 Ayelén Ivana Pesce Viglietti
Ayelén Ivana Pesce Viglietti Franco Agustín Sviercz
Franco Agustín Sviercz Cinthya Alicia Marcela López
Cinthya Alicia Marcela López Rosa Nicole Freiberger
Rosa Nicole Freiberger Jorge Quarleri
Jorge Quarleri María Victoria Delpino*
María Victoria Delpino*- Instituto de Investigaciones Biomédicas en Retrovirus y Sida (INBIRS), Universidad de Buenos Aires, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Buenos Aires, Argentina
Kingella kingae is an emerging pathogen that causes septic arthritis, osteomyelitis, and bacteremia in children from 6 to 48 months of age. The presence of bacteria within or near the bone is associated with an inflammatory process that results in osteolysis, but the underlying pathogenic mechanisms involved are largely unknown. To determine the link between K. kingae and bone loss, we have assessed whether infection per se or through the genesis of a pro-inflammatory microenvironment can promote osteoclastogenesis. For that purpose, we examined both the direct effect of K. kingae and the immune-mediated mechanism involved in K. kingae-infected macrophage-induced osteoclastogenesis. Our results indicate that osteoclastogenesis is stimulated by K. kingae infection directly and indirectly by fueling a potent pro-inflammatory response that drives macrophages to undergo functional osteoclasts via TNF-α and IL-1β induction. Such osteoclastogenic capability of K. kingae is counteracted by their outer membrane vesicles (OMV) in a concentration-dependent manner. In conclusion, this model allowed elucidating the interplay between the K. kingae and their OMV to modulate osteoclastogenesis from exposed macrophages, thus contributing to the modulation in joint and bone damage.
Introduction
Kingella kingae is a common etiology agent of septic arthritis, osteomyelitis, and bacteremia in children from 6 to 48 months of age. This bacteria is an emerging pediatric pathogen characterized by asymptomatic pharyngeal colonization that could be followed by bacterial translocation across the pharyngeal epithelial barrier with dissemination to distant locations, including infective endocarditis, bacteremia, septic arthritis, and osteomyelitis (1, 2). With the notorious exception of endocardial invasion, many patients are afebrile or have a mildly elevated body temperature and are in good general condition, and the yield of blood cultures is low, suggesting that the bacteremic stage of the disease is transient (1–3).
The bacterial infection localized in bones or contiguous tissue is a serious complication that causes bone loss. The presence of bacteria within or near the bone is associated with an inflammatory process that results in osteolysis, and K. kingae appears not to be an exception (4–6). The number of children colonized by this bacteria is more than those with clinical disease (7). Its infection can produce occult bacteremia, lower respiratory tract manifestations, and ocular infections. However, the major manifestation of K. kingae disease is represented by skeletal system infections (8). Patients treated with antibiotics without surgical intervention have frequently favorable outcomes, and even for those who were not treated at all, the need for extended intravenous treatment or surgical intervention seems very low (9). Self-limited involvement of the joints and/or bones in the course of a K. kingae bacteremic episode has also been reported, suggesting an abortive clinical course (2, 9–12).
A different virulence factor has been associated to K. kingae disease (8). Among these, K. kingae secretes the RtxA toxin that belongs to the family of Repeats in ToXin (RTX) cytotoxins that are produced by bacteria (13, 14). This is the main virulence factor identified, and its role as a virulence factor has been demonstrated in animal models of infection (13, 15).
K. kingae is protected from host immune response due to the presence of a capsule, which facilitates mucosal colonization and survival during bloodstream invasion (16–18). The attachment of the bacteria to the respiratory tract is possible due to the presence of type IV pili and KnnH adhesion (16). Bacteria use specialized secretion systems to deliver virulence factors (19). However, in addition to these systems, some bacteria release outer membrane vesicles (OMV) that act as a mechanism for the delivery of virulence factors to the host. In previous studies, it has been demonstrated that K. kingae produced OMV that could play a role in bacterial pathogenesis (4). The role of this OMV in bone pathology started to be elucidated. These studies revealed that OMV could be internalized by osteoblasts and synoviocytes, inducing the secretion of inflammatory mediators (4).
It is now recognized that the formation of osteoclasts is centered on the key osteoclastogenic cytokine, the receptor activator of NF-κB ligand (RANKL). The balance between RANKL and osteoprotegerin (its decoy receptor) in osteoblast determines the differentiation of new osteoclast under physiological conditions (20), but the cellular recruitment in chronic inflammatory bone disease, in particular B and T lymphocytes, neutrophils, dendritic cells, and macrophages, contributes to bone resorption. In this setting, not only RANKL but also proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6, are important in the development of the disease and bone loss (20–25). Bone resorption is accelerated by RANKL and cytokines produced by T and B lymphocytes (26–28). During inflammation, macrophages can contribute to bone resorption through the production of proinflammatory cytokines and also by differentiating into osteoclasts (28, 29).
To determine a link between K. kingae infection and loss of bone, we tested the hypothesis that the infection might create a microenvironment that would promote the generation of osteoclasts, the only cells known so far to be able to degrade bone. For that purpose, we examined the immune-mediated mechanisms of osteoclastogenesis triggered by K. kingae-infected macrophages. Moreover, as K. kingae effects on eukaryotic cells can be caused by a combination of virulence factors, we aim to investigate the direct effect and global impact of K. kingae on osteoclastogenesis.
This model allowed us to elucidate, at the single-cell-type level, the ability of K. kingae to modulate the osteoimmune response contributing to the observed bone damage and also to bacterial persistence.
Materials and Methods
Bacterial Culture
K. kingae strain was isolated from the joint fluid of a patient attended to at the Hospital de Clínicas “José de San Martín”, Buenos Aires, Argentina. It was grown on chocolate agar (Chocolate II Agar; BD, Franklin Lakes, NJ) or Brain Heart Infusion broth (BHI) with 5% of hemoglobin at 37°C with 5% CO2. The stocks were stored at -80°C in BHI with 30% of glycerol. The bacterial number in culture was estimated by comparing optical densities at 600 nm with a standard curve obtained in our laboratory.
To obtain the standard curve, the spectrophotometer was calibrated using BHI as the blank reference. A single colony of K. kingae was inoculated into 1 ml of BHI and then incubated at 37°C with 5% CO2 for 48 h. Then, serial dilutions of the inoculum were performed and used to determine the OD600, and another aliquot was plated on chocolate agar plates and incubated overnight at 37°C with 5% CO2 to determine the colony-forming unit counts.
To prepare the inoculum, cultures were diluted in sterile phosphate-buffered saline (PBS) to the desired bacterial concentration.
To obtain heat-killed K. kingae (HKKk), the bacteria were washed five times for 10 min each in sterile PBS, heat-killed at 60°C for 1 h, aliquoted, and stored at -70°C until they were used. The total absence of K. kingae viability after heat killing was verified by the absence of bacterial growth on chocolate agar.
All experiments were performed in BSL-2 and BSL-3 facilities at the INBIRS.
Isolation of Outer Membrane Vesicles
K. kingae was grown on chocolate agar plates for 72 h, and the bacteria were scraped from plates and harvested by centrifugation at 6,000 × g. The cell-free supernatant was sterilized through a 0.22-µm-pore-size filter. Then, the supernatant was ultracentrifuged at 150,000 × g for 6 h at 4°C to pellet the vesicles. The supernatant was removed, and the pellets were resuspended in PBS with 20% glycerol and 2 mM CaCl2. The protein concentration was measured using a bicinchoninic acid assay (Pierce). The OMV were stored at -80°C. Between 9 to 14 µg of OMV was obtained from 1 × 1010 bacteria.
Cellular Infection and HKKk Stimulation
Unless otherwise specified, all experiments were performed at 37°C in 5% CO2 atmosphere. THP-1 cells (human monocytic cell line) were obtained from the American Type Culture Collection (Manassas, VA) and grown in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (Gibco-BRL, Life Technologies, Grand Island, NY), 100 U/ml of penicillin, and 100 mg/ml of streptomycin (complete medium). To induce maturation, the cells were cultured in the presence of 150 U/ml of 1,25-dihydroxyvitamin D3 (Endogen) for 72 h. For the experiments, 5 × 105 cells/ml were stimulated with HKKk at a final concentration of 1 × 105 and 1 × 106 bacteria/ml or infected with K. kingae at multiplicities of infection of 1 and 10 for 2 h in medium without antibiotics. Then, the cells were extensively washed to remove extracellular bacteria, and the infected cells were maintained for an additional 24 h in a complete medium with 50 μg/ml of gentamicin. The supernatants were harvested and stored at -80°C for the determination of cytokine and osteoclastogenesis experiments. The viability of THP-1 cells after the addition of K. kingae (measured using trypan blue exclusion test) was preserved between 90 to 95% irrespective of the inoculum used.
Measurement of Cytokine Concentrations in Culture Supernatants
The secretion of human and mouse IL-1β, IL-6, and TNF-α in the supernatants was quantified with an ELISA kit (BD PharMingen, San Diego, CA, USA) and the production of human and mouse RANKL by an ELISA kit (R&D Systems Minneapolis, MN, USA).
Osteoclast Formation Assay
RAW 264.7 cells (mouse macrophage), at a concentration of 5 × 104 cells/well, were cultured onto glass coverslips in 24-well plates for 7 days and cultured in a complete medium containing 30 ng/ml macrophage colony-stimulating factor (M-CSF) and 0.2 ml of culture supernatants from K. kingae-infected THP-1 cells. As positive controls of osteoclast formation, the RAW 264.7 cell cultures received 50 ng/ml murine RANKL. On day 3, the culture medium and all reagents were replaced. To identify osteoclasts, the cells were fixed in 4% paraformaldehyde and stained for tartrate-resistant acid phosphatase (TRAP) (Sigma-Aldrich, St. Louis, MO, USA). TRAP-positive, multinucleated (more than three nuclei) cells were defined as osteoclasts, and the number was determined by microscopic counts. For each well, TRAP-positive multinucleated cells were calculated by counting their number in five microscopic fields (×20).
