 Katiane Roversi1,2,3,4
Katiane Roversi1,2,3,4 Natalia Callai-Silva1,2,3Karine Roversi1
Natalia Callai-Silva1,2,3Karine Roversi1 May Griffith2,3Christos Boutopoulos2,3
May Griffith2,3Christos Boutopoulos2,3 Rui Daniel Prediger4*
Rui Daniel Prediger4* Sébastien Talbot1*
Sébastien Talbot1*- 1Département de Pharmacologie et Physiologie, Université de Montréal, Montréal, QC, Canada
- 2Centre de Recherche Hôpital Maisonneuve-Rosemont, Montréal, QC, Canada
- 3Département d’Ophtalmologie, Université de Montréal, Montréal, QC, Canada
- 4Departamento de Farmacologia, Universidade Federal de Santa Catarina, Florianópolis, Brazil
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, affecting 1–2% of the population aged 65 and over. Additionally, non-motor symptoms such as pain and gastrointestinal dysregulation are also common in PD. These impairments might stem from a dysregulation within the gut-brain axis that alters immunity and the inflammatory state and subsequently drives neurodegeneration. There is increasing evidence linking gut dysbiosis to the severity of PD’s motor symptoms as well as to somatosensory hypersensitivities. Altogether, these interdependent features highlight the urgency of reviewing the links between the onset of PD’s non-motor symptoms and gut immunity and whether such interplays drive the progression of PD. This review will shed light on maladaptive neuro-immune crosstalk in the context of gut dysbiosis and will posit that such deleterious interplays lead to PD-induced pain hypersensitivity.
Pain
Pain is defined as an unpleasant sensory and emotional experience associated with real or potential injury (1). Physiologically, pain serves as a protective mechanism, alerting the host to environmental danger. The sensation results from the integration of complex neurobiological systems that detect, integrate, and coordinate protective responses to noxious stimuli that threaten the host’s homeostasis and survival (2).
Nociceptors express various ion-channel receptors that are specialized to respond to threats posed by pathogens, allergens, and pollutants. Nociception is then initiated upon sensing these noxious stimuli by first-order neurons which, once activated by their cognate ligands, allow the influx of cations (Na+, Ca2+) leading to the generation of an action potential. These electrical signals are then propagated through the length of small, unmyelinated C or myelinated Aδ fibers to the spinal cord, where they synapse with second-order neurons (3). These electrical signals are then modulated—either amplified or blunted—by local immune cells or descending neurons.
The descending pathways originate from supraspinal structures such as the rostral ventromedial medulla (RVM), the dorsolateral pontomesencephalic tegmentum, and the periaqueductal gray matter (PAG). The descending pathways inhibit pain by releasing monoamines, such as dopamine, norepinephrine, and serotonin into the dorsal horn. Additionally, endogenous opioids exert descending inhibition of nociception (4).
Once modulated in the spinal cord, the nociceptive signal is processed in the supraspinal region and recognized as pain. The lateral pain system comprises the spinothalamic tract, which projects through the lateral thalamus and toward the sensory cortical areas, and is primarily involved in processing sensory discrimination, localization, and pain intensity. In contrast, the medial pain system processes the motivational-affective and cognitive-evaluative aspects of pain (e.g., unpleasantness, suffering) and projects through the medial thalamic nuclei toward the anterior cingulate cortex (5).
Chronic Pain
Chronic pain affects approximately 20% of the general population and individual of any age. It negatively impacts the patient’s quality of life and is also associated with mood and sleep disorders. It is considered the chief debilitating symptom of a variety of diseases, ranging from cancer to multiple sclerosis (6–8). While acute pain serves a physiological purpose, chronic pain is rarely self-resolving and remains resistant to pharmacological treatment (9). This pain can be either perceived more severely, a phenomenon known as hyperalgesia, or it can be generated by non-noxious stimuli, a condition known as allodynia.
Chronic pain results from persistent and repeated stimuli which can lead to peripheral and/or central sensitization of nociceptor neurons. Specifically, central sensitization is characterized by the persistent hyperexcitability of the central nervous system (CNS) circuitry triggered by excessive neuronal activity resulting from peripheral tissue inflammation or neuropathic injury. Central sensitization can result from i) modifications of glutamatergic receptors; ii) the upregulation of proteins involved in maintaining synaptic strength; iii) Aβ neuron sprouting; iv) decreased inhibitory control by GABAergic interneurons; and v) increased expression of activator ion-channel or neuropeptide receptors (10).
Secondary to the actions of pro-inflammatory cytokines released by microglia and astrocytes, these modifications increase the synaptic activity between first- and second-order neurons by altering the biophysical properties of danger-detecting ion-channel receptors and promoting the trafficking of these receptors to the synaptic membrane. Functionally, the sensitization is reflected by a long-term potentiation of the synaptic transmission between primary and second-order sensory neurons, rendering the CNS hypersensitive to normal (or previously innocuous) inputs. Clinically, this higher level of excitability maintains chronic pain (10–12).
Immunity Drives Pain
Along with detecting adverse temperature, pressure, and chemicals, nociceptor neurons express specific receptors for numerous immunoglobulins, cytokines, and chemokines. The nociceptor neurons are tuned to detect and respond to mediators derived from immunocytes (13–16). Typically, the binding of these sensitizing molecules generates intracellular signaling via tyrosine kinase or G-protein-coupled receptors (15, 17, 18). The second messengers downstream of these receptors trigger i) the phosphorylation and membrane expression of ion-channel receptors and voltage-gated sodium channels; and ii) the overproduction of neuropeptides and neurotransmitters (19–21).
A few well-known examples of immunocyte-releasing pain-sensitizing mediators include the action of interleukin 1 beta (IL-1β) (22–24), tumor necrosis factor (TNF-α) (25, 26), prostaglandin E2 (PGE2) (27), and nerve growth factor (NGF) (24). For example, PGE2 stimulates nociceptor-expressed DP2, which leads to an increase in protein kinase A (PKA) and protein kinase C (PKC) activity, which, in turn, phosphorylates transient receptor potential vanilloid 1 (TRPV1). Consequently, PGE2 increases capsaicin-induced currents, as found in cultured rat dorsal root ganglion (DRG) neurons (28, 29). In the case of NGF, when binding to its cognate receptor TrkA, it triggers PI3K/Src kinase activation which also leads to the phosphorylation of TRPV1 (18, 30).
These mechanisms stem from work done on models of rodents with nerve injury or auto-immune diseases. While no particular neuro-immune cascade appears to be the primary driver of the pain sensation, data show the involvement of central and peripheral innate (i.e. macrophages) and adaptive (i.e. T cells) immune cells. While typically studied in isolation, peripheral and central mechanisms should be studied concurrently. This is exemplified by the data showing that monocytes and microglia synergize in driving neuropathic pain in mice with nerve injuries. Thus, both types of cells need to be eliminated to alleviate pain. However, when used separately, monocyte depletion using clodronate liposomes or CX3CR1+ microglia ablation failed to impact the course of the disease (31).
In a model of chronic constriction injury (CCI), T cells are recruited to the sciatic nerve and induce mechanical allodynia and thermal hyperalgesia via the production of IL-17A and IFN-γ (32). Athymic nude male rats, which have no T cells, were protected. Upon nerve injury, T cells infiltrate the DRG and release leukocyte elastase (LE) which promotes mechanical allodynia (33–35) while spinal nerve transection-induced neuropathic pain was found to be mediated by TH1 cells released IFN-γ, TNF-α, and GM-CSF (36). Interestingly, when comparing the spinal dorsal horn gene profile of spared nerve injury (SNI) animals, T cells and glia seem predominately impacted in adult mice rather than younger animals. These data may indicate age-dependent neuromodulation by immune cells, which could explain why hypersensitivity seems to increase with age.
Neuro-immune crosstalks are not limited to the site of injury, as shown in models of chemotherapy and sciatic nerve ligation. Immunocytes (i.e., macrophages, monocytes, neutrophils, and T cells) infiltrate the DRG in a TLR2- or CCL-2-mediated fashion (37, 38). Once in the tissue, they release IL-1β and TNF-α, which can cause thermal hyperalgesia (39–44). In contrast, the targeted depletion of IL-10-producing monocytes and macrophages delayed pain resolution (45).
Aside from these traditional mechanisms, antibodies produced in auto-immune diseases can initiate pain. For instance, the injection of autoantibodies against citrullinated proteins (ACPA) purified from animals with rheumatoid arthritis promotes pain-like behavior without inflammation. This is achieved by acting on osteoclasts and inducing CXCL1, a human analog of IL-8 (46). In addition, IgG from patients with complex regional pain syndrome (CRPS) prolonged postsurgical hypersensitivity to mechanical, cold, and heat stimuli. Finally, skin-saphenous nerve preparations from tCRPS mice show increase sensitivity to auto-antibodies (47), which impairs the function of the potassium channel Kv1.2 and promotes mechanical hypersensitivity (48).
Another novel mechanism of pain modulation was described by Chen et al., who found that by binding to peripheral sensory neuron-expressed PD-1, the immune checkpoint ligand PD-L1 triggers the phosphorylation of SHP-1 and the downstream modulation of sodium and potassium channels. Consequently, PD-L1 suppressed excitatory synaptic transmission (sEPSC) in lamina II neurons of the spinal cord which trigger analgesia (49).
Beyond the commonly known Pattern recognition receptor (PRR) typically expressed by immune cells, Stimulator of interferon genes protein (STING1) was recently found to be abundant in TRPV1+ neurons (50, 51). Its activation by IFN-I ligands led to long-lasting analgesia by suppressing the excitability of nociceptors through the modulation of sodium and calcium channel function (51).
Microbes Induce Pain
The Gastrointestinal (GI) tract is innervated by intrinsic neurons from the enteric nervous system (ENS) and by the axons of extrinsic sympathetic, parasympathetic, and visceral afferent neurons (52, 53). The ENS is organized into two major neuronal networks—the myenteric plexus, and the submucosal plexus—and also comprises connective interneurons and various types of supporting glial cells. The ENS sympathetic (noradrenergic) neurons control blood vessel vasocontraction, while the parasympathetic (cholinergic) neurons control gut contraction (54).
The gut’s extrinsic innervation is made up of neurons originating from lumbar (DRG) and nodose (ND) ganglia. These neurons work to monitor GI volume and intestinal contents, while the gut hormones regulate the digestive physiology (55). The DRG neurons project along the mesenteric arteries, while the ND ganglion neurons project from the vagus nerve.
The vagus, which consists of ~2,300 sensory neurons, projects to half of the large intestine. The GI tract innervation accounts for ~20% of its terminals (56). Most of these extrinsic fibers (DRG and ND) express sensory neuron markers such as TRP channels (TRPV1) (57), voltage-gated sodium channels (NaV1.8) (58), and mechanosensitive channels (Piezo2) (59). These sensory neurons are designed to limit tissue damage by detecting and initiating protective reflexes (60, 61).
Under homeostatic conditions, the lumen of the intestine is not directly innervated, meaning that there is no direct neuron sensing of the luminal content (50, 62). The signals are sent by intestinal enteroendocrine cells which expressed glutamate receptors and can release a few neuropeptides (i.e., cholecystokinin, peptide YY (PYY)), consequently enabling the perception of the luminal content by vagal neurons (63). Upon penetrating the epithelial barrier, as in the case of a lesion or an infection, the proteases, reactive oxygen species, or cytokines produced by mucosa-resident immune cells may stimulate the ENS neurons (64, 65).
Different making in gut pathogens, as found in dysbiosis, is associated with headaches, chemotherapy-induced neuropathic pain, and abdominal pain. Staphylococcus aureus heightens sensory hypersensitivity via membrane-bound N-formylated peptides or by releasing various pore-forming toxins (i.e., alpha-hemolysin, phenol-soluble modulins, leukocidin). Conversely, gut nociceptor-released Calcitonin gene-related peptide (CGRP) regulates M cell density, limiting pathogen entry into the GI tract (66). TRPV1+ neurons also appear to be associated with mucosal resistance against Candida albicans. They do so by increasing CD301b+ dDCs release of IL-23 and subsequent production of IL-17A from γδ T cells (67). Finally, the recognition of soluble bacterial products by ENS axonal termini, as found in the microfluidic gut model, drive RORλ+ Treg induction and immunosuppression (68).
