2020) 227:8–27. doi: 10.1016/j.imlet.2020.07.008
69. Kuchen S, Resch W, Yamane A, Kuo N, Li Z, Chakraborty T, et al. Regulation of microRNA Expression and Abundance During Lymphopoiesis. Immunity (2010) 32:828–39. doi: 10.1016/j.immuni.2010.05.009
70. Park H, Huang X, Lu C, Cairo MS, Zhou X. MicroRNA-146a and microRNA-146b Regulate Human Dendritic Cell Apoptosis and Cytokine Production by Targeting TRAF6 and IRAK1 Proteins. J Biol Chem (2015) 290:2831–41. doi: 10.1074/jbc.M114.591420
71. Kawai T, Akira S. Signaling to NF-κB by Toll-Like Receptors. Trends Mol Med (2007) 13:460–9. doi: 10.1016/j.molmed.2007.09.002
72. Muroi M, Tanamoto K. TRAF6 Distinctively Mediates MyD88- and IRAK-1-Induced Activation of NF-κB. J Leukoc Biol (2008) 83:702–7. doi: 10.1189/jlb.0907629
73. Hata A, Chen YG. TGF-β Signaling From Receptors to Smads. Cold Spring Harb Perspect Biol (2016) 8:a022061. doi: 10.1101/cshperspect.a022061
74. Massague J, Chen YG. Controlling TGF-β Signaling. Genes Dev (2000) 14:627–44. doi: 10.1101/gad.14.6.627
75. Davis BN, Hilyard AC, Lagna G, Hata A. SMAD Proteins Control DROSHA-Mediated microRNA Maturation. Nature (2008) 454:56–61. doi: 10.1038/nature07086
76. Blahna MT, Hata A. Smad-Mediated Regulation of microRNA Biosynthesis. FEBS Lett (2012) 586:1906–12. doi: 10.1016/j.febslet.2012.01.041
77. Guo L, Zhang Y, Zhang L, Huang F, Li J, Wang S. MicroRNAs, TGF-β Signaling, and the Inflammatory Microenvironment in Cancer. Tumour Biol (2016) 37:115–25. doi: 10.1007/s13277-015-4374-2
78. Mu Y, Gudey SK, Landstrom M. Non-Smad Signaling Pathways. Cell Tissue Res (2012) 347:11–20. doi: 10.1007/s00441-011-1201-y
79. Yang Y, Palm NW. Immunoglobulin A and the Microbiome. Curr Opin Microbiol (2020) 56:89–96. doi: 10.1016/j.mib.2020.08.003
80. Sutherland DB, Suzuki K, Fagarasan S. Fostering of Advanced Mutualism With Gut Microbiota by Immunoglobulin A. Immunol Rev (2016) 270:20–31. doi: 10.1111/imr.12384
81. Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, Chou WC, et al. Gut Microbiota Utilize Immunoglobulin A for Mucosal Colonization. Science (2018) 360:795–800. doi: 10.1126/science.aaq0926
82. Macpherson AJ, Yilmaz B, Limenitakis JP, Ganal-Vonarburg SC. IgA Function in Relation to the Intestinal Microbiota. Annu Rev Immunol (2018) 36:359–81. doi: 10.1146/annurev-immunol-042617-053238
83. Catanzaro JR, Strauss JD, Bielecka A, Porto AF, Lobo FM, Urban A, et al. IgA-Deficient Humans Exhibit Gut Microbiota Dysbiosis Despite Secretion of Compensatory IgM. Sci Rep (2019) 9:13574. doi: 10.1038/s41598-019-49923-2
84. Huus KE, Petersen C, Finlay BB. Diversity and Dynamism of IgA-Microbiota Interactions. Nat Rev Immunol (2021) 21:514–25. doi: 10.1038/s41577-021-00506-1
85. Kubinak JL, Round JL. Do Antibodies Select a Healthy Microbiota? Nat Rev Immunol (2016) 16:767–74. doi: 10.1038/nri.2016.114
86. Schofield WB, Palm NW. Gut Microbiota: IgA Protects the Pioneers. Curr Biol (2018) 28:R1117–9. doi: 10.1016/j.cub.2018.08.019
87. Yang C, Mogno I, Contijoch EJ, Borgerding JN, Aggarwala V, Li Z, et al. Fecal IgA Levels Are Determined by Strain-Level Differences in Bacteroides Ovatus and Are Modifiable by Gut Microbiota Manipulation. Cell Host Microbe (2020) 27:467–75.e6. doi: 10.1016/j.chom.2020.01.016
88. Copetti V, Pastore S, De Pieri C, Radillo O, Taddio A, Ventura A, et al. Clinical Significance of Hyper-IgA in a Paediatric Laboratory Series. Arch Dis Child (2014) 99:1114–6. doi: 10.1136/archdischild-2014-306607
89. Rawla P, Joseph N. IgA Deficiency. In: StatPearls. Treasure Island (FL): StatPearls Publishing (2021).
90. Kamata T, Nogaki F, Fagarasan S, Sakiyama T, Kobayashi I, Miyawaki S, et al. Increased Frequency of Surface IgA-Positive Plasma Cells in the Intestinal Lamina Propria and Decreased IgA Excretion in Hyper IgA (HIGA) Mice, a Murine Model of IgA Nephropathy With Hyperserum IgA. J Immunol (2000) 165:1387–94. doi: 10.4049/jimmunol.165.3.1387
Keywords: AID, B cell, class switch DNA recombination (CSR), epigenetics, IgA, miR-146a, microRNA, Smad2/Smad3/Smad4
Citation: Casali P, Li S, Morales G, Daw CC, Chupp DP, Fisher AD and Zan H (2021) Epigenetic Modulation of Class-Switch DNA Recombination to IgA by miR-146a Through Downregulation of Smad2, Smad3 and Smad4. Front. Immunol. 12:761450. doi: 10.3389/fimmu.2021.761450
Received: 19 August 2021; Accepted: 19 October 2021;
Published: 16 November 2021.
Edited by:
Deborah K. Dunn-Walters, University of Surrey, United KingdomReviewed by:
Egest James Pone, University of California, Irvine, United StatesLori Ruth Covey, Rutgers, The State University of New Jersey - Busch Campus, United States
Copyright © 2021 Casali, Li, Morales, Daw, Chupp, Fisher and Zan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paolo Casali, cGNhc2FsaUB1dGhzY3NhLmVkdQ==; Hong Zan, emFuQHV0aHNjc2EuZWR1
†SL was an exchange student from the Department of Immunology, School of Basic Medical Sciences, Central South University, Changsha, China