Direct Effect of K. kingae and Its OMV on Osteoclastogenesis
RAW264.7 cells were infected with K. kingae at multiplicity of infection (MOI) = 1 or stimulated with HKKk (1 × 105 bacteria/ml) in a complete medium without antibiotics containing 30 ng/ml M-CSF. Alternatively, RAW264.7 cells were exposed to the bacteria or HKKk during 2 h, then extensively washed with culture medium to remove extracellular bacteria, and incubated in complete medium containing 30 ng/ml M-CSF with 50 μg/ml of gentamicin. To determine the role of OMV, osteoclast differentiation was performed in the presence of HKKk (1 × 105 bacteria/ml) and different amounts of OMV (10, 1, and 0.1 μg/ml.). The presence of TRAP-positive multinucleated cells was determined as was previously described.
Pit Formation Assay
Osteoclast differentiation assay was performed on hydroxyapatite disks (BD BioCoat Osteologic (BD Biosciences, San Diego, CA, USA) for 9 days. The media and all reagents were replaced every day to avoid the acidification of the medium. After culturing with cells, the hydroxyapatite discs were washed with 1 M NH4OH to remove adherent cells. After rinsing with water, the hydroxyapatite discs were visually examined by light microscopy, and the area of resorption was calculated using the NIH Image J analysis software (http://rsbweb.nih.gov/ij/). The results were expressed as the percentage of the total area of the well that appears resorbed.
Neutralization Experiments
Neutralization experiments were performed using anti-TNF-α-neutralizing antibody (clone MAb1) or its isotype control (both from BD Biosciences, San Diego, CA) and anti-IL-1β-neutralizing antibody (clone AS10) or its isotype control (BD Biosciences). In the neutralization experiments with anti-TNF-α antibody or anti-IL-1β antibody, the conditioned medium was preincubated with the corresponding antibody (or isotype control) for 1 h at 37°C before use.
MTT Colorimetric Assay
Cell proliferation/viability was measured by MTT colorimetric assay (Sigma-Aldrich, Argentina) at 7 days post-infection or stimulation with HKKk and OMV, according to the instructions of the manufacturer. The absorbance was measured by using a microplate reader at 570 nm. The OD of each well was quantified as a percentage compared with the untreated cells. All experiments were carried out in triplicate.
Statistical Analysis
Statistical analysis was performed with one-way ANOVA. Multiple comparisons between all pairs of groups were made with Tukey’s post-test, and those against two groups were done with Student᾽s t-test and Mann–Whitney test. To determine normality, the Shapiro–Wilk normality test was used. Graphical and statistical analyses were performed with GraphPad Prism 7.0 software. Each experiment was performed in triplicate with different culture preparations on three independent occasions. Data were represented as mean ± SD measured in triplicate from three individual experiments.
Results
K. kingae Infection Induces Proinflammatory Cytokine Secretion
It is known that some proinflammatory cytokines could have the ability to promote or inhibit osteoclast differentiation directly or via the RANKL/OPG system (30). Thus, the production of IL-6, TNF-α, IL-1β, and RANKL in culture supernatants from THP-1 monocytes in response to K. kingae infection was examined. Infection of human monocytes with K. kingae performed at MOIs of 1 and 10 elicited the secretion of IL-6, TNF-α, and IL-1β at 24 h post-infection in an inoculum-dependent manner (Figure 1A). On the contrary, RANKL secretion was not detected in monocytes infected with K. kingae.
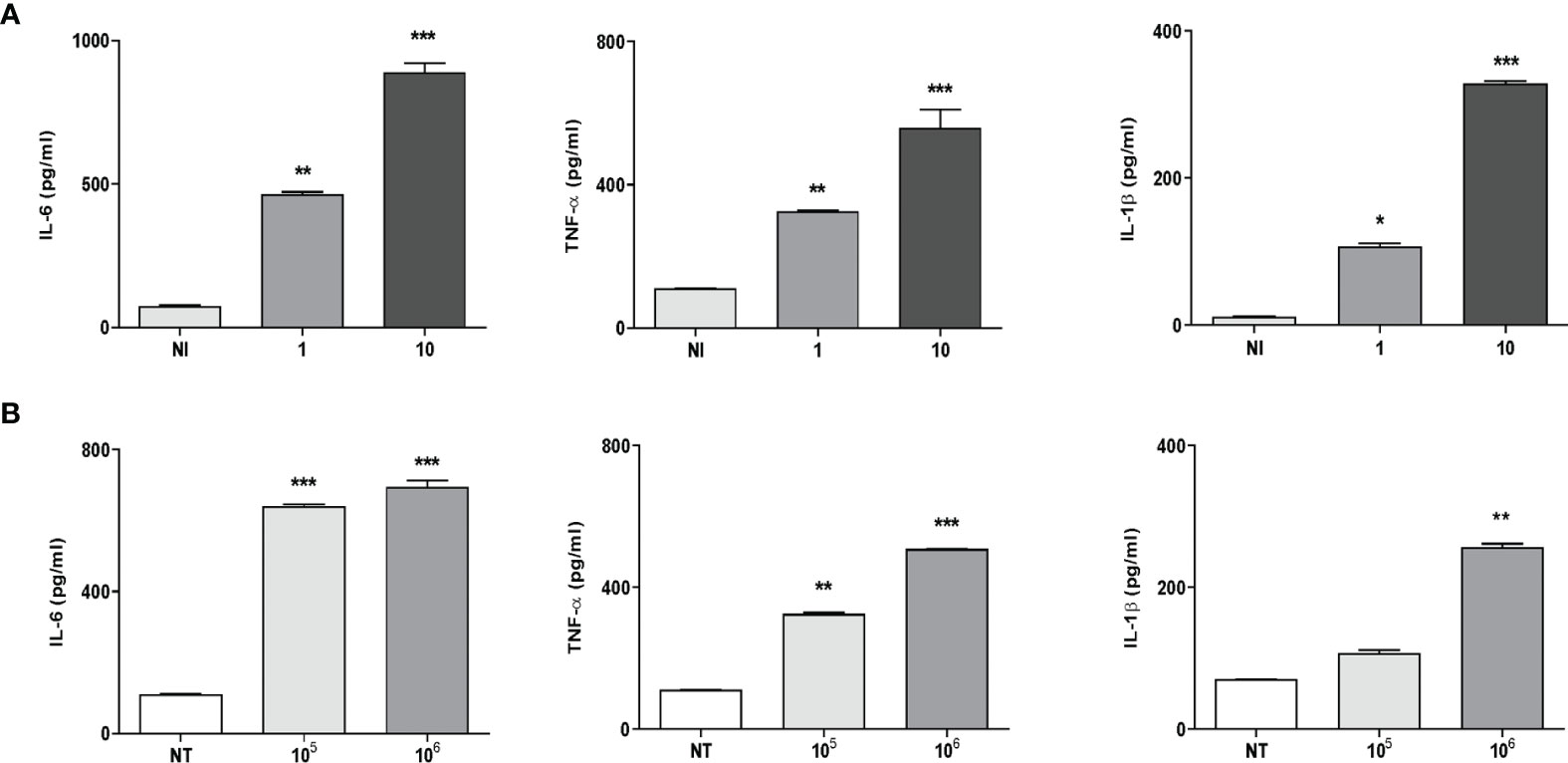
Figure 1 Monocytes produce proinflammatory cytokines in response to Kingella kingae infection. THP-1 cells were infected with K. kingae at different multiplicities of infection 1 and 10 (A) or stimulated with HKKk (1 × 105 and 1 × 106 bacteria/ml (B). IL-6, TNF-α, and IL-1β were determined in culture supernatants by ELISA at 24 h post-infection. NI, non-infected; NT, non-treated. The graphics are showing values obtained from three independent experiments. Data are given as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 versus control (NI and NT).
To elucidate whether viable bacteria were necessary to induce proinflammatory cytokine secretion, monocytes were stimulated with HKKk. The production of IL-6, TNF-α, and IL-1β was also markedly increased in culture supernatants from monocytes that were stimulated with HKKk when compared with unstimulated cells (Figure 1B). Additionally, the levels of cytokines induced by HKKk were comparable to those produced by similar amounts of live bacteria (Figure 1). These results indicate that human monocytes exposed to K. kingae can produce proinflammatory cytokines but do not produce RANKL. The ability of HKKk to produce proinflammatory cytokines also suggests that the inflammatory response could be induced by a structural component of the bacteria.
Culture Supernatants From K. kingae-Infected Monocytes Induce Osteoclast Differentiation Mediated by TNF-α and IL-1β
Osteoclasts originate from the fusion of precursors from monocyte/macrophage lineage in the bone marrow (31, 32) and play a key role in bone resorption. The differentiation process could involve soluble mediators belonging to inflammatory cells, such as monocytes/macrophages, in combination with M-CSF (33, 34). The role of IL-6 in the induction of osteoclast differentiation appears to be controversial and mainly indirectly mediated through osteoblast stimulation (30, 35–37). On the contrary, TNF-α and IL-1β have been implicated in RANKL-independent osteoclast formation (30, 34). To determine if soluble mediators secreted by K. kingae-infected human monocytes could stimulate osteoclast differentiation from RAW 264.7, these cells were stimulated with M-CSF in the presence of culture supernatants from K. kingae-infected human monocytes, and osteoclastogenesis was evaluated by the generation of TRAP-positive multinucleated cells. RANKL was used as a positive control. The production of osteoclast-like cells was generated by culture supernatants from K. kingae-infected human monocytes but not by those from uninfected cells.
To determine the role of TNF-α and IL-1β in the osteoclastogenesis elicited by K. kingae, stimulation experiments were performed as described above but in the presence of an anti-TNF-α- or an anti-IL-1β-neutralizing antibody. The TNF-α blocking antibody—but not the isotype control—completely inhibits osteoclastogenesis induced by culture supernatants from monocytes infected by K. kingae (Figure 2). Osteoclastogenesis was likewise significantly reduced in experiments performed with culture supernatants treated with IL-1β-blocking antibody in which the presence of binucleated cells could also be detected. These results indicate that monocyte-secreted TNF-α and IL-1β induced by K. kingae could be involved in the bone resorption observed in patients.