PD-Induced Pain
Pain is a non-motor symptom present in 60–85% of PD patients (6, 69, 70). PD-induced pain negatively impacts a patient’s quality of life (8) and may exacerbate other non-motor PD symptoms such as depression and sleep disorders (7). In approximately one-third of PD sufferers, pain precedes the onset of PD motor symptoms by several years (71, 72). Peripheral neuropathic pain is also twice as frequent in PD patients (73, 74). Conversely, patients experiencing chronic pain are at increased risk of developing PD (75).
Pain often manifests as a musculoskeletal hypersensitivity affecting the neck, arms, or paravertebral muscles. It is believed to be a consequence of PD motor symptoms. PD can also trigger visceral pain, which affects the internal organs and results from the activation of nociceptors localized in the thoracic and pelvic organs. Visceral pain is associated with gastrointestinal dysfunction, as seen in PD patients (76). Finally, neuropathic pain is observed in 4%–10% of PD patients (77–79) and is typically associated with lesions in the central or peripheral nervous system (80). Clinically, it presents as burning, cramping, aching, numbness, tingling, vibrating, or lancinating sensations. This type of pain may be associated with autonomic manifestations, and it often stems from the face, head, pharynx, epigastrium, abdomen, pelvis, rectum, and genitalia. This type of pain does not correlate with the severity of motor impairments and often precedes their onset (79, 81–84).
Despite its clinical relevance and predictive value, pain is often neglected or misdiagnosed in PD patients and remains poorly managed (85). The contribution of maladaptive peripheral (Peripheral Mechanisms of PD-Induced Pain) and central (Central Mechanisms of PD-Induced Pain) neuro-immune interplays as well as gut-brain axis dysregulation (sections GI Dysfunction in PD-Induced Pain and Dysbiosis in PD-Induced Pain) (86) to the induction of PD-induced pain will now be discussed.
Immunity in PD
Based on the overall role that neuro-immunity plays in sustaining sensory hypersensitivity (Immunity Drives Pain), we posit that neuronal loss or damage leads to the local recruitment of immune cells. It is well established that inflammation drives PD progression (87), involves innate and adaptive immune cells, and can occur in the peripheral or CNS. An indication that the immune system is responding to tissue damage stems from increased TNF-α, IL-1β, IL-2, IL-6, IFN-γ, and CCL2 levels observed in the blood and cerebrospinal fluid (CSF) of PD patients (88–91). These increases correlate with disease progression. For example, higher serum levels of TNF-α were linked to motor dysfunction, while raised levels of IL-1β and IL-2 were associated with cognitive decline (92).
This increase in circulating cytokines was secondary to a rise in the number of immunocytes in a patient’s bloodstream. As expected, the severity of the disease correlates with lower levels of naïve CD4+ T cells (93–95) but increased levels of blood-circulating Treg, activated CD4+ T cells, IL-17-producing TH17 cells, and IL-6-producing monocytes (Figure 1) (96–101). While this may sound counterintuitive, a study reported a decreased capability of PD-isolated Treg to suppress the activity of effector T cells in vitro (96, 97). In fact, the levels of blood cytokines found in PD patients correlate with an increase in sensory hypersensitivity. Thus, CD4+ T cells in patients experiencing pain have a lower IL-6/IL-10 ratio, while CD8+ T cells display a higher TNF-α/IL-10 ratio (102).
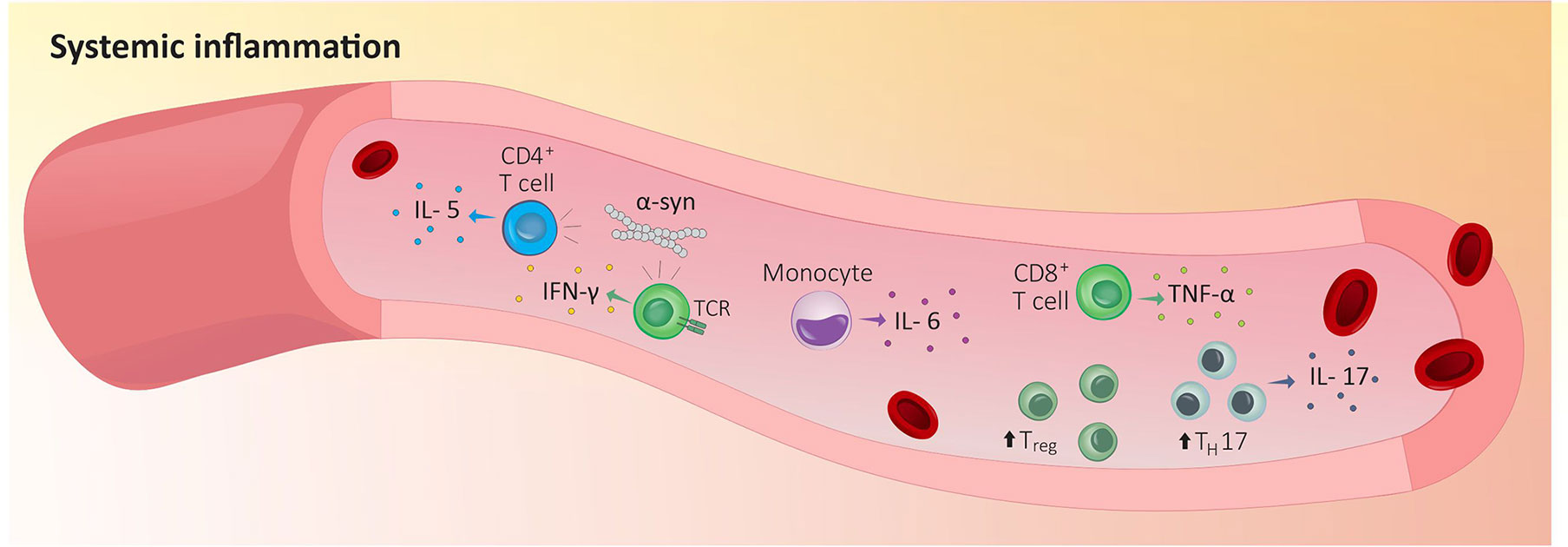
Figure 1 Peripheral inflammation in Parkinson’s disease. Indicators that the immune system is responding to tissue damage stems from increased blood-circulating Treg, activated CD4+ T cells, IL-17-producing TH17 cells and IL-6-producing monocytes in PD patients’ blood. In addition, circulating α-syn-reactive CD4 T cells are expanded in PD patients’ blood and blood purified CD4+ and CD8+ T cells from PD patients recognize and respond to α-syn which increase the production of IL-5 or IFN-γ. IFN-γ, interferon γ; IL-5, interleukin 5; IL-6, interleukin 6; IL-17, interleukin 17; TH17, T helper 17 cells; TCR, T cell receptor; TNF-α, tumor necrosis factor; Treg, regulatory T cells; α-syn, α-synuclein.
Peripheral Mechanisms of PD-Induced Pain
Compared with ~5% of the general population, 20%–60% of PD patients show large- and small-fiber PN (73, 103). Interestingly, the severity of large-fiber neuropathy is also a marker of PD severity (104). Skin biopsies of PD patients with sensory hypersensitivity revealed that α-synuclein aggregates in cutaneous sensory nerves and leads to their degeneration (105–109). In a study of 72 PD patients, damage found in Aδ sensory fibers correlated with the level of sensory hypersensitivity they experienced. As a functional indication for abnormal pain fiber inputs, hypersensitive PD patients have a lower pain threshold to electrical stimuli as well as a higher current perception threshold than their normosensitive counterparts (110).
By contrast, Nolano et al. found that PD patients (n=18 subjects) were generally hyposensitive. They explained these findings as the loss of epidermal nerve fibers and Meissner corpuscles, which translated into an increase in tactile and thermal thresholds (105). A more recent study by the same authors analyzed skin innervation in 85 PD patients and found a significant reduction in intraepidermal nerve fiber density. This phenotype was correlated with a decrease in the perception of mechanical pain (107). However, further clinical studies are needed to clarify any putative correlation between PD pain thresholds, peripheral nerve degeneration, levels of dermal α-syn, and immunocyte infiltration.
Circulating α-syn-reactive CD4+ T cells are expanded in PD patients’ blood (Figure 1) (111). When injected into the gastrocnemius muscle of PD mice (M83 model), α-Syn preformed fibrils (PFF) aggregate in the dorsal nerve roots and lumbar DRG sensory neurons, as well as the lumbar spinal cord, the midbrain PAG matter, and the thalamus. When α-Syn aggregates in sensory neurons, it decreases nerve conduction velocity, drives small- and medium-sized myelinated fiber pathology, and induces mechanical allodynia (112). Importantly, these alterations were observed in the absence of motor dysfunction.
Injecting 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP; i.p. 11 mg/kg, daily for five days), a pro-neurotoxin used to model PD, led to an increase in sensory neuron expression of NaV1.1, NaV1.7, NaV1.9. These alterations occurred 12 days after the first MPTP challenge and were accompanied by thermal hypersensitivity (confirmed by the hot plate and tail-flick tests) and extensive loss of striatal dopamine (113). In reserpine-injected rats presenting with mechanical hyperalgesia, neuron-profiling data showed increased expression of the acid-sensing ion channel ASIC3. Its specific blockade reversed reserpine-induced pain. This effect was accompanied by sustained spinal dorsal horn microglial activation, whose inhibition with minocycline reversed mechanical hyperalgesia (114).
Using a PD animal model, these studies demonstrate the presence of functional and transcription alterations in the somatosensory nervous system. They also provide a mechanistic link between the expression of prototypical pain-associated ion channels receptors and the onset of PD non-motor symptoms. Further research is necessary to test whether targeting these changes would, along with stopping PD-induced pain, alleviate CNS alteration by delaying or preventing the onset of PD motor symptoms.
Central Mechanisms of PD-Induced Pain
In PD patients suffering from chronic pain, positron emission tomography (PET) studies showed increased neuronal activity in the prefrontal cortex, the primary somatosensory cortex, the posterior insula, and the anterior cingulate cortex (84, 115). At resting-state, magnetic resonance imaging (MRI) analysis of the connectivity between the right nucleus accumbens and the left hippocampus showed that it was reduced in PD patients experiencing pain compared with pain-free PD patients (116, 117). Before there is any noticeable degeneration in the substantia nigra and the onset of motor symptoms, early signs of PD neuropathology are first found in the locus coeruleus and raphe nuclei. Notably, these two supraspinal regions are typically associated with pain processing (118).
α-syn aggregates are present in the regions associated with pain processing including the lamina I of the spinal cord, the preganglionic neurons of the vagal nerve, and the sympathetic preganglionic neurons as well as in the coeliac ganglion (119). Blood-purified CD4+ and CD8+ T cells from PD patients recognize and respond to α-syn, which leads to the production of IL-5 or IFN-γ (Figure 1) (120). Brain circulating α-syn-specific CD4+ and CD8+ T cells can recognize specific peptides bound to MHC-II on microglia and MHC-I on dopaminergic neurons (111). In addition, effector CD8 T cells expressing the immune checkpoint receptors lymphocyte activation gene-3 (LAG3) bind with pathogenic α-syn, which favors its endocytosis and central dissemination (Figure 2) (121).