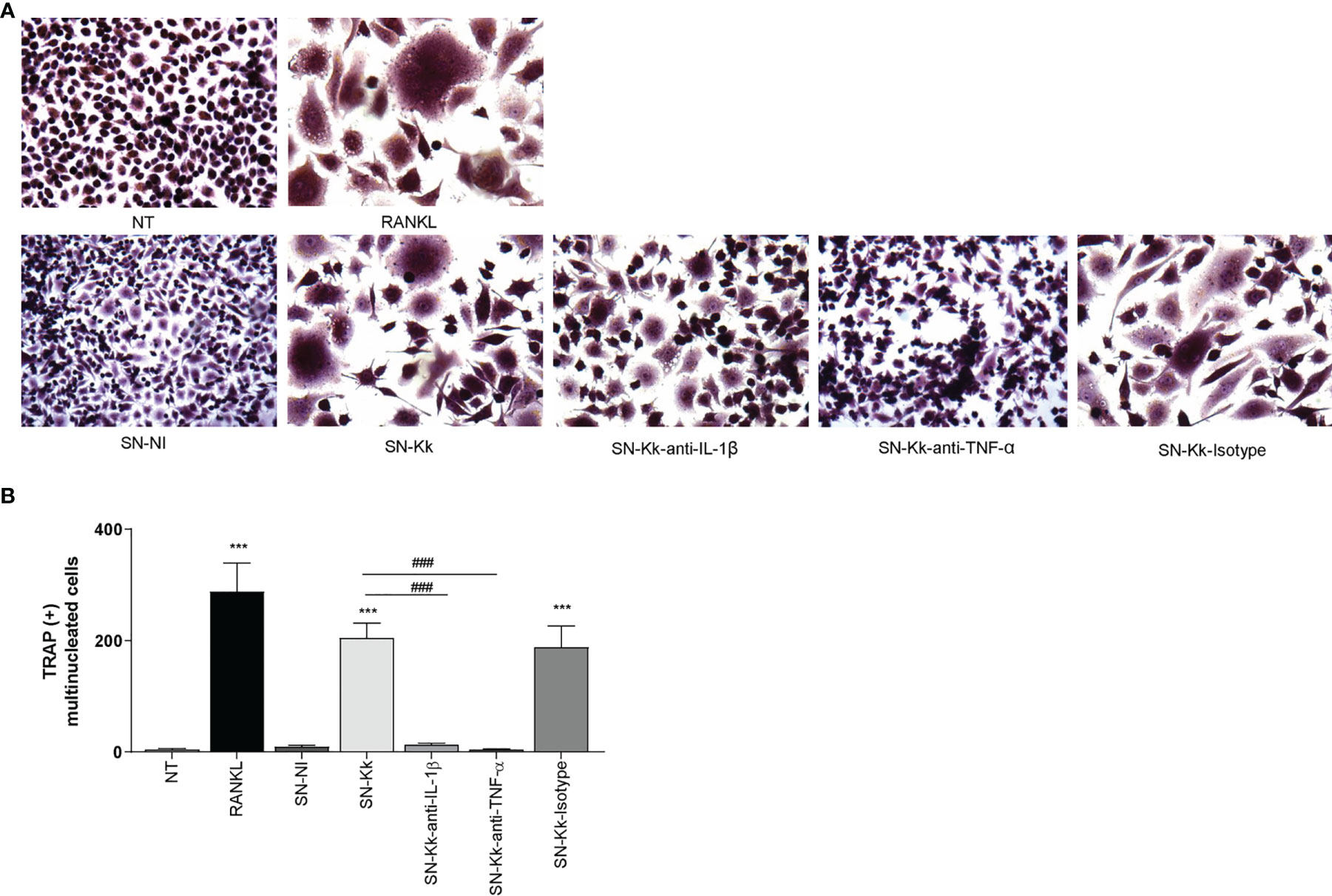
Figure 2 Culture supernatants from Kingella kingae-infected monocytes induce osteoclast differentiation mediated by TNF-α and IL-1β. Supernatants from K. kingae-infected THP-1 monocytes (multiplicity of infection = 1) induce RAW 264.7-derived osteoclastogenesis. RAW 264.7 cells were either untreated or stimulated with culture supernatants from K. kingae-infected THP-1 monocytes (SN-Kk) or with culture supernatants from non-infected THP-1 monocytes (SN-NI) added at a 1:2 proportion in conjunction with macrophage colony-stimulating factor. To inhibit TNF-α and IL-1β, experiments were conducted in the presence of neutralizing antibodies. After 7 days, osteoclastogenesis was determined by the generation of TRAP-positive multinucleated cells (more than three nuclei). Representative digital images were taken by light microscopy (A), and TRAP-positive multinucleated cells were identified and counted (B). RANKL was used as a positive control. NT, non-treated. The graphics are showing values obtained from three independent experiments. Data are given as mean ± SD. ***P < 0.001 versus cells treated with SN-NI or NT. ###P < 0.001.
TNF-α and IL-1β Secreted by K. kingae-Infected Monocytes Determine Proinflammatory Cytokine Secretion by Osteoclast
To better understand the osteoimmunological features involved in K. kingae-induced osteoclastogenesis, experiments were conducted to evaluate the ability of RAW 264.7-derived osteoclast to secrete cytokines in response to culture supernatants from K. kingae-infected THP-1 monocytes in the presence or not of neutralizing antibodies. Our results indicated that the addition of culture supernatants from K. kingae-infected human monocytes to uninfected murine osteoclast precursors induced a significant secretion of TNF-α, IL-1β, and IL-6 compared with unstimulated cells or cells stimulated with culture supernatants from uninfected monocytes. In addition, blocking of TNF-α and IL-1β significantly reduced the ability of culture supernatants from K. kingae-infected human monocytes to induce proinflammatory cytokine secretion, whereas the isotype control had no effect (Figure 3). Considering the species specificity of the antibodies used in the ELISA kit for cytokine measurement, the number of secreted factors present in culture supernatants from stimulated murine osteoclast precursors did not cross-react with human cytokines (not shown). These results indicate that the culture supernatants from K. kingae-infected monocytes activate proinflammatory cytokine production on osteoclast precursors.
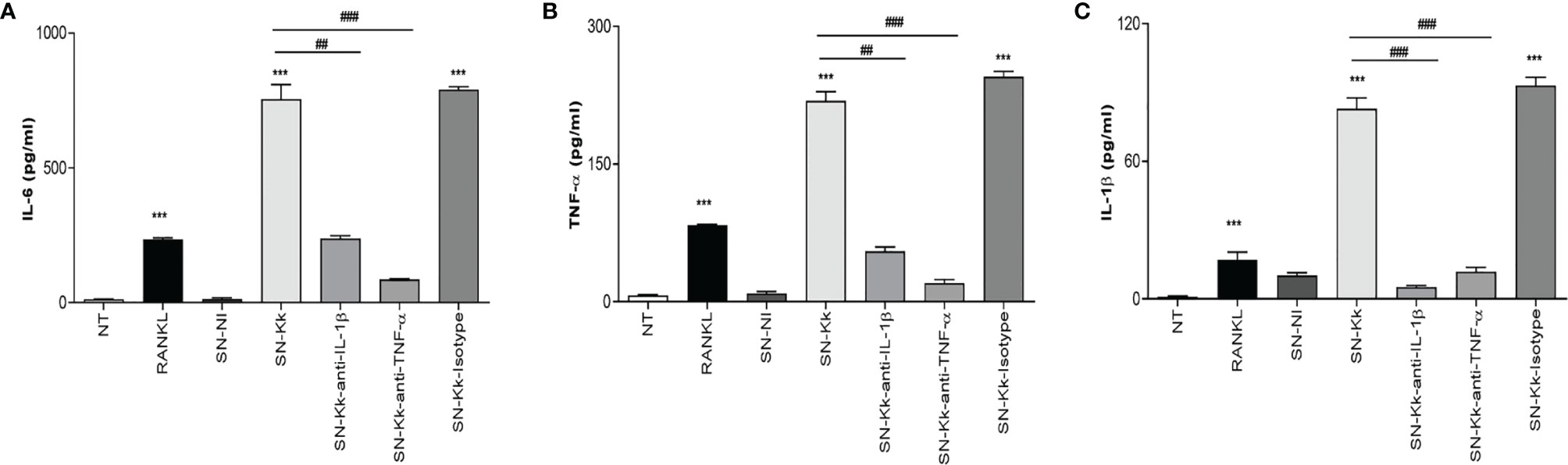
Figure 3 TNF-α and IL-1β secreted by Kingella kingae-infected monocytes determine proinflammatory cytokine secretion by osteoclast precursors. Culture supernatants from K. kingae-infected THP-1 monocytes at multiplicity of infection = 1 (SN-Kk), pre-incubated or not with anti-TNF-α or anti-IL-1β-neutralizing antibody or its isotype control, were used to stimulate osteoclast precursor RAW 264.7 cells. IL-6 (A), TNF-α (B), and IL-1β (C) secretion was determined in culture supernatants by ELISA. NT, non-treated. The graphics are showing values obtained from three independent experiments. Data are given as mean ± SD. ***P < 0.001 versus NT. ##P < 0.01; ###P < 0.001.
K. kingae Induces Functional Osteoclast Cells
To elucidate whether K. kingae infection can generate a microenvironment favoring osteoclastogenesis and bone loss, we have assessed the functional activity of K. kingae-induced osteoclast-like cells by their ability to resorb hydroxyapatite. The K. kingae-infected human monocytes were able to induce significant hydroxyapatite resorption. In contrast, culture supernatants from uninfected cells did not (Figure 4). Additionally, the resorption of hydroxyapatite was significantly inhibited either by anti-TNF-α antibody or by anti-IL-1β antibody but not by the isotype control. These results together indicate that K. kingae-infected monocytes released cytokines, such as TNF-α and IL-1β, that promote functional osteoclast formation.
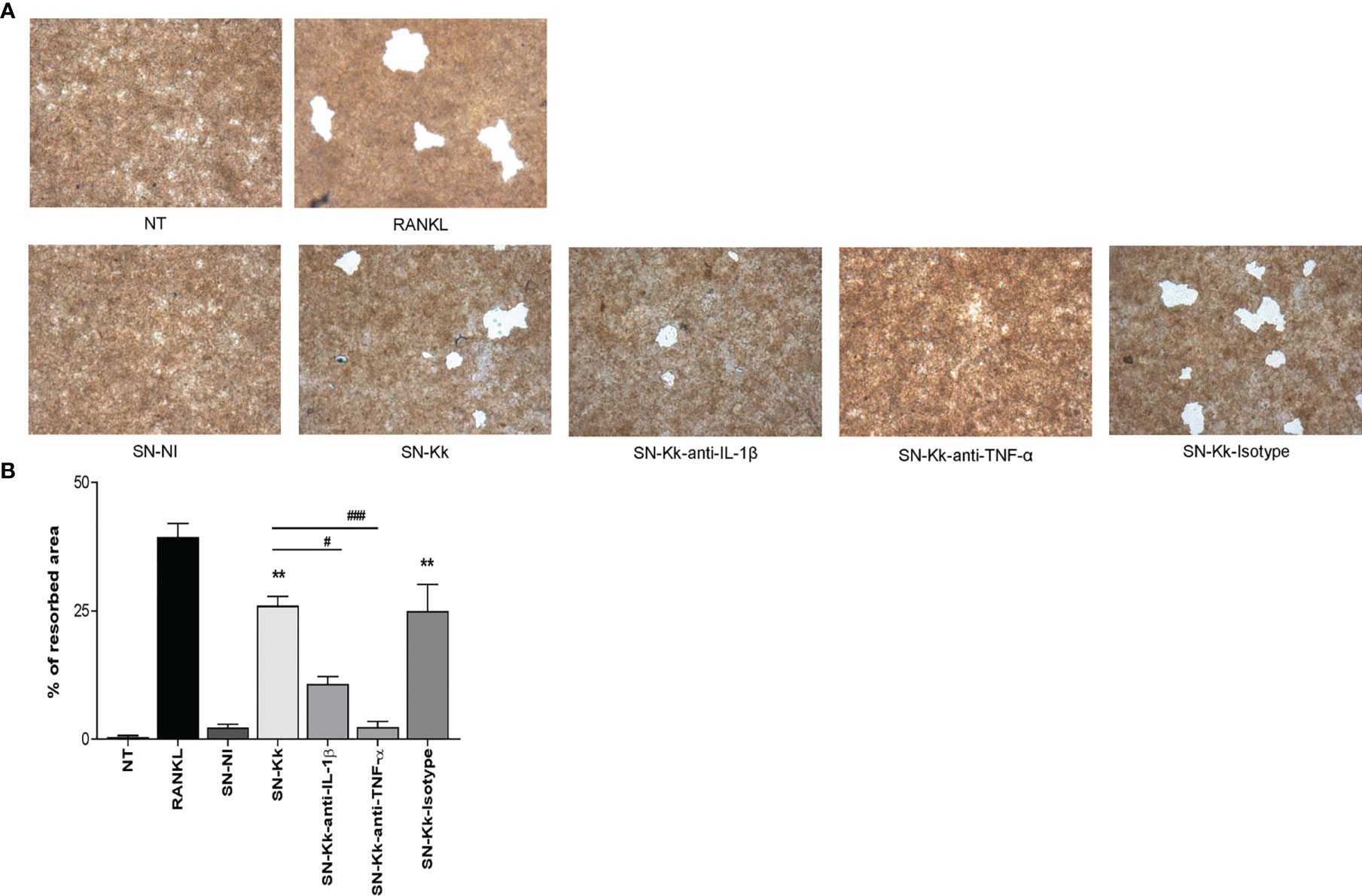
Figure 4 Kingella kingae induces functional osteoclasts cells. The functional activity of K. kingae-induced osteoclasts was determined by their ability to resorb hydroxyapatite. RAW 264.7 cells were cultured on hydroxyapatite disks under the same conditions as described above. After 7 days, the cells were removed, hydroxyapatite resorption was determined by light microscopy (A), and the resorbed area was quantified (% of resorbed area) (B). The graphics are showing values obtained from three independent experiments. Data are given as mean ± SD. *P < 0.05; **P < 0.01 versus cells treated with SN-NI or NT. ##P < 0.01; ###P < 0.001.
Pretreatment With K. kingae-Derived OMV Decreases the Cytokine Response of Monocytes to Kingella Infection
OMV from K. kingae have been previously implicated in the ability to induce cytokine production from osteoblast and synovial fibroblasts (4). Therefore, experiments were conducted to assess whether stimulation with K. kingae-released OMV can modulate the cytokine responses of human monocytes. To this end, cytokine production was measured in culture supernatants from THP-1 cells exposed to 0.1, 1, and 10 μg/ml of OMV. Our results indicated that THP-1 cells exposed to OMV during 4 and 24 h did not induce IL-1β, TNF-α, and IL-6 secretion (Figure 5).
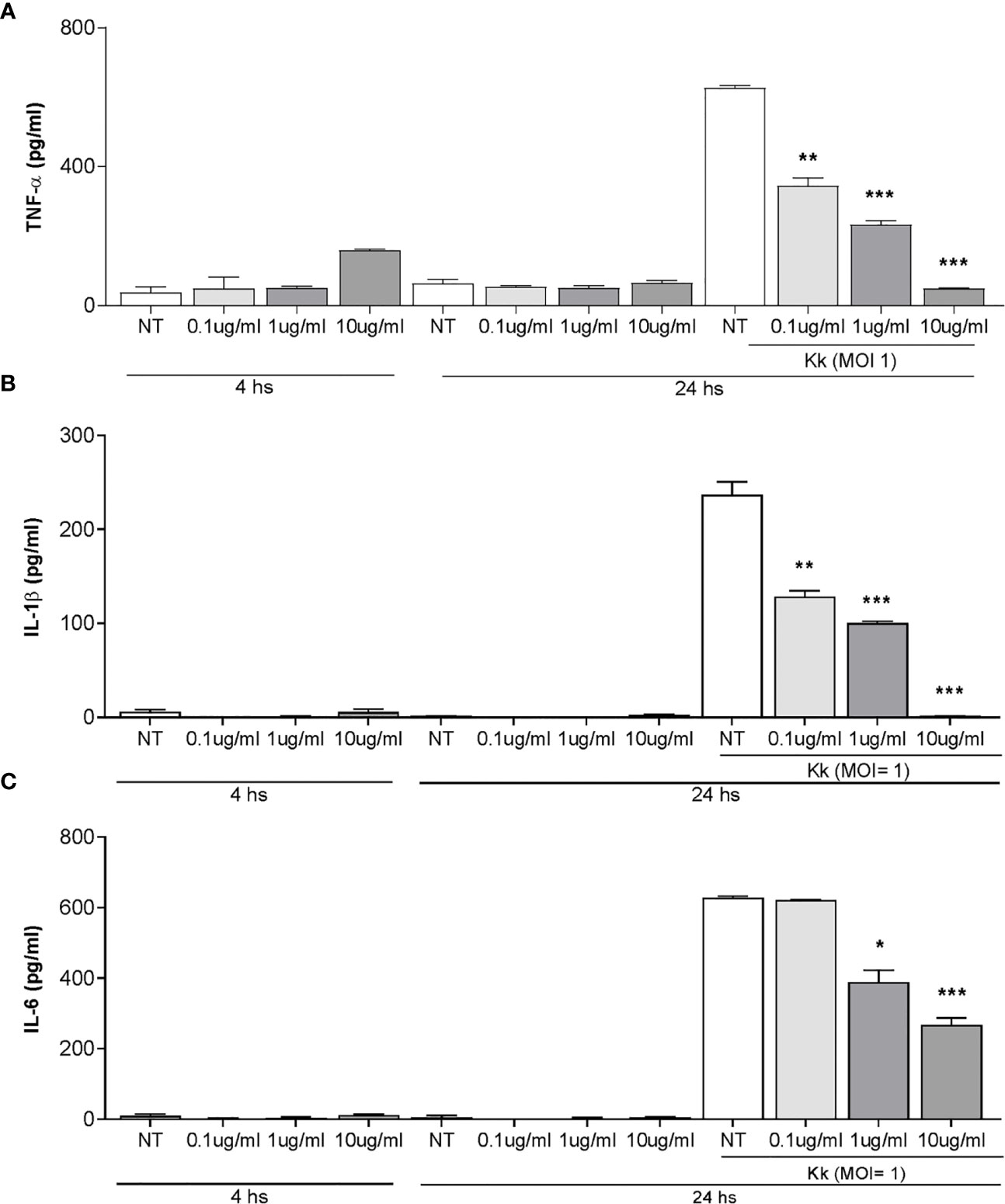
Figure 5 Pretreatment with Kingella kingae outer membrane vesicles (OMV) decreases the cytokine response of THP-1 cells to K. kingae infection. THP-1 cells were incubated with OMV at a concentration of 0.1, 1, and 10 µg/ml for 4 h as control cells were non-treated. Then, these cells were infected or not with K. kingae (multiplicity of infection = 1). Culture supernatants were collected at 4 h post-stimulation with OMV and at 24 h post-infection. TNF-α (A), IL-1β (B), and IL-6 (C) were measured by commercial ELISAs in the mentioned culture supernatants. The graphics are showing values obtained from three independent experiments. Data are given as the mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 versus NT.
OMV have a range of immunomodulatory outcomes to stimulate or suppress immune cell responses through their direct effects on host cells (38). Therefore, experiments were conducted to assess whether pre-exposure with K. kingae-derived OMV could modulate cytokine release from human monocytes in response to K. kingae infection. As shown in Figure 5, the level of TNF-α, IL-1β, and IL-6 released from THP-1 cells pre-treated with OMV was significantly lower than from untreated cells. Such inhibitory effects of OMV appear to be dose dependent (Figure 5C). In conjunction, these results indicated that OMV from K. kingae could inhibit proinflammatory response monocytes to K. kingae infection.