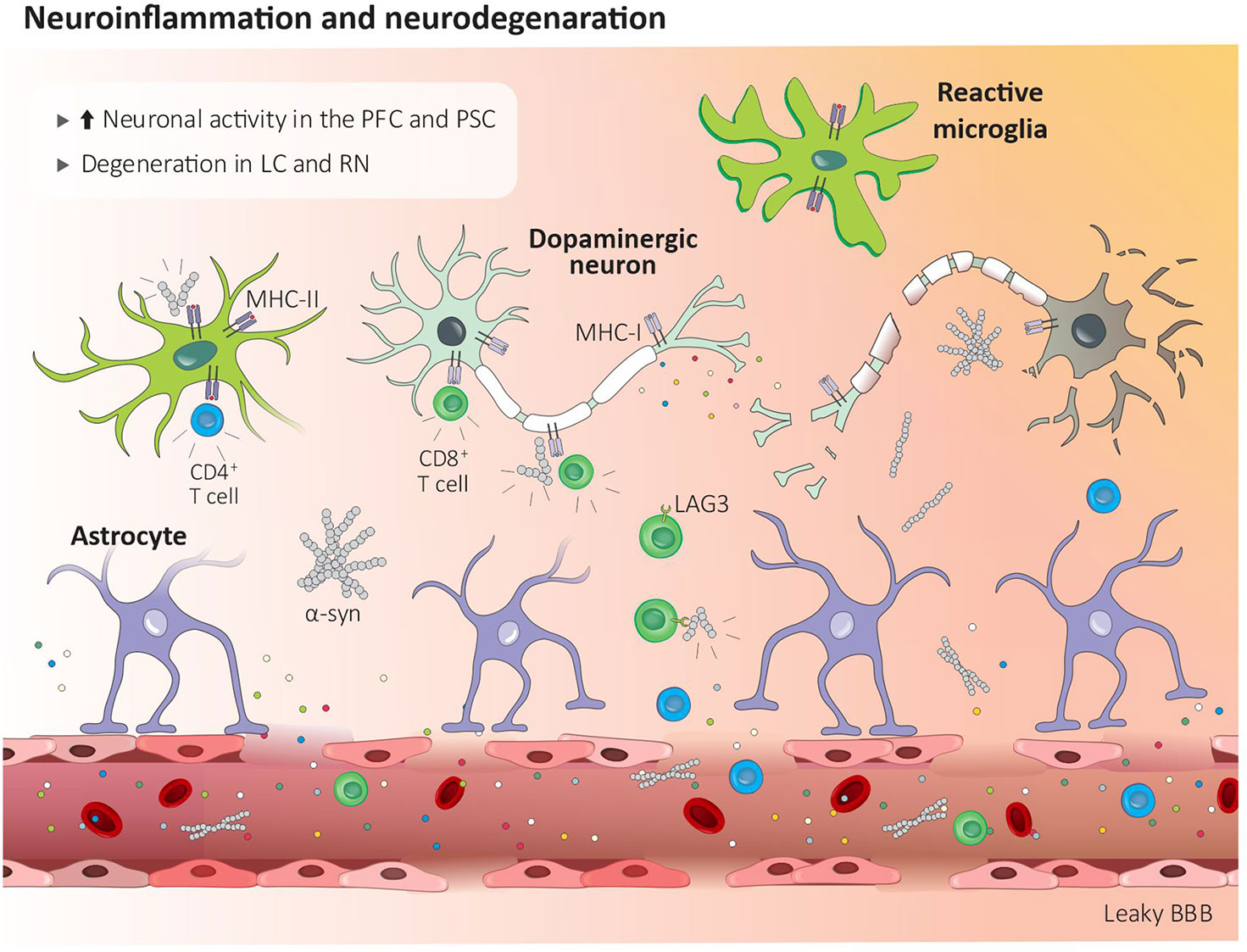
Figure 2 Central inflammation in Parkinson’s disease. Peripheral inflammation increases the blood brain barrier permeability which facilitates the infiltration of CD4+ and CD8+ T cells. Lag3+ CD8 T cells help disseminate α-syn centrally, while brain circulating α-syn-reactive CD4+ and CD8+ T cells recognize MHC-II bound peptides on microglia and MHC-I on dopaminergic neurons. Modified α-synuclein acts as a damage-associated molecular pattern, and via its action on receptors found on microglia and macrophages, induces cytokines release and neurodegeneration. BBB, blood brain barrier; LAG3, lymphocyte activation gene-3; LC, locus coeruleus; MHC-I, major histocompatibility complex I; MHC-II, major histocompatibility complex II; PFC, prefrontal cortex; PSC, primary somatosensory cortex; RN, raphe nuclei; α-syn, α-synuclein.
Along with these changes, peripheral inflammation also increases the permeability of the blood-brain barrier (BBB), which facilitates the infiltration of pathogenic lymphocytes in the CNS. CD4+ and CD8+ T cells were found in the brain parenchyma of PD patients as well as in different PD animal models. Although the mechanisms are not clearly understood, there is evidence indicating that infiltrated T cells drive neurodegeneration through the release and action of cytokines on their cognate receptors, which are found on CNS neurons (Figure 2). For instance, CD8 cytotoxic T cells’ TCR recognize MHCI-expressed mitochondrial antigens expressed by dopaminergic neurons and were posited to drive their elimination by a mechanism that is not fully understood (Figure 2) (122). Another example of T cell-mediated neurodegeneration stems from IL-17-derived from CD3+ T cells, which, when co-cultured, eliminate PD patients’ iPSC-derived midbrain dopaminergic neurons (98).
GI Dysfunction in PD-Induced Pain
Gastrointestinal (GI) alterations are found in up to 80% of PD patients (123–125), with symptoms ranging from constipation to nausea, dyspepsia, and dysphagia (126). Constipation might precede the onset of PD motor symptoms by several decades (127, 128) and has been considered a prodromal hallmark of PD (126). In PD patients, severe GI symptoms are predictive of impaired cognitive performance.
α-syn, which is typically found in CNS of PD patients, is also present in the colon, the neurons of the ENS, and the vagus nerve (129, 130). These findings were phenocopy in animal models of PD, in which aggregates of α-syn were found in the GI tract (Figure 3A) (131–134). As such, a growing number of investigators argue that PD might start in the gut and spread to the CNS through the vagus nerve (Figure 3A). This hypothesis is supported by animal studies showing that exogenous α-syn injected into the gut wall migrated to the brain via the vagus nerve at a rate estimated to be 5–10 mm/day in rats (135), while a patient who underwent a truncal vagotomy showed a decreased risk of developing PD later in life (136, 137). Although the cause of these disruptions and their role in PD pathogenesis remains unclear, the presence of α-syn in the ENS is sufficient to induce colonic dysmotility in the Gl tract, which correlates with the severity of motor impairment in some animal models (138, 139).
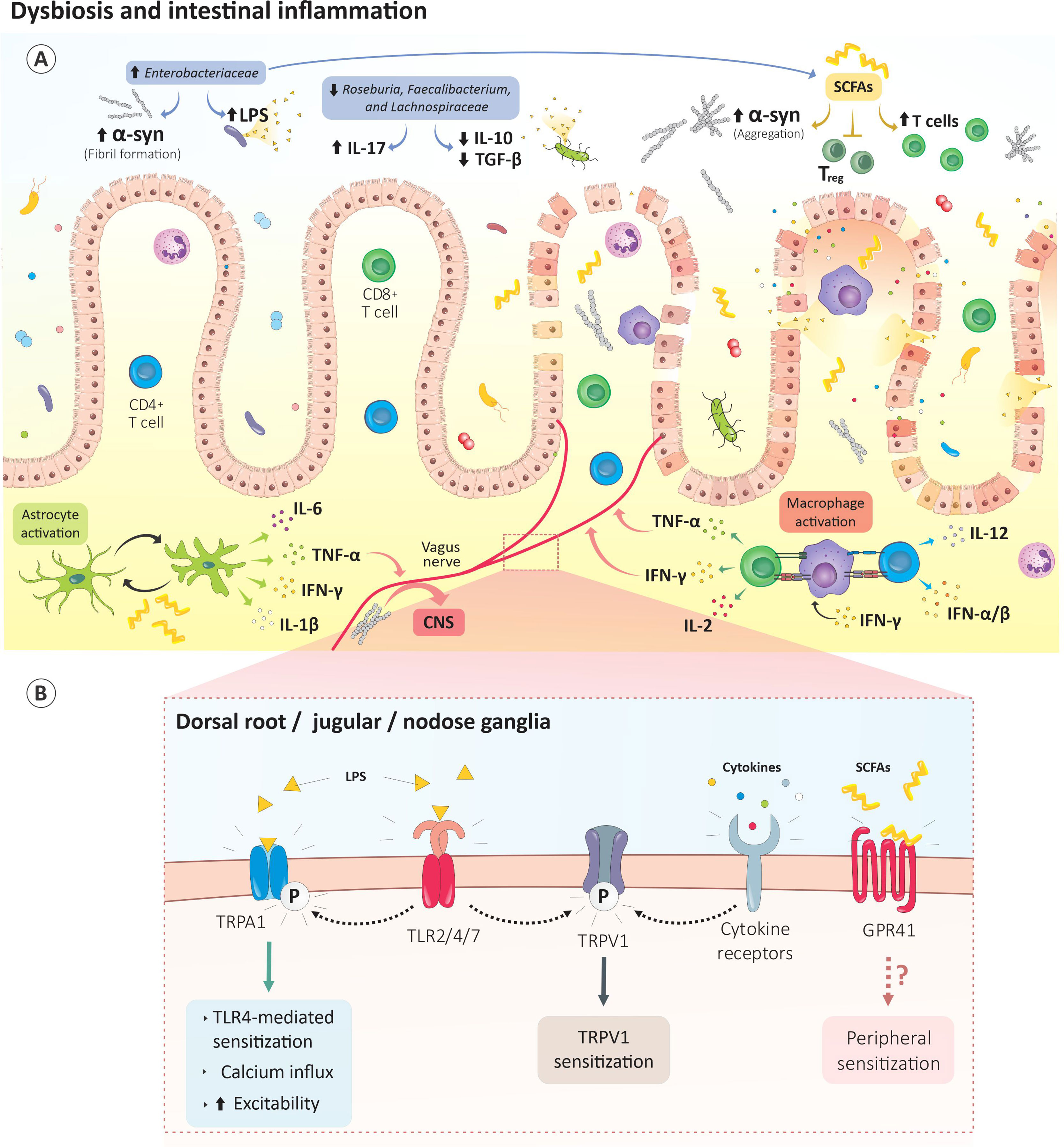
Figure 3 Gut dysbiosis drives PD-induced pain. (A) The gut dysbiosis in PD is characterized by enhances Enterobacteriaceae content. These bacteria increase LPS levels which subsequently promote gut permeability. Raises in Enterobacteriaceae metabolites, such as short-chain fatty acids, promote α‐syn fibril formation, microglia activation and blunt Treg-mediated immunosuppression. Proteolyzed α-syn by neurons and antigen presenting cells is then presented by the MHC machinery and leads to activation of autoreactive CD8 and CD4 T cells. (B) The dysbiotic bacteria and their metabolites directly activate vagal or ENS neurons. For instance, LPS activates nociceptor neurons-expressed TLR2/4/7 while SCFAs sensitize GPR41-expressing neurons. Bacteria-activated or α-syn-autoreactive immunocytes released cytokines can also activate their cognate receptors on sensory neurons. Subsequently, intracellular mechanisms are triggered in these neurons and culminate in TRP channels phosphorylation, neurons sensitization and sensory hypersensitivity. CNS, central nervous system; GPR41, G protein-coupled receptor 41; IFN-γ, interferon γ; IL-1β, interleukin 1 beta; IL-2, interleukin 2; IL-6, interleukin 6; IL-10, interleukin 10; IL-12, interleukin 12; IL-17, interleukin 17; LPS, lipopolysaccharide; TGFβ, transforming growth factor beta; TLR2/4/7, Toll-like receptor 2/4/7; Treg, regulatory T cells TRPA1, transient receptor potential ankyrin-like 1; TRPV1, transient receptor potential cation channel subfamily V; TNF-α, tumor necrosis factor; SCFAs, short-chain fatty acids; α-syn, α-synuclein.
PD patients experiencing constipation showed increased infiltration of CD4+ T cells into the colonic mucosa as well as elevated circulating TH17 and Treg cells (140). In addition, PINK1 or Parkin deficient mice exposed to bacterial intestinal infection showed an increase in BBB permeability, which facilitates the influx of cytotoxic CD8 T cells into the CNS. These pro-inflammatory cytotoxic CD8 T cells target the host’s mitochondrial antigens. Such auto-immune reactions can lead to the elimination of dopaminergic neurons in the striatum and subsequent motor impairments (141). Finally, histologic data showed increases in the immunoreactivity of the astrocytic marker GFAP in the colon of PD patients as well as increases in TNF-α, IFN-γ, IL-6, and IL-1β levels (142) (Figure 3A). These mediators were elevated in the early stages of the disease and were negatively correlated with disease duration (142). Given that enteric and central glial cells respond to IL-6, and IL-1β, and that their upregulation is associated with inflammatory pain (143), it is conceivable that the influx of cytotoxic CD8 T cells heightens pain transmission centrally or within the gut wall.