Pretreatment With OMV Decreases Osteoclastogenesis Induced by Supernatants From Monocytes Infected With K. kingae
Our results indicate that OMV are crucial for inflammatory responses induced by K. kingae. To assess the role of OMV in subsequent osteoclastogenesis induction in response to K. kingae infection, osteoclast differentiation was performed in the presence of M-CSF and culture supernatants from K. kingae-infected monocytes. Osteoclastogenesis was induced by culture supernatants obtained from K. kingae-infected human monocytes, but it was abrogated when cultured human monocytes were exposed to OMV before K. kingae infection (Figure 6). Additionally, culture supernatants from uninfected monocytes treated with OMV were unable to induce osteoclastogenesis (Figure 6). Collectively, these results indicate that cytokine response from human monocytes to K. kingae exposure may be conditioned by the OMV coexistence, even modulating its effects on osteoclastogenesis.
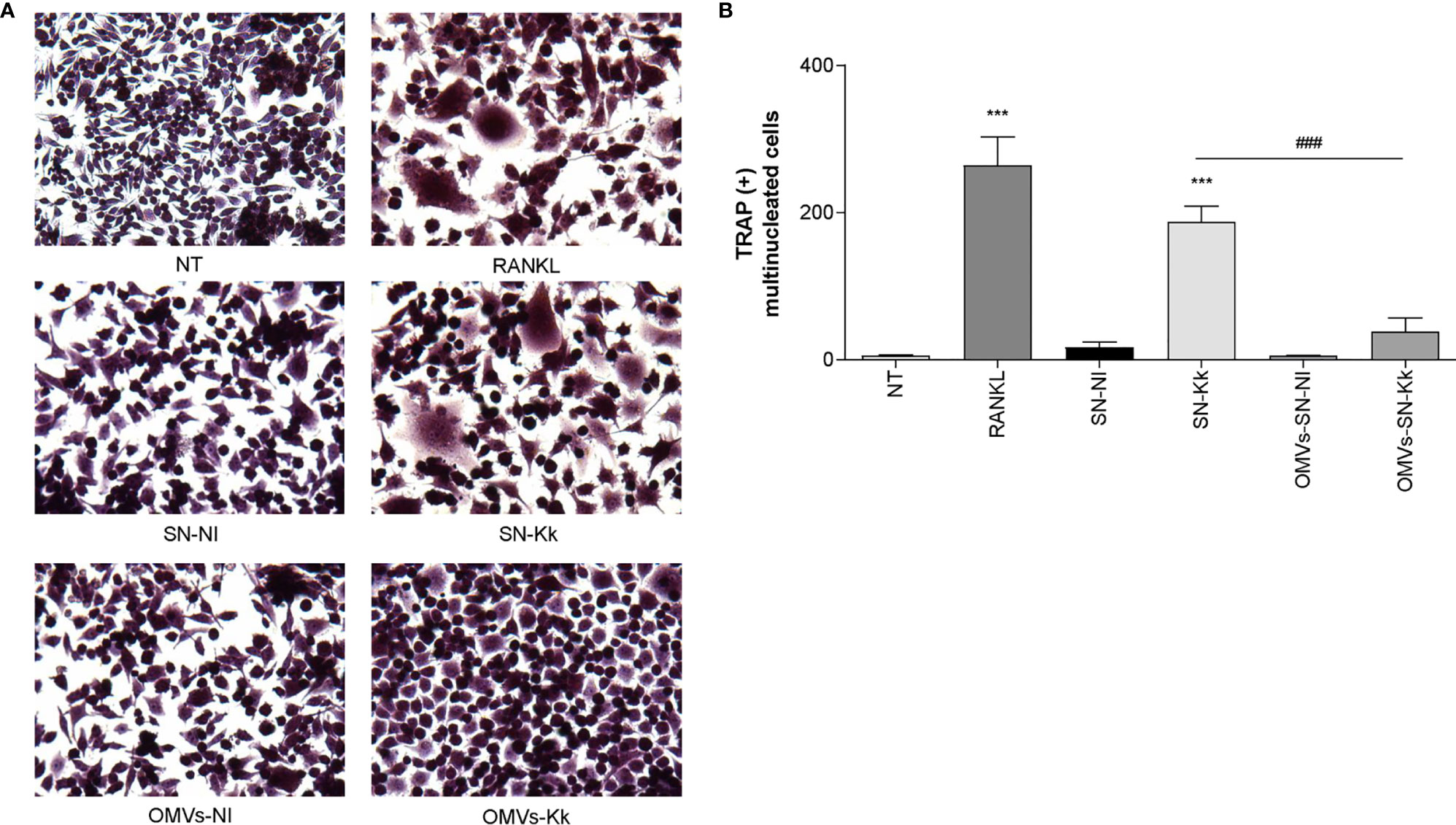
Figure 6 Pretreatment with outer membrane vesicles (OMV) decreases the osteoclastogenesis induced by supernatants from monocytes infected with Kingella kingae. RAW 264.7 cells were stimulated with culture supernatants from K. kingae-infected THP-1 monocytes (multiplicity of infection = 1) pretreated with OMV (10 µg/ml; OMV-SN-Kk) or with culture supernatants from non-infected THP-1 monocytes pretreated with OMV (OMV-SN-NI) or with supernatants non-pretreated with OMV (SN-Kk and SN-NI, respectively) added at a 1:2 proportion in conjunction with macrophage colony-stimulating factor. After 7 days, osteoclastogenesis was determined by the generation of TRAP-positive multinucleated cells (more than three nuclei). Representative digital images were taken by light microscopy (A), and TRAP-positive multinucleated cells were identified and counted (B). RANKL was used as a positive control. NT, non-treated. The graphics are showing values obtained from three independent experiments. Data are given as mean ± SD. ***P < 0.001 versus cells treated with SN-NI or NT. ###P < 0.001.
An Increase of OMV/Bacteria Ratio Is Related to the Reduction of Proinflammatory Microenvironment and Inhibition of Osteoclast Differentiation
Live bacteria could contribute to OMV release during in vitro infection experiments. To measure the OMV capability to downregulate the osteoclastogenesis, human monocytes were incubated with different doses of OMV prior to HKKk exposure, and then the supernatants were collected for osteoclastogenesis experiments.
As shown in Figure 7, the osteoclastogenesis was lower as the OMV dose was higher. In concordance, when monocytes were incubated with OMV before HKKk treatment, the secretion of IL-1β, TNF-α, and IL-6 was significantly lower than cells not pretreated with OMV. These results together indicate that K. kingae infection could have an opposite action on osteoclast differentiation which is dependent on the bacteria–OMV ratio.
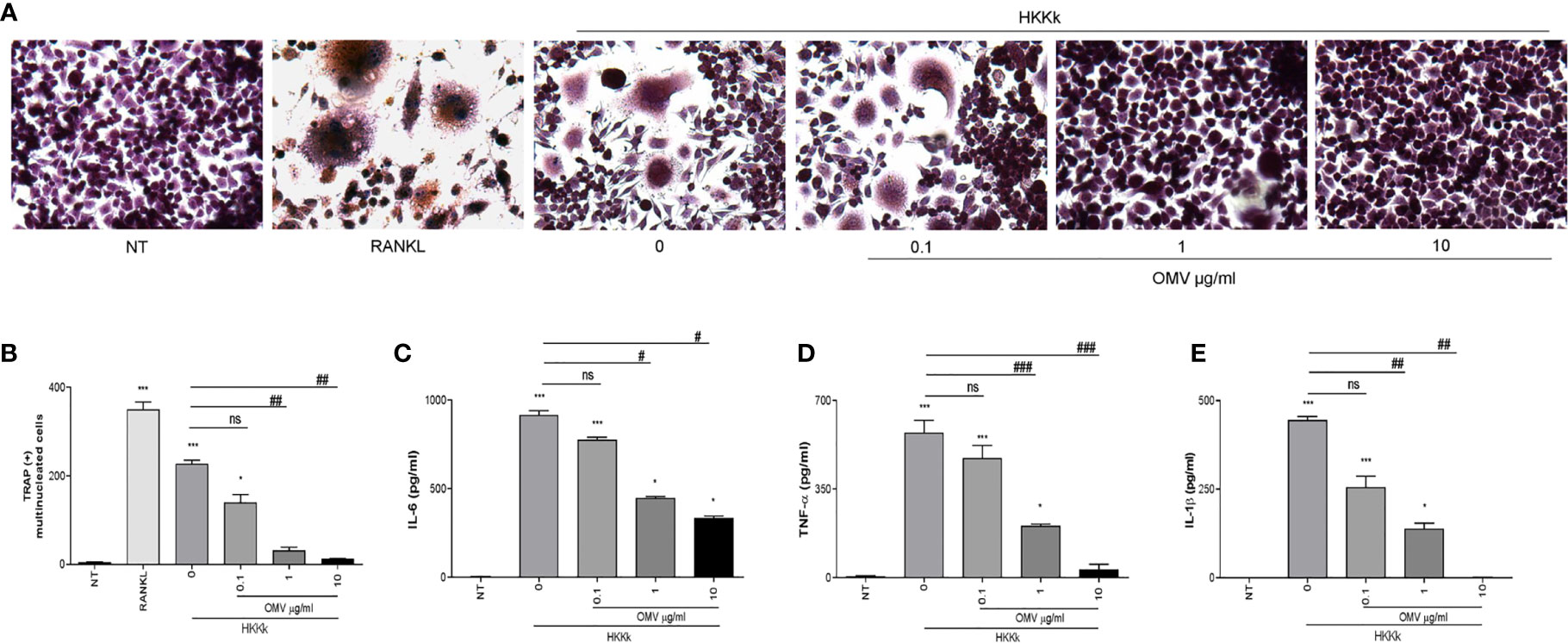
Figure 7 Outer membrane vesicles (OMV) pretreatment decrees osteoclastogenesis induced by HKKk in a dose-dependent manner. RAW 264.7 cells were stimulated with culture supernatants from HKKk-stimulated THP-1 monocytes (1 × 106 bacteria/ml) pretreated with OMV (0, 0.1, 1, and 10 µg/ml) added at a 1:2 proportion in conjunction with macrophage colony-stimulating factor. After 7 days, osteoclastogenesis was determined by the generation of multinucleated TRAP-positive cells (more than three nuclei). Representative digital images were taken by light microscopy (A), and TRAP-positive multinucleated cells were identified and counted (B). RANKL was used as a positive control. NT, non-treated. IL-6 (C), TNF-α (D), and IL-1β (E) were measured by commercial ELISAs in culture supernatants from THP-1 cells collected at 24 h post-stimulation. Graphics are showing values obtained from three independent experiments. Data are given as mean ± SD. *P < 0.05; ***P < 0.001 versus NT. #P < 0.05; ##P < 0.01; ###P < 0.001. ns, non-significant.
K. kingae Infection Induces Osteoclast Differentiation
The osteoarticular damage associated to K. kingae infection may also involve direct pathogenic mechanisms. To elucidate this contribution to osteoclastogenesis, the following experiments were carried out. First, cultured cells were infected with live K. kingae or stimulated with HKKk and maintained in the presence of M-CSF, without antibiotics. Second, the cultured cells were challenged as previously but during 2 h and then were washed and maintained in the presence of antibiotics. In both conditions, K. kingae infection was able to induce osteoclast differentiation (Figure 8). However, when osteoclast differentiation was performed in the absence of antibiotics, the number of osteoclasts from cells treated with HKKk was significantly higher than those produced by live bacteria. The RAW 264.7 cell viability of K. kingae-infected ones or from those treated with HKKk was well preserved (80–90%). These results show that K. kingae can directly promote osteoclastogenesis beyond its viability. Paradoxically, the osteoclastogenic capability of live bacteria is lower than that of the heat-killed ones, thus insinuating that viable bacteria-released factor/s may downregulate it.
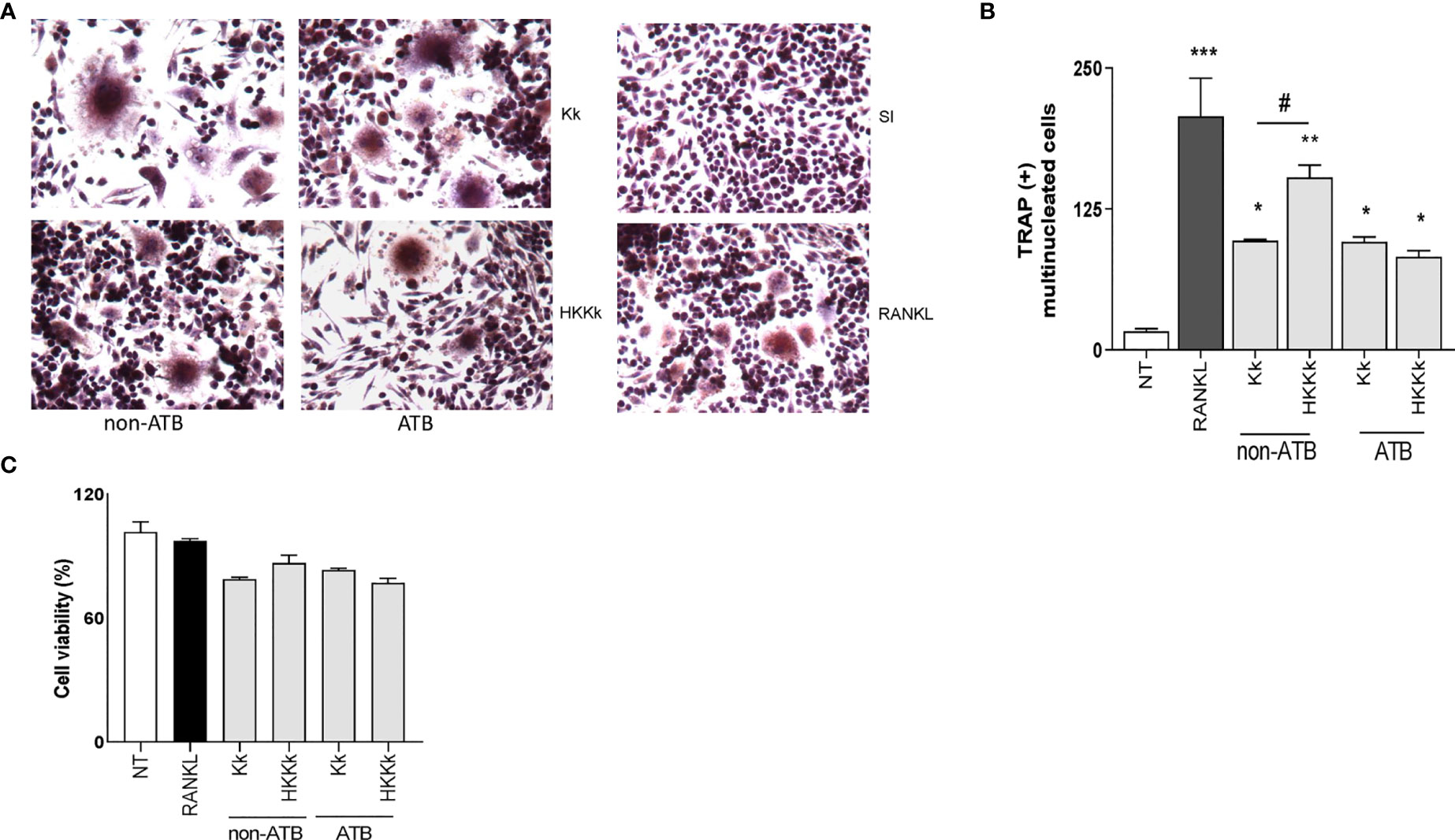
Figure 8 Kingella kingae induces osteoclast differentiation. RAW 264.7 cells were infected with K. kingae (multiplicity of infection = 1) and HKKk (1 × 105 bacteria/ml) added in conjunction with macrophage colony-stimulating factor without antibiotics (non-ATB). In some wells, the cells were washed, and new culture medium containing antibiotics was added after 2 h (ATB). After 7 days, osteoclastogenesis was determined by the generation of TRAP-positive multinucleated cells (more than three nuclei). Representative digital images were taken by light microscopy (A), and TRAP-positive multinucleated cells were identified and counted (B). Cell viability was measured by MTT colorimetric assay; results were expressed as percent of control (NT cells) (C). RANKL was used as a positive control. NT, non-treated. Graphics are showing values obtained from three independent experiments. Data are given as mean ± SD. *P < 0.05; ***P < 0.001 versus NT. #P < 0.05.
An Increase of OMV/Bacteria Ratio Reduces Osteoclastogenesis
Since HKKk induces higher levels of osteoclastogenesis than live bacteria, we have examined the role of OMV in such osteoclast differentiation. To this aim, RAW264.7 cells were pretreated with different doses of OMV and then treated with HKKk. The OMV diminished osteoclastogenesis in a concentration-dependent manner (Figure 9), but they were unable to induce osteoclastogenesis (not shown). These results together insinuate that K. kingae-mediated osteoclastogenesis may be counteracted directly by their OMV in a dose-dependent manner. The viability of RAW 264.7 cells exposed to HKKk and treated with OMV was preserved (80–90%).
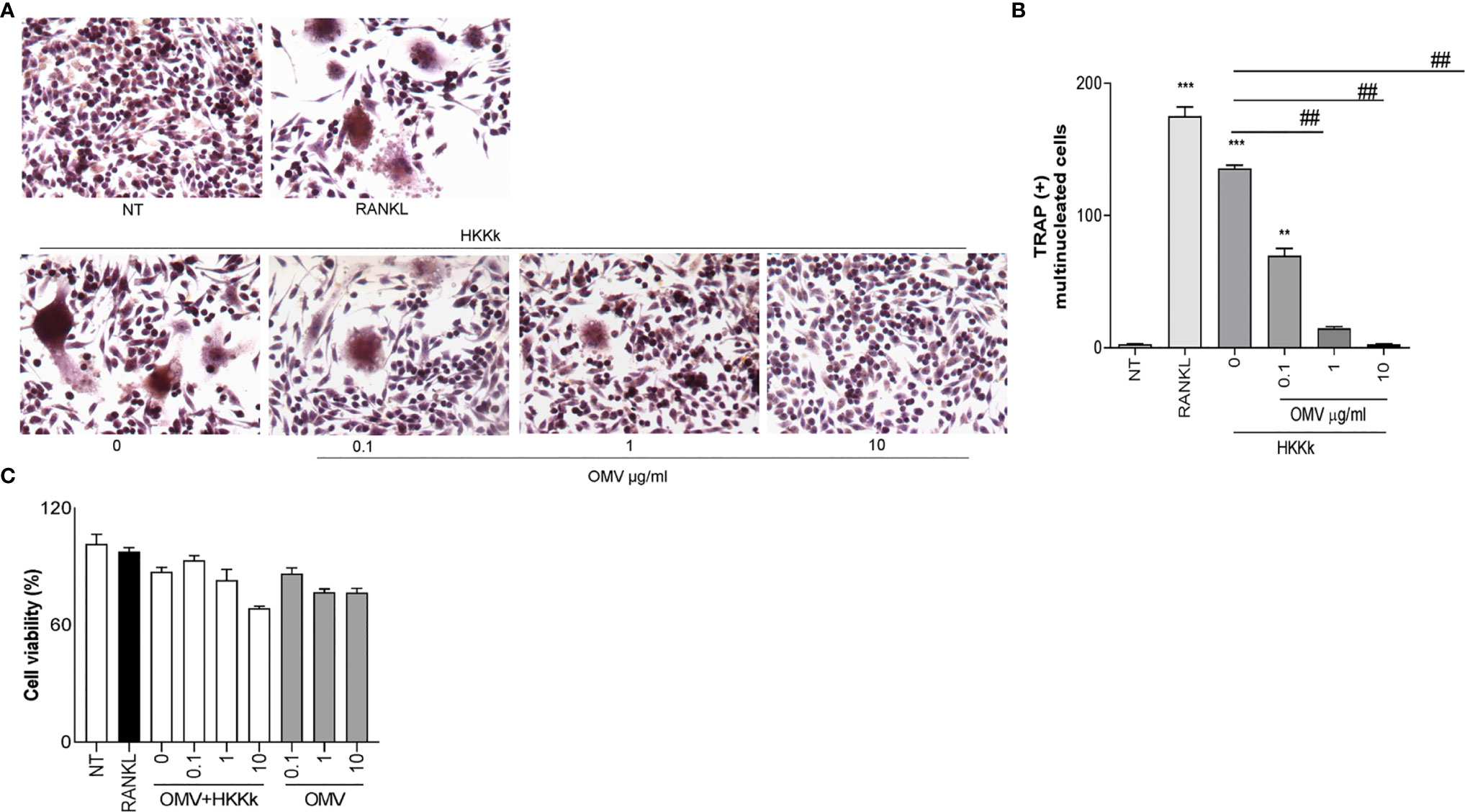
Figure 9 Outer membrane vesicles (OMV) inhibits osteoclast differentiation induced by HKKk. RAW 264.7 cells were stimulated HKKk (1 × 105 bacteria/ml) pretreated with OMV (0, 0.1, 1, and 10 µg/ml) in conjunction with macrophage colony-stimulating factor. After 7 days, osteoclastogenesis was determined by the generation of multinucleated TRAP-positive cells (more than three nuclei). Representative digital images were taken by light microscopy (A), and TRAP-positive multinucleated cells were identified and counted (B). Cell viability was measured by MTT colorimetric assay; results were expressed as percent of control (NT cells) (C). RANKL was used as a positive control. NT, non-treated. The graphics are showing values obtained from three independent experiments. Data are given as mean ± SD. **P < 0.01; ***P < 0.001 versus NT. ##P < 0.01.