Dysbiosis in PD-Induced Pain
In conjunction with gastrointestinal dysfunction and inflammation, gut dysbiosis may contribute to PD progression by increasing the permeability of the blood-gut barrier and BBB and facilitating the transport of peripheral α-syn to the brain (138). Similarly, microbes can directly activate sensory neurons to trigger pain hypersensitivity (see Immunity in PD). As such, PD-induced dysbiotic bacteria and their metabolites may activate vagal or ENS neurons directly, or indirectly via activated immunocyte-released cytokines (Figure 3B) (21, 144).
Such dysbiosis is characterized by increased levels of Enterobacteriaceae, Akkermansia spp., Catabacter spp., and Akkermansiaceae and a decreased level of Roseburia spp., Faecalibacterium spp., and Lachnospiraceae (145, 146). While the function of these bacteria is diverse and is likely to be context-dependent, some patterns are evident. Roseburia spp. and Faecalibacterium spp. are typically known for their anti-inflammatory properties. Faecalibacterium spp. would blunt CD4 differentiation to TH17 cells and promote differentiation to Treg (147–149). Roseburia spp. and Faecalibacterium spp. would also downregulate IL-17 expression (150), and promote the release of the anti-inflammatory cytokines IL-10 and TGF-β (Figure 3A) (151, 152).
Roseburia spp. was found in increased levels in the blood and stool samples of fibromyalgia patients (153) and in stool samples of obese patients with back pain (154). When supplemented, Roseburia spp. alleviates stress-related visceral pain (155). It is therefore conceivable that reduced levels of Roseburia spp. and Faecalibacterium spp. exacerbates TH17 activity and/or limits the Treg which promotes PD-induced pain.
In the stools of PD patients, Enterobacteriaceae levels correlate with motor symptoms (146) and are associated with increased lipopolysaccharide (LPS) levels and α‐syn fibril formation (Figure 3A) (156). While no direct link exists between Enterobacteriaceae and PD-induced pain, LPS is known to activate nociceptor neurons expressed by TLR2, -4 and -7 (157) and, possibly, TRPA1, TRPM3, TRPM8, and TRPV1 (158, 159). Enterobacteriaceae-mediated increases in LPS levels may therefore lead to the sensitization of gut-innervating nociceptor neurons (Figure 3B).
Bacteria produce various short-chain fatty acid (SCFA) metabolites including acetate, propionate, butyrate, and valeric acid. In the gut of SPF mice that underwent a CCI, these SCFAs drive hippocampal microglia polarization and IL-1β and TNF-α release. These cytokines, in turn, mediate mechanical and thermal hyperalgesia (160). When administered to germ-free α-syn overexpressing mice, SCFAs increased α-syn aggregation and microglia activation and contributed to motor dysfunction (161). SCFAs were also shown to modulate microglia activation during viral infections (162) (Figure 3A). Therefore, SFCA-mediated microglial activation may drive the central sensitization of pain pathways.
SCFAs blunt Treg-mediated immunosuppression and increase T cell density (163–165) (Figure 3A). By activating GPR43 and GPR41, SCFA inhibits the leukocytes’ histone deacetylase (HDAC) which, in turn, increase the leukocyte chemotaxis, chemokine production, and the expression of adhesion molecules (166). Given that sodium butyrate, an HDAC inhibitor diminishes CCI-induced TNF-α release and pain (167), it can be surmised that PD-induced gut SCFAs can raise circulating cytokine levels (i.e., TNF-α) and promote sensory hypersensitivities by increasing the immunomodulatory action of HDAC in leukocytes Finally, SCFA were found to sensitize GPR41-expressing lumbar and vagal neurons (Figure 3B) (168).
Fecal microbiota transplantation (FMT) was found to rescue gut dysbiosis, decrease SCFAs levels, alleviate physical impairment, and increase striatal DA and 5-HT content in PD animal models. Microglia and astrocyte activation was diminished in the substantia nigra, and neuroinflammation was suppressed by reducing TLR4/TNF-α signaling (169). In PD patients, FMT reduced constipation and, albeit transiently, leg tremors (170, 171). A preliminary study with 15 PD patients reported that colonic FMT administration alleviates motor and non-motor symptoms, while improving anxiety, depression, and sleep quality scores (172). In another prospective study, 11 PD patients who underwent FMT had reduced constipation and improved postural instability and gait (173).
Modulating pain by rescuing a healthy microbiome has also been postulated (174). Preclinical and clinical studies indicate that probiotic consumption alleviates visceral pain (175). Based on these findings, along with the fact that PD is characterized by dysbiosis, and that bacteria and their metabolites modulate sensory neuron function, we hypothesize that FMT may improve PD-induced pain and delay the onset of motor symptoms.
Conclusion
Along with constipation, sensory hypersensitivity precedes the onset of motor symptoms in PD. These physiological alterations are accompanied by gut dysbiosis, altered peripheral and central immunity, and increased local (i.e., gut) and systemic cytokine content, as well as increased gut-brain barrier and BBB permeability. We posit that gut dysbiosis leads to systemic inflammation, which drives sensory hypersensitivity (Figure 4). Via local pro-inflammatory loops, these hypersensitized nociceptor neurons are likely to amplify immune responses and speed up central neurodegeneration. Alleviating constipation, rescuing microbiota via fecal matter transplant, blocking leukocyte activity and cytokine action on neurons using targeted antibodies, and limiting BBB and gut hyperpermeability all constitute potential ways of preventing neuro-immune and microbe-neuron interplays and subsequent pain hypersensitivity. Further studies should investigate how nociceptor neurons increase activity, would, in turn, modulate dysbiosis and central neurodegeneration. Should pain constitute an early driver of PD pathophysiology, monitoring and alleviating this symptom may constitute a novel biomarker and therapeutic target to slow the progression of PD.
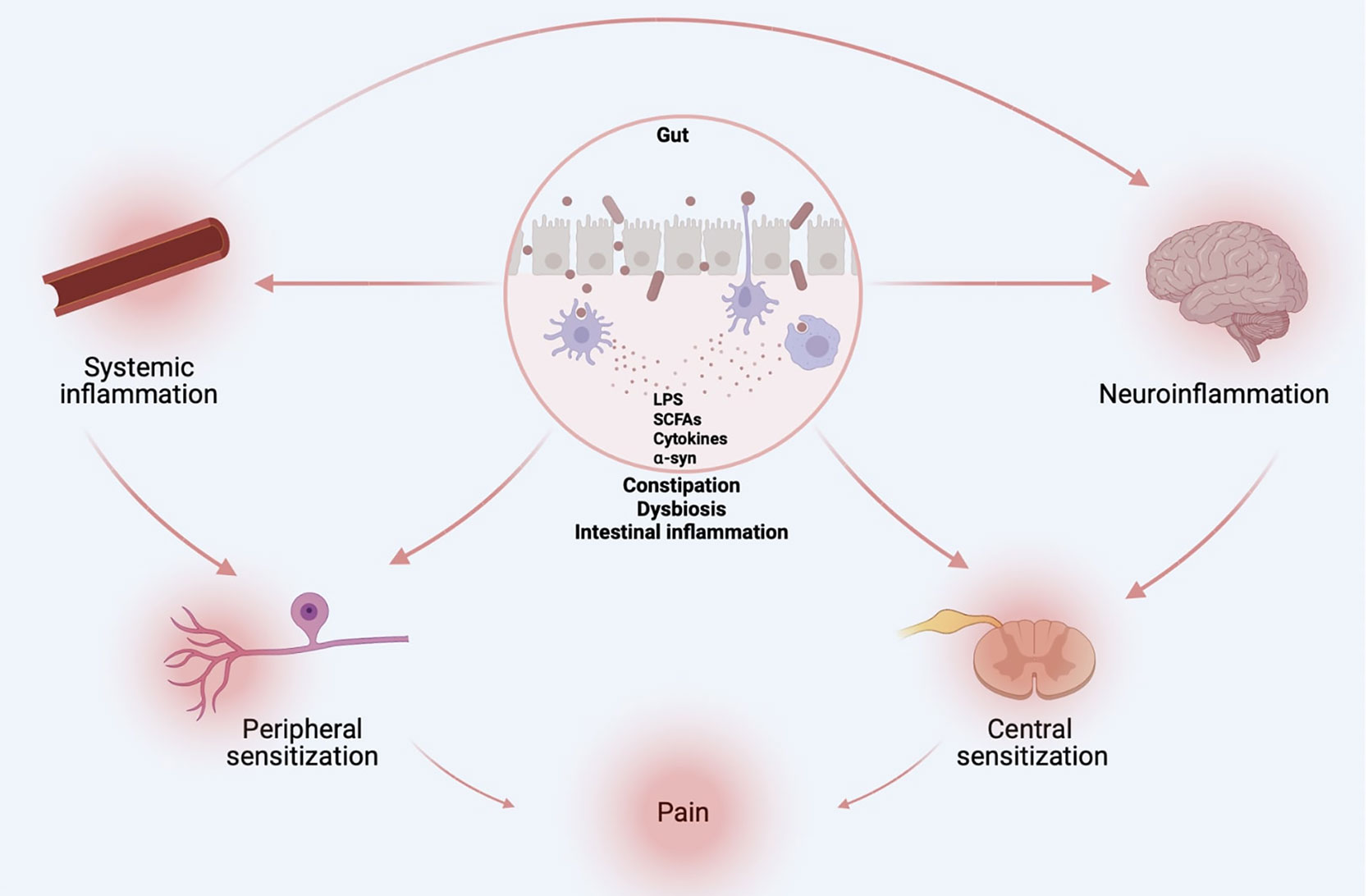
Figure 4 Origins of PD-induced pain. Gut dysbiosis, altered peripheral and central immunity, and increased local (i.e., gut) and systemic cytokine content, as well as increased gut-brain barrier and BBB permeability are likely drivers of sensory hypersensitivity in PD. Figure created with biorender.com.
Author Contributions
KR and ST designed the study. KR, NC-S, KrR, MG, CB, RP, and ST wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
ST is financed by Canada Research Chair program (#950-231859), Canadian Institutes of Health Research (#162211, #461274, #461275), the Canadian Foundation for Innovation (#37439), and Natural Sciences and Engineering Research Council of Canada (#RGPIN-2019- 06824), as well as the Fonds de recherche du Québec – Santé and Centre interdisciplinaire de recherche sur le cerveau et l'apprentissage (CIRCA). KR holds postdoctoral fellowships from the Fonds de recherche du Québec - Nature et technologies (FRQNT; #289949), CIRCA and Fonds de recherche en ophtalmologie - Université de Montréal (FROUM). KrR holds postdoctoral fellowships from CIRCA and FROUM. NCS holds a Merit scholarship from the Faculty of Medicine of the Université de Montréal.
Conflict of Interest
ST has an equity stake in Nocion Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ripamonti CI. Pain Management. Ann Oncol (2012) 23(Suppl 10):x294–301. doi: 10.1093/annonc/mds360
2. Melzack R. From the Gate to the Neuromatrix. Pain (1999) Suppl 6:S121–6. doi: 10.1016/s0304-3959(99)00145-1
3. Tesfaye S, Kempler P. Painful Diabetic Neuropathy. Diabetologia (2005) 48(5):805–7. doi: 10.1007/s00125-005-1721-7
4. Ossipov MH, Porreca F. Challenges in the Development of Novel Treatment Strategies for Neuropathic Pain. NeuroRx (2005) 2(4):650–61. doi: 10.1602/neurorx.2.4.650
5. Cameron AA, Khan IA, Westlund KN, Cliffer KD, Willis WD. The Efferent Projections of the Periaqueductal Gray in the Rat: A Phaseolus Vulgaris-Leucoagglutinin Study. I. Ascending Projections. J Comp Neurol (1995) 351(4):568–84. doi: 10.1002/cne.903510407
6. Politis M, Wu K, Molloy S, GB P, Chaudhuri KR, Piccini P. Parkinson's Disease Symptoms: The Patient's Perspective. Mov Disord (2010) 25(11):1646–51. doi: 10.1002/mds.23135
7. Santos-Garcia D, Abella-Corral J, Aneiros-Diaz A, Santos-Canelles H, Llaneza-Gonzalez MA, Macias-Arribi M. Pain in Parkinson's Disease: Prevalence, Characteristics, Associated Factors, and Relation With Other non Motor Symptoms, Quality of Life, Autonomy, and Caregiver Burden. Rev Neurol (2011) 52(7):385–93. doi: 10.33588/rn.5207.2010558
8. Skorvanek M, Martinez-Martin P, Kovacs N, Zezula I, Rodriguez-Violante M, Corvol JC, et al. Relationship Between the MDS-UPDRS and Quality of Life: A Large Multicenter Study of 3206 Patients. Parkinsonism Relat Disord (2018) 52:83–9. doi: 10.1016/j.parkreldis.2018.03.027
9. Anwar K. Pathophysiology of Pain. Dis Mon (2016) 62(9):324–9. doi: 10.1016/j.disamonth.2016.05.015
10. Latremoliere A, Woolf CJ. Central Sensitization: A Generator of Pain Hypersensitivity by Central Neural Plasticity. J Pain (2009) 10(9):895–926. doi: 10.1016/j.jpain.2009.06.012
11. Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and Central Sensitization in Chronic and Widespread Pain. Anesthesiology (2018) 129(2):343–66. doi: 10.1097/ALN.0000000000002130
12. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and Molecular Mechanisms of Pain. Cell (2009) 139(2):267–84. doi: 10.1016/j.cell.2009.09.028
13. Cook AD, Christensen AD, Tewari D, McMahon SB, Hamilton JA. Immune Cytokines and Their Receptors in Inflammatory Pain. Trends Immunol (2018) 39(3):240–55. doi: 10.1016/j.it.2017.12.003
14. Austin PJ, Moalem-Taylor G. The Neuro-Immune Balance in Neuropathic Pain: Involvement of Inflammatory Immune Cells, Immune-Like Glial Cells and Cytokines. J Neuroimmunol (2010) 229(1-2):26–50. doi: 10.1016/j.jneuroim.2010.08.013
15. Ji RR, Xu ZZ, Gao YJ. Emerging Targets in Neuroinflammation-Driven Chronic Pain. Nat Rev Drug Discov (2014) 13(7):533–48. doi: 10.1038/nrd4334
16. Romero-Sandoval EA, Horvath RJ, DeLeo JA. Neuroimmune Interactions and Pain: Focus on Glial-Modulating Targets. Curr Opin Investig Drugs (2008) 9(7):726–34.
17. Julius D, Basbaum AI. Molecular Mechanisms of Nociception. Nature (2001) 413(6852):203–10. doi: 10.1038/35093019
18. Amaya F, Izumi Y, Matsuda M, Sasaki M. Tissue Injury and Related Mediators of Pain Exacerbation. Curr Neuropharmacol (2013) 11(6):592–7. doi: 10.2174/1570159x11311060003
19. Imamachi N, Park GH, Lee H, Anderson DJ, Simon MI, Basbaum AI, et al. TRPV1-Expressing Primary Afferents Generate Behavioral Responses to Pruritogens via Multiple Mechanisms. Proc Natl Acad Sci USA (2009) 106(27):11330–5. doi: 10.1073/pnas.0905605106
20. Fitzgerald EM, Okuse K, Wood JN, Dolphin AC, Moss SJ. cAMP-Dependent Phosphorylation of the Tetrodotoxin-Resistant Voltage-Dependent Sodium Channel SNS. J Physiol (1999) 516( Pt 2):433–46. doi: 10.1111/j.1469-7793.1999.0433v.x
21. Qi J, Buzas K, Fan H, Cohen JI, Wang K, Mont E, et al. Painful Pathways Induced by TLR Stimulation of Dorsal Root Ganglion Neurons. J Immunol (2011) 186(11):6417–26. doi: 10.4049/jimmunol.1001241
22. Ferreira SH, Lorenzetti BB, Bristow AF, Poole S. Interleukin-1 Beta as a Potent Hyperalgesic Agent Antagonized by a Tripeptide Analogue. Nature (1988) 334(6184):698–700. doi: 10.1038/334698a0
23. Ebbinghaus M, Uhlig B, Richter F, von Banchet GS, Gajda M, Brauer R, et al. The Role of Interleukin-1beta in Arthritic Pain: Main Involvement in Thermal, But Not Mechanical, Hyperalgesia in Rat Antigen-Induced Arthritis. Arthritis Rheum (2012) 64(12):3897–907. doi: 10.1002/art.34675
24. Safieh-Garabedian B, Poole S, Allchorne A, Winter J, Woolf CJ. Contribution of Interleukin-1 Beta to the Inflammation-Induced Increase in Nerve Growth Factor Levels and Inflammatory Hyperalgesia. Br J Pharmacol (1995) 115(7):1265–75. doi: 10.1111/j.1476-5381.1995.tb15035.x
25. Richter F, Natura G, Loser S, Schmidt K, Viisanen H, Schaible HG. Tumor Necrosis Factor Causes Persistent Sensitization of Joint Nociceptors to Mechanical Stimuli in Rats. Arthritis Rheum (2010) 62(12):3806–14. doi: 10.1002/art.27715
26. Cunha TM, Verri WA Jr, Silva JS, Poole S, Cunha FQ, Ferreira SH. A Cascade of Cytokines Mediates Mechanical Inflammatory Hypernociception in Mice. Proc Natl Acad Sci USA (2005) 102(5):1755–60. doi: 10.1073/pnas.0409225102
27. Maier JA, Hla T, Maciag T. Cyclooxygenase is an Immediate-Early Gene Induced by Interleukin-1 in Human Endothelial Cells. J Biol Chem (1990) 265(19):10805–8.
28. Pitchford S, Levine JD. Prostaglandins Sensitize Nociceptors in Cell Culture. Neurosci Lett (1991) 132(1):105–8. doi: 10.1016/0304-3940(91)90444-x
29. Moriyama T, Higashi T, Togashi K, Iida T, Segi E, Sugimoto Y, et al. Sensitization of TRPV1 by EP1 and IP Reveals Peripheral Nociceptive Mechanism of Prostaglandins. Mol Pain (2005) 1:3. doi: 10.1186/1744-8069-1-3
30. Zhang X, Huang J, McNaughton PA. NGF Rapidly Increases Membrane Expression of TRPV1 Heat-Gated Ion Channels. EMBO J (2005) 24(24):4211–23. doi: 10.1038/sj.emboj.7600893
31. Peng J, Gu N, Zhou L, BE U, Murugan M, Gan WB, et al. Microglia and Monocytes Synergistically Promote the Transition From Acute to Chronic Pain After Nerve Injury. Nat Commun (2016) 7:12029. doi: 10.1038/ncomms12029
32. Kleinschnitz C, Hofstetter HH, Meuth SG, Braeuninger S, Sommer C, Stoll G. T Cell Infiltration After Chronic Constriction Injury of Mouse Sciatic Nerve is Associated With Interleukin-17 Expression. Exp Neurol (2006) 200(2):480–5. doi: 10.1016/j.expneurol.2006.03.014
33. Vicuna L, Strochlic DE, Latremoliere A, Bali KK, Simonetti M, Husainie D, et al. The Serine Protease Inhibitor SerpinA3N Attenuates Neuropathic Pain by Inhibiting T Cell-Derived Leukocyte Elastase. Nat Med (2015) 21(5):518–23. doi: 10.1038/nm.3852
34. Bali KK, Kuner R. Therapeutic Potential for Leukocyte Elastase in Chronic Pain States Harboring a Neuropathic Component. Pain (2017) 158(11):2243–58. doi: 10.1097/j.pain.0000000000001032
35. Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, et al. T-Cell Infiltration and Signaling in the Adult Dorsal Spinal Cord Is a Major Contributor to Neuropathic Pain-Like Hypersensitivity. J Neurosci (2009) 29(46):14415–22. doi: 10.1523/JNEUROSCI.4569-09.2009
36. Draleau K, Maddula S, Slaiby A, Nutile-McMenemy N, De Leo J, Cao L. Phenotypic Identification of Spinal Cord-Infiltrating CD4(+) T Lymphocytes in a Murine Model of Neuropathic Pain. J Pain Relief (2014) Suppl 3:3. doi: 10.4172/2167-0846.S3-003
37. Lotz M, Vaughan JH, Carson DA. Effect of Neuropeptides on Production of Inflammatory Cytokines by Human Monocytes. Science (1988) 241(4870):1218–21. doi: 10.1126/science.2457950
38. Sun J, Ramnath RD, Zhi L, Tamizhselvi R, Bhatia M. Substance P Enhances NF-kappaB Transactivation and Chemokine Response in Murine Macrophages via ERK1/2 and P38 MAPK Signaling Pathways. Am J Physiol Cell Physiol (2008) 294(6):C1586–96. doi: 10.1152/ajpcell.00129.2008
39. Hackel D, Pflucke D, Neumann A, Viebahn J, Mousa S, Wischmeyer E, et al. The Connection of Monocytes and Reactive Oxygen Species in Pain. PLoS One (2013) 8(5):e63564. doi: 10.1371/journal.pone.0063564
40. Zelenka M, Schafers M, Sommer C. Intraneural Injection of Interleukin-1beta and Tumor Necrosis Factor-Alpha Into Rat Sciatic Nerve at Physiological Doses Induces Signs of Neuropathic Pain. Pain (2005) 116(3):257–63. doi: 10.1016/j.pain.2005.04.018
41. Kim D, You B, Lim H, Lee SJ. Toll-Like Receptor 2 Contributes to Chemokine Gene Expression and Macrophage Infiltration in the Dorsal Root Ganglia After Peripheral Nerve Injury. Mol Pain (2011) 7:74. doi: 10.1186/1744-8069-7-74
42. Miller RE, Tran PB, Das R, Ghoreishi-Haack N, Ren D, Miller RJ, et al. CCR2 Chemokine Receptor Signaling Mediates Pain in Experimental Osteoarthritis. Proc Natl Acad Sci USA (2012) 109(50):20602–7. doi: 10.1073/pnas.1209294110
43. Lourenco C, Babloyantz A. Control of Spatiotemporal Chaos in Neuronal Networks. Int J Neural Syst (1996) 7(4):507–17. doi: 10.1142/s012906579600049x
44. Trevisan G, Benemei S, Materazzi S, De Logu F, De Siena G, Fusi C, et al. TRPA1 Mediates Trigeminal Neuropathic Pain in Mice Downstream of Monocytes/Macrophages and Oxidative Stress. Brain (2016) 139(Pt 5):1361–77. doi: 10.1093/brain/aww038
45. Willemen HL, Eijkelkamp N, Garza Carbajal A, Wang H, Mack M, Zijlstra J, et al. Monocytes/Macrophages Control Resolution of Transient Inflammatory Pain. J Pain (2014) 15(5):496–506. doi: 10.1016/j.jpain.2014.01.491
46. Wigerblad G, Bas DB, Fernades-Cerqueira C, Krishnamurthy A, Nandakumar KS, Rogoz K, et al. Autoantibodies to Citrullinated Proteins Induce Joint Pain Independent of Inflammation via a Chemokine-Dependent Mechanism. Ann Rheum Dis (2016) 75(4):730–8. doi: 10.1136/annrheumdis-2015-208094
47. Cuhadar U, Gentry C, Vastani N, Sensi S, Bevan S, Goebel A, et al. Autoantibodies Produce Pain in Complex Regional Pain Syndrome by Sensitizing Nociceptors. Pain (2019) 160(12):2855–65. doi: 10.1097/j.pain.0000000000001662
48. Dawes JM, Weir GA, Middleton SJ, Patel R, Chisholm KI, Pettingill P, et al. Immune or Genetic-Mediated Disruption of CASPR2 Causes Pain Hypersensitivity Due to Enhanced Primary Afferent Excitability. Neuron (2018) 97(4):806–22.e10. doi: 10.1016/j.neuron.2018.01.033
49. Chen G, Kim YH, Li H, Luo H, Liu DL, Zhang ZJ, et al. PD-L1 Inhibits Acute and Chronic Pain by Suppressing Nociceptive Neuron Activity via PD-1. Nat Neurosci (2017) 20(7):917–26. doi: 10.1038/nn.4571
50. Donnelly CR, Chen O, Ji RR. How Do Sensory Neurons Sense Danger Signals? Trends Neurosci (2020) 43(10):822–38. doi: 10.1016/j.tins.2020.07.008
51. Donnelly CR, Jiang C, Andriessen AS, Wang K, Wang Z, Ding H, et al. STING Controls Nociception via Type I Interferon Signalling in Sensory Neurons. Nature (2021) 591(7849):275–80. doi: 10.1038/s41586-020-03151-1
52. Gershon MD. The Enteric Nervous System. Annu Rev Neurosci (1981) 4:227–72. doi: 10.1146/annurev.ne.04.030181.001303
53. Kunze WA, Furness JB. The Enteric Nervous System and Regulation of Intestinal Motility. Annu Rev Physiol (1999) 61:117–42. doi: 10.1146/annurev.physiol.61.1.117
54. Goyal RK, Hirano I. The Enteric Nervous System. N Engl J Med (1996) 334(17):1106–15. doi: 10.1056/NEJM199604253341707
55. Ratcliffe EM. Molecular Development of the Extrinsic Sensory Innervation of the Gastrointestinal Tract. Auton Neurosci (2011) 161(1-2):1–5. doi: 10.1016/j.autneu.2010.11.003
56. Williams EK, Chang RB, Strochlic DE, Umans BD, Lowell BB, Liberles SD. Sensory Neurons That Detect Stretch and Nutrients in the Digestive System. Cell (2016) 166(1):209–21. doi: 10.1016/j.cell.2016.05.011
57. Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The Capsaicin Receptor: A Heat-Activated Ion Channel in the Pain Pathway. Nature (1997) 389(6653):816–24. doi: 10.1038/39807
58. Kerr BJ, Souslova V, McMahon SB, Wood JN. A Role for the TTX-Resistant Sodium Channel Nav 1.8 in NGF-Induced Hyperalgesia, But Not Neuropathic Pain. Neuroreport (2001) 12(14):3077–80. doi: 10.1097/00001756-200110080-00019
59. Nonomura K, Woo SH, Chang RB, Gillich A, Qiu Z, Francisco AG, et al. Piezo2 Senses Airway Stretch and Mediates Lung Inflation-Induced Apnoea. Nature (2017) 541(7636):176–81. doi: 10.1038/nature20793
60. Talbot S, Foster SL, Woolf CJ. Neuroimmunity: Physiology and Pathology. Annu Rev Immunol (2016) 34:421–47. doi: 10.1146/annurev-immunol-041015-055340
61. Foster SL, Seehus CR, Woolf CJ, Talbot S. Sense and Immunity: Context-Dependent Neuro-Immune Interplay. Front Immunol (2017) 8:1463. doi: 10.3389/fimmu.2017.01463
62. Bellono NW, Bayrer JR, Leitch DB, Castro J, Zhang C, O'Donnell TA, et al. Enterochromaffin Cells Are Gut Chemosensors That Couple to Sensory Neural Pathways. Cell (2017) 170(1):185–98.e16. doi: 10.1016/j.cell.2017.05.034
63. Kaelberer MM, Buchanan KL, Klein ME, Barth BB, Montoya MM, Shen X, et al. A Gut-Brain Neural Circuit for Nutrient Sensory Transduction. Science (2018) 361(6408):1219–27. doi: 10.1126/science.aat5236
64. Chiu IM, Pinho-Ribeiro FA, Woolf CJ. Pain and Infection: Pathogen Detection by Nociceptors. Pain (2016) 157(6):1192–3. doi: 10.1097/j.pain.0000000000000559
65. Blake KJ, Baral P, Voisin T, Lubkin A, Pinho-Ribeiro FA, Adams KL, et al. Staphylococcus Aureus Produces Pain Through Pore-Forming Toxins and Neuronal TRPV1 That is Silenced by QX-314. Nat Commun (2018) 9(1):37. doi: 10.1038/s41467-017-02448-6
66. Lai NY, Musser MA, Pinho-Ribeiro FA, Baral P, Jacobson A, Ma P, et al. Gut-Innervating Nociceptor Neurons Regulate Peyer's Patch Microfold Cells and SFB Levels to Mediate Salmonella Host Defense. Cell (2020) 180(1):33–49.e22. doi: 10.1016/j.cell.2019.11.014
67. Kashem SW, Riedl MS, Yao C, Honda CN, Vulchanova L, Kaplan DH. Nociceptive Sensory Fibers Drive Interleukin-23 Production From CD301b+ Dermal Dendritic Cells and Drive Protective Cutaneous Immunity. Immunity (2015) 43(3):515–26. doi: 10.1016/j.immuni.2015.08.016
68. Yissachar N, Zhou Y, Ung L, Lai NY, Mohan JF, Ehrlicher A, et al. An Intestinal Organ Culture System Uncovers a Role for the Nervous System in Microbe-Immune Crosstalk. Cell (2017) 168(6):1135–48.e12. doi: 10.1016/j.cell.2017.02.009
69. Silverdale MA, Kobylecki C, Kass-Iliyya L, Martinez-Martin P, Lawton M, Cotterill S, et al. A Detailed Clinical Study of Pain in 1957 Participants With Early/Moderate Parkinson's Disease. Parkinsonism Relat Disord (2018) 56:27–32. doi: 10.1016/j.parkreldis.2018.06.001
70. Beiske AG, Loge JH, Ronningen A, Svensson E. Pain in Parkinson's Disease: Prevalence and Characteristics. Pain (2009) 141(1-2):173–7. doi: 10.1016/j.pain.2008.12.004
71. Skogar O, Fall PA, Hallgren G, Bringer B, Carlsson M, Lennartsson U, et al. Parkinson's Disease Patients' Subjective Descriptions of Characteristics of Chronic Pain, Sleeping Patterns and Health-Related Quality of Life. Neuropsychiatr Dis Treat (2012) 8:435–42. doi: 10.2147/NDT.S34882
72. Ha AD, Jankovic J. Pain in Parkinson's Disease. Mov Disord (2012) 27(4):485–91. doi: 10.1002/mds.23959
73. Zis P, Grunewald RA, Chaudhuri RK, Hadjivassiliou M. Peripheral Neuropathy in Idiopathic Parkinson's Disease: A Systematic Review. J Neurol Sci (2017) 378:204–9. doi: 10.1016/j.jns.2017.05.023
74. Adewusi JK, Hadjivassiliou M, Vinagre-Aragon A, O'Connor KR, Khan A, Grunewald RA, et al. Peripheral Neuropathic Pain in Idiopathic Parkinson's Disease: Prevalence and Impact on Quality of Life; a Case Controlled Study. J Neurol Sci (2018) 392:3–7. doi: 10.1016/j.jns.2018.06.022
75. Lin CH, Wu RM, Chang HY, Chiang YT, Lin HH. Preceding Pain Symptoms and Parkinson's Disease: A Nationwide Population-Based Cohort Study. Eur J Neurol (2013) 20(10):1398–404. doi: 10.1111/ene.12197
76. Allen NE, Wong CM, Canning CG, Moloney N. The Association Between Parkinson's Disease Motor Impairments and Pain. Pain Med (2016) 17(3):456–62. doi: 10.1111/pme.12898
77. Negre-Pages L, Regragui W, Bouhassira D, Grandjean H, Rascol O, DoPaMi PSG. Chronic Pain in Parkinson's Disease: The Cross-Sectional French DoPaMiP Survey. Mov Disord (2008) 23(10):1361–9. doi: 10.1002/mds.22142
78. Lee MA, Walker RW, Hildreth TJ, Prentice WM. A Survey of Pain in Idiopathic Parkinson's Disease. J Pain Symptom Manage (2006) 32(5):462–9. doi: 10.1016/j.jpainsymman.2006.05.020
79. Defazio G, Berardelli A, Fabbrini G, Martino D, Fincati E, Fiaschi A, et al. Pain as a Nonmotor Symptom of Parkinson Disease: Evidence From a Case-Control Study. Arch Neurol (2008) 65(9):1191–4. doi: 10.1001/archneurol.2008.2
80. Blanchet PJ, Brefel-Courbon C. Chronic Pain and Pain Processing in Parkinson's Disease. Prog Neuropsychopharmacol Biol Psychiatry (2018) 87(Pt B):200–6. doi: 10.1016/j.pnpbp.2017.10.010
81. Schestatsky P, Kumru H, Valls-Sole J, Valldeoriola F, Marti MJ, Tolosa E, et al. Neurophysiologic Study of Central Pain in Patients With Parkinson Disease. Neurology (2007) 69(23):2162–9. doi: 10.1212/01.wnl.0000295669.12443.d3
82. Mylius V, Engau I, Teepker M, Stiasny-Kolster K, Schepelmann K, Oertel WH, et al. Pain Sensitivity and Descending Inhibition of Pain in Parkinson's Disease. J Neurol Neurosurg Psychiatry (2009) 80(1):24–8. doi: 10.1136/jnnp.2008.145995
83. Djaldetti R, Yust-Katz S, Kolianov V, Melamed E, Dabby R. The Effect of Duloxetine on Primary Pain Symptoms in Parkinson Disease. Clin Neuropharmacol (2007) 30(4):201–5. doi: 10.1097/wnf.0b013e3180340319
84. Brefel-Courbon C, Payoux P, Thalamas C, Ory F, Quelven I, Chollet F, et al. Effect of Levodopa on Pain Threshold in Parkinson's Disease: A Clinical and Positron Emission Tomography Study. Mov Disord (2005) 20(12):1557–63. doi: 10.1002/mds.20629
85. Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. Prediagnostic Presentations of Parkinson's Disease in Primary Care: A Case-Control Study. Lancet Neurol (2015) 14(1):57–64. doi: 10.1016/S1474-4422(14)70287-X
86. Wasner G, Deuschl G. Pains in Parkinson Disease–Many Syndromes Under One Umbrella. Nat Rev Neurol (2012) 8(5):284–94. doi: 10.1038/nrneurol.2012.54
87. Reale M, Iarlori C, Thomas A, Gambi D, Perfetti B, Di Nicola M, et al. Peripheral Cytokines Profile in Parkinson's Disease. Brain Behav Immun (2009) 23(1):55–63. doi: 10.1016/j.bbi.2008.07.003
88. Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 Beta and Interleukin-6 are Elevated in the Cerebrospinal Fluid of Alzheimer's and De Novo Parkinson’s Disease Patients. Neurosci Lett (1995) 202(1-2):17–20. doi: 10.1016/0304-3940(95)12192-7
89. Grozdanov V, Bliederhaeuser C, Ruf WP, Roth V, Fundel-Clemens K, Zondler L, et al. Inflammatory Dysregulation of Blood Monocytes in Parkinson's Disease Patients. Acta Neuropathol (2014) 128(5):651–63. doi: 10.1007/s00401-014-1345-4
90. Mogi M, Harada M, Narabayashi H, Inagaki H, Minami M, Nagatsu T. Interleukin (IL)-1 Beta, IL-2, IL-4, IL-6 and Transforming Growth Factor-Alpha Levels are Elevated in Ventricular Cerebrospinal Fluid in Juvenile Parkinsonism and Parkinson's Disease. Neurosci Lett (1996) 211(1):13–6. doi: 10.1016/0304-3940(96)12706-3
91. Qin XY, Zhang SP, Cao C, Loh YP, Cheng Y. Aberrations in Peripheral Inflammatory Cytokine Levels in Parkinson Disease: A Systematic Review and Meta-Analysis. JAMA Neurol (2016) 73(11):1316–24. doi: 10.1001/jamaneurol.2016.2742
92. Williams-Gray CH, Wijeyekoon R, Yarnall AJ, Lawson RA, Breen DP, Evans JR, et al. Serum Immune Markers and Disease Progression in an Incident Parkinson's Disease Cohort (ICICLE-Pd). Mov Disord (2016) 31(7):995–1003. doi: 10.1002/mds.26563
93. Bas J, Calopa M, Mestre M, Mollevi DG, Cutillas B, Ambrosio S, et al. Lymphocyte Populations in Parkinson's Disease and in Rat Models of Parkinsonism. J Neuroimmunol (2001) 113(1):146–52. doi: 10.1016/s0165-5728(00)00422-7
94. Calopa M, Bas J, Callen A, Mestre M. Apoptosis of Peripheral Blood Lymphocytes in Parkinson Patients. Neurobiol Dis (2010) 38(1):1–7. doi: 10.1016/j.nbd.2009.12.017
95. Stevens CH, Rowe D, Morel-Kopp MC, Orr C, Russell T, Ranola M, et al. Reduced T Helper and B Lymphocytes in Parkinson's Disease. J Neuroimmunol (2012) 252(1-2):95–9. doi: 10.1016/j.jneuroim.2012.07.015
96. Rosenkranz D, Weyer S, Tolosa E, Gaenslen A, Berg D, Leyhe T, et al. Higher Frequency of Regulatory T Cells in the Elderly and Increased Suppressive Activity in Neurodegeneration. J Neuroimmunol (2007) 188(1-2):117–27. doi: 10.1016/j.jneuroim.2007.05.011
97. Saunders JA, Estes KA, Kosloski LM, Allen HE, Dempsey KM, Torres-Russotto DR, et al. CD4+ Regulatory and Effector/Memory T Cell Subsets Profile Motor Dysfunction in Parkinson's Disease. J Neuroimmune Pharmacol (2012) 7(4):927–38. doi: 10.1007/s11481-012-9402-z
98. Sommer A, Marxreiter F, Krach F, Fadler T, Grosch J, Maroni M, et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human iPSC-Based Model of Parkinson's Disease. Cell Stem Cell (2018) 23(1):123–31.e6. doi: 10.1016/j.stem.2018.06.015
99. Chen S, Liu Y, Niu Y, Xu Y, Zhou Q, Xu X, et al. Increased Abundance of Myeloid-Derived Suppressor Cells and Th17 Cells in Peripheral Blood of Newly-Diagnosed Parkinson's Disease Patients. Neurosci Lett (2017) 648:21–5. doi: 10.1016/j.neulet.2017.03.045
100. Chen Y, Qi B, Xu W, Ma B, Li L, Chen Q, et al. Clinical Correlation of Peripheral CD4+cell Subsets, Their Imbalance and Parkinson's Disease. Mol Med Rep (2015) 12(4):6105–11. doi: 10.3892/mmr.2015.4136
101. Kustrimovic N, Comi C, Magistrelli L, Rasini E, Legnaro M, Bombelli R, et al. Parkinson's Disease Patients Have a Complex Phenotypic and Functional Th1 Bias: Cross-Sectional Studies of CD4+ Th1/Th2/T17 and Treg in Drug-Naive and Drug-Treated Patients. J Neuroinflamm (2018) 15(1):205. doi: 10.1186/s12974-018-1248-8
102. Santos RMS. Doença De Parkinson: Há Associação Entre Dor E Resposta Imunologica? Minas Gerais: Universidade Federal de Minas Gerais (2017).
103. Hanewinckel R, Drenthen J, van Oijen M, Hofman A, van Doorn PA, Ikram MA. Prevalence of Polyneuropathy in the General Middle-Aged and Elderly Population. Neurology (2016) 87(18):1892–8. doi: 10.1212/wnl.0000000000003293
104. Merola A, Rosso M, Romagnolo A, Comi C, Fasano A, Zibetti M, et al. Peripheral Neuropathy as Marker of Severe Parkinson's Disease Phenotype. Mov Disord (2017) 32(8):1256–8. doi: 10.1002/mds.27025
105. Nolano M, Provitera V, Estraneo A, Selim MM, Caporaso G, Stancanelli A, et al. Sensory Deficit in Parkinson's Disease: Evidence of a Cutaneous Denervation. Brain (2008) 131(Pt 7):1903–11. doi: 10.1093/brain/awn102
106. Jeziorska M, Atkinson A, Kass-Iliyya L, Kobylecki C, Gosal D, Marshall A, et al. Small Fibre Neuropathy in Parkinson's Disease: Comparison of Skin Biopsies From the More Affected and Less Affected Sides. J Parkinsons Dis (2019) 9(4):761–5. doi: 10.3233/jpd-191697
107. Nolano M, Provitera V, Manganelli F, Iodice R, Stancanelli A, Caporaso G, et al. Loss of Cutaneous Large and Small Fibers in Naive and L-Dopa-Treated PD Patients. Neurology (2017) 89(8):776–84. doi: 10.1212/wnl.0000000000004274
108. Wang N, Gibbons CH, Lafo J, Freeman R. Alpha-Synuclein in Cutaneous Autonomic Nerves. Neurology (2013) 81(18):1604–10. doi: 10.1212/WNL.0b013e3182a9f449
109. Rodriguez-Leyva I, Calderon-Garciduenas AL, Jimenez-Capdeville ME, Renteria-Palomo AA, Hernandez-Rodriguez HG, Valdes-Rodriguez R, et al. Alpha-Synuclein Inclusions in the Skin of Parkinson's Disease and Parkinsonism. Ann Clin Transl Neurol (2014) 1(7):471–8. doi: 10.1002/acn3.78
110. Chen Y, Mao CJ, Li SJ, Wang F, Chen J, Zhang HJ, et al. Quantitative and Fiber-Selective Evaluation of Pain and Sensory Dysfunction in Patients With Parkinson's Disease. Parkinsonism Relat Disord (2015) 21(4):361–5. doi: 10.1016/j.parkreldis.2015.01.008
111. Lodygin D, Hermann M, Schweingruber N, Flugel-Koch C, Watanabe T, Schlosser C, et al. Beta-Synuclein-Reactive T Cells Induce Autoimmune CNS Grey Matter Degeneration. Nature (2019) 566(7745):503–8. doi: 10.1038/s41586-019-0964-2
112. Ferreira N, Goncalves NP, Jan A, Jensen NM, van der Laan A, Mohseni S, et al. Trans-Synaptic Spreading of Alpha-Synuclein Pathology Through Sensory Afferents Leads to Sensory Nerve Degeneration and Neuropathic Pain. Acta Neuropathol Commun (2021) 9(1):31. doi: 10.1186/s40478-021-01131-8
113. Ding H, Wang Q, Liu J, Qian W, Wang W, Wang J, et al. Alterations of Gene Expression of Sodium Channels in Dorsal Root Ganglion Neurons of Estrogen Receptor Knockout (ERKO) Mice Induced by 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP). Endocrine (2012) 42(1):118–24. doi: 10.1007/s12020-012-9637-8
114. Taguchi T, Katanosaka K, Yasui M, Hayashi K, Yamashita M, Wakatsuki K, et al. Peripheral and Spinal Mechanisms of Nociception in a Rat Reserpine-Induced Pain Model. Pain (2015) 156(3):415–27. doi: 10.1097/01.j.pain.0000460334.49525.5e
115. Brefel-Courbon C, Ory-Magne F, Thalamas C, Payoux P, Rascol O. Nociceptive Brain Activation in Patients With Neuropathic Pain Related to Parkinson's Disease. Parkinsonism Relat Disord (2013) 19(5):548–52. doi: 10.1016/j.parkreldis.2013.02.003
116. Polli A, Weis L, Biundo R, Thacker M, Turolla A, Koutsikos K, et al. Anatomical and Functional Correlates of Persistent Pain in Parkinson's Disease. Mov Disord (2016) 31(12):1854–64. doi: 10.1002/mds.26826
117. Antonini A, Tinazzi M, Abbruzzese G, Berardelli A, Chaudhuri KR, Defazio G, et al. Pain in Parkinson's Disease: Facts and Uncertainties. Eur J Neurol (2018) 25(7):917–e69. doi: 10.1111/ene.13624
118. Hawkes CH, Del Tredici K, Braak H. A Timeline for Parkinson's Disease. Parkinsonism Relat Disord (2010) 16(2):79–84. doi: 10.1016/j.parkreldis.2009.08.007
119. Braak H, Sastre M, Bohl JR, de Vos RA, Del Tredici K. Parkinson's Disease: Lesions in Dorsal Horn Layer I, Involvement of Parasympathetic and Sympathetic Pre- and Postganglionic Neurons. Acta Neuropathol (2007) 113(4):421–9. doi: 10.1007/s00401-007-0193-x
120. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T Cells From Patients With Parkinson's Disease Recognize Alpha-Synuclein Peptides. Nature (2017) 546(7660):656–61. doi: 10.1038/nature22815
121. Mao X, Ou MT, Karuppagounder SS, Kam TI, Yin X, Xiong Y, et al. Pathological Alpha-Synuclein Transmission Initiated by Binding Lymphocyte-Activation Gene 3. Science (2016) 353(6307):1513–24. doi: 10.1126/science.aah3374
122. Cebrian C, Zucca FA, Mauri P, Steinbeck JA, Studer L, Scherzer CR, et al. MHC-I Expression Renders Catecholaminergic Neurons Susceptible to T-Cell-Mediated Degeneration. Nat Commun (2014) 5:3633. doi: 10.1038/ncomms4633
123. Cersosimo MG, Raina GB, Pecci C, Pellene A, Calandra CR, Gutierrez C, et al. Gastrointestinal Manifestations in Parkinson's Disease: Prevalence and Occurrence Before Motor Symptoms. J Neurol (2013) 260(5):1332–8. doi: 10.1007/s00415-012-6801-2
124. Sung HY, Park JW, Kim JS. The Frequency and Severity of Gastrointestinal Symptoms in Patients With Early Parkinson's Disease. J Mov Disord (2014) 7(1):7–12. doi: 10.14802/jmd.14002
125. Stankovic I, Petrovic I, Pekmezovic T, Markovic V, Stojkovic T, Dragasevic-Miskovic N, et al. Longitudinal Assessment of Autonomic Dysfunction in Early Parkinson's Disease. Parkinsonism Relat Disord (2019) 66:74–9. doi: 10.1016/j.parkreldis.2019.07.008
126. Martinez-Martin P. The Importance of non-Motor Disturbances to Quality of Life in Parkinson's Disease. J Neurol Sci (2011) 310(1-2):12–6. doi: 10.1016/j.jns.2011.05.006
127. Pont-Sunyer C, Hotter A, Gaig C, Seppi K, Compta Y, Katzenschlager R, et al. The Onset of Nonmotor Symptoms in Parkinson's Disease (the ONSET PD Study). Mov Disord (2015) 30(2):229–37. doi: 10.1002/mds.26077
128. Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD, et al. Frequency of Bowel Movements and the Future Risk of Parkinson's Disease. Neurology (2001) 57(3):456–62. doi: 10.1212/wnl.57.3.456
129. Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric Alpha-Synuclein Immunoreactive Inclusions in Meissner's and Auerbach's Plexuses in Cases Staged for Parkinson's Disease-Related Brain Pathology. Neurosci Lett (2006) 396(1):67–72. doi: 10.1016/j.neulet.2005.11.012
130. Shannon KM, Keshavarzian A, Mutlu E, Dodiya HB, Daian D, Jaglin JA, et al. Alpha-Synuclein in Colonic Submucosa in Early Untreated Parkinson's Disease. Mov Disord (2012) 27(6):709–15. doi: 10.1002/mds.23838
131. Kelly LP, Carvey PM, Keshavarzian A, Shannon KM, Shaikh M, Bakay RA, et al. Progression of Intestinal Permeability Changes and Alpha-Synuclein Expression in a Mouse Model of Parkinson's Disease. Mov Disord (2014) 29(8):999–1009. doi: 10.1002/mds.25736
132. Drolet RE, Cannon JR, Montero L, Greenamyre JT. Chronic Rotenone Exposure Reproduces Parkinson's Disease Gastrointestinal Neuropathology. Neurobiol Dis (2009) 36(1):96–102. doi: 10.1016/j.nbd.2009.06.017
133. Natale G, Kastsiushenka O, Fulceri F, Ruggieri S, Paparelli A, Fornai F. MPTP-Induced Parkinsonism Extends to a Subclass of TH-Positive Neurons in the Gut. Brain Res (2010) 1355:195–206. doi: 10.1016/j.brainres.2010.07.076
134. Hallett PJ, McLean JR, Kartunen A, Langston JW, Isacson O. Alpha-Synuclein Overexpressing Transgenic Mice Show Internal Organ Pathology and Autonomic Deficits. Neurobiol Dis (2012) 47(2):258–67. doi: 10.1016/j.nbd.2012.04.009
135. Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Bjorklund T, et al. Direct Evidence of Parkinson Pathology Spread From the Gastrointestinal Tract to the Brain in Rats. Acta Neuropathol (2014) 128(6):805–20. doi: 10.1007/s00401-014-1343-6
136. Svensson E, Horvath-Puho E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, et al. Vagotomy and Subsequent Risk of Parkinson's Disease. Ann Neurol (2015) 78(4):522–9. doi: 10.1002/ana.24448
137. Hui KY, Fernandez-Hernandez H, Hu J, Schaffner A, Pankratz N, Hsu NY, et al. Functional Variants in the LRRK2 Gene Confer Shared Effects on Risk for Crohn's Disease and Parkinson's Disease. Sci Transl Med (2018) 10(423). doi: 10.1126/scitranslmed.aai7795
138. Manfredsson FP, Luk KC, Benskey MJ, Gezer A, Garcia J, Kuhn NC, et al. Induction of Alpha-Synuclein Pathology in the Enteric Nervous System of the Rat and non-Human Primate Results in Gastrointestinal Dysmotility and Transient CNS Pathology. Neurobiol Dis (2018) 112:106–18. doi: 10.1016/j.nbd.2018.01.008
139. Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, et al. Intrastriatal Injection of Pre-Formed Mouse Alpha-Synuclein Fibrils Into Rats Triggers Alpha-Synuclein Pathology and Bilateral Nigrostriatal Degeneration. Neurobiol Dis (2015) 82:185–99. doi: 10.1016/j.nbd.2015.06.003
140. Chen Y, Yu M, Liu X, Qu H, Chen Q, Qian W, et al. Clinical Characteristics and Peripheral T Cell Subsets in Parkinson's Disease Patients With Constipation. Int J Clin Exp Pathol (2015) 8(3):2495–504.
141. Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, et al. Intestinal Infection Triggers Parkinson's Disease-Like Symptoms in Pink1(-/-) Mice. Nature (2019) 571(7766):565–9. doi: 10.1038/s41586-019-1405-y
142. Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, et al. Colonic Inflammation in Parkinson's Disease. Neurobiol Dis (2013) 50:42–8. doi: 10.1016/j.nbd.2012.09.007
143. Morales-Soto W, Gulbransen BD. Enteric Glia: A New Player in Abdominal Pain. Cell Mol Gastroenterol Hepatol (2019) 7(2):433–45. doi: 10.1016/j.jcmgh.2018.11.005
144. Liu T, Gao YJ, Ji RR. Emerging Role of Toll-Like Receptors in the Control of Pain and Itch. Neurosci Bull (2012) 28(2):131–44. doi: 10.1007/s12264-012-1219-5
145. Nishiwaki H, Ito M, Ishida T, Hamaguchi T, Maeda T, Kashihara K, et al. Meta-Analysis of Gut Dysbiosis in Parkinson's Disease. Mov Disord (2020) 35(9):1626–35. doi: 10.1002/mds.28119
146. Lin CH, Chen CC, Chiang HL, Liou JM, Chang CM, Lu TP, et al. Altered Gut Microbiota and Inflammatory Cytokine Responses in Patients With Parkinson's Disease. J Neuroinflamm (2019) 16(1):129. doi: 10.1186/s12974-019-1528-y
147. Zhu J, Paul WE. CD4 T Cells: Fates, Functions, and Faults. Blood (2008) 112(5):1557–69. doi: 10.1182/blood-2008-05-078154
148. Martin R, Chain F, Miquel S, Lu J, Gratadoux JJ, Sokol H, et al. The Commensal Bacterium Faecalibacterium Prausnitzii is Protective in DNBS-Induced Chronic Moderate and Severe Colitis Models. Inflamm Bowel Dis (2014) 20(3):417–30. doi: 10.1097/01.MIB.0000440815.76627.64
149. Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium Prausnitzii is an Anti-Inflammatory Commensal Bacterium Identified by Gut Microbiota Analysis of Crohn Disease Patients. Proc Natl Acad Sci U S A (2008) 105(43):16731–6. doi: 10.1073/pnas.0804812105
150. Zhu C, Song K, Shen Z, Quan Y, Tan B, Luo W, et al. Roseburia Intestinalis Inhibits Interleukin17 Excretion and Promotes Regulatory T Cells Differentiation in Colitis. Mol Med Rep (2018) 17(6):7567–74. doi: 10.3892/mmr.2018.8833
151. Patterson AM, Mulder IE, Travis AJ, Lan A, Cerf-Bensussan N, Gaboriau-Routhiau V, et al. Human Gut Symbiont Roseburia Hominis Promotes and Regulates Innate Immunity. Front Immunol (2017) 8:1166. doi: 10.3389/fimmu.2017.01166
152. Shen Z, Zhu C, Quan Y, Yang J, Yuan W, Yang Z, et al. Insights Into Roseburia Intestinalis Which Alleviates Experimental Colitis Pathology by Inducing Anti-Inflammatory Responses. J Gastroenterol Hepatol (2018) 33(10):1751–60. doi: 10.1111/jgh.14144
153. Clos-Garcia M, Andres-Marin N, Fernandez-Eulate G, Abecia L, Lavin JL, van Liempd S, et al. Gut Microbiome and Serum Metabolome Analyses Identify Molecular Biomarkers and Altered Glutamate Metabolism in Fibromyalgia. EBioMedicine (2019) 46:499–511. doi: 10.1016/j.ebiom.2019.07.031
154. Dekker Nitert M, Mousa A, Barrett HL, Naderpoor N, de Courten B. Altered Gut Microbiota Composition Is Associated With Back Pain in Overweight and Obese Individuals. Front Endocrinol (Lausanne) (2020) 11:605. doi: 10.3389/fendo.2020.00605
155. Zhang J, Song L, Wang Y, Liu C, Zhang L, Zhu S, et al. Beneficial Effect of Butyrate-Producing Lachnospiraceae on Stress-Induced Visceral Hypersensitivity in Rats. J Gastroenterol Hepatol (2019) 34(8):1368–76. doi: 10.1111/jgh.14536
156. Bhattacharyya D, Mohite GM, Krishnamoorthy J, Gayen N, Mehra S, Navalkar A, et al. Lipopolysaccharide From Gut Microbiota Modulates Alpha-Synuclein Aggregation and Alters Its Biological Function. ACS Chem Neurosci (2019) 10(5):2229–36. doi: 10.1021/acschemneuro.8b00733
157. Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS Sensitizes TRPV1 via Activation of TLR4 in Trigeminal Sensory Neurons. J Dent Res (2011) 90(6):759–64. doi: 10.1177/0022034511400225
158. Meseguer V, Alpizar YA, Luis E, Tajada S, Denlinger B, Fajardo O, et al. TRPA1 Channels Mediate Acute Neurogenic Inflammation and Pain Produced by Bacterial Endotoxins. Nat Commun (2014) 5:3125. doi: 10.1038/ncomms4125
159. Boonen B, Alpizar YA, Sanchez A, Lopez-Requena A, Voets T, Talavera K. Differential Effects of Lipopolysaccharide on Mouse Sensory TRP Channels. Cell Calcium (2018) 73:72–81. doi: 10.1016/j.ceca.2018.04.004
160. Zhou F, Wang X, Han B, Tang X, Liu R, Ji Q, et al. Short-Chain Fatty Acids Contribute to Neuropathic Pain via Regulating Microglia Activation and Polarization. Mol Pain (2021) 17:1744806921996520. doi: 10.1177/1744806921996520
161. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson's Disease. Cell (2016) 167(6):1469–80.e12. doi: 10.1016/j.cell.2016.11.018
162. Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, et al. Host Microbiota Constantly Control Maturation and Function of Microglia in the CNS. Nat Neurosci (2015) 18(7):965–77. doi: 10.1038/nn.4030
163. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic Treg Cell Homeostasis. Science (2013) 341(6145):569–73. doi: 10.1126/science.1241165
164. Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, et al. Metabolites Produced by Commensal Bacteria Promote Peripheral Regulatory T-Cell Generation. Nature (2013) 504(7480):451–5. doi: 10.1038/nature12726
165. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal Microbe-Derived Butyrate Induces the Differentiation of Colonic Regulatory T Cells. Nature (2013) 504(7480):446–50. doi: 10.1038/nature12721
166. Vinolo MA, Rodrigues HG, Nachbar RT, Curi R. Regulation of Inflammation by Short Chain Fatty Acids. Nutrients (2011) 3(10):858–76. doi: 10.3390/nu3100858
167. Kukkar A, Singh N, Jaggi AS. Attenuation of Neuropathic Pain by Sodium Butyrate in an Experimental Model of Chronic Constriction Injury in Rats. J Formos Med Assoc (2014) 113(12):921–8. doi: 10.1016/j.jfma.2013.05.013
168. Nohr MK, Egerod KL, Christiansen SH, Gille A, Offermanns S, Schwartz TW, et al. Expression of the Short Chain Fatty Acid Receptor GPR41/FFAR3 in Autonomic and Somatic Sensory Ganglia. Neuroscience (2015) 290:126–37. doi: 10.1016/j.neuroscience.2015.01.040
169. Sun MF, Zhu YL, Zhou ZL, Jia XB, Xu YD, Yang Q, et al. Neuroprotective Effects of Fecal Microbiota Transplantation on MPTP-Induced Parkinson's Disease Mice: Gut Microbiota, Glial Reaction and TLR4/TNF-Alpha Signaling Pathway. Brain Behav Immun (2018) 70:48–60. doi: 10.1016/j.bbi.2018.02.005
170. Vendrik KEW, Ooijevaar RE, de Jong PRC, Laman JD, van Oosten BW, van Hilten JJ, et al. Fecal Microbiota Transplantation in Neurological Disorders. Front Cell Infect Microbiol (2020) 10:98. doi: 10.3389/fcimb.2020.00098
171. Huang H, Xu H, Luo Q, He J, Li M, Chen H, et al. Fecal Microbiota Transplantation to Treat Parkinson's Disease With Constipation: A Case Report. Med (Baltimore) (2019) 98(26):e16163. doi: 10.1097/MD.0000000000016163
172. Xue LJ, Yang XZ, Tong Q, Shen P, Ma SJ, Wu SN, et al. Fecal Microbiota Transplantation Therapy for Parkinson's Disease: A Preliminary Study. Med (Baltimore) (2020) 99(35):e22035. doi: 10.1097/MD.0000000000022035
173. Kuai XY, Yao XH, Xu LJ, Zhou YQ, Zhang LP, Liu Y, et al. Evaluation of Fecal Microbiota Transplantation in Parkinson's Disease Patients With Constipation. Microb Cell Fact (2021) 20(1):98. doi: 10.1186/s12934-021-01589-0
174. Roman P, Abalo R, Marco EM, Cardona D. Probiotics in Digestive, Emotional, and Pain-Related Disorders. Behav Pharmacol (2018) 29(2 and 3-Spec Issue):103–19. doi: 10.1097/FBP.0000000000000385
Keywords: Parkinson’s disease, pain, nociceptor neurons, neuro-immunity, microbiota, dysbiosis, gut-brain axis
Citation: Roversi K, Callai-Silva N, Roversi K, Griffith M, Boutopoulos C, Prediger RD and Talbot S (2021) Neuro-Immunity and Gut Dysbiosis Drive Parkinson’s Disease-Induced Pain. Front. Immunol. 12:759679. doi: 10.3389/fimmu.2021.759679
Received: 16 August 2021; Accepted: 21 October 2021;
Published: 18 November 2021.
Edited by:
Fabienne Brilot, The University of Sydney, AustraliaReviewed by:
Hikoaki Fukaura, Saitama Medical University, JapanMaria F Cano-Abad, Autonomous University of Madrid, Spain
Copyright © 2021 Roversi, Callai-Silva, Roversi, Griffith, Boutopoulos, Prediger and Talbot. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sébastien Talbot, c2ViYXN0aWVuLnRhbGJvdEB1bW9udHJlYWwuY2E=; Rui Daniel Prediger, cnVpLnByZWRpZ2VyQHVmc2MuYnI=