Discussion
K. kingae mainly produces osteoarticular infections, including septic osteomyelitis and arthritis which cause bone and joint damage (2). While the clinical and imaging features of the osteoarticular disease produced by K. kingae have been widely described (8), the pathogenic mechanisms of bone loss have not been elucidated at the cellular and molecular levels. Different cell types are involved in bone resorption, including macrophages, which are pivotal ones not only by the secretion of proinflammatory cytokines but also by differentiating into osteoclasts (33).
Here we report a dual contribution of Kingella kingae to promote osteoclastogenesis using direct and indirect mechanisms. About the former, we have observed that K. kingae can directly propitiate macrophage differentiation to osteoclasts after an infection or even after exposure, thus inferring that bacteria viability is not an indispensable condition.
Under pathological conditions, the increase of proinflammatory cytokines and RANKL—a key molecule involved in osteoclast differentiation—could contribute to bone injury, encouraging osteoclastogenesis (20–29, 33, 39, 40). Therefore, such differentiation toward osteoclasts was also promoted indirectly by the secreted inflammatory mediators released by bacteria-challenged macrophages. The osteoclasts generated from precursor cells were phenotypically characterized by the generation of TRAP-positive multinucleated cells as well as functionally by the capability of these cells to induce hydroxyapatite resorption.
Interestingly, the two observed osteoclastogenic K. kingae-mediated pathways are counteracted by the bacteria-released OMV in a dose-dependent manner. Hence, it is tempting to speculate that in vivo, when environmental conditions propitiate OMV production, the tissue damage will be probably limited, while those that diminish OMV will likely increase the tissue damage by enhancing the osteoclastogenic activity. OMV also inhibit proinflammatory cytokine secretion by infected macrophages. It is in contrast with the previous report, in which K. kingae-derived OMV can promote a proinflammatory microenvironment by releasing IL-6 and granulocyte–M-CSF secretion from fibroblast, synoviocytes, and osteoblasts (4). The plausible dual OMV role in opposite outcome has been extensively reported for other Gram-negative bacteria (41–46). However, differences in K. kingae strain and/or in the preparation of OMV could also be implicated (4).
Both direct and indirect mechanisms of K. kingae-mediated osteoclastogenesis do not require bacterial viability, suggesting that osteoarticular damage could involve several virulence factors, such as structural bacterial components or even the RTX (RtxA) toxin. RtxA appears to be secreted in the extracellular environment in a soluble form and probably as a component of the OMV (4). RtxA toxin could participate in bacterial pathogenesis during extravasation of the airway epithelial respiratory barrier and could also contribute to synovium damage and the production of inflammatory mediators (13). The toxic effect of RtxA on THP-1 cells was demonstrated in previous studies (5); however, a non-significant toxic effect was detected in this study. This may be explained by the several differences that display monocyte and macrophage differentiated from THP-1 cells and the time of evaluation of cell death after K. kingae infection.
Our data revealed that K. kingae is capable of inducing bone destruction mediated by osteoclast independently of the osteoblast contribution. Nevertheless, other models deserve to be developed to determine the role of each bone cell type in bone damage during the infection. The interaction of bone cells during in vivo bone loss is not reached in our in vitro model. This is considered a limitation of our data. The development of a juvenile rat model was a significant contribution for examining K. kingae virulence (15); however, no in vivo septic arthritis model is able to determine the contribution of each cell type, the bacteria, and its OMV in the promotion of bone destruction.
In summary, here the results presented a novel pathogenic mechanism of osteoarticular disease triggered by the interplay between the direct and indirect effects of K. kingae on osteoclastogenesis and bacteria-released OMV in the opposite direction.
Finally, the present study constitutes the first analysis on osteoclastogenesis alterations during K. kingae infection. The role of OMV as a modulator of direct infection and the inflammatory response provide an initial background for more detailed studies as a possible target of therapeutic interventions.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
AP, FS, CL, and RF performed the experiments. All authors analyzed the data. MD wrote the article. MD and JQ designed the experiments, revised the article, and obtained research funding. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from Agencia Nacional of Promoción Científica y Tecnológica (ANPCYT, Argentina): PICT 2014-1111, PICT 2015-0316, and PICT 2017-2859 to MD and PICT-2015-1921 to JQ. The funding agency had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dubnov-Raz G, Ephros M, Garty BZ, Schlesinger Y, Maayan-Metzger A, Hasson J, et al. Invasive Pediatric Kingella Kingae Infections: A Nationwide Collaborative Study. Pediatr Infect Dis J (2010) 29(7):639–43. doi: 10.1097/INF.0b013e3181d57a6c
2. Yagupsky P. Kingella Kingae Infections of the Skeletal System in Children: Diagnosis and Therapy. Expert Rev Anti Infect Ther (2004) 2(5):787–94. doi: 10.1586/14789072.2.5.787
3. Dubnov-Raz G, Scheuerman O, Chodick G, Finkelstein Y, Samra Z, Garty BZ. Invasive Kingella Kingae Infections in Children: Clinical and Laboratory Characteristics. Pediatrics (2008) 122(6):1305–9. doi: 10.1542/peds.2007-3070
4. Maldonado R, Wei R, Kachlany SC, Kazi M, Balashova NV. Cytotoxic Effects of Kingella Kingae Outer Membrane Vesicles on Human Cells. Microb Pathog (2011) 51(1-2):22–30. doi: 10.1016/j.micpath.2011.03.005
5. Srikhanta YN, Fung KY, Pollock GL, Bennett-Wood V, Howden BP, Hartland EL. Phasevarion-Regulated Virulence in the Emerging Pediatric Pathogen Kingella Kingae. Infect Immun (2017) 85(12):1–15. doi: 10.1128/IAI.00319-17
6. Coulin B, Demarco G, Spyropoulou V, Juchler C, Vendeuvre T, Habre C, et al. Osteoarticular Infection in Children. Bone Joint J (2021) 103-B(3):578–83. doi: 10.1302/0301-620X.103B3.BJJ-2020-0936.R2
7. Ceroni D, Dubois-Ferriere V, Anderson R, Combescure C, Lamah L, Cherkaoui A, et al. Small Risk of Osteoarticular Infections in Children With Asymptomatic Oropharyngeal Carriage of Kingella Kingae. Pediatr Infect Dis J (2012) 31(9):983–5. doi: 10.1097/INF.0b013e31825d3419
8. Yagupsky P. Kingella Kingae: Carriage, Transmission, and Disease. Clin Microbiol Rev (2015) 28(1):54–79. doi: 10.1128/CMR.00028-14
9. Lebel E, Rudensky B, Karasik M, Itzchaki M, Schlesinger Y. Kingella Kingae Infections in Children. J Pediatr Orthop B (2006) 15(4):289–92. doi: 10.1097/01202412-200607000-00011
10. Birgisson H, Steingrimsson O, Gudnason T. Kingella Kingae Infections in Paediatric Patients: 5 Cases of Septic Arthritis, Osteomyelitis and Bacteraemia. Scand J Infect Dis (1997) 29(5):495–8. doi: 10.3109/00365549709011861
11. Caballero Rabasco MA, Gonzalez Cuevas A, Martinez Roig A. Isolated Bacteraemia Caused by Kingella Kingae. Pediatr (Barc) (2010) 72(1):89–90. doi: 10.1016/j.anpedi.2009.09.005
12. Dodman T, Robson J, Pincus D. Kingella Kingae Infections in Children. J Paediatr Child Health (2000) 36(1):87–90. doi: 10.1046/j.1440-1754.2000.00447.x
13. Kehl-Fie TE, St Geme JW 3rd. Identification and Characterization of an RTX Toxin in the Emerging Pathogen Kingella Kingae. J Bacteriol (2007) 189(2):430–6. doi: 10.1128/JB.01319-06
14. Osickova A, Balashova N, Masin J, Sulc M, Roderova J, Wald T, et al. Cytotoxic Activity of Kingella Kingae RtxA Toxin Depends on Post-Translational Acylation of Lysine Residues and Cholesterol Binding. Emerg Microbes Infect (2018) 7(1):178. doi: 10.1038/s41426-018-0179-x
15. Chang DW, Nudell YA, Lau J, Zakharian E, Balashova NV. RTX Toxin Plays a Key Role in Kingella Kingae Virulence in an Infant Rat Model. Infect Immun (2014) 82(6):2318–28. doi: 10.1128/IAI.01636-14
16. Porsch EA, Kehl-Fie TE, St Geme JW 3rd. Modulation of Kingella Kingae Adherence to Human Epithelial Cells by Type IV Pili, Capsule, and a Novel Trimeric Autotransporter. mBio (2012) 3(5):1–9. doi: 10.1128/mBio.00372-12
17. Starr KF, Porsch EA, Heiss C, Black I, Azadi P, St Geme JW 3rd. Characterization of the Kingella Kingae Polysaccharide Capsule and Exopolysaccharide. PloS One (2013) 8(9):e75409. doi: 10.1371/journal.pone.0075409
18. Starr KF, Porsch EA, Seed PC, Heiss C, Naran R, Forsberg LS, et al. Kingella Kingae Expresses Four Structurally Distinct Polysaccharide Capsules That Differ in Their Correlation With Invasive Disease. PloS Pathog (2016) 12(10):e1005944. doi: 10.1371/journal.ppat.1005944
19. Green ER, Mecsas J. Bacterial Secretion Systems: An Overview. Microbiol Spectr (2016) 4(1):1–19. doi: 10.1128/microbiolspec.VMBF-0012-2015
20. Suda T, Kobayashi K, Jimi E, Udagawa N, Takahashi N. The Molecular Basis of Osteoclast Differentiation and Activation. Novartis Found Symp (2001) 232:235–47. doi: 10.1002/0470846658.ch16
21. Haynes DR. Bone Lysis and Inflammation. Inflamm Res (2004) 53(11):596–600. doi: 10.1007/s00011-004-1303-z
22. Merkel KD, Erdmann JM, McHugh KP, Abu-Amer Y, Ross FP, Teitelbaum SL. Tumor Necrosis Factor-Alpha Mediates Orthopedic Implant Osteolysis. Am J Pathol (1999) 154(1):203–10. doi: 10.1016/s0002-9440(10)65266-2
23. Nair SP, Meghji S, Wilson M, Reddi K, White P, Henderson B. Bacterially Induced Bone Destruction: Mechanisms and Misconceptions. Infect Immun (1996) 64(7):2371–80. doi: 10.1128/IAI.64.7.2371-2380.1996
24. Wei S, Kitaura H, Zhou P, Ross FP, Teitelbaum SL. IL-1 Mediates TNF-Induced Osteoclastogenesis. J Clin Invest (2005) 115(2):282–90. doi: 10.1172/JCI23394
25. Kotake S, Sato K, Kim KJ, Takahashi N, Udagawa N, Nakamura I, et al. Interleukin-6 and Soluble Interleukin-6 Receptors in the Synovial Fluids From Rheumatoid Arthritis Patients Are Responsible for Osteoclast-Like Cell Formation. J Bone Miner Res (1996) 11(1):88–95. doi: 10.1002/jbmr.5650110113
26. Revell PA, Mayston V, Lalor P, Mapp P. The Synovial Membrane in Osteoarthritis: A Histological Study Including the Characterisation of the Cellular Infiltrate Present in Inflammatory Osteoarthritis Using Monoclonal Antibodies. Ann Rheum Dis (1988) 47(4):300–7. doi: 10.1136/ard.47.4.300
27. Smith MD, O’Donnell J, Highton J, Palmer DG, Rozenbilds M, Roberts-Thomson PJ. Immunohistochemical Analysis of Synovial Membranes From Inflammatory and Non-Inflammatory Arthritides: Scarcity of CD5 Positive B Cells and IL2 Receptor Bearing T Cells. Pathology (1992) 24(1):19–26. doi: 10.3109/00313029209063615
28. Chu CQ, Field M, Allard S, Abney E, Feldmann M, Maini RN. Detection of Cytokines at the Cartilage/Pannus Junction in Patients With Rheumatoid Arthritis: Implications for the Role of Cytokines in Cartilage Destruction and Repair. Br J Rheumatol (1992) 31(10):653–61. doi: 10.1093/rheumatology/31.10.653
29. Adamopoulos IE, Sabokbar A, Wordsworth BP, Carr A, Ferguson DJ, Athanasou NA. Synovial Fluid Macrophages Are Capable of Osteoclast Formation and Resorption. J Pathol (2006) 208(1):35–43. doi: 10.1002/path.1891
30. Zupan J, Jeras M, Marc J. Osteoimmunology and the Influence of Pro-Inflammatory Cytokines on Osteoclasts. Biochem Med (Zagreb) (2013) 23(1):43–63. doi: 10.11613/bm.2013.007
31. Ibbotson KJ, Roodman GD, McManus LM, Mundy GR. Identification and Characterization of Osteoclast-Like Cells and Their Progenitors in Cultures of Feline Marrow Mononuclear Cells. J Cell Biol (1984) 99(2):471–80. doi: 10.1083/jcb.99.2.471
32. Scheven BA, Visser JW, Nijweide PJ. In Vitro Osteoclast Generation From Different Bone Marrow Fractions, Including a Highly Enriched Haematopoietic Stem Cell Population. Nature (1986) 321(6065):79–81. doi: 10.1038/321079a0
33. Ukai T, Yumoto H, Gibson FC 3rd, Genco CA. Macrophage-Elicited Osteoclastogenesis in Response to Bacterial Stimulation Requires Toll-Like Receptor 2-Dependent Tumor Necrosis Factor-Alpha Production. Infect Immun (2008) 76(2):812–9. doi: 10.1128/IAI.01241-07
34. Feng W, Guo J, Li M. RANKL-Independent Modulation of Osteoclastogenesis. J Oral Biosci (2019) 61(1):16–21. doi: 10.1016/j.job.2019.01.001
35. Yoshitake F, Itoh S, Narita H, Ishihara K, Ebisu S. Interleukin-6 Directly Inhibits Osteoclast Differentiation by Suppressing Receptor Activator of NF-kappaB Signaling Pathways. J Biol Chem (2008) 283(17):11535–40. doi: 10.1074/jbc.M607999200
36. Duplomb L, Baud’huin M, Charrier C, Berreur M, Trichet V, Blanchard F, et al. Interleukin-6 Inhibits Receptor Activator of Nuclear Factor kappaB Ligand-Induced Osteoclastogenesis by Diverting Cells Into the Macrophage Lineage: Key Role of Serine727 Phosphorylation of Signal Transducer and Activator of Transcription 3. Endocrinology (2008) 149(7):3688–97. doi: 10.1210/en.2007-1719
37. Wu Q, Zhou X, Huang D, Ji Y, Kang F. IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity. In Vitro Cell Physiol Biochem (2017) 41(4):1360–9. doi: 10.1159/000465455
38. Kaparakis-Liaskos M, Ferrero RL. Immune Modulation by Bacterial Outer Membrane Vesicles. Nat Rev Immunol (2015) 15(6):375–87. doi: 10.1038/nri3837
39. Delpino MV, Barrionuevo P, Macedo GC, Oliveira SC, Genaro SD, Scian R, et al. Macrophage-Elicited Osteoclastogenesis in Response to Brucella Abortus Infection Requires TLR2/MyD88-Dependent TNF-Alpha Production. J Leukoc Biol (2012) 91(2):285–98. doi: 10.1189/jlb.04111185
40. Mendoza Bertelli A, Delpino MV, Lattar S, Giai C, Llana MN, Sanjuan N, et al. Staphylococcus Aureus Protein A Enhances Osteoclastogenesis via TNFR1 and EGFR Signaling. Biochim Biophys Acta (2016) 1862(10):1975–83. doi: 10.1016/j.bbadis.2016.07.016
41. Jung AL, Hoffmann K, Herkt CE, Schulz C, Bertrams W, Schmeck B. Legionella Pneumophila Outer Membrane Vesicles: Isolation and Analysis of Their Pro-Inflammatory Potential on Macrophages. J Vis Exp (2017) 120):1–6. doi: 10.3791/55146
42. Alaniz RC, Deatherage BL, Lara JC, Cookson BT. Membrane Vesicles Are Immunogenic Facsimiles of Salmonella Typhimurium That Potently Activate Dendritic Cells, Prime B and T Cell Responses, and Stimulate Protective Immunity In Vivo. J Immunol (2007) 179(11):7692–701. doi: 10.4049/jimmunol.179.11.7692
43. Ismail S, Hampton MB, Keenan JI. Helicobacter Pylori Outer Membrane Vesicles Modulate Proliferation and Interleukin-8 Production by Gastric Epithelial Cells. Infect Immun (2003) 71(10):5670–5. doi: 10.1128/iai.71.10.5670-5675.2003
44. Bauman SJ, Kuehn MJ. Purification of Outer Membrane Vesicles From Pseudomonas Aeruginosa and Their Activation of an IL-8 Response. Microbes Infect (2006) 8(9-10):2400–8. doi: 10.1016/j.micinf.2006.05.001
45. Zhang J, Zhao J, Li J, Xia Y, Cao J. Outer Membrane Vesicles Derived From Hypervirulent Klebsiella Pneumoniae Stimulate the Inflammatory Response. Microb Pathog (2021) 154:104841. doi: 10.1016/j.micpath.2021.104841
Keywords: inflammation, osteoclastogenesis and bone loss, Kingella kingae, TNF-α, IL-1β
Citation: Pesce Viglietti AI, Sviercz FA, López CAM, Freiberger RN, Quarleri J and Delpino MV (2021) Proinflammatory Microenvironment During Kingella kingae Infection Modulates Osteoclastogenesis. Front. Immunol. 12:757827. doi: 10.3389/fimmu.2021.757827
Received: 12 August 2021; Accepted: 12 November 2021;
Published: 02 December 2021.
Edited by:
Teun J. De Vries, VU University Amsterdam, NetherlandsReviewed by:
Ineke Jansen, VU University Amsterdam, NetherlandsDeborah Veis, Washington University School of Medicine in St. Louis, United States
Copyright © 2021 Pesce Viglietti, Sviercz, López, Freiberger, Quarleri and Delpino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Victoria Delpino, bWRlbHBpbm9AZmZ5Yi51YmEuYXI=; orcid.org/0000-0003-2077-